
Charge storage mechanism of sonochemically prepared MnO 2 as supercapacitor electrode: Effects of...
8
Available online at www.sciencedirect.com Electrochimica Acta 53 (2008) 4607–4614 Charge storage mechanism of sonochemically prepared MnO 2 as supercapacitor electrode: Effects of physisorbed water and proton conduction M. Ghaemi a,∗ , F. Ataherian b , A. Zolfaghari c , S.M. Jafari d a Department of Chemistry, Science Faculty, Golestan University, P.O. Box 49138-15739, Gorgan, Iran b Department of Chemistry, School of Sciences, Tarbiat Modares University, P.O. Box 14115-175, Tehran, Iran c Chemistry and Chemical Engineering Research Center of Iran, Iran d Department of Mathematics, Science Faculty, Golestan University, P.O. Box 49138-15739, Gorgan, Iran Received 26 September 2007; received in revised form 18 November 2007; accepted 13 December 2007 Available online 23 December 2007 Abstract The charge storage mechanism of nanostructured hydrated manganese dioxide, as a supercapacitor electrode, was investigated with respect to the role of amount of hydrates on the electrolyte cations diffusion. The MnO 2 materials (- and layered types), prepared by a novel ultrasonic aided procedure. Thermal gravimetric analysis (TGA), differential scanning calorimetry (DSC) and Fourier transform infrared (FT-IR) spectroscopy were employed to characterize the water content of the samples. The samples then were heat-treated in air for 2 h at 70, 100 and 150 ◦ C to prepare different - and L-series of electrodes with various amounts of hydrates. To determine the role of the protonic conduction on the charge storage mechanism, the electrochemical properties of the electrodes were investigated in two different electrolyte pH values of 3.3 and 6. Compared to 25, the higher specific capacitance of L25, especially in more acidic electrolytes, is attributed to the higher amount of physically adsorbed water molecules and their contribution in diffusion process. Furthermore, it is clearly demonstrated that the total electrochemical performance of the systems under consideration is also influenced by the crystalline structure of the prepared electrodes, especially when the size of the tunnels limits the intercalation of cations. Analyzing the results of cyclic voltammetry and electrochemical impedance spectroscopy for both series of the electrodes, revealed that, increasing the heat-treatment temperature makes the charge-transfer resistance increase and the Warburg impedance decrease. This effect can be attributed to the more amount of surface physisorbed water lost by the higher heat-treatment temperatures. © 2007 Elsevier Ltd. All rights reserved. Keywords: Manganese dioxide; Supercapacitor; Charge storage mechanism; Physisorbed water; Cation diffusion 1. Introduction During the last few years, electrochemical capacitors are extensively studied as a new kind of accumulators for use in devices that require peak power pulses [1]. Among those, redox pseudocapacitors have shown excellent capacity, due to occur- ring the redox-type reactions not only at the surface but also inside the electrode [2,3]. Recently, manganese oxide electrode has been extensively used as an electrode material in the redox pseudocapacitor because of abundance, low cost and environmental friendliness ∗ Corresponding author. Tel.: +98 171 4427173; fax: +98 171 4427040. E-mail address: Ghaemi [email protected] (M. Ghaemi). of manganese. Literature survey reveals that the electrochem- ical properties of MnO 2 samples are highly influenced by the synthetic methods applied. Various procedures such as ther- mal decomposition [4], co-precipitation [5], sol–gel process [7,8], electrochemical deposition [9,10], solution-based chem- ical routes [11] and solid state reaction route [12] have been introduced and characterized in this respect. The specific capac- itance of the manganese oxides, prepared by the above addressed methods, generally ranges between 150 and 400 Fg −1 [4–12]. In our previous work [13], a novel ultrasonic aided proce- dure was introduced to prepare excellent capacitance manganese dioxide with different crystalline structures: -MnO 2 (labeled as 25) and a rather layered structure (labeled as L25). These crys- talline structures of manganese dioxide have been extensively described by Brousse et al. [14]. 0013-4686/$ – see front matter © 2007 Elsevier Ltd. All rights reserved. doi:10.1016/j.electacta.2007.12.040
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Charge storage mechanism of sonochemically prepared MnO 2 as supercapacitor electrode: Effects of...

A
tpwdm
apo
ia©
K
1
edpri
ub
0d
Available online at www.sciencedirect.com
Electrochimica Acta 53 (2008) 4607–4614
Charge storage mechanism of sonochemically prepared MnO2 assupercapacitor electrode: Effects of physisorbed water
and proton conduction
M. Ghaemi a,∗, F. Ataherian b, A. Zolfaghari c, S.M. Jafari d
a Department of Chemistry, Science Faculty, Golestan University, P.O. Box 49138-15739, Gorgan, Iranb Department of Chemistry, School of Sciences, Tarbiat Modares University, P.O. Box 14115-175, Tehran, Iran
c Chemistry and Chemical Engineering Research Center of Iran, Irand Department of Mathematics, Science Faculty, Golestan University, P.O. Box 49138-15739, Gorgan, Iran
Received 26 September 2007; received in revised form 18 November 2007; accepted 13 December 2007Available online 23 December 2007
bstract
The charge storage mechanism of nanostructured hydrated manganese dioxide, as a supercapacitor electrode, was investigated with respect tohe role of amount of hydrates on the electrolyte cations diffusion. The MnO2 materials (�- and layered types), prepared by a novel ultrasonic aidedrocedure. Thermal gravimetric analysis (TGA), differential scanning calorimetry (DSC) and Fourier transform infrared (FT-IR) spectroscopyere employed to characterize the water content of the samples. The samples then were heat-treated in air for 2 h at 70, 100 and 150 ◦C to prepareifferent �- and L-series of electrodes with various amounts of hydrates. To determine the role of the protonic conduction on the charge storageechanism, the electrochemical properties of the electrodes were investigated in two different electrolyte pH values of 3.3 and 6.Compared to �25, the higher specific capacitance of L25, especially in more acidic electrolytes, is attributed to the higher amount of physically
dsorbed water molecules and their contribution in diffusion process. Furthermore, it is clearly demonstrated that the total electrochemicalerformance of the systems under consideration is also influenced by the crystalline structure of the prepared electrodes, especially when the size
f the tunnels limits the intercalation of cations.Analyzing the results of cyclic voltammetry and electrochemical impedance spectroscopy for both series of the electrodes, revealed that,ncreasing the heat-treatment temperature makes the charge-transfer resistance increase and the Warburg impedance decrease. This effect can bettributed to the more amount of surface physisorbed water lost by the higher heat-treatment temperatures.
2007 Elsevier Ltd. All rights reserved.
ysiso
oism[iii
eywords: Manganese dioxide; Supercapacitor; Charge storage mechanism; Ph
. Introduction
During the last few years, electrochemical capacitors arextensively studied as a new kind of accumulators for use inevices that require peak power pulses [1]. Among those, redoxseudocapacitors have shown excellent capacity, due to occur-ing the redox-type reactions not only at the surface but alsonside the electrode [2,3].
Recently, manganese oxide electrode has been extensivelysed as an electrode material in the redox pseudocapacitorecause of abundance, low cost and environmental friendliness
∗ Corresponding author. Tel.: +98 171 4427173; fax: +98 171 4427040.E-mail address: Ghaemi [email protected] (M. Ghaemi).
m
dd�td
013-4686/$ – see front matter © 2007 Elsevier Ltd. All rights reserved.oi:10.1016/j.electacta.2007.12.040
rbed water; Cation diffusion
f manganese. Literature survey reveals that the electrochem-cal properties of MnO2 samples are highly influenced by theynthetic methods applied. Various procedures such as ther-al decomposition [4], co-precipitation [5], sol–gel process
7,8], electrochemical deposition [9,10], solution-based chem-cal routes [11] and solid state reaction route [12] have beenntroduced and characterized in this respect. The specific capac-tance of the manganese oxides, prepared by the above addressed
ethods, generally ranges between 150 and 400 Fg−1 [4–12].In our previous work [13], a novel ultrasonic aided proce-
ure was introduced to prepare excellent capacitance manganese
ioxide with different crystalline structures: �-MnO2 (labeled as25) and a rather layered structure (labeled as L25). These crys-alline structures of manganese dioxide have been extensivelyescribed by Brousse et al. [14].

4 imica
ug
o
cNaIttfb
gprfHp
sFpw
2
pseiMtts
bgu[[
mQiaCB
ouTsL(gti
pm8m(sMiTpiItcgwmlgsm
ucAKg
TS
S
�
L
608 M. Ghaemi et al. / Electroch
Many excellent investigations have provided the fundamentalnderstanding of the charge storage mechanism of the man-anese oxides as supercapacitor electrode [6,7,15,16].
The proposed charge/discharge cycles of hydrous manganesexide for capacitance measurements is as following [6,7]:
MnOa(OH)b + δM+ + δe− ↔ MnOa−δ(OM)b+δ
M = H or alkaline ion(1)
Recently, Belanger and co-workers [15] have reported theharge storage mechanism of a MnO2 electrode in 0.1 Ma2SO4 electrolyte by X-ray photoelectron spectroscopy (XPS)
nd showed that the manganese oxidation state varies from III toV for the reduced and oxidized states of the electrodes duringhe charging/discharging process, respectively. They deducedhat the pseudocapacitive behaviors of MnO2 thin films resultedrom the reversible insertion/extraction of electrolyte cations toalance the charge during reduction/oxidation of Mn3+/Mn4+.
It has been well recognized that the water content of man-anese dioxide is one of the key factors in the electrochemicalerformance of MnO2 [10,17,18]. Fundamentally, the hydrousegions in the electrode provide the kinetically facile sites neededor the charge-transfer reaction and cation diffusion [19,20].owever, the impact of the physisorbed water on the capacitiveroperties has not been well documented.
In this study, the charge storage mechanism of the preparedamples was explored as a function of the physisorbed water.urthermore, the influences of the crystalline structures of therepared electrodes on the total electrochemical performanceere considered as well.
. Experimental
The manganese dioxide samples of the present study wererepared by a novel chemical oxidation technique using ultra-onic irradiation. The detailed preparation procedure wasxpensively described in our previous work [13]. As briefly listedn Table 1, two different concentrations of aqueous solutions of
nSO4 and KBrO3 (both from Merck, research grade), by theotal volume of 200 mL, were irradiated by ultrasonic radia-ion at constant temperature of 40 ◦C. The obtained as-preparedamples were labeled as �25 and L25.
The total amount of potassium in the samples was determinedy flame photometry (Schrown 410) and was found to be negli-
ible. The chemical composition of the samples was determinedsing the potentiometric titration approach of Vetter and Jaeger21], based on the cation vacancy model proposed by Ruetschi22]. The specific surface area and pore size distribution werewet(
able 1ynthesis condition, results of BET measurements and chemical composition analysi
ample Synthesis condition Mn1−x−y+4 Mny
+3
[MnSO4] (M) [KBrO3] (M)
25 0.125 0.250 Mn0.838+4 Mn0.01
25 0.250 0.500 Mn0.762+4 Mn0.06
Acta 53 (2008) 4607–4614
easured by Brunauer–Emmett–Teller (BET) analysis using auantasorb, Quantachrom apparatus. The results are presented
n Table 1. Thermal Gravimetric Analysis was carried out usingTGA Q50 V6.3 Build 189 analyzer. Differential Scanningalorimetry results were obtained by using a DSC Q100 V9.0uild 275 analyzer.
To prepare hydrous manganese oxide with different amountf water contents, the both prepared samples were heat treatednder the same conditions at 70, 100, and 150 ◦C for 2 h in air.he resulting samples were labeled as �70, �100 and �150. Theame procedure was applied for L25 to obtain L70, L100 and150. The crystallographic structures and FT-IR experiments
between 400 and 4000 Cm−1) of all the samples were investi-ated using a diffractometer (Philips PC-APD with a Cu anodehat generated Cu K� radiation, λ = 1.5406 A) and a Shimadzunfrared apparatus, respectively.
For electrochemical measurements, the samples were pre-ared as electrodes by mixing 60 wt.% MnO2 powder as activeaterial, 10 wt.% acetylene black (Alfa Aesar, >99.9%, S.A.
0 m2/g), 25 wt.% graphite (Alfa Aesar, conducting grade, 200esh), and 5 wt.% poly (tetrafluoroethylene) dried powder
Merck). Graphite improves the conductivity of the overallystem, covering the high resistance of manganese dioxide.
oreover, due to its platelet structure and lubricating properties,t may help the prepared films to be more stable mechanically.he main role of acetylene black is to increase the mass trans-ort through the film, adding additional mesopores. However,t results in better electronic conductivity of the film as well.t is worth nothing that the contribution of these additives tohe total electrochemical properties is negligible. The three firstonstituents were primarily mixed together to obtain a homo-eneous black powder. The poly (tetrafluoroethylene) powderas then added with a few drops of ethanol to this composite toake a more homogeneous mixture. This resulted in a rubber-
ike paste that was rolled into a film (100–200 �m thick) on a flatlass surface. A piece of film (1 cm2) was cut and pressed ontotainless steel grid current collector. The weight of the activeaterial in all prepared electrodes equals to 3 mg.An Autolab, Eco Chemie. B.V. potentiostat/galvanostat was
sed for electrochemical measurements. A beaker type electro-hemical cell equipped with a MnO2-based working electrode,g/AgCl reference electrode (Metrohm AG 9101 Herisau, 3 MCl, 0.207 V versus SHE at 25 ◦C) and a 1 cm2 stainless palatinerid counter electrode was used. A Luggin capillary faced the
orking electrode at a distance of 2 mm, was used to minimizerrors due to iR drop in the electrolyte. The solution tempera-ure was maintained at 25 ◦C by means of a water thermostatRE 104 Ecoline, LAUDA).
s of initial samples
On−4x−y2− OH4x−y
− SBET (m2 g−1) Dav (A)
7+3 O1.403
2− OH0.597− 301 46.0
6+3 O1.246
2− OH0.754− 161 69.1

imica Acta 53 (2008) 4607–4614 4609
tewtwsMif
3
3d
emoA
M. Ghaemi et al. / Electroch
It has been well demonstrated, in our previous report [13], thathe proton mobility has a significant role on the capacitive prop-rties. To consider this effect, the electrochemical measurementsere performed in aqueous electrolyte of 0.5 M Na2SO4 solu-
ion with two different pH values of 3.3 and 6. The electrolytesere degassed with purified argon before the measurement. The
pecific capacitance is reported in unit of Farads per mass ofnO2 (Fg−1 MnO2). Impedance measurements were conducted
n a constant voltage mode (0.5 V versus Ag/AgCl) by sweepingrequencies from 10 mHz to100 kHz.
. Results and discussion
.1. Influence of heat treatment on IR pattern and X-rayiffraction
The FT-IR spectra of all the samples are shown in Fig. 1. Gen-−1
rally, bands in the region between 400 and 800 cm , whichay reveal information about manganese oxide lattice (MnO6ctahedral), can be assigned to Mn O stretching vibrations [23].s deduced from Fig. 1, the bands of the samples in this region
Fig. 1. IR spectra of (a) �-series and (b) L-series.
Fig. 2. XRD patterns of MnO2 powder of as-prepared materials �25 andLL
atacovam
wMdtl
25 and after thermal treatment for 2 h at 70–150 ◦C, (a) �-series and (b)-series.
re not affected by the heat-treatment temperature. The vibra-ions in the interval 1400–1450 and 1630–1670 cm−1 can bessigned to bending vibrations of OH group of adsorbed and/orrystal water molecules [23]. It should be noticed that IR spectraf both sample series exhibit a slight red shift in OH bendingibrations by increasing the heat-treatment temperature which isrisen from decreasing the abundance of loosely adsorbed waterolecules.Fig. 2 presents the X-ray patterns of the samples. Compared
ith the as-prepared samples, the patterns of the heat-treated
nO2 powders do not show significant changes. The broad andiffused peak in the XRD patterns of �-series indicates thathe oxide has a low level of crystalline due to the absence ofong–range order of the MnO6 octahedral framework (Fig. 2a).
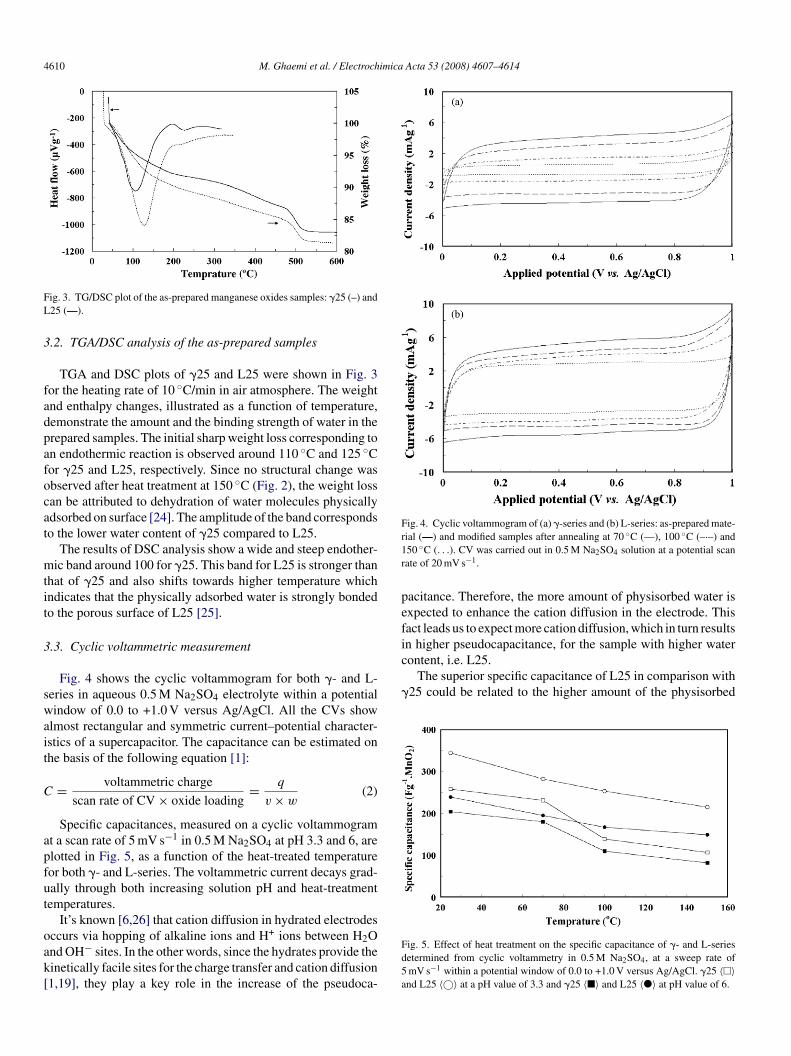
4610 M. Ghaemi et al. / Electrochimica Acta 53 (2008) 4607–4614
FL
3
fadpafocat
mtit
3
swait
C
apfut
oak[
Fig. 4. Cyclic voltammogram of (a) �-series and (b) L-series: as-prepared mate-r1r
peficontent, i.e. L25.
The superior specific capacitance of L25 in comparison with�25 could be related to the higher amount of the physisorbed
ig. 3. TG/DSC plot of the as-prepared manganese oxides samples: �25 (–) and25 (—).
.2. TGA/DSC analysis of the as-prepared samples
TGA and DSC plots of �25 and L25 were shown in Fig. 3or the heating rate of 10 ◦C/min in air atmosphere. The weightnd enthalpy changes, illustrated as a function of temperature,emonstrate the amount and the binding strength of water in therepared samples. The initial sharp weight loss corresponding ton endothermic reaction is observed around 110 ◦C and 125 ◦Cor �25 and L25, respectively. Since no structural change wasbserved after heat treatment at 150 ◦C (Fig. 2), the weight lossan be attributed to dehydration of water molecules physicallydsorbed on surface [24]. The amplitude of the band correspondso the lower water content of �25 compared to L25.
The results of DSC analysis show a wide and steep endother-ic band around 100 for �25. This band for L25 is stronger than
hat of �25 and also shifts towards higher temperature whichndicates that the physically adsorbed water is strongly bondedo the porous surface of L25 [25].
.3. Cyclic voltammetric measurement
Fig. 4 shows the cyclic voltammogram for both �- and L-eries in aqueous 0.5 M Na2SO4 electrolyte within a potentialindow of 0.0 to +1.0 V versus Ag/AgCl. All the CVs show
lmost rectangular and symmetric current–potential character-stics of a supercapacitor. The capacitance can be estimated onhe basis of the following equation [1]:
= voltammetric charge
scan rate of CV × oxide loading= q
v × w(2)
Specific capacitances, measured on a cyclic voltammogramt a scan rate of 5 mV s−1 in 0.5 M Na2SO4 at pH 3.3 and 6, arelotted in Fig. 5, as a function of the heat-treated temperatureor both �- and L-series. The voltammetric current decays grad-ally through both increasing solution pH and heat-treatmentemperatures.
It’s known [6,26] that cation diffusion in hydrated electrodes
ccurs via hopping of alkaline ions and H+ ions between H2Ond OH− sites. In the other words, since the hydrates provide theinetically facile sites for the charge transfer and cation diffusion1,19], they play a key role in the increase of the pseudoca-Fd5a
ial (—) and modified samples after annealing at 70 ◦C (—), 100 ◦C (–·–) and50 ◦C (. . .). CV was carried out in 0.5 M Na2SO4 solution at a potential scanate of 20 mV s−1.
acitance. Therefore, the more amount of physisorbed water isxpected to enhance the cation diffusion in the electrode. Thisact leads us to expect more cation diffusion, which in turn resultsn higher pseudocapacitance, for the sample with higher water
ig. 5. Effect of heat treatment on the specific capacitance of �- and L-seriesetermined from cyclic voltammetry in 0.5 M Na2SO4, at a sweep rate ofmV s−1 within a potential window of 0.0 to +1.0 V versus Ag/AgCl. �25 〈�〉nd L25 〈©〉 at a pH value of 3.3 and �25 〈�〉 and L25 〈�〉 at pH value of 6.
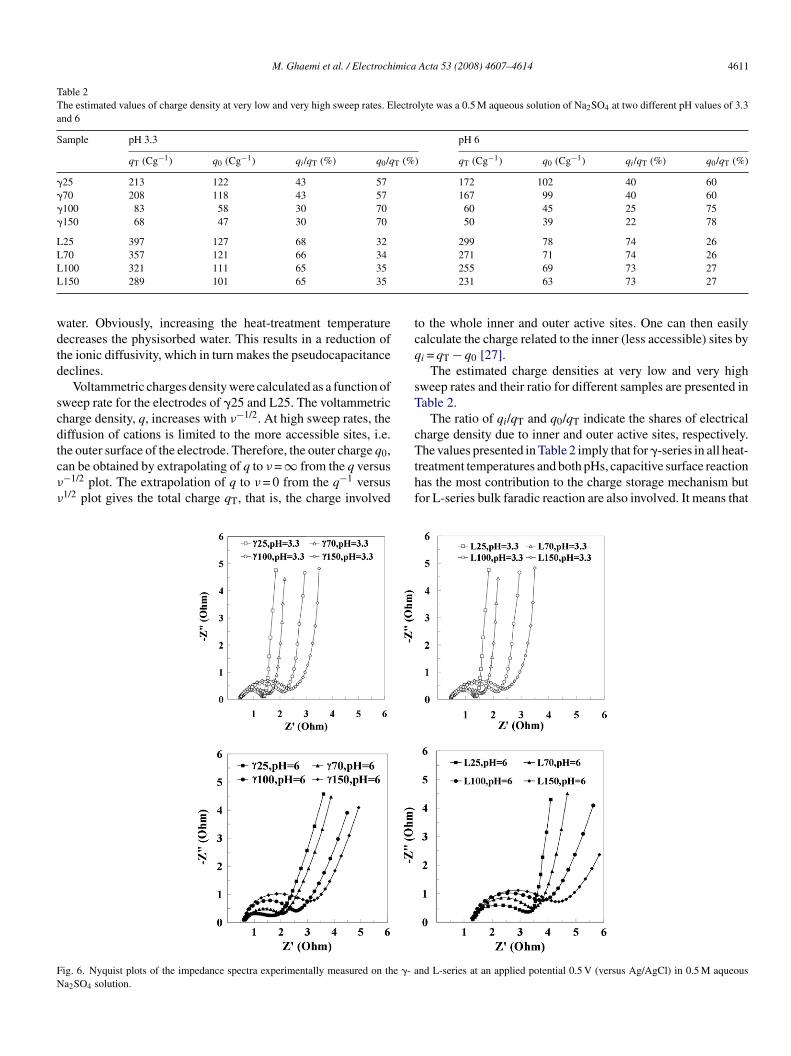
M. Ghaemi et al. / Electrochimica Acta 53 (2008) 4607–4614 4611
Table 2The estimated values of charge density at very low and very high sweep rates. Electrolyte was a 0.5 M aqueous solution of Na2SO4 at two different pH values of 3.3and 6
Sample pH 3.3 pH 6
qT (Cg−1) q0 (Cg−1) qi/qT (%) q0/qT (%) qT (Cg−1) q0 (Cg−1) qi/qT (%) q0/qT (%)
�25 213 122 43 57 172 102 40 60�70 208 118 43 57 167 99 40 60�100 83 58 30 70 60 45 25 75�150 68 47 30 70 50 39 22 78
L25 397 127 68 32 299 78 74 26LLL
wdtd
scdtcν
ν
tcq
sT
c
FN
70 357 121 66 34100 321 111 65 35150 289 101 65 35
ater. Obviously, increasing the heat-treatment temperatureecreases the physisorbed water. This results in a reduction ofhe ionic diffusivity, which in turn makes the pseudocapacitanceeclines.
Voltammetric charges density were calculated as a function ofweep rate for the electrodes of �25 and L25. The voltammetricharge density, q, increases with ν−1/2. At high sweep rates, theiffusion of cations is limited to the more accessible sites, i.e.
he outer surface of the electrode. Therefore, the outer charge q0,an be obtained by extrapolating of q to ν = ∞ from the q versus−1/2 plot. The extrapolation of q to ν = 0 from the q−1 versus1/2 plot gives the total charge qT, that is, the charge involvedTthf
ig. 6. Nyquist plots of the impedance spectra experimentally measured on the �- aa2SO4 solution.
271 71 74 26255 69 73 27231 63 73 27
o the whole inner and outer active sites. One can then easilyalculate the charge related to the inner (less accessible) sites byi = qT − q0 [27].
The estimated charge densities at very low and very highweep rates and their ratio for different samples are presented inable 2.
The ratio of qi/qT and q0/qT indicate the shares of electricalharge density due to inner and outer active sites, respectively.
he values presented in Table 2 imply that for �-series in all heat-reatment temperatures and both pHs, capacitive surface reactionas the most contribution to the charge storage mechanism butor L-series bulk faradic reaction are also involved. It means that
nd L-series at an applied potential 0.5 V (versus Ag/AgCl) in 0.5 M aqueous

4 imica Acta 53 (2008) 4607–4614
fsAtbttc
3
eaNiieatwsAb
tie�dthttp
Ccfibac
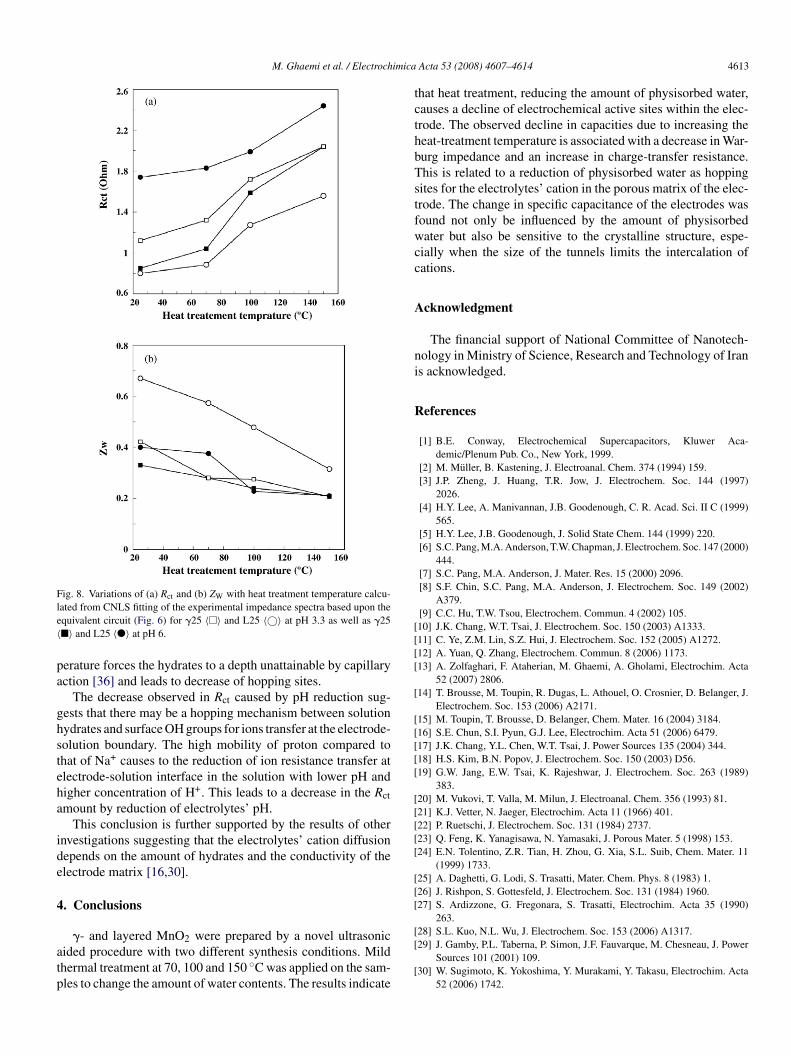
Fig. 7. Variations of (a) Rct and (b) ZW with heat-treatment temperature calcu-lew
wecReratprCsmfi
[fLtpd(oadbe
TT
S
�
�
�
�
LLLL
E
612 M. Ghaemi et al. / Electroch
or the layered compounds, partial intercalation of cations in thetructure probably governs the charge storage process [14,28].
drop of qi/qT is observed for both sample series by increasinghe heat-treatment temperature. This suggests that pores coulde sealed due to heat treatment and leads to make uneasy massransfer into and out of the inner sites. These results suggest thathe crystalline structure of samples has influence on the specificapacitance.
.4. Electrochemical impedance investigation
Fig. 6 describes Nyquist plots of the impedance spectraxperimentally measured on the MnO2 electrode specimenst an applied potential of 0.5 V (versus Ag/AgCl) in 0.5 Ma2SO4 solutions with pH of 3.3 and 6.0. All the measured
mpedance spectra are similar in shape. The high-frequencympedance arc is attributed to the processes occurring at thelectrode–electrolyte interface [29]. Following the impedancerc, a curve with its slope gradually changing from ca. 45◦o 90◦ represents the finite-length diffusion Warburg behaviorhich ascribes the impedance due to electrolytes’ cation diffu-
ion within the electrode in the medium frequency region [30].t the low frequency, a vertical line representing a capacitiveehavior is clearly found.
L-sample series exhibit a line with a slope close to 90◦ alonghe imaginary axis (Z′′) in the low-frequency region, whichs characteristic of a porous matrix, composed of pores withqual dimensions. Whereas capacitive line in Nyquist plots of-sample series exhibit a deviation from the vertical line. Thisifference between the theoretical behavior and the experimen-al one can be explained based on a mathematical model whichave been recently reported by Song et al. [31]. They showed thathe wider distribution of the pore size of a porous matrix leadso more frequency dispersion at the low frequency in Nyquistlot.
The measured impedance spectra were analyzed using theNLS fitting method [1,32] on the basis of the equivalent cir-uit, which is given in Fig. 7. The model circuit comprised of
ve elements: the internal resistance (RS) which includes theulk electrolyte solution resistance, the intrinsic resistance ofctive material, and the electron transfer resistance at currentollector/electrode boundary [33]. The Rs element is in seriesp
sm
able 3he calculated values of Cdl, Rct, ZW and CF through CNLS fitting of the experiment
ample pH 3.3
Cdl (mF) Rct (Ohm) Zw CF (F
25 0.28 1.12 0.45 0.5170 0.26 1.32 0.28 0.42100 0.23 1.72 0.27 0.33150 0.21 2.04 0.21 0.29
25 0.39 0.80 0.67 0.5970 0.36 0.88 0.57 0.50100 0.33 1.27 0.48 0.44150 0.29 1.56 0.32 0.35
lectrolyte was a 0.5 M aqueous solution of Na2SO4 at two different pH values of 3.
ated from CNLS fitting of the experimental impedance spectra based upon thequivalent circuit (inside graph of Fig. 6) for �25 〈�〉, L25 〈©〉 at pH 3.3 asell as �25 〈�〉 and L25 〈�〉 at pH 6.
ith the electrical double layer capacitance at the interface oflectrode and electrolyte (Cdl) [1]. Cdl is in parallel with theharge-transfer resistance (Rct) and Warburg impedance (ZW).ct denotes either the kinetic resistance to ions transfer at thelectrode-solution boundary [34] or intrinsic charge-transferesistance of porous electrode [35]. Warburg impedance (ZW)ssociates with the semi-infinite diffusion of cation in the elec-rode [1]. The above set is in series with a CF element as aseudocapacitance, which was used to account for the faradiceaction. The values of Rct, ZW and CF were calculated fromNLS fitting of the experimental impedance spectra and pre-
ented in Table 3. These results are well reproducible. Also, theean error of modulus being smaller than 1% represents a goodtting of the experimental data.
The surface of a hydrated oxide is covered by OH groups24]. Therefore, a surface hopping mechanism can be envisagedor proton diffusion. The higher Warburg impedance value of25 originates from higher amount hopping site resulted from
he higher amount of surface strongly physisorbed water com-ared to �25. Increasing the heat-treatment temperature causes aecrease in the Warburg impedance values of both �-and L-seriesFig. 8a). This can be attributed to a shallower diffusion depthf proton or electrolyte’s cations in the electrode with the lessmounts of hydrates. Exactly like in solution, the mobility of H+
uring hopping between OH− sites on the surface is expected toe considerably higher than those of the other cations. This canxplain why the Warburg impedance values increase at higher
Hs, i.e. at lower proton concentrations.An augmentation in Rct could be observed for both �- and L-eries with increasing heat-treatment temperature (Fig. 8b). Thisay be related to the fact that increasing the heat-treatment tem-
al impedance spectra based upon the proposed equivalent circuit
pH 6
) Cdl (mF) Rct (Ohm) Zw CF (F)
0.24 0.85 0.33 0.490.17 1.04 0.29 0.390.15 1.59 0.24 0.280.11 2.04 0.21 0.21
0.27 1.74 0.40 0.580.19 1.83 0.38 0.440.17 1.99 0.23 0.340.13 2.44 0.21 0.29
3 and 6.
M. Ghaemi et al. / Electrochimica
Fig. 8. Variations of (a) Rct and (b) ZW with heat treatment temperature calcu-le〈
pa
ghsteha
ide
4
atp
tcthbTstfwcc
A
ni
R
[[[[
[
[[[[[
[[[[[
[[[
ated from CNLS fitting of the experimental impedance spectra based upon thequivalent circuit (Fig. 6) for �25 〈�〉 and L25 〈©〉 at pH 3.3 as well as �25�〉 and L25 〈�〉 at pH 6.
erature forces the hydrates to a depth unattainable by capillaryction [36] and leads to decrease of hopping sites.
The decrease observed in Rct caused by pH reduction sug-ests that there may be a hopping mechanism between solutionydrates and surface OH groups for ions transfer at the electrode-olution boundary. The high mobility of proton compared tohat of Na+ causes to the reduction of ion resistance transfer atlectrode-solution interface in the solution with lower pH andigher concentration of H+. This leads to a decrease in the Rctmount by reduction of electrolytes’ pH.
This conclusion is further supported by the results of othernvestigations suggesting that the electrolytes’ cation diffusionepends on the amount of hydrates and the conductivity of thelectrode matrix [16,30].
. Conclusions
�- and layered MnO2 were prepared by a novel ultrasonicided procedure with two different synthesis conditions. Mildhermal treatment at 70, 100 and 150 ◦C was applied on the sam-les to change the amount of water contents. The results indicate
[[
[
Acta 53 (2008) 4607–4614 4613
hat heat treatment, reducing the amount of physisorbed water,auses a decline of electrochemical active sites within the elec-rode. The observed decline in capacities due to increasing theeat-treatment temperature is associated with a decrease in War-urg impedance and an increase in charge-transfer resistance.his is related to a reduction of physisorbed water as hoppingites for the electrolytes’ cation in the porous matrix of the elec-rode. The change in specific capacitance of the electrodes wasound not only be influenced by the amount of physisorbedater but also be sensitive to the crystalline structure, espe-
ially when the size of the tunnels limits the intercalation ofations.
cknowledgment
The financial support of National Committee of Nanotech-ology in Ministry of Science, Research and Technology of Irans acknowledged.
eferences
[1] B.E. Conway, Electrochemical Supercapacitors, Kluwer Aca-demic/Plenum Pub. Co., New York, 1999.
[2] M. Muller, B. Kastening, J. Electroanal. Chem. 374 (1994) 159.[3] J.P. Zheng, J. Huang, T.R. Jow, J. Electrochem. Soc. 144 (1997)
2026.[4] H.Y. Lee, A. Manivannan, J.B. Goodenough, C. R. Acad. Sci. II C (1999)
565.[5] H.Y. Lee, J.B. Goodenough, J. Solid State Chem. 144 (1999) 220.[6] S.C. Pang, M.A. Anderson, T.W. Chapman, J. Electrochem. Soc. 147 (2000)
444.[7] S.C. Pang, M.A. Anderson, J. Mater. Res. 15 (2000) 2096.[8] S.F. Chin, S.C. Pang, M.A. Anderson, J. Electrochem. Soc. 149 (2002)
A379.[9] C.C. Hu, T.W. Tsou, Electrochem. Commun. 4 (2002) 105.10] J.K. Chang, W.T. Tsai, J. Electrochem. Soc. 150 (2003) A1333.11] C. Ye, Z.M. Lin, S.Z. Hui, J. Electrochem. Soc. 152 (2005) A1272.12] A. Yuan, Q. Zhang, Electrochem. Commun. 8 (2006) 1173.13] A. Zolfaghari, F. Ataherian, M. Ghaemi, A. Gholami, Electrochim. Acta
52 (2007) 2806.14] T. Brousse, M. Toupin, R. Dugas, L. Athouel, O. Crosnier, D. Belanger, J.
Electrochem. Soc. 153 (2006) A2171.15] M. Toupin, T. Brousse, D. Belanger, Chem. Mater. 16 (2004) 3184.16] S.E. Chun, S.I. Pyun, G.J. Lee, Electrochim. Acta 51 (2006) 6479.17] J.K. Chang, Y.L. Chen, W.T. Tsai, J. Power Sources 135 (2004) 344.18] H.S. Kim, B.N. Popov, J. Electrochem. Soc. 150 (2003) D56.19] G.W. Jang, E.W. Tsai, K. Rajeshwar, J. Electrochem. Soc. 263 (1989)
383.20] M. Vukovi, T. Valla, M. Milun, J. Electroanal. Chem. 356 (1993) 81.21] K.J. Vetter, N. Jaeger, Electrochim. Acta 11 (1966) 401.22] P. Ruetschi, J. Electrochem. Soc. 131 (1984) 2737.23] Q. Feng, K. Yanagisawa, N. Yamasaki, J. Porous Mater. 5 (1998) 153.24] E.N. Tolentino, Z.R. Tian, H. Zhou, G. Xia, S.L. Suib, Chem. Mater. 11
(1999) 1733.25] A. Daghetti, G. Lodi, S. Trasatti, Mater. Chem. Phys. 8 (1983) 1.26] J. Rishpon, S. Gottesfeld, J. Electrochem. Soc. 131 (1984) 1960.27] S. Ardizzone, G. Fregonara, S. Trasatti, Electrochim. Acta 35 (1990)
263.
28] S.L. Kuo, N.L. Wu, J. Electrochem. Soc. 153 (2006) A1317.29] J. Gamby, P.L. Taberna, P. Simon, J.F. Fauvarque, M. Chesneau, J. PowerSources 101 (2001) 109.30] W. Sugimoto, K. Yokoshima, Y. Murakami, Y. Takasu, Electrochim. Acta
52 (2006) 1742.

4 imica
[
[[
614 M. Ghaemi et al. / Electroch
31] H.K. Song, H.Y. Hwang, K.H. Lee, L.H. Dao, Electrochim. Acta 45 (2000)2241.
32] B.E. Conway, J. Electrochem. Soc. 142 (1991) 1539.33] Q.F. Wu, K.X. He, H.Y. Mi, X.G. Zhang, Mater. Chem. Phys. 101 (2007)
367.
[[
[
Acta 53 (2008) 4607–4614
34] X. Ren, P.G. Pickup, J. Electroanal. Chem. 420 (1997) 251.35] C. Ehrenbeck, K. Juttner, S. Ludwig, G. Paasch, Electrochim. Acta 43
(1998) 2781.36] S.W. Donne, J.H. Kennedey, J. Appl. Electrochem. 34 (2004) 159.




















