
Asporin and the Mineralization Process in Fluoride-Treated Rats
27
1 Original Article Asporin and the Mineralization Process in Fluoride-Treated Rats † Sophia Houari 1, 2, 3, 4 (DDS, Ph.D), Tilmann Wurtz 1, 2, 3, 4 (Ph.D), Didier Ferbus 1, 2, 3, 4 (Ph.D), Danielle Chateau 1, 2, 3, 5 (Ph.D), Arnaud Dessombz 1, 2, 3, 4 (Ph.D), Ariane Berdal 1, 2, 3, 4 (DDS, Ph.D), Sylvie Babajko* 1, 2, 3, 4 (Ph.D) 1. Centre de Recherche des Cordeliers, INSERM UMRS 872, Team 5, Laboratory of Molecular Oral Pathophysiology, Paris, F-75006 France 2. Université Paris-Descartes, Paris, F-75006 France 3. Université Pierre et Marie Curie-Paris, Paris, F-75006 France 4. Université Paris- Diderot, UFR d’Odontologie, F-75006, Paris, France 5. Centre de Recherche des Cordeliers, INSERM UMRS 872, Team 9, Pathophysiology of sugars transport and detection, Paris, F-75006 France * Address correspondence to: Sylvie Babajko. Centre de Recherche des Cordeliers, INSERM UMRS872 Team 5, Laboratory of Molecular Oral Pathophysiology, 15/21 rue de l'école de Médecine, 75006 Paris, France. E-mail: [email protected] Grants: This work was funded by the University Paris-Diderot, the French National Institute of Health and Medical Research (INSERM). SH is a fellow of DIM-SeNT Ile-de-France (Domaine d’Intérêt Majeur en Santé, Environnement et Toxicologie). DIM-SeNT † This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: [10.1002/jbmr.2153] Additional Supporting Information may be found in the online version of this article. Initial Date Submitted August 1, 2013; Date Revision Submitted November 8, 2013; Date Final Disposition Set November 21, 2013 Journal of Bone and Mineral Research © 2013 American Society for Bone and Mineral Research DOI 10.1002/jbmr.2153
-
Upload
univ-paris-diderot -
Category
Documents
-
view
3 -
download
0
Transcript of Asporin and the Mineralization Process in Fluoride-Treated Rats

1
Original Article
Asporin and the Mineralization Process in Fluoride-Treated Rats†
Sophia Houari1, 2, 3, 4 (DDS, Ph.D), Tilmann Wurtz1, 2, 3, 4 (Ph.D), Didier Ferbus1, 2, 3, 4 (Ph.D), Danielle
Chateau 1, 2, 3, 5 (Ph.D), Arnaud Dessombz1, 2, 3, 4 (Ph.D), Ariane Berdal1, 2, 3, 4 (DDS, Ph.D), Sylvie
Babajko*1, 2, 3, 4 (Ph.D)
1. Centre de Recherche des Cordeliers, INSERM UMRS 872, Team 5, Laboratory of Molecular Oral
Pathophysiology, Paris, F-75006 France
2. Université Paris-Descartes, Paris, F-75006 France
3. Université Pierre et Marie Curie-Paris, Paris, F-75006 France
4. Université Paris- Diderot, UFR d’Odontologie, F-75006, Paris, France
5. Centre de Recherche des Cordeliers, INSERM UMRS 872, Team 9, Pathophysiology of sugars transport
and detection, Paris, F-75006 France
* Address correspondence to: Sylvie Babajko. Centre de Recherche des Cordeliers, INSERM UMRS872
Team 5, Laboratory of Molecular Oral Pathophysiology, 15/21 rue de l'école de Médecine, 75006 Paris, France.
E-mail: [email protected]
Grants: This work was funded by the University Paris-Diderot, the French National Institute of Health and
Medical Research (INSERM). SH is a fellow of DIM-SeNT Ile-de-France (Domaine d’Intérêt Majeur en Santé,
Environnement et Toxicologie).
DIM-SeNT
†This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: [10.1002/jbmr.2153] Additional Supporting Information may be found in the online version of this article. Initial Date Submitted August 1, 2013; Date Revision Submitted November 8, 2013; Date Final Disposition Set November 21, 2013
Journal of Bone and Mineral Research © 2013 American Society for Bone and Mineral Research
DOI 10.1002/jbmr.2153

2
Disclosure Page
We acknowledge that we have read and understand the Disclosure Guide of the Journal of Bone and Mineral
Research.
All authors state that they have no conflicts of interest.
All authors state that there are no restrictions to full access for all authors to all raw data, statistical analyses,
and materials used in the study.
All authors state that they have no conflicts of interes
Relationship
t
Yes No If yes, identify
organization or
other entity
Board membership: Have you served as a paid board
member with any entity related to the work reported in the
manuscript?
X
Consulting: Have you received
consulting fees for any entity related to the work reported in
the manuscript?
X
Employment: Have you been
employed by any entity related to the work reported in the
manuscript?
X
Expert testimony: Have you served as an expert witness or
advisor in legal proceedings for any entity related to the work
reported in the manuscript?
X
Honoraria/Royalties: Have you
received honoraria or royalties
connected to any entity related to the work reported in the
manuscript?
X
Grants and Contracts: Have you
received grants for research from any
entity related to the work reported in the manuscript?
X
Spokesperson/Speakers Bureau:
Have you served as a paid
spokesperson for any entity related to the work reported in
the manuscript?
X

3
Abstract
Microarray analysis of odontoblastic cells treated with sodium fluoride has identified the asporin gene as a
fluoride target. Asporin is a member of the small leucine-rich repeat proteoglycan/protein (SLRP) family which
is believed to be important in the mineralization process. In this study, asporin expression and distribution were
investigated by systematic analysis of dentin and enamel, with and without fluoride treatment. Specific
attention was focused on a major difference between the two mineralized tissues: the presence of a collagenous
scaffold in dentin, and its absence in enamel. Normal and fluorotic, continually growing incisors from Wistar
rats treated with 2.5 to 7.5 mM sodium fluoride (NaF) were studied by immunochemistry, in situ hybridization,
western blotting and RT-qPCR. Asporin was continuously expressed in odontoblasts throughout dentin
formation as expected. Asporin was also found, for the first time, in dental epithelial cells, particularly in
maturation-stage ameloblasts. NaF decreased asporin expression in odontoblasts whereas it enhanced it in
ameloblasts both in vivo and in vitro. The inverse response in the two cell types suggests that the effector,
fluoride, is a trigger that elicits a cell-type specific reaction. Confocal and ultrastructural immunohistochemistry
evidenced an association between asporin and the type 1 collagen in the pericellular non-mineralized
compartments of both bone and dentin. In addition, transmission electron microscopy revealed asporin in the
microenvironment of all cells observed. Thus, asporin is produced by collagen- and non collagen-matrix-
forming cells but may have different effects on the mineralization process. A model is proposed that predicts
impaired mineral formation associated with the deficiency and excess of asporin.
Key words: asporin, mineralization, enamel, dentin, fluorosis

4
Introduction Asporin mRNA has been identified as a gene transcript that is repressed in fluoride-treated odontoblastic
cells.(1) Asporin, also known as periodontal ligament-associated protein 1 (PLAP1), is an extracellular matrix
protein that belongs to the class I small leucine-rich repeat proteoglycan/protein (SLRP) family that also
includes decorin and biglycan.(2) Asporin lacks consensus sequences for glycosaminoglycan attachment in its
N-terminus part, and is not considered to be a true proteoglycan.(2) Some SLRPs (asporin, decorin,
fibromodulin and lumican) can be associated with collagen fibrils and may be regulatory elements in the
formation of collagen-based matrices.(3)
Asporin contains a unique aspartic acid (D) repeat at its N-terminus that interacts with calcium. This D-repeat
is responsible for a major functional difference between asporin and the homologous decorin and biglycan.(4)
Polyaspartate sequences in proteins can regulate hydroxyapatite deposition and oxalate crystallisation in vitro.(5-
7) The asporin sequence also contains a high-affinity collagen-binding domain in the C-terminal part.(4) It has
been suggested that these two domains may work in concert to trigger biomineralization of collagen.(4) Asporin
has also been detected in globular calcification regions at the junction between predentin and dentin, associated
with initial calcium deposition.(8) In vitro knock-down experiments suggest that it has an important role in
predentin mineralization.(8) Although it is not detected in fully mineralized dentin, its presence is required for
odontoblastic differentiation and dentin mineralization. However, asporin has also been identified as a negative
regulator of biomineralization in the periodontal ligament (PDL)(9,10) and articular cartilage.(11) There is thus no
simple relation between the presence of asporin and particular events in the mineralization process.
The tooth is a suitable model for studying the role of asporin in biomineralization because dentin and enamel
develop in parallel and both incorporate mineral; dentin contains a collagen scaffold whereas enamel does not.
In dentin, as well as in bone and cementum, apatite crystals form directly on collagen fibrils which are actively
involved in the mineral formation process. There are no such collagen fibrils in enamel, and the precise site of
crystal initiation is still unknown.(12) The enamel scaffold is provided by enamel matrix proteins, including
amelogenins and ameloblastin, secreted by ameloblasts during the secretion stage.(12-14) Although enamel
mineralization begins during the secretion stage, it essentially takes place during the ensuing maturation phase.
The continually growing rodent incisor provides a good model in which all stages of ameloblastic
differentiation and enamel mineralization are aligned in a continuous sequence which remains similar

5
throughout adulthood.(15) It is consequently a convenient system for studying the formation of dental tissues
independently of age. Moreover, dentin and enamel formation are linked and can be studied in parallel.
The asporin expression pattern was investigated in dentin and enamel, both mineral-containing tissues, during
their formation in the rat incisor. It was also studied in the context of pathophysiological alterations of enamel
and dentin mineralization caused by fluoride treatment. Fluoride is widely used because, at low concentrations
(<1mg/day for human according to Dean’s score) it protects against caries by generating fluoridated apatite and
inhibiting bacterial enolase activity;(16,17) however, long-term chronic exposure to fluoride (>2mg/day) can also
cause enamel hypomineralization(18-20) and impact bone mineralization (10-40 mg/day).(21, 22) At higher doses
(>125mg/day), fluoride has various toxic cellular effects in many organs, the severity of which is directly
related to dose and duration of exposure.(18,23) The mechanisms of action of fluoride have not been clearly
established, but seem to involve both intracellular and extracellular events(18,24-27) and to depend on the genetic
background.(21)
Materials and methods
Animals and biological samples
Six week-old Wistar Han rats were purchased from Charles River (France). All animals were maintained in
accordance with the French Ministry of Agriculture guidelines for care and use of laboratory animals (A-75-06-
12).
Forty Wistar rats were allocated to three experimental groups and one control group: sodium fluoride (NaF)
was included in the drinking water of each experimental group for 6 weeks at different concentrations (2.5, 5.0
and 7.5 mM NaF; 50 to 150 ppm). The fourth control group of rats was not treated with NaF. To obtain similar
effects in rodents and in humans much higher levels of fluoride in the drinking water (10–20 times) are required
for rodents:(20) the NaF concentrations used here for rats thus correspond to those measured in some drinking
waters causing dental fluorosis in humans.
Five animals from each group were randomly selected for histological analysis, anesthetized by isofurane
inhalation and perfused with 4% paraformaldehyde (PFA) (Sigma-Aldrich, St. Louis, MO) in phosphate-
buffered saline (PBS; pH 7.4). The remaining rats were immediately killed and the incisors were dissected out

6
of the alveolar bone. Epithelial and mesenchymal tissues were microdissected and RNA extracted as described
previously.(15,28) Cellular proteins were also extracted concomitantly.
Cell culture
HAT-7 cells derived from a dental epithelial cell line originating from the apical bud of a rat incisor(29) were
grown in Dulbecco’s modified Eagle’s medium/F-12 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal
bovine serum (FBS) and penicillin (100 units/ml)/streptomycin (100 μg/ml).
705IH8 cells are mesenchymal cells of odontoblastic origin isolated from the dental pulp of transgenic mice
expressing the SV40 T antigen under the control of the E1A adenoviral promoter.(30) They were harvested in
Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% FBS and penicillin
(100 units/ml)/streptomycin (100 μg/ml).
Forty-eight hours after addition of 0.5 mM NaF (Sigma-Aldrich, St. Louis, MO) to the culture medium, cells
were collected for RNA and protein extractions.
RNA extraction and RT-qPCR analysis
Total RNAs were extracted with Tri-Reagent (Euromedex, Paris, France) according to the manufacturer’s
instructions. RNA concentrations were determined by spectrophotometry at 260 nm (NanoDrop 1000;
ThermoScientific, Villebon-sur-Yvette, France). Reverse transcription was carried out with 1µg total RNA for
45 minutes at 42ºC, using a primer mix (oligodT random primers) according to the manufacturer’s instructions
(Superscript II® - Invitrogen, Carlsbad, CA). Real-time quantitative PCR was performed using the Opticon
Monitor device (Bio-Rad Laboratories, Hercules, CA). Each PCR was repeated in triplicate and the results were
normalized against those for rs15 and gapdh. Details of the primers and the corresponding amplicon sizes are
presented in Table 1. RNA quantities were calculated by the method of standard curves; similar results were
obtained when ΔΔCt method was used.
Western Blotting
Total cellular proteins were extracted using Tri-Reagent (Euromedex, France) according to the manufacturer’s
instructions. Proteins were suspended in SDS-containing buffer by boiling in the presence of DTT. Protein
concentrations were measured using the Pierce protein assay according to the manufacturer’s instructions
(Pierce®660nm protein assay 22660, ThermoScientific, Villebon-sur-Yvette, France). Forty micrograms of
extracted proteins were subjected to 12% SDS-PAGE and electrotransferred onto PVDF membranes

7
(ThermoScientific, Villebon-sur-Yvette, France). Loading and electro-transfer efficiency were checked by
staining membranes for total proteins with Ponceau red. Membranes were first blocked with 5% milk for 2
hours, then incubated at 37° C for 120 minutes with a rabbit polyclonal anti-asporin antibody (abcam ab58741)
diluted 1:400 and subsequently for 1 hour with a goat polyclonal anti-rabbit IgG antibody coupled to
horseradish peroxidase (Sigma-Aldrich, St Louis, MO) diluted 1:2000.
Immuno-reactivity was detected by chemiluminescence (Luminata Crescendo, Millipore, MA) and visualized
by charge-couple device (CCD) imaging system (Fuji, Rochester, NY) or with a bioimager (ImageQuant LAS
4000; Uppsala, Sweden). The membrane was stripped and incubated again with a mouse monoclonal antibody
specific for the ubiquitously expressed 38 kD gapdh protein (Millipore, Temecula, CA). All experiments were
performed at least three times.
In situ hybridization (ISH) assays
Rat hemi-mandibles were fixed overnight in 4% PFA (Sigma-Aldrich, St. Louis, MO), and decalcified in
4.13% EDTA (pH 7.4)/0.2% PFA for three months. The decalcification solution was changed twice a week.
After decalcification, samples were immersed in 30% sucrose for 3 hours and slowly frozen at −80 ◦C. The
hemi-mandibles were embedded in Freeze Gel (Labonord SASA, Templemars, France), and sectioned
longitudinally such that all stages of incisor ameloblast differentiation were represented. Cryosections of 10 µm
were subjected to ISH as previously described.(31,32) Briefly, sections on “superfrost plus” slides were immersed
in ethanol for 5 minutes, dried for 2 hours at 52°C in air, re-hydrated, treated with 20 µg/ml proteinase K for 30
minutes at 37°C and hydrochloric acid, and hybridized to digoxigenin (DIG)-labeled RNA antisense probes.
The asporin probe was obtained by inserting RT-PCR fragments (using the same primers as for RTqPCR
experiments, table 1) into the pGEM-T vector.(1) After washing and RNase treatment,(31) the presence of DIG
probe was revealed using an anti-DIG antibody (Roche Diagnostics, Indianapolis, IN), coupled to alkaline
phosphatase (Vecta-Blue,Vector Laboratories, Inc. Burlingame, CA). Sections hybridized with a probe for
tachykinin mRNA, not detected in dental tissues, served as negative controls. All probes were subjected to gel
electrophoresis followed by northern blotting and detection with anti-DIG antibody, to confirm that probe
synthesis had yielded the specific product of the expected size.
Immunohistochemistry assays
Rat hemi-mandibles were post-fixed with 4% PFA (Sigma-Aldrich, St. Louis, MO) overnight at 4°C and

8
decalcified in 4.13% EDTA (pH 7.4) / 0.2% PFA for three months. The decalcification solution was changed
twice a week. After extensive washing in PBS, the samples were dehydrated in increasing concentrations of
ethanol and Safe solv (LaboNord, Templemars, France) and were then embedded in paraffin (Paraplast Plus,
Sigma-Aldrich, St. Louis, MO). Serial 8 µm-thick sagittal sections were cut using a microtome (RM 2145;
Leica, Rueil-Malmaison, France), deparaffinized and rehydrated. The sections were immersed in 0.3%
hydrogen peroxide for 15 minutes at room temperature, and immersed twice in 10 mM Na citrate, then
subjected to microwave treatment at 90°C for 20 minutes, and rinsed in PBS. The sections were blocked with
10% normal goat serum for 30 minutes at room temperature, and incubated with a rabbit polyclonal anti-
asporin antibody (ab58741, abcam) diluted 1:400 in 4% normal goat serum for 90 minutes at room temperature.
The slides were then incubated with a goat polyclonal anti-rabbit IgG antibody coupled to horseradish
peroxidase (Sigma-Aldrich, St. Louis, MO) diluted 1:150 for 60 minutes at room temperature.
Immunoreactivity was visualized using a NovaRED Peroxidase Substrate Kit (Vector laboratories).
Similar procedures were used for asporin and type 1 collagen double-labeling experiments, by sequentially
applying a rabbit polyclonal anti-asporin antibody (ab58741, abcam) diluted 1:400, a donkey anti-rabbit IgG
antibody coupled to Alexa Fluor 488 (A-11072; Life Technologies, Carlsbad, CA) (1:500), a mouse
monoclonal anti-collagen 1A1 antibody (ab6308, abcam) diluted (1:100), and a donkey anti-mouse IgG
antibody coupled to Alexa Fluor 594 (life technologies Carlsbad, CA) (1:500). After rinsing with PBS, sections
were immersed in DAPI (Sigma-Aldrich, St. Louis, MO) for one minute and finally mounted with aquamount
(Lerner Laboratories, Pittsburgh, PA).
Transmission Electron Microscopy (TEM)
Combined electron microscopy and immunocytochemical analyses of rat incisors were carried out on frontal
ultrathin sections. Samples were fixed in 4% PFA (Sigma-Aldrich), decalcified, dehydrated by standard
procedures, and embedded in LR White resin (Sigma-Aldrich).
Ultrathin sections (70 nm thick) were incubated with a rabbit polyclonal anti-asporin antibody (ab58741,
abcam) diluted 1:10, followed by protein A coupled to 15 nm gold beads (gift from Dr. Raposo, Institut Curie,
Paris, France) (diluted 1:60). For double labeling, this step was followed by incubation with a mouse
monoclonal anti-type I collagen antibody (ab6308, abcam) diluted 1:50 and a donkey anti-mouse antibody

9
(Jackson ImmunoResearch Laboratories, West Grove, PA) coupled to 6 nm gold beads (diluted 1:60). Sections
were examined using a Philips CM 12 electron microscope.
Statistical analysis
In vivo results are reported as means + standard deviation (SD). Measurements of asporin mRNA and protein
concentrations for samples from various groups after NaF treatment were compared to those for controls by one
way ANOVA (ANOVA) followed by Bonferroni’s post-test. For in vitro experiments, data from three series of
independent experiments are presented as means + SD and the two-tailed non-parametric Mann-Whitney test
was used for comparisons. All data were analyzed with GraphPad Prism software version 4.0. Values were
considered significantly different (*) when p ≤ 0.05 , and highly significantly different (**) when p≤0.01 or
(***) p≤0.001.
Results
Fluorosis in rat incisors
Four groups of ten rats each were administered 0 (control), 2.5, 5.0 and 7.5 mM NaF (0 to 150 ppm) orally (in
drinking water) for 6 weeks. This period was chosen to ensure the complete impregnation of the growing
incisor with NaF. These teeth grow continuously such that none of the tooth is ever more than 40-50 days old.
Control incisor enamel was homogeneously colored yellow to orange (Fig. 1). Consistent with previously
described fluorosis models,(33,34) treated rats presented discolored enamel incisors. At 2.5 mM NaF treatment
(50 ppm), there were faint white bands that were more pronounced in the 5 mM NaF (100 ppm) group. At 7.5
mM NaF (150 ppm), the orange coloring completely disappeared and the entire incisor was white and opaque.
NaF modulates asporin expression in rat dental tissues
Immunohistochemical staining for asporin in control rat sections resulted in a specific strong signal in all
odontoblasts, especially in the apical part of the cells (Fig. 2B). NaF treatment decreased the asporin signal in a
dose-dependent manner (Fig. 2B). In the ameloblast layer, asporin appeared mostly during the maturation stage
with the signal being very weak in the secretion stage; again, NaF treatment increased the signal strength in a
dose-dependent manner (Fig. 2A).
Western blotting of proteins extracted from microdissected mesenchymal and epithelial tissues confirmed the
presence of asporin in dental epithelium and the dose-dependent response of asporin levels to NaF observed by

10
immunohistochemistry (Fig. 2C and 2D). Asporin protein level in the epithelium was 2.1-fold higher in the 7.5
mM NaF group than in controls (SD ± 0.2, p≤0.05) whereas in the mesenchyme of the same group of rats,
asporin was 2.1-fold less abundant than in controls (SD ± 0.2, p≤ 0.01) (Fig. 2E).
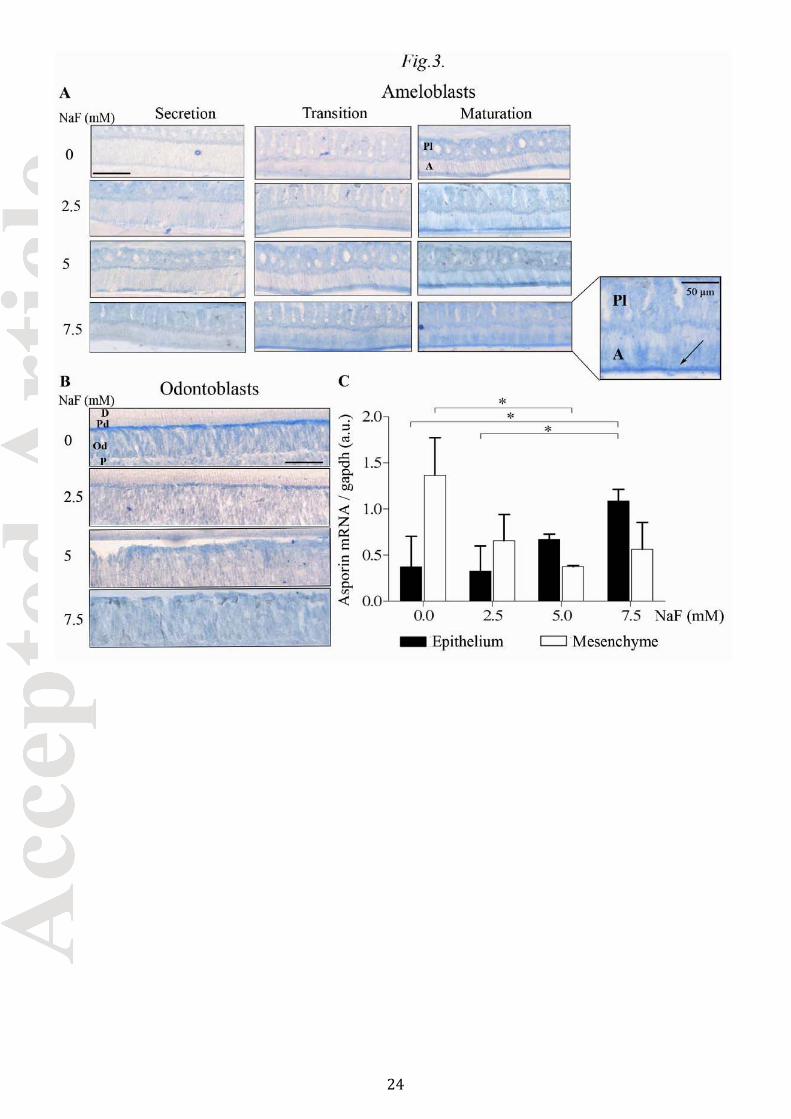
The results of ISH and RT-qPCR for asporin mRNA were consistent with the findings for the protein (Fig. 3)
and suggested a fluoride effect on asporin gene expression rather than a post-translational effect. Relative to
controls, NaF decreased the asporin mRNA level in odontoblasts by 1.7 fold at 2.5 mM and 3.1 fold at 7.5 mM
NaF (p≤0.05, Fig. 3B and 3C). Conversely, in maturation stage ameloblasts, the abundance of asporin mRNA
was increased by NaF (Fig. 3A): RT-qPCR quantification showed a significant 3.0 fold induction in rats treated
with 7.5 mM NaF (p≤0.05, Fig.3C).
Asporin in cultured cell lines
Cells of the ameloblastic line HAT-7 and of the odontoblastic line 705IH8 were treated with 0.5 mM NaF for
48 hours. Preliminary experiments showed toxic effects of 1mM NaF on HAT-7 cells whereas 0.1 mM NaF
provoked only a slight response. The full and optimal NaF concentration for both cell lines was established as
0.5 mM.
Both cell lines expressed asporin at the protein and mRNA levels (Fig. 4). As in vivo, NaF significantly
decreased asporin protein (p≤0.05) and mRNA (p≤0.001) levels in mesenchymal 705IH8 cells (Fig. 4B, C, D).
By contrast, asporin expression in epithelial HAT-7 cells was significantly increased by 0.5 mM NaF (p≤0.05)
(Fig. 4A, C, D).
Asporin localization in rat dental tissues
The distribution of asporin in odontoblasts, predentin and dentin was analyzed by confocal microscopy.
Asporin was present in odontoblasts and accumulated in predentin, the non-mineralized dentin (Fig. 5A). The
signal for type I collagen was detected in both dentin and predentin (Fig. 5B). Superposition of the two signals
revealed co-localization (yellow) of type I collagen (red) and asporin (green) in predentin and predominantly at
the edges of odontoblast processes (Fig. 5D).
Transmission electron microscopy (TEM) analysis clearly evidenced large gold grains (15 nm-diameter)
specific for asporin, and small dots (6 nm-diameter representing type I collagen in the odontoblastic pericellular
space in the apical part of the cells (Fig. 5E). The asporin signal was intertwined with the collagen signal,

11
strongly suggesting an interaction between the two proteins. The small gold beads coupled to anti-collagen
antibody were also observed in the dentinal matrix, as expected.
Careful and systematic analysis of the distribution of asporin in the dental mesenchyme showed the presence of
the protein in the pericellular environment, essentially around the apical part of odontoblastic cells and, to a
lesser extent, along cellular processes (Fig. 6A). Similarly, asporin accumulated around the osteocytes of rat
mandibular bone (Fig. 6C) and also around mature ameloblasts in NaF-treated rat incisors (Fig. 6B). Asporin
was also detected in enamel space (ES), especially in fluorotic enamel (Fig. 6B).
Discussion
Asporin expression and distribution were investigated in the context of extracellular matrix formation and
mineralization. Our principal findings are:
1. Asporin is not only present in tissues with a collagen scaffold, such as dentin, but also in enamel, a
mineralized tissue without collagen
2. Fluoride increases asporin expression in ameloblasts, but reduces it in odontoblasts.
3. In mineralized tissues, asporin accumulates in the non-mineralized pericellular space.
NaF modulates asporin expression oppositely in different cell types
Asporin expression has been found in diverse tissues synthesizing collagen including PDL, dentin, bone and
cartilage.(8,9,35-38) The present study demonstrates, for the first time, the presence of asporin in dental epithelial
cells. This was evidenced in vivo by immunostaining and ISH, by biochemical analysis of microdissected
tissues, and in vitro, in the rat HAT-7 ameloblastic cell line. In control conditions, this expression was weaker
than that in odontoblasts, but it was induced by fluoride especially in the ameloblasts during the maturation
stage. Most changes caused by fluoride are related to cell/matrix/mineral interactions, especially during the
maturation stage of amelogenesis,(20,24,25,39) precisely when asporin is most strongly expressed. Apatite mineral
may locally enhance fluoride concentration due to inorganic fluoride-mineral interactions.
Asporin expression was differently regulated depending on the cell type. Fluoride decreased asporin expression
in odontoblasts, and enhanced it in dental epithelial cells. As already demonstrated for Mmp20, fluoride does
not have only inhibitory effects on enamel gene expression.(40) The opposite responses in the two cell types
suggests that the effector, fluoride, is a trigger that elicits a cell-type specific reaction. Although the mechanism

12
of action has not been elucidated, our previous and present in vitro data suggest that NaF acts directly on dental
cells, irrespective of the presence of absence of a mineral phase.(1) Several signaling pathways have been
reported which may differ depending on the cell type considered.(40-45) In vivo, fluoride can have additional
effects on enamel formation and can modulate protease and enolase activities.(16-20) The mechanisms affected
by long-term chronic exposure to fluoride are likely to differ from those affected by acute exposure.(18,20) Some
targets affected by chronic exposure to lower fluoride levels, resulting in enamel fluorosis, are likely to be
specific to this uniquely mineralizing tissue, whereas others may be common to diverse cells and tissues.(20)
These various observations are consistent with fluoride having a selective influence on gene expression
depending on the cell type, but not on rodent age. The NaF effects reported here in the continually growing rat
incisor where amelogenesis and odontogenesis are permanent are expected to be independent of age, at least
during adulthood.
Dual effects of asporin: inhibitor and promoter of mineralization
TEM analysis clearly showed that the highest asporin concentration was found in the pericellular space close to
mineralized matrix. Similar observations were made in bone around osteocytes and in dentin around
odontoblasts where asporin accumulated in the apical part and progressively decreased in abundance along the
odontoblastic processes. Thus, asporin was concentrated in the predentin and barely detectable in mineralized
dentin. This observation was reproduced with techniques including electron and confocal microscopy, and
colorimetric and fluorescent immunohistochemistry. Asporin was also evidenced in enamel around the
ameloblasts of the maturation stage and around the cells of the papillary layer (data not shown). Asporin was
present immediately around matrix-forming cells, i.e. regions where mineral formation must be avoided.
Therefore, asporin has been perceived to be a negative regulator of mineral formation. Indeed, it has been
previously reported to be associated with the suppression of mineralization in the PDL and in cartilage, where it
may prevent the actions of BMP2 and TGFβ.(9,10,38,46) Similarly, asporin may prevent the action of BMP2
previously pointed out in ameloblast differentiation and enamel mineralization.(47) This inhibitory effect may be
stronger in fluorotic enamel, due to the chronic induction of asporin expression. The mechanism of asporin
action in enamel and ameloblasts is still unknown, but considering its inhibitory effect on mineralization and its
induction of expression by NaF, it may contribute to the enamel hypomineralization described previously in
fluorosis(18, 20) and reported here (supplemental figure 1).

13
Conversely, asporin can also promote mineralization in human dental pulp stem cells, in dentin(8) and in
osteoblast cell cultures.(4) Asporin, but not other SLRPs, induces collagen mineralization driven by an
osteosarcoma cell line.(4) Type I collagen deposition(48) and asporin expression(1) can be modulated by fluoride,
thus generating a regulatory loop of mineralization. Like other SLRPs, asporin was found co-localized with
collagen in collagen-containing matrices. Earlier data suggested that SLRP shape and collagen fibril geometry
are associated.(3) Although a direct interaction between asporin and collagen was only suggested by our
immunogold colocalization experiments, this possibility is strongly supported by in vitro binding assays.(4) In
this context, asporin may act as a promoter of mineralization. Consequently, dentin hypomineralization could
be expected as reported by others(18) and here (supplemental figure 1). However, there are apparently
contradictory data in the literature showing dentin hypermineralization in human dental fluorosis.(49) Thus, the
role of asporin in dentin mineralization in vivo needs to be investigated further. Asporin KO mouse models
could be generated to elucidate the details of asporin action in vivo in the various mineralized tissues in normal
and pathological conditions.
The effects of asporin on mineralization may depend on its localization and its concentration
As reported earlier, asporin apparently may have two opposite effects in the mineralization process: it may be
either a promoter of mineralization, or an inhibitor.(4,9) One major difference between the two dental extra-
cellular matrices considered here is the presence of collagen scaffold which may organize asporin distribution
and which may be necessary for the pro-mineralizing effect of asporin. The second main difference between
these two matrices is the water content and circulation of extracellular fluids which may be highly significant in
the mineralization process(50) and the local concentrations of asporin and calcium.(51) Our data suggest that
asporin may interact with collagen in dedicated space depending on its concentration. Thus, asporin may be
involved as a cofactor in calcium transport, and its binding to collagen may constitute an element that directs
the calcium to the site of mineralization, similarly to other polyaspartate containing proteins as SIBLINGS
(which can also function as promoters or inhibitors of mineralization).(50, 52) In this model, the mineralized
extracellular matrix would not form normally if there was either an excess or a deficit of asporin; this may
explain, at least in part, the contradictory findings for the effects of fluoride on dentin mineralization. The
preferential localization of asporin in the pericellular space is in accordance with this model in which free
asporin may block docking sites and lead to inefficient mineralization. Low asporin content may render calcium

14
transport less efficient, and in addition, may disable the routing of the calcium carrier to the mineralization site,
thus allowing mineral formation at ectopic sites. This model can explain the different effects of fluoride: the
hampered mineral formation in enamel, the compromised tissue structure in dentin, and the spurious mineral
formation in the periodontal ligament.
In conclusion, asporin is expressed in mineralizing matrices, and is the only SLRP to be modulated by
fluoride(1) and able to interact with calcium and collagen;(4) it appears to be a key factor in the mineralization
process. It seems relevant to fluorosis and probably other pathologies affecting mineralizing matrix tissues.
Indeed, numerous epidemiological studies have described associations between polymorphism of the asporin
polyaspartate sequence and osteoarthritis of disc, knee, hand or hip.(53-55) Further studies are required to
characterize the activities of asporin in relation to calcium, BMP-2, collagen and possibly enamel matrix
proteins in diverse tissues in both physiological and pathological conditions.
Acknowledgments
We are indebted to Prof. Anne Poliard and Prof. Odile Kellermann (Université Paris-Descartes, France) for
705IH8 cells and Prof. Hidemitsu Harada (Iwate Medical University, Japan) for HAT-7 cells. We thank Sophia
Loiodice and Ali Nassif for their technical help and Christophe Klein for his assistance with confocal
microscopy.
Authors’ roles
Study design: SH, TW, AB and SB.
Study conduct: AB and SB.
Data collection: SH, DF and DC.
Data analysis: SH, TW, DF, DC, AD, AB and SB.
Data interpretation: SH, TW, DF, AD, AB, and SB.
Drafting manuscript: SH, TW, AB and SB.
Revising manuscript content: SH, TW, AB and SB.
Approving final version of manuscript: SH, TW, DF, DC, AD, AB, and SB.
SH and SB take responsibility for the integrity of the data analysis.

15
References
1. Wurtz T, Houari S, Mauro N, MacDougall M, Peters H, Berdal A. Fluoride at non-toxic dose affects odontoblast gene expression in vitro. Toxicology. 2008;249(1):26-34.
2. Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D. Identification and
characterization of asporin. a novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. The Journal of biological chemistry. 2001;276(15):12201-11.
3. Kalamajski S, Oldberg A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix
biology : journal of the International Society for Matrix Biology. 2010;29(4):248-53. 4. Kalamajski S, Aspberg A, Lindblom K, Heinegard D, Oldberg A. Asporin competes with decorin for
collagen binding, binds calcium and promotes osteoblast collagen mineralization. The Biochemical journal. 2009;423(1):53-9.
5. Addison WN, Nakano Y, Loisel T, Crine P, McKee MD. MEPE-ASARM peptides control extracellular
matrix mineralization by binding to hydroxyapatite: an inhibition regulated by PHEX cleavage of ASARM. J Bone Miner Res. 2008;23(10):1638-49.
6. Boskey AL, Christensen B, Taleb H, Sørensen ES. Post-translational modification of osteopontin: effects on
in vitro hydroxyapatite formation and growth. Biochem Biophys Res Commun. 2012;419(2):333-8. 7. Qiu SR, Wierzbicki A, Orme CA, et al. Molecular modulation of calcium oxalate crystallization by
osteopontin and citrate. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):1811-5.
8. Lee EH, Park HJ, Jeong JH, et al. The role of asporin in mineralization of human dental pulp stem cells.
Journal of cellular physiology. 2011;226(6):1676-82. 9. Yamada S, Tomoeda M, Ozawa Y, et al. PLAP-1/asporin, a novel negative regulator of periodontal
ligament mineralization. The Journal of biological chemistry. 2007;282(32):23070-80. 10. Leong NL, Hurng JM, Djomehri SI, Gansky SA, Ryder MI, Ho SP. Age-related adaptation of bone-PDL-
tooth complex: Rattus-Norvegicus as a model system. PloS one. 2012;7(4):e35980. 11. Kou I, Nakajima M, Ikegawa S. Expression and regulation of the osteoarthritis-associated protein asporin.
The Journal of biological chemistry. 2007;282(44):32193-9. 12. Wald T, Osickova A, Sulc M, et al. Intrinsically Disordered Enamel Matrix Protein Ameloblastin Forms
Ribbon-like Supramolecular Structures via an N-Terminal Segment Encoded by Exon 5. The Journal of biological chemistry. 2013.
13. Margolis HC, Beniash E, Fowler CE. Role of macromolecular assembly of enamel matrix proteins in
enamel formation. Journal of dental research. 2006;85(9):775-93. 14. Bartlett JD, Ganss B, Goldberg M, et al. 3. Protein-protein interactions of the developing enamel matrix.
Current topics in developmental biology. 2006;74:57-115. 15. Berdal A, Hotton D, Pike JW, Mathieu H, Dupret JM. Cell- and stage-specific expression of vitamin D
receptor and calbindin genes in rat incisor: regulation by 1,25-dihydroxyvitamin D3. Developmental biology. 1993;155(1):172-9.
16. Hamilton IR. Biochemical effects of fluoride on oral bacteria. J Dent Res. 1990;69 Spec No:660-7;
discussion 682-3. Review.

16
17. Moreno EC, Kresak M, Zahradnik RT. Fluoridated hydroxyapatite solubility and caries formation. Nature.
1974;247(5435):64-5. 18. Fejerskov O, Larsen MJ, Richards A, Baelum V. Dental tissue effects of fluoride. Advances in dental
research. 1994;8(1):15-31. 19. Aoba T, Fejerskov O. Dental fluorosis: chemistry and biology. Critical reviews in oral biology and
medicine : an official publication of the American Association of Oral Biologists. 2002;13(2):155-70. 20. Denbesten P, Li W. Chronic fluoride toxicity: dental fluorosis. Monographs in oral science. 2011;22:81-96. 21. Everett ET, McHenry MA, Reynolds N, et al. Dental fluorosis: variability among different inbred mouse
strains. Journal of dental research. 2002;81(11):794-8. 22. Chachra D, Limeback H, Willett TL, Grynpas MD. The long-term effects of water fluoridation on the
human skeleton. Journal of dental research. 2010;89(11):1219-23. 23. Carvalho JG, Leite Ade L, Peres-Buzalaf C, et al. Renal proteome in mice with different susceptibilities to
fluorosis. PloS one. 2013;8(1):e53261. 24. Robinson C, Connell S, Kirkham J, Brookes SJ, Shore RC, Smith AM. The effect of fluoride on the
developing tooth. Caries research. 2004;38(3):268-76. 25. Bronckers AL, Bervoets TJ, Woltgens JH, Lyaruu DM. Effect of calcium, given before or after a fluoride
insult, on hamster secretory amelogenesis in vitro. European journal of oral sciences. 2006;114 Suppl 1:116-22; discussion 27-9, 380.
26. Bartlett JD, Dwyer SE, Beniash E, Skobe Z, Payne-Ferreira TL. Fluorosis: a new model and new insights.
Journal of dental research. 2005;84(9):832-6. 27. Bronckers AL, Lyaruu DM, DenBesten PK. The impact of fluoride on ameloblasts and the mechanisms of
enamel fluorosis. Journal of dental research. 2009;88(10):877-93. 28. Jedeon K, De la Dure-Molla M, Brookes SJ, et al. Enamel defects reflect perinatal exposure to bisphenol a.
The American journal of pathology. 2013;183(1):108-18. 29. Kawano S, Morotomi T, Toyono T, et al. Establishment of dental epithelial cell line (HAT-7) and the cell
differentiation dependent on Notch signaling pathway. Connective tissue research. 2002;43(2-3):409-12. 30. Priam F, Ronco V, Locker M, et al. New cellular models for tracking the odontoblast phenotype. Archives
of oral biology. 2005;50(2):271-7. 31. Liao H, Brandsten C, Lundmark C, Christersson C, Wurtz T. Osteonectin RNA and collagen alpha1(I)
RNA in the developing rat maxilla. European journal of oral sciences. 1998;106 Suppl 1:418-23. 32. Hotton D, Davideau JL, Bernaudin JF, Berdal A. In situ hybridization of calbindin-D 28 k transcripts in
undecalcified sections of the rat continuously erupting incisor. Connective tissue research. 1995;32(1-4):137-43.
33. Den Besten PK. Effects of fluoride on protein secretion and removal during enamel development in the rat.
Journal of dental research. 1986;65(10):1272-7. 34. Kato K, Nakagaki H, Sakakibara Y, Weatherell JA, Robinson C. The dissolution rate of enamel in acid in
developing rat incisors. Archives of oral biology. 1988;33(9):657-60.

17
35. Henry SP, Takanosu M, Boyd TC, et al. Expression pattern and gene characterization of asporin. a newly discovered member of the leucine-rich repeat protein family. The Journal of biological chemistry. 2001;276(15):12212-21.
36. Park ES, Cho HS, Kwon TG, et al. Proteomics analysis of human dentin reveals distinct protein expression
profiles. Journal of proteome research. 2009;8(3):1338-46. 37. Devarajan-Ketha H, Craig TA, Madden BJ, Robert Bergen H 3rd, Kumar R. The sclerostin-bone protein
interactome. Biochemical and biophysical research communications. 2012;417(2):830-5. 38. Kou I, Nakajima M, Ikegawa S. Binding characteristics of the osteoarthritis-associated protein asporin.
Journal of bone and mineral metabolism. 2010;28(4):395-402. 39. Richards A, Kragstrup J, Josephsen K, Fejerskov O. Dental fluorosis developed in post-secretory enamel.
Journal of dental research. 1986;65(12):1406-9. 40. Zhang Y, Li W, Chi HS, Chen J, Den-besten PK. JNK/c-Jun signaling pathway mediates the fluoride-
induced downregulation of MMP-20 in vitro. Matrix Biol. 2007; 26:633–641. 41. Peng L, Li Y, Shusterman K, Kuehl M, Gibson CW. Wnt-RhoA signaling is involved in dental enamel
development. European journal of oral sciences. 2011;119 Suppl 1:41-9. 42. Pei J, Li B, Gao Y, et al. Fluoride decreased osteoclastic bone resorption through the inhibition of NFATc1
gene expression. Environmental toxicology. 2012. 43. Manduca P, Marchisio S, Astigiano S, et al. FMS*Calciumfluor specifically increases mRNA levels and
induces signaling via MAPK 42,44 and not FAK in differentiating rat osteoblasts. Cell biology international. 2005;29(8):629-37.
44. Bourgoin SG, Harbour D, Poubelle PE. Role of protein kinase C alpha, Arf, and cytoplasmic calcium
transients in phospholipase D activation by sodium fluoride in osteoblast-like cells. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1996;11(11):1655-65.
45. Zerwekh JE, Morris AC, Padalino PK, Gottschalk F, Pak CY. Fluoride rapidly and transiently raises
intracellular calcium in human osteoblasts. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1990;5 Suppl 1:S131-6.
46. Tomoeda M, Yamada S, Shirai H, Ozawa Y, Yanagita M, Murakami S. PLAP-1/asporin inhibits activation
of BMP receptor via its leucine-rich repeat motif. Biochemical and biophysical research communications. 2008;371(2):191-6.
47. Lee HK, Park JT, Cho YS, Bae HS, Cho MI, Park JC. Odontogenic ameloblasts-associated protein
(ODAM), via phosphorylation by bone morphogenetic protein receptor type IB (BMPR-IB), is implicated in ameloblast differentiation. J Cell Biochem. 2012;113(5):1754-65.
48. Maciejewska I, Spodnik JH, Domaradzka-Pytel B, Sidor-Kaczmarek J, Bereznowski Z. Fluoride alters type
I collagen expression in the early stages of odontogenesis. Folia morphologica. 2006;65(4):359-66. 49. Rojas-Sánchez F, Alaminos M, Campos A, Rivera H, Sánchez-Quevedo MC. Dentin in severe fluorosis: a
quantitative histochemical study. J Dent Res. 2007 Sep;86(9):857-61. 50. Veis A, Dorvee JR. Biomineralization Mechanisms: A New Paradigm for Crystal Nucleation in Organic
Matrices. Calcified tissue international. 2012.

18
51. Woltgens JH, Lyaruu DM, Bronckers AL, Bervoets TJ, Van Duin M. Biomineralization during early stages of the developing tooth in vitro with special reference to secretory stage of amelogenesis. The International journal of developmental biology. 1995;39(1):203-12.
52. George A, Veis A. Phosphorylated proteins and control over apatite nucleation, crystal growth, and
inhibition. Chemical reviews. 2008;108(11):4670-93. 53. Mayer JE, Iatridis JC, Chan D, Qureshi SA, Gottesman O, Hecht AC. Genetic polymorphisms associated
with intervertebral disc degeneration. The spine journal : official journal of the North American Spine Society. 2013;13(3):299-317.
54. Bijsterbosch J, Kloppenburg M, Reijnierse M, et al. Association study of candidate genes for the
progression of hand osteoarthritis. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society. 2013;21(4):565-9.
55. Loughlin J. Knee osteoarthritis, lumbar-disc degeneration and developmental dysplasia of the hip--an
emerging genetic overlap. Arthritis research & therapy. 2011;13(2):108.

19
Figure legends
Fig.1. Phenotypes of Wistar rats treated with fluoride: Fluorosis rat model
Untreated control rats presented incisor enamel homogeneously colored yellow to orange. Following 2.5 mM
NaF treatment, faint white bands appeared, and such bands were larger in animals treated with 5 mM NaF. At
7.5 mM NaF, severe fluorosis was observed, and the whole incisor appeared completely white and opaque.
Fig.2. Distribution of the asporin protein in mesenchymal and epithelial dental tissues
(A) Immunohistochemical analysis revealing asporin protein preferentially in ameloblasts at the maturation
stage (the signal was detected at the transition stage and was clearly evidenced during the maturation stage).
The signal for asporin protein during the transition and the maturation stages increased with the concentration
of fluoride in the drinking water. A: ameloblast; Pl: papillary layer. (B) Immunohistochemistry showing asporin
protein at the apical pole of rat incisor odontoblasts. The signal corresponding to asporin protein decreased with
increasing concentrations of fluoride in the drinking water. Pd: predentin; Od: odontoblast; P: pulp cells. (C,
D). Western blotting analysis showing asporin in the upper blot and gapdh in the lower blot for reference. (E)
Western blotting quantification (each bar corresponds to the mean of three independent experiments). Asporin
protein is down-regulated in the mesenchyme and up- regulated in the dental epithelium as the dose of fluoride
increases. Scale bar: (A) 100 µm, (B) 50 µm.
Fig.3. Asporin mRNA localization and expression in NaF-treated rat incisor
Asporin mRNA was evidenced by in situ hybridization (ISH). Longitudinal sections were hybridized to
digoxigenin (DIG)-labeled asporin RNA antisense probes which presence was revealed using an anti-DIG
antibody coupled to alkaline phosphatase. (A) Asporin mRNA was evidenced in the dental epithelium where it
was mainly present in the papillary layer and in the ameloblasts during the maturation stage. The signal
corresponding to asporin mRNA increased with increasing concentrations of fluoride. A: ameloblast; Pl:
papillary layer. (B) Asporin mRNA was located at the apical pole of the odontoblasts. The signal decreased
with increasing concentrations of fluoride. D: dentin; Pd: predentin; Od: odontoblast; P: pulp cells. (C) RT-
qPCR quantification of asporin mRNA levels in the microdissected epithelium and mesenchyme. Asporin

20
expression dose-dependently increased in the dental epithelium and decreased in the mesenchyme as the
concentration of fluoride increased. Scale bar: (A) 100 µm, (B) 50 µm.
Fig.4. Asporin expression in odontoblastic and ameloblastic cell lines
(A, B) Western blot analysis of asporin protein in the epithelial HAT-7 (A) and mesenchymal 705IH8 (B) cells
treated with 0.5 mM NaF; analysis for gapdh protein is presented in the lower blots for reference. (C)
Quantification of asporin protein levels by western blotting in the HAT-7 ameloblastic and 705IH8
odontoblast-like cell extracts. (D) Quantification of asporin mRNA levels by RT-qPCR in the HAT-7 and
705IH8 cell lines.
Fig.5. Asporin and type I collagen co-localization by immunofluorescence confocal microscopy and
transmission electron microscopy (TEM)
(A) Asporin protein was found at the apical pole of odontoblasts and in the predentin, mostly at the edges of
odontoblast processes. (B) Type I collagen was detected in the predentin and dentin. (C) DAPI-stained
odontoblast nuclei. (D) Merge of the two images revealed the co-localization (yellow) of asporin (green) and
type I collagen (red) mainly in predentin and in the edges of odontoblast processes. D: dentin; Pd: predentin;
Od: odontoblast. (E) TEM analysis of asporin (large dots) and type I collagen (small dots) using specific
antibodies directed against the two proteins sequentially. Colloidal gold (black dots) immunocytochemistry.
Ultrathin sections (70 nm) were sequentially incubated first with rabbit anti-asporin polyclonal antibody and
protein A coupled to 15 nm-diameter gold beads, and then with mouse anti-collagen 1 monoclonal antibody and
secondary antibody coupled to 6 nm-diameter beads.
Fig.6. Asporin localization in predentin and odontoblasts by transmission electron microscopy (TEM)
(A) Samples were labeled with asporin-specific antibody and protein A coupled to 15 nm-diameter gold beads.
The signal was restricted to the odontoblastic pericellular environment, in the apical part of the odontoblastic
cells and along the processes. OP: Odontoblast process. (B) Asporin in matured ameloblasts. ES: Enamel space.
(C) Asporin in the osteocytic pericellular environment.

21
Tables
Table 1. Primer sequences used for RT-qPCR assays
cDNA Amplicon size
(pb)
Primer sequences
asporin 464
5’-ACCAGGGGCATTTGAAGGGGTGA-3’ 5’-CAACGCAGCGAAACGTTGCAGG-3’
gapdh 260 5’ -GACCCCTTCATTGACCTCAACTAC-3’ 5’-AAGTTGTCATGGATGACCTTGGCC-3’
rs15 315 5’-GGCTTGTAGGTGATGGAGAA-3’ 5’-CTTCCGCAAGTTCACCTACC-3’

22

23
24

25

26

27




















