
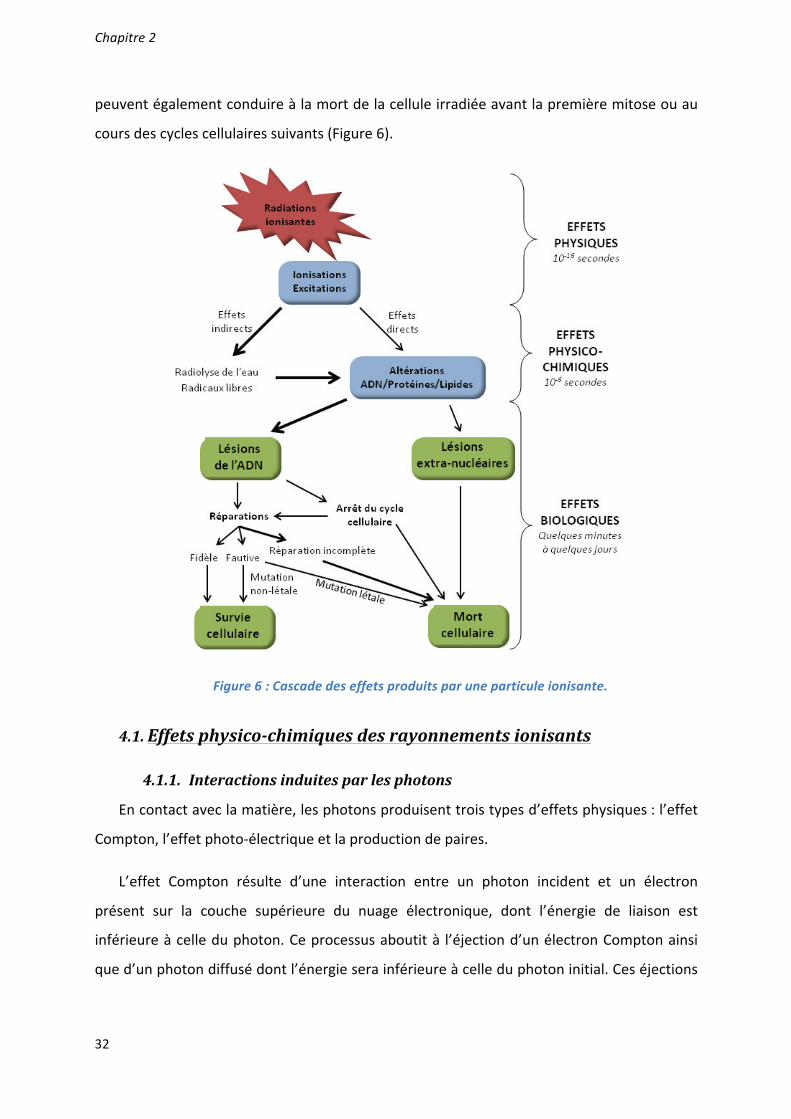
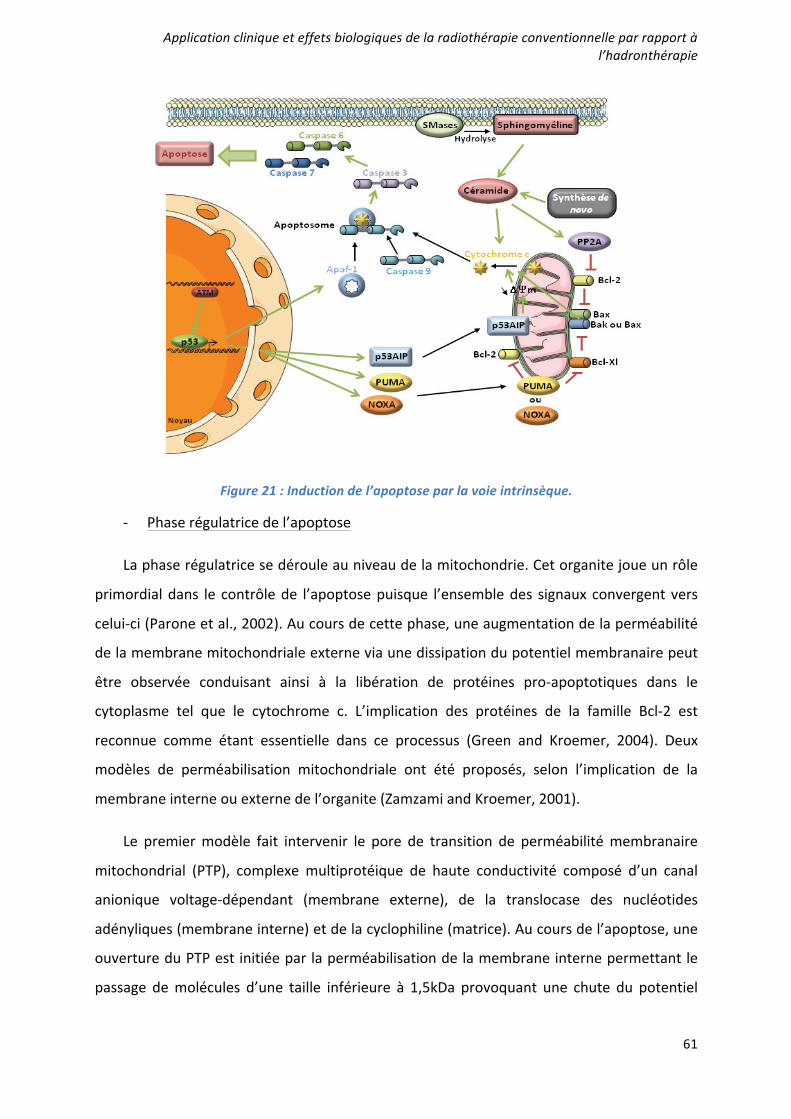
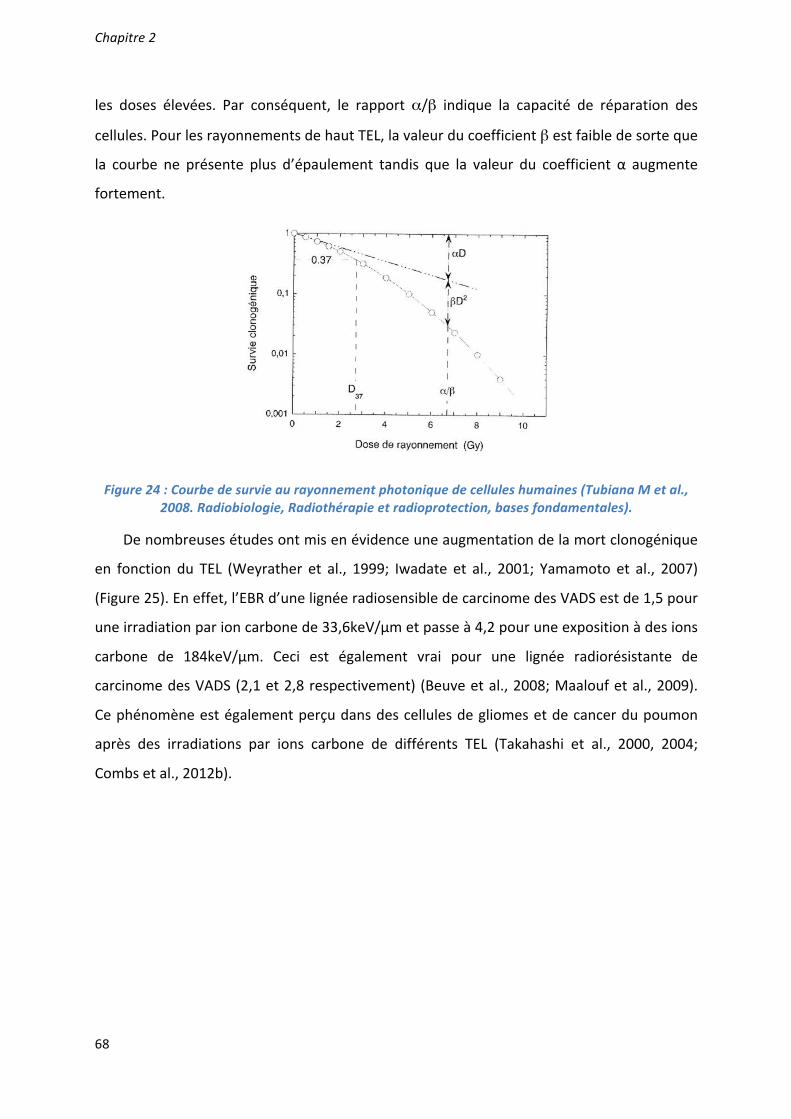
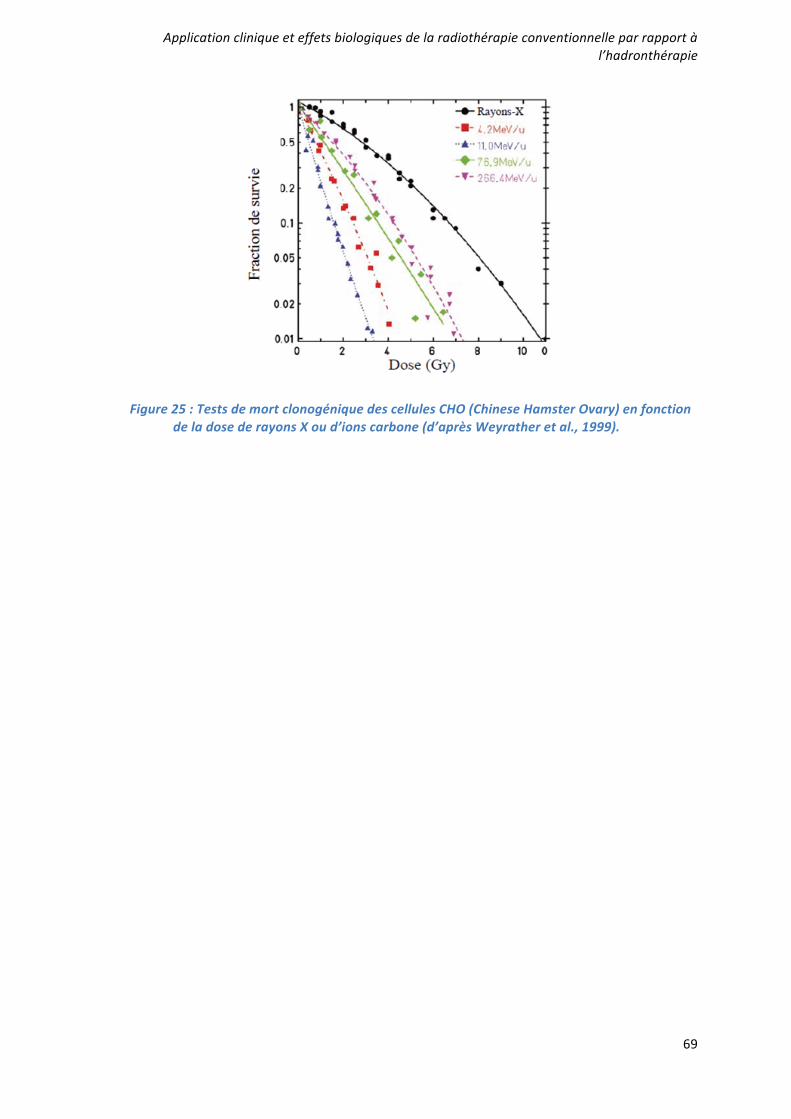
Caractérisation et ciblage thérapeutique d'une - TEL - Thèses
301
HAL Id: tel-01175903 https://tel.archives-ouvertes.fr/tel-01175903 Submitted on 13 Jul 2015 HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci- entific research documents, whether they are pub- lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers. L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés. Caractérisation et ciblage thérapeutique d’une sous-population de cellules souches cancéreuses dans un modèle cellulaire de carcinome épidermoïde de la tête et du cou résistant à l’irradiation par photon et ions carbone Gérald Bertrand To cite this version: Gérald Bertrand. Caractérisation et ciblage thérapeutique d’une sous-population de cellules souches cancéreuses dans un modèle cellulaire de carcinome épidermoïde de la tête et du cou résistant à l’irradiation par photon et ions carbone. Biologie cellulaire. Université Claude Bernard - Lyon I, 2013. Français. NNT: 2013LYO10118. tel-01175903
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of Caractérisation et ciblage thérapeutique d'une - TEL - Thèses

HAL Id: tel-01175903https://tel.archives-ouvertes.fr/tel-01175903
Submitted on 13 Jul 2015
HAL is a multi-disciplinary open accessarchive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come fromteaching and research institutions in France orabroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, estdestinée au dépôt et à la diffusion de documentsscientifiques de niveau recherche, publiés ou non,émanant des établissements d’enseignement et derecherche français ou étrangers, des laboratoirespublics ou privés.
Caractérisation et ciblage thérapeutique d’unesous-population de cellules souches cancéreuses dans unmodèle cellulaire de carcinome épidermoïde de la tête et
du cou résistant à l’irradiation par photon et ionscarbone
Gérald Bertrand
To cite this version:Gérald Bertrand. Caractérisation et ciblage thérapeutique d’une sous-population de cellules souchescancéreuses dans un modèle cellulaire de carcinome épidermoïde de la tête et du cou résistant àl’irradiation par photon et ions carbone. Biologie cellulaire. Université Claude Bernard - Lyon I,2013. Français. �NNT : 2013LYO10118�. �tel-01175903�









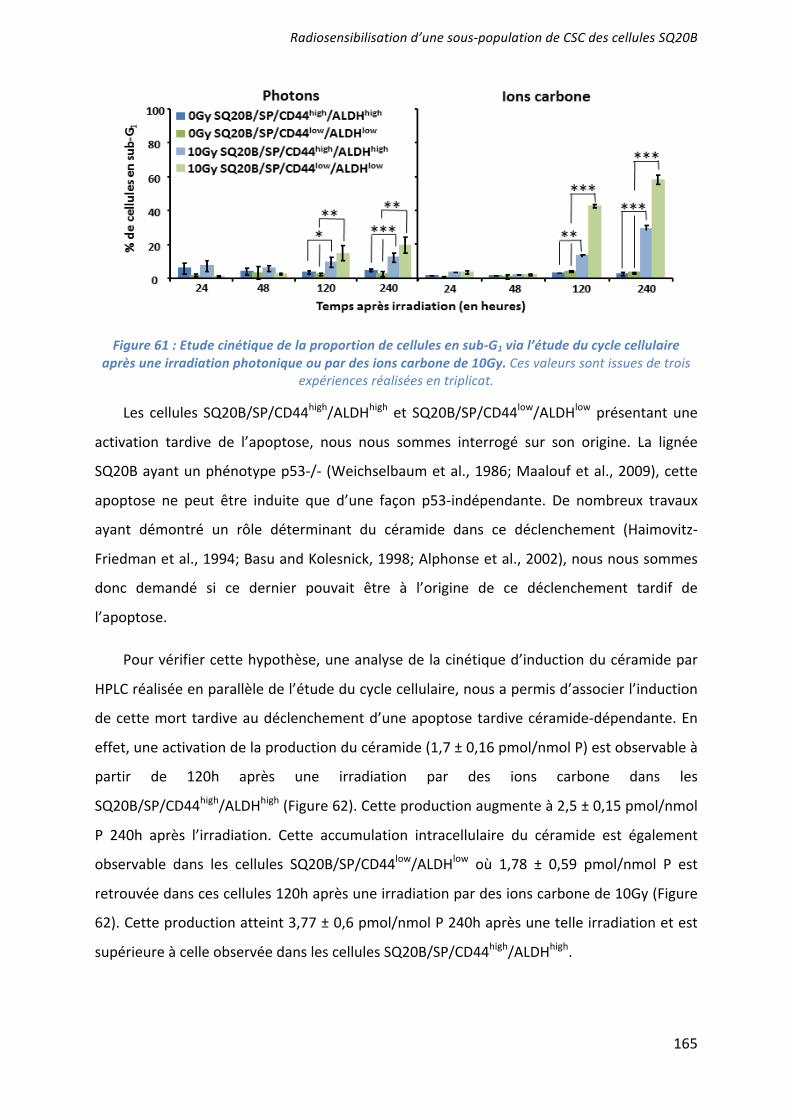
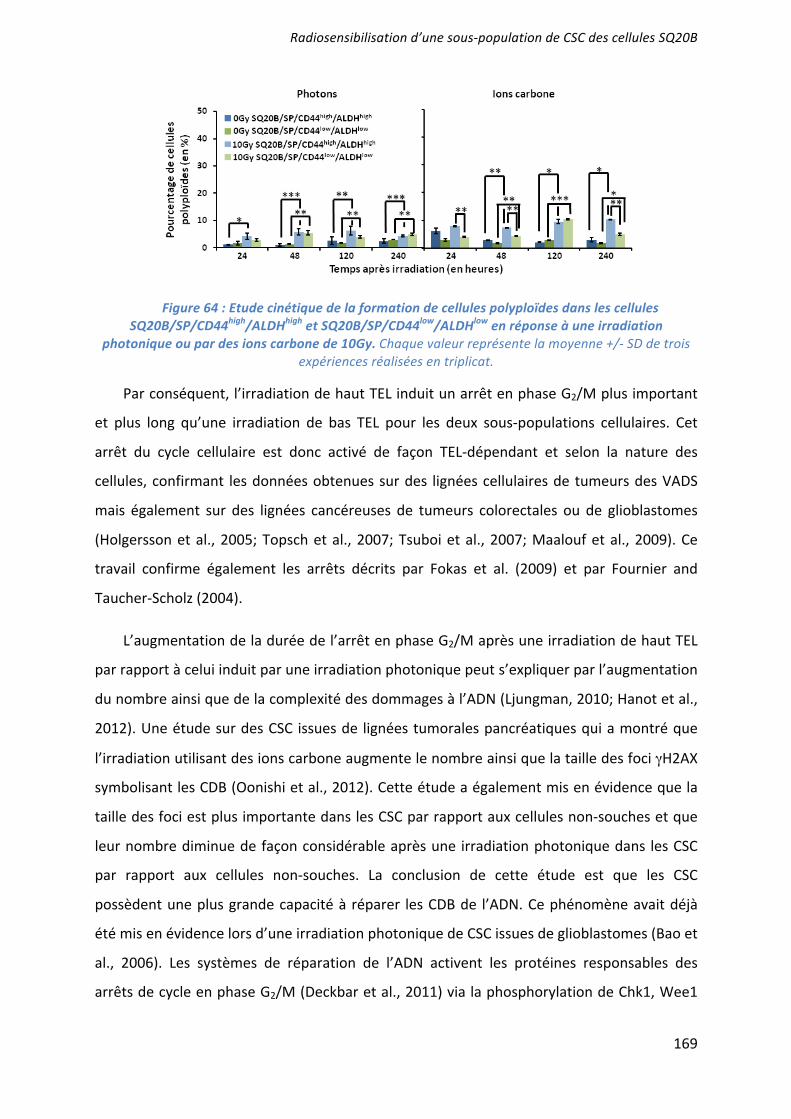
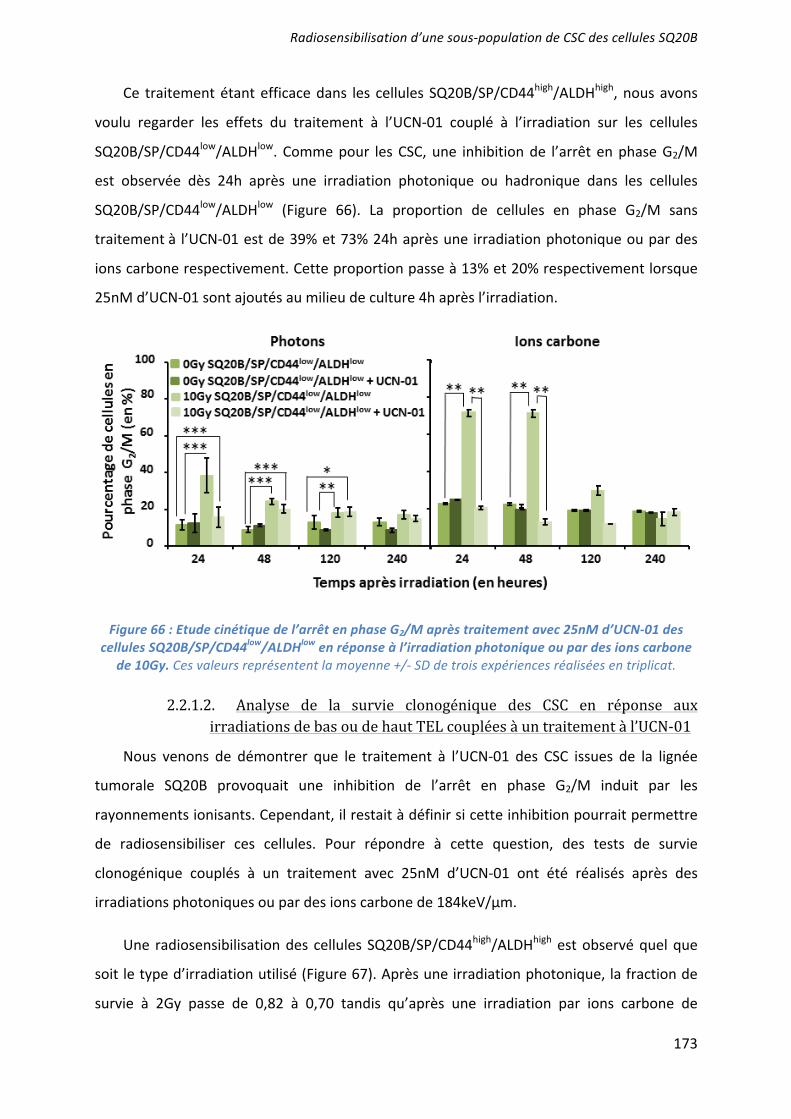
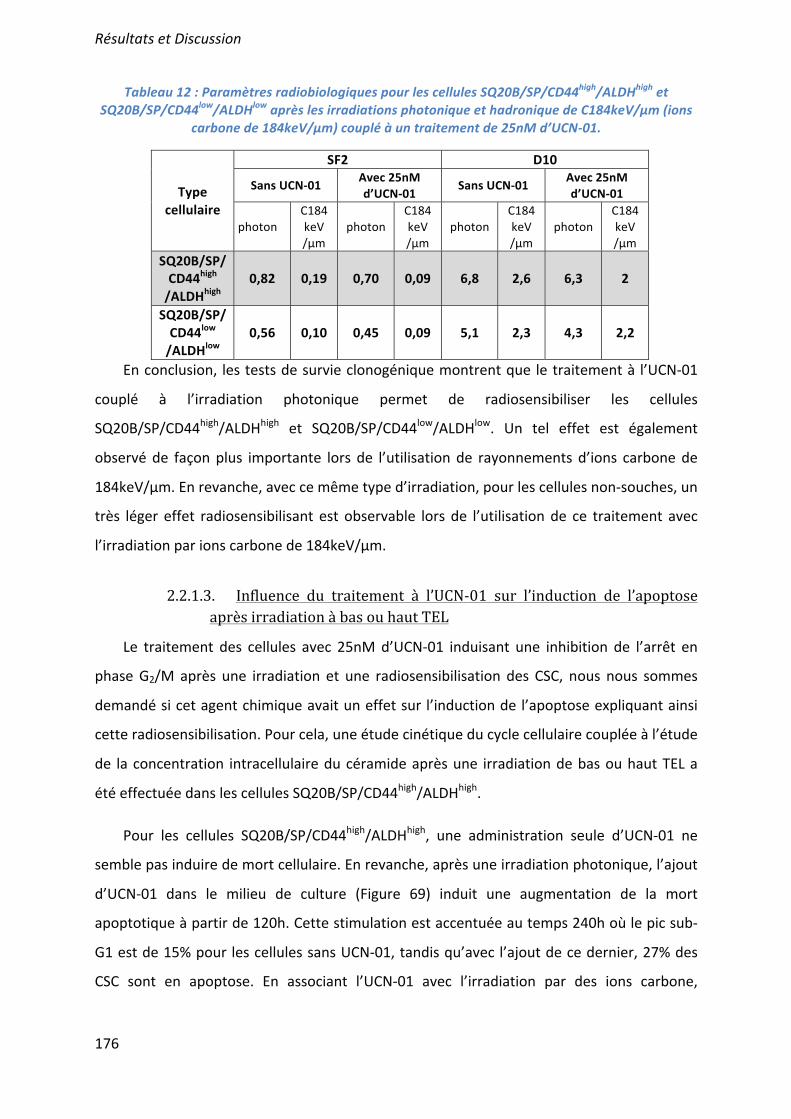
L’effet radiosensibilisant de 3 molécules ciblant les CSC a été démontré : la mort apoptotique induite par l’UCN-01 en inhibant l’arrêt en phase G2/M ; les capacités prolifératives ciblées par l’acide trans-rétinoïque (ATRA) induisant la différenciation ; et la voie de l’autorenouvellement Bmi-1 inhibée par l’artésunate. Seules ou associées (UCN-01 + ATRA), elles agissent en synergie avec une irradiation par photons ou ions carbone. Des études pré-clinique, puis clinique, devraient confirmer l’intérêt du ciblage des CSC dans le contrôle de l’échappement de ces cancers radiorésistants. ___________________________________________________________________________ Characterization and therapeutic targeting of a cancer stem cell subpopulation in a head and neck squamous cell carcinoma resistant to photon and carbon ion irradiation. ___________________________________________________________________________
The radiosensitizing effects of 3 molecules targeting the CSC has been demonstrated: an induction of apoptotic cell death by the inhibition of the G2/M phase arrest after a treatment with UCN01; an inhibition of proliferative capacities using the all-trans-retinoic acid (ATRA) which induce their differentiation; and an inhibition of Bmi1 by artesunate. These treatments, alone or in combination (UCN01+ATRA), have a synergistic effect with photon or carbon ion irradiation to overcome CSC radioresistance. Preclinical and clinical studies should confirm the benefit of targeting CSC and improve the control of tumor escape in patients with radiorésistant HNSCC cancers. ___________________________________________________________________________ DISCIPLINE : Aspect moléculaire et cellulaire de la biologie __________________________________________________________________________ MOTS-CLES : cellules souches cancéreuses, irradiation photonique, irradiation par ions carbone, radiosensibilisation, tumeurs des voies aéro-digestives supérieures. ___________________________________________________________________________ INTITULE ET ADRESSE DE L'U.F.R. OU DU LABORATOIRE :




ββ
ββ
ββ
β




β



γ




κ

β ββ β
β







( )


σ


α

α

(







(




(
)


γ

θ








(

α
α


α α


δ

λ

γ


δ


β β






α


ξ κ
α






α β
α β
α
β
α β
β





κ
β







β

β

β

β
β

β





γ
κ
κ

γ


β

κ


ββ
β
β
β
β
β
β
β

β
ββ
β β
β
β
β
β
β
β
β

β

βββ
β
β
β
β
β

βββ β
β
β
β
β β
β
β
β

ββ
β
β
β
β

β
β
β
κ
β
β
β




β


β
β
ββ
β

β
β β β
β
β
β
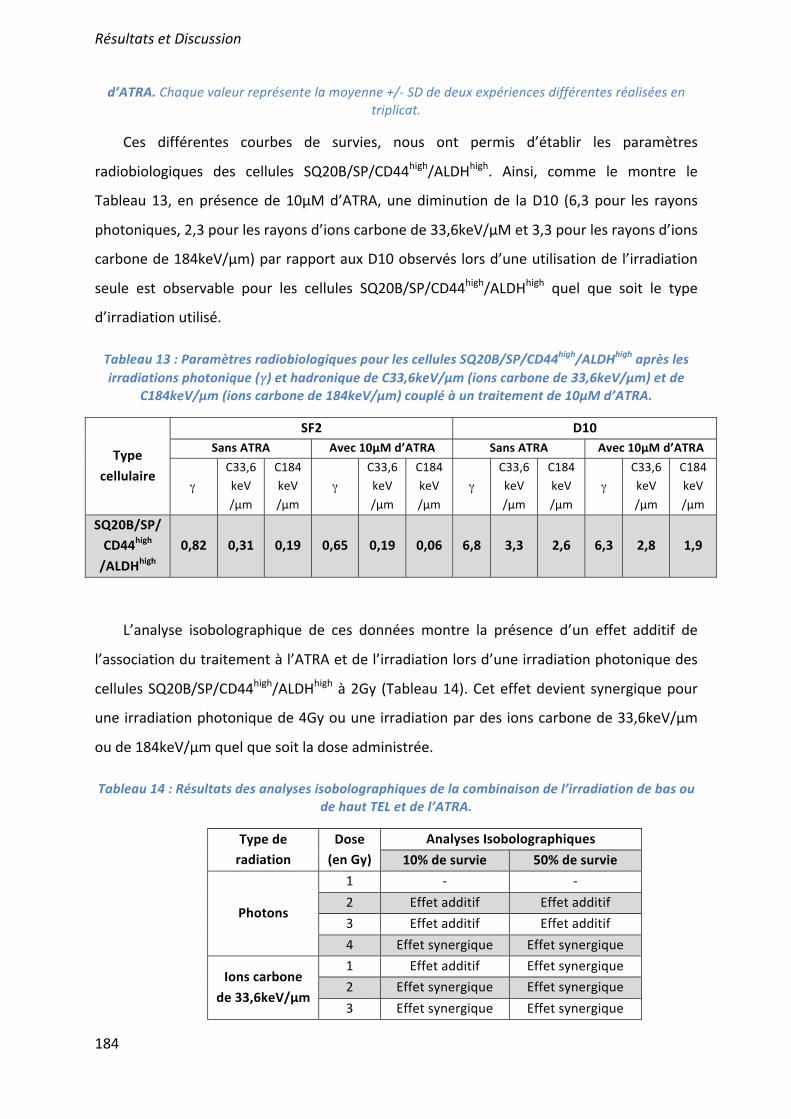
β
β

ε







β



•
•
•
•




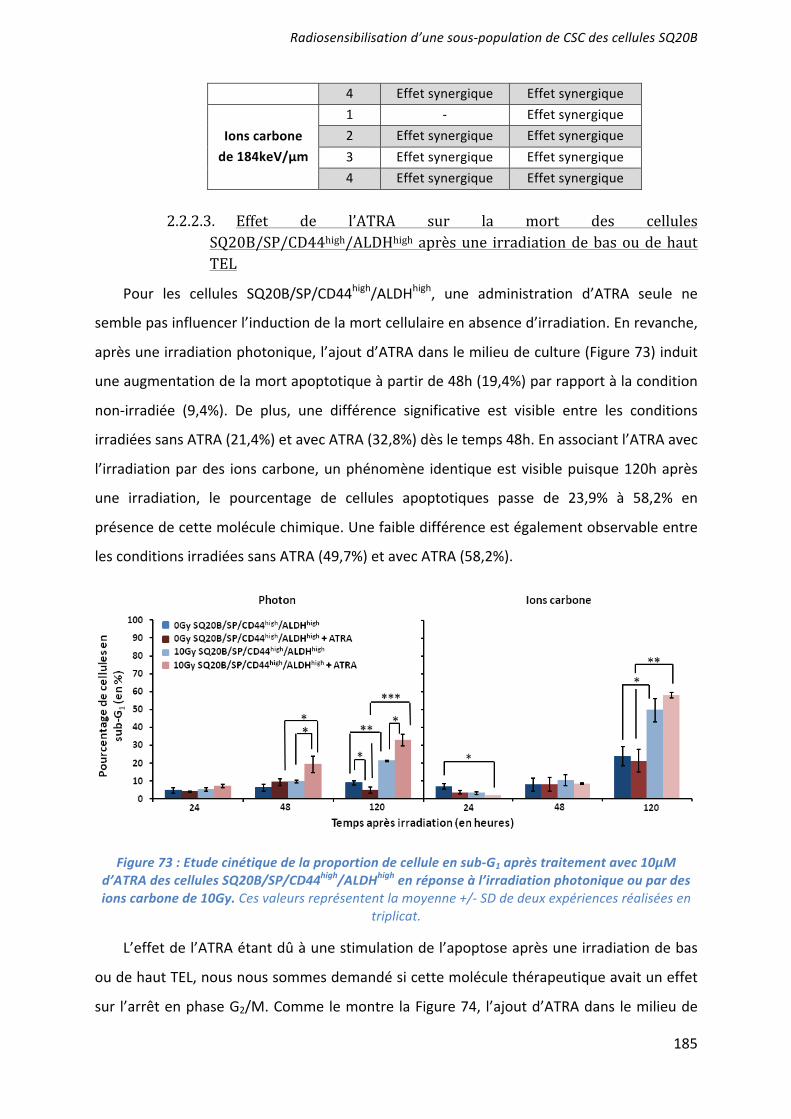
αβα/β






�
�
�


α β
α
β
α β
α β






β


β
β
ΔΔ

β

� β
�
�








β
β
β
β

β
β






β





γ

γ












γγ
γ γ γ γ

β

β








κ

















































1
Targeting head and neck cancer stem cells to overcome resistance to photon and carbon ion radiation Gérald Bertrand MS*,a,b, Mira Maalouf PhD*,a,b, Antony Boivin PhDa,b, Priscillia Battiston-Montagne BSca,b, Michael Beuve PhDa,e, Antonin Levy MDd, Patrice Jalade PhDc, Claudia Fournier PhDf, Dominique Ardail PhDa,b, Nicolas Magné MD PhDa,b, Gersende Alphonse PhDb,c and Claire Rodriguez-Lafrasse PharmD PhDa,b,c aUniversité de Lyon, F-69622, Lyon, France; Université Lyon I, Faculté de Médecine-Lyon-Sud, F-69921 Oullins, France bLaboratoire de Radiobiologie-Cellulaire-et-Moléculaire, EMR3738, F-69921 Oullins, France cHospices-Civils-de-Lyon, CHLS, Pierre-Bénite, F-69495, France dDépartement de radiothérapie oncologique, IGR, Université ParisXI, Villejuif, France eIPNL, LIRIS, CNRS, Villeurbanne, F-69622, France fGSI Helmholtz Center for HeavyIonResearch, Biophysics Department, 64291 Darmstadt, Germany *G. Bertrand and M. Maalouf contributed equally to this work and should be considered as first authors. No conflit of interest ABSTRACT
Although promising new radiation therapy techniques such as hadrontherapy are currently being evaluated in the treatment of head and neck malignancies, local control of head and neck squamous cell carcinoma (HNSCC) remains low. Here, we investigated the involvement of cancer stem-like cells (CSCs) in a radioresistant HNSCC cell line (SQ20B). Stem-like cells SQ20B/SP/CD44+/ALDHhigh were more resistant to both photon and carbon ion irradiation compared with non-CSCs. This was confirmed by a BrdU labeling experiment, which suggests that CSCs were able to proliferate and to induce tumorigenicity after irradiation. SQ20B/SP/CD44+/ALDHhigh were capable of an extended G2/M arrest phase in response to photon or carbon ion irradiation compared with non-CSCs. Moreover, our data strongly suggest that resistance of CSCs may result from an imbalance between exacerbated self-renewal and proliferative capacities and the decrease in apoptotic cell death triggering. In order to modulate these processes, two targeted pharmacological strategies were tested. Firstly, UCN-01, a checkpoint kinase (Chk1) inhibitor, induced the relapse of G2/M arrest and radiosensitization of SQ20B-CSCs. Secondly, all-trans retinoic acid (ATRA) resulted in an inhibition of ALDH activity, and induction of the differentiation and radiosensitization of SQ20B/SP/CD44+/ALDHhigh cells. The combination of ATRA and UCN-01 treatments with irradiation drastically decreased the surviving fraction at 2Gy of SQ20B-CSCs from 0.85 to 0.38 after photon irradiation, and from 0.45 to 0.21 in response to carbon ions. Taken together, our results suggest that the combination of UCN-01 and ATRA represent a promising pharmacological-targeted strategy that significantly sensitizes CSCs to photon or carbon ion radiation. Keywords: Hadrontherapy, Photon irradiation, Carbon ion irradiation, Radiosensitization, Cancer stem cells, HNSCC

2
INTRODUCTION
Despite progress in current treatments, the prognosis of advanced Head and Neck Squamous Cell Carcinoma (HNSCC) remains poor. The overall survival rate is less than 50% and has remained relatively unchanged over the last decades. Radiation therapy is an essential treatment modality for HNSCC. However, even with the use of promising new radiation techniques such as hadrontherapy that demonstrated interesting clinical results on skull base tumors, soft-tissue sarcoma, or adenocarcinoma of the head and neck, control rates remain low for HNSCC1–3. Recent studies attribute local recurrence and distant metastasis to the survival of cancer stem cells (CSCs) following anticancer therapies4. Although there is accumulating evidence supporting the existence of stem-like cancer cells in primary tumors, the functional and mechanistic impact of these cells in mediating resistance to therapy remains poorly understood. It has nevertheless been proposed that CSCs are more resistant to photon radiation and chemotherapy treatments than the bulk of tumor cells5,6. Numerous factors have been proposed to explain the resistance of CSCs to radiation, such as propensity to quiescence, enhanced DNA repair, upregulated cell cycle control mechanisms and free-radical scavenging5,7. Moreover, several oncogenic molecular pathways may be specifically increased in CSCs8. Interestingly, Cui et al. demonstrated that hadrontherapy may have a biological advantage over conventional radiotherapy through improved targeting of putative colon cancer-stem-like-cells9. In HNSCC, stem-like tumor-initiating cells have been isolated from patient biopsies and cell lines based on a CD44+ phenotype and/or high aldehyde-dehydrogenase (ALDH) activity10,11.
In a previous study12, we demonstrated that despite the efficiency of carbon ions in inducing mitotic catastrophe and delayed apoptosis in radioresistant SQ20B cells, a subpopulation could reenter the cell cycle and proliferate. In order to obtain more insight into the mechanisms of re-proliferation, we have investigated the role of cancer stem-like cells obtained from the SQ20B HNSCC cell line in the radioresistance to photons and carbon ions. According to the literature13, we have shown that their resistance may result, among other factors, from an imbalance between exacerbated self-renewal and proliferative capacities and the decrease in apoptotic cell death triggering. Attempts to pharmacologically modulate these processes in combination with irradiation seem therefore to be promising therapeutic strategies5. Among possible strategies, we firstly investigated the effect of UCN-01, an inhibitor of the G2/M arrest, that allowed us to demonstrate that untreated irradiated CSCs do not undergo early apoptosis because of an extended arrest in the G2/M phase. According to recent successful data obtained with leukemia patients14, we used, in a second step, all-trans retinoic acid (ATRA) in order to induce differentiation of CSC before irradiation. These two pharmacological approaches were investigated separately or in combination and allowed us to demonstrate that pharmacological adjuvant treatments targeting either the inhibition of survival/self-renewal pathways or the triggering of apoptosis strongly sensitize cells exposed to radio- or hadron- therapy. MATERIAL AND METHODS Cell culture
The parental HNSCC SQ20B human cell line and the SQ20B/SP/CD44-/ALDHlow
cells were maintained at 37 C with 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM) and Glutamax I (0.86 mg/ml) supplemented with 10% heat-inactivated fetal calf serum (FCS), 100 units/ml penicillin, 100 g/ml streptomycin, and 0.4 mg/ml hydrocortisone as described in 12. SQ20B/SP/CD44+/ALDHhigh cells were cultured in DMEM:F12 (3:1) (v:v) supplemented with 5% FCS, 100 U/ml penicillin, 100 mg/ml streptomycin, 0.04 mg/l

3
hydrocortisone, and 20 ng/ml epidermal growth factor (EGF) according to15. The doubling time for SQ20B/SP/CD44+/ALDHhigh cells was 22hours compared to 27hours for SQ20B/SP/ CD44-/ALDHlow cells.
For tumor sphere formation, cells were grown in serum-free DMEM:F12(3:1) medium containing 20ng/ml of EGF. Irradiation procedures and cell drug exposures
Sixteen hours before irradiation, the cells were seeded on 25cm² flasks or 6-well plates at a concentration varying between 3.104 to 2.106 depending on the kinetics and the dose of irradiation as previously described12.
250kV photon irradiation was performed using an X-RAD320 irradiator (PrecisionX-ray Inc., NorthBranford, USA). 75MeV/n carbon ion (LET=33.6keV/ m) and 11.4MeV/n carbon ion (LET=184keV/ m) irradiations were performed at Grand-Accélérateur-National-Ion-Lourds (France) and Helmholtz Center for Heavy Ion Research (Germany), respectively, as previously described12,16.
UCN-01 25nM (7-hydroxystaurosporine), a Chk1 phosphorylation inhibitor, was added to the culture medium 4h after radiation. ATRA (all-trans retinoic acid), a derivative of vitamin A which induced differentiation, was added to the cancer stem-like cell population 7days before radiation at a final concentration of 10 M.
When both drugs were applied to cultured cells, the previously described procedure was performed for each drug, i.e. ATRA 7days before and UCN-01 4h after radiation. Markers of stemness
Flow cytometry analysis for Hoechst efflux After trypsinization, SQ20B cells were adjusted to 106 cells/ml in PBS and incubated for
90 min at 37 C with 5 g/ml Hoechst 33342 dye. Cells were then washed with PBS and resuspended at a concentration of 107 cells/ml in PBS. For dead cell discrimination, propidium iodide solution was added at a final concentration of 1 g/ml. In order to analyze the effect of Hoechst efflux inhibition, 50 M verapamil, an inhibitor of ABC transporters, was added 10 min before Hoechst staining to a parallel set of samples. Hoechst efflux, distinctive of side population (SP) cells, was analyzed and sorted using a FACS-LSRII and FACS Vantage SE, respectively. The Hoechst dye was excited by an ultraviolet laser at 350 nm, and its fluorescence was measured at two wavelengths using 405/30 (Hoechst blue) and 570/20 (Hoechst red) band-pass filters. Propidium iodide fluorescence was excited by a laser at 488 nm and detected after passing through a 630/22 band-pass filter. Propidium iodide-positive dead cells and debris were excluded. The SP and non-SP cells were sorted.
Flow cytometry analysis for CD44 determination Twenty to 240h after 10Gy photon or carbon ion irradiation, cells were trypsinized and
centrifuged. Cell pellets were washed with PBS and resuspended at a concentration of 107
cells/ml in a PBS buffer containing 0.5% BSA 2 mM EDTA. Cells were then incubated for 10 min at 4 C with an anti-CD44-FITC mouse monoclonal antibody (1/100, BD BioScience). The samples were then washed in PBS, incubate in propidium iodide 1 g/ml to exclude death cells and analyzed with a FACS-LSRII flow cytometer. For FACS cell sorting, SQ20B/SP cells were stained and sorted using a FACS Vantage SE. The purity of sorted cells (SQ20B/SP/CD44+ and SQ20B/SP/CD44–) was estimated to be more than 98%.
Flow cytometry analysis of ALDH activity ALDH activity was determined using an ALDEFLUOR-kit (StemCellTechnologies,
Grenoble, France) according to the manufacturer’s instructions. For cell sorting, cells were also labeled with an anti-CD44-APC mouse monoclonal antibody and then sorted using a FACS VantageSE.

4
RT-qPCR of Bmi-1
After extraction of the RNA with the RNeasy® Micro Kit (Qiagen), reverse transcription was performed with the Quantitect® Reverse Transcription Kit (Qiagen) as decribed in17. Then, a qPCR was proceeded with using the Quantitect® SYBR green PCR Master Mix (Qiagen) in order to quantify Bmi-1 expression on a Stratagene Mx3000 system (Agilent technologies). The geometrical mean of 3 reference genes (GAPDH, β-actin and RPL13) was used for gene expression normalization. β-actin: S: 5’-CCAACCGCGAGAAGATGA-3’, AS: 5’-CCAGAGGCGTACAGGGATAG-3’; Bmi-1: S : 5’-CCAGGGCTTTTCAAAAATGA-3’, AS: 5’-CCGATCCAATCTGTTCTGGT-3’; GAPDH: S: 5’-GAGTCAACGGATTTGGTCGT-3’, AS: 5’-TTGATTTTGGAGGGATCTCG-3’; RPL13: S: 5’-CTGGACCGTCTCAAGGTGTT-3’, AS: 5’-TGGTACTTCCAGCCAACCTC-3’. Evaluation of in vivo tumorigenicity after subcutaneous injection of sorted cells
Tumorigenicity was tested in NOD-SCID mice. After cell sorting, SQ20B/SP/CD44+/ALDHhigh versus SQ20B/SP/CD44–/ALDHlow cells were suspended in PBS at three different concentrations (104-105-106 cells/ml) and 100μl (so 103-104-105 cells) were injected subcutaneously. Injected mice were followed for up to 10 weeks. Survival curves
Cell survival following irradiation was realized using a colony-forming assay as previously described12,16. Briefly, 10 to 16 hours before irradiation, cells were seeded in 25 cm² flasks at different densities, depending on the radiation dose. Cells were irradiated at room temperature at doses of 1, 2, 3, 4, or 5 Gy, delivered at a dose rate of 2 Gy/min. After 1 week, which corresponds to the time required for six cell divisions, colonies were fixed with ethanol 95% and stained with Giemsa 1/20. The number of colonies containing at least 50 cells was counted using a Coltcount (Optronix, United Kingdom) and the surviving fraction was calculated using the formula S(Dose)=e-( D+ D²). Cell cycle distribution
The percentage of cells in each phase of the cell cycle was quantified by flow cytometry using propidium iodide labelling as described by12. The number of cells in sub-G1 phase in the course of the cell cycle was taken as an index of apoptosis. Cells were pelleted and fixed in ice-cold 70% ethanol for at least 24 h after 2 washes with cold PBS. The cells were then washed with PBS and incubated for 15 min in the dark at room temperature in RNase 500 μg/ml, and propidium iodide 100 μg/ml. A minimum of 10.000 cells was analysed by flow cytometry (BD LSRII flow cytometer). Cellular proliferation
The proliferative capacity of cells was determined using a BrdU labeling kit (Roche, Meylan, France) according to the manufacturer’s instructions after a 10Gy photon or carbon ion irradiation. This labeling was conjugated with CD44 labeling using an anti-CD44-APC mouse monoclonal antibody. A minimum of 500 cells were scored on two slides and the ratio CD44 positive-BrdU positive cells over to the whole population labeled with DAPI. Statistical analysis Dose-response interactions between radiation and drugs were evaluated using the classical isobolographic method described by Steel and Peckham18. For a given level of efficacy (% survival), an ‘envelope of additivity’ curve was calculated from the dose effect curves of the drug (UCN-01 or ATRA or ATRA/UCN-01), and from the dose effect curves for radiation alone. The coordinates of the experimental point are the drug concentration and the radiation dose, which, when combined, give the level of efficacy. If the experimental point falls above, beyond, or under the limits of the envelope of additivity, the combination of drugs and radiation gives rise to antagonistic (ant), additive (+), or synergistic (syn) effects, respectively.

5
Student’s t test was used to compare differences between groups.
RESULTS Enrichment of SQ20B-CSCs after low (photon) and high (184keV/ m carbon ions) LET radiation
In order to determine whether the SQ20B cell line contains CSCs responsible for tumor formation and radioresistance, we measured the expression of CD44, a surface marker of HNSCC stem cells10 after exposure to both radiation treatments.
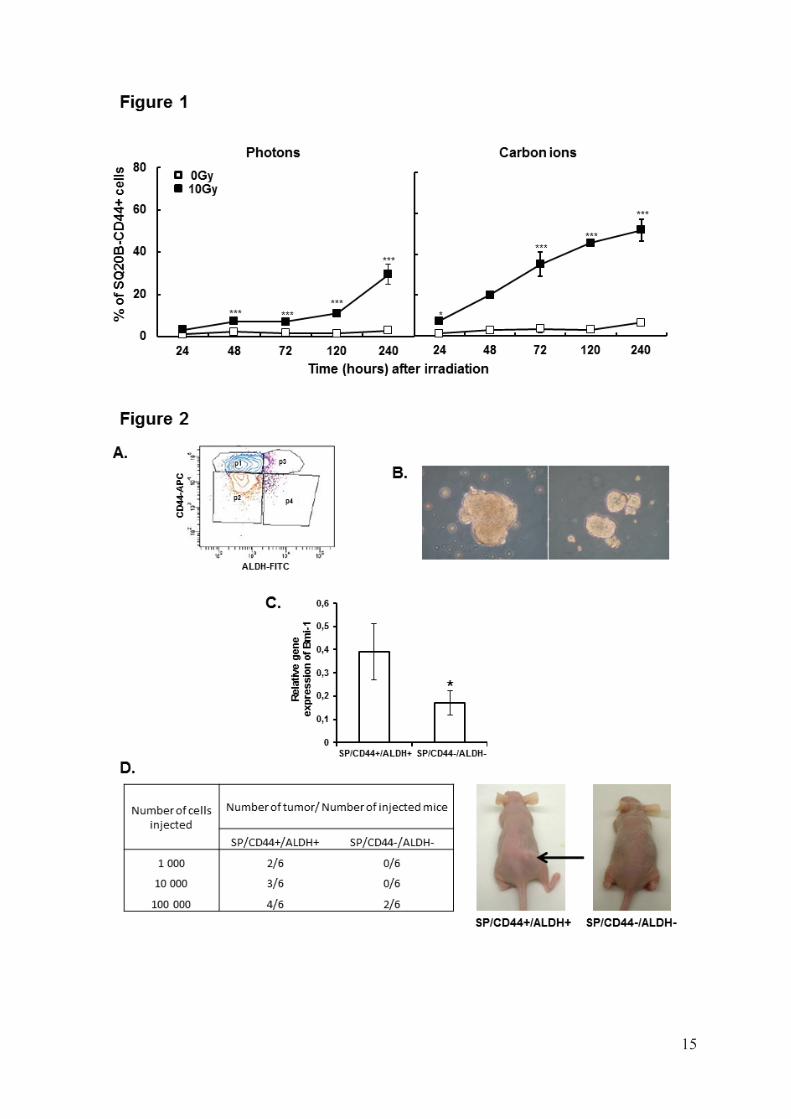
A kinetic study of SQ20B cells expressing CD44 after 10Gy photon or carbon ion exposure was performed. The basal percentage of cells expressing CD44 (CD44+) was less than 1%, whereas after exposure to 10Gy photon or carbon radiation, the number of CD44+ cells increased with time to reach, after 10 days 30% (p<0.001) and 55% (p<0.001), respectively (Fig1). These results suggest that low or high-LET radiation preferentially kills non-CSCs, and that the cells surviving after irradiation are enriched in CD44+cells. Determination of SQ20B-CSCs properties
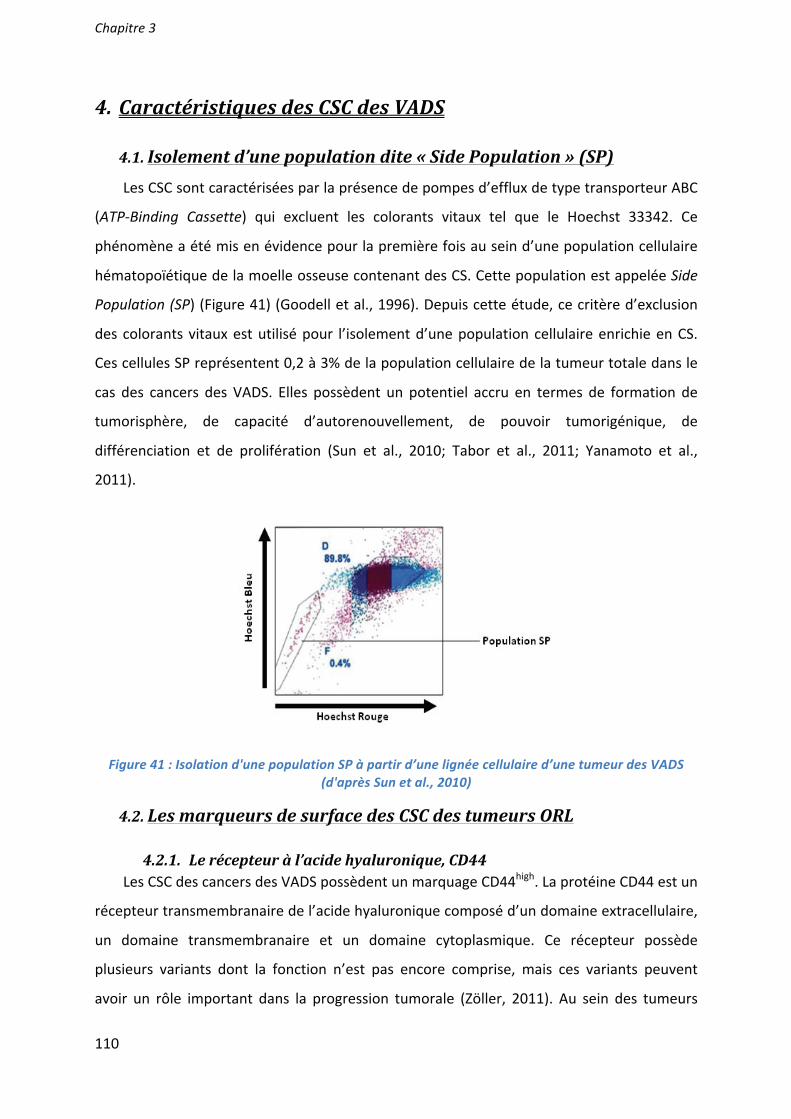
Two populations, SQ20B/SP/CD44+/ALDHhigh and SQ20B/SP/CD44–/ALDHlow, were isolated after two consecutive cell sortings. The first sorting with Hoechst labeling led to isolation of the Side Population (SP) whereas the second one performed on this sorted SP population used double labeling CD44/ALDH (Fig2A).
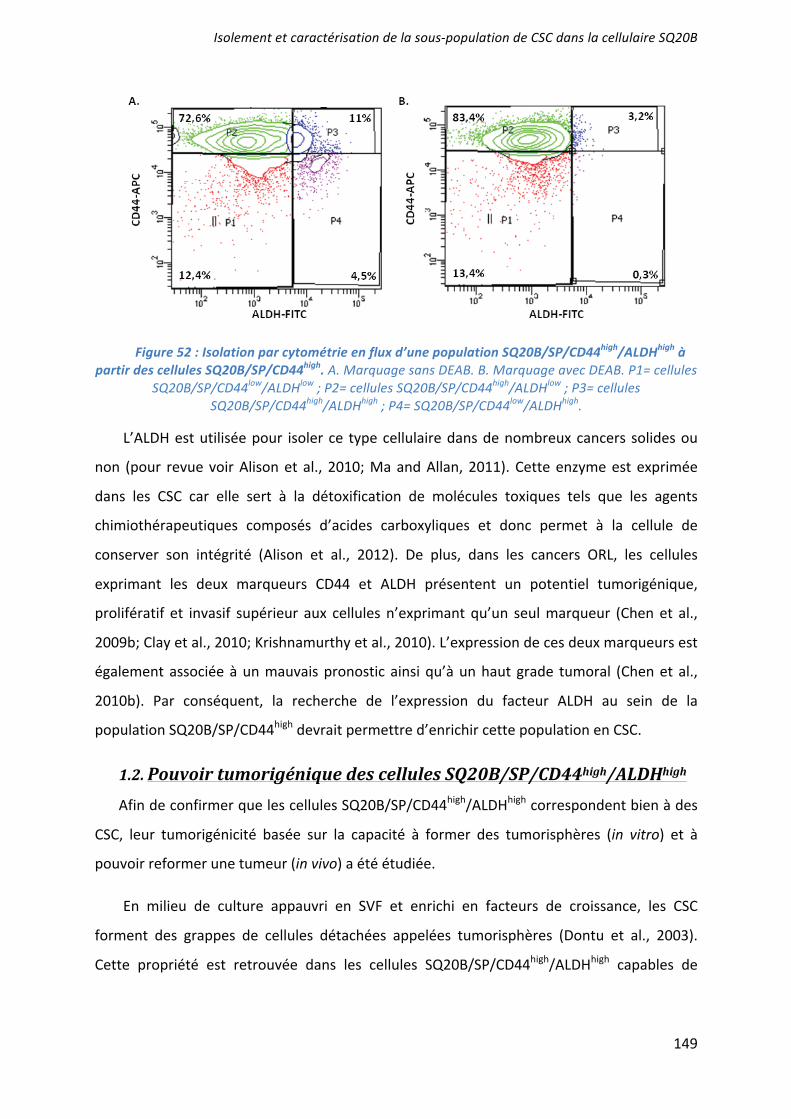
The in vitro cancer stem-like cell properties of SQ20B/SP/CD44+/ALDHhigh cells was firstly determined using a sphere formation assay. SQ20B/SP/CD44+/ALDHhigh cells showed enhanced tumor sphere formation in terms of number and size, when grown for 2 weeks in serum-free DMEM:F12 (Fig2B). In contrast, SQ20B/SP/CD44–/ALDHlow cells did not form any tumor sphere. In order to confirm that SQ20B/SP/CD44+/ALDHhigh population are putative stem-like cells, different stemness markers, specific to HNSCC CSCs (Oct4, Notch,
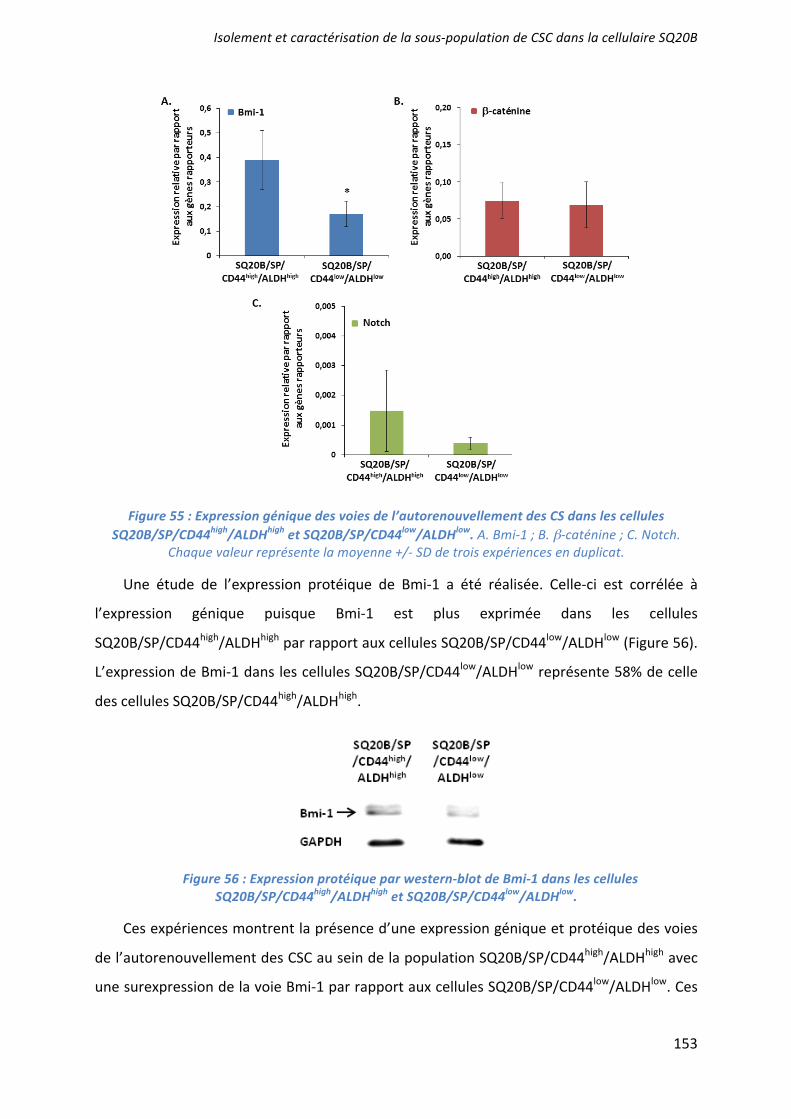
-catenin and Bmi1), were investigated10,11,13. Among all the different genes studied, only Bmi1 (Fig2C) was significantly (p<0,05) more expressed in the CSCs population compared with SQ20B/SP/CD44–/ALDHlow cells.
In order to evaluate the tumorigenicity of SQ20B/SP/CD44+/ALDHhigh cells in vivo, we injected different amounts of SQ20B/SP/CD44+/ALDHhigh or SQ20B/SP/CD44–/ALDHlow
cells into the leg of NOD-SCID-mice. SQ20B/SP/CD44–/ALDHlow cells gave rise to new tumors when at least 105 cells were injected, but failed at lower doses. In contrast, SQ20B/SP/CD44+/ALDHhigh cells were able to give rise to tumors even when 103 cells were injected thus demonstrating their much higher tumorigenicity (Fig2D). Radiosensitivity of SQ20B/SP cells according to their CD44/ALDH status
In order to evaluate the sensitivity of CSCs and non-CSCs subpopulations in response to photon and carbon ion radiation (33.6 or 184keV/ m), survival curves were then established. Two parameters were calculated from these curves : the Surviving Fraction at 2Gy (SF2), used as an index of radiosensitivity and Relative Biological Efficiency (RBE) which is the ratio of the dose of carbon ion radiation relative to those of photons at 10% survival. The RBE is then directly related to the efficiency of carbon ion irradiation. The SQ20B/SP/CD44+/ALDHhigh cells were found to be more resistant to photons than SQ20B/SP/CD44–/ALDHlow cells, as evidenced by their SF2 of 0.82 and 0.56, respectively (p<0.001) (Fig3). The SF2 of SQ20B/SP/CD44–/ALDHlow decreased to 0.17 (p<0.001) and 0.09 (p<0.001) after a respective 33.6 and 184keV/ m carbon irradiation (Table1). In contrast, SQ20B/SP/CD44+/ALDHhigh remained more resistant than SQ20B/SP/CD44–
/ALDHlow, with SF2 values of 0.28 and 0.19 respectively (p<0.001). RBE values were almost the same for the two subpopulations; around 2 for 33.6keV/μm and 2.5 for 184keV/μm carbon ion irradiation (Table1).

6
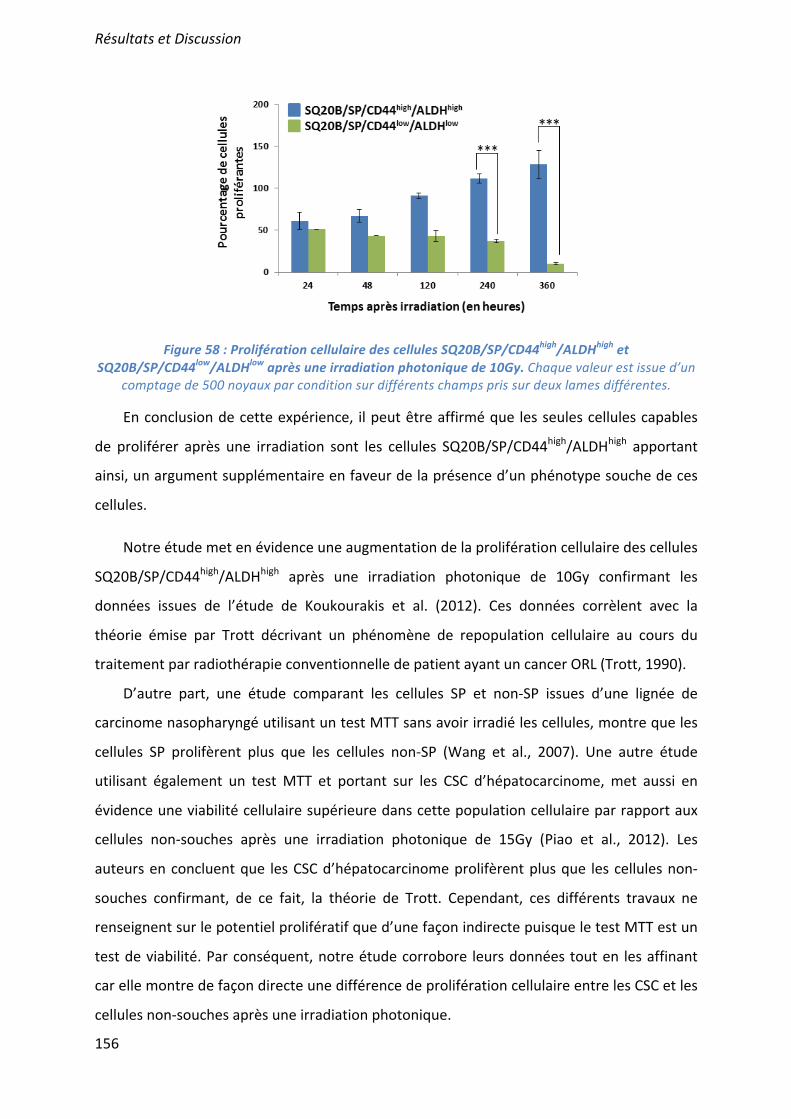
Characteristics of SQ20B/SP/CD44+/ALDHhigh radioresistance In a previous study12, we demonstrated that, after irradiation, a subpopulation of SQ20B cells was able to escape mitotic catastrophe and continued to proliferate. In order to show that these proliferating cells correspond to the SQ20B/SP/CD44+/ALDHhigh subpopulation, a BrdU proliferating test was performed. As depicted in Figure 4A, the proliferation of this subpopulation was greatly reduced from 24 to 120 hours (p<0.001) after photon or carbon ion exposure. A significant delay in the regrowth of SQ20B/SP/CD44+/ALDHhigh cells was observed after carbon ion irradiation when compared with photons. Less than 6% of the SQ20B/SP/CD44–/ALDHlow cells were able to proliferate after irradiation. As we previously reported that the parental SQ20B cell line radioresistance was related to a lack of apoptosis and a G2/M cell arrest12, we therefore investigated the cell cycle distribution of the SQ20B/SP/CD44+/ALDHhigh and SQ20B/SP/CD44–/ALDHlow cells after irradiation. As depicted in Figure 4B, the percentage of sub-G1 cells increased in a TEL-dependent manner in both subpopulations, starting from 5 days after irradiation. Thus, the percentage of SQ20B/SP/CD44–/ALDHlow cells in sub-G1 phase was about 40% at 5 days and peaked to 60% 10 days after carbon ion exposure. For the SQ20B/SP/CD44+/ALDHhigh cells, this percentage was lower, with a maximum of 30% of sub-G1 cells 10 days after irradiation. Regarding the G2/M phase (Fig 4B), only a small and transient arrest of the SQ20B/SP/CD44–/ALDHlow cells (30% of cells) was observed 24h after photon or carbon ion exposure and maintained until 48h. Higher percentage of SQ20B/SP/CD44+/ALDHhigh cells were found to be arrested in the G2/M phase up to 48h (40% of cells still arrested at 48h) following photon exposure. This percentage was higher (80% of cells at 24 h) and extended up to 120h (more than 60% of cells arrested) after carbon ion exposure demonstrating that CSCs are capable of prolonged G2/M phase arrest in response to carbon or photon irradiation. Radiosensitization of SQ20B cancer stem-like cells by inhibition of the G2/M arrest
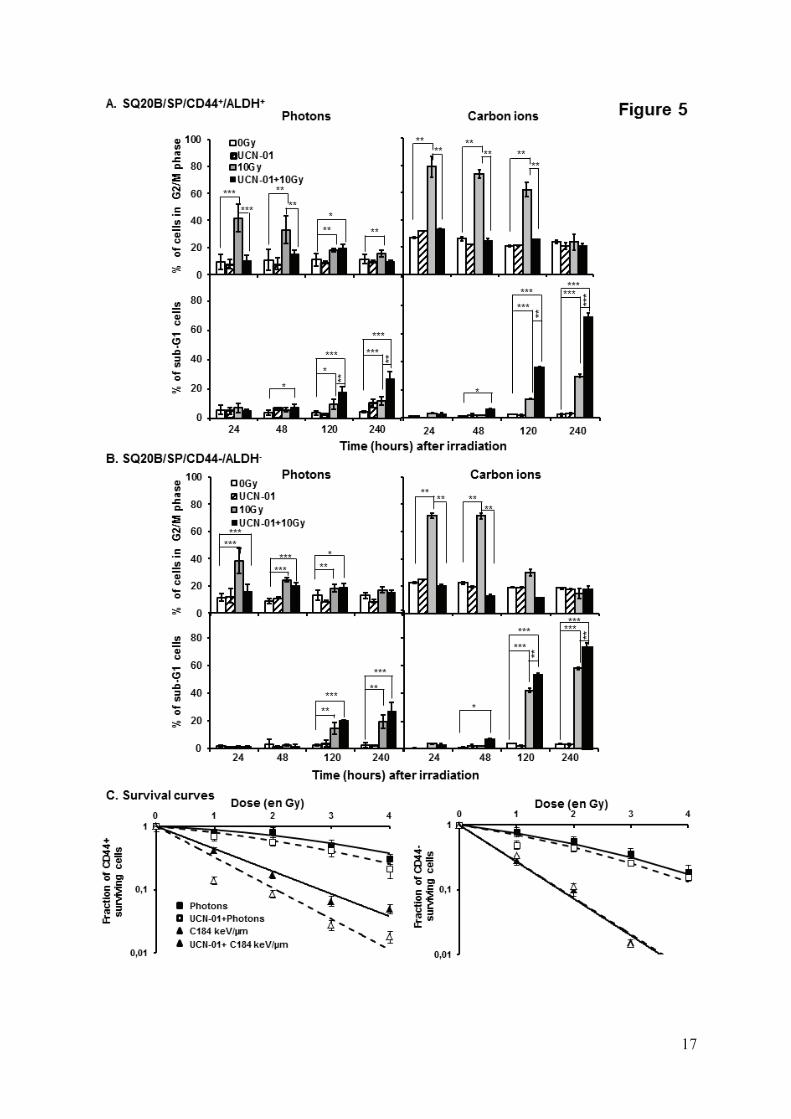
In light of these results, we used the Chk1 inhibitor UCN-0119 in order to investigate whether the inhibition of G2/M arrest could sensitize the SQ20B/SP/CD44+/ALDHhigh cells to high and low LET radiation. Exposure of SQ20B/SP/CD44+/ALDHhigh-treated cells to both irradiation modalities induced a significant inhibition of the G2/M phase arrest (Fig5A). As a consequence, the percentage of untreated SQ20B/SP/CD44+/ALDHhigh cells in sub-G1 was less than 30% at 240h and increased to more than 70% after treatment with UCN-01 in combination with carbon ion exposure (p<0.001). Moreover, the combination of UCN-01 with carbon ion irradiation resulted in a very significant enhancement of cell killing compared to photons (25% of the SQ20B/SP/CD44+/ALDHhigh treated cells in sub-G1 after photons compared with 70% after carbon ions). Moreover, all the effects obtained after treatment of cells with UCN-01 were found to be much more pronounced in the SQ20B/SP/CD44+/ALDHhigh subpopulation when compared to SQ20B/SP/CD44–/ALDHlow
cells (Fig5B). Under these experimental conditions, survival curves were also established (Fig5C) which allowed us to demonstrate a significant radiosensitization of SQ20B/SP/CD44+/ALDHhigh subpopulation. At the opposite, UCN-01 do not induced such a radiosensitization in the SQ20B/SP/CD44–/ALDHlow cells.
Isobolographic analyses of radiation and UCN-01 interactions are summarized in Table 2. The resulting effects for UCN-01 were additive or synergistic to radiotherapy, whatever the final level of cytotoxic efficiency (50 and 10% survival), the applied dose of radiation (ranging from 1 to 4Gy), and the type of radiation. Enhanced radiosentization of SQ20B cancer stem-like cells by induction of differentiation combined or not with inhibition of G2/M phase arrest.
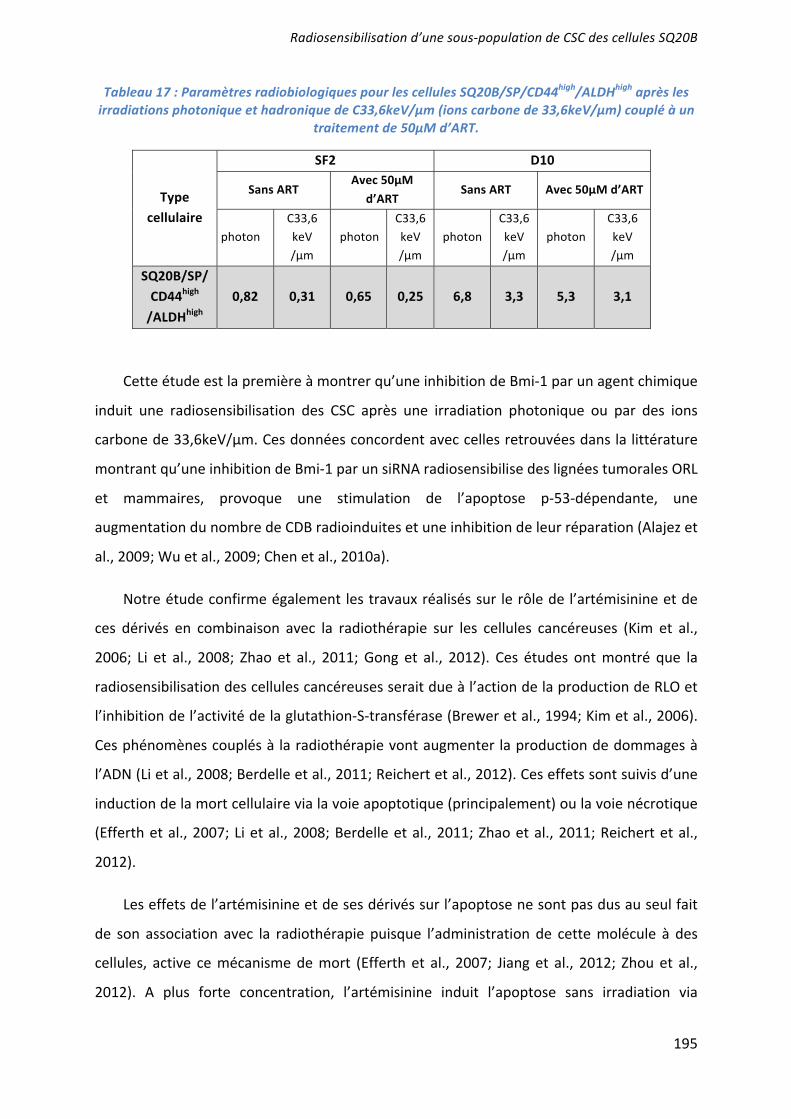
We then evaluated whether the radioresistance of SQ20B/SP/CD44+/ALDHhigh cells may also be partly related to their high endogenous ALDH activity. ATRA was used to inhibit this activity and to induce SQ20B-CSCs differentiation. Pretreatment of

7
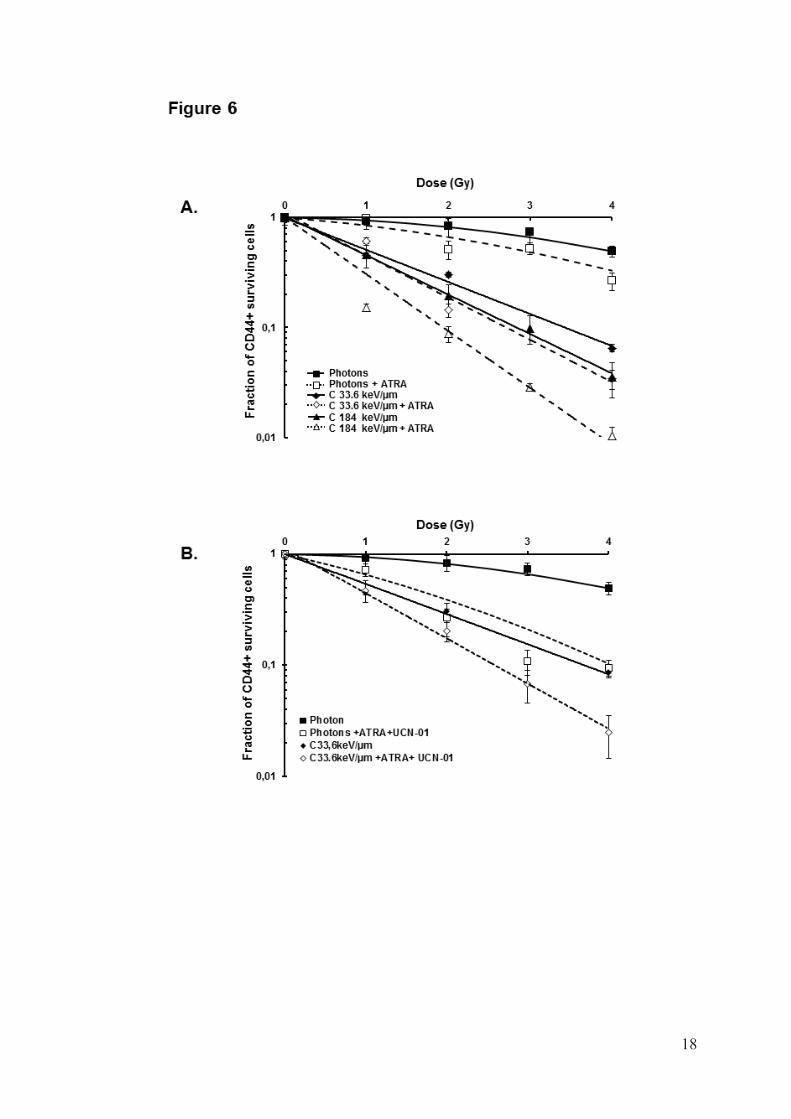
SQ20B/SP/CD44+/ALDHhigh cells with ATRA induced a decrease in ALDH activity, and consequently the number of CD44+ cells decreased to less than 20% compared with control cells (data not shown). Clonogenic survival curves were established after photon and carbon ion exposure (Fig6A). For all radiation qualities, SQ20B/SP/CD44+/ALDHhigh cells treated with ATRA were more sensitive than untreated cells. As an example, the SF2 of SQ20B/SP/CD44+/ALDHhigh cells after exposure to 184keV/ m carbon ions was 0.06 for ATRA-pretreated cells versus 0.19 for untreated cells. All isobolographic analyses of radiation and ATRA interactions are summarized in Table 2. The resulting effects for ATRA were additive or synergistic to radiotherapy, whatever the final level of cytotoxic efficiency, the applied dose of radiation, and the radiation modality.
After demonstrating that CSCs radioresistance could be significantly decreased by targeting the G2/M phase arrest with UCN-01 or the proliferation capacity with ATRA, the combination of both drugs on SQ20B/SP/CD44+/ALDHhigh cells was then tested. Treatment with both drugs resulted in a very significant radiosensitization of CSC after irradiation with either photons or carbon ions. The SF2 of SQ20B/SP/CD44+/ALDHhigh cells initially observed at 0.42 decreased to 0.19 after 33.6keV/μm carbon irradiation (Fig6B). The combination of UCN-01 and ATRA also resulted in additive or synergistic effects (table 2) independently of the final level of cytotoxic efficiency, the applied dose of radiation and the radiation modality. DISCUSSION
In HNSCC, stem-like tumor-initiating cells have been isolated from patient biopsies and have been shown to usually express a CD44+ phenotype and/or high ALDH activity 10,11. Moreover, it was demonstrated that cells expressing both CD44 and ALDH markers have higher tumorigenic, proliferative and invasive potential than those expressing only one marker11,20,21. The expression of these two markers is also associated with a poor prognosis and a high tumor grade22. In our study, while the analysis of CD44 by flow cytometry revealed that less than 1% of the parental SQ20B cell line expressed this marker, photon or carbon ion exposure drastically increase this percentage thus suggesting that high and low-LET irradiation kill CD44- cells, which results in an enrichment of CD44+ in the surviving cell population (Fig1). The mechanism of enrichment of CSCs in response to X-ray irradiation was previously reported in various tumor subtypes such as HNSCC10 or glioblastoma5. Cui et al.9 have shown that, at a biological isodose, colon cancer stem-like cells were more highly enriched after irradiation with X-rays than carbon ions in the surviving population. However, they also reported in the same work, in agreement with our study, that the proportion of cancer stem-like cells was highly enriched after carbon ion irradiation compared with photons when delivered at a physical isodose of 4 or 6Gy.
Although Bao et al.5 were the first to suggest that cancer stem cells contribute to glioma radioresistance, our work clearly demonstrates, using cell survival curve assays, that irradiation with carbon ions of 33.6 or 184keV/ m induces higher clonogenic cell death compared to X-rays in both SQ20B/SP/CD44+/ALDHhigh and SQ20B/SP/CD44-
/ALDHlowcells. The decrease in the SF2 in a TEL-dependent manner confirms previous studies that showed a greater effectiveness of high LET radiation in tumor cell killing12,23,24. In a more recent study that compared the effect of carbon ion and photons exposure of pancreatic cancer stem cell-like cells, Oonishi et al.25 reported that carbon ions killed more CSCs and prolonged the induction of DNA damage. Other authors have also evaluated the biological effects of carbon ion beam treatment on CD133+CD44+/ESA+ colon cancer stem-like cells compared to photon exposure. Similarly, they showed that carbon irradiation induced a lower CSCs survival fraction than X-ray irradiation, both in vitro and in vivo9.

8
The suggested common mechanisms involved in the resistance of CSCs to ionizing radiation include a propensity to quiescence, resistance to apoptosis, enhanced capacity for DNA repair, upregulation of cell cycle control mechanisms, free-radical scavenging and specific interactions with the stromal microenvironment5,7,10. In our work, we show that SQ20B/SP/CD44+/ALDHhigh radioresistant cells have the ability to self-renew in vitro. The increase in CSCs proliferation obtained after 10Gy photon exposure confirms the data from Koukourakis et al.26. It may be explained by the need to recreate an ideal environment for cells as suggested by Trott27 which showed that a phenomenon of cell repopulation occurred in head and neck cancer patients treated by conventional radiotherapy. Our present study also shows a higher tumorigenic potential of SQ20B/SP/CD44+/ALDHhigh cells in vivo compared with SQ20B/SP/CD44-/ALDHlow cells, a result which is in accordance with previous works performed on HNSCC11,20,21. Moreover, the time needed for tumor growth obtained after injection of SQ20B/SP/CD44+/ALDHhigh cells is in complete accordance with that previously reported in the literature25. Resistance to apoptosis was also demonstrated in the SQ20B/SP/CD44+/ALDHhigh subpopulation since high and low-LET irradiation induced late apoptosis preferentially in the non-cancer stem-like cells population (Fig4B). Our study is the first to show a resistance in radiation-induced apoptosis of CSCs after irradiation with carbon ions. In response to photon exposure, it was previously shown that hepatocarcinoma stem cells are more resistant to radiation-induced apoptosis compared with non-CSCs, CSCs expressing more Bcl-2 and enabling less cleavage of PARP compared to non-stem cells28. This phenomenon was also found in CSCs from head and neck tumors or glioblastoma5,22. In glioblastoma tumoral cell lines, it has been demonstrated that resistance to apoptotic cell death of CSC could be linked to a defect in cell cycle checkpoint control5. In our work, cell cycle analysis revealed that irradiation with photons or carbon ions induced an arrest of both populations in the G2/M phase but this was more extended for the SQ20B/SP/CD44+/ALDHhigh subpopulation (Fig4B). This could, among other factors, be explained by the increasing amount and complexity of DNA damage29,30. Indeed, Oonishi et al.25 showed that carbon ion irradiation of CSCs from pancreatic tumor cell lines increases the number and size of the H2AX foci. Our results also suggest that an upregulation in the cell cycle checkpoints may be partly involved in the radioresistance of SQ20B/SP/CD44+/ALDHhigh cells. Indeed, the family of Chk1/2 kinases is highly expressed in CSCs and could therefore activate an arrest of the cell cycle to enable DNA repair after ionizing stress5. Among the different stemness properties of cells, we have also shown that the resistance of SQ20B/SP/CD44+/ALDHhigh cells may result from an imbalance between exacerbated self-renewal and proliferative capacities and a decrease in apoptotic cell death triggering.
As there are still a relatively high number of recurrences after primary radiotherapy of HNSCC due to surviving CSCs, adjuvant treatments targeting either the inhibition of survival/self-renewal pathways or the triggering of apoptosis are essential to improve the efficiency of radio- or hadron-therapy. In a previous study12, we demonstrated that the G2/M phase arrest in the parental SQ20B cell line depends on Chk1 phosphorylation. Furthermore, other authors have reported that the use of Chk1/2 inhibitors such as UCN-01 are able to reverse radioresistance of colorectal or human lung cancer cell lines19,31. Here, we demonstrate that UCN-01, an inhibitor of Chk1 phosphorylation, induces a significant inhibition of the G2/M phase arrest after both irradiation modalities, increased the sub-G1 peak and significantly enhanced the effect of carbon ions not only in the parental SQ20B population but more specifically on SQ20B/SP/CD44+/ALDHhigh cells (Fig5). The data obtained after photon irradiation confirm those found in the literature showing that the combination of UCN-01 and radiotherapy can sensitize tumor cells5,19,31 leading to pioneering clinical trials32. While the exact cell fate following checkpoint abrogation remains to be

9
defined, it is likely that mitotic catastrophe is a key point in cell death mechanisms19. This is why a number of studies have investigated the enhancement of radiation-induced cytotoxicity in vivo and in vitro through Chk1 inhibition with UCN-01 but also other specific inhibitors such as SAR-02010633, Gö697634 or AZD624435. These studies and our own have demonstrated a radiosensitization in various types of tumor cell lines, which could be the first step towards an improvement in treatment.
Because high ALDH activity has been shown to enhance cancer cell resistance to both chemotherapy and radiotherapy36, we postulated that ATRA could radiosensitize the SQ20B/SP/CD44+/ALDHhigh cells by inducing their differentiation. Under our experimental conditions, ATRA effectively sensitized the SQ20B/SP/CD44+/ALDHhigh cell population to high and low LET radiation in a synergistic manner (Fig6A, Table2). This radiosensitization should probably involve an activation of apoptosis since ATRA was previously shown to induce both apoptosis and repression of Bcl-2 in myeloma cells37. As ALDH activity is known to play a well-characterized role in differentiation through its action in the retinoic acid pathway38, we therefore postulate that the differentiation induced by ATRA may allow the sensitization of this specific CSC subpopulation to both photon and carbon ion irradiation as reported by Lim et al.39 for the chemosensitization to cisplatin. The use of ATRA can therefore be considered as a very promising strategy since an early clinical study has demonstrated that the use of 13-cis-retinoic acid was potentially active in the treatment of patients with HNSCC40.
As the use of ATRA does not induce a complete differentiation, the addition of UCN-01 has been considered (Fig.6B). This combination induced a strong radiosensitization both after photon or carbon irradiation. To date the standard adjuvant chemotherapy associated with radiation is based especially on DNA intercalating agents that are not very specific for cancer cells41. Here we have designed a pharmacological treatment that targets specifically most of the cancer stem cells, with the use of ATRA and the remaining radioresistant cells, using UCN-01. This combination of both drugs could then represent a promising adjuvant treatment to overcome radioresistance in HNSCC cancer stem cells.
In conclusion, although the exact underlying molecular mechanisms of SQ20B/SP/CD44+/ALDHhigh radioresistance could not be totally elucidated, our study demonstrates that the undifferentiated state of cancer stem-like cells associated with an upregulated control of the cell cycle checkpoints might be intimately involved in the radioresistant phenotype of this subpopulation of HNSCC. Moreover, we provide evidence that UCN-01 and ATRA could potentially be used in combination as a powerful radiosensitizing strategy. To date, other drugs associations such as cisplatin and 5-Fluorouracil are commonly used to optimize radiotherapy but their toxicity remained very high and recurrence still takes place. Given these poor results, the development of novel adjuvant therapies specifically targeting cancer stem cells remains therefore a constant challenge in the management of HNSCC in order to improve the local control of radioresistant tumors even after hadrontherapy. ACKNOWLEDGMENTS We thank all those who participated in the experiments at GANIL and GSI. We acknowledge the contribution of the flow cytometry platform of UFR BioSciences Gerland-Lyon-Sud (UMS3444/US8). This work was achieved within the scientific framework of ETOILE and Labex-PRIMES. It was supported by the Contrat-Plan-Etat-Region and the Ligue contre le Cancer (Ain).

10
REFERENCES 1. Jingu K, Tsujii H, Mizoe J-E, et al. Carbon ion radiation therapy improves the prognosis
of unresectable adult bone and soft-tissue sarcoma of the head and neck. Int J Radiat Oncol Biol Phys 2012;82(5):2125–31.
2. Mizoe J-E, Tsujii H, Kamada T, et al. Dose escalation study of carbon ion radiotherapy for locally advanced head-and-neck cancer. Int J Radiat Oncol Biol Phys 2004;60(2):358–64.
3. Mizoe J-E, Hasegawa A, Jingu K, et al. Results of carbon ion radiotherapy for head and neck cancer. Radiother Oncol 2012;103(1):32–7.
4. Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer 2008;8(7):545–54.
5. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444(7120):756–60.
6. Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 2006;5:67.
7. Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009;458(7239):780–3.
8. Moncharmont C, Levy A, Gilormini M, et al. Targeting a cornerstone of radiation resistance: Cancer stem cell. Cancer Lett 2012;322(2):139–47.
9. Cui X, Oonishi K, Tsujii H, et al. Effects of Carbon Ion Beam on Putative Colon Cancer Stem Cells and Its Comparison with X-rays. Cancer Res 2011;71(10):3676–87.
10. Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA 2007;104(3):973–8.
11. Chen Y-C, Chen Y-W, Hsu H-S, et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem Biophys Res Commun 2009;385(3):307–13.
12. Maalouf M, Alphonse G, Colliaux A, et al. Different mechanisms of cell death in radiosensitive and radioresistant p53 mutated head and neck squamous cell carcinoma cell lines exposed to carbon ions and x-rays. Int J Radiat Oncol Biol Phys 2009;74(1):200–9.
13. Facompre N, Nakagawa H, Herlyn M, Basu D. Stem-like cells and therapy resistance in squamous cell carcinomas. Adv Pharmacol 2012;65:235–65.
14. Mi J. Current treatment strategy of acute promyelocytic leukemia. Front Med 2011;5(4):341–7.
15. Boivin A, Hanot M, Malesys C, et al. Transient alteration of cellular redox buffering before irradiation triggers apoptosis in head and neck carcinoma stem and non-stem cells. PLoS ONE 2011;6(1):e14558.
16. Beuve M, Alphonse G, Maalouf M, et al. Radiobiologic parameters and local effect model predictions for head-and-neck squamous cell carcinomas exposed to high linear energy transfer ions. Int J Radiat Oncol Biol Phys 2008;71(2):635–42.
17. Ferrandon S, Saultier P, Carras J, et al. Telomere profiling: toward glioblastoma personalized medicine. Mol Neurobiol 2013;47(1):64–76.
18. Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy: the concept of additivity. Int J Radiat Oncol Biol Phys 1979;5(1):85–91.
19. Playle LC, Hicks DJ, Qualtrough D, Paraskeva C. Abrogation of the radiation-induced G2 checkpoint by the staurosporine derivative UCN-01 is associated with radiosensitisation in a subset of colorectal tumour cell lines. Br J Cancer 2002;87(3):352–8.

11
20. Clay MR, Tabor M, Owen JH, et al. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck 2010;32(9):1195–201.
21. Krishnamurthy S, Dong Z, Vodopyanov D, et al. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res 2010;70(23):9969–78.
22. Chen Y-C, Chang C-J, Hsu H-S, et al. Inhibition of tumorigenicity and enhancement of radiochemosensitivity in head and neck squamous cell cancer-derived ALDH1-positive cells by knockdown of Bmi-1. Oral Oncol 2010;46(3):158–65.
23. Iwadate Y, Mizoe J, Osaka Y, Yamaura A, Tsujii H. High linear energy transfer carbon radiation effectively kills cultured glioma cells with either mutant or wild-type p53. Int J Radiat Oncol Biol Phys 2001;50(3):803–8.
24. Combs SE, Zipp L, Rieken S, et al. In vitro evaluation of photon and carbon ion radiotherapy in combination with chemotherapy in glioblastoma cells. Radiat Oncol 2012;7:9.
25. Oonishi K, Cui X, Hirakawa H, et al. Different effects of carbon ion beams and X-rays on clonogenic survival and DNA repair in human pancreatic cancer stem-like cells. Radiother Oncol 2012;
26. Koukourakis MI, Giatromanolaki A, Tsakmaki V, Danielidis V, Sivridis E. Cancer stem cell phenotype relates to radio-chemotherapy outcome in locally advanced squamous cell head-neck cancer. Br J Cancer 2012;106(5):846–53.
27. Trott KR. Cell repopulation and overall treatment time. Int J Radiat Oncol Biol Phys 1990;19(4):1071–5.
28. Piao LS, Hur W, Kim T-K, et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett 2012;315(2):129–37.
29. Ljungman M. The DNA damage response--repair or despair? Environ Mol Mutagen 2010;51(8-9):879–89.
30. Hanot M, Boivin A, Malésys C, et al. Glutathione depletion and carbon ion radiation potentiate clustered DNA lesions, cell death and prevent chromosomal changes in cancer cells progeny. PLoS ONE 2012;7(11):e44367.
31. Kim Y-M, Jeong I-H, Pyo H. Celecoxib enhances the radiosensitizing effect of 7-hydroxystaurosporine (UCN-01) in human lung cancer cell lines. Int J Radiat Oncol Biol Phys 2012;83(3):e399–407.
32. Jimeno A, Rudek MA, Purcell T, et al. Phase I and pharmacokinetic study of UCN-01 in combination with irinotecan in patients with solid tumors. Cancer Chemother Pharmacol 2008;61(3):423–33.
33. Chung EJ, Brown AP, Asano H, et al. In vitro and in vivo radiosensitization with AZD6244 (ARRY-142886), an inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 kinase. Clin Cancer Res 2009;15(9):3050–7.
34. Feng Z, Xu S, Liu M, Zeng Y-X, Kang T. Chk1 inhibitor Gö6976 enhances the sensitivity of nasopharyngeal carcinoma cells to radiotherapy and chemotherapy in vitro and in vivo. Cancer Lett 2010;297(2):190–7.
35. Borst GR, McLaughlin M, Kyula JN, et al. Targeted Radiosensitization by the Chk1 Inhibitor SAR-020106. Int J Radiat Oncol Biol Phys 2012;
36. Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDH(hi)CD44 (+) human breast cancer cells. Breast Cancer Research and Treatment [Internet] 2011 [cited 2011 Oct 21];Available from: http://www.ncbi.nlm.nih.gov/pubmed/21818590

12
37. Otsuki T, Sakaguchi H, Hatayama T, Wu P, Takata A, Hyodoh F. Effects of all-trans retinoic acid (ATRA) on human myeloma cells. Leuk Lymphoma 2003;44(10):1651–6.
38. Ginestier C, Wicinski J, Cervera N, et al. Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle 2009;8(20):3297–302.
39. Lim YC, Kang HJ, Kim YS, Choi EC. All-trans-retinoic acid inhibits growth of head and neck cancer stem cells by suppression of Wnt/β-catenin pathway. European journal of cancer (Oxford, England: 1990) [Internet] 2012 [cited 2012 Jun 14];Available from: http://www.ncbi.nlm.nih.gov/pubmed/22640830
40. Recchia F, Lalli A, Lombardo M, et al. Ifosfamide, cisplatin, and 13-Cis retinoic acid for patients with advanced or recurrent squamous cell carcinoma of the head and neck: a phase I-II study. Cancer 2001;92(4):814–21.
41. Price KAR, Cohen EE. Current treatment options for metastatic head and neck cancer. Curr Treat Options Oncol 2012;13(1):35–46.
LEGEND TO THE FIGURES
Figure 1: Kinetics of CD44 induction in the SQ20B cell line after a 10Gy photon irradiation (left panel) or a 10Gy carbon ion 184 keV/μm irradiation (right panel). Values represent the mean±SD of two independent experiments performed in triplicate. *p<0.05, **p<0.01, and ***p<0.001 compared to controls. Figure 2: A: Sorting of SQ20B/SP cells: p1=SQ20B/SP/CD44+/ALDHlow cells, p2=SQ20B/SP/CD44–/ALDHlow cells, p3=SQ20B/SP/CD44+/ALDHhigh cells, p4=SQ20B/SP/CD44–/ALDHhigh cells. B: Tumor sphere formation of SB20B/SP/CD44+/ALDHhigh cells. C: Relative gene expression of Bmi-1 in SB20B/SP/CD44+/ALDHhigh and SB20B/SP/CD44–/ALDHlow cells. D: Tumor formation ability of sorted SB20B/SP/CD44+/ALDHhigh cells: number of tumors formed/number of injections. Figure 3: Dose–response curves for cell killing of SB20B/SP/CD44+/ALDHhigh and SB20B/SP/CD44-/ALDHlow cells following X-ray or carbon ion exposures. Values represent the mean±SD of three independent experiments. Figure 4: A: Kinetics of BrdU incorporation in SB20B/SP/CD44+/ALDHhigh cell line after irradiation with 10Gy photons or with 33.6 or 184keV/ m carbon ions. Values represent the mean±SD of two independent experiments. B: Kinetic study of the percentage of cells in sub-G1 and G2/M phase in SB20B/SP/CD44+/ALDHhighand SB20B/SP/CD44-/ALDHlow cells irradiated with 10Gy photons or with 184keV/ m carbon ions. Values represent the mean±SD of three independent experiments realized in triplicate. *p<0.05, **p<0.01, and ***p<0.001 compared to controls. Figure 5: A: Kinetics of G2/M phase and apoptosis, quantified as the percentage of cells in sub-G1, in SB20B/SP/CD44+/ALDHhigh cells treated or not with UCN-01 25nM and irradiated with 10Gy photons or with 184keV/ m carbon ions. Values represent the mean±SD of three independent experiments realized in triplicate. *p<0.05, **p<0.01, and ***p<0.001 compared to control cells or 10 Gy irradiated. B: Kinetics of G2/M phase and apoptosis, quantified as the percentage of cells in sub-G1 phase, in SB20B/SP/CD44-/ALDHlow cells treated or not with UCN-01 and irradiated with 10Gy photons or with 184keV/ m carbon ions. Values represent the mean±SD of three independent experiments realized in triplicate. *p<0.05, **p<0.01, and ***p<0.001 compared to control cells or 10 Gy irradiated. C: Dose–response curves for cell killing of SB20B/SP/CD44+/ALDHhigh cells treated or not with UCN-

13
01 25nM following photon or carbon ion irradiation. Values represent the mean±SD of three independent experiments. Isobolographic statistical analysis was realized and show in table 2 Figure 6 : Dose–response curves for cell killing of SB20B/SP/CD44+/ALDHhigh treated or not with 10μM ATRA (A) or with both 25nM UCN-01and 10μM ATRA (B)following photon or carbon ion exposure. Values represent the mean±SD of two independent experiments. Isobolographic statistical analysis is presented in table 2. Table 1: Radiobiological parameters for SQ20B/SP/CD44+/ALDH+ and SQ20B/SP/CD44-
/ALDH- cells.
SF2 D10 RBE
Photon C33.6
keV/μm C184
keV/μm Photon C33.6
keV/μm C184
keV/μm Photon C33.6
keV/μm C184
keV/μm SQ20B/SP/CD44+/
ALDH+ 0.82 0.28 0.19 5.1 2.6 2.1 - 1.9 2.4 SQ20B/SP/CD44-
/ALDH- 0.56 0.17 0.09 6.8 3.3 2.6 - 2.1 2.6

14
Table 2. Results of isobolographic analyses of radiation and UCN-01 (7-hydroxystaurosporine, a specific inhibitor of Chk1 phosphorylation) or ATRA (all-trans retinoic acid) or the combined UCN-01/ATRA in SQ20B-SP-CD44+ALDH+ stem cells obtained from SQ20B human parental head and neck squamous cell line Type of radiation Radiation dose Isobolographic analyses Gy 10% surv 50% surv UCN-01 Photon 1 + + 2 + + 3 + Syn 4 Syn Syn Carbon ions 1 + Syn 184 keV/μm 2 Syn Syn 3 Syn Syn 4 Syn Syn ATRA Photon 1 Ant Ant 2 + + 3 + + 4 Syn Syn Carbon ions 1 + Syn 33.6 keV/μm 2 Syn Syn 3 Syn Syn 4 Syn Syn Carbon ions 1 Ant Syn 184 keV/μm 2 Syn Syn 3 Syn Syn 4 Syn Syn UCN-01/ATRA Photon 1 + + 2 Syn Syn 3 Syn Syn 4 Syn Syn Carbon ions 1 + + 33.6 keV/μm 2 Syn Syn 3 Syn Syn 4 Syn Syn surv = survival; Syn = synergistic effect; Ant = antagonistic effect; + = additive effect
15

16
17
18

Mini-review
Targeting a cornerstone of radiation resistance: Cancer stem cell
Coralie Moncharmont a, Antonin Levy b, Marion Gilormini a, Gérald Bertrand a, Cyrus Chargari c,Gersende Alphonse a, Dominique Ardail a, Claire Rodriguez-Lafrasse a, Nicolas Magné a,⇑a Laboratoire de Radiobiologie Cellulaire et Moléculaire, Faculté de Médecine Lyon-Sud, Université de Lyon 1, Oullins cedex, FrancebDepartment of Radiation Oncology, Institut Gustave Roussy, Université Paris XI, Villejuif, FrancecDepartment of Radiation Oncology, Hôpital Val de Grâce, Paris, France
a r t i c l e i n f o
Article history:Received 9 February 2012Received in revised form 18 March 2012Accepted 21 March 2012
Keywords:Cancer stem cellsRadioresistanceHypoxiaRadiation therapy
a b s t r a c t
In radiation oncology, cancer stem cells (CSCs) have become an important research field. In fact, itappears that most cancer types contain populations of cells that exhibit stem-cell properties. CSCs havethe ability to renew indefinitely, which can drive tumor development and metastatic invasion. As thosecells are classically resistant to conventional chemotherapy and to radiation therapy, they may contributeto treatment failure and relapse. Over past decades, preclinical research has highlighted that variations inthe CSCs content within tumor could affect their radiocurability by interfering with mechanisms of DNArepair, redistribution in the cell cycle, tumor cells repopulation, and hypoxia. It is now possible to isolateparticular cells expressing specific surface markers and thus better investigating CSCs pathways. Numer-ous inhibitory agents targeting these specific signaling pathways, such as Notch and Wnt/B-catenin, arecurrently evaluated in early clinical trials. By targeting CSCs, tumor radioresistance could be potentiallyovercome to improve outcome for patients with solid malignancies. Radiation therapy using ion particles(proton and carbon) may be also more effective than classic photon on CSCs. This review presents themajor pathophysiological mechanisms involved in CSCs radioresistance and recent developments for tar-geted strategies.
� 2012 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
For over a half a century, cancer stem cells (CSCs) have been animportant area of radiation research. It appears that most cancerscontain populations of cells that exhibit stem-cell properties.Those are characterized by their ability to self-renew and may pro-duce a much larger set of cells with very limited proliferative po-tential that are named the bulk. The definition of a CSC requiresthree characteristics: (i) a selective capacity to initiate tumor anddrive neoplastic proliferation, (ii) an ability to generate endlesscopies of themselves through self-renewal, and (iii) the potentialto give rise to more differentiated non-stem cell cancer progeny[1].
Since CSCs have been described for first time in leukaemia bythe Dick laboratory in 1994, other groups have demonstrated thepresence of CSCs in numerous solid tumors (glioblastoma, colorec-tal cancer, prostate, head and neck, liver, melanoma, etc.), suggest-ing that the majority of malignancies are dependent on such a cellcompartment. Anyhow, properties of CSCs in different tumors were
not similar, and the proportion of stem cells in tumors was differ-ent [2]. Moreover, the origin of CSCs remains unclear and manyworks tried to identify this. Barker et al. [3], Zhu et al. [4], andAlcantara Llaguno et al. [5] demonstrated that CSCs were derivedfrom normal stem cells if oncogenes were activated. But, there isevidence that more differentiated cancer cell populations may ac-quire CSC properties through epithelial mesenchymal transition(EMT) [6,7]. Chaffer et al. [8] recently observed in a subpopulationof basal-like human mammary epithelial cells that spontaneouslydedifferentiated into stem-like cells. Then, some non-stem cancercells gave rise to CSCs. Classically, the definition of adult stem cellimplicates that those are capable to generate full cellular compo-nents of the relevant organ from a single stem cell. This could berevealed by in vivo functional assays, which consists in implantinga progressively smaller number of tumor cells in immunodeficientanimals. The number of cells could be an appropriate surrogatewhen analyzing the frequency of proper cancer stem cells. Now,through the development of immunofluorescence tools, it is possi-ble to more easily isolate CSCs using their surface proteins (e.g.CD133, CD44, CD29).
Cancer stem cells account for a minor fraction of a tumor popu-lation, but those might be particularly involved in tumor initiation,proliferation, or in metastatic process. CSCs are believed to sharemany properties with normal stem cells that render them relatively
0304-3835/$ - see front matter � 2012 Elsevier Ireland Ltd. All rights reserved.http://dx.doi.org/10.1016/j.canlet.2012.03.024
⇑ Corresponding author. Address: Laboratoire de Radiobiologie Cellulaire etMoléculaire, UFR Médicale Lyon-Sud, BP 12, 69921 Oullins cedex, France. Tel.: +33(0) 4 26 23 59 58; fax: +33 (0) 4 26 23 59 66.
E-mail address: [email protected] (N. Magné).
Cancer Letters 322 (2012) 139–147
Contents lists available at SciVerse ScienceDirect
Cancer Letters
journal homepage: www.elsevier .com/locate /canlet

insensitive to conventional cytotoxic agents, such as chemotherapyand to radiation therapy. This is an advocated factor of treatmentfailure and relapse. The mechanisms through which solid tumorsbecome resistant to conventional therapy are only partially under-stood. Those involve numerous factors implicated in the resistanceof CSC to ionizing radiation: quiescence propensity, enhanced DNArepair, upregulated cell cycle control mechanisms, mechanisms offree-radical scavenging, and specific interaction with stromalmicroenvironnement [9,10]. From a radiobiological point of view,four factors are classically involved in the efficiency of ionizingradiation: repair of DNA damage systems, redistribution of the cellcycle, tumor cells repopulation, and level of intratumor hypoxia.Although the identity of cancer cell clonogens has not beendefinitively resolved, the understanding of CSCs pathways contin-ues to grow and new potential molecular therapeutic targets haverecently emerged. Numerous inhibitory agents targeting thesespecific signaling pathways, such as Notch and Wnt/B-catenin, arecurrently evaluated in early clinical trials (http://clinicaltrials.gov:NCT01122901, NCT01351103). Overcoming radioresistance by con-currently targeting cancer stem cells pathways may potentially im-prove outcome for patients with localized solid tumors. Radiationtherapy using heavy particles (proton and carbon)may be alsomoreeffective than classic photon on CSCs [11,12]. The aim of this paperis to report the various intracellular pathways involved in CSCsradioresistance for which potential intracellular target and high-light strategies for enhancing antitumor effectiveness.
2. Mechanism of radiation resistance
Although debated, CSCs’ radioresistance could give explanationfor their capacity to cause tumor recurrence. Few clinical data havecorroborated this hypothesis. In 2006, Bao et al. [13] determined asubpopulation of glioblastoma, which contributes to radioresis-tance. This fraction of tumor cells expressed CD133 and had a low-er sensitivity to radiation-induced apoptosis. Authors also reportedthat their frequency was 2- to 4- fold higher after radiation in bothprimary tumors and xenografts. The relative enrichment of CD133positive cells was accompanied by a maintained ability to self-renewal and tumor progression. The same year, Phillips et al.[14] isolated CSCs from two cell-lines (MCF-7 and MDA-MB-231)by selecting cells which were CD24�/low/CD44+. The resultsin vitro (clonogenic assays evaluating mean surviving fraction at2 Gy) confirmed that CSCs were more radioresistant than their par-ent monolayer cultures MCF-7 (p = 0.026), but not with MDA-MB-231 (p = 0.09). In the same time, fractionated doses of irradiationincreased the percentage of the CSCs in MCF-7 monolayer cultures.Woodward et al. [15] also worked on MCF-7 cell lines. Authorshypothesized that mammary gland progenitors may be resistantto radiation, involving the b-catenin stem cell survival signalingpathway. In two selected populations, side population progenitors(Sca1+) and the more primitive stem cells (lin�CD24+CD29+), irra-diation induced less double-strand DNA breaks than in non-progenitor cells. Later, Parikh et al. [16] exposed two NSCLC cell
lines (H460 and A549) to a single fraction dose of 5 Gy and culti-vated them under stem cell selective conditions. The cells thenformed spheres, which expressed vascular endothelial growth fac-tor (VEGF)-receptor 1, CD117, CD133, CXCR1, CXCR2, and CXCR4.As the radioresistant cells had the same profile as stem cells, theauthors concluded that those were CSCs.
Marker systems used to identify CSC in various solid cancers ispresented Table 1. It is important to notify that (i) this list is notcomplete and is till increasing (2) reliability of such surface markermay not be sufficient to discriminate cancer stem and non-stemcell subpopulations [17].
2.1. Quiescence propensity: targeting repopulation and redistribution
In a high quality review, Pajonk et al. [9] have precisely exam-ined how CSCs might become radioresistant through repopulationand redistribution mechanisms. Briefly, accelerated repopulationof CSCs is also believed to occur in human tumors during radio-therapy. As in normal stem cells, CSCs could be able to employdevelopmental signaling pathways like the Notch, Wnt, and Sonichedgehog which are described below. The choice of which partnerbeta-catenin associates with could contribute to determinewhether cells divide symmetrically and thus maintain their levelof potency or make asymmetric divisions. While CBP/catenin-mediated transcription is associated with a non-differentiativeprocess, p300/catenin-driven transcription generates a differentia-tion process [18].
Redistribution in cell cycle refers to the fact that cells change intheir radiosensitivity as they traverse the division cycle. In fact, mi-totic cells are particularly sensitive to ionizing radiation, whilethose in late S-phase would be most resistant [19]. It is believedthat both normal tissue stem cells and CSCs are usually in the qui-escent phase of the cell cycle [20]. Wnt/b-catenin pathway mayalso be implicated in CSCs cell cycle regulation. Kendziorra et al. re-ported an overexpression of the Wnt transcription factor T cell fac-tor (TCF4) in rectal cancers cell lines that were resistant tochemoradiotherapy. Silencing of TCF4 induced a G2/M phase arrestand caused a significant sensitization of cells to clinically relevantdoses of X-rays. This effect was observed only in tumor cells withhigh TCF4 reporter activity, in a b-catenin-independent manner[21].
Mechanisms of CSC radioresistance are illustrated Fig. 1.
2.2. Toward mechanistic explanations: enhanced repair of DNA
The biological efficacy of ionizing radiation depends on its abil-ity to produce unrepairable DNA lesions, mainly double-strandbreaks (DSBs) within tumor cell. Beyond these lethal damages,there are also sublethal DNA damages which accumulate and con-tribute to tumor lethality. Repair of sublethal damage betweenradiation fractions is exploited in radiation therapy because criticalnormal tissues and tumors often differ in their ability to repairsuch damages [9]. Bao et al. have highlighted that CSCs had a great-er activation of DNA damage checkpoint in response to radiation,and repaired radiation-induced DNA damage [13], leading to radi-ation resistance. The family of checkpoint kinases 1/2 (Chek1/2 ki-nases) is activated after ionizing stress and arrest the cell cycle toallow DNA repair. These kinases are expressed at higher basal andinducible levels in CSCs than in non-stem cells. Furthermore, theradioresistance can be partially reversed through inhibition ofthe Chek1 and Chek2 checkpoint kinases. Other authors have ob-served that radiation induced similar levels of DNA damages inCD133+ and CD133� cells, but CD133+ cells repaired more effi-ciently and underwent less in apoptosis. CD133+ cells showed ba-sal activity of one of protein of DNA damage checkpoint, rad17,suggesting that CSCs are primed to act in response to genotoxic
Table 1Marker systems used to identify CSC in various solid cancers.
CSC marker Location Refs.
CD133+CD44+/ESA+ Colon [12]CD133+ Brain [13]CD24�/low/CD44+ Breast [14]CD117 NSCLC [16]CD44+ALDH1+ HNSCC [76]CD133+ HNSCC [94]CD133+ Liver [93]
Abbreviations; NSCLC: non-small cell lung cancer; HNSCC: head and neck squa-mous cell carcinoma.
140 C. Moncharmont et al. / Cancer Letters 322 (2012) 139–147

stresses. In response to DSB, several kinases were activated such asataxia telangiectasia mutated (ATM) and Chek1/2. Furthermore,the use of Chek1/2 inhibitors reversed CSCs resistance [22]. Simi-larly, Yin et al. showed that ATM inhibitor decreased the radiationresistance of CD44(+)/CD24(-or low) subset in both from breastcancer cell lines and primary human breast cancer cells. Authorsfound that CD44(+)/CD24(-or low) cells had an increased activationof ATM signaling compared to non- CD44(+)/CD24. The increasedradiation resistance was not dependent on the result of alterednon-homologous end joining (NHEJ). Indeed, NHEJ activity and
expression of the various proteins involved in this repair DNA re-pair system were not significantly different between theCD44(+)/CD24(-or low) and non- CD44(+)/CD24(-or low) subsets[23].
More recently, several teams worked on BMI-1, an E3-ubiquitinligase which is upregulated in some normal stem cells and in CSCs[24–26]. BMI-1 was enriched by chromatin after exposure to irra-diation, revealing an important role in early DSB damage response.Its co-factors are multiple, including ATM kinase, histonegammaH2AX, DNA-dependent protein kinase catalytic subunit
Fig. 1. Mechanisms of CSC radioresistance. (A) Redistribution. Cells change in their radiosensitivity as they traverse the division cycle. Mitotic cells are particularly sensitiveto ionizing radiation, while those in late S-phase would be most resistant. CSCs are usually in the quiescent phase of the cell cycle. (B) Enhanced repair of DNA. Non-homologous end joining (NHEJ) is the main mechanism of reparation implicated after DNA double-strand breaks (DSBs) induced by ionizing radiation are principally repairedby NHEJ. NHEJ involves recognition and reparation by several molecules such as ATM, DNA-PKcs, XRCC4 and Ligase 4. It is suggested that CSCs may have an increasedactivation of ATM signaling compared to non-tumoral stem cells. (C) Upregulated cell cycle control mechanisms. The family of checkpoint kinases 1/2 (Chek1/2 kinases) isactivated after ionizing stress and arrest the cell cycle to allow DNA repair. In response to DSB, ATM and ATR may be activated by Chek1/2. These kinases are expressed athigher basal and inducible levels in CSCs than in non-stem cells (D) Reactive Oxygen Species (ROSs) and free radical scavenging. CSCs reside in a perivascular niche and areexposed to oxygen and high ROS levels. A higher activity of ROS-scavenging enzymes, such as superoxide dismutase (SOD) and glutathione (GSH) in CSCs could lower levels ofROS (and then DNA damage) after irradiation. Prolonged exposure to ROS might also lead to a differentiation process and/or growth arrest. Redox-sensitive transcriptionfactors, such as nuclear factor j-B (NF-j-B) and Nrf2 are activated and are lead to an increase of ROS-scavenging enzymes and cancer promotion. (E) Interaction with stromalenvironment. Irradiation cause changes in gene expression in the resident fibroblasts and the release of transforming growth factor–b (TGF-b) that may be implicated CSCsepithelial mesenchymal transition (EMT). CD44 is a extracellular matrix (ECM) receptor abundantly expressed at the CSCs surface and is associated with malignancy.
C. Moncharmont et al. / Cancer Letters 322 (2012) 139–147 141

(DNA-PKcs), PARP-1, and histone H1. Moreover, BMI-1 was associ-ated with accumulation of BRCA1 near DSB damages. In order toexplore the role of this protein, Facchino et al. induced BMI-1 defi-ciency cells and observed an increased DSB, resulting in increasedradiosensitivity. At the opposite, BMI-1 overexpression in normalCSCs enhanced radioresistance. Recently, polycomb group pro-teins, including BMI-1 have been implied in the radioresistancein both normal stem cells and CSCs. Improving our understandingof the contribution of polycomb group proteins to the DNA damageresponses pathways may lead to the identification of potential no-vel target to sensitize CSCs to radiation [26].
2.3. Upregulated cell cycle control mechanisms
The maintenance of DNA represents a primary and uninter-rupted challenge to all cells. Main cell response mechanisms toADN lesion are active arrest of cell cycle or induction of massiveapoptosis or senescence [13]. Chromosomal instability is a factorinvolved in the inactivation of pro-apoptotic pathways. Thus, geno-mic instability and apoptosis are linked phenomena, with impor-tant implications when investigating the pathophysiology ofcancer. Furthermore, irradiation may induce an important celldeath period that may ultimately lead to generate chromosomalinstability. Radiation may decrease the normal stem-cell poolthrough apoptosis and senescence. Concomitantly, it could poten-tially increase the growth advantage of resistant CSCs. Conse-quently, CSCs could be involved in the transmission of genomicinstability [27,28]. Bensimon et al. demonstrated that, radiationlead to the emergence of chromosomal unstable cells duringmore than 35 population doublings. Then, they showed thatCD24(-/low), a CSC marker, correlated with the transmission ofgenomic instability. In fact, delayed chromosomal instability wasonly expressed by CD24(+) cells, but was transmitted by stablesurviving CD24(-/low) cells [29].
As ionizing radiation, chemotherapies or targeted therapiesmay also affect other way of cell death, as autophagy. Autophagysubstantially differs from apoptosis because autophagosomes andautolysosomes are involved, and the nucleus remains intact inthe cell. Moreover, it is independent of phagocytes. Although apop-tosis is usually considered as being the most common radiation-induced cell death, autophagy was involved in anti-neoplasic ef-fects of radiation therapy [30,31]. Interestingly, there are biologicallinkages between autophagy and apoptosis. In response to theblockade of one pathway, the alternative type of cell death willbe activated. In addition, recent studies suggested that autophagycould also precede and have some regulatory influence on senes-cence [32]. Zhuang et al. [33] tried to understand molecular mod-ifications in autophagy, and showed that DNA-PKcs played a majorrole in the repair of dsDNA breaks and in cell radiosensitization.Plasmids encoding shRNA which targeted DNA-PKcs were con-structed. These plasmids were transfected into glioma stem cells.Ionizing radiation induced massive autophagy in transfected cells,but rarely apoptosis.
2.4. Hypoxia, reactive oxygen species and free radical scavenging
Given its crucial role in biological processes, oxygen concentra-tion is accurately regulated in cells. While hyperoxia can inducethe formation of Reactive Oxygen Species (ROSs) (which may resultin cell death), hypoxia has a major role in the transcriptional acti-vation of pro-apoptotic and pro-angiogenic molecular pathways.Lower tumoral oxygen level results from several mechanisms,including anarchic vasculature, low oxygen diffusion, and irregularblood flow. Hypoxia is implicated in radioresistance and cells lo-cated in areas of low oxygen tensions are classically relatively pro-tected from radiation effects [34]. Oxygen plays a crucial role in
radiotherapy induce cell-death through the ROS. It has been re-ported that CSCs resided in a perivascular niche [35], so that thosewere exposed to oxygen and high ROS levels. However, it has beenshown that central nervous system stem cells contain lower levelsof ROS as well as epithelial tissue stem cells (CD24medCD49fhighLin�
cells) and mammary gland CSCs (CD44+CD24-/lowLin� cells). [36–39]. Interestingly, CSCs are less likely to develop DNA damage afterirradiation, compared to non-tumorigenic cells, while this differ-ence was not shown in untreated cells. Consistent with this find-ing, it could be hypothesized that a higher activity of ROSscavenging pathways in CSCs could lower levels of DNA damageafter irradiation, and thus antitumor effect. ROS levels have alsobeen explored by Phillips et al. in breast CSCs from two cell lines:MCF-7 and MDA-MB-231 [14]. Without any radiation, MCF-7cellmammospheres (culture of cells in non-adherent non-differentiat-ing conditions which contains CSCs) had a lower level of ROS thanMCF-7 monolayer cell cultures. After exposure to 10 Gy, the levelof ROS increased in the MCF-7 monolayer cell cultures. This effectwas not reported in the MCF-7 cell mammospheres, suggestinghigh levels of free radical scavengers in CSCs as a possible explana-tion of radioresistance. Indeed, adaptive stress responses to ROSmay lead the activation of redox-sensitive transcription factors,such as nuclear factor j-B (NF-j-B) and Nrf2. Downstream activa-tion of such pathways may ultimately include an increase in theexpression of ROS-scavenging enzymes, such as superoxide dismu-tase and glutathione. Additional mechanism for stress adaptationcan make some cells more resistant to high levels of ROS. As NF-j-B and Nrf2 also interact with proliferation, senescence evasion,angiogenesis and metastasis, the redox adaptation processes couldbe involved in cancer promotion and tumor development [40].Remarkably, ROS level may also directly or indirectly play anessential role in cell cycle progression. While, low levels of ROSmay result in increased cell cycle progression, prolonged exposuremight lead to a differentiation process and/or growth arrest. Expla-nation may include the effects of different levels of ROS on the cy-clin kinase inhibitor protein p21. In fact, at higher but not directlylethal level of ROS, p21 might be able to induce a partial or perma-nent cell cycle arrest [41].
Other regulatory mechanisms are mediated by Hypoxia Induc-ible Factors (HIFs). The enhanced level of HIFs in tumor can be cor-related with the level of oxygen within the tumor and has beenshown to correlate to tumor radiation resistance [42,43]. In cancer,two isoforms of HIF are important: HIF1a and HIF2a. While HIF1ais regulated by oxygen tension, it is ubiquitously expressed in nor-mal tissue. In contrast, HIF2a expression is restricted to endothe-lial cells of vascular organs. Both share transcriptional targetssuch as VEGF and VEGF-R1 and are regulators of migration anderythropoiesis. The majority of tumor cells responded to hypoxiaby stabilizing the HIF1a protein. In many cases, HIF2a expressionwas no different between hypoxia and normoxia. However, Liet al. have demonstrated differential protein and mRNA expressionof HIF1a and HIF2a between non-stem and CSCs [44]. In this study,HIF2a was significantly present in the CSCs population, whileHIF1a was present in both populations and was stabilized in se-vere hypoxic conditions. This finding suggested differential rolesof the two isoforms in CSCs. CSCs that had a lower HIF activity wereunable to form tumors and had reduced survival in vitro. Knock-down experiments concluded that HIFs were required to CSCs sur-vival and tumor progression. More, HIF2a seemed to promote astem cell phenotype. McCord et al. [45] showed that hypoxia pro-moted tumorosphere formation and stem cell marker expression.Using microarray techniques, two genes correlating with stem cellfunctions, Sox2 and Oct4, were overexpressed in cells under severehypoxia, only when HIF2a was stabilized. More, HIF2a transcrip-tionally regulated Oct4 [46] suggesting a specific role of HIF2a inthe plasticity of phenotype in cancer cells [47].
142 C. Moncharmont et al. / Cancer Letters 322 (2012) 139–147

2.5. Interaction with stromal environment
The central role of perivascular niche was mentioned above.Additionally, most of tumor cells interact with their microenviron-ment (stromal fibroblasts, adipocytes, endothelial cells, and theextracellular matrix). One of the chief components of the extracel-lular matrix, hyaluronan, contributes significantly to cell prolifera-tion and migration, and may be involved in tumor progression.CD44 is a major hyaluronan receptor abundantly expressed atthe CSCs surface and is also associated with malignancy [48]. Be-sides, it is likely that CSCs are influenced by their stromal environ-ment, including immune cells and inflammatory responses. Asmatter of fact, irradiation to tissue may cause changes in geneexpression in the resident fibroblasts and the release of transform-ing growth factor-b (TGF-b) which may be implicated in inflamma-tion pathway and tumorigenesis [49]. In some circumstances, CSCsin solid tumors might show an EMT. EMT is critical for metastasisand colonization at distant sites may directly be influenced by TGF-b. Gregory et al. reported that EMT transcription factors, such asZeb1 and -2 and Twist were inhibited by TGFb. Although ionizingradiation may adversely increase tumor motility signals, their re-sults suggest novel targets that may permit new approach forimproving the therapeutic strategy [50].
Moreover, microenvironment interactions could be underesti-mated since stromal cell influence may not necessarily be obvious.As an example, in estrogen receptor negative breast cancer cells,hormones have been shown to protect cell against radiation. Toinvestigate this phenomenon, Gupta et al. used a xenograft mousemodel of parturition-induced breast carcinoma formation, inwhich the tumors that rose following pregnancy lacked the expres-sion of nuclear hormone receptors. Authors described that estro-gen indirectly caused the tumor growth by increasing hostangiogenesis and recruiting of bone marrow-derived stromal cells[51].
3. Therapeutical strategies to target CSCs
Several studies have investigated the possibility of specificallytarget CSC’s molecular pathways for overcoming radiationresistance.
3.1. Wnt/b-catenin signaling
Studies have suggested that Wnt and b-catenin signaling maycontribute to radioresistance of breast CSCs [15,52]. Both pathwayshave been implicated in normal stem cell survival [53–56]. Themechanism of cell survival is unclear but aberrant Wnt and b-cate-nin signaling can link to chromosomal instability through regula-tion of the mitotic spindle [57], leading to tolerance of DNAdamage even if repair was not successful. In fact, Wnt pathwayactivated a DNA damage response [58] and genomic instabilitymay drive the malignant transformation of non-tumorigenic stemcells to CSCs [59]. Furthermore, it had been observed that in radi-oresistant mammosphere cells, the DNA damages were more rap-idly resolved than in monolayer culture cells. This suggest a roleof Wnt-b-catenin downstream signal transduction and of moreeffective DNA repair in cells with stem cell characteristics afterirradiation [15].
Che et al. described the implication of Wnt pathway in oesoph-ageal cancer. This team evaluated the radiosensitizing effect of aCOX-2 inhibitor, NS398, on radioresistant esophageal cancerEca109R50 Gy cells. Eca109R50 Gy cells were obtained throughfractional irradiation from Eca109 cells. NS398 enhanced radiosen-sitivity of Eca109R50 Gy cells. The underlying effects includedredistribution of cell cycle, inhibition of DNA-dependent protein
kinase catalytic subunit expression and finally induction of tumorcells apoptosis. Authors also reported that NS398 reduced phos-pho-AKT level, upregulated expression of proapoptotic genes (pro-caspase-3, active caspase-3), while downregulated expression ofantiapoptotic Bcl-2. These effects were partly reversed when cellswere exposed to insulin-like growth factor-1 [60]. The same teamreported that, in vivo xenograft tumorigenicity assay Eca109R50 Gycells had CSCs properties (proliferation, colony-formation) andtumorigenic ability. At the molecular level, escalating doses wasaccompanied by an increase in b-catenin. While the intake ofNS398 enhanced the radiosensitivity of Eca109R50 Gy cells, it de-creased expression of b-catenin [61].
In rectal cancers cell lines that were resistant to chemoradio-therapy, Kendziorra et al. reported overexpression of the Wnt tran-scription factor T cell factor (TCF4). Moreover, silencing of TCF4caused a significant sensitization of cells to clinically relevantdoses of X-rays. This effect was restricted to tumor cells with highTCF4 reporter activity, in a b-catenin-independent manner. Radio-sensitization was the consequence of (i) a transcriptional deregula-tion of Wnt/TCF4 target genes, (ii) a silencing-induced G2/M phasearrest, (iii) an impaired ability to adequately halt cell cycle progres-sion after radiation and (iv) a compromised DNA double strandbreak repair as assessed by cH2AX staining. TCF4 is a promisingmolecular target to sensitize resistant tumor cells to (chemo-)radiotherapy [21]. RO4929097, another Wnt pathway inhibitor, isnow evaluated in a phase I trial recruiting breast cancer patients(http://clinicaltrials.gov: NCT01351103).
3.2. PI3k-mTOR pathway
PI3K pathway is another pathway explored in breast CSCs. ThePI3K are activated by receptor tyrosine kinase, such as epidermalgrowth factor receptor (EGFR), or by G- protein coupled receptors.Among the proteins activated by PI3K, the serine/threonin kinaseAkt target Mammalian target of rapamycin (mTOR), a serine/thre-onine kinase playing a key role in regulation of basic cellular func-tions including growth, cell proliferation and survival by inhibitionof apoptosis [62,63]. Zhan et al. [64] used 8 Gy-irradiated MCF-7mammosphere cells (CD24�/low/CD44+) and MCF-7 cells (D24+/CD44+). Akt1 and Akt2 proteins were higher expressed in mammo-sphere cells than in more differentiated cells. More, Akt inhibitorradiosensitized mammosphere cells but not monolayer cells.Although PI3K is an interesting target and inhibitors are yet avail-able, other studies should confirm this result.
3.3. Notch cell signaling system
Rich and Eyler [65] synthesized the works on brain stem cells inhis laboratory by working on L1CAM (CD171), Phosphatidylinositol3-kinases (PI3K) pathway, bone morphogenetic protein (BMP),Notch, sonic hedgehog and EGFR. Using inhibitors of each targetled to reduced neurospheres formation (which contains CSCs),invasion, and then tumor initiation [66–71]. Notch pathway wasfirst described by Phillips et al. in mammospheres [14]. Notchactivity increased in response to radiation. More, targeting Notchcleavage by gamma-secretase inhibitor has proved to be an effec-tive antitumor therapy by medulloblastoma cell lines [68]. The ef-fect was a reduced CD133+ cell fraction, by increasing apoptosis.Notch pathway was reported to be implicated in radioresistancein glioma stem cells [72]. The use of gamma-secretase inhibitorsrendered the glioma stem cells more sensitive to radiation, butdid not have effect on the non-stem glioma cells. Gamma-secretaseinhibitors reduced Akt activity and Mcl-1 levels. Knockdown ofNotch1 or Notch2 also sensitized glioma stem cells to radiationand impaired xenograft tumor formation, indicating that CSCs inbrain tumors seem to be selectively vulnerable to Notch pathway
C. Moncharmont et al. / Cancer Letters 322 (2012) 139–147 143

inhibitor. A phase II trial evaluating RO4929097, a new notchinhibitor, is now ongoing for patients with recurrent or progres-sive glioblastoma (http://clinicaltrials.gov: NCT01122901).
3.4. Targeting JAK/STAT
JAK/STAT signaling in CSCs is a promising pathway on HNSCCmodels. This pathway involves a family of proteins with tyrosinekinase activity, the JAK, which in turn regulate transcription ofmany genes. It was shown that activation of the EGF receptor acti-vated STAT through JAK or Src tyrosine kinase. After its phosphor-ylation by JAK, STAT proteins migrate to the nucleus to bind to thepromoters of their target genes to activate their transcription.STAT3 is a strongly activated protein in solid tumor implicated inthe formation of nitric oxide involved in cell survival. STAT3 is alsoconcerned in proliferation and cell differentiation by activating theexpression of anti-apoptotic Bcl-2 proteins [73–76]. Chen et al. [77]observed that patients CD44+ALDH1+ and phosphorylated STAT3had a worse prognosis with higher grade of HNSCC. CD44+ALDH1+
cells isolated showed greater tumorigenicity and radioresistance.Interestingly, cucurbitacin 1, which is a STAT3 inhibitor, wasshown to effectively inhibit in vitro capacities for tumorigenicity,sphere formation, resistance to radiation, and Bcl-2 expression. Italso activated apoptosis in CD44+ALDH1+ cells. Xenotransplantexperiments revealed that cucurbitacin I combined with radiother-apy significantly suppressed tumorigenesis and lung metastasisand further improved the survival rate in HNSCC-CD44+ALDH1+
transplanted immunodeficient mice.JAK/STAT pathways have also been evaluated in non-small cell
lung cancer (NSCLC) [78]. Hsu et al. isolated radioresistantCD133+ cells expressing high expression of Oct-4, Nanog homoe-box, and especially high p-STAT3 levels. Two cell pools (CD133+
and CD133�) were treated with the STAT3 inhibitor, cucurbitacin1. Same results as in the HNSCC were observed: decreased levelof p-STAT3, loss of stemness (e.g. cells became CD133�), and highlevel of apoptosis. Xenotransplantation in immunocompromisedmice revealed a loss of tumorigenesis of CD133+ cells when micewere treated by association of cucurbitacin 1 and radiotherapy.
3.5. Interference with ROS cell response (use of oxygen activators)
Glutathione (GSH) is an intracellular reducing molecule withantioxidant activity. Diehn et al. reported that two GSH-relatedgenes could be overexpressed in CSCs: Gclm and Gss. Those encodethe regulatory subunit of the enzyme (glutamate-cysteine ligase)that was found to catalyze GSH synthesis. The use of ButhionineSulfoximine (BSO), a glutamate-cysteine ligase inhibitor, resultedin decrease of colony forming ability in CSCs. Moreover, BSO-pre-treated CSCs were radiosensitized, demonstrating the importanceof low ROS levels and antioxidant defense to CSCs survival andradiosensitivity in these tumors [36].
3.6. Others implicated molecular pathways
The hedgehog signaling pathway lead to a cascade of activationof transcription factors such as Gli proteins, that activate transcrip-tion of several genes involved in cell proliferation such as cyclins,c-Myc, epidermal growth factor (EGF), and VEGF. Breast CSCsshowed increased expression of Ptch1, Gli1, and Gli2 which sug-gested an important role for hedgehog signaling in stem cell prolif-eration. In addition, inhibition of Gli1 induced decreasedproliferation of breast CSCs [79].
Another described target is c-Met. This protein is overexpressedin CSCs in glioblastoma [80]. Ellsworth et al. evaluated Mp470, amulti-targeted tyrosine kinase inhibitor as radiosensitizing mole-cule, [81]. Mp470 activity was compared with temozolomide
(TMZ). Results showed that MP470 associated with radiotherapywas more efficient than TMZ and radiotherapy in clonogenic as-says. Apoptosis was the main cause of cell death and its levelwas twice more important in radiation combined with MP470 thanwith TMZ-radiotherapy.
Insulin-like growth factor (IGF) pathway could likewise be aninteresting target in NSCLC-CSCs Sun et al. [82] explored a che-mo-radioresistant cell line that expressed CSCs characteristics:expression of markers (CD133 and ALDH1), and self renewing.Authors used DNA microarrays to find out which pathways wereinvolved in the CSCs in comparison to H460 NSCLC cell lines. Ofthe 180 RNA whose expression varied between the two cell popu-lations, IGFBP3 to be the most interesting. The use of IGF-1R siRNAinhibitor gave back significant radiosensitization of initially radio-resistant cells. Interestingly, the inhibition of the transcription ofgluconeogenesis genes has also been shown to reduce the risk ofseveral cancers in diabetic patients [83]. These results are sup-ported by the selective effects of Metformin, a standard drug fortype II diabetes, on CSCs. In fact, metformin was reported torepress key drivers of the EMT genetic program (e.g. ZEB1 andTGF-b) [84]. Also, Jung et al. showed that metformin reduced thesize and number of human breast carcinoma cell line grown inmammospheres. Furthermore, metformin reduced the expressionof OCT4, used as a stem cell marker, in mammospheres exposedto tumor promoting chemicals [85].
Finally, bortezomib is a therapeutic proteasome inhibitor. It wasapproved for treatment of haematological malignancies such as re-lapsed multiple myeloma and mantle cell lymphoma. It has re-cently been reported that proteasome inhibitor sensitized gliomacells and glioma stem cells to tumor necrosis factor-related apop-tosis-inducing ligand induced apoptosis. (TRAIL) in vitro andin vivo. Moreover, TRAIL and the proteasome inhibitors decreasedthe expression of AKT and XIAP in a PKCe-dependent manner, sug-gesting a rationale for targeting malignant glioma CSCs [86].
3.7. Relationships between diverse molecular pathways
Putative complex signaling cross-talks between differentgrowth factor cascades may regulate the cancer development.EGFR, constitutively activated EGFRvIII mutant and sonic hedgehogpathways play all a pivotal role in the development of brain tu-mors, and more particularly when focusing on initiating cells(CSCs) in aggressive and recurrent glioblastoma and medulloblas-tomas. Mimeault et al. suggested that those pathways could inter-act with other signaling system that provide survival advantagesduring disease progression such as Wnt/b-catenin, Notch, plate-let-derived growth factor (PDGF)/PDGF receptors (PDGFRs), hepa-tocyte growth factor (HGF)/c-Met receptor and VEGF/VEGFreceptors (VEGFRs). Confirmation of these results may constitutethe rationale for the use of pan-inhibitor targeted therapies to in-crease the probability of eradicating brain CTCs and then reachinghigher efficacy for aggressive and recurrent primary brain tumors.
Relationships exist between notch, met, PI3K pathways andautophagy. It was described that Notch signaling and the PI3K-AKT pathway synergized and that mutational loss of PTEN (Phos-phatase and Tensin homolog, that physiologically inhibit thePI3K-AKT pathway) induces resistance to NOTCH1 inhibition[87]. Similarly, c-Met-dependent gene expression is also regulatedby PTEN [88]. Because autophagy is negatively regulated by PI3K-AKT-mTOR, Notch and c-Met could also decrease activation ofautophagy. Finally, CD44v6 contains a binding site for hepatocytegrowth factor (HGF) that binds c-Met and VEGF. Then, CD44v6 inconcert with extra cellular matrix may activate the PI3K-Akt path-way, regulate Met transcription and play on autophagy [89].
Fig. 2 illustrates main molecular pathways and potential cross-talks implicated in CSCs.
144 C. Moncharmont et al. / Cancer Letters 322 (2012) 139–147

3.8. Ions therapy as a tool for overcoming CSCs radioresistance
Many assays demonstrate the chemo- and radio-resistance ofCSCs. Chang et al. hypothesized that protons may be more effectivethan photons to eliminate CSCs [11]. Authors worked on twochemoresistant (CR) NSCLC cell lines: (H460/CR and A549/CR)and three radioresistant (RR) cell lines (H460/RR, A549/RR andSeg-1/RR). Both CR and RR cell lines expressed higher percentagesof ‘side population’ cells (i.e., those showing CSC phenotype) thandid parental cells. The control cell line was normal bronchial epi-thelial cells. All cells were irradiated with the same doses relativebiological effectiveness of protons or photons. Authors distin-guished parental cells and CSCs using flow cytometry and analyzedthe following parameters: growth, apoptosis, invasion, and intra-cellular concentrations of ROS. Compared to control, protons killedmore pathologic cells, increased apoptosis, reduced metastatic po-tential than photons in CSCs. Whereas CSCs contained initially lessthan half ROS levels of the parental cells or the normal bronchialepithelial cells, protons induced higher increase of ROS levels thanphotons in CR/RR. Their main findings were that protons preferen-tially target CSCs. When focusing on treatment-resistant CSCs, theincrease in ROS levels was higher after protons exposure, com-pared to photons for the same dose. However, protons and photonsproduced equivalent effects in normal bronchial epithelial cells,suggesting no higher toxicity toward normal tissue. These attrac-tive results should be confirmed in vivo.
Another way of irradiation is the use of carbon ion. Cui et al.[12] had focused on the biological effects of carbon ion beam treat-ment on CD133+CD44+/ESA+ human colon cancer cells comparedwith photon beam treatment. The in vitro relative biological effec-tiveness values of a carbon ion beam relative to X-rays at D10 (doserequired to reduce the surviving fraction to 10%) values were 1.63–1.74. Results showed that carbon ion irradiation induced lowerCSCs fraction than with X-ray irradiation. A colony assay was per-formed in order to confirm this result, and the relative biological
effectiveness values at D10 levels were 2.05–2.28. These resultswere also observed in xenograft assay. This suggests that carbonion beam therapy could have higher biological efficacy, as com-pared to classical photon beam therapy. One explanation couldbe a higher biological efficacy against CSCs.
4. Conclusion
Despite substantial research and important progress in the anti-cancer therapeutic arsenal, about 50% of cancer patients will expe-rience local or distant tumor relapse despite treatment. The factthat most current strategies did not target CSCs but rather moredifferentiated tumor cells may have a putative role in the failureof numerous intent-to-cure strategies. Since local tumor seems tobe potentially related to CSCs control, radiobiological researchshould focus in radiation effects on cancer stem cells. Many newmolecular pathways involved in self-renewal, tumor growth, andtumor cell differentiation have also been explored [15,71,77].Although preclinical models can be debated because of intrinsiclimitations, and particularly the use of immunodeficient animalsassays [90], new irradiation modalities could be promising toenhance the effects against CSCs. Since CSCs are a heterogeneouspopulation which has different resistance profiles, a single targetedmolecular therapy should however not be sufficient. Subsequently,the combination of cancer stem cell-targeting agents with neweffective cytotoxic agents and/or radiation therapy may allowtargeting both CSCs and bulk population. Ion therapy might alsoenhance the efficacy/toxicity ratio by preferentially targeting CSCswith rather limited effects on normal stem cells [11,12] and normaltissues. As reviewed by Supiot and colleagues, dose-painting radio-therapy may be applied for a heterogeneous delivery of radiationdose within the tumor volume by preferentially targeting radiore-sistant areas according to data from functional imaging [91]. With-in gross tumor volume, it is possible to define one or more target
Fig. 2. Main molecular pathways implicated in CSCs. While Wnt bind its receptor (frizzled) and induce b-catenin signaling, various cytokines may activate JAK-signaltransducer and activator of transcription (STAT) that result in cell proliferation, growth and survival. Interactions exist between Notch, Phosphatidylinositol 3-kinases-AKT-mTOR (PI3K) and Hepatocyte growth factor (HGF)/cMET pathways. While cMET downstream may trigger AKT, Notch activation is able to inhibit PTEN that may result indiminished autophagy through mTOR activation. Extra cellular matrix (ECM) may interact with CD44v6 that can provoke the dimerization of receptor tyrosine kinases such asvascular endothelial growth factor receptor (VEGFR) or bind cMET and play on PI3K-AKT signaling.
C. Moncharmont et al. / Cancer Letters 322 (2012) 139–147 145

volumes based on biology (biological target volume [92]. BecauseCSCs metabolic status may vary from the common tumor cells,functional and metabolic imaging could allow better identifyingthe niches with hypoxic areas and/or high CSCs numbers are fac-tors of radiation resistance. New studies including such biologi-cal-guided radiation devices combined to modern cytotoxicagents will be a promising search area for optimizing strategiestargeting CSCs.
References
[1] T. Schatton, N.Y. Frank, M.H. Frank, Identification and targeting of cancer stemcells, BioEssays 10 (2009) 1038–1049.
[2] R.P. Hill, L. Milas, The proportion of stem cells in murine tumours, Int. J. Radiat.Oncol. Biol. Phys. 16 (1989) 513–518.
[3] N. Barker, R.A. Ridgway, J.H. van Es, M. van de Wetering, H. Begthel, M. van denBorn, E. Danenberg, A.R. Clarke, O.J. Sansom, H. Clevers, Crypts stem cells as thecells-of-origin of intestinal cancer, Nature 457 (2009) 608–611.
[4] L. Zhu, P. Gibson, D.S. Currle, Y. Tong, R.J. Richardson, I.T. Bayazitov, H.Poppleton, S. Zakharenko, D.W. Ellison, R.J. Gilbertson, Prominin 1 marksintestinal stem cells that are susceptible to neoplastic transformation, Nature45 (2009) 603–607.
[5] S. Alcantara Llaguno, J. Chen, C.H. Kwon, E.L. Jackson, Y. Li, D.K. Burns, A.Alvarez-Buylla, L.F. Parada, Malignant astrocytomas originate from neuralstem/progenitor cells in a somatic tumor suppressor mouse model, Cancer Cell15 (2009) 45–56.
[6] S.A. Mani, W. Guo, M.J. Liao, E.N. Eaton, A. Ayyanan, A.Y. Zhou, M. Brooks, F.Reinhard, C.C. Zhang, M. Shipitsin, L.L. Campbell, K. Polyak, C. Brisken, J. Yang,R.A. Weinberg, The epithelial–mesenchymal transition generates cells withproperties of stem cells, Cell 133 (2008) 704–715.
[7] K. Polyak, R.A. Weinberg, Transitions between epithelial and mesenchymalstates: acquisition of malignant and stem cell traits, Nat. Rev. Cancer 9 (2009)265–273.
[8] C.L. Chaffer, I. Brueckmann, C. Scheel, A.J. Kaestli, P.A. Wiggins, L.O. Rodrigues,M. Brooks, F. Reinhardt, Y. Su, K. Polyak, L.M. Arendt, C. Kuperwasser, B. Bierie,R.A. Weinberg, Normal and neoplastic nonstem cells can spontaneouslyconvert to a stem-like state, Proc. Natl. Acad. Sci. USA 108 (2011) 7950–7955.
[9] F. Pajonk, E. Vlashi, W.H. McBride, Radiation resistance of cancer stem cells:the 4 R’s of radiobiology revisited, Stem Cells 28 (2010) 639–648.
[10] L. Mils, W.N. Hittelman, Cancer stem cells and tumour response to therapy:current problems and future prospects, Semin. Radiat. Oncol. 19 (2009) 96–105.
[11] J.Y. Chang, X. Zhang, O. Vassiliev, M. Gillin, R. Mohan, et al., Proton therapytargets cancer stem cells in treatment-resistant non-small cell lung cancer, Int.J. Radiat. Oncol. Biol. Phys. 78 (2010) S644.
[12] X. Cui, K. Oonishi, H. Tsujii, T. Yasuda, Y. Matsumoto, Y. Frusawa, M. Akashi, T.Kamada, R. Okayasu, Effects of carbon ion beam on putative colon cancer stemcells and its comparison with X-rays, Cancer Res. 71 (2011) 3676–3687.
[13] S. Bao, Q. Wu, R.E. McLendon, Y. Hao, Q. Shi, A.B. Hjelmeland, M.W. Dewhirst,D.D. Bigner, J.N. Rich, Glioma stem cells promote radioresistance bypreferential activation of the DNA damage response, Nature 444 (2006)756–760.
[14] T.M. Phillips, W.H. McBride, F. Pajonk, The response of CD24(-/low)/CD44+breast cancer-initiating cells to radiation, J. Natl Cancer Inst. 98 (2006) 1777–1785.
[15] W.A. Woodward, M.S. Chen, F. Behbod, M.P. Alfaro, T.A. Buchholz, J.M. Rosen,WNT/b-catenin mediates radiation resistance of mouse mammary progenitorcells, Proc. Natl. Acad. Sci. USA 104 (2007) 618–623.
[16] S.D. Parikh, V. Levina, T. Wang, M.K. Gibson, A.E. Lokshin, Radioresistance ofnon-small cell lung cancer stem cells, Int. J. Radiat. Oncol. Biol. Phys. 75 (2009)S542–S543.
[17] B. Campos, C.C. Herold-Mende, Insight into the complex regulation of CD133 inglioma, Int. J. Cancer 128 (2011) 501–510.
[18] T. Miyabayashi, J.L. Teo, M. Yamamoto, M. McMillan, C. Nguyen, M. Kahn, Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cellpluripotency, Proc. Natl. Acad. Sci. USA 104 (2007) 5668–5673.
[19] T.M. Pawlik, K. Keyomarsi, Role of cell cycle in mediating sensitivity toradiotherapy, Int. J. Radiat. Oncol. Biol. Phys. 59 (2004) 928–942.
[20] A. Wilson, E. Laurenti, G. Oser, et al., Hematopoietic stem cells reversiblyswitch from dormancy to self-renewal during homeostasis and repair, Cell 135(2008) 1118–1129.
[21] E. Kendziorra, K. Ahlborn, M. Spitzner, M. Rave-Fränk, G. Emons, J. Gaedcke, F.Kramer, H.A. Wolff, H. Becker, T. Beissbarth, R. Ebner, B.M. Ghadimi, T. Pukrop,T. Ried, M. Grade, Silencing of the Wnt transcription factor TCF4 sensitizescolorectal cancer cells to (chemo-) radiotherapy, Carcinogenesis 32 (2011)1824–1831.
[22] J.N. Rich, Cancer stem cells in radiation resistance, Cancer Res. 67 (2007) 8980–8984.
[23] H. Yin, J. Glass, The phenotypic radiation resistance of CD44+/CD24(-or low)breast cancer cells is mediated through the enhanced activation of ATMsignaling, PLoS ONE 6 (2011) e24080.
[24] R. Cao, Y. Tsukada, Y. Zhang, Role of Bmi-1 and Ring1A in H2A ubiquitylationand Hox gene silencing, Mol. Cell 20 (2005) 845–854.
[25] S. Facchino, M. Abdouh, W. Chatoo, G. Bernier, BMI confers radioresistance tonormal and cancerous neural stem cells through recruitment of the DNAdamage response machinery, J. Neurosci. 30 (2010) 10096–10111.
[26] R.S. Gieni, I.H. Ismail, S. Campbell, M.J. Hendzel, Polycomb group proteins inthe DNA damage response: a link between radiation resistance and‘‘stemness’’, Cell Cycle 10 (2011) 883–894.
[27] C.T. Jordan, M.L. Guzman, M. Noble, Cancer stem cells, N. Engl. J. Med. 355(2006) 1253–1261.
[28] B. Zhivotovsky, G. Kroemer, Apoptosis and genomic instability, Nat. Rev. Mol.Cell Biol. 5 (2007) 752–762.
[29] J. Bensimon, S. Altmeyer-Morel, H. Benjelloun, S. Chevillard, J. Lebeau, CD24(-/low) stem-like breast cancer marker defines the radiation-resistant cellsinvolved in memorization and transmission of radiation-induced genomicinstability, Oncogene, 2012. doi: http://dx.doi.org/10.1038/onc.2012.31 (Epubahead of print).
[30] S. Paglin, T. Hollister, T. Delohery, N. Hackett, M. McMahill, E. Sphicas, D.Domingo, J. Yahalom, A novel response of cancer cells to radiation involvesautophagy and formation of acidic vesicles, Cancer Res. 61 (2001) 439–444.
[31] M.J. Abedin, D. Wang, M.A. McDonnell, U. Lehmann, A. Kelekar, Autophagydelays apoptotic death in breast cancer cells following DNA damage, CellDeath Differ. 14 (2007) 500–510.
[32] D.A. Gewirtz, M.L. Hilliker, E.N. Wilson, Promotion of autophagy as amechanism for radiation sensitization of breast tumor cells, Radiother.Oncol. 92 (2009) 323–328.
[33] W. Zhuang, B. Li, L. Long, L. Chen, Q. Huang, Z.Q. Liang, Knockdown of the DNA-dependent protein kinase catalytic subunit radiosensitizes glioma-initiatingcells by inducing autophagy, Brain Res. 1371 (2011) 7–15.
[34] J.M. Brown, W.R. Wilson, Exploiting tumour hypoxia in cancer treatment, Nat.Rev. Cancer 4 (2004) 437–447.
[35] C. Calabrese, H. Poppleton, M. Kocak, T.L. Hogg, C. Fuller, B. Hamner, E.Y. Oh,M.W. Gaber, D. Kinklestein, M. Allen, A. Frank, I.T. Bayazitov, S.S. Zakharenko,A. Gajjar, A. Davidoff, R.J. Gilbertson RJ, A perivascular niche for brain tumorstem cells, Cancer Cell 11 (2007) 69–82.
[36] M. Diehn, R.W. Cho, N.A. Lobo, T. Kalisky, M.J. Dorie, A.N. Kulp, D. Qian, J.S. Lam,L.E. Ailles, M. Wong, B. Joshua, M.J. Kaplan, I. Wapnir, F.M. Dirbas, G. Somlo, C.Garberoglio, B. Paz, J. Shen, S.K. Lau, S.R. Quake, J.M. Brown, I.L. Weissman, M.F.Clarke, Association of reactive oxygen species levels and radioresistance incancer stem cells, Nature 458 (2009) 780–783.
[37] J. Smith, E. Ladi, M. Mayer-Proschel, M. Noble, Redox state is a centralmodulator of the balance between self-renewal and differentiation in adividing glial precursor cell, Proc. Natl. Acad. Sci. USA 97 (2000) 10032–10037.
[38] M. Tsatmali, E.C. Walcott, K.L. Crossin, Newborn neurons acquire high levels ofreactive oxygen species and increased mitochondrial proteins upondifferentiation from progenitors, Brain Res. 1040 (2005) 137–150.
[39] R.A. Riley, Free radicals in biology: oxidative stress and the effects of ionizingradiation, Int. J. Radiat. Biol. 65 (1994) 27–33.
[40] D. Trachootham, J. Alexandre, P. Huang, Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?, Nat Rev. Drug. Discov.8 (2009) 579–591.
[41] J. Boonstra, J.A. Post, Molecular events associated with reactive oxygen speciesand cell cycle progression in mammalian cells, Gene 337 (2004) 1–13.
[42] B.J. Moeller, Y. Cao, C.Y. Li, M.W. Dewhirst, Radiation activates HIF1 regulatevascular radiosensitivity in tumors: role of reoxygenation, free radicals, andstress granules, Cancer Cell 5 (2004) 429–441.
[43] M.W. Dewhirst, Y. Cao, B. Moeller, Cycling hypoxia and free radicals regulateangiogenesis and radiotherapy response, Nat. Rev. Cancer 8 (2008) 425–437.
[44] Z. Li, S. Bao, Q. Wu, H. Wang, C. Eyler, S. Sathornsumetee, Q. Shi, Y. Cao, J. Lathia,R.E. McLendon, A.B. Hjelmeland, J.N. Rich, Hypoxia-inducible factors regulatetumorigenic capacity of glioma stem cells, Cancer Cell 15 (2009) 501–513.
[45] A.M. McCord, M. Jamal, U.T. Shankavarum, F.F. Lang, K. Camphausen, P.J.Tofilon, Physiologic oxygen concentration enhances the stem-like propertiesof CD133+ human glioblastoma cells in vitro, Mol. Cancer Res. 7 (2009) 489–497.
[46] K.L. Covello, J. Kehler, H. Yu, J.D. Gordan, A.M. Arsham, C.J. Hu, P.A. Labosky,M.C. Simon, B. Keith, HIF-2alpha regulates Oct-4: effects of hypoxia on stemcell function, embryonic development, and tumor growth, Genes Dev. 20(2006) 557–570.
[47] A. Pietras, D. Gisselsson, I. Ora, R. Noguera, S. Beckman, S. Navarro, S. Pahlman,High levels of HIF-2alpha highlight an immature neural crest-likeneuroblastoma cell cohort located in a perivascular niche, J. Pathol. 214(2008) 482–488.
[48] B.P. Toole, Hyaluronan-CD44 Interactions in cancer: paradoxes andpossibilities, Clin. Cancer Res. 15 (2009) 7462–7468.
[49] M.J. Bissell, M.A. Labarge, Context, tissue plasticity, and cancer: are tumor stemcells also regulated by the microenvironment?, Cancer Cell 7 (2005) 17–23
[50] P.A. Gregory, A.G. Bert, E.L. Paterson, S.C. Barry, A. Tsykin, G. Farshid, M.A.Vadas, Y. Khew-Goodall, G.J. Goodall, The miR-200 family and miR-205regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1, Nat.Cell Biol. 10 (2008) 593–601.
[51] P.B. Gupta, D. Proia, O. Cingoz, J. Weremowicz, S.P. Naber, R.A. Weinberg, C.Kuperwasser, Systemic stromal effects of estrogen promote the growth ofestrogen receptor-negative cancers, Cancer Res. 67 (2007) 2062–2071.
[52] Y. Li, B. Welm, K. Podsypanina, S. Huang, M. Chamorro, X. Zhang, T. Rowlands,M. Egeblad, P. Cowin, Z. Werb, L.K. Tan, J.M. Rosen, H.E. Varmus, Evidence thattransgenes encoding components of the Wnt signaling pathway preferentially
146 C. Moncharmont et al. / Cancer Letters 322 (2012) 139–147

induce mammary cancers from progenitor cells, Proc. Natl. Acad. Sci. USA 100(2003) 15853–15858.
[53] M.S. Chen, W.A. Woodward, F. Behbod, S. Peddibhotla, M.P. Alfaro, T.A.Buchholz, J.M. Rosen, Wnt/beta-catenin mediates radiation resistance of Sca1+progenitors in an immortalized mammary gland cell line, J. Cell Sci. 120 (2007)468–477.
[54] P. Polakis, Wnt signaling and cancer, Genes Dev. 14 (2000) 1837–1851.[55] N.A. Theodosiou, C.J. Tabin, Wnt signaling during development of the
gastrointestinal tract, Dev. Biol. 15 (2003) 258–271.[56] J. Taipale, P.A. Beachy, The hedgehog and Wnt signalling pathways in cancer,
Nature 411 (2001) 349–354.[57] M.V. Hadjihannas, M. Bruckner, B. Jerchow, W. Birchmeier, W. Dietmaier, J.
Behrens, Aberrant Wnt/b-catenin signalling can induce chromosomalinstability in colon cancer, Proc. Natl. Acad. Sci. USA 103 (2006) 10747–10752.
[58] A. Ayyanan, G. Civenni, L. Ciarloni, C. Morel, N. Mueller, K. Lefort, A.Mandinova, W. Raffoul, M. Fiche, G.P. Dotto, C. Brisken, Increased Wntsignaling triggers oncogenic conversion of human breast epithelial cells by aNotch-dependent mechanism, Proc. Natl. Acad. Sci. USA 103 (2006) 3799–3804.
[59] A. Shiras, S.T. Chettiar, V. Shepal, G. Rajendran, G.R. Prasad, P. Shastry,Spontaneous transformation of human adult nontumorigenic stem cells tocancer stem cells is driven by genomic instability in a human model ofglioblastoma, Stem Cells 25 (2007) 1478–1489.
[60] S.M. Che, X.Z. Zhang, L. Hou, T.B. Song, Cyclooxygenase-2 inhibitor NS398enhances radiosensitivity of radioresistant esophageal cancer cells byinhibiting AKT activation and inducing apoptosis, Cancer Invest. 28 (2010)679–688.
[61] S.M. Che, X.Z. Zhang, X.L. Liu, X. Chen, L. Hou, The radiosensitization effect ofNS398 on esophageal cancer stem cell-like radioresistant cells, Dis. Esophagus24 (2011) 265–273.
[62] M. Osaki, M. Oshimura, H. Ito, PI3K-Akt pathway: its functions and alterationsin human cancer, Apoptosis 9 (2004) 667–676.
[63] J.A. Fresno Vara, E. Casado, J. de Castro, P. Cejas, C. Belda-Iniesta, M. González-Barón, PI3K/Akt signalling pathway and cancer, Cancer Treat. Rev. 30 (2004)193–204.
[64] J.F. Zhan, L.P. Wu, L.H. Chen, Y.W. Yuan, G.Z. Xie, A.M. Sun, Y. Liu, Z.X. Chen,Pharmacological inhibition of AKT sensitizes MCF-7 human breast cancer-initiating cells to radiation, Cell Oncol. (Dordr) 34 (2011) 451–456.
[65] J.N. Rich, C.E. Eyler, Cancer stem cells in brain tumor biology, Cold Spring Harb.Symp. Quant. Biol. 73 (2008) 411–420.
[66] E.E. Bar, A. Chaudhry, A. Lin, X. Fan, K. Schreck, W. Matsui, S. Piccirillo, A.L.Vescovi, F. DiMeco, A. Olivi, C.G. Eberhart, Cyclopamine-mediated hedgehogpathway inhibition depletes stem-like cancer cells in glioblastoma, Stem Cells25 (2007) 2524–2533.
[67] V. Clement, P. Sanchez, N. de Tribolet, I. Radovanovic, I. Ruiz, A. Altaba,HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cellself-renewal, and tumorigenicity, Curr. Biol. 17 (2007) 165–172.
[68] X. Fan, W. Matsui, L. Khaki, D. Stearns, J. Chun, Y.M. Li, C.G. Eberhart, Notchpathway inhibition depletes stem-like cells and blocks engraftment inembryonal brain tumors, Cancer Res. 66 (2006) 7445–7452.
[69] J. Lee, M.J. Son, K. Woolard, N.M. Donin, A. Li, C.H. Cheng, S. Kotliarova, Y.Kotliarov, J. Walling, S. Ahn, M. Kim, M. Totonchy, T. Cusack, C. Ene, H. Ma, Q.Su, J.C. Zenklusen, W. Zhang, D. Maric, F.A. Fine, Epigenetic-mediateddysfunction of the bone morphogenetic protein pathway inhibitsdifferentiation of glioblastoma-initiating cells, Cancer Cell 13 (2008) 69–80.
[70] S.G. Piccirillo, B.A. Reynolds, N. Zanetti, G. Lamorte, E. Binda, G. Broggi, H. Brem,A. Olivi, F. DiMeco, A.L. Vescovi, Bone morphogenetic proteins inhibit thetumorigenic potential of human brain tumour-initiating cells, Nature 444(2006) 761–765.
[71] A. Soeda, A. Inagaki, N. Oka, Y. Ikegame, H. Aoki, S. Yoshimura, S. Nakashima, T.Kunisada, T. Iwama, Epidermal growth factor plays a crucial role in mitogenicregulation of human brain tumor stem cells, J. Biol. Chem. 283 (2008) 10958–10966.
[72] J. Wang, T.P. Wakeman, J.D. Lathia, A.B. Hjelmeland, X.F. Wang, R.R. White, J.N.Rich, B.A. Sullenger, Notch promotes radioresistance of glioma stem cells, StemCells 28 (2010) 17–28.
[73] A. Stephanou, B.K. Brar, R.A. Knight, D.S. Latchman, Opposing actions of STAT-1and STAT-3 on the Bcl-2 and Bcl-x promoters, Cell Death Differ. 7 (2000) 329–330.
[74] S.O. Rahaman, P.C. Harbor, O. Chernova, G.H. Barnett, M.A. Vogelbaum, S.J.Haque, Inhibition of constitutively active Stat3 suppresses proliferation and
induces apoptosis in glioblastoma multiforme cells, Oncogene 21 (2002)8404–8413.
[75] A. Iwamaru, S. Szymanski, E. Iwado, H. Aoki, T. Yokoyama, I. Fokt, K. Hess, C.Conrad, T. Madden, R. Sawaya, S. Kondo, W. Priebe, Y. Kondo, A novel inhibitorof the STAT3 pathway induces apoptosis in malignant glioma cells both in vitroand in vivo, Oncogene 26 (2007) 2435–2444.
[76] H.W. Lo, S.C. Hsu, M. Ali-Seyed, M. Gunduz, W. Xia, Y. Wei, G. Bartholomeusz,J.Y. Shih, M.C. Hung, Nuclear interaction of EGFR and STAT3 in the activation ofthe iNOS/NO pathway, Cancer Cell 7 (2005) 575–589.
[77] Y.W. Chen, K.H. Chen, P.I. Huang, Y.C. Chen, G.Y. Chiou, W.L. Lo, L.M. Tseng, H.S.Hsu, K.W. Chang, S.H. Chiou, Cucurbitacin 1 suppressed stem-like property andenhanced radiation-induced apoptosis in head and neck squamous carcinoma–derived CD44(+)ALDH1(+) cells, Mol. Cancer Ther. 11 (2010) 2879–2892.
[78] H.S. Hsu, P.I. Huang, Y.L. Chang, C. Tzao, Y.W. Chen, H.C. Shih, S.C. Hung, Y.C.Chen, L.M. Tseng, S.H. Chiou, Cucurbitacin I inhibits tumorigenic ability andenhances radiochemosensitivity in nonsmall cell lung cancer-derived CD133-positive cells, Cancer 117 (2011) 2970–2985.
[79] N.P. Nguyen, F.S. Almeida, A. Chi, L.M. Nguyen, D. Cohen, U. Karisson, V. Vinh-Hung, Molecular biology of breast cancer stem cells: potential clinicalapplications, Cancer Treat. Rev. 36 (2010) 485–491.
[80] H.S. Günther, N.O. Schmidt, H.S. Phillips, D. Kemming, S. Kharbanda, R. Soriano,Z. Modrusan, H. Meissner, M. Westphal, K. Lamszus, Gli lastoma-derived stemcell-enriched cultures from distinct subgroups according to molecular andphenotypic criteria, Oncogene 27 (2008) 2897–2899.
[81] R. Ellsworth, J. Welsh, D. Mahadevan, D. Bearrs, D. Hsieh, K. Fjerstad, M.Marsella, A. Sanan, M. Badrudojja, B. Slea, The radiosensitizing effects of anovel tyrosine kinase inhibitor, Mp470 in glioblastoma multiforme stem cells,Int. J. Radiat. Oncol. Biol. Phys. 72 (2008) S714–S715.
[82] Y. Sun, S. Zheng, C. Speirs, P. Kopsombut, Z. Zhao, B. Lu, Gene profiling of cancerstem cells-like cells reveals IGF-1R and IGFBP3 pathway as a promisingtherapeutic target for lung cancer, Int. J. Radiat. Oncol. Biol. Phys. 78 (2010)S131.
[83] J.M. Evans, L.A. Donnelly, A.M. Emslie-Smith, D.R. Alessi, A.D. Morris,Metformin and reduced risk of cancer in diabetic patients, BMJ 330 (2005)1304–1305.
[84] S. Cufí, A. Vazquez-Martin, C. Oliveras-Ferraros, B. Martin-Castillo, J. Joven, J.A.Menendez, Metformin against TGFb-induced epithelial-to-mesenchymaltransition (EMT): from cancer stem cells to aging-associated fibrosis, CellCycle 15 (2010) 4461–4468.
[85] J.W. Jung, S.B. Park, S.J. Lee, M.S. Seo, J.E. Trosko, K.S. Kang, Metformin repressesself-renewal of the human breast carcinoma stem cells via inhibition ofestrogen receptor-mediated OCT4 expression, PLoS ONE 6 (2011) e28068.
[86] S. Kahana, S. Finniss, S. Cazacu, C. Xiang, H.K. Lee, S. Brodie, R.S. Goldstein, V.Roitman, S. Slavin, T. Mikkelsen, C. Brodie, Proteasome inhibitors sensitizeglioma cells and glioma stem cells to TRAIL-induced apoptosis by PKCe-dependent downregulation of AKT and XIAP expressions, Cell. Signal. 23(2011) 1348–1357.
[87] T. Palomero, M.L. Sulis, M. Cortina, P.J. Real, K. Barnes, M. Ciofani, E. Caparros, J.Buteau, K. Brown, S.L. Perkins, G. Bhagat, A.M. Agarwal, G. Basso, M. Castillo, S.Nagase, C. Cordon-Cardo, R. Parsons, J.C. Zúñiga-Pflücker, M. Dominguez, A.A.Ferrando, Mutational loss of PTEN induces resistance to NOTCH1 inhibition inT-cell leukemia, Nat. Med. 13 (2007) 1203–1210.
[88] R. Abounader, T. Reznik, C. Colantuoni, F. Martinez-Murillo, E.M. Rosen, J.Laterra, Regulation of c-Met-dependent gene expression by PTEN, Oncogene23 (2004) 9173–9182.
[89] M. Zöller, CD44: can a cancer-initiating cell profit from an abundantlyexpressed molecule?, Nat Rev. Cancer 11 (2011) 254–2567.
[90] M. Baumann, M. Krause, R. Hill, Exploring the role of cancer stem cells inradioresistance, Nat. Rev. Cancer 8 (2008) 545–554.
[91] S. Supiot, A. Lisbona, F. Paris, D. Azria, P. Fenoglietto, ‘‘Dose-painting’’: myth orreality?, Cancer Radiother 14 (2010) 554–562.
[92] C.C. Ling, J. Humm, S. Larson, H. Amols, Z. Fuks, S. Leibel, J.A. Koutcher, Towardsmultidimensional radiotherapy (MD-CRT): biological imaging and biologicalconformality, Int. J. Radiat. Oncol. Biol. Phys. 47 (2000) 551–560.
[93] L.S. Piao, W. Hur, T.K. Kim, S.W. Hong, S.W. Kim, J.E. Choi, P.S. Sung, M.J. Song,B.C. Lee, D. Hwang, S.K. Yoon, CD133+ liver cancer stem cells modulateradioresistance in human hepatocellular carcinoma, Cancer Lett. 315 (2012)129–137.
[94] Q. Zhang, S. Shi, Y. Yen, J. Brown, J.Q. Ta, A.D. Le, A subpopulation of CD133(+)cancer stem-like cells characterized in human oral squamous cell carcinomaconfer resistance to chemotherapy, Cancer Lett. 289 (2010) 151–160.
C. Moncharmont et al. / Cancer Letters 322 (2012) 139–147 147

doi:
10.1
684/
bdc.
2012
.166
6
Bull Cancer vol. 99 • N◦ 12 • décembre 2012 1153
SynthèseGeneral review
Volume 99 • N◦ 12 • décembre 2012©John Libbey Eurotext
Article recu le 10 octobre2012,accepté le 31 octobre 2012Tirés à part : N. Magné
Facteurs de radiorésistance des cellulessouches cancéreuses et perspectivesde radiosensibilisation : l’exempledu glioblastomeCancer stem cells, cornerstone of radioresistance and perspectivesfor radiosensitization: glioblastoma as an exampleCyrus Chargari1,2, Coralie Moncharmont3,4, Antonin Lévy6, Jean-Baptiste Guy3, Gérald Bertrand4,Matthieu Guilbert5, Claire Rousseau1, Lionel Védrine1, Gersende Alphonse4, Robert-Alain Toillon5,Claire Rodriguez-Lafrasse4, Éric Deutsch1,6, Nicolas Magné3,4
1 Hôpital d’instruction des armées du Val-de-Grâce, service d’oncologie-radiothérapie, 74, boulevard de Port-Royal,75230 Paris cedex, France2 Institut Gustave-Roussy, Inserm 1030, département de radiothérapie moléculaire, 39, rue Camille-Desmoulins, 94805Villejuif cedex, France3 Institut de cancérologie Lucien-Neuwirth, département de radiothérapie, 108 bis, avenue Albert-Raimond, BP 60008,42271 Saint-Priest-en-Jarez cedex, France<[email protected]>4 Faculté de médecine Lyon-Sud, laboratoire de radiobiologie cellulaire et moléculaire, EMR3738, BP 12, 69921 Oullins,France5 University Lille-1, Inserm U908, Growth factor signalling in breast cancer, functional proteomics, IFR-147, 59000Villeneuve-d’Ascq, France6 Institut Gustave-Roussy, département de radiothérapie, 39, rue Camille-Desmoulins, 94805 Villejuif cedex, France
Pour citer cet article : Chargari C, Moncharmont C, Lévy A, Guy JB, Bertrand G, Guilbert M, Rousseau C, Védrine L,Alphonse G, Toillon RA, Rodriguez-Lafrasse C, Deutsch É, Magné N. Facteurs de radiorésistance des cellules souchescancéreuses et perspectives de radiosensibilisation : l’exemple du glioblastome. Bull Cancer 2012 ; 99 : 1153-60.doi : 10.1684/bdc.2012.1666.
Résumé. Les cellules souches cancéreuses sontl’objet d’un intérêt croissant. En particulier, plu-sieurs données suggèrent leur implication dans lesmécanismes de radiorésistance tumorale, pouvantainsi expliquer les échappements thérapeutiques aprèsradiothérapie. De par son pronostic péjoratif et sontaux élevé de rechutes après irradiation, le glioblas-tome constitue un modèle très souvent étudié dansla recherche de nouveaux radiosensiblisants. Il existeplusieurs données précliniques suggérant que les cel-lules souches cancéreuses pourraient constituer unecible thérapeutique potentielle dans la perspectived’améliorer l’efficacité biologique de l’irradiation. Àtravers l’exemple du glioblastome, nous effectuons unerevue des principales voies de signalisation intervenantdans les mécanismes de radiorésistance des cellulessouches cancéreuses et pour lesquelles un ciblagepharmacologique laisse entrevoir des perspectives deradiosensibilisation. �
Abstract. Cancer stem cells are a subject ofincreasing interest in oncology. In particular, severaldata suggest that cancer stem cells are involved inthe mechanisms of tumor radioresistance, and mayexplain the therapeutic failures after radiotherapy.Because of its poor prognosis and high recurrencerate after irradiation, glioblastoma model is oftenstudied in the search for new radiosensitizers. Thereare several preclinical data suggesting that cancerstem cells could be a potential therapeutic targetfor improving the biological effectiveness of radia-tion therapy. Through the example of glioblastoma,we review the main signaling pathways involved inthe mechanisms of radiation resistance of cancerstem cells and for which pharmacological targetingcould potentially enhance tumor radiosensitivity. �
Mots clés : cellules souches cancéreuses, radiorésistance,hypoxie, radiothérapie
Key words: cancer stem cells, radioresistance, hypoxia,radiation therapy

1154 Bull Cancer vol. 99 • N◦ 12 • décembre 2012
C. Chargari, et al.
IntroductionPlusieurs travaux suggèrent l’existence de populationscellulaires possédant certaines propriétés des cellulessouches dans de nombreux types tumoraux et caracté-risées par leurs capacités [1] :– à initier un processus néoplasique ;– à générer sans limite des copies d’elles-mêmes parauto-renouvellement ;– à générer des cellules plus différenciées non auto-renouvelantes.En particulier, la présence en quantité variable decellules souches cancéreuses a été décrite dans lesmodèles de glioblastomes [2]. Bien que leur originesoit incertaine, plusieurs auteurs ont montré que lescellules souches cancéreuses pouvaient dériver de cel-lules normales dès lors que certains oncogènes étaientactivés ou par transition épithélio-mésenchymateuse[3-8].Bien qu’elles ne représentent qu’une fraction mino-ritaire des contingents cellulaires présents dans latumeur, les cellules souches cancéreuses jouent unrôle important dans l’initiation tumorale, la prolifé-ration ou le processus métastatique. Par ailleurs, ilexisterait un lien entre les échecs de la radiothéra-pie et la radiorésistance de cette population cellulaire.Les mécanismes sous-jacents sont nombreux et variés(redistribution dans le cycle, réparation de l’acidedésoxyribonucléique [ADN], mécanismes de protec-tion contre les radicaux libres, relation particulière avecle microenvironnement tumoral. . .) [9, 10]. Le ciblagedes cellules souches constitue donc une perspectivedans l’amélioration de l’efficacité de la radiothérapie.À travers l’exemple du glioblastome, tumeur carac-térisée par sa radiorésistance et son pronosticparticulièrement péjoratif, nous effectuons une revuedes perspectives des principales voies de signalisationpréférentiellement activées dans les cellules souches etdont le ciblage pharmacologique pourrait augmenter laradiosensibilité tumorale.
Rationnel au ciblagedes cellules souches
Impact de l’irradiation sur la populationdes cellules souchesLa glycoprotéine CD133 est exprimée dans plusieursmodèles de cellules souches normales, mais égale-ment de progéniteurs endothéliaux, de cellules souchesgliales ou de glioblastomes. Au sein d’une popula-tion tumorale de glioblastomes, certains sous-typescellulaires exprimant le marqueur CD133 présententun profil de radiosensibilité particulier, marqué parune sensibilité moindre à l’apoptose et aux lésionsde l’ADN radio-induites. L’importance relative decette fraction de cellules augmente après irradiation,
de même que leur capacité à l’auto-renouvellement[11]. Des résultats corroborant ont été rapportés dansd’autres modèles, en particulier de cancers mam-maires. Ainsi, la radiothérapie fractionnée augmentesous certaines conditions le pourcentage de cellulessouches observables in vitro [12]. Kim et al. ont mon-tré que la kinase LCK (lymphocyte-specific proteintyrosine kinase) était impliquée dans les phénomènesd’expansion du pool de cellules souches cancé-reuses après exposition à une radiothérapie fractionnéeet donc de radiorésistance secondaire. L’activité dece récepteur de la famille src était augmentée parl’irradiation. L’inhibition par transfection de siRNAde l’expression de LCK permettait de bloquer effi-cacement l’expansion de la population de cellulesCD133(+) et de diminuer l’expression de Notch-2 et Sox2, impliquées dans l’auto-renouvellementcellulaire [14].
Redistribution dans le cycleet repopulationPlusieurs données suggèrent que des mécanismesde redistribution dans le cycle et de repopulationcontribuent à la radiorésistance des cellules souchescancéreuses, celles-ci étant habituellement en phasequiescente du cycle, classiquement plus radiorésis-tante [14, 15]. L’impact de la repopulation et de laredistribution dans le cycle a été souligné par Pajonket al. [9]. Bien que l’existence de niches de cel-lules souches cancéreuses soit débattue, il semble queles cellules souches de gliomes siègent préférentiel-lement à proximité des vaisseaux. L’exposition à uneradiothérapie fractionnée de ces cellules quiescentesentraîne une repopulation accélérée avec augmenta-tion de la proportion des cellules en cycle, phaseplus radiosensible [16]. Cette observation suggère unintérêt potentiel à remobiliser les cellules souchescancéreuses pour les rendre plus radiosensibles. Àl’instar des cellules souches normales, les cellulessouches cancéreuses activent certaines voies de signa-lisation lors de leur passage d’une phase quiescente àune phase cyclique, dont certaines sont exploitablesà des fins de radiosensibilisation. Ainsi, l’activationde Notch-1 dans les cellules souches cancéreusescontribuerait à la repopulation tumorale et à la redistri-bution dans le cycle des cellules quiescentes. Outrece rôle, les ligands du récepteur transmembranaireNotch-1 sont impliqués dans le développement neuro-nal, les interactions cellules-cellules, la différenciationgliale, l’angiogenèse et l’expansion du pool de cellulessouches hématopoïétiques. L’activité de Notch-1 aug-mente également en réponse à l’irradiation. Le ciblagede Notch-1 par inhibition de la gamma-secrétase (uneprotéine membranaire impliquée dans son activation)exerce un effet antitumoral sur les lignées de médullo-blastome [17] associé à une réduction de la fraction des

Bull Cancer vol. 99 • N◦ 12 • décembre 2012 1155
Facteurs de radiorésistance des cellules souches cancéreuses
cellules CD133(+), à une augmentation de l’apoptoseet à une réduction de l’activité Akt et de l’expressionde Mcl-1. Dans l’expérience de Wang et al., la voieNotch semblait impliquée dans la radiorésistance descellules souches gliales [18] et l’utilisation d’inhibiteursde la gamma-secrétase permettait de radiosensibiliserles cellules gliales souches, mais pas les cellules glialesCD133(-) plus différenciées et non auto-renouvelantes.Alors que l’activation constitutive des domaines intra-cellulaires de Notch-1 et Notch-2 protégeait les cellulessouches de gliome de l’irradiation, l’inactivation de cescibles par transfection permettait de radiosensibiliserles cellules souches gliales et d’inhiber la tumorigenèsein vivo, suggérant que les cellules souches cancé-reuses sont sélectivement sensibles aux inhibiteurs deNotch. Une étude de phase II évalue actuellement leRO4929097, un inhibiteur de Notch chez des patientsprésentant un glioblastome récidivant ou progressif(http://clinicaltrials.gov : NCT01122901).
Mécanismes de réparation de l’acidedésoxyribonucléiqueL’efficacité biologique de la radiothérapie dépend desa capacité à produire des cassures double brins etdes dommages sublétaux cumulatifs. Cette dernièrecomposante des lésions radio-induites est l’objet d’unintérêt croissant dans la recherche d’un effet différen-tiel car les cellules normales les réparent mieux que lescellules tumorales [10]. Bao et al. ont montré que lescellules souches cancéreuses de gliomes présentaientune activation accrue des points de contrôle du cycleimpliqués dans la réparation des dommages de l’ADNaprès exposition aux rayonnements ionisants, et quecette activation était facteur de radiorésistance [12].Ainsi, les protéines kinases Chk1 et Chk2 impliquéesdans ces points de contrôle et activées après irradia-tion entraînent des arrêts dans le cycle et permettentl’initiation de la réparation. Leurs niveaux d’expressionbasal et radio-induit sont supérieurs dans les cellulespossédant des propriétés de cellules souches can-céreuses. Les caractéristiques de radiorésistance descellules souches sont reversées après traitement par desinhibiteurs pharmacologiques de Chk1 et/ou de Chk2[19]. En réalité, la différence entre cellules souchescancéreuses et cellules non progénitrices est quan-titative mais également qualitative. Ainsi, lorsque leniveau d’induction des dommages de l’ADN est simi-laire entre cellules CD133(+) et CD133(-), c’est leurefficacité à réparer les cassures de l’ADN et à entreren apoptose qui diffère. Les cellules souches semblentdonc « programmées » pour mieux se défendreface à l’exposition aux rayonnements ionisants ouà tout autre stress génotoxique, en témoigne leurniveau basal accru d’expression de certaines protéinesimpliquées dans les arrêts du cycle et la réparation[19, 20].
L’E3 ubiquitine ligase BIM-1 est surexprimée dans lescellules souches normales et tumorales [20-22]. Aprèsexposition aux rayonnements ionisants de cellules deglioblastomes, BIM-1 se concentre au niveau de lachromatine et se colocalise avec ataxia telangectasiamutated (ATM) et H2AX, suggérant un rôle importantdans la prise en charge précoce des cassures doublebrins de l’ADN. Outre ATM et l’histone H2AX, sesautres cofacteurs sont nombreux et incluent la DNA-dependent protein kinase (DNA-PK), poly-ADP-ribosepolymerase-1 (PARP-1), et histone H1. Facchino et al.ont montré que la déplétion en BIM-1 augmentait laradiosensibilité et le taux de cassures double brins. Aucontraire, sa surexpression était un facteur de radioré-sistance dans des cellules souches cancéreuses [22].L1 cell adhesion molecule (L1CAM) régule les points decontrôle du cycle cellulaire après dommages de l’ADN.Cette protéine transmembranaire d’adhésion impliquéedans la migration cellulaire joue également un rôledans la radiosensibilité des cellules souches de glio-blastomes. Ainsi, L1CAM régule l’expression de NBS1,un composant clé du complexe MRE11-RAD50-NBS1(complexe MRN) qui permet l’activation d’ATM aprèsexposition aux rayonnements ionisants. En particulier,la translocation nucléaire de L1-ICD (domaine intracel-lulaire de L1CAM) active NBS1 via le proto-oncogènec-MYC. Ainsi, L1CAM pourrait augmenter le niveaubasal d’activation des points de contrôle impliqués dansla réparation des lésions de l’ADN radio-induite en acti-vant la voie de signalisation via le complexe MRN [23].Enfin, Sarcar et al. ont montré que le MK-1775, inhi-biteur de Wee-1, permettait d’inhiber les arrêts enG2 du cycle cellulaire induits par la radiothérapie etd’augmenter la radiosensibilité de cellules de glioblas-tomes cultivées in vitro ou implantées chez l’animal.Wee-1 est l’inhibiteur de Cdc2, nécessaire au pas-sage cycline-dépendant des points de contrôle ducycle cellulaire, en particulier du point de restrictionG2/M qui empêche l’initiation de la mitose lorsqu’ilpersiste des lésions non réparées de l’ADN. Cepen-dant, cet effet du MK-1775 sur le cycle était plusmodéré et moins prolongé lors du traitement de cel-lules souches de glioblastomes, sans traduction sur laradiosensibilité [24].
Impact du microenvironnementLes différences qui existent entre le faible taux deréponse tumorale chez les patients et la radiosensi-bilité des cellules de glioblastome in vitro suggèreun impact du microenvironnement dans la radiorésis-tance de ces tumeurs. Lorsque des xénogreffes sontréalisées à partir de cellules souches de glioblastome,l’étude des cellules constituant les tumeurs montrequ’il coexiste des cellules CD133(+) et des cellulesplus différenciées CD133(-). Jamal et al. ont comparéla réponse à l’irradiation des cellules CD133(+) et

1156 Bull Cancer vol. 99 • N◦ 12 • décembre 2012
C. Chargari, et al.
CD133(-). In vitro, il n’était pas retrouvé de différencesignificative entre ces deux populations tumorales entermes d’induction ou d’élimination des foci H2AX.In vivo, les auteurs montraient que les xénogreffesissues des cellules CD133(+) présentaient moins defoci H2AX que les cellules CD133(-) et cette diffé-rence se traduisait par un retard de croissance de septjours et une augmentation de la proportion des cellulesCD133(+) au sein des xénogreffes issues de ces cel-lules CD133(+) [25]. Ces résultats montrent l’impactdu microenvironnement sur la réponse à l’irradiation,et tout particulièrement sur la radiosensibilité des cel-lules souches. Comme Rich et al. l’ont souligné [19],les cellules souches de glioblastome sont un acteur dela néoangiogenèse tumorale par une expression accruede vascular endothelial growth factor (VEGF) compa-rativement aux cellules souches non cancéreuses, etle bévacizumab exerce une activité anti-angiogéniquesur ces cellules souches de glioblastome, alorsqu’elle est marginale sur les cellules souches noncancéreuses [26].Plusieurs données indiquent que les cellules souchescancéreuses se situent préférentiellement à proximitédes vaisseaux, en zone oxygénée et donc théorique-ment radiosensible [27]. Pourtant, il semble qu’ellesprésentent moins de radicaux libres après exposi-tion à la radiothérapie que des cellules souchesnon tumorales. Le glutathion est un réducteur intra-cellulaire aux propriétés antioxydantes et protecteurvis-à-vis des radicaux libres de l’oxygène. Les cellulessouches cancéreuses surexpriment les gènes Gclm etGss codant respectivement pour la sous-unité régu-latrice de la glutamate-cystéine ligase et pour laglutathion-synthase. L’exposition à un inhibiteur de laglutamate-cystéine ligase permet de réduire la capacitédes cellules souches cancéreuses à former des colo-nies et de les radiosensibiliser. Ces résultats montrentl’implication de mécanismes de défense vis-à-vis desradicaux libres de l’oxygène dans la protection descellules souches vis-à-vis des rayonnements ionisants[28].Hypoxia-inducible factor (HIF)-1 est un régulateur cen-tral de l’hypoxie et un facteur de radiorésistance. Ilexiste une différence de transcription et d’expressiondes isoformes HIF-1� et HIF-2� entre les cellules tumo-rales souches et les cellules plus différenciées nonauto-renouvelantes [29]. Ainsi, HIF-2� est préférentiel-lement exprimé dans la population de cellules souchescancéreuses, alors que HIF-1� l’est dans les deux typesde populations cellulaires et se stabilise en situationd’hypoxie. L’inhibition des gènes de l’hypoxie montreque ces isoformes sont nécessaires à la survie des cel-lules souches et à la progression tumorale. McCordet al. ont par ailleurs montré que l’hypoxie activait la for-mation de tumorosphères et l’expression de marqueursde cellules souches à la surface des cellules tumorales[30].
Autres perspectivesde radiosensibilisationpharmacologiqueOutre les particularités liées à leurs protections vis-à-vis des lésions de l’ADN et les spécificités relatives àleur microenvironnement, les cellules souches cancé-reuses se distinguent par l’activation de nombreusesvoies de signalisation impliquées dans les mécanismesde leur radiorésistance. Ces voies incluent JAK/STAT,Notch, Wnt/�-caténine, Hedgehog, phosphatidylinosi-tol 3-kinase (PI3k)/Akt, insulin growth factor (IGF)-1 etla cycloxygénase 2 (Cox-2) [9, 31]. Ainsi, Rich etEyler ont étudié l’impact du ciblage de plusieurs voiesde signalisation sur la radiosensibilité des cellulessouche tumorales [32]. Ils ont utilisé des inhibiteursde L1CAM (CE171), de PI3k, du bone morphoge-netic protein (BMP), de Notch, de sonic Hedgehog,et du récepteur de l’epidermal growth factor (REGF).Le ciblage de chacune de ces cibles permettait deréduire la formation des gliomasphères contenant lescellules souches cancéreuses, ainsi que l’invasion,et l’initiation tumorale [33-37]. Nous présentonsci-dessous les principales cibles des stratégies deradiosensibilisation.
Cycloxygénase 2Surexprimée dans de nombreux types de tumeurs etassociée à un mauvais pronostic, y compris dans desmodèles de glioblastomes CD133(+), Cox-2 convertitl’acide arachidonique en prostaglandines impliquéesdans la néovascularisation tumorale, l’inflammation, lacarcinogenèse, l’inhibition de l’apoptose, et l’invasiontumorale. Ma et al. ont étudié le celecoxib, un inhi-biteur sélectif de Cox-2, en association à l’irradiation,dans des cellules de glioblastomes isolés par leur mar-quage CD133(+) en cytométrie de flux, et dont il étaitmontré qu’elles exprimaient Cox-2 à un niveau basalsupérieur aux cellules CD133(-). Il était mis en évi-dence un effet radiosensibilisant corrélé à l’inductiond’arrêts en G2/M. In vivo, la tumorigénicité des cellulesCD133(+) était inhibée par le celecoxib qui augmentaità la fois l’effet antitumoral de l’irradiation et la surviedes souris [38].
STAT3Le facteur de transcription STAT3 est anormalementactivé dans plusieurs modèles tumoraux. Son rôle estpeu connu et il existe des rapports contradictoiresquant à son caractère oncogénique ou suppresseurde tumeur, qui semble dépendre du profil muta-tionnel de la tumeur, y compris dans des modèlesde tumeurs gliales. STAT3 est cependant impliquédans les propriétés souches de certains types tumo-raux. Chang et al. ont montré que la cucurbitacinI, un inhibiteur de STAT3, permettait de réprimer

Bull Cancer vol. 99 • N◦ 12 • décembre 2012 1157
Facteurs de radiorésistance des cellules souches cancéreuses
le phénotype souches de cellules de médulloblas-tomes CD133(+). Par ailleurs, ce traitement permettaitd’augmenter la sensibilité à l’irradiation de ces celluleset d’inhiber la tumorigénicité des cellules CD133(+)lorsqu’elles étaient inoculées in vivo, cet effet étantsynergique avec l’irradiation [39]. Yang et al. ont mon-tré que l’inhibition de STAT3 par des shRNA-STAT3,ou l’exposition à l’AG490 ou au resveratrol inhibaitles propriétés souches et la radiorésistance des cellulesde glioblastomes CD133(+). Chez l’animal, l’inhibitionde STAT3 augmentait la réponse à la radiothérapie decellules souches de glioblastomes [40].
TGF-�TGF-�, classiquement considérée comme antiproli-férative et compétitive de la voie Notch, intervientdans différents processus biologiques, dont la différen-ciation endothéliale, la prolifération et la croissancecellulaire, le contrôle de l’apoptose, la transitionépithélio-mésenchymateuse et la synthèse de la matriceextracellulaire, avec des effets variables en fonctionsdes conditions de stress cellulaire. Il est activé parl’exposition aux rayonnements ionisants et constitue unimportant acteur de la réponse aux rayonnements ioni-sants, en particulier dans les mécanismes de la fibroseradio-induite. Zhang et al. ont montré que l’inhibitiondu récepteur tyrosine kinase de type 1 de TGF-� (TGF-�R1) par le LY2109761 permettait de réduire la clo-nogénicité et d’augmenter la sensibilité de lignées deglioblastomes et de cellules souches de gliomes à uneradiothérapie fractionnée, avec un effet supra additifin vivo. D’un point de vue fonctionnel, l’inhibitionde TGF-� R1 inhibait l’effet activateur de l’irradiationsur l’invasion tumorale, exercait un effet antivasculaireet diminuait la transition épithélio-mésenchymateuse[41].
c-METc-MET est le récepteur à tyrosine kinase de l’hepatocytegrowth factor (HGF), impliqué dans de nombreux voiesde signalisation impliquées dans la réponse à la radio-thérapie, dont la migration cellulaire radio-induite,la prolifération cellulaire, la néoangiogenèse tumo-rale et la réponse tumorale en situation d’hypoxie.C’est un facteur de mauvais pronostic, de radiorésis-tance et d’échappement métastatique. En particulier,c-MET est surexprimée dans les cellules souches can-céreuses de glioblastomes [42, 43]. Ellsworth et al.ont évalué le Mp470, un inhibiteur multicibles desrécepteurs à tyrosine kinase qui inhibe en parti-culier MET, en association à l’irradiation [44]. Lesauteurs montraient que l’association du Mp470 et dela radiothérapie était plus efficace que celle du témo-zolomide et de la radiothérapie pour inhiber la survieclonogénique. L’étude des mécanismes sous-jacentsmontrait que l’apoptose était la principale cause de
létalité cellulaire, et qu’elle était deux fois plus acti-vée par l’association Mp470-radiothérapie que parl’association témozolomide-radiothérapie.
L’autophagieCaractérisée par la formation d’autophagosomes,d’autolysosomes et le maintien de l’intégrité nucléaire,l’autophagie émerge comme un acteur important duprocessus de mort cellulaire radio-induite [45, 46].Zhuang et al. [47] ont souligné le rôle de l’autophagiedans les mécanismes de radiorésistance des cellulessouches cancéreuses. Ils montraient que la DNA-PK jouait un rôle important dans la réparation descassures double brins de l’ADN et que la transfec-tion de plasmides codant pour des shRNA ciblant laDNA-PK dans des cellules gliales initiatrices entraî-nait l’induction d’une autophagie massive, l’apoptosen’étant que peu modifiée. Lomonaco et al. ont exa-miné l’effet du cilengitide, un inhibiteur des intégrines,sur la radiosensibilité de cellules de gliomes et decellules souches de gliomes. Les auteurs montraientque le cilengitide permettait d’induire un détache-ment cellulaire, de réduire la viabilité cellulaire eninduisant le processus d’autophagie, et de réduire lacapacité d’auto-renouvellement des cellules souchesde gliomes. L’effet du cilengitide était contrecarré parl’utilisation d’inhibiteurs de l’autophagie. Un prétraite-ment par cilengitide, augmentait l’effet de l’irradiationsur l’autophagie. L’effet du cilengitide sur l’autophagieet sur la radiosensibilité tumorale était maintenu in vivo[48].
REGF/PI3k/AktAlors que le REGF est fréquemment surexprimé dansles glioblasomes, l’activation de la voie de signali-sation d’aval PI3k/Akt est un facteur de progressiondans le cycle (active la cycline D1), d’inhibition del’apoptose mitochondriale (lève le contrôle de BADsur l‘antiapoptotique bcl2) ou caspase-indépendante(inhibe FAS ligand) et d’inhibition de la croissance cel-lulaire (activation de mTOR). Kang et al. ont comparé laradiosensibilité de cellules souches cancéreuses céré-brales avec celle de cellules plus différenciées et nonauto-renouvelantes et étudié l’impact d’un traitementpar géfitinib sur leur radiosensibilité. L’irradiation seuleexercait plusieurs effets sur les cellules CD133(-) :inhibition de la survie clonogénique, arrêts en G2/M,augmentation de l’immunomarquage H2AX, mar-queurs de cassures double brins. Ces effets n’étaientpas reproduits dans les cellules souches organisées engliomasphères. Un co-traitement par géfitinib permet-tait d’augmenter sélectivement les effets de l’irradiationdans les cellules des gliomasphères en inhibant l’axeREGF-Akt et l’expression de la DNA-PK tout en aug-mentant la fréquence des cassures double brins del’ADN. Les cellules gliales plus différenciées n’étaient

1158 Bull Cancer vol. 99 • N◦ 12 • décembre 2012
C. Chargari, et al.
pas affectées. Cet effet différentiel était corrélé à uneinhibition de cibles spécifiques par le géfitinib dépen-dant du type de lignée étudié [49]. Hambardzumyanet al. ont montré que l’irradiation de cellules proliférantde médulloblastomes entraînait leur apoptose p53-dépendante. Cependant, les cellules souches exprimantla nestine et situées à proximité des vaisseaux sur-vivaient à l’irradiation, activaient la voie PI3k/Akt,et subissaient des arrêts dans le cycle dépendant dep53 avant de reprendre leur cycle 72 h plus tard. Cetteinduction de p53 était dépendante du gène suppres-seur de tumeur PTEN (phosphatase and tensin). Lesauteurs montraient que l’inhibition de Akt permettaitde sensibiliser les cellules des régions périvasculairesà l’apoptose radio-induite [50]. Li et al. ont infectédes cellules précurseurs neurales PTEN -/- avec unrétrovirus codant pour le récepteur constitutionnel-lement activé REGFvIII. Les cellules ainsi obtenuesformaient des tumeurs très mitotiques, avec régionsnécrotiques, présentant un important pléomorphismenucléaire et des marqueurs de glioblastome, une pro-lifération accrue, des amplifications du centrosome,ainsi que des caractéristiques de cellules souches : auto-renouvellement, expression du CD133 et résistancerelative vis-à-vis du stress oxydatif et des rayonnementsionisants. La transfection de REGFvIII activait la voie desMAPkinases et PI3k/Akt. Par ailleurs, le régulateur despoints de restriction Chk1 était phosphorylé par Akt,empêchant son activation en réponse à l’irradiation.Lorsque ces cellules étaient injectées chez l’animal, lesauteurs observaient une fréquence accrue d’anomalieschromosomiques [51].
Wnt/�-caténineLa voie Wnt/�-caténine est impliquée dans plusieursétapes de l’oncogenèse. Ainsi, des cellules épithélialesmammaires transformées pour présenter une activitéectopique de Wnt-1 ont une activité télomérase aug-mentée et des altérations fonctionnelles de p53 et pRb,poussent en sphères lorsqu’elles sont mises en suspen-sion et forment chez l’animal des tumeurs proches descarcinomes médullaires mammaires [52]. Wnt inter-vient dans le maintien de la pluripotence des cellulessouches embryonnaires, l’instabilité chromosomique,participe au contrôle du cycle cellulaire et active la sur-vivine en réponse aux signaux proapoptotiques [53].Il peut donc jouer un rôle important comme proto-oncogène. Wnt régule l’activité de la �-caténine, ellemême régule la croissance cellulaire et les adhésionsintercellulaires, en particulier via les jonctions adhé-rentes. La voie de signalisation Wnt/�-caténine estimpliquée dans la réponse aux rayonnements ioni-sants, car la signalisation de Wnt active la réponseaux dommages de l’ADN. Kendziorra et al. ont ainsimontré que certaines lignées de cancers rectaux résis-tants à la radiochimiothérapie surexprimaient TCF4, un
facteur de transcription régulé par Wnt. L’inhibitionde TCF4 arrêtait les cellules en G2/M et permettait deradiosensibiliser les cellules tumorales [54].
HedgehogLa voie de signalisation Hedgehog est impliquée dansla régulation de l’embryogenèse et la morphogenèse.Hedgehog et son effecteur d’aval Gli1 sont impliquésdans la régulation des cellules souches, en particu-lier hématopoïétiques, mammaires et neurales, maissont également des facteurs de tumorigénicité et derésistance à la radiothérapie ou à la chimiothérapielorsqu’ils sont anormalement activés [55, 56]. Bar et al.ont étudié l’impact du blocage de la voie Hedgehogsur les cellules souches de glioblastomes. Son inhi-bition par la cyclopamine réduisait de 40 à 60 % lacroissance des lignées de gliomes exprimant Gli1. Lasubstance était sans effet sur les lignées tumorales neprésentant pas d’activation de cette voie. Les auteursont ensuite traité des neurosphères dérivées de glioblas-tomes par la cyclopamine et les ont ensuite dissociéeset mises en culture dans un milieu sans inhibiteur. Iln’était pas observé de formation de nouvelle neuro-sphère, suggérant aux auteurs que la population descellules clonogéniques souches avait disparu. Aprèsirradiation des neurosphères de glioblastomes, le pour-centage de cellules souches augmentait, suggérant quela radiothérapie cible préférentiellement les cellulesbien différenciées. Lorsque les auteurs réalisaient desmodèles orthotopiques de glioblastomes chez sourisnude, les cellules pour lesquelles la voie Hedgehogavait été inhibée perdaient leur capacité à former destumeurs [33]. D’autres auteurs ont montré qu’il exis-tait dans les cellules de glioblastomes une coopérationentre la voie Hedgehog et la voie de l’IGF dans lemaintien d’un phénotype de type cellules souches.L’inhibition des voies Hedgehog et IGF permettaitd’inhiber la résistance intrinsèque et acquise des cel-lules souches de gliomes au témozolomide [57].
Hadronthérapie et irradiationdes niches de cellules souchesIl semble que les photons ne soient pas les parti-cules les plus efficaces pour éradiquer les cellulessouches cancéreuses. Ainsi, Chang et al. ont montréque des lignées de cancers bronchiques non à petitescellules chimiorésistantes et radiorésistantes présen-taient également le pourcentage plus élevé de celluless’approchant phénotypiquement des cellules souches.Lorsque les auteurs irradiaient les cellules souches can-céreuses par un rayonnement de photon ou de protonsd’efficacité biologique équivalente, ils observaient queles faisceaux de protons tuaient davantage de cellules,activaient davantage d’apoptose, et réduisaient mieuxle risque métastatique que les faisceaux de photons. Les

Bull Cancer vol. 99 • N◦ 12 • décembre 2012 1159
Facteurs de radiorésistance des cellules souches cancéreuses
protons permettaient également d’induire davantagede radicaux libres que les photons. Ces résultats invitro suggèrent un ciblage préférentiel des cellulessouches cancéreuses par les protons [58]. D’autresauteurs ont également rapporté des résultats encoura-geants sur des cellules souches cancéreuses de cancerscoliques avec une irradiation à base d’ions carbones,peut-être plus efficace qu’une irradiation à base dephotons en raison d’une efficacité biologique rela-tive supérieure [59]. Une autre stratégie séduisanteest le ciblage spécifique des régions périventriculairesoù se trouvent des niches de cellules souches. Ainsi,dans une cohorte rétrospective de 40 patients pris encharge pour un glioblastome, Gupta et al. ont rap-porté une corrélation entre la dose délivrée à la zonepériventriculaire homolatérale et la survie globale. Cesdonnées sont discutables car une dose supérieure pour-rait également signifier une meilleure couverture desvolumes cibles. Elles posent cependant la questiond’une irradiation spécifique de certaines régions céré-brales [60], rendue possible par le développement detechniques d’imagerie permettant d’identifier les zonesriches en cellules souches, par exemple, en utilisantle fait qu’elles présentent une activité réduite du 26Sprotéasome [61].
ConclusionLes cellules souches cancéreuses constituent un axesérieux de recherche en oncologie-radiothérapie. Plu-sieurs voies de signalisation anormalement activéescontribuent à leurs caractéristiques phénotypiques,représentant autant de cibles potentielles, en particulierdans des modèles de tumeurs gliales. Si la littérature estriche en études précliniques suggérant le bénéfice d’unciblage des cellules souches cancéreuses en associa-tion à l’irradiation, il n’existe à ce jour pas de donnéeclinique apportant la preuve de l’intérêt de ce conceptchez les patients. L’autre piste de recherche repose surl’escalade de dose ciblée sur les zones riches en cel-lules souches cancéreuses. Bien que séduisant, cettestratégie n’a pas non plus été validée chez les patients.L’avenir est peut-être à l’association de ces deuxstratégies. �Conflits d’intérêts : aucun.
Références1. Schatton T, Frank NY, Frank MH. Identification and targeting ofcancer stem cells. Bioessays 2009 ; 10 : 1038-49.2. Hill RP, Milas L. The proportion of stem cells in murine tumours.Int J Radiat Oncol Biol Phys 1989 ; 16 : 513-8.3. Barker N, Ridgway RA, van Es JH, et al. Crypts stem cells as thecells-of-origin of intestinal cancer. Nature 2009 ; 457 : 608-11.4. Zhu L, Gibson P, Currle DS, et al. Prominin 1 marks intestinalstem cells that are susceptible to neoplastic transformation. Nature2009 ; 45 : 603-7.
5. Alcantara Llaguno S, Chen J, Kwon CH, et al. Malignant astrocy-tomas originate from neural stem/progenitor cells in a somatic tumorsuppressor mouse model. Cancer Cell 2009 ; 15 : 45-56.6. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymaltransition generates cells with properties of stem cells. Cell2008 ; 13 : 704-15.7. Polyak K, Weinberg RA. Transitions between epithelial and mesen-chymal states: acquisition of malignant and stem cell traits. Nat RevCancer 2009 ; 9 : 265-73.8. Chaffer CL, Brueckmann I, Scheel C, et al. Normal and neoplasticnonstem cells can spontaneously convert to a stem-like state. ProcNatl Acad Sci U S A 2011 ; 108 : 7950-5.9. Pajonk F, Vlashi E, McBride WH. Radiation resistance of can-cer stem cells: the 4 R’s of radiobiology revisited. Stem Cells2010 ; 28 : 639-48.10. Mils L, Hittelman WN. Cancer stem cells and tumour response totherapy: current problems and future prospects. Semin Radiat Oncol2009 ; 19 : 96-105.11. Baumann M, Krause M, Hill R. Exploring the role of cancer stemcells in radioresistance. Nat Rev Cancer 2008 ; 8 : 545-54.12. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promoteradioresistance by preferential activation of the DNA damage res-ponse. Nature 2006 ; 444 : 756-60.13. Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl CancerInst 2006 ; 98 : 1777-85.14. Kim RK, Yoon CH, Hyun KH, et al. Role of lymphocyte-specificprotein tyrosine kinase (LCK) in the expansion of glioma-initiatingcells by fractionated radiation. Biochem Biophys Res Commun2010 ; 402 : 631-6.15. Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensiti-vity to radiotherapy. Int J Radiat Oncol Biol Phys 2004 ; 59 : 928-42.16. Wilson A, Laurenti E, Oser G, et al. Hematopoietic stem cellsreversibly switch from dormancy to self-renewal during homeostasisand repair. Cell 2008 ; 135 : 1118-29.17. Fan X, Matsui W, Khaki L, et al. Notch pathway inhibitiondepletes stem-like cells and blocks engraftment in embryonal braintumors. Cancer Res 2006 ; 66 : 7445-52.18. Wang J, Wakeman TP, Lathia JD, et al. Notch promotes radiore-sistance of glioma stem cells. Stem Cells 2010 ; 28 : 17-28.19. Rich JN. Cancer stem cells in radiation resistance. Cancer Res2007 ; 67 : 8980-4.20. Cheng L, Bao S, Rich JN. Potential therapeutic implica-tions of cancer stem cells in glioblastoma. Biochem Pharmacol2010 ; 80 : 654-65.21. Cao R, Tsukada Y, Zhang Y. Role of BMI-1 and Ring1A in H2Aubiquitylation and Hox gene silencing. Mol Cell 2005 ; 20 : 845-54.22. Facchino S, Abdouh M, Chatoo W, et al. BMI confers radio-resistance to normal and cancerous neural stem cells throughrecruitment of the DNA damage response machinery. J Neurosci2010 ; 30 : 10096-111.23. Cheng L, Wu Q, Huang Z, et al. L1CAM regulates DNA damagecheckpoint response of glioblastoma stem cells through NBS1. EMBOJ 2011 ; 30 : 800-13.24. Sarcar B, Kahali S, Prabhu AH, et al. Targeting radiation-inducedG(2) checkpoint activation with the Wee-1 inhibitor MK-1775 inglioblastoma cell lines. Mol Cancer Ther 2011 ; 10 : 2405-14.25. Jamal M, Rath BH, Tsang PS, et al. The brain microenvironmentpreferentially enhances the radioresistance of CD133(+) glioblastomastem-like cells. Neoplasia 2012 ; 14 : 150-8.26. Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cellspromote tumor angiogenesis through vascular endothelial growthfactor. Cancer Res 2006 ; 66 : 7843-8.27. Calabrese C, Poppleton H, Kocak M, et al. A perivascular nichefor brain tumor stem cells. Cancer Cell 2007 ; 11 : 69-82.28. Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxy-gen species levels and radioresistance in cancer stem cells. Nature2009 ; 458 : 780-3.29. Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumo-rigenic capacity of glioma stem cells. Cancer Cell 2009 ; 15 : 501-13.30. McCord AM, Jamal M, Shankavarum UT, et al. Physiologic oxy-gen concentration enhances the stem-like properties of CD133+human glioblastoma cells in vitro. Mol Cancer Res 2009 ; 7 : 489-97.

1160 Bull Cancer vol. 99 • N◦ 12 • décembre 2012
C. Chargari, et al.
31. Moncharmont C, Levy A, Gilormini M, et al. Targeting a cor-nerstone of radiation resistance: cancer stem cell. Cancer Lett2012 ; 322 : 139-47.32. Rich JN, Eyler CE. Cancer stem cells in brain tumor biology. ColdSpring Harb Symp Quant Biol 2008 ; 73 : 411-20.33. Bar EE, Chaudhry A, Lin A, et al. Cyclopamine-mediatedhedgehog pathway inhibition depletes stem-like cancer cells in glio-blastoma. Stem Cells 2007 ; 25 : 2524-33.34. Clement V, Sanchez P, de Tribolet N, et al. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stemcell self-renewal, and tumorigenicity. Curr Biol 2007 ; 17 :165-72.35. Lee J, Son MJ, Woolard K, et al. Epigenetic-mediated dysfunctionof the bone morphogenetic protein pathway inhibits differentiationof glioblastoma-iniating cells. Cancer Cell 2008 ; 13 : 69-80.36. Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogeneticproteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006 ; 444 : 761-5.37. Soeda A, Inagaki A, Oka N, et al. Epidermal growth factor plays acrucial role in mitogenic regulation of human brain tumor stem cells.J Biol Chem 2008 ; 283 : 10958-66.38. Ma HI, Chiou SH, Hueng DY, et al. Celecoxib and radioresistantglioblastoma-derived CD133+ cells: improvement in radiothera-peutic effects. Laboratory investigation. J Neurosurg 2011 ; 114 :651-62.39. Chang CJ, Chiang CH, Song WS, et al. Inhibition of phos-phorylated STAT3 by cucurbitacin I enhances chemoradiosensitivityin medulloblastoma-derived cancer stem cells. Childs Nerv Syst2012 ; 28 : 363-73.40. Yang YP, Chang YL, Huang PI, et al. Resveratrol suppressestumorigenicity and enhances radiosensitivity in primary glioblastomatumor initiating cells by inhibiting the STAT3 axis. J Cell Physiol2012 ; 227 : 976-93.41. Zhang M, Kleber S, Röhrich M, et al. Blockade of TGF-�signaling by the TGF�R-I kinase inhibitor LY2109761 enhances radia-tion response and prolongs survival in glioblastoma. Cancer Res2011 ; 71 : 7155-67.42. Günther HS, Schmidt NO, Phillips HS, et al. Glioblastoma-derived stem cell-enriched cultures from distinct subgroupsaccording to molecular and phenotypic criteria. Oncogene2008 ; 27 : 2897-9.43. Joo KM, Jin J, Kim E, et al. MET signaling regulates glioblastomastem cells. Cancer Res 2012 ; 72 : 3828-38.44. Ellsworth R, Welsh J, Mahadevan D, et al. The radiosensitizingeffects of a novel tyrosine kinase inhibitor, Mp470 in glioblas-toma multiforme stem cells. Int J Radiat Oncol Biol Phys 2008 ; 72 :714-5.45. Paglin S, Hollister T, Delohery T, et al. A novel response of cancercells to radiation involves autophagy and formation of acidic vesicles.Cancer Res 2001 ; 61 : 439-44.
46. Abedin MJ, Wang D, McDonnell MA, et al. Autophagy delaysapoptotic death in breast cancer cells following DNA damage. CellDeath Differ 2007 ; 14 : 500-10.47. Zhuang W, Li B, Long L, et al. Knockdown of the DNA-dependentprotein kinase catalytic subunit radiosensitizes glioma-initiating cellsby inducing autophagy. Brain Res 2011 : 13717-815.48. Lomonaco SL, Finniss S, Xiang C, et al. Cilengitide inducesautophagy-mediated cell death in glioma cells. Neuro Oncol2011 ; 13 : 857-65.49. Kang KB, Zhu C, Wong YL, et al. Gefitinib radiosensitizes stem-like glioma cells: inhibition of epidermal growth factor receptor-Akt-DNA-PK signaling, accompanied by inhibition of DNA double-strandbreak repair. Int J Radiat Oncol Biol Phys 2012 ; 83 : 43-52.50. Hambardzumyan D, Becher OJ, Rosenblum MK, et al. PI3K path-way regulates survival of cancer stem cells residing in the perivascularniche following radiation in medulloblastoma in vivo. Genes Dev2008 ; 22 : 436-48.51. Li L, Dutra A, Pak E, et al. EGFRvIII expression and PTENloss synergistically induce chromosomal instability and glial tumors.Neuro Oncol 2009 ; 11 : 9-21.52. Ayyanan A, Civenni G, Ciarloni L, et al. Increased Wnt signa-ling triggers oncogenic conversion of human breast epithelial cellsby a Notch-dependent mechanism. Proc Natl Acad Sci U S A2006 ; 103 : 3799-804.53. Polakis P. Wnt signaling and cancer. Genes Dev 2000 ; 14 : 1837-51.54. Kendziorra E, Ahlborn K, Spitzner M, et al. Silencing of the Wnttranscription factor TCF4 sensitizes colorectal cancer cells to (chemo-) radiotherapy. Carcinogenesis 2011 ; 32 : 1824-31.55. Huang FT, Zhuan-Sun YX, Zhuang YY, et al. Inhibition of hedge-hog signaling depresses self-renewal of pancreatic cancer stem cellsand reverses chemoresistance. Int J Oncol 2012 ; 4 : 1707-14.56. Santini R, Vinci MC, Pandolfi S, et al. HEDGEHOG-GLI signalingdrives self-renewal and tumorigenicity of human melanoma-initiatingcells. Stem Cells 2012 ; 30 : 1808-18.57. Hsieh A, Ellsworth R, Hsieh D. Hedgehog/GLI1 regulates IGFdependent malignant behaviors in glioma stem cells. J Cell Physiol2011 ; 226 : 1118-27.58. Chang JY, Zhang X, Vassiliev O, et al. Proton therapy targetscancer stem cells in treatment-resistant non-small cell lung cancer.Int J Radiat Oncol Biol Phys 2010 ; 78 : 644.59. Cui X, Oonishi K, Tsujii H, et al. Effects of carbon ion beamon putative colon cancer stem cells and its comparison with X-rays.Cancer Res 2011 ; 71 : 3676-87.60. Gupta T, Nair V, Paul SN, et al. Can irradiation of potentialcancer stem-cell niche in the subventricular zone influence survi-val in patients with newly diagnosed glioblastoma? J Neurooncol2012 ; 109 : 195-203.61. Vlashi E, Kim K, Dealla DL, et al. In-vivo imaging, tracking, andtargeting of cancer stem cells. J Natl Cancer Inst 2009 ; 101 : 350-9.

Caractérisation et ciblage thérapeutique d’une sous-population de cellules souches cancéreuses dans un modèle cellulaire de carcinome épidermoïde de la tête et du cou
résistant à l’irradiation par photons et ions carbone
Les carcinomes épidermoïdes de la tête et du cou sont souvent de mauvais pronostic, en raison de leur résistance aux traitements suivie de récidives loco-régionales, voire de métastases. Ce travail s’est focalisé sur le rôle des cellules souches cancéreuses (CSC) dans la radiorésistance d’un modèle cellulaire de cancer du larynx, SQ20B, ainsi que sur leur ciblage thérapeutique en association avec la radiothérapie photonique ou par ions carbone.
Une sous-population a été isolée à partir de la lignée SQ20B par tris cellulaires successifs selon 3 critères spécifiques des CSC de tumeurs ORL : exclusion du Hoechst 33342, expression de CD44 et activité élevée de l’aldéhyde déshydrogénase (ALDH). Les cellules SQ20B/SP/CD44high/ALDHhigh présentent bien les caractéristiques de CSC (tumorisphères, tumorigénèse, radiorésistance).
La résistance des CSC aux 2 types d’irradiation, par rapport aux cellules « non souche » SQ20B/SP/CD44low/ALDHlow, implique une diminution de la mort par apoptose, une augmentation des capacités prolifératives ainsi qu’une surexpression de la voie de l’autorenouvellement Bmi1.
L’effet radiosensibilisant de 3 molécules ciblant les CSC a été démontré : la mort apoptotique induite par l’UCN-01 en inhibant l’arrêt en phase G2/M ; les capacités prolifératives ciblées par l’acide trans-rétinoïque (ATRA) induisant la différenciation ; et la voie de l’autorenouvellement Bmi-1 inhibée par l’artésunate. Seules ou associées (UCN-01 + ATRA), elles agissent en synergie avec une irradiation par photons ou ions carbone. Des études pré-clinique, puis clinique, devraient confirmer l’intérêt du ciblage des CSC dans le contrôle de l’échappement de ces cancers radiorésistants.
Mots clés : cellules souches cancéreuses, irradiation photonique, irradiation par ions carbone,
radiosensibilisation, tumeurs des voies aéro-digestives supérieures.
Characterization and therapeutic targeting of a cancer stem cell subpopulation in a head and
neck squamous cell carcinoma resistant to photon and carbon ion irradiation.
Head and neck squamous cell carcinomas (HNSCC) have a poor prognosis, due to their resistance to standard treatments. In most cases, locoregional recurrence or metastases occur. This study has focused on the role of cancer stem cells (CSC) in the radioresistance of the SQ20B HNSCC cell line and their therapeutic targeting in association with photon or carbon ions irradiation.
A subpopulation of SQ20B-CSC has been isolated by cell sorting based on 3 specific characteristics of HNSCC-CSC: Hoechst 33342 exclusion, CD44 expression and high aldehyde dehydrogenase activity (ALDH). SQ20B/SP/CD44high/ALDHhigh cells show the CSC characteristics (in vitro and in vivo tumorigenesis, high radioresistance).
The response of CSC to both types of irradiation was compared to the non-“stem cells” SQ20B/SP/CD44low/ALDHlow sub-population. The observed radioresistance involves a decrease in apoptotic cell death, an increase in proliferative capacities and an overexpression of the Bmi1 self-renewing signaling pathway.
The radiosensitizing effects of 3 molecules targeting the CSC has been demonstrated: an induction of apoptotic cell death by the inhibition of the G2/M phase arrest after a treatment with UCN01; an inhibition of proliferative capacities using the all-trans-retinoic acid (ATRA) which induce their differentiation; and an inhibition of Bmi1 by artesunate. These treatments, alone or in combination (UCN01+ATRA), have a synergistic effect with photon or carbon ion irradiation to overcome CSC radioresistance. Preclinical and clinical studies should confirm the benefit of targeting CSC and improve the control of tumor escape in patients with radiorésistant HNSCC cancers.




















