
Assessment of theN-oxidation of deprenyl, methamphetamine, and amphetamine enantiomers by chiral...
10
Éva Szöko ˝ 1 Tamás Tábi 1 Tímea Borbás 2 Balázs Dalmadi 2 Károly Tihanyi 2 Kálmán Magyar 1, 3 1 Department of Pharmacodynamics, Semmelweis University 2 In vitro Metabolism Laboratory, Richter Gedeon Ltd. 3 Neurochemical Research Unit of the Hungarian Academy of Sciences, Budapest, Hungary Assessment of the N-oxidation of deprenyl, methamphetamine, and amphetamine enantiomers by chiral capillary electrophoresis: An in vitro metabolism study A chiral capillary electrophoresis method using hydroxypropyl-b-cyclodextrin as chiral selector was developed and validated for the quantification of the N-oxygenated me- tabolites of deprenyl, methamphetamine, and amphetamine enantiomers, formed in vitro. The influence of various parameters (selector concentration, buffer pH, tempera- ture, polymer additive, etc.) on the simultaneous separation of the optical isomers of the parent drugs and their metabolites has been evaluated. The buffer pH had the greatest impact on the separation selectivity of the N-oxygenated compounds. Linear calibration curves were obtained over the concentration range of 2.5–50 mM for the enantiomers of amphetamine-hydroxylamine, methamphetamine-hydroxylamine, and deprenyl-N-oxide. The inter- and intra-assay precision and accuracy varied by less than 15% for all analytes at concentrations of 5, 10, and 30 mM, and less than 20% at the lower limit of quantitation (2.5 mM). The sample extraction recovery ranged between 109 and 129% at the three concentration levels. The drug enantiomers were incubated with recombinant human flavin-containing monooxygenase enzymes (FMO3 and FMO1), and human liver microsomes, respectively. The enantioselectivity of the sub- strate preference, as well as the stereoselective formation of the new chiral center upon the oxidation of the prochiral tertiary nitrogen of deprenyl were assessed. FMO1, the extrahepatic form of the enzyme in man, was shown to be more active in the N-oxygenation of both deprenyl and methamphetamine isomers than FMO3. Deprenyl enantiomers and S-methamphetamine were substrates of human recombi- nant FMO3. Conversion of amphetamine to its hydroxylamine derivative could not be observed on incubation with either FMO1 or FMO3. Formation of the new chiral center on the nitrogen, during N-oxidation of the tertiary amine deprenyl, was found stereo- selective. The two FMO isoforms have shown opposite preference in the formation of this chiral center. Methamphetamine-hydroxylamine formed from methamphetamine was further transformed by FMO, amphetamine-hydroxylamine was identified as the product of a demethylation reaction. Keywords: Chiral capillary electrophoresis / Deprenyl / In vitro drug metabolism / Methampheta- mine / N-Oxidation DOI 10.1002/elps.200406023 1 Introduction In vitro metabolism studies, performed in the early phase of drug development, have gained importance to reveal the potential metabolites, allowing the elucidation of their tox- icity as well as understanding and avoidance of some adverse drug reactions. The fundamental metabolizing organ is the liver, thus human liver microsomal preparations and hepatocytes are usually used in such experiments. Additionally, the main metabolic routes can be identified by the use of single isoforms of metabolic enzymes, provid- ing information about the influence of genetic polymor- phism on the metabolic pathway and the probability of drug interactions. The majority of these studies focus on the various cytochrome P450 isoforms (CYP450), the main oxidizing enzymes, although the involvement of flavin-con- taining monooxygenase (FMO) enzymes in drug metabo- lism has also been well established. Correspondence: Professor Éva Szöko ˝ , Department of Pharma- codynamics, Semmelweis University, Nagyvárad tér 4., Buda- pest, H-1089 Hungary E-mail: [email protected] Fax: 136-1-210-4411 Abbreviations: CYP450, cytochrome P450; FMO, flavin-con- taining monooxygenase; HPBCD, 2-(hydroxy-propyl)-b-cyclo- dextrin 2866 Electrophoresis 2004, 25, 2866–2875 2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Assessment of theN-oxidation of deprenyl, methamphetamine, and amphetamine enantiomers by chiral...

Éva Szöko1
Tamás Tábi1
Tímea Borbás2
Balázs Dalmadi2
Károly Tihanyi2
Kálmán Magyar1, 3
1Department ofPharmacodynamics,Semmelweis University
2In vitro Metabolism Laboratory,Richter Gedeon Ltd.
3Neurochemical Research Unitof the Hungarian Academyof Sciences,Budapest, Hungary
Assessment of the N-oxidation of deprenyl,methamphetamine, and amphetamine enantiomersby chiral capillary electrophoresis:An in vitro metabolism study
A chiral capillary electrophoresis method using hydroxypropyl-b-cyclodextrin as chiralselector was developed and validated for the quantification of the N-oxygenated me-tabolites of deprenyl, methamphetamine, and amphetamine enantiomers, formed invitro. The influence of various parameters (selector concentration, buffer pH, tempera-ture, polymer additive, etc.) on the simultaneous separation of the optical isomers ofthe parent drugs and their metabolites has been evaluated. The buffer pH had thegreatest impact on the separation selectivity of the N-oxygenated compounds. Linearcalibration curves were obtained over the concentration range of 2.5–50 mM for theenantiomers of amphetamine-hydroxylamine, methamphetamine-hydroxylamine, anddeprenyl-N-oxide. The inter- and intra-assay precision and accuracy varied by lessthan 15% for all analytes at concentrations of 5, 10, and 30 mM, and less than 20% atthe lower limit of quantitation (2.5 mM). The sample extraction recovery ranged between109 and 129% at the three concentration levels. The drug enantiomers were incubatedwith recombinant human flavin-containing monooxygenase enzymes (FMO3 andFMO1), and human liver microsomes, respectively. The enantioselectivity of the sub-strate preference, as well as the stereoselective formation of the new chiral centerupon the oxidation of the prochiral tertiary nitrogen of deprenyl were assessed.FMO1, the extrahepatic form of the enzyme in man, was shown to be more active inthe N-oxygenation of both deprenyl and methamphetamine isomers than FMO3.Deprenyl enantiomers and S-methamphetamine were substrates of human recombi-nant FMO3. Conversion of amphetamine to its hydroxylamine derivative could not beobserved on incubation with either FMO1 or FMO3. Formation of the new chiral centeron the nitrogen, during N-oxidation of the tertiary amine deprenyl, was found stereo-selective. The two FMO isoforms have shown opposite preference in the formation ofthis chiral center. Methamphetamine-hydroxylamine formed from methamphetaminewas further transformed by FMO, amphetamine-hydroxylamine was identified as theproduct of a demethylation reaction.
Keywords: Chiral capillary electrophoresis / Deprenyl / In vitro drug metabolism / Methampheta-mine / N-Oxidation DOI 10.1002/elps.200406023
1 Introduction
In vitro metabolism studies, performed in the early phase ofdrug development, have gained importance to reveal thepotential metabolites, allowing the elucidation of their tox-
icity as well as understanding and avoidance of someadverse drug reactions. The fundamental metabolizingorgan is the liver, thus human liver microsomal preparationsand hepatocytes are usually used in such experiments.Additionally, the main metabolic routes can be identifiedby the use of single isoforms of metabolic enzymes, provid-ing information about the influence of genetic polymor-phism on the metabolic pathway and the probability ofdrug interactions. The majority of these studies focus onthe various cytochrome P450 isoforms (CYP450), the mainoxidizing enzymes, although the involvement of flavin-con-taining monooxygenase (FMO) enzymes in drug metabo-lism has also been well established.
Correspondence: Professor Éva Szöko, Department of Pharma-codynamics, Semmelweis University, Nagyvárad tér 4., Buda-pest, H-1089 HungaryE-mail: [email protected]: 136-1-210-4411
Abbreviations: CYP450, cytochrome P450; FMO, flavin-con-taining monooxygenase; HPBCD, 2-(hydroxy-propyl)-b-cyclo-dextrin
2866 Electrophoresis 2004, 25, 2866–2875
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim

Electrophoresis 2004, 25, 2866–2875 Chiral CE for in vitro metabolism study 2867
FMO participates in the oxygenation of nucleophilicnitrogen-, sulfur-, phosphorus, and other heteroatom-containing drugs, xenobiotics and endogenous com-pounds (for recent reviews see [1–3]). Currently, sixgenes of FMO are known [2] and the species- and tis-sue-specific expression of the various FMO isoforms iswell recognized. In humans, the main isoform found inadult liver microsomal preparation is FMO3, while FMO1is abundant in the kidney, and is also present in theintestine [4, 5]. Genetic polymorphisms and functionalvariation of human FMO3 and FMO2 have been estab-lished [2, 6]. The human FMO isoforms differ in their sub-strate specificity, and usually stereoselectively introducea new stereogenic center during oxidation of prochiralheteroatoms [4, 7–9]. As amine compounds are con-cerned, human FMO3 is assumed to catalyze N-oxy-genation of primary, secondary, and tertiary amines aswell, although it is sensitive to the steric features of theamines [1]. The preferred substrates for human FMO1are the tertiary amine compounds, and their principalproducts are N-oxides. Hydroxylamines, formed fromprimary or secondary amine containing drug are un-stable, and are readily converted further by FMOthrough intermediate compounds to oxime and thelabile nitrone (imine-N-oxide) derivatives, respectively[9–14], and these products can still be further trans-formed. Oxidative metabolism of drugs catalyzed byFMO or CYP450 can lead to the same products.
N-Oxide formation, as a metabolic conversion of depre-nyl, has recently been recognized. Deprenyl-N-oxidewas identified in human [15, 16] and rat urine [17, 18] inin vivo metabolism studies. However, the contribution ofthe various FMO or CYP450 isoforms to this metabolicpathway has not yet been clarified. Stereospecific deal-kylation of deprenyl is well-established as its main meta-bolic transformation, the products are methampheta-mine, amphetamine, and desmethyldeprenyl. Metabolicconversion of methamphetamine and amphetamine byhuman FMO3 has been assessed by Cashman et al.[14]. Amphetamine-hydroxylamine and methampheta-mine-hydroxyalmine were identified to be produced byFMO3, which products by themselves were substratesof FMO3 and were converted through various intermedi-ate compounds to phenylpropanone oxime and phenyl-propanone, respectively [14]. The in vivo formation ofphenylpropanone during deprenyl metabolism has alsobeen shown by Kalász and co-workers [19]. Previously,demethylation of methamphetamine-hydroxylamine toamphetamine-hydroxylamine by guinea-pig liver micro-somes, and its further reduction to amphetamine hasbeen reported [13]. Two independent metabolic path-ways of methamphetamine N-demethylation to am-phetamine have been well recognized via either C-hydro-
xylation or N-hydroxylation, catalyzed mainly by CYP450and FMO enzymes, respectively. In the latter path-way conversion of methamphetamine-hydroxylamine toamphetamine-hydroxylamine and its further reduction toamphetamine was assumed. Components of a reduc-tase system, termed benzamidoxime reductase, respon-sible for the reduction of N-hydroxylated derivatives havebeen identified [20, 21]. The system contains benzami-doxime reductase, which is a CYP450 enzyme, cyto-chrome b5, and NADH-cytochrome b5-reductase, and issuggested to be involved both in the retroreduction ofthe N-oxygenated metabolites to the parent compounds,and in demethylation processes [20, 21]. Figure 1 sum-marizes the FMO-catalyzed metabolic transformations ofdeprenyl, methamphetamine, and amphetamine.
The enantiomers of chiral drugs are usually distinguishedby the metabolic enzymes, and the product formationcan also show stereospecificity or stereoselectivity.Chiral CE methods can readily be used to identifiy thestereochemistry of the enzymatic transformation, sincesimultaneous separation of several enantiomeric pairswith similar chemical structure can be achieved becauseof the high efficiency provided by these methods.Recently, several papers on in vitro drug metabolismstudies using chiral CE have been published [22–25].The first CE method for the analysis of N-oxide enantio-
Figure 1. Suggested FMO-catalyzed metabolic path-ways of deprenyl, methamphetamine, and amphetamine.BOR, benzamidoxime reductase.
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
CE
and
CE
C

2868 E. Szöko et al. Electrophoresis 2004, 25, 2866–2875
mers formed by the FMO catalyzed oxidation of prochiraltertiary amines was reported by Hadley et al. [23]. Theyhave found a highly stereoselective N-oxygenation of par-gyline.
In our present study, a chiral CE method using hydroxy-propyl-b-cyclodextrin (HPBCD) was developed andapplied to compare the in vitro metabolism of the enantio-mers of deprenyl, methamphetamine, and amphetamineby recombinant human FMO1 and FMO3, respectively.In the case of deprenyl enantiomers, the contributionof CYP450 enzymes to deprenyl-N-oxide formation hasalso been evaluated. Our method allows the simulta-neous enantioselective analysis of the N-oxygenatedmetabolites of primary, secondary, and tertiary phenyl-ethylamines, as well as their parent compounds.
2 Materials and methods
2.1 Chemicals
Chemicals used were of the highest purity availablefrom commercial sources. Human liver microsomeswere obtained from Xenotech LLC (Kansas City, KS,USA). Microsomes containing recombinant humanFMO1 and FMO3 from baculovirus-infected insectcells (supersomes) were purchased from BD Gentest(Woburn, MA, USA). Glucose-6-phosphate and glu-cose-6-phosphate dehydrogenase were supplied bySigma-Aldrich (St. Louis, MO, USA). Sodium pyro-phosphate, magnesium chloride, NADPH, perchloricacid, Tris, sodium bicarbonate, phosphoric acid, andacetic acid were obtained from Reanal (Budapest,Hungary). Methanol was purchased from Merck (Darm-stadt, Germany) and was HPLC grade. The ultrapurewater used in the CE experiments was obtained froma MilliQ Water system (Millipore, Bedford, MA, USA).Standard compounds, hydrochloride salts of S- andR-methamphetamine, S- and R-amphetamine, S- andR-deprenyl, S- and R-methamphetamine-hydroxyl-amine, S- and R-amphetamine-hydroxylamine, 1R,NR-and 1S, NS-deprenyl-N-oxide, and the diastereomericmixture (1:1) of 1R,NR- and 1R,NS- and 1S,NS- and1S,NR-deprenyl-N-oxide, and S-para-fluorodesmethyl-deprenyl (internal standard, IS) were kindly provided byChinoin Pharmaceutical and Chemical Works, a mem-ber of the Sanofi-Synthelabo Group (Budapest, Hun-gary). Standard stock solutions (1022 and 1023 M) ofall compounds were prepared in distilled water, werestored frozen (2207C), and diluted just prior to use.CE-grade 2-(hydroxy-propyl)-b-cyclodextrin (degree ofsubstitution 3; HPBCD) was supplied by BeckmanCoulter (Fullerton, CA, USA). Hydroxypropylmethylcel-lulose (viscosity of 2% solution is 80–120 cP at 207C)
was obtained from Sigma. Discovery C18 Lt cartridges(100 mg/1 mL; Supelco, Bellefonte, PA, USA) wereused for solid-phase extraction of the samples.
2.2 Incubation conditions
All incubation mixtures contained 6 mM sodium pyrophos-phate, 5 mM magnesium chloride, 5 mM glucose-6-phos-phate, 1 U mL21 glucose-6-phosphate dehydrogenase in afinal volume of 0.5 mL of 0.1 M Tris-HCl buffer, pH 7.4. Theconcentration of the substrates was 1 mM. In human livermicrosome incubations, the mixture contained 1 mg?mL21
microsomal protein and the reaction was initiated by addi-tion of NADPH (0.5 mM final concentration), and carried outin a shaking water bath at 377C for 30 min. In incubations,containing insect cell-expressed recombinant FMOs, theenzyme concentration was 100 nM (FMO1 or FMO3). Sub-strates were used at a concentration of 1 mM. The reactionswere initiated by the addition of supersomes and incubatedin a water bath at 377C without shaking, in accordance withthe manufacturer’s recommendation. Microsomespreparedfrom wild-type baculovirus-infected cells were used as con-trol. The reactions were stopped with an equal volume of0.4 M cold perchloric acid, centrifuged for 10 min at10 0006g. The supernatants were stored at 2207C untiluse (not longer than a week).
2.3 Sample preparation
The sample pH was adjusted to 8–8.5 by addition of satu-rated sodium bicarbonate solution (400–800 mL superna-tant). The resultant solutions were applied to solid-phaseextraction cartridges (prewashed successively with 1 mLmethanol, 1 mL distilled water, 1 mL sodium bicarbonate),washed with 1 mL distilled water, and eluted with 1.5 mLmethanol. The extracts were acidified with 5 M hydrochlo-ric acid (30 mL) and evaporated to dryness at 407C undervacuum. The residue was redissolved in 80 mL distilledwater and used for CE analysis.
2.4 Instrumentation
All experiments were performed with a P/ACE MDQ CE sys-tem controlled by 32 Karat software Versions 5.0 (BeckmanCoulter). The instrument was equipped with a UV detector,set at 200 nm. Separations were carried out in fused-silicacapillaries, 75 mm ID, 365 mm OD, 50.2 cm total length,40 cm to the detector (Polymicro Technologies, Phoenix,AZ, USA). The capillary was washed successively with 0.1 M
phosphoric acid, water, and the separation buffer at thebeginning of all workdays, and the separation buffer be-tween runs. Samples were introduced into the capillary by
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim

Electrophoresis 2004, 25, 2866–2875 Chiral CE for in vitro metabolism study 2869
pressure (5 s, 0.5 psi), the sample volume corresponding toabout 1.25% of the capillary volume. The separations werecarried out applying a constant voltage of 25 kV, and thetemperature was maintained at 157C if not noted otherwise.
3 Results and discussion
3.1 Chiral CE method for the separation of theparent compounds and their respectivemetabolites
Low-pH (2.7) phosphate buffer was chosen for theseparation of the parent amine compounds and theiroxygenated metabolites deprenyl-N-oxide, metham-phetamine-hydroxylamine, and amphetamine-hydroxyl-amine, where all the analytes are positively charged. Inour recent studies, cyclodextrin derivatives containinghydrophilic substituents (HPBCD and carboxymethyl-b-cyclodextrin) have been found appropriate chiral selec-tors for the separation of deprenyl-N-oxide enantiomers[17, 26]. Both of them have also shown good enantio-selectivity toward methamphetamine-hydroxylamine andamphetamine-hydroxylamine in the present study (datanot shown). Because of the shorter analysis time, lesszone dispersion, and lower baseline noise, HPBCD waschosen as the chiral selector throughout this study.The optimum concentration for the chiral separationof amphetamine, methamphetamine, deprenyl, amphet-amine-hydroxylamine, methamphetamine-hydroxylamine,and deprenyl-N-oxide isomers was observed between10 and 20 mM HPBCD. Because of the shorter analysistime 10 mM selector concentration was used in the furtherexperiments to determine the FMO products of ampheta-mine, methamphetamine, and deprenyl enantiomers. Elec-tropherograms of standards of the parent drugs and theirsupposed oxygenated metabolites are shown in Fig. 2.
The method was validated for the quantitative determina-tion of amphetamine-hydroxylamine, methamphetamine-hydroxylamine, and deprenyl-N-oxide isomers. Systemsuitability studies performed at three concentration levelsof the individual stereoisomers (5, 10, and 30 mM) haveshown good reproducibilty of the migration times, peakarea, and height. The migration times of the eight metabo-lites were between 11 and 15 min, and their RSD valueswere less than 0.44%. The RSD of the peak area werebetween 1–12%, 3–10%, and 4–8% at 5, 10, and 30 mM
concentrations of the enantiomers, respectively. TheRSD of the peak height were between 2–8%, 1–5%, and1–6% at the three concentration levels, respectively. Cali-bration curves were constructed in the 2.5–50 mM concen-tration range of the individual stereoisomers of the ana-lytes. Peak height was used for the quantification of the
Figure 2. Electropherograms of standards of the parentdrugs and the corresponding N-oxygenated metabolites(A: deprenyl, B: methamphetamine, C: amphetamine).Separation conditions: 20 mM Tris-phosphate buffer,pH 2.7, containing 10 mM HPBCD. Voltage, 25 kV; current,35 mA; temperature, 157C. D: deprenyl; DNO, deprenyl-N-oxide; MA, methamphetamine; MAOH, methampheta-mine-hydroxylamine; A, amphetamine; AOH, ampheta-mine-hydroxylamine; IS, internal standard.
analytes, because it has better reproducibility comparedto peak area. The equations and r values of the calibrationlines for each analyte are summarized in Table 1. The sen-sitivity of the assay was also evaluated; the limit of detec-tion (LOD) at a signal-to-noise ratio of 3:1 was 1 mM for thestereoisomers of the metabolites. The lower limit of quan-titation (LLOQ), where the accuracy was within 80–120%and the precision was less than 20% expressed as RSD,
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
2870 E. Szöko et al. Electrophoresis 2004, 25, 2866–2875
Table 1. Equations and r values of the calibration lines forthe analytes
Compound Equation r
R-Amphetamine-hydroxylamine
y = 0.0095x 1 0.1133 0.9971
S-Amphetamine-hydroxylamine
y = 0.0089x – 0.3237 0.9958
R-Methamphetamine-hydroxylamine
y = 0.0084x – 0.5462 0.9966
S-Methamphetamine-hydroxylamine
y = 0.0076x – 0.0450 0.9960
1R,NR-Deprenyl-N-oxide y = 0.0082x – 0.5493 0.9954
1S,NS-Deprenyl-N-oxide y = 0.0084x – 0.3931 0.9951
1R,NS-Deprenyl-N-oxide y = 0.0084x – 0.6705 0.9955
1S,NR-Deprenyl-N-oxide y = 0.0092x – 0.7794 0.9955
y is the concentration of the analyte (in mM) and x is thepeak height. Calibration curves were constructed in the2.5–50 mM concentration range.
was determined to be 2.5 mM for each enantiomer. Duringsample pretreatment, fivefold concentration of the ana-lytes was performed, thus 0.5 mM analyte in the originalsample could be determined. The sample extractionrecovery for the analyte enantiomers was assessed atthree concentration levels, and ranged between 109 and129%. No stereoselectivity of the sample cleanup proce-dure was observed.
3.2 Simultaneous separation of the studieddrugs and their metabolites
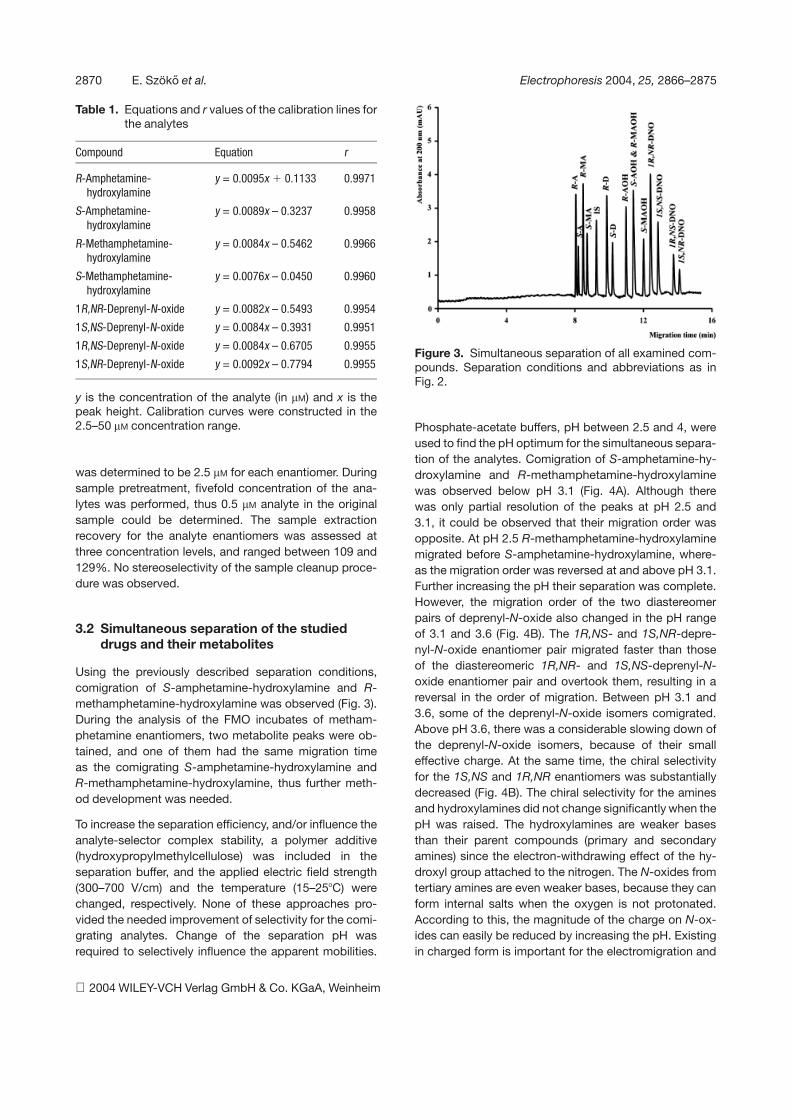
Using the previously described separation conditions,comigration of S-amphetamine-hydroxylamine and R-methamphetamine-hydroxylamine was observed (Fig. 3).During the analysis of the FMO incubates of metham-phetamine enantiomers, two metabolite peaks were ob-tained, and one of them had the same migration timeas the comigrating S-amphetamine-hydroxylamine andR-methamphetamine-hydroxylamine, thus further meth-od development was needed.
To increase the separation efficiency, and/or influence theanalyte-selector complex stability, a polymer additive(hydroxypropylmethylcellulose) was included in theseparation buffer, and the applied electric field strength(300–700 V/cm) and the temperature (15–257C) werechanged, respectively. None of these approaches pro-vided the needed improvement of selectivity for the comi-grating analytes. Change of the separation pH wasrequired to selectively influence the apparent mobilities.
Figure 3. Simultaneous separation of all examined com-pounds. Separation conditions and abbreviations as inFig. 2.
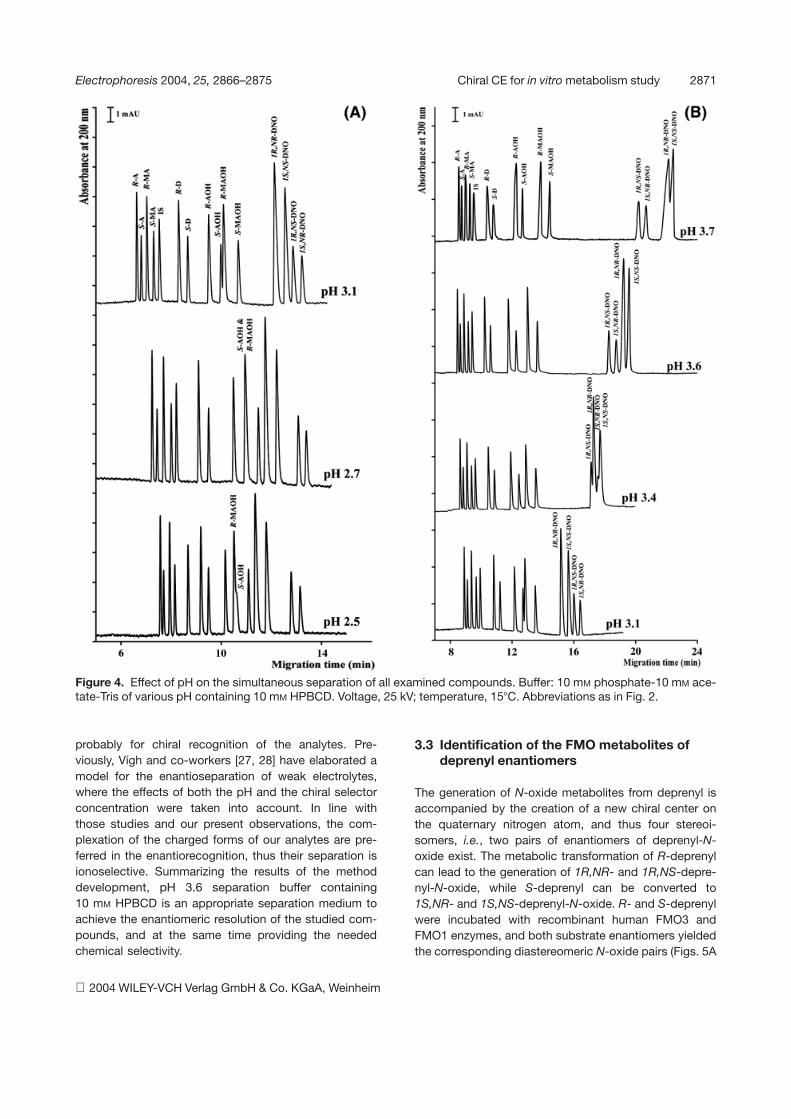
Phosphate-acetate buffers, pH between 2.5 and 4, wereused to find the pH optimum for the simultaneous separa-tion of the analytes. Comigration of S-amphetamine-hy-droxylamine and R-methamphetamine-hydroxylaminewas observed below pH 3.1 (Fig. 4A). Although therewas only partial resolution of the peaks at pH 2.5 and3.1, it could be observed that their migration order wasopposite. At pH 2.5 R-methamphetamine-hydroxylaminemigrated before S-amphetamine-hydroxylamine, where-as the migration order was reversed at and above pH 3.1.Further increasing the pH their separation was complete.However, the migration order of the two diastereomerpairs of deprenyl-N-oxide also changed in the pH rangeof 3.1 and 3.6 (Fig. 4B). The 1R,NS- and 1S,NR-depre-nyl-N-oxide enantiomer pair migrated faster than thoseof the diastereomeric 1R,NR- and 1S,NS-deprenyl-N-oxide enantiomer pair and overtook them, resulting in areversal in the order of migration. Between pH 3.1 and3.6, some of the deprenyl-N-oxide isomers comigrated.Above pH 3.6, there was a considerable slowing down ofthe deprenyl-N-oxide isomers, because of their smalleffective charge. At the same time, the chiral selectivityfor the 1S,NS and 1R,NR enantiomers was substantiallydecreased (Fig. 4B). The chiral selectivity for the aminesand hydroxylamines did not change significantly when thepH was raised. The hydroxylamines are weaker basesthan their parent compounds (primary and secondaryamines) since the electron-withdrawing effect of the hy-droxyl group attached to the nitrogen. The N-oxides fromtertiary amines are even weaker bases, because they canform internal salts when the oxygen is not protonated.According to this, the magnitude of the charge on N-ox-ides can easily be reduced by increasing the pH. Existingin charged form is important for the electromigration and
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Electrophoresis 2004, 25, 2866–2875 Chiral CE for in vitro metabolism study 2871
Figure 4. Effect of pH on the simultaneous separation of all examined compounds. Buffer: 10 mM phosphate-10 mM ace-tate-Tris of various pH containing 10 mM HPBCD. Voltage, 25 kV; temperature, 157C. Abbreviations as in Fig. 2.
probably for chiral recognition of the analytes. Pre-viously, Vígh and co-workers [27, 28] have elaborated amodel for the enantioseparation of weak electrolytes,where the effects of both the pH and the chiral selectorconcentration were taken into account. In line withthose studies and our present observations, the com-plexation of the charged forms of our analytes are pre-ferred in the enantiorecognition, thus their separation isionoselective. Summarizing the results of the methoddevelopment, pH 3.6 separation buffer containing10 mM HPBCD is an appropriate separation medium toachieve the enantiomeric resolution of the studied com-pounds, and at the same time providing the neededchemical selectivity.
3.3 Identification of the FMO metabolites ofdeprenyl enantiomers
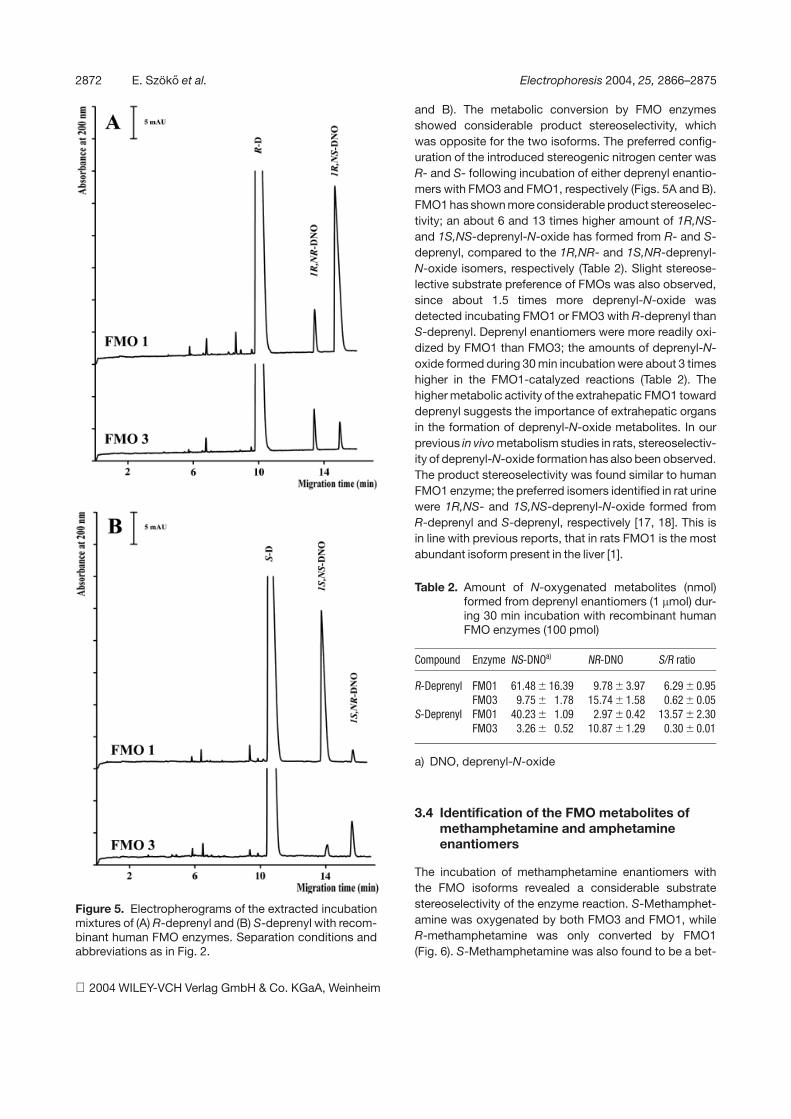
The generation of N-oxide metabolites from deprenyl isaccompanied by the creation of a new chiral center onthe quaternary nitrogen atom, and thus four stereoi-somers, i.e., two pairs of enantiomers of deprenyl-N-oxide exist. The metabolic transformation of R-deprenylcan lead to the generation of 1R,NR- and 1R,NS-depre-nyl-N-oxide, while S-deprenyl can be converted to1S,NR- and 1S,NS-deprenyl-N-oxide. R- and S-deprenylwere incubated with recombinant human FMO3 andFMO1 enzymes, and both substrate enantiomers yieldedthe corresponding diastereomeric N-oxide pairs (Figs. 5A
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
2872 E. Szöko et al. Electrophoresis 2004, 25, 2866–2875
Figure 5. Electropherograms of the extracted incubationmixtures of (A) R-deprenyl and (B) S-deprenyl with recom-binant human FMO enzymes. Separation conditions andabbreviations as in Fig. 2.
and B). The metabolic conversion by FMO enzymesshowed considerable product stereoselectivity, whichwas opposite for the two isoforms. The preferred config-uration of the introduced stereogenic nitrogen center wasR- and S- following incubation of either deprenyl enantio-mers with FMO3 and FMO1, respectively (Figs. 5A and B).FMO1 has shown more considerable product stereoselec-tivity; an about 6 and 13 times higher amount of 1R,NS-and 1S,NS-deprenyl-N-oxide has formed from R- and S-deprenyl, compared to the 1R,NR- and 1S,NR-deprenyl-N-oxide isomers, respectively (Table 2). Slight stereose-lective substrate preference of FMOs was also observed,since about 1.5 times more deprenyl-N-oxide wasdetected incubating FMO1 or FMO3 with R-deprenyl thanS-deprenyl. Deprenyl enantiomers were more readily oxi-dized by FMO1 than FMO3; the amounts of deprenyl-N-oxide formed during 30 min incubation were about 3 timeshigher in the FMO1-catalyzed reactions (Table 2). Thehigher metabolic activity of the extrahepatic FMO1 towarddeprenyl suggests the importance of extrahepatic organsin the formation of deprenyl-N-oxide metabolites. In ourprevious in vivo metabolism studies in rats, stereoselectiv-ity of deprenyl-N-oxide formation has also been observed.The product stereoselectivity was found similar to humanFMO1 enzyme; the preferred isomers identified in rat urinewere 1R,NS- and 1S,NS-deprenyl-N-oxide formed fromR-deprenyl and S-deprenyl, respectively [17, 18]. This isin line with previous reports, that in rats FMO1 is the mostabundant isoform present in the liver [1].
Table 2. Amount of N-oxygenated metabolites (nmol)formed from deprenyl enantiomers (1 mmol) dur-ing 30 min incubation with recombinant humanFMO enzymes (100 pmol)
Compound Enzyme NS-DNOa) NR-DNO S/R ratio
R-Deprenyl FMO1 61.486 16.39 9.786 3.97 6.296 0.95FMO3 9.756 1.78 15.746 1.58 0.626 0.05
S-Deprenyl FMO1 40.236 1.09 2.976 0.42 13.576 2.30FMO3 3.266 0.52 10.876 1.29 0.306 0.01
a) DNO, deprenyl-N-oxide
3.4 Identification of the FMO metabolites ofmethamphetamine and amphetamineenantiomers
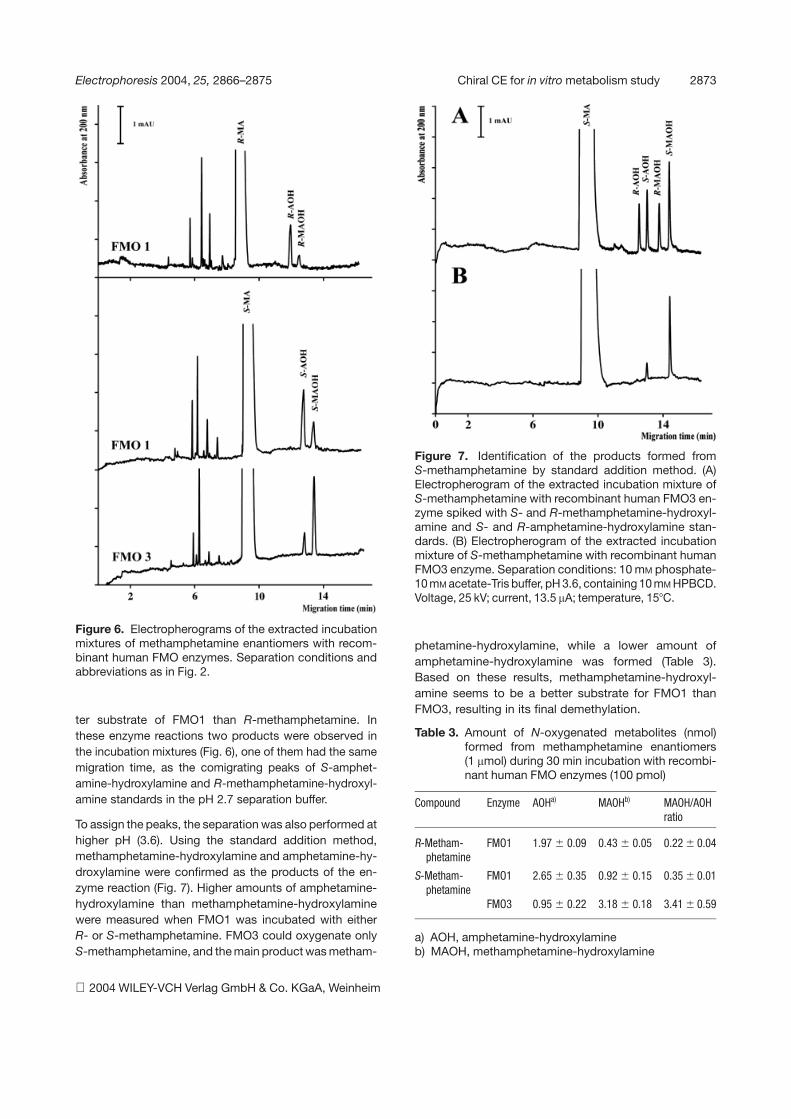
The incubation of methamphetamine enantiomers withthe FMO isoforms revealed a considerable substratestereoselectivity of the enzyme reaction. S-Methamphet-amine was oxygenated by both FMO3 and FMO1, whileR-methamphetamine was only converted by FMO1(Fig. 6). S-Methamphetamine was also found to be a bet-
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Electrophoresis 2004, 25, 2866–2875 Chiral CE for in vitro metabolism study 2873
Figure 6. Electropherograms of the extracted incubationmixtures of methamphetamine enantiomers with recom-binant human FMO enzymes. Separation conditions andabbreviations as in Fig. 2.
ter substrate of FMO1 than R-methamphetamine. Inthese enzyme reactions two products were observed inthe incubation mixtures (Fig. 6), one of them had the samemigration time, as the comigrating peaks of S-amphet-amine-hydroxylamine and R-methamphetamine-hydroxyl-amine standards in the pH 2.7 separation buffer.
To assign the peaks, the separation was also performed athigher pH (3.6). Using the standard addition method,methamphetamine-hydroxylamine and amphetamine-hy-droxylamine were confirmed as the products of the en-zyme reaction (Fig. 7). Higher amounts of amphetamine-hydroxylamine than methamphetamine-hydroxylaminewere measured when FMO1 was incubated with eitherR- or S-methamphetamine. FMO3 could oxygenate onlyS-methamphetamine, and the main product was metham-
Figure 7. Identification of the products formed fromS-methamphetamine by standard addition method. (A)Electropherogram of the extracted incubation mixture ofS-methamphetamine with recombinant human FMO3 en-zyme spiked with S- and R-methamphetamine-hydroxyl-amine and S- and R-amphetamine-hydroxylamine stan-dards. (B) Electropherogram of the extracted incubationmixture of S-methamphetamine with recombinant humanFMO3 enzyme. Separation conditions: 10 mM phosphate-10 mM acetate-Tris buffer, pH 3.6, containing 10 mM HPBCD.Voltage, 25 kV; current, 13.5 mA; temperature, 157C.
phetamine-hydroxylamine, while a lower amount ofamphetamine-hydroxylamine was formed (Table 3).Based on these results, methamphetamine-hydroxyl-amine seems to be a better substrate for FMO1 thanFMO3, resulting in its final demethylation.
Table 3. Amount of N-oxygenated metabolites (nmol)formed from methamphetamine enantiomers(1 mmol) during 30 min incubation with recombi-nant human FMO enzymes (100 pmol)
Compound Enzyme AOHa) MAOHb) MAOH/AOHratio
R-Metham-phetamine
FMO1 1.97 6 0.09 0.43 6 0.05 0.22 6 0.04
S-Metham-phetamine
FMO1 2.65 6 0.35 0.92 6 0.15 0.35 6 0.01
FMO3 0.95 6 0.22 3.18 6 0.18 3.41 6 0.59
a) AOH, amphetamine-hydroxylamineb) MAOH, methamphetamine-hydroxylamine
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim

2874 E. Szöko et al. Electrophoresis 2004, 25, 2866–2875
Amphetamine-hydroxylamine can be formed frommethamphetamine-hydroxylamine by a multistep reac-tion; the first oxygenation reaction is catalyzed by FMO.This finding is in accordance with previous results of Babaet al. [13]. They have observed, that the demethylation ofmethamphetamine-hydroxylamine to amphetamine-hy-droxylamine in guinea pig microsomes was catalyzedmainly by FMO, based on experiments using variousFMO and CYP450enzyme inhibitors.The purified FMO en-zyme also showed high catalytic activity. Demethylation ofmethamphetamine-hydroxylamine to amphetamine-hy-droxylamine was suggested to proceed via the formationof a nitrone intermediate by FMO (Fig. 1). This intermediatecan be converted to amphetamine-hydroxylamine by theloss of formaldehyde. In another study by Cashman andco-workers [14], similar conversion of methampheta-mine-hydroxylamine to an isomeric nitrone by recombi-nant human FMO3 has been suggested. Further transfor-mation of this intermediate to the ultimate product phenyl-propanone was identified. However, the conversion ofmethamphetamine-hydroxylamine to amphetamine-hy-droxylamine has not been reported in their study. Basedon previous results and our present experiments, it can bespeculated, that methamphetamine-hydroxylamine canbe converted through a nitrone intermediate to both am-phetamine-hydroxylamine and phenylpropanone (Fig. 1).The product formation preferentially catalyzed by FMOisoforms may be different. The nitrone isomers are pro-duced by water loss at different sites of the molecule, andFMO isoforms might preferentially catalyze one or theother way of this step. The nitrone formed might determinethe ultimate product, namely amphetamine-hydroxyl-amine or phenylpropanone. This may also explain the dif-ferent ratio of amphetamine-hydroxylamine and metham-phetamine-hydroxylamine production from methamphe-tamine by FMO1 and FMO3, which was found in ourexperiments. (The formation of phenylpropanone was notinvestigated.) In other studies, a further metabolic trans-formation of hydroxylamines, namely the reduction ofamphetamine-hydroxylamine to amphetamine, and meth-amphetamine-hydroxylamine to methamphetamine by acytochrome P (CYP)-dependent metabolic pathway, havealso been reported [20, 21].
When amphetamine enantiomers were incubated withFMO3 and FMO1, respectively, no products of the enzymereactions were detected. Contrary to our findings, the con-version of amphetamine to amphetamine-hydroxylamineand its further stereoselective transformation to phenyl-propanone oxime by recombinant human FMO3 enzymehas been reported [14]. Two major polymorphic forms ofhuman FMO3 were studied, and the results suggestedpreferential N-oxygenation of amphetamine by only oneof the two enzymes, differing in their sequence by one
amino acid. The rate of amphetamine conversion by hu-man FMO3 isoforms was found considerably lower thanthat of phenylethylamine or tyramine, thus the high sensi-tivity of human FMO3 to substrate steric features was sug-gested [14]. Human FMO1 is known to N-oxygenate pri-mary amines at an exceedingly low level, if at all [1].
Comparing the results of the recombinant human FMOisoenzymes catalyzed reactions of deprenyl, metham-phetamine, and amphetamine, FMO1 was found morecapable of oxygenating the tertiary and secondaryamines (deprenyl and methamphetamine) than FMO3.The primary amine amphetamine was not converted tohydroxylamine by either of the two human FMO isoforms.The tertiary amine deprenyl was a better substrate forboth FMO3 and FMO1 than the secondary aminemethamphetamine.
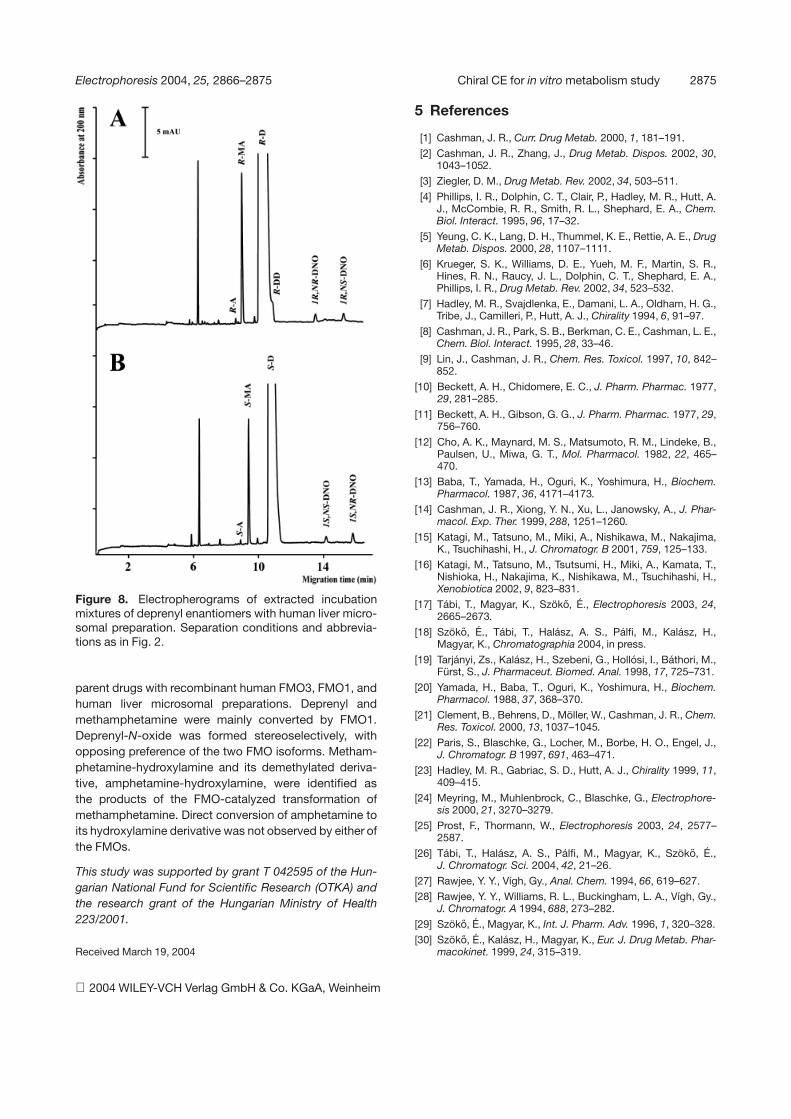
3.5 Identification of the metabolites of deprenylenantiomers incubated with human livermicrosomes
Deprenyl enantiomers were incubated with human livermicrosomal preparations. In the case of both enantio-mers, the dealkylated and the N-oxide metabolites couldbe identified in the incubation mixture. Although the quan-tification of the dealkylated derivatives was not the aim ofthis study, we could observe that methamphetamine wasthe main metabolite for both enantiomers, amphetaminewas formed in a much lower concentration, and a con-siderable amount of desmethyldeprenyl could also bedetected (Fig. 8). The formation of these metabolites wasstereospecific with respect to the configuration of thea-carbon, in accordance with our previous findings [29,30]. Similar quantities of N-oxides were detected formedfrom both deprenyl enantiomers. No stereoselectivity inthe creation of the new chiral center of the N-oxides wasobserved, although stereoselectivity of the recombinanthuman FMO3 catalyzed N-oxygenation has been foundin our experiments (3.3- and 1.6-fold excess of 1S,NR-and 1R,NR-deprenyl-N-oxide compared to 1S,NS and1R,NS isomers, Table 2). The lack of stereoselective for-mation of deprenyl-N-oxides in the microsome prepara-tion may indicate that beside FMO3, CYP enzymes mayalso contribute to the N-oxygenation reaction.
4 Concluding remarks
A chiral CE method, using a low-pH separation buffercontaining HPBCD as chiral selector, has been developedand used for the quantification of deprenyl-N-oxide,methamphetamine-hydroxylamine, and amphetamine-hydroxylamine enantiomers in in vitro incubates of the
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Electrophoresis 2004, 25, 2866–2875 Chiral CE for in vitro metabolism study 2875
Figure 8. Electropherograms of extracted incubationmixtures of deprenyl enantiomers with human liver micro-somal preparation. Separation conditions and abbrevia-tions as in Fig. 2.
parent drugs with recombinant human FMO3, FMO1, andhuman liver microsomal preparations. Deprenyl andmethamphetamine were mainly converted by FMO1.Deprenyl-N-oxide was formed stereoselectively, withopposing preference of the two FMO isoforms. Metham-phetamine-hydroxylamine and its demethylated deriva-tive, amphetamine-hydroxylamine, were identified asthe products of the FMO-catalyzed transformation ofmethamphetamine. Direct conversion of amphetamine toits hydroxylamine derivative was not observed by either ofthe FMOs.
This study was supported by grant T 042595 of the Hun-garian National Fund for Scientific Research (OTKA) andthe research grant of the Hungarian Ministry of Health223/2001.
Received March 19, 2004
5 References
[1] Cashman, J. R., Curr. Drug Metab. 2000, 1, 181–191.
[2] Cashman, J. R., Zhang, J., Drug Metab. Dispos. 2002, 30,1043–1052.
[3] Ziegler, D. M., Drug Metab. Rev. 2002, 34, 503–511.
[4] Phillips, I. R., Dolphin, C. T., Clair, P., Hadley, M. R., Hutt, A.J., McCombie, R. R., Smith, R. L., Shephard, E. A., Chem.Biol. Interact. 1995, 96, 17–32.
[5] Yeung, C. K., Lang, D. H., Thummel, K. E., Rettie, A. E., DrugMetab. Dispos. 2000, 28, 1107–1111.
[6] Krueger, S. K., Williams, D. E., Yueh, M. F., Martin, S. R.,Hines, R. N., Raucy, J. L., Dolphin, C. T., Shephard, E. A.,Phillips, I. R., Drug Metab. Rev. 2002, 34, 523–532.
[7] Hadley, M. R., Svajdlenka, E., Damani, L. A., Oldham, H. G.,Tribe, J., Camilleri, P., Hutt, A. J., Chirality 1994, 6, 91–97.
[8] Cashman, J. R., Park, S. B., Berkman, C. E., Cashman, L. E.,Chem. Biol. Interact. 1995, 28, 33–46.
[9] Lin, J., Cashman, J. R., Chem. Res. Toxicol. 1997, 10, 842–852.
[10] Beckett, A. H., Chidomere, E. C., J. Pharm. Pharmac. 1977,29, 281–285.
[11] Beckett, A. H., Gibson, G. G., J. Pharm. Pharmac. 1977, 29,756–760.
[12] Cho, A. K., Maynard, M. S., Matsumoto, R. M., Lindeke, B.,Paulsen, U., Miwa, G. T., Mol. Pharmacol. 1982, 22, 465–470.
[13] Baba, T., Yamada, H., Oguri, K., Yoshimura, H., Biochem.Pharmacol. 1987, 36, 4171–4173.
[14] Cashman, J. R., Xiong, Y. N., Xu, L., Janowsky, A., J. Phar-macol. Exp. Ther. 1999, 288, 1251–1260.
[15] Katagi, M., Tatsuno, M., Miki, A., Nishikawa, M., Nakajima,K., Tsuchihashi, H., J. Chromatogr. B 2001, 759, 125–133.
[16] Katagi, M., Tatsuno, M., Tsutsumi, H., Miki, A., Kamata, T.,Nishioka, H., Nakajima, K., Nishikawa, M., Tsuchihashi, H.,Xenobiotica 2002, 9, 823–831.
[17] Tábi, T., Magyar, K., Szöko, É., Electrophoresis 2003, 24,2665–2673.
[18] Szöko, É., Tábi, T., Halász, A. S., Pálfi, M., Kalász, H.,Magyar, K., Chromatographia 2004, in press.
[19] Tarjányi, Zs., Kalász, H., Szebeni, G., Hollósi, I., Báthori, M.,Fürst, S., J. Pharmaceut. Biomed. Anal. 1998, 17, 725–731.
[20] Yamada, H., Baba, T., Oguri, K., Yoshimura, H., Biochem.Pharmacol. 1988, 37, 368–370.
[21] Clement, B., Behrens, D., Möller, W., Cashman, J. R., Chem.Res. Toxicol. 2000, 13, 1037–1045.
[22] Paris, S., Blaschke, G., Locher, M., Borbe, H. O., Engel, J.,J. Chromatogr. B 1997, 691, 463–471.
[23] Hadley, M. R., Gabriac, S. D., Hutt, A. J., Chirality 1999, 11,409–415.
[24] Meyring, M., Muhlenbrock, C., Blaschke, G., Electrophore-sis 2000, 21, 3270–3279.
[25] Prost, F., Thormann, W., Electrophoresis 2003, 24, 2577–2587.
[26] Tábi, T., Halász, A. S., Pálfi, M., Magyar, K., Szöko, É.,J. Chromatogr. Sci. 2004, 42, 21–26.
[27] Rawjee, Y. Y., Vígh, Gy., Anal. Chem. 1994, 66, 619–627.
[28] Rawjee, Y. Y., Williams, R. L., Buckingham, L. A., Vígh, Gy.,J. Chromatogr. A 1994, 688, 273–282.
[29] Szöko, É., Magyar, K., Int. J. Pharm. Adv. 1996, 1, 320–328.
[30] Szöko, É., Kalász, H., Magyar, K., Eur. J. Drug Metab. Phar-macokinet. 1999, 24, 315–319.
2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim




















