
Canonical Transient Receptor Potential 3 Channel Triggers Vascular Endothelial Growth Factor-Induced...
20
ORIGINAL RESEARCH REPORTS Canonical Transient Receptor Potential 3 Channel Triggers Vascular Endothelial Growth Factor-Induced Intracellular Ca 2 + Oscillations in Endothelial Progenitor Cells Isolated from Umbilical Cord Blood Silvia Dragoni, 1 Umberto Laforenza, 2 Elisa Bonetti, 3 Francesco Lodola, 1 Cinzia Bottino, 2 Germano Guerra, 4 Alessandro Borghesi, 5 Mauro Stronati, 6 Vittorio Rosti, 3 Franco Tanzi, 1 and Francesco Moccia 1 Endothelial colony-forming cells (ECFCs) are the only endothelial progenitor cells (EPCs) that are capable of acquiring a mature endothelial phenotype. ECFCs are mainly mobilized from bone marrow to promote vas- cularization and represent a promising tool for cell-based therapy of severe ischemic diseases. Vascular endo- thelial growth factor (VEGF) stimulates the proliferation of peripheral blood-derived ECFCs (PB-ECFCs) through oscillations in intracellular Ca 2 + concentration ([Ca 2 + ] i ). VEGF-induced Ca 2 + spikes are driven by the interplay between inositol-1,4,5-trisphosphate (InsP 3 )-dependent Ca 2 + release and store-operated Ca 2 + entry (SOCE). The therapeutic potential of umbilical cord blood-derived ECFCs (UCB-ECFCs) has also been shown in recent studies. However, VEGF-induced proliferation of UCB-ECFCs is faster compared with their peripheral counterpart. Unlike PB-ECFCs, UCB-ECFCs express canonical transient receptor potential channel 3 (TRPC3) that mediates diacylglycerol-dependent Ca 2 + entry. The present study aimed at investigating whether the higher proliferative potential of UCB-ECFCs was associated to any difference in the molecular underpinnings of their Ca 2 + response to VEGF. We found that VEGF induces oscillations in [Ca 2 + ] i that are patterned by the interaction between InsP 3 -dependent Ca 2 + release and SOCE. Unlike PB-ECFCs, VEGF-evoked Ca 2 + oscillations do not arise in the absence of extracellular Ca 2 + entry and after pharmacological (with Pyr3 and flufenamic acid) and genetic (by employing selective small interference RNA) suppression of TRPC3. VEGF-induced UCB-ECFC proliferation is abrogated on inhibition of the intracellular Ca 2 + spikes. Therefore, the Ca 2 + response to VEGF in UCB-ECFCs is shaped by a different Ca 2 + machinery as compared with PB-ECFCs, and TRPC3 stands out as a promising target in EPC-based treatment of ischemic pathologies. Introduction E ndothelial progenitor cells (EPCs) are mobilized from bone marrow (BM) in response to an ischemic insult to recapitulate the injured vascular network and restore blood perfusion [1–6]. Moreover, circulating EPCs replace either injured or senescent endothelial cells that are detached from the vascular wall into peripheral blood (PB), thereby pre- venting the establishment of conditions which are prone to atherosclerosis and other vascular pathologies [6–8]. As a consequence, the transplantation of autologous EPCs holds major promise as an alternative tool in the stem cell therapy of peripheral artery disease (PAD) and myocardial infarction (MI) [1–3,9–12]. The term EPC fails to refer to a unique pop- ulation of BM-derived mononuclear cells (MNCs) due to the absence of a cell-specific panel of surface antigens or gene expression profiles [3,6,13]. The so-called endothelial colony- forming cells (ECFCs), which may be mobilized from both BM and arterial vessel walls, stand amid the different subsets of EPCs described in the literature, for the following reasons: (1) They truly belong to an endothelial lineage and display a clonal proliferative potential (ie, may undergo approximately 30 population doublings without senescence and replate into secondary and tertiary colonies); (2) they possess the rare ability to form bidimensional tubular networks in vitro; and (3) they may form patent vessels that anastomize with the 1 Department of Biology and Biotechnology ‘‘Lazzaro Spallanzani,’’ 2 Department of Molecular Medicine, University of Pavia, Pavia, Italy. 3 Laboratory of Biotechnology, Center for the Study of Myelofibrosis, Foundation IRCCS Policlinico San Matteo, Pavia, Italy. 4 Department of Health Sciences, University of Molise, Campobasso, Italy. 5 Neonatal Intensive Care, Foundation IRCCS Policlinico San Matteo, Pavia, Italy. 6 Laboratory of Neonatal Immunology, Foundation IRCCS Policlinico San Matteo, Pavia, Italy. STEM CELLS AND DEVELOPMENT Volume 22, Number 19, 2013 ȑ Mary Ann Liebert, Inc. DOI: 10.1089/scd.2013.0032 2561
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Canonical Transient Receptor Potential 3 Channel Triggers Vascular Endothelial Growth Factor-Induced...

ORIGINAL RESEARCH REPORTS
Canonical Transient Receptor Potential 3 Channel TriggersVascular Endothelial Growth Factor-Induced Intracellular
Ca2 + Oscillations in Endothelial Progenitor CellsIsolated from Umbilical Cord Blood
Silvia Dragoni,1 Umberto Laforenza,2 Elisa Bonetti,3 Francesco Lodola,1 Cinzia Bottino,2 Germano Guerra,4
Alessandro Borghesi,5 Mauro Stronati,6 Vittorio Rosti,3 Franco Tanzi,1 and Francesco Moccia1
Endothelial colony-forming cells (ECFCs) are the only endothelial progenitor cells (EPCs) that are capable ofacquiring a mature endothelial phenotype. ECFCs are mainly mobilized from bone marrow to promote vas-cularization and represent a promising tool for cell-based therapy of severe ischemic diseases. Vascular endo-thelial growth factor (VEGF) stimulates the proliferation of peripheral blood-derived ECFCs (PB-ECFCs)through oscillations in intracellular Ca2 + concentration ([Ca2 + ]i). VEGF-induced Ca2 + spikes are driven by theinterplay between inositol-1,4,5-trisphosphate (InsP3)-dependent Ca2 + release and store-operated Ca2 + entry(SOCE). The therapeutic potential of umbilical cord blood-derived ECFCs (UCB-ECFCs) has also been shown inrecent studies. However, VEGF-induced proliferation of UCB-ECFCs is faster compared with their peripheralcounterpart. Unlike PB-ECFCs, UCB-ECFCs express canonical transient receptor potential channel 3 (TRPC3)that mediates diacylglycerol-dependent Ca2 + entry. The present study aimed at investigating whether the higherproliferative potential of UCB-ECFCs was associated to any difference in the molecular underpinnings of theirCa2 + response to VEGF. We found that VEGF induces oscillations in [Ca2 + ]i that are patterned by the interactionbetween InsP3-dependent Ca2 + release and SOCE. Unlike PB-ECFCs, VEGF-evoked Ca2 + oscillations do notarise in the absence of extracellular Ca2 + entry and after pharmacological (with Pyr3 and flufenamic acid) andgenetic (by employing selective small interference RNA) suppression of TRPC3. VEGF-induced UCB-ECFCproliferation is abrogated on inhibition of the intracellular Ca2 + spikes. Therefore, the Ca2 + response to VEGF inUCB-ECFCs is shaped by a different Ca2 + machinery as compared with PB-ECFCs, and TRPC3 stands out as apromising target in EPC-based treatment of ischemic pathologies.
Introduction
Endothelial progenitor cells (EPCs) are mobilizedfrom bone marrow (BM) in response to an ischemic insult
to recapitulate the injured vascular network and restore bloodperfusion [1–6]. Moreover, circulating EPCs replace eitherinjured or senescent endothelial cells that are detached fromthe vascular wall into peripheral blood (PB), thereby pre-venting the establishment of conditions which are prone toatherosclerosis and other vascular pathologies [6–8]. As aconsequence, the transplantation of autologous EPCs holdsmajor promise as an alternative tool in the stem cell therapy ofperipheral artery disease (PAD) and myocardial infarction
(MI) [1–3,9–12]. The term EPC fails to refer to a unique pop-ulation of BM-derived mononuclear cells (MNCs) due to theabsence of a cell-specific panel of surface antigens or geneexpression profiles [3,6,13]. The so-called endothelial colony-forming cells (ECFCs), which may be mobilized from both BMand arterial vessel walls, stand amid the different subsets ofEPCs described in the literature, for the following reasons: (1)They truly belong to an endothelial lineage and display aclonal proliferative potential (ie, may undergo approximately30 population doublings without senescence and replate intosecondary and tertiary colonies); (2) they possess the rareability to form bidimensional tubular networks in vitro; and(3) they may form patent vessels that anastomize with the
1Department of Biology and Biotechnology ‘‘Lazzaro Spallanzani,’’ 2Department of Molecular Medicine, University of Pavia, Pavia, Italy.3Laboratory of Biotechnology, Center for the Study of Myelofibrosis, Foundation IRCCS Policlinico San Matteo, Pavia, Italy.4Department of Health Sciences, University of Molise, Campobasso, Italy.5Neonatal Intensive Care, Foundation IRCCS Policlinico San Matteo, Pavia, Italy.6Laboratory of Neonatal Immunology, Foundation IRCCS Policlinico San Matteo, Pavia, Italy.
STEM CELLS AND DEVELOPMENT
Volume 22, Number 19, 2013
� Mary Ann Liebert, Inc.
DOI: 10.1089/scd.2013.0032
2561

systemic vasculature of the host when implanted into im-munodeficient mice [1–3,14–17]. Unfortunately, the frequencyof circulating ECFCs in PB is scarce, that is, about 0.05–0.2cells/mL [14], a feature that does not render them amenablefor cell therapy purposes. Indeed, the estimated volume ofblood required to achieve therapeutically relevant numbers ofcells is equal to *12 L [9]. An additional disadvantage is thedecreased number and impaired functional capability of adultstem cells in a patient population of advanced age burdenedwith significant comorbidity [3,9,18]. It has, however, beendemonstrated that ECFCs are far more abundant in humanumbilical cord blood (UCB), where they attain a concentrationof about 2–5 cells/mL [14]. In addition, UCB-derived ECFCs(UCB-ECFCs) exhibit a much higher proliferation potential(approximately 100 population doublings) and a greater tel-omerase activity than their peripheral counterparts [14].When considering that UCB-ECFCs give rise to functionalcapillary networks in vivo [16,17], they represent a more at-tractive solution for regenerative medicine. The mechanisticdifferences between UCB- and PB-derived ECFCs have neverbeen elucidated. Vascular endothelial growth factor (VEGF) isthe most potent pro-angiogenic cytokine driving ECFC pro-liferation, recruitment to target organs, and physical assemblyinto perfused vascular networks. Assessing whether VEGFstimulation activates distinct signal transduction pathways inthe two cell types might outline novel molecular targets todevelop more efficacious EPCs-based strategies.
An increase in intracellular Ca2 + concentration ([Ca2 + ]i)represents a key signal in the activation of mature endothe-lium [19–21], as well as of human ECFCs [1–3]. A growingbody of evidence demonstrated that store-operated Ca2 +
entry (SOCE) controls ECFC expansion in vitro [22–24]. Thephysiological stimulus for the induction of SOCE in PB-ECFCs is provided by VEGF [24,25], which binds to itscognate tyrosine kinase receptor, VEGFR-2 (KDR/Flk1), tophosphorylate and engage phospholipase C-g (PLCg) [3,26].PLCg, in turn, cleaves the membrane-bound phosphatidyli-nositol-4,5-bisphosphate (PIP2) to generate the intracellularsecond messengers, inositol-1,4,5-trisphosphate (InsP3) anddiacylglycerol (DAG). Then, InsP3 diffuses throughout thecytoplasm, where it acts on specific InsP3 receptors (InsP3R)to release Ca2 + from the endoplasmic reticulum (ER) reser-voir [3,26]. The following drop in ER Ca2 + content is de-tected by an intralumenal Ca2 + sensor, termed StromalInteraction Molecule-1 (Stim1), which communicates theamount of stored Ca2 + to store-operated Ca2 + channels onthe plasma membrane (PM) [3,21,27,28]. Stim1 is stimulatedby the InsP3-dependent Ca2 + discharge to associate intomultimers that rapidly redistribute to peripheral ER sites inclose proximity to PM. Here, Stim1 clusters aggregate intomultiple puncta, interact with and gate Orai1 and canonicaltransient receptor potential 1 (TRPC1) channel, the Ca2 + -permeable routes that conduct Ca2 + into human ECFCs[3,22,24,25,27–29]. The interplay between InsP3-inducedCa2 + release and SOCE leads to asynchronous oscillations in[Ca2 + ]i, which encode the information necessary to triggerthe program of gene expression driving PB-ECFC prolifera-tion and tubulogenesis [2,3,24,25,30,31]. Therefore, Ca2 +
entry is not required to initiate the spiking response to VEGFin PB-ECFCs, but is essential to maintain Ca2 + oscillationsover time by replenishing the InsP3-sensitive Ca2 + pool [25].In addition to SOCE, mature endothelial cells are endowed
with alternative pathways for Ca2 + influx that are activatedby second messengers, such as DAG [20,21]. For instance,TRPC3 and TRPC6 support the pro-angiogenic Ca2 + signaltriggered by VEGF in human microvascular endothelial cells[32,33], as well as TRPC6 drives VEGF-induced proliferationand tube formation in human umbilical vein endothelial cells(HUVECs) [34]. Surprisingly, PB-ECFCs lack DAG-operatedCa2 + channels, whereas UCB-express TRPC3 [23]. TRPC3-mediated Ca2 + entry has recently been associated to the se-lective enlisting of a number of Ca2 + -sensitive transcriptionfactors, including nuclear factor of activated T-cells (NFAT)[35,36] and nuclear factor kappaB (NF-kB) [37], as well as tothe sustained activation of extracellular signal-regulated ki-nase (ERK), which may also impact gene expression [38]. Therole of TRPC3 in the pro-angiogenic action of VEGF on UCB-ECFCs is, however, yet to be elucidated.
The present investigation aimed at elucidating TRPC3involvement in the mechanistic differences of VEGF signal-ing in human PB- and UCB-ECFC by combining Ca2 + im-aging measurements with a gene silencing approach. Wefound that, similar to PB-ECFCs, VEGF triggers repeated[Ca2 + ]i transients that were not synchronous between adja-cent cells. However, the oscillatory mechanism was strik-ingly different as, in UCB-ECFCs, the interplay betweenInsP3-dependent Ca2 + release and SOCE was triggered byTRPC3-mediated Ca2 + inflow. In addition, TRPC3 openingwas necessary throughout the duration of the stimulation tomaintain the spiking signal. Consistent with these data, thegenetic suppression of TRPC3 prevented VEGF-inducedCa2 + oscillations, as well as the entry of Ca2 + induced by 1-oleoyl-2-acetyl-sn-glycerol (OAG), a membrane-permeantanalog of DAG. Therefore, the selective recruitment ofTRPC3 by VEGF in UCB-ECFCs, but not in PB-ECFCs, rep-resents the first molecular difference ever described betweenthe two cell types and might open a new avenue in the de-sign of alternative strategies for EPCs-based therapies.
Materials and Methods
Isolation and cultivation of ECFCs
Blood samples (40 mL) were obtained from healthy humanvolunteers aged from 20 to 48 years old (n = 12). The In-stitutional Review Board at ‘‘Istituto di Ricovero e Cura aCarattere Scientifico Policlinico San Matteo Foundation’’ inPavia approved all protocols and specifically approved thisstudy. Informed written consent was obtained according tothe Declaration of Helsinki of 1975 as revised in 2008. Wefocussed on the so-called ECFCs [22,23,25], a subgroup ofEPCs that are found in the CD34 + CD45 - fraction of circu-lating MNCs, exhibit robust proliferative potential, and formcapillary-like structures in vitro [13–15]. To isolate ECFCs,MNCs were separated from either PB or UCB by densitygradient centrifugation on lymphocyte separation mediumfor 30 min at 400 g and washed twice in endothelial basalmedium (EBM)-2 with 2% fetal calf serum. A median of36 · 106 MNCs (range 18–66) was plated on collagen-coatedculture dishes (BD Biosciences) in the presence of the endo-thelial cell growth medium EGM-2 MV Bullet Kit (Lonza)containing EBM-2, 5% fetal bovine serum, recombinant hu-man (rh) EGF, rhVEGF, rhFGF-B, rhIGF-1, ascorbic acid, andheparin, and maintained at 37�C in 5% CO2 and a humidifiedatmosphere. Discarding of non-adherent cells was performed
2562 DRAGONI ET AL.

after 2 days; thereafter, medium was changed thrice a week.The outgrowth of ECs from adherent MNCs was character-ized by the formation of a cluster of cobblestone-appearingcells [13–15,22,23,25]. That ECFC-derived colonies belongedto endothelial lineage was confirmed as described in [23] and[22]. In greater detail, EPC-derived colonies were stainedwith anti-CD31, anti-CD105, anti-CD144, anti-CD146, anti-vWf, anti-CD45, and anti-CD14 monoclonal antibodies andby assessment of capillary-like network formation in an invitro Matrigel assay.
For our experiments, we have mainly used endothelialcells obtained from early passage ECFC (P1-3, which roughlyencompasses a 15–18 day period) with the purpose ofavoiding (or maximally reducing) any potential bias due tocell differentiation. However, in order to make sure that thephenotype of the cells did not change throughout the ex-periments, in preliminary experiments, we tested the im-munophenotype of ECFCs at different passages and wefound no differences, as shown in [22]. We also testedwhether functional differences occurred when early- (P2) andlate- (P6) passage ECFCs were used by testing the in vitrocapacity of capillary network formation in a Matrigel assayand found no differences between early- and late-passageECFC-derived cells.
Solutions
Physiological salt solution (PSS) had the following com-position (in mM): 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10Glucose, and 10 Hepes. In Ca2 + -free solution (0Ca2 + ), Ca2 +
was substituted with 2 mM NaCl, and 0.5 mM EGTA wasadded. Solutions were titrated to pH 7.4 with NaOH. Thehigh-K + extracellular solution was prepared by replacing100 mM NaCl with an equimolar amount of KCl. The solu-tion was then titrated to pH 7.4 with KOH. The osmolality ofthe extracellular solution, as measured with an osmometer(Wescor 5500), was 338 mmol/kg.
[Ca2 + ]i measurements
ECFCs were loaded with 4mM fura-2 acetoxymethyl ester(fura-2/AM; 1 mM stock in dimethyl sulfoxide) in PSS for 1 hat room temperature. After washing in PSS, the coverslipwas fixed to the bottom of a Petri dish and the cells wereobserved by an upright epifluorescence Axiolab microscope(Carl Zeiss), which was usually equipped with a Zeiss · 40Achroplan objective (water immersion, 2.0 mm workingdistance, and 0.9 numerical aperture). ECFCs were excited
alternately at 340 and 380 nm, and the emitted light wasdetected at 510 nm. A first neutral density filter (1 or 0.3optical density) reduced the overall intensity of the excitationlight, and a second neutral density filter (optical densi-ty = 0.3) was coupled to the 380 nm filter to approach theintensity of the 340 nm light. A round diaphragm was usedto increase the contrast. The excitation filters were mountedon a filter wheel (Lambda 10, Sutter Instrument). Customsoftware, working in the LINUX environment, was used todrive the camera (Extended-ISIS Camera, Photonic Science)and the filter wheel, and to measure and plot online thefluorescence from 10 to100 rectangular ‘‘regions of interest’’(ROI). Each ROI was identified by a number. Since cellborders were not clearly identifiable, an ROI may not includethe whole cell or may include a part of an adjacent cell.Adjacent ROIs never superimposed. [Ca2 + ]i was monitoredby measuring, for each ROI, the ratio of the mean fluores-cence emitted at 510 nm when exciting alternatively at 340and 380 nm (shortly termed ‘‘ratio’’). An increase in [Ca2 + ]i
causes an increase in the ratio [22,23,25]. Ratio measurementswere performed and plotted online every 3 s. The experi-ments were performed at room temperature (22�C).
RNA isolation and real-time RT-PCR (qRT-PCR)
Total RNA was extracted from the EPCs using the QIAzolLysis Reagent (QIAGEN). Single cDNA was synthesizedfrom RNA (1mg) using random hexamers and M-MLV Re-verse Transcriptase (Invitrogen S.R.L.). Reverse transcriptionwas always performed in the presence or absence (negativecontrol) of the reverse transcriptase enzyme. qRT-PCR wasperformed in triplicate using 1 mg cDNA and specific primers(intron-spanning primers) for InsP3R1, InsP3R2, and InsP3R3and for TRPC3 (Table 1), as previously described elsewhere[22,23,25]. GoTaq qPCR Mastermix (Promega) was used ac-cording to the manufacturer’s instructions, and qRT-PCRwas performed using Rotor Gene 6000 (Corbett, Concorde).The conditions were as follows: initial denaturation at 95�Cfor 5 min; 40 cycles of denaturation at 95�C for 30 s; an-nealing at 58�C for 30 s; and elongation at 72�C for 40 s. TheqRT-PCR reactions were normalized using b-actin as ahousekeeping gene. Melting curves were generated to detectthe melting temperatures of specific products immediatelyafter the PCR run. The triplicate threshold cycles (Ct) valuesfor each sample were averaged, resulting in mean Ct valuesfor both the gene of interest and the housekeeping gene b-actin. Relative mRNA levels were determined by compara-tive quantitation (Corbett), and the results were expressed as
Table 1. Primer Sequences Used for Real-Time Reverse Transcription/Polymerase Chain Reaction
Gene Primer sequences Size (bp) Accession number
InsP3R1 Forward 5¢-TCAACAAACTGCACCACGCT-3¢ 180 ENSG00000150995Reverse 5¢-CTCTCATGGCATTCTTCTCC-3¢
InsP3R2 Forward 5¢-ACCTTGGG GTTAGTGGATGA-3¢ 158 ENSG00000123104Reverse 5¢-CCTTGTTTGGCTTGCTTTGC-3¢
InsP3R3 Forward 5¢-TGGCTTCATCAGCACTTTGG-3¢ 173 ENSG00000096433Reverse 5¢-TGTCCTGCTTAGTCTGCTTG-3¢
TRPC3 Forward 5¢-GGAGATCTGGAATCAGCAGA-3¢ 336 NM_001130698.1 variant 1NM_003305.2 variant 2Reverse 5¢-AAGCAGACCCAGGAAGATGA-3¢
b-actin Hs_ACTB_1_SG, QuantiTect Primer Assay QT00095431, Qiagen 146 NM_001101
VEGF-INDUCED CA21 OSCILLATIONS IN CORD BLOOD-DERIVED EPCS 2563

fold change. PCR products were also separated with agarosegel electrophoresis and stained with ethidium bromide.
Sample preparation and immunoblotting
UCB-ECFCs were homogenized by using a Dounce ho-mogenizer in a solution containing 250 mM Sucrose, 1 mMEDTA, 10 mM Tris-HCl, pH 7.6, 0.1 mg/mL PMSF, 100 mMb-mercaptoethanol, and Protease Inhibitor Cocktail (P8340;Sigma). The homogenates were solubilized in Laemmli buf-fer [22,23], and 20mg proteins were separated on 10% sodiumdodecyl sulfate-polyacrilamide gel electrophoresis andtransferred to the Hybond-P polyvinyl difluoride Membrane(GE Healthcare) by electroelution. After 1 h blocking withTris buffered saline (TBS) containing 3% bovine serum al-bumin (BSA) and 0.1% Tween (blocking solution), themembranes were incubated for 3 h at room temperature withthe TRPC3/6/7 (N-18) (Santa Cruz Biotechnology, Inc.; sc-15056) goat polyclonal antibody or TRPC3 rabbit polyclonalantibody (Acris Antibodies GmbH) diluted 1:200 in the TBSand 0.1% Tween. The membranes were washed and incu-bated for 1 h with peroxidase-conjugated goat anti-rabbit IgG(Chemicon, AP132P) or mouse anti-goat IgG (Santa CruzBiotechnology, Inc.; sc-2354) diluted 1:120,000 in blockingsolution. The bands were detected with ECL� Advancewestern blotting detection system (GE Healthcare EuropeGmbH). Control experiments were performed as describedin [23] and [22]. Prestained molecular-weight markers(SDS7B2, Sigma) were used to estimate the molecularweight of the bands. Blots were stripped with the methoddescribed in [23] and re-probed with anti b-actin rabbitantibody as loading control (Rockland Immunochemicalsfor Research, U.S.A.; code, 600-401-886). The antibody wasdiluted 1:2,000 in the TBS and 0.1% Tween. Blots were ac-quired with the Image Master VDS (Amersham BiosciencesEurope). Densitometric analysis of the bands was per-formed by the Total Lab V 1.11 computer program(Amersham), and the results were expressed as a percent-age of the gene/b-actin densitometric ratio.
Protein content
Protein contents of all the samples were determined by theBradford’s method using BSA as standard [22,23].
Gene silencing
siRNA targeting TRPC3 was purchased by Thermo sci-entific-Dharmacon (ON-TARGET plus SMART pool HumanTRPC3). Scrambled siRNA were used as a negative control.As recently depicted elsewhere [22], once the monolayer cellshad reached 50% confluency, the medium was removed andthe cells were added with Opti-MEM I reduced serum mediumwithout antibiotics (Life Technologies). siRNAs (120 nM finalconcentration) were diluted in Opti-MEM I reduced serummedium and mixed with Lipofectamine� RNAiMAX trans-fection reagent (Invitrogen) pre-diluted in Opti-MEM), ac-cording to the manufacturer’s instructions. After 20 minincubation at room temperature, the mixes were added to thecells and incubated at 37�C for 5 h. Transfection mixes werethen completely removed, and fresh culture media were ad-ded. The effectiveness in silencing was determined by immu-noblotting, and the KO-cells were used 72 h after transfection.
Proliferation assays
As described elsewhere [25], a total of 1 · 105 UCB-ECFC-derived cells (first passage) were plated in 30-mm collagen-treated dishes in EBM medium (Lonza) in the presenceof 10 ng/mL VEGF (PeproTech) and with or without 30mM1,2-bis(o-aminophenoxy)ethane-N,N,N¢,N¢-tetracetic acid(BAPTA), 10 mM ethyl-1-(4-(2,3,3-trichloroacrylamide)phe-nyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate (Pyr3),and 20 mM of N-(4-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide (BTP-2).Cultures were incubated at 37�C, 5% CO2 and cell growthwas assessed every day until confluence was reached in thecontrol dishes, usually after 3–4 days. Cells were then re-covered by trypsinization, and their number was assessed bycounting in a hemocytometer. The percentage of growthstimulation (by VEGF) or inhibition [by Pyr3, flufenamic acid(FFA), BAPTA, and BTP-2] was calculated by dividing thetotal number of cells obtained in the presence of the afore-mentioned compounds by the number of cells in controlexperiments and by multiplying the ratio by 100.
Statistics
All the Ca2 + data have been collected from PB-ECFCs iso-lated from PB of at least three healthy volunteers (for OAG-induced Ca2 + signals) or from UCB-ECFCs harvested from atleast three different cord blood samples. Pooled data are givenas mean – SE, and statistical significance (P < 0.05) was evalu-ated by the Student’s t-test for unpaired observations. As de-scribed elsewhere [25,39], the interspike interval (ISI) analyzedin Fig. 2E was defined as the peak-to-peak interval betweensuccessive Ca2 + transients. Only cells displaying at least threeCa2 + oscillations were included in the analysis depicted in Fig.2B–F. The statistical analysis was performed on cells chal-lenged with VEGF for 1 h, which was chosen as an arbitrarytime interval to compare the Ca2 + activity of different ECFCsboth from the same coverslip and from different batches.
As for mRNA analysis, all data are expressed as mean –SE. The significance of the differences of the means wasevaluated with Student’s t test. In the proliferation assays,results are expressed as percentage ( – SD) of growth com-pared with controls (given as 100% growth), obtained fromthree different sets of experiments, each performed in du-plicate. Differences were assessed by the Student’s t-test forunpaired values. All statistical tests were carried out withGraphPad Prism 4.
Chemicals
EBM and EGM-2 were purchased from Clonetics (CellSystem). Fura-2/AM was obtained from Molecular Probes(Molecular Probes Europe BV). BTP-2 was purchased fromCalbiochem. VEGF was provided by PeproTech. All otherchemicals were obtained from Sigma Chemical Co.
Results
VEGF induces asynchronous Ca2 + oscillationsin UCB-ECFCs
We have recently shown that 10 ng/mL is the most suit-able dose for VEGF to induce oscillations in [Ca2 + ]i in human
2564 DRAGONI ET AL.
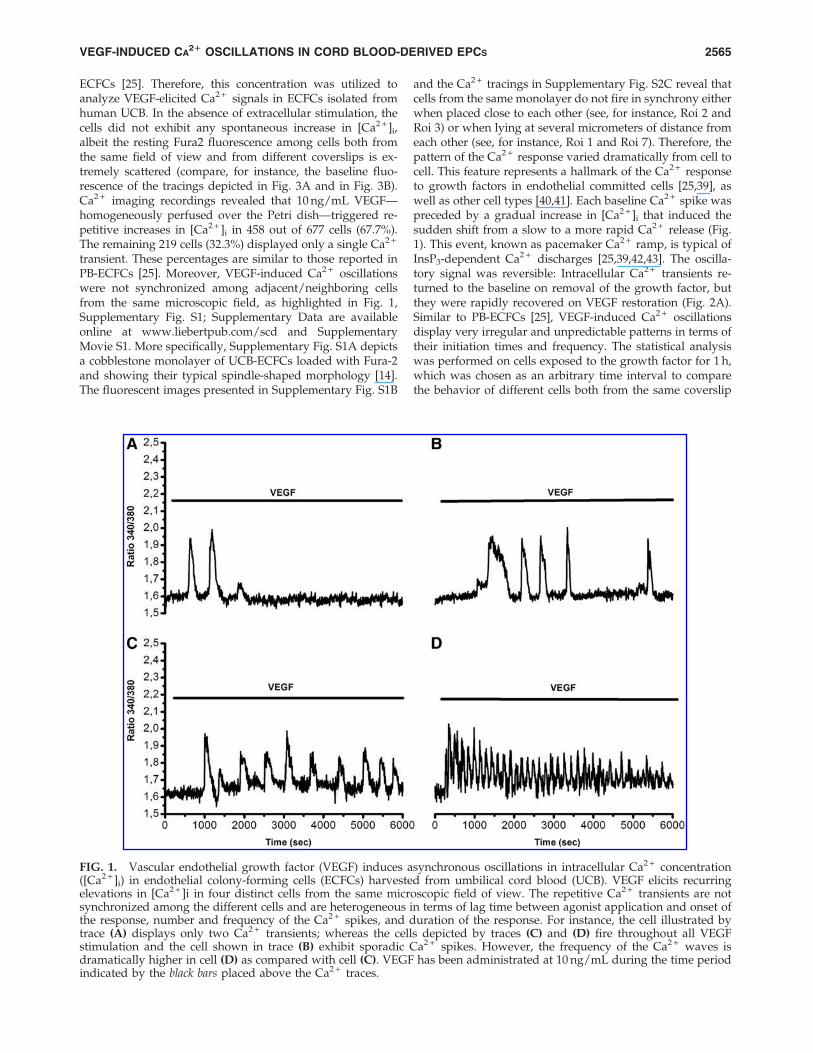
ECFCs [25]. Therefore, this concentration was utilized toanalyze VEGF-elicited Ca2 + signals in ECFCs isolated fromhuman UCB. In the absence of extracellular stimulation, thecells did not exhibit any spontaneous increase in [Ca2 + ]i,albeit the resting Fura2 fluorescence among cells both fromthe same field of view and from different coverslips is ex-tremely scattered (compare, for instance, the baseline fluo-rescence of the tracings depicted in Fig. 3A and in Fig. 3B).Ca2 + imaging recordings revealed that 10 ng/mL VEGF—homogeneously perfused over the Petri dish—triggered re-petitive increases in [Ca2 + ]i in 458 out of 677 cells (67.7%).The remaining 219 cells (32.3%) displayed only a single Ca2 +
transient. These percentages are similar to those reported inPB-ECFCs [25]. Moreover, VEGF-induced Ca2 + oscillationswere not synchronized among adjacent/neighboring cellsfrom the same microscopic field, as highlighted in Fig. 1,Supplementary Fig. S1; Supplementary Data are availableonline at www.liebertpub.com/scd and SupplementaryMovie S1. More specifically, Supplementary Fig. S1A depictsa cobblestone monolayer of UCB-ECFCs loaded with Fura-2and showing their typical spindle-shaped morphology [14].The fluorescent images presented in Supplementary Fig. S1B
and the Ca2 + tracings in Supplementary Fig. S2C reveal thatcells from the same monolayer do not fire in synchrony eitherwhen placed close to each other (see, for instance, Roi 2 andRoi 3) or when lying at several micrometers of distance fromeach other (see, for instance, Roi 1 and Roi 7). Therefore, thepattern of the Ca2 + response varied dramatically from cell tocell. This feature represents a hallmark of the Ca2 + responseto growth factors in endothelial committed cells [25,39], aswell as other cell types [40,41]. Each baseline Ca2 + spike waspreceded by a gradual increase in [Ca2 + ]i that induced thesudden shift from a slow to a more rapid Ca2 + release (Fig.1). This event, known as pacemaker Ca2 + ramp, is typical ofInsP3-dependent Ca2 + discharges [25,39,42,43]. The oscilla-tory signal was reversible: Intracellular Ca2 + transients re-turned to the baseline on removal of the growth factor, butthey were rapidly recovered on VEGF restoration (Fig. 2A).Similar to PB-ECFCs [25], VEGF-induced Ca2 + oscillationsdisplay very irregular and unpredictable patterns in terms oftheir initiation times and frequency. The statistical analysiswas performed on cells exposed to the growth factor for 1 h,which was chosen as an arbitrary time interval to comparethe behavior of different cells both from the same coverslip
FIG. 1. Vascular endothelial growth factor (VEGF) induces asynchronous oscillations in intracellular Ca2 + concentration([Ca2 + ]i) in endothelial colony-forming cells (ECFCs) harvested from umbilical cord blood (UCB). VEGF elicits recurringelevations in [Ca2 + ]i in four distinct cells from the same microscopic field of view. The repetitive Ca2 + transients are notsynchronized among the different cells and are heterogeneous in terms of lag time between agonist application and onset ofthe response, number and frequency of the Ca2 + spikes, and duration of the response. For instance, the cell illustrated bytrace (A) displays only two Ca2 + transients; whereas the cells depicted by traces (C) and (D) fire throughout all VEGFstimulation and the cell shown in trace (B) exhibit sporadic Ca2 + spikes. However, the frequency of the Ca2 + waves isdramatically higher in cell (D) as compared with cell (C). VEGF has been administrated at 10 ng/mL during the time periodindicated by the black bars placed above the Ca2 + traces.
VEGF-INDUCED CA21 OSCILLATIONS IN CORD BLOOD-DERIVED EPCS 2565
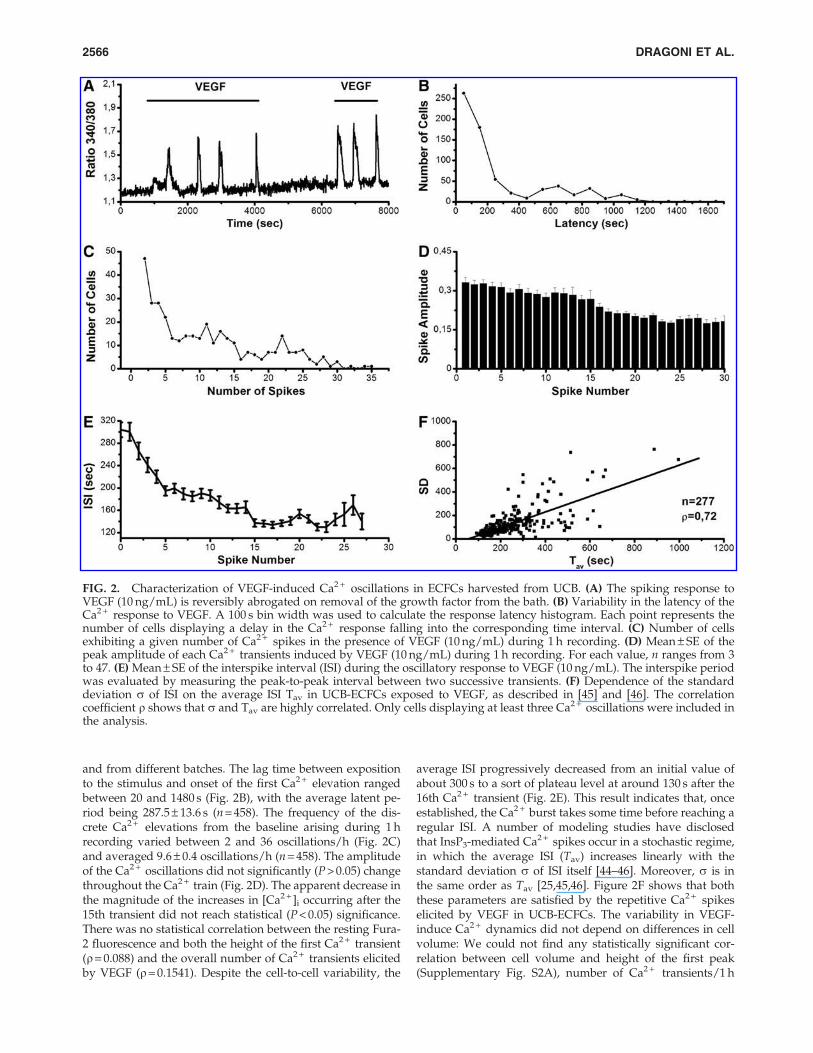
and from different batches. The lag time between expositionto the stimulus and onset of the first Ca2 + elevation rangedbetween 20 and 1480 s (Fig. 2B), with the average latent pe-riod being 287.5 – 13.6 s (n = 458). The frequency of the dis-crete Ca2 + elevations from the baseline arising during 1 hrecording varied between 2 and 36 oscillations/h (Fig. 2C)and averaged 9.6 – 0.4 oscillations/h (n = 458). The amplitudeof the Ca2 + oscillations did not significantly (P > 0.05) changethroughout the Ca2 + train (Fig. 2D). The apparent decrease inthe magnitude of the increases in [Ca2 + ]i occurring after the15th transient did not reach statistical (P < 0.05) significance.There was no statistical correlation between the resting Fura-2 fluorescence and both the height of the first Ca2 + transient(r= 0.088) and the overall number of Ca2 + transients elicitedby VEGF (r = 0.1541). Despite the cell-to-cell variability, the
average ISI progressively decreased from an initial value ofabout 300 s to a sort of plateau level at around 130 s after the16th Ca2 + transient (Fig. 2E). This result indicates that, onceestablished, the Ca2 + burst takes some time before reaching aregular ISI. A number of modeling studies have disclosedthat InsP3-mediated Ca2 + spikes occur in a stochastic regime,in which the average ISI (Tav) increases linearly with thestandard deviation s of ISI itself [44–46]. Moreover, s is inthe same order as Tav [25,45,46]. Figure 2F shows that boththese parameters are satisfied by the repetitive Ca2 + spikeselicited by VEGF in UCB-ECFCs. The variability in VEGF-induce Ca2 + dynamics did not depend on differences in cellvolume: We could not find any statistically significant cor-relation between cell volume and height of the first peak(Supplementary Fig. S2A), number of Ca2 + transients/1 h
FIG. 2. Characterization of VEGF-induced Ca2 + oscillations in ECFCs harvested from UCB. (A) The spiking response toVEGF (10 ng/mL) is reversibly abrogated on removal of the growth factor from the bath. (B) Variability in the latency of theCa2 + response to VEGF. A 100 s bin width was used to calculate the response latency histogram. Each point represents thenumber of cells displaying a delay in the Ca2 + response falling into the corresponding time interval. (C) Number of cellsexhibiting a given number of Ca2 + spikes in the presence of VEGF (10 ng/mL) during 1 h recording. (D) Mean – SE of thepeak amplitude of each Ca2 + transients induced by VEGF (10 ng/mL) during 1 h recording. For each value, n ranges from 3to 47. (E) Mean – SE of the interspike interval (ISI) during the oscillatory response to VEGF (10 ng/mL). The interspike periodwas evaluated by measuring the peak-to-peak interval between two successive transients. (F) Dependence of the standarddeviation s of ISI on the average ISI Tav in UCB-ECFCs exposed to VEGF, as described in [45] and [46]. The correlationcoefficient r shows that s and Tav are highly correlated. Only cells displaying at least three Ca2 + oscillations were included inthe analysis.
2566 DRAGONI ET AL.
(Supplementary Fig. S2B), and average ISI (SupplementaryFig. S2C). In conclusion, our findings indicate that the modeof the Ca2 + response is similar in ECFCs irrespective of theirsource; that is, human peripheral or cord blood, and it mainlyconsists of asynchronous Ca2 + oscillations that are charac-terized by a large variability in both the onset and the fre-quency of the Ca2 + spikes.
Ca2 + entry is required to initiate and maintainVEGF-induced Ca2 + oscillations in UCB-ECFCs
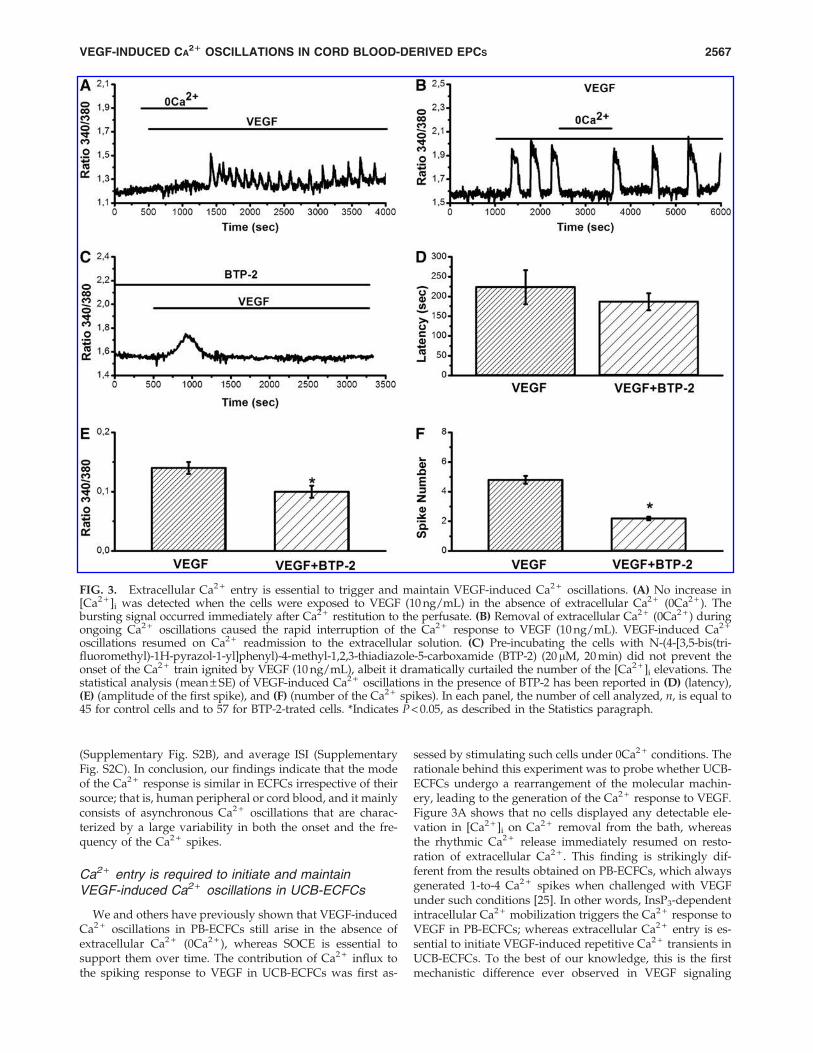
We and others have previously shown that VEGF-inducedCa2 + oscillations in PB-ECFCs still arise in the absence ofextracellular Ca2 + (0Ca2 + ), whereas SOCE is essential tosupport them over time. The contribution of Ca2 + influx tothe spiking response to VEGF in UCB-ECFCs was first as-
sessed by stimulating such cells under 0Ca2 + conditions. Therationale behind this experiment was to probe whether UCB-ECFCs undergo a rearrangement of the molecular machin-ery, leading to the generation of the Ca2 + response to VEGF.Figure 3A shows that no cells displayed any detectable ele-vation in [Ca2 + ]i on Ca2 + removal from the bath, whereasthe rhythmic Ca2 + release immediately resumed on resto-ration of extracellular Ca2 + . This finding is strikingly dif-ferent from the results obtained on PB-ECFCs, which alwaysgenerated 1-to-4 Ca2 + spikes when challenged with VEGFunder such conditions [25]. In other words, InsP3-dependentintracellular Ca2 + mobilization triggers the Ca2 + response toVEGF in PB-ECFCs; whereas extracellular Ca2 + entry is es-sential to initiate VEGF-induced repetitive Ca2 + transients inUCB-ECFCs. To the best of our knowledge, this is the firstmechanistic difference ever observed in VEGF signaling
FIG. 3. Extracellular Ca2 + entry is essential to trigger and maintain VEGF-induced Ca2 + oscillations. (A) No increase in[Ca2 + ]i was detected when the cells were exposed to VEGF (10 ng/mL) in the absence of extracellular Ca2 + (0Ca2 + ). Thebursting signal occurred immediately after Ca2 + restitution to the perfusate. (B) Removal of extracellular Ca2 + (0Ca2 + ) duringongoing Ca2 + oscillations caused the rapid interruption of the Ca2 + response to VEGF (10 ng/mL). VEGF-induced Ca2 +
oscillations resumed on Ca2 + readmission to the extracellular solution. (C) Pre-incubating the cells with N-(4-[3,5-bis(tri-fluoromethyl)-1H-pyrazol-1-yl]phenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide (BTP-2) (20mM, 20 min) did not prevent theonset of the Ca2 + train ignited by VEGF (10 ng/mL), albeit it dramatically curtailed the number of the [Ca2 + ]i elevations. Thestatistical analysis (mean – SE) of VEGF-induced Ca2 + oscillations in the presence of BTP-2 has been reported in (D) (latency),(E) (amplitude of the first spike), and (F) (number of the Ca2 + spikes). In each panel, the number of cell analyzed, n, is equal to45 for control cells and to 57 for BTP-2-trated cells. *Indicates P < 0.05, as described in the Statistics paragraph.
VEGF-INDUCED CA21 OSCILLATIONS IN CORD BLOOD-DERIVED EPCS 2567

between the two cell types. To determine whether the os-cillatory dynamics became independent on Ca2 + entry afterits onset, extracellular Ca2 + was depleted from the perfusateafter the onset of the signal. Figure 3B shows that this ma-neuver reversibly abolished the Ca2 + train elicited by VEGFin 73 out of 79 cells (92.4%).
SOCE is activated by emptying of the InsP3-sensitive Ca2 +
pool in UCB-ECFCs [23] and sustains VEGF-induced Ca2 +
oscillations in PB-ECFCs [25]. It has been proposed thatSOCE may be activated by the initial drop in the ER Ca2 +
content that occurs within the more peripheral organellesheets, which are located in close proximity to PM. Ca2 + en-tering the cells through such a pathway is sequestered back bySarco/Endoplasmatic Reticulum Ca2 + -ATPase (SERCA), there-by loading up the store and sensitizing from within theInsP3Rs to initiate the first Ca2 + spike [47,48]. We hypothe-sized that an InsP3-dependent Ca2 + release event restrictedto the sub-plasmalemmal domain might contribute towardactivating SOCE and initiating the spiking signal. Therefore,VEGF was administered to UCB-ECFCs pre-incubated in thepresence of BTP-2 (20 mM, 20 min), a selective blocker ofstore-dependent Ca2 + influx in human ECFCs [1,3,22,23,25],as well as all other cell types [31]. This treatment did notinhibit the onset of the intracellular Ca2 + waves (Fig. 3C),and the latency of the Ca2 + signal was not significantly(P < 0.05) different as compared with control cells (Fig. 3D).The magnitude of the first Ca2 + transient elicited by VEGFwas slightly (P < 0.05) smaller in the presence of BTP-2 (Fig.3E); in addition, it was followed by no more than 1–3 spikes(Fig. 3C, F and Supplementary Fig. S3). Collectively, theseresults suggest that SOCE does not initiate the oscillatorysignal, but is required to support it over time. Therefore, astore-independent membrane pathway is responsible for theonset of the Ca2 + response to VEGF in UCB-ECFCs.
The role of PLCc and InsP3 receptorsin the oscillatory response to VEGF
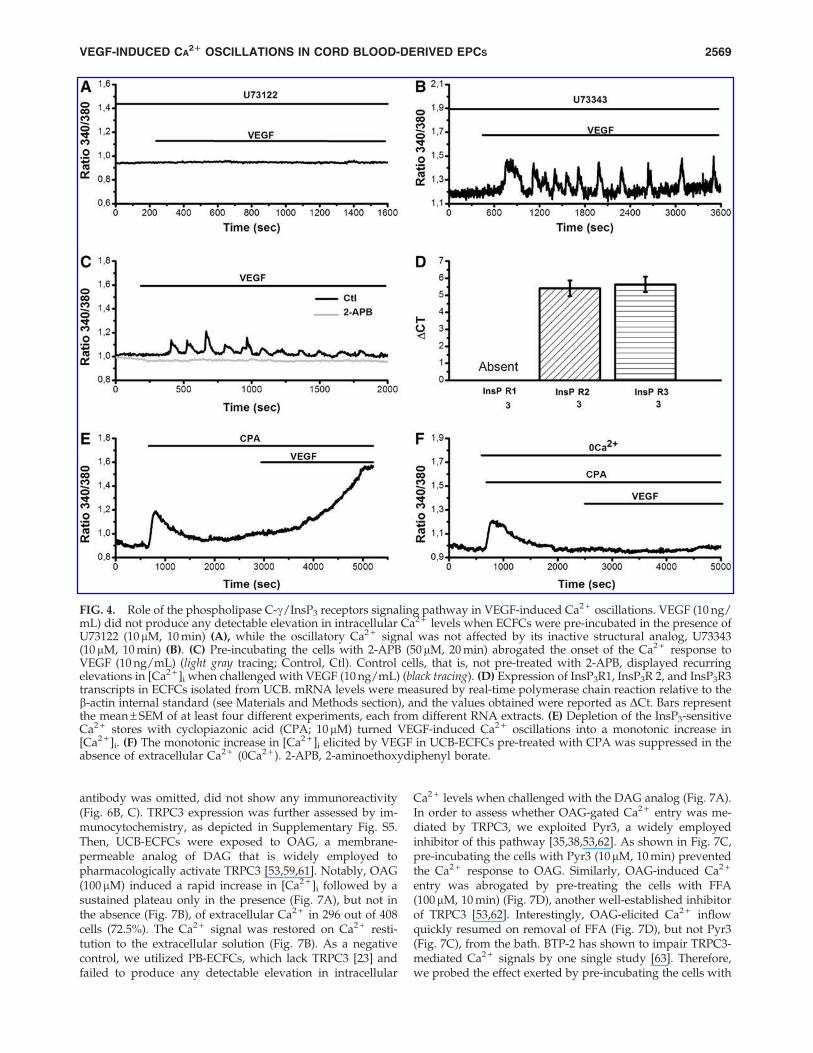
The activation of VEGFR-2 by VEGF leads to the synthesisof the two intracellular second messengers, InsP3 and DAG,which is mediated by the effector enzyme, PLCg [3,26]. Therole served by PLCg in establishing VEGF-induced Ca2 +
oscillations was probed by pre-incubating the cells withU73122 (10 mM, 10 min), an aminosteroid that blocks PLCg inhuman ECFCs [23,25], as well as other cell types [40,49–52].This treatment prevented the onset of VEGF-elicited Ca2 +
oscillations in 44 out of 46 (95.7%) cells (Fig. 4A), whereasthe inactive analogue, U73343 (10 mM, 10 min), was withouteffect in 64 out of 66 cells (96.7%) (Fig. 4B and SupplementaryFig. S4). Therefore, either InsP3 or DAG (or both) wererequired for the onset of the oscillatory response to VEGFin UCB-ECFCs. The involvement of the InsP3-sensitiveCa2 + pool was evaluated by pre-incubating the cells with 2-aminoethoxydiphenyl borate (2-APB) (50mM, 20 min), a well-known InsP3R inhibitor [25,49]. As shown in Fig. 4C, 2-APBsuppressed the repetitive Ca2 + transients evoked by VEGF in105 out of 111 cells (94.6%). It should, however, be noted that2-APB may also affect a number of plasmalemmal TRPchannels [3,53], including TRPC3, thereby potentially inter-fering with the influx of Ca2 + that triggers Ca2 + signals inUCB-ECFCs. In the absence of other membrane-permeableInsP3R blockers devoid of side-effects, we sought to elucidate
the contribution of the intracellular reservoir by first asses-sing the expression of InsP3Rs. Unlike PB-ECFCs [25], UCB-ECFCs lack InsP3R1, whereas they display both InsP3R2 andInsP3R3 mRNAs (Fig. 4D), the qRT-PCR products being ofthe expected size. Negative controls were performed byomitting the reverse transcriptase (not shown). The transcriptlevels of InsP3R2 and InsP3R3 in UCB-ECFCs were not sta-tistically different (P < 0.05). Subsequently, we depleted theInsP3-dependent Ca2 + pool by pre-treating the cells withcyclopiazonic acid (CPA; 10 mM) [23,25], which preventsSERCA from counterbalancing the passive and activity-de-pendent Ca2 + fluxes from ER to the cytosol. Figure 4E showsthat CPA caused an immediate elevation in [Ca2 + ]i due topassive Ca2 + efflux from the intracellular reservoir followedby a decay phase to a plateau level slightly higher than thebaseline, due to SOCE activation [23]. When added at 30 minafter the exposition to CPA, a time interval that is sufficientto fully empty the InsP3-sensitive Ca2 + pool [23,25], VEGFgenerated a monotonic elevation in [Ca2 + ]i rather than anoscillatory signal (Fig. 4E). This pattern of Ca2 + signal hasbeen well associated to a VEGF-induced store-independentCa2 + entry from the extracellular space [32,54,55]. Indeed,under these conditions (ie, after 30 min of stimulation withCPA), the ER Ca2 + store has already been depleted andSOCE has attained a plateau [56,57]. Accordingly, no Ca2 +
response occurred when CPA was perfused in the absence ofextracellular Ca2 + (Fig. 4F). Overall, these results indicatethat (1) the onset of VEGF-elicited Ca2 + oscillations in UCB-ECFCs lies downstream the signaling pathway triggered byPLCg; (2) the initial Ca2 + response to VEGF consists of theactivation of a Ca2 + entry pathway, which is not regulatedby the ER Ca2 + content and is likely gated by an intracellularsecond messenger; and (3) InsP3-evoked intracellular Ca2 +
release drives the Ca2 + spikes triggered by VEGF-inducedCa2 + influx. The role of InsP3-dependent Ca2 + mobilizationwas further investigated by interfering with ER refillingduring the ongoing variations in [Ca2 + ]i induced by VEGF[25,39]. The acute addition of CPA (10mM) rapidly inhibitedVEGF-induced Ca2 + oscillations in 54 out of 56 cells (96.4%)(Fig. 5A), whereas thapsigargin (2mM) blocked the Ca2 +
response to VEGF in 45 out of 48 cells (93.7%) (Fig. 5B). Thesedata demonstrate that SERCA-mediated refilling of theInsP3-sensitive Ca2 + is essential to maintain the Ca2 + train.
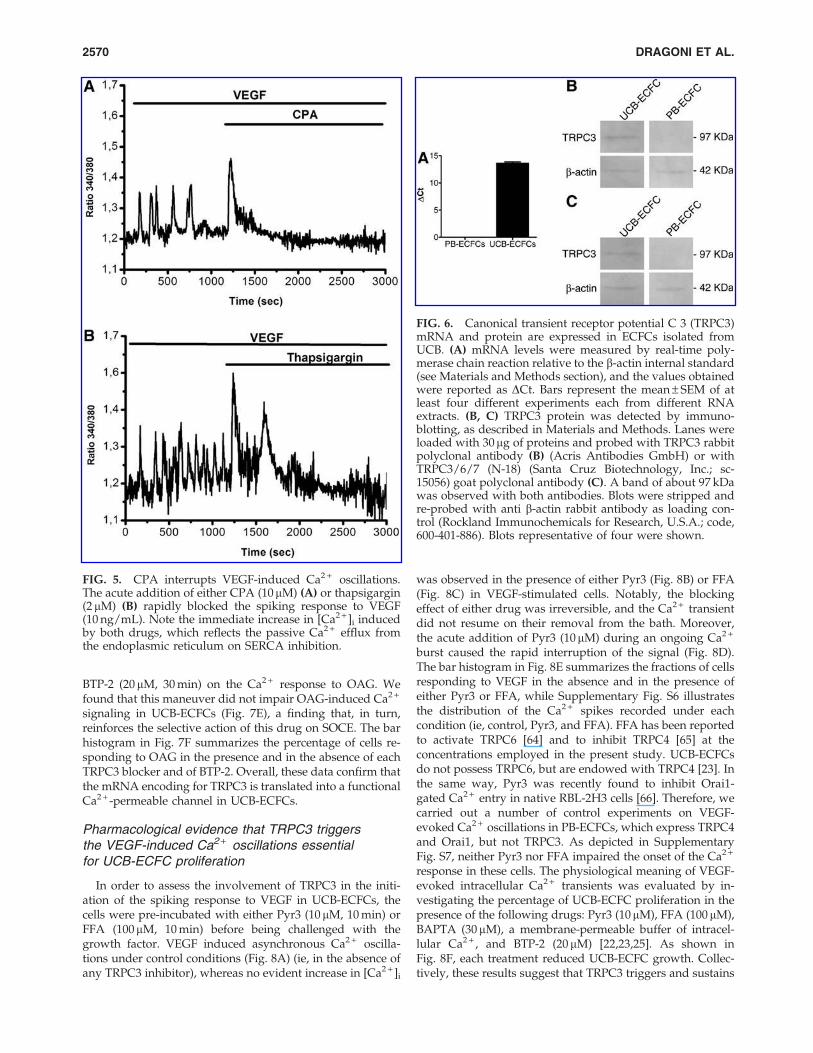
Evidence for TRPC3 expression in UCB-ECFCs
The influx of Ca2 + necessary for the onset of the oscilla-tory response to VEGF in UCB-ECFCs could be provided byCa2 + -permeable membrane channel that is sensitive to DAG,an intracellular second messenger that is synthesized byPLCg along with InsP3 [3,58]. Unlike PB-ECFCs [23] (see alsoFig. 6A), these cells are endowed with a transcript encodingfor the DAG-dependent TRPC3 channel [23] (see also Fig.6A), which provides the pathway for VEGF-induced Ca2 +
entry in mature endothelium [32,33]. Therefore, we sought tounveil the role served by TRPC3 in the initiation of VEGF-evoked Ca2 + oscillations in UCB-ECFCs. First, we evaluatedthe translation of TRPC3 mRNA into a functional protein byimmunoblotting. We utilized two different antibodies, pur-chased from two distinct companies, which recognized asingle band of about 97 kDa, the expected molecular weightfor TRPC3 [59,60] (Fig. 6B, C). The blanks, where the primary
2568 DRAGONI ET AL.
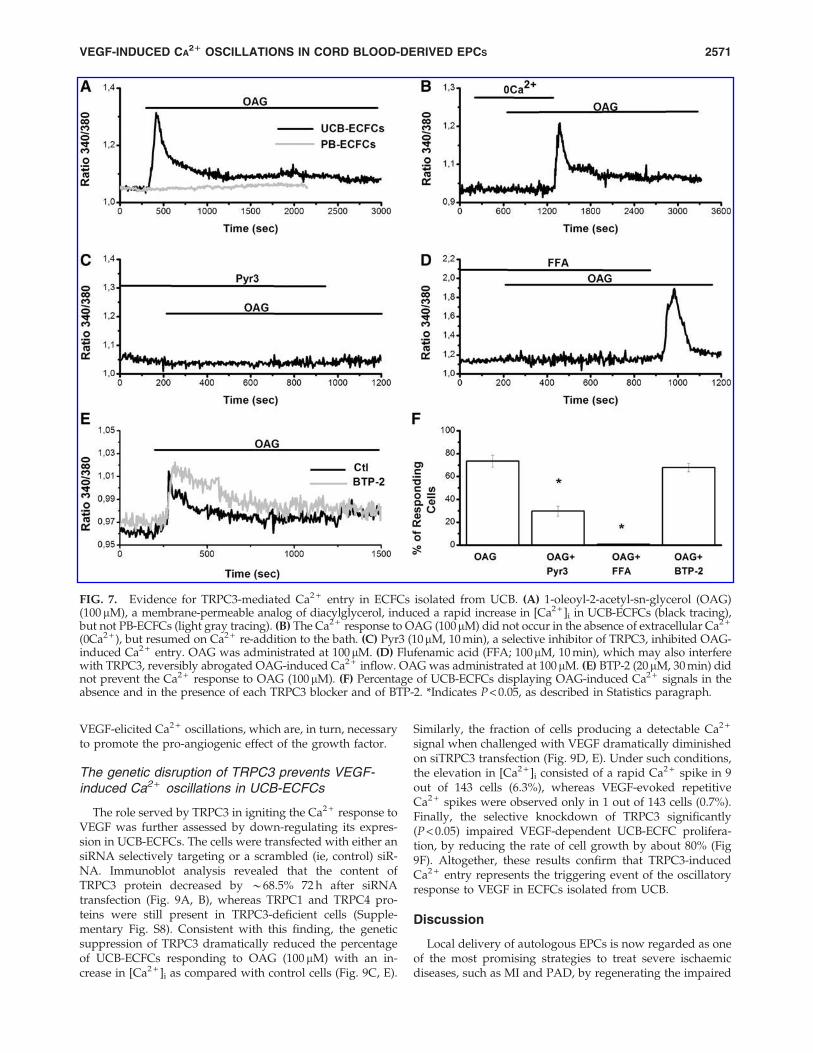
antibody was omitted, did not show any immunoreactivity(Fig. 6B, C). TRPC3 expression was further assessed by im-munocytochemistry, as depicted in Supplementary Fig. S5.Then, UCB-ECFCs were exposed to OAG, a membrane-permeable analog of DAG that is widely employed topharmacologically activate TRPC3 [53,59,61]. Notably, OAG(100 mM) induced a rapid increase in [Ca2 + ]i followed by asustained plateau only in the presence (Fig. 7A), but not inthe absence (Fig. 7B), of extracellular Ca2 + in 296 out of 408cells (72.5%). The Ca2 + signal was restored on Ca2 + resti-tution to the extracellular solution (Fig. 7B). As a negativecontrol, we utilized PB-ECFCs, which lack TRPC3 [23] andfailed to produce any detectable elevation in intracellular
Ca2 + levels when challenged with the DAG analog (Fig. 7A).In order to assess whether OAG-gated Ca2 + entry was me-diated by TRPC3, we exploited Pyr3, a widely employedinhibitor of this pathway [35,38,53,62]. As shown in Fig. 7C,pre-incubating the cells with Pyr3 (10mM, 10 min) preventedthe Ca2 + response to OAG. Similarly, OAG-induced Ca2 +
entry was abrogated by pre-treating the cells with FFA(100mM, 10 min) (Fig. 7D), another well-established inhibitorof TRPC3 [53,62]. Interestingly, OAG-elicited Ca2 + inflowquickly resumed on removal of FFA (Fig. 7D), but not Pyr3(Fig. 7C), from the bath. BTP-2 has shown to impair TRPC3-mediated Ca2 + signals by one single study [63]. Therefore,we probed the effect exerted by pre-incubating the cells with
FIG. 4. Role of the phospholipase C-g/InsP3 receptors signaling pathway in VEGF-induced Ca2 + oscillations. VEGF (10 ng/mL) did not produce any detectable elevation in intracellular Ca2 + levels when ECFCs were pre-incubated in the presence ofU73122 (10mM, 10 min) (A), while the oscillatory Ca2 + signal was not affected by its inactive structural analog, U73343(10 mM, 10 min) (B). (C) Pre-incubating the cells with 2-APB (50mM, 20 min) abrogated the onset of the Ca2 + response toVEGF (10 ng/mL) (light gray tracing; Control, Ctl). Control cells, that is, not pre-treated with 2-APB, displayed recurringelevations in [Ca2 + ]i when challenged with VEGF (10 ng/mL) (black tracing). (D) Expression of InsP3R1, InsP3R 2, and InsP3R3transcripts in ECFCs isolated from UCB. mRNA levels were measured by real-time polymerase chain reaction relative to theb-actin internal standard (see Materials and Methods section), and the values obtained were reported as DCt. Bars representthe mean – SEM of at least four different experiments, each from different RNA extracts. (E) Depletion of the InsP3-sensitiveCa2 + stores with cyclopiazonic acid (CPA; 10 mM) turned VEGF-induced Ca2 + oscillations into a monotonic increase in[Ca2 + ]i. (F) The monotonic increase in [Ca2 + ]i elicited by VEGF in UCB-ECFCs pre-treated with CPA was suppressed in theabsence of extracellular Ca2 + (0Ca2 + ). 2-APB, 2-aminoethoxydiphenyl borate.
VEGF-INDUCED CA21 OSCILLATIONS IN CORD BLOOD-DERIVED EPCS 2569
BTP-2 (20 mM, 30 min) on the Ca2 + response to OAG. Wefound that this maneuver did not impair OAG-induced Ca2 +
signaling in UCB-ECFCs (Fig. 7E), a finding that, in turn,reinforces the selective action of this drug on SOCE. The barhistogram in Fig. 7F summarizes the percentage of cells re-sponding to OAG in the presence and in the absence of eachTRPC3 blocker and of BTP-2. Overall, these data confirm thatthe mRNA encoding for TRPC3 is translated into a functionalCa2 + -permeable channel in UCB-ECFCs.
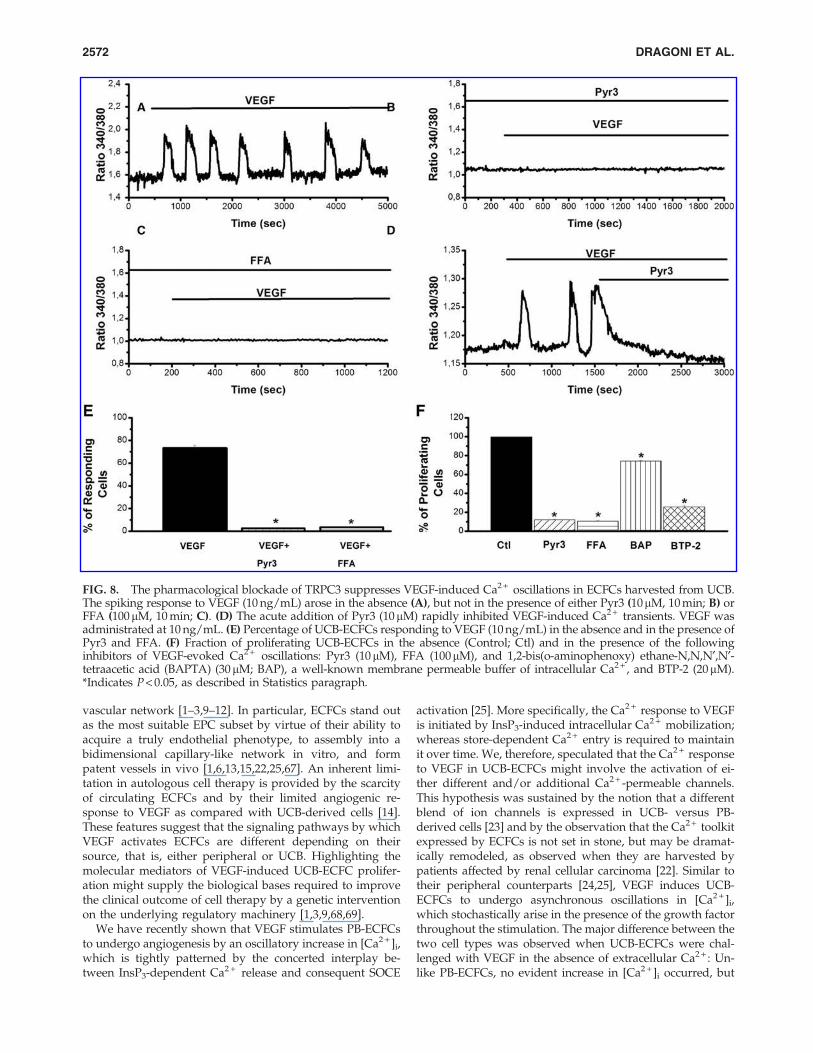
Pharmacological evidence that TRPC3 triggersthe VEGF-induced Ca2 + oscillations essentialfor UCB-ECFC proliferation
In order to assess the involvement of TRPC3 in the initi-ation of the spiking response to VEGF in UCB-ECFCs, thecells were pre-incubated with either Pyr3 (10 mM, 10 min) orFFA (100mM, 10 min) before being challenged with thegrowth factor. VEGF induced asynchronous Ca2 + oscilla-tions under control conditions (Fig. 8A) (ie, in the absence ofany TRPC3 inhibitor), whereas no evident increase in [Ca2 + ]i
was observed in the presence of either Pyr3 (Fig. 8B) or FFA(Fig. 8C) in VEGF-stimulated cells. Notably, the blockingeffect of either drug was irreversible, and the Ca2 + transientdid not resume on their removal from the bath. Moreover,the acute addition of Pyr3 (10mM) during an ongoing Ca2 +
burst caused the rapid interruption of the signal (Fig. 8D).The bar histogram in Fig. 8E summarizes the fractions of cellsresponding to VEGF in the absence and in the presence ofeither Pyr3 or FFA, while Supplementary Fig. S6 illustratesthe distribution of the Ca2 + spikes recorded under eachcondition (ie, control, Pyr3, and FFA). FFA has been reportedto activate TRPC6 [64] and to inhibit TRPC4 [65] at theconcentrations employed in the present study. UCB-ECFCsdo not possess TRPC6, but are endowed with TRPC4 [23]. Inthe same way, Pyr3 was recently found to inhibit Orai1-gated Ca2 + entry in native RBL-2H3 cells [66]. Therefore, wecarried out a number of control experiments on VEGF-evoked Ca2 + oscillations in PB-ECFCs, which express TRPC4and Orai1, but not TRPC3. As depicted in SupplementaryFig. S7, neither Pyr3 nor FFA impaired the onset of the Ca2 +
response in these cells. The physiological meaning of VEGF-evoked intracellular Ca2 + transients was evaluated by in-vestigating the percentage of UCB-ECFC proliferation in thepresence of the following drugs: Pyr3 (10mM), FFA (100 mM),BAPTA (30 mM), a membrane-permeable buffer of intracel-lular Ca2 + , and BTP-2 (20 mM) [22,23,25]. As shown inFig. 8F, each treatment reduced UCB-ECFC growth. Collec-tively, these results suggest that TRPC3 triggers and sustains
FIG. 5. CPA interrupts VEGF-induced Ca2 + oscillations.The acute addition of either CPA (10mM) (A) or thapsigargin(2 mM) (B) rapidly blocked the spiking response to VEGF(10 ng/mL). Note the immediate increase in [Ca2 + ]i inducedby both drugs, which reflects the passive Ca2 + efflux fromthe endoplasmic reticulum on SERCA inhibition.
FIG. 6. Canonical transient receptor potential C 3 (TRPC3)mRNA and protein are expressed in ECFCs isolated fromUCB. (A) mRNA levels were measured by real-time poly-merase chain reaction relative to the b-actin internal standard(see Materials and Methods section), and the values obtainedwere reported as DCt. Bars represent the mean – SEM of atleast four different experiments each from different RNAextracts. (B, C) TRPC3 protein was detected by immuno-blotting, as described in Materials and Methods. Lanes wereloaded with 30mg of proteins and probed with TRPC3 rabbitpolyclonal antibody (B) (Acris Antibodies GmbH) or withTRPC3/6/7 (N-18) (Santa Cruz Biotechnology, Inc.; sc-15056) goat polyclonal antibody (C). A band of about 97 kDawas observed with both antibodies. Blots were stripped andre-probed with anti b-actin rabbit antibody as loading con-trol (Rockland Immunochemicals for Research, U.S.A.; code,600-401-886). Blots representative of four were shown.
2570 DRAGONI ET AL.
VEGF-elicited Ca2 + oscillations, which are, in turn, necessaryto promote the pro-angiogenic effect of the growth factor.
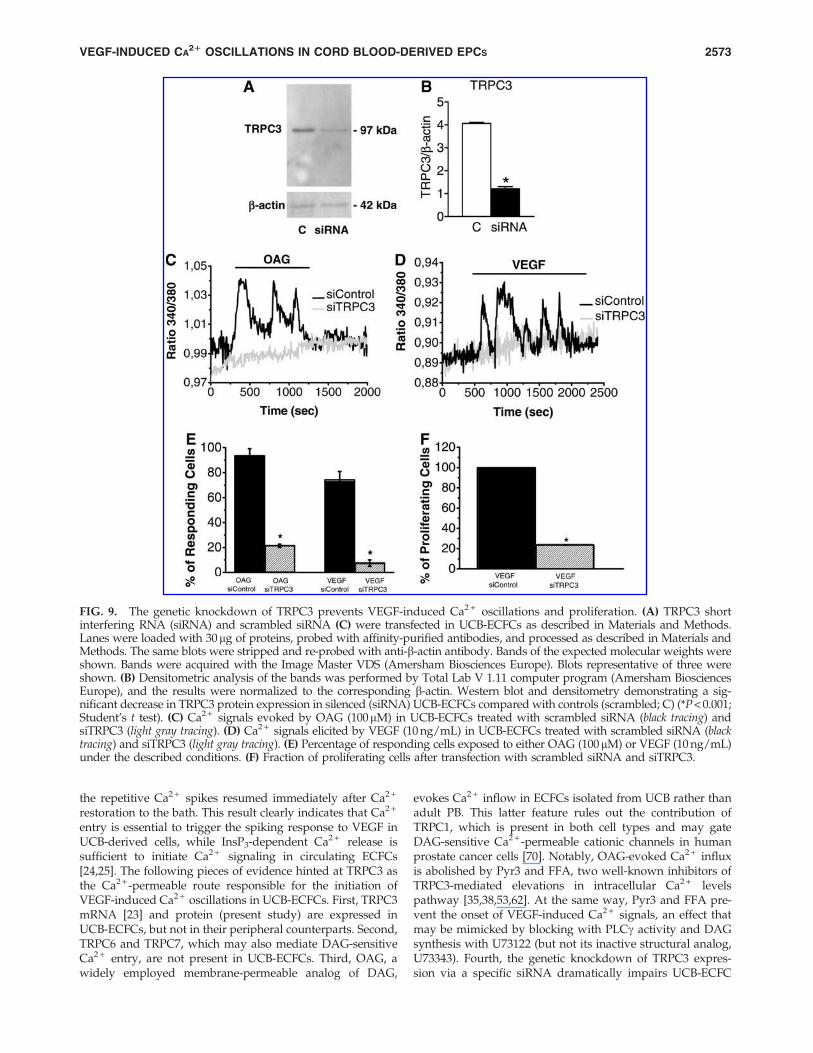
The genetic disruption of TRPC3 prevents VEGF-induced Ca2 + oscillations in UCB-ECFCs
The role served by TRPC3 in igniting the Ca2 + response toVEGF was further assessed by down-regulating its expres-sion in UCB-ECFCs. The cells were transfected with either ansiRNA selectively targeting or a scrambled (ie, control) siR-NA. Immunoblot analysis revealed that the content ofTRPC3 protein decreased by *68.5% 72 h after siRNAtransfection (Fig. 9A, B), whereas TRPC1 and TRPC4 pro-teins were still present in TRPC3-deficient cells (Supple-mentary Fig. S8). Consistent with this finding, the geneticsuppression of TRPC3 dramatically reduced the percentageof UCB-ECFCs responding to OAG (100 mM) with an in-crease in [Ca2 + ]i as compared with control cells (Fig. 9C, E).
Similarly, the fraction of cells producing a detectable Ca2 +
signal when challenged with VEGF dramatically diminishedon siTRPC3 transfection (Fig. 9D, E). Under such conditions,the elevation in [Ca2 + ]i consisted of a rapid Ca2 + spike in 9out of 143 cells (6.3%), whereas VEGF-evoked repetitiveCa2 + spikes were observed only in 1 out of 143 cells (0.7%).Finally, the selective knockdown of TRPC3 significantly(P < 0.05) impaired VEGF-dependent UCB-ECFC prolifera-tion, by reducing the rate of cell growth by about 80% (Fig9F). Altogether, these results confirm that TRPC3-inducedCa2 + entry represents the triggering event of the oscillatoryresponse to VEGF in ECFCs isolated from UCB.
Discussion
Local delivery of autologous EPCs is now regarded as oneof the most promising strategies to treat severe ischaemicdiseases, such as MI and PAD, by regenerating the impaired
FIG. 7. Evidence for TRPC3-mediated Ca2 + entry in ECFCs isolated from UCB. (A) 1-oleoyl-2-acetyl-sn-glycerol (OAG)(100 mM), a membrane-permeable analog of diacylglycerol, induced a rapid increase in [Ca2 + ]i in UCB-ECFCs (black tracing),but not PB-ECFCs (light gray tracing). (B) The Ca2 + response to OAG (100mM) did not occur in the absence of extracellular Ca2 +
(0Ca2 + ), but resumed on Ca2 + re-addition to the bath. (C) Pyr3 (10mM, 10 min), a selective inhibitor of TRPC3, inhibited OAG-induced Ca2 + entry. OAG was administrated at 100mM. (D) Flufenamic acid (FFA; 100mM, 10 min), which may also interferewith TRPC3, reversibly abrogated OAG-induced Ca2 + inflow. OAG was administrated at 100mM. (E) BTP-2 (20mM, 30 min) didnot prevent the Ca2 + response to OAG (100mM). (F) Percentage of UCB-ECFCs displaying OAG-induced Ca2 + signals in theabsence and in the presence of each TRPC3 blocker and of BTP-2. *Indicates P < 0.05, as described in Statistics paragraph.
VEGF-INDUCED CA21 OSCILLATIONS IN CORD BLOOD-DERIVED EPCS 2571
vascular network [1–3,9–12]. In particular, ECFCs stand outas the most suitable EPC subset by virtue of their ability toacquire a truly endothelial phenotype, to assembly into abidimensional capillary-like network in vitro, and formpatent vessels in vivo [1,6,13,15,22,25,67]. An inherent limi-tation in autologous cell therapy is provided by the scarcityof circulating ECFCs and by their limited angiogenic re-sponse to VEGF as compared with UCB-derived cells [14].These features suggest that the signaling pathways by whichVEGF activates ECFCs are different depending on theirsource, that is, either peripheral or UCB. Highlighting themolecular mediators of VEGF-induced UCB-ECFC prolifer-ation might supply the biological bases required to improvethe clinical outcome of cell therapy by a genetic interventionon the underlying regulatory machinery [1,3,9,68,69].
We have recently shown that VEGF stimulates PB-ECFCsto undergo angiogenesis by an oscillatory increase in [Ca2 + ]i,which is tightly patterned by the concerted interplay be-tween InsP3-dependent Ca2 + release and consequent SOCE
activation [25]. More specifically, the Ca2 + response to VEGFis initiated by InsP3-induced intracellular Ca2 + mobilization;whereas store-dependent Ca2 + entry is required to maintainit over time. We, therefore, speculated that the Ca2 + responseto VEGF in UCB-ECFCs might involve the activation of ei-ther different and/or additional Ca2 + -permeable channels.This hypothesis was sustained by the notion that a differentblend of ion channels is expressed in UCB- versus PB-derived cells [23] and by the observation that the Ca2 + toolkitexpressed by ECFCs is not set in stone, but may be dramat-ically remodeled, as observed when they are harvested bypatients affected by renal cellular carcinoma [22]. Similar totheir peripheral counterparts [24,25], VEGF induces UCB-ECFCs to undergo asynchronous oscillations in [Ca2 + ]i,which stochastically arise in the presence of the growth factorthroughout the stimulation. The major difference between thetwo cell types was observed when UCB-ECFCs were chal-lenged with VEGF in the absence of extracellular Ca2 + : Un-like PB-ECFCs, no evident increase in [Ca2 + ]i occurred, but
FIG. 8. The pharmacological blockade of TRPC3 suppresses VEGF-induced Ca2 + oscillations in ECFCs harvested from UCB.The spiking response to VEGF (10 ng/mL) arose in the absence (A), but not in the presence of either Pyr3 (10mM, 10 min; B) orFFA (100mM, 10 min; C). (D) The acute addition of Pyr3 (10mM) rapidly inhibited VEGF-induced Ca2 + transients. VEGF wasadministrated at 10 ng/mL. (E) Percentage of UCB-ECFCs responding to VEGF (10 ng/mL) in the absence and in the presence ofPyr3 and FFA. (F) Fraction of proliferating UCB-ECFCs in the absence (Control; Ctl) and in the presence of the followinginhibitors of VEGF-evoked Ca2 + oscillations: Pyr3 (10mM), FFA (100mM), and 1,2-bis(o-aminophenoxy) ethane-N,N,N¢,N¢-tetraacetic acid (BAPTA) (30mM; BAP), a well-known membrane permeable buffer of intracellular Ca2 + , and BTP-2 (20mM).*Indicates P < 0.05, as described in Statistics paragraph.
2572 DRAGONI ET AL.
the repetitive Ca2 + spikes resumed immediately after Ca2 +
restoration to the bath. This result clearly indicates that Ca2 +
entry is essential to trigger the spiking response to VEGF inUCB-derived cells, while InsP3-dependent Ca2 + release issufficient to initiate Ca2 + signaling in circulating ECFCs[24,25]. The following pieces of evidence hinted at TRPC3 asthe Ca2 + -permeable route responsible for the initiation ofVEGF-induced Ca2 + oscillations in UCB-ECFCs. First, TRPC3mRNA [23] and protein (present study) are expressed inUCB-ECFCs, but not in their peripheral counterparts. Second,TRPC6 and TRPC7, which may also mediate DAG-sensitiveCa2 + entry, are not present in UCB-ECFCs. Third, OAG, awidely employed membrane-permeable analog of DAG,
evokes Ca2 + inflow in ECFCs isolated from UCB rather thanadult PB. This latter feature rules out the contribution ofTRPC1, which is present in both cell types and may gateDAG-sensitive Ca2 + -permeable cationic channels in humanprostate cancer cells [70]. Notably, OAG-evoked Ca2 + influxis abolished by Pyr3 and FFA, two well-known inhibitors ofTRPC3-mediated elevations in intracellular Ca2 + levelspathway [35,38,53,62]. At the same way, Pyr3 and FFA pre-vent the onset of VEGF-induced Ca2 + signals, an effect thatmay be mimicked by blocking with PLCg activity and DAGsynthesis with U73122 (but not its inactive structural analog,U73343). Fourth, the genetic knockdown of TRPC3 expres-sion via a specific siRNA dramatically impairs UCB-ECFC
FIG. 9. The genetic knockdown of TRPC3 prevents VEGF-induced Ca2 + oscillations and proliferation. (A) TRPC3 shortinterfering RNA (siRNA) and scrambled siRNA (C) were transfected in UCB-ECFCs as described in Materials and Methods.Lanes were loaded with 30mg of proteins, probed with affinity-purified antibodies, and processed as described in Materials andMethods. The same blots were stripped and re-probed with anti-b-actin antibody. Bands of the expected molecular weights wereshown. Bands were acquired with the Image Master VDS (Amersham Biosciences Europe). Blots representative of three wereshown. (B) Densitometric analysis of the bands was performed by Total Lab V 1.11 computer program (Amersham BiosciencesEurope), and the results were normalized to the corresponding b-actin. Western blot and densitometry demonstrating a sig-nificant decrease in TRPC3 protein expression in silenced (siRNA) UCB-ECFCs compared with controls (scrambled; C) (*P < 0.001;Student’s t test). (C) Ca2 + signals evoked by OAG (100mM) in UCB-ECFCs treated with scrambled siRNA (black tracing) andsiTRPC3 (light gray tracing). (D) Ca2 + signals elicited by VEGF (10 ng/mL) in UCB-ECFCs treated with scrambled siRNA (blacktracing) and siTRPC3 (light gray tracing). (E) Percentage of responding cells exposed to either OAG (100mM) or VEGF (10 ng/mL)under the described conditions. (F) Fraction of proliferating cells after transfection with scrambled siRNA and siTRPC3.
VEGF-INDUCED CA21 OSCILLATIONS IN CORD BLOOD-DERIVED EPCS 2573

sensitivity to OAG and suppresses the oscillatory response toVEGF. Fifth, both the pharmacological (Pyr3 and FFA) andthe genetic (siRNA) inhibition of TRPC3-gated Ca2 + entrystrongly affects VEGF-induced UCB-ECFC proliferation.Sixth, the blockade of SOCE by pre-incubating the cells withBTP-2 does not affect the onset of the Ca2 + response, albeit itdramatically reduces the number of the following Ca2 +
transients. As a consequence, the specific expression ofTRPC3 in ECFCs harvested from UCB does not change thekinetics of the Ca2 + response, that is, asynchronous Ca2 +
spikes, to VEGF as compared with their peripheral counter-parts. Nevertheless, it profoundly alters its underlyingmechanisms; whereas TRPC3 is absolutely required both totrigger the signal and to maintain Ca2 + transients over time.Indeed, the acute addition of Pyr3 during an ongoing re-sponse causes the rapid interruption of the [Ca2 + ]i elevation.This finding concurs with previous studies, which docu-mented the role served by TRPC3 as a stimulator of intra-cellular Ca2 + spiking in a variety of cell types. For instance,TRPC3 drives the spontaneous Ca2 + oscillations arising inhuman umbilical vein-derived endothelial cell line EA.hy926plated on Matrigel [71] and the periodic increases in [Ca2 + ]i
elicited by B-cell receptor engagement [38,72]. In addition,TRPC3-dependent Ca2 + entry triggers thrombin-evoked re-petitive Ca2 + spikes in 1321N1 human astrocytoma cells [73],as well as antigen-induced intracellular Ca2 + waves in ratbasophilic leukemia and BM-derived rat mast cells [74]. Fi-nally, TRPC3 activation causes cyclic oscillations in [Ca2 + ]i inrat type I astrocytes [59] and is likely to underpin OAG-induced repetitive Ca2 + spikes in human myometrial cells[75]. Conversely, the monotonic Ca2 + entry mediated byTRPC3 in CD133 + progenitor cells isolated from human ad-ipose stroma and exposed to VEGF does not lead to an in-tracellular Ca2 + spike [55]. A caveat in the interpretation ofthe data generated by the pharmacological inhibition ofTRPC3 is represented by the lack of selectivity of the inhibi-tors that we employed. FFA may also affect TRPC6, which ishowever absent in UCB-ECFCs [23], and TRPC4 [65]; whilePyr3 has been recently reported to interfere with Orai1-mediated SOCE [66]. Similarly, BTP-2 has previously been re-ported to affect TRPC3-dependent Ca2 + entry in heterologousexpression systems [63]. However, the Ca2 + response to OAG isnot affected by BTP-2 in UCB-ECFCs (present study); whereas itis sensitive to both Pyr3 and FFA. These results suggest that theside-effects of Ca2 + channels blockers may be absent in thesecells and related to ancillary subunits that are not expressed inECFCs. Consistently, control experiments conducted on PB-ECFCs, which express TRPC1, TRPC4, and Orai1 but lackTRPC3, revealed that the Ca2 + response to VEGF still occurredand adopted an oscillatory pattern in the presence of either Pyr3or FFA. Finally, immunoblotting analysis disclosed thatTRPC3-deficient cells do not manifest evident off-target effectson the Ca2 + machinery, such as a drop in TRPC1 and TRPC4proteins. These data strongly indicate that Pyr3 and FFA areunlikely to affect SOCE in UCB-ECFCs, but caution should bewarranted when these compounds are utilized in the absence ofgene silencing as a confirmatory approach.
The Ca2 + signaling machinery placed downstreamTRPC3-mediated Ca2 + entry in ECFCs isolated from UCB issomehow similar to that recruited by VEGF in their periph-eral counterparts [22–25,76]. Accordingly, TRPC3-initiatedCa2 + oscillations are shaped by the rhythmic Ca2 + release
through InsP3Rs and sustained over time by store-dependentCa2 + influx accomplished by Stim1, Orai1, and TRPC1. Thisscenario is corroborated by four pieces of evidence. First, thepharmacological inhibition of InsP3-dependent signalingwith either U73122 or 2-APB, a well-known blocker ofInsP3Rs, prevents the onset of the Ca2 + response to VEGF inUCB-ECFCs, as well as in their peripheral counterparts[24,25]. Unfortunately, 2-APB is not a selective drug, as itmay also interfere with Stim1, Orai1, and TRPC1, all ofwhich contribute to SOCE in human ECFCs [22,23,76], andTRPC3 at the concentration employed in the present study(ie, 50mM) [3,53]. Since 2-APB could not be probed in theabsence of extracellular Ca2 + , which is required for thegeneration of the Ca2 + oscillations, UCB-ECFCs were ex-posed to VEGF on depletion of the InsP3-sensitive Ca2 + poolwith either CPA or thapsigargin [23,25]. Under these condi-tions, VEGF causes a monotonic elevation in [Ca2 + ]i, whichis a hallmark of VEGF-induced DAG-activated Ca2 + entry inmature endothelium [32–34,54]. This result indicates thatTRPC3-mediated Ca2 + entry controls InsP3-dependent Ca2 +
mobilization and, as a consequence of the drop in ER Ca2 +
levels, it ultimately leads to SOCE activation. Second, UCB-ECFCs express the transcripts encoding for two of the threehitherto identified InsP3R isoforms, namely InsP3R2 andInsP3R3, whereas they lack InsP3R1. This feature adds to thegrowing list of molecular differences in the Ca2 + machineryreported in PB- versus UCB-derived ECFCs, as the formerpresent all the three isotypes [25]. It is, however, worth ofnoting that InsP3R2 is the isoform that is more strictly re-quired for both the onset and the maintenance of Ca2 + os-cillations, due to its peculiar sensitivity to both InsP3 andCa2 + [77]. Third, the slow pacemaker-like increase in [Ca2 + ]i
which precedes the Ca2 + spikes, is a hallmark of InsP3-dependent Ca2 + release [25,39,42,43], as well as the sto-chastic sequence of the intracellular Ca2 + spikes elicited byVEGF has been faithfully modeled by the random openingof InsP3R within the ER membrane [44–46]. Fourth, the in-hibition of SOCE, which is activated on depletion of theInsP3-dependent Ca2 + store in ECFCs [3,22,23], dramaticallycurtails the duration of the oscillatory response induced byVEGF in the presence of BTP-2. In particular, SOCE isrequired to recharge the ER Ca2 + reservoir in an SERCA-dependent manner during cell stimulation. This same func-tion, that is, refilling the InsP3-dependent Ca2 + pool in ago-nist-solicited cells, has long been linked to store-dependentCa2 + inflow [78], albeit the signaling role served by SOCE inthe context of gene expression modulation has been recentlyunveiled [30,44,78]. SOCE in human ECFCs is mediated bythe interaction of Stim1, the ER Ca2 + -sensor, with Orai1 andTRPC1, both of which contribute to the Ca2 + -permeable poreon the PM. It is, however, yet to be elucidated whether Stim1,Orai1, and TRPC1 assemble into a heteromeric ternary com-plex, as described in human platelets [28], or whether TRPC1and Orai1 constitute two distinct store-operated pathways,each of which are activated by Stim1 [79]. While TRPC3 isonly expressed by UCB-ECFCs [23], Stim1, Orai1, and TRPC1are present in ECFCs isolated from both UCB and PB[22,23,25]. This feature highlights the central role served bySOCE-related proteins in maintaining the oscillatory responseto VEGF over time in different types of ECFCs.
The question then arises about the mechanistic link be-tween TRPC3 activation and the ignition of the repetitive
2574 DRAGONI ET AL.

intracellular Ca2 + waves in UCB-ECFCs. Three alternative,albeit not mutually exclusive, scenarios might be envisaged.The first model is based on the well-known increase inInsP3R sensitivity to InsP3 on elevation of both luminal andcytosolic Ca2 + , the latter mechanisms referred to as Ca2 + -induced Ca2 + release (CICR) [47,48,58]. If either the luminalCa2 + load and/or the ambient levels of Ca2 + are too low toenable InsP3-dependent Ca2 + mobilization, TRPC3-gatedCa2 + inflow might provide the bolus of Ca2 + required tosensitize the receptors to their ligand, that is, InsP3, and ac-tivate the release. This hypothesis is supported by the findingthat extracellular Ca2 + influx may control the frequency ofCa2 + oscillations by triggering InsP3-dependent Ca2 + dis-charges in a number of cell types [80,81]. Nevertheless, thereis no report of direct InsP3R modulation by TRPC3-mediatedCa2 + inflow. Moreover, Pyr3 caused an instantaneous ter-mination of ongoing Ca2 + transients, which suggests thatTRPC3-induced Ca2 + influx stimulates the spiking responseas long as VEGF is administrated to the cells. Under suchconditions, however, the cytosol is turned into an excitablemedium by the rhythmic Ca2 + release, and the ER is con-tinuously refurnished with Ca2 + by SOCE, so that it is hardto conceive that InsP3R is not sensitive to InsP3. Therefore,TRPC3 should be more tightly linked to InsP3R activation.According to the second model, TRPC3 might induce Ca2 +
release by mediating the selective interaction betweenInsP3R1 and the receptor for activated C-kinase-1 (RACK1)[82,83]. However, this mechanism implies the formation of aternary complex formed by TRPC3, InsP3R1 and RACK1 andis not consistent with our observations that Ca2 + inflowthrough TRPC3, rather than the channel protein itself, is es-sential to trigger the oscillatory response and that InsP3R1 isabsent in UCB-ECFCs. Moreover, the physical recruitment ofInsP3R1 by TRPC3 leads to a sustained increase in [Ca2 + ]i
rather than to periodic Ca2 + pulses [82], as observed underour conditions. Conversely, according to the third model,TRPC3-induced Ca2 + entry might promote InsP3 synthesisby enhancing the rate of PLCg activation. Indeed, TRPC3elicits translocation toward the PM and subsequent activa-tion of the Ca2 + -sensitive PLCg in antigen-stimulated DT40B lymphocytes [72], a process that is suppressed by eitherPyr3 [35] or TRPC3 down-regulation by siRNA [72]. Wesuggest that the amount of InsP3 produced immediately afterVEGFR-2 activation is not sufficient to initiate the intracel-lular Ca2 + spikes, either because of scarce PLCg recruitmentto the PM or rapid InsP3 metabolism. However, DAG, whichis generated along with InsP3, gates TRPC3 to provide thesource of Ca2 + that is necessary to further stimulate PLCgand trigger the first Ca2 + pulse in an InsP3-sensitive manner.The following drop in the ER Ca2 + content will lead to Stim1heteromerization and translocation beneath the PM, where itrecruits Orai1 and TRPC1 to mediate SOCE, thereby re-charging the intracellular Ca2 + store. This positive feedbackbetween PLCg, DAG, TRPC3, InsP3R, and SOCE occursthroughout the oscillatory signal, as suggested by its strongsensitivity to Pyr3. This hypothesis is supported by the ob-servation that PLCd, another PLC isoform tightly regulatedby sub-membranal Ca2 + levels, is recruited by Ca2 + entry tosustain InsP3-dependent Ca2 + oscillations in damaged en-dothelium [8,84]. This model implies that VEGF-inducedInsP3 synthesis in PB-ECFCs is well beyond the thresholdrequired to ignite a robust intracellular Ca2 + discharge, so
that the spiking response occurs even in the absence of ex-tracellular Ca2 + entry.
The Ca2 + responses generated by adjacent/neighboringCB-ECFCs to VEGF differ among each other in terms oflatent period, number, and period of the Ca2 + spikes.Variability in the pattern of Ca2 + signals is the hallmark ofgrowth factors-evoked elevations in [Ca2 + ]i in mature en-dothelial cells [21,39]. Moreover, we have recently found thatVEGF-evoked Ca2 + oscillations are highly heterogeneous inPB-ECFCs as well [25]. The molecular underpinnings of thediversity in the spatiotemporal dynamics of VEGF-inducedCa2 + events are far from being fully understood. A combi-nation of experimental and computational studies has dis-closed that such a feature is not peculiar to Ca2 + signaling,but is an intrinsic property of tyrosine kinase receptor acti-vation. For instance, the differential activation of jagged1 andDII4 in the Notch signaling pathway determines the distinctfate (tip vs. stalk) of two adjacent endothelial cells exposed toa VEGF gradient during the angiogenic process [85]. A recentstudy has further demonstrated that the mitogen-activatedprotein kinase pathway is triggered by VEGF in individualmicrovascular endothelial cells, whereas their adjoining cellsare silent [86]. The dissimilar response of neighboring cells toVEGF might be linked to the wide variation in VEGFR-2levels, as only about 60% of UCB-ECFCs express it (unpub-lished data from our group). This feature might drive theobserved differences in the signal strength, as originally re-ported for the epidermal growth factor receptor [87]. Theheterogeneous number of VEGFR-2 molecules on the endo-thelial membrane quantitatively and qualitatively activatesdistinct intracellular pathways within the same cell mono-layer [88]. The heterogeneity in VEGF signaling is exacer-bated by the internalization of VEGFR-2 [89], whichcontributes toward coordinating the complementary func-tions of adjoining cells during tissue morphogenesis, and itsregulation by binding partners, such as VEGFR1 and neu-ropilin-1, that are differentially distributed within the endo-thelial network [26]. An additional source of variability isprovided by the release of the soluble, ligand-sequesteringform of VEGFR1 (sFLT-1), that engenders a difference in theamount of VEGF sensed by individual cells, albeit placed inclose proximity to each other [90]. Alternatively, the specificCa2 + signatures evoked by VEGF in UCB-ECFCs might in-volve a difference in the sub-cellular distribution of the un-derlying signaling pathways, such as InsP3Rs [44–46]. Thestochastic behavior of the periodic fluctuations in [Ca2 + ]i
evoked by VEGF in UCB-ECFCs corroborates this hypothe-sis. VEGF-dependent Ca2 + oscillations arise randomly, andthe standard deviation of their ISI is of the same order ofmagnitude as the average value. A number of computationalstudies have revealed the mechanistic link between thisfeature and the topographical hierarchy of InsP3Rs [25–27].InsP3Rs are spatially arranged into clusters of 1-to-15 chan-nels, which are scattered within the ER membrane withdistances of 1–7mm. The coupling between adjoining chan-nels and clusters is accomplished by Ca2 + diffusion throughthe CICR process. As a consequence, InsP3 locally synthe-sized by PLCg primes all the channels in the cluster foractivation by Ca2 + and originates a Ca2 + puff, the elemen-tary Ca2 + event. The probability that a single Ca2 + initiatesCa2 + release from an adjacent site is rather low, as Ca2 + is apoorly diffusible messenger for distances £ 2 mm [45,46,91].
VEGF-INDUCED CA21 OSCILLATIONS IN CORD BLOOD-DERIVED EPCS 2575

Conversely, if, by chance, a supercritical number of InsP3Rclusters open in synchrony, the CICR couples all the unitaryCa2 + puffs and ignites a global Ca2 + spike [25–26]. Theprobability that such a cell wide event (ie, an intracellularCa2 + wave) occurs depends on several parameters, that is,strength of the spatial coupling, intracellular Ca2 + buffering,and puff duration, and is responsible for the cell-to-cell vari-ability reported by Ca2 + imaging measurements [44–46,91].Mathematical modeling unveiled that the heterogeneity in thepattern of Ca2 + oscillations arising within adjacent/neigh-boring cells is independent of morphological parameters,such as cell volume, ER volume, and shape [91,92]. Thisfinding was supported by lack of any statistically relevantcorrelation between variability in cell volume and intracel-lular Ca2 + dynamics in VEGF-stimulated UCB-ECFCs. It is,however, worth noting that earlier computational studiespointed at a cross-talk between the pattern of Ca2 + oscilla-tions and cellular morphology [93]. For instance, a significantenlargement in cell surface area may lead to a decrease in theISI [93]. More sophisticated experiments should thus be de-voted to further addressing this intriguing mechanistic issue.
The biological meaning of VEGF-induced Ca2 + oscilla-tions is to drive UCB-ECFC proliferation, as shown by thesensitivity of this process to Pyr3, FFA, BAPTA, BTP-2, andsiTRPC3. In addition, our preliminary results indicate thatthe Ca2 + response to VEGF regulates UCB-ECFCs assemblyinto capillary-like networks in vitro. These results are similarto our previous report on PB-ECFCs [25]. The intracellularCa2 + transients are generated by the rhythmic Ca2 + releasefrom InsP3Rs, while SOCE is essential to refill the ER Ca2 +
pool during prolonged stimulation. Therefore, InsP3 is likelyto mediate the pro-angiogenic effect of VEGF-induced Ca2 +
burst. Consistently, InsP3 was found to drive VEGF-elicitedmigration and proliferation in HUVEC [94,95], in bovineretinal endothelial cells [49], and in human choroidal endo-thelial cells [96]. More in general, InsP3-dependent Ca2 +
mobilization has been linked to cell division, survival, andmotility in a variety of cell types [41,48,97], and a remodelingof the InsP3 signaling machinery has been described inhighly proliferating and metastatic tumor cells [98,99].SOCE, however, does not only fulfil the function to replenishemptied stores, but it also controls a number of vital cellularprocesses, including gene expression, migration, and nitricoxide synthesis, via the physical coupling between Orai1 anda number of Ca2 + -sensitive decoders [3,30,31]. Therefore, thepossibility that the biological message encoded by the re-petitive Ca2 + spikes, that is, UCB-ECFC proliferation, is notonly delivered by InsP3Rs, but is also contributed by SOCE,should not be ruled out.
It has long been known that the pro-angiogenic responseto VEGF, as evaluated in terms of in vitro proliferation andtubulogenic rates, is dramatically higher in ECFCs isolatedfrom UCB rather than from PB [14]. This notion was notaccompanied by the knowledge of any relevant difference inthe signal transduction pathways downstream of VEGF inthe two cell types. In this context, the involvement of TRPC3in VEGF-induced Ca2 + oscillations and proliferation in UCB-ECFCs gains particular interest in the light of the selectivecoupling between TRPC3 and a number of Ca2 + -sensitiveregulators of angiogenesis, such as NF-kB, NFAT, and ERK[100,101]. For instance, in DT40 B cells stimulated with ananti-B cell receptor antibody, TRPC3 recruits to the PM
protein kinase Cb (PKCb), which, in turn, phosphorylatesand stimulates ERK [38]. Moreover, TRPC3-gated Ca2 + in-flux controls NF-kB signaling in human coronary artery en-dothelial cells [37], whereas Ca2 + entry via TRPC3, ratherthan through L-type (CaV1.2) Ca2 + channels, is selectivelyconveyed to calcineurin to induce the nuclear translocationof NFAT in HL-1 atrial myocytes [36]. In agreement withthese findings, TRPC3 induces the expression of hypertro-phy-associated genes in response to hypertrophic stimuli inrat ventricular myocytes, while it does not interfere withCaV1.2-paced cell contraction [102]. Finally, it has beendemonstrated that TRPC3-induced Ca2 + influx is essentialfor endothelial tube formation and proliferation in EA.hy926cells [71] and for vascular cell adhesion molecule-1 expres-sion in human coronary artery endothelial cells [37]. Thetight coupling between TRPC3-mediated Ca2 + inflow andVEGF-induced proliferation is highlighted by the dramaticeffect on UCB-ECFC growth exerted by Pyr3, FFA, andsiTRPC3. Indeed, all of these treatments blocked the prolif-erative response to VEGF to a much higher extent thansimply buffering intracellular Ca2 + with BAPTA. As out-lined in a recent review [103], the weak effect of BAPTAindicates that the Ca2 + -sensitive decoder(s) should be placedat a distance much shorter than 10–100 nm from the channelpore; whereas the inhibition is significantly higher when theCa2 + source is abrogated by either pharmacological or ge-netic interventions. It, thus, appears that TRPC3 has thepotential to increase the repertoire of Ca2 + -sensitive pro-angiogenic pathways that are activated during VEGF-in-duced Ca2 + oscillations. In this view, it is tempting tospeculate that the selective expression of a functional TRPC3protein in UCB-ECFCs contributes to render these cells moresensible to VEGF as compared with PB-derived cells. Ex-periments are under way in our laboratory to assess whetherTRPC3 expression in PB-ECFCs will enhance their angio-genic response to VEGF in vitro.
Conclusion
The present investigation described the molecular under-pinnings of VEGF-induced proliferation in ECFCs, the onlyEPC subset committed to acquire a truly mature endothelialphenotype, isolated from UCB. We found that the VEGFactivates UCB-ECFCs by inducing oscillatory changes in[Ca2 + ]i which are not synchronized between adjacent cellsand are shaped by the concerted interaction between InsP3-dependent Ca2 + release and SOCE activation. These featuresare similar to those described in their peripheral counter-parts, but a major difference is represented by the absoluterequirement for TRPC3-mediated Ca2 + entry to initiate andsustain the spiking response. It has long been known thatEPCs are more sensitive to VEGF stimulation when har-vested from UCB rather than from PB. This property alsoapplies to ECFCs; as a consequence, UCB-ECFCs appearmore amenable for the purposes of cell-based therapy ofischemic diseases than PB-derived cells. The selective con-tribution of TRPC3 to VEGF-induced Ca2 + signals in UCB-ECFCs hints at this channel as a potential candidate toachieve a genetic improvement of PB-ECFC functionality byvirtue of its selective coupling to a number of Ca2 + -sensitivesignaling pathways involved in endothelial proliferation,migration, and differentiation.
2576 DRAGONI ET AL.

Author Disclosure statement
The authors declare no conflict of interest.
References
1. Moccia F, F Lodola, S Dragoni, E Bonetti, C Bottino, GGuerra, U Laforezan, V Rosti and F Tanzi. (2012). Ca2 +
signalling in endothelial progenitor cells: a novel means toimprove cell-based therapy and impair tumour vascular-isation. Curr Vasc Pharmacol, under press.
2. Moccia F, E Bonetti, S Dragoni, J Fontana, F Lodola, R BerraRomani, U Laforenza, V Rosti and F Tanzi. (2012). Hema-topoietic progenitor and stem cells circulate by surfing onintracellular Ca2 + waves: a novel target for cell-basedtherapy and anti-cancer treatment? Curr Signal TransdTher 7:161–176.
3. Moccia F, S Dragoni, F Lodola, E Bonetti, C Bottino, GGuerra, U Laforenza, V Rosti and F Tanzi. (2012). Store-dependent Ca2 + entry in endothelial progenitor cells as aperspective tool to enhance cell-based therapy and adversetumour vascularization. Curr Med Chem 19:5802–2818.
4. Yoder MC. (2010). Defining human endothelial progenitorcells. Exp Hematol 38:S111–S111.
5. Critser PJ and MC Yoder. (2010). Endothelial colony-forming cell role in neoangiogenesis and tissue repair. CurrOpin Organ Transplant 15:68–72.
6. Yoder MC. (2012). Human endothelial progenitor cells.Cold Spring Harb Perspect Med 2:a006692.
7. Yoder M and S Rounds. (2011). Bad blood, bad endotheli-um: ill fate? Blood 117:3479–3480.
8. Moccia F, JE Avelino-Cruz, Y Sanchez-Hernandez and FTanzi. (2010). Ca2 + signalling in damaged endothelium: doconnexin hemichannels aid in filling the gap? Curr DrugTher 5:277–287.
9. Asahara T. (2007). Cell therapy and gene therapy usingendothelial progenitor cells for vascular regeneration.Handb Exp Pharmacol 180:181–194.
10. Pompilio G, MC Capogrossi, M Pesce, F Alamanni, C Di-Campli, F Achilli, A Germani and P Biglioli. (2009). En-dothelial progenitor cells and cardiovascular homeostasis:clinical implications. Int J Cardiol 131:156–167.
11. Germani A, C Di Campli, G Pompilio, P Biglioli and MCCapogrossi. (2009). Regenerative therapy in peripheral ar-tery disease. Cardiovasc Ther 27:289–304.
12. Passier R, LW van Laake and CL Mummery. (2008). Stem-cell-based therapy and lessons from the heart. Nature 453:322–329.
13. Hirschi KK, DA Ingram and MC Yoder. (2008). Assessingidentity, phenotype, and fate of endothelial progenitorcells. Arterioscler Thromb Vasc Biol 28:1484–1595.
14. Ingram DA, LE Mead, H Tanaka, V Meade, A Fenoglio, KMortell, K Pollok, MJ Ferkowicz, D Gilley and MC Yoder.(2004). Identification of a novel hierarchy of endothelialprogenitor cells using human peripheral and umbilical cordblood. Blood 104:2752–2760.
15. Mead LE, D Prater, MC Yoder and DA Ingram. (2008).Isolation and characterization of endothelial progenitorcells from human blood. Curr Protoc Stem Cell BiolChapter 2:Unit 2C.1.
16. Melero-Martin JM, ME De Obaldia, S-Y Kang, ZA Khan, LYuan, P Oettgen and J Bischoff. (2008). Engineering robustand functional vascular networks in vivo with human adultand cord blood-derived progenitor cells. Circ Res 103:194–202.
17. Melero-Martin JM, ZA Khan, A Picard, X Wu, S Paruchuriand J Bischoff. (2007). In vivo vasculogenic potential ofhuman blood-derived endothelial progenitor cells. Blood109:4761–4768.
18. Ben-Shoshan J and J George. (2007). Endothelial progenitorcells as therapeutic vectors in cardiovascular disorders:from experimental models to human trials. Pharmacol Ther115:25–36.
19. Munaron L and AF Pla. (2009). Endothelial calciummachinery and angiogenesis: understanding physiology tointerfere with pathology. Curr Med Chem 16:4691–4703.
20. Pla AF, D Avanzato, L Munaron and IS Ambudkar. (2012).Ion channels and transporters in cancer. 6. Vascularizingthe tumor: TRP channels as molecular targets. Am J PhysiolCell Physiol 302:C9–C15.
21. Moccia F, R Berra-Romani and F Tanzi. (2012). Update onvascular endothelial Ca2 + signalling: a tale of ion channels,pumps and transporters. World J Biol Chem 3:127–158.
22. Lodola F, U Laforenza, E Bonetti, D Lim, S Dragoni, CBottino, HL Ong, G Guerra, C Ganini, et al. (2012). Store-operated Ca2 + entry is remodelled and controls in vitroangiogenesis in endothelial progenitor cells isolated fromtumoral patients. PLoS One 7:e42541.
23. Sanchez-Hernandez Y, U Laforenza, E Bonetti, J Fontana,S Dragoni, M Russo, JE Avelino-Cruz, S Schinelli, D Testa,et al. (2010). Store-operated Ca2 + entry is expressed inhuman endothelial progenitor cells. Stem Cells Dev 19:1967–1981.
24. Li J, RM Cubbon, LA Wilson, MS Amer, L McKeown, BHou, Y Majeed, S Tumova, VAL Seymour, et al. (2011).Orai1 and CRAC channel dependence of VEGF-activatedCa2 + entry and endothelial tube formation. Circ Res 108:1190–1198.
25. Dragoni S, U Laforenza, E Bonetti, F Lodola, C Bottino, RBerra-Romani, GC Bongio, MP Cinelli, G Guerra, et al.(2011). Vascular endothelial growth factor stimulatesendothelial colony forming cells proliferation and tubulo-genesis by inducing oscillations in intracellular Ca2 + con-centration. Stem Cells 29:1898–1907.
26. Koch S and L Claesson-Welsh. (2012). Signal transductionby vascular endothelial growth factor receptors. ColdSpring Harb Perspect Med 2:a006502.
27. Salido GM, SO Sage and JA Rosado. (2009). Biochemicaland functional properties of the store-operated Ca2 +
channels. Cell Signal 21:457–461.28. Jardin I, JJ Lopez, GM Salido and JA Rosado. (2008). Orai1
mediates the interaction between STIM1 and hTRPC1 andregulates the mode of activation of hTRPC1-forming Ca2 +
channels. J Biol Chem 283:25296–25304.29. Ong HL and IS Ambudkar. (2011). The dynamic com-
plexity of the TRPC1 channelosome. Channels 5:424–431.30. Parekh AB. (2011). Decoding cytosolic Ca2 + oscillations.
Trends Biochem Sci 36:78–87.31. Parekh AB. (2010). Store-operated CRAC channels: func-
tion in health and disease. Nat Rev Drug Discov 9:399–410.32. Cheng HW, AF James, RR Foster, JC Hancox and DO Bates.
(2006). VEGF activates receptor-operated cation channelsin human microvascular endothelial cells. ArteriosclerThromb Vasc Biol 26:1768–1776.
33. Hamdollah Zadeh MA, CA Glass, A Magnussen, JCHancox and DO Bates. (2008). VEGF-mediated elevatedintracellular calcium and angiogenesis in human micro-vascular endothelial cells in vitro are inhibited by dominantnegative TRPC6. Microcirculation 15:605–614.
VEGF-INDUCED CA21 OSCILLATIONS IN CORD BLOOD-DERIVED EPCS 2577

34. Ge RL, YL Tai, YY Sun, KC Zhou, SL Yang, TL Cheng, QFZou, F Shen and YZ Wang. (2009). Critical role of TRPC6channels in VEGF-mediated angiogenesis. Cancer Lett283:43–51.
35. Kiyonaka S, K Kato, M Nishida, K Mio, T Numaga, Y Sa-waguchi, T Yoshida, M Wakamori, E Mori, et al. (2009).Selective and direct inhibition of TRPC3 channels underliesbiological activities of a pyrazole compound. Proc NatlAcad Sci U S A 106:5400–5405.
36. Poteser M, H Schleifer, M Lichtenegger, M Schernthaner, TStockner, CO Kappe, TN Glasnov, C Romanin and KGroschner. (2011). PKC-dependent coupling of calciumpermeation through transient receptor potential canonical 3(TRPC3) to calcineurin signaling in HL-1 myocytes. ProcNatl Acad Sci U S A 108:13876–13878.
37. Smedlund K, J-Y Tano and G Vazquez. (2010). The consti-tutive function of native TRPC3 channels modulates vas-cular cell adhesion molecule-1 expression in coronaryendothelial cells through nuclear factor kappa B signaling.Circ Res 106:1479–1488.
38. Numaga T, M Nishida, S Kiyonaka, K Kato, M Katano, EMori, T Kurosaki, R Inoue, M Hikida, JW Putney, Jr. and YMori. (2010). Ca2 + influx and protein scaffolding viaTRPC3 sustain PKC beta and ERK activation in B cells. JCell Sci 123:927–938.
39. Moccia F, R Berra-Romani, S Tritto, S Signorelli, V Tagliettiand F Tanzi. (2003). Epidermal growth factor induces in-tracellular Ca2 + oscillations in microvascular endothelialcells. J Cell Physiol 194:139–150.
40. Foreman MA, J Smith and SJ Publicover. (2006). Char-acterisation of serum-induced intracellular Ca2 + oscilla-tions in primary bone marrow stromal cells. J Cell Physiol206:664–671.
41. Lipskaia L and AM Lompre. (2004). Alteration in temporalkinetics of Ca2 + signaling and control of growth and pro-liferation. Biol Cell 96:55–68.
42. Tovey SC, P de Smet, P Lipp, D Thomas, KW Young, LMissiaen, H De Smedt, JB Parys, MJ Berridge, J Thuring, AHolmes and MD Bootman. (2001). Calcium puffs are ge-neric InsP(3)-activated elementary calcium signals and aredownregulated by prolonged hormonal stimulation to in-hibit cellular calcium responses. J Cell Sci 114:3979–3989.
43. Bootman MD, MJ Berridge and P Lipp. (1997). Cookingwith calcium: the recipes for composing global signals fromelementary events. Cell 91:367–373.
44. Dupont G, L Combettes, GS Bird and JW Putney. (2011).Calcium oscillations. Cold Spring Harb Perspect Biol3:a004226.
45. Skupin A, H Kettenmann, U Winkler, M Wartenberg, HSauer, SC Tovey, CW Taylor and M Falcke. (2008). Howdoes intracellular Ca2 + oscillate: by chance or by the clock?Biophys J 94:2404–2411.
46. Skupin A and M Falcke. (2007). Statistical properties andinformation content of calcium oscillations. Genome Inform18:44–53.
47. Berridge MJ. (2007). Inositol trisphosphate and calciumoscillations. In: Cell Biology of Inositol Lipids and Phosphates.Wakelam MJO, ed. Portland Press Ltd, London. pp 1–7.
48. Berridge MJ. (2009). Inositol trisphosphate and calciumsignalling mechanisms. Biochim Biophys Acta 1793:933–940.
49. Banumathi E, A O’Connor, S Gurunathan, DA Simpson, JGMcGeown and TM Curtis. (2011). VEGF-induced retinal an-giogenic signaling is critically dependent on Ca2 + signaling
by Ca2 +/Calmodulin-dependent protein kinase II. InvestOphthalm Vis Sci 52:3103–3111.
50. Berra-Romani R, A Raqeeb, JE Avelino-Cruz, F Moccia, AOldani, F Speroni, V Taglietti and F Tanzi. (2008). Ca2 +
signaling in injured in situ endothelium of rat aorta. CellCalcium 44:298–309.
51. Romani RB, A Raqeeb, U Laforenza, MF Scaffino, F Moccia,JE Avelino-Cruz, A Oldani, D Coltrini, V Milesi, V Tagliettiand F Tanzi. (2009). Cardiac microvascular endothelial cellsexpress a functional Ca2 + -sensing receptor. J Vasc Res46:73–82.
52. Moccia F, GA Nusco, D Lim, K Kyozuka and L Santella.(2006). NAADP and InsP3 play distinct roles at fertilizationin starfish oocytes. Dev Biol 294:24–38.
53. Harteneck C and M Gollasch. (2011). Pharmacologicalmodulation of diacylglycerol-sensitive TRPC3/6/7 chan-nels. Curr Pharm Biotechnol 12:35–41.
54. Pupo E, AF Pla, D Avanzato, F Moccia, J-EA Cruz, F Tanzi,A Merlino, D Mancardi and L Munaron. (2011). Hydrogensulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Rad Biol Med 51:1765–1773.
55. Poteser M, A Graziani, P Eder, A Yates, H Maechler, CRomanin and K Groschner. (2008). Identification of a raresubset of adipose tissue-resident progenitor cells, whichexpress CD133 and TRPC3 as a VEGF-regulated Ca2 + entrychannel. FEBS Lett 582:2696–2702.
56. Kwan H-Y, K-T Cheng, Y Ma, Y Huang, NL Tang, S Yu andX Yao. (2010). CNGA2 contributes to ATP-induced non-capacitative Ca2 + influx in vascular endothelial cells. J VascRes 47:148–156.
57. Leung P-C, K-T Cheng, C Liu, W-T Cheung, H-Y Kwan,K-L Lau, Y Huang and X Yao. (2006). Mechanism of non-capacitative Ca2 + influx in response to bradykinin in vas-cular endothelial cells. J Vasc Res 43:367–376.
58. Berridge MJ, MD Bootman and HL Roderick. (2003). Cal-cium signalling: dynamics, homeostasis and remodelling.Nat Rev Mol Cell Biol 4:517–529.
59. Grimaldi M, M Maratos and A Verma. (2003). Transientreceptor potential channel activation causes a novel form ofCa2 + i oscillations and is not involved in capacitative Ca2 +
entry in glial cells. J Neurosci 23:4737–4745.60. Smedlund K and G Vazquez. (2008). Involvement of native
TRPC3 proteins in ATP-dependent expression of VCAM-1and monocyte adherence in coronary artery endothelialcells. Arterioscler Thromb Vasc Biol 28:2049–2055.
61. Vazquez G, J-Y Tano and K Smedlund. (2010). On the po-tential role of source and species of diacylglycerol inphospholipase-dependent regulation of TRPC3 channels.Channels 4:232–240.
62. Smedlund K, M Bah and G Vazquez. (2012). On the role ofendothelial TRPC3 channels in endothelial dysfunction andcardiovascular disease. Cardiovasc Hematol Agents MedChem 10:265–274.
63. He LP, T Hewavitharana, J Soboloff, MA Spassova and DL Gill.(2005). A functional link between store-operated and TRPCchannels revealed by the 3,5-bis(trifluoromethyl)pyrazolederivative, BTP2. J Biol Chem 280:10997–11006.
64. Foster RR, GI Welsh, SC Satchell, RD Marlow, MD Wher-lock, D Pons, PW Mathieson, DO Bates and MA Saleem.(2010). Functional distinctions in cytosolic calcium regula-tion between cells of the glomerular filtration barrier. CellCalcium 48:44–53.
65. Jiang H, B Zeng, G-L Chen, D Bot, S Eastmond, SEElsenussi, SL Atkin, AN Boa and S-Z Xu. (2012). Effect of
2578 DRAGONI ET AL.

non-steroidal anti-inflammatory drugs and new fenamateanalogues on TRPC4 and TRPC5 channels. Biochem Phar-macol 83:923–931.
66. Schleifer H, B Doleschal, M Lichtenegger, R Oppenrieder, IDerler, I Frischauf, TN Glasnov, CO Kappe, C Romaninand K Groschner. (2012). Novel pyrazole compounds forpharmacological discrimination between receptor-operatedand store-operated Ca2 + entry pathways. Brit J Pharmacol167:1712–1722.
67. Critser PJ, SL Voytik-Harbin and MC Yoder. (2011). Iso-lating and defining cells to engineer human blood vessels.Cell Prolif 44:15–21.
68. Kalka C and I Baumgartner. (2008). Gene and stem celltherapy in peripheral arterial occlusive disease. Vasc Med13:157–172.
69. Penn MS and AA Mangi. (2008). Genetic enhancement ofstem cell engraftment, survival, and efficacy. Circ Res 102:1471–1482.
70. Sydorenko V, Y Shuba, S Thebault, M Roudbaraki, G Le-page, N Prevarskaya and R Skryma. (2003). Receptor-cou-pled, DAG-gated Ca2 + -permeable cationic channels inLNCaP human prostate cancer epithelial cells. J Physiol548:823–836.
71. Antigny F, N Girardin and M Frieden. (2012). Transientreceptor potential canonical channels are required for invitro endothelial tube formation. J Biol Chem 287:5917–5927.
72. Nishida M, K Sugimoto, Y Hara, E Mori, T Morii, T Kur-osaki and Y Mori. (2003). Amplification of receptor sig-nalling by Ca2 + entry-mediated translocation andactivation of PLC gamma 2 in B lymphocytes. EMBO J 22:4677–4688.
73. Nakao K, H Shirakawa, A Sugishita, I Matsutani, T Nii-dome, T Nakagawa and S Kaneko. (2008). Ca2 + mobiliza-tion mediated by transient receptor potential canonical 3 isassociated with thrombin-induced morphological changesin 1321N1 human astrocytoma cells. J Neurosci Res 86:2722–2732.
74. Cohen R, A Torres, H-T Ma, D Holowka and B Baird.(2009). Ca2 + waves initiate antigen-stimulated Ca2 + re-sponses in mast cells. J Immunol 183:6478–6488.
75. Shlykov SG and BA Sanborn. (2004). Stimulation of intra-cellular Ca2 + oscillations by diacylglycerol in humanmyometrial cells. Cell Calcium 36:157–164.
76. Beech DJ. (2012). Orai1 calcium channels in the vasculature.Pflugers Arch 463:635–647.
77. Taylor CW and SC Tovey. (2010). IP(3) receptors: towardunderstanding their activation. Cold Spring Harb PerspectBiol 2:a004010.
78. Parekh AB and JW Putney. (2005). Store-operated calciumchannels. Physiol Rev 85:757–810.
79. Cheng KT, X Liu, HL Ong, W Swaim and IS Ambudkar.(2011). Local Ca2 + entry via Orai1 regulates plasma mem-brane recruitment of TRPC1 and controls cytosolic Ca2 +
signals required for specific cell functions. PLos Biol 9:e1001025.
80. Bootman MD, KW Young, JM Young, RB Moreton and MJBerridge. (1996). Extracellular calcium concentration con-trols the frequency of intracellular calcium spiking inde-pendently of inositol 1,4,5-trisphosphate production inHeLa cells. Biochem J 314:347–354.
81. Shuttleworth TJ and JL Thompson. (1996). Ca2 + entrymodulates oscillation frequency by triggering Ca2 + release.Biochem J 313:815–819.
82. Bandyopadhyay BC, HL Ong, TP Lockwich, X Liu, BCParia, BB Singh and IS Ambudkar. (2008). TRPC3 controlsagonist-stimulated intracellular Ca2 + release by mediatingthe interaction between inositol 1,4,5-trisphosphate recep-tor and RACK1. J Biol Chem 283:32821–32830.
83. Woodard GE, JJ Lopez, I Jardin, GM Salido and JA Rosado.(2010). TRPC3 regulates agonist-stimulated Ca2 + mobili-zation by mediating the interaction between type I inositol1,4,5-trisphosphate receptor, RACK1, and Orai1. J BiolChem 285:8045–8053.
84. Mi LY, DS Ettenson and ER Edelman. (2008). Phospholi-pase C-delta extends intercellular signalling range and re-sponses to injury-released growth factors in non-excitablecells. Cell Prolif 41:671–690.
85. Benedito R, C Roca, I Sorensen, S Adams, A Gossler, MFruttiger and RH Adams. (2009). The Notch ligands Dll4and Jagged1 have opposing effects on angiogenesis. Cell137:1124–1135.
86. Akeson A, A Herman, D Wiginton and J Greenberg. (2010).Endothelial cell activation in a VEGF-A gradient: relevanceto cell fate decisions. Microvasc Res 80:65–74.
87. Cheyette TE and DJ Gross. (1991). Epidermal growth fac-tor-stimulated calcium ion transients in individual A431cells: initiation kinetics and ligand concentration depen-dence. Cell Regul 2:827–840.
88. Mac Gabhann F, MT Yang and AS Popel. (2005). MonteCarlo simulations of VEGF binding to cell surface receptorsin vitro. Biochim Biophys Acta 1746:95–107.
89. Nakayama M, A Nakayama, M van Lessen, H Yamamoto,S Hoffmann, HCA Drexler, N Itoh, T Hirose, G Breier, et al.(2013). Spatial regulation of VEGF receptor endocytosis inangiogenesis. Nat Cell Biol 15:249–260.
90. Kappas NC, G Zeng, JC Chappell, JB Kearney, S Hazarika,KG Kallianos, C Patterson, BH Annex and VL Bautch.(2008). The VEGF receptor Flt-1 spatially modulates Flk-1signaling and blood vessel branching. J Cell Biol 181:847–858.
91. Thurley K, A Skupin, R Thul and M Falcke. (2012). Fun-damental properties of Ca2 + signals. Biochim Biophys Acta1820:1185–1194.
92. Skupin A, H Kettenmann and M Falcke. (2010). Calciumsignals driven by single channel noise. PLoS Comput Biol6:e1000870.
93. Kraus M and B Wolf. (1996). Crosstalk between cellularmorphology and calcium oscillation patterns—insightsfrom a stochastic computer model. Cell Calcium 19:461–472.
94. Faehling M, J Kroll, KJ Fohr, G Fellbrich, U Mayr, GTrischler and J Waltenberger. (2002). Essential role ofcalcium in vascular endothelial growth factor A-inducedsignaling: mechanism of the antiangiogenic effect of car-boxyamidotriazole. FASEB J 16:1805–1807.
95. Bhattacharya R, J Kwon, X Li, E Wang, S Patra, JP Bida, ZBajzer, L Claesson-Welsh and D Mukhopadhyay. (2009).Distinct role of PLC beta 3 in VEGF-mediated directionalmigration and vascular sprouting. J Cell Sci 122:1025–1034.
96. McLaughlin AP and GW De Vries. (2001). Role of PLCgamma and Ca2 + in VEGF- and FGF-induced choroidalendothelial cell proliferation. Am J Physiol Cell Physiol281:C1448–C1456.
97. Wilkerson MK, TJ Heppner, AD Bonev and MT Nelson.(2006). Inositol trisphosphate receptor calcium release isrequired for cerebral artery smooth muscle cell prolifera-tion. Am J Physiol Heart Circ Physiol 290:H240–H247.
VEGF-INDUCED CA21 OSCILLATIONS IN CORD BLOOD-DERIVED EPCS 2579

98. Shibao K, MJ Fiedler, J Nagata, N Minagawa, K Hirata, YNakayama, Y Iwakiri, MH Nathanson and K Yamaguchi.(2010). The type III inositol 1,4,5-trisphosphate receptor isassociated with aggressiveness of colorectal carcinoma. CellCalcium 48:315–323.
99. Szatkowski C, JB Parys, H Ouadid-Ahidouch and F Mati-fat. (2010). Inositol 1,4,5-trisphosphate-induced Ca2 + sig-nalling is involved in estradiol-induced breast cancerepithelial cell growth. Mol Cancer 9:156.
100. Hofer E and B Schweighofer. (2007). Signal transductioninduced in endothelial cells by growth factor receptors in-volved in angiogenesis. Thromb Haemost 97:355–363.
101. Mancini M and A Toker. (2009). NFAT proteins: emergingroles in cancer progression. Nat Rev Cancer 9:810–820.
102. Brenner JS and RE Dolmetsch. (2007). TrpC3 regulateshypertrophy-associated gene expression without affectingmyocyte beating or cell size. PLoS One 2:e802.
103. Eggermann E, I Bucurenciu, SP Goswami and P Jonas.(2012). Nanodomain coupling between Ca2 + channels andsensors of exocytosis at fast mammalian synapses. Nat RevNeurosci 13:7–21.
Address correspondence to:Dr. Francesco Moccia
Department of Biology and Biotechnology ‘‘Lazzaro Spallanzani’’University of Pavia
Via Forlanini 6, 27100PaviaItaly
E-mail: [email protected]
Received for publication January 15, 2013Accepted after revision May 17, 2013
Prepublished on Liebert Instant Online May 17, 2013
2580 DRAGONI ET AL.




















