
AMPK activation enhances PPARα activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling...
7
AMPK activation enhances PPARa activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling pathway Rongsen Meng a,b,d,⇑,1 , Zhaohui Pei c,1 , Aixia Zhang a,b,1 , Yutian Zhou d , Xingming Cai a,b , Baolin Chen a,b , Guanjie Liu e , Weiyi Mai a,b , Jianrui Wei e,⇑ , Yugang Dong a,b,⇑ a Department of Cardiology, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou 510080, PR China b The Key Laboratory on Assisted Circulation of the Ministry of Health, Gaungzhou 510089, PR China c Department of Cardiology, The Third Hospital of Nanchang, Jiangxi 330009, PR China d Department of Cadre Healthcare, Sichuan People’s Hospital, Sichuan 610072, PR China e Department of Cardiology, The Red-cross Hospital of Guangzhou, Guangzhou 510220, PR China article info Article history: Received 20 January 2011 and in revised form 13 April 2011 Available online 21 April 2011 Keywords: AMPK PPARa ERK1/2 p38 Cardiac hypertrophy abstract Activation of adenosine monophosphate-activated protein kinase (AMPK) has been shown to inhibit car- diac hypertrophy through peroxisome proliferators-activated receptor-a (PPARa) signaling pathway, but the detailed mechanism remains unclear. A rat model of cardiac hypertrophy created by transaortic con- striction (TAC) was used to investigate the mechanism involved in regulation of PPARa activity by AMPK. It was observed that treatment with AICAR (5-aminoimidazole 1 carboxamide ribonucleoside), an AMPK activator, significantly inhibited cardiac hypertrophy in vivo and in vitro. Phosphorylated extracellular signal regulated protein kinase (phospho-ERK1/2) and phospho-p38 mitogen-activated protein kinase (MAPK) protein levels were significantly up-regulated, while PPARa protein level was down-regulated in TAC rats. AICAR treatment reversed the changes of PPARa and phospho-ERK1/2, but increased phos- pho-p38 MAPK protein level in TAC rats. Similar changes of PPARa and phospho-ERK1/2 protein levels were observed in the hypertrophied cardiomyocytes induced by phenylephrine treatment. Epidermal growth factor (EGF, ERK1/2 activator), but not SB203580 (p38 inhibitor) blocked the up-regulation of PPARa protein level induced by AICAR. Luciferase assay showed that AICAR increased PPARa transcrip- tional activity which was abrogated by EGF, but not by SB203580. These results demonstrate that AMPK activation enhances the activity of PPARa to inhibit cardiac hypertrophy through ERK1/2, but not p38 MAPK, signaling pathway. Ó 2011 Elsevier Inc. All rights reserved. Introduction Cardiac hypertrophy is one of the major pathological changes associated with cardiovascular diseases. Cardiac hypertrophy is characterized by an increase in myocardial cell size, a higher de- gree of sarcomeric organization, re-activation of the fetal gene pro- gram, and changes in gene transcription and translation resulting in enhanced protein synthesis [1,2]. Adenosine monophosphate (AMP)-activated protein kinase (AMPK) 2 is a serine/threonine kinase that can be activated by cellular stresses and ATP depletion. AMPK has been identified as a key regulator of cellular energy homeostasis [3]. It has been shown that activation of AMPK inhibits protein synthesis and the development of cardiac hypertrophy via a number of pathways, such as eukaryotic elongation factor-2 (eEF2), p70S6 kinase, and mammalian target of rapamycin (mTOR) [4]. Peroxisome proliferators-activated receptor-a (PPARa) is a ligand- activated transcription factor belonging to the nuclear hormone receptor superfamily and considered as a critical regulator of myo- cardial metabolism. Studies have demonstrated that the occurrence of cardiac hypertrophy is associated with the deactivation of PPARa [5,6]. Our recent study showed that AMPK activation inhibits cardiac hypertrophy through reactivation of the PPARa signaling pathway 0003-9861/$ - see front matter Ó 2011 Elsevier Inc. All rights reserved. doi:10.1016/j.abb.2011.04.010 ⇑ Corresponding authors. Addresses: Department of Cardiology, The First Affil- iated Hospital of Sun Yat-sen University, and Ministry of Health Key Laboratory of Assisted Circulation, 74 Zhongshan Road II, Guangzhou 510080, PR China. Fax: +86 20 87755766 8151 (R. Meng, Y. Dong), Department of Cardiology, The Red-cross Hospital of Guangzhou, 396 Tongfuzhong Road, Guangzhou 510220, PR China. Fax: +86 20 34403819 (J. Wei). E-mail addresses: [email protected] (R. Meng), [email protected] (J. Wei), [email protected] (Y. Dong). 1 These authors contributed equally to this work. 2 Abbreviations used: AMPK, adenosine monophosphate-activated protein kinase; PPARa, peroxisome proliferators-activated receptor-a; TAC, transaortic constriction; EGF, epidermal growth factor; mTOR, mammalian target of rapamycin; JNK, c-Jun NH 2 -terminal protein kinase; LV, left ventricle; RV, right ventricle; eEF2, eukaryotic elongation factor-2; PE, phenylephrine. Archives of Biochemistry and Biophysics 511 (2011) 1–7 Contents lists available at ScienceDirect Archives of Biochemistry and Biophysics journal homepage: www.elsevier.com/locate/yabbi
Transcript of AMPK activation enhances PPARα activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling...

Archives of Biochemistry and Biophysics 511 (2011) 1–7
Contents lists available at ScienceDirect
Archives of Biochemistry and Biophysics
journal homepage: www.elsevier .com/ locate/yabbi
AMPK activation enhances PPARa activity to inhibit cardiac hypertrophy via ERK1/2MAPK signaling pathway
Rongsen Meng a,b,d,⇑,1, Zhaohui Pei c,1, Aixia Zhang a,b,1, Yutian Zhou d, Xingming Cai a,b, Baolin Chen a,b,Guanjie Liu e, Weiyi Mai a,b, Jianrui Wei e,⇑, Yugang Dong a,b,⇑a Department of Cardiology, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou 510080, PR Chinab The Key Laboratory on Assisted Circulation of the Ministry of Health, Gaungzhou 510089, PR Chinac Department of Cardiology, The Third Hospital of Nanchang, Jiangxi 330009, PR Chinad Department of Cadre Healthcare, Sichuan People’s Hospital, Sichuan 610072, PR Chinae Department of Cardiology, The Red-cross Hospital of Guangzhou, Guangzhou 510220, PR China
a r t i c l e i n f o
Article history:Received 20 January 2011and in revised form 13 April 2011Available online 21 April 2011
Keywords:AMPKPPARaERK1/2p38Cardiac hypertrophy
0003-9861/$ - see front matter � 2011 Elsevier Inc. Adoi:10.1016/j.abb.2011.04.010
⇑ Corresponding authors. Addresses: Department oiated Hospital of Sun Yat-sen University, and MinistryAssisted Circulation, 74 Zhongshan Road II, Guangzho20 87755766 8151 (R. Meng, Y. Dong), DepartmentHospital of Guangzhou, 396 Tongfuzhong Road, Guan+86 20 34403819 (J. Wei).
E-mail addresses: [email protected] (R. Meng),[email protected] (Y. Dong).
1 These authors contributed equally to this work.
a b s t r a c t
Activation of adenosine monophosphate-activated protein kinase (AMPK) has been shown to inhibit car-diac hypertrophy through peroxisome proliferators-activated receptor-a (PPARa) signaling pathway, butthe detailed mechanism remains unclear. A rat model of cardiac hypertrophy created by transaortic con-striction (TAC) was used to investigate the mechanism involved in regulation of PPARa activity by AMPK.It was observed that treatment with AICAR (5-aminoimidazole 1 carboxamide ribonucleoside), an AMPKactivator, significantly inhibited cardiac hypertrophy in vivo and in vitro. Phosphorylated extracellularsignal regulated protein kinase (phospho-ERK1/2) and phospho-p38 mitogen-activated protein kinase(MAPK) protein levels were significantly up-regulated, while PPARa protein level was down-regulatedin TAC rats. AICAR treatment reversed the changes of PPARa and phospho-ERK1/2, but increased phos-pho-p38 MAPK protein level in TAC rats. Similar changes of PPARa and phospho-ERK1/2 protein levelswere observed in the hypertrophied cardiomyocytes induced by phenylephrine treatment. Epidermalgrowth factor (EGF, ERK1/2 activator), but not SB203580 (p38 inhibitor) blocked the up-regulation ofPPARa protein level induced by AICAR. Luciferase assay showed that AICAR increased PPARa transcrip-tional activity which was abrogated by EGF, but not by SB203580. These results demonstrate that AMPKactivation enhances the activity of PPARa to inhibit cardiac hypertrophy through ERK1/2, but notp38 MAPK, signaling pathway.
� 2011 Elsevier Inc. All rights reserved.
Introduction
Cardiac hypertrophy is one of the major pathological changesassociated with cardiovascular diseases. Cardiac hypertrophy ischaracterized by an increase in myocardial cell size, a higher de-gree of sarcomeric organization, re-activation of the fetal gene pro-gram, and changes in gene transcription and translation resultingin enhanced protein synthesis [1,2]. Adenosine monophosphate
ll rights reserved.
f Cardiology, The First Affil-of Health Key Laboratory of
u 510080, PR China. Fax: +86of Cardiology, The Red-crossgzhou 510220, PR China. Fax:
[email protected] (J. Wei),
(AMP)-activated protein kinase (AMPK)2 is a serine/threoninekinase that can be activated by cellular stresses and ATP depletion.AMPK has been identified as a key regulator of cellular energyhomeostasis [3]. It has been shown that activation of AMPK inhibitsprotein synthesis and the development of cardiac hypertrophy via anumber of pathways, such as eukaryotic elongation factor-2 (eEF2),p70S6 kinase, and mammalian target of rapamycin (mTOR) [4].Peroxisome proliferators-activated receptor-a (PPARa) is a ligand-activated transcription factor belonging to the nuclear hormonereceptor superfamily and considered as a critical regulator of myo-cardial metabolism. Studies have demonstrated that the occurrenceof cardiac hypertrophy is associated with the deactivation of PPARa[5,6]. Our recent study showed that AMPK activation inhibits cardiachypertrophy through reactivation of the PPARa signaling pathway
2 Abbreviations used: AMPK, adenosine monophosphate-activated protein kinase;PPARa, peroxisome proliferators-activated receptor-a; TAC, transaortic constriction;EGF, epidermal growth factor; mTOR, mammalian target of rapamycin; JNK, c-JunNH2-terminal protein kinase; LV, left ventricle; RV, right ventricle; eEF2, eukaryoticelongation factor-2; PE, phenylephrine.

2 R. Meng et al. / Archives of Biochemistry and Biophysics 511 (2011) 1–7
[7]. However, the detailed mechanism(s) by which AMPK activationmodulate the PPARa signaling pathway to inhibit cardiac hypertro-phy is still unclear.
Mitogen-activated protein kinase (MAPK) cascades are highlyconserved signal transduction pathways which, coupled with var-ious extracellular and intracellular signals, play important roles forthe organisms to adapt, survive, and maintain homeostasis. In themammalian heart tissue, MAPK signaling pathways are believed toregulate myocyte growth in response to developmental signals orphysiologic and pathologic stimuli [8–10]. The MAP kinase familyconsists of extracellular signal regulated protein kinase (ERK), c-Jun NH2-terminal protein kinase (JNK), and p38 MAPK. Previousstudies by others have demonstrated that both ERK1/2 activityand p38 expression were increased in cardiac hypertrophy[11,12], suggesting that ERK and p38 MAPK may participate in reg-ulation of myocardial hypertrophy development. PPARa activityhas been shown to be modulated by ERK–MAPK and p38 MAPK[5,13–15]. Since AMPK is an important factor upstream to ERKand p38 MAPK [16–19], it is possible that activation of AMPKmay regulate the activation of PPARa and control the developmentof cardiac hypertrophy through p38 and ERK1/2 MAPK signalingpathway. In the present study, we tested this hypothesis with ratmodel of cardiac hypertrophy and revealed the important rolesof p38 and ERK1/2 MAPK signaling pathway in reactivation ofPPARa by AMPK.
Materials and methods
Materials
Polyclonal antibodies against ERK1/2, phospho-ERK1/2, p38,and phospho-p38 were purchased from Cell Signaling Technology(Beverly, MA, USA). Anti-PPARa and anti-GAPDH antibodies wereobtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).[3H]-leucine was purchased from Perkin Eimer (Boston, MA,USA). Epidermal growth factor (EGF) was from Accurate Chemical(Westbury, NY, USA) and SB203580 was purchased from Upstate(Temecula, CA). Unless otherwise indicated, all other chemicalsand reagents were from Sigma (St. Louis, MO).
Animal models of cardiac hypertrophy
Animal experimentations were carried out in compliance withthe Guide for the Care and Use of Laboratory Animals by the Na-tional Institutes of Health (USA) and the guidelines of the AnimalCare and Use Committees of Sun Yat-sen University. Following a3-day acclimation period, 40 male Sprague–Dawley rats (8-week-old, 230–250 g) were randomly assigned into the following groups:(1) sham; (2) sham with AICAR treatment; (3) transaortic constric-tion (TAC); (4) TAC with AICAR treatment. TAC was created using a5–0 suture tied twice around the abdominal aorta in which a 21-gauge needle was inserted. The needle was then retracted yieldinga 70–80% constriction with an outer aortic diameter of approxi-mately 0.8 mm. In the sham control group, the same surgery wasperformed as described above except that the aorta was not con-stricted. The animals were injected subcutaneously with 0.5 mg AI-CAR/g body wt daily for 8 weeks as previously described [20].Control animals were injected with the same volume of 0.9% NaCldaily.
Neonatal rat cardiomyocyte culture
Neonatal rat cardiomyocytes were prepared from 1- to 3-day-old Sprague–Dawley rats as described previously [21]. The cellswere pre-plated in 6-well plates and cultured at 37 �C for 90 min
and unattached cardiomyocytes were collected, counted andseeded in 6-well culture plates at a density of 3 � 105/well. Bromo-deoxyuridine (0.1 mM) was added to the culture for the first 72 hto deplete fibroblasts and culture medium was changed every48 h. The cardiomyocytes were treated with 0.1 mM AICAR,0.1 lM EGF, 10 lM SB203580 and stimulated with 0.5 lM phenyl-ephrine (PE) at the indicated time. The cultured cardiomyocyteswere randomly divided into the following groups for 24–48 hin vitro drug treatment: (1) control; (2) AICAR treatment; (3) PEtreatment; (4) PE and AICAR treatment; (5) PE, AICAR and EGFtreatment; and (6) PE, AICAR and SB203580 treatment.
Cardiac hypertrophy
Cardiac hypertrophy was measured by septal and posterior wallthicknesses, the ratio of heart to body weight, mean cardiomyocytearea and [3H]-leucine incorporation. Rats were anesthetized withintraperitoneal injection of pentobarbital (50 mg/kg) before sur-gery. Eight weeks after surgery, transthoracic echocardiographywas performed on each rat. Septal and posterior wall thicknesseswere measured at the end-diastolic stage and analyzed by echocar-diographic system (ATL-HDI5000) equipped with a 10-MHz imag-ing transducer. All Echocardiography measurements were carriedout for 10 consecutive cardiac cycles and completed within 24 h.Eight weeks after surgery, rats were sacrificed by decapitationand hearts were perfused with 10 ml of phosphate buffered salineto wash out the blood. The heartbeat was stopped at diastole byperfusion with 14 mM KCl and the left ventricle (LV, free wall plusseptum) was obtained after removal of both atria and the free wallof the right ventricle (RV). The cardiac chambers were weighed andrelative cardiac mass (LV weight/body weight in mg/g) was deter-mined to evaluate cardiac hypertrophy. Subsequently, The LV tis-sues were cut into tow parts, one was stored at �80 �C formolecular biological analysis and the other was embedded in par-affin. Paraffin fixed heart tissues were sectioned at 4-mm thick-ness, and stained with hematoxylin and eosin for morphologyexamination. Myocyte cross-sectional area was calculated by mea-suring 100 cells from sections stained with hematoxylin and eosin.The surface area of cultured cardiomyocytes was determined afterthe cells were incubated with AICAR, PE for 48 h. The surface areaof 100 cells from randomly selected fields was examined for eachgroup. Cell images captured by CCD Camera (Olympus, Tokyo, Ja-pan) were traced and analyzed with Image-pro Plus 5.1 (MediaCyberneties, USA).
[3H]-leucine incorporation was measured as described previ-ously [21]. Briefly, cardiomyocytes were stimulated with PE andAICAR in the presence of [3H]-leucine (1 lCi/mL) for 48 h. The cellswere harvested, washed with Hanks’ solution and treated with 10%trichloroacetic acid at 4 �C for 60 min. The precipitates were dis-solved in NaOH (0.4 N) and aliquots were counted by scintillationcounting.
Western blotting
Protein levels were determined by Western blotting. Heart tis-sue and cells were lysed in RIPA buffer (50 mM Tris–HCl (pH7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X100, 1% Na-deoxycho-late, 0.1% SDS, 1 mM PMSF, 1 lg/ml Aprotinin, and 1 lg/ml Leu-peptin). Equal amount of protein (50 lg) from each sample wasseparated on 12% SDS–PAGE gels and transferred to PVDF mem-branes (Millipore). The membranes were blocked with blockingbuffer (1� TBS, 0.1% Tween 20 with 5% w/v fat-free dry milk),washed and incubated with primary antibodies at 4 �C for 12 h.The membranes were subsequently washed with TBS-T (10 mMTris–HCl, pH 8.0, 150 mM NaCl, and 0.1% Tween 20) and incubatedwith horseradish peroxidase-conjugated anti-mouse or anti-rabbit
R. Meng et al. / Archives of Biochemistry and Biophysics 511 (2011) 1–7 3
IgG antibodies (Santa Cruz Biotechnology) at 37 �C for 1 h. The im-mune complex was visualized with an ECL system (Cell SignalingTechnology). Films were analyzed by densitometry.
Reporter assays
PPARa transcription activity was determined by luciferasereporter assays. Briefly, cardiomyocytes were transiently co-transfected with a plasmid containing the luciferase gene underthe control of three tandem PPAR response elements (PPRE)(pGL3-PPRE TK-luciferase), a PPARa expression plasmid (providedby Yong-xue Liu, Institute of Military Medical Sciences, Beijing,China) and an internal control plasmid DNA using 10 ll ofLipofectamine 2000 reagent (Invitrogen). Twenty-four hours aftertransfection, the cells were treated with AICAR, EGF, SB203580 orPE for 24 h and the PPARa transcriptional activity was measuredaccording to the manufacturer’s instructions.
Statistical analysis
Data are expressed as means ± SD. Significance of differencesbetween groups were tested by one-way ANOVA or paired Stu-dent’s t-test using the SPSS statistical software (Chicago, IL, USA).Differences between groups were considered significant whenthe p < 0.05.
Results
Activation of AMPK inhibits cardiac hypertrophy in vivo and in vitro
To determine whether AMPK activation modulates the develop-ment of cardiac hypertrophy, we analyzed the effects of AICAR
Table 1The end-diastolic intraventricular septal thickness (IVSd) and the end-diastolicposterior wall thickness (LVDd) of rats in different groups.
IVSd (mm) LVDd (mm)
BS AS BS AS
Sham 1.81 ± 0.26 1.86 ± 0.20 1.73 ± 0.26 1.83 ± 0.18Sham + AICAR 1.83 ± 0.25 1.87 ± 0.22 1.77 ± 0.24 1.81 ± 0.19TAC 1.83 ± 0.23 2.39 ± 0.33⁄⁄## 1.76 ± 0.20 2.35 ± 0.29⁄⁄##
TAC + AICAR 1.82 ± 0.24 2.09 ± 0.20⁄#$$ 1.73 ± 0.29 1.99 ± 0.22⁄$$
Data are expressed as means ± SD, n = 10. BS, before surgery; AS, 8 weeks aftersurgery. TAC, transaortic constriction. ⁄p < 0.05 and ⁄⁄p < 0.01, in comparison withBS in the same group (paired Student’s t-test); #p < 0.05 and ##p < 0.01, in com-parison with sham group; $$p < 0.01, in comparison with TAC only group (one-wayANOVA).
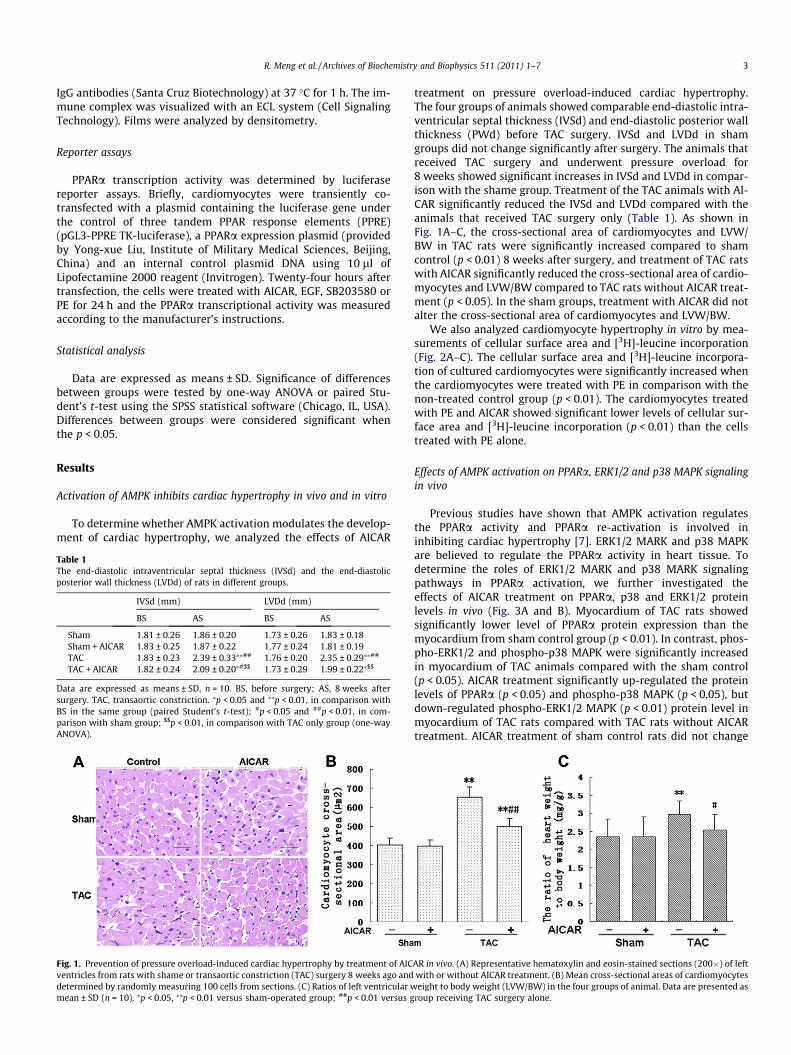
Fig. 1. Prevention of pressure overload-induced cardiac hypertrophy by treatment of AICventricles from rats with shame or transaortic constriction (TAC) surgery 8 weeks ago anddetermined by randomly measuring 100 cells from sections. (C) Ratios of left ventricular wmean ± SD (n = 10). ⁄p < 0.05, ⁄⁄p < 0.01 versus sham-operated group; ##p < 0.01 versus g
treatment on pressure overload-induced cardiac hypertrophy.The four groups of animals showed comparable end-diastolic intra-ventricular septal thickness (IVSd) and end-diastolic posterior wallthickness (PWd) before TAC surgery. IVSd and LVDd in shamgroups did not change significantly after surgery. The animals thatreceived TAC surgery and underwent pressure overload for8 weeks showed significant increases in IVSd and LVDd in compar-ison with the shame group. Treatment of the TAC animals with AI-CAR significantly reduced the IVSd and LVDd compared with theanimals that received TAC surgery only (Table 1). As shown inFig. 1A–C, the cross-sectional area of cardiomyocytes and LVW/BW in TAC rats were significantly increased compared to shamcontrol (p < 0.01) 8 weeks after surgery, and treatment of TAC ratswith AICAR significantly reduced the cross-sectional area of cardio-myocytes and LVW/BW compared to TAC rats without AICAR treat-ment (p < 0.05). In the sham groups, treatment with AICAR did notalter the cross-sectional area of cardiomyocytes and LVW/BW.
We also analyzed cardiomyocyte hypertrophy in vitro by mea-surements of cellular surface area and [3H]-leucine incorporation(Fig. 2A–C). The cellular surface area and [3H]-leucine incorpora-tion of cultured cardiomyocytes were significantly increased whenthe cardiomyocytes were treated with PE in comparison with thenon-treated control group (p < 0.01). The cardiomyocytes treatedwith PE and AICAR showed significant lower levels of cellular sur-face area and [3H]-leucine incorporation (p < 0.01) than the cellstreated with PE alone.
Effects of AMPK activation on PPARa, ERK1/2 and p38 MAPK signalingin vivo
Previous studies have shown that AMPK activation regulatesthe PPARa activity and PPARa re-activation is involved ininhibiting cardiac hypertrophy [7]. ERK1/2 MARK and p38 MAPKare believed to regulate the PPARa activity in heart tissue. Todetermine the roles of ERK1/2 MARK and p38 MARK signalingpathways in PPARa activation, we further investigated theeffects of AICAR treatment on PPARa, p38 and ERK1/2 proteinlevels in vivo (Fig. 3A and B). Myocardium of TAC rats showedsignificantly lower level of PPARa protein expression than themyocardium from sham control group (p < 0.01). In contrast, phos-pho-ERK1/2 and phospho-p38 MAPK were significantly increasedin myocardium of TAC animals compared with the sham control(p < 0.05). AICAR treatment significantly up-regulated the proteinlevels of PPARa (p < 0.05) and phospho-p38 MAPK (p < 0.05), butdown-regulated phospho-ERK1/2 MAPK (p < 0.01) protein level inmyocardium of TAC rats compared with TAC rats without AICARtreatment. AICAR treatment of sham control rats did not change
AR in vivo. (A) Representative hematoxylin and eosin-stained sections (200�) of leftwith or without AICAR treatment. (B) Mean cross-sectional areas of cardiomyocyteseight to body weight (LVW/BW) in the four groups of animal. Data are presented as
roup receiving TAC surgery alone.
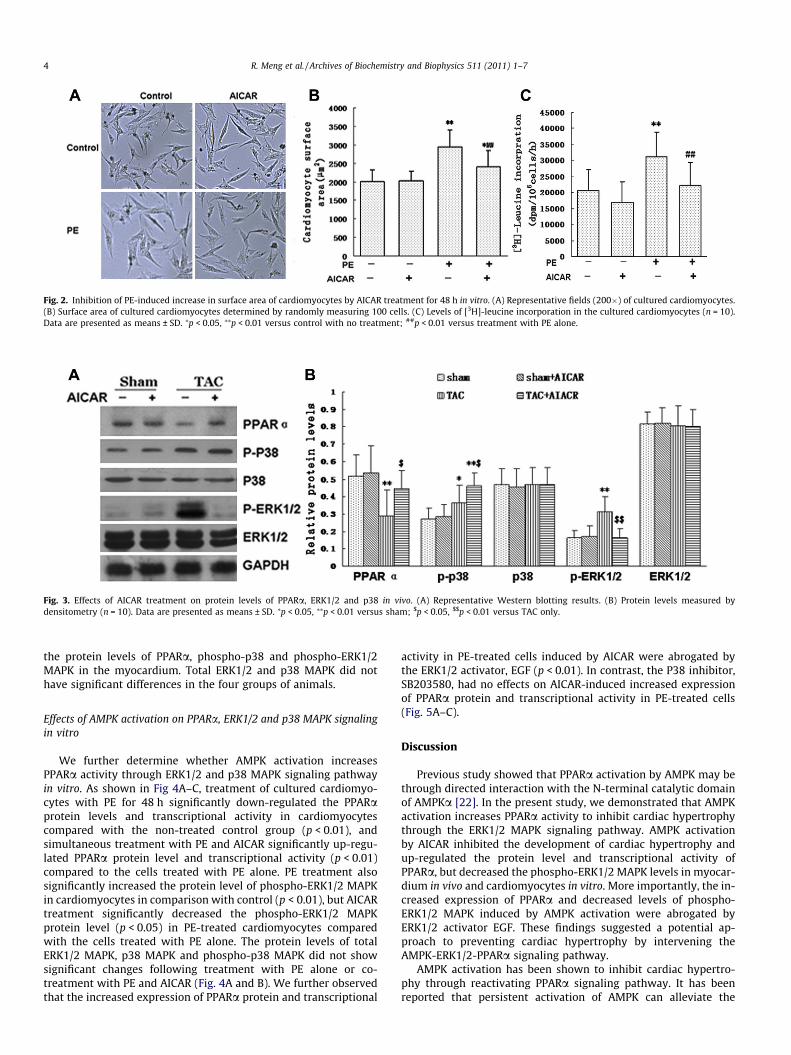
Fig. 2. Inhibition of PE-induced increase in surface area of cardiomyocytes by AICAR treatment for 48 h in vitro. (A) Representative fields (200�) of cultured cardiomyocytes.(B) Surface area of cultured cardiomyocytes determined by randomly measuring 100 cells. (C) Levels of [3H]-leucine incorporation in the cultured cardiomyocytes (n = 10).Data are presented as means ± SD. ⁄p < 0.05, ⁄⁄p < 0.01 versus control with no treatment; ##p < 0.01 versus treatment with PE alone.
Fig. 3. Effects of AICAR treatment on protein levels of PPARa, ERK1/2 and p38 in vivo. (A) Representative Western blotting results. (B) Protein levels measured bydensitometry (n = 10). Data are presented as means ± SD. ⁄p < 0.05, ⁄⁄p < 0.01 versus sham; $p < 0.05, $$p < 0.01 versus TAC only.
4 R. Meng et al. / Archives of Biochemistry and Biophysics 511 (2011) 1–7
the protein levels of PPARa, phospho-p38 and phospho-ERK1/2MAPK in the myocardium. Total ERK1/2 and p38 MAPK did nothave significant differences in the four groups of animals.
Effects of AMPK activation on PPARa, ERK1/2 and p38 MAPK signalingin vitro
We further determine whether AMPK activation increasesPPARa activity through ERK1/2 and p38 MAPK signaling pathwayin vitro. As shown in Fig 4A–C, treatment of cultured cardiomyo-cytes with PE for 48 h significantly down-regulated the PPARaprotein levels and transcriptional activity in cardiomyocytescompared with the non-treated control group (p < 0.01), andsimultaneous treatment with PE and AICAR significantly up-regu-lated PPARa protein level and transcriptional activity (p < 0.01)compared to the cells treated with PE alone. PE treatment alsosignificantly increased the protein level of phospho-ERK1/2 MAPKin cardiomyocytes in comparison with control (p < 0.01), but AICARtreatment significantly decreased the phospho-ERK1/2 MAPKprotein level (p < 0.05) in PE-treated cardiomyocytes comparedwith the cells treated with PE alone. The protein levels of totalERK1/2 MAPK, p38 MAPK and phospho-p38 MAPK did not showsignificant changes following treatment with PE alone or co-treatment with PE and AICAR (Fig. 4A and B). We further observedthat the increased expression of PPARa protein and transcriptional
activity in PE-treated cells induced by AICAR were abrogated bythe ERK1/2 activator, EGF (p < 0.01). In contrast, the P38 inhibitor,SB203580, had no effects on AICAR-induced increased expressionof PPARa protein and transcriptional activity in PE-treated cells(Fig. 5A–C).
Discussion
Previous study showed that PPARa activation by AMPK may bethrough directed interaction with the N-terminal catalytic domainof AMPKa [22]. In the present study, we demonstrated that AMPKactivation increases PPARa activity to inhibit cardiac hypertrophythrough the ERK1/2 MAPK signaling pathway. AMPK activationby AICAR inhibited the development of cardiac hypertrophy andup-regulated the protein level and transcriptional activity ofPPARa, but decreased the phospho-ERK1/2 MAPK levels in myocar-dium in vivo and cardiomyocytes in vitro. More importantly, the in-creased expression of PPARa and decreased levels of phospho-ERK1/2 MAPK induced by AMPK activation were abrogated byERK1/2 activator EGF. These findings suggested a potential ap-proach to preventing cardiac hypertrophy by intervening theAMPK-ERK1/2-PPARa signaling pathway.
AMPK activation has been shown to inhibit cardiac hypertro-phy through reactivating PPARa signaling pathway. It has beenreported that persistent activation of AMPK can alleviate the

Fig. 4. Effects of AICAR treatment on protein levels of PPARa, ERK1/2 and p38, and on transcriptional activity of PPARa in cardiomyocytes in vitro. (A) Representative Westernblotting results. (B) Protein levels determined by densitometry (n = 10). (C) Effect of AICAR treatment on PPARa transcriptional activation (n = 6). The relative luciferaseactivities are expressed as fold increase over vehicle control. Data are presented as means ± SD. ⁄⁄p < 0.01 versus control with no treatment; $p < 0.05, $$p < 0.01 versus PEtreatment alone.
R. Meng et al. / Archives of Biochemistry and Biophysics 511 (2011) 1–7 5
pressure-overload-induced cardiac hypertrophy [20]. Activationof AMPK by AICAR inhibits cardiac hypertrophy by blocking theprotein synthesis and suppressing cardiac fibroblast growth andproliferation [3,23,24]. Recently, we reported that AMPK activa-tion inhibits cardiac hypertrophy which is associated with reacti-vation of PPARa and improvement of myocardium energymetabolism [7]. In the present study, we showed that activationof AMPK by AICAR treatment significantly inhibited cardiachypertrophy as evidenced by reduced cross-sectional area ofcardiomyocytes, IVSd, PWd and LVW/BW in TAC rats and the cel-lular surface area and [3H]-leucine incorporation in PE-treatedcardiomyocytes. These results are in agreement with the obser-vations reported previously by others [20,25]. We also observedthat AICAR treatment did not have any effects on IVSd, PWdand LVW/BW in sham rats and [3H]-leucine incorporation andsurface area in the cultured cardiomyocytes without PE treat-ment. These results demonstrated that AMPK activation did notchange ventricular wall thicknesses of normal heart. The reasonsfor lack of effect of AMPK activation on normal heart tissues arenot clear. It is possible that AMPK activation do not change thecardiac metabolic model under normal condition, and AMPKactivation prevents cardiac hypertrophy by improving themyocardial energy metabolic shift through PPARa signaling path-way [7,26].
PPARa, a member of nuclear hormone receptor superfamily is acritical regulator of myocardial energy metabolism. The develop-ment of cardiac hypertrophy is associated with deactivation ofPPARa [5]. Consistent with previous studies, our results showedthat the protein level and transcriptional activity of PPARa wasdown-regulated in pressure overload-induced hypertrophic heartsand PE-induced hypertrophied cardiomyocytes. AICAR treatmentrestored the protein level and transcriptional activity of PPARa
and inhibited cardiac hypertrophy. These data demonstrated thatAMPK activation prevented the occurrence of cardiac hypertrophyby enhancing the activity of PPARa signaling pathway. PPARa is aphosphoprotein and its transcriptional activity is affected by phos-phatases. It has been shown that activation of p38 induces PPARaphosphorylation [27] and aldose reductase can suppress PPARaactivity by ERK1/2 signaling pathway [28]. Other studies havedemonstrated that AMPK is an up-regulator of ERK1/2 and p38MAPK [16,17]. These findings suggest that AMPK enhances theactivity of PPARa through ERK1/2 and p38 MAPK signaling path-way. Our study revealed that TAC rats showed decreased level ofPPARa protein and increased expression of phospho-ERK1/2 andphospho-p38 MAPK. These results are consistent with the observa-tions made by others [12,18,29–31]. We further demonstrated thatAICAR treatment enhanced the expression of PPARa and phospho-p38 MAPK, but decreased expression of phospho-ERK1/2 MAPK inTAC rats, suggesting that AMPK activation regulates PPARa activitythrough p38 and ERK1/2 signaling pathway. To further verify thispossibility, we performed in vitro study and observed that PE treat-ment of cardiomyocytes significantly down-regulated PPARa pro-tein level and transcriptional activity, while AICAR treatment ofthe PE-exposed cells restored the PPARa protein level and tran-scriptional activity. Phospho-ERK1/2 MAPK protein level was sig-nificantly increased in cardiomyocytes treated with PE, andAICAR reduced the phospho-ERK1/2 MAPK protein level in thePE-treated cells. These results strongly suggest that AICAR regu-lates PPARa signaling pathway in cardiac hypertrophy throughERK1/2, but not p38, signaling pathway. This signaling pathwaycascade was confirmed by our observation that the increasedPPARa protein level and transcriptional activity induced by AICARwere blocked by the ERK1/2 activator EGF, but the P38 inhibitor,SB203580, had no effect.
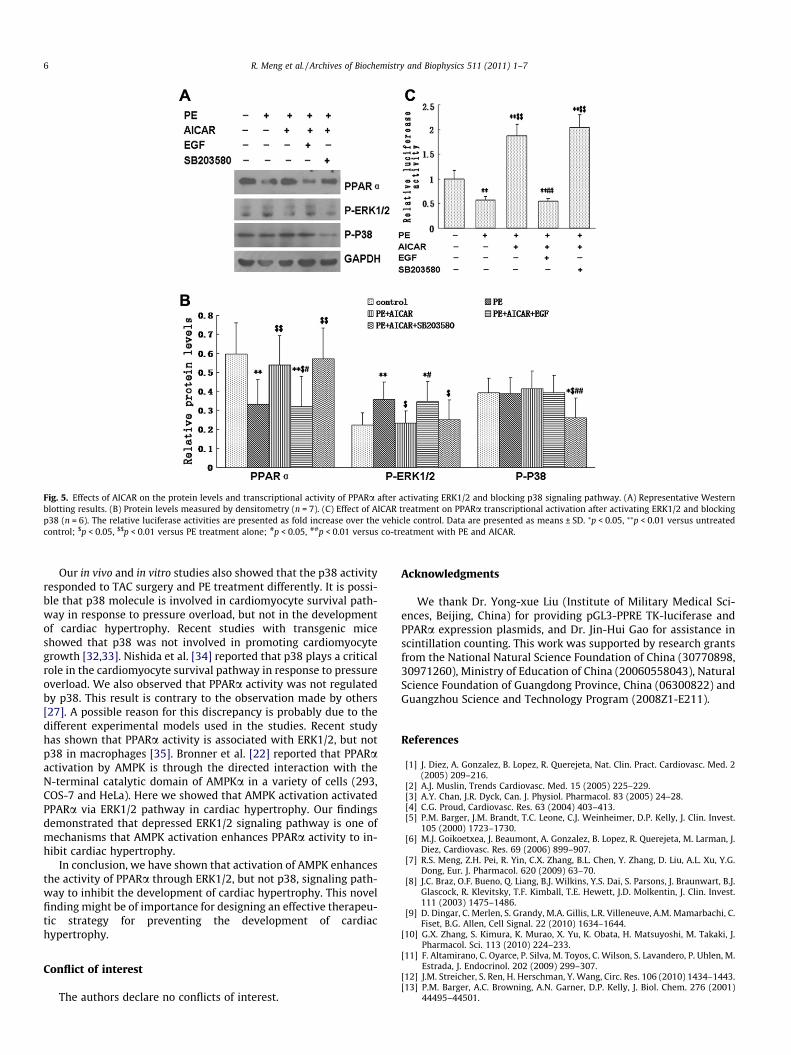
Fig. 5. Effects of AICAR on the protein levels and transcriptional activity of PPARa after activating ERK1/2 and blocking p38 signaling pathway. (A) Representative Westernblotting results. (B) Protein levels measured by densitometry (n = 7). (C) Effect of AICAR treatment on PPARa transcriptional activation after activating ERK1/2 and blockingp38 (n = 6). The relative luciferase activities are presented as fold increase over the vehicle control. Data are presented as means ± SD. ⁄p < 0.05, ⁄⁄p < 0.01 versus untreatedcontrol; $p < 0.05, $$p < 0.01 versus PE treatment alone; #p < 0.05, ##p < 0.01 versus co-treatment with PE and AICAR.
6 R. Meng et al. / Archives of Biochemistry and Biophysics 511 (2011) 1–7
Our in vivo and in vitro studies also showed that the p38 activityresponded to TAC surgery and PE treatment differently. It is possi-ble that p38 molecule is involved in cardiomyocyte survival path-way in response to pressure overload, but not in the developmentof cardiac hypertrophy. Recent studies with transgenic miceshowed that p38 was not involved in promoting cardiomyocytegrowth [32,33]. Nishida et al. [34] reported that p38 plays a criticalrole in the cardiomyocyte survival pathway in response to pressureoverload. We also observed that PPARa activity was not regulatedby p38. This result is contrary to the observation made by others[27]. A possible reason for this discrepancy is probably due to thedifferent experimental models used in the studies. Recent studyhas shown that PPARa activity is associated with ERK1/2, but notp38 in macrophages [35]. Bronner et al. [22] reported that PPARaactivation by AMPK is through the directed interaction with theN-terminal catalytic domain of AMPKa in a variety of cells (293,COS-7 and HeLa). Here we showed that AMPK activation activatedPPARa via ERK1/2 pathway in cardiac hypertrophy. Our findingsdemonstrated that depressed ERK1/2 signaling pathway is one ofmechanisms that AMPK activation enhances PPARa activity to in-hibit cardiac hypertrophy.
In conclusion, we have shown that activation of AMPK enhancesthe activity of PPARa through ERK1/2, but not p38, signaling path-way to inhibit the development of cardiac hypertrophy. This novelfinding might be of importance for designing an effective therapeu-tic strategy for preventing the development of cardiachypertrophy.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgments
We thank Dr. Yong-xue Liu (Institute of Military Medical Sci-ences, Beijing, China) for providing pGL3-PPRE TK-luciferase andPPARa expression plasmids, and Dr. Jin-Hui Gao for assistance inscintillation counting. This work was supported by research grantsfrom the National Natural Science Foundation of China (30770898,30971260), Ministry of Education of China (20060558043), NaturalScience Foundation of Guangdong Province, China (06300822) andGuangzhou Science and Technology Program (2008Z1-E211).
References
[1] J. Diez, A. Gonzalez, B. Lopez, R. Querejeta, Nat. Clin. Pract. Cardiovasc. Med. 2(2005) 209–216.
[2] A.J. Muslin, Trends Cardiovasc. Med. 15 (2005) 225–229.[3] A.Y. Chan, J.R. Dyck, Can. J. Physiol. Pharmacol. 83 (2005) 24–28.[4] C.G. Proud, Cardiovasc. Res. 63 (2004) 403–413.[5] P.M. Barger, J.M. Brandt, T.C. Leone, C.J. Weinheimer, D.P. Kelly, J. Clin. Invest.
105 (2000) 1723–1730.[6] M.J. Goikoetxea, J. Beaumont, A. Gonzalez, B. Lopez, R. Querejeta, M. Larman, J.
Diez, Cardiovasc. Res. 69 (2006) 899–907.[7] R.S. Meng, Z.H. Pei, R. Yin, C.X. Zhang, B.L. Chen, Y. Zhang, D. Liu, A.L. Xu, Y.G.
Dong, Eur. J. Pharmacol. 620 (2009) 63–70.[8] J.C. Braz, O.F. Bueno, Q. Liang, B.J. Wilkins, Y.S. Dai, S. Parsons, J. Braunwart, B.J.
Glascock, R. Klevitsky, T.F. Kimball, T.E. Hewett, J.D. Molkentin, J. Clin. Invest.111 (2003) 1475–1486.
[9] D. Dingar, C. Merlen, S. Grandy, M.A. Gillis, L.R. Villeneuve, A.M. Mamarbachi, C.Fiset, B.G. Allen, Cell Signal. 22 (2010) 1634–1644.
[10] G.X. Zhang, S. Kimura, K. Murao, X. Yu, K. Obata, H. Matsuyoshi, M. Takaki, J.Pharmacol. Sci. 113 (2010) 224–233.
[11] F. Altamirano, C. Oyarce, P. Silva, M. Toyos, C. Wilson, S. Lavandero, P. Uhlen, M.Estrada, J. Endocrinol. 202 (2009) 299–307.
[12] J.M. Streicher, S. Ren, H. Herschman, Y. Wang, Circ. Res. 106 (2010) 1434–1443.[13] P.M. Barger, A.C. Browning, A.N. Garner, D.P. Kelly, J. Biol. Chem. 276 (2001)
44495–44501.

R. Meng et al. / Archives of Biochemistry and Biophysics 511 (2011) 1–7 7
[14] M.J. Yoon, G.Y. Lee, J.J. Chung, Y.H. Ahn, S.H. Hong, J.B. Kim, Diabetes 55 (2006)2562–2570.
[15] L. Jing, W.M. Li, L.J. Zhou, S. Li, J.J. Kou, J. Song, Alcohol. Clin. Exp. Res. 32 (2008)1999–2007.
[16] K.J. Sweadner, Am. J. Physiol. Cell Physiol. 295 (2008) C588–C589.[17] J.H. Han, Y.H. Ahn, K.Y. Choi, S.H. Hong, J. Cell. Physiol. 218 (2009) 104–112.[18] J. Li, E.J. Miller, J. Ninomiya-Tsuji, R.R. Russell 3rd, L.H. Young, Circ. Res. 97
(2005) 872–879.[19] Y. Motobayashi, Y. Izawa-Ishizawa, K. Ishizawa, S. Orino, K. Yamaguchi, K.
Kawazoe, S. Hamano, K. Tsuchiya, S. Tomita, T. Tamaki, Hypertens. Res. 32(2009) 188–193.
[20] H.L. Li, R. Yin, D. Chen, D. Liu, D. Wang, Q. Yang, Y.G. Dong, J. Cell. Biochem. 100(2007) 1086–1099.
[21] H.L. Li, Y. Huang, C.N. Zhang, G. Liu, Y.S. Wei, A.B. Wang, Y.Q. Liu, R.T. Hui, C.Wei, G.M. Williams, D.P. Liu, C.C. Liang, Free Radic. Biol. Med. 40 (2006) 1756–1775.
[22] M. Bronner, R. Hertz, J. Bar-Tana, Biochem. J. 384 (2004) 295–305.[23] J. Du, T. Guan, H. Zhang, Y. Xia, F. Liu, Y. Zhang, Biochem. Biophys. Res.
Commun. 368 (2008) 402–407.[24] A.Y. Chan, V.W. Dolinsky, C.L. Soltys, B. Viollet, S. Baksh, P.E. Light, J.R. Dyck, J.
Biol. Chem. 283 (2008) 24194–24201.[25] B.J. Stuck, M. Lenski, M. Bohm, U. Laufs, J. Biol. Chem. 283 (2008) 32562–
32569.
[26] H. Yamashita, K.G. Bharadwaj, S. Ikeda, T.S. Park, I.J. Goldberg, Am. J. Physiol.295 (2008) E705–713.
[27] C. Diradourian, C. Le May, M. Cauzac, J. Girard, A.F. Burnol, J.P. Pegorier,Biochim. Biophys. Acta 1781 (2008) 239–244.
[28] L. Qiu, X. Wu, J.F. Chau, I.Y. Szeto, W.Y. Tam, Z. Guo, S.K. Chung, P.J. Oates, S.S.Chung, J.Y. Yang, J. Biol. Chem. 283 (2008) 17175–17183.
[29] M. Capano, M. Crompton, Biochem. J. 395 (2006) 57–64.[30] C. Nunn, M.X. Zou, A.J. Sobiesiak, A.A. Roy, L.A. Kirshenbaum, P. Chidiac, Cell
Signal. 22 (2010) 1231–1239.[31] J.H. Du, N. Xu, Y. Song, M. Xu, Z.Z. Lu, C. Han, Y.Y. Zhang, Biochem. Biophys. Res.
Commun. 337 (2005) 1139–1144.[32] P. Liao, D. Georgakopoulos, A. Kovacs, M. Zheng, D. Lerner, H. Pu, J. Saffitz, K.
Chien, R.P. Xiao, D.A. Kass, Y. Wang, Proc. Natl. Acad. Sci. USA 98 (2001)12283–12288.
[33] S. Zhang, C. Weinheimer, M. Courtois, A. Kovacs, C.E. Zhang, A.M. Cheng, Y.Wang, A.J. Muslin, J. Clin. Invest. 111 (2003) 833–841.
[34] K. Nishida, O. Yamaguchi, S. Hirotani, S. Hikoso, Y. Higuchi, T. Watanabe, T.Takeda, S. Osuka, T. Morita, G. Kondoh, Y. Uno, K. Kashiwase, M. Taniike, A.Nakai, Y. Matsumura, J. Miyazaki, T. Sudo, K. Hongo, Y. Kusakari, S. Kurihara,K.R. Chien, J. Takeda, M. Hori, K. Otsu, Mol. Cell. Biol. 24 (2004) 10611–10620.
[35] K. Taketa, T. Matsumura, M. Yano, N. Ishii, T. Senokuchi, H. Motoshima, Y.Murata, S. Kim-Mitsuyama, T. Kawada, H. Itabe, M. Takeya, T. Nishikawa, K.Tsuruzoe, E. Araki, J. Biol. Chem. 283 (2008) 9852–9862.




















