
Chronic Cardiac-Specific Thyrotoxicosis Increases Myocardial Adrenergic Responsiveness
10
Chronic Cardiac-Specific Thyrotoxicosis Increases Myocardial -Adrenergic Responsiveness SUZY D. CARVALHO-BIANCO, BRIAN W. KIM, JULIE X. ZHANG, JOHN W. HARNEY, ROGE ´ RIO S. RIBEIRO, BALAZS GEREBEN, ANTONIO C. BIANCO, ULRIKE MENDE, AND P. REED LARSEN Thyroid Section, Division of Endocrinology, Diabetes and Hypertension (S.D.C.-B., B.W.K., J.W.H., R.S.R., A.C.B., P.R.L.); Cardiology Division of the Brigham and Women’s Hospital (J.X.Z., U.M.); Harvard Medical School, Boston, Massachusetts 02115; and Department of Endocrine- and Behavioral Neurobiology (B.G.), Institute of Experimental Medicine, Hungarian Academy of Sciences, Budapest H-1083, Hungary Whereas many cardiac symptoms of thyrotoxico- sis resemble those of the hyperadrenergic state, circulating catecholamines are reduced or normal in this condition. To test the hypothesis that the thyrotoxic heart is hypersensitive to catechol- amines, we studied -adrenergic signaling in a transgenic (TG) mouse in which the human type 2 iodothyronine deiodinase (D2) gene is expressed in myocardium. Because D2 converts T 4 to T 3 , the active form of thyroid hormone, the D2 TG mouse exhibits mild, chronic thyrotoxicosis that is limited to the myocardium. In the current study, we deter- mined that cAMP accumulation in response to ei- ther norepinephrine or forskolin treatment was in- creased in isolated ventricular myocardiocytes and membrane-enriched fractions prepared from these D2 TG hearts as compared with wild type. This increase in adenylyl cyclase (AC) V max could not be explained by changes in AC isoform expression or changes in the long or short forms of stimulatory G-protein Gs, which were approximately 10% de- creased in D2 TG membranes. However, Western analysis and ADP-ribosylation studies suggest that the increase in AC V max is mediated by a decrease in the expression of inhibitory G proteins (Gi-3 and/or Go). These data suggest that cardiac thy- rotoxicosis leads to increased -adrenergic re- sponsiveness of cardiomyocytes via alterations in the regulatory G-protein elements of the AC membrane complex. (Molecular Endocrinology 18: 1840–1849, 2004) W HEREAS THE MOST prominent cardiac mani- festations of thyrotoxicosis are also seen in the hyperadrenergic state, circulating catecholamine lev- els in thyrotoxicosis are normal or low. Based on these observations, it has been hypothesized that thyrotox- icosis increases myocardial sensitivity or responsive- ness to -adrenergic stimuli (1). Studies documenting the effectiveness of -adrenergic blockade in amelio- rating the cardiac symptoms of thyrotoxicosis have lent credence to this hypothesis (2, 3). However, con- flicting data exist as to whether patients with thyro- toxicosis have augmented increases in heart rate or systolic blood pressure in response to catecholamine administration, and attempts to prove that adrenergic sensitization occurs in thyrotoxic animals have also met with mixed results (4–6). In spite of numerous studies showing changes in the elements of the ad- enylyl cyclase (AC) complex in response to thyroid hormone, it remains unclear whether cardiac -adren- ergic responsiveness is increased in thyrotoxicosis. Several experimental factors have limited the inter- pretation of previous work in this field. The adminis- tration of pharmacological, rather than physiological, quantities of thyroid hormone has been an experimen- tal paradigm. Such treatments often result in an acutely negative caloric balance, with associated mus- cle wasting, significant weight loss, and/or cardiac hypertrophy (7). These changes are not typical of hu- man thyrotoxicosis, which typically develops over many months. It has also become clear that the choice of endpoint used to assess -adrenergic responsive- ness must be considered, because certain features of the thyrotoxic cardiac phenotype, contractility being the most notable, have been shown to be augmented by thyrotoxicosis through nonadrenergic pathways (8). Finally, and most importantly, few studies have been able to distinguish between the direct effects of thy- roid hormone on myocardial tissue and the indirect cardiovascular effects that result from systemic thyro- toxicosis. These indirect, compensatory effects occur as the heart responds to a decrease in systemic vas- cular resistance and to changes in neurohormonal in- put from the central nervous system (6, 9). Only the Abbreviations: AC, Adenylyl cyclase; -AR, -adrenergic receptor; BDM, butane-dione monoxime; D2, type 2 iodothy- ronine deiodinase; IBMX, 3-isobutyl-1-methylxanthine; MHC, myosin heavy chain; NAD, nicotinamide adenine dinucle- otide; NE, norepinephrine; PKA, protein kinase A; PTX, per- tussis toxin; TG, transgenic. Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving the endocrine community. 0888-8809/04/$15.00/0 Molecular Endocrinology 18(7):1840–1849 Printed in U.S.A. Copyright © 2004 by The Endocrine Society doi: 10.1210/me.2003-0125 1840
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Chronic Cardiac-Specific Thyrotoxicosis Increases Myocardial Adrenergic Responsiveness

Chronic Cardiac-Specific Thyrotoxicosis IncreasesMyocardial �-Adrenergic Responsiveness
SUZY D. CARVALHO-BIANCO, BRIAN W. KIM, JULIE X. ZHANG, JOHN W. HARNEY,ROGERIO S. RIBEIRO, BALAZS GEREBEN, ANTONIO C. BIANCO, ULRIKE MENDE, AND
P. REED LARSEN
Thyroid Section, Division of Endocrinology, Diabetes and Hypertension (S.D.C.-B., B.W.K., J.W.H.,R.S.R., A.C.B., P.R.L.); Cardiology Division of the Brigham and Women’s Hospital (J.X.Z., U.M.);Harvard Medical School, Boston, Massachusetts 02115; and Department of Endocrine- andBehavioral Neurobiology (B.G.), Institute of Experimental Medicine, Hungarian Academy of Sciences,Budapest H-1083, Hungary
Whereas many cardiac symptoms of thyrotoxico-sis resemble those of the hyperadrenergic state,circulating catecholamines are reduced or normalin this condition. To test the hypothesis that thethyrotoxic heart is hypersensitive to catechol-amines, we studied �-adrenergic signaling in atransgenic (TG) mouse in which the human type 2iodothyronine deiodinase (D2) gene is expressed inmyocardium. Because D2 converts T4 to T3, theactive form of thyroid hormone, the D2 TG mouseexhibits mild, chronic thyrotoxicosis that is limitedto the myocardium. In the current study, we deter-mined that cAMP accumulation in response to ei-ther norepinephrine or forskolin treatment was in-creased in isolated ventricular myocardiocytes andmembrane-enriched fractions prepared from these
D2 TG hearts as compared with wild type. Thisincrease in adenylyl cyclase (AC) Vmax could not beexplained by changes in AC isoform expression orchanges in the long or short forms of stimulatoryG-protein Gs�, which were approximately 10% de-creased in D2 TG membranes. However, Westernanalysis and ADP-ribosylation studies suggest thatthe increase in AC Vmax is mediated by a decreasein the expression of inhibitory G proteins (Gi�-3and/or Go�). These data suggest that cardiac thy-rotoxicosis leads to increased �-adrenergic re-sponsiveness of cardiomyocytes via alterationsin the regulatory G-protein elements of the ACmembrane complex. (Molecular Endocrinology 18:1840–1849, 2004)
WHEREAS THE MOST prominent cardiac mani-festations of thyrotoxicosis are also seen in the
hyperadrenergic state, circulating catecholamine lev-els in thyrotoxicosis are normal or low. Based on theseobservations, it has been hypothesized that thyrotox-icosis increases myocardial sensitivity or responsive-ness to �-adrenergic stimuli (1). Studies documentingthe effectiveness of �-adrenergic blockade in amelio-rating the cardiac symptoms of thyrotoxicosis havelent credence to this hypothesis (2, 3). However, con-flicting data exist as to whether patients with thyro-toxicosis have augmented increases in heart rate orsystolic blood pressure in response to catecholamineadministration, and attempts to prove that adrenergicsensitization occurs in thyrotoxic animals have alsomet with mixed results (4–6). In spite of numerousstudies showing changes in the elements of the ad-
enylyl cyclase (AC) complex in response to thyroidhormone, it remains unclear whether cardiac �-adren-ergic responsiveness is increased in thyrotoxicosis.
Several experimental factors have limited the inter-pretation of previous work in this field. The adminis-tration of pharmacological, rather than physiological,quantities of thyroid hormone has been an experimen-tal paradigm. Such treatments often result in anacutely negative caloric balance, with associated mus-cle wasting, significant weight loss, and/or cardiachypertrophy (7). These changes are not typical of hu-man thyrotoxicosis, which typically develops overmany months. It has also become clear that the choiceof endpoint used to assess �-adrenergic responsive-ness must be considered, because certain features ofthe thyrotoxic cardiac phenotype, contractility beingthe most notable, have been shown to be augmentedby thyrotoxicosis through nonadrenergic pathways (8).Finally, and most importantly, few studies have beenable to distinguish between the direct effects of thy-roid hormone on myocardial tissue and the indirectcardiovascular effects that result from systemic thyro-toxicosis. These indirect, compensatory effects occuras the heart responds to a decrease in systemic vas-cular resistance and to changes in neurohormonal in-put from the central nervous system (6, 9). Only the
Abbreviations: AC, Adenylyl cyclase; �-AR, �-adrenergicreceptor; BDM, butane-dione monoxime; D2, type 2 iodothy-ronine deiodinase; IBMX, 3-isobutyl-1-methylxanthine; MHC,myosin heavy chain; NAD, nicotinamide adenine dinucle-otide; NE, norepinephrine; PKA, protein kinase A; PTX, per-tussis toxin; TG, transgenic.
Molecular Endocrinology is published monthly by TheEndocrine Society (http://www.endo-society.org), theforemost professional society serving the endocrinecommunity.
0888-8809/04/$15.00/0 Molecular Endocrinology 18(7):1840–1849Printed in U.S.A. Copyright © 2004 by The Endocrine Society
doi: 10.1210/me.2003-0125
1840
heterotopically transplanted rat heart model (the un-loaded heart model) has directly addressed this ob-stacle (10). However, this method required surgicalintervention, and the transplanted unloaded hearts ex-hibited a reduction in cell protein compared with thenative euthyroid hearts (11). Considering that most ofthe published data are limited in regard to one or moreof the above considerations, it is perhaps not surpris-ing that the fundamental question of the nature of thechanges in myocardial �-adrenergic responsivenessin thyrotoxicosis remains controversial.
The murine model of cardiac thyrotoxicosis used forthe current study is one model that overcomes all ofthese limitations. Unlike humans, rodents do not ex-press the type 2 iodothyronine deiodinase (D2) in theirmyocardium (12, 13). D2 converts T4 to T3, and tissuesthat express D2 may therefore be more sensitive tochanges in circulating T4. We took advantage of thisinterspecies difference to create a transgenic (TG)mouse model of cardiac-specific thyrotoxicosis, i.e. amouse with cardiac thyrotoxicosis mediated by TGexpression of D2, but with normal serum thyroid hor-mones and thus systemic euthyroidism (14). Briefly,D2 is overexpressed in the myocardium under thecontrol of the �-myosin heavy chain (MHC) promoter,leading to a modest, but chronic, increase in myocar-dial thyroid status reflected by an increase in thyroidhormone-regulated genes such as HCN2 (hyperpolar-ization-activated cyclic nucleotide-gated channel) (14,15). Whereas the cardiac phenotype of the D2 TGmouse is not apparent in vivo, examination of theisolated perfused D2 TG hearts reveals findings typi-cally seen in thyrotoxicosis, such as tachycardia and adecrease in both creatine and phosphocreatine (16,17). This suggests that the cardiac-specific thyrotox-
icosis seen in this model can be compensated for byautonomic regulation and/or other factors.
In the current study, we used the D2 TG murinemodel of cardiac-specific thyrotoxicosis to investigatethe direct effects of thyrotoxicosis on cardiac �-adrenergic signaling. We compared the function of theAC complex in D2 TG and wild-type mice, studyingcAMP accumulation in isolated cardiomyocytes andventricular membranes. In addition to AC function, weexamined the expression of the critical postreceptorsignaling elements including AC isoforms and both thestimulatory (Gs�) and inhibitory (Gi� and Go�) hetero-trimeric G proteins.
RESULTS
cAMP Accumulation in Fresh Cardiomyocytesfrom D2 TG vs. Wild-Type Mice
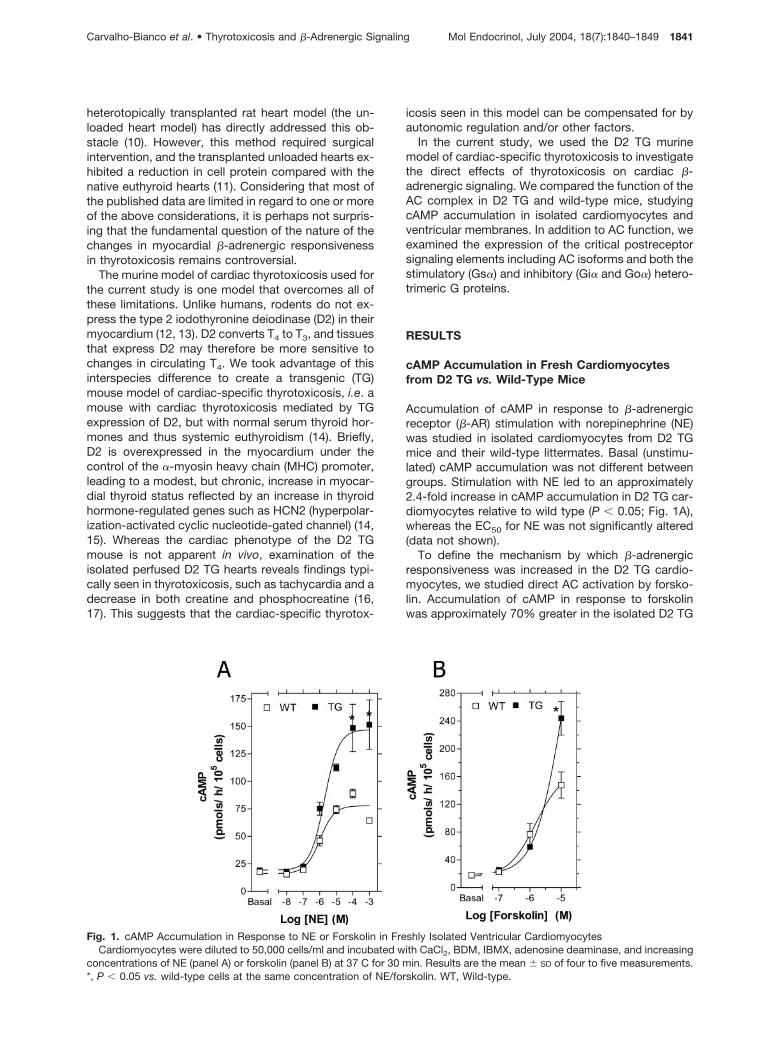
Accumulation of cAMP in response to �-adrenergicreceptor (�-AR) stimulation with norepinephrine (NE)was studied in isolated cardiomyocytes from D2 TGmice and their wild-type littermates. Basal (unstimu-lated) cAMP accumulation was not different betweengroups. Stimulation with NE led to an approximately2.4-fold increase in cAMP accumulation in D2 TG car-diomyocytes relative to wild type (P � 0.05; Fig. 1A),whereas the EC50 for NE was not significantly altered(data not shown).
To define the mechanism by which �-adrenergicresponsiveness was increased in the D2 TG cardio-myocytes, we studied direct AC activation by forsko-lin. Accumulation of cAMP in response to forskolinwas approximately 70% greater in the isolated D2 TG
Fig. 1. cAMP Accumulation in Response to NE or Forskolin in Freshly Isolated Ventricular CardiomyocytesCardiomyocytes were diluted to 50,000 cells/ml and incubated with CaCl2, BDM, IBMX, adenosine deaminase, and increasing
concentrations of NE (panel A) or forskolin (panel B) at 37 C for 30 min. Results are the mean � SD of four to five measurements.*, P � 0.05 vs. wild-type cells at the same concentration of NE/forskolin. WT, Wild-type.
Carvalho-Bianco et al. • Thyrotoxicosis and �-Adrenergic Signaling Mol Endocrinol, July 2004, 18(7):1840–1849 1841

cardiomyocytes at 10�5 M forskolin (P � 0.05; Fig. 1B),suggesting that a postreceptor change might beresponsible for the enhanced responsiveness tocatecholamines.
Characterization of AC Kinetics in VentricularMembranes of D2 TG vs. Wild-Type Mice
To further characterize possible differences in the�-adrenergic signaling elements in D2 TG cardiomyo-cytes, forskolin-stimulated cAMP accumulation wasstudied in membrane-enriched fractions preparedfrom ventricular tissue. Such preparations allow ma-nipulation of the cellular components that might alterAC activity such as ATP, GTP, Mg2�, and Ca2�, whilealso virtually eliminating effects on cAMP accumula-tion mediated via cross-talk by cytosolic proteins (18).This system also allows for the determination of ap-parent Michaelis-Menten constant (Km) and maximumvelocity (Vmax) of the AC complex in response to ATP.
Under the assay conditions, cAMP production waslinear with the amount of membrane protein used andtime of incubation (Fig. 2, A and B). Accumulation ofcAMP in response to increasing concentrations of for-skolin was enhanced in the D2 TG membranes (Fig.2C), consistent with the results from intact cardiomy-ocytes. At every forskolin concentration there was asignificantly higher cAMP accumulation in the D2 TGmembranes, and the maximal response (10�3 M fors-kolin) was approximately 75% higher in the D2 TGmembranes. This difference was similar in two differ-ent lines of D2 TG mice and was observed in bothmales and females (data not shown).
The greater cAMP accumulation in membranes fromD2 TG ventricles was also present when the mem-branes were stimulated with 0.1 mM forskolin � in-creasing concentrations of ATP (Fig. 3A). cAMP accu-mulation was approximately 1.8-fold higher in the D2TG membranes (P � 0.05) at 1 mM ATP, the con-centration resulting in a maximal response. TheLineweaver-Burk replot of the data resulted in a Vmax
that was approximately 70% greater in the D2 TGmembranes (P � 0.05; Fig. 3B), whereas the Km valueswere not significantly different in the two groups.Again, the effect was not gender specific (data notshown).
Characterization of Ca2� Sensitivity of ACActivity in Ventricular Membranes of D2 TG vs.Wild-Type Mice
The existence of nine AC isoforms sharing similar to-pology but differing in regulatory properties, tissuedistribution, and even in relative abundance indicatesthat this system has the potential for extreme plasticity(19, 20). These isoforms are differentially regulated byGs, Gi, Ca2�, protein kinase A (PKA), protein kinase C,and other cellular components. Five isoforms of AC(IV–VII and IX) have been found in mouse heart (21),and isoforms V and VI have been reported to be pre-dominant (22).
AC isoforms V and VI are directly inhibited by mi-cromolar concentrations of Ca2�, whereas other iso-forms are significantly less sensitive (or not sensitive)to Ca2� (23). We took advantage of this fact to try tofurther discriminate as to which AC isoforms were
Fig. 2. cAMP Accumulation in Response to Forskolin in Ventricular MembranesA, Aliquots of pooled ventricular membranes from wild-type (WT) and D2 TG mice were incubated at 30 C for 30 min. cAMP
production was stimulated by 10�5 M forskolin, in a 10 mM Tris-HCl buffer (pH 8.0), containing 10 mM propranolol, 1 mM IBMX,0.4 U/ml adenosine deaminase, 1 mM GTP, 5 mM MgCl2, and 0.5 mM ATP. B, Same assay conditions as in panel A except that5 �g ventricular membrane protein were employed and the incubation time varied up to 90 min. C, Assays were performed with5 �g of the membrane-enriched fractions incubated in Tris buffer, pH 8.0, at 30 C for 30 min with IBMX, adenosine deaminase,MgCl2, GTP, ATP, propranolol, BSA, protease inhibitors � increasing concentrations of forskolin. For panels A–C, results are themean � SD of four to five measurements. *, P � 0.05 vs. wild-type membranes at the same concentration of forskolin.
1842 Mol Endocrinol, July 2004, 18(7):1840–1849 Carvalho-Bianco et al. • Thyrotoxicosis and �-Adrenergic Signaling

affected by D2 transgene expression. If the increasedAC Vmax in D2 TG ventricular membranes involved ACV and/or VI, these membranes should exhibit height-ened sensitivity to Ca2� inhibition. To test this hy-pothesis, we compared the Ca2� sensitivity of forsko-lin-stimulated cAMP accumulation in D2 TG andwild-type membranes. For the 0 free Ca2� (baseline)samples, EGTA was added to the forskolin-stimulatedcAMP accumulation assay mixture to chelate any re-sidual Ca2� in the membrane preparation. Addition ofEGTA led to an increase in the absolute value of cAMPaccumulation in response to 10�5 M forskolin in bothD2 TG and wild-type membranes (compare 0 freeCa2� samples in Fig. 4 vs. 10�5 M forskolin samples inFig. 2). Whereas cAMP accumulation in the wild-typemembranes was increased approximately 3.5-fold bythe addition of EGTA, the increase was about 5.9-foldin the D2 TG membranes, suggesting increased cal-cium sensitivity in the D2 TG membranes. As ex-pected, addition of defined free Ca2� concentrationsto the EGTA-containing buffer decreased forskolin-stimulated cAMP accumulation in both D2 TG andwild-type membranes (Fig. 4). At 200 �M free Ca2�,cAMP accumulation fell to approximately 40% in thewild-type group, whereas it fell to about 25% in the D2TG group (P � 0.001). These data indicate thatforskolin-stimulated cAMP accumulation in D2 TGmembranes was significantly more sensitive to cal-cium inhibition than that in wild-type membranes. Thisis consistent with the hypothesis that the contributionof Ca2�-sensitive AC isoforms (V and VI) to cAMPgeneration is increased in the D2 TG membranes.
Expression of AC Isoforms and G Proteins inPurified Membranes
The simplest explanation for the increased AC Vmax
and increased Ca2� sensitivity would be an increase inAC V/VI expression in the D2 TG ventricular mem-branes. This was tested by real-time PCR, which wasused to quantitate AC IV, V, VI, and VII isoform mRNAsin the D2 TG and wild-type myocardial tissue. �-Actinand cyclophilin A were used as housekeeping genes.
Fig. 3. cAMP Accumulation in Response to ATP in Ventricular MembranesA, Same as in Fig. 2 except that incubations were performed in the presence of 10�4 M forskolin � increasing concentrations
of ATP. B, Lineweaver-Burk replot of the data in panel A. Results are the mean � SD of four to five measurements. *, P � 0.05vs. wild-type samples at the same ATP concentrations.
Fig. 4. Forskolin-Stimulated cAMP Accumulation in Re-sponse to Ca2� in Ventricular Membranes
Same as in Fig. 3 except that incubations were performedwith 1 mM EGTA and in the presence of 10�5 M forskolin �increasing concentrations of CaCl2. The total amount ofCaCl2 to yield the exact desired free Ca2� was calculated bythe Chelator software developed by Theo J. M. Schoenmak-ers (University of Nijmegen, The Netherlands) and available atBiotechniques Software Library (http://www.biotechniques.com/). Results are the mean � SD of four to five measure-ments. *, P � 0.05 vs. 0 free calcium. WT, Wild-type.
Carvalho-Bianco et al. • Thyrotoxicosis and �-Adrenergic Signaling Mol Endocrinol, July 2004, 18(7):1840–1849 1843

Regardless of which housekeeping gene was used,there was no difference in mRNA for AC isoforms(Table 1). Although changes less than 0.5-fold in mag-nitude may be difficult to demonstrate using this tech-nique, the data suggest that the increase in AC activityis mediated via posttranscriptional changes or throughmodulation of proteins that affect AC activity. To in-vestigate these possibilities, Western analysis wasperformed using specific antiserum against AC V/VIisoforms, the two most abundant AC isoforms ex-pressed in the myocardium. There was no statisticallysignificant difference between the two groups (Fig.5A).
Next we sought to investigate whether the amountsof the AC-modulating G proteins Gs� and Gi�/Go�were altered in the D2 TG hearts. Surprisingly, Westernanalysis revealed that there were small but significantdecreases in both the long (�10%; P � 0.05) and short(�10%; P � 0.05) forms of Gs� protein in D2 TG
membranes (Fig. 5B). The Gi subfamily has at leastthree distinct gene products, Gi�-1, Gi�-2, and Gi�-3(24), which were assessed by Western analysis. Therewas a decrease of approximately 30% in Gi�-3/Go� inthe D2 TG membranes (P � 0.05; Fig. 6A), whereasGi�-1 and Gi�-2 isoforms were not altered (Fig. 6).Further evidence for reduced expression of the Gi�/Go� protein in the D2 TG membranes was obtained byevaluating the levels of heterotrimeric Gi�/Go� in themyocardium by pertussis toxin (PTX)-catalyzed ADPribosylation in purified membranes (Fig. 6) (25, 26).The 40-kDa [32P]nicotinamide adenine dinucleotide(NAD)� PTX-ribosylated substrate was decreased byapproximately 50% in myocardial membranes from D2TG mice, suggesting a reduced availability of Gi�/Go�
(Fig. 6). Note that G� levels are not changed in these
Fig. 5. Quantification of Ventricular AC-V/VI and Gs�Purified ventricular membranes were processed for West-
ern analysis with antibodies against ACV/VI (panel A) or Gs�(panel B). The signal intensity of the indicated bands (left grayarrow) was quantified by direct chemiluminescent imaging.The results are shown below each gel and the values are themean � SD. Both experiments were repeated three times withsimilar results. *, P � 0.05 vs. wild type.
Fig. 6. Quantification of Ventricular Gi�-1,2 and Gi�-3/Go�Purified ventricular membranes were processed for West-
ern analysis with antibodies against Gi�-1,2, Gi�-3/Go�, orG� proteins (panel A). The signal intensity of the indicatedbands (left gray arrow) was quantified by direct chemilumi-nescent imaging. The results are shown below each gel, andthe values are the mean � SD. The experiment was repeatedthree times with similar results. *, P � 0.02 vs. wild-typesamples. B, SDS-PAGE of purified D2 TG or wild-type ven-tricular membrane proteins after ADP ribosylation using PTXand [32P]NAD�. As indicated, two different amounts of ven-tricular membrane protein were used. The Gi�/Go� band isindicated by a left gray arrow. After exposure to the film, thegel region corresponding to the band of interest was excisedand counted directly in a �-counter. The results are shownbelow the gel, and the values are the mean � SD. The exper-iment was repeated four times with similar results. *, P � 0.05vs. wild-type membranes.
Table 1. Myocardial Levels of AC mRNA
AC Isoform D2-TG/Wild Type n
AC IV 1.2 � 0.4 3AC V 1.1 � 0.4 6AC VI 0.8 � 0.2 4AC VII 1.3 � 0.2 4
Values are the mean � SD of the specific AC subtype mRNA/�-actin mRNA ratio observed in the indicated number of D2TG/wild-type pairs. Similar results were obtained when cy-clophilin A was used as the housekeeping gene.
1844 Mol Endocrinol, July 2004, 18(7):1840–1849 Carvalho-Bianco et al. • Thyrotoxicosis and �-Adrenergic Signaling

membranes, excluding the possibility that the reducedlabeling represents a decrease in heterotrimer.
To discriminate between the Gi� and Go� compo-nents of the adrenergic signaling pathway, real-timePCR was used to evaluate Go� mRNA levels, with thefinding that there was no significant difference be-tween the wild-type and D2 TG groups (ratio of D2TG/wild type, 0.96 � 0.12; similar results using cyclo-philin A and �-actin housekeeping genes). Specificprimers for Gi�-3 could not be designed based onexisting sequence information using Beacon Designersoftware.
DISCUSSION
Our results indicate that D2 TG ventricular cardiomy-ocytes exhibit an increase in �-adrenergic responsive-ness, defined as an increased AC Vmax, which is likelymediated by changes in the expression of regulatory Gproteins. The novelty of these results is heightened bythe fact that the chronically thyrotoxic D2 TG cardio-myocytes were harvested from systemically euthyroidmice, such that the increase in AC Vmax must stemdirectly from the effects of intracellularly generated T3.The current study therefore represents the first as-sessment of the direct effects of chronic, tissue-specific thyrotoxicosis on the �-adrenergic signalingpathway in the heart.
The increase in forskolin-stimulated cAMP accumu-lation was similar in magnitude to that observed withNE, suggesting that the major changes in D2 TG �-ad-renergic signaling occur at a postreceptor level (Fig. 1).This is not surprising considering that increases in�-AR number have not consistently been found inexperimental thyrotoxicosis (27–29), and in one studythe acute increase in �-AR number did not persist inlonger-term thyrotoxicosis (30). Furthermore, changesin �-AR number do not necessarily correlate with cat-echolamine-stimulated cAMP accumulation (31–33). Apostreceptor mechanism is also indirectly supportedby the observations that mice overexpressing AC iso-forms V (34) or VI (35) in their hearts have a propor-tional increase in catecholamine-stimulated myocar-dial cAMP accumulation in an otherwise normalmyocardium. On the other hand, cardiac hypertrophyor heart failure has been observed with overexpressionof �-AR or Gs� isoforms (36–39). These deleteriouschanges, which do not occur in the D2 TG hearts (14),may result because increases in �-AR cause consti-tutive activation of the signaling pathway, whereaspostreceptor changes may not abrogate the liganddependence of the system.
An increase in the amount of AC would have been aplausible mechanism for increased cardiac �-adren-ergic responsiveness. Such a mechanism has beensuggested to occur in brain (40) and brown fat (41, 42).Whereas it is possible that thyrotoxicosis might alterthe expression of various AC isoforms selectively,
there were no significant differences in AC IV-VIImRNA (Table 1) or in AC V and VI protein levels (Fig.5A) in D2 TG vs. wild-type animals. It is notable, how-ever, that forskolin-stimulated cAMP accumulation inthe D2 TG ventricular membranes was significantlymore sensitive to inhibition by Ca2� (Fig. 4), indicatingan even more pronounced functional predominance ofisoforms AC V and VI in the D2 TG group. These dataindicate that regulation of AC, rather than AC itself,is the critical target in cardiac-specific chronicthyrotoxicosis.
Gi is the major inhibitory pathway with respect to ACfunction (43, 44), and in cardiomyocytes, Gi�-2 is be-lieved to be the isoform primarily linked to the musca-rinic receptors (45, 46). Gi�-2 protein is not increasedin the D2 TG membranes (Fig. 6A), suggesting that thecardiac muscarinic pathway is not a major target ofthyroid hormone.
The finding of a decrease in the amount of theinhibitory Gi�-3/Go� proteins in the D2 TG ventricularmembranes, together with the finding of no change inGo� mRNA, suggests that a decrease in Gi�-3 is themechanism by which adrenergic responsiveness isincreased in the D2 TG hearts. Such a decrease inGi�-3 could explain the increased sensitivity of the ACcomplex to both NE and forskolin. The fact that thepredominant isoforms of AC in the heart, AC V and VI,are also the most sensitive to inhibition by the Gi�pathway (23) lends further support to this hypothesis.
The proposed effect of thyroid hormone (D2 expres-sion) on the Gi� pathway can explain why differencesin cAMP generation between D2 TG and wild-typecardiomyocytes are only seen with higher amounts ofadrenergic stimulators (NE or forskolin) (Fig. 1).Whereas cardiac �-1 adrenergic receptors are pre-dominantly coupled to G�s and AC VI (47), �-2 adren-ergic receptors can be coupled to either G�s or to Gi�,depending on the degree of receptor phosphorylationby PKA (48). Under basal conditions, �-2 adrenergicreceptors are predominantly coupled to G�s. How-ever, as cAMP-stimulated PKA activity increases,phosphorylation of the �-2 adrenergic receptors shiftstheir coupling predominance to the inhibitory Gi�pathways, thus providing feedback inhibition. Accord-ing to our model, the major effect of D2-mediatedcardiac thyrotoxicosis is to decrease the efficacy ofthis feedback mechanism by decreasing the amountof Gi�. However, the data do not allow us to evaluatethe possibility that the effects of thyroid hormone mayalso affect feedback inhibition via Gi� at the level ofconstitutively active Gi�-coupled receptors. In eithercase, the difference between D2 TG and wild-typecardiomyocytes is unmasked only when the cAMPgeneration system is maximally activated.
Several previous studies using pharmacologicaldoses of thyroid hormones and acute treatment pro-tocols have examined the relationship between regu-latory G protein levels in the heart and thyroid status.Rats treated with L-T4 in their drinking water for 18 dhad no difference in Gs�, Gi�-2 and Gi�-3 protein, or
Carvalho-Bianco et al. • Thyrotoxicosis and �-Adrenergic Signaling Mol Endocrinol, July 2004, 18(7):1840–1849 1845

mRNA compared with untreated rats, as well as noincrease in forskolin-stimulated AC activity (49). In aseparate study of rats made thyrotoxic with injectionof T3 for 5 d, there was no correlation between thyroidstatus and Gs� (the major target of cholera toxin-catalyzed [32P]ADP ribosylation) (50). The only previ-ous study in which Gi� was found to be reduced bythyrotoxicosis involved immature rats treated with T3
during 2–21 d postpartum. However, in these animalsthere was a decrease in the amounts of Gi�-2 andGi�-3 and an increase in the long form of Gs� (29).
Given that none of these studies showed a decreasein Gs�, it is intriguing that the D2 TG mice had anapproximately 10% decrease in the long and shortforms of Gs� (Fig. 5B). One could speculate that thisdecrease represents a compensatory mechanism. It isalso possible that the intensity or duration of thyrotox-icosis, or the maturity of the animal at the time of onsetof thyrotoxicosis, is critical with regard to G proteinexpression. In any case, the decrease in Gs� clearlydoes not prevent the thyroid hormone-induced in-crease in AC Vmax.
Because D2 is normally expressed in the humanheart, the current data have implications for both nor-mal human cardiac physiology and the cardiac mani-festations of systemic thyrotoxicosis. It is conceivablethat when adrenergic input to the heart increases,such as during stress, the expression of the cAMP-responsive D2 gene increases, therefore increasingthe local conversion of T4 to T3. This increase in car-diac thyroid status would, in turn, amplify the �-adrenergic responsiveness of the heart, operatingin a positive feed back loop such as the one recentlydescribed in brown adipocytes (51).
In conclusion, the current data indicate that chronicthyrotoxicosis directly increases the �-adrenergic re-sponsiveness of cardiomyocytes via alterations in theregulatory elements of the AC complex, specifically bydecreasing the Gi� pathway-mediated feedback inhi-bition of the AC complex.
MATERIALS AND METHODS
Animals
All aspects of animal care and experimentation were ap-proved by the Institutional Animal Care and Use Committeeof the Beth Israel Deaconess Medical Center and the Brighamand Women’s Hospital. Animals were maintained on a 12-hlight/12-h dark schedule (light on at 0600 h) and fed labora-tory chow and water ad libitum if not otherwise indicated. Theexpression of the human D2 gene transgene was directed tothe heart by placing it under the control of the �-MHC pro-moter (14). The genotyping was determined by PCR using a21-bp sequence complementary to a region of the �-MHCpromoter next to the insertion point as the upstream primerand a 19-bp fragment complementary to a carboxyl terminussequence of the D2 cDNA as the downstream primer. Tailingfor genotyping is performed before the animals are 21 d old.Genotyping was confirmed by measuring D2 activity in theventricles used in the experiments as has been previouslydescribed (52). Cardiomyocytes were isolated from both
2-month-old and 1-yr-old animals. Other experiments wereperformed with 2- to 3-month-old animals.
Isolation of Ventricular Cardiomyocytes and Stimulationof cAMP Accumulation (See Below)
Male or female mice were anesthetized by ethrane inhalationand heparinized (�5000 U ip). The chests were opened,hearts were quickly removed, and the aorta was cannulatedfor retrograde (Langendorff) perfusion of the coronary arterieswith Tyrode’s buffer containing 126 mM NaCl, 4.4 mM KCl, 1mM MgCl2, 4 mM NaHCO3, 30 mM 2,3-butane-dione mon-oxime (BDM), 10 mM HEPES, and 11 mM Glucose (pH 7.3).After the blood had been washed out, the hearts were di-gested with Tyrode’s buffer containing 0.4 mg/ml collage-nase II (Worthington Biochemical Corp., Freehold, NJ; 266U/mg) and 0.3 mg/ml hyaluronidase II (Sigma Chemical Co.,St. Louis, MO) for 15 min. The atria were trimmed away, andthe ventricles were minced and transferred to 10 ml Tyrode’sbuffer containing 0.02 mg/ml trypsin IX, 0.02 mg/ml deoxyri-bonuclease I, and 0.3 mM CaCl2. After 10 min incubation in a37 C shaking water bath, myocytes were released by gentlecentrifugation. The resulting supernatant containing the cellsuspension was transferred to another tube containing 10 mlof a 1:1 mixture of Tyrode’s solution and DMEM plus 0.9 mM
CaCl2 and 2% FBS. If needed, the trypsin digestion wasrepeated until the majority of the ventricular myocytes werereleased. The cell suspensions were pooled and centrifugedat 50 � g for 3 min. The pellet was then carefully resuspendedwith 10 ml of Tyrode’s solution/ DMEM and cells were al-lowed to settle by gravity for 10–15 min. The final pellet offreshly isolated cardiomyocytes was then resuspended inTyrode’s buffer (0.45 mM CaCl2 and 15 mM BDM), counted,adjusted to a final density of 40,000–50,000 cells/ml, andincubated at 37 C for 30 min in a 5% CO2 controlled envi-ronment in the presence of the phosphodiesterase inhibitorIBMX (1 mM), adenosine deaminase (0.5 U/ml), and increas-ing concentrations of NE or forskolin. cAMP accumulationwas stopped by the addition of 1.5% perchloric acid.
Isolation of Membrane-Enriched Fractions andStimulation of cAMP Accumulation
Membrane-enriched fractions from WT and TG mice ventri-cles were isolated as described previously (31, 42). Micehearts were excised, and the ventricle was separated fromthe atria and immediately frozen in liquid nitrogen. Four to fivefrozen heart ventricles were pooled in a cold pestle andhomogenized in 5 mM Tris-HCl (pH 8.0) buffer containing 0.25M sucrose, 1 mM EDTA, and protease inhibitors (10 �g/mlaprotinin, 10 �g/ml leupeptin, and 50 �g/ml PMSF). Homog-enates were centrifuged at 15,000 � g for 15 min. The pelletswere washed in the same buffer, and final pellets were re-suspended in assay buffer (10 mM Tris-HCl, pH 8.0, contain-ing 0.25 M sucrose, 1 mM EGTA, and protease inhibitors asabove). Membrane fractions were frozen at �70 C in smallaliquots. For each experiment, an aliquot was defrosted anddiluted to the appropriate protein concentration in assaybuffer. One milliliter of this membrane solution was incubatedat 30 C for 30–60 min with 1 mM IBMX, 0.4 U/ml adenosinedeaminase, 5 mM MgCl2, 0.1% BSA, 0.1 �M GTP, and 10 �M
propranolol. cAMP accumulation was stimulated by increas-ing concentrations of forskolin or ATP. When studying thedose response to forskolin, 1 mM ATP was also included inthe incubation solution. When studying AC kinetic properties,concentrations of ATP varied from 0–2 mM, and 0.1 mM
forskolin was present in the incubation solution. In the exper-iments with Ca2�, membranes were diluted in assay bufferwithout EGTA, and an exact final concentration of 1 mM EGTAwas added to the assay tube. Forskolin (10�5 M) was presentin the incubation solution. The total CaCl2 added to obtaineach exact desired concentration of free Ca2� was calcu-
1846 Mol Endocrinol, July 2004, 18(7):1840–1849 Carvalho-Bianco et al. • Thyrotoxicosis and �-Adrenergic Signaling

lated by a computer program (The Chelator). cAMP accumu-lation was stopped by the addition of 3% perchloric acid.
Quantification of cAMP Content in Cells andMembranes
cAMP in all samples was quantified by a solid-phase RIA Kit(NEK-033) from New England Nuclear (Boston, MA). De-frosted samples were briefly centrifuged, pellets were dis-carded, and the pH in supernatants was adjusted to 5–6 with15% KHCO3. Solutions (10–50 �l) were incubated overnightat 4 C with [125I]cAMP and anti-cAMP antibody. The cAMP-antibody complexes were precipitated and counted. cAMPconcentration was estimated from the standard curves. Re-sults are expressed as cAMP content/h�105 cells or cAMPcontent/ h�mg of membrane protein.
Real-Time PCR of AC Isoform mRNA
For total RNA extraction, animals were euthanized via CO2,and ventricular tissue was immediately harvested and frozenin liquid nitrogen. Frozen hearts were pulverized in liquidnitrogen, and the resulting frozen fragments were homoge-nized in Trizol Reagent (Invitrogen, Carlsbad CA) using aBrinkmann homogenizer (Brinkmann Instruments, Inc., West-bury, NY). RNA was extracted from homogenized tissue viathe Trizol protocol supplied by the manufacturer. RNA pelletswere treated with deoxyribonuclease I (Invitrogen) accordingto the instructions of the manufacturer, and the samples wererepurified using Trizol. RNA concentration was estimated bymeasuring the absorption at 260 nm (A260), with purity beingestimated via A260/A280 ratio between 1.8–2.1 and degrada-tion being assessed via Tris-acetate-EDTA agarose electro-phoresis. cDNA synthesis was performed using the Super-Script First-Strand Synthesis System for RT-PCR (Invitrogen)according to the instructions of the manufacturer. RNA (5 �g)was reverse transcribed using oligo dT primers to generatereal-time PCR template cDNA. cDNA content was quantifiedand checked for purity and condition via spectrophotometryand gel electrophoresis. Real-time PCR was performed usingthe iCycler iQ real-time PCR detection system (Bio-Rad Lab-oratories, Inc., Hercules CA) and iQ SYBR green supermix(Bio-Rad) according to the instructions of the manufacturer atthe 30 � scale. Data analysis was performed using the iCyclersystem software, with fold change of a given target gene fromTG samples to wild-type samples being expressed relative to ahousekeeping gene (�-actin and cyclophilin A were used in thisstudy). Melt curve analysis was performed for each primer set.Specific primer sets for murine AC IV (sense, 5�-CTGGACA-CTGGTGATGCTAAG; antisense, 5�-AGGCTGCGTAGTATTT-GAAGG), V (sense, 5�-GCCTGCTCCGTGTTCCTG; antisense,5�-CCGTTGTTGCTGAAGTCTATGG), VI (sense, 5�-CCTCCTG-GTTCCCAAAGTG; antisense, 5�-GGTGGCTCCGCATTCTTG),VII (sense, 5�-TGCTGCTCCAAGTCTGATG; antisense, 5�-CACAGGCGAAGTCGTAGC), G-protein �-subunit o (sense,5�-GGACAGACTGACCACCATC; antisense, 5�-AGAGGAAG-GATTGCCAACC0). �-actin (sense, 5�-TTTGTTTTGGCGCTTT-TGACTC; antisense, 5�-TGGGAGGGTGAGGGACTTC), and cy-clophilin A (sense, 5�-CGGCAGGTCCATCTACGG; antisense,5�-CCATCCAGCCATTCAGTCTTG) were designed usingpublished nucleotide sequences and Beacon Designer ver-sion 2.0 software (Premier Biosoft, Palo Alto, CA). To gener-ate standard curves, serial 5-fold dilutions of a cDNA stock(cDNA was prepared from TG ventricular tissue as described)were prepared for use as templates. Amplification was per-formed with housekeeping or target gene-specific primers.Standard curves were determined for housekeeping and tar-get genes on every experimental plate, with each samplebeing run in triplicate (standard curve points) or two tripli-cates (unknown quantity points). Results were expressed asmean of fold change � SD.
Quantitation of AC, Gs�, Gi�, and G� Proteins byWestern Blotting
Western blot analysis was used to investigate changes in thetotal amount of isoforms V/VI of AC, the long and shortisoforms of the Gs� protein, and isoforms 2 and 3 of the Gi�proteins. This was done using ventricular membranes iso-lated from TG and WT mouse ventricles that were furtherpurified through a sucrose cushion, as described for theADP-ribosylation assay. Equal amounts of protein (10–30�g/lane) were size fractionated by SDS-PAGE, followed bytransfer to polyvinylidene fluoride membranes (PVDF, Immo-bilon-P by Millipore Corp., Bedford, MA). Immunoblots wereperformed using rabbit antibodies as described below.AC V/VI: a 1:200 Dilution of C-17 from Santa CruzBiotechnology, Inc. (Santa Cruz, CA). This is an affinitypurified polyclonal antibody raised against a peptide map-ping at the C terminus of mouse AC V/VI that partially crossreacts with AC I (not present in the myocardium) but not otherAC isoforms).Gi�-1 and Gi�-2: a 1:1000 Dilution of 371723 from Calbio-chem (La Jolla, CA). This is a Protein A affinity-purifiedpolyclonal antibody raised against a synthetic peptide map-ping at the C terminus of Gi�-1 and Gi�-2, the specificity ofwhich was confirmed with lysates from bacterially producedrecombinant G proteins.Gi�-3 and Go�: a 1:1000 Dilution of Calbiochem 371726,an Antiserum that Was Raised against a Synthetic Pep-tide Mapping at the C Terminus of Gi�-3, but Which AlsoRecognizes Go� because Both Share the Terminal FourResidues. The specificity of this antiserum was confirmedusing bacterial lysates containing recombinant G proteins.G�s: a 1:500 Dilution of C-18 from Santa Cruz. This is anaffinity purified polyclonal antibody raised against a peptidemapping at the C terminus of rat G�s. It also reacts withmouse G�s, but not other G� subunits.G�: a 1:500 Dilution of SC25413 from Santa Cruz. Thisis a rabbit polyclonal antibody raised against a recombinantprotein corresponding to amino acids 1–300 mapping at theamino terminus of G�. Membranes were incubated for 1 h atroom temperature, washed, and incubated for 1 h with horse-radish peroxidase-labeled antirabbit IgG (Roche MolecularBiochemicals, Mannheim, Germany). The antibodies werethen detected by chemiluminescence (ECL�Plus, AmershamBiosciences, Piscataway NJ) and visualized by exposure tophotographic film. The membranes were also scanned bydirect chemiluminescent imaging using the high-perfor-mance gel and blot imager Typhoon 9410 (Amersham Bio-sciences), and the intensity of the bands of interest wasquantified using ImageQuant 5.2 software (Molecular Dynam-ics, Inc., Sunnyvale, CA/Amersham). In preliminary ex-periments, we ascertained that the signal obtained was inthe linear range. Blots were stripped and reprobed forconsistency.
Functional State of Gi Proteins by ADP Ribosylation
The functional state of Gi/Go protein was assayed based onthe susceptibility of membrane proteins to ADP ribosylationby PTX with the presence of [32P]NAD�. The ribosylatedGi/Go was quantified through SDS-PAGE and reflects thetotal amount of the Gi/Go capable of activation. Ventricularmembranes used for this assay were obtained as describedabove, and then purified through a sucrose cushion as fol-lows: 500 �l of the membrane-enriched fraction were loadedover a 2-ml layer of 0.67 M sucrose buffer (10 mM Tris-HCl, pH7.4; 0.67 M sucrose; and 1 mM EDTA) and then centrifuged at20,000 � g for 20 min at 4 C. The membrane interface wascollected and centrifuged again at 100,000 � g for 1 h at 4 C.The pellets were resuspended in 10 mM Tris-HCl, pH 8.0,containing 0.25 M sucrose, 1 mM EGTA, and protease inhib-itors. Before the assay, the membranes were incubated with
Carvalho-Bianco et al. • Thyrotoxicosis and �-Adrenergic Signaling Mol Endocrinol, July 2004, 18(7):1840–1849 1847

0.6% lubrol for 30 min at 4 C to solubilize the G proteins.Immediately before ribosylation, the ADP-ribosyltransferaseactivity in the S1 subunit of the PTX was activated by incu-bation in 25 mM Tris-HCl buffer, pH 8.0, containing 20 mM
dithiothreitol, 0.12% sodium dodecyl sulfate, and 15 �g BSAfor 20 min at 30 C. Then, the activated toxin was transferredto a 25 mM Tris, pH 8.0, buffer containing 15–25 �g mem-brane protein, 1 mM EDTA, 10 mM thymidine, 1 mM GTP, 0.5mM ATP, and 5–10 �M [32P]NAD� (�4–5 � 106 cpm/tube).After 30 min at 30 C, the reaction was stopped by the additionof 150 mM NaCl, followed by 1 ml of ice-cold acetone. After10 min on ice, samples were centrifuged at 2,500 rpm for 30min at 4 C and supernatants were discarded. Pellets werewashed with 1 ml of ice-cold 20% TCA and centrifuged at2500 rpm for 30 min at 4 C. Pellets were extracted withice-cold ethyl ether and centrifuged again. Supernatantswere aspirated, and remaining ether was evaporated at roomtemperature (RT). The pellets were then dissolved in loadingbuffer with 1% sodium dodecyl sulfate and applied to a 12%SDS-PAGE. The dried gels were exposed to photographicfilm, and later the gel region corresponding to the band ofinterest was excised and counted directly in a �-counter.Results are expressed as arbitrary units.
Statistical Analysis
Experiments containing multiple groups were analyzed byANOVA followed by the Bonferroni test for multiple compar-isons. Student’s t test was used when experiments containedonly two groups. The 5% threshold was used to reject the nullhypothesis.
Acknowledgments
Received April 8, 2003. Accepted April 16, 2004.Address all correspondence and requests for reprints to:
P. Reed Larsen, M.D., Division of Endocrinology, Diabetesand Hypertension, Brigham & Women’s Hospital, 77 AvenueLouis Pasteur, Room 550, Boston, Massachusetts 02115.
This work was supported by National Institutes of HealthGrants DK44128 and HL52320 and Grant 9930032N from theAmerican Heart Association. B.W.K. was a recipient of agrant from the Endocrine Fellows Foundation and DK07529training grant.
REFERENCES
1. Dillmann WH 2002 Cellular action of thyroid hormone onthe heart. Thyroid 12:447–452
2. Steinbock M, Grossman W 1971 Tachycardia in thyro-toxicosis. Ann Intern Med 75:973
3. Wiener L, Stout BD, Cox JW 1969 Influence of � sym-pathetic blockade (propranolol) on the hemodynamics ofhyperthyroidism. Am J Med 46:227–233
4. Rutherford JD, Vatner SF, Braunwald E 1979 Adrenergiccontrol of myocardial contractility in conscious hyperthy-roid dogs. Am J Physiol 237:H590–H596
5. Hoit BD, Khoury SF, Shao Y, Gabel M, Liggett SB, WalshRA 1997 Effects of thyroid hormone on cardiac �-adren-ergic responsiveness in conscious baboons. Circulation96:592–598
6. Klein I, Levey GS 2000 The cardiovascular system inthyrotoxicosis. In: Braverman LE, Utiger RD, eds. Wernerand Ingbar’s the thyroid: a fundamental and clinical text,8th ed. Baltimore: Lippincott, Williams & Wilkins;586–604
7. Hohl CM, Wetzel S, Fertel RH, Wimsatt DK, Brierley GP,Altschuld RA 1989 Hyperthyroid adult rat cardiomyo-
cytes. I. Nucleotide content, �- and �-adrenoreceptors,and cAMP production. Am J Physiol 257:C948–C956
8. Ojamaa K, Kenessey A, Klein I 2000 Thyroid hormoneregulation of phospholamban phosphorylation in the ratheart. Endocrinology 141:2139–2144
9. Silva JE 2000 Catecholamines and the sympathoadrenalsystem in hypothyroidism. In: Braverman LE, Utiger RD,eds. Werner and Ingbar’s the thyroid: a fundamental andclinical text. Baltimore: Lippincott Williams & Wilkins;820–823
10. Ojamaa K, Samarel AM, Kupfer JM, Hong C, Klein I 1992Thyroid hormone effects on cardiac gene expressionindependent of cardiac growth and protein synthesis.Am J Physiol 263:E534–E540
11. Klein I, Hong C 1986 Effects of thyroid hormone oncardiac size and myosin content of the heterotopicallytransplanted rat heart. J Clin Invest 77:1694–1698
12. Croteau W, Davey JC, Galton VA, St Germain DL 1996Cloning of the mammalian type II iodothyronine deiodi-nase. A selenoprotein differentially expressed and regu-lated in human and rat brain and other tissues. J ClinInvest 98:405–417
13. Salvatore D, Bartha T, Harney JW, Larsen PR 1996 Mo-lecular biological and biochemical characterization of thehuman type 2 selenodeiodinase. Endocrinology 137:3308–3315
14. Pachucki J, Hopkins J, Peeters R, Tu H, Carvalho SD,Kaulbach H, Abel ED, Wondisford FE, Ingwall JS, LarsenPR 2001 Type 2 iodothyronine deiodinase transgene ex-pression in the mouse heart causes cardiac-specific thy-rotoxicosis. Endocrinology 142:13–20
15. Pachucki J, Burmeister LA, Larsen PR 1999 Thyroidhormone regulates hyperpolarization-activated cyclicnucleotide-gated channel (HCN2) mRNA in the ratheart. Circ Res 85:498–503
16. Lortet S, Heckmann M, Ray A, Rossi A, Aussedat J,Grably S, Zimmer HG 1995 Energy metabolism responseto calcium activation in isolated rat hearts during devel-opment and regression of T3-induced hypertrophy. MolCell Biochem 151:99–106
17. Bak MI, Ingwall JS 1998 Regulation of cardiac AMP-specific 5�-nucleotidase during ischemia mediates ATPresynthesis on reflow. Am J Physiol 274:C992–C1001
18. Kessler PD, Cates AE, Van Dop C, Feldman AM 1989Decreased bioactivity of the guanine nucleotide-bindingprotein that stimulates adenylate cyclase in hearts fromcardiomyopathic Syrian hamsters. J Clin Invest 84:244–252
19. Iyengar R 1993 Molecular and functional diversity ofmammalian Gs-stimulated adenylyl cyclases. FASEB J7:768–775
20. Taussig R, Tang WJ, Hepler JR, Gilman AG 1994 Distinctpatterns of bidirectional regulation of mammalian adeny-lyl cyclases. J Biol Chem 269:6093–6100
21. Defer N, Best-Belpomme M, Hanoune J 2000 Tissuespecificity and physiological relevance of various iso-forms of adenylyl cyclase. Am J Physiol 279:F400–F416
22. Feldman AM 2002 Adenylyl cyclase: a new target forheart failure therapeutics. Circulation 105:1876–1878
23. Patel TB, Du Z, Pierre S, Cartin L, Scholich K 2001Molecular biological approaches to unravel adenylylcyclase signaling and function. Gene 269:13–25
24. Hadcock JR, Malbon CC 1993 Agonist regulation of geneexpression of adrenergic receptors and G proteins.J Neurochem 60:1–9
25. Ribeiro-Neto F, Mattera R, Grenet D, Sekura RD, Birn-baumer L, Field JB 1987 Adenosine diphosphate ribosy-lation of G proteins by pertussis and cholera toxin inisolated membranes. Different requirements for and ef-fects of guanine nucleotides and Mg2�. Mol Endocrinol1:472–481
1848 Mol Endocrinol, July 2004, 18(7):1840–1849 Carvalho-Bianco et al. • Thyrotoxicosis and �-Adrenergic Signaling

26. Kopf GS, Woolkalis MJ 1991 ADP-ribosylation of Gproteins with pertussis toxin. Methods Enzymol 195:257–266
27. Williams LT, Lefkowitz RJ, Watanabe AM, Hathaway DR,Besch Jr HR 1977 Thyroid hormone regulation of �-ad-renergic receptor number. J Biol Chem 252:2787–2789
28. Stiles GL, Lefkowitz RJ 1981 Thyroid hormone modula-tion of agonist-�-adrenergic receptor interactions in therat heart. Life Sci 28:2529–2536
29. Novotny J, Bourova L, Malkova O, Svoboda P, Kolar F1999 G proteins, �-adrenoreceptors and �-adrenergicresponsiveness in immature and adult rat ventricularmyocardium: influence of neonatal hypo- and hyperthy-roidism. J Mol Cell Cardiol 31:761–772
30. Zwaveling J, Batink HD, Taguchi K, de Jong S, MichelMC, Pfaffendorf M, van Zwieten A 1998 Thyroid statusaffects the rat cardiac �-adrenoceptor system transientlyand time-dependently. J Auton Pharmacol 18:1–11
31. Zolk O, Kilter H, Flesch M, Mansier P, Swynghedauw B,Schnabel P, Bohm M 1998 Functional coupling of over-expressed �1-adrenoceptors in the myocardium oftransgenic mice. Biochem Biophys Res Commun 248:801–805
32. Heubach JF, Trebess I, Wettwer E, Himmel HM, MichelMC, Kaumann AJ, Koch WJ, Harding SE, Ravens U 1999L-type calcium current and contractility in ventricularmyocytes from mice overexpressing the cardiac �2-adrenoceptor. Cardiovasc Res 42:173–182
33. Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD,Mitarai S, Yatani A, Dorn II GW 2000 Early and delayedconsequences of �(2)-adrenergic receptor overexpres-sion in mouse hearts: critical role for expression level.Circulation 101:1707–1714
34. Tepe NM, Lorenz JN, Yatani A, Dash R, Kranias EG, DornII GW, Liggett SB 1999 Altering the receptor-effectorratio by transgenic overexpression of type V adenylylcyclase: enhanced basal catalytic activity and functionwithout increased cardiomyocyte �-adrenergic signal-ling. Biochemistry 38:16706–16713
35. Gao M, Ping P, Post S, Insel PA, Tang R, Hammond HK1998 Increased expression of adenylylcyclase typeVI proportionately increases �-adrenergic receptor-stimulated production of cAMP in neonatal rat cardiacmyocytes. Proc Natl Acad Sci USA 95:1038–1043
36. Iwase M, Bishop SP, Uechi M, Vatner DE, Shannon RP,Kudej RK, Wight DC, Wagner TE, Ishikawa Y, Homcy CJ,Vatner SF 1996 Adverse effects of chronic endogenoussympathetic drive induced by cardiac GS � overexpres-sion. Circ Res 78:517–524
37. Iwase M, Uechi M, Vatner DE, Asai K, Shannon RP, KudejRK, Wagner TE, Wight DC, Patrick TA, Ishikawa Y,Homcy CJ, Vatner SF 1997 Cardiomyopathy induced bycardiac Gs � overexpression. Am J Physiol 272:H585–H589
38. Engelhardt S, Hein L, Wiesmann F, Lohse MJ 1999 Pro-gressive hypertrophy and heart failure in �1-adrenergicreceptor transgenic mice. Proc Natl Acad Sci USA 96:7059–7064
39. Gao MH, Lai NC, Roth DM, Zhou J, Zhu J, Anzai T, DaltonN, Hammond HK 1999 Adenylylcyclase increases re-
sponsiveness to catecholamine stimulation in transgenicmice. Circulation 99:1618–1622
40. Wagner JP, Seidler FJ, Lappi SE, McCook EC, Slotkin TA1994 Role of thyroid status in the ontogeny of adrenergiccell signaling in rat brain: � receptors, adenylate cyclase,ornithine decarboxylase and c-fos protooncogene ex-pression. J Pharmacol Exp Ther 271:472–483
41. Granneman JG 1995 Expression of adenylyl cyclase sub-types in brown adipose tissue: neural regulation of typeIII. Endocrinology 136:2007–2012
42. Carvalho SD, Bianco AC, Silva JE 1996 Effects of hypo-thyroidism on brown adipose tissue adenylyl cyclaseactivity. Endocrinology 137:5519–5529
43. Katada T, Bokoch GM, Northup JK, Ui M, Gilman AG1984 The inhibitory guanine nucleotide-binding regula-tory component of adenylate cyclase. Properties andfunction of the purified protein. J Biol Chem 259:3568–3577
44. Cerione RA, Staniszewski C, Gierschik P, Codina J,Somers RL, Birnbaumer L, Spiegel AM, Caron MG,Lefkowitz RJ 1986 Mechanism of guanine nucleotideregulatory protein-mediated inhibition of adenylate cy-clase. Studies with isolated subunits of transducin in areconstituted system. J Biol Chem 261:9514–9520
45. Rudolph U, Spicher K, Birnbaumer L 1996 Adenylyl cy-clase inhibition and altered G protein subunit expressionand ADP-ribosylation patterns in tissues and cells fromGi2 ��/� mice. Proc Natl Acad Sci USA 93:3209–3214
46. Nagata K, Ye C, Jain M, Milstone DS, Liao R, MortensenRM 2000 G�(i2) but not G�(i3) is required for muscarinicinhibition of contractility and calcium currents in adultcardiomyocytes. Circ Res 87:903–909
47. Ostrom RS, Violin JD, Coleman S, Insel PA 2000 Selec-tive enhancement of �-adrenergic receptor signaling byoverexpression of adenylyl cyclase type 6: colocalizationof receptor and adenylyl cyclase in caveolae of cardiacmyocytes. Mol Pharmacol 57:1075–1079
48. Zamah AM, Delahunty M, Luttrell LM, Lefkowitz RJ 2002Protein kinase A-mediated phosphorylation of the �2-adrenergic receptor regulates its coupling to Gs and Gi.Demonstration in a reconstituted system. J Biol Chem277:31249–31256
49. Levine MA, Feldman AM, Robishaw JD, Ladenson PW,Ahn TG, Moroney JF, Smallwood PM 1990 Influence ofthyroid hormone status on expression of genes encodingG protein subunits in the rat heart. J Biol Chem 265:3553–3560
50. Krawietz W, Werdan K, Erdmann E 1982 Effect of thyroidstatus on �-adrenergic receptor, adenylate cyclase ac-tivity and guanine, nucleotide regulatory unit in rat car-diac and erythrocyte membranes. Biochem Pharmacol31:2463–2469
51. de Jesus LA, Carvalho SD, Ribeiro MO, Schneider M,Kim SW, Harney JW, Larsen PR, Bianco AC 2001 Thetype 2 iodothyronine deiodinase is essential for adaptivethermogenesis in brown adipose tissue. J Clin Invest108:1379–1385
52. Steinsapir J, Harney J, Larsen PR 1998 Type 2 iodothy-ronine deiodinase in rat pituitary tumor cells is inacti-vated in proteasomes. J Clin Invest 102:1895–1899
Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremostprofessional society serving the endocrine community.
Carvalho-Bianco et al. • Thyrotoxicosis and �-Adrenergic Signaling Mol Endocrinol, July 2004, 18(7):1840–1849 1849




















