
BDNF‐mediated modulation of GABA and glycine release in dorsal horn lamina II from postnatal rats
16
BDNF-Mediated Modulation of GABA and Glycine Release in Dorsal Horn Lamina II from Postnatal Rats Rita Bardoni, 1 Alessia Ghirri, 1 Chiara Salio, 2 Massimiliano Prandini, 1 Adalberto Merighi 2,3 1 Department of Biomedical Sciences, University of Modena and Reggio Emilia, Modena, Italy 2 Department of Veterinary Morphophysiology, University of Turin, Turin, Italy 3 Istituto Nazionale di Neuroscienze, Turin, Italy Received 19 September 2006; revised 6 December 2006; accepted 13 January 2007 ABSTRACT: Recent studies show that excitatory glutamatergic transmission is potentiated by BDNF in superficial dorsal horn, both at the pre- and the postsy- naptic site. The role of BDNF in modulating GABA and glycine-mediated inhibitory transmission has not been fully investigated. To determine whether the neurotrophin is effective in regulating the spontaneous release of the two neurotransmitters, we have recorded miniature inhib- itory postsynaptic currents (mIPSCs) in lamina II of post- natal rats. We show that application of BDNF enhanced the spontaneous release of GABA and glycine, in presence of tetrodotoxin. The effect was blocked by the trk-recep- tor inhibitor k-252a. Amplitude and kinetics of mIPSCs were not altered. Evoked GABA and glycine IPSCs (eIPSCs) were depressed by BDNF and the coefficient of variation of eIPSC amplitude was significantly increased. By recording glycine eIPSCs with the paired-pulse proto- col, an increase of paired-pulse ratio during BDNF appli- cation was observed. We performed parallel ultrastruc- tural studies to unveil the circuitry involved in the effects of BDNF. These studies show that synaptic interactions between full length functional trkB receptors and GABA- containing profiles only occur at non peptidergic synaptic glomeruli of types I and II. Expression of trkB in pre- synaptic vesicle-containing dendrites originating from GABAergic islet cells, indicates these profiles as key struc- tures in the modulation of inhibitory neurotransmission by the neurotrophin. Our results thus describe a yet uncharacterized effect of BDNF in lamina II, giving fur- ther strength to the notion that the neurotrophin plays an important role in pain neurotransmission. ' 2007 Wiley Periodicals, Inc. Develop Neurobiol 67: 960–975, 2007 Keywords: neurotrophin; inhibitory transmission; spinal cord; pain; presynaptic modulation INTRODUCTION The neurotrophin BDNF is involved in neuronal dif- ferentiation and survival during development and in synaptic transmission and plasticity, in immature and mature stages. BDNF has been reported to regulate both excitatory and inhibitory synapses in several areas of central nervous system (CNS), by acting on presynaptic terminals (Frerking et al., 1998; McLean Bolton et al., 2000; Xu et al., 2000) or by modulating postsynaptic receptor expression and activity (Levine et al., 1995, 1998; Tanaka et al., 1997; Bru ¨nig et al., 2001; Pezet and McMahon, 2006). In spinal cord dorsal horn, BDNF has been impli- cated in the control of nociceptive neurotransmission on behavioral, physiological, and pharmacological grounds. Behavioral studies have shown complex effects of both endogenous and exogenous BDNF. Endogenous BDNF, derived from a sub-population of Correspondence to: R. Bardoni; ([email protected]) Contract grant sponsor: Italian MIUR Contract grant sponsor: Compagnia di San Paolo, Turin, Italy Contract grant sponsor: Fondazione CRT, Turin, Italy ' 2007 Wiley Periodicals, Inc. Published online 28 February 2007 in Wiley InterScience (www. interscience.wiley.com). DOI 10.1002/dneu.20401 960
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of BDNF‐mediated modulation of GABA and glycine release in dorsal horn lamina II from postnatal rats

BDNF-Mediated Modulation of GABA and GlycineRelease in Dorsal Horn Lamina II from Postnatal Rats
Rita Bardoni,1 Alessia Ghirri,1 Chiara Salio,2 Massimiliano Prandini,1
Adalberto Merighi2,3
1 Department of Biomedical Sciences, University of Modena and Reggio Emilia, Modena, Italy
2 Department of Veterinary Morphophysiology, University of Turin, Turin, Italy
3 Istituto Nazionale di Neuroscienze, Turin, Italy
Received 19 September 2006; revised 6 December 2006; accepted 13 January 2007
ABSTRACT: Recent studies show that excitatory
glutamatergic transmission is potentiated by BDNF in
superficial dorsal horn, both at the pre- and the postsy-
naptic site. The role of BDNF in modulating GABA and
glycine-mediated inhibitory transmission has not been
fully investigated. To determine whether the neurotrophin
is effective in regulating the spontaneous release of the
two neurotransmitters, we have recorded miniature inhib-
itory postsynaptic currents (mIPSCs) in lamina II of post-
natal rats. We show that application of BDNF enhanced
the spontaneous release of GABA and glycine, in presence
of tetrodotoxin. The effect was blocked by the trk-recep-
tor inhibitor k-252a. Amplitude and kinetics of mIPSCs
were not altered. Evoked GABA and glycine IPSCs
(eIPSCs) were depressed by BDNF and the coefficient of
variation of eIPSC amplitude was significantly increased.
By recording glycine eIPSCs with the paired-pulse proto-
col, an increase of paired-pulse ratio during BDNF appli-
cation was observed. We performed parallel ultrastruc-
tural studies to unveil the circuitry involved in the effects
of BDNF. These studies show that synaptic interactions
between full length functional trkB receptors and GABA-
containing profiles only occur at non peptidergic synaptic
glomeruli of types I and II. Expression of trkB in pre-
synaptic vesicle-containing dendrites originating from
GABAergic islet cells, indicates these profiles as key struc-
tures in the modulation of inhibitory neurotransmission
by the neurotrophin. Our results thus describe a yet
uncharacterized effect of BDNF in lamina II, giving fur-
ther strength to the notion that the neurotrophin plays an
important role in pain neurotransmission. ' 2007 Wiley
Periodicals, Inc. Develop Neurobiol 67: 960–975, 2007
Keywords: neurotrophin; inhibitory transmission; spinal
cord; pain; presynaptic modulation
INTRODUCTION
The neurotrophin BDNF is involved in neuronal dif-
ferentiation and survival during development and in
synaptic transmission and plasticity, in immature and
mature stages. BDNF has been reported to regulate
both excitatory and inhibitory synapses in several
areas of central nervous system (CNS), by acting on
presynaptic terminals (Frerking et al., 1998; McLean
Bolton et al., 2000; Xu et al., 2000) or by modulating
postsynaptic receptor expression and activity (Levine
et al., 1995, 1998; Tanaka et al., 1997; Brunig et al.,
2001; Pezet and McMahon, 2006).
In spinal cord dorsal horn, BDNF has been impli-
cated in the control of nociceptive neurotransmission
on behavioral, physiological, and pharmacological
grounds. Behavioral studies have shown complex
effects of both endogenous and exogenous BDNF.
Endogenous BDNF, derived from a sub-population of
Correspondence to: R. Bardoni; ([email protected])Contract grant sponsor: Italian MIURContract grant sponsor: Compagnia di San Paolo, Turin, ItalyContract grant sponsor: Fondazione CRT, Turin, Italy
' 2007 Wiley Periodicals, Inc.Published online 28 February 2007 in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/dneu.20401
960

nociceptors, can be released following high frequencystimulation of nociceptive primary afferent fibers(Lever et al., 2001), and plays an important role inregulating inflammatory pain thresholds and second-ary hyperalgesia (Thompson et al., 1999; Zhao et al.,2006). On the other hand exogenously applied BDNFcauses thermal analgesia and attenuates mechanicalallodynia (Siuciak et al., 1994; Cejas et al., 2000).
At the cellular level, the neurotrophin is able topotentiate glutamatergic synaptic transmission be-tween primary afferent fibers and dorsal horn neu-rons, by modulating postsynaptic NMDA receptors(Slack et al., 2004; Garraway et al., 2003). Recentevidence has been provided that BDNF, acting ontrkB receptors, causes an increase of spontaneous glu-tamate release in dorsal horn lamina II, both in con-trol animals and during inflammation (Bardoni et al.,2004; Matayoshi et al., 2005).
Nonetheless, a modulatory action of BDNF on in-
hibitory synaptic transmission in dorsal horn has also
been reported: BDNF released from microglia cells
causes a modulation of Cl-transporter expression in
lamina I neurons, and a shift of chloride equilibrium
potential to positive values (Coull et al., 2005). More-
over, the application of BDNF to the isolated adult
rat spinal cord increases KCl-evoked GABA content
in the collected superfusate (Pezet et al., 2002).
The role of BDNF in regulating GABAergic andglycinergic transmission in dorsal horn, however, hasnot been fully investigated. Furthermore, although thedistribution of full length (fl) functional trkB receptorshas recently been investigated in rat and mouse laminaII (Salio et al., 2005), the presence of these receptorson inhibitory interneurons in dorsal horn has not beenreported yet. In this study, we tested the hypothesisthat BDNF can control the release of GABA and gly-cine in superficial dorsal horn. Since local GABAergicand glycinergic inhibition is critical for gating of noci-ceptive transmission (Malcangio and Bowery, 1996),any effect of BDNF on inhibitory synapses could havea great impact on pain signaling, especially in patho-logical conditions of persistent pain. By using thepatch-clamp technique, we have studied the modula-tory effects of BDNF on spontaneous and evokedrelease of GABA and glycine in lamina II neuronsfrom neonatal rats. Furthermore, by performing ultra-structural immunocytochemical studies, we havedetermined the pattern of expression of fl-trkB recep-tors at GABAergic synapses in this lamina.
METHODS
Animals
Experiments were carried out in Sprague-Dawley postnatal
rats, ranging in age from 8 (P8) to 30 days (P30). Patch-
clamp experiments were carried out in P8–P16 animals,
whereas histology was performed in young-adult rats (P25–
P30). All experimental procedures were in strict accordance
with the Italian and EU regulation on animal welfare, and
previously authorized by the Italian Ministry of Health.
Electrophysiological Recordings
Slice Preparation. Postnatal Sprague-Dawley rats were
anesthetized with halothane and decapitated, the spinal cord
and vertebrae were rapidly removed and placed in ice-cold
Krebs’ solution (composition in mM: 125 NaCl, 2.5 KCl, 1.25
NaH2PO4, 26 NaHCO3, 25 glucose, 1 MgCl2, and 2 CaCl2, pH
7.4, 320 mOsm), bubbled with carboxygen (95% O2, 5%
CO2). The lumbar part of the spinal cord was isolated, laid
down on an agarose block, and transverse slices (500 lm thick)
were obtained using a vibrating microtome (Campden Instru-
ments, UK). Slices were incubated in oxygenated Krebs’ solu-
tion at 358C for 1 h and used for recording within 6–7 h.
Patch-Clamp Recording and Focal Synaptic Stimula-tion. Patch-clamp recording in whole-cell configuration was
performed on visually identified lamina II neurons at room
temperature. Neurons were visualized by using a Zeiss Axio-
skop microscope, fitted with Nomarski optics and connected
to a camera (Dage-MTI, USA). Slices were perfused at 1–
2 mL/min. Thick-walled borosilicate pipettes, having a resist-
ance of 3–5 mO, were filled with a solution having the fol-
lowing composition (in mM): 130 Cs-gluconate, 10 CsCl, 11
EGTA, 1 CaCl2, 10 HEPES, and 2 Mg2þ -ATP, pH adjusted
to 7.2 with NaOH, osmolarity adjusted to 305 with sucrose.
Junction potential was corrected after recording.
Data were recorded and acquired using a Multiclamp
700A amplifier and pClamp 9 software (Molecular Devices,
USA). Sampling rate was 10 kHz, and data were filtered at
2 kHz. Both miniature inhibitory postsynaptic currents
(mIPSCs) and evoked inhibitory postsynaptic currents (eIPSCs)
were recorded in Krebs’ solution, in constant presence of
10 lM NBQX and 50 lM D-APV; only for mIPSCs recording,
0.5 lM tetrodotoxin (TTX) was added to the bath solution.
Evoked IPSCs were obtained by focally stimulating the
tissue around the recorded cell with a patch-clamp pipette,
filled with Krebs’ solution. The second pole was repre-
sented by a silver wire placed close to the slice. Stimuli had
intensities of 0.1–1 mA and duration of 0.05–0.1 ms.
Evoked IPSCs with constant latency and no failures during
1 Hz stimulation were considered monosynaptic and tested
for BDNF.
Data Analysis and Statistics. Miniature IPSC amplitudes,
frequencies, and kinetics were determined using Minianaly-
sis software (Synaptosoft, Decatur, USA). Detection thresh-
old for mIPSCs was set at 8–10 pA. Selection of mIPSCs
was performed manually and events with rise-time higher
than 3 ms were usually discarded (the exclusion of slower
events did not affect the results of the analysis). Analysis of
mIPSCs was performed on the same time window (10 min)
during control and when the BDNF effect was stabilized.
BDNF Modulates GABA and Glycine in Lamina II 961
Developmental Neurobiology. DOI 10.1002/dneu

Effects of BDNF on mIPSC inter-event intervals and ampli-
tudes were assessed for each cell by comparing control and
test plots in cumulative probability histograms, using the
Kolmogorov–Smirnov 2-sample test. In neurons were an
insufficient number of events was detected (<100), the
unpaired t-test was applied. Decays of isolated GABA or
glycine mIPSCs were interpolated with a single exponential
function, by using Minianalysis software. Events that did
not return to the baseline or composed by multiple peaks
were not included in the kinetic analysis. Kinetic parame-
ters (10–90% rise-time and decay time constants) were
compared by using the unpaired t-test. Values of p < 0.05
were considered significant.
Analysis of peak amplitudes and kinetics of evoked
IPSCs was performed by using Clampfit (pClamp9, Molec-
ular Devices, USA). Graphs and statistical analysis were
obtained by using Sigmaplot 8 (SPSS, USA). Evoked IPSC
peaks were manually detected for each trace, in the same
2 ms window. Decay phase fitting was obtained by interpo-
lating the 10–90% portion of current decay with a two ex-
ponential function. Statistical analysis on amplitudes and
kinetic parameters was performed by using the unpaired t-test. Coefficient of variation (CV) of eIPSC amplitudes was
determined as standard deviation/mean, considering at least
30 traces in control and in BDNF, at the time when the
effect had fully developed. For paired pulse experiments,
the analysis of the second peak was determined by extrapo-
lating the fitting of the first response decay phase to the
time of the second peak and subtracting this value from the
peak amplitude of the second response. Mean paired pulse
ratio (PPR) was determined by averaging PPRs deriving
from 20–30 traces in each condition. Comparison of CV
and PPR in control and BDNF were obtained by applying
paired t-tests. p values <0.05 were considered significant.
Data were expressed as mean value 6 S.E.
Drugs. All chemicals were obtained from Sigma Aldrich
(St. Louis, MO), except the following: bicuculline, NBQX,
and TTX were obtained from Tocris Cookson (UK),
D-APV from Ascent scientific (UK), BDNF from Peprotech
(UK), and k-252a from Calbiochem (USA).
Immunocytochemistry
Tissue Preparation. Under deep pentobarbital anesthesia
(60 mg/100 g), animals were injected intracardially with
1 mL heparin (5000 U/mL) and perfused through the de-
scending aorta with Ringer solution followed by cold fixa-
tive solution (4% paraformaldehyde þ 0.5% glutaraldehyde
in sodium cacodylate buffer 0.05 M, pH 7.4). Segments
from lumbar spinal cord were carefully dissected out, and
postfixed for 2 additional hours in the same aldehyde mix-
ture. Coronal sections were cut on a vibratome at a thick-
ness of 50 lm.
Immunolabeling. Dual labeling experiments were carried
out simultaneously to localize high affinity functional
BDNF receptor (fl-trkB receptor) and GABA immunoreac-
tivities at the ultrastructural level.
The immunolocalization of fl-trkB receptors was carried
out by a pre-embedding protocol with ultra small gold
(Salio et al., 2005). Briefly, free-floating spinal cord vibra-
tome sections were incubated overnight with a chicken
anti-fl-trkB primary antibody (Promega Corporation, USA),
followed by anti-chicken IgY biotinylated secondary anti-
body (Vector, USA), and finally by AlexaFluor 488-Fluoro-
nanogoldTM-Streptavidin (Nanoprobes, USA). Sections
were postfixed in osmium ferrocyanide, dehydrated in
increasing concentrations of ethanol, and embedded in
Araldite. FluoronanogoldTM particles were intensified
directly on grid by a Gold Enhancement Kit (Nanoprobes).
Fl-trkB-labeled ultrathin sections were subsequently
stained for GABA following a post-embedding immuno-
staining protocol on grid (Merighi and Polak, 1993).
Briefly, after sodium metaperiodate etching sections were
placed on drops of an anti-GABA antiserum (Merighi et al.,
1989) and incubated overnight. After extensive rinsing in
buffer, they were transferred in goat anti-rabbit IgGs conju-
gated with 10-nm gold (BioCell, UK). Section were then
fixed in 2.5% glutaraldehyde in cacodylate buffer, 0.05 M,
pH 7.4, and finally washed in distilled water.
After routine counterstaining, grids were observed with
a Philips CM10 electron microscope.
RESULTS
BDNF Causes the Increase of MiniatureGABA and Glycine IPSC Frequency
Patch-clamp recordings were obtained from 72 neu-
rons in rat dorsal horn lamina II, by using the volt-
age-clamp modality and holding the recorded cell at
0/þ10 mV. To test whether BDNF can regulate
GABA and glycine release in lamina II, we first
recorded miniature GABA and glycine IPSCs
(mIPSCs), in continuous presence of TTX (0.5 lM)
and the glutamatergic receptor antagonists NBQX
(10 lM) and D-APV (50 lM). In most cells both
GABA and glycine mIPSCs were present, as revealed
by blocking mIPSCs with the GABAA receptor antag-
onist bicuculline (10 lM) and the glycine receptor an-
tagonist strychnine (3 lM) at the end of the experi-
ment. In one neuron mIPSCs were mediated by
GABA receptors only, since they were completely
blocked by bicuculline. This cell was excluded from
this first set of experiments and added to the group of
neurons where GABA mIPSCs were pharmacologi-
cally isolated (see later).
Application of 100 ng/mL BDNF caused a signifi-
cant increase of GABA and glycine mIPSC frequency
in 9 out of 13 lamina II neurons tested [Kolmogorov–
Smirnov test, p < 0.01; Fig. 1(A,B,D)]. The effect
962 Bardoni et al.
Developmental Neurobiology. DOI 10.1002/dneu
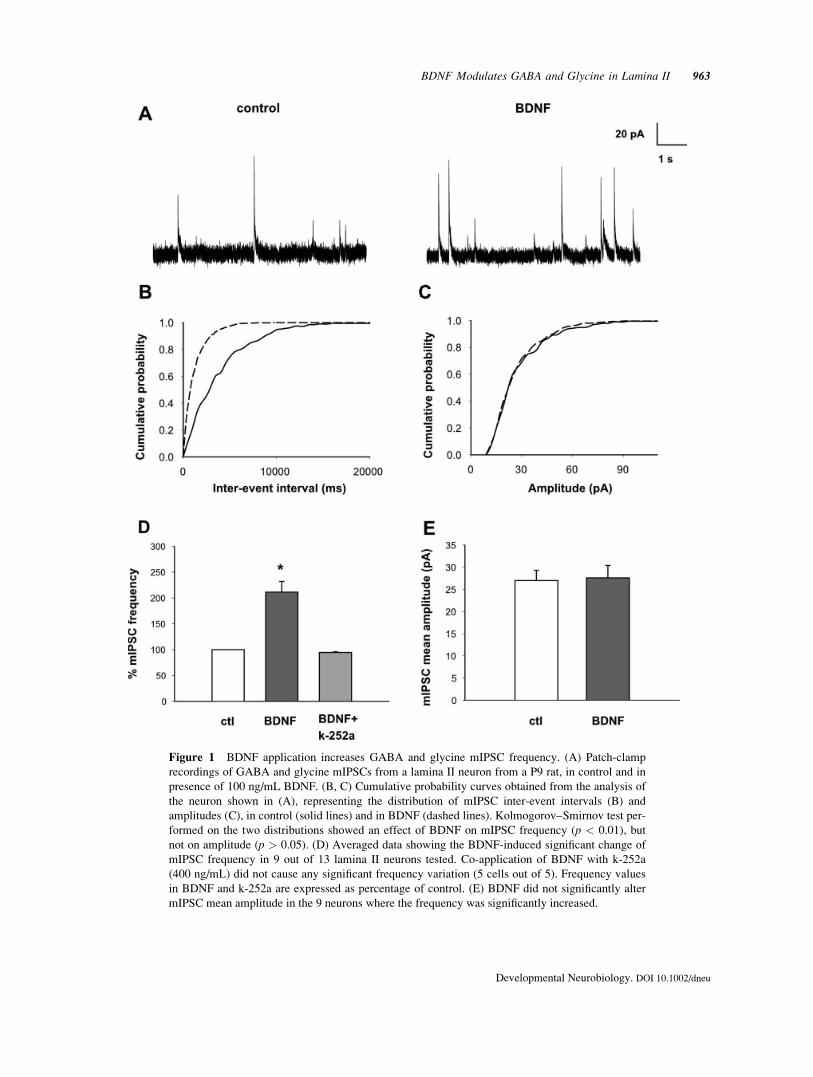
Figure 1 BDNF application increases GABA and glycine mIPSC frequency. (A) Patch-clamp
recordings of GABA and glycine mIPSCs from a lamina II neuron from a P9 rat, in control and in
presence of 100 ng/mL BDNF. (B, C) Cumulative probability curves obtained from the analysis of
the neuron shown in (A), representing the distribution of mIPSC inter-event intervals (B) and
amplitudes (C), in control (solid lines) and in BDNF (dashed lines). Kolmogorov–Smirnov test per-
formed on the two distributions showed an effect of BDNF on mIPSC frequency (p < 0.01), but
not on amplitude (p > 0.05). (D) Averaged data showing the BDNF-induced significant change of
mIPSC frequency in 9 out of 13 lamina II neurons tested. Co-application of BDNF with k-252a
(400 ng/mL) did not cause any significant frequency variation (5 cells out of 5). Frequency values
in BDNF and k-252a are expressed as percentage of control. (E) BDNF did not significantly alter
mIPSC mean amplitude in the 9 neurons where the frequency was significantly increased.
BDNF Modulates GABA and Glycine in Lamina II 963
Developmental Neurobiology. DOI 10.1002/dneu

started after 15 min of BDNF application and usually
persisted during removal of the neurotrophin. In three
cells a partial recovery was observed after about
20 min wash out. The mean frequency of mIPSCs in
control was 0.19 6 0.04 Hz, in BDNF was 0.42 6
0.1. Co-application of BDNF with the trk receptor an-
tagonist k-252a (400 ng/mL) did not cause any signif-
icant change of mIPSC frequency in all 5 neurons
tested [Kolmogorov–Smirnov test, p > 0.05; Fig.
1(D)]. Miniature IPSC amplitude was not signifi-
cantly altered during BDNF application in 8 of the 9
cells responsive to the neurotrophin [Kolmogorov–
Smirnov test, p > 0.05; Fig. 1(C,E)]. One cell exhib-
ited a bimodal distribution of mIPSC amplitudes in
control; in BDNF, the mIPSC population of larger
amplitude was preferentially potentiated, causing a
significant change of mean mIPSC amplitude.
To determine whether BDNF acts on both GABA
and glycine mediated mIPSCs, we performed experi-
ments where a single population was isolated by
application of 10 lM bicuculline or 3 lM strychnine
(Fig. 2). A significant increase of mIPSC frequency
in BDNF was observed for both GABA and glycine
mIPSCs, respectively in 5 out of 9 and 5 out of 8
tested neurons [Kolmogorov–Smirnov test, p < 0.01;
Fig. 2(A,B)]. Mean GABA mIPSC frequency was
0.22 6 0.09 Hz in control and 0.45 6 0.14 in BDNF;
mean glycine mIPSC frequency increased from 0.20
6 0.09 in control to 0.65 6 0.3 Hz in BDNF. The
effects of BDNF on GABA and glycine mIPSC fre-
quency are summarized in Figure 2(C).
BDNF did not affect the peak amplitude of pure
GABA and glycine mIPSCs [Kolmogorov–Smirnov
test, p > 0.05; Fig. 2(B,D)]. GABA and glycine
mIPSC 10–90% rise time values and decay time con-
stants (obtained by fitting the mIPSC decay with a
single exponential function) were also not altered by
BDNF [t-test, p > 0.05; Fig. 2(E)]. The increase of
mIPSC frequency produced by BDNF, without any
significant effect on mIPSC amplitude or kinetics,
would suggest a presynaptic action of the neurotro-
phin on GABAergic and glycinergic synapses in su-
perficial dorsal horn.
Evoked IPSCs Are Depressed inPresence of BDNF
To further investigate the role of BDNF in regulating
GABAergic and glycinergic transmission in lamina
II, we have studied the effects of the neurotrophin on
evoked GABA and glycine IPSCs (eIPSCs), obtained
by focally stimulating the superficial dorsal horn.
Mixed GABA and glycine IPSCs were recorded in
presence of 10 lM NBQX and 50 lM D-APV, at the
holding potential of 0 mV; stimulation intensity was
set right above the threshold and the stimulus was
delivered at the frequency of 0.03 Hz. Experiments
performed in control conditions showed that stimula-
tion at this frequency did not produce by itself any
significant depression of eIPSC during long time
recordings (45–60 min) (data not shown).
BDNF (100 ng/mL) caused a significant depres-
sion of eIPSC peak amplitude in 9 out of 16 lamina II
neurons tested [t-test, p < 0.05; Fig. 3(A,B)]. The
time course of BDNF effect was similar to that
observed during mIPSC recordings, the onset of
effect occurring after about 15 min from the begin-
ning of application and the maximum of depression
developing after 20–30 min. Only in three cells a par-
tial recovery was obtained during BDNF wash out.
At the end of the experiments, application of 10 lMbicuculline blocked a substantial part of eIPSCs (67.4
6 8.1% of peak amplitude). Addition of 3 lM strych-
nine to bicuculline caused the complete block of
eIPSCs, showing that both GABAA and glycine
receptors contributed to the generation of inhibitory
currents.
Incubation of slices in the trk receptor antagonist
k-252a (400 ng/mL) for at least 20 min before BDNF
application and during BDNF prevented any depress-
ing action of the neurotrophin on eIPSCs in all lamina
II neurons tested, suggesting a BDNF-induced activa-
tion of trkB receptors [t-test, p > 0.05, n ¼ 5; Fig.
3(B)]. Incubation of slices in k-252a before BDNF
application did not cause any significant alteration of
eIPSC amplitude, indicating that endogenous BDNF
was not able to modulate inhibitory synaptic trans-
mission in our experimental conditions.
Since recordings were performed in whole cell
configuration, a shift of chloride equilibrium potential
(ECl) was unlikely. Nevertheless, an incomplete diffu-
sion of the intracellular solution into the cytoplasm
could have partially preserved the intracellular chlo-
ride concentration. In this case, a shift of ECl towards
more positive values could determine a depression of
eIPSCs at 0 mV and an increase of IPSC amplitude at
potentials more negative than ECl. In our experimen-
tal conditions ECl was �60.7 mV. Evoked IPSCs
recorded at �90 mV in three neurons showed a sig-
nificant depression in presence of BDNF, excluding
an alteration of intracellular chloride concentration
due to BDNF (data not shown).
The depressing action of BDNF on eIPSCs could
be due to a presynaptic inhibition of GABA and gly-
cine release or rather to a direct modulation of post-
synaptic GABA and glycine receptors. To assess
whether the neurotrophin modulates inhibitory synap-
964 Bardoni et al.
Developmental Neurobiology. DOI 10.1002/dneu

Figure 2 (See legend on following page)
BDNF Modulates GABA and Glycine in Lamina II 965
Developmental Neurobiology. DOI 10.1002/dneu

Figure 2 BDNF increases the frequency of pharmacologically isolated GABA and glycine mIPSCs.
(A) Recordings of mIPSCs mediated by GABA (left) and by glycine (right) receptors, obtained from
two different lamina II neurons, in control and in 100 ng/mL BDNF. The mIPSCs were recorded in
presence of 3 lM strychnine or 10 lM bicuculline, respectively. (B) Cumulative probability histo-
grams obtained from the analysis of the two neurons in (A), representing the effect of BDNF on inter-
event interval and amplitude distributions (control: solid lines, BDNF: dashed lines). BDNF caused a
significant decrease of inter-event intervals (Kolmogorov–Smirnov test, p < 0.01), leaving mIPSC am-
plitude unaffected (Kolmogorov–Smirnov test, p > 0.05). (C) Averaged data demonstrating BDNF
effect on isolated GABA and glycine mIPSC frequency. BDNF caused a significant increase of fre-
quency of both GABA (5 positive cells out of 9) and glycine mIPSCs (5 positive neurons out of 8).
(D) Mean amplitudes of GABA and glycine mIPSCs were not significantly affected by BDNF; (E)
BDNF did not significantly change 10–90% rise time and decay time of GABA and glycine mIPSCs.
Decay time was determined as the time constant of the single exponential function fitting the mIPSC
decay phase. Analysis was performed on the cells responsive to BDNF.
Figure 3 Evoked IPSCs are depressed by BDNF (A) Individual traces of GABA and glycine
mediated evoked IPSCs (eIPSCs), recorded from a lamina II neuron (from a P9 rat) and evoked by
focal stimulation of lamina II. BDNF effect started after about 15 min. (B) Summary of BDNF
effects on eIPSC amplitude: the neurotrophin induced a significant depression in 9 out of 16 lamina
II neurons tested. Peak amplitudes are expressed as percentage of control. Co-application of BDNF
and k-252 did not cause any significant depression of evoked IPSC amplitude (n ¼ 5). (C) BDNF
altered coefficient of variation (CV) of eIPSC amplitudes, causing a significant increase (n ¼ 8).
CV is represented as percentage of control. No significant increase of CV was observed in cells
negative to BDNF (n ¼ 7).
966 Bardoni et al.
Developmental Neurobiology. DOI 10.1002/dneu

ses at the presynaptic site, we determined the effect
of BDNF on the eIPSC variability (expressed as CV
of eIPSC amplitudes, CV). In a subpopulation of
lamina II cells positive to BDNF, the CV of peak am-
plitude was calculated on 30–40 traces in control and
during the test. BDNF caused a significant increase of
CV in all neurons [paired t-test, p < 0.05, n ¼ 8; Fig.
3(C)]: the mean value of CV was 0.16 6 0.03 in con-
trol and 0.26 6 0.04 in BDNF. In the three neurons
where a recovery of peak amplitude was observed in
wash, CV returned close to the values in control. No
significant changes in CV were observed in the cells
where BDNF did not cause a significant depression of
eIPSC amplitude (paired t-test, p > 0.05, n ¼ 7).
The depression of eIPSC amplitude was not
accompanied by a change of decay kinetics (Fig. 4).
Evoked eIPSCs were usually fitted by a double expo-
nential function. Neither the fast nor the slow decay
time constants (tau1 and tau2) were significantly
affected in cells responsive to BDNF [t-test, p >0.05, n ¼ 9; Fig. 4(B)]. The relative amplitudes of the
fast (A1) and slow components (A2) were not altered
in presence of the neurotrophin [t-test, p > 0.05, n ¼9; Fig. 4(C)].
To further confirm the presynaptic nature of
BDNF inhibition of eIPSCs, paired pulse protocol
was applied to glycinergic eIPSCs, recorded in pres-
ence of 10 lM bicuculline to block the GABA medi-
ated component [Fig. 5(A)]. Glycinergic eIPSCs were
used because of their fast decay kinetics: in most
cells, at the inter-pulse interval of 50 ms, the first
response produced only a negligible contamination of
the second peak. Paired pulse protocol caused the
facilitation of the second peak in the majority of cells
tested; in some traces, where the first peak had large
amplitude, the second peak was depressed. Applica-
tion of BDNF caused a significant depression of the
first peak in 9 cells out of 16 tested [t-test, p < 0.05;
Fig. 5(B)], whereas the second peak was either
depressed or unchanged. In all positive cells BDNF
increased the amount of facilitation produced by the
paired pulse protocol, expressed as a significant
Figure 4 BDNF does not alter evoked IPSC decay kinetics. (A) (left and center) Averaged traces
of GABA/glycine eIPSCs recorded from a lamina II neuron (P12 rat). BDNF caused a strong
depression of eIPSC amplitude. Right: The graph, obtained by normalizing the trace in BDNF to
that in control and by superimposing the two traces, shows that BDNF does not alter the eIPSC
kinetics. (B, C) Fast and slow decay time constants (tau1 and tau2, B) and the relative contribution
of the fast and slow components (C) are not significantly altered by BDNF. The analysis was per-
formed on the 9 neurons where eIPSCs were depressed by BDNF.
BDNF Modulates GABA and Glycine in Lamina II 967
Developmental Neurobiology. DOI 10.1002/dneu

increase of the PPR in presence of the neurotrophin
[paired t-test, p < 0.05, n ¼ 9; Fig. 5(C)]. These
results provide additional evidence of BDNF-medi-
ated modulation of glycine release in dorsal horn
lamina II.
Localization of trkB Receptors on DorsalHorn Inhibitory Interneurons
The ultrastructural distribution of fl-trkB receptors in
rat and mouse lamina II has been recently described
in details on both qualitative and quantitative bases
(Salio et al., 2005). Here we have performed a series
of dual labeling experiments to combine together
fl-trkB and GABA immunolocalization to assess
whether the two labels were expressed at the same
synapses, and, in the affirmative, if they coexisted
within individual profiles. According to the parame-
ters employed in the gold intensification procedure
for rendering the ultra-small 1-nm gold particles of
the FluoroNanogoldTM easily visible and recogniz-
able under the electronic beam, the positive signal
appeared in the form of very electrondense particles
of relatively large sizes (25–30 nm) and irregular
shapes, that were very easily distinguishable from the
unintensified 10-nm colloidal gold particles used to
visualize GABA immunoreactive profiles (Figs. 6
and 7).
In our double labeled preparations, synaptic inter-
actions between fl-trkB and GABA immunoreactive
Figure 5 BDNF application changes the paired-pulse ratio of evoked glycinergic IPSCs. (A)
Averaged traces of evoked glycinergic IPSCs, recorded using the paired-pulse protocol (lamina II
neuron from a P10 rat). The IPSCs are represented in control, in presence of 100 ng/mL BDNF and
superimposed (the trace in BDNF is normalized to that in control and represented as dashed line).
BDNF causes a depression of both peaks, but the amount of depression was larger for the first
peak. (B) Average amplitude of the first and the second peak in BDNF, expressed as percentage of
control, for 9 cells responsive to BDNF (out of 16 neurons tested). BDNF causes a large and signif-
icant depression of the first peak in all 9 cells, while the second peak is less affected. (C) PPR (2nd
peak/1st peak) is significantly increased in BDNF (n ¼ 9).
968 Bardoni et al.
Developmental Neurobiology. DOI 10.1002/dneu

profiles were consistently observed only at the level
of synaptic glomeruli, although they represent a mi-
nority (about 5%) of the synapses in lamina II. Syn-
aptic glomeruli are multi-synaptic complexes whose
core, commonly referred to as C bouton, is formed by
an axon terminal derived from a primary afferent
fiber. Two main types of glomeruli, named type I and
II, have been described in rat (see Ribeiro-da-Silva,
2004). Type I glomeruli may have a particularly
electron-dense C bouton filled by small agranular
Figure 6 Dual labeling immunocytochemistry of fl-trkB receptors and GABA in rat spinal cord
lamina II. (A, B) Synaptic glomeruli of the Ia (nonpeptidergic) type displaying peripheral labeling
for fl-trkB (gold-intensified FluoronanogoldTM) and GABA (10 nm gold). Note that fl-trkB has a
postsynaptic localization in one of the ‘‘plain’’ dendrites engaged in the glomerulus (D), as well as
a presynaptic localization in one vesicle-containing or presynaptic dendrite (V1). All V1 dendrites
are immunoreactive for GABA, but not for fl-trkB (A). The area indicated by the rectangle in B is
shown at higher magnification in the bottom insert at down right corner. A symmetric synaptic spe-
cialization is clearly visible between the V1 profile and the central bouton of the glomerulus. (C) A
synaptic glomerulus of the IIa type displaying a peripheral V1 profile immunolabeled for trkB
(gold-intensified FluoronanogoldTM) and GABA (10 nm gold). The area indicated by the rectangle
is shown at higher magnification in the top insert at down right corner. Scale bars ¼ 500 nm; inserts
¼ 100 nm.
BDNF Modulates GABA and Glycine in Lamina II 969
Developmental Neurobiology. DOI 10.1002/dneu

synaptic vesicles and usually display one V1 (presyn-
aptic dendrite) and one V2 (axon) peripheral profiles
(type Ia or type I nonpeptidergic), whereas others
show a C bouton enriched with a number of peptide-
containing large granular vesicles (type Ib or type I
peptidergic). Type II glomeruli have a larger, lighter,
and less irregular C bouton, particularly rich in
mitochondria. Type II glomeruli also exist in two
subcategories: type IIa, with a C bouton devoid of
neurofilaments; and type IIb, with a C bouton rich of
neurofilaments.
Concurrent presence of trkB and GABA immunor-
eactivities was observed at glomeruli of the Ia [Fig.
6(A,B)], IIa [Fig. 6(C)] and IIb (Fig. 7) types. Irre-
spectively of the glomerular type, fl-trkB receptors
were identified in plain dendrites (D), and vesicle-
containing presynaptic dendrites (V1). Fl-trkB immu-
nolabeled plain dendrites were never positive for
GABA, whereas certain V1 profiles were dual labeled
for fl-trkB and GABA, the remaining ones being
only singularly immunostained by the anti-GABA
antiserum.
It seems noteworthy to remark here that, although
axons constituted the 33.2% of fl-trkB immunoreac-
tive profiles in rat lamina II (Salio et al., 2005), we
never observed fl-trkB immunopositive axons in con-
tact with dendritic or axonal profiles that stained posi-
tively for GABA.
DISCUSSION
In this article we show, for the first time, that exoge-
nous application of BDNF modulates the release of
both the inhibitory neurotransmitters GABA and gly-
cine onto lamina II neurons. In a subpopulation of
lamina II neurons the neurotrophin caused the
increase of GABA and glycine spontaneous mIPSC
frequency, leaving mIPSC amplitude and kinetics
unchanged. Action potential-dependent release of the
two neurotransmitters was also affected by BDNF:
the depression of eIPSCs was accompanied by
changes of CV and PPR, usually considered as indi-
cators of presynaptic modulation. Ultrastructural
studies, showing the co-localization of fl-trkB recep-
tors and GABA at presynaptic V1 profiles in synaptic
glomeruli, give further support to the role of BDNF
as a modulator of GABA and glycine release in spinal
cord dorsal horn.
Modulation of GABAergic andGlycinergic Miniature IPSCs
In our recordings most neurons exhibited both ‘‘pure
mIPSCs’’, mediated by a single type of receptor,
exhibiting monophasic decay and small amplitude,
and ‘‘mixed mIPSCs’’, mediated by both receptors,
larger and biphasic, with partial time constants simi-
lar to those of GABAergic and glycinergic mIPSCs.
This was not surprising, since GABA and glycine are
often co-released in dorsal horn, whereas a minority
of inhibitory synapses is mediated by GABA only
(Todd and Sullivan, 1990). Moreover, our recordings
were obtained from rats in the second postnatal week:
at this developmental stage mIPSCs can be mediated
by GABA or glycine or by both neurotransmitters, as
shown by previous studies (Keller et al., 2001). Fre-
quency of pure and mixed mIPSCs was increased by
BDNF, while the amplitude was not affected, sug-
gesting a presynaptic effect of the neurotrophin. The
frequency of pharmacologically isolated GABA and
glycine mIPSCs was also significantly increased, con-
firming a modulatory action of BDNF on both neuro-
transmitters. Moreover, the lack of effect on both
GABA and glycine mIPSC amplitude and kinetics
would likely exclude a modulation of the postsynap-
tic receptor function.
While evidence of a postsynaptic modulation of
glycine receptors by BDNF in CNS has not been pro-
vided to our knowledge, several studies performed in
different areas of the brain have shown that BDNF
can rapidly alter the functionality of postsynaptic
GABAA receptors, causing inhibition and receptor
Figure 7 Dual labeling immunocytochemistry for fl-trkB
receptors and GABA in rat spinal cord lamina II. Synaptic
glomerulus of the IIb type displaying two peripheral vesi-
cle-containing presynaptic dendrites (V1) immunoreactive
for GABA (10 nm gold). The V1 profile at right is also pos-
itively stained for fl-trkB (gold-intensified Fluoronano-
goldTM). The area indicated by the rectangle is shown at
higher magnification in the insert. Scale bars ¼ 500 nm;
insert ¼ 100 nm.
970 Bardoni et al.
Developmental Neurobiology. DOI 10.1002/dneu

internalization (Tanaka et al., 1997; Brunig et al.,
2001; Hennebeger et al., 2002; Cheng and Yeh, 2003;
Jovanovic et al., 2004). Differently from our data, in
these studies a significant depression of mIPSC
amplitude was usually observed, which was rarely
accompanied by a change of mIPSC frequency.
However, studies in several CNS areas have dem-
onstrated that BDNF can modulate GABAergic syn-
apses by regulating transmitter release. Thus, in
agreement with our results, facilitatory effects on
spontaneous GABA release have usually been re-
ported after acute (10–20 min) and chronic (h or
days) application of BDNF (McLean Bolton et al.,
2000; Wardle and Poo, 2003; Henneberger et al.,
2005; Baldelli et al., 2005).
In spinal cord dorsal horn, recent studies have pre-
viously provided evidence for BDNF-mediated con-
trol of neurotransmitter release. We have shown that
the acute application (10 min) of BDNF increases
AMPA mediated mEPSC frequency in neonatal rats,
without altering their amplitude or decay time (Bar-
doni et al., 2004). Matayoshi et al. (2005) report that
a very short application of BDNF (2 min) is sufficient
to increase mEPSC frequency in lamina II neurons
from adult rats, but only after induction of peripheral
inflammation. A facilitatory action of BDNF on GABA
release has been previously reported by Pezet et al.
(2002), showing that acute BDNF application induces
the increase of KCl-elicited GABA release in the
superfusate collected from the isolated spinal cord in
adult rats.
Effect of BDNF on GABAergic andGlycinergic Evoked IPSCs
While BDNF caused a facilitation of spontaneous
release of GABA and glycine, evoked IPSCs
recorded at 0 mV were depressed by the neurotro-
phin. Time course of BDNF action was similar to that
observed for mIPSCs (about 15–20 min) and wash
out was difficult to obtain. Depression of eIPSCs was
likely not simply due to the stimulation protocol,
since pre-incubation of the slices in the trk receptor
antagonist k-252a prevented the amplitude decrease
over the same time period. BDNF caused an increase
of synaptic variability of GABA/Glycine eIPSC peak
amplitude, expressed by the increase of CV, indicat-
ing a possible presynaptic modulatory site. Further-
more, the experiments performed on glycinergic
IPSCs applying the paired pulse protocol showed that
the PPR increases, suggesting a decrease of probabil-
ity of glycine release due to BDNF. Similar results
have been reported by Frerking et al. (1998) in CA1
pyramidal neurons, where BDNF causes the depres-
sion of GABAergic eIPSCs, inducing changes in CV
and PPR. Although the depressing action of BDNF
on eIPSCs is seemingly in contrast with the facilita-
tion of mIPSCs, a differential regulation of spontane-
ous and evoked release by BDNF has been already
reported. In cultured hippocampal neurons, at specific
synaptic types, BDNF causes an increase in mEPSC
frequency, which is not accompanied by a change of
evoked synaptic responses (Schinder et al., 2000).
Furthermore and in keeping with our results, Wardle
and Poo (2003) have observed in the same neuronal
type that BDNF causes a depression of eIPSCs, while
mIPSC frequency is increased. Moreover, examples
of opposite modulation of spontaneous and evoked
GABA and glycine release have also been observed
in dorsal horn neurons, involving ionotropic and
metabotropic presynaptic receptors (Jang et al., 2001;
Kerchner et al., 2001; Engelman et al., 2006). Cellu-
lar mechanisms regulating spontaneous, TTX resist-
ant, and action potential-dependent evoked release
can be different. The first can be regulated by pre-
synaptic receptors expressed close to the terminal,
controlling the intracellular calcium concentration.
Evoked release can also be affected by mechanisms
modulating action potential generation and propaga-
tion in the presynaptic neuron. Moreover, presynaptic
proteins can also contribute to the independent regu-
lation of spontaneous and evoked neurotransmitter
vesicular release: lack of functional synaptotagmin,
for instance, reduces evoked transmitter release but
increases mEPSC frequency (Broadie et al., 1994;
Geppert et al., 1994). These considerations indicate
that the final outcome of the BDNF modulatory
action on GABA and glycine release in dorsal horn
results by a complex interaction of multiple factors,
such as the cellular type(s) involved in synaptic activ-
ity, the subcellular localization of trkB receptors, and
the specific intracellular pathway(s) that are activated
by the neurotrophin.
Circuitry and Functional Significance
The ultrastructural studies presented in this article
show that co-localization of fl-trkB receptors and
GABA is present only at the level of V1 profiles in
any type of glomeruli, with the exception of type Ib
(peptidergic). In addition fl-trkB and GABA immuno-
reactive profiles were never engaged together in any
type of synaptic arrangement outside of glomeruli,
i.e. axo-dendritic or axo-somatic synapses. These
synaptic contacts, which represent the majority of
profiles in lamina II (Ribeiro-da-Silva, 2004), did not
BDNF Modulates GABA and Glycine in Lamina II 971
Developmental Neurobiology. DOI 10.1002/dneu

display the two labels together as pre and postsynap-
tic partners in any possible configuration. Although
we have performed these studies only on GABA
expression, it has been suggested that virtually all
glycinergic neurons in laminae I-III are also
GABAergic (Todd, 1990, 1996; Todd and Sullivan,
1990). This would suggest that co-localization of gly-
cine with trkB receptors follows a similar pattern.
Figure 8 summarizes in a schematic fashion one
possible circuital arrangement that is compatible with
the results of the present work.
V1 profiles in glomeruli (in red) are presynaptic
vesicular dendrites that are known to belong to
GABAergic islet cells, which often also contain gly-
cine (Spike and Todd, 1992). Fl-trkB receptor expres-
sion was detected not only in this type of profiles at
glomeruli, but also on plain dendrites, lacking of
GABA vesicles (D–in yellow): since it appears that
most GABAergic interneurons possess GABA-con-
taining vesicles in their dendrites (Spike and Todd,
1992), we can safely assume that these D profiles
belong to neurons other than the islet cells.
Our electron microscopy analysis shows that fl-
trkB receptors can be localized at V1 presynaptic ve-
sicular dendrites that contain, and likely release
GABA/glycine (Carton and Hayes, 1991; Spike and
Todd, 1992) and provide support for a presynaptic
action of BDNF on spontaneous release of these neu-
rotransmitters. Involvement of these presynaptic den-
drites in the regulation of evoked release is more dif-
ficult to explain, since they do not seem to be capable
to antidromically conduct action potentials (Safronov,
1999). In olfactory bulb mitral cells, release of
GABA from presynaptic dendrites has been reported
to be caused by calcium entry through NMDA recep-
tors and by membrane depolarization and subsequent
activation of voltage-dependent calcium channels
(Schoppa et al., 1998; Isaacson, 2001). The mecha-
nisms responsible for dendritic transmitter release in
dorsal horn glomeruli are still unknown: in our exper-
imental conditions, membrane depolarization of pre-
synaptic dendrites could be generated by focal stimu-
lation, evoking the release of the two inhibitory neu-
rotransmitters. Further studies are needed to confirm
this hypothesis.
The lack of expression of fl-trkB receptors on
GABAergic extraglomerular axo-dendritic and axo-
somatic synapses would exclude a direct and local
modulation of BDNF at these presynaptic terminals.
However, control of GABA and glycine release
(spontaneous and evoked) could result from the inter-
action between BDNF and fl-trkB on the dendrites,
triggering intracellular signals that rapidly propagate
up to the axon terminals or the modulation of ionic
Figure 8 Circuitry involved in the response of lamina II
neurons to BDNF. Schematic drawing representing a sim-
plified neuronal circuitry that may be responsible for the in-
hibitory response to BDNF of lamina II neurons. The cir-
cuitry is derived from the electrophysiological and ultrastruc-
tural experiments described in the result section. A
GABAergic islet cell (red) expressing fl-trkB receptors (blue
dots) at its V1 dendrites in glomeruli releases GABA at ter-
minal making axo-dendritic and/or axo-somatic synapses
onto the recorded neuron (orange). V1 profiles at glomeruli
are known to be bi-directionally connected (blue/red arrows)
with the central bouton (light blue). Dendro-dendritic synap-
ses between V1 and D profiles at glomeruli are also
described (see Ribeiro-da-Silva, 2004). The red arrows indi-
cate the putative sites of GABA release from V1 profiles.
Modulation at this level may be also linked to activation of
metabotropic glutamate receptors (not represented) that have
been described to be specifically localized at this type of glo-
merular profiles in non-peptidergic glomeruli. D profiles
expressing fl-trkB receptors (yellow–blue dots) are unlikely
to participate to modulation of inhibitory neurotransmission.
They may belong to excitatory neurons, whose influence
could be minimal in our experimental conditions of this
study, since ionotropic glutamate receptors are blocked (see
text).
972 Bardoni et al.
Developmental Neurobiology. DOI 10.1002/dneu

channels affecting action potential conduction (see
Blum and Konnerth, 2005).
Finally and strictly related to our present findings,
ultrastructural studies provided evidence for expres-
sion of metabotropic glutamate receptors specifically
at V1 profiles in non-peptidergic glomeruli (Jia et al.,
1999): this suggests that BDNF-mediated modulation
of inhibitory synaptic transmission could also involve
more indirect mechanisms, such as glutamate release.
BDNF is an important modulator of pain transmis-
sion in dorsal horn, exerting, in most cases, a pronoci-
ceptive action (see Merighi et al., 2004; Pezet and
McMahon, 2006). Our data are the first functional
demonstration that the neurotrophin can also exert
complex effects on inhibitory synaptic transmission
in lamina II. It is generally hold that peptidergic noci-
ceptors express low basal levels of BDNF, which are
upregulated following inflammation and peripheral
NGF stimulation. However, we have recently pro-
vided evidence that a subpopulation of these nocicep-
tors possesses substantial levels of the neurotrophin
also under basal conditions (Salio et al., 2005), and
that the neurotrophin is anterogradely transported to
terminals that form the C bouton in peptidergic type
Ib glomeruli (Salio et al., 2007). Interestingly, this
glomerular type does not have V1 profiles colocaliz-
ing fl-trkB receptors and GABA. One is therefore
tempted to speculate that in normal conditions of pain
transmission, low levels of BDNF are locally released
at type Ib glomeruli and do not significantly affect
inhibitory synaptic transmission. Upregulation of
BDNF in inflammatory and neuropathic pain condi-
tions would lead to a more abundant synaptic and/or
extrasynaptic release of the neurotrophin that could
diffuse to nearby glomeruli with fl-trkB/GABA con-
taining V1 profiles, and regulate, on a rapid time
scale (min), GABAergic and glycinergic transmission
in this population of islet cells. In particular, the con-
trol of evoked release could determine a general
depression of inhibition, compatible with the previ-
ously reported pronociceptive effects of BDNF (see
Pezet and McMahon, 2006), whereas the regulation
of spontaneous release could exert a local modulation
of the inhibitory tone and the control of primary
afferent terminal function at the glomerular level.
REFERENCES
Baldelli P, Hernandez-Guijo JM, Carabelli V, Carbone E.
2005. Brain-derived neurotrophic factor enhances GABA
release probability and nonuniform distribution of N- and
P/Q-type channels on release sites of hippocampal inhibi-
tory synapses. J Neurosci 25:3358–3368.
Bardoni R, Salio C, Prandini M, Merighi A. 2004. Func-
tional and histological evidence for pre-synaptic TrkB
autoreceptor modulation of glutamate neurotransmission
in dorsal horn spinal cord. Society For Neuroscience
34th Annual meeting Abstracts: 852.17
Blum R, Konnerth A. 2005. Neurotrophin-mediated rapid
signaling in the central nervous system: Mechanisms and
functions. Physiology 20:70–78.
Broadie K, Bellen HJ, DiAntonio A, Littleton JT, Schwarz
TL. 1994. Absence of synaptotagmin disrupts excitation-
secretion coupling during synaptic transmission. Proc
Natl Acad Sci USA 91:10727–10731.
Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM.
2001. BDNF reduces miniature inhibitory postsynaptic
currents by rapid downregulation of GABAA receptor
surface expression. Eur J Neurosci 13:1320–1328.
Carlton SM, Hayes ES. 1991. GABAergic vesicle-contain-
ing dendrites and spines: A critical element in processing
sensory input in the monkey dorsal horn. Neurosci Lett
121:40–42.
Cejas PJ, Martinez M, Karmally S, McKillop M, McKillop
J, Plunkett JA, Oudega M, et al. 2000. Lumbar transplant
of neurons genetically modified to secrete brain-derived
neurotrophic factor attenuates allodynia and hyper-
algesia after sciatic nerve constriction. Pain 86:195–
210.
Cheng Q, Yeh HH. 2003. Brain-derived neurotrophic factor
attenuates mouse cerebellar granule cell GABA(A) re-
ceptor-mediated responses via postsynaptic mechanisms.
J Physiol 548:711–721.
Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue
K, Gravel C, et al. 2005. BDNF from microglia causes
the shift in neuronal anion gradient underlying neuro-
pathic pain. Nature 438:1017–1021.
Engelman HS, Anderson RL, Daniele C, MacDermott AB.
2006. Presynaptic a-amino-3-hydroxy-5-methyl-4-isoxa-
zolepropionic acid (AMPA) receptors modulate release
of inhibitory amino acids in rat spinal cord dorsal horn.
Neuroscience 139:539–553.
Frerking M, Malenka RC, Nicoll RA. 1998. Brain-derived
neurotrophic factor (BDNF) modulates inhibitory, but
not excitatory, transmission in the CA1 region of the hip-
pocampus. J Neurophysiol 80:3383–3386.
Garraway SM, Petruska JC, Mendell LM. 2003. BDNF
sensitizes the response of lamina II neurons to high thresh-
old primary afferent inputs. Eur J Neurosci 18:2467–
2476.
Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Ste-
vens CF, Sudhof TC. 1994. Synaptotagmin I: A major
Ca2þ sensor for transmitter release at a central synapse.
Cell 79:717–727.
Henneberger C, Juttner R, Rothe T, Grantyn R. 2002. Post-
synaptic action of BDNF on GABAergic synaptic trans-
mission in the superficial layers of the mouse superior
colliculus. J Neurophysiol 88:595–603.
Henneberger C, Kirischuk S, Grantyn R. 2005. Brain-
derived neurotrophic factor modulates GABAergic syn-
aptic transmission by enhancing presynaptic glutamic
acid decarboxylase 65 levels, promoting asynchronous
BDNF Modulates GABA and Glycine in Lamina II 973
Developmental Neurobiology. DOI 10.1002/dneu

release and reducing the number of activated postsynap-
tic receptors. Neuroscience 135:749–763.
Isaacson JS. 2001. Mechanisms governing dendritic c-ami-
nobutyric acid (GABA) release in the rat olfactory bulb.
Proc Natl Acad Sci USA 98:337–342.
Jang IS, Rhee JS, Kubota H, Akaike N, Akaike N. 2001.
Developmental changes in P2X purinoceptors on glyci-
nergic presynaptic nerve terminals projecting to rat
substantia gelatinosa neurones. J Physiol 536:505–
519.
Jia H, Rustioni A, Valtshanoff JG. 1999. Metabotropic glu-
tamate receptors in superficial laminae of the rat dorsal
horn. J Comp Neurol 410:627–642.
Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ.
2004. Brain-derived neurotrophic factor modulates fast
synaptic inhibition by regulating GABA(A) receptor
phosphorylation, activity, and cell-surface stability.
J Neurosci 24:522–530.
Keller AF, Coull JAM, Chery N, Poisbeau P, De Koninck
Y. 2001. Region-specific developmental specialization of
GABA-glycine cosynapses in laminas I-II of the rat spi-
nal dorsal horn. J Neurosci 21:7871–7880.
Kerchner GA, Wang GD, Qiu CS, Huettner JE, Zhuo M.
2001. Direct presynaptic regulation of GABA/glycine
release by kainate receptors in the dorsal horn: An iono-
tropic mechanism. Neuron 32:477–488.
Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW,
Jones MG, McMahon SB, Marvizon JCG, et al. 2001.
Brain-derived neurotrophic factor is released in the dor-
sal horn by distinctive patterns of afferent fiber stimula-
tion. J Neurosci 21:4469–4477.
Levine ES, Crozier RA, Black IB, Plummer MR. 1998.
Brain-derived neurotrophic factor modulates hippocam-
pal synaptic transmission by increasing N-methyl-D-
aspartic acid receptor activity. Proc Natl Acad Sci USA
95:10235–10239.
Levine ES, Dreyfus CF, Black IB, Plummer MR. 1995.
Brain-derived neurotrophic factor rapidly enhances syn-
aptic transmission in hippocampal neurons via postsy-
naptic tyrosine kinase receptors. Proc Natl Acad Sci
USA 92:8074–8077.
Malcangio M, Bowery NG. 1996. GABA and its receptors
in the spinal cord. Trends Pharmacol Sci 17:457–62.
Matayoshi S, Jiang N, Katafuchi T, Koga K, Furue H,
Yasaka T, Nakatsuka T, et al. 2005. Actions of brain-
derived neurotrophic factor on spinal nociceptive trans-
mission during inflammation in the rat. J Physiol
569:685–695.
McLean Bolton M, Pittman AJ, Lo DC. 2000. Brain-
derived neurotrophic factor differentially regulates exci-
tatory and inhibitory synaptic transmission in hippocam-
pal cultures. J Neurosci 20:3221–3232.
Merighi A, Carmignoto G, Gobbo S, Lossi L, Salio C,
Vergnano AM, Zonta M. 2004. Neurotrophins in spinal
cord nociceptive pathways. Prog Brain Res 146:291–
321.
Merighi A, Polak JM. 1993. Postembedding immunogold
staining. In: Cuello AC, editor. Immunocytotchemistrty
II. New York: Wiley, pp 229–264
Merighi A, Polak JM, Fumagalli G, Theodosis DT. 1989.
Ultrastructural localization of neuropeptides and GABA
in rat dorsal horn: A comparison of different immuno-
gold labeling techniques. J Histochem Cytochem 37:
529–540.
Pezet S, Cunningam J, Patel J, Grist J, Gavazzi I, Lever IJ,
Malcangio M. 2002. BDNF modulates sensory neuron
synaptic activity by a facilitation of GABA transmission
in the dorsal horn. Mol Cell Neurosci 21:51–62.
Pezet S, McMahon SB. 2006. Neurotrophins: Mediators
and modulators of pain. Annu Rev Neurosci 29:507–
538.
Ribeiro da Silva A. 2004. Substantia gelatinosa of spinal
cord. In: Paxinos G, editor. The Rat Nervous System.
New York: Academic Press.
Safronov BV. 1999. Spatial distribution of Naþ and Kþ
channels in spinal dorsal horn neurones: Role of the
soma, axon and dendrites in spike generation. Prog Neu-
robiol 593:217–241.
Salio C, Lossi L, Ferrini F, Merighi A. 2005. Ultrastructural
evidence for a pre- and postsynaptic localization of full-
length trkB receptors in substantia gelatinosa (lamina II)
of rat and mouse spinal cord. Eur J Neurosci 22:1951–
1966.
Salio C, Averill S, Priestley JV, Merighi A. 2007. Costor-
age of BDNF and neuropeptides within individual dense-
core vesicles in central and peripheral neurons. Develop
Neurobiol 67:326–338.
Schinder AF, Berninger B, Poo M. 2000. Postsynaptic
target specificity of neurotrophin-induced presynaptic
potentiation. Neuron 25:151–163.
Schoppa NE, Kinzie JM, Sahara Y, Segerson TP, West-
brook GL. 1998. Dendrodendritic inhibition in the olfac-
tory bulb is driven by NMDA receptors. J Neurosci
18:6790–6802.
Siuciak JA, Altar CA, Wiengand SJ, Lindsay RM. 1994.
Antinociceptive effect of brain derived neurotrophic fac-
tor and neurotrophin-3. Brain Res 633:326–330.
Slack SE, Pezet S, McMahon SB, Thompson SWN, Mal-
cangio M. 2004. Brain-derived neurotrophic factor indu-
ces NMDA receptor subunit one phosphorylation via
ERK and PKC in the rat spinal cord. Eur J Neurosci
20:1769–1778.
Spike RC, Todd AJ. 1992. Ultrastructural and immunocyto-
chemical study of lamina II islet cells in rat spinal dorsal
horn. J Comp Neurol 323:359–369.
Tanaka T, Saito H, Matsuki N. 1997. Inhibition of GABAA
synaptic responses by brain-derived neurotrophic factor
(BDNF) in rat hippocampus. J Neurosci 17:2959–2966.
Thompson SWN, Bennett DLH, Kerr BJ, Bradbury EJ,
McMahon SB. 1999. Brain-derived neurotrophic factor is
an endogenous modulator of nociceptive responses in the
spinal cord. Proc Natl Acad Sci USA 96:7714–7718.
Todd AJ. 1990. An electron microscope study of glycine-
like immunoreactivity in laminae I-III of the spinal dor-
sal horn of the rat. Neuroscience 39:398–394.
Todd AJ. 1996. GABA and glycine in synaptic glomeruli
of the rat spinal dorsal horn. Eur J Neurosci 8:2492–
2498.
974 Bardoni et al.
Developmental Neurobiology. DOI 10.1002/dneu

Todd AJ, Sullivan AC. 1990. Light microscope study of the
coexistence of GABA-like and glycine-like immunoreac-
tivities in the spinal cord of the rat. J Comp Neurol
296:496–505.
Wardle RA, Poo MM. 2003. Brain-derived neurotrophic
factor modulation of GABAergic synapses by postsynap-
tic regulation of chloride transport. J Neurosci 23:8722–
8732.
Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang
K, Wang D, et al. 2000. The role of brain-derived neuro-
throphic factor receptors in the mature hippocampus:
Modulation of long-term potentiation through a presyn-
aptic mechanism involving TrkB. J Neurosci 20:6888–
6897.
Zhao J, Seereeram A, Nassar MA, Levato A, Pezet S,
Hathaway G, Morenilla-Palao C, et al. London Pain
Consortium. 2006. Nociceptor-derived brain-derived
neurotrophic factor regulates acute and inflammatory
but not neuropathic pain. Mol Cell Neurosci 31:539–
548.
BDNF Modulates GABA and Glycine in Lamina II 975
Developmental Neurobiology. DOI 10.1002/dneu




















