
Catalytic combustion of volatile organic compounds on gold/cerium oxide catalysts
14
Applied Catalysis A: General 234 (2002) 65–78 Catalytic combustion of volatile organic compounds on Au/CeO 2 /Al 2 O 3 and Au/Al 2 O 3 catalysts M.A. Centeno a,∗ , M. Paulis b , M. Montes b , J.A. Odriozola a a Instituto de Ciencia de Materiales de Sevilla, Centro Mixto CSIC-Universidad de Sevilla, Avda Americo Vespuccio s/n, 41092 Sevilla, Spain b Grupo de Ingenier´ ıa Qu´ ımica, Departmento Qu´ ımica Aplicada, Facultad Ciencias Qu´ ımicas de San Sebastián, UPV/EHU, Paseo Manuel de Lardizábal 3, 20009 San Sebastián, Spain Received 29 October 2001; received in revised form 12 March 2002; accepted 13 March 2002 Abstract Catalytic oxidation of n-hexane, benzene and 2-propanol was investigated on Au/CeO 2 /Al 2 O 3 and Au/Al 2 O 3 catalysts prepared from the deposition–precipitation method and characterised by XRD, scanning electron microscopy (SEM), trans- mission electron microscopy (TEM), UV–VIS and X-ray photoelectron spectroscopy (XPS) techniques. It is shown that ceria enhances the fixation and final dispersion of gold particles, leading to stabilise them in lower crystallite sizes. Catalytic results show that ceria improves the activity of gold particles in the oxidation of the tested volatile organic compounds (VOCs), probably by increasing the mobility of the lattice oxygen and controlling and maintaining the adequate oxidation state of the active gold particles. © 2002 Elsevier Science B.V. All rights reserved. Keywords: Volatile organic compounds; Gold catalysts; Catalytic combustion; CeO 2 /Al 2 O 3 catalysts; n-Hexane; Benzene; 2-Propanol 1. Introduction Gold has been until recently considered as one of the least catalytically useful metal because of its chemical inertness and the difficulty to obtain a high dispersion on common support materials [1]. However, in the last decade it has been widely proved that it is possible to prepare gold nanoparticles deposited on metal-oxide supports [2–6] and, in such conditions, gold exhibits high catalytic activity towards low temperature CO oxidation [7–11]. The catalytic performance of gold strongly depends on the particle size, in such a way that smaller particles produce higher activities. On the ∗ Corresponding author. Tel.: +34-954489543; fax: +34-954460665. E-mail address: [email protected] (M.A. Centeno). other hand, the nature of the support is also a key fac- tor: first of all, the different wetting ability of the sup- port during calcination determines the final size of the gold particles. Secondly, small metallic particles are unstable with respect to massive metal, so it is neces- sary that the support presents a suitable high surface area which keeps them apart and a strong interaction between the gold particles and the surface of the sup- port that stabilises them and prevents their sinterisa- tion. This interaction may produce some changes in the properties and behaviour of these gold particles [12]. Thirdly, up to now it has not been well estab- lished if the catalytic reaction proceeds exclusively on metallic gold particles [2,13] or it may happen prefer- entially at the edges of gold particles involving either sites on the adjacent support or gold atoms influenced by the support [14]. Moreover, it has been suggested 0926-860X/02/$ – see front matter © 2002 Elsevier Science B.V. All rights reserved. PII:S0926-860X(02)00214-4
Transcript of Catalytic combustion of volatile organic compounds on gold/cerium oxide catalysts

Applied Catalysis A: General 234 (2002) 65–78
Catalytic combustion of volatile organic compoundson Au/CeO2/Al2O3 and Au/Al2O3 catalysts
M.A. Centenoa,∗, M. Paulisb, M. Montesb, J.A. Odriozolaaa Instituto de Ciencia de Materiales de Sevilla, Centro Mixto CSIC-Universidad de Sevilla,
Avda Americo Vespuccio s/n, 41092 Sevilla, Spainb Grupo de Ingenierıa Quımica, Departmento Quımica Aplicada, Facultad Ciencias Quımicas de San Sebastián,
UPV/EHU, Paseo Manuel de Lardizábal 3, 20009 San Sebastián, Spain
Received 29 October 2001; received in revised form 12 March 2002; accepted 13 March 2002
Abstract
Catalytic oxidation ofn-hexane, benzene and 2-propanol was investigated on Au/CeO2/Al2O3 and Au/Al2O3 catalystsprepared from the deposition–precipitation method and characterised by XRD, scanning electron microscopy (SEM), trans-mission electron microscopy (TEM), UV–VIS and X-ray photoelectron spectroscopy (XPS) techniques. It is shown that ceriaenhances the fixation and final dispersion of gold particles, leading to stabilise them in lower crystallite sizes. Catalytic resultsshow that ceria improves the activity of gold particles in the oxidation of the tested volatile organic compounds (VOCs),probably by increasing the mobility of the lattice oxygen and controlling and maintaining the adequate oxidation state of theactive gold particles.© 2002 Elsevier Science B.V. All rights reserved.
Keywords:Volatile organic compounds; Gold catalysts; Catalytic combustion; CeO2/Al2O3 catalysts;n-Hexane; Benzene; 2-Propanol
1. Introduction
Gold has been until recently considered as one of theleast catalytically useful metal because of its chemicalinertness and the difficulty to obtain a high dispersionon common support materials[1]. However, in the lastdecade it has been widely proved that it is possible toprepare gold nanoparticles deposited on metal-oxidesupports[2–6] and, in such conditions, gold exhibitshigh catalytic activity towards low temperature COoxidation [7–11]. The catalytic performance of goldstrongly depends on the particle size, in such a waythat smaller particles produce higher activities. On the
∗ Corresponding author. Tel.:+34-954489543;fax: +34-954460665.E-mail address:[email protected] (M.A. Centeno).
other hand, the nature of the support is also a key fac-tor: first of all, the different wetting ability of the sup-port during calcination determines the final size of thegold particles. Secondly, small metallic particles areunstable with respect to massive metal, so it is neces-sary that the support presents a suitable high surfacearea which keeps them apart and a strong interactionbetween the gold particles and the surface of the sup-port that stabilises them and prevents their sinterisa-tion. This interaction may produce some changes inthe properties and behaviour of these gold particles[12]. Thirdly, up to now it has not been well estab-lished if the catalytic reaction proceeds exclusively onmetallic gold particles[2,13] or it may happen prefer-entially at the edges of gold particles involving eithersites on the adjacent support or gold atoms influencedby the support[14]. Moreover, it has been suggested
0926-860X/02/$ – see front matter © 2002 Elsevier Science B.V. All rights reserved.PII: S0926-860X(02)00214-4

66 M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78
that the reaction may occur only on the support, in-volving adsorbed CO molecules migrated from thegold particles by spillover and oxygen species of thesupport[7]. Evidently, these mechanisms may coex-ist. In the three cases, the interface between the goldparticles and the support plays a determinant role inthe catalytic activity of the sample.
A large number of oxidic supports has been reportedin literature. Among them�-Al2O3 has been widelyused[15–20]. However, to our knowledge, no reportson the use of CeO2/Al2O3 as gold support have beenpublished. Alumina-supported ceria is one of the sup-ports of three way catalysts used for the eliminationof pollutants in automobile exhausts onto which pre-cious metals (typically Rh, Pt and/or Pd) are dispersed[21]. It is well known that ceria is a structural promot-ing component. It enhances the metal dispersion andparticipates in the stabilisation of the alumina supportagainst thermal sintering[22,23]. On the other hand,the addition of ceria to�-Al2O3 induces a modifica-tion of the electronic density of the aluminium andlanthanide cations, implying a change in the acid–baseproperties of the support surface[24]. Besides this,the well established catalytic and redox properties ofthe Ce3+/Ce4+ pair, makes CeO2/Al2O3 solids a verypromising material for obtaining well dispersed andvery active gold catalysts.
Apart from the low temperature CO oxidationreaction, gold catalysts have been successfully testedin many other catalytic reactions, such as hydro-genation of carbon oxides and hydrocarbons, NOreduction, hydrochlorination of ethylene and theoxidation of volatile organic compounds (VOCs)[12,25]. VOCs are a major contributor to air pollu-tion because of their toxic and malodorous natureand its contribution to ozone and smog formation[26]. Mainly, mobile sources and industrial processesare responsible for the VOCs emissions. When therecovery of these compounds is not desired, theyare usually destroyed by deep oxidation. Catalyticcombustion is preferred to thermal one due to thelower temperature required and to its more selectiv-ity, which implies a considerable saving of energy[27].
In this paper, we study the oxidation of some rep-resentative VOCs (n-hexane, benzene and 2-propanol)on a series of Au/Al2O3 and Au/CeO2/Al2O3 catalystsprepared by deposition precipitation. The aim of this
work is to understand the role of gold and the supportin the catalytic combustion of VOCs.
2. Experimental
2.1. Catalysts
An impregnation method was followed to preparethe CeO2/Al2O3 support. The adequate amount ofCeO2 (Sigma 99.9% pure) was dissolved in the small-est possible volume of 4 M HNO3. Then, a slurryof Al2O3 powder (Degussa, type C) in 200 ml ofdeionised water was added. The slurry thus preparedwas taken dried by continuos stirring and heating(60–70◦C). The solid was then kept overnight at120◦C in the oven, crushed in an agate mortar andcalcined for 4 h at 600◦C.
The gold containing catalysts were prepared by de-position precipitation[3]. The adequate amount ofHAuCl4·3H2O (Alfa, 99.99% pure) was dissolved in150 ml of deionised water and the pH of the solutionadjusted to 7.0 by addition of NaOH 0.1 M. The so-lution was heated to 70◦C and then, the support (asreceived Al2O3 or prepared CeO2/Al2O3) was addedand kept under continuous stirring for 1 h. After cool-ing to room temperature, the obtained solids were sep-arate from the solution by centrifugation and washedwith deionised water several times (until the disappear-ance of chloride and Na+), then dried during 7 days inan oven at 30◦C and finally calcined for 2 h at 500◦C.
2.2. Physico–chemical characterisation
BET specific surface area was measured by nitrogenadsorption measurements at liquid nitrogen tempera-ture in a Micromeritics ASAP 2000 apparatus. Beforeanalysis, the samples were degassed for 2 h at 150◦Cin vacuum. The Ce, Al and Au contents of the sam-ples were determined by an ICP-AES Philips PV8250spectrometer.
XRD analysis was performed on a Siemens D 5000powder X-ray diffractometer. Diffraction patternswere recorded with detector-sided Ni-filtered Cu K�radiation (40 mA, 40 kV) over a 2θ range of 22–74◦and a position-sensitive detector using a step size of0.010◦ and a step time of 2.5 s. The mean crystallitesizes were estimated using the Scherrer equation[28]and the selected reflection peaks in the diffraction

M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78 67
pattern were fitted by a Gaussian function. A peakbroadening due to the instrumental broadening of2θ = 0.08◦ was taken into account.
Scanning electron microscopy (SEM) analyses wereperformed with a JEOL JSM 5400 microscope usinga back-scattered electrons detector (BSE).
Transmission electron microscopy (TEM) observa-tions were carried out by a Philips CM200 microscopeoperating at 200 kV. The samples were dispersed inethanol by sonication and dropped on a copper gridcoated with a carbon film.
Diffuse reflectance UV–VIS spectra of the solidsdiluted in BaSO4 were recorded at room temperatureon a Shimadzu UV-2101PC spectrometer equippedwith an integrating sphere and using BaSO4 as ref-erence. They are presented in Kubelka–Munk modewithout any other transformation.
The X-ray photoelectron spectroscopy (XPS)analyses were performed with an SSI X-Probe(SSX-100/206) photoelectron spectrometer (Fisons),using a monochromatised Al K� X-ray source(1486±1 eV), at a pressure around 10−9 Torr (1 Torr=133.3 N m2). A flood gun set at 8 eV and a nickel gridplaced 3 mm above the sample surface were used forcharge compensation. Before analyses, the sampleswere degassed overnight (10−7 Torr). The bindingenergy was calculated with respect to the C–(C, H)component of the C 1s adventitious carbon fixed at284.8 eV. The spectra were decomposed accordingto the least-squares fitting procedure (Fisons) witha Gaussian/Lorentzian ratio of 85/15 and after sub-tracting a Shirley-type nonlinear baseline. The atomicratios were calculated from the relative intensity cor-rected by the elemental sensitivity factors providedby the manufacturer.
2.3. Activity measurement
n-Hexane, benzene and 2-propanol were chosen asrepresentative VOC reactants. Preliminary tests werecarried out at different gas linear velocity and partialsize to discard diffusional limitations. Forn-hexaneand benzene oxidation measurements the catalytictests were carried out in a conventional continuousflow U-shape glass reactor at atmospheric pressure.In order to avoid intrareactor gradients, the catalystpowder (100 mg) was diluted in 7 cm3 of inert glassbeads (φ = 0.2–0.315 mm). The 120 ppm ofn-hexane
and 250 ppm of benzene in air were used as reactantsat a total flow of 100 cm3/min (W/QAo = 8.33 and4.00 gcatalystmin/cm3
VOC NPT, respectively, beingWthe catalysts weight andQAo the flow of the reactantVOC). The catalysts were first activated “in situ” at150◦C during 90 min under oxygen (50 cm3/min)and then stabilised by letting the reaction proceedfor 16 h at 150◦C. The conversion was measuredduring this period, at the end of which it reachedan almost constant value. Then the temperature wasdecreased to 100◦C where the ignition curve startedwhen increasing the temperature by steps of 20 or10◦C. The conversion was then again measured at150◦C to verify that the conversion at the end of thetest was the same as after the preliminary stabilisa-tion period. Reactants and products were separatedby on-line gas chromatography (GC, Chrompack col-umn #773, Wcott fused silica 50 mm× 0.32 mm)and then quantified using a flame ionisation detector(FID).
2-Propanol complete oxidation reactions were car-ried out in a tubular fixed-bed reactor at atmosphericpressure. Mass flow controllers (Brooks 5850TR)were used to prepare the feed mixture. 2-Propanolwas pumped with a HPLC pump (Jasco, PU-1585)and evaporated in a dry air stream. This stream wasfurther mixed with O2/He (Praxair, 99.999%, 21%O2 and 79% He) in order to set the feed stream com-position at 500 ppmv (parts per million by volume)of 2-propanol and passed through the catalyst placedon the top of a carborundum bed. This configura-tion was designed to premix and preheat the streamentering the reactor, to obtain a homogeneous tem-perature, measured by a thermocouple placed just atthe beginning of the catalyst bed. The reactor wassurrounded by an electrical furnace equipped witha temperature programmer. 2-Propanol oxidationover the Au supported catalysts was carried out ata W/QAo ratio of 0.39 gcatalystmin/cm3
2-propanol NPT,with 100 mg of catalyst and a total gas flow rate of510 cm3/min (GHSV 9700 h−1). The ignition curveswere constructed following both the VOC disappear-ance and the CO2 appearance at a controlled heatingrate of 2.5◦C/min. Analyses were done by on-lineGC (Hewlett-Packard 5890, TR-WAX 30 m column)equipped with a thermal conductivity detector (TCD).The CO2 concentration was continuously moni-tored by means of an IR analyser (Digital Control
68 M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78
Systems Model 300). The catalysts were treatedunder 100 cm3/min of O2/He (21/79) at 300◦C for1 h before reaction and then cooled down in air to thestarting reaction temperature.
Additionally, the 2-propanol oxidation reactionwas followed by on-line mass spectrometry (Om-nistar, Balzers). A bargraph analysis of the reactoroutlet composition was carried out measuring them/zsignals from 1 to 100.
3. Results
3.1. Catalysts
Table 1shows the code and the physical propertiesof the final solids. The gold deposition procedureinduces a very low decrease of the surface BET areaof the starting supports (<10% for alumina,<4%for CeO2/Al2O3). SEM images of the gold catalysts(not shown) do not detect any gold particles, thusmeaning that their diameters are below the SEMdetection limit. For CeO2/Al2O3 samples, SEM mi-crographs (not shown) reveal the existence of a largenumber of particles bigger than 0.5�m which containcerium.
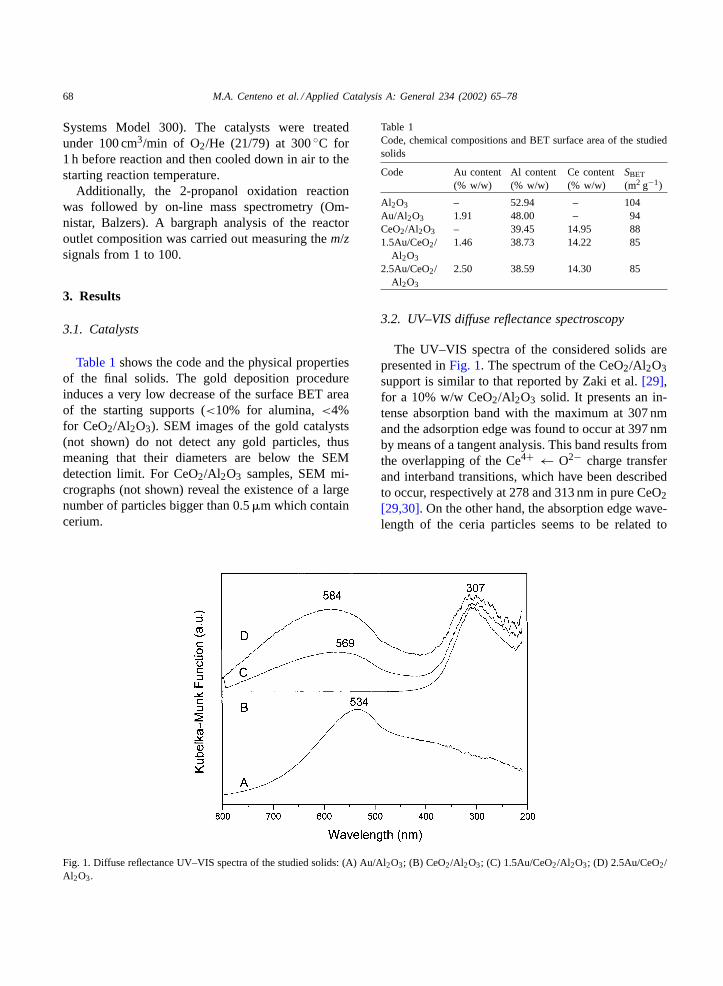
Fig. 1. Diffuse reflectance UV–VIS spectra of the studied solids: (A) Au/Al2O3; (B) CeO2/Al2O3; (C) 1.5Au/CeO2/Al2O3; (D) 2.5Au/CeO2/Al2O3.
Table 1Code, chemical compositions and BET surface area of the studiedsolids
Code Au content(% w/w)
Al content(% w/w)
Ce content(% w/w)
SBET
(m2 g−1)
Al2O3 – 52.94 – 104Au/Al2O3 1.91 48.00 – 94CeO2/Al2O3 – 39.45 14.95 881.5Au/CeO2/
Al2O3
1.46 38.73 14.22 85
2.5Au/CeO2/Al2O3
2.50 38.59 14.30 85
3.2. UV–VIS diffuse reflectance spectroscopy
The UV–VIS spectra of the considered solids arepresented inFig. 1. The spectrum of the CeO2/Al2O3support is similar to that reported by Zaki et al.[29],for a 10% w/w CeO2/Al2O3 solid. It presents an in-tense absorption band with the maximum at 307 nmand the adsorption edge was found to occur at 397 nmby means of a tangent analysis. This band results fromthe overlapping of the Ce4+ ← O2− charge transferand interband transitions, which have been describedto occur, respectively at 278 and 313 nm in pure CeO2[29,30]. On the other hand, the absorption edge wave-length of the ceria particles seems to be related to

M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78 69
their crystallite size[29,31]: the smaller the crystal-lite size, the lower the absorption edge wavelength.The absorption edge value observed (397 nm) coin-cides with that reported for pure ceria, consisting ofcrystallites of average size≥20 nm[29,32,33].
Besides the contribution of the support, the UV–VISspectra of the gold catalysts show a new band due tothe surface plasmon resonance of the Au metal fineparticles. This band is ascribed to a collective oscil-lation of conduction electrons in response to opticalexcitation[34] and is affected by the dispersed metalparticle size, the shape of the particle and the dielec-tric properties of the surrounding material[35,36].For the Au/CeO2/Al2O3 catalysts, the plasmon bandincreases in relative intensity and shifts toward longerwavelengths from 569 to 584 nm with an increaseof the gold content. In the Au/Al2O3 catalyst, theplasmon band is centered at a lower wavelength,534 nm.
3.3. X-ray photoelectron spectroscopy (XPS)
Table 2shows the binding energy values obtainedfor all the considered solids. The detected O 1s, Al 2p,Ce 3d and Au 4f levels did not show any appreciableshift in the binding energies from one catalyst to an-other. The complex shape of the 3d cerium spectra ofthe CeO2/Al2O3 support (Fig. 2), arising from multi-plet effects such as hybridisation of Ce 4f orbitals withO 2p levels and final state effects[37], is characteris-tic of CeO2. As it can be seen, it is not modified uponthe introduction of gold. This indicates that there is nochange in the electronic density of the cerium atoms,which remains as Ce4+ [24]. In a similar way, Au 4fpeaks positions coincide with those of bulk gold, in-dicating that the gold particles were in a metallic stateunder high vacuum conditions.
Table 2Binding energy values (eV) referred to as C 1s (284.8 eV) of thestudied solids
Code O 1s Al 2p Au 4f7 Au 4f5
Al2O3 531.1 74.3 – –Au/Al2O3 531.1 74.4 83.6 87.3CeO2/Al2O3 531.2 74.4 83.6 87.31.5Au/CeO2/Al2O3 531.0 74.3 83.6 87.32.5Au/CeO2/Al2O3 531.0 74.2 83.7 87.2
Fig. 2. XPS spectra at Ce 3d region of the CeO2/Al2O3 sup-port (A) and gold catalysts supported on CeO2/Al2O3: (B)1.5Au/CeO2/Al2O3; (C) 2.5Au/CeO2/Al2O3.
The surface composition of the catalysts (atomicand weight percentage) determined by XPS and someatomic relationships between the elements implied, arepresented inTables 3 and 4. When gold is deposited onAl2O3, the surface Al content increases. This fact hasbeen attributed to the no participation of aluminiumatoms in the anchoring procedure of the gold parti-cles, in which oxygen atoms and hydroxyl groups ofthe support seems to play a predominant role. In thecase of gold deposition on CeO2/Al2O3, the atomicpercentage of aluminium increases and those of oxy-gen and cerium decrease. In this case cerium atomsseem to be involved in the gold deposition process,as it can also be deduced from the increment in theAl/Ce ratio when increasing the gold content.
The comparison of the surface and the bulk com-position of the different elements in the differentsamples can also be done, in order to obtain some in-formation on the dispersion of CeO2 and Au (Tables 1and 3). On the one hand, it can be seen that the sur-face proportion of cerium is much lower than that ofthe bulk (3% instead of 14%). This fact could implythe low dispersion of CeO2 on Al2O3 or a dipping ofthe cerium phase in the alumina. On the other hand,comparing the Au weight percentages (the bulk ones)with the surface percentages obtained by XPS, it canbe seen a better dispersion of gold over the cerium

70 M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78
Table 3Surface composition (atomic and weight percent) of the studied solids, determined from XPS results
Code Atomic (%) Weight (%)
O Al Au Ce O Al Au Ce
Al2O3 63.89 36.11 – – 51.20 48.80 – –Au/Al2O3 61.11 38.80 0.09 – 47.87 51.26 0.87 –CeO2/Al2O3 61.40 38.09 – 0.51 47.19 49.37 – 3.431.5Au/CeO2/Al2O3 60.21 39.25 0.10 0.45 45.76 50.31 0.94 3.002.5Au/CeO2/Al2O3 59.27 40.03 0.25 0.45 44.30 50.45 2.30 2.95
Table 4Some XPS Atomic ratios for the studied solids
Code Al/O Al/Au Al/Ce Ce/Au O/Au
Al2O3 0.565 – – – –Au/Al2O3 0.635 431.1 – – 679CeO2/Al2O3 0.620 – 74.69 – –1.5Au/CeO2/Al2O3 0.652 392.5 87.22 4.5 602.12.5Au/CeO2/Al2O3 0.675 160.1 88.95 1.8 237.08
doped support. The surface/bulk ratio varies from 45%in Au/Al2O3 to 64% in 1.5Au/CeO2/Al2O3 and to92% in 2.5Au/CeO2/Al2O3. This better dispersion onCeO2/Al2O3 can be attributed to the positive role ofthe cerium atoms in the fixation and dispersion of thegold particles, more evident in the 2.5Au/CeO2/Al2O3sample.
Fig. 3. TEM micrographs of catalysts: Au/Al2O3 (A); 1.5Au/CeO2/Al2O3 (B); 2.5Au/CeO2/Al2O3 (C).
3.4. TEM
Typical TEM micrographs of the gold catalystsare shown inFig. 3. The gold particles are regu-larly distributed on the whole support. The parti-cle morphology is similar in the three solids. Thehigh atomic weight of cerium atoms makes difficultthe contrast differentiation of the gold particles onAu/CeO2/Al2O3 samples, although the more spheri-cal shape of the gold particles helps. In the same way,particles with a very low diameter (typically, below1 nm) are also difficult to be detected due to the lowmass and diffraction contrast. These factors probablydrive to the main detection of the larger particles,mainly on 1.5Au/CeO2/Al2O3 catalysts. Keepingin mind these events, the mean gold particle size,

M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78 71
Fig. 4. XRD patterns of the studied solids: (A) Al2O3; (B) Au/Al2O3; (C) CeO2/Al2O3; (D) 1.5Au/CeO2/Al2O3; (E) 2.5Au/CeO2/Al2O3
((�), CeO2; (∗), �-Al2O3; (�), gold).
calculated from the particle size distribution in theTEM micrographs, results to be 8.2 nm on Au/Al2O3and 3.8 nm on both Au/CeO2/Al2O3 samples.
3.5. XRD
The XRD patterns for the studied solids are shownin Fig. 4. The XRD-pattern for Au/Al2O3 presents,besides the characteristic peaks of the original (δ+γ )structure of the Al2O3 support, the reflections due topolycrystalline gold at 2θ = 38.2, 44.4 and 64.6◦.Gold crystallite sizing, based on diffraction peakbroadening according to Scherrer’s equation andassuming the spherical shape of the gold particles,reveals a crystallite size of about 8.9 nm.
The XRD results for the CeO2/Al2O3 support showonly peaks corresponding to fluorite-structured CeO2and (δ + γ ) Al2O3 and no crystalline bulk structuresof interaction species involving ceria and aluminacan be observed. The high amorphous character ofthe samples can be deduced from the high level ofnoise detected in the patterns. The size of the ceriacrystallites is calculated to be about 8 nm. This value
agrees with that reported early for CeO2/Al2O3 sam-ples with a similar ceria content[29,38]. The intro-duction of gold induces the appearance of small peaksof polycrystalline gold. These peaks are very weakand broad and only that at 38.2◦ is discernible. Aftersubtraction of the XRD pattern of the support, thesepeaks emerge. The size of the gold crystallites, de-duced from the broadening of these reflection peaks isbetween 4.5 and 5.6 nm for both Au/CeO2/Al2O3 sam-ples. No changes in the structure of the CeO2/Al2O3support due to the introduction of gold are detected.
3.6. Catalytic tests
Fig. 5 shows the conversion ofn-hexane and ben-zene as function of temperature on the consideredcatalysts. In all cases, the presence of gold stronglyenhances the rate of VOCs oxidation. Gold supportedon CeO2/Al2O3 samples present considerably higheractivities than that supported on pure alumina. For thesame CeO2/Al2O3 support, the higher gold contentof the sample seems to play a weak positive role onthe activity, as deduced from the low increase on the

72 M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78
Fig. 5. Conversion of representative VOCs on the considered catalysts: (�) CeO2/Al2O3; (�) 1.5Au/CeO2/Al2O3; (�) 2.5Au/CeO2/Al2O3;(�) Au/Al2O3.

M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78 73
Fig. 6. Conversion of 2-propanol (�) and products distribution (water (×); acetone (�) and CO2 (�)) on the studied catalysts: (A)Au/Al2O3; (B) 1.5Au/CeO2/Al2O3; (C) 2.5Au/CeO2/Al2O3.

74 M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78
Fig. 7. Products distribution of the 2-propanol oxidation reaction followed by MS spectrometry on Al2O3 and Au/Al2O3 solids:m/z = 41,propene (�); m/z = 45, 2-propanol (+); m/z = 43, acetone (�) and m/z = 44, CO2 (�).

M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78 75
VOCs conversion on 2.5Au/CeO2/Al2O3 comparedto 1.5Au/CeO2/Al2O3. For the most active sample(2.5Au/CeO2/Al2O3), the complete oxidation of ben-zene andn-hexane are obtained at 300◦C with a T50of 265◦C and at 370◦C with aT50 of 305◦C, respec-tively. The CeO2/Al2O3 support itself presents a veryweak activity in the benzene oxidation. However,it is comparable to that of Au/Al2O3 on n-hexaneoxidation.
Fig. 6 shows the conversion of 2-propanol andproducts distribution (acetone and CO2) as a func-tion of reaction temperature. As it can be seen, theoxidation of 2-propanol produces acetone as an inter-mediate at low temperatures over the three catalysts.However, at higher temperatures complete oxidationof 2-propanol to CO2 is obtained. This completeoxidation of 2-propanol to CO2 is produced at lowertemperatures on gold supported CeO2/Al2O3 cata-lysts, showing the favourable effect of CeO2. 100%oxidation of 2-propanol to CO2 is obtained at 380◦Con 2.5Au/CeO2/Al2O3 catalyst, with aT50 of 280◦C.The production of the partial oxidation intermedi-ate (acetone) in the Au/CeO2/Al2O3 catalysts passesthrough a maximum at about 230◦C and it is higherwhen the amount of gold increases. Above this tem-perature the complete oxidation to CO2 seems to beactivated, producing sharper increases in the H2O andCO2 signals and a decrease in the acetone. Similarbehaviour has been reported on Au/Fe2O3 [39]. ForAu/Al2O3 sample, two maximum of acetone pro-duction are detected, one at 160◦C and the other at348◦C. The disappearance of 2-propanol and its con-version into acetone is very fast in this catalyst (100%conversion at 280◦C) pointing out the high dehydro-genating activity of the sample. However, completeoxidation to CO2 is achieved only at 430◦C. From 160to 280◦C, the acetone outlet concentration decreaseswhile 2-propanol conversion increases continuouslyuntil 100%. Around 280◦C, total oxidation to CO2begins and acetone production increases again untila maximum is reached at 348◦C. In order to explainthe observed behaviour of Au/Al2O3 sample, the2-propanol oxidation reaction was additionally fol-lowed by on-line spectrometry (Fig. 7). The resultedCO2 (m/z = 44), 2-propanol (m/z = 45) and acetone(m/z = 43) profiles are similar to those obtained byon-line GC (Fig. 5). Two peaks of acetone produc-tion are detected again and the formation of CO2
begins with the high temperature one. Besides this,mass spectrometry detects the production of propene(m/z = 41) in the range of temperatures between thesetwo peaks. The analysis of the 2-propanol oxidationreaction over the Al2O3 support itself, reveals a simi-lar production of propene. In fact, the only differencebetween the results obtained for the Au/Al2O3 andAl2O3 samples, is the presence of the low temperaturepeak of production of acetone in the gold catalyst.
4. Discussion
TEM and XRD analyses agree in the determinationof the average gold particle size in all catalysts. In theabsence of cerium, gold is deposited in particles ofaround 8 nm. The low surface concentration of goldfound by XPS (0.87% w/w) compared to that presentin the bulk (1.91% w/w), points out that either thedispersion is not very high or an important precip-itation of gold has occurred away from the supportsurface during the synthesis. Over the CeO2/Al2O3support, gold particles of lower diameter (around4 nm) are obtained. TEM and XRD have detectedno significant differences in the gold crystalline sizebetween 1.5Au/CeO2/Al2O3 and 2.5Au/CeO2/Al2O3samples. However, XPS has shown that at highergold content the difference between surface andbulk gold concentration is reduced (0.94/1.91%w/w for 1.5Au/CeO2/Al2O3, 2.30/2.50% w/w for2.5Au/CeO2/Al2O3), indicating a better dispersion ofgold in the more loaded catalysts.
On the contrary, the increase in intensity and shifttoward longer wavelengths detected on the gold plas-mon band in the UV–VIS spectra of Au/CeO2/Al2O3catalysts when increasing the gold content (Fig. 1)could be indicative of the increase of the gold par-ticle size when increasing the gold loading[40,41].The apparent contradiction can be solved taking intoaccount the limitations of each technique. First of all,particles with a very low diameter (typically, below1 nm) are difficult to be detected by TEM due to thelow mass and diffraction contrast, leading to the de-tection essentially of the larger particles. This effectmainly affects the 1.5Au/CeO2/Al2O3 sample andcould drive to an erroneous calculation of the meanparticle size. A similar limitation can be consideredfor XRD, where crystallites with diameter lower than

76 M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78
a few nanometers escape XRD detection. On the otherhand, the plasmon band observed in the UV–VISspectra is strongly affected by the dielectric propertiesof the surrounding material. Although no modifica-tions in the structure of the CeO2/Al2O3 support aredetected as a function of the gold content, we cannotfully discard the changes in the plasmon band due tothe better dispersion of gold in 2.5Au/CeO2/Al2O3compared with 1.5Au/CeO2/Al2O3. Gold has an ab-sorption coefficient higher than the support and ahigher gold covering leads to a larger fraction of theoxide not interacting with the radiation[36], varyingthe plasmon position. In the case of Au/Al2O3, mainlythe different dielectric properties of the alumina ascompared with the CeO2/Al2O3 ones, are responsi-ble for the differences observed in the plasmon bandbetween Au/Al2O3 and Au/CeO2/Al2O3 catalysts.Therefore, no conclusions can be extracted concern-ing the gold particle size in Au/Al2O3 catalyst.
As a conclusion of the gold particles size, charac-terisation results seem to show that the presence ofcerium atoms exerts a positive influence on the fixa-tion and final dispersion of gold on the alumina sup-port. In the synthesis conditions used, the anchoringof gold particles on the CeO2/Al2O3 support surfaceseems to proceed mainly over the CeO2 phase, as de-duced from XPS results. This is not surprising, sincethe deposition–precipitation method requires a signif-icant interaction between the solution containing goldand the support to be carried out at an acceptablerate[42]. Commercial aluminas present unfavourableproperties for this kind of deposition due to their longand narrow pores which make extremely difficult toapply an active precursor uniformly[42].
In any case, it seems that the gold content does notinfluence strongly the final mean gold crystalline size,although a better gold dispersion is obtained over theCeO2/Al2O2 support for the high gold content used.In spite of the participation of cerium atoms in thefixation process of the gold particles, no modificationis detected in the structure of the CeO2/Al2O3 sup-port after gold deposition. In this sense, no changes inthe oxidation state of cerium atoms, electronic densityof Al or O, or optical properties of ceria phase havebeen seen. The low surface cerium content (Table 3) ascompared with the one presented in the bulk (Table 1)is indicative of the low dispersion of the ceria phase.In this sense, the UV–VIS results are similar to those
reported for pure CeO2, where the average crystal-lite size is higher than 20 nm. In good agreement withthese results, SEM images of the CeO2/Al2O3 samples(not shown) reveal the existence of a large number ofparticles higher than 0.5�m that contain cerium. Theamorphous nature of such big particles can be postu-lated from XRD results (Fig. 4), where an importantamorphous contribution in the CeO2/Al2O3 pattern isdeduced and the calculated average CeO2 crystallitessize was very low (around 8 nm). From the above con-siderations, it seems clearly that when improving thedispersion and decreasing particle size of the ceriaphase, an enhanced gold dispersion and particle sizecould be achieved.
Catalytic results have shown that the oxidationrate of VOCs is strongly enhanced by the presenceof gold, even in the case of the CeO2/Al2O3 supportwhere some catalytic activity due to the presence ofcerium atoms could be attained. For the same support(CeO2/Al2O3), the increasing amount of gold playsalso a positive role. This effect is more evident in theoxidation of linear VOCs (n-hexane and 2-propanol)than of aromatic ones (benzene) and could be relatedto the accessibility of gold particles to the reactantmolecules. In this sense, gold particles seem to beassociated to the ceria phase, which presents low dis-persion and high particle size. In this situation, goldparticles may be placed close to each other, beingmore accessible to a linear molecule than to a morevoluminous aromatic.
From the results reported inFig. 5, the reactivityof the different VOCs can be compared. 2-Propanolis the most easily destroyed, followed by benzene andbeingn-hexane is the least reactive. This sequence ofreactivity is in good agreement with that reported byTichenor and Palazzolo[43]. They found that usinga noble metal catalyst, the sequence of reactivity ofnonhalogenated VOCs were: alcohols, aldehydes, aro-matics, ketones, acetates and alkanes.
Catalytic activity is strongly dependent on thenature of the support. The Au/CeO2/Al2O3 sam-ples are always more active for deep oxidation thanAu/Al2O3, even having a lower gold content. One ofthe factors that determine this is the lower crystallinesize of the gold particles obtained (4 nm instead of8 nm). The higher dispersion of gold, which drivesto a larger number of catalytic sites disposable, is animportant factor to be taken into account. However,

M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78 77
the difference in activity between cerium doped andundoped alumina gold catalysts, is higher enough tosuggest a positive influence of the ceria phase. As theCeO2/Al2O3 support itself does not present an impor-tant catalytic activity, this must be ascribed to the roleof gold/ceria interface. In this sense, a strong synergis-tic effect between gold and vanadia has been recentlydescribed in Au-V2O5/TiO2 and Au-V2O5/ZrO2 cat-alysts for the complete benzene oxidation reactionand ascribed to the effect of gold in the vanadiumoxide reducibility, resulting in a considerable lower-ing of the temperature of the V5+ → V3+ transition[44]. On the other hand, the redox properties of ceriacan be claimed as one of those mainly responsiblefor such effect. Indeed, it is well known that ceriaenhances the oxygen storage capacity of the catalysts,due to its particular ability to undergo deep and rapidreduction/oxidation cycles according to the reactionCeO2 ↔ CeO2−x + (x/2)O2 upon interaction withreducing or oxidising agents present in the reactionconditions[23,45,46]. In this way, ceria can controland maintain the adequate oxidation state of goldactive sites and enhance the lattice oxygen mobility,which leads to an increase in the catalytic activity. Inthis sense, it has been proposed that partially oxidisedgold is more active than metallic gold in CO oxidation[15] and it has been described that low temperaturecatalytic combustion of VOCs involves surface lat-tice oxygen species via Mars–van Krevelen cycles[39,47,48]. The detection of acetone as intermediatein 2-propanol oxidation can support this mechanism,since it suggests that carbonyl compounds are thefirst oxidation products of alcohols and they can beintermediate adsorbed species involved in the totaloxidation reaction[47,49]. Indeed, the mechanism ofthe 2-propanol oxidation seems to begin with the ad-sorption of gaseous 2-propanol as 2-propoxide surfacespecies[49]. The presence of gold favours the alco-hol dehydrogenation to give acetone in the gas phase,even at low temperatures. The Au/Al2O3 presents avery high dehydrogenating activity as can be deducedfrom the total conversion of 2-propanol to acetoneat low temperature (280◦C), without any productionof CO2 (Fig. 6). In the case of this catalyst and pureAl2O3, the decrease observed in the acetone produc-tion between 160 and 348◦C is associated with thedetection of propene in the gas phase (Fig. 7). Thisbehaviour has been previously reported for pure and
metal-modified aluminas at 300–400◦C and related tothe exposition, due to a moderate dehydroxilation ofthe surface, of acid–base site pairs capable of dehy-drating 2-propanol to propene[49]. At higher temper-atures the total oxidation to CO2 begins. As proposedabove, the role of ceria in Au/CeO2/Al2O3 catalystsmay be the modification of the oxidation state anddispersion of gold atoms, but it can also affect in someway the catalytic activity due to its high lattice oxy-gen mobility. Therefore, the ceria doped catalysts canfavour the deep oxidation of the intermediate speciesadsorbed on the support to CO2. In good agreementwith this postulate, Au/CeO2/Al2O3 catalysts start toproduce CO2 at lower temperatures than Au/Al2O3(Fig. 6) and no production of propene is detected.
5. Conclusions
CeO2/Al2O3 materials have been shown as a moreadequate support than alumina for preparing activegold catalysts for VOCs catalytic combustion. Ceriaparticipates in the fixation of gold and enhances itsfinal dispersion, thus leading to the stabilisation ofgold particles with lower crystallites size. Besides theincrease of the activity due to this, the redox prop-erties of ceria improve the catalytic behaviour of thecatalysts, probably by increasing the mobility of thelattice oxygen and by controlling the adequate oxida-tion state of gold.
Acknowledgements
M.A.C. gratefully acknowledges a fellowshipgranted by the European Union. XPS and cat-alytic studies withn-hexane and benzene were per-formed in the “Unité de Catalyse et Chimie desMateriaux Divisés”, Université Catholique de Lou-vain, Louvain-la-Neuve, Belgium.
References
[1] J. Schwank, Gold. Bull. 16 (1983) 103.[2] M. Haruta, Catal. Today 36 (1997) 153.[3] S. Tsubota, D.A. Cunningham, Y. Bando, M. Haruta, in:
G. Poncelet, P. Grange, P.A. Jacobs (Eds.), Preparation ofCatalysts VI, Elsevier, Amsterdam, 1995, p. 227.

78 M.A. Centeno et al. / Applied Catalysis A: General 234 (2002) 65–78
[4] M. Haruta, T. Kobayashi, H. Sano, N. Yamada, Chem. Lett.405 (1997).
[5] D.A. Cunningham, T. Kobayashi, N. Kamijo, M. Haruta,Catal. Lett. 25 (1994) 257.
[6] M. Haruta, N. Yamada, T. Kobayashi, S. Ijima, J. Catal. 115(1989) 301.
[7] M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M.J.Genet, B. Delmon, J. Catal. 144 (1993) 175.
[8] J. Grunwaldt, C. Kiener, C. Wögerbauer, A. Baiker, J. Catal.181 (1999) 223.
[9] M.A. Bollinger, N.A. Vannice, Appl. Catal. B 8 (1996) 417.[10] M. Daté, M. Haruta, J. Catal. 201 (2001) 221.[11] J. Grunwaldt, M. Maciejewski, O.S. Becker, P. Fabrizioli, A.
Baiker, J. Catal. 186 (1999) 458.[12] G.C. Bond, D.T. Thompson, Catal. Rev. Sci. Eng. 41 (1999)
319.[13] F. Bocuzzi, A. Chiorino, S. Tsubota, M. Haruta, J. Phys.
Chem. 100 (1996) 3625.[14] S.D. Lin, M. Bollinger, M.A. Vannice, Catal. Lett. 17 (1993)
245.[15] E.D. Park, J.S. Lee, J. Catal. 186 (1999) 1.[16] S. Galvagno, G. Parravano, J. Catal. 55 (1978) 178.[17] P.A. Sermon, G.C. Bond, P.B. Wells, J. Chem. Soc., Faraday
Trans. I 75 (1979) 385.[18] G.C. Bond, P.A. Sermón, Gold. Bull. 6 (1973) 102.[19] A. Ueda, M. Haruta, Appl. Catal. B 18 (1998) 115.[20] F. Porta, L. Prati, M. Rossi, S. Coluccia, G. Martra, Catal.
Today 61 (2000) 165.[21] A. Trovarelli, Catal. Rev. Sci. Eng. 38 (1996) 439.[22] R. Dictor, S. Roberts, J. Phys. Chem. 93 (1989) 5846.[23] A. Martinez-Arias, M. Fernández-Garcıa, L.N. Salamanaca,
R.X. Valenzuela, J.C. Conesa, J. Soria, J. Phys. Chem. B 104(2000) 4038.
[24] M.A. Centeno, P. Malet, I. Carrizosa, J.A. Odriozola, J. Phys.Chem. B 104 (2000) 3310.
[25] H. Sakurai, M. Haruta, Appl. Catal. A 127 (1995) 93.[26] J.A. Horsley, Catalytica Environmental Report E4, Catalytica
Studies Division, Mountain View, CA, USA, 1993.
[27] M. McGraph, Appl. Catal. B 5 (1995) N25.[28] P. Scherrer, Gott. Nachr. 2 (1918) 98.[29] M.I. Zaki, G.A.M. Hussein, S.A.A. Mansour, H.M. Ismail,
G.A.H. Mekhemer, Colloids and Surfaces A 127 (1997) 47.[30] A. Bensalem, J.C. Muller, P. Bozon-Verduraz, J. Chem. Soc.,
Faraday Trans. 88 (1992).[31] A. Bensalem, P. Bozon-Verduraz, M. Delamar, G. Bugli,
Appl. Catal. 121 (1995) 81.[32] K.B. Sundaram, P. Wahid, Phys. Rev. B 161 (1990) 163.[33] C.A. Hogarth, Z.T. Aldham, Phys. Rev. B 137 (1986) 151.[34] P. Mulvaey, Langmuir 12 (1996) 788.[35] S. Deki, Y. Aoi, H. Yanagimoto, K. Ishii, K. Akamatsu, M.
Mizuhata, A. Kajinami, J. Mater. Chem. 6 (1996) 1879.[36] F. Bocuzzi, G. Cerrato, F. Pinna, G. Strukul, J. Phys. Chem.
B 102 (1998) 5733.[37] F. Le Normand, L. Hilarie, K. Kili, G. Krill, G. Maire, J.
Phys. Chem. 92 (1988) 2561.[38] P.J. Schmitz, R.K. Usmen, C.R. Peters, G.W. Graham, R.W.
McCabe, Appl. Surf. Sci. 72 (1993) 181.[39] S. Minicò, S. Scirè, C. Crisafulli, R. Maggiore, S. Calvagno,
Appl. Catal. B 28 (2000) 245.[40] S. Chen, K. Kimura, Langmuir 15 (1999) 1075.[41] M. Alvarez, J. Khoury, T. Schaaff, M. Shafigullin, I. Vezmar,
R.L. Whetten, J. Phys. Chem. B 101 (1997) 3706.[42] J.W. Geuss, Stud. Surf. Sci. Catal. 16 (1983) 1.[43] B.A. Tichenor, M.A. Palazzolo, Environ. Progr. 6 (1987) 172.[44] D. Andreeva, T. Tabakova, L. Ilieva, A. Naydenov, D.
Mehanjiev, M.V. Abrashev, Appl. Catal. A 209 (2001) 291.[45] H.C. Yao, Y.F. Yao, J. Catal. 86 (1984) 254.[46] T. Miki, T. Ogawa, M. Haneda, N. Kakuta, A. Ueno, S.
Tateishi, S. Matsuura, S. Sato, J. Phys. Chem. 94 (1990)6464.
[47] M. Baldi, E. Finocchio, F. Milella, G. Busca, Appl. Catal. B16 (1998) 43.
[48] E.M. Cordi, P.J. O’Neill, J.L. Falconer, Appl. Catal. B 14(1997) 23.
[49] M.I. Zaki, M.A. Hasan, L. Pasupulety, Langmuir 17 (2001)4025.




















