
Calpain activity contributes to the control of SNAP-25 levels in neurons
10
Calpain activity contributes to the control of SNAP-25 levels in neurons Carlotta Grumelli a , Paul Berghuis b , Davide Pozzi a , Matteo Caleo c , Flavia Antonucci c , Giambattista Bonanno d , Giorgio Carmignoto e , Marton Benedek Dobszay b , Tibor Harkany b,f , Michela Matteoli a,g , Claudia Verderio a, ⁎ a Istituto CNR di Neuroscienze, Dipartimento di Farmacologia Medica, Universita' di Milano, Italy b Division of Molecular Neurobiology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden c Istituto CNR di Neuroscienze, Pisa, Italy d Department of Experimental Medicine, Center of Excellence for Biomedical Research, University of Genova, Italy e Istituto CNR di Neuroscienze, Dipartimento di Scienze Biomediche Sperimentali, Università di Padova, Italy f Institute of Medical Sciences, College of Life Sciences and Medicine, Foresterhill, University of Aberdeen, Scotland, United Kingdom g IRCCS Fondazione Don Gnocchi, Milano, Italy abstract article info Article history: Received 25 February 2008 Revised 4 July 2008 Accepted 8 July 2008 Available online 29 July 2008 Calpains are a family of calcium-dependent proteases with abundant expression in the CNS, and potent in cleaving some synaptic components. Assessment of calpain activity by its fluorescent substrate, Boc-Leu- Met-CMAC, revealed that cultured neurons display a significant level of constitutive enzyme activity. Notably, calpain activity differs in distinct neuronal populations, with a significantly higher level of activity in GABAergic cells. Using selectively-enriched cultures of fast-spiking GABAergic interneurons, we show that calpain activity partially contributes to the post-translational down regulation of SNAP-25, a calpain substrate, in differentiated GABA cells. In addition, we demonstrate that SNAP-25 is cleaved by calpain in response to acute seizures induced by intraperitoneal kainate injection in vivo. These data indicate that calpains in neurons are active even at physiological calcium concentrations and that different levels of calpain activation in selected neuron subtypes may contribute to the pattern of synaptic protein expression. © 2008 Elsevier Inc. All rights reserved. Introduction Calpains are a family of calcium-dependent neutral proteases which cleave several cytosolic, membrane or cytoskeleton-associated proteins. Proteolysis by calpain is likely to change the integrity, localization, and/or activity of endogenous proteins, and results in either the activation or the inhibition of substrate functions. Despite the fact that more than 100 proteins have been identified as calpain substrates, the sequential/structural determinants of calpain recogni- tion are not completely understood. Various enzymatic determinants, such as amino acid preference, secondary structure, and PEST segments, have been shown to correlate with the cleavage site selected by calpain (Tompa et al., 2004). At least three ubiquitous calpains, calpain-1 (μ-calpain), calpain-2 (m-calpain), and calpain-10 are highly expressed in the central nervous system along with the preferential localization of several calpain substrates, including voltage-gated calcium channels (Hell et al., 1996), NMDA receptor subunits (Guttmann et al., 2001), postsynaptic density proteins (Lu et al., 2000), kinases, and phosphatases, to synaptic compartments in neurons (Wu and Lynch, 2006). Recent data suggest that some components of the fusion machinery, in particular the SNARE proteins SNAP-25 and SNAP-23 are substrates of various members of the calpain family in neurons (Ando et al., 2005), as previously described in alveolar epithelial cells (Zimmerman et al., 1999), platelets (Lai and Flaumenhaft, 2003; Rutledge and Whiteheart, 2002) and pancreatic islets (Marshall et al., 2005). Whether the cleavage of these substrates occurs only under pathological (Xu et al., 2007) or physiological conditions is still ambiguous. SNAP-25 and SNAP-23 are plasma membrane proteins which together with their partner SNAREs, the membrane protein syntaxin and the synaptic vesicle protein synaptobrevin/VAMP, form the fusion complex mediating regulated exocytosis. The calpain members proteolitically cleaving SNARE proteins include calpain-1 (Rutledge and Whiteheart, 2002) and calpain-10 (Marshall et al., 2005), an atypical calpain, which lacks the calcium/calmodulin domain (Ma et al., 2001). Analysis of SNAP-23 cleavage products suggests that the calpain cleavage site(s) is situated in the C-terminal third of the molecule, potentially between the cysteine-rich acyl attachment sites and the C-terminal coiled-coil domain (Rutledge and Whiteheart, 2002). The calpain cleavage site in SNAP-25 has been instead localized to the N-terminal third of the molecule in cultured cerebellar granule cells (Ando et al., 2005) and to a region adjacent to the N-terminus in insulinoma INS-1 cells (Marshall et al., 2005). Molecular and Cellular Neuroscience 39 (2008) 314–323 ⁎ Corresponding author. via Vanvitelli 32 20129 Milano, Italy. Fax: +39 027490574. E-mail address: [email protected] (C. Verderio). 1044-7431/$ – see front matter © 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.mcn.2008.07.011 Contents lists available at ScienceDirect Molecular and Cellular Neuroscience journal homepage: www.elsevier.com/locate/ymcne
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Calpain activity contributes to the control of SNAP-25 levels in neurons

Molecular and Cellular Neuroscience 39 (2008) 314–323
Contents lists available at ScienceDirect
Molecular and Cellular Neuroscience
j ourna l homepage: www.e lsev ie r.com/ locate /ymcne
Calpain activity contributes to the control of SNAP-25 levels in neurons
Carlotta Grumelli a, Paul Berghuis b, Davide Pozzi a, Matteo Caleo c, Flavia Antonucci c,Giambattista Bonanno d, Giorgio Carmignoto e, Marton Benedek Dobszay b, Tibor Harkany b,f,Michela Matteoli a,g, Claudia Verderio a,⁎a Istituto CNR di Neuroscienze, Dipartimento di Farmacologia Medica, Universita' di Milano, Italyb Division of Molecular Neurobiology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Swedenc Istituto CNR di Neuroscienze, Pisa, Italyd Department of Experimental Medicine, Center of Excellence for Biomedical Research, University of Genova, Italye Istituto CNR di Neuroscienze, Dipartimento di Scienze Biomediche Sperimentali, Università di Padova, Italyf Institute of Medical Sciences, College of Life Sciences and Medicine, Foresterhill, University of Aberdeen, Scotland, United Kingdomg IRCCS Fondazione Don Gnocchi, Milano, Italy
⁎ Corresponding author. via Vanvitelli 32 20129 MilanE-mail address: [email protected] (C. Verderio).
1044-7431/$ – see front matter © 2008 Elsevier Inc. Aldoi:10.1016/j.mcn.2008.07.011
a b s t r a c t
a r t i c l e i n f oArticle history:
Calpains are a family of cal Received 25 February 2008Revised 4 July 2008Accepted 8 July 2008Available online 29 July 2008cium-dependent proteases with abundant expression in the CNS, and potent incleaving some synaptic components. Assessment of calpain activity by its fluorescent substrate, Boc-Leu-Met-CMAC, revealed that cultured neurons display a significant level of constitutive enzyme activity. Notably,calpain activity differs in distinct neuronal populations, with a significantly higher level of activity inGABAergic cells. Using selectively-enriched cultures of fast-spiking GABAergic interneurons, we show thatcalpain activity partially contributes to the post-translational down regulation of SNAP-25, a calpainsubstrate, in differentiated GABA cells. In addition, we demonstrate that SNAP-25 is cleaved by calpain inresponse to acute seizures induced by intraperitoneal kainate injection in vivo. These data indicate thatcalpains in neurons are active even at physiological calcium concentrations and that different levels ofcalpain activation in selected neuron subtypes may contribute to the pattern of synaptic protein expression.
© 2008 Elsevier Inc. All rights reserved.
Introduction
Calpains are a family of calcium-dependent neutral proteaseswhich cleave several cytosolic, membrane or cytoskeleton-associatedproteins. Proteolysis by calpain is likely to change the integrity,localization, and/or activity of endogenous proteins, and results ineither the activation or the inhibition of substrate functions. Despitethe fact that more than 100 proteins have been identified as calpainsubstrates, the sequential/structural determinants of calpain recogni-tion are not completely understood. Various enzymatic determinants,such as amino acid preference, secondary structure, and PESTsegments, have been shown to correlate with the cleavage site selectedby calpain (Tompa et al., 2004). At least three ubiquitous calpains,calpain-1 (μ-calpain), calpain-2 (m-calpain), and calpain-10 are highlyexpressed in the central nervous system along with the preferentiallocalization of several calpain substrates, including voltage-gatedcalcium channels (Hell et al., 1996), NMDA receptor subunits(Guttmann et al., 2001), postsynaptic density proteins (Lu et al.,2000), kinases, and phosphatases, to synaptic compartments in
o, Italy. Fax: +39 02 7490574.
l rights reserved.
neurons (Wu and Lynch, 2006). Recent data suggest that somecomponents of the fusion machinery, in particular the SNARE proteinsSNAP-25 and SNAP-23 are substrates of variousmembers of the calpainfamily in neurons (Ando et al., 2005), as previously described inalveolar epithelial cells (Zimmerman et al., 1999), platelets (Lai andFlaumenhaft, 2003; Rutledge and Whiteheart, 2002) and pancreaticislets (Marshall et al., 2005). Whether the cleavage of these substratesoccurs only under pathological (Xu et al., 2007) or physiologicalconditions is still ambiguous.
SNAP-25 and SNAP-23 are plasma membrane proteins whichtogether with their partner SNAREs, the membrane protein syntaxinand the synaptic vesicle protein synaptobrevin/VAMP, form the fusioncomplex mediating regulated exocytosis. The calpain membersproteolitically cleaving SNARE proteins include calpain-1 (Rutledgeand Whiteheart, 2002) and calpain-10 (Marshall et al., 2005), anatypical calpain, which lacks the calcium/calmodulin domain (Ma et al.,2001). Analysis of SNAP-23 cleavage products suggests that thecalpain cleavage site(s) is situated in the C-terminal third of themolecule, potentially between the cysteine-rich acyl attachment sitesand the C-terminal coiled-coil domain (Rutledge and Whiteheart,2002). The calpain cleavage site in SNAP-25 has been instead localizedto the N-terminal third of the molecule in cultured cerebellar granulecells (Ando et al., 2005) and to a region adjacent to the N-terminus ininsulinoma INS-1 cells (Marshall et al., 2005).
315C. Grumelli et al. / Molecular and Cellular Neuroscience 39 (2008) 314–323
Controversial results have been reported on calpain-mediatedSNARE degradation and secretion: Lai and Flaumenhaft (2003) foundthat ATP-dependent alpha-granule secretion from platelets is inhi-bited under conditions producing SNAP-23 cleavage, indicating thatcalpain-dependent SNARE degradation impairs secretion. In contrast,calpain-mediated SNARE cleavage may not affect vesicle fusion as ithas been reported to occur after granule exocytosis (Rutledge andWhiteheart, 2002), and calpain activity may even be necessary forsecretion since calpain inhibitors inhibit exocytosis (Marshall et al.,2005). The inhibition of insulin secretion by calpain inhibitors isconsistent with the possibility that calpain-10 acts as a calcium sensorwhich partially proteolyses SNAP-25 upon calcium influx thustriggering rearrangement of the SNARE complex and granule fusion(Marshall et al., 2005). Alternatively, calpain inhibitors could suppressinsulin secretion via inhibition of μ-calpain-mediated proteolysis ofthe secretory granule protein ICA512, which is important for granulemobilization from the cytoskeleton to the cell surface (Ort et al., 2001).The sole study performed in neurons indicates that SNAP-25 cleavageby calpain results in an inhibitory effect on neurotransmitter release in
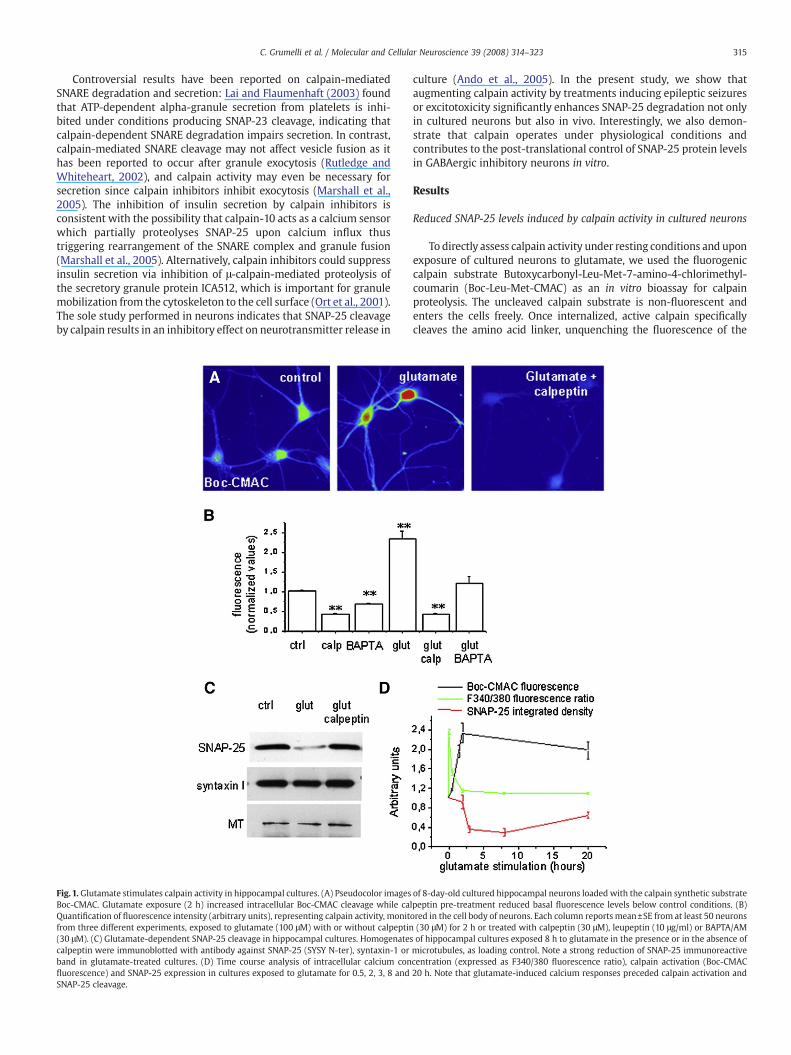
Fig. 1. Glutamate stimulates calpain activity in hippocampal cultures. (A) Pseudocolor imagesBoc-CMAC. Glutamate exposure (2 h) increased intracellular Boc-CMAC cleavage while caQuantification of fluorescence intensity (arbitrary units), representing calpain activity, monitfrom three different experiments, exposed to glutamate (100 μM) with or without calpeptin(30 μM). (C) Glutamate-dependent SNAP-25 cleavage in hippocampal cultures. Homogenatescalpeptin were immunoblotted with antibody against SNAP-25 (SYSY N-ter), syntaxin-1 orband in glutamate-treated cultures. (D) Time course analysis of intracellular calcium confluorescence) and SNAP-25 expression in cultures exposed to glutamate for 0.5, 2, 3, 8 andSNAP-25 cleavage.
culture (Ando et al., 2005). In the present study, we show thataugmenting calpain activity by treatments inducing epileptic seizuresor excitotoxicity significantly enhances SNAP-25 degradation not onlyin cultured neurons but also in vivo. Interestingly, we also demon-strate that calpain operates under physiological conditions andcontributes to the post-translational control of SNAP-25 protein levelsin GABAergic inhibitory neurons in vitro.
Results
Reduced SNAP-25 levels induced by calpain activity in cultured neurons
To directly assess calpain activity under resting conditions and uponexposure of cultured neurons to glutamate, we used the fluorogeniccalpain substrate Butoxycarbonyl-Leu-Met-7-amino-4-chlorimethyl-coumarin (Boc-Leu-Met-CMAC) as an in vitro bioassay for calpainproteolysis. The uncleaved calpain substrate is non-fluorescent andenters the cells freely. Once internalized, active calpain specificallycleaves the amino acid linker, unquenching the fluorescence of the
of 8-day-old cultured hippocampal neurons loaded with the calpain synthetic substratelpeptin pre-treatment reduced basal fluorescence levels below control conditions. (B)ored in the cell body of neurons. Each column reports mean±SE from at least 50 neurons(30 μM) for 2 h or treated with calpeptin (30 μM), leupeptin (10 μg/ml) or BAPTA/AMof hippocampal cultures exposed 8 h to glutamate in the presence or in the absence ofmicrotubules, as loading control. Note a strong reduction of SNAP-25 immunoreactivecentration (expressed as F340/380 fluorescence ratio), calpain activation (Boc-CMAC20 h. Note that glutamate-induced calcium responses preceded calpain activation and
316 C. Grumelli et al. / Molecular and Cellular Neuroscience 39 (2008) 314–323
coumarin fluorophore. Treatment of primary hippocampal cultures,mainly containing glutamatergic neurons, with 100 μM glutamate (for2 h) increased the fluorescence intensity of neurons loaded with thecalpain substrate, thus indicating an increase in calpain activity. Pre-incubationwith the cell permeable calpain inhibitor, calpeptin (30 μM),abolished glutamate-induced calpain activation and reduced thefluorescence intensity of neurons below control levels (Figs. 1A and B).Pre-treatmentof neuronal cultureswith calpeptin reducedbasal calpainactivity (Fig. 1B). Treatment of neurons with the cell-permeant calciumchelator BAPTA/AM (30 μM) revealed that constitutive activity ofcalpain was dependent on intracellular calcium given that thefluorescent intensity of Boc-CMAC decreased significantly belowcontrol levels after treatment with this calcium chelator (Fig. 1B). Pre-treatment with BAPTA/AM also strongly prevented calpain activationinduced by 1.5 h glutamate exposure (mean fluorescent intensitynormalized to control: 196±11.8%, glutamate-treated vs. 121±15.9%,BAPTA pre-treated).
The plasma membrane SNARE protein SNAP-25 has been describedto represent a calpain substrate (Ando et al., 2005). To confirm thisevidence, homogenates of low density hippocampal neurons, exposedto 100 μM glutamate, were analyzed by western blotting withantibodies directed against SNAP-25. As shown in Figs. 1C and D, theamount of SNAP-25 was decreased by 8, 64 and 70% in cultures treatedfor 2, 3 and 8 h respectively with glutamate. Such reductionwas largelypreventedwhen cultureswere exposed to glutamate in the presence of30 μMcalpeptin (integrated intensityof SNAP-25 immunoreactive band
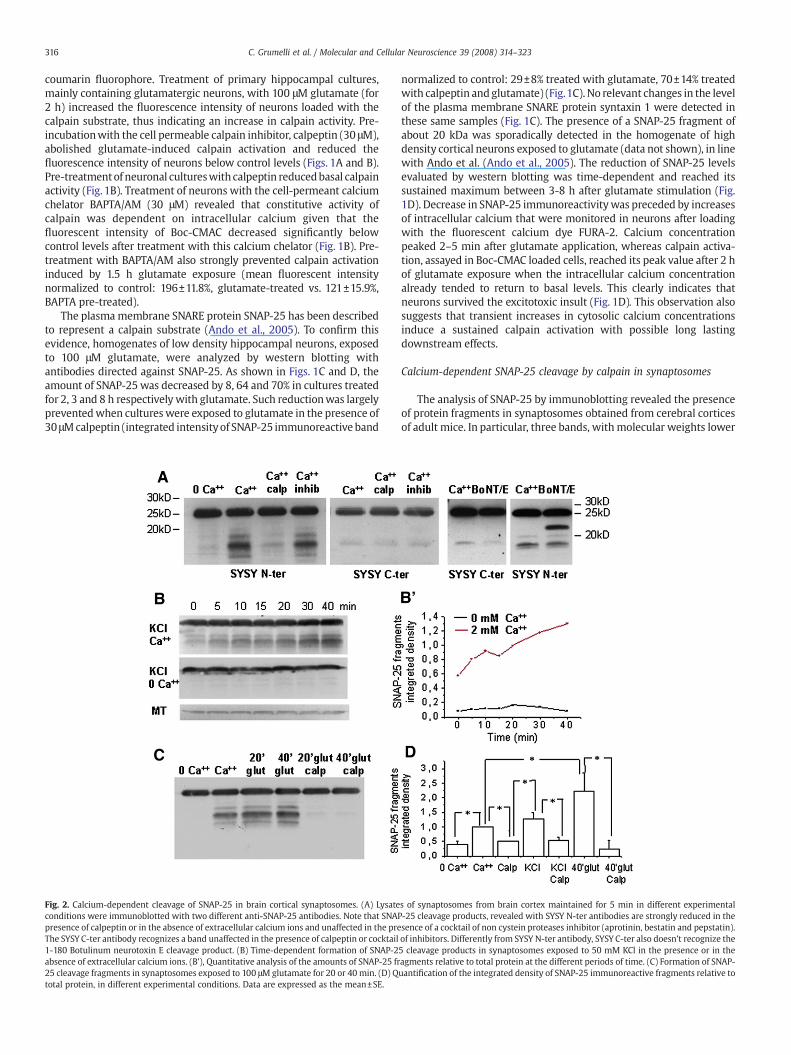
Fig. 2. Calcium-dependent cleavage of SNAP-25 in brain cortical synaptosomes. (A) Lysateconditions were immunoblotted with two different anti-SNAP-25 antibodies. Note that SNAPpresence of calpeptin or in the absence of extracellular calcium ions and unaffected in the preThe SYSY C-ter antibody recognizes a band unaffected in the presence of calpeptin or cocktail1-180 Botulinum neurotoxin E cleavage product. (B) Time-dependent formation of SNAP-2absence of extracellular calcium ions. (B'), Quantitative analysis of the amounts of SNAP-25 fr25 cleavage fragments in synaptosomes exposed to 100 μMglutamate for 20 or 40 min. (D) Qtotal protein, in different experimental conditions. Data are expressed as the mean±SE.
normalized to control: 29±8% treated with glutamate, 70±14% treatedwith calpeptin andglutamate) (Fig.1C). No relevant changes in the levelof the plasma membrane SNARE protein syntaxin 1 were detected inthese same samples (Fig. 1C). The presence of a SNAP-25 fragment ofabout 20 kDa was sporadically detected in the homogenate of highdensity cortical neurons exposed to glutamate (data not shown), in linewith Ando et al. (Ando et al., 2005). The reduction of SNAP-25 levelsevaluated by western blotting was time-dependent and reached itssustained maximum between 3-8 h after glutamate stimulation (Fig.1D). Decrease in SNAP-25 immunoreactivitywas preceded by increasesof intracellular calcium that were monitored in neurons after loadingwith the fluorescent calcium dye FURA-2. Calcium concentrationpeaked 2–5 min after glutamate application, whereas calpain activa-tion, assayed in Boc-CMAC loaded cells, reached its peak value after 2 hof glutamate exposure when the intracellular calcium concentrationalready tended to return to basal levels. This clearly indicates thatneurons survived the excitotoxic insult (Fig. 1D). This observation alsosuggests that transient increases in cytosolic calcium concentrationsinduce a sustained calpain activation with possible long lastingdownstream effects.
Calcium-dependent SNAP-25 cleavage by calpain in synaptosomes
The analysis of SNAP-25 by immunoblotting revealed the presenceof protein fragments in synaptosomes obtained from cerebral corticesof adult mice. In particular, three bands, withmolecular weights lower
s of synaptosomes from brain cortex maintained for 5 min in different experimental-25 cleavage products, revealed with SYSY N-ter antibodies are strongly reduced in thesence of a cocktail of non cystein proteases inhibitor (aprotinin, bestatin and pepstatin).of inhibitors. Differently from SYSY N-ter antibody, SYSY C-ter also doesn't recognize the5 cleavage products in synaptosomes exposed to 50 mM KCl in the presence or in theagments relative to total protein at the different periods of time. (C) Formation of SNAP-uantification of the integrated density of SNAP-25 immunoreactive fragments relative to
317C. Grumelli et al. / Molecular and Cellular Neuroscience 39 (2008) 314–323
than 20 kDa were detected when using three different antibodies: amonoclonal antibody directed against the N-terminus of SNAP-25(Synaptic Systems, N-terminal, Fig. 2A) and two monoclonal anti-bodies (MAB331 and SMI 81, not shown) produced against the wholeprotein. SNAP-25 fragments were due to calpain activation, since noobvious fragments were observed in synaptosomes pre-treated withcalpeptin (30 μM, Fig. 2A), and less fragmentation was detected inleupeptin-treated synaptosomes (10 μg/ml, not shown). SNAP-25immunopositive bands were instead detectable in synaptosomestreated with a cocktail of inhibitors: aprotinin (100 U/ml), a serineproteinase inhibitor, bestatin (10 μg/ml), an aminopeptidase inhibitor,and pepstatin (10 μg/ml), an inhibitor of aspartic proteinases (Fig. 2A).Also the C-terminal antibody (Synaptic Systems, C-terminal) recognizeda band of low molecular weight, which was however insensitive tocalpeptin or protease inhibitors (Fig. 2A Synaptic Systems [SYSY C-ter]).Calpain-dependent cleavage products were not detectable in synapto-somes maintained in the absence of extracellular calcium ions, indi-cating that calpain activity requires extracellular calcium (Figs. 2A–D).Formation of SNAP-25 fragments occurred in the absence of synapto-some stimulation, weakly increased under synaptosome depolariza-tion or glutamate exposure (Figs. 2C, D). Appearance of SNAP-25fragments increased progressively in a time-dependent manner in thepresence of extracellular calcium and either 55 mM KCl or 100 μMglutamate (Figs. 2B–D). Different from primary cultures, SNAP-25cleavage in synaptosomes was already detectable 5–10 min afterdepolarisation (Fig. 2B), possibly due to a substantial lack of de novoprotein synthesis in isolated terminals. Fragments generated by calpainwere clearly distinct from that produced by Botulinum toxin E (Fig. 2A,
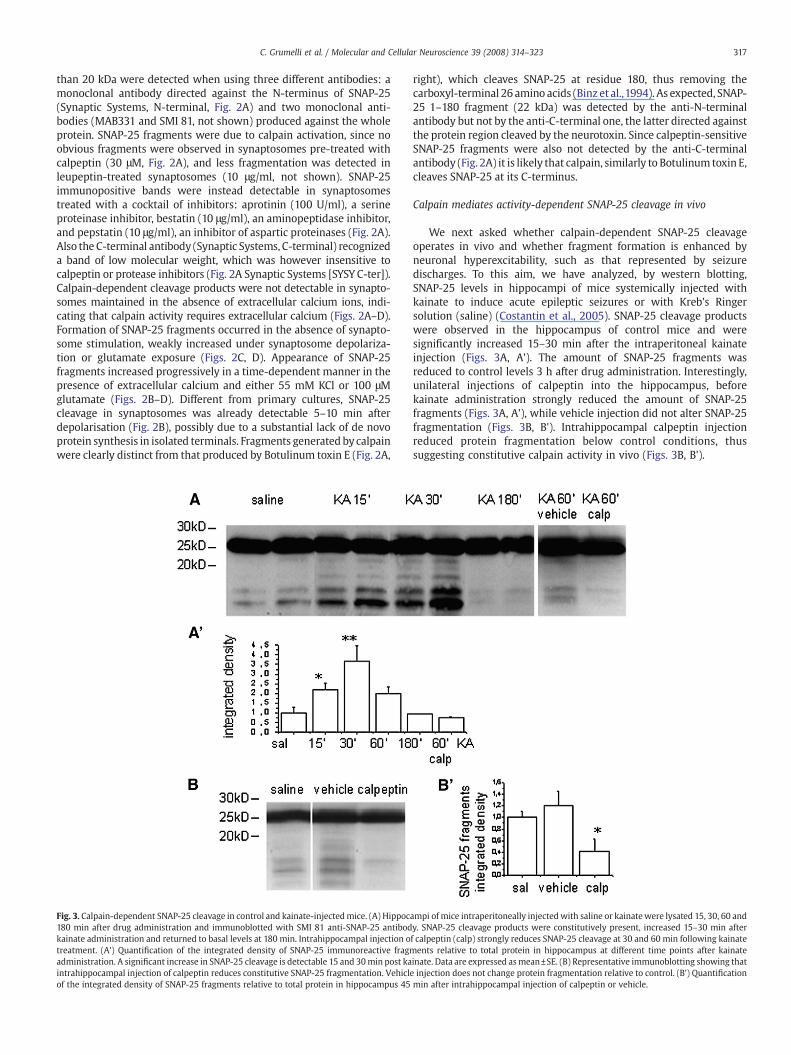
Fig. 3. Calpain-dependent SNAP-25 cleavage in control and kainate-injectedmice. (A) Hippoc180 min after drug administration and immunoblotted with SMI 81 anti-SNAP-25 antibodkainate administration and returned to basal levels at 180 min. Intrahippocampal injection otreatment. (A') Quantification of the integrated density of SNAP-25 immunoreactive fragmadministration. A significant increase in SNAP-25 cleavage is detectable 15 and 30min post kaintrahippocampal injection of calpeptin reduces constitutive SNAP-25 fragmentation. Vehiclof the integrated density of SNAP-25 fragments relative to total protein in hippocampus 45
right), which cleaves SNAP-25 at residue 180, thus removing thecarboxyl-terminal 26 amino acids (Binz et al.,1994). As expected, SNAP-25 1–180 fragment (22 kDa) was detected by the anti-N-terminalantibody but not by the anti-C-terminal one, the latter directed againstthe protein region cleaved by the neurotoxin. Since calpeptin-sensitiveSNAP-25 fragments were also not detected by the anti-C-terminalantibody (Fig. 2A) it is likely that calpain, similarly to Botulinum toxin E,cleaves SNAP-25 at its C-terminus.
Calpain mediates activity-dependent SNAP-25 cleavage in vivo
We next asked whether calpain-dependent SNAP-25 cleavageoperates in vivo and whether fragment formation is enhanced byneuronal hyperexcitability, such as that represented by seizuredischarges. To this aim, we have analyzed, by western blotting,SNAP-25 levels in hippocampi of mice systemically injected withkainate to induce acute epileptic seizures or with Kreb's Ringersolution (saline) (Costantin et al., 2005). SNAP-25 cleavage productswere observed in the hippocampus of control mice and weresignificantly increased 15–30 min after the intraperitoneal kainateinjection (Figs. 3A, A'). The amount of SNAP-25 fragments wasreduced to control levels 3 h after drug administration. Interestingly,unilateral injections of calpeptin into the hippocampus, beforekainate administration strongly reduced the amount of SNAP-25fragments (Figs. 3A, A'), while vehicle injection did not alter SNAP-25fragmentation (Figs. 3B, B'). Intrahippocampal calpeptin injectionreduced protein fragmentation below control conditions, thussuggesting constitutive calpain activity in vivo (Figs. 3B, B').
ampi of mice intraperitoneally injectedwith saline or kainatewere lysated 15, 30, 60 andy. SNAP-25 cleavage products were constitutively present, increased 15–30 min afterf calpeptin (calp) strongly reduces SNAP-25 cleavage at 30 and 60 min following kainateents relative to total protein in hippocampus at different time points after kainate
inate. Data are expressed asmean±SE. (B) Representative immunoblotting showing thate injection does not change protein fragmentation relative to control. (B') Quantificationmin after intrahippocampal injection of calpeptin or vehicle.

318 C. Grumelli et al. / Molecular and Cellular Neuroscience 39 (2008) 314–323
Calpain activity in glutamatergic and GABAergic neurons
As calpeptin reduces Boc-CMAC fluorescence below control levels(Fig. 1B), the possibility of constitutive calpain activation in restingneurons emerges. The major population of cells present in primarycultures of hippocampal neurons is excitatory glutamatergic neurons.Neurons displaying GABA immunoreactivity represent about 6–10% ofthe total number of cultured neurons. Most GABAergic cells havefusiform or polygonal shaped somata, non-spiny and less-taperingdendrites. Moreover, GABA cells appear more phase-dense than nonGABAergic cells thus allowing reliable identification by morphologicalcriteria (Benson et al.,1994). Bymonitoring basal and glutamate-evokedcalpain activity in cultures loaded with Boc-CMAC, GABAergic neuronsdisplayed a higher fluorescence signal (data not shown). Boc-CMACfluorescence measurements were also performed on fixed cultures,where the GABAergic or glutamatergic nature of neurons could beunequivocally identified by immunostaining for the neurotransmitterGABA (Fig. 4A). Quantitative analysis of Boc-CMAC fluorescence at thesoma of excitatory or inhibitory neurons revealed that the fluorescence
Fig. 4. Calpain activity in glutamatergic and GABAergic neurons. (A) Pseudocolor imagesantibodies directed against the neurotransmitter GABA. Note the higher signal of Boc-CMAC(B) Quantification of Boc-CMAC fluorescence, an index of calpain activity in glutamatergicstimulated with glutamate (GABA stim, third bar) and GABAergic neurons stimulated with glmean±SE and normalized to mean average intensity of glutamatergic neurons in the samconcentration, and Boc-CMAC fluorescence in either control neurons (black dots) or neuronsintracellular calcium concentrations result in increasing calpain activity. Data represent meglutamatergic and GABAergic cultured neurons. Data represent mean±SE and are normalizedistribution of 405/485 ratio changes evoked by 30 mM KCl in glutamatergic and GABAergic nselected based on the size of their somata (interneurons: diameter<11 μm; pyramidal cells
signal in GABAergic cells exceeded by 1.5 and 2 fold that detected inglutamatergic cells. These differences were stably present both undercontrol conditions and after glutamate exposure (Fig. 4B). To evaluatecalcium dynamics in glutamatergic and GABAergic neurons, neuronalcultures or hippocampal slices were loaded with the calcium sensitivedyes Fura-2 or INDO-1, respectively, and exposed to 30 mM KCl in thepresence of the NMDA receptor blocker APV (50 μM) and the Na+
channel blocker TTX (1 μM). Higher calpain activity in GABAergicneurons was associated with higher calcium responses to 30 mM KCl,both in hippocampal cultures (glutamatergic neurons: 0.999±0.014n=115; GABAergic neurons: 1.24±0.07 n=50, p<0.001; Verderio et al.,2004) and in brain slices (glutamatergic neurons: 0.36±0.013 n=87;GABAergic neurons: 0.428±0.017 n=29, p=0.0094) (Figs. 4D, E).
Reduced SNAP-25 expression in differentiated GABAergic interneurons
It has previously been shown that mature GABAergic neurons donot display detectable levels of SNAP-25 immunoreactivity, althoughthey are able to synthesize this protein (Verderio et al., 2004; Frassoni
of 8-day-old hippocampal cultures loaded with Boc-CMAC, fixed and stained withfluorescent products in GABAergic neurons (arrows) as compared to glutamatergic cells.neurons (Glut, first bar), GABAergic neurons (GABA, second bar), GABAergic neurons
utamate in the presence of calpeptin (GABA stim+calp, fourth bar). Data are expressed ase conditions. (C) Correlation analysis of 340/380 ratios, an index of cytosolic calciumtreated with the calcium ionophore A23187 (red and green dots) reveals that increasingan±SE. (D) Frequency distribution of 340/380 ratio changes evoked by 30 mM KCl ind to mean value of glutamatergic neurons recorded in the same coverslip. (E) Frequencyeurons in hippocampal slices. Interneurons and pyramidal glutamatergic neurons were>12 μm) and the absence of a clear apical dendrite in GABAergic cells.
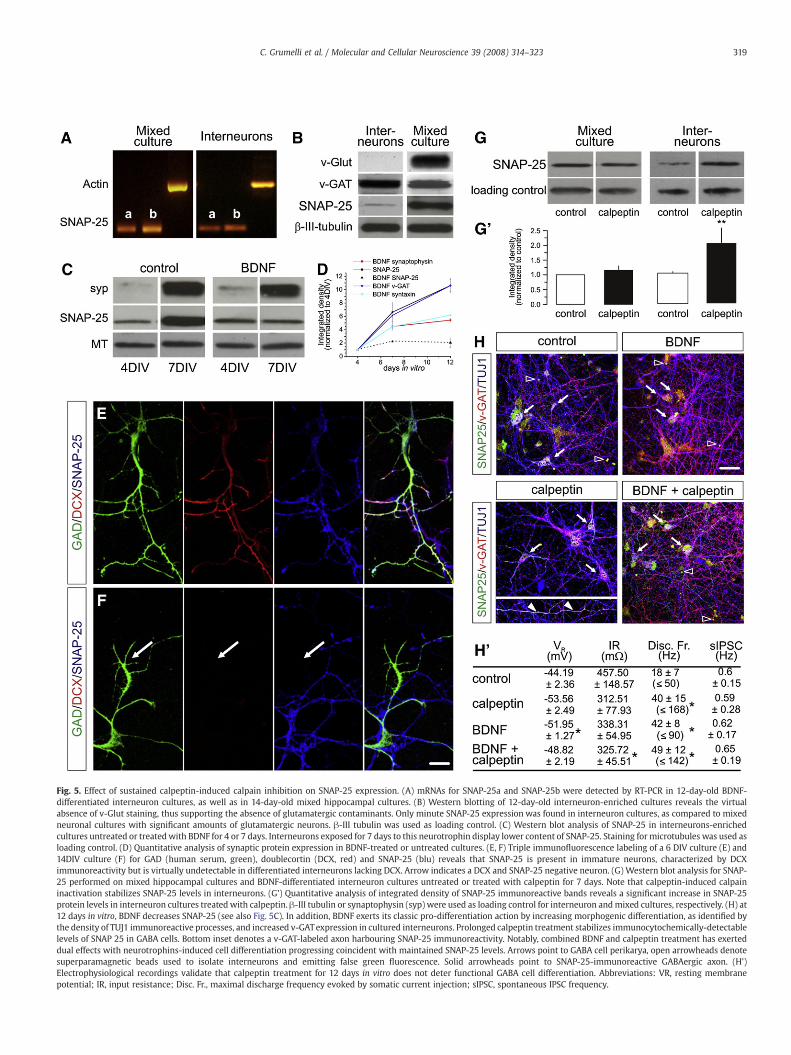
Fig. 5. Effect of sustained calpeptin-induced calpain inhibition on SNAP-25 expression. (A) mRNAs for SNAP-25a and SNAP-25b were detected by RT-PCR in 12-day-old BDNF-differentiated interneuron cultures, as well as in 14-day-old mixed hippocampal cultures. (B) Western blotting of 12-day-old interneuron-enriched cultures reveals the virtualabsence of v-Glut staining, thus supporting the absence of glutamatergic contaminants. Only minute SNAP-25 expression was found in interneuron cultures, as compared to mixedneuronal cultures with significant amounts of glutamatergic neurons. β-III tubulin was used as loading control. (C) Western blot analysis of SNAP-25 in interneurons-enrichedcultures untreated or treatedwith BDNF for 4 or 7 days. Interneurons exposed for 7 days to this neurotrophin display lower content of SNAP-25. Staining for microtubules was used asloading control. (D) Quantitative analysis of synaptic protein expression in BDNF-treated or untreated cultures. (E, F) Triple immunofluorescence labeling of a 6 DIV culture (E) and14DIV culture (F) for GAD (human serum, green), doublecortin (DCX, red) and SNAP-25 (blu) reveals that SNAP-25 is present in immature neurons, characterized by DCXimmunoreactivity but is virtually undetectable in differentiated interneurons lacking DCX. Arrow indicates a DCX and SNAP-25 negative neuron. (G) Western blot analysis for SNAP-25 performed on mixed hippocampal cultures and BDNF-differentiated interneuron cultures untreated or treated with calpeptin for 7 days. Note that calpeptin-induced calpaininactivation stabilizes SNAP-25 levels in interneurons. (G') Quantitative analysis of integrated density of SNAP-25 immunoreactive bands reveals a significant increase in SNAP-25protein levels in interneuron cultures treated with calpeptin. β-III tubulin or synaptophysin (syp) were used as loading control for interneuron andmixed cultures, respectively. (H) at12 days in vitro, BDNF decreases SNAP-25 (see also Fig. 5C). In addition, BDNF exerts its classic pro-differentiation action by increasing morphogenic differentiation, as identified bythe density of TUJ1 immunoreactive processes, and increased v-GATexpression in cultured interneurons. Prolonged calpeptin treatment stabilizes immunocytochemically-detectablelevels of SNAP 25 in GABA cells. Bottom inset denotes a v-GAT-labeled axon harbouring SNAP-25 immunoreactivity. Notably, combined BDNF and calpeptin treatment has exerteddual effects with neurotrophins-induced cell differentiation progressing coincident with maintained SNAP-25 levels. Arrows point to GABA cell perikarya, open arrowheads denotesuperparamagnetic beads used to isolate interneurons and emitting false green fluorescence. Solid arrowheads point to SNAP-25-immunoreactive GABAergic axon. (H')Electrophysiological recordings validate that calpeptin treatment for 12 days in vitro does not deter functional GABA cell differentiation. Abbreviations: VR, resting membranepotential; IR, input resistance; Disc. Fr., maximal discharge frequency evoked by somatic current injection; sIPSC, spontaneous IPSC frequency.
319C. Grumelli et al. / Molecular and Cellular Neuroscience 39 (2008) 314–323

320 C. Grumelli et al. / Molecular and Cellular Neuroscience 39 (2008) 314–323
et al., 2005; Bragina et al., 2007; Garbelli et al., 2008). Two SNAP-25variants, SNAP-25a and b, are generated from a single, highlyconserved gene through alternative splicing (Bark, 1993). In linewith the ability of GABAergic interneurons to synthesize SNAP-25(Frassoni et al., 2005), we here show by RT-PCR the presence of bothSNAP-25a and SNAP-25b mRNAs in phenotypically-differentiatedcultures fast-spiking GABAergic interneurons (Berghuis et al., 2004),similarly to mixed hippocampal cultures (Fig. 5A). We also confirmed,by western blotting, minute levels of SNAP-25 protein in interneuroncultures treated with BDNF relative to mixed primary cultures ofsimilar ages in vitro, predominantly containing glutamatergic neurons(Fig. 5B). The assumed proportions of inhibitory and excitatorycomponents in the different cultures were evaluated by stainingwith antibodies against v-Glut and v-GAT (Fig. 5B). In line with thedevelopmentally-regulated reduction of SNAP-25 expression in inter-neurons (Frassoni et al., 2005), differentiation of fast-spiking GABAer-gic cells by BDNFwas associatedwith a specific downregulation of thisSNARE protein (Figs. 5C, D). BDNF-induced interneuron differentiationwas confirmed by increased v-GAT, synaptophysin and syntaxinexpression (Fig. 5D and Berghuis et al., 2004) and by loss ofdoublecortin (DCX), a microtubule-associated protein expressedalmost exclusively in neuronal progenitors and immature neurons(Figs. 5E, F and Brown et al., 2003). These data indicate that correctdifferentiation of GABAergic neurons is associated with post-transla-tional reduction of SNAP-25 expression.
Calpain contributes to reduced SNAP-25 expression in differentiatedGABAergic interneurons
The higher activity of calpain in GABAergic neurons (Figs. 4A and B)prompted us to study whether calpain levels directly contribute to theregulation of SNAP-25 protein levels in inhibitory interneurons.Therefore, mixed and interneuron-enriched cultures were maintainedin the presence of calpeptin. We then analyzed the amounts of SNAP-25 by western blotting. No effects were detectable after a 4-daytreatment. Treatment of the cultures with calpeptin for 7 days did notsignificantly affect SNAP-25 expression in mixed primary culturespredominantly containing glutamatergic neurons either (Figs. 5G, G').In contrast, exposure of interneurons to calpeptin for 7 dayssignificantly increased the amount of SNAP-25 protein (Figs. 5G, G').We next tested by multiple label immunocytochemistry whetherprolonged calpeptin treatment affects SNAP-25 expression in GABAer-gic interneuron cultures. BDNF markedly decreased SNAP-25 levels,while enhancing v-GAT expression, in interneurons at 12 days in vitro(Fig. 5H). In contrast, calpain blockade by calpeptin led to themaintenance of SNAP-25 in GABA cells with SNAP-25 distributedalong their axons. Combined treatmentwith BDNF and calpeptin had aclear dual action: BDNF increased v-GAT expression and morphogenicdifferentiation, whilst calpeptin maintained SNAP-25 expression. Wethen used whole-cell patch recordings to determine whethercalpeptin affects functional interneuron differentiation. As shownin Fig. 5H', both BDNF and calpeptin increased the restingmembrane potential of interneurons together with a significantincrease in their average discharge frequencies. These data confirmthat calpeptin per se did not compromise the acquiring of a GABAergicphenotype. Overall, our data suggest that calpain activity in GABAergicneurons contributes to the adjustment of intracellular SNAP-25 levels.
Discussion
The major finding of the present study is that cultured neuronsdisplay a constitutive level of calpain activity, which can be reduced by75% upon treatment with the calpain inhibitor calpeptin. This findingclearly indicates that calpains in neurons are active even atphysiological calcium concentrations (100–300 nM) and displayreduced calcium requirements relative to calpain activation in vitro
(5–50 μM for μ-calpain and 200–1000 μM form-calpain), as previouslysuggested (Strobl et al., 2000). In support of this concept, smallcalcium changes (±0.1/0.2 340/380 ratios), falling into the physiolo-gical range, can already result in significant variations of calpainactivity. Taken together, these results suggest that physiologicaldifferences in resting calcium among distinct neuronal populationsmay impact neuronal functions controlled by calpain substrates. Toprove this hypothesis, calcium concentrations were evaluated inexcitatory and inhibitory interneurons, from both hippocampalcultures and slice preparations. Significant differences in GABAergicand glutamatergic responsiveness to depolarisation were detected(Verderio et al., 2004 and this study). In line with the strictdependency of calpain on calcium concentrations also in thephysiological range, evaluation of calpain activity revealed a higheractivation of the enzyme in inhibitory interneurons, indicating thatdifferences in calcium homeostasis between distinct neuronalphenotypes are reflected in changes of calpain activity. Among thefunctional consequences that the higher calpain activity can cause inGABAergic interneurons, we here show that increased enzyme activitycontributes to the post-translational control of the synaptic proteinSNAP-25. Indeed chronic inhibition of calpain specifically increasesthe level of this SNARE protein in cultured interneurons, whichexpress mRNA for both embryonic and adult isoforms, but containnegligible levels of SNAP-25 (Frassoni et al., 2005; Bragina et al., 2007;Garbelli et al., 2008; but see Tafoya et al., 2006). Although we cannotexclude the contribution of other mechanisms, such as differences inmRNA translation, in protein turnover, or in axonal transport, ourresults demonstrate that post-translational proteolysis by calpain is atleast one of the mechanisms which control the level of SNAP-25 inGABAergic neurons. The post-translational control of SNAP-25 bycalpain also suggests that calcium homeostasis and calpain activitymay contribute to the phenotype specification of neurons, bymodulating the expression of synaptic components which, likeSNAP-25, are differently expressed in excitatory and inhibitoryneurons.
Calpains are upregulated in a wide range of pathophysiologicalconditions characterized by dysregulation of neuronal calcium home-ostasis, including stroke, epilepsy, traumatic brain injury and neuro-degenerative disorders (Sun et al., 2004;Wu and Lynch, 2006) and theyare, indeed, recognized to be important mediators of excitotoxicneuronal damage (Neumar et al., 2003). At the basis of calpain actionis the cleavage of different substrates, including theNa+/Ca++ exchanger(Bano et al., 2005) and NMDA receptors (Simpkins et al., 2003), thusenhancing calcium overload and triggering neurotoxicity. Proteolyticinactivation of calcium extrusion driven by the Na+/Ca++ exchanger isresponsible for a delayed Ca++ deregulation while NMDA receptorcleavage gives rise to an active form of the receptor, present on the cellsurface, which contributes to excitotoxicity. In addition, by truncatingthe mGluR1α, calpain disrupts the neuroprotective PI3K-Akt signallingpathway that is linked to the activation ofmGluR1α signalling (Xu et al.,2007). Finally, calpain also promotes cell-death by degrading anti-apoptotic proteins, i.e. NFk-B (Scholzke et al., 2003) and cytoskeletalconstituents such as spectrin (Hu and Bennett, 1991). In line with thewell-described sensitivity of calpain to calcium overload, we heredemonstrate for the first time that SNAP-25 proteolysis by calpainoccurs in vivo and is enhanced in response to acute seizure activity.Interestingly, SNAP-25 fragments are also detectable in humancortexofepileptic patients, thus suggesting that mechanisms of SNAP-25cleavage are active in humans (Rita Garbelli, Carolina Frassoni, ClaudiaVerderio and Michela Matteoli, unpublished observations). It has beenshown (Ando et al., 2005) that the calpain-mediated SNAP-25fragmentation correlates with a reduction of the SNARE function andinhibition of neurotransmitter release. Therefore, SNAP-25 calpain-mediated cleavage can represent a neuroprotective mechanismpotently counteracting excitotoxicity that transiently operates duringacute seizures thus limiting neuronal network excitability.

321C. Grumelli et al. / Molecular and Cellular Neuroscience 39 (2008) 314–323
Experimental methods
Hippocampal and cortical cell cultures
Primary cultures of hippocampal or cortical neurons wereprepared from 18-day-old rat fetuses as previously described(Verderio et al., 1999). Cells were maintained in MEM without sera,supplemented with 1% N2 and 1 mg/ml BSA or in neurobasal mediumsupplemented with 2% B27 (Brewer et al., 1993). The cultures werefixed with 4% paraformaldehyde and 4% sucrose in 0.12 M phosphatebuffer for 20 min at 37 °C and immunofluorescence stainings werecarried out as described (Verderio et al., 1999).
High purity interneurons cultures
Primary interneuron cultures were prepared from the hippo-campi and neocortices of 18-day-old rat embryos as described inBerghuis et al. (Berghuis et al., 2004, 2005).
Synaptosome preparation
The purification of synaptosomes from rat forebrain was carriedout as described (Huttner et al., 1983). Samples with a protein contentof 80 μg were pelleted and resuspended in Krebs Ringer solution withor without 2.5 mM Ca 2+ containing 55 mM KCl, or protease-inhibitorcocktail if needed. The samples were kept for 5–40 min at 37 °C.Treatment with 80 nM Botulinum toxin E was carried out for 2 h at37 °C. At the end of the incubation, samples were mixed with SDScontaining sample buffer for western blotting analysis.
Animals
Animals were housed with a 12 h light/dark cycle with food andwater available ad libitum. All experimental procedures conformed tothe European Communities Council Directive 86/609/EEC.
Calpain activity measurements
Single cell calpain activity was measured in primary neuronalcultures plated on glass coverslips. Control cells or cells pre-treatedwith calpeptin, leupeptin or BAPTAwere loaded for 20minwith 50 μMBoc-Leu-Met-CMAC in culture medium and then exposed to gluta-mate or the ionophore A23187 for different periods of time. Thecalpain substrate is retained within the cells by its progressiveconjugation with intracellular thiol groups. Cleavage of the substrateresults in intracellular retention of the chloromethyl aminocoumarinmoiety of the molecule thus leading to increased fluorescence(Glading et al., 2000). Images of Boc-Leu-Met-CMAC loaded neuronswere acquired with a PCO SuperVGA SensiCam camera (AxonInstruments, Forest City, CA) at excitation and emission wavelengthof 370 and 500 nm, respectively. Polichrome IV (TiLL Photonics,Germany) was used as a light source. The image exposure settingswere maintained identical within each experiment but did varybetween experiments. The average intensity of Boc-CMAC-loadedneurons in the somatodendritic regions was measured by META-MORPH IMAGING Series 6.1 software (Universal Imaging Corporation).In a set of experiments, images were also acquired from fixed culturesstained with antibodies direct against GABA, to unequivocally identifyGABAergic neurons.
Intracellular calcium measurements in cultures
Cultures were loaded with the ratiometric calcium dye fura-2-pentacetoxymethylester (2 μM) in Krebs–Ringer solution for 45 min at37 °C (Verderio et al., 2004). Polychrome IV (TILL Photonics, Grafelfing,Germany) was used as a light source. Fura-2 fluorescence images were
collected with a PCO Super VGA SensiCam (Axon Instruments, ForestCity, CA, USA) and analysed with Axon Imaging Workbench 2.2software (Axon Instruments). Images were acquired at 1–4 Hz. Afterexcitation at 340 and 380 nm wavelengths, the emitted light wasacquired at 505 nm. Calcium concentration are expressed as F340/380fluorescence ratio. “Ratio changes” indicate the amplitude of peakcalcium responses evoked by 30 mM KCl stimulation.
Confocal calcium microscopy in hippocampal slices
Digital fluorescence microscopy (Nikon, RCM8000) was used tomonitor the change in Indo-1 emission after cell loadingwith Indo-1 AM(Molecular Probes, Invitrogen) as previously described (Pasti et al.,1997).After excitation at a wavelength of 351 nm, the emitted light wasseparated into its two components (405 and 485 nm), and the INDO-1fluorescence ratio R (R405/485, the ratio of the intensity of the lightemitted at the two wavelengths 405 nm and 485 nm) was used toexpress intracellular calcium changes on a pseudocolor scale. Duringexperiments, cultured cellswereperfused continuously (1.5–3mlmin−1)with anextracellular solution consistingof (mM): 145NaCl, 5KCl,1CaCl2,1MgCl2,1Na3PO4, 5.5 glucose,10HEPES and0.2 sulfinpyrazone, at pH7.4with NaOH, at 32 °C. The sampling rate was 0.5 Hz. Somaticdiameters<11 μm and the absence of a clear apical dendrite were usedto distinguish interneurons from pyramidal neurons. (Alpar et al., 2004).Interneuronswere distinguished fromother small cells such as astroglialcells on the basis of the distinct kinetics of their calcium response to K+stimulation, as previously reported (Pasti et al., 1997).
Western blotting — detection of cleaved SNAP-25 in primary culturesand brain synaptosomes
Homogenates of primary cultures and lysates of cortical synapto-someswere separated by electrophoresis, blotted on PVDFmembrane,and SNAP-25 cleavage products were detected by incubating filters inanti-SNAP-25 N-terminal monoclonal (directed against the epitopecontaining amino acid (AA) residues 1–20) and C-terminal polyclonalantibodies (directed against AAs 192–206) (1:10,000; SynapticSystems, Goettingen, Germany), monoclonal SMI 81 antibody(1:200,000; Sternberger, Monoclonals Inc., Baltimore, MD), or mono-clonal MAB331 antibody (1:1000; Chemicon, Temecula, CA, USA). Themonoclonal antibodies didn't produce any staining in brain homo-genates from SNAP-25 knock out mice (data not shown).
Detection of cleaved SNAP-25 following epileptic seizures in vivo
C57Bl/6mice (n=15) were injected with a convulsive dose of kainicacid (30 mg/kg, i.p.; Ocean Produce International, Shelburne, NS,Canada) or with Kreb's Ringer solution (saline) (n=7). Effectiveness ofkainate was confirmed in all mice by behavioral observation.Hippocampi were dissected 15, 30, 60 and 180 min after kainateexposure. Protein extracts (25 μg) were separated by electrophoresis,blotted, and filters were incubated with anti-SNAP-25 SMI 81antibody. In a set of experiments addressing the effect of calpeptinon SNAP-25 cleavage, mice (n=11) were anesthetized with avertin andcraniotomy was performed in the left hemisphere. A guide cannulawas positioned on top of the dura for the intrahippocampal injectionof calpeptin or its vehicle (0.25% DMSO in saline). Coordinates forcannula implantationwere as follows: A–P −2.0, M–L 1.5 (in mm frombregma). The cannulawas secured to the skull by acrylic dental cement.Threedays after surgery,micewere split into twogroups andunilaterallyinjected with either calpeptin (0.3 nM; n=6) or vehicle (0.25% DMSO insaline; n=5). Injection was performed in freely moving mice using aneedle protruding 1.8 mm below the guide cannula. Kainic acid (30mg/kg, i.p.) was given 15 min after the intrahippocampal injection. Micewere killed 30–60min after kainate exposure and the left hippocampuswas dissected for the analysis of SNAP-25 cleavage products.

322 C. Grumelli et al. / Molecular and Cellular Neuroscience 39 (2008) 314–323
Western blot quantitative analysis
Immunoreactive bands were acquired with an Image Scanner(Amershampharmacia biotech, Uppsala, Sweden) with LabScan 3.00software converted to TIFF format, and analyzed with Image Jsoftware. The integrated density of immunopositive bands wasdetermined relative to background and results were analyzed withStudent's paired two-sample t test for statistically significantdifferences of the means. Integrated density parameter was chosenfor analysis assuming that the strength of the signal depends on boththe intensity and the width of the stained band.
Whole-cell patch clamp electrophysiology
Experiments were performed at 12 days in vitro according topublished protocols (Berghuis et al., 2004). In brief, the extracellularsolution was continuously oxygenated and contained: 125 mM NaCl,2.5 mMKCl, 25mM glucose, 25mMNaHCO3,1.25mMNaH2PO4, 2 mMCaCl2, and 1 mM MgCl2. The pipette solution contained: 115 mM K-gluconate, 4 mM ATP-Mg, 10 mM Na-phosphocreatine, 0.3 mM GTP,20 mM KCl, and 10 mM HEPES (pH 7.3; 310 mOsm/l). Microelectrodeswere pulled from borosilicate glass (uncoated, PG165T-10; HarvardApparatus, Edenbridge, Kent, UK) and had a resistance of 3.5–5.0 MΩ.Whole-cell voltage and current-clamp recordings were obtained at32–35 °C. Input resistance (IR) plots were constructed after incre-menting current injections in neuronal soma. sIPSC recordings wereperformed at a −70 mV holding potential. Data were analyzed off-lineusing IGOR Pro (v. 4.0, WaveMetrics Inc., Lake Oswego, OR, USA) withcustom-written routines.
Extraction of mRNA and isoform-specific RT-PCR
Total mRNA from 7- and 12-day-old interneurons cultures wasextracted using Micro-FastTrack 2.0 Kit (Invitrogen, Milan, Italy).Isoform-specific RT-PCR was performed using SNAP-25a and SNAP-25b-specific primers (Grant et al., 1999). RT-PCR was performed byusing SuperScript One-Step RT-PCR System (Invitrogen). For eachreaction 5 ng of mRNA were used, and a total of 40 cycles were run.The products of amplification were analyzed by 1.5% agarose gelelectrophoresis.
Reagents and antibodies
Polyclonal anti-GABA antibodies, monoclonal anti-microtubule,and anti-syntaxin 1 antibodies, FURA-2/AM, BAPTA/AM, A23187 werepurchased from Sigma (Milan, Italy). Boc-Leu-Met-CMAC was fromMolecular Probes (Invitrogen). The monoclonal antibodies anti-β-IIItubulin was from Promega (Milan, Italy). Calpeptin was from MerckChemicals Ltd (Nottingham, UK).
Secondary antibodies were from Jackson Immunoresearch Labora-tories. Recombinant human BDNF was obtained from Regeneron(Regeneron Pharmaceuticals, Inc., Tarrytown, NY, USA). Human serafrom patients affected by Stiff-man syndrome and specificallyrecognizing GAD, kind gift of Dr. M. Solimena, Dresden, Germany(Solimena et al., 1990).
Statistical analysis
Results are presented as means±SEM. Data were statisticallycompared using Student's t-test. Differences were considered sig-nificant if p<0.05 (single asterisk), p<0.01 (double asterisks).
Acknowledgments
We thank Yuri Bozzi (Pisa), Micaela Zonta and Debora Crippa(Padova), Marco Milanese (Genova), Irene Corradini and Federica Torri
(Milano) for help in some experiments.We also thank Carolina Frassoni(Milano) and Elisabetta Menna (Milano) for discussion and criticallyreading the manuscript. This work was supported by EU-SynapseIntegrated Project (LSHM-CT-2005-019055), by Telethon GP04196,CARIPLO 2006.0779/109251, contribution of Ministero della Salute perla realizzazione dell progetto Ex art. 56 (2005) toM.M., and the SwedishMedical Research Council and the Alzheimer's Association to T.H.
References
Alpar, A., Seeger, G., Hartig, W., Arendt, T., Gartner, U., 2004. Adaptive morphologicalchanges of neocortical interneurons in response to enlarged and more complexpyramidal cells in p21H-Ras(Val12) transgenic mice. Brain Res. Bull. 62, 335–343.
Ando, K., Kudo, Y., Takahashi, M., 2005. Negative regulation of neurotransmitter releaseby calpain: a possible involvement of specific SNAP-25 cleavage. J. Neurochem. 94,651–658.
Bano, D., Young, K.W., Guerin, C.J., Lefeuvre, R., Rothwell, N.J., Naldini, L., Rizzuto, R.,Carafoli, E., Nicotera, P., 2005. Cleavage of the plasma membrane Na+/Ca2+exchanger in excitotoxicity. Cell 120, 275–285.
Bark, I.C., 1993. Structure of the chicken gene for SNAP-25 reveals duplicated exonencoding distinct isoforms of the protein. J. Mol. Biol. 233, 67–76.
Benson, D.L., Watkins, F.H., Steward, O., Banker, G., 1994. Characterization of GABAergicneurons in hippocampal cell cultures. J. Neurocytol. 23, 279–295.
Berghuis, P., Dobszay, M.B., Sousa, K.M., Schulte, G., Mager, P.P., Hartig, W., Gorcs, T.J.,Zilberter, Y., Ernfors, P., Harkany, T., 2004. Brain-derived neurotrophic factorcontrols functional differentiation andmicrocircuit formation of selectively isolatedfast-spiking GABAergic interneurons. Eur. J. Neurosci. 20, 1290–1306.
Berghuis, P., Dobszay, M.B., Wang, X., Spano, S., Ledda, F., Sousa, K.M., Schulte, G.,Ernfors, P., Mackie, K., Paratcha, G., Hurd, Y.L., Harkany, T., 2005. Endocannabinoidsregulate interneuron migration and morphogenesis by transactivating the TrkBreceptor. Proc. Natl. Acad. Sci. U. S. A. 102, 19115–19120.
Binz, T., Blasi, J., Yamasaki, S., Baumeister, A., Link, E., Sudhof, T.C., Jahn, R., Niemann, H.,1994. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J. Biol. Chem.269, 1617–1620.
Bragina, L., Candiracci, C., Barbaresi, P., Giovedi, S., Benfenati, F., Conti, F., 2007.Heterogeneity of glutamatergic and GABAergic release machinery in cerebralcortex. Neuroscience 146, 1829–1840.
Brewer, G.J., Torricelli, J.R., Evege, E.K., Price, P.J., 1993. Optimized survival ofhippocampal neurons in B27-supplemented Neurobasal, a new serum-freemedium combination. J. Neurosci. Res. 35, 567–576.
Brown, J.P., Couillard-Despres, S., Cooper-Kuhn, C.M., Winkler, J., Aigner, L., Kuhn, H.G.,2003. Transient expression of doublecortin during adult neurogenesis. J. Comp.Neurol. 467, 1–10.
Costantin, L., Bozzi, Y., Richichi, C., Viegi, A., Antonucci, F., Funicello, M., Gobbi, M.,Mennini, T., Rossetto, O., Montecucco, C., Maffei, L., Vezzani, A., Caleo, M., 2005.Antiepileptic effects of botulinum neurotoxin E. J. Neurosci. 25, 1943–1951.
Frassoni, C., Inverardi, F., Coco, S., Ortino, B., Grumelli, C., Pozzi,D., Verderio, C.,Matteoli,M.,2005. Analysis of SNAP-25 immunoreactivity in hippocampal inhibitory neuronsduring development in culture and in situ. Neuroscience 131, 813–823.
Garbelli, R., Inverardi, F., Medici, V., Amadeo, A., Verderio, C., Matteoli, M., Frassoni, C.,2008. Heterogeneous expression of SNAP-25 in rat and human brain. J. Comp.Neurol. 506, 373–386.
Glading, A., Chang, P., Lauffenburger, D.A., Wells, A., 2000. Epidermal growth factorreceptor activation of calpain is required for fibroblast motility and occurs via anERK/MAP kinase signaling pathway. J. Biol. Chem. 275, 2390–2398.
Grant, N.J., Hepp, R., Krause, W., Aunis, D., Oehme, P., Langley, K., 1999. Differentialexpression of SNAP-25 isoforms and SNAP-23 in the adrenal gland. J. Neurochem.72, 363–372.
Guttmann, R.P., Baker, D.L., Seifert, K.M., Cohen, A.S., Coulter, D.A., Lynch, D.R., 2001.Specific proteolysis of the NR2 subunit at multiple sites by calpain. J. Neurochem.78, 1083–1093.
Hell, J.W., Westenbroek, R.E., Breeze, L.J., Wang, K.K., Chavkin, C., Catterall, W.A., 1996.N-methyl-D-aspartate receptor-induced proteolytic conversion of postsynapticclass C L-type calcium channels in hippocampal neurons. Proc. Natl. Acad. Sci. U. S. A.93, 3362–3367.
Hu, R.J., Bennett, V., 1991. In vitro proteolysis of brain spectrin by calpain I inhibitsassociation of spectrin with ankyrin-independent membrane binding site(s). J. Biol.Chem. 266, 18200–18205.
Huttner, W.B., Schiebler, W., Greengard, P., De Camilli, P., 1983. Synapsin I (protein I), anerve terminal-specific phosphoprotein. III. Its association with synaptic vesiclesstudied in a highly purified synaptic vesicle preparation. J. Cell. Biol. 96, 1374–1388.
Lai, K.C., Flaumenhaft, R., 2003. SNARE protein degradation upon platelet activation:calpain cleaves SNAP-23. J. Cell. Physiol. 194, 206–214.
Lu, X., Rong, Y., Baudry, M., 2000. Calpain-mediated degradation of PSD-95 indeveloping and adult rat brain. Neurosci. Lett. 286, 149–153.
Ma, H., Fukiage, C., Kim, Y.H., Duncan, M.K., Reed, N.A., Shih, M., Azuma, M., Shearer, T.R.,2001. Characterization and expression of calpain 10. A novel ubiquitous calpainwith nuclear localization. J. Biol. Chem. 276, 28525–28531.
Marshall, C., Hitman, G.A., Partridge, C.J., Clark, A., Ma, H., Shearer, T.R., Turner, M.D.,2005. Evidence that an isoform of calpain-10 is a regulator of exocytosis inpancreatic beta-cells. Mol. Endocrinol. 19, 213–224.
Neumar, R.W., Xu, Y.A., Gada, H., Guttmann, R.P., Siman, R., 2003. Cross-talk betweencalpain and caspase proteolytic systems during neuronal apoptosis. J. Biol. Chem.278, 14162–14167.

323C. Grumelli et al. / Molecular and Cellular Neuroscience 39 (2008) 314–323
Ort, T., Voronov, S., Guo, J., Zawalich, K., Froehner, S.C., Zawalich, W., Solimena, M., 2001.Dephosphorylation of beta2-syntrophin and Ca2+/mu-calpain-mediated cleavageof ICA512 upon stimulation of insulin secretion. Embo J. 20, 4013–4023.
Pasti, L., Volterra, A., Pozzan, T., Carmignoto, G., 1997. Intracellular calcium oscillations inastrocytes: a highly plastic, bidirectional form of communication between neuronsand astrocytes in situ. J. Neurosci. 17, 7817–7830.
Rutledge, T.W., Whiteheart, S.W., 2002. SNAP-23 is a target for calpain cleavage inactivated platelets. J. Biol. Chem. 277, 37009–37015.
Scholzke, M.N., Potrovita, I., Subramaniam, S., Prinz, S., Schwaninger, M., 2003.Glutamate activates NF-kappaB through calpain in neurons. Eur. J. Neurosci. 18,3305–3310.
Simpkins, K.L., Guttmann, R.P., Dong, Y., Chen, Z., Sokol, S., Neumar, R.W., Lynch, D.R., 2003.Selective activation induced cleavage of the NR2B subunit by calpain. J. Neurosci. 23,11322–11331.
Solimena, M., Folli, F., Aparisi, R., Pozza, G., De Camilli, P., 1990. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N. Engl. J. Med. 322,1555–1560.
Strobl, S., Fernandez-Catalan, C., Braun,M., Huber, R.,Masumoto, H., Nakagawa, K., Irie, A.,Sorimachi, H., Bourenkow, G., Bartunik, H., Suzuki, K., Bode, W., 2000. The crystalstructure of calcium-free human m-calpain suggests an electrostatic switchmechanism for activation by calcium. Proc. Natl. Acad. Sci. U. S. A. 97, 588–592.
Sun, D.A., Sombati, S., Blair, R.E., DeLorenzo, R.J., 2004. Long-lasting alterations inneuronal calcium homeostasis in an in vitro model of stroke-induced epilepsy. Cell.Calcium 35, 155–163.
Tafoya, L.C., Mameli, M., Miyashita, T., Guzowski, J.F., Valenzuela, C.F., Wilson, M.C., 2006.Expression and function of SNAP-25 as a universal SNARE component in GABAergicneurons. J. Neurosci. 26, 7826–7838.
Tompa, P., Buzder-Lantos, P., Tantos, A., Farkas, A., Szilagyi, A., Banoczi, Z., Hudecz, F.,Friedrich, P., 2004. On the sequential determinants of calpain cleavage. J. Biol. Chem.279, 20775–20785.
Verderio, C., Coco, S., Bacci, A., Rossetto, O., De Camilli, P., Montecucco, C., Matteoli, M.,1999. Tetanus toxin blocks the exocytosis of synaptic vesicles clustered at synapsesbut not of synaptic vesicles in isolated axons. J. Neurosci. 19, 6723–6732.
Verderio, C., Pozzi, D., Pravettoni, E., Inverardi, F., Schenk,U., Coco, S., Proux-Gillardeaux, V.,Galli, T., Rossetto, O., Frassoni, C., Matteoli, M., 2004. SNAP-25 modulation of calciumdynamics underlies differences in GABAergic and glutamatergic responsiveness todepolarization. Neuron 41, 599–610.
Wu, H.Y., Lynch, D.R., 2006. Calpain and synaptic function. Mol. Neurobiol. 33, 215–236.Xu,W., Wong, T.P., Chery, N., Gaertner, T., Wang, Y.T., Baudry, M., 2007. Calpain-mediated
mGluR1alpha truncation: a key step in excitotoxicity. Neuron 53, 399–412.Zimmerman, U.J., Malek, S.K., Liu, L., Li, H.L., 1999. Proteolysis of synaptobrevin,
syntaxin, and SNAP-25 in alveolar epithelial type II cells. IUBMB Life 48, 453–458.




















