
Cholinergic signaling in the hippocampus regulates social stress resilience and anxiety- and...
11
Cholinergic signaling in the hippocampus regulates social stress resilience and anxiety- and depression-like behavior Yann S. Mineur, Adetokunbo Obayemi 1 , Mattis B. Wigestrand 1 , Gianna M. Fote, Cali A. Calarco, Alice M. Li, and Marina R. Picciotto 2 Department of Psychiatry, Yale University School of Medicine, New Haven, CT 06508 Edited by Floyd Bloom, The Scripps Research Institute, La Jolla, CA, and approved January 11, 2013 (received for review November 15, 2012) Symptoms of depression can be induced in humans through block- ade of acetylcholinesterase (AChE) whereas antidepressant-like ef- fects can be produced in animal models and some clinical trials by limiting activity of acetylcholine (ACh) receptors. Thus, ACh signaling could contribute to the etiology of mood regulation. To test this hypothesis, we administered the AChE inhibitor physostigmine to mice and demonstrated an increase in anxiety- and depression-like behaviors that was reversed by administration of nicotinic or mus- carinic antagonists. The behavioral effects of physostigmine were also reversed by administration of the selective serotonin reuptake inhibitor fluoxetine. Administration of fluoxetine also increased AChE activity throughout the brain, with the greatest change in the hippocampus. To determine whether cholinergic signaling in the hippocampus could contribute to the systemic effects of cholinergic drugs, we infused physostigmine or virally delivered shRNAs target- ing AChE into the hippocampus. Both pharmacological and molecular genetic decreases in hippocampal AChE activity increased anxiety- and depression-like behaviors and decreased resilience to repeated stress in a social defeat paradigm. The behavioral changes due to shRNA-mediated knockdown of AChE were rescued by coinfusion of an shRNA-resistant AChE transgene into the hippocampus and re- versed by systemic administration of fluoxetine. These data demon- strate that ACh signaling in the hippocampus promotes behaviors related to anxiety and depression. The sensitivity of these effects to fluoxetine suggests that shRNA-mediated knockdown of hippocam- pal AChE represents a model for anxiety- and depression-like pheno- types. Furthermore, abnormalities in the cholinergic system may be critical for the etiology of mood disorders and could represent an endophenotype of depression. psychosocial stress | affective disorders D epression affects one in six individuals worldwide, resulting in a substantial personal and economic burden. Although the precise etiology of depression is not known, a constellation of findings suggests that predisposing factors, including individual genetic makeup, interacting with stressful life events, can pre- cipitate depressive disorders. Currently available antidepressants are effective at treating both depression and anxiety, but the therapeutic response remains variable. Only 30% of patients achieve remission during the first line of treatment with a selec- tive serotonin reuptake inhibitor (SSRI) such as fluoxetine, and many patients show only limited improvements. Thus, it is crit- ical to identify the neurobiological factors that predispose indi- viduals to stress susceptibility and depression to understand the etiology and develop therapeutics for the disorder. A recent human imaging study has suggested that acetylcholine (ACh) levels are elevated in patients who are actively depressed, as measured by occupancy of nicotinic receptors throughout the brain, and remain high in patients who have a history of depression (1). In addition, despite the recent failure of a large clinical trial, other clinical and preclinical studies have shown that blockers of cho- linergic (both muscarinic and nicotinic) receptors can induce an- tidepressant-like responses (2). These observations have renewed interest (3, 4) in the adrenergic-cholinergic balance hypothesis of mania and depression (5), which states that heightened cholinergic tone and decreased noradrenergic tone could lead to depressive symptoms (6). In these human studies, physostigmine, an inhibitor of acetylcholinesterase (AChE: the primary enzyme regulating ACh levels in the extracellular space), increased depressive symp- toms in individuals with or without a history of depression. Simi- larly, hypersensitivity of the cholinergic system has been connected to the induction of depression-like behavior induced by various stressors in rodents (7). ACh levels and activity can be modulated by stress [one of the main triggers of depression (8)] in several brain regions (9). Taken together, these observations suggest that hy- peractivity of brain cholinergic systems can contribute to the pathophysiology of depression; however, the mechanisms under- lying this relationship are largely unknown, and it is not clear whether there is a causal relationship between ACh regulation and mood or whether these observations are solely correlative. Previous studies have attempted to alter AChE activity ge- netically, but broad peripheral effects of AChE knockout result in early death and severe motor abnormalities, whereas broad overexpression results in compensatory alterations in AChE ac- tivity (10, 11). Other studies have manipulated expression of a specific, alternatively spliced isoform of AChE (AChE-R) that can be induced by stress, that results in hypersensitivity of hip- pocampal neurons through neurite reorganization (12), and that may protect against stress-induced neurodegeneration (13). Mice overexpressing AChE-R show heightened anxiety-like behavior (14), but it remains difficult to determine whether this is a primary consequence of altering AChE activity or whether the behavioral change is due to compensation that can occur in genetically ma- nipulated animals during development. In this study, we investigated the role of cholinergic signaling in anxiety- and mood-related behaviors, using pharmacological and molecular genetic techniques to alter AChE levels or activity in adulthood. We tested the hypothesis that ACh dynamics in the hippocampus are involved in anxiety- and depression-like behav- ior, as well as susceptibility to stress in the social defeat paradigm. Results Acute Treatment with Physostigmine Induces a Depression-Like Phenotype in the Tail Suspension Test. As expected, acute adminis- tration of physostigmine significantly decreased activity of AChE across brain regions (P’s < 0.05) (Fig. S1). Acute physostigmine (0–0.5 mg/kg) administration also induced an increase in immo- bility in the tail suspension test, suggesting that AChE blockade Author contributions: Y.S.M., A.O., M.B.W., and M.R.P. designed research; Y.S.M., A.O., M.B.W., G.M.F., C.A.C., and A.M.L. performed research; Y.S.M. contributed new reagents/ analytic tools; Y.S.M., A.O., M.B.W., and C.A.C. analyzed data; and Y.S.M. and M.R.P. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. 1 A.O. and M.B.W. contributed equally to this work. 2 To whom correspondence should be addressed. E-mail: [email protected]. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1219731110/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1219731110 PNAS Early Edition | 1 of 6 NEUROSCIENCE
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Cholinergic signaling in the hippocampus regulates social stress resilience and anxiety- and...

Cholinergic signaling in the hippocampus regulatessocial stress resilience and anxiety- anddepression-like behaviorYann S. Mineur, Adetokunbo Obayemi1, Mattis B. Wigestrand1, Gianna M. Fote, Cali A. Calarco, Alice M. Li,and Marina R. Picciotto2
Department of Psychiatry, Yale University School of Medicine, New Haven, CT 06508
Edited by Floyd Bloom, The Scripps Research Institute, La Jolla, CA, and approved January 11, 2013 (received for review November 15, 2012)
Symptoms of depression can be induced in humans through block-ade of acetylcholinesterase (AChE) whereas antidepressant-like ef-fects can be produced in animal models and some clinical trials bylimiting activity of acetylcholine (ACh) receptors. Thus, ACh signalingcould contribute to the etiology of mood regulation. To test thishypothesis, we administered the AChE inhibitor physostigmine tomice and demonstrated an increase in anxiety- and depression-likebehaviors that was reversed by administration of nicotinic or mus-carinic antagonists. The behavioral effects of physostigmine werealso reversed by administration of the selective serotonin reuptakeinhibitor fluoxetine. Administration of fluoxetine also increasedAChE activity throughout the brain, with the greatest change inthe hippocampus. To determine whether cholinergic signaling in thehippocampus could contribute to the systemic effects of cholinergicdrugs, we infused physostigmine or virally delivered shRNAs target-ingAChE into the hippocampus. Bothpharmacological andmoleculargenetic decreases in hippocampal AChE activity increased anxiety-and depression-like behaviors and decreased resilience to repeatedstress in a social defeat paradigm. The behavioral changes due toshRNA-mediated knockdown of AChEwere rescued by coinfusion ofan shRNA-resistant AChE transgene into the hippocampus and re-versed by systemic administration of fluoxetine. These data demon-strate that ACh signaling in the hippocampus promotes behaviorsrelated to anxiety and depression. The sensitivity of these effects tofluoxetine suggests that shRNA-mediated knockdown of hippocam-pal AChE represents a model for anxiety- and depression-like pheno-types. Furthermore, abnormalities in the cholinergic system may becritical for the etiology of mood disorders and could represent anendophenotype of depression.
psychosocial stress | affective disorders
Depression affects one in six individuals worldwide, resultingin a substantial personal and economic burden. Although
the precise etiology of depression is not known, a constellation offindings suggests that predisposing factors, including individualgenetic makeup, interacting with stressful life events, can pre-cipitate depressive disorders. Currently available antidepressantsare effective at treating both depression and anxiety, but thetherapeutic response remains variable. Only 30% of patientsachieve remission during the first line of treatment with a selec-tive serotonin reuptake inhibitor (SSRI) such as fluoxetine, andmany patients show only limited improvements. Thus, it is crit-ical to identify the neurobiological factors that predispose indi-viduals to stress susceptibility and depression to understand theetiology and develop therapeutics for the disorder.A recent human imaging study has suggested that acetylcholine
(ACh) levels are elevated in patients who are actively depressed, asmeasured by occupancy of nicotinic receptors throughout the brain,and remain high in patients who have a history of depression (1). Inaddition, despite the recent failure of a large clinical trial, otherclinical and preclinical studies have shown that blockers of cho-linergic (both muscarinic and nicotinic) receptors can induce an-tidepressant-like responses (2). These observations have renewedinterest (3, 4) in the adrenergic-cholinergic balance hypothesis of
mania and depression (5), which states that heightened cholinergictone and decreased noradrenergic tone could lead to depressivesymptoms (6). In these human studies, physostigmine, an inhibitorof acetylcholinesterase (AChE: the primary enzyme regulatingACh levels in the extracellular space), increased depressive symp-toms in individuals with or without a history of depression. Simi-larly, hypersensitivity of the cholinergic system has been connectedto the induction of depression-like behavior induced by variousstressors in rodents (7). ACh levels and activity can be modulatedby stress [one of themain triggers of depression (8)] in several brainregions (9). Taken together, these observations suggest that hy-peractivity of brain cholinergic systems can contribute to thepathophysiology of depression; however, the mechanisms under-lying this relationship are largely unknown, and it is not clearwhether there is a causal relationship between ACh regulation andmood or whether these observations are solely correlative.Previous studies have attempted to alter AChE activity ge-
netically, but broad peripheral effects of AChE knockout resultin early death and severe motor abnormalities, whereas broadoverexpression results in compensatory alterations in AChE ac-tivity (10, 11). Other studies have manipulated expression ofa specific, alternatively spliced isoform of AChE (AChE-R) thatcan be induced by stress, that results in hypersensitivity of hip-pocampal neurons through neurite reorganization (12), and thatmay protect against stress-induced neurodegeneration (13). Miceoverexpressing AChE-R show heightened anxiety-like behavior(14), but it remains difficult to determine whether this is a primaryconsequence of altering AChE activity or whether the behavioralchange is due to compensation that can occur in genetically ma-nipulated animals during development.In this study, we investigated the role of cholinergic signaling in
anxiety- and mood-related behaviors, using pharmacological andmolecular genetic techniques to alter AChE levels or activity inadulthood. We tested the hypothesis that ACh dynamics in thehippocampus are involved in anxiety- and depression-like behav-ior, as well as susceptibility to stress in the social defeat paradigm.
ResultsAcute Treatment with Physostigmine Induces a Depression-LikePhenotype in the Tail Suspension Test. As expected, acute adminis-tration of physostigmine significantly decreased activity of AChEacross brain regions (P’s < 0.05) (Fig. S1). Acute physostigmine(0–0.5 mg/kg) administration also induced an increase in immo-bility in the tail suspension test, suggesting that AChE blockade
Author contributions: Y.S.M., A.O., M.B.W., and M.R.P. designed research; Y.S.M., A.O.,M.B.W., G.M.F., C.A.C., and A.M.L. performed research; Y.S.M. contributed new reagents/analytic tools; Y.S.M., A.O., M.B.W., and C.A.C. analyzed data; and Y.S.M. and M.R.P.wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1A.O. and M.B.W. contributed equally to this work.2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1219731110/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1219731110 PNAS Early Edition | 1 of 6
NEU
ROSC
IENCE

has a prodepressant-like effect (15) [treatment: F(3, 54) = 3.68,P = 0.017] (Fig. 1A, Left). Post hoc analyses demonstrated thatphysostigmine concentrations above 0.25 mg/kg were effective(0.25 mg/kg: P = 0.005; 0.5 mg/kg: P = 0.005) without affectinglocomotor activity (Fig. 1B, F < 1). Physostigmine administra-tion induced a dose-dependent decrease in brain AChE activity(Fig. 1A, Right) [treatment: F(3, 148) = 10.1, P < 0. 0001]. Posthoc analyses revealed that concentrations inducing a significantdecrease in AChE activity (0.25 mg/kg, P = 0.0002; 0.5 mg/kg,P = 0.0007) were effective in the tail suspension test.
Acute Effects of Physostigmine Are Reversed by ACh ReceptorAntagonists or Treatment with the Antidepressant Fluoxetine. Block-ade of AChE results in increased ACh signaling at its targets, bothmuscarinic and nicotinic acetylcholine receptors (AChRs). Acuteadministration of scopolamine (a broad muscarinic antagonist;0.5 mg/kg), mecamylamine (a broad nicotinic antagonist; 1 mg/kg),or a combination of both drugs blocked the effects of physo-stigmine in the tail suspension test, suggesting that physo-stigmine’s effects are due to downstream activation of AChRs(F < 1) (Fig. 1C). In addition, administration of the antidepressantfluoxetine (10 mg/kg) also reversed the prodepressant-like effectsof physostigmine observed in the tail suspension test (Fig. 2A)(saline + saline vs. physostigmine + fluoxetine: F < 1), suggestingthat an antidepressant used in human depressed patients wasalso effective in reversing physostigmine-induced depression-like behavior.
Chronic Treatmentwith Fluoxetine Increases Hippocampal AChE Activity.Because fluoxetine reversed the effects of AChE blockade, we
hypothesized that antidepressant administration might also alterAChE activity. We therefore treated animals with fluoxetine for15 d, tested them in the tail suspension test to verify fluoxetine’santidepressant-like effect, and measured AChE activity in brainslices. Fluoxetine induced a brain region-dependent increase inAChE activity [treatment × brain region: F(9, 130) = 4.16, P <0.0001 (Fig. 2B)]. Post hoc analyses revealed that significance wasreached only in the hippocampus (P = 0.04). To control for anyeffects of the behavioral assay on AChE activity, we repeated theevaluation of AChE activity in mice treated chronically with flu-oxetine with no behavioral testing. Brain regions were micro-dissected and AChE activity was measured in homogenates,confirming the increase of hippocampal AChE activity followingchronic fluoxetine treatment (P = 0.008) (Fig. S2). In contrast,chronic administration of physostigmine resulted in a paradoxicalup-regulation of AChE activity, suggesting that there was com-pensation at the transcriptional level and that pharmacologicalantagonism cannot be used to induce a long-term increase in brainacetylcholine levels (Fig. S3).
Local Infusion of Physostigmine into the Hippocampus Induces aDepression-Like Phenotype in the Tail Suspension Test. To deter-mine whether the change in hippocampal AChE activity couldbe causally related to behavior in the tail suspension test, wemicroinfused physostigmine into the hippocampus. Acute AChEantagonism in the hippocampus resulted in a significant increasein immobility in the tail suspension test [F(1, 22) = 4.71, P = 0.04],suggesting that increased ACh signaling in this brain structure issufficient to induce depression-like behavior (Fig. 2C).
Knockdown of AChE in the Hippocampus Increases Anxiety- andDepression-Like Behaviors and Susceptibility to Social Stress. Todetermine whether a local, long-term change in ACh dynamics inthe hippocampus could result in altered anxiety- and depression-like behavior, we generated adeno-associated virus (AAV) car-rying shRNAs targeting the regions common to all isoforms ofAChE (shAChE). The ability of these vectors to decrease AChEactivity was validated in vitro (Fig. S4) and in vivo (Fig. 3 A andB). AAV-shAChEs were infused into the hippocampus, and ac-curacy of targeting was determined based on expression of GFP(Fig. 3C). Off-target effects of shRNAs are an important concernin in vivo knockdown studies. To verify that the behavioral effectsof AAV-shAChE infusion were due to AChE knockdown spe-cifically, we used the human AChE cDNA (hAChE) to design anAChE expression construct that would not be recognized by theAAV-shAChE construct. We infused AAV-hAChE into thehippocampus alone or coinfused with AAV-shAChE and verifiedthat hAChE was expressed even when the shAChE (Fig. 3D) waspresent, demonstrating that hAChE was resistant to knockdownby shAChE.Hippocampal AChE knockdown resulted in a significant de-
crease in time spent in the open arms in the elevated plus maze[T(18) = 7.19, P < 0.001 (Fig. 4A, Left)] compared with hippo-campal infusion of a scrambled control vector (AAV-Scr). Similarly,in the light-dark test, mice that received infusions of the AAV-
Fig. 1. Time spent immobile in the tail suspension testafter administration of the cholinesterase antagonist phy-sostigmine 30 min before testing (A, Right). AChE activity inwhole brain after administration of physostigmine (A, Left).Locomotor activity over 20 min following physostigmineinjection (B). Time spent immobile in the tail suspensiontest after treatment with physostigmine with, or without, anicotinic (mecamylamine) and/or muscarinic (scopolamine)antagonist (C). n = 8–12 per group. **P < 0.01; ***P < 0.001.All data are expressed as mean ± SEM.
Fig. 2. (A) Time spent immobile in the tail suspension test after physo-stigmine-induced immobility and its reversal by fluoxetine; n = 9–12 pergroup. (B) AChE activity in microdissected brain regions following chronictreatment with fluoxetine; n = 5 per group. (C) Time spent immobile in thetail suspension test following an acute bilateral infusion of physostigmineinto the hippocampus; n = 7–12 per group. #P = 0.07; *P < 0.05; ***P < 0.001.All data are expressed as mean ± SEM.
2 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1219731110 Mineur et al.

shAChE into the hippocampus spent less time in the light side of thebox [T(20) = 2.75, P < 0.05 (Fig. 4A, Right)] compared with micethat received AAV-Scr infusions. In both tests, no differences in thenumber of entries and overall activity were detected betweengroups, suggesting that these changes were due to increased anxiety-like behavior and not to changes in locomotor activity.In both the tail suspension and the forced-swim tests, AAV-
shAChE infusion into the hippocampus resulted in increasedimmobility time [tail suspension: T(20) = 3.19, P < 0.01; forcedswim: T(19) = 2.35, P < 0.05 (Fig. 4B)] compared with AAV-Scrcontrols. No difference was observed in locomotor activity be-tween these groups (Fig. 4C), suggesting that knockdown ofhipopocampal AChE increased depression-like behaviors. Con-versely, coinfusion of AAV-hAChE prevented the behavioraleffects of AAV-shAChE infusion in the tail suspension test andthe forced-swim test.
Although these latter tests have been useful as tests of antide-pressant efficacy, human depression generally results from emo-tional stress rather than physical stress. To determine whetherACh dynamics in the hippocampus alters the response to sociallyinduced stressors, we evaluated behavior in the social defeatmodel. As a first step, we confirmed that mice that experiencerepeated aggression (social defeat) exhibit social avoidance (16).Mice were subjected to 10 d of social defeat stress, followed 24 hlater by the social interaction test. As expected, mice exposed tochronic social defeat stress were significantly more likely to avoida target mouse during social interaction testing compared withnonstressed controls [T(16) = 3.79, P < 0.01 (Fig. 4D)].We then tested whether hippocampal AChE knockdown al-
tered stress susceptibility using a submaximal version of socialdefeat, which does not induce social avoidance in wild-typemice. AAV-shAChE or AAV-Scr vectors were infused into the
Fig. 3. AAV2-mediated delivery of AChE shRNAs(AAV-shAChE) into the hippocampus of mice.Representative photomicrograph showing hippo-campus-specific decrease in AChE activity afterhippocampal infusion of AAV-shRNA by stereo-taxic surgery (A). Real-time PCR showed knock-down of AChE mRNA levels in shAChE-infectedhippocampus compared with scrambled controls;n = 6 per group (B). GFP expression after infusionof AAV in the hippocampus demonstrating thespecificity of the localization and the spread of in-fusion (C ). Representative activity-based stainingof AChE showing hippocampal AChE overex-pression following infusion of the AAV-hAChEconstruct (D). A control group infused with AAV-GFP shows baseline AChE activity (D, Top), whereas the group infused with the human esterase constructshows increased AChE activity (D, Middle), and the overexpression is not decreased by coinfusion of the AAV-shRNA (D, Bottom). Data are expressed asmean ± SEM. *P < 0.05.
Fig. 4. AChE knockdown in the hip-pocampus promotes anxiety-like be-havior inmice. Infusion of shAChE intothe hippocampus (n = 10 per group)decreased both time spent in openarms in the elevated plus maze test (A,Left) and time spent in the light com-partment of the light–dark box (A,Right). AChE knockdown in the hip-pocampus increased depression-likebehavior in mice. Infusion of shAChEinto the hippocampus increased im-mobility time in the tail suspension (B,Left) and forced-swim (B, Right) tests,with no effects on locomotor activity(C). AChE knockdown in the hippo-campus promotes stress susceptibilityin mice. Ten days of social defeat stressdecreased social interaction in WTmice (D). Infusion of shAChE into thehippocampus decreased social in-teraction after submaximal defeatstress compared with mice receivingscrambled shRNA infusion (E). Expres-sion of hAChE in the hippocampusprevents AChE knockdown and res-cues the behavioral effect of shAChEinfusion in mice. Infusion of AAV-hAChE in the hippocampus preventedprodepressive effects of hippocampalshAChE delivery in the tail suspensionand forced-swim tests, as well as in thesubmaximal social defeat paradigm.Fluoxetine reversed the behavioral effects of hippocampal shRNA-mediated AChE knockdown. Acute administration of fluoxetine (10 mg/kg) reversed the effectsof AChE knockdown in the hippocampus in the tail suspension test (F, Left). Chronic administration offluoxetine (10mg/kg) reversed the effects of shAChE infusionin the social defeat paradigm (F, Right); n = 8–10 per group. All data are expressed as mean ± SEM. *P < 0.05; **P < 0.01, ***P < 0.001.
Mineur et al. PNAS Early Edition | 3 of 6
NEU
ROSC
IENCE

hippocampus, mice were allowed to recover for 4 wk, and ani-mals were then subjected to three social defeat episodes in 1 d.Twenty-four hours later, social avoidance was measured in thesocial interaction test. As expected, control animals that weresubjected to submaximal defeat stress displayed levels of socialinteraction that were comparable to nondefeated mice (Fig. 4E).In contrast, mice that received AAV-shAChE infusions into thehippocampus displayed significant social avoidance (P < 0.05)after submaximal defeat stress. Importantly, AAV-shAChE in-fusion did not influence social behavior in the absence of stress,indicating that hippocampal AChE knockdown increased thesusceptibility to social defeat stress rather than altering behavioron its own. ANOVA for interaction scores detected a significantmain effect of AChE knockdown [F(1,31) = 4.17, P < 0.05] withno significant main effect of stress [F(1,31) = 0.59, P = 0.45], aswell as a significant knockdown X stress interaction [F(1,31) =5.13, P < 0.05]. Bonferroni post hoc comparison further revealedsignificant social avoidance in AChE knockdown mice comparedwith controls that was present only after social defeat stress (P <0.05), with no significant effect of knockdown in naive non-stressed mice. Coinfusion of AAV-hAChE prevented the be-havioral effects of AAV-shAChE infusion in the submaximalsocial defeat paradigm (Fig. 4E), whereas locomotor activity wasunaffected. Although infusion of AAV-hAChE into the hippo-campus increased AChE activity, the overexpression construct hadno effect on any of the behavioral paradigms tested (Fig. S5).
Behavioral Effects of AChE Knockdown Can Be Reversed withFluoxetine Treatment. To determine whether the prodepressant-like effects of hippocampal AChE knockdown were sensitive toantidepressant administration, mice infused with shAChE into thehippocampus were treated with fluoxetine (10 mg/kg) and testedin the tail suspension test or social defeat models. Acute admin-istration of fluoxetine (10 mg/kg) decreased immobility in micewith hippocampal shAChE infusions to a level similar to controlanimals [treatment: F(2, 26) = 8.17, P = 0.0018) (Fig. 4F)].Unlike the tail suspension test, social defeat behavior is sensitive
to chronic, but not acute, treatment with antidepressants that areeffective in human depressed subjects (17, 18). Chronic treatmentwith fluoxetine (10 mg/kg for 15 d) prevented the effects of hip-pocampal AAV-shAChE infusion on submaximal social defeatbehavior [saline vs. fluoxetine: F(1, 18) = 67.1, P < 0.001) (Fig. 4F)].
DiscussionThe ability of ACh signaling to alter mood in human subjects wasfirst discovered several decades ago (5, 6). However, the brainregions involved in cholinergic control of anxiety- and depression-like symptoms have remained unknown, mainly due to the com-plex nature of mood regulation and the neuromodulatory role ofACh (see ref. 19 for review). The effects of ACh are dependent onthe site, pattern, and timescale of release. ACh release can beinduced by environmental stressors in many brain areas, includingthe prefrontal cortex and the hippocampus, two regions known tobe involved in depression and mood regulation (20). ACh sig-naling can coordinate the response of neuronal networks in-volving these stress-sensitive brain areas, all of which could berelevant for behaviors related to anxiety and depression. AChE isthe key enzyme in ACh breakdown and is extremely efficient atmodulating extracellular ACh levels (21). AChE is also the targetof drugs used as treatments for Alzheimer’s disease and is alsotargeted by nerve agents, and insecticides. Therefore, it is of greatinterest to determine whether alterations of AChE activity andcholinergic tone may mediate stress-induced changes leading toanxiety- and depression-related behaviors.In the current set of experiments, C57BL/6J male mice injected
with physostigmine showed increased immobility in the tail sus-pension test, replicating human studies showing that a pharma-cological increase in cholinergic tone can precipitate symptoms ofdepression (5, 6). Recently, human imaging using a nicotinictracer that can be displaced by endogenous ACh showed thatthere is decreased nicotinic receptor availability in depressed
subjects, with no change in receptor number as measured post-mortem (1), further suggesting that heightened cholinergic tonecould be associated with symptoms of depression. In the currentstudy, we determined that depression-like consequences of AChEantagonism were due to signaling through its downstream re-ceptors because both muscarinic and nicotinic AChR antagonistsreversed the effects of physostigmine in the tail suspension test.Both clinical and preclinical studies have shown that AChRblockers can have antidepressant-like effects (22, 23), suggestingthat reversal of a hypercholinergic state could result in an anti-depressant response. Finally, the SSRI fluoxetine also abolishedphysostigmine-induced increases in immobility in the tail sus-pension test, validating the interpretation that these effects ofphysostigmine are depression-like.Although acute administration of physostigmine is a useful way
to test the hypothesis that increased ACh signaling can lead todepression-like symptoms, it has limitations as a mouse model ofdepression and has limited utility for mechanistic studies of ACheffects on circuits related to depression-like behavior. Whereasacute administration of physostigmine results in increased AChsignaling, chronic administration of the drug results in increasedtranscription and translation of the AChE gene that can compen-sate for pharmacological blockade (24). shAChE infusions there-fore represent a useful model of chronic increased ACh signaling inthe hippocampus. The current set of experiments demonstratesthat shAChE infusion results in long-term decreases in AChE ac-tivity and in increases in anxiety- and depression-like behavior.Thus, hippocampal AChE knockdown is a useful model of cho-linergic dysfunction leading to anxiety- and depression-like behavior.The pharmacological and molecular genetic studies described
here show that increased ACh signaling in the hippocampus issufficient to recapitulate the consequences of peripheral physo-stigmine administration and to identify a brain region involved inACh-mediated regulation of anxiety- and depression-like behav-iors. The hippocampus, amygdala, and prefrontal cortex receivea high level of cholinergic input from the basal forebrain complex,and in particular, from themedial septum and nucleus basalis (25).Several studies have shown that stress increases ACh release ina brain region-specific manner (20). Importantly, although in-creasing cholinergic tone globally or in the hippocampus inducesdepression-like symptoms, a recent study also showed that de-creasing cholinergic tone in the striatum can lead to depression-likesymptoms, likely through interneuron-dependent disinhibition ofstriatal neurons (26). This highlights the fact that cholinergic sig-naling is not homogenous throughout the brain and that the hip-pocampus may be a critical node mediating cholinergic effects onstress-related behaviors. In the current study, we found thatchronic administration with fluoxetine up-regulated AChE spe-cifically in the hippocampus. Previous studies have demonstratedchanges in AChE activity, a switch in expression of particularAChE isoforms, and increased ACh release in the hippocampusfollowing exposure to stress (27, 28). ACh signals through mus-carinic and nicotinic receptors to modulate the dynamic propertiesof the hippocampus, generating a range of stable oscillatory net-work states (29) that may mediate the stress response.Despite the clear effects of stress on hippocampal activity,
stress-induced activity may also lead to a compensatory increasein hippocampal AChE activity, reducing extracellular acetyl-choline levels and suppressing cholinergic neurotransmission(27). Psychological stress (27) and various environmental stimulisuch as AChE inhibitors (30) and head injury (31) all increaseAChE transcription. These findings clearly demonstrate thatstress can alter cholinergic signaling at different levels; however,it was not known whether changes in hippocampal cholinergictransmission could alter the behavioral response to stress (32). Inthe current study, we showed that facilitating cholinergic sig-naling during social defeat stress by knockdown of AChE activityin the hippocampus precipitated significant social avoidance inmice. AChE knockdown in the absence of exposure to defeat stressdid not change social behavior at baseline. Our results furthersuggest that maintaining high ACh levels in the hippocampus
4 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1219731110 Mineur et al.

during stress may intensify the stress response and alter copingskills in adverse situations. Supporting this conclusion, pharma-cological inhibition of AChE during elevated stress was recentlyshown to increase anxiety-like behavior in mice without similareffects in nonstressed controls (33). Similarly, rats with increasedcholinergic sensitivity are more susceptible to the immobility-inducing effects of mild stressors (34). Consistent with the resultsshown here, acute pharmacological AChE inhibition by physo-stigmine has also been shown to increase immobility in the forced-swim test in rats (35).Studies of the cholinergic influence on anxiety have yielded
more mixed results than studies of depression-like behavior. Forexample, acute injection of high doses of nicotine into the hippo-campus has anxiogenic effects that may be mediated by activationof α7 nicotinic acetylcholine receptors (nAChRs) (36), whereaslower doses of nicotine tend to produce anxiolytic effects (37).Interestingly, the anxiogenic effects of nicotine in the hippocam-pus were induced only under experimental conditions involving anunfamiliar arena or high light (38), which indicates that nAChRstimulation can produce distinct behavioral outcomes dependingon the level of stress generated by the test itself. It follows that,under experimental conditions that generate low levels of anxiety,infusions of either a nicotinic or a muscarinic cholinergic receptorantagonist into the hippocampus can have anxiogenic effects (32,38). Together with our findings, these studies indicate that theeffect of cholinergic tone on anxiety-like behavior may depend onlevels of stress.In summary, the results presented here identify a cholinergic
mechanism in the hippocampus that regulates behavioral sus-ceptibility to stress and to anxiety- and depression-like behaviorsin mice. In particular, this study shows that whereas other brainnuclei are also essential for regulating mood and anxiety, main-taining hippocampal ACh at homeostatic levels is critical forregulation of emotional behaviors. Furthermore, increasing hip-pocampal ACh signaling is sufficient to induce behaviors relatedto anxiety and depression. The current dataset also suggests thatthe ability of physostigmine to induce symptoms of depression inhumans may be due to increases in hippocampal ACh levels. Inaddition, knockdown of AChE in the hippocampus appears tobe an animal model of depression-like behavior with relevantpredictive validity for human antidepressant response due to itsreversibility by chronic fluoxetine administration and good con-struct validity based on data suggesting that individuals who areactively depressed may have increased brain ACh levels (1). Thecurrent study builds on previous studies of the Flinders sensitiveline of rats in which alterations in cholinergic signaling contributeto depression-like phenotypes (34). The experiments presented herealso identify the hippocampus as one area critical for the effects ofACh signaling on anxiety and depression, providing a manipulationin adulthood that bypasses potential developmental alterations.
MethodsAnimals. C57BL/6J male mice (10–12 wk of age) were obtained from JacksonLaboratory and housed under standard laboratory conditions (21 ± 2 °C, 12 hlight–dark cycle with food and water available ad libitum). Upon arrival,mice were split into groups of five mice and randomly assigned to experi-mental groups. Mice were allowed to acclimate to the laboratory for 1–2 wkbefore experiments. Male CD1 mice (20–40 wk of age) obtained fromCharles River were used as aggressors in the social defeat paradigm andhoused under the same conditions, unless specified otherwise.
Drugs. Chemicals were obtained from Sigma-Aldrich, and injectable solutionswere diluted in phosphate buffered saline (PBS; 1 mM KH2PO4, 155 mMNaCl,3 mM Na2HPO4, pH 7.4). Thirty minutes before behavioral testing, solutionswere injected i.p. unless stated otherwise (local infusions). For experimentsthat used two drugs (Fig. 2A), mice were injected with saline or physostig-mine followed by saline or fluoxetine 15 min later, and tail suspensiontesting was performed 30 min after the last injection.
Measurement of AChE Activity. In brain slices, AChE activity was measured aspreviously described (39, 40) and based on colorimetric reaction (see SIMethods for details). In microdissected brain nuclei, enzymatic activity was
determined using a similar method modified for microassays using DACE-100-QuantiChrom Acetylcholinesterase Assay kit following the manu-facturer’s recommendations. Each measurement was performed in triplicate.
AChE Knockdown by AAV-shRNAs. Three shRNAs directed against AChE wereconstructedusingpublishedmethods (41)by selectingunique24base sequencesand tested in vitro in N2A cells (see Fig. S4 for details). To achieve long-term invivo knockdown, efficacious shRNA-AChEs (shAChE) or a scrambled construct(Scr) were incorporated into AAV-2. AAV-shAChE or AAV-Scr (a scrambled se-quence with no known target) was infused bilaterally by stereotaxic surgeryinto the hippocampus. Mice were left to recover for 21 d to allow for knock-down, and efficacy of AAV infusions was further evaluated both by measuringAChE activity in sections and by quantifying mRNA levels in microdissectedtissue (see SI Methods for details). At the end of behavioral experiments, ani-mals were perfused and brain sections were examined with fluorescence mi-croscopy to validate the infusion and infection sites by visualization of GFP.
Behavioral Testing. The sequence of behavioral tests following AAV infusionsstarted with the elevated plus maze, followed by the light–dark test, tailsuspension test, and the forced-swim test. Tests were performed 48–72 hapart to limit any effects of one test on the next.
At least 3wk after the last behavioral test in the battery,micewere exposedto either chronic or submaximal social defeat episodes, followed by the socialinteraction test. To validate that submaximal social defeat measures wereunaffected by preceding behavioral testing, we also tested mice naive to anyprior behavioral testing. Naivemice displayed similar levels of social avoidanceafter AChE knockdown compared with mice that had been tested in thebehavioral battery. For all procedures, mice were habituated to testing roomsfor at least 30 min before behavioral measures were taken, and testing tookplace between 1000 and 1800 hours. All procedures were approved by theYale University Animal Care and Use Committee and conformed to thestandards of the National Institutes of Health Guide for the Care and Use ofLaboratory Animals.
Elevated Plus Maze. The elevated plus maze was made of black plexiglas, hadfour 30- × 5-cm arms, and was elevated 50 cm above the floor. Two armswere enclosed by 15-cm walls; the other two arms had a 3-mm edge toprevent slipping, and all arms were illuminated equally. A 5- × 5-cm platformat the center was considered a neutral area. One hour before the experi-ment, animals were placed in the testing room. At the beginning of the test,mice were placed in the center of the maze facing an open arm and wereallowed to explore the maze for 5 min. The percentage of time spent inthe open arms [100 * (time in open arms/(total time – time in the center)] wasused as the primary measure of anxiety-like behavior. Number of entries intoeach arm was also recorded to detect any unforeseen change in global activity.
Light–Dark Test. The light–dark test was performed as described previously (23).The apparatus consisted of two opaque Plexiglas compartments of the samesize connected by a central opening (18- × 10- × 13-cm dimensions: light com-partment illuminated by a 60-W desk lamp through a transparent Plexiglascover). Mice were placed into the dark compartment facing away from theopening and observed for 5min after thefirst cross wasmade. Number of entriesinto the dark side and time spent in the dark compartment were measured.
Tail Suspension Test. The tail suspension test is used routinely for antide-pressant screening and to identify depression-like behavior in mice (15). Micewere gently suspended by the tail, and videotapes were scored for time spentimmobile over 6 min. Immobility was defined as no movement except forrespiration. Subjects were returned to their home cage at the end of the test.
Forced-Swim Test.Mice were placed in clear glass beakers (18 cm in diameter)filled with 15 cm water (∼25 °C). Videotapes were scored for time spentimmobile over a 15-min period as described previously (42). Immobility wasdefined as the minimal amount of movement made by the mouse to stayafloat. Care was taken not to put the nose of the mouse below the waterlevel when initially placed in the water, and mice that appeared to be indistress were removed immediately. Subjects were returned to their homecage at the end of the test.
Social Defeat Stress. Suprathreshold defeat paradigm. Mice were subjected toa social defeat stress protocol adapted from ref. 43 and based on previouswork showing that chronic social defeat can induce depression-like endo-phenotypes (44). During each defeat episode, a C57BL/6J test mouse wasplaced in the home cage of an unfamiliar, aggressive CD1 mouse for 10 min,
Mineur et al. PNAS Early Edition | 5 of 6
NEU
ROSC
IENCE

during which time the C57BL/6J mouse displayed subordinate posturing. Forthe chronic social defeat paradigm, defeated mice were housed for 24 h withthe aggressive CD1 mouse separated only by a metal grid, and this process ofsocial defeat and cohousing was repeated daily for 10 consecutive days.Submaximal defeat paradigm. To test whether manipulations of AChE couldpotentiate an animal’s susceptibility to psychosocial stress induced by re-peated defeat, we also used the submaximal defeat paradigm as reportedpreviously (43). For the submaximal social defeat paradigm, C57BL/6J micewere subjected to three social defeat episodes in 1 d, and each defeat epi-sode was separated by 15 min of rest.
Before defeat episodes, CD1 mice (6–12 mo of age) were screened foraggressive behavior, and only CD1s with attack latency shorter than 1 minwere used in defeat episodes (45). Immediately after the 10th defeat of thechronic social defeat paradigm, or the third defeat of the submaximal socialdefeat paradigm, C57BL/6J mice were singly housed over night, and the nextday a social interaction test was performed in a different room between1000 and 1400 hours, unless stated otherwise.Social interaction.At the end of either the 10-d or the submaximal social defeatparadigm, approach–avoidance behavior toward an unfamiliar social targetwas measured as described previously (16). Experimental mice were placedwithin a novel white plastic open field (39 × 39 cm) in a dark environmentwith dimmed red light for two consecutive sessions of 2.5 min. In the firstsession (no target), the open field contained an empty metal grid cage (10 ×
6 cm) at one end of the arena. In the second session (target present), con-ditions were similar, but the metal grid cage contained an unfamiliar CD1mouse. In between the two sessions, the experimental mouse was returnedto its home cage for 1–2 min. Videotapes were scored for time spent by theexperimental mouse in the interaction zone (an 8-cm wide corridor aroundthe metal grid cage) and for time spent in the open-field corners oppositethe cage. The interaction score was calculated as 100 * (interaction time,target present)/(interaction time, no target).
Locomotor Activity. Mice were placed in a clean Plexiglas cage (48 × 22 × 18cm) for 20 min, and locomotor activity was recorded using the OptiMaxsystem (Columbus Instruments). Subjects were returned to their home cageat the end of the test.
Statistical Analysis. For comparisons of two groups, we used two-tailedStudent t test, and ANOVA (with “drug treatment” or “virus” and/or“stress” as between-subject factors) and the post hoc t test with Bonferronicorrections were used when relevant. For analysis of AChE mRNA levels aftershAChE infusions, we used a one-tailed Student t test. Values of P < 0.05were considered to be significant.
ACKNOWLEDGMENTS. This work was supported by National Institutes ofHealth Grants MH077681 and DA033945.
1. Saricicek A, et al. (2012) Persistent β2*-nicotinic acetylcholinergic receptor dysfunctionin major depressive disorder. Am J Psychiatry 169(8):851–859.
2. Furey ML, Drevets WC (2006) Antidepressant efficacy of the antimuscarinic drugscopolamine: A randomized, placebo-controlled clinical trial. Arch Gen Psychiatry63(10):1121–1129.
3. Janowsky DS (2011) Serendipity strikes again: Scopolamine as an antidepressantagent in bipolar depressed patients. Curr Psychiatry Rep 13(6):443–445.
4. Mineur YS, Picciotto MR (2010) Nicotine receptors and depression: Revisiting andrevising the cholinergic hypothesis. Trends Pharmacol Sci 31(12):580–586.
5. Janowsky DS, el-Yousef MK, Davis JM, Sekerke HJ (1972) A cholinergic-adrenergichypothesis of mania and depression. Lancet 2(7778):632–635.
6. Risch SC, et al. (1981) Physostigmine induction of depressive symptomatology innormal human subjects. Psychiatry Res 4(1):89–94.
7. Dilsaver SC, Peck JA, Overstreet DH (1992) The Flinders Sensitive Line exhibits en-hanced thermic responsiveness to nicotine relative to the Sprague-Dawley rat. Phar-macol Biochem Behav 41(1):23–27.
8. Caspi A, et al. (2003) Influence of life stress on depression: Moderation by a poly-morphism in the 5-HTT gene. Science 301(5631):386–389.
9. Meerson A, et al. (2010) Changes in brain MicroRNAs contribute to cholinergic stressreactions. J Mol Neurosci 40(1–2):47–55.
10. Hrabovska A, Duysen EG, Sanders JD, Murrin LC, Lockridge O (2005) Delivery of hu-man acetylcholinesterase by adeno-associated virus to the acetylcholinesteraseknockout mouse. Chem Biol Interact 157–158:71–78.
11. Camp S, et al. (2010) Contributions of selective knockout studies to understandingcholinesterase disposition and function. Chem Biol Interact 187(1–3):72–77.
12. Meshorer E, et al. (2002) Alternative splicing and neuritic mRNA translocation underlong-term neuronal hypersensitivity. Science 295(5554):508–512.
13. Sternfeld M, et al. (2000) Excess “read-through” acetylcholinesterase attenuates butthe “synaptic” variant intensifies neurodeterioration correlates. Proc Natl Acad SciUSA 97(15):8647–8652.
14. Salas R, et al. (2008) Nicotine relieves anxiogenic-like behavior in mice that over-express the read-through variant of acetylcholinesterase but not in wild-type mice.Mol Pharmacol 74(6):1641–1648.
15. Cryan JF, Mombereau C, Vassout A (2005) The tail suspension test as a model forassessing antidepressant activity: Review of pharmacological and genetic studies inmice. Neurosci Biobehav Rev 29(4–5):571–625.
16. Berton O, et al. (2006) Essential role of BDNF in the mesolimbic dopamine pathway insocial defeat stress. Science 311(5762):864–868.
17. Vialou V, et al. (2010) DeltaFosB in brain reward circuits mediates resilience to stressand antidepressant responses. Nat Neurosci 13(6):745–752.
18. Krishnan V, Nestler EJ (2010) Linking molecules to mood: New insight into the biologyof depression. Am J Psychiatry 167(11):1305–1320.
19. Picciotto MR, Higley MJ, Mineur YS (2012) Acetylcholine as a neuromodulator: Cho-linergic signaling shapes nervous system function and behavior. Neuron 76(1):116–129.
20. Mark GP, Rada PV, Shors TJ (1996) Inescapable stress enhances extracellular acetyl-choline in the rat hippocampus and prefrontal cortex but not the nucleus accumbensor amygdala. Neuroscience 74(3):767–774.
21. Sarter M, Parikh V, Howe WM (2009) Phasic acetylcholine release and the volumetransmission hypothesis: Time to move on. Nat Rev Neurosci 10(5):383–390.
22. George TP, Sacco KA, Vessicchio JC, Weinberger AH, Shytle RD (2008) Nicotinic an-tagonist augmentation of selective serotonin reuptake inhibitor-refractory majordepressive disorder: A preliminary study. J Clin Psychopharmacol 28(3):340–344.
23. Mineur YS, Somenzi O, Picciotto MR (2007) Cytisine, a partial agonist of high-affinitynicotinic acetylcholine receptors, has antidepressant-like properties in male C57BL/6Jmice. Neuropharmacology 52(5):1256–1262.
24. Kaufer D, Friedman A, Seidman S, Soreq H (1999) Anticholinesterases induce multi-genic transcriptional feedback response suppressing cholinergic neurotransmission.Chem Biol Interact 119–120:349–360.
25. Mesulam MM (1995) Cholinergic pathways and the ascending reticular activatingsystem of the human brain. Ann N Y Acad Sci 757:169–179.
26. Warner-Schmidt JL, et al. (2012) Cholinergic interneurons in the nucleus accumbensregulate depression-like behavior. Proc Natl Acad Sci USA 109(28):11360–11365.
27. Kaufer D, Friedman A, Seidman S, Soreq H (1998) Acute stress facilitates long-lastingchanges in cholinergic gene expression. Nature 393(6683):373–377.
28. Gilad GM (1987) The stress-induced response of the septo-hippocampal cholinergicsystem. A vectorial outcome of psychoneuroendocrinological interactions. Psycho-neuroendocrinology 12(3):167–184.
29. Cobb SR, Davies CH (2005) Cholinergic modulation of hippocampal cells and circuits. JPhysiol 562(Pt 1):81–88.
30. Soreq H, Seidman S (2001) Acetylcholinesterase: New roles for an old actor. Nat RevNeurosci 2(4):294–302.
31. Shohami E, et al. (2000) Antisense prevention of neuronal damages following headinjury in mice. J Mol Med (Berl) 78(4):228–236.
32. Smythe JW, Bhatnagar S, Murphy D, Timothy C, Costall B (1998) The effects of in-trahippocampal scopolamine infusions on anxiety in rats as measured by the black-white box test. Brain Res Bull 45(1):89–93.
33. Martinowich K, et al. (2012) Roles of p75(NTR), long-term depression, and cholinergictransmission in anxiety and acute stress coping. Biol Psychiatry 71(1):75–83.
34. Overstreet DH (1993) The Flinders sensitive line rats: A genetic animal model of de-pression. Neurosci Biobehav Rev 17(1):51–68.
35. Hasey G, Hanin I (1991) The cholinergic-adrenergic hypothesis of depression re-examined using clonidine, metoprolol, and physostigmine in an animal model. BiolPsychiatry 29(2):127–138.
36. File SE, Kenny PJ, Cheeta S (2000) The role of the dorsal hippocampal serotonergic andcholinergic systems in themodulation of anxiety. Pharmacol BiochemBehav 66(1):65–72.
37. Tucci S, Cheeta S, Seth P, File SE (2003) Corticotropin releasing factor antagonist, al-pha-helical CRF(9-41), reverses nicotine-induced conditioned, but not unconditioned,anxiety. Psychopharmacology (Berl) 167(3):251–256.
38. File SE, Gonzalez LE, Andrews N (1998) Endogenous acetylcholine in the dorsal hip-pocampus reduces anxiety through actions on nicotinic and muscarinic1 receptors.Behav Neurosci 112(2):352–359.
39. Andrä J, Lojda Z (1986) A histochemical method for the demonstration of acetylcho-linesterase activity using semipermeable membranes.Histochemistry 84(4–6):575–579.
40. Karnovsky MJ, Roots L (1964) A “direct-coloring” thiocholine method for chol-inesterases. J Histochem Cytochem 12:219–221.
41. Mineur YS, et al. (2011) Nicotine decreases food intake through activation of POMCneurons. Science 332(6035):1330–1332.
42. Mineur YS, et al. (2011) α4β2 nicotinic acetylcholine receptor partial agonists with lowintrinsic efficacy have antidepressant-like properties. Behav Pharmacol 22(4):291–299.
43. Krishnan V, et al. (2007) Molecular adaptations underlying susceptibility and re-sistance to social defeat in brain reward regions. Cell 131(2):391–404.
44. Kudryavtseva NN, Bakshtanovskaya IV, Koryakina LA (1991) Social model of de-pression in mice of C57BL/6J strain. Pharmacol Biochem Behav 38(2):315–320.
45. Wilkinson MB, et al. (2011) A novel role of the WNT-dishevelled-GSK3β signalingcascade in the mouse nucleus accumbens in a social defeat model of depression.J Neurosci 31(25):9084–9092.
6 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1219731110 Mineur et al.

Supporting InformationMineur et al. 10.1073/pnas.1219731110SI MethodsMeasurement of Acetylcholinesterase Activity. Brain sections werepreincubated in 10 mL medium [7.4 mL 0.1 M Tris-maleatebuffer, pH 5.1; 1.0 mL 0.4 M sodium citrate; 1.0 mL 0.12 Mcopper sulfate; 0.5 mL 0.16M potassium ferricyanide; and 0.1 mL10–3 M iso-tetraisopropylpyrophosphoramide (CAS#: 513-00-8;OMPA)] for 20 min at room temperature. Sections were thenincubated in the same medium containing 2 mM acetylthiocholinefor 1 h at 37 °C. Brain sections were rinsed in medium, mounted onslides, and coverslipped. Specificity of acetylcholinesterase (AChE)measurements were confirmed by including inhibitors of AChE[either 40 μM A9013 (Sigma-Aldrich) or 10 μM physostigmine] orby omitting acetylthiocholine from the incubation medium.
shRNA Design. Three sequences targeting AChE were chosen inthe consensus coding region of the mRNA encoding the differentisoforms of AChE (GenBank accession NM_009599.3) usingmethods described previously (1): 5′–3′—GAGTTGAGTGAA-GACTGCCTGTAT; GAGCTGATAGCCTGCTTGAGGACA;and GAGCCTGAACCTGAAGCCCTTAGA).For the scrambled shRNA, we selected a random sequence of
24 bases with no similarity to any known mRNA. Synthetic oli-gonucleotide duplexes were designed by adding antisensesequences directed against the selected mRNA region, followedby a miR23 loop of 10 nucleotides (CTTCCTGTCA) to the 5′ endof the sequences above and by adding overhanging ends identicalto those created by SapI and XbaI restriction enzyme digestion.Annealed oligonucleotides were ligated into a pAAV-EGFP-shRNA vector as described previously (1), and positive cloneswere verified by sequencing. In vitro efficacy of AChE knock-down was assessed in N2a cells (see below).
In Vitro Efficacy of AChE Knockdown in N2a Cells. N2a cells weretransfected with plasmids carrying AChE-shRNA-1, -2, or -3 aloneor in combination. Five days after transfection, AChE activity incell homogenates was measured as described for brain sectionsafter rapid fixation with paraformaldehyde (PFA). Cells trans-fected with shRNA-AChE plasmids showed greatly reducedAChE activity (Fig. S4), verifying that shRNA-mediated AChEknockdown was effective. A 1:1 mixture of AChE shRNA-2 and -3induced a significant decrease in AChE activity and was selectedfor subsequent knockdown experiments in vivo. A human ACHEcDNA (refered to as “hAChE” in the main text) was kindlyprovided by Oksana Lockridge (University of Nebraska MedicalCenter, Omaha, NE) (2).
AAV Production. Viral production was achieved by triple trans-fection with 135 μg each of pAAV2-shRNA, pHelper, andpAAV-RC plasmids using calcium phosphate in HEK 293 cells(3). Cells were harvested 72 h after transfection, resuspended infreezing buffer (0.15 M NaCl and 50 mM Tris, pH 8.0), subjectedto three freeze/thaw cycles, and treated with 50 U/mL of ben-zonase for 30 min at 37 °C. After centrifugation (3,700 × g for20 min), the supernatant was carefully added to a centrifuge tubecontaining a 15%, 25%, 40%, and 60% (vol/vol) iodixanol stepgradient. The iodixanol gradient was centrifuged (50,000 × g for200 min at 10 °C) before the 40% fraction was carefully col-lected, diluted in PBS-MK (1× PBS, 1 mM MgCl2, 2.5 mM KCl),and concentrated and purified with a Centricon Plus-20 (100K)filter device. Purified viruses were stored at 4 °C.
Stereotaxic Surgery.C57BL/6J mice (10–12 wk old) were placed ina stereotaxic frame (Kopf Instruments) under isoflurane anes-thesia. For surgeries requiring cannulation (pharmacologicalinfusion and rescue experiments), a double-guide cannula (3-mmpedestal; designed to receive cannulas of gauge 33; Plastic One)was implanted targeting the hippocampus (from bregma: −2.5 mmanteroposterior, ±1.5 mm lateral, 2.7 mm dorsoventral). Aftercannulation, mice were allowed to recover for 1 wk before infusion.For infusion of adenoassociated virus (AAV) constructs, pu-
rified high-titer AAVs were injected bilaterally into the hippo-campus (from bregma: −2.5 mm anteroposterior, ±1.5 mmlateral, 2.7 mm dorsoventral) over 15 min using a 26 gauge blunt-tipped Hamilton syringe. For AChE knockdown experiments,injection volumes were 1 μL of AAV-shRNA-AChE (shAChE)or AAV-shRNA-Scrambled (Scr). For hAChE expression ex-periments, infusion volumes were 1.5 μL of AAV-hAChE orAAV-EGFP (control). To reduce endogenous AChE mRNAlevels while expressing knockdown-resistant hAChE, we injected2 μL of a 1:3 mixture of AAV-hAChE and shAChE or 2 μL ofa 1:3 mixture of AAV-EGFP and Scr. Infusion speed was ∼0.2μL/min, and the needle was kept in place for an additional 5 minbefore it was slowly withdrawn. Following stereotaxic surgery,mice were allowed to recover for at least 3–5 wk before behav-ioral assays to allow for expression or knockdown of AChE.
Quantitative Real-Time PCR for in Vivo Knockdown. Quantitation ofmRNA was performed as described previously (4). Followingviral infusions, mice were decapitated and the hippocampus wasquickly removed before total RNA was extracted using theRNeasy Lipid tissue kit (Qiagen). Primer design, reverse tran-scription, and quantitative real-time PCR (qPCR) was per-formed exactly as described previously (1). β-Glucuronidase(Gusb) was used to normalize across samples, and amplificationproducts were quantified using the ΔΔCt method. Primer se-quences were as follows: AChE (NM_009599.3)—forward, GC-AATGACACCGAGCTGATA; reverse, CTACCACAGGCAC-GAAGGA; Gusb (NM_010368.1)—forward, AACCTCTGGT-GGCCTTACCT; reverse, TCCCGATAGGAAGGGTGTAG;EGFP (EU056362.1)—forward, CGGCATCAAGGTGAACTT;reverse, TCACGAACTCCAGCAGGAC.Only samples with detectable EGFP expression were included
in the analysis. qPCR identified a significant reduction in AChEmRNA levels in hippocampus of AAV-shAChE–injected com-pared with AAV-Scr–injected mice [T(9) = 1.89, P < 0.05], whichcould be observed visually in brain slices (Fig. 3) . In contrast, nogross morphological differences were observed between neuronsinfected with AAV and controls.
Validation of AAV Infusion Site. Mice were anesthetized withchloral hydrate and perfused intracardially with PBS (∼75 mL)followed by 4% (wt/vol) PFA in PBS (∼75 mL) for 5 min each.Brains were then removed, postfixed for 24 h in 4% PFA at 4 °C,then placed in 30% (wt/vol) sucrose in PBS for cryoprotection,and stored at 4 °C. Brain sections (40 μm) were cut witha sliding microtome and placed in PBS before injection siteswere examined by visual confirmation of EGFP expression usinga Nikon fluorescence microscope.
Mineur et al. www.pnas.org/cgi/content/short/1219731110 1 of 5

1. Mineur YS, et al. (2011) Nicotine decreases food intake through activation of POMCneurons. Science 332(6035):1330–1332.
2. Hrabovska A, Duysen EG, Sanders JD, Murrin LC, Lockridge O (2005) Delivery of humanacetylcholinesterase by adeno-associated virus to the acetylcholinesterase knockoutmouse. Chem Biol Interact 157–158:71–78.
3. Hommel JD, Sears RM, Georgescu D, Simmons DL, DiLeone RJ (2003) Local geneknockdown in the brain using viral-mediated RNA interference.NatMed 9(12):1539–1544.
4. Wigestrand MB, et al. (2011) Decreased α4β2 nicotinic receptor number in the absenceof mRNA changes suggests post-transcriptional regulation in the spontaneouslyhypertensive rat model of ADHD. J Neurochem 119(1):240–250.
Fig. S1. AChE activity measured in brain slices following acute physostigmine administration. The y axis represents integrated density of the colorimetricreaction used to measure AChE activity. n = 8–10 per group. Data are expressed as mean ± SEM. *P < 0.05; ***P < 0.001
Mineur et al. www.pnas.org/cgi/content/short/1219731110 2 of 5

Fig. S2. AChE activity measured in brain slices following 2 wk of chronic treatment with fluoxetine. The y axis represents integrated density of the colorimetricreaction used to measure AChE activity. n = 8–10 per group. Data are expressed as mean ± SEM. **P < 0.01.
Fig. S3. AChE activity measured in brain slices following 2 wk of chronic treatment with physostigmine. The y axis represents integrated density of thecolorimetric reaction used to measure AChE activity. n = 8–10 per group. Data are expressed as mean ± SEM. *P < 0.05.
Mineur et al. www.pnas.org/cgi/content/short/1219731110 3 of 5

Fig. S4. In vitro knockdown of AChE in N2a cells. Representative histochemical staining of AChE activity in N2A cells (mouse neuroblastoma) showing reducedAChE activity in cells transfected with plasmids expressing shRNA-AChE-GFP. Three days after transfection, cells were evaluated with the same colorometricassay used to detect AChE activity in tissue (observed as black staining). The merged picture shows that the cells transfected with the shRNA construct (green)had decreased AChE activity compared with nontransfected cells. Insets highlight the decreased AChE activity in a transfected cell (Inset 1), relative toa nontransfected cell (Inset 2).
Mineur et al. www.pnas.org/cgi/content/short/1219731110 4 of 5
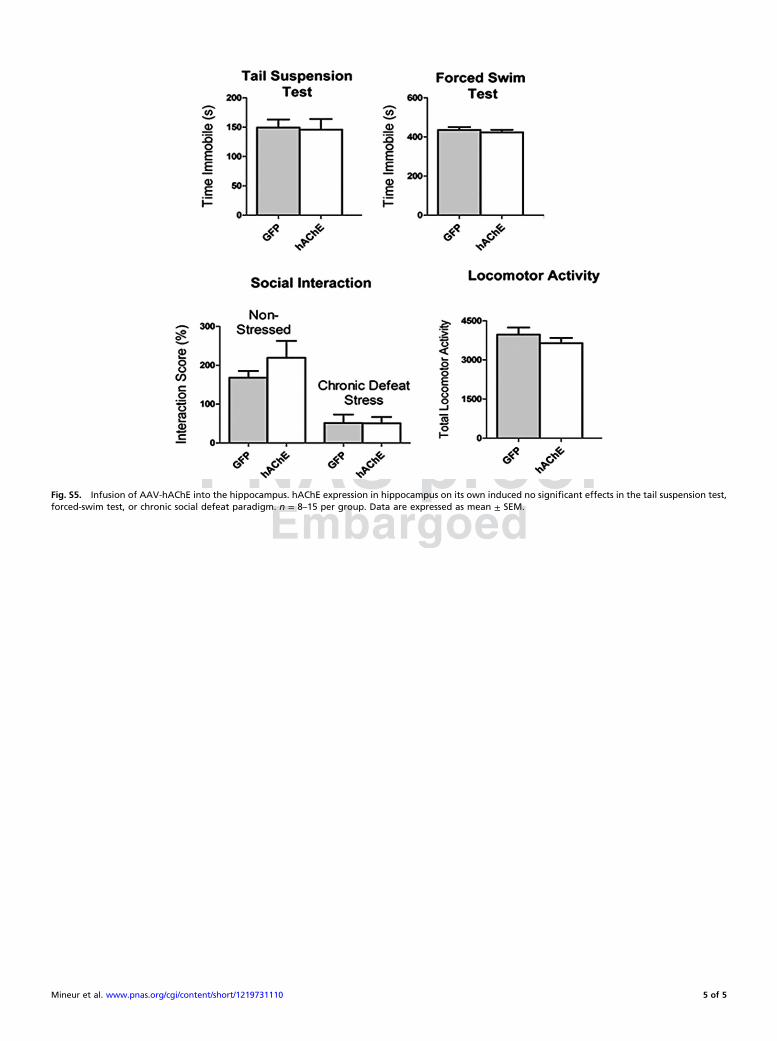
Fig. S5. Infusion of AAV-hAChE into the hippocampus. hAChE expression in hippocampus on its own induced no significant effects in the tail suspension test,forced-swim test, or chronic social defeat paradigm. n = 8–15 per group. Data are expressed as mean ± SEM.
Mineur et al. www.pnas.org/cgi/content/short/1219731110 5 of 5




















