
CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile...
12
doi:10.1182/blood-2007-07-100651 Prepublished online Dec 5, 2007; 2008 111: 2765-2775 and Giorgio Inghirami Hunter, Hugh Zhao, Antonino Neri, Antonio Palumbo, Celia Berkers, Huib Ovaa, Alberto Bernareggi Kathryn Ilaria Roato, Riccardo Ferracini, Benedetta Bussolati, Giovanni Camussi, Susan Jones-Bolin, Nicoletta Pescalli, Mara Cassin, Stefano di Giovine, Paola Nicoli, Paola de Feudis, Ivan Strepponi, Valentina Giai, Marta Coscia, Silvia Peola, Massimo Massaia, Gabriella Pezzoni, Cecilia Allievi, Roberto Piva, Bruce Ruggeri, Michael Williams, Giulia Costa, Ilaria Tamagno, Dario Ferrero, tumor-selective pharmacologic profile competitive with bortezomib CEP-18770: A novel, orally active proteasome inhibitor with a http://bloodjournal.hematologylibrary.org/cgi/content/full/111/5/2765 Updated information and services can be found at: (4131 articles) Neoplasia collections: Blood Articles on similar topics may be found in the following http://bloodjournal.hematologylibrary.org/misc/rights.dtl#repub_requests Information about reproducing this article in parts or in its entirety may be found online at: http://bloodjournal.hematologylibrary.org/misc/rights.dtl#reprints Information about ordering reprints may be found online at: http://bloodjournal.hematologylibrary.org/subscriptions/index.dtl Information about subscriptions and ASH membership may be found online at: . Hematology; all rights reserved Copyright 2007 by The American Society of 200, Washington DC 20036. semimonthly by the American Society of Hematology, 1900 M St, NW, Suite Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.org From
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile...

doi:10.1182/blood-2007-07-100651 Prepublished online Dec 5, 2007;2008 111: 2765-2775
and Giorgio Inghirami Hunter, Hugh Zhao, Antonino Neri, Antonio Palumbo, Celia Berkers, Huib Ovaa, Alberto Bernareggi
KathrynIlaria Roato, Riccardo Ferracini, Benedetta Bussolati, Giovanni Camussi, Susan Jones-Bolin, Nicoletta Pescalli, Mara Cassin, Stefano di Giovine, Paola Nicoli, Paola de Feudis, Ivan Strepponi,Valentina Giai, Marta Coscia, Silvia Peola, Massimo Massaia, Gabriella Pezzoni, Cecilia Allievi, Roberto Piva, Bruce Ruggeri, Michael Williams, Giulia Costa, Ilaria Tamagno, Dario Ferrero,
tumor-selective pharmacologic profile competitive with bortezomibCEP-18770: A novel, orally active proteasome inhibitor with a
http://bloodjournal.hematologylibrary.org/cgi/content/full/111/5/2765Updated information and services can be found at:
(4131 articles)Neoplasia � collections: BloodArticles on similar topics may be found in the following
http://bloodjournal.hematologylibrary.org/misc/rights.dtl#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/misc/rights.dtl#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/subscriptions/index.dtlInformation about subscriptions and ASH membership may be found online at:
. Hematology; all rights reservedCopyright 2007 by The American Society of 200, Washington DC 20036.semimonthly by the American Society of Hematology, 1900 M St, NW, Suite Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

NEOPLASIA
CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selectivepharmacologic profile competitive with bortezomibRoberto Piva,1-3 Bruce Ruggeri,4 Michael Williams,4 Giulia Costa,1 Ilaria Tamagno,1 Dario Ferrero,5 Valentina Giai,5
Marta Coscia,1,5 Silvia Peola,1,5 Massimo Massaia,1,5 Gabriella Pezzoni,6 Cecilia Allievi,6 Nicoletta Pescalli,6 Mara Cassin,6
Stefano di Giovine,6 Paola Nicoli,6 Paola de Feudis,6 Ivan Strepponi,6 Ilaria Roato,7 Riccardo Ferracini,7
Benedetta Bussolati,1,8 Giovanni Camussi,1,8 Susan Jones-Bolin,4 Kathryn Hunter,4 Hugh Zhao,4 Antonino Neri,9
Antonio Palumbo,5 Celia Berkers,10 Huib Ovaa,10 Alberto Bernareggi,11 and Giorgio Inghirami1-3
1Center for Experimental Research and Medical Studies (CeRMS) and 2Department of Biomedical Sciences and Human Oncology, University of Torino, Torino,Italy; 3Department of Pathology and New York University Cancer Center, New York University School of Medicine, New York; 4Discovery Research, Cephalon,West Chester, PA; 5Hematology Division, University of Torino, Torino, Italy; 6Sede Secondaria Della Cell Therapeutics, Bresso, Italy; 7Orthopedic andTraumatology, San Giovanni Battista of Torino, Torino, Italy; 8Department of Internal Medicine, University of Torino, Torino, Italy; 9Laboratory of ExperimentalHematology and Molecular Genetics, Ospedale Maggiore Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Milano, Italy; 10Netherlands CancerInstitute, Amsterdam, the Netherlands; and 11RBM, Brescia, Italy
Modulating protein ubiquitination via pro-teasome inhibition represents a promis-ing target for cancer therapy, because ofthe higher sensitivity of cancer cells tothe cytotoxic effects of proteasome inhibi-tion. Here we show that CEP-18770 is anovel orally-active inhibitor of thechymotrypsin-like activity of the protea-some that down-modulates the nuclearfactor-�B (NF-�B) activity and the expres-sion of several NF-�B downstream effec-tors. CEP-18770 induces apoptotic celldeath in multiple myeloma (MM) cell lines
and in primary purified CD138-positiveexplant cultures from untreated andbortezomib-treated MM patients. In vitro,CEP-18770 has a strong antiangiogenicactivity and potently represses RANKL–induced osteoclastogenesis. Importantly,CEP-18770 exhibits a favorable cytotoxic-ity profile toward normal human epithelialcells, bone marrow progenitors, and bonemarrow–derived stromal cells. Intrave-nous and oral administration of CEP-18770 resulted in a more sustained phar-macodynamic inhibition of proteasome
activity in tumors relative to normal tis-sues, complete tumor regression of MMxenografts and improved overall mediansurvival in a systemic model of humanMM. Collectively, these findings provideevidence for the utility of CEP-18770 as anovel orally active proteasome inhibitorwith a favorable tumor selectivity profilefor the treatment of MM and other malig-nancies responsive to proteasome inhibi-tion. (Blood. 2008;111:2765-2775)
© 2008 by The American Society of Hematology
Introduction
The ubiquitin-proteasome pathway plays a key role in proteinprocessing and degradation, regulating crucial transduction path-ways for cell growth and survival, including cell-cycle control,transcriptional regulation, cellular stress responses, and antigenpresentation.1,2 Numerous investigators have demonstrated thatproteasome inhibition results in the disruption of normal homeo-static mechanisms, and that cancer cells are more likely to undergoapoptosis after proteasome inhibition compared with normal cells.3-6
It is known that proteasome inhibition can reverse the aberrantchanges that perpetuate proliferation and block apoptosis pathwaysin cancer cells. In fact, treatment of cancer cells with proteasomeinhibitors results in the stabilization of p21 and p27,7,8 as well asinhibition of nuclear factor-kappaB (NF-�B)–mediated transcrip-tion.9,10 The NF-�B pathway is activated in a variety of tumor typesand often deregulated in chemotherapy-resistant cells, suggestingthat NF-�B activation plays a role in cancer cell survival andchemoresistance, and that NF-�B inhibition via proteasome inhibi-tion can sensitize transformed cells to apoptosis.11-13
The prevalent sensitivity of transformed cells to proteasomeinhibitors and the successful design of clinical protocols, with
tolerable although relatively narrow therapeutic indices, have madethe proteasome a novel and promising target for cancer treat-ment.2,6 Clinical validation for the application of proteasomeinhibition as a therapeutic strategy was achieved with bortezomib(Velcade; Millennium Pharmaceuticals, Cambridge, MA), a modi-fied dipeptidyl boronic acid, intravenously administered, which is aslowly reversible inhibitor of the chymotrypsin-like activity of the26S mammalian proteasome.4,5,14 On the basis of pivotal clinicaltrials, bortezomib has proven efficacious as single agent for thetreatment of multiple myeloma (MM), non-Hodgkin, and mantlecell lymphoma patients.15-17 Although the toxicity profile ofbortezomib is quite well controlled in clinical settings, its sideeffects include peripheral neuropathy, orthostatic hypotension,pyrexia, cardiac and pulmonary disorders, gastrointestinal adverseevents, myelosuppression, thrombocytopenia, asthenia, and pain.18,19
Consequently, there is a need for the identification of proteasomeinhibitors with enhanced tolerability and safety profiles. Salinospo-ramide A (NPI-0052) extracted from a marine actinomycetebacteria is a novel orally active proteasome inhibitor shown toeffectively induce apoptosis in tumor cells of MM20 and chronic
Submitted July 11, 2007; accepted November 2, 2007. Prepublished online as BloodFirst Edition paper, December 5, 2007; DOI 10.1182/blood-2007-07-100651.
R.P. and B.R. contributed equally to this manuscript.
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 USC section 1734.
© 2008 by The American Society of Hematology
2765BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

lymphocytic leukemia (CLL), while displaying a lower toxicactivity to bone marrow-derived stromal cells (BMSC) comparedwith bortezomib.21 An irreversible epoxy-ketone peptidyl inhibitorof all 3 proteasome proteolytic sites (PR-171) is currently inclinical evaluation (Phase 1) for advanced solid tumors andrefractory hematologic malignancies.22
In the present study, we describe the in vitro and in vivobiologic characterization of CEP-18770, a novel reversible P2threonine boronic acid proteasome inhibitor (B. Dorsey et al,manuscript submitted, 2007). We demonstrate that CEP-18770potently promotes apoptosis in human MM cell lines and patient-derived cells; inhibits endothelial cell survival, vasculogenesis, andosteoclastogenesis in vitro; and displays a favorable cytotoxicityprofile toward normal cells. In addition, we show that intravenousor oral administration of CEP-18770 is well tolerated and results insustained pharmacodynamic and antitumor activity and survivalbenefits in xenograft and systemic models of human MM.
Methods
Enzyme and cellular biochemical activity profiles against themammalian proteasome
The proteasome inhibitory activity of CEP-18770 and bortezomib wereevaluated in an isolated human erythrocyte proteasome fluorimetric kineticassay of chymotrypsin-like proteasome activity.23 Experiments were per-formed in triplicate.
Cell lines and primary cell cultures and treatments
Human multiple myeloma (MM) cell lines CMA-03, JJN3, H929, KMM1,KMS11, KMS18, KMS27, SKMM1, U266, RPMI-8226, human chronicmyelogenous leukemia cell line K562, T-cell leukemic line MOLT-4,human anaplastic lymphoma ALK positive TS cells were used.24,25 MMmyeloid and T-cell lines were grown at 37°C in 5% CO2 humidified air inRPMI 1640 medium (Cambrex, Verviers, Belgium) or in Iscove modifiedDulbecco medium (IMDM; Cambrex), respectively, all supplemented with10% fetal calf serum (FCS), without growth factors or alternatively with10 ng/mL IL-6 (CMA-03).26 Cells were seeded at 5 � 105 cells/mL for theproteasome inhibitor treatments. Normal human microvascular endothelialcells from derma (HMEC) were immortalized by infection with a replication-defective adeno-5/SV40 virus as previously described.27 The tumor-derivedendothelial cells (TEC) have previously been described.28 HMEC and TECcell lines were established and maintained in culture in endothelial cellbasal medium (EBM) supplemented with epidermal growth factor(10 ng/mL), hydrocortisone (1 mg/mL), bovine brain extract (CambrexBioscience, Baltimore, MD), and 10% FCS.
Peripheral blood mononuclear cells (PBMC) were obtained fromhealthy individuals after Ficoll-Hypaque density separation. Bone marrowmononuclear cells (BMNCs) were used to establish long-term BMSCcultures from multiple myeloma patients (7) and from noncancenerousindividuals (5). Briefly, BMNC (2 � 106 cells) were suspended in IMDMcontaining 20% FCS and plated onto 12-well plates. Nonadherent cellswere subsequently removed after 24 hours of culture. Adherent BMSCshowing fibroblast morphology were grown until they reached confluenceand then expanded. BMSC were seeded at 30% confluence in 6-well platesand treated with proteasome inhibitors for 6 days.
CD138� plasma cells (PCs) were isolated from BM aspirates of15 patients with MM (PC � 10%) and enriched by magnetic bead basedpositive selection (CD138 MicroBeads; Miltenyi Biotech, Bergisch Glad-bach, Germany). Purified PC were seeded (106 cells/mL) on a 50%confluent BMSC monolayer in 6-well plates and maintained in IMDMsupplemented with 20% FCS and 10 ng/mL IL-6. PCs were then treatedwith proteasome inhibitors for 72 hours.
Flow cytometric analyses of apoptosis
Apoptosis was measured by flow cytometry with the mitochondrion-permeable, voltage sensitive dye tetrametylrodamine methyl ester (TMRM;Molecular Probes, Eugene, OR) as described.24
Proliferation and viability assays
The effect of proteasome inhibitors on mononuclear cell viability was measuredusing a methylthiazolyldiphenyl-tetrazolium bromide (MTT) based assay asdetailed using the following formula: percentage cell viability � (OD of theexperimental samples / mean OD of the control) � 100.29
Western blotting
The following primary antibodies were used for Western blotting aspreviously described30: rabbit anti–phospho IKB� (Ser32), anti–cleavedcaspase 3, anti–cleaved caspase 7, and anti–cleaved caspase 9 from CellSignaling Technology (Beverly, MA); rabbit anti-PARP (H-250), anti-IKB�from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit anti-XIAP fromR&D Systems (Minneapolis, MN); mouse anti-ubiquitin from ZymedLaboratories (South San Francisco, CA) or Invitrogen (Carlsbad, CA); andmouse anti–�-tubulin (B-5-1-1) from Sigma-Aldrich (St Louis, MO).
Electrophoretic mobility shift assay
Aliquots of total extracts (12 �g protein/sample) in 0.1% Triton X-100 lysisbuffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Triton X-100, 5 mMEDTA, 1 mM Na3VO4, and 1 mM PMSF and protease inhibitors) wereincubated with [32P]-labeled �B DNA probe in binding buffer (10 mMTris-HCl, pH 7.5, 1mM EDTA, 5% glycerol) for 30 minutes at roomtemperature. Quantitative evaluation of NF-�B–DNA probe complexformation was determined as previously described.30
Reverse transcription–polymerase chain reaction
Total RNA from BMSC cultured in 10-cm plates (passage 4 to 6) and aftertreatment with proteasome inhibitors (60 hours) were extracted using theRNAeasy Mini Kit (Qiagen, Hilden, Germany). Reverse transcription–polymerase chain reactions (RT-PCRs) were performed (1 �g RNA)according to the manufacturer’s protocols (Invitrogen). The oligonucleotideprimer pairs and PCR conditions for RT-PCR are available upon request.
Culture assay for CFU-GM and BFU-E
BMMC derived from 16 patients with MM (PC � 10%) were seeded inIMDM supplemented with 10% FCS at 5 � 105 cells/mL and treated withproteasome inhibitors for 48 hours. Cells were then washed and cultured forCFU-GM and BFU-E assay. Briefly, 5 � 104 proteasome inhibitor-treatedor control BMMC were cultured in 35-mm Petri dishes containing completemethylcellulose medium (Methocult GF H4434 with recombinant cyto-kines; Stem Cell Technologies; with: IMDM, FCS, methylcellulose,2-mercaptoethanol, l-glutammine, IL-3, GM-CSF, “stem cell factor,”erythropoietin). Colony growth from early (day 14) CFU-GM and BFU-Ewas scored after 14 days of incubation at 37°C in a 5% CO2 atmosphere.The differences in colony growth from control and proteasome inhibitors-treated cells were analyzed by the Student t test.
Endothelial cell proliferation, apoptosis, and tubulogenesisassays
HMEC and TEC cells were seeded into 24-well plates at a density of10 000 cells/well in DMEM supplemented with 5% FCS. After incubationwith proteasome inhibitors (48 hours), cells were washed, air dried, andstained with crystal violet as described.28 Cell number was determined,with duplicate samples, on the basis of a standard curve obtained withknown cell numbers. All experiments were performed in triplicate. Invitro formation of capillary-like structures was studied on cells(4 � 104 cells/well in 24-well plates) seeded onto Matrigel-coated wells inDMEM containing 0.25% BSA. HMEC and TEC cells (5 � 103 per well),suspended in 200 �L DMEM with 5% FCS (positive control), serum-free
2766 PIVA et al BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

medium (negative control), were layered onto the Matrigel surface in thepresence or absence of proteasome inhibitors. Cells were observed with aNikon inverted microscope and experimental results were then recordedafter a 6-hour incubation at 37°C. Data were analyzed, as the mean( 1 SD) of total length of capillary-like structures, by the Micro-Imagesystem (Casti Imaging, Mira, Italy) and expressed as mm/field by thecomputer analysis system in 5 different files at 100� magnification induplicated wells of 4 different experiments.
Osteoclast formation
PBMC (2 � 106 cells/well) were cultured for 10 days in the presence of25 ng/mL recombinant human M-CSF (R&D Systems, Abingdon, UnitedKingdom). Osteoclasts were then activated by 30 ng/mL RANKL (Alexix,San Diego, CA), and simultaneously treated with proteasome inhibitors for5 days, to reach the maximal inhibition and to avoid possible irreversibleeffects of commitment osteoclastogenesis induced by RANKL signaling.Culture media were supplemented with fresh media every 3 to 4 days. At theend of the culture period, cells were fixed and stained for tartrate-resistantacid phosphatase (TRAP Kit; Sigma Diagnostics, St Louis, MO). Matureosteoclasts were identified as TRAP positive multinucleated cells, contain-ing 3 or more nuclei and quantified.31
Animals
Female athymic nu/nu nude mice, severe combined immunodeficiency(SCID) mice, and nonobese diabetic (NOD)/SCID mice (6-8 weeks old;Charles River, Wilmington, MA) were maintained on standard sterilizedmicroisolator units (Teklad Labchow, Madison, WI) under humidity andtemperature controlled conditions. Experiments were approved by theInstitutional Animal Care and Use Committee of Cephalon.
Subcutaneous tumor xenograft models
RPMI 8226 cell suspensions (6 � 106 cells) were injected subcutaneouslyinto the flank of SCID mice. Treatment started after establishing a palpablesubcutaneous tumor (100-140 mm3) usually 40 days after implantation afterrandomization. CEP-18770 was administered to tumor-bearing animalsintravenously, twice a week for 4 weeks (2q7d�8 injections) at doses of1.0 to 6.0 mg/kg, and orally in a solution of 3% DMSO, 10% Solutol, and87% sterile NaCl 0.9% as multiple administrations, twice-a-week injectionsfor 4 weeks at doses of 7.8, 10, 13 mg/kg in a volume of 20 mL/kg bodyweight of mouse. Bortezomib was given intravenously in sterile NaCl0.9% at doses of 0.8, 1, 1.2 and 1.6 mg/kg, twice-a-week injections for4 weeks in a volume of 10 mL/kg body weight of mouse.
Systemic human multiple myeloma model
ARP-1 human MM cells (gift from Joshua Epstein, University of Arkansas,Little Rock, AR) were intravenously injected (5 � 106) into NOD/SCIDmice. Approximately 6 weeks after injections, mice were bled to detectpositive human IgA levels as measured by enzyme-linked immunosorbentassay (ELISA; Bethyl Laboratories, Montgomery, TX), determining humanmyelomatous tumor burden. Randomization of animals into treatmentgroups was as follows: CEP-18770, 3.0 mg/kg intravenously, once weeklyfor 8 doses (1q7d�8 injections); CEP-18770, 3.0 mg/kg, intravenously,twice weekly for 8 doses (2q7d � 8 injections); CEP-18770, 4.0 mg/kg,intravenously, once weekly for 8 doses; CEP-18770, 4.0 mg/kg, intrave-nously, twice weekly for 8 doses; bortezomib, 1.2 mg/kg, intravenously,once weekly for 8 doses; bortezomib, 1.2 mg/kg, intravenously, twiceweekly for 8 doses; high-dose dexamethasone, 1.5 mg/kg, intraperitoneally,once daily; melphalan, 10 mg/kg, intraperitoneally, every 3 weeks, andthalidomide, 100 mg/kg, intraperitoneally, once daily. The doses ofCEP-18770 and bortezomib were chosen based on maximally tolerateddose (MTD) and 0.75 MTD with chronic administration in mice. Thetreatment efficacy of each protocol were determined by log-rank statisticalanalyses and plotted by the Kaplan-Meier method as required for datasetsusing SAS 8.2 (SAS Institute, Cary, NC). Additional statistical analyseswere performed using Mann-Whitney rank sum test and ANOVA.
Results
Enzyme and cellular biochemical activity profiles against theubiquitin-proteasome complex
The proteasome inhibitory activity of CEP-18770 was first evalu-ated in comparison with bortezomib in the isolated human erythro-cytes. CEP-18770 and bortezomib showed comparable potencyagainst chymotrypsin-like proteasome activity, cellular inhibitoryactivity (IC50) values of 3.8 ( 1.0) nM and 3.8 ( 0.4) nM,respectively, but demonstrated marginal inhibition of the trypticand peptidyl glutamyl activities of the proteosome (Figure 1).Analyses of the IC50 values of proteasome chymotryptic (5) andcaspase-like (1) catalytic activities in MM and HeLa carcinomacell lysates for CEP-18770 and bortezomib are shown in Figure 1B.The IC50 values of CEP-18770 were similar to those of bortezomib,with the chymotryptic and caspase-like activities being inhibited atlow-nanomolar concentrations. CEP-18770 and several structur-ally related inhibitors not advancing into clinical development werealso tested for their ability to inhibit the ubiquitin-proteasomepathway in several MM and in the chronic myelogenous leukemiacell line, K562 (and data not shown). Cells were treated withincreasing concentrations (5, 10, 20, 40 nM) of the selectedproteasome inhibitors, and the accumulation of ubiquitinatedproteins measured by Western blotting. In this experimental setting,CEP-18770 induced an accumulation of ubiquitinated proteins over4 to 8 hours with a profile similar to that observed after bortezomibtreatment (Figure 1C,D).
CEP-18770 targets NF-�B activity
The most compelling rationale for the therapeutic use of protea-some inhibitors in oncology relies on their ability to inhibit thenuclear factor-�B (NF-�B) transcriptional activity resulting in thereduced expression of several target genes.11-13 Because protea-some inhibitors modulate the activation of NF-�B primarily byblocking I�B� degradation,9 the ability of CEP-18770 to inhibit theTNF�-stimulated activation of NF-�B was examined. RPMI-8226cells were pretreated with 20 nM CEP-18770 for 8 hours andsubsequently stimulated by TNF� (10 ng/mL). I�B� serine 32 to36 phosphorylation was rapidly induced (5 minutes) by TNF�,however, I�B� degradation was completely blocked by pretreat-ment with CEP-18770 (Figure 2A). The effect of CEP-18770 orbortezomib treatment on NF-�B DNA-binding activity revealedthat both compounds significantly inhibited high levels of NF-�Bactivity in both RPMI-8226 and U266 cells. In contrast, low basalactivity of NF-�B was not appreciably modified in K562 cells(Figure 2B). The time- and concentration-dependent inhibition ofNF-kB DNA-binding activity in MM cell lines by CEP-18770resulted in a reduction of several NF-�B-modulated genes mediat-ing the growth and survival of tumor cells including IkB� itself, theX-chromosome-linked inhibitor-of-apoptosis protein (XIAP),the pro-inflammatory cytokines TNF-� and interleukin-1 (IL-1),the intracellular adhesion molecule (ICAM1), and the pro-angiogeneic factor vascular endothelial growth factor (VEGF;Figure 2C,D). Recent studies indicate that the expression of theseNF-�B–mediated genes and their modulation by bortezomib areassociated with more favorable clinical responsiveness to thisagent,32 highlighting their potential prognostic value in response toCEP-18770 exposure.
CEP-18770, AN ORALLY-ACTIVE PROTEASOME INHIBITOR 2767BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

CEP-18770 induces apoptosis of human MM cell lines andprimary human MM explant cultures
The pharmacologic evaluation of CEP-18 770 in vitro demon-strated that CEP-18 770 treatment resulted in a progressive appear-ance of cleaved caspases-3, -7, and -9 between 12 and 24 hours’exposure in the human MM cell lines, RPMI-8226, and U266, witha kinetic profile comparable with that of bortezomib. Activation ofcaspases was further confirmed by the cleavage of the poly(ADP-ribose) polymerase (PARP) protein (Figure 3A) and induction ofapoptosis was further supported by TMRM staining. No activationof caspases or apoptosis was however observed in the K562 cell
line after proteasome inhibitor treatment (Figure 3B). To furtherextend these findings, 8 additional MM cell lines were treated withCEP-18770 or control vehicle. Overall, 9 of 10 MM cell lines weresensitive to the apoptogenic action of CEP-18770 (Figure 3C), withthe only exception of JJN3. The antiproliferative activity ofCEP-18770 and bortezomib in vitro was then evaluated in apanel of human hematologic and solid tumor cell lines. CEP-18770had a 2- to 11-fold lower cytotoxic potency compared withbortezomib against solid tumor cell lines, comparable potencyagainst 2 hematologic tumor cell lines, and a similar spectrum ofantiproliferative activity with IC50 values for both compounds of
Figure 1. Structure of CEP-18 770 and analyses of CEP boronic acid proteasome inhibitors in human multiple myeloma (MM) cells. (A) Structure of CEP-18770;(B) Cellular proteasome inhibitory profile in cell lysates obtained from RPMI-8226 MM incubated with increasing concentrations of bortezomib, CEP-18770, and a relatedanalog, compound 19 (Dorsey et al, manuscript submitted). Inhibition was measured using fluorogenic substrates for the different catalytic activities of the proteasome. Resultswere plotted as percentage inhibition compared with nontreated lysates. Experiments were performed in triplicate. Error bars represent SD. (C) Ubiquitination assay:RPMI-8226 cells were treated for 24 hours with compounds 17, 24, 19 described previously (Dorsey et al, 2007) as well as CEP-18770 and bortezomib at the indicatedconcentrations; the accumulation of ubiquitinated proteins induced by proteasome inhibitors was assayed by Western blotting (WB) with antiubiquitin antibody. �-tubulinprotein expression was included for protein loading normalization. (D) RPMI-8226, U266, and K562 cells were treated for different times with 20 nM CEP-18770 and 10 nMbortezomib (BOR); accumulation of ubiquitinated proteins was assayed by WB as described.
2768 PIVA et al BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

Figure 2. CEP-18 770 modulates the NF-�B signal-ing pathway in human MM cell lines. (A) RPMI-8226cells were pretreated with control solvent (dimethylsulfoxide; DMSO) or CEP-18770 (20 nM) for 6 hoursbefore stimulation with TNF-� (10 ng/mL) for theindicated times. Whole cell extracts were immunoblot-ted with anti-I�B� and phosphorylated I�B� (P-I�B�) toassess IKK-mediated NF-�B activation. �-tubulin immu-noblotting was included for protein loading normaliza-tion. (B) Whole-cell extracts were analyzed for NF-�Bactivation by electrophoretic mobility shift assay(EMSA). A section of the fluorogram is shown.(C) RPMI-8226, U266, and K562 cells were treatedwith CEP-18770 (20 nM) or BOR (10 nM) and immuno-blotted with the specified antibodies to detect levels oftotal I�B�, P-I�B� and XIAP proteins. (D) U266 cellswere treated with control diluent, CEP-18770 (20 nM),or BOR (10 nM) for 12 hours. Levels of ICAM1, TNF-�,IL1, VEGF, and 2-microglobulin mRNA were ana-lyzed by semiquantitative RT-PCR.
Figure 3. CEP-18770 induces apoptosis in humanMM cell lines and in purified primary human MMexplant cultures. (A) RPMI-8226, U266, and K562cells were treated with CEP-18770 (20 nM) or BOR(10 nM) for the indicated times. Levels of cleavedfragments of caspase 3, caspase 7, and caspase 9(CF) and of full-length (115 kDa) and cleaved fragmentof PARP (85 kDa) were determined by immunoblottingon whole-cell lysates with the indicated antibodies;�-tubulin protein expression was included as loadingcontrol. (B) Apoptosis was measured by flow cytometrywith the mitochondrion-permeable dye TMRM in RPMI-8226, U266, and K562 cells after 12 and 24 hours ofCEP-18770 (20 nM) or BOR (10 nM) treatment. Histo-grams are representative of the percentage of apopto-tic cells in one of 3 experiments (C) Human MM celllines were treated with control diluent or CEP-18770(20 nM) for 24 hours, percentages of apoptosis weremeasured after TMRM staining by flow cytometry.Histograms are representative of the percentage ofapoptotic cells in one of 3 experiments (D) CD138� PC,obtained from 2 patients with MM, were separated bypositive selection and PC were seeded on a BMSCmonolayer and treated with CEP-18770 or inactivedeboronated CEP-18770 for 72 hours at different con-centrations. The percentage of purified MM cells under-going apoptosis as measured by staining with TMRMand flow cytometry is indicated. (E) CD138� PC ob-tained from 8 patients with MM (PC � 10%) as de-scribed above, were treated with CEP-18870 (20 nM)or bortezomib (BOR) (10 nM) for 72 hours. Apoptosiswas measured by flow cytometry after TMRM staining.Histograms represent the percentage of viable cellsnormalized versus control samples.
CEP-18770, AN ORALLY-ACTIVE PROTEASOME INHIBITOR 2769BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

less than 35 nM (Table 1). To determine whether CEP-18770similarly affected the viability of primary neoplastic PCs, theconcentration-related proapoptotic activity of both inhibitors wereevaluated against CD138� MM cells obtained from the bonemarrow aspirates of 10 MM patients, who had relapsed aftermultiple therapeutic protocols. These studies demonstrated compa-rable concentration-dependent proapoptotic effects of CEP-18770and bortezomib ex vivo after 48 hours exposure as measured bymitochondrial depolarization assays, with complete MM cell deathobserved at 20 nM for both CEP-18770 and bortezomib (Figure3D). Similar findings were observed for CEP-18770 with primaryMM biopsies cultured ex vivo from 3 bortezomib-resistant MMpatients (Figure 3E). These data indicate that the proapoptoticactivity of CEP-18770 against MM is not limited solely totumor-derived MM cell lines, but extends to primary MM explantsfrom relapsed or refractory patients including those previouslytreated with bortezomib.
Cytotoxicity profile of CEP-18770 in normal versus tumorhuman cells
The potential tumor-selective cytotoxic activity of CEP-18770against MM cells as compared with normal human cells wasexamined, studying the susceptibility to these compounds ofnormal BMMCs and PBMCs obtained from healthy volunteers orfrom MM ex vivo. CEP-18770 and bortezomib resulted in asignificant and comparable decrease of cell viability after 48 hours’treatment in both type of mononuclear cells (Figure 4A,B).
Because the interaction of MM cells with the bone marrowmicroenvironment elicits complex signals sustaining the paracrinegrowth of tumor cells and protecting MM cells against drug-induced apoptosis,33,34 the effects of CEP-18770 on the viability ofBMSCs was also examined. Treatment of BMSCs (from 5 healthydonors and 7 MM patients) with CEP-18770 (20 nM) wassignificantly less cyototoxic (P � .05) compared with bortezomib(Figure 4C). Importantly, CEP-18770 exposure decreased theexpression of interleukins IL-6, IL-1, vascular cell adhesionmolecule (VCAM1), and VEGF comparably in these cells. ICAMlevels remained largely unchanged (Figure 4D). To compare furtherthe potential hematotoxicity of CEP-18770 and bortezomib onhuman bone marrow progenitors, proteasome treated (48 hours)BMMC derived from 16 MM patients were plated on methylcellu-lose in the presence of myelopoietic or erythtropoietic cytokines.These conditions enabled the assessment of the effects of protea-some inhibitor treatment on bone marrow progenitor cells ofboth myeloid and erythroid lineages, respectively, as assayed bygranulocyte-macrophage (CFU-GM) and erythroid (BFU-E) colonyformation. Suppression of the clonogenic potential of BMMC wasobserved after both treatments. However, the percentage inhibitionof CFU-GM and CFU-E colony formation was significant onlyafter bortezomib treatment (P � .008 and P � .001, respectively),versus control cultures, but not upon exposure to CEP-18770.Bortezomib was also 3 times more potent in inhibiting the growthof BFU-E colonies compared with CEP-18770 treatment (P � .01)
(Figure 4F). Finally, CEP-18770 exhibited reduced cytotoxicity(P � .05) compared with bortezomib against normal human intesti-nal epithelium cells (CCD-18Co; data not shown).
Table 1. Cytotoxicity profile of CEP-18770 and bortezomib in representative human solid and hematological tumor cell lines
CompoundA2780
ovarian CAPC3
prostate CAH460 SClung CA
LoVocolon CA
RPMI8226multiple myeloma
HS-Sultananaplastic NHL
CEP-18770 13.7 2.3 22.2 2.5 34.2 5 11.3 1.7 5.6 3.2 8.2 3.3
Bortezomib 1.7 0.4 4.8 1.2 4.8 0.5 0.97 0.2 2.7 0.4 4.1 0.4
Data are mean IC50 values (nM) plus or minus SD.CA indicate cancer; and NHL, non-Hodgkin lymphoma.
Figure 4. Cytotoxicity profile of CEP-18870 against normal human cells.(A,B) BMMCs and PBMCs were treated with control diluent, CEP-18770 (20 nM) orBOR (10 nM) for 48 hours. PBMCs were derived from 5 nonmyelomatous donors;BMMCs from 2 nonmyelomatous donors and from 7 patients with MM; cell viabilitywas measured by MTT assay. Cells were incubated in triplicate for each condition.Histograms represent means ( SD, error bars) of data derived from differentpatients, as indicated above in this paragraph. (C) BMMCs derived from 5 nonmyelo-matous donors and from 7 patients with MM were used to establish long-term BMSCcultures. BMSCs were treated with control diluent, CEP-18770 (20 nM), or BOR(10 nM) for 6 days. Apoptosis was measured by flow cytometry after TMRM staining.Histograms represent the means ( SD, error bars). (D) CEP-18870 inhibits theexpression of inflammatory cytokines and adhesion molecules in BMSCs. BMSCcultures, obtained as described above, were expanded for 4 to 6 passages andtreated with control diluent, CEP-18870 (20 nM), or bortezomib (BOR) (10 nM) for60 hours. Levels of VCAM1, ICAM1, IL6, IL1, VEGF isoforms, and 2-microglobulinmRNA were analyzed by semiquantitative RT-PCR. Fragment length is indicated;275 base pairs (bp) and 407 bp amplification fragments correspond to the knownVEGF isoforms of 121 and of 165 amino acids, respectively. BMSCs were derivedfrom 1 patient with MM (BMSC 1) and from 2 nonmyelomatous donors (BMSC 2 and3). (E,F) Hematotoxicity of CEP-18770 and BOR on bone marrow progenitors.BMMC derived from 16 patients with MM (PC � 10%) were pretreated with controldiluent, CEP-18870 (20 nM), or BOR (10 nM) for 48 hours. Cells were washed andcultured in cytokine-containing methylcellulose medium for granulocyte-macro-phages (CFU-GM) or erythroid (BFU-E) colony formation. (E) Early CFU-GM and(F) BFU-E-derived colonies were scored after 14 days of incubation. Histogramsrepresent the means ( SD; error bars). *P � .05; **P � .01 as analyzed byStudent t test.
2770 PIVA et al BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom
Collectively these results indicate that CEP-18770 is lesscytotoxic compared with bortezomib against normal human epithe-lial cells, bone marrow progenitors, and BMSCs at concentrationseffective in inducing apoptosis in a panel of human MM cell linesand primary human MM explants.
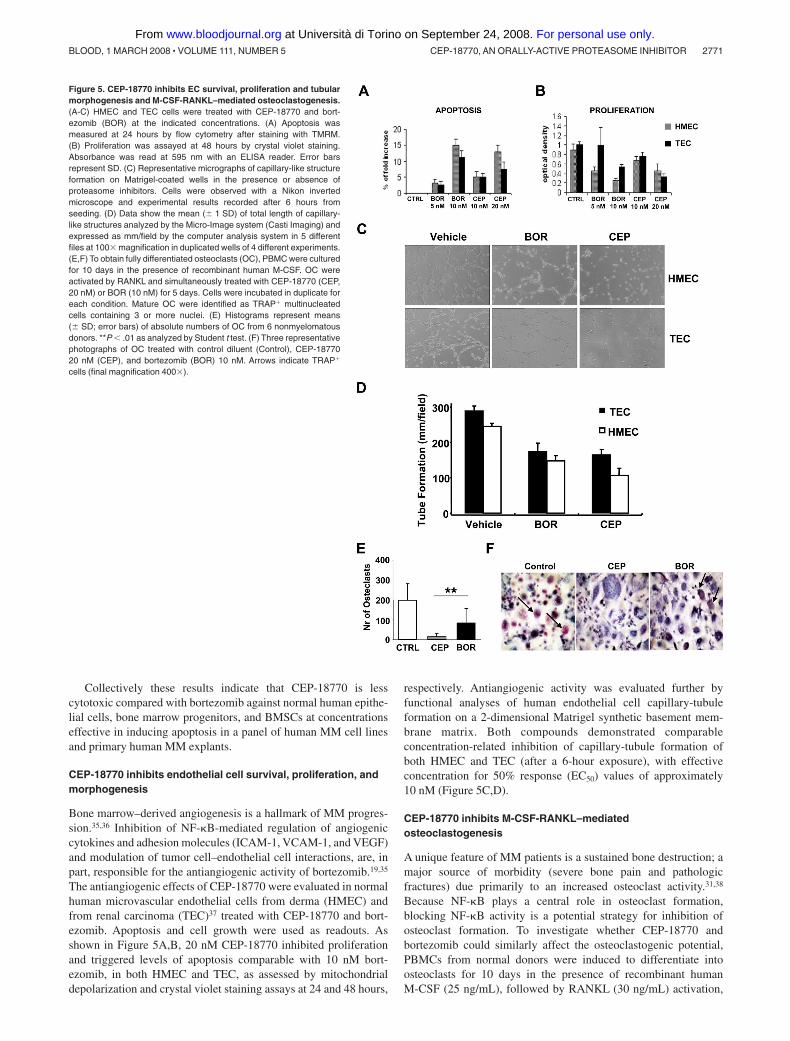
CEP-18770 inhibits endothelial cell survival, proliferation, andmorphogenesis
Bone marrow–derived angiogenesis is a hallmark of MM progres-sion.35,36 Inhibition of NF-�B-mediated regulation of angiogeniccytokines and adhesion molecules (ICAM-1, VCAM-1, and VEGF)and modulation of tumor cell–endothelial cell interactions, are, inpart, responsible for the antiangiogenic activity of bortezomib.19,35
The antiangiogenic effects of CEP-18770 were evaluated in normalhuman microvascular endothelial cells from derma (HMEC) andfrom renal carcinoma (TEC)37 treated with CEP-18770 and bort-ezomib. Apoptosis and cell growth were used as readouts. Asshown in Figure 5A,B, 20 nM CEP-18770 inhibited proliferationand triggered levels of apoptosis comparable with 10 nM bort-ezomib, in both HMEC and TEC, as assessed by mitochondrialdepolarization and crystal violet staining assays at 24 and 48 hours,
respectively. Antiangiogenic activity was evaluated further byfunctional analyses of human endothelial cell capillary-tubuleformation on a 2-dimensional Matrigel synthetic basement mem-brane matrix. Both compounds demonstrated comparableconcentration-related inhibition of capillary-tubule formation ofboth HMEC and TEC (after a 6-hour exposure), with effectiveconcentration for 50% response (EC50) values of approximately10 nM (Figure 5C,D).
CEP-18770 inhibits M-CSF-RANKL–mediatedosteoclastogenesis
A unique feature of MM patients is a sustained bone destruction; amajor source of morbidity (severe bone pain and pathologicfractures) due primarily to an increased osteoclast activity.31,38
Because NF-�B plays a central role in osteoclast formation,blocking NF-�B activity is a potential strategy for inhibition ofosteoclast formation. To investigate whether CEP-18770 andbortezomib could similarly affect the osteoclastogenic potential,PBMCs from normal donors were induced to differentiate intoosteoclasts for 10 days in the presence of recombinant humanM-CSF (25 ng/mL), followed by RANKL (30 ng/mL) activation,
Figure 5. CEP-18770 inhibits EC survival, proliferation and tubularmorphogenesis and M-CSF-RANKL–mediated osteoclastogenesis.(A-C) HMEC and TEC cells were treated with CEP-18770 and bort-ezomib (BOR) at the indicated concentrations. (A) Apoptosis wasmeasured at 24 hours by flow cytometry after staining with TMRM.(B) Proliferation was assayed at 48 hours by crystal violet staining.Absorbance was read at 595 nm with an ELISA reader. Error barsrepresent SD. (C) Representative micrographs of capillary-like structureformation on Matrigel-coated wells in the presence or absence ofproteasome inhibitors. Cells were observed with a Nikon invertedmicroscope and experimental results recorded after 6 hours fromseeding. (D) Data show the mean ( 1 SD) of total length of capillary-like structures analyzed by the Micro-Image system (Casti Imaging) andexpressed as mm/field by the computer analysis system in 5 differentfiles at 100� magnification in duplicated wells of 4 different experiments.(E,F) To obtain fully differentiated osteoclasts (OC), PBMC were culturedfor 10 days in the presence of recombinant human M-CSF. OC wereactivated by RANKL and simultaneously treated with CEP-18770 (CEP,20 nM) or BOR (10 nM) for 5 days. Cells were incubated in duplicate foreach condition. Mature OC were identified as TRAP� multinucleatedcells containing 3 or more nuclei. (E) Histograms represent means( SD; error bars) of absolute numbers of OC from 6 nonmyelomatousdonors. **P � .01 as analyzed by Student t test. (F) Three representativephotographs of OC treated with control diluent (Control), CEP-1877020 nM (CEP), and bortezomib (BOR) 10 nM. Arrows indicate TRAP�
cells (final magnification 400�).
CEP-18770, AN ORALLY-ACTIVE PROTEASOME INHIBITOR 2771BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

for additional 5 days. Numerous, large tartrate-resistant acidphosphatase (TRAP)–positive osteoclasts were reproducibly gener-ated (osteoclasts averaged 195 127 per well). Consistent withearlier studies,39,40 both CEP-18770 or bortezomib added simulta-neously with RANKL dramatically suppressed osteoclastogenesis(Figure 5E,F). Importantly, CEP-18770 resulted in a more pro-nounced inhibition of osteoclast formation (13 8; P � .003 vsbortezomib) compared with that of bortezomib (83 100; P � .06vs controls). These findings indicate that CEP-18770 is a potentinhibitor of RANKL-mediated osteoclastogenesis in vitro.
Efficacy profile of CEP-18770 in human MM models in mice
The in vivo antitumor efficacy of CEP-18770 compared withbortezomib was examined first in a series of studies using thehuman MM RPMI 8226 subcutaneous xenograft model in SCIDmice after repeated intravenous or oral administration. CEP-18770 exhibited sustained and dose-related (from 1.5 to 4 mg/kgintravenously, 2q7d�8 injections) relative tumor weight inhibi-tion (RTWI � 100% at all tested doses, P � .001 vs untreatedgroups). Bortezomib at doses of 0.8, 1.0 and 1.2 mg/kg (2q7d�8injections) was similarly efficacious in inhibiting MM growth(RTWI from 93% to 98%, P � .001 vs untreated group) with acomparable tumor growth delay profile. CEP-18770 administra-tion, however, resulted in dose-related induction of completetumor regressions (CR from 78% to 100%), as compared withbortezomib treatment, which resulted in a 50% incidence of CRat its MTD of 1.2 mg/kg intravenously (Figure 6A, and data notshown). In contrast to bortezomib, CEP-18770 exhibited dose-related increases in the incidence of tumor-free mice by thecompletion of these studies (120 days after tumor transplanta-tion); within the 3- and 4-mg/kg intravenous treatment groups,89% and 80%, respectively, of CEP-18770–treated mice weretumor-free and treatment was well tolerated. Oral administrationof CEP-18770 resulted in a significant reduction of tumorweight (P � .001) and notable dose-related incidence ofcomplete tumor regression (75% incidence of CR and 25%
tumor-free mice at 10 mg/kg orally; 100% incidence of CRand tumor-free mice at 13 mg/kg orally) with minimal changesin animal body weight over the course of 120 day studies(Figure 6B).
To compare CEP-18770 and bortezomib inhibition of protea-some activity in tumors and peripheral normal host tissues, their exvivo proteasome pharmacodynamic (PD) inhibition profiles werecharacterized in RPMI 8266 MM tumor-bearing SCID mice.Relative to bortezomib, equiactive doses of CEP-18770 demon-strated a greater and more sustained dose-related inhibition oftumor proteasome activity, corresponding temporally with maxi-mum induction of caspase-3 and 7 activity (Figure 6C,D, and datanot shown). The maximum apoptotic signal was 2.5 fold greater forCEP-18770 versus bortezomib. In contrast, proteasome inhibitionprofiles of CEP-18870 and bortezomib were comparable in thenormal peripheral mouse tissues examined (liver, lungs, wholeblood, and brain [no activity]), in both their magnitude and theirduration. No proteasome inhibition was detected in brain tissue atany time point for either compound.
The antitumor efficacy profile of CEP-18770 relative to bort-ezomib was further evaluated in a ARP-1 systemic model of humanMM. Female NOD/SCID mice were implanted intravenously withARP-1 human MM cells and myelomatous mice (confirmed basedupon plasma human IgA levels) were randomized and administeredCEP-18770 or bortezomib (2q7d�8 injections) using dosingregimens comparable with those described in subcutaneous MMxenograft studies. Analyses of median survival data obtained withCEP-18770, bortezomib, and standard-of-care therapeutic agentsrelative to vehicle-treated mice are summarized in Table 2. Thesedata indicate comparable and similarly robust median survivalbenefits for twice-weekly administration of CEP-18770 (at 3 and4 mg/kg intravenously) and bortezomib (at 1.2 mg/kg intrave-nously), as well as high-dose dexamethasone administration.Melphalan treatment was associated with the greatest survivalbenefit relative to vehicle control mice (P � .001).
Figure 6. Antitumor efficacy and pharmacodynamicprofile of proteasome inhibition of CEP-18770 andbortezomib in MM tumor xenografts. RPMI 8226human MM xenografts were implanted subcutaneouslyin SCID mice. Treatment started after establishing apalpable subcutaneous tumor (100-140 mm3). Represen-tative intravenous and oral efficacy data are shown.(A) CEP-18770 and bortezomib were administered intrave-nously, twice a week for 4 weeks (2q7d�8 injections) at thedoses indicated as a solution of 10% Solutol HS15/87%buffered saline/3% DMSO in a volume of 10 mL/kg bodyweight mouse. (B) CEP-18770 was administered orally (p.o.)in a solution of 3% DMSO, 10% Solutol, and 87% sterile NaCl0.9% twice a week for 4 weeks at the indicated doses in avolume of 20 mL/kg body weight mouse. Tumor parameterswere measured and analyzed as detailed in “Subcutaneoustumor xenograft models.” (C,D) Pharmacodynamic profile ofproteasome inhibition in RPMI 8226 xenografts and normalperipheral tissues of mice at the maximum tolerated doses(MTD) of CEP-18770 and bortezomib administered intrave-nously in SCID mice. Time-course proteasome inhibition aftera single intravenous administration was measured by an exvivo fluorimetric kinetic assay of chymotrypsin-like activity.Proteasome percentage inhibition (PI %) in sample tissuelysates from treated versus vehicle-treated control mice wascalculated as: (Slope of control tissues � slope of treatedtissues) / (Slope of control tissues � 100) after normalizationfor protein content of tissue and hemoglobin content of wholeblood. Results shown are means ( standard deviation) of3 independent experiments (3 mice per time point).
2772 PIVA et al BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

Discussion
The ubiquitin proteasome pathway represents a promising targetfor cancer therapy, because of the higher sensitivity of cancer cellsto cytotoxic effects of proteasome inhibition compared with normalcells.6,19 To date, bortezomib is the only proteasome inhibitorcurrently approved for human use, despite relevant side effects,including peripheral neuropathy, orthostatic hypotension, pyrexia,gastrointestinal symptoms, thrombocytopenia, asthenia, andpain.14,15 Because targeting the ubiquitin-proteasome system isanticipated as a powerful strategy for the treatment of a largespectrum of human malignancies, the discovery of new antiprotea-some drugs with a more favorable profile and enhanced tolerabilityis recommended.20,22,41
A primary rationale for the therapeutic use of proteasomeinhibitors in oncology relies on their ability to inhibit NF-�Bactivity. The inhibition of NF-�B activity reduces the expression ofseveral target genes regulating cell proliferation and survival ofcancer cells.11-13 Therefore, the modulation of NF-�B activityoffers significant therapeutic results in numerous malignancies,including MM, lymphomas, and several solid tumors. Notably,NF-�B also mediates important cellular functions in normalstromal and lymphoid cells, key players regulating the growth,survival, and apoptosis of tumor cells, as well as in initiatingspecific antitumor immune responses in MM patients. This reportdescribes the pharmacologic and activity profile of the novel P2threonine boronic acid proteasome inhibitor, CEP-18770, a potentinhibitor of both constitutive and TNF-� triggered NF-�B activa-tion. Of note in these studies was the observation that CEP-18770and bortezomib exhibit a comparable cytotoxic profile against apanel of human MM cell lines in vitro and in explant cultures ofpurified CD138 positive cells derived from bone marrow aspiratesof MM subjects.
Given that protein degradation and proteasome activity arerequired for normal cell survival and proliferation, it was of interestto compare the effects of CEP-18770 in normal human cells.CEP-18770 demonstrated a marked reduction in toxicity towardhuman bone marrow progenitors, bone marrow stromal cells, andnormal human intestinal cells compared with bortezomib despitethe ability of CEP-18770 to efficiently inhibit the secretion ofBMSC-derived growth factors, cytokines, and adhesion molecules(IL-6, IL1-, and VCAM1). The down-modulation of thesemolecules by CEP-18770 may affect the interaction of tumor cellswith their microenvironment, impacting tumor growth, survival,and immune surveillance. Similarly, CEP-18770 abrogates VEGFproduction in both MM and BMSC cells, suggesting an inhibitoryeffect on both MM cell migration and vasculogenesis fromendothelial progenitors. Inhibition of angiogenesis is also sup-ported by a direct effect of CEP-18770 on endothelial cellproliferation, survival, and capillary tubular morphogenesis. Collec-tively, these data support the view that CEP-18770 exerts bothdirect and an indirect antiangiogenic activity on endothelial cells,defining a supplementary mechanism that may contribute to itsanti-MM activity.
A hallmark in the clinical presentation of MM patients is lyticbone disease, a consequence of enhanced differentiation andactivity of osteoclasts. Osteoclastogenesis can be driven directly bycell–cell contact with tumor cells, or by a paracrine stimulationinduced by circulating factors such as IL-1, IL-6, PTHrP, MIP-1�,and RANKL, produced by tumor and/or surrounding stromalcells.42,43 Remarkably, CEP-18770 was significantly more effectiveTa
ble
2.E
ffec
tso
fCE
P-1
8770
,bo
rtez
om
ib(B
OR
),h
igh
-do
sed
exam
eth
aso
ne
(HD
Dex
),an
dm
elp
hal
an(M
LP
)on
surv
ival
inth
esy
stem
icA
RP
-1h
um
anm
ult
iple
mye
lom
am
od
el
Su
rviv
alV
ehic
leg
rou
p
CE
P-1
8770
3m
g/k
gin
trav
eno
usl
yo
nce
wee
kly
CE
P-1
8770
3m
g/k
gin
trav
eno
usl
ytw
ice
wee
kly
CE
P-1
8770
4m
g/k
gin
trav
eno
usl
yo
nce
wee
kly
CE
P-1
8770
4m
g/k
gin
trav
eno
usl
ytw
ice
wee
kly
BO
R1.
2m
g/k
gin
trav
eno
usl
yo
nce
wee
kly
BO
R1.
2m
g/k
gin
trav
eno
usl
ytw
ice
wee
kly
HD
Dex
1.5
mg
/kg
intr
aper
ito
nea
llyo
nce
dai
ly
ML
P10
mg
/kg
intr
aper
ito
nea
llyo
nce
dai
ly
Mea
n,d
34.8
60.4
75.8
54.5
63.6
58.3
58.7
56.2
90.5
Med
ian,
d34
5161
.550
6547
5957
100
SE
Mof
med
ian
surv
ival
,d5.
510
.513
.710
.411
.311
.26.
75.
79.
3
Pva
lues
vers
usve
hicl
e—
.06
.02
.14
.05
.09
.02
.02
�.0
01
Sur
viva
ldat
afr
omK
apla
n-M
eier
anal
yses
ofdi
ffere
ntdo
sing
sche
dule
sof
CE
P-1
8770
and
bort
ezom
ibre
lativ
eto
that
achi
eved
with
first
-lin
est
anda
rd-o
f-ca
reth
erap
ies
used
fort
hetr
eatm
ento
fMM
.Det
ails
and
stat
istic
alan
alys
esof
data
are
desc
ribed
in“S
yste
mic
hum
anm
ultip
lem
yelo
ma
mod
el.”
—in
dica
tes
nota
pplic
able
.
CEP-18770, AN ORALLY-ACTIVE PROTEASOME INHIBITOR 2773BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

than bortezomib in inhibiting RANKL-induced osteoclast differen-tiation in vitro. These findings could indicate a potential benefit ofCEP-18770 in managing the osteolytic-related morbidity andmortality associated with the progression of MM.
The proteasome pharmacodynamic profile demonstrated that inMM tumor xenografts CEP-18770 achieved a superior and persis-tent dose-related inhibition of proteasome activity after intravenousadministration, compared with normal peripheral murine tissues.Moreover, the tumor proteasome inhibition profile and induction oftumor apoptosis achieved by the intravenous MTD of CEP-18770was comparable with that achieved after single-dose oral adminis-tration of CEP-18770 at 10 and 13 mg/kg (oral MTD in mice).These PD effects achieved with intravenous or oral administrationof CEP-18770 were directly related to significant and sustainedantitumor efficacy and median survival benefits in subcutaneousand systemic models of human MM, respectively, upon chronicadministration.
In summary our data show that the novel, water-soluble andorally bioavailable proteasome inhibitor CEP-18770 exhibits(1) low-nanomolar inhibition of chymotrypsin-like proteasomeactivity in silico and in vitro; (2) suppression of NF-�B–mediatedsignaling pathways and transcriptional targets of NF-�B;(3) concentration-related induction of apoptotic cell death inhuman MM and tumor-derived cell lines and in primary purifiedCD138 positive MM cells in the absence of intrinsic DNAintercalating or topoisomerase inhibitory activities; (4) a favorablecytotoxicity profile toward normal human endothelial cells, bonemarrow progenitors, and BMSCs relative to bortezomib;(5) inhibition of endothelial cell survival and morphogenesis invitro; (6) potent inhibition of RANKL-induced osteoclastogenesiscompared with bortezomib; and (7) significant and sustained tumorproteasome inhibition and antitumor efficacy and survival benefitsin preclinical models of human MM. The in vitro and in vivoantitumor and anticlastogenic pharmacologic profile of CEP-18870and its diminished cytotoxicity against a variety normal human cell
lineages compared with tumor cells provide the rationale for furtherstudies evaluating its preclinical and clinical efficacy in MM andother hematologic malignancies.
Acknowledgments
The discovery and development of CEP-18770 was supported byCephalon, West Chester, PA, in collaboration with Cell TherapeuticsEurope, Bresso, Italy. Additional support for the studies described wasprovided by Ministero dell’Universita e Ricerca Scientifica (MIUR),Regione Piemonte, Compagnia di San Paolo, Torino (Progetto Oncolo-gia), andAssociazione Italiana per la Ricerca sul Cancro (AIRC). R.P. issupported by the MIUR program “Incentivazione alla mobilita distudiosi residenti all’estero.”
Authorship
Contribution: R.P., G.C., I.T., D.F., V.G., M. Coscia, S.P., M.M.,G.P., C.A., N.P., M. Cassin, S.d.G., P.N., P.d.F., I.S., I.R., R.F.,B.B., G.C., S.J.-B., K.H., H.Z., A.N., A.P., C.B., H.O., and A.B.designed, performed, and analyzed experimental data; and R.P.,B.R., M.W., and G.I. oversaw the direction of all experimentalstudies and wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests. Cephalon, Sede Secondaria Della CellTherapeutics, and RBM have no commercial products that arediscussed in the text of this manuscript. The compound CEP-18770is in clinical development as an oncology agent but is not acommercial or marketed product now.
Correspondences: Giorgio Inghirami, Center for ExperimentalResearch and Medical Studies (CeRMS), Department of Biomedi-cal Sciences and Human Oncology, University of Torino, ViaSantena 5, 10126 Torino, Italy; e-mail: [email protected].
References
1. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction forthe sake of construction. Physiol Rev. 2002;82:373-428.
2. Rajkumar SV, Richardson PG, Hideshima T,Anderson KC. Proteasome inhibition as a noveltherapeutic target in human cancer. J Clin Oncol.2005;23:630-639.
3. Hideshima T, Richardson P, Chauhan D, et al.The proteasome inhibitor PS-341 inhibits growth,induces apoptosis, and overcomes drug resis-tance in human multiple myeloma cells. CancerRes. 2001;61:3071-3076.
4. Adams J. The development of proteasome inhibi-tors as anticancer drugs. Cancer Cell. 2004;5:417-421.
5. Cavo M. Proteasome inhibitor bortezomib for thetreatment of multiple myeloma. Leukemia. 2006;20:1341-1352.
6. Nalepa G, Rolfe M, Harper JW. Drug discovery inthe ubiquitin-proteasome system. Nat Rev DrugDiscov. 2006;5:596-613.
7. Pagano M, Tam SW, Theodoras AM, et al. Role ofthe ubiquitin-proteasome pathway in regulatingabundance of the cyclin-dependent kinase inhibi-tor p27. Science. 1995;269:682-685.
8. Bloom J, Amador V, Bartolini F, DeMartino G, Pa-gano M. Proteasome-mediated degradation ofp21 via N-terminal ubiquitinylation. Cell. 2003;115:71-82.
9. Karin M, Ben-Neriah Y. Phosphorylation meets
ubiquitination: the control of NF-[kappa]B activity.Annu Rev Immunol. 2000;18:621-663.
10. Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB incancer: from innocent bystander to major culprit.Nat Rev Cancer. 2002;2:301-310.
11. Tergaonkar V, Pando M, Vafa O, Wahl G, VermaI. p53 stabilization is decreased upon NFkappaBactivation: a role for NFkappaB in acquisition ofresistance to chemotherapy. Cancer Cell. 2002;1:493-503.
12. Nakanishi C, Toi M. Nuclear factor-kappaB inhibi-tors as sensitizers to anticancer drugs. Nat RevCancer. 2005;5:297-309.
13. Piva R, Belardo G, Santoro MG. NF-kappaB: astress-regulated switch for cell survival. AntioxidRedox Signal. 2006;8:478-486.
14. Dispenzieri A. Bortezomib for myeloma – muchado about something. N Engl J Med. 2005;352:2546-2548.
15. Richardson PG, Sonneveld P, Schuster MW, etal. Bortezomib or high-dose dexamethasone forrelapsed multiple myeloma. N Engl J Med. 2005;352:2487-2498.
16. O’Connor OA. Marked clinical activity of the pro-teasome inhibitor bortezomib in patients with fol-licular and mantle-cell lymphoma. Clin Lym-phoma Myeloma. 2005;6:191-199.
17. Fisher RI, Bernstein SH, Kahl BS, et al. Multi-center phase II study of bortezomib in patientswith relapsed or refractory mantle cell lymphoma.J Clin Oncol. 2006;24:4867-4874.
18. Richardson PG, Barlogie B, Berenson J, et al. A
phase 2 study of bortezomib in relapsed, refrac-tory myeloma. N Engl J Med. 2003;348:2609-2617.
19. Ludwig H, Khayat D, Giaccone G, Facon T. Pro-teasome inhibition and its clinical prospects in thetreatment of hematologic and solid malignancies.Cancer. 2005;104:1794-1807.
20. Chauhan D, Catley L, Li G, et al. A novel orallyactive proteasome inhibitor induces apoptosis inmultiple myeloma cells with mechanisms distinctfrom Bortezomib. Cancer Cell. 2005;8:407-419.
21. Ruiz S, Krupnik Y, Keating M, Chandra J, Palla-dino M, McConkey D. The proteasome inhibitorNPI-0052 is a more effective inducer of apoptosisthan bortezomib in lymphocytes from patientswith chronic lymphocytic leukemia. Mol CancerTher. 2006;5:1836-1843.
22. Demo E, Frush D, Gottfried M, et al. Glycogenstorage disease type III-hepatocellular carcinomaa long-term complication? J Hepatol. 2007;46:492-498.
23. Berkers CR, Verdoes M, Lichtman E, et al. Activ-ity probe for in vivo profiling of the specificity ofproteasome inhibitor bortezomib. Nat Methods.2005;2:357-362.
24. Piva R, Chiarle R, Manazza AD, et al. Ablation ofoncogenic ALK is a viable therapeutic approachfor anaplastic large-cell lymphomas. Blood. 2006;107:689-697.
25. Lombardi L, Poretti G, Mattioli M, et al. Molecularcharacterization of human multiple myeloma celllines by integrative genomics: insights into the
2774 PIVA et al BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom

biology of the disease. Genes ChromosomesCancer. 2007;46:226-238.
26. Verdelli D, Mattioli M, Fabris S, et al. Molecularand biological characterization of three novelinterleukin-6-dependent human myeloma celllines. Haematologica. 2005;90:1541-1548.
27. Conaldi PG, Serra C, Mossa A, et al. Persistentinfection of human vascular endothelial cells bygroup B coxsackieviruses. J Infect Dis. 1997;175:693-696.
28. Bussolati B, Deambrosis I, Russo S, DeregibusMC, Camussi G. Altered angiogenesis and sur-vival in human tumor-derived endothelial cells.Faseb J. 2003;17:1159-1161.
29. Sun X, Gulyas M, Hjerpe A, Dobra K. Proteasomeinhibitor PSI induces apoptosis in human me-sothelioma cells. Cancer Lett. 2006;232:161-169.
30. Piva R, Gianferretti P, Ciucci A, Taulli R, BelardoG, Santoro MG. 15-Deoxy-delta 12,14-prosta-glandin J2 induces apoptosis in human malignantB cells: an effect associated with inhibition of NF-kappa B activity and down-regulation of antiapo-ptotic proteins. Blood. 2005;105:1750-1758.
31. Berenson JR. Advances in the biology and treat-
ment of myeloma bone disease. Semin Oncol.2002;29:11-16.
32. Mulligan G, Mitsiades C, Bryant B, et al. Geneexpression profiling and correlation with outcomein clinical trials of the proteasome inhibitor bort-ezomib. Blood. 2007;109:3177-3188.
33. Klein B, Zhang XG, Jourdan M, et al. Paracrinerather than autocrine regulation of myeloma-cellgrowth and differentiation by interleukin-6. Blood.1989;73:517-526.
34. Chauhan D, Uchiyama H, Akbarali Y, et al. Mul-tiple myeloma cell adhesion-induced interleukin-6expression in bone marrow stromal cells involvesactivation of NF-kappa B. Blood. 1996;87:1104-1112.
35. Roccaro AM, Hideshima T, Raje N, et al. Bort-ezomib mediates antiangiogenesis in multiplemyeloma via direct and indirect effects on endo-thelial cells. Cancer Res. 2006;66:184-191.
36. Cavo M, Baccarani M. The changing landscapeof myeloma therapy. N Engl J Med. 2006;354:1076-1078.
37. Bruno S, Bussolati B, Grange C, et al. CD133�renal progenitor cells contribute to tumor angio-genesis. Am J Pathol. 2006;169:2223-2235.
38. Terpos E, Dimopoulos MA. Myeloma bone dis-ease: pathophysiology and management. AnnOncol. 2005;16:1223-1231.
39. Shishodia S, Gutierrez AM, Lotan R, AggarwalBB. N-(4-hydroxyphenyl)retinamide inhibits inva-sion, suppresses osteoclastogenesis, and po-tentiates apoptosis through down-regulation ofI(kappa)B(alpha) kinase and nuclear factor-kappaB-regulated gene products. Cancer Res.2005;65:9555-9565.
40. Zavrski I, Krebbel H, Wildemann B, et al. Protea-some inhibitors abrogate osteoclast differentia-tion and osteoclast function. Biochem BiophysRes Commun. 2005;333:200-205.
41. Williamson MJ, Blank JL, Bruzzese FJ, et al.Comparison of biochemical and biological effectsof ML858 (salinosporamide A) and bortezomib.Mol Cancer Ther. 2006;5:3052-3061.
42. Boyle WJ, Simonet WS, Lacey DL. Osteoclastdifferentiation and activation. Nature. 2003;423:337-342.
43. Sezer O, Heider U, Zavrski I, Kuhne CA, Hof-bauer LC. RANK ligand and osteoprotegerin inmyeloma bone disease. Blood. 2003;101:2094-2098.
CEP-18770, AN ORALLY-ACTIVE PROTEASOME INHIBITOR 2775BLOOD, 1 MARCH 2008 � VOLUME 111, NUMBER 5
For personal use only. at Università di Torino on September 24, 2008. www.bloodjournal.orgFrom




















