
Effect of flow on endothelial endocytosis of nanocarriers targeted to ICAM-1
Upload
independentCategory
view
3download
0
Kidney International, Vol. 58 (2000), pp. 1121–1134
Antibodies to both ICAM-1 and LFA-1 do not protect thekidney against toxic (HgCl2) injury
MANUELA GHIELLI, WALTER A. VERSTREPEN, KATHLEEN E.J. DE GREEF, MARK H. HELBERT,DIRK K. YSEBAERT, ETIENNE J. NOUWEN,1 and MARC E. DE BROE
Department of Nephrology-Hypertension, University of Antwerp, Belgium
an identical antibody treatment protocol. Resolving that con-Antibodies to both ICAM-1 and LFA-1 do not protect thetroversy should bring better insight in fundamental processeskidney against toxic (HgCl2) injury.underlying different types of ARF, and will be the subject ofBackground. The role of inflammatory leukocytes in acutefurther study.renal failure (ARF) remains controversial and appears largely
uninvestigated in toxic (in contrast to ischemic) ARF.Methods. Female Wistar rats were injected with monoclonal
antibodies (mAbs) directed to both the leukocyte function-Despite its high vulnerability for the exposure to toxicassociated antigen 1 (LFA-1) and the intercellular adhesion
agents or to an episode of oxidative stress, the kidneymolecule 1 (ICAM-1). Doses (6 mg/kg of each mAb) weregiven 24 hours prior to the induction of acute tubular necrosis demonstrates an impressive regeneration capacity [1–4].(ATN) by mercuric chloride administration (2 mg/kg, subcuta- A protective/regenerative or in contrast detrimental orneously, day 0) and subsequently every 48 hours. Control rats mere clean-up potential has been proposed for the numer-similarly received either control antibody (12 mg/kg) or vehicle
ous inflammatory leukocytes always present in the kid-prior to and following the induction of ATN. Renal functionney during the process of injury and/or subsequent regen-was also measured from male Lewis rats that were similarly
treated with anti-adhesion antibodies during exposure to 30 eration [5–10]. Macrophages and also both subsets of Tminutes of unilateral renal ischemia. lymphocytes remain localized predominantly in the zone
Results. Injected antibodies were demonstrated on periph- of most severe injury for as long as the process of restor-eral blood leukocytes (flow cytometrical detection of mouseing renal tubular morphology and function lasts [11, 12].anti–LFA-1) and on endothelium (immunohistochemical stain-Polymorphonuclear cells, but also macrophages, indeeding of mouse anti–ICAM-1) and were measured in serum (en-
zyme-linked immunosorbent assay). Macrophages and T cells can release damaging agents such as oxygen radicals andwere prominent in the kidney of control treatment rats after nitric oxide [13]. Moreover, a leukocyte infiltrate is wellHgCl2 injection, but anti-adhesion treatment clearly had pre-
equipped to produce and secrete vasoactive substancesvented their infiltration. Notwithstanding, renal tubular injurythat, in turn, may functionally influence the kidney [14].was equally pronounced in all mercuric chloride treatment groups
and so was the decline in renal function (serum creatinine, pro- Activated mononuclear cells, on the other hand, canteinuria). Tubular epithelial cell proliferation seemed slightly produce variable kinds of growth factors able to promoteless pronounced and delayed in anti-adhesion treated rats. Kid- epithelialization, angiogenesis, and/or extracellular ma-neys from ischemia exposed rats were, however, functionally
trix turnover [15, 16].protected by identical anti–ICAM-1/anti–LFA-1 treatment.Different molecular partners of leukocytes and endo-Conclusion. Prevention of cellular infiltration by mAbs to
LFA-1 and ICAM-1 has no effect on renal morphology, func- thelial cells involved in adhesion, a crucial step towardtion, or regeneration following mercuric chloride-induced ARF inflammation, are becoming more readily known, andin the rat. This result contrasts with the functional protection
preventing the interaction between them may indeed keepof the rat kidney to ischemia/reperfusion injury by virtue ofleukocytes from proceeding to endothelial diapedesis[17, 18]. Of the leukocyte integrins that appear most com-monly in leukocyte-endothelial adhesion, only CD11a/1 Present address: Laboratory of Neurobiology and Neuroendocrinoim-
munology, University of Antwerp, Belgium. CD18 [leukocyte function-associated antigen 1 (LFA-1)]is constitutively expressed by all leukocytes [19–24]. ItsKey words: acute renal failure, adhesion molecules, mercuric chloride,
nephrotoxicity. major ligand, the Ig-like intercellular adhesion molecule 1(ICAM-1) [25, 26], is constitutively expressed at low
Received for publication December 29, 1999levels by endothelial cells, and its expression is markedlyand in revised form March 23, 2000
Accepted for publication March 27, 2000 increased upon exposure to cytokines such as tumor ne-crosis factor (TNF) and interleukin-1 (IL-1). Adhesion 2000 by the International Society of Nephrology
1121

Ghielli et al: Antibodies to adhesion molecules in toxic ARF1122
molecules have been implicated in ischemia/reperfusion were subsequently conjugated either with fluoresceineisothyocyanate (FITC) [44] or with biotin (b) [45], ac-(I/R) injury of the kidney [27] as well as the heart [28]
and the liver [29, 30]: these organs became successfully cording to a standard protocol.protected by a variety of direct/indirect methods targeting
Experimental designICAM-1 (anti–ICAM-1 antibodies [27], ICAM-1 knock-out mice [31], antisense oligonucleotides for ICAM-1 Female Wistar rats (180 to 200 g) were individually
housed in metabolic cages one day prior to the start of[32], etc.). Studies investigating the importance of adhe-sion molecules in toxic ARF are limited to a very recent the experiment, and particular attention was given to the
cleanness of their cages and to their handling. Standardone involving cisplatin [33].We investigated whether the combined treatment with rat chow and fresh tap water were supplied ad libitum,
and a 12-hour light/dark cycle was respected during themonoclonal antibodies (mAbs) directed to CD11a/CD18(LFA-1) and its major ligand, ICAM-1, could influence entire experimental period. Rats were injected intraperi-
toneally with sterile solutions of: (1) mAb WT1 (mousethe infiltration behavior of different leukocyte subsetsin our previously optimized toxic model of mercuric chlo- IgG2a) directed against LFA-1 (6 mg/kg) and mAb 1A29
(mouse IgG1) reactive with ICAM-1 (6 mg/kg); (2) con-ride (HgCl2)-induced acute renal failure (ARF) in therat [34]. Disease progress and subsequent recovery after trol mAb (7E8, antihuman placental alkaline phospha-
tase or h-PLAP; mouse IgG1; 12 mg/kg); or (3) vehicleHgCl2 injection were compared at renal functional andmorphological level between rats treated with antibodies (PBS). Injections were performed 24 hours prior to (day 1)
induction of acute tubular necrosis (ATN; day 0) by ato adhesion molecules and those treated with a controlantibody or with vehicle. single administration of mercuric chloride (HgCl2, 2 mg/kg
subcutaneously) and were repeated every 48 hours. Se-rum titers of antibody were measured throughout the
METHODSexperiment. During the subsequent 10-day observation
Production, purification, and quality assurance of mAbs period, four rats of each group were sacrificed at differenttime intervals (days 1, 2, 4, 6, and 10). At each of theseExcept for the hybridoma producing mAb 7E8 (anti-
human placental alkaline phosphatase) [35], hybridoma time points, one additional rat exposed to treatments 1through 3 that had, however, not been given mercuriccell lines producing mAbs (antirat mAbs: WT.1,LFA-1,
1A29,ICAM-1, OX-35,CD4) were obtained from the chloride was sacrificed and served as noninjured control.All rats were weighed daily. Blood samples were ob-European Collection of Animal Cell Culture. mAbs
PC10,human proliferating cell nuclear antigen-PCNA tained by tail vein bleeding. Serum creatinine concentra-tion was determined colorimetrically by Jaffe’s method(Dako, Carpinteria, CA, USA), ED1,rat macrophages
(Serotec, Oxford, UK) and also OX-8 (Caltag, Bur- [46]. Protein concentration in 24-hour urine samples wasdetermined colorimetrically by Bradford’s method [47].lingame, CA, USA) were purchased [36–41]. Hybridoma
cells were grown in RPMI 1640 buffered by 25 mmol/L In an additional experiment, male Lewis rats (250 g)were treated similarly with 6 mg/kg of both mAb WT1HEPES and supplemented with 300 mg/mL l-glutamine
(Life Technologies, Grand Island, NY, USA). The initial (anti–LFA-1) and mAb 1A29 (anti–ICAM-1) at the timeof exposure of one kidney to 30 minutes of ischemiagrowth medium supplement of 15% fetal calf serum
(FCS; Life Technologies) was lowered to 5% prior to (intravenous, day 0), and subsequently every 48 hours.transfer of the cells from customary culture flasks to a
Flow cytometric analysis of peripheral blood leukocytesspecialized roller bottle (mini-PERM; Heraeus Instru-ments, Ofterode am Harz, Germany) [42] in order to Leukocytes in 25 mL of heparinized peripheral blood
obtained from rats between subsequent antibody injec-increase the antibody production rate. Secreted antibod-ies were purified from culture supernatant by affinity tions were double labeled to detect bound injected anti-
body and free LFA-1. After red blood cell lysis by thechromatography with protein A (Bioprocessing, Consett,UK) and dialyzed against phosphate-buffered saline addition of 10 volumes of lysis buffer [163 mmol/L NH4Cl,
10.5 mmol/L KHCO3, 105 mmol/L ethylenediaminetetra-(PBS). Denaturing sodium dodecyl sulfate-polyacryl-amide gel electrophoresis (SDS-PAGE) in reducing con- acetic acid (EDTA); centrifugation during 7 minutes at
120 g; room temperature] and subsequent washing (PBS 1ditions confirmed the absence of contamination of anti-body solutions with non-IgG proteins (.90% pure 0.1% bovine serum albumin 1 0.05% sodium azide; cen-
trifugation during 7 minutes at 120 g), leukocytes weremAb), and protein concentration was determined spec-trophotometrically at 280 nm. Pure antibody solutions incubated with biotinylated rabbit-anti-mouse immuno-
globulins (RAM-b; Dako) that in our case would bindmeant for in vivo application were assured to containless than 20 endotoxin units [43] per mg protein (LAL to cells that had bound the injected antibodies. RAMb was
visualized by the addition of phycoerythrin-conjugatedcolorimetric test, end-point method; bio-Whittacker,Walkersville, MD, USA). Some of the purified mAbs streptavidin (SA-PE; Becton Dickinson, Mountain View,

Ghielli et al: Antibodies to adhesion molecules in toxic ARF 1123
CA, USA). Next, the remaining binding sites on RAM-b Histologic evaluation of renal tubular damagefor mouse IgG were blocked by normal mouse serum. Four micrometer sections from methacarn-fixed, par-Finally, FITC-conjugated anti–LFA-1 (WT.1-FITC) was affin-embedded tissue were stained with periodic acid-added to assure that LFA-1 molecules on leukocytes were Schiff (PAS) reagent and methyl green (nuclei). To eval-saturated with injected anti–LFA-1 (WT.1). Incubations uate tubular epithelial cell injury, both in the renal cortexwere performed cold (48C) and in the dark, and were and outer stripe of outer medulla (OSOM) of each ani-separated by washing steps. Labeled cells were kept in mal, 30 proximal and distal tubules were assigned a scorewashing buffer (dark, 48C) until analysis. indicating intact tubules, tubules with damaged or ne-
Individual leukocytes were interrogated in a FACStar crotic cells, and completely necrotic tubules. Cells show-PLUS flow-cytometer from Becton Dickinson (Immuno- ing excessive vacuolization, swelling, or membrane irreg-cytometry Systems) using a 488 nm light beam originat- ularities were considered damaged. Cells showing aing from an Argon-ion laser set at 40 mW power. Data ruptured cell membrane or an extruded nucleus wereacquisition was assisted by the lysis II software package. considered necrotic. The tubules were scored at a magni-The number of white blood cells was counted by an fication of at least 3320.automated blood cell analyzer (Technicon H1; Bayer,
Immunohistochemical stainingsBrussels, Belgium).Four micrometer sections from paraffin embedded tis-
Serum titers of injected mouse antibodies sue or 6 mm air-dried frozen sections were washed inSerum titers of injected mouse IgG1 (1A29 5 anti-rat TSB (0.01 mol/L Tris-HCl, pH 7.6, in 0.9% NaCl) supple-
ICAM-1 or 7E8 5 anti-human PLAP) and IgG2a (WT1 5 mented with 0.1% Triton X-100 and 1% bovine serumanti-rat LFA-1) were monitored by enzyme-linked im- albumin, respectively, and were subsequently exposedmunosorbent assay (ELISA) throughout the experi- to 2 3 5 minutes of microwave heating (0.01 mol/L Nament. For this purpose, immunoplates were coated with citrate). As evidenced by appropriate control experi-a rabbit polyclonal antibody recognizing mouse immuno- ments, this treatment denatured injected mouse mAbsglobulins (RAM) from which cross-reactivity with rat IgG (anti–LFA-1 and anti–ICAM-1 or anti–h-PLAP) presenthad been previously removed (Dako, Glostrup, Denmark). in the kidney that could otherwise also lead to staining.Secondary biotinylated polyclonal antibodies raised in Then sections were blocked by normal horse serum (onegoat were directed to mouse IgG1 or to mouse IgG2a, fifth in TSB). Following overnight incubation with pri-respectively (Pharmingen, San Diego, CA, USA). After mary mouse antibody (OX8, ED1, PC10, or 1A29), wash-amplification by a complex of avidin and biotinylated ing in TSB, and rinsing in distilled water, endogenoushorse radish peroxidase, color was developed (OD405nm) peroxidase activity was suppressed in frozen sections byby the addition of 2,29azino-di (3-ethyl benstialozine sul- a 15-minute incubation in methanol, rinsing in water,fonate) (ABTS) [6] and hydrogen peroxide at pH 4.3 and a 30-minute incubation with 0.03% H2O2 in TSB.(5.5 mg ABTS in 10 mL phosphate/citrate buffer and After additional washing (TSB), the sections were incu-200 mL 1% H2O2). Mouse mAbs directed to the hapten bated for 30 minutes with biotinylated horse anti-mousetrinitrophenol (TNP) of either the IgG1 or IgG2a isotype IgG and were washed (TSB), followed by a 60-minuteserved as standards and were purchased from Phar- incubation with the avidin-biotin complex (Vectorstain;mingen. Vector Laboratories, Burlingame, CA, USA). Washing
(3 3 TSB) preceded the preparation for color develop-Animal sacrifice and renal tissue preparation ment. Then the peroxidase substrate solution [either 20 mg
Rats were sacrificed under ether anesthesia by cardiac 3-amino-ethyl-carbazole (Sigma, St. Louis, MO, USA)puncture. Left kidneys were decapsulated, rinsed in 0.9% in 24 mL dimethyl sulfoxide, 200 mL acetate, pH 5 toNaCl, and weighed. They were cut into approximately 5.2, and 4 mL 0.3% H2O2; or 0.5 mg/mL 3,39-diaminoben-2 mm thick slices and were either immediately fixed in zidine (tetrahydrochloride; Sigma) in 0.1 mol/L Tris-HCl,methacarn (60% methanol, 10% acetic acid, 30% trichlor- pH 7.6, with 0.03% H2O2] was added. The sections wereethane, 4 h at room temperature) or formol-calcium fixa- lightly counterstained with hematoxylin and covered intive (4% formaldehyde—BDH—in 0.1 mol/L Na-caco- glycerin-gelatin or in DPX (BDH) mounting medium,dylate, pH 7.4, with 1% CaCl2; 1.5 h at room temperature). respectively.Formol calcium-fixed slices were rinsed in buffer without As the CD4 molecule could no longer be recognized byformol and were subsequently frozen between gold- diverse mAbs (OX-35, OX-38, W3/25) after microwaveplated copper blocks at liquid nitrogen temperature. treatment, CD4-positive cells were immunohistochemi-Methacarn-fixed tissue was rinsed in 70% ethanol and cally stained by means of a biotinylated anti-CD4 mAbwas dehydrated and embedded in low-melting point par- (OX-35-b). Reduction of signal amplification by omis-
sion of incubation with secondary anti-mouse polyclonalaffin (528C, BDH).

Ghielli et al: Antibodies to adhesion molecules in toxic ARF1124
antibody, however, makes this procedure much less sen- serum titers of mouse IgG1 (1A29 or 7E8) and IgG2a(WT1) throughout the experiment.sitive. Immunohistochemical detection of injected anti–
ICAM-1 bound to endothelium was established on sec- Figure 1 shows a flow cytometric analysis of PBL la-beled either with FITC-conjugated anti–LFA-1 (WT1-tions that had not been placed in the microwave but that
were pretreated with trypsin (5-min exposure to 0.003% FITC) alone prior to antibody injection at day 21 (Fig.1A) or dual labeled with biotinylated rabbit-anti-mousetrypsin type III) instead and that were not incubated
with a primary mAb. immunoglobulins (RAM-b 1 SA-PE) and with FITC-conjugated anti–LFA-1 mAb (WT1-FITC) after the first
Tubular epithelial cell proliferation and interstitial antibody injection at day 0 (Fig. 1 B–E), as explained inleukocyte accumulation the Methods section. In control conditions (Fig. 1A), three
Immunohistochemical staining for PCNA on 4 mm sec- subsets of cells differentially expressing the LFA-1 anti-tions of methacarn-fixed paraffin-embedded tissue com- gen can be distinguished. These weakly, intermediately,bined with the PAS staining permitted counting of posi- or highly LFA-1–positive cells previously were shown totive cells with the distinction made between proximal and be lymphocytes, polymorphonuclear cells, and mono-distal tubular cross-sections and the interstitium. The num- cytes, respectively [48]. After injection with control mAbber of positive cells in the OSOM (25 microscopic fields, 7E8 (Fig. 1 B, C), no positive cells could be detected bymagnification 3320, total surface 2.68 mm2) is expressed mere (in vitro) addition of rabbit-anti-mouse polyclonalper square millimeter. Staining for macrophages (mAb antibody (RAM-b 1 SA-PE; Fig. 1B). Labeling withED1) was performed on 4 mm sections of methacarn- FITC conjugated WT.1 (anti–LFA-1; Fig. 1C), on thefixed, paraffin-embedded tissue. In the OSOM (30 micro- other hand, resulted as expected in the distinction ofscopic fields, magnification 3320, total surface 3.22 mm2), three cell subsets similar to those seen in Figure 1A.the number of ED1-positive cells per square millimeter In contrast, injection of the anti–ICAM-1/anti–LFA-1was counted. CD8 (Ts/c and NK cells)-positive cells and antibody mixture (Fig. 1 D, E) resulted in three subsetsCD4-positive cells were stained on formol calcium-fixed, of PBL becoming detectable after in vitro addition offrozen sections by mAbs OX-8 and W3/25, respectively, “RAM-b 1 SA-PE” (Fig. 1D), which is in accordanceand were quantitated according to the procedure de- with the normal expression of LFA-1 on rat PBL (Fig.scribed for macrophages (45 microscopic fields, magnifi- 1A). These RAM-binding cells were nearly saturatedcation 3320, total surface 4.82 mm2). Hematoxylin and with injected anti-LFA-1 (WT.1) since hardly any of ineosin staining was performed to estimate polymorphonu- vitro added FITC-conjugated WT.1 could bind (Fig. 1E).clear neutrophil (PMN) presence. A comparison of renal tissue sections originating from
rats injected in vivo with anti–ICAM-1/anti–LFA-1 (Fig.Statistical analysis 2A) or control mAb 7E8 (Fig. 2B) and sacrificed at day 4
Statistical analysis was performed with SYSTAT 7.0 immunohistochemically stained with horse-anti-mousefor Windows, and significant differences are reported in polyclonal antibody shows intense staining of renal vas-the Results section. Significant increases (P , 0.007) culature in the former (Fig. 2A), while the latter (Fig. 2B)within one treatment group of serum creatinine and uri- sections were negative. This staining pattern in kidneysnary protein excretion were demonstrated by paired t-tests from anti–ICAM-1/anti–LFA-1 treated rats corresponds(days 21, 1, 2, 4, 6, 8, and 10 vs. day 0). Leukocyte infiltra- to immunohistochemical staining for ICAM-1 on renaltion data and PCNA positivity obtained at different times tissue from non–antibody-treated HgCl2 ARF rats at dayof sacrifice (days 1, 2, 4, 6, and 10) within each group were 4 (Fig. 2C) and thus presumably represents ICAM-1compared with untreated non-HgCl2–injected animals by expressing endothelial cells to which injected mouse-Mann–Whitney U-tests. Furthermore, at each time point, antirat ICAM-1 (mAb 1A29) is bound.it was tested by Mann–Whitney U-test whether anti– The administration protocol appeared sufficient toICAM-1/anti–LFA-1 treated groups differed from either provide a constant excess of circulating mAbs so thatcontrol mAb or vehicle-treated groups. Significance lev- both ICAM-1 and LFA-1 could be maximally blockedels (P , 0.05) needed not to be adjusted since multiple by 1A29 (IgG1) and WT1 (IgG2a), respectively. Serumcomparisons with the same data set were not performed. titers of injected mAbs (specific as well as control) in-
creased with first (day 21) and second (day 0) injections,after which subsequent injections maintained a plateau.RESULTSConcentrations of control antibody (12 mg/kg/injection)
Efficacy of antibody treatment remained somewhat higher than this of anti-ICAM-1 1Antibody targeting. Successful antibody injection was anti-LFA-1 [(6 mg/kg 1 6 mg/kg)/injection], since the
demonstrated by: (1) flow-cytometric demonstration of former does not bind to rat antigens (data not shown).anti–LFA-1 bound to peripheral blood leukocytes (PBLs); Interstitial leukocyte infiltration. In both control groups(2) immunohistochemical detection of anti–ICAM-1 (control antibody 5 7E8; vehicle 5 PBS), renal intersti-
tial macrophages (ED1 stained) reached numbers as highbound to endothelium; and (3) monitoring (ELISA) of
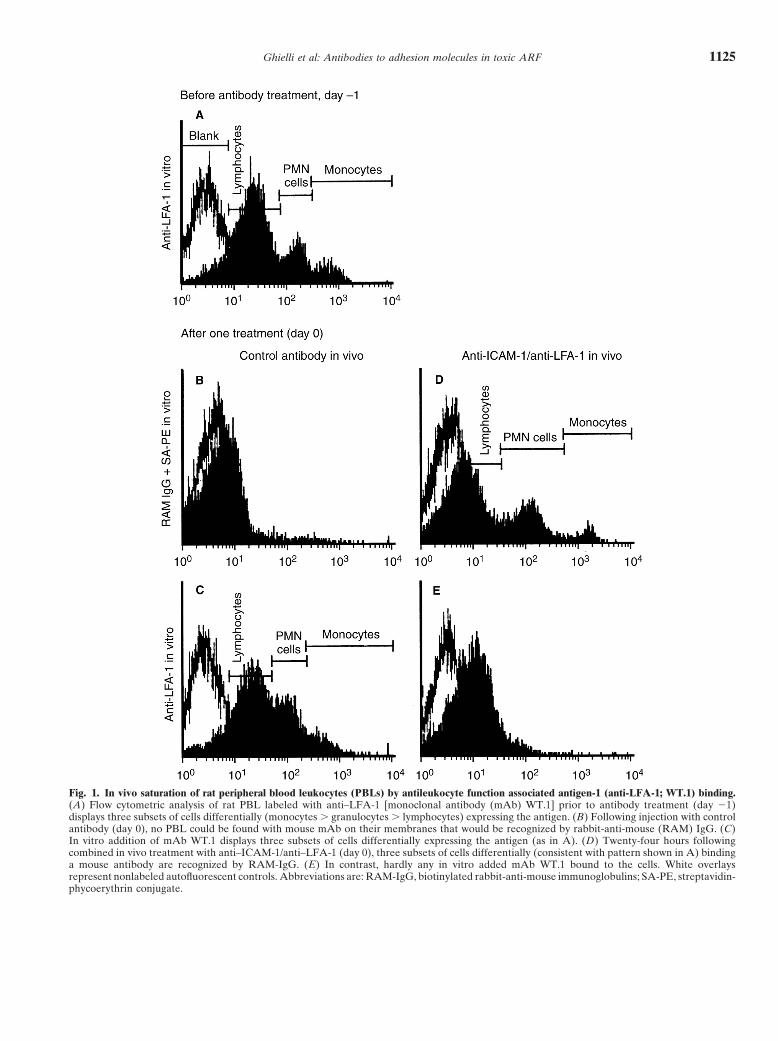
Ghielli et al: Antibodies to adhesion molecules in toxic ARF 1125
Fig. 1. In vivo saturation of rat peripheral blood leukocytes (PBLs) by antileukocyte function associated antigen-1 (anti-LFA-1; WT.1) binding.(A) Flow cytometric analysis of rat PBL labeled with anti–LFA-1 [monoclonal antibody (mAb) WT.1] prior to antibody treatment (day 21)displays three subsets of cells differentially (monocytes . granulocytes . lymphocytes) expressing the antigen. (B) Following injection with controlantibody (day 0), no PBL could be found with mouse mAb on their membranes that would be recognized by rabbit-anti-mouse (RAM) IgG. (C)In vitro addition of mAb WT.1 displays three subsets of cells differentially expressing the antigen (as in A). (D) Twenty-four hours followingcombined in vivo treatment with anti–ICAM-1/anti–LFA-1 (day 0), three subsets of cells differentially (consistent with pattern shown in A) bindinga mouse antibody are recognized by RAM-IgG. (E) In contrast, hardly any in vitro added mAb WT.1 bound to the cells. White overlaysrepresent nonlabeled autofluorescent controls. Abbreviations are: RAM-IgG, biotinylated rabbit-anti-mouse immunoglobulins; SA-PE, streptavidin-phycoerythrin conjugate.
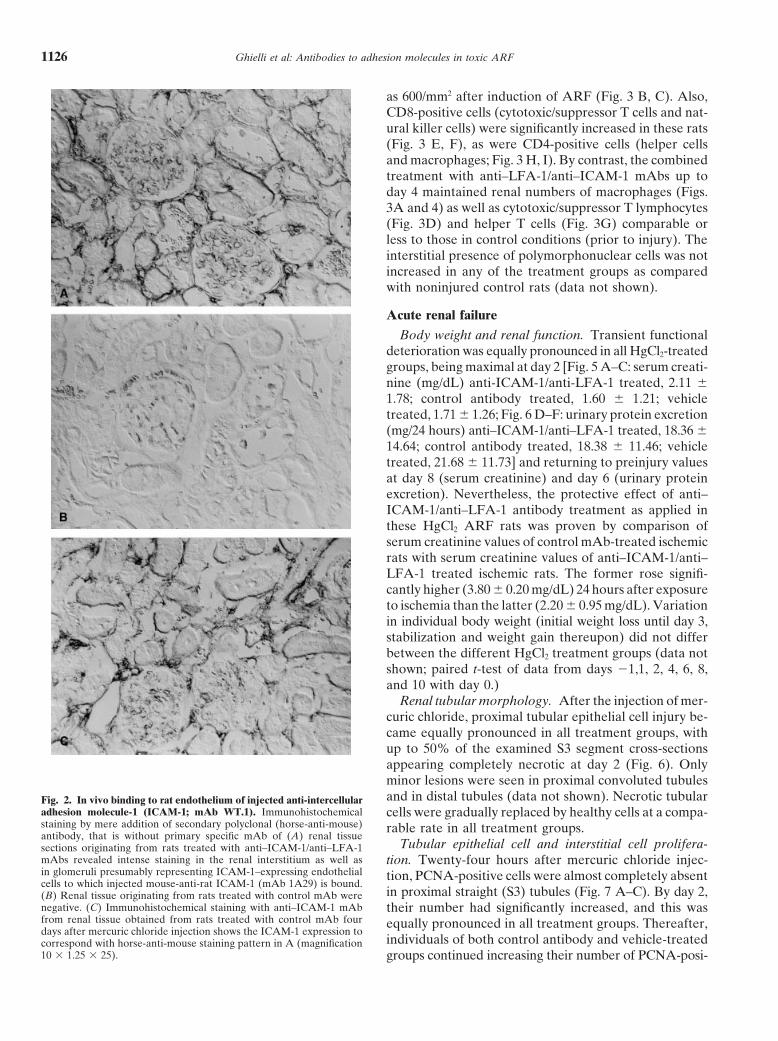
Ghielli et al: Antibodies to adhesion molecules in toxic ARF1126
as 600/mm2 after induction of ARF (Fig. 3 B, C). Also,CD8-positive cells (cytotoxic/suppressor T cells and nat-ural killer cells) were significantly increased in these rats(Fig. 3 E, F), as were CD4-positive cells (helper cellsand macrophages; Fig. 3 H, I). By contrast, the combinedtreatment with anti–LFA-1/anti–ICAM-1 mAbs up today 4 maintained renal numbers of macrophages (Figs.3A and 4) as well as cytotoxic/suppressor T lymphocytes(Fig. 3D) and helper T cells (Fig. 3G) comparable orless to those in control conditions (prior to injury). Theinterstitial presence of polymorphonuclear cells was notincreased in any of the treatment groups as comparedwith noninjured control rats (data not shown).
Acute renal failure
Body weight and renal function. Transient functionaldeterioration was equally pronounced in all HgCl2-treatedgroups, being maximal at day 2 [Fig. 5 A–C: serum creati-nine (mg/dL) anti-ICAM-1/anti-LFA-1 treated, 2.11 61.78; control antibody treated, 1.60 6 1.21; vehicletreated, 1.71 6 1.26; Fig. 6 D–F: urinary protein excretion(mg/24 hours) anti–ICAM-1/anti–LFA-1 treated, 18.36 614.64; control antibody treated, 18.38 6 11.46; vehicletreated, 21.68 6 11.73] and returning to preinjury valuesat day 8 (serum creatinine) and day 6 (urinary proteinexcretion). Nevertheless, the protective effect of anti–ICAM-1/anti–LFA-1 antibody treatment as applied inthese HgCl2 ARF rats was proven by comparison ofserum creatinine values of control mAb-treated ischemicrats with serum creatinine values of anti–ICAM-1/anti–LFA-1 treated ischemic rats. The former rose signifi-cantly higher (3.80 6 0.20 mg/dL) 24 hours after exposureto ischemia than the latter (2.20 6 0.95 mg/dL). Variationin individual body weight (initial weight loss until day 3,stabilization and weight gain thereupon) did not differbetween the different HgCl2 treatment groups (data notshown; paired t-test of data from days 21,1, 2, 4, 6, 8,and 10 with day 0.)
Renal tubular morphology. After the injection of mer-curic chloride, proximal tubular epithelial cell injury be-came equally pronounced in all treatment groups, withup to 50% of the examined S3 segment cross-sectionsappearing completely necrotic at day 2 (Fig. 6). Onlyminor lesions were seen in proximal convoluted tubulesand in distal tubules (data not shown). Necrotic tubularFig. 2. In vivo binding to rat endothelium of injected anti-intercellular
adhesion molecule-1 (ICAM-1; mAb WT.1). Immunohistochemical cells were gradually replaced by healthy cells at a compa-staining by mere addition of secondary polyclonal (horse-anti-mouse) rable rate in all treatment groups.antibody, that is without primary specific mAb of (A) renal tissue
Tubular epithelial cell and interstitial cell prolifera-sections originating from rats treated with anti–ICAM-1/anti–LFA-1mAbs revealed intense staining in the renal interstitium as well as tion. Twenty-four hours after mercuric chloride injec-in glomeruli presumably representing ICAM-1–expressing endothelial tion, PCNA-positive cells were almost completely absentcells to which injected mouse-anti-rat ICAM-1 (mAb 1A29) is bound.
in proximal straight (S3) tubules (Fig. 7 A–C). By day 2,(B) Renal tissue originating from rats treated with control mAb werenegative. (C) Immunohistochemical staining with anti–ICAM-1 mAb their number had significantly increased, and this wasfrom renal tissue obtained from rats treated with control mAb four equally pronounced in all treatment groups. Thereafter,days after mercuric chloride injection shows the ICAM-1 expression to
individuals of both control antibody and vehicle-treatedcorrespond with horse-anti-mouse staining pattern in A (magnification10 3 1.25 3 25). groups continued increasing their number of PCNA-posi-

Ghielli et al: Antibodies to adhesion molecules in toxic ARF 1127
Fig. 3. Interstitial infiltration of mononuclear cells. The number of positive cells upon immunohistochemical staining of renal tissue sections with(A–C ) mAb ED1 (macrophages), (D–F ) mAb OX-8 (CD8-positive cells), and (G–I ) mAb OX-35 (CD4-positive cells). Whereas control antibody(B, E, and H) and vehicle (C, F, and I)-treated rats showed marked interstitial numbers of these three leukocyte subsets, infiltration was suppressedin anti–ICAM-1/anti–LFA-1 (A, D, and G) treated rats. Nevertheless, macrophages were apparent at day 6 in the anti-adhesion treated group.*P , 0.05 as compared with non–HgCl2-injected control within one treatment group. #P , 0.05 between anti-adhesion and control mAb-treatedgroups. 8P , 0.05 between anti-adhesion and vehicle-treated groups.

Ghielli et al: Antibodies to adhesion molecules in toxic ARF1128
Fig. 4. Macrophages in the rat kidney afterHgCl2 injection. Immunohistochemical stain-ing of renal tissue sections obtained at day 4from (A) control antibody-treated rats and(B) anti–ICAM-1/anti–LFA-1 treated rats.Microwave treatment 1 mAb ED1 (magnifi-cation 10 3 1.25 3 25).
tive S3 segment cells to a maximum at day 4. By day 10, flammatory leukocytes to a site of injury. The initiationproximal tubular cell proliferation had returned closely of these events is mostly due to tissue damage, whichback to normal in each group. Also, distal tubular cells can trigger the release of early inflammatory cytokinesshowed generally increased PCNA positivity, which in such as IL-1 and TNF-a, and thereby induce the expres-both control antibody and vehicle-treated groups started sion of a number of chemokines as well as the up-regula-and peaked already at day 2, while the anti–ICAM-1/ tion of adhesion molecules. Injection of 2 mg/kg mercuricanti–LFA-1 treated group showed pronounced distal tu- chloride in the rat results in rapid renal tubular epithelialbular cell proliferation only at day 6 (Fig. 7 D–F). Some cell necrosis, which is maximal already after 24 hoursinterstitial cells were PCNA positive in all three groups [49, 50] and which mainly affects the proximal straight(Mann–Whitney U-test, *P , 0.05 as compared with tubule (PST). Backleak of tubular fluid caused by ob-non-HgCl2 injected control within one treatment group; struction and tubuloglomerular feedback, both causingFig. 7 G–I). an increase of serum creatinine concentration (used as
measure of renal function), are secondary to the necroticDISCUSSION lesions [51]. In the present experiment, injection of rats
with HgCl2 characteristically induced severe renal tubu-Local expression of chemoattractant cytokines arethought to be responsible for the accumulation of in- lar epithelial cell necrosis in the PST, and this was equally

Ghielli et al: Antibodies to adhesion molecules in toxic ARF 1129
Fig. 5. Alterations in renal function. (A–C ) Serum creatinine concentration and (D–F ) urinary protein excretion increased after mercuric chlorideinjection to a maximum at day 2, after which both these parameters for renal function recovered. The alterations were equivalent in all treatmentgroups. (Paired t-test between data from days 21, 1, 2, 4, 6, 8, and 10, and day 0.)
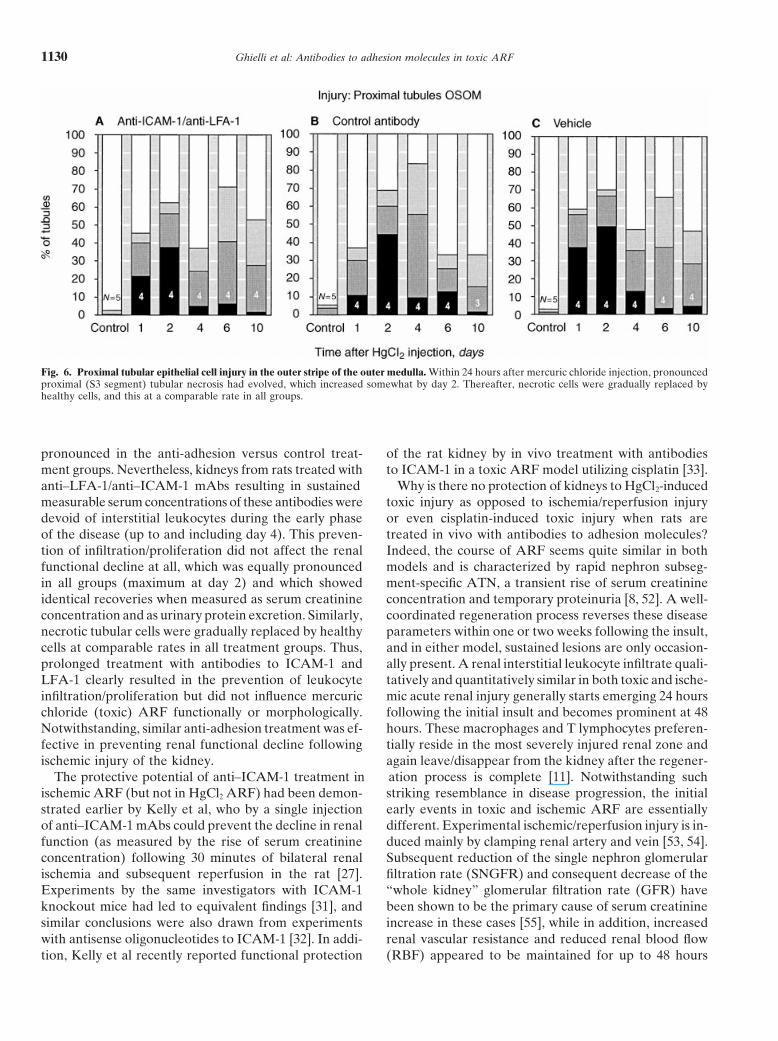
Ghielli et al: Antibodies to adhesion molecules in toxic ARF1130
Fig. 6. Proximal tubular epithelial cell injury in the outer stripe of the outer medulla. Within 24 hours after mercuric chloride injection, pronouncedproximal (S3 segment) tubular necrosis had evolved, which increased somewhat by day 2. Thereafter, necrotic cells were gradually replaced byhealthy cells, and this at a comparable rate in all groups.
pronounced in the anti-adhesion versus control treat- of the rat kidney by in vivo treatment with antibodiesto ICAM-1 in a toxic ARF model utilizing cisplatin [33].ment groups. Nevertheless, kidneys from rats treated with
anti–LFA-1/anti–ICAM-1 mAbs resulting in sustained Why is there no protection of kidneys to HgCl2-inducedtoxic injury as opposed to ischemia/reperfusion injurymeasurable serum concentrations of these antibodies were
devoid of interstitial leukocytes during the early phase or even cisplatin-induced toxic injury when rats aretreated in vivo with antibodies to adhesion molecules?of the disease (up to and including day 4). This preven-
tion of infiltration/proliferation did not affect the renal Indeed, the course of ARF seems quite similar in bothmodels and is characterized by rapid nephron subseg-functional decline at all, which was equally pronounced
in all groups (maximum at day 2) and which showed ment-specific ATN, a transient rise of serum creatinineconcentration and temporary proteinuria [8, 52]. A well-identical recoveries when measured as serum creatinine
concentration and as urinary protein excretion. Similarly, coordinated regeneration process reverses these diseaseparameters within one or two weeks following the insult,necrotic tubular cells were gradually replaced by healthy
cells at comparable rates in all treatment groups. Thus, and in either model, sustained lesions are only occasion-ally present. A renal interstitial leukocyte infiltrate quali-prolonged treatment with antibodies to ICAM-1 and
LFA-1 clearly resulted in the prevention of leukocyte tatively and quantitatively similar in both toxic and ische-mic acute renal injury generally starts emerging 24 hoursinfiltration/proliferation but did not influence mercuric
chloride (toxic) ARF functionally or morphologically. following the initial insult and becomes prominent at 48hours. These macrophages and T lymphocytes preferen-Notwithstanding, similar anti-adhesion treatment was ef-
fective in preventing renal functional decline following tially reside in the most severely injured renal zone andagain leave/disappear from the kidney after the regener-ischemic injury of the kidney.
The protective potential of anti–ICAM-1 treatment in ation process is complete [11]. Notwithstanding suchstriking resemblance in disease progression, the initialischemic ARF (but not in HgCl2 ARF) had been demon-
strated earlier by Kelly et al, who by a single injection early events in toxic and ischemic ARF are essentiallydifferent. Experimental ischemic/reperfusion injury is in-of anti–ICAM-1 mAbs could prevent the decline in renal
function (as measured by the rise of serum creatinine duced mainly by clamping renal artery and vein [53, 54].Subsequent reduction of the single nephron glomerularconcentration) following 30 minutes of bilateral renal
ischemia and subsequent reperfusion in the rat [27]. filtration rate (SNGFR) and consequent decrease of the“whole kidney” glomerular filtration rate (GFR) haveExperiments by the same investigators with ICAM-1
knockout mice had led to equivalent findings [31], and been shown to be the primary cause of serum creatinineincrease in these cases [55], while in addition, increasedsimilar conclusions were also drawn from experiments
with antisense oligonucleotides to ICAM-1 [32]. In addi- renal vascular resistance and reduced renal blood flow(RBF) appeared to be maintained for up to 48 hourstion, Kelly et al recently reported functional protection

Ghielli et al: Antibodies to adhesion molecules in toxic ARF 1131
Fig. 7. Proliferating cell nuclear antigen (PCNA) expression as an indicator of cell proliferation. PCNA positivity was highest in proximal tubules(A–C ), although some distal tubular (D–F ) as well as interstitial (G–I ) cell proliferation also was apparent. Peak proliferation in proximal tubules(day 4) was higher in control antibody (B) and vehicle (C)-treated rats as compared with anti–ICAM-1/anti–LFA-1 (A)-treated rats. Also, distaltubular proliferation started sooner (day 2) in control antibody (E) and vehicle (F) treated rats as compared with anti-adhesion treated rats (D).*P , 0.05 as compared with non-HgCl2 injected control within one treatment group.

Ghielli et al: Antibodies to adhesion molecules in toxic ARF1132
after ischemia. Toxic ARF is experimentally induced when an increase of MPO activity and serum creatinineare already reversing and when morphological injury noby an injection of a compound that becomes directly
tubulotoxic [49]. Conger and Falk demonstrated that the longer augments [8, 11]. Clearly, there seems to be asingle nephron glomerular filtration rate (SNGFR) after dissociation between the time sequence of leukocyte in-HgCl2 injection was unchanged when measured at Bow- filtration and increase of MPO activity. One could sug-man’s space, but when measured in the late proximal gest that MPO activity serves more adequately as atubule, SNGFR was significantly reduced [51]. More- marker for leukocyte activation rather than as a measureover, a reduction of the glomerular ultrafiltration coeffi- for leukocyte number. Therefore, one could argue thatcient in this case was not associated with alterations in PMN cells and/or other leukocytes act during ischemiaglomerular plasma flow and glomerular capillary pres- by means of a “hit and run” phenomenon, during whichsure. The authors concluded that backleak of tubular they do not infiltrate, but only attach to the endotheliumfluid secondary to the necrotic lesions is the primary long enough either to release highly reactive enzymescause of decreased GFR and rise of serum creatinine in and oxygen species, or to support molecular interactionsHgCl2 ARF, while alterations in renal hemodynamics involving ICAM-1 presumably inducing intracellular sig-seemed less important in this model. For a matter of naling events controlling vascular tonus or to simplycompleteness, it should be mentioned that when mercu- block vascular flow in the capillary network [65, 66]. Thatric chloride was administered to rats at extremely high ICAM-1 must be an important target at an early stage indoses (5 to 12 mg/kg), some authors no longer could the disease process of renal ischemic/reperfusion injury isexclude a vascular component as contributing to the re- indeed indicated by the fact that anti–ICAM-1 mAbsduction of whole kidney GFR [56–58]. Finally, direct were effective in Kelly et al’s experiment when giventoxicity to renal tubular epithelial cells associated with before, during, or two hours after ischemia; however,vascular congestion is said to contribute to renal dysfunc- when given eight hours after ischemia, the antibody ef-tion following exposure to cisplatin [59–61]. fect was abrogated. That such initial renal vascular alter-
Kelly et al interpreted the results obtained by ischemic ations cannot be demonstrated in mercuric chloride ARFmodels as supportive for the widely accepted concept but are important in cisplatin-induced renal pathologythat PMNs, which are thought to be the major source [59–61] may explain why antibodies to ICAM-1 also pre-of myeloperoxidase (MPO), are important mediators of vent increased serum creatinine in the latter model, butischemic/reperfusion injury. Indeed, they found that remain ineffective in HgCl2 ARF.MPO activity in renal tissue homogenates from anti– In conclusion, treatment with anti–LFA-1/anti–ICAM-1ICAM-1-treated rats was reduced as compared with this mAbs clearly prevented leukocyte influx/proliferation infrom control mAb-treated rats, the latter of which was the HgCl2-injured kidney when compared with controlmarkedly increased as early as 4 hours after release of antibody treatments. Transient functional deteriorationclamps, rising until 24 hours, but decreasing thereafter. and regeneration were nevertheless equally pronouncedAnti–ICAM-1 mAbs were thus considered to have inhib- in all HgCl2 ARF groups, whereas anti–ICAM-1/anti–ited PMNs from adhering/infiltrating the ischemic kidney LFA-1 treatment resulted in functional protection of ratand from releasing their highly reactive substances (pro- kidneys from ischemia. Hence, treatment with mAbs di-teolytic enzymes, oxygen radicals, etc.), which would oth- rected against ICAM-1 and LFA-1 does not lessen or ag-erwise have caused significant damage and an associated gravate mercuric chloride-induced ARF in the rat. Therise in serum creatinine. It has been insufficiently investi- absence of an anti–ICAM-1/anti–LFA-1 antibody treat-gated, however, which cells are actually responsible for ment effect in HgCl2 as opposed to ischemic ARF or tothis early increased renal MPO activity. Bradley et al toxic (cisplatin) ARF associated with hemodynamic alter-emphasized that MPO measurement indicates neutro- ations reflects the relative unimportance of the vascularphils as well as macrophages, since both leukocyte sub- component leading to reduced GFR in the HgCl2 ARF.sets contain the enzyme [62]. Other methods frequentlyapplied to demonstrate PMN cells in tissue, including ACKNOWLEDGMENTS“antineutrophil serum,” monoclonal, and polyclonal an-
Dr. Ghielli is the recipient of a grant of the “Vlaams instituut voortibodies, and some enzymatic stains also appear to bede bevordering van het wetenschappelijk-technologisch onderzoek in
nonspecific and unable to distinguish between PMNs and de industrie (IWT).” This work could not have been presented withoutthe excellent technical assistance of Ms. Simonne Dauwe, Ms. Hildemacrophages [63, 64]. We and others could not, however,Geryl, Ms. Rita Marijnissen, and Mr. August Van Laer. Rats wereobserve neutrophils in the kidney within either a fewcared for by Mr. Fernand Dierckx, Ms. Lieven Ruysschaert and Ms.
hours or several days following ischemia on hematoxylin Maria Vanderheijden (university animal lab). We thank Mr. GeertBehets for his aid with proper injection of the animals. The hematologyand eosin-stained renal tissue sections (cell nuclei stilllaboratory kindly permitted the use of their blood cell analyzer (Tech-serving as the golden standard for recognizing PMNs)nicon H1). Some metabolic cages were provided by Dr. Paul Dierckx
[8, 61]. On the other hand, macrophages have only been (Instituut voor Hygiene en Epidemiologie, labo Toxicologie, Brussels,Belgium). Graphics design has been the responsibility of Mr. Dirk deshown to start infiltrating the ischemic kidney at a time

Ghielli et al: Antibodies to adhesion molecules in toxic ARF 1133
Weerdt. Mr. Erik Snelders, Mr. Eddy Van Hout, and Ms. Diane Kint 17. Adams DH, Shaw S: Leukocyte-endothelial interactions and regu-lation of leukocyte migration. Lancet 343:831–836, 1994are thanked for their secretarial work. Part of this work was presented
at the 1998 conference of the European Dialysis and Transplantation 18. Brady HR: Leukocyte adhesion molecules and kidney diseases.Kidney Int 45:1285–1300, 1994Association (EDTA) in Rimini, Italy, and was published in abstract
form (Nephrol Dial Transplant 13:A51, 1998). 19. Brady HR: Leukocyte adhesion molecules, in Molecular Nephrol-ogy: Kidney Function in Health and Disease, edited by Schlon-dorff D, Bonventre JV, New York, Marcel Dekker, 1995, ppReprint requests to Dr. Marc E. De Broe, Department of Nephrology-
Hypertension, University of Antwerp, p/a University Hospital, Wilrijk- 717–74020. Hebert M-J, Brady HR: Leukocyte adhesion, in Immunologicstraat 10, B-2650 Edegem, Belgium.
E-mail: [email protected] Renal Disease, edited by Neilson EG, Couser WG, Philadelphia,Lippincott-Raven Publishers, 1997, pp 519–546
21. Carlos TM, Harlan JM: Leukocyte-endothelial adhesion mole-cules. Blood 84:2068–2101, 1994APPENDIX
22. Arnaout MA: Structure and function of leukocyte adhesion mole-cules CD11/CD18. Blood 75:1037–1050, 1990Abbreviations used in this article are: ABTS, 2,29azino-di (3-ethyl
benstialozine sulfonate); ARF, acute renal failure; ATN, acute tubular 23. Arnaout MA: Leukocyte adhesion molecules deficiency: Its struc-tural basis, pathophysiology and implications for modulating thenecrosis; FCS, fetal calf serum; FITC, fluoresceine isothyocyanate;
h-PLAP, anti-human placental alkaline phosphatase; ICAM-1, intercel- inflammatory response. Immunol Rev 114:145–180, 199024. Springer TA: Traffic signals for lymphocyte recirculation and leu-lular adhesion molecule-1; IL-1, interleukin-1; I/R, ischemia/reperfu-
sion; LFA-1, leukocyte function associated antigen-1; mAb, mono- kocyte emigration: The multistep paradigm. Cell 76(2):301–413,1994clonal antibody; MPO, myeloperoxidase; OSOM, outer stripe of the
outer medulla; PBL, peripheral blood leukocyte; PBS, phosphate-buf- 25. Wuthrich RP: Intercellular adhesion molecules and vascular celladhesion molecule-1 and the kidney. J Am Soc Nephrol 3:1201–fered saline; PCNA, proliferating cell nuclear antigen; PST, proximal
straight tubule; RAM-b, biotinylated rabbit-anti-mouse immunoglobu- 1211, 199226. Gahmberg CG: Leukocyte adhesion: CD11/CD18 integrins andlins; SA-PE, phycoerythrin conjugated streptavidin; SDS-PAGE, so-
dium dodecyl sulfate–polyacrylamide gel electrophoresis; TNF, tumor intercellular adhesion molecules. Curr Opin Cell Biol 9:643–650,1997necrosis factor; TNP, trinitrophenol.
27. Kelly KJ, Winfred WW, Colvin RB, Bonventre JV: Antibodyto intercellular adhesion molecule 1 protects the kidney against
REFERENCES ischemic injury. Proc Natl Acad Sci USA 91:812–816, 199428. Hawkins HK, Entman ML, Zhu JY, Youker KA, Berens K,1. Toback FG: Regeneration after acute tubular necrosis. Kidney Int
Dore M, Smith CW: Acute inflammatory reaction after myocardial41:226–246, 1992ischemic injury and reperfusion. Am J Pathol 148:1957–1969, 19962. Liu S, Humes HD: Cellular and molecular aspects of renal repair
29. Farhood A, McGuire GM, Manning AM, Miyasaka M, Smithin acute renal failure. Curr Opin Nephrol Hypertens 2:618–624,WC, Jaeschke H: Intercellular adhesion molecule 1 expression1993and its role in neutrophil-induced ischemia-reperfusion injury in3. Wolf G, Neilson EG: Molecular mechanisms of tubulointerstitialrat liver. J Leukoc Biol 57:368–374, 1995hypertrophy and hyperplasia. Kidney Int 39:401–420, 1991
30. Oshiro Y, Marubayashi S, Maede T, Asahara T, Fukuda Y,4. Fish EM, Molitoris BA: Alterations in epithelial polarity and theYamada K, Koyama S, Ito H, Miyasaka M, Dohi K: Contributionpathogenesis of disease states. N Engl J Med 330:1580–1588, 1994of neutrophils and effect of monoclonal antibodies to adhesion5. Neilson EG: Pathogenesis and therapy of interstitial nephritis.molecules on ischemia and reperfusion injury in rat livers. Trans-Kidney Int 35:1257–1270, 1989plant Proc 27:743–744, 19956. Eddy AA, Michael AF: Acute tubulointerstitial nephritis associ-
31. Kelly KJ, Williams WW, Colvin RB, Meehan SM, Springerated with aminonucleoside nephrosis. Kidney Int 33:14–23, 1988TA, Gutierrez-Ramos JC, Bonventre JV: Intercellular adhesion7. Verstrepen WA: Acute renal failure in the rat: Temporal andmolecule-1 deficient mice are protected against renal ischemia.spatial distribution patterns of tubular injury, cell proliferation andJ Clin Invest 97:1056–1063, 1996infiltration in relation to endogenous growth factor expression.
32. Haller H, Dragun D, Miethke A, Park JK, Weis A, LippolstPhD Thesis, Universitaire Instelling Antwerpen, 1995A, Grob V, Luft FC: Antisense oligonucleotides for ICAM-18. Takada M, Nadeau KC, Shaw GD, Marquette KA, Tilney NL:attenuate reperfusion injury and renal failure in the rat. KidneyThe cytokine-adhesion molecule cascade in ischemia/reperfusionInt 50:473–480, 1996injury of the rat kidney: Inhibition by a soluble P-selectin ligand.
33. Kelly KJ, Meehan SH, Colvin RB, Williams WW, BonventreJ Clin Invest 99:2682–2690, 1997JV: Protection from toxicant-mediated renal injury in the rat with9. Schreiner GF, Harris KPG, Purkerson ML, Klahr S: Immuno-anti-CD54 antibody. Kidney Int 56:922–931, 1999logical aspects of acute ureteral obstruction: Immune cell infiltrate
34. Verstrepen WA, Nouwen EJ, Zhu MQ, Ghielli M, De Broein the kidney. Kidney Int 34:487–493, 1988ME: Time course of growth factor expression in mercuric chloride10. Eddy A, McCulloch L, Adams J, Liu E: Interstitial nephritisacute renal failure. Nephrol Dial Transplant 10:1361–1371, 1995induced by protein-overload proteinuria. Am J Pathol 135:719–733,
35. Hendrix PG, Hoylaerts MF, Nouwen EJ, De Broe ME: Enzyme1989immunoassay of human placental and germ-cell alkaline phospha-11. Ghielli M, Verstrepen WA, Nouwen EJ, De Broe ME: Regener-tase in serum. Clin Chem 36:1793–1799, 1990ation processes in the kidney after acute injury: Role of infiltrating
36. Tamatani T, Kotani M, Miyasaka M: Characterization of the ratcells. Exp Nephrol 6:502–507, 1998leukocyte integrin, CD11/CD18, by use of LFA-1 subunit-specific12. Ghielli M, Verstrepen WA, Nouwen EJ, De Broe ME: Inflam-monoclonal antibodies. Eur J Immunol 21:627–633, 1991matory cells in renal regeneration. Ren Fail 18:255–275, 1996
37. Tamatani T, Miyasaka M: Identification of monoclonal antibodies13. Kashem A, Endoh M, Yano N, Yamanchi F, Nanoto Y, Sakaireactive with the rat homologue of ICAM-1, and evidence for aH: Expression of inducible-NOS in human glomerulonephritis: Thedifferential involvement of ICAM-1 in the adherence of restingpossible source is infiltrating monocytes/macrophages. Kidney Intversus activated lymphocytes to high endothelial cells. Int Immunol50:392–399, 19962:165–171, 199014. Schreiner GF, Kohan DE: Regulation of renal transport processes
38. Brideau JR, Philip BC, McMaster WR, Mason DW, Williamsand hemodynamics by macrophages and lymphocytes. Am J Phys-AF: Two subsets of rat T lymphocytes defined with monoclonaliol 27:F761–F767, 1990antibodies. Eur J Immunol 10:609–615, 198015. Kunt TK: Basic principles of wound healing. J Trauma 30(Suppl
39. Jefferies WA, Green JR, Williams AF: Authentic T helper CD412):S122–S128, 1990(W3/25) on rat peritoneal macrophages. J Exp Med 162:117–127,16. Strutz F, Neilson EG: The role of lymphocytes in the progression
of interstitial disease. Kidney Int 45(Suppl 45):S106–S110, 1994 1985

Ghielli et al: Antibodies to adhesion molecules in toxic ARF1134
40. Garcia RL, Coltrera MD, Gown AM: Analysis of proliferative mercuric chloride induced acute renal failure in the rat. ExpNephrol 5:69–81, 1997grade using anti-PCNA/cyclin monoclonal antibodies in fixed, em-
53. Lieberthal W: Biology of ischemic and toxic renal tubular cellbedded tissues: Comparison with flow cytometric analysis. Am Jinjury: Role of nitric oxide and the inflammatory response. CurrPathol 134:733–739, 1989Opin Nephrol Hypertens 7:289–295, 199841. Dijkstra CD, Dopp EA, Joling P, Kraal G: The heterogeneity of
54. Lauriat S, Linas SL: The role of neutrophils in acute renal failure.mononuclear phagocytes in lymphoid organs: Distinct macrophageSemin Nephrol 18:498–504, 1998subpopulations in the rat recognized by monoclonal antibodies
55. Kon V, Yoshioka T, Fogo A, Ichikawa I: Glomerular actions ofED1, ED2 and ED3. Immunology 54:589–599, 1985endothelin in vivo. J Clin Invest 83:1762–1767, 198942. Falkenberg FW, Weichert H, Krane M, Bartels I, Nagels HO,
56. Flanigan WJ, Oken DE: Renal micropuncture study of the devel-Fiebig H: In vitro production of monoclonal antibodies in highopment of anuria in the rat with mercury-induced acute renalconcentration in a new and easy to handle modular minifermenter.failure. J Clin Invest 44:449–457, 1965J Immunol Methods 179:13–29, 1995 57. Flamenbaum W, McDonald FD, DiBona GF, Oken DE: Micro-
43. Savelkoul HF, Vossen AC, Breedland EG, Tibbe GJ: Semi- puncture study of renal tubular factors in low dose mercury poison-preparative purification and validation of monoclonal antibodies ing. Nephron 8:221–234, 1971for immunotherapy in mice. J Immunol Methods 172:33–42, 1994 58. Vanholder R, Praet M, Pattyn P, Vakaet L, Ringoir S, Lameire
44. Coligan JE, Kruisbeek AM, Marguilies DH, Shevach EM, N: Role of early renal vasoconstriction in the pathophysiology ofStrober W: Current Protocols in Immunology. New York, Wiley, acute renal failure induced by mercuric chloride, in Acute Renal1994, p 5.3.5 Failure, edited by Eliahou HE, New York, John Libbey and Co.,
45. Coligan JE, Kruisbeek AM, Marguilies DH, Shevach EM, 1982, pp 206–209Strober W: Current Protocols in Immunology. New York, Wiley, 59. Safirstein R, Winston J, Guttenplan J: Cisplatin nephrotoxicity:1994, p 5.3.6 Physiological and biochemical aspects, in Biochemical Mechanisms
46. Seelig HP: The Jaffe reaction with creatinine. Reaction product of Platinum Antitumor Drugs, edited by McBrien DCH, SlaterTF, Oxford, IRL Press, 1986, pp 271–306and general reaction conditions. Z Klin Chem Klin Biochem 7:248–
60. Luke DR, Vadei K, Lopez-Berestein G: Role of vascular conges-254, 1969tion in cisplatin-induced acute renal failure in the rat. Nephrol47. Bradford M: A rapid and sensitive method for the quantitationDial Transplant 7:1–7, 1992of microgram quantities of protein utilizing the principle of protein-
61. Dos Santos OF, Boim MA, Barros EJ, Schor N: Role of plateletdye binding. Anal Biochem 72:248–254, 1976activating factor in gentamicin and cisplatin nephrotoxicity. Kidney48. Ghielli M, Verstrepen WA, Helbert M, Verhulst A, NouwenInt 40:742–747, 1991EJ, De Broe ME: Phenotypical characterization of rat inflamma-
62. Bradley PP, Priebat DA, Christensen RD, Rothstein G: Mea-tory leukocytes by immuno-flowcytometry and immuno-histo-surement of cutaneous inflammation: Estimation of neutrophil con-chemsitry, in Role of Inflammatory Cells in Injury and Regenerationtent with an enzyme marker. J Invest Dermatol 78:206–209, 1982of the Kidney, PhD Thesis, Universitairte Instelling Antwerpen,
63. De Greef KE, Ysebaert DK, Ghielli M, Vercauteren S,1999Nouwen EJ, Eyskens EJ, De Broe ME: Neutrophils and acute49. Diamond GL, Zalups RK: Understanding renal toxicity of heavy ischemia-reperfusion injury. J Nephrol 11:110–122, 1998
metals. Toxicol Pathol 26:92–103, 1998 64. Ysebaert DK, De Greef KE, Vercauteren SR, Ghielli M,50. Zalups RK, Knutson KL, Schnellmann RG: In vitro analysis of Nouwen EJ, Verpooten GA, Eyskens EJ, De Broe ME: Leuko-
the accumulation and toxicity of inorganic mercury in segments cyte infiltration in relation to damage and regeneration of the ratof the proximal tubule isolated from the rabbit kidney. Toxicol kidney after severe warm ischemia/reperfusion injury. Kidney IntAppl Pharmacol 119:221–227, 1993 56:601–611, 1999
51. Conger JD, Falk SA: Glomerular and tubular dynamics in mercu- 65. Bonventre JV, Colvin RB: Adhesion molecules and renal disease.ric chloride-induced acute renal failure. J Lab Clin Med 107:281– Curr Opin Nephrol Hypertens 5:254–261, 1996289, 1986 66. Rabb H, Postler G: Leukocyte adhesion molecules in ischaemic
52. Ghielli M, Verstrepen WA, Dauwe S, Nouwen EJ, De Broe renal injury: Kidney specific paradigms? Clin Exp Pharmacol Phys-iol 25:286–291, 1998ME: Selective depletion of CD8-positive leukocytes does not alter
Copyright © 2022 FDOKUMEN




















