
Botulinum Neurotoxin E-Insensitive Mutants of SNAP25 Fail to Bind VAMP but Support Exocytosis
10
Botulinum Neurotoxin E-Insensitive Mutants of SNAP-25 Fail to Bind VAMP but Support Exocytosis *Philip Washbourne, *Nicola Bortoletto, ‡Margaret E. Graham, ²Michael C. Wilson, ‡Robert D. Burgoyne, and *Cesare Montecucco *Centro CNR Biomembrane and Department of Biomedical Sciences, University of Padova, Padova, Italy; ² Department of Neurosciences, University of New Mexico, Albuquerque, New Mexico, U.S.A.; and ‡Physiological Laboratory, University of Liverpool, Liverpool, England Abstract: Neurotransmitter release from synaptic vesicles is mediated by complex machinery, which includes the v- and t-SNAP receptors (SNAREs), vesicle-associated mem- brane protein (VAMP), synaptotagmin, syntaxin, and synap- tosome-associated protein of 25 kDa (SNAP-25). They are essential for neurotransmitter exocytosis because they are the proteolytic substrates of the clostridial neurotoxins tet- anus neurotoxin and botulinum neurotoxins (BoNTs), which cause tetanus and botulism, respectively. Specifically, SNAP-25 is cleaved by both BoNT/A and E at separate sites within the COOH-terminus. We now demonstrate, using toxin-insensitive mutants of SNAP-25, that these two toxins differ in their specificity for the cleavage site. Following modification within the COOH-terminus, the mutants com- pletely resistant to BoNT/E do not bind VAMP but were still able to form a sodium dodecyl sulfate-resistant complex with VAMP and syntaxin. Furthermore, these mutants retain function in vivo, conferring BoNT/E-resistant exocytosis to transfected PC12 cells. These data provide information on structural requirements within the C-terminal domain of SNAP-25 for its function in exocytosis and raise doubts about the significance of in vitro binary interactions for the in vivo functions of synaptic protein complexes. Key Words: SNAP-25—Syntaxin—Vesicle-associated membrane pro- tein—SNAP receptor—Botulinum neurotoxin—Exocytosis. J. Neurochem. 73, 2424 –2433 (1999). Neuroexocytosis results from the fusion of synaptic vesicles with the presynaptic plasma membrane, leading to the discharge of the vesicle cargo of neurotransmitter into the synaptic cleft. This is accomplished by a highly regulated Ca 21 -dependent process, requiring a great number of proteins both in the vesicle and plasma mem- branes and in the cytoplasm (Su ¨dhof, 1995; Calakos and Scheller, 1996). It has been proposed that the presynaptic plasma membrane proteins syntaxin and synaptosome- associated protein of 25 kDa (SNAP-25), also collec- tively called t-SNAREs, engage with the synaptic vesicle membrane proteins vesicle-associated membrane protein (VAMP) and synaptotagmin (v-SNAREs) (Schiavo et al., 1997) to dock the vesicles at the plasma membrane (Chapman et al., 1994). Accordingly, the SNARE com- plex is thought to act as a scaffold to assemble other components necessary for fusion, such as NSF and a/b/x SNAPs (Sollner et al., 1993; Rothman and Warren, 1994). It has recently been suggested that the complex of these SNARE proteins could directly act to bring about membrane fusion and consequently neurotransmitter re- lease (Weber et al., 1998). The identification of SNAP-25, syntaxin, and VAMP as specific targets of the clostridial neurotoxins has illus- trated the essential role of these proteins in neuroexocy- tosis. These toxins are zinc-dependent proteases that completely block neuroexocytosis and thus give rise to the paralytic symptoms of both tetanus and botulism (Schiavo et al., 1992a–c). VAMP is proteolyzed by tet- anus neurotoxin and botulinum neurotoxins (BoNTs) B, D, F, and G (Schiavo et al., 1992a,b, 1993b,c, 1994). Syntaxin is cleaved by BoNT/C (Blasi et al., 1993; Schiavo et al., 1995), whereas SNAP-25 is cleaved by BoNT/A, C, and E (Schiavo et al., 1993a; Foran et al., 1996; Osen Sand et al., 1996; Vaidyanathan et al., 1999). The specificity of these toxins for their substrates has recently been shown to rely on recognition of the nona- peptide SNARE motif, which can lie a great distance from the cleavage sites in the substrates (Rossetto et al., 1994; Pellizzari et al., 1996, 1997). In particular, both BoNT/A and E have been shown to recognize and cleave Received May 17, 1999; revised manuscript received July 28, 1999; accepted July 30, 1999. Address correspondence and reprint requests to Dr. P. Washbourne at his present address: Department of Neurosciences, University of New Mexico, Basic Medical Science Building, 915 Camino de Salud, Albuquerque, NM 87131, U.S.A. Abbreviations used: BoNT, botulinum neurotoxin; GST, glutathione S-transferase; hGH, human growth hormone; NSF, N-ethylmaleimide- sensitive fusion protein; SDS, sodium dodecyl sulfate; SNAP, soluble NSF attachment protein; SNAP-25, synaptosome-associated protein of 25 kDa; SNARE, SNAP receptor; VAMP, vesicle-associated mem- brane protein. 2424 Journal of Neurochemistry Lippincott Williams & Wilkins, Inc., Philadelphia © 1999 International Society for Neurochemistry
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Botulinum Neurotoxin E-Insensitive Mutants of SNAP25 Fail to Bind VAMP but Support Exocytosis

Botulinum Neurotoxin E-Insensitive Mutants of SNAP-25 Failto Bind VAMP but Support Exocytosis
*Philip Washbourne, *Nicola Bortoletto, ‡Margaret E. Graham, †Michael C. Wilson,‡Robert D. Burgoyne, and *Cesare Montecucco
*Centro CNR Biomembrane and Department of Biomedical Sciences, University of Padova, Padova, Italy;†Department ofNeurosciences, University of New Mexico, Albuquerque, New Mexico, U.S.A.; and‡Physiological Laboratory,
University of Liverpool, Liverpool, England
Abstract: Neurotransmitter release from synaptic vesiclesis mediated by complex machinery, which includes the v-and t-SNAP receptors (SNAREs), vesicle-associated mem-brane protein (VAMP), synaptotagmin, syntaxin, and synap-tosome-associated protein of 25 kDa (SNAP-25). They areessential for neurotransmitter exocytosis because they arethe proteolytic substrates of the clostridial neurotoxins tet-anus neurotoxin and botulinum neurotoxins (BoNTs), whichcause tetanus and botulism, respectively. Specifically,SNAP-25 is cleaved by both BoNT/A and E at separate siteswithin the COOH-terminus. We now demonstrate, usingtoxin-insensitive mutants of SNAP-25, that these two toxinsdiffer in their specificity for the cleavage site. Followingmodification within the COOH-terminus, the mutants com-pletely resistant to BoNT/E do not bind VAMP but were stillable to form a sodium dodecyl sulfate-resistant complexwith VAMP and syntaxin. Furthermore, these mutants retainfunction in vivo, conferring BoNT/E-resistant exocytosis totransfected PC12 cells. These data provide information onstructural requirements within the C-terminal domain ofSNAP-25 for its function in exocytosis and raise doubtsabout the significance of in vitro binary interactions for the invivo functions of synaptic protein complexes. Key Words:SNAP-25—Syntaxin—Vesicle-associated membrane pro-tein—SNAP receptor—Botulinum neurotoxin—Exocytosis.J. Neurochem. 73, 2424–2433 (1999).
Neuroexocytosis results from the fusion of synapticvesicles with the presynaptic plasma membrane, leadingto the discharge of the vesicle cargo of neurotransmitterinto the synaptic cleft. This is accomplished by a highlyregulated Ca21-dependent process, requiring a greatnumber of proteins both in the vesicle and plasma mem-branes and in the cytoplasm (Su¨dhof, 1995; Calakos andScheller, 1996). It has been proposed that the presynapticplasma membrane proteins syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25), also collec-tively called t-SNAREs, engage with the synaptic vesiclemembrane proteins vesicle-associated membrane protein(VAMP) and synaptotagmin (v-SNAREs) (Schiavo etal., 1997) to dock the vesicles at the plasma membrane
(Chapman et al., 1994). Accordingly, the SNARE com-plex is thought to act as a scaffold to assemble othercomponents necessary for fusion, such as NSF anda/b/xSNAPs (Sollner et al., 1993; Rothman and Warren,1994). It has recently been suggested that the complex ofthese SNARE proteins could directly act to bring aboutmembrane fusion and consequently neurotransmitter re-lease (Weber et al., 1998).
The identification of SNAP-25, syntaxin, and VAMPas specific targets of the clostridial neurotoxins has illus-trated the essential role of these proteins in neuroexocy-tosis. These toxins are zinc-dependent proteases thatcompletely block neuroexocytosis and thus give rise tothe paralytic symptoms of both tetanus and botulism(Schiavo et al., 1992a–c). VAMP is proteolyzed by tet-anus neurotoxin and botulinum neurotoxins (BoNTs) B,D, F, and G (Schiavo et al., 1992a,b, 1993b,c, 1994).Syntaxin is cleaved by BoNT/C (Blasi et al., 1993;Schiavo et al., 1995), whereas SNAP-25 is cleaved byBoNT/A, C, and E (Schiavo et al., 1993a; Foran et al.,1996; Osen Sand et al., 1996; Vaidyanathan et al., 1999).The specificity of these toxins for their substrates hasrecently been shown to rely on recognition of the nona-peptide SNARE motif, which can lie a great distancefrom the cleavage sites in the substrates (Rossetto et al.,1994; Pellizzari et al., 1996, 1997). In particular, bothBoNT/A and E have been shown to recognize and cleave
Received May 17, 1999; revised manuscript received July 28, 1999;accepted July 30, 1999.
Address correspondence and reprint requests to Dr. P. Washbourneat his present address: Department of Neurosciences, University ofNew Mexico, Basic Medical Science Building, 915 Camino de Salud,Albuquerque, NM 87131, U.S.A.
Abbreviations used:BoNT, botulinum neurotoxin; GST, glutathioneS-transferase; hGH, human growth hormone; NSF,N-ethylmaleimide-sensitive fusion protein; SDS, sodium dodecyl sulfate; SNAP, solubleNSF attachment protein; SNAP-25, synaptosome-associated protein of25 kDa; SNARE, SNAP receptor; VAMP, vesicle-associated mem-brane protein.
2424
Journal of NeurochemistryLippincott Williams & Wilkins, Inc., Philadelphia© 1999 International Society for Neurochemistry

SNAP-25 as long as one of its four SNARE motifs ispresent (Washbourne et al., 1997).
SNAP-25 and its nonneuronal homologues have beencloned from a wide variety of eukaryotic organisms,ranging from yeast and plants to sea urchin and mam-mals (Bark, 1993; Risinger et al., 1993; Bark and Wil-son, 1994a; Brennwald et al., 1994; Schulz et al., 1998).The protein sequence displays great conservation, espe-cially among vertebrates such as chicken, mouse, andhuman, which share complete identity. The nonneuronalhomologue SNAP-23 has been found in mammals,where it is expressed in most other exocytotic cells suchas adipocytes and hepatocytes and is thought to have afunction similar to SNAP-25 in slow exocytosis (Rav-ichandran et al., 1996; Gaisano et al., 1997). Recently,another homologue of SNAP-25, SNAP-29, has beenfound (Steegmaier et al., 1998), underlining the fact thatwe do not know the complete number of isoforms andsplice variants present in mammals. In neurons,SNAP-25 has been suggested to play a role in neuriteextension and thus in the development of the nervoussystem (Osen Sand et al., 1993, 1996). In fact, SNAP-25is present in two isoforms, a and b, the former beingpresent during fetal development and the synthesis of theb isoform increased postnatally during the period ofsynapse stabilization (Bark and Wilson, 1994a,b; Bark etal., 1995). This suggests that SNAP-25 isoforms and itshomologues play a variety of roles that may influence thecharacteristics of distinct fusion events in different cellsand stages of development (Bark and Wilson, 1994a,b).
To examine the specificity of BoNT recognition ofcleavage sites, we have generated BoNT-resistant mu-tants of SNAP-25. We demonstrate that whereas bothBoNT/A and E have previously been shown to havesimilar substrate recognition of SNARE motifs present inSNAP-25 (Washbourne et al., 1997), the recognition ofthe cleavage sites by the two toxins differs. AlthoughBoNT/A is rendered inactive by changing an arginine totryptophan in the cleavage site of SNAP-25, completeinsensitivity to BoNT/E can be achieved only by alsomoving a negative charge into the cleavage site. Thissuggests that the binding requirements for the catalyti-cally active sites of BoNT/A and E differ considerably.Immunoprecipitation experiments with these mutants re-vealed that the mutations that make SNAP-25 com-pletely resistant to BoNT/E also abolish the binary asso-ciation of SNAP-25 with VAMP2. The mutants arenonetheless able to form sodium dodecyl sulfate (SDS)-resistant complexes with VAMP and syntaxin. Further-more, the mutants are active in exocytosis in vivo, res-cuing the exocytosis block caused by BoNT/E in PC12cells. This provides new information on the importanceof specific C-terminal residues of SNAP-25 for exocy-tosis.
EXPERIMENTAL PROCEDURES
Vectors and strainsAll glutathioneS-transferase (GST)-fusion proteins were pro-
duced from pGEX 4T3 (Pharmacia) containing the SNAP-25
fragments and expressed inEscherichia coliBL21. The transfec-tions of mammalian cells were carried out with the SNAP-25fragments in pcDNA/HA1. This vector was produced by anneal-ing the two oligonucleotides 59-GCTAAGCTTATGTACCCAT-ACGACGTCCCAGACTACGCTGGATCCCGGC and 59-GCCGGATCCAGCGTAGTCTGGGACGTCGTATGGGT-ACATAAGCTTAGC to each other, cutting withHindIII andBamHI, and then insertion into the polylinker of pcDNA3 (In-vitrogen). Site-directed mutagenesis was performed on SNAP-25bin pBluescript SK1 (Invitrogen). All subcloning and mutagenesissteps were performed in XL-1 Blue or JM 101.
Site-directed mutagenesisThe SNAP-25b A mutant Am(R198W) was produced by PCR
using the primers SBf, 59-CGGGATCCCCCACCACTACCAT-GGCCGAGGAC, and SBAm, 59-GCGGAATTCTTAACCACT-TCCCAGCATCTTTGTTGCCCATTGGTTGGCTTC, withSNAP-25b as a template, cutting withEcoRI and BamHI, andinsertion into pcDNA/HA1. The obtained clone was subclonedinto pGEX4T3. The SNAP-25 E mutants Em1(I178L,R180W)and Em2(R180W,I181E,E183I) were produced by the mega-primer PCR method (Barik, 1996). The megaprimers were createdusing the reverse primer SBr, 59-CGGAATTCTTAACCACTTC-CCAGCATCTTTGT, and the mutagenic primers SBEm1,59-ACCCAGAATCGCCAGCTGGACTGGATCATGGAGA-AGGCT, and SBEm2, 59-CGCCAGATTGACTGGGAGAT-GATAAAGGCTGACTCC. The PCR products were then purifiedfrom the excess nucleotides and primers and used as reverseprimers in conjunction with SBf for the second PCR step. Theproducts were purified from agarose, cut withBamHI andEcoRI,inserted into pcDNA/HA1, and treated as SNAP-25b Am. Thedouble-resistance mutant AEm2(R180W,I181E,W183I,R198W)was created by performing PCR using the primers SBf and SBAmwith the SNAP-25 Em2 DNA clone as a template. This wastreated as above and also inserted into pcDNA/HA1. All mutantsobtained by PCR mutagenesis were sequenced using a femtomolesequencing kit (Pharmacia) with T7 and Sp6 primers (GIBCO).
Proteolytic cleavageGST fusion proteins of all mutants and SNAP-25b wild type
were produced as described previously (Pellizzari et al., 1996).They were assayed for their sensitivity to BoNT/A and E byincubating the fusion proteins (0.2 mg/ml final concentration)with 100 nM preactivated toxin for various times from 0 to 120min as before (Washbourne et al., 1997). The samples werethen electrophoresed, blotted, and probed with an anti-C-ter-minal antibody for SNAP-25. The blots were visualized withenhanced chemiluminescence blotting reagents and a peroxi-dase-conjugated secondary antibody. Cleavage is expressed aspercent reduction of immunoreactivity compared with the 0time point.
Immunoprecipitation of SNAP-25/VAMP2/syntaxincomplexes
COS 7 cells were grown overnight at a density of 0.253 106
cells/well in six-well plates. For transfection, the medium wasremoved and replaced with OptiMEM (GibcoBRL) containing0.875mg of VAMP2/pcDNA3 and/or syntaxin 1a/pcDNA3 and0.875 mg of either pcDNA3 or the SNAP-25 constructs inpcDNA/HA and 3.8 mg of pBluescript with 15ml of Lipo-fectamine reagent (GibcoBRL) that had been preincubated for 30min at room temperature in a volume of 300ml and then diluted1:5. The cells were left for 6 h, and then 1 ml of Dulbecco’smodified Eagle’s medium with 20% fetal calf serum was added.The medium was completely changed after a further 12 h. After
J. Neurochem., Vol. 73, No. 6, 1999
2425BoNT-INSENSITIVE SNAP-25 MUTANTS PROMOTE EXOCYTOSIS

24 h, the cells were trypsinized, collected, washed in phosphate-buffered saline, and lysed in lysis solution (150 mM NaCl, 20 mMTris-HCl, 2 mM EDTA, 1% Triton X-100, 0.5% deoxycholate,0.05% SDS, 1 mM phenylmethylsulfonyl fluoride, pH 8.0). Thelysate was centrifuged at 10,000g for 5 min, and the supernatantwas incubated at room temperature for 1 h with protein G–Sepha-rose (Pharmacia) to which had previously been conjugated amonoclonal anti-SNAP-25 antibody. The resin was washed threetimes with lysis solution, and the bound proteins were eluted withLaemmli sample buffer at 50°C (1% SDS, 60 mM Tris-HCl, 10%glycerol, 20 mM dithiothreitol, 0.05% bromophenol blue). Toassay SDS-resistant complexes, samples were divided into two,and one aliquot was boiled for 10 min. The samples were thenelectrophoresed in a 15% polyacrylamide gel, transferred to nitro-cellulose, and probed with polyclonal antibodies to SNAP-25 andVAMP and monoclonal antibodies to SNAP-25 (SMI-81; Stern-berger Monoclonals) and syntaxin (HPC-1; Sigma) at opportunedilutions. The blots were visualized with ECL1 (Amersham) anda Storm phosphoimager. The VAMP immunoreactivity was com-pared with that of SNAP-25 and the VAMP levels precipitatedwith wild-type SNAP-25 normalized to 100%.
Gradient fractionation of SDS-resistant complexCOS 7 cells were grown overnight at a density of 106
cells/plate in 10-cm culture dishes and were transfected withVAMP2/pcDNA3, syntaxin 1a/pcDNA3, and either SNAP-25b/HA/pcDNA or SNAP-25b-Em2/HA/pcDNA, as describedabove. After 48 h, the cells were harvested with trypsin andlysed in 1 ml of lysis solution. After pelleting insoluble matter,the lysate was split into two; half was heated to 50°C and theother to 95°C. The lysates were then loaded on 10–35% glyc-erol gradients and centrifuged as described by Sollner et al.(1993). The gradients were fractionated, and the aliquots wereprecipitated with 80% acetone (final volume) at220°C for 2 h.The fractions were then submitted to 15% polyacrylamide gelelectrophoresis and western blotting and then probed as men-tioned above. Ovalbumin (44 kDa) andg-globulin (150 kDa)were used as markers, sedimenting at fractions 3–4 and 7–9,respectively.
Expression and purification of recombinantHis6-tagged toxin light chains
Plasmids encoding BoNT/A and BoNT/E light chains asHis6-tagged constructs were a gift from Dr. Heiner Niemann(Hannover, Germany). Recombinant protein expression wasinduced with 0.5 mM isopropyl-1-thio-b-D-galactopyranosideat 37°C for 2.5–5 h and His6-tagged proteins purified from thecytosolic fraction of M15 (pREP4) cells on Ni-NTA-agarose.His6-tagged proteins were eluted from the Ni-NTA-agarose byapplying a 50–500 mM imidazole gradient. All chromatogra-phy was performed at 4°C using a Pharmacia FPLC system.Peak fractions containing toxin light chains were identified bySDS–polyacrylamide gel electrophoresis, and the pooled stocksolutions were stored at270°C in aliquots.
Transfection of PC12 cells and assay of growthhormone release
The method of Wick et al. (1993) for transient co-transfec-tion of PC12 cells and assay of growth hormone secretion as amarker of transfected cells was adapted (Graham et al.,1997a,b). PC12 cells were grown overnight in collagen-coated24-well trays at a density of 0.853 106/well (Graham et al.,1997a). For transfection, the medium was removed from cellsand replaced with RPMI 1640 medium containing 4mg of thegrowth hormone plasmid pXGH5 and 2mg of additional plas-
mid consisting of control pcDNA3 or the SNAP-25 constructsand 20ml of Lipofectamine reagent that had been preincubatedfor 45 min in 200ml of medium and then diluted 1:5. The cellswere left for 5 h, the medium was replaced with fresh serum-containing medium containing 10mM CdCl2 to induce humangrowth hormone (hGH) expression, and the cells were main-tained in culture for 72 h. For use in permeabilization assays,the cells were washed twice in a Krebs–Ringer buffer (Grahamet al., 1997a). The cells were permeabilized by addition of 300ml/well of permeabilization buffer [139 mM potassium gluta-mate, 20 mM piperazine-N,N9-bis(2-ethanesulfonic acid), 5mM EGTA, 2 mM ATP, 2 mM MgCl2 pH 6.5] containing 20mM digitonin for 6 min. After removal of the buffer, the cellswere incubated for an additional 40 min in permeabilizationbuffer with or without added toxin light chain (400 nM forBoNT/A and 14 nM for BoNT/E light chain) and then chal-lenged by the addition of permeabilization buffer containing 0Ca21 or with 10 mM Ca21. Buffer samples and the cells wereprocessed and assayed for hGH levels using an enzyme-linkedimmunosorbent kit according to the manufacturer’s instructions(Boehringer-Mannheim). In all experiments, the amount ofhGH release is expressed as a percentage of total cellularcontent of hGH.
RESULTS
Proteolytic cleavage of SNAP-25 mutantsTo probe the cleavage site requirements of BoNT/A
and E, site-directed mutagenesis was carried out on therespective cleavage sites. We considered that a largehydrophobic residue such as tryptophan in place of thepositively charged amino acid arginine at the P91 or P1position would sterically hinder access to the peptidebond to be cleaved. Consequently, the SNAP-25b Amutant, Am(R198W), and the SNAP-25b E mutant,Em1(I178L,R180W), were cloned and expressed in bac-teria as GST fusion proteins (see Fig. 1). As shown inFig. 2A, Am was resistant to BoNT/A cleavage in toxinexcess. In contrast, mutation of the BoNT/E cleavage sitedid not influence proteolysis by BoNT/A. On the otherhand, cleavage of Em1 by BoNT/E was much reducedwith respect to the wild type, suggesting that partialinhibition of BoNT/E had been achieved (Fig. 2B).
To further reduce cleavage of SNAP-25 by BoNT/E, weexamined the exchange of the negatively charged glutamateresidue in the P93 position (E183) with the hydrophobicisoleucine residue in the P91 position (I181; see Fig. 1),resulting in mutant Em2(R180W,I181E,E183I). This sub-stitution caused no change in the overall charge or aminoacid content of the protein but moved a negative chargecloser to the cleavage site. As can be appreciated from Fig.2B, this resulted in a total abolition of proteolysis, alsoobserved after prolonged periods of incubation (data notshown). In addition, a fourth mutant was made in which themutations for insensitivity to BoNT/A and BoNT/E werecombined into one SNAP-25 molecule. As was expected,the SNAP-25 AEm2(R180W,I181E,E183I,R198W) pro-tein was not cleaved by either toxin even after prolongedperiods of incubation (data not shown).
J. Neurochem., Vol. 73, No. 6, 1999
2426 P. WASHBOURNE ET AL.

Co-immunoprecipitation of binary complexes withVAMP and syntaxin
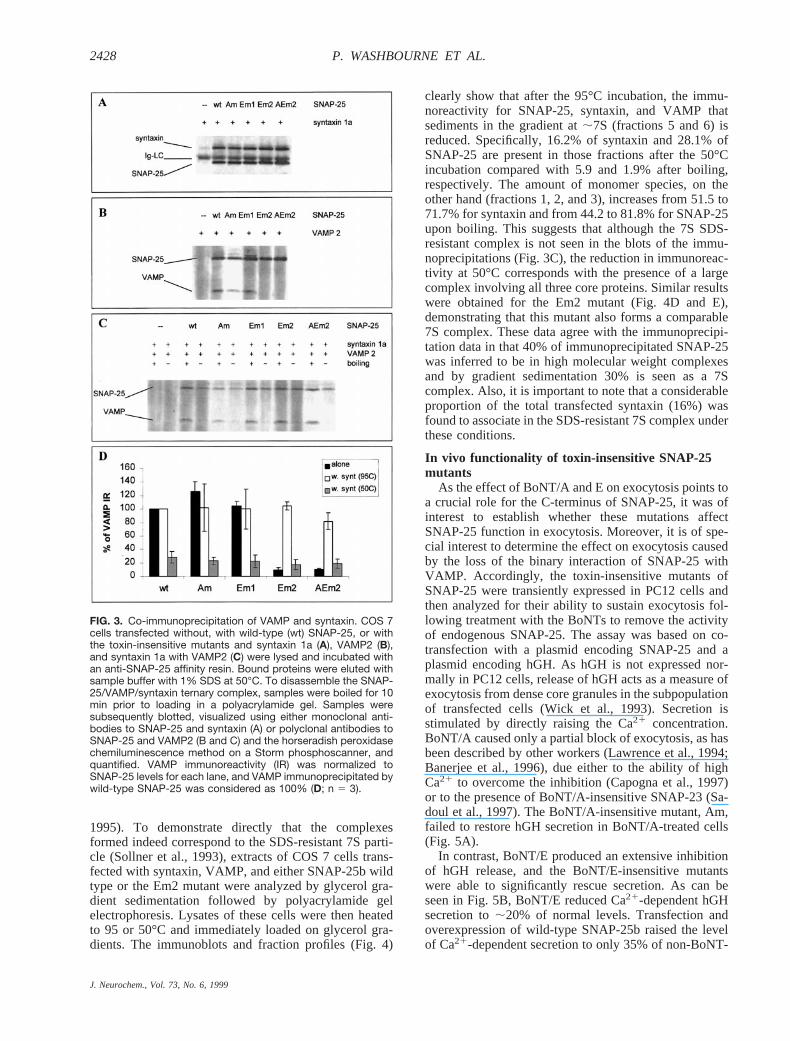
The mutations we introduced at the BoNT cleavagesites lie within the carboxy-terminal region of SNAP-25,which has been identified to bind VAMP and has con-sequently been implicated as critical for the formation ofthe ternary SDS-resistant complex (Hayashi et al., 1994).It was therefore of interest to determine whether themutations alter the interaction with two proteins of thecore complex, syntaxin 1a and VAMP2. To assess this,we co-transfected these proteins in COS 7 cells andevaluated their ability to associate by co-immunoprecipi-tating with an anti-SNAP-25 affinity resin. Wild-typeSNAP-25 forms stable binary complexes with both theseproteins. As shown in Fig. 3A, the C-terminal mutationshad no effect on the ability of SNAP-25 to co-precipitatesyntaxin, whose binding sites have been mapped to theN-terminal portion of the SNAP-25 protein. In contrast,the mutations in Em2, which completely abolished cleav-age by BoNT/E, totally abrogated the binding ofSNAP-25 to VAMP2 (Fig. 3B and D). The mutants Amand Em1 showed no reduction in affinity for VAMP2.
Formation of SDS-resistant ternary complexesIn the presence of SDS, the ternary core complex
formed by SNAP-25, syntaxin, and VAMP requires boil-ing for dissociation (Hayashi et al., 1994). BecauseBoNT cleavage of SNARE proteins prevents the forma-tion of the ternary complex, the ability of these proteinsto form a heat-stable, SDS-resistant complex has beenconsidered a measure of their tight association and prob-able cooperative function in exocytosis. When eitherwild-type or mutant forms of SNAP-25 were co-ex-pressed with syntaxin and VAMP, equivalent amounts ofVAMP were co-immunoprecipitated with SNAP-25 an-tibodies if the samples were boiled prior to loading on apolyacrylamide gel. However, if the immunoprecipitate
samples were heated only to 50°C, to release SNAP-25from the antibody, but not boiled before loading, theSNARE proteins were retained in the ternary complex(not distinguished by our antibodies). The amount ofVAMP detected as a separately migrating protein wasreduced to;20% compared with boiled samples (Fig.3C and D). The amount of SNAP-25, on the other hand,was reduced to between 48 and 73% of the boiled sam-ples for all SNAP-25 constructs (average 59%; data notshown). This suggests that under these conditions,;80% of the VAMP immunoprecipitated associates in ahigher molecular mass, SDS-resistant complex, whereasonly 40% of the SNAP-25 immunoprecipitated partakesin the SDS-resistant complex. Thus, 20% of total VAMPis involved in non-SDS-resistant complexes. Surpris-ingly, the Em2 and AEm2 mutants, which are unable tobind VAMP directly, in the presence of syntaxin arecapable not only of co-precipitating VAMP but also offorming an SDS-resistant complex that withstands tem-peratures up to 50°C (Fig. 3C and D).
Immunoblotting of the 50°C-treated samples afterSDS–polyacrylamide gel electrophoresis failed to effec-tively identify 7S complexes, even on 4–20% gradientgels (not shown), possibly because of the small amountsand masking of the relevant epitopes in the complex, anobservation previously made by others (Pellegrini et al.,
FIG. 1. The C-terminal portion of SNAP-25. The diagram showsthe BoNT cleavage sites and the mutations introduced by site-directed mutagenesis. The I178L mutation in Em1 was insertedfor screening purposes but is not considered to be of importancefor protein structure. The a–g labeling refers to the heptad re-peats of the coiled coil motif and the underlined residues to theamino acids whose side chains are oriented to the inner core ofthe SDS-resistant four-helix bundle (from Sutton et al., 1998).Am, R198W; Em1, I178L,R180W; Em2, R180W,I181E,E183I;AEm2, R180W,I181E,E183I,R198W.
FIG. 2. The proteolysis by BoNT/A and E of the cleavage sitemutants of SNAP-25. All mutants were incubated in 100 nMBoNT/A (A) and 100 nM BoNT/E (B). All time point samples werethen submitted to electrophoresis and western blot. The blotswere visualized using an anti-C-terminal antibody to SNAP-25and the horseradish peroxidase chemiluminescence methodand were quantified by densitometric scanning (n 5 3).
J. Neurochem., Vol. 73, No. 6, 1999
2427BoNT-INSENSITIVE SNAP-25 MUTANTS PROMOTE EXOCYTOSIS
1995). To demonstrate directly that the complexesformed indeed correspond to the SDS-resistant 7S parti-cle (Sollner et al., 1993), extracts of COS 7 cells trans-fected with syntaxin, VAMP, and either SNAP-25b wildtype or the Em2 mutant were analyzed by glycerol gra-dient sedimentation followed by polyacrylamide gelelectrophoresis. Lysates of these cells were then heatedto 95 or 50°C and immediately loaded on glycerol gra-dients. The immunoblots and fraction profiles (Fig. 4)
clearly show that after the 95°C incubation, the immu-noreactivity for SNAP-25, syntaxin, and VAMP thatsediments in the gradient at;7S (fractions 5 and 6) isreduced. Specifically, 16.2% of syntaxin and 28.1% ofSNAP-25 are present in those fractions after the 50°Cincubation compared with 5.9 and 1.9% after boiling,respectively. The amount of monomer species, on theother hand (fractions 1, 2, and 3), increases from 51.5 to71.7% for syntaxin and from 44.2 to 81.8% for SNAP-25upon boiling. This suggests that although the 7S SDS-resistant complex is not seen in the blots of the immu-noprecipitations (Fig. 3C), the reduction in immunoreac-tivity at 50°C corresponds with the presence of a largecomplex involving all three core proteins. Similar resultswere obtained for the Em2 mutant (Fig. 4D and E),demonstrating that this mutant also forms a comparable7S complex. These data agree with the immunoprecipi-tation data in that 40% of immunoprecipitated SNAP-25was inferred to be in high molecular weight complexesand by gradient sedimentation 30% is seen as a 7Scomplex. Also, it is important to note that a considerableproportion of the total transfected syntaxin (16%) wasfound to associate in the SDS-resistant 7S complex underthese conditions.
In vivo functionality of toxin-insensitive SNAP-25mutants
As the effect of BoNT/A and E on exocytosis points toa crucial role for the C-terminus of SNAP-25, it was ofinterest to establish whether these mutations affectSNAP-25 function in exocytosis. Moreover, it is of spe-cial interest to determine the effect on exocytosis causedby the loss of the binary interaction of SNAP-25 withVAMP. Accordingly, the toxin-insensitive mutants ofSNAP-25 were transiently expressed in PC12 cells andthen analyzed for their ability to sustain exocytosis fol-lowing treatment with the BoNTs to remove the activityof endogenous SNAP-25. The assay was based on co-transfection with a plasmid encoding SNAP-25 and aplasmid encoding hGH. As hGH is not expressed nor-mally in PC12 cells, release of hGH acts as a measure ofexocytosis from dense core granules in the subpopulationof transfected cells (Wick et al., 1993). Secretion isstimulated by directly raising the Ca21 concentration.BoNT/A caused only a partial block of exocytosis, as hasbeen described by other workers (Lawrence et al., 1994;Banerjee et al., 1996), due either to the ability of highCa21 to overcome the inhibition (Capogna et al., 1997)or to the presence of BoNT/A-insensitive SNAP-23 (Sa-doul et al., 1997). The BoNT/A-insensitive mutant, Am,failed to restore hGH secretion in BoNT/A-treated cells(Fig. 5A).
In contrast, BoNT/E produced an extensive inhibitionof hGH release, and the BoNT/E-insensitive mutantswere able to significantly rescue secretion. As can beseen in Fig. 5B, BoNT/E reduced Ca21-dependent hGHsecretion to;20% of normal levels. Transfection andoverexpression of wild-type SNAP-25b raised the levelof Ca21-dependent secretion to only 35% of non-BoNT-
FIG. 3. Co-immunoprecipitation of VAMP and syntaxin. COS 7cells transfected without, with wild-type (wt) SNAP-25, or withthe toxin-insensitive mutants and syntaxin 1a (A), VAMP2 (B),and syntaxin 1a with VAMP2 (C) were lysed and incubated withan anti-SNAP-25 affinity resin. Bound proteins were eluted withsample buffer with 1% SDS at 50°C. To disassemble the SNAP-25/VAMP/syntaxin ternary complex, samples were boiled for 10min prior to loading in a polyacrylamide gel. Samples weresubsequently blotted, visualized using either monoclonal anti-bodies to SNAP-25 and syntaxin (A) or polyclonal antibodies toSNAP-25 and VAMP2 (B and C) and the horseradish peroxidasechemiluminescence method on a Storm phosphoscanner, andquantified. VAMP immunoreactivity (IR) was normalized toSNAP-25 levels for each lane, and VAMP immunoprecipitated bywild-type SNAP-25 was considered as 100% (D; n 5 3).
J. Neurochem., Vol. 73, No. 6, 1999
2428 P. WASHBOURNE ET AL.

treated cells. In contrast, the BoNT/E-insensitiveSNAP-25 mutants provided significantly more secretionthan the transfected wild-type SNAP-25, with the Em2and AEm2 constructs restoring normal secretion levels.Close to complete restoration of Ca21-dependent exocy-tosis was seen with the Em2 construct in nine separatetransfections. Also, transfection of all SNAP-25 con-structs did not alter secretion from non-toxin-treatedcells in any way. Localization of the constructs in im-munofluorescence experiments utilizing an antibodyagainst the HA1 tag revealed that the transfected con-structs were localized primarily in the plasma membrane,whereas cells transfected with the vector pCDNA/HA1showed cytoplasmic staining (data not shown).
It was considered that the toxin-insensitive mutantsEm2 and AEm2 might act as inhibitors of the proteolyticactivity of BoNT/E by binding tightly to the active site.
This would thus allow exocytosis to proceed as thoughno toxin had been added to the cells. This possibility wasinvestigated by cleaving wild-type SNAP-25 withBoNT/E in the presence of varying amounts of the mu-tant Em2. Even when the mutant protein was in largeexcess compared with the wild-type form, the toxin wasable to efficiently cleave SNAP-25 (data not shown). Wethus conclude that the protection of the exocytotic blockby the toxin-insensitive mutants is afforded by theirability to function at the vesicle/plasma membrane inter-face and not because they act as potent inhibitors ofBoNT/E.
DISCUSSION
The role of SNAP-25 in the orchestration of mem-brane trafficking and fusion required for regulated re-
FIG. 4. Glycerol gradient fractionation of theSDS-resistant complex. COS 7 cells trans-fected with syntaxin, VAMP, and SNAP-25b orEm2 were lysed, heated to 50°C (A and D) or95°C (B and E), and loaded on 10–35% glyc-erol gradients. Fractions collected were precip-itated with acetone, loaded on a 15% poly-acrylamide gel, and immunoblotted. Proteinswere visualized using monoclonal antibodies tosyntaxin and SNAP-25 and a polyclonal anti-body to VAMP2 and the horseradish peroxi-dase chemiluminescence method on a Stormphosphoscanner and quantified (C). The dataare displayed as a percentage of the total syn-taxin or SNAP-25. wt, wild type.
J. Neurochem., Vol. 73, No. 6, 1999
2429BoNT-INSENSITIVE SNAP-25 MUTANTS PROMOTE EXOCYTOSIS

lease of neurotransmitter from neurons or other neuro-secretory cells has yet to be fully resolved. Unlike theother v- and t-SNAREs, SNAP-25 is not an intrinsicmembrane protein but is thought to be required tostrengthen the association between VAMP and syntaxin(Sutton et al., 1998; Weber et al., 1998). SNAP-25 maytherefore be viewed as serving as an accessory proteinthat facilitates or increases the efficacy of those proteinmechanics that actively promote vesicular fusion. HowSNAP-25 functions to provide that service and how itmay influence the divergent vesicular fusion processesused in fast synaptic transmission, secretion of neuropep-tides, and axonal outgrowth during development (Barkand Wilson, 1994a,b) may be therefore evaluatedthrough how it associates with other proteins of theexocytotic machinery. The cleavage of SNAP-25 byeither BoNT/A or E, which effectively blocks neuro-transmitter release (Schiavo et al., 1993a) and excises aregion of the protein thought to directly bind VAMP(Hayashi et al., 1994), therefore provides a probe into thesequence and binding requirements for the physiologi-cally relevant function of SNAP-25.
The functional analysis of the mutations that renderSNAP-25 insensitive to BoNT/A and E suggests that theactive site pockets of these toxins are considerably dif-ferent. Removal of a positive charge and insertion of thelarge hydrophobic tryptophan residue block proteolytic
cleavage by BoNT/A, suggesting either that access to thepeptide bond is sterically hindered or that the positivelycharged amino acid arginine 198 is important for bindingof the toxin to the cleavage site. The second possibility iscorroborated by the recent crystal structure of BoNT/A,which shows a high density of negative charge in theactive site pocket (Lacy et al., 1998). In contrast, cleav-age by BoNT/E is dramatically reduced, but not entirelyabolished, by the analogous arginine-to-tryptophan sub-stitution in the BoNT/E cleavage site. Complete inhibi-tion was achieved by switching nearby glutamate andisoleucine residues. The cleavage of Em1, despite thepresence of the tryptophan residue, suggests thatBoNT/E has a larger, more accommodating active sitecleft.
It is interesting to note that the mutations to theBoNT/E cleavage site also progressively affect the rateof proteolysis of BoNT/A, although being at a distance of;18 amino acids. This may reflect a possible change inthe secondary or tertiary structure of the C-terminus,resulting in a reduced affinity or cleavage efficacy byBoNT/A. On the other hand, this may suggest a largeinteraction interface between BoNT/A and its targetpolypeptide.
The effects of the mutations on the gross secondarystructure of the mutant proteins were probed by analyz-ing their interaction with syntaxin-1a and VAMP. It wasexpected that the interaction between SNAP-25 and syn-taxin would not be affected, as the region of SNAP-25that interacts with syntaxin is the N-terminal half of theprotein (Hayashi et al., 1994). It has been well docu-mented that the removal of the carboxy-terminus ofSNAP-25 by BoNT/A and E prevents binding of VAMP(Hayashi et al., 1994). The co-immunoprecipitation as-says from transfected cells demonstrated that whereasthe Am mutation had no effect on the ability of SNAP-25to bind VAMP, mutations at the BoNT/E cleavage sitedid greatly reduce VAMP binding. The results suggestthat the arginine-to-tryptophan mutations at either theBoNT/A or E cleavage sites do not have a deleteriouseffect on the secondary/tertiary structure, allowingVAMP to bind with normal affinity. In contrast, theresidues immediately C-terminal to the BoNT/E cleav-age site seem critical for the binary interaction ofSNAP-25 with VAMP.
As SNAP-25 may act principally to reinforce the in-teraction of syntaxin and VAMP when those moleculesare positioned on opposing membranes, we examinedwhether the SNAP-25 mutants could support the forma-tion of the heat-stable, SDS-resistant complex. It is worthnoting that these co-immunoprecipitations were carriedout with proteins not expressed as bacterial recombinantfusion proteins, but synthesized, posttranslationally mod-ified, and possibly assembled into complexes in mam-malian cells. Surprisingly, toxin-insensitive SNAP-25proteins that cannot bind VAMP were able to recruitVAMP into a ternary complex capable of resisting 50°C.The fact that;80% of the immunoprecipitated VAMPforms a part of SDS-resistant complexes, regardless of
FIG. 5. Ca21-dependent hGH release from PC12 cells. Trans-fected PC12 cells were permeabilized for 6 min and then incu-bated for 40 min without (control) or with 400 nM BoNT/A (A) or4 nM BoNT/E (B). In all cases, the controls were transfected withpcDNA3 plasmid. The cells were then challenged by the additionof buffer containing 0 mM Ca21 (open columns) or 10 mM Ca21
(filled columns). Buffer samples and cells were processed andassayed for hGH levels and hGH release expressed as a per-centage of total cell content (A, n 5 3; B, n 5 5).
J. Neurochem., Vol. 73, No. 6, 1999
2430 P. WASHBOURNE ET AL.

which SNAP-25 mutant is used to immunoprecipitate,suggests that the mutations made in the carboxy-termi-nus are not detrimental for the integrity of the SDS-resistant complex. Surprisingly, isoleucine residue 181,which is mutated to glutamate in the BoNT/E-resistantmutants, maps to the surface of the amphipathic helixthat lies toward the inner core of the four-helix bundlewithin the complex (Sutton et al., 1998). On the basis ofthe crystal structure, the authors suggest that the hydro-phobic core of the four-helix bundle plays a major role inits stability, thus predicting a destabilizing effect of ourEm2 and AEm2 mutants. We observed no effect of theseSNAP-25 variants in the formation of the SNARE com-plex and its SDS-resistant properties. From a mutationalstudy of the hydrophobic core residues (Chen et al.,1999), it is clear that at least two contiguous core layersmust be disrupted to reduce thermostability and function-ality of the SDS-resistant complex. This, together withour results, suggests that the binary interaction of VAMPand SNAP-25 is stabilized considerably by the formationof a four-helix bundle with syntaxin and the N-terminalportion of SNAP-25.
The ability of BoNT/A and BoNT/E to inhibit exocy-tosis by cleavage of SNAP-25 close to its C-terminusimplicates this region of SNAP-25 as functionally im-portant in the exocytotic machinery. For this reason, thenonconservative mutations produced in this study, toprobe the cleavage site requirements of BoNT/A and E,were assayed for function in PC12 cells. The hGH se-cretion assay has been used extensively to probe struc-tural requirements of proteins of the exocytotic apparatus(Barnard et al., 1997; Graham et al., 1997a,b). Introduc-tion of the recombinant light chains of BoNT/A and Ereduced exocytosis to different extents. In the case ofBoNT/A, although this toxin causes a strong block inintact tissues and cells, in this preparation only partialinhibition was seen, as previously reported for PC12cells (Banerjee et al., 1996). This may reflect the obser-vation that BoNT/A toxicity can be overcome by expo-sure to high Ca21 (Molgo et al., 1990; Lawrence et al.,1994; Banerjee et al., 1996; Capogna et al., 1997). On theother hand, this partial inhibition could be attributed tothe presence of SNAP-23, which, in mice, is cleaved atvery low rates by BoNT/A (Vaidyanathan et al., 1999)and has been shown to sustain exocytosis from insuli-noma cells (Sadoul et al., 1997).
In contrast, BoNT/E treatment resulted in a verystrong inhibition under these conditions, almost reducingsecretion to the basal Ca21-independent levels. Interest-ingly, introduction of exogenous BoNT/E-resistantSNAP-25 by transient transfection overcame the blockby BoNT/E. Overexpression of wild-type SNAP-25causes only a small increase of Ca21-dependent secre-tion, probably due to incomplete proteolysis of SNAP-25by BoNT/E in those cells. Expression of any of theBoNT/E-insensitive mutants, on the other hand, dramat-ically increased secretion. The extent of sensitivity toBoNT/E proteolysis of the SNAP-25 mutants is reflectedin the amount to which secretion in BoNT-treated cells is
regained; that is, the completely insensitive mutant, Em2,completely restores normal levels of hGH Ca21-depen-dent exocytosis, whereas the partially resistant mutant,Em1, restores around one third of the activity beyondthat of wild-type SNAP-25. Also, the double-resistancemutant, AEm2, is able to restore secretion levels afterBoNT/E treatment to those of toxin-free cells. This sug-gests that the inability to detect whether the Am mutantcan effectively restore secretion in the presence ofBoNT/A reflects the inefficiency of BoNT/A in produc-ing a block in secretion in this cellular model. Ourobservation that the Am mutation (R198W) does notaffect function of the SDS-resistant complex is corrobo-rated by recent data from Chen et al. (1999). The samearginine residue (198) was mutated to alanine, causingno appreciable change in thermal stability or function ofthe complex.
Our results provide an initial characterization of thefunctional effects for exocytosis of amino acid substitu-tions close to the C-terminus of SNAP-25. Cleavage ofSNAP-25 by BoNT/A or BoNT/E not only inhibits exo-cytosis but has also been shown to prevent VAMP bind-ing to SNAP-25 and assembly of the SDS-resistant corecomplex (Hayashi et al., 1994), implicating this domaincleaved by the toxins in key protein–protein interactions.The data presented here show that residues of the C-terminus of SNAP-25 around the BoNT/E cleavage site(specifically isoleucine 181 and glutamate 183) are cru-cial for SNAP-25 binding of VAMP, while not destabi-lizing protein–protein interactions made by the protein inthe SDS-resistant complex and during exocytosis. Theobservation that the BoNT/E-resistant SNAP-25 mutantsdo restore secretion, while being unable to bind VAMPin a binary complex, is not surprising considering thesemutants can participate in forming a heat-stable, SDS-resistant complex. Because the BoNT/E-insensitive Em2and AEm2 mutants are functional, it does not seem likelythat the mutations carried out at the BoNT/E cleavagesite significantly affect the secondary structure of themolecule and therefore that secondary and tertiary struc-ture changes are not prerequisites for toxin insensitivityor the loss of VAMP binding. The ability of SNAP-25 toco-immunoprecipitate VAMP, in the presence of syn-taxin, is probably due to formation of a stable four-helixbundle (Sutton et al., 1998), for which the presence of theSNAP-25 C-terminus is necessary and not weakened bythe mutations used in this study. It is likely that thepurely binary interaction of SNAP-25 with VAMP is notcrucial for vesicle fusion.
In conclusion, we have produced a functionalSNAP-25 molecule that cannot be cleaved by two sero-types of BoNT. The results presented here, therefore,help redefine the essential interactions required for theformation of the SDS-resistant core complex and lendweight to the functional importance of this protein as-sembly as a physiological determinant necessary for exo-cytosis. Our approach, together with the report byGonelle-Gispert et al. (1999), demonstrates that toxin-insensitive mutants can be used to reconstitute SNAP-25
J. Neurochem., Vol. 73, No. 6, 1999
2431BoNT-INSENSITIVE SNAP-25 MUTANTS PROMOTE EXOCYTOSIS

function and exocytosis in cells depleted of endogenousSNAP-25 by BoNTs. This provides a novel strategy toprobe further the key roles played by specific regionswithin the SNAP-25 critically required for functionalprotein interactions in exocytosis.
Acknowledgment: We thank G. Schiavo, R. Pellizzari, andW. Shuttleworth for critical reading of the manuscript, A. Klipfor the gift of the VAMP polyclonal antibody, and S. Censinifor the production of oligonucleotides. This work was sup-ported by Telethon grant 1068 and EC grant BMH4-CT97-2410 to C.M., a Wellcome Trust grant to R.D.B., and USPHSgrant MH48989 to M.C.W.
REFERENCES
Banerjee A., Kowalchyk J. A., Dasgupta B. R., and Martin T. F. J.(1996) SNAP-25 is required for a late postdocking step in Ca21
dependent exocytosis.J. Biol. Chem.271,20227–20230.Barik S. (1996) Site-directed mutagenesis in vitro by megaprimer PCR.
Methods Mol. Biol.57, 203–215.Bark I. C. (1993) Structure of the chicken gene for SNAP-25 reveals
duplicated exon encoding distinct isoforms of the protein.J. Mol.Biol. 233,67–76.
Bark I. C. and Wilson M. C. (1994a) Human cDNA clones encodingtwo different isoforms of the nerve terminal protein SNAP-25.Gene139,291–292.
Bark I. C. and Wilson M. C. (1994b) Regulated vesicular fusion inneurons: snapping together the details.Proc. Natl. Acad. Sci. USA91, 4621–4624.
Bark I. C., Hahn K. M., Ryabinin A. E., and Wilson M. C. (1995)Differential expression of SNAP-25 protein isoforms during di-vergent vesicle fusion events of neural development.Proc. Natl.Acad. Sci. USA92, 1510–1514.
Barnard R. J., Morgan A., and Burgoyne R. D. (1997) Stimulation ofNSF ATPase activity by alpha-SNAP is required for SNAREcomplex disassembly and exocytosis.J. Cell Biol.139,875–883.
Blasi J., Chapman E. R., Yamasaki S., Binz T., Niemann H., and JahnR. (1993) Botulinum neurotoxin C1 blocks neurotransmitter re-lease by means of cleaving HPC-1/syntaxin.EMBO J.12, 4821–4828.
Brennwald P., Kearns B., Champion K., Keranen S., Bankaitis V., andNovick P. (1994) Sec9 is a SNAP-25-like component of a yeastSNARE complex that may be the effector of Sec4 function inexocytosis.Cell 79, 245–258.
Calakos N. and Scheller R. H. (1996) Synaptic vesicle biogenesis,docking and fusion: a molecular description.Physiol. Rev.76,1–29.
Capogna M., McKinney R. A., O’Connor V., Gahwiler B. H., andThompson S. M. (1997) Ca21 or Sr21 partially rescues synaptictransmission in hippocampal cultures treated with botulinum toxinA and C, but not tetanus toxin.J. Neurosci.17, 7190–7202.
Chapman E. R., An S., Barton N., and Jahn R. (1994) SNAP-25, a TSnare which binds to both syntaxin and synaptobrevin via do-mains that may form coiled coils.J. Biol. Chem.269, 27427–27432.
Chen Y. A., Scales S. J., Patel S. M., Doung Y., and Scheller R. H.(1999) SNARE complex formation is triggered by Ca21 anddrives membrane fusion.Cell 97, 165–174.
Foran P., Lawrence G. W., Shone C. C., Foster K. A., and Dolly J. O.(1996) Botulinum neurotoxin C1 cleaves both syntaxin andSNAP-25 in intact and permeabilized chromaffin cells: correlationwith its blockade of catecholamine release.Biochemistry35,2630–2636.
Gaisano H. Y., Sheu L., Wong P. P., Klip A., and Trimble W. S. (1997)SNAP-23 is located in the basolateral plasma membrane of ratpancreatic acinar cells.FEBS Lett.414,298–302.
Gonelle-Gispert C., Halban P. A., Niemann H., Palmer M., Catsicas S.,and Sadoul K. (1999) SNAP-25a and -25b isoforms are both
expressed in insulin-producing cells and can function in insulinsecretion.Biochem. J.339,159–165.
Graham M. E., Gerke V., and Burgoyne R. D. (1997a) Modification ofannexin II expression in PC12 cell lines does not affect Ca21-dependent exocytosis.Mol. Biol. Cell 8, 431–442.
Graham M. E., Sudlow A. W., and Burgoyne R. D. (1997b) Evidenceagainst an acute inhibitory role of nSec-1 (munc-18) in late stepsof regulated exocytosis in chromaffin and PC12 cells.J. Neuro-chem.69, 2369–2377.
Hayashi T., McMahon H., Yamasaki S., Binz T., Hata Y., Sudhof T. C.,and Niemann H. (1994) Synaptic vesicle membrane fusion com-plex: action of clostridial neurotoxins on assembly.EMBO J.13,5051–5061.
Lacy D. B., Tepp W., Cohen A. C., DasGupta B. R., and Stevens R. C.(1998) Crystal structure of botulinum neurotoxin type A andimplications for toxicity.Nat. Struct. Biol.5, 898–902.
Lawrence G. W., Weller U., and Dolly J. O. (1994) Botulinum A andthe light chain of tetanus toxins inhibit distinct stages of Mg-ATP-dependent catecholamine exocytosis from permeabilised chromaf-fin cells.Eur. J. Biochem.222,325–333.
Molgo J., Comella J. X., Angaut-Petit D., Pecot-Dechavassine M.,Tabti N., Faille L., Mallart A., and Thesleff S. (1990) Presynapticactions of botulinal neurotoxins at vertebrate neuromuscular junc-tions.J. Physiol. (Paris)84, 152–166.
Osen Sand A., Catsicas M., Staple J. K., Jones K. A., Ayala G.,Knowles J., Grenningloh G., and Catsicas S. (1993) Inhibition ofaxonal growth by SNAP-25 antisense oligonucleotides in vitroand in vivo.Nature364,445–448.
Osen Sand A., Staple J. K., Naldi E., Schiavo G., Rossetto O., Petit-pierre S., Malgaroli A., Montecucco C., and Catsicas S. (1996)Common and distinct fusion proteins in axonal growth and trans-mitter release.J. Comp. Neurol.367,222–234.
Pellegrini L. L., O’Connor V., Lottspeich F., and Betz H. (1995)Clostridial neurotoxins compromise the stability of a low energySNARE complex mediating NSF activation of synaptic vesiclefusion.EMBO J.14, 4705–4713.
Pellizzari R., Rossetto O., Lozzi L., Giovedi S., Johnson E., ShoneC. C., and Montecucco C. (1996) Structural determinants of thespecificity for synaptic vesicle associated membrane protein/syn-aptobrevin of tetanus and botulinum type B and G neurotoxins.J. Biol. Chem.271,20353–20358.
Pellizzari R., Mason S., Shone C. C., and Montecucco C. (1997) Theinteraction of synaptic vesicle-associated membrane protein/syn-aptobrevin with botulinum neurotoxins D and F.FEBS Lett.409,339–342.
Ravichandran V., Chawla A., and Roche P. A. (1996) Identification ofa novel syntaxin and synaptobrevin/VAMP binding protein,SNAP-23, expressed in non neuronal tissues.J. Biol. Chem.271,13300–13303.
Risinger C., Blomqvist A. G., Lundell I., Lambertsson A., Nassel D.,Pieribone V. A., Brodin L., and Larhammar D. (1993) Evolution-ary conservation of synaptosome-associated protein 25 kDa(SNAP-25) shown byDrosophila and Torpedo cDNA clones.J. Biol. Chem.268,24408–24414.
Rossetto O., Schiavo G., Montecucco C., Poulain B., Deloye F., LozziL., and Shone C. C. (1994) SNARE motif and neurotoxins.Nature372,415–416.
Rothman J. E. and Warren G. (1994) Implications of the SNAREhypothesis for intracellular membrane topology and dynamics.Curr. Biol. 4, 220–233.
Sadoul K., Berger A., Niemann H., Weller U., Roche P. A., Klip A.,Trimble W. S., Regazzi R., Catsicas S., and Halban P. A. (1997)SNAP-23 is not cleaved by botulinum neurotoxin E and canreplace SNAP-25 in the process of insulin secretion.J. Biol.Chem.272,33023–33027.
Schiavo G., Benfenati F., Poulain B., Rossetto O., Polverino de LauretoP., DasGupta B. R., and Montecucco C. (1992a) Tetanus andbotulinum-B neurotoxins block neurotransmitter release by pro-teolytic cleavage of synaptobrevin.Nature359,832–835.
Schiavo G., Poulain B., Rossetto O., Benfenati F., Tauc L., and Mon-tecucco C. (1992b) Tetanus toxin is a zinc protein and its inhibi-
J. Neurochem., Vol. 73, No. 6, 1999
2432 P. WASHBOURNE ET AL.

tion of neurotransmitter release and protease activity depend onzinc. EMBO J.11, 3577–3583.
Schiavo G., Rossetto O., Santucci A., DasGupta B. R., and MontecuccoC. (1992c) Botulinum neurotoxins are zinc proteins.J. Biol.Chem.267,23479–23483.
Schiavo G., Santucci A., Dasgupta B. R., Mehta P. P., Jontes J.,Benfenati F., Wilson M. C., and Montecucco C. (1993a) Botuli-num neurotoxins serotypes A and E cleave SNAP-25 at distinctCOOH-terminal peptide bonds.FEBS Lett.335,99–103.
Schiavo G., Shone C. C., Rossetto O., Alexander F. C., and Mon-tecucco C. (1993b) Botulinum neurotoxin serotype F is a zincendopeptidase specific for VAMP/synaptobrevin.J. Biol. Chem.268,11516–11519.
Schiavo G., Malizio C., Trimble W. S., Polverino de Laureto P., MilanG., Sugiyama H., Johnson E. A., and Montecucco C. (1994)Botulinum G neurotoxin cleaves VAMP/synaptobrevin at a singleAla-Ala peptide bond.J. Biol. Chem.269,20213–20216.
Schiavo G., Shone C. C., Bennett M. K., Scheller R. H., and Mon-tecucco C. M. (1995) Botulinum neurotoxin type C cleaves asingle Lys-Ala bond within the carboxyl terminal region of syn-taxins.J. Biol. Chem.270,10566–10570.
Schiavo G., Stenbeck G., Rothman J. E., and Sollner T. H. (1997)Binding of the synaptic vesicle v-SNARE, synaptotagmin, to theplasma membrane t-SNARE, SNAP-25, can explain docked ves-icles at neurotoxin-treated synapses.Proc. Natl. Acad. Sci. USA94, 997–1001.
Schulz J. R., Sasaki J. D., and Vacquier V. D. (1998) Increasedassociation of synaptosome-associated protein of 25 kDA withsyntaxin and vesicle-associated membrane protein following ac-rosomal exocytosis of sea-urchin sperm.J. Biol. Chem.273,24355–24359.
Sollner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H.,Geromanos S., Tempst P., and Rothman J. E. (1993) SNAPreceptors implicated in vesicle targeting and fusion.Nature362,318–324.
Steegmaier M., Yang B., Yoo J.-S., Huang B., Shen M., Yu S., Luo Y.,and Scheller R. H. (1998) Three novel proteins of the syntaxin/SNAP-25 family.J. Biol. Chem.273,34171–34179.
Sudhof T. C. (1995) The synaptic vesicle cycle: a cascade of protein–protein interactions.Nature375,645–653.
Sutton R. B., Fasshauer D., Jahn R., and Brunger A. T. (1998) Crystalstructure of a SNARE complex involved in synaptic exocytosis at2.4 A resolution.Nature395,347–353.
Vaidyanathan V. V., Yoshino K., Jahnz M., Do¨rries C., Bade S.,Nauenburg S., Niemann H., and Binz T. (1999) Proteolysis ofSNAP-25 isoforms by botulinum neurotoxin types A, C, and E:domains and amino acid residues controlling the formation ofenzyme–substrate complexes and cleavage.J. Neurochem.72,327–337.
Washbourne P., Pellizzari R., Baldini G., Wilson M. C., and Mon-tecucco C. (1997) Botulinum neurotoxin types A and E requirethe SNARE motif in SNAP-25 for proteolysis.FEBS Lett.418,1–5.
Weber T., Zemelman B. V., McNew J. A., Westermann B., GmachlM., Parlati F., Sollner T. H., and Rothman J. E. (1998) SNARE-pins: minimal machinery for membrane fusion.Cell 92, 759 –772.
Wick P. F., Senter R. A., Parsels L. A., Uhler M. D., and Holz R. W.(1993) Transient transfection studies of secretion in bovine chro-maffin cells and PC12 cells. Generation of kainate-sensitive chro-maffin cells.J. Biol. Chem.268,10983–10989.
J. Neurochem., Vol. 73, No. 6, 1999
2433BoNT-INSENSITIVE SNAP-25 MUTANTS PROMOTE EXOCYTOSIS




















