
Frequent detection of human cytomegalovirus in neuroblastoma: A novel therapeutic target?
Upload
independentCategory
view
0download
0
This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955
Available online at www.sciencedirect.com
Cell type-specific activation of the cytomegalovirus promoterby dimethylsulfoxide and 5-Aza-2′-deoxycytidine
Prakash Radhakrishnan a,1, Hesham Basma a,1, David Klinkebiel a,Judith Christman a,c, Pi-Wan Cheng a,b,c,∗
a Department of Biochemistry and Molecular Biology, College of Medicine, University of Nebraska Medical Center,Omaha, NE 68198-5870, USA
b Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center,Omaha, NE 68198-5870, USA
c Eppley Institute for Research in Cancer and Allied Diseases, University of NebraskaMedical Center, Omaha, NE 68198-5870, USA
Received 20 October 2007; received in revised form 17 January 2008; accepted 10 February 2008Available online 7 March 2008
Abstract
The cytomegalovirus promoter is a very potent promoter commonly used for driving the expression of transgenes, though itgradually becomes silenced in stably transfected cells. We examined the methylation status of the cytomegalovirus promoter intwo different cell lines and characterized its mechanisms of activation by dimethylsulfoxide and 5-Aza-2′-deoxycytidine. Thecytomegalovirus promoter stably transfected into Chinese hamster ovary cells is suppressed by DNA methylation-independentmechanisms, which is different from the rat embryonic cardiomyoblast H9c2-Fluc.3 cells in which the cytomegalovirus promoteris silenced by methylation. Dimethylsulfoxide and 5-Aza-2′-deoxycytidine can activate the cytomegalovirus promoter in both celltypes by overlapping mechanisms. Dimethylsulfoxide activates the cytomegalovirus promoter in Chinese hamster ovary cells bypromoting histone acetylation and the activation of p38 mitogen-activated protein kinase and nuclear factor �B (NF�B) signalingpathways, while 5-Aza-2′-deoxycytidine increases histone acetylation and activates the nuclear factor �B but not the p38 mitogen-activated protein kinase pathway. In H9c2-Fluc.3 cells, both agents promote demethylation of the cytomegalovirus promoter, andenhance its activity exclusively through activation of the nuclear factor �B pathway and to a lesser extent of the p38 mitogen-activated protein kinase pathway. Our findings suggest that suppression and activation of the cytomegalovirus promoter are celltype-specific. These results may be used for developing strategies to enhance the expression of transgenes and the production ofrecombinant proteins encoded by transgenes controlled by a cytomegalovirus promoter.© 2008 Elsevier Ltd. All rights reserved.
Keywords: Cytomegalovirus promoter; Dimethylsulfoxide; 5-Aza-2′-deoxycytidine; NF�B; p38 MAP kinase
∗ Corresponding author at: Department of Biochemistry and Molec-ular Biology, College of Medicine, University of Nebraska MedicalCenter, Omaha, NE 68198-5870, USA. Tel.: +1 402 559 5776;fax: +1 402 559 6650.
E-mail address: [email protected] (P.-W. Cheng).1 They contributed equally to this study.
1. Introduction
The immediate/early promoter enhancer ofcytomegalovirus (CMV promoter) is one of themost commonly used in vitro and in vivo promotersfor driving the expression of transgenes in mammaliancells because of its strong promoter activity. However,
1357-2725/$ – see front matter © 2008 Elsevier Ltd. All rights reserved.doi:10.1016/j.biocel.2008.02.014

Author's personal copy
P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955 1945
many transgenes under the control of viral promoters,including the CMV promoter, have been found tobe gradually silenced over a period of months inculture (Choi, Basma, Singh, & Cheng, 2005; Knust,Bruggemann, & Doerfler, 1989; Palmer, Rosman,Osborne, & Miller, 1991; Verma & Somia, 1997). Thegene silencing mechanism of these promoters is notclear, although DNA methylation, histone hypoacety-lation, activation of signaling molecules and alteredlevels of transcriptional factors have been suggested(Swindle & Klug, 2002). In eukaryotes, particularlyin mammals, methylation is involved in the formationof transcriptionally repressive chromatin states andmaintenance of genome stability. The repression is oftenbrought about by methylation of CpG dinucleotidesclustered to form CpG Islands which are usually locatedin the promoter and the 5′ exon/intron region of agene. Proposed mechanisms for inhibition of geneexpression due to promoter methylation include directinterference of methyl group in binding of transcriptionfactors due to steric hindrance, methyl groups mediatingthe binding of certain proteins to DNA by forming aprotein complex that blocks the binding of transcriptionfactors and methyl groups affecting DNA–histoneinteraction, resulting in chromatin compaction, therebypreventing transcription (Esteller, Corn, Baylin, &Herman, 2001; Esteller & Herman, 2002; Singal &Ginder, 1999). The acetylation status of lysine sidechains in histones also can regulate gene expression.Hypoacetylation of histones leads to the condensa-tion of DNA through interactions of the free lysineresidues in histones and DNA backbones, resulting insuppression of gene expression. Measurement of thestatus of DNA methylation and histone acetylation inthe promoter regions can serve as an indicator of thetranscription activity of a specific gene (Grassi et al.,2003).
We observed a several-fold higher �-galactosidase(�Gal) activity in the pCMVLacZ stably transfectedCHO cells (CHO-LacZ) freshly prepared from frozenstocks when compared with same cells maintained inculture. Upon subculture of the freshly recovered cells,the �-Gal activity returned to the same level as cellswhich had been maintained in culture. The only dif-ference between these two culture conditions was thepresence of 2.5% dimethylsulfoxide (DMSO) in the cul-ture medium containing freshly recovered cells. Thisamount of DMSO was carry over from the 10% DMSOused for cryopreservation of the cells. This observationled to the current study.
DMSO is a water-soluble organic solvent routinelyused in cell biology. Its application includes cryopro-
tection of cells (Karlsson et al., 1993), enhancementof stem cell differentiation in cardiac-, smooth- andskeletal-muscle, blood and neural cell lineages (Bonser,Siegel, McConnell, & Cuatrecasas, 1981; Dinsmore,Ratliff, Jacoby, Wunderlich, & Lindberg, 1998; Pruitt,1994). Some DMSO actions, such as cryopreservationof cells, may be attributed to its capability to stabi-lize the liquid matrix of cell membranes (Yu & Quinn,1994). Mechanisms of other DMSO actions includingour initial finding are not well understood. Our obser-vation that the CMV promoter in CHO cells was notmethylated, coupled with the finding that this promoterwas activated by DMSO and 5-Aza-2′-deoxycytidine(5-Aza CdR), an agent known to inhibit DNA methy-lation (Krishnan et al., 2006), prompted us to study themechanisms of the activation of unmethylated CMV pro-moter. To get a better understanding of the mechanismsof the activation of the CMV promoter in general, wealso examined the mode of action of these two agents inrat embryonic cardiomyoblast H9c2-Fluc.3 cells. Thesecells contain a luciferase gene controlled by the CMVpromoter, which was inactivated by methylation after 66passages (Krishnan et al., 2006). The results of the stud-ies suggest that suppression and activation of the CMVpromoter are cell type-specific.
2. Materials and methods
2.1. Cell culture
CHO cells were grown in F12 Ham medium supple-mented with 7.5% heat-inactivated fetal bovine serum(FBS), 50 U/mL penicillin and 50 �g/mL streptomycinand 2 mM l-glutamine. The rat embryonic cardiomy-oblast H9c2-Fluc.3 cells (passage 66) which wereoriginally prepared by stable transfection with CMV-luciferase gene was a kind gift from Dr. Joseph C.Wu, Stanford University School of Medicine, Stanford,CA. The passage 66 H9c2-Fluc.3 cells, which had theCMV promoter inactivated by methylation (Krishnanet al., 2006) were grown in DMEM medium, supple-mented with 5% fetal bovine serum (FBS), 10 units/mLpenicillin and 10 �g/mL streptomycin. All cell culturereagents were from GIBCOBRL (Carlsbad, CA).
2.2. Construction of the expression vector andgeneration of stable cell clones
Bovine C2GnT-1(L) (Li, Adler, & Cheng, 1998)was cloned into the mammalian expression vec-tor, pcDNA3/Zeocin (Beum, Singh, Burdick,Hollingsworth, & Cheng, 1999), which contained

Author's personal copy
1946 P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955
a CMV promoter and a bovine growth hormonepolyadenylation signal (Invitrogen, Carlsbad, CA,USA) to generate pCMV-C2GnT-L. PCMV-C2GnT-Lwas transfected into CHO cells to generate stableclones, CHO-C2GnT-L, selected for resistance tozeocin (300 �g/mL). CHO cells stably transfectedwith pCMVLacZ (CHO-LacZ) were generated bytransfection with pcDNA6-LacZ/bsd (Invitrogen) andselected with blasticidin (10 �g/mL).
2.3. Quantitation of LacZ expression after transienttransfection
Transient transfections were performed as describedpreviously except for the following modification (Seki,Iwakawa, Cheng, & Cheng, 2002). Each well of the48-well plate was exposed to a transfection solution con-taining 5 �g Maackia amurensis agglutinin (EY Lab, Inc,San Mateo, CA) ligand, 3 �g DMRIE-cholesterol lipo-some (Invitrogen, Carlsbad, CA) and 1 �g pCMVLacZDNA. �-Gal activity in the transfected cells was mea-sured after 24 h culture.
2.4. β-Gal assay
pCMV CHO-LacZ #3 cells were treated with vari-ous agents, including DMSO, 5-Aza CdR (Sigma, USA),SB203580 (SB) (EMD Calbiochem, USA) and Wedelo-lactone (WL) (EMD Calbiochem). After 24 h treatmentwith DMSO or 5-Aza CdR, cells were measured for �-Gal activity using a Mammalian �-Gal Assay ReagentKit (Pierce) as described previously (Yanagihara, Cheng,& Cheng, 2000). A UVmax microplate reader was usedfor measuring the optical density at 404 nm (Molec-ular Devices, Sunnyvale, CA), which was convertedto the amounts of �-Gal using Escherichia coli �-Galenzyme as the standard. For inhibitor treatment, the cellswere incubated with a MAP kinase inhibitor (SB203580,30 �M) or IKK � and � kinase inhibitor (Wedelolactone,100 �M) for 1 h prior to DMSO treatment. The enzymeactivity was expressed as nanogram �-Gal activity/�gprotein.
2.5. β1-6 N-Acetylglucosaminyltransferase(C2GnT) assay
C2GnT activity was assayed using total cell lysatesprepared from T25 flasks as described previously(Ropp, Little, & Cheng, 1991). Aliquots of thehomogenates were measured for enzyme activity usingUDP-[3H]GlcNAc (∼3500 dpm/nmol, American Radio-labeled Chemicals, St. Louis, MO) as the sugar donor and
2 mM Gal�1,3GalNAc�-O-benzyl (Toronto ResearchChemicals Inc. Downsview, Ontario, Canada) as theacceptor. The products were isolated with C18 car-tridges and quantitated using a Liquid ScintillationCounter (Packard, Meriden, CT). Protein concentra-tion was measured with Coomassie Plus Protein AssayReagent (Pierce, Rockford, IL) using bovine serumalbumin as the standard. Enzyme activity was calcu-lated by subtracting the endogenous activity measuredwithout exogenous acceptor from total activity andexpressed as nmol of sugar donor transferred/h/mg pro-tein.
2.6. Luciferase assay
H9c2 Fluc.3 cells were grown in 48-well plates.After treatment with DMSO or 5-Aza CdR for 24 h,the cells were measured for luciferase activity using aLuciferase assay system with a Reporter Lysis buffer kitas described by manufacturer protocol (Promega corpo-ration, Madison, WI). The light produced was measuredon a TD-20/20 Luminometer (Turner Designs, Sunny-vale, CA). Protein was measured as described above.Data were expressed as relative light units per �g pro-tein. The inhibitor study was performed as describedabove.
2.7. Western blot analysis
CHO-LacZ cells were grown in T-25 flasks untilthey were 80% confluent, and then treated with 2–4%DMSO for 24 h. Cell lysates were resolved on SDS-polyacrylamide gel (4% stacking and 7.5% separatinggels) electrophoresis. Proteins were electroblotted ontoa PVDF membrane (Immobilon-P, 0.45 �m, Millipore,Bedford, MA) overnight at 4 ◦C and 100 mA, thenblocked with 5% non-fat milk in Tris buffered saline(TBS: 0.9% NaCl, 10 mM Tris, pH 7.5) at room tem-perature for 1 h. The membrane was then incubated for1 h at room temperature with anti-V5 monoclonal anti-body (1:2000) in 5% non-fat milk. The membrane waswashed with 5% non-fat milk in TBS for 15 min followedby two additional 5 min washes. The membrane wasincubated with peroxidase-conjugated goat anti-mouseIgG secondary antibodies (1:10,000) in TBS contain-ing 5% non-fat milk at room temperature for 1 h. Themembrane was rinsed 3× with TBS as described above.ECL reagents (Pierce, Rockford, IL) were applied perthe manufacturer instruction and the blots were exposedto ECL-sensitive film (Amersham Pharmacia Biotech,Uppsala, Sweden).

Author's personal copy
P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955 1947
2.8. Northern blot analysis
Total RNA from 0% to 4% DMSO-treated CHO-LacZ cells cultured in T25 flasks was isolated usingTri Reagent (MRC Inc., Cincinnati, OH). Twenty-five micrograms of total RNA was fractionated byformaldehyde-1% agarose gel electrophoresis andtransferred to a nitrocellulose membrane and UV cross-linked. The membrane was prehybridized in 50%formamide, 5× saline sodium citrate (SSC), 5× Den-hardt’s solution, 5 mM EDTA, 0.1% SDS and 100 �g/mLdenatured salmon sperm DNA at 42 ◦C for 4 h. Openreading frames (ORFs) of both glyceraldehyde 3-phosphate dehydrogensae (GAPDH) and LacZ wereprepared from the respective plasmids by EcoRI diges-tion and used as hybridization probes. These probeswere radiolabeled with [�-32P]dCTP (ICN, Costa Mesa,CA) using Prime-It II Random Primer DNA LabelingKit (Stratagene, Cedar Creek, TX) and added to a pre-hybridization solution containing 10% dextran sulfatefollowed by incubation at 42 ◦C for 14 h. The membranewas washed with 0.1% SDS in 2× SSC at room temper-ature for 15 min and twice with 0.1% SDS in 0.2× SSCat 58 ◦C for 15 min and exposed to X-OMAT AR film(Kodak, Rochester, NY) with an intensifying screen at−80 ◦C for 48–72 h.
2.9. Bisulfite modification and methylation-specificPCR
Bisulfite treatment of 2 �g genomic DNA wascarried out using the EZ DNA methylation-goldkit (Zymo Research, Orange, CA). Bisulfite treat-ment deaminates unmethylated cytosines to convertthem to uracils leaving methylated cytosine residuesunchanged. Methylation-specific PCR (MSPCR) wasperformed to determine the methylation status ofthe CMV promoter in CHO-LacZ and H9c2 Fluc.3cells. Two hundred picograms of bisulfite-modifiedDNA was used as template. MSPCR primers spe-cific for methylated CpG sequences were: (M) senseprimer 5′ CGTTTTTTATTGACGTTAATGACGG3′;antisense primer 5′ TACGTCAATAAAACGAAAT-TATTACGAC 3′. Primers specific for unmethylatedCpG sequences were: (U) sense primer 5′TGTTTTT-TATTGATGTTAATGATGG 3′; antisense primerTACATCAATAAAACAAAATTATTA CAAC 3′. ThePCR reactions (40 cycles) were performed in a totalvolume of 25 �L using standard conditions, with denat-uration at 95 ◦C, annealing at 60–64 ◦C and extensionat 72 ◦C. The CMV promoter-containing pcDNA6-LacZ/bsd vector, methylated using M. SssI (CpG) DNA
methyltransferase (New England Biolabs, Ipswich,MA), was used as the positive, fully methylated control.The untreated vector served as unmethylated controland no-template DNA served as negative controls. Allprocedures followed the manufacturer’s instructions.The PCR products were electrophoresed on 2% agarosegel, stained with ethidium bromide and visualized undera UV illuminator. Detection of a discrete visible bandof PCR product with methylation-specific primers wasconsidered positive. All experiments were carried out induplicate.
2.10. Formaldehyde cross-linking and chromatinimmunoprecipitation
The formaldehyde cross-linking and chromatinimmunoprecipitation assays of cultured cells were per-formed as described (Christenson, Stouffer, & Strauss,2001; Kuo & Allis, 1999). Briefly, the CHO-LacZ cellswere pretreated with 2–4% DMSO, 5-Aza CdR or 2%DMSO + 5-Aza CdR (1 �g/mL). After a 12 h incuba-tion, 37% formaldehyde (Fisher Scientific) was addeddirectly to the cell culture medium to cross-link DNAand its associated proteins. Immunoprecipitation wascarried out overnight at 4 ◦C using 10 �g of anti-acetylhistone (H3) antibody specific for the acetylated form oflysine-K9 and K14 of histone (H3) (Upstate cell signal-ing solutions, Charlottesville, VA). Non-immune rabbitserum-treated sample served as the negative control.Immunoprecipitation, washing and elution of immunecomplexes were carried out as described in the ChIPassay kit (Upstate cell signaling solutions). Before thefirst wash, 10% of the supernatant from the sampleswithout antibody for each condition was saved for deter-mination of total input chromatin and processed withthe eluted immunoprecipitates beginning at the cross-link reversal step. After final ethanol precipitation, boththe samples and the inputs were eluted with 30 �L ofhigh grade nuclease free water (Promega) (Kuo & Allis,1999).
The PCR reaction contained 2 �L of samples(IPs) or 2 �L of diluted input (1:100 diluted input-chromatin) and 2× PCR master mixes (Promega) in25 �L of total reaction volume. After 32–34 cyclesof amplification, 10–12 �L of the PCR products weresubjected to electrophoresis on a 4% agarose gel, andDNA was stained with ethidium bromide and visu-alized under UV light. The signals (intensity) werequantitated by Alpha imager I (3.3d). Fold changesin acetylation were calculated by first dividing thesample signal intensity by the input-signal intensity(to control for varying levels of chromatin in each
Author's personal copy
1948 P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955
sample) of CHO LacZ cells. The primers used forPCR analysis were: CMV promoter sense primer5′TCCGCGTTACATAACTTACGG3′; antisense primer5′TGCCAAGTGGGCAGTTTAC3′; CHO cell GAPDHsense primer 5′ACAT CAAATGGGGTGATGCT3′;antisense primer 5′CTTGGTTCACACCCATCACA3′.
2.11. Statistical analysis
The difference in means between two experimen-tal groups was analyzed by the Student’s t-test whilestatistical analysis of multiple samples was carried outusing one way analysis of variance (ANOVA). A p-valueof less than 0.05 was considered statistically signifi-cant.
3. Results
3.1. DMSO enhanced CMV promoter activity inCHO-LacZ cells
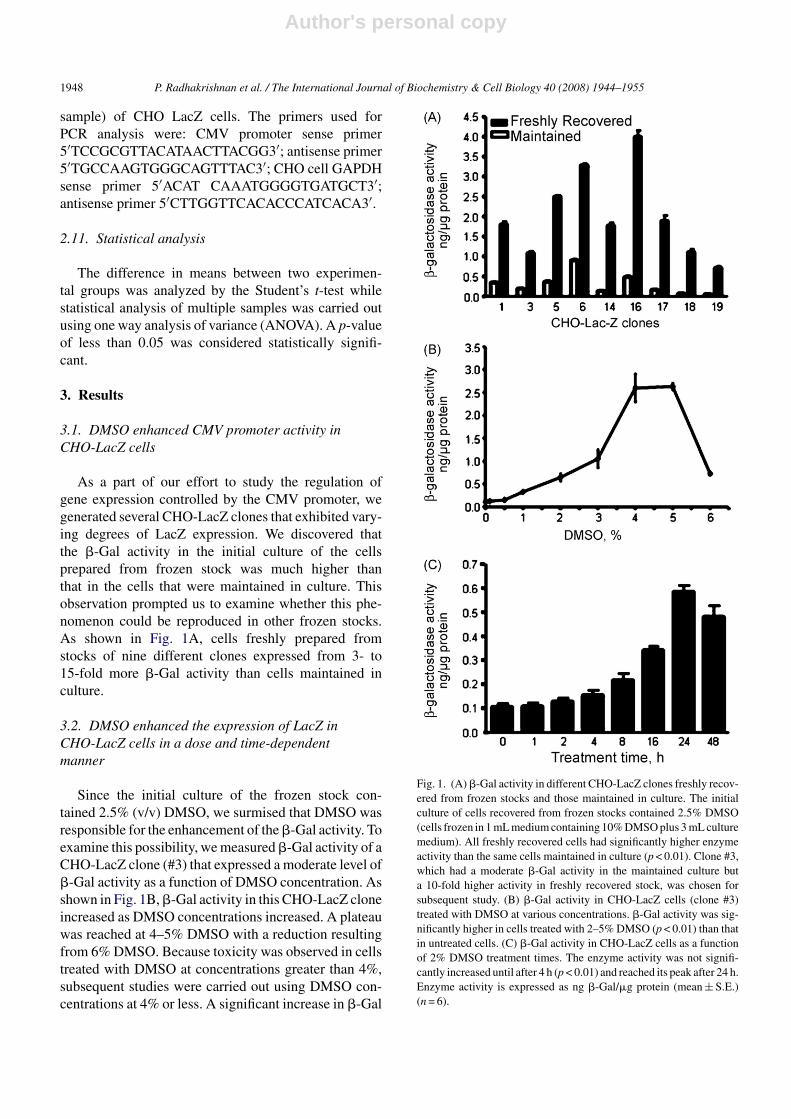
As a part of our effort to study the regulation ofgene expression controlled by the CMV promoter, wegenerated several CHO-LacZ clones that exhibited vary-ing degrees of LacZ expression. We discovered thatthe �-Gal activity in the initial culture of the cellsprepared from frozen stock was much higher thanthat in the cells that were maintained in culture. Thisobservation prompted us to examine whether this phe-nomenon could be reproduced in other frozen stocks.As shown in Fig. 1A, cells freshly prepared fromstocks of nine different clones expressed from 3- to15-fold more �-Gal activity than cells maintained inculture.
3.2. DMSO enhanced the expression of LacZ inCHO-LacZ cells in a dose and time-dependentmanner
Since the initial culture of the frozen stock con-tained 2.5% (v/v) DMSO, we surmised that DMSO wasresponsible for the enhancement of the �-Gal activity. Toexamine this possibility, we measured �-Gal activity of aCHO-LacZ clone (#3) that expressed a moderate level of�-Gal activity as a function of DMSO concentration. Asshown in Fig. 1B, �-Gal activity in this CHO-LacZ cloneincreased as DMSO concentrations increased. A plateauwas reached at 4–5% DMSO with a reduction resultingfrom 6% DMSO. Because toxicity was observed in cellstreated with DMSO at concentrations greater than 4%,subsequent studies were carried out using DMSO con-centrations at 4% or less. A significant increase in �-Gal
Fig. 1. (A) �-Gal activity in different CHO-LacZ clones freshly recov-ered from frozen stocks and those maintained in culture. The initialculture of cells recovered from frozen stocks contained 2.5% DMSO(cells frozen in 1 mL medium containing 10% DMSO plus 3 mL culturemedium). All freshly recovered cells had significantly higher enzymeactivity than the same cells maintained in culture (p < 0.01). Clone #3,which had a moderate �-Gal activity in the maintained culture buta 10-fold higher activity in freshly recovered stock, was chosen forsubsequent study. (B) �-Gal activity in CHO-LacZ cells (clone #3)treated with DMSO at various concentrations. �-Gal activity was sig-nificantly higher in cells treated with 2–5% DMSO (p < 0.01) than thatin untreated cells. (C) �-Gal activity in CHO-LacZ cells as a functionof 2% DMSO treatment times. The enzyme activity was not signifi-cantly increased until after 4 h (p < 0.01) and reached its peak after 24 h.Enzyme activity is expressed as ng �-Gal/�g protein (mean ± S.E.)(n = 6).
Author's personal copy
P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955 1949
activity in CHO-LacZ cells was detected as early as 4 hafter adding 2% DMSO to the culture (Fig. 1C). �-Galactivity in DMSO-treated cells was about 2.5-fold higherthan the activity found in the control cells after 8 h, andabout 10-fold higher after 24 h.
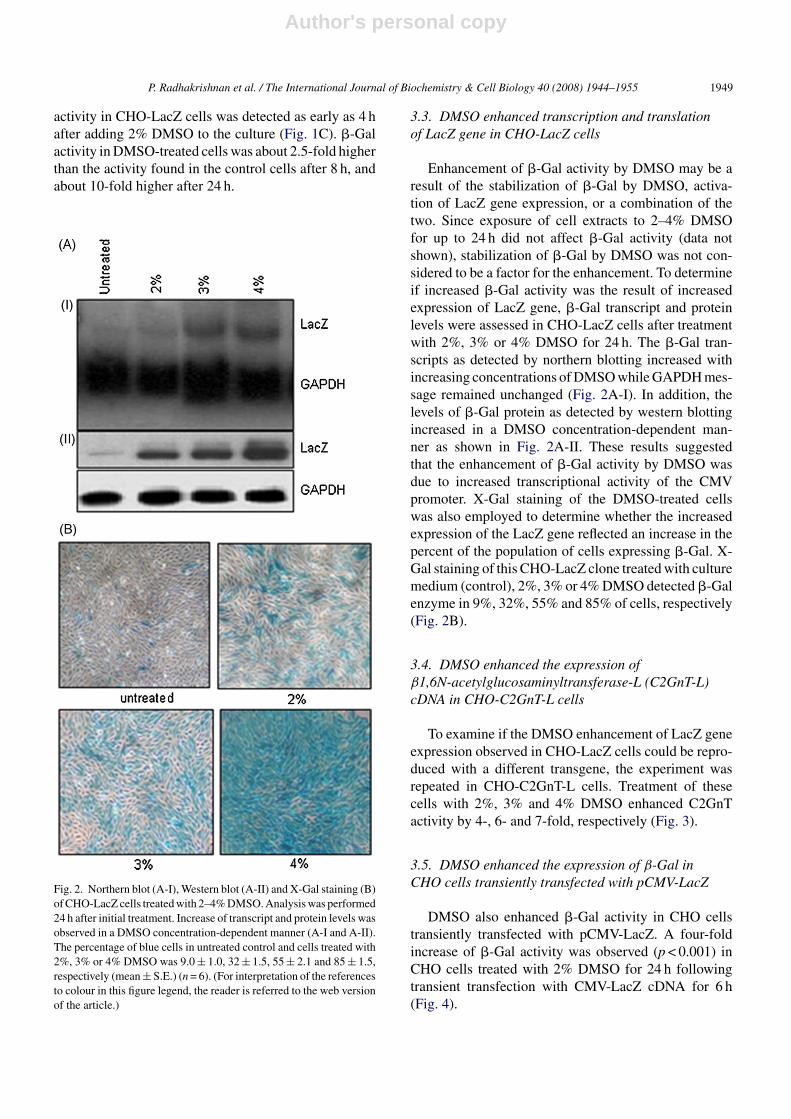
Fig. 2. Northern blot (A-I), Western blot (A-II) and X-Gal staining (B)of CHO-LacZ cells treated with 2–4% DMSO. Analysis was performed24 h after initial treatment. Increase of transcript and protein levels wasobserved in a DMSO concentration-dependent manner (A-I and A-II).The percentage of blue cells in untreated control and cells treated with2%, 3% or 4% DMSO was 9.0 ± 1.0, 32 ± 1.5, 55 ± 2.1 and 85 ± 1.5,respectively (mean ± S.E.) (n = 6). (For interpretation of the referencesto colour in this figure legend, the reader is referred to the web versionof the article.)
3.3. DMSO enhanced transcription and translationof LacZ gene in CHO-LacZ cells
Enhancement of �-Gal activity by DMSO may be aresult of the stabilization of �-Gal by DMSO, activa-tion of LacZ gene expression, or a combination of thetwo. Since exposure of cell extracts to 2–4% DMSOfor up to 24 h did not affect �-Gal activity (data notshown), stabilization of �-Gal by DMSO was not con-sidered to be a factor for the enhancement. To determineif increased �-Gal activity was the result of increasedexpression of LacZ gene, �-Gal transcript and proteinlevels were assessed in CHO-LacZ cells after treatmentwith 2%, 3% or 4% DMSO for 24 h. The �-Gal tran-scripts as detected by northern blotting increased withincreasing concentrations of DMSO while GAPDH mes-sage remained unchanged (Fig. 2A-I). In addition, thelevels of �-Gal protein as detected by western blottingincreased in a DMSO concentration-dependent man-ner as shown in Fig. 2A-II. These results suggestedthat the enhancement of �-Gal activity by DMSO wasdue to increased transcriptional activity of the CMVpromoter. X-Gal staining of the DMSO-treated cellswas also employed to determine whether the increasedexpression of the LacZ gene reflected an increase in thepercent of the population of cells expressing �-Gal. X-Gal staining of this CHO-LacZ clone treated with culturemedium (control), 2%, 3% or 4% DMSO detected �-Galenzyme in 9%, 32%, 55% and 85% of cells, respectively(Fig. 2B).
3.4. DMSO enhanced the expression ofβ1,6N-acetylglucosaminyltransferase-L (C2GnT-L)cDNA in CHO-C2GnT-L cells
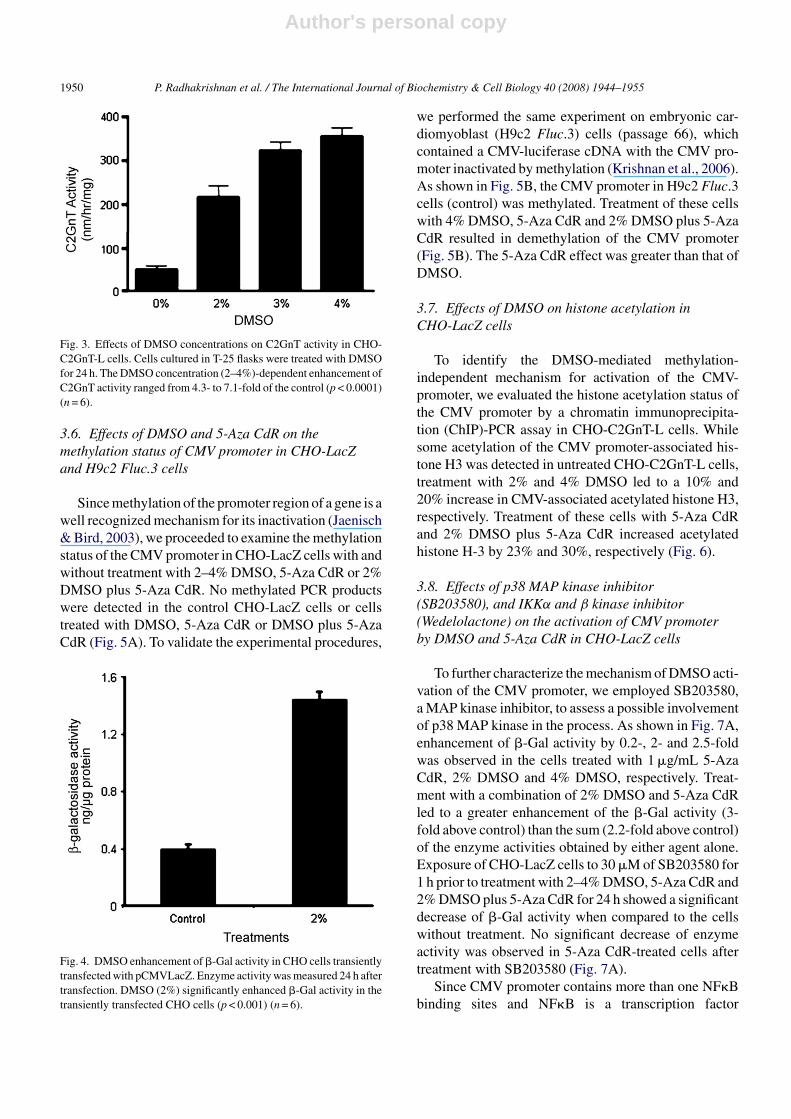
To examine if the DMSO enhancement of LacZ geneexpression observed in CHO-LacZ cells could be repro-duced with a different transgene, the experiment wasrepeated in CHO-C2GnT-L cells. Treatment of thesecells with 2%, 3% and 4% DMSO enhanced C2GnTactivity by 4-, 6- and 7-fold, respectively (Fig. 3).
3.5. DMSO enhanced the expression of β-Gal inCHO cells transiently transfected with pCMV-LacZ
DMSO also enhanced �-Gal activity in CHO cellstransiently transfected with pCMV-LacZ. A four-foldincrease of �-Gal activity was observed (p < 0.001) inCHO cells treated with 2% DMSO for 24 h followingtransient transfection with CMV-LacZ cDNA for 6 h(Fig. 4).
Author's personal copy
1950 P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955
Fig. 3. Effects of DMSO concentrations on C2GnT activity in CHO-C2GnT-L cells. Cells cultured in T-25 flasks were treated with DMSOfor 24 h. The DMSO concentration (2–4%)-dependent enhancement ofC2GnT activity ranged from 4.3- to 7.1-fold of the control (p < 0.0001)(n = 6).
3.6. Effects of DMSO and 5-Aza CdR on themethylation status of CMV promoter in CHO-LacZand H9c2 Fluc.3 cells
Since methylation of the promoter region of a gene is awell recognized mechanism for its inactivation (Jaenisch& Bird, 2003), we proceeded to examine the methylationstatus of the CMV promoter in CHO-LacZ cells with andwithout treatment with 2–4% DMSO, 5-Aza CdR or 2%DMSO plus 5-Aza CdR. No methylated PCR productswere detected in the control CHO-LacZ cells or cellstreated with DMSO, 5-Aza CdR or DMSO plus 5-AzaCdR (Fig. 5A). To validate the experimental procedures,
Fig. 4. DMSO enhancement of �-Gal activity in CHO cells transientlytransfected with pCMVLacZ. Enzyme activity was measured 24 h aftertransfection. DMSO (2%) significantly enhanced �-Gal activity in thetransiently transfected CHO cells (p < 0.001) (n = 6).
we performed the same experiment on embryonic car-diomyoblast (H9c2 Fluc.3) cells (passage 66), whichcontained a CMV-luciferase cDNA with the CMV pro-moter inactivated by methylation (Krishnan et al., 2006).As shown in Fig. 5B, the CMV promoter in H9c2 Fluc.3cells (control) was methylated. Treatment of these cellswith 4% DMSO, 5-Aza CdR and 2% DMSO plus 5-AzaCdR resulted in demethylation of the CMV promoter(Fig. 5B). The 5-Aza CdR effect was greater than that ofDMSO.
3.7. Effects of DMSO on histone acetylation inCHO-LacZ cells
To identify the DMSO-mediated methylation-independent mechanism for activation of the CMV-promoter, we evaluated the histone acetylation status ofthe CMV promoter by a chromatin immunoprecipita-tion (ChIP)-PCR assay in CHO-C2GnT-L cells. Whilesome acetylation of the CMV promoter-associated his-tone H3 was detected in untreated CHO-C2GnT-L cells,treatment with 2% and 4% DMSO led to a 10% and20% increase in CMV-associated acetylated histone H3,respectively. Treatment of these cells with 5-Aza CdRand 2% DMSO plus 5-Aza CdR increased acetylatedhistone H-3 by 23% and 30%, respectively (Fig. 6).
3.8. Effects of p38 MAP kinase inhibitor(SB203580), and IKKα and β kinase inhibitor(Wedelolactone) on the activation of CMV promoterby DMSO and 5-Aza CdR in CHO-LacZ cells
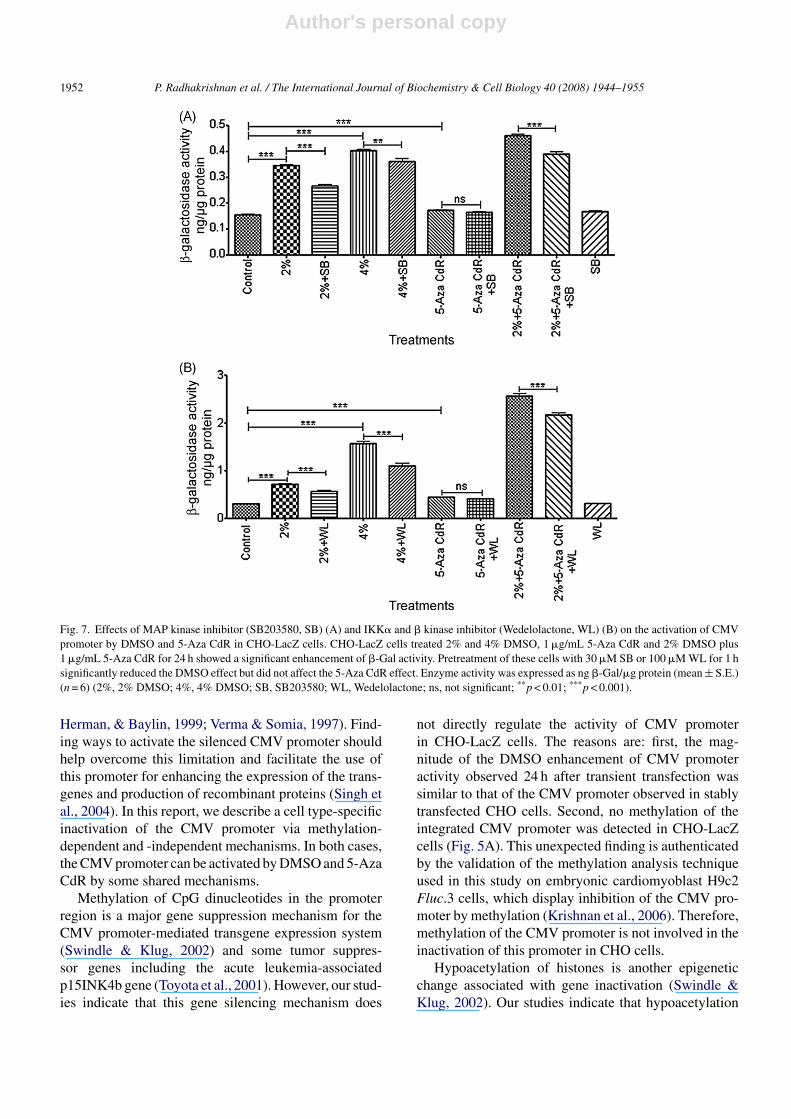
To further characterize the mechanism of DMSO acti-vation of the CMV promoter, we employed SB203580,a MAP kinase inhibitor, to assess a possible involvementof p38 MAP kinase in the process. As shown in Fig. 7A,enhancement of �-Gal activity by 0.2-, 2- and 2.5-foldwas observed in the cells treated with 1 �g/mL 5-AzaCdR, 2% DMSO and 4% DMSO, respectively. Treat-ment with a combination of 2% DMSO and 5-Aza CdRled to a greater enhancement of the �-Gal activity (3-fold above control) than the sum (2.2-fold above control)of the enzyme activities obtained by either agent alone.Exposure of CHO-LacZ cells to 30 �M of SB203580 for1 h prior to treatment with 2–4% DMSO, 5-Aza CdR and2% DMSO plus 5-Aza CdR for 24 h showed a significantdecrease of �-Gal activity when compared to the cellswithout treatment. No significant decrease of enzymeactivity was observed in 5-Aza CdR-treated cells aftertreatment with SB203580 (Fig. 7A).
Since CMV promoter contains more than one NF�Bbinding sites and NF�B is a transcription factor

Author's personal copy
P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955 1951
Fig. 5. Methylation-specific PCR analysis of CMV promoter containing CpG dinucleotides in CHO-LacZ (A) and H9c2 Fluc.3 (B) cells. Primer setsused for amplification are designated as unmethylated (U) and methylated (M). No methylated PCR products were detected in untreated CHO-LacZcells (control) and same cells treated with 2% DMSO, 4% DMSO, 5-Aza CdR and 2% DMSO + 5-Aza CdR. Methylated PCR products weredetected in untreated H9c2 Fluc.3 cells (control) and same cells treated with 2% DMSO, 4% DMSO, 5-Aza CdR and 2% DMSO + 5-Aza CdR.Unmethylated PCR products were only detected in H9c2 Fluc.3 cells treated with 4% DMSO, 5-Aza CdR and 2% DMSO + 5-Aza CdR. MethylatedpcDNA6-LacZ/bsd was used as a positive control vector (MV) and non-methylated pcDNA6-LacZ/bsd was used as a negative control vector (NV).No-template DNA was used as a reaction control (L, 100 bp ladder).
downstream to p38 MAP kinase signaling pathway(Madrid, Mayo, Reuther, & Baldwin, 2001; Sambucetti,Cherrington, Wilkinson, & Mocarski, 1989; Wong,Zhang, Ip, & Lam, 2002), we proceeded to examineif NF�B was involved in the activation of the CMV
Fig. 6. Chromatin immunoprecipitation (ChIP)-PCR assay for CMVpromoter sequence from CHO-LacZ. Chromatin DNA was immuno-precipitated with antibodies specific for acetylated histone H3(acetyl-H3). DNA fragments were amplified with primers specific forCMV promoter and control GAPDH. ChIP-PCR products for CMVpromoter (IP) (161 bp): Lane 1, 1 kb + ladder; Lane 2, no DNA con-trol; Lane 3, negative control; Lane 4, no treatment control; Lane 5,2% DMSO; Lane 6, 4% DMSO; Lane 7, 5-Aza CdR; Lane 8, 2%DMSO + 5-Aza CdR. ChIP-PCR products obtained for CMV promoterinputs under various conditions as described above are designatedas (Input) (161 bp). ChIP-PCR products obtained for GAPDH con-trol under various conditions as described above are designated as(GAPDH) (162 bp). The fold changes in signal intensity were mea-sured by the ratio of IP/inputs to control GAPDH genes as describedunder Section 2. Increased signal intensity observed in CHO-LacZ cellstreated with various agents was: 2% DMSO, 10%; 4% DMSO, 20%;5-Aza CdR, 23%; 5-Aza CdR + 2% DMSO, 30% when compared withthat of the untreated control cells.
promoter by DMSO. Exposure of CHO-LacZ cells to100 �M Wedelolactone for 1 h prior to treatment with2% DMSO, 4% DMSO or 2% DMSO plus 5-Aza CdRfor 24 h significantly reduced �-Gal activity as comparedto the cells without Wedelolactone pretreatment. WL didnot significantly reduce �-Gal activity in cells treatedwith 5-Aza CdR (Fig. 7B).
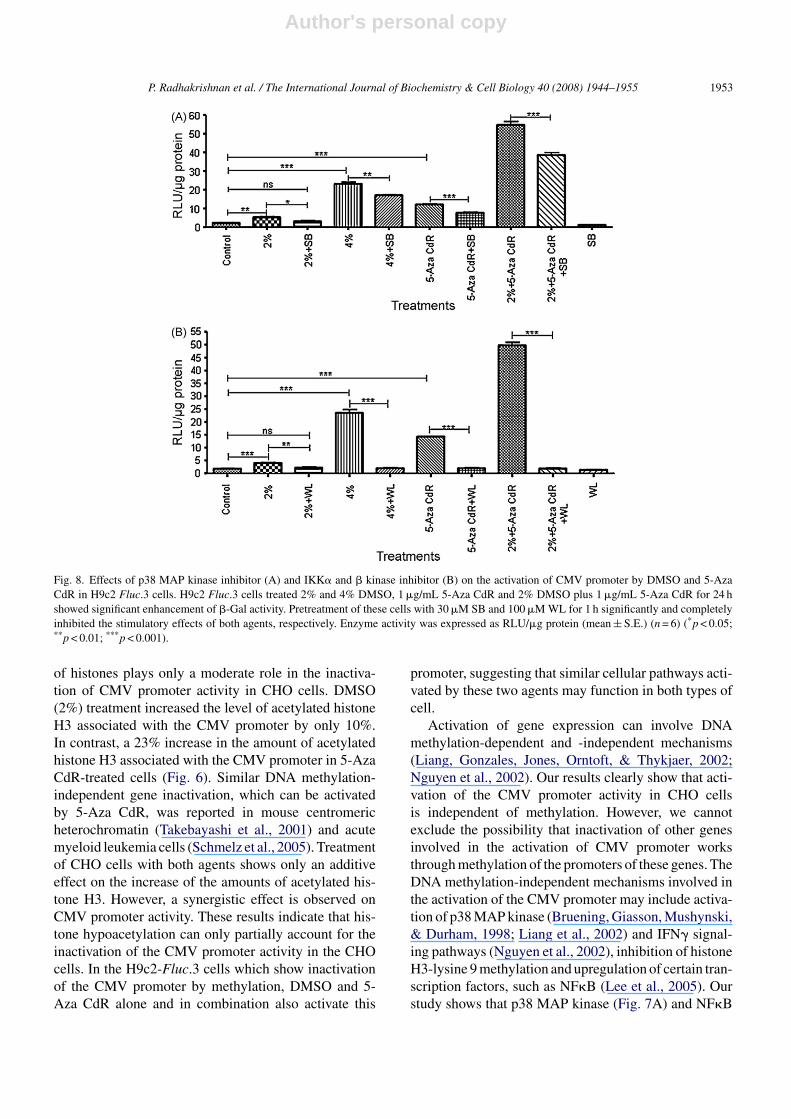
3.9. Effects of p38 MAP kinase inhibitor(SB203580), and IKKα and β kinase inhibitor(Wedelolactone) on the activation of CMV promoterby DMSO and 5-Aza CdR in H9c2 Fluc.3 cells
H9c2 Fluc.3 cells treated with 2% DMSO, 4%DMSO, 5-Aza CdR and 2% DMSO plus 5-Aza CdRfor 24 h showed 1.3-, 9.3-, 4.4- and 23-fold enhance-ment of luciferase activity. Pretreatment of these cellswith SB203580 significantly reduced luciferase activityin the cells treated with 2% DMSO, 4% DMSO, 5-AzaCdR and 2% DMSO plus 5-Aza CdR as compared tothe cells treated with each agent alone (Fig. 8A). Also,pretreatment of H9c2 Fluc.3 cells with Wedelolactonecompletely prevented the activation of the CMV pro-moter by 2% DMSO, 4% DMSO, 5-Aza CdR and 2%DMSO plus 5-Aza CdR (Fig. 8B).
4. Discussion
CMV promoter is a powerful promoter widely usedfor driving transgene expression in mammalian cells.However, it becomes inactivated over time after insertioninto the host genome (Cameron, Bachman, Myohanen,
Author's personal copy
1952 P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955
Fig. 7. Effects of MAP kinase inhibitor (SB203580, SB) (A) and IKK� and � kinase inhibitor (Wedelolactone, WL) (B) on the activation of CMVpromoter by DMSO and 5-Aza CdR in CHO-LacZ cells. CHO-LacZ cells treated 2% and 4% DMSO, 1 �g/mL 5-Aza CdR and 2% DMSO plus1 �g/mL 5-Aza CdR for 24 h showed a significant enhancement of �-Gal activity. Pretreatment of these cells with 30 �M SB or 100 �M WL for 1 hsignificantly reduced the DMSO effect but did not affect the 5-Aza CdR effect. Enzyme activity was expressed as ng �-Gal/�g protein (mean ± S.E.)(n = 6) (2%, 2% DMSO; 4%, 4% DMSO; SB, SB203580; WL, Wedelolactone; ns, not significant; **p < 0.01; ***p < 0.001).
Herman, & Baylin, 1999; Verma & Somia, 1997). Find-ing ways to activate the silenced CMV promoter shouldhelp overcome this limitation and facilitate the use ofthis promoter for enhancing the expression of the trans-genes and production of recombinant proteins (Singh etal., 2004). In this report, we describe a cell type-specificinactivation of the CMV promoter via methylation-dependent and -independent mechanisms. In both cases,the CMV promoter can be activated by DMSO and 5-AzaCdR by some shared mechanisms.
Methylation of CpG dinucleotides in the promoterregion is a major gene suppression mechanism for theCMV promoter-mediated transgene expression system(Swindle & Klug, 2002) and some tumor suppres-sor genes including the acute leukemia-associatedp15INK4b gene (Toyota et al., 2001). However, our stud-ies indicate that this gene silencing mechanism does
not directly regulate the activity of CMV promoterin CHO-LacZ cells. The reasons are: first, the mag-nitude of the DMSO enhancement of CMV promoteractivity observed 24 h after transient transfection wassimilar to that of the CMV promoter observed in stablytransfected CHO cells. Second, no methylation of theintegrated CMV promoter was detected in CHO-LacZcells (Fig. 5A). This unexpected finding is authenticatedby the validation of the methylation analysis techniqueused in this study on embryonic cardiomyoblast H9c2Fluc.3 cells, which display inhibition of the CMV pro-moter by methylation (Krishnan et al., 2006). Therefore,methylation of the CMV promoter is not involved in theinactivation of this promoter in CHO cells.
Hypoacetylation of histones is another epigeneticchange associated with gene inactivation (Swindle &Klug, 2002). Our studies indicate that hypoacetylation
Author's personal copy
P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955 1953
Fig. 8. Effects of p38 MAP kinase inhibitor (A) and IKK� and � kinase inhibitor (B) on the activation of CMV promoter by DMSO and 5-AzaCdR in H9c2 Fluc.3 cells. H9c2 Fluc.3 cells treated 2% and 4% DMSO, 1 �g/mL 5-Aza CdR and 2% DMSO plus 1 �g/mL 5-Aza CdR for 24 hshowed significant enhancement of �-Gal activity. Pretreatment of these cells with 30 �M SB and 100 �M WL for 1 h significantly and completelyinhibited the stimulatory effects of both agents, respectively. Enzyme activity was expressed as RLU/�g protein (mean ± S.E.) (n = 6) (*p < 0.05;**p < 0.01; ***p < 0.001).
of histones plays only a moderate role in the inactiva-tion of CMV promoter activity in CHO cells. DMSO(2%) treatment increased the level of acetylated histoneH3 associated with the CMV promoter by only 10%.In contrast, a 23% increase in the amount of acetylatedhistone H3 associated with the CMV promoter in 5-AzaCdR-treated cells (Fig. 6). Similar DNA methylation-independent gene inactivation, which can be activatedby 5-Aza CdR, was reported in mouse centromericheterochromatin (Takebayashi et al., 2001) and acutemyeloid leukemia cells (Schmelz et al., 2005). Treatmentof CHO cells with both agents shows only an additiveeffect on the increase of the amounts of acetylated his-tone H3. However, a synergistic effect is observed onCMV promoter activity. These results indicate that his-tone hypoacetylation can only partially account for theinactivation of the CMV promoter activity in the CHOcells. In the H9c2-Fluc.3 cells which show inactivationof the CMV promoter by methylation, DMSO and 5-Aza CdR alone and in combination also activate this
promoter, suggesting that similar cellular pathways acti-vated by these two agents may function in both types ofcell.
Activation of gene expression can involve DNAmethylation-dependent and -independent mechanisms(Liang, Gonzales, Jones, Orntoft, & Thykjaer, 2002;Nguyen et al., 2002). Our results clearly show that acti-vation of the CMV promoter activity in CHO cellsis independent of methylation. However, we cannotexclude the possibility that inactivation of other genesinvolved in the activation of CMV promoter worksthrough methylation of the promoters of these genes. TheDNA methylation-independent mechanisms involved inthe activation of the CMV promoter may include activa-tion of p38 MAP kinase (Bruening, Giasson, Mushynski,& Durham, 1998; Liang et al., 2002) and IFN� signal-ing pathways (Nguyen et al., 2002), inhibition of histoneH3-lysine 9 methylation and upregulation of certain tran-scription factors, such as NF�B (Lee et al., 2005). Ourstudy shows that p38 MAP kinase (Fig. 7A) and NF�B

Author's personal copy
1954 P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955
(Fig. 7B) are involved in the activation of the CMV pro-moter by DMSO. This is consistent with our observationthat DMSO activation of a silenced CMV promoter inCHO cells is independent of the methylation status of theCMV promoter, and only moderately associated withhistone acetylation. Activation of the CMV promoterby DMSO is correlated with activation of p38 MAPkinase-NF�B signaling pathways. However, the activa-tion of the methylation-independent CMV promoter by5-Aza CdR in CHO cells does not involve p38 MAPkinase-NF�B pathway. In the cells (H9c2 Fluc.3), whichshow inactivation of the CMV promoter by methylation(Krishnan et al., 2006), treatment with DMSO and/or5-Aza CdR results in demethylation. MAP treatmentkinase is involved to some extent while NF�B pathwayplays an essential role in the activation of this promoterby DMSO and 5-Aza CdR.
These findings raise an important data interpretationissue when DMSO is used as a solvent in biologicalapplication. Because DMSO can activate cellular genesand transgenes (Ishiguro & Sartorelli, 2004; Zhu, Smith,Ansah, & Gooderham, 2005), some of these gene prod-ucts may affect the outcome of the experiment. In thiscase, the effect observed by the test agent dissolved inDMSO may not represent its true action rather a resultof the interactive actions of the test agent and DMSO.Therefore, the experimental results should be validatedby alternative approaches when DMSO is used in anexperiment.
In conclusion, we show that the mechanisms for theinactivation of the CMV promoter are cell type-specificand can operate in either methylation-dependent and -independent pathways. Both types of CMV promoterinactivation can be activated by DMSO and 5-Aza CdR.Mechanistically, histone acetylation, P38-MAP kinaseand NF�B each plays some moderate role in the activa-tion of the CMV promoter in CHO cells while P38-MAPkinase plays some moderate role and NF�B plays anessential role in the activation of the CMV promoter in amethylation-dependent H9c2 Fluc.3 cell line (Krishnanet al., 2006). For generation of a gene product con-trolled by a CMV promoter, activation of p38 MAPkinase-NF�B pathway may be employed to enhance theproduction of the recombinant protein.
Acknowledgements
The authors wish to acknowledge the research sup-port by NIH RO1 HL48282, State of Nebraska CancerGlycobiology Program and LB506, and Cystic FibrosisFoundation CFF-NIH grant, and expert technical assis-tance of Ms. Helen Cheng and Ms. Lin Tang. The authors
also wish to thank Dr. Joseph Wu at the Stanford Uni-versity for providing the rat embryonic cardiomyoblastH9c2-Fluc.3 cells.
References
Beum, P. V., Singh, J., Burdick, M., Hollingsworth, M. A.,& Cheng, P. W. (1999). Expression of core 2 beta-1,6-N-acetylglucosaminyltransferase in a human pancreatic cancer cellline results in altered expression of MUC1 tumor-associated epi-topes. Journal of Biological Chemistry, 274, 24641–24648.
Bonser, R. W., Siegel, M. I., McConnell, R. T., & Cuatrecasas, P.(1981). The appearance of phospholipase and cyclo-oxygenaseactivities in the human promyelocytic leukemia cell line HL60 dur-ing dimethyl sulfoxide-induced differentiation. Biochemical andBiophysical Research Communications, 98, 614–620.
Brooks, A. R., Harkins, R. N., Wang, P., Qian, H. S., Liu, P., &Rubanyi, G. M. (2004). Transcriptional silencing is associated withextensive methylation of the CMV promoter following adenoviralgene delivery to muscle. The Journal of Gene Medicine, 6, 395–404.
Bruening, W., Giasson, B., Mushynski, W., & Durham, H. D. (1998).Activation of stress-activated MAP protein kinases up-regulatesexpression of transgenes driven by the cytomegalovirus immedi-ate/early promoter. Nucleic Acids Research, 26, 486–489.
Cameron, E. E., Bachman, K. E., Myohanen, S., Herman, J. G.,& Baylin, S. B. (1999). Synergy of demethylation and histonedeacetylase inhibition in the re-expression of genes silenced incancer. Nature Genetics, 21, 103–107.
Cheng, P. W. (1996). Receptor ligand-facilitated gene transfer:Enhancement of liposome-mediated gene transfer and expressionby transferring. Human Gene Therapy, 7, 275–282.
Choi, K. H., Basma, H., Singh, J., & Cheng, P. W. (2005).Activation of CMV promoter-controlled glycosyltransferase andbeta-galactosidase glycogenes by butyrate, tricostatin A, and 5-aza-2′-deoxycytidine. Glycoconjugate Journal, 22, 63–69.
Christenson, L. K., Stouffer, R. L., & Strauss, J. F., III. (2001). Quanti-tative analysis of the hormone-induced hyperacetylation of histoneH3 associated with the steroidogenic acute regulatory protein genepromoter. Journal of Biological Chemistry, 276, 27392–27399.
Dinsmore, J., Ratliff, J., Deacon, T., Pakzaban, P., Jacoby, D., Galpern,W., et al. (1996). Embryonic stem cells differentiated in vitro as anovel source of cells for transplantation. Cell transplantation, 5,131–143.
Dinsmore, J., Ratliff, J., Jacoby, D., Wunderlich, M., & Lindberg, C.(1998). Embryonic stem cells as a model for studying regulationof cellular differentiation. Theriogenology, 49, 145–151.
Esteller, M., Corn, P. G., Baylin, S. B., & Herman, J. G. (2001). A genehypermethylation profile of human cancer. Cancer Research, 61,3225–3229.
Esteller, M., & Herman, J. G. (2002). Cancer as an epigenetic disease:DNA methylation and chromatin alterations in human tumours.The Journal of Pathology, 196, 1–7.
Grassi, G., Maccaroni, P., Meyer, R., Kaiser, H., D’Ambrosio, E.,Pascale, E., et al. (2003). Inhibitors of DNA methylation and his-tone deacetylation activate cytomegalovirus promoter-controlledreporter gene expression in human glioblastoma cell line U87.Carcinogenesis, 24, 1625–1635.
Ishiguro, K., & Sartorelli, A. C. (2004). Activation of transientlytransfected reporter genes in 3T3 swiss cells by the induc-ers of differentiation/apoptosis–dimethylsulfoxide, hexamethylene

Author's personal copy
P. Radhakrishnan et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 1944–1955 1955
bisacetamide and trichostatin A. European Journal of Biochem-istry/FEBS, 271, 2379–2390.
Jaenisch, R., & Bird, A. (2003). Epigenetic regulation of gene expres-sion: How the genome integrates intrinsic and environmentalsignals. Nature Genetics, 33 Suppl, 245–254.
Karlsson, J. O., Cravalho, E. G., Borel, R. I., Tompkins, R. G., Yarmush,M. L., & Toner, M. (1993). Nucleation and growth of ice crys-tals inside cultured hepatocytes during freezing in the presence ofdimethyl sulfoxide. Biophysical Journal, 65(6), 2524–2536.
Knust, B., Bruggemann, U., & Doerfler, W. (1989). Reactivation ofa methylation-silenced gene in adenovirus-transformed cells by5-azacytidine or by E1A transactivation. Journal of Virology, 63,3519–3524.
Krishnan, M., Park, J. M., Cao, F., Wang, D., Paulmurugan, R., Tseng,J. R., et al. (2006). Effects of epigenetic modulation on reportergene expression: Implications for stem cell imaging. The FASEBJournal, 20, 106–108.
Kuo, M. H., & Allis, C. D. (1999). In vivo cross-linking and immuno-precipitation for studying dynamic protein:DNA associations in achromatin environment. Methods (San Diego, Calif.), 19, 425–433.
Lee, Y. R., Shim, H. J., Yu, H. N., Song, E. K., Park, J., Kwon, K.B., et al. (2005). Dimethylsulfoxide induces upregulation of tumorsuppressor protein PTEN through nuclear factor-kappaB activationin HL-60 cells. Leukemia Research, 29, 401–405.
Li, C. M., Adler, K. B., & Cheng, P. W. (1998). Mucin biosyn-thesis: Molecular cloning and expression of bovine lung mucincore 2N-acetylglucosaminyltransferase cDNA. American Journalof Respiratory Cell and Molecular Biology, 18, 343–352.
Liang, G., Gonzales, F. A., Jones, P. A., Orntoft, T. F., & Thykjaer,T. (2002). Analysis of gene induction in human fibroblasts andbladder cancer cells exposed to the methylation inhibitor 5-aza-2′-deoxycytidine. Cancer Research, 62, 961–966.
Madrid, L. V., Mayo, M. W., Reuther, J. Y., & Baldwin, A. S., Jr.(2001). Akt stimulates the transactivation potential of the RelA/p65subunit of NF-kappa B through utilization of the ikappa B kinaseand activation of the mitogen-activated protein kinase p38. Journalof Biological Chemistry, 276, 18934–18940.
Nguyen, C. T., Weisenberger, D. J., Velicescu, M., Gonzales, F. A., Lin,J. C., Liang, G., et al. (2002). Histone H3-lysine 9 methylationis associated with aberrant gene silencing in cancer cells and israpidly reversed by 5-aza-2′-deoxycytidine. Cancer Research, 62,6456–6461.
Palmer, T. D., Rosman, G. J., Osborne, W. R., & Miller, A. D. (1991).Genetically modified skin fibroblasts persist long after transplanta-tion but gradually inactivate introduced genes. Proceedings of theNational Academy of Sciences of the United States of America, 88,1330–1334.
Pruitt, S. C. (1994). Discrete endogenous signals mediate neural com-petence and induction in P19 embryonal carcinoma stem cells.Development (Cambridge, England), 120, 3301–3312.
Ropp, P. A., Little, M. R., & Cheng, P. W. (1991). Mucinbiosynthesis: Purification and characterization of a mucin beta 6N-
acetylglucosaminyltransferase. Journal of Biological Chemistry,266, 23863–23871.
Sambucetti, L. C., Cherrington, J. M., Wilkinson, G. W., & Mocarski,E. S. (1989). NF-kappa B activation of the cytomegalovirusenhancer is mediated by a viral transactivator and by T cell stimu-lation. The EMBO Journal, 8, 4251–4258.
Schmelz, K., Sattler, N., Wagner, M., Lubbert, M., Dorken, B.,& Tamm, I. (2005). Induction of gene expression by 5-aza-2′-deoxycytidine in acute myeloid leukemia (AML) andmyelodysplastic syndrome (MDS) but not epithelial cells by DNA-methylation-dependent and -independent mechanisms. Leukemia,19, 103–111.
Seki, M., Iwakawa, J., Cheng, H., & Cheng, P. W. (2002). p53and PTEN/MMAC1/TEP1 gene therapy of human prostatePC-3 carcinoma xenograft, using transferrin-facilitated lipofec-tion gene delivery strategy. Human Gene Therapy, 13, 761–773.
Singal, R., & Ginder, G. D. (1999). DNA methylation. Blood, 93,4059–4070.
Singh, J., Khan, G. A., Kinarsky, L., Cheng, H., Wilken, J., Choi, K. H.,et al. (2004). Identification of disulfide bonds among the nine core2 N-acetylglucosaminyltransferase-M cysteines conserved in themucin beta6-N-acetylglucosaminyltransferase family. The Journalof Biological Chemistry, 279, 38969–38977.
Swindle, C. S., & Klug, C. A. (2002). Mechanisms that regulatesilencing of gene expression from retroviral vectors. Journal ofHematotherapy & Stem Cell Research, 11, 449–456.
Takebayashi, S., Nakao, M., Fujita, N., Sado, T., Tanaka, M., Taguchi,H., et al. (2001). 5-aza-2′-deoxycytidine induces histone hyper-acetylation of mouse centromeric heterochromatin by a mechanismindependent of DNA demethylation. Biochemical and BiophysicalResearch Communications, 288, 921–926.
Toyota, M., Kopecky, K. J., Toyota, M. O., Jair, K. W., Willman, C.L., & Issa, J. P. (2001). Methylation profiling in acute myeloidleukaemia. Blood, 97, 2823–2829.
Verma, I. M., & Somia, N. (1997). Gene therapy—Promises, problemsand prospects. Nature, 389, 239–242.
Wong, C. K., Zhang, J. P., Ip, W. K., & Lam, C. W. (2002).Activation of p38 mitogen-activated protein kinase and nuclearfactor-kappaB in tumour necrosis factor-induced eotaxin release ofhuman eosinophils. Clinical and Experimental Immunology, 128,483–489.
Yanagihara, K., Cheng, H., & Cheng, P. W. (2000). Effects of epidermalgrowth factor, transferrin, and insulin on lipofection efficiency inhuman lung carcinoma cells. Cancer Gene Therapy, 7, 59–65.
Yu, Z. W., & Quinn, P. J. (1994). Dimethyl sulphoxide: A reviewof its applications in cell biology. Bioscience reports, 14, 259–281.
Zhu, H., Smith, C., Ansah, C., & Gooderham, N. J. (2005). Responsesof genes involved in cell cycle control to diverse DNA damagingchemicals in human lung adenocarcinoma A549 cells. Cancer CellInternational, 5, 28.
Copyright © 2022 FDOKUMEN




















