
Alternative Strategies for Toxicity Testing of Species-Specific Biopharmaceuticals
25
http://ijt.sagepub.com International Journal of Toxicology DOI: 10.1177/1091581809337262 2009; 28; 230 Int J Toxicol Beyer and Christopher Horvath Jeanine L. Bussiere, Pauline Martin, Michelle Horner, Jessica Couch, Meghan Flaherty, Laura Andrews, Joseph Alternative Strategies for Toxicity Testing of Species-Specific Biopharmaceuticals http://ijt.sagepub.com/cgi/content/abstract/28/3/230 The online version of this article can be found at: Published by: http://www.sagepublications.com On behalf of: American College of Toxicology can be found at: International Journal of Toxicology Additional services and information for http://ijt.sagepub.com/cgi/alerts Email Alerts: http://ijt.sagepub.com/subscriptions Subscriptions: http://www.sagepub.com/journalsReprints.nav Reprints: http://www.sagepub.com/journalsPermissions.nav Permissions: at American College of Toxicology on July 17, 2009 http://ijt.sagepub.com Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Alternative Strategies for Toxicity Testing of Species-Specific Biopharmaceuticals

http://ijt.sagepub.com
International Journal of Toxicology
DOI: 10.1177/1091581809337262 2009; 28; 230 Int J Toxicol
Beyer and Christopher Horvath Jeanine L. Bussiere, Pauline Martin, Michelle Horner, Jessica Couch, Meghan Flaherty, Laura Andrews, Joseph
Alternative Strategies for Toxicity Testing of Species-Specific Biopharmaceuticals
http://ijt.sagepub.com/cgi/content/abstract/28/3/230 The online version of this article can be found at:
Published by:
http://www.sagepublications.com
On behalf of: American College of Toxicology
can be found at:International Journal of Toxicology Additional services and information for
http://ijt.sagepub.com/cgi/alerts Email Alerts:
http://ijt.sagepub.com/subscriptions Subscriptions:
http://www.sagepub.com/journalsReprints.navReprints:
http://www.sagepub.com/journalsPermissions.navPermissions:
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

Alternative Strategies for Toxicity Testingof Species-Specific Biopharmaceuticals
Jeanine L. Bussiere, Pauline Martin, Michelle Horner,Jessica Couch, Meghan Flaherty, Laura Andrews,Joseph Beyer, and Christopher Horvath
Although toxicology studies should always be conductedin pharmacologically relevant species, the specificity ofmany biopharmaceuticals can present challenges inidentification of a relevant species. In certain cases,that is, when the clinical product is active only inhumans or chimpanzees, or if the clinical candidateis active in other species but immunogenicity limitsthe ability to conduct a thorough safety assessment,alternative approaches to evaluating the safety of a bio-pharmaceutical must be considered. Alternativeapproaches, including animal models of disease, gene-tically modified mice, or use of surrogate molecules,
may improve the predictive value of preclinical safetyassessments of species-specific biopharmaceuticals,although many caveats associated with these modelsmust be considered. Because of the many caveats thatare discussed in this article, alternative approachesshould only be used to evaluate safety when the clinicalcandidate cannot be readily tested in at least one rele-vant species to identify potential hazards.
Keywords: alternate strategies; biologics; biopharma-ceuticals; genetically modified mice; homologous pro-teins; surrogate molecules
Although toxicology studies should always beconducted in pharmacologically relevant spe-cies, the high degree of species specificity of
many biopharmaceuticals can present certain chal-lenges in identification of a relevant species. Thisspecificity also implies that the toxicity is generallydue to on-target activity (based on the intended phar-macology) rather than off-target effects (nonspecificbinding) as is often the case for traditional smallmolecule pharmaceuticals. ICH S61 defines a phar-macologically relevant species as ‘‘one in which thetest material is pharmacologically active due toexpression of the receptor or an epitope (in the case
of monoclonal antibodies).’’ In some cases, it maynot be possible to evaluate the toxicity of a clinicalproduct in animals (for example, when the productis active only in humans or chimpanzees, or if immu-nogenicity limits the ability to conduct a thoroughsafety assessment). Alternative approaches to evalu-ating the safety of a biopharmaceutical should beconsidered in these cases. ICH S6 has identifiedthe following as potentially viable alternativeapproaches: animal models of disease, transgenic/knock-out models, and surrogate molecules (Table 1).Each of these has unique advantages and disadvan-tages that must be considered in the developmentof preclinical safety assessment strategies (Table 2).Multiple approaches to understanding the toxicitiesof a development compound may be equally valid,provided that they can be scientifically justified asrelevant to understanding the potential safety ofthe molecule. This article will discuss some of thebenefits and limitations of each approach and willprovide a series of examples from approved productsas well as current development programs on how
From Amgen Inc, Thousand Oaks, California; Centocor Research& Development, Inc, Radnor, Pennsylvania; Genzyme,Framingham, Massachusetts; Genentech Inc, South SanFrancisco, California; and Taligen Therapeutics, Cambridge,Massachusetts.
Please address correspondence to Jeanine L. Bussiere, PhD,DABT, Scientific Executive Director, Toxicology, Amgen Inc,One Amgen Center Dr, MS 29-2-A, Thousand Oaks, CA91320-1799; e-mail: [email protected].
International
Journal of Toxicology
Volume 28 Number 3
May/June 2009 230-253
# 2009 The Author(s)
10.1177/1091581809337262
http://ijt.sagepub.com
hosted at
http://online.sagepub.com
230
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

these alternative approaches have been used.Although most of the discussion will focus onprotein biopharmaceuticals (eg, recombinant pro-teins, monoclonal antibodies, and fusion proteins),the principles for alternative testing approaches arealso relevant for nonprotein biopharmaceuticals(eg, peptides, antisense oligonucleotides, small inter-fering RNAs, and aptamers) and could potentially beapplied to some new chemical entities (eg, smallmolecules showing restricted species specificity).
Animal Models of Human Disease
One challenge in identifying a pharmacologicallyrelevant species for biopharmaceutical toxicity
testing is that, although the target may be expressedin different animal species and may even havecomparable function as in humans, the target maynot be recognized (pharmacologically modulated)by the clinical candidate. Alternatively, the therapeu-tic target may be expressed or upregulated only incertain disease settings. Examples include nonhostproteins such as viral or bacterial antigens that arenot normally present in healthy animals, or hostproteins that are expressed only in certain diseasestates such as unique cancer antigens, Rh factor, oraltered (mutated) host proteins. Because such tar-gets are unlikely to be present in normal animals,these represent perhaps the most challenging targetsfor which to assess the pharmacologic and toxicolo-gic effects of the clinical candidate.
Table 2. General Issues With Nonclinical Development Programs for Biopharmaceuticals LackingCross-Reactivity in Traditional Species (ie, Rat and Dog)
Evaluate Human Test Article(Clinical Candidate) in
Cross-Reactive NonhumanPrimate Species
Evaluate Surrogate Rodent Homologous TestArticle in Rodent Species
(Mouse or Rat)
Evaluate Effect of GeneticDeletion of Target in
Knockout Mice
Preference May be preferable because usingactual test article
May be less preferable because not evaluatingactual test article
Relevance Relevance to humans is assumedinitially
Relevance to humans is questioned initially
Acceptance Historically well accepted(becoming ‘‘traditional’’)
Historically of varying acceptance (remainsnontraditional)
Design andtiming
May require nonstandard studydesigns, cost structures andtimelines
Standard study designs modeled after traditionalstudies with standard cost structures ($, Personnel)and standard timelines
Expenses Standard costs for materials,assays, reagents; often serve as‘‘dry run’’ for clinical trials
Requires dual development of clinicalcandidate and homologue; additional costfor surrogate materials, assays, reagents
Additional costs for breeding,genotyping, assays and reagents,licensing fees, etc
Otheroptions
Sometimes the ‘‘relevant’’species may not be primate(eg, pigs)
Sometimes the surrogate homologue may betested in a non-rodent species (eg, primate)
Sometimes the clinical candidate maybe tested in rodent knockout micewith human ‘‘knockin’’ target
Table 1. Alternative Approaches Identified in ICH S6
Approach Advantages Limitations
Animal modelsof disease
More closely mimics effects in humanpatients
Lack of historical data; limited life span; confounding effects ofdisease
Geneticallymodifiedanimals
Can gather data in species where nocross-reactivity exists
Can do additional studies in a morestandard toxicology model
Lack of historical data; limited number of animals; limited life span;technical challenges
Surrogatemolecules
Can gather data in species where nocross-reactivity exists
Can do additional studies in a morestandard toxicology model
Different product (production process, impurities, mechanism);technical challenges, pharmacology differences (ie, potentialdifferences in signaling based on the epitope that the surrogatebinds)
231
Alternative Strategies for Biopharmaceuticals / Bussiere et al 231
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

Animal models of human disease have been usednot only to facilitate compound selection and allowfor an early understanding of mechanism of actionbut also to assess safety. The use of animal modelsof disease to assess in vivo activity as well as toxicitycan provide a better understanding of the therapeuticindex and therefore improve clinical dose selection.2
Animal models of human disease may exist spon-taneously (eg, dogs with factor VIII deficiency)or may be experimentally developed for preclinicaltesting. Common examples of the latter include theuse of virally challenged animals to test the efficacyof a vaccine or a therapeutic directed against viralproteins, mice inoculated with xenogenic (human)tumors expressing the target antigen, micechallenged with infectious agents, or geneticallymodified animals that develop spontaneous disease.As with any model, there are both advantages anddisadvantages to this approach (Table 1). One keyconsideration is that the immune system may be verydifferent between rodents and humans.3 Forexample, animal models of many inflammatory dis-eases such as multiple sclerosis, where complexmulticomponent processes are involved, may differsubstantially between mice and humans.4 In addi-tion, animal models of disease are generally not wellcharacterized to understand what the ‘‘background’’lesions are relative to what may be a compound-related effect.
Genetically Modified Animals
Genetically modified (knock-out, knock-in, trans-genic) mice are rapidly gaining acceptance as toolsfor mechanistic research and validation of targetbiology and have considerable potential as specificmodels of toxicological importance. Gene-targetedor knock-out (KO) animals have been created usingmolecular and cellular genetic engineering tech-niques to produce animals that specifically lack anendogenous gene and therefore fail to express therelated protein(s), whereas transgenic (Tg) mice areengineered to overexpress a target protein.5 ManyKO and Tg mice appear morphologically normalalthough functional abnormalities may be apparent;in other cases, these mice may exhibit a normalphenotype. Pharmacological challenges or otherphysiological stressors may also unmask subtle phe-notypes (functional and/or morphologic changesresulting from the genetic engineering event).6,7 In
addition, alteration of a target that has a significantrole in prenatal growth and development may havesignificant adverse effects (eg, may be embryolethal)that are not relevant when inhibiting the target in anadult. Embryolethal effects can be avoided throughdevelopment of conditional KO animals in which tar-get disruption is limited to a specific developmentalstage and/or tissues of interest.8
Knock-out mice have been used to assess drugspecificity, investigate mechanisms of toxicity, andscreen for mutagenic and carcinogenic activities oftherapeutic candidates. Similarly, the effect of targetblockade by novel therapeutic candidates can beestimated in KO mice; for example, generation ofviable and fertile animals with null mutations for apotential target protein may suggest that pharmaco-logical inhibition of the molecule in vivo is unlikelyto elicit major adverse effects on normal physiologi-cal functions.9 As such, an apparent lack of a deleter-ious phenotype could be used as supportive evidencefor the safety of inhibiting the intended target follow-ing extensive characterization of the KO mouse.
However, because KO mice may have uncharac-terized compensatory mechanisms or redundantpathways that are not readily apparent to replace thefunction of the absent protein(s) or target,10-12 theiruse in assessing safety will likely be supportive ratherthan providing definitive safety data. One possiblealternative to avoid this issue could be directedtoward utilizing conditional KO mice, which permitevaluation of effects resulting from inhibition of aparticular gene product during only the relevantstage of life. Finally, when considering animal mod-els of disease using a complete or partial gene KO, itis important to consider the genetic structure andfunction of the gene, because genetic mutationsunderlying human disease may have a different phy-siological effect in the rodent because of differencesin gene structure and/or function between species.13
‘‘Humanized’’ knock-in (KI) animals, in whichthe human gene is inserted into the mouse genome(either independently or in conjunction with aknock-out of the endogenous mouse gene), are ofparticular utility in evaluating the efficacy andtoxicity of human biopharmaceuticals that are notpharmacologically active in normal rodents.14,15 Onecriticism of this approach is that humanized micemay express one or a few human proteins of interest,but other proteins that interact with the humanmolecules are still of mouse origin. The physiologicaleffects of human-mouse protein interactions may
232
232 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

differ slightly—or substantially—from that of thenormal human-human association (eg, Toll-likereceptors16,17). It is important to note that in severalcases, KI mice have been used to support appro-priate function of the human KI gene.13,15 Prior touse, comprehensive studies must be conducted todefine the biology of mouse-human protein interac-tions to allow validation of the human gene KI miceas appropriate models. As transgenic insertions maynot be targeted to a specific site in the genome,some of the regulatory sequences around them mayfunction differently than those of the endogenousgene, which may affect both spatiotemporal patternsas well as overall levels of protein expression. Inaddition, administration of the clinical candidate inthese animals may still be immunogenic thus limit-ing long-term studies in KI mice.
With the increasing number of biopharmaceuti-cals on the market, data from studies in geneticallyengineered mice will become important to demon-strate that they have utility as a viable alternative totesting in nonhuman primates and that findings ofefficacy and toxicity obtained using these models arerelevant to humans. This will take time, because theultimate validation will not occur until there areclinical data to compare with relevant preclinicalmodels.
Despite their potential utility, the use of KO/Tgmice for toxicity studies may be technically challen-ging because of the activity of the target (ie, may bedifferent in mice and humans) and/or early embryoniclethality. In cases of embryonic lethality, developmentof alternative models, such as the BRCA1 andBRCA2 conditional KO animals, may enable a moreappropriate evaluation of target inhibition in theadult animal.8
The technical and feasibility challenges associ-ated with using KO/Tg mice may significantly affectprogram time lines, and consequently, the potentialuse of KO/Tg mice in a safety assessment programmust be considered very early in development. Aspreviously discussed, standard KO mice that lack atarget throughout embryogenesis and developmentmay not accurately reflect disease or pharmacologi-cal interventions in the clinical scenario, whereprotein function may be nullified only during adult-hood. Another important consideration when usinga KO/Tg mouse is that they are often generatedon different background strains (which may producedifferent phenotypes) or on strains that havenot typically been used in toxicity assessments
(eg, C57Bl/6 and S129), and therefore less historicaldata may be available. Multigenerational studies orincreased numbers of KO/Tg control animals maytherefore be required for interpretation of safetyresults in the absence of adequate historical data.This may be particularly pertinent in reproductivetoxicity assessments, where fetal variations orabnormalities may be strain dependent. For lesswell-characterized strains of mice, it may be neces-sary to rely on the characterization of the wild-typemice as well as the genetically modified mice. Asimilar situation exists for pharmacology or diseasemodels, which are notoriously specific for certainstrains of mice. Immune function in these strains isgenerally not well characterized and specific straindifferences are known to exist, especially in theimmune response.18 Therefore, the pharmacologicactivity of the biotherapeutic must be evaluated inthe KO/Tg animal prior to committing to its use forsafety assessment. Last, genetically modified miceare often developed in discovery or academicresearch settings, where the incidence of latent oropportunistic pathogenic infections tends to behigher, and may necessitate rederivation of the strainprior to scaling up their production. This couldpotentially result in a 1- to 2-year delay before themodel would be ready to use in a safety assessmentsetting. During the rederivation process andthroughout the use of a KO/Tg model, it may benecessary to genotype each animal to ensure thathetero- or homozygosity and/or copy number areappropriate for the desired genetic modification.There are also certain KO/Tg mice that are sterileor not viable as homozygotes, so the animals mustbe generated from heterozygotes and therefore mustbe genotyped each time. Finally, as all of thesemodels have strengths and limitations, the scientificrationale for their use must be justified andthoroughly discussed (generation of the geneticallymodified animal, backcrossing stage, strain, etc).
Surrogate Molecules
Considerations for Use of a SurrogateMolecule for Safety Assessment
For the purpose of this discussion, a surrogate mole-cule is one that is selected for use in place of thedevelopment compound to evaluate pharmacologyand/or toxicology parameters in a species that is
233
Alternative Strategies for Biopharmaceuticals / Bussiere et al 233
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

believed to be biologically relevant to humans. Thesemolecules have also been referred to as analogous orhomologous compounds. However, until the clinicalcandidate has been evaluated in humans, the extentto which the surrogate molecule is truly homologousor analogous to the clinical candidate cannot becompletely understood. Of course, the same maybe said with respect to the biological relevance of theanimal species selected for testing of the clinicalcandidate. Although all analogous or homologouscompounds are surrogates for the clinical candidate,not all surrogates will be proven to be analogous orhomologous (ie, a monoclonal antibody [mAb] to theanimal target may be a completely different mole-cule). These caveats should be kept in mind wheninterpreting the results of any alternative testingstrategy.
Surrogate molecules may enable an evaluation ofthe consequences of modulating the target-mediatedpharmacologic activity of the drug in cases when anevaluation of the clinical candidate is limited by spe-cies specificity. The surrogate molecule shouldresemble the clinical candidate as much as possiblewith regard to pharmacologic activity (ie, target epi-tope recognized, binding affinity, potency, etc) if itis to provide relevant and useful safety information.Because the formulation, production process, rangeof impurities/contaminants, glycosylation patterns,and many other factors could also influence the find-ings on a study, one should also understand theessential quality characteristics of the molecule. Thismay be challenging and will require significantresources to characterize a molecule that will neverbe a drug candidate.
Although in some cases surrogate molecules haveemerged as scientifically accepted tools for the safetyassessment of biopharmaceuticals, surrogate mole-cules inherently differ from the clinical candidatesthey are designed to represent. As such, several ques-tions regarding the validity of a surrogate moleculefor toxicity assessments, as well as the overalladvantages and disadvantages of using a surrogatemolecule over the clinical candidate, should be takeninto consideration. Both in vivo and in vitro assess-ments should be used to characterize the relativesimilarity of the surrogate molecule to the clinicalcandidate. An important consideration for certainmonoclonal antibodies is use of the appropriateisotype for the surrogate, particularly for those withanticipated ADCC or CDC activity as part of themechanism of toxicity or pharmacologic action.
In vitro assessments should include pharmacolo-gic activity, potency, affinity/avidity, as well asgeneral physicochemical characterization. In addi-tion, consideration must be given to the appropriateisotype to understand the similarity of ADCC orCDC activity of the Fc region of the antibody.Similarities in tissue binding and specificity mayalso be useful in assessing the appropriateness ofthe surrogate molecule. In vivo evaluations of thepharmacokinetics and pharmacodynamics of thesurrogate molecule should also be conducted, withcomparisons drawn to the clinical candidate wherepossible. Investigations into biomarkers of thesurrogate molecule’s activity and/or application ofthe surrogate molecule in models of efficacy may alsoaid in further characterization.
Limitations in the utility of surrogate moleculesare dictated by the nature of the molecule itself. Thesurrogate molecule should be comparable inpharmacologic activity as well as structure, to theclinical candidate. However, in some cases, the sur-rogate molecule may be structurally distinct from theclinical candidate but have similar pharmacody-namic effects in vivo. This is often a requirement ifsignificant interspecies variability exists in the targetmolecule structure, but may also occur when thedevelopment of the surrogate molecule is conductedeither separately from the clinical candidate or usingdifferent manufacturing methods. In cases where ananimal model lacks the human target, a surrogatemolecule may be developed to mimic biologicallyequivalent pharmacologic effects rather than specifictarget recognition. An example of this might be amolecule targeting murine keratinocyte (KC) proteinas a surrogate for a clinical candidate directedagainst human IL-8.19,20
Additional limitations of the surrogate moleculemay exist with respect to the magnitude or specificityof its biological effect, if the intended pharmacologi-cal effect is either amplified or reduced relative to theclinical candidate. It is important to be aware thatsurrogate molecules may elicit different downstreamsignaling cascades that could yield potentiallysignificant, yet irrelevant, effects. Differences ineither pharmacologic effect or molecular structurecan also affect the pharmacokinetics of the surrogatemolecule. Thus, when the pharmacokinetic profile ofthe clinical candidate is known in a relevant animalspecies, relative comparisons between the surrogatemolecule and the clinical candidate should be made,taking into consideration species differences in
234
234 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

affinity, target distribution, or differences in otherclearance processes (ie, Fc-mediated clearance) thatmay impact pharmacokinetics. When the pharmaco-kinetic profile of the clinical candidate in an animalmodel is unknown due to species specificity, thesimilarity in pharmacologic effects between mole-cules (assessed using in vitro or in vivo data) can beused to justify comparability.
When a surrogate molecule believed to be repre-sentative of the clinical candidate is identified, and adecision to use it to support development of the clin-ical candidate is made, the sponsor has effectivelyassumed the responsibility of codeveloping 2 mole-cules, only one of which will be tested in the clinic.Several factors should be considered when assessingthe feasibility of surrogate molecule codevelopmentwith the clinical candidate, including the require-ments for development, manufacture, and character-ization of the surrogate molecule. The extent towhich the surrogate molecule will be manufacturedto Good Manufacturing Practice (GMP) guidelinesor tested in safety studies compliant with GoodLaboratory Practice (GLP) guidelines will need tobe decided upon, with appropriate justification(s).Development, including validation (or qualification),of different assays to detect the surrogate moleculeand to monitor the effects of immunogenicity (ie,anti-product antibodies) is also necessary. Asproduction processes associated with manufacturinga surrogate molecule can be different from theclinical product, attention should be given to anypotential impact these changes may have on thepharmacologic activity and/or the range of impuri-ties. Manufacture of the surrogate molecule willrequire additional resources to develop the process,formulation, and packaging of the material. In casesof limited manufacturing capabilities or when clinicalprocesses are utilized in the manufacturing of the sur-rogate molecule, additional pressure may be placedon manufacturing infrastructure. Additional assaysare needed for characterization (aggregates, clips ofthe intact protein and impurities such as host cellprotein, endotoxin, etc) and stability testing to sup-port the duration of the toxicology studies.
Similar to the use of KO/Tg animals in preclinicalsafety evaluations, the use of surrogate moleculesshould be undertaken with an understanding of thetime, cost, and effort of surrogate molecule develop-ment, in context with the applicability and relativebenefits toward assessing clinical safety. Ultimately,a clear understanding and characterization of the
pharmacological and biological behavior of thesurrogate molecule is critical not only to the designof a relevant toxicity program, but also to the deter-mination of the overall risk-to-benefit ratio forhumans. As long-term repeated dosing of a humanbiopharmaceutical in rodent species is often not fea-sible due to immunogenicity and development ofneutralizing antibodies that reduce exposure, a mur-ine surrogate molecule may be developed on theassumption that it would be less immunogenic inmice. However, just as a human protein can beimmunogenic in humans, the homologous proteincan also be immunogenic in rodents. Therefore, thepotential for immunogenicity from a surrogate mole-cule should be evaluated as early as feasible. Theintended application of a surrogate molecule in anonclinical safety program should also be considered(ie, for developmental and reproductive toxicology[DART], for chronic toxicology, etc), as it will definethe extent of in vitro and in vivo studies required foradequate surrogate molecule characterization. Forexample, if a surrogate molecule is intended for usein DART assessments, in vitro placental transferstudies should be considered at an early stage ofsurrogate molecule development to characterize therelevance of the surrogate molecule to the clinicalcandidate in this respect and demonstrate that thefetus will be exposed to the drug.21 Finally, determi-nation of clinically relevant starting doses frompreclinical toxicity evaluations using a surrogatemolecule should involve careful consideration ofrelevant biological and pharmacokinetic differencesand their potential impact on extrapolation of asafe dose.
Potential Advantages/Disadvantages ofUsing a Surrogate Molecule
In situations where the biopharmaceutical ispharmacologically active only in humans and/orchimpanzees, the use of a surrogate molecule couldbe helpful for determining potential hazards associ-ated with the use of the product. Although limitednonterminal safety studies can be conducted inchimpanzees, the small group sizes, the heterogene-ity of the animals that are available, previous drugexposure, and lack of histopathological endpointslimit the use of chimpanzees to an assessment ofacute tolerability rather than an assessment of toxi-city. Because of this, many companies now screencandidates and select lead molecules that have
235
Alternative Strategies for Biopharmaceuticals / Bussiere et al 235
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

pharmacologic activity in at least one toxicologicallyrelevant species. However, when a suitable speciescannot be identified, a full single-species toxicologyassessment can be conducted using a surrogatemolecule. To date, the molecular classes that haveshown the greatest degree of species specificity arethe monoclonal antibodies and the interferons.Surrogate monoclonal antibodies and homologousinterferons have been generated for some species-restricted products and have been used in nonclini-cal safety assessments.21-24 For the monoclonalantibodies, surrogate antibodies could potentiallybe developed for any species, but the most commonsurrogate molecules are rat anti-mouse antibodies.These rat antibodies can then be chimerized (ratFab-mouse Fc) or engineered to replace certain ratsequences with murine sequences. One consider-ation for surrogate antibodies or Fc-fusion proteinsis that the Fc region in the murine surrogate mole-cule must match the appropriate effector functionof the Fc in humans (ie, the IgG2a isotype in miceto match the effector function of an IgG1 isotypein humans).25,26
One of the advantages of developing a murinesurrogate for toxicology evaluation is that theoutbred CD-1 mouse is a common toxicological spe-cies and therefore standard, well-validated toxicologyprotocols and endpoints can be applied to these stud-ies. For immunomodulatory biopharmaceuticals,many of the standard immunotoxicity endpoints havebeen well established in mice, and many reagents areavailable for evaluating immunological endpoints.An added advantage of using the mouse is that manywell-established disease models have been created inmice, allowing both pharmacology and toxicology tobe conducted in the same species and providing apotential margin of safety in that species. A disadvan-tage of using mice is the limited blood volume forvarious endpoints and thus the relatively large num-ber of animals needed for each study.
With the availability of an appropriate murinesurrogate molecule, acute, subchronic, and chronictoxicity studies can potentially be conducted,depending on the immunogenicity of the surrogatemolecule. Additional endpoints can be incorporatedinto the studies based on the pharmacology of themolecule, such as immunotoxicity endpoints forimmune modulating biopharmaceuticals as men-tioned previously. It is important to evaluate toxico-kinetic parameters to demonstrate that exposure tothe surrogate molecule is maintained throughout the
dosing period and that antibodies that may developagainst the surrogate molecule do not limit theexposure. However, if mice are used for toxicologyevaluations, the limited blood volume does not allowthe toxicokinetic and immune response to be mea-sured in the main toxicology animals. Consequently,satellite animals must be included for the toxicoki-netic and immune response evaluations, makingdirect correlations between exposure, immuneresponse, and toxicity difficult.
For biopharmaceuticals that require an evaluationof potential effects on reproduction and/or develop-ment, DART studies in rodents using a surrogatemolecule can be an acceptable option. This approachfor DART studies can be used for molecules that donot cross-react with relevant toxicology species, or canbe used to reduce the use of nonhuman primates forthose molecules that show cross-reactivity only tohumans and nonhuman primates. However, becauseof the caveats discussed previously, it may be moreappropriate for developmental studies to evaluate theclinical candidate in nonhuman primates than to usea surrogate molecule that may not be truly representa-tive. With the use of a surrogate molecule in rodents,all aspects of the DART evaluation as outlined in ICHS5 (R2)27 can be evaluated using standard well-established protocols. Numerous testing facilitieshave the experience and capabilities of conductingDART studies in rodents, and large historical data-bases are available for comparison with normal back-ground findings.
Rodent studies may be particularly useful for theevaluation of fertility (ie, the number of successfulpregnancies). Mating behavior and number of suc-cessful pregnancies cannot be evaluated practicallyin nonhuman primates because they have a naturallylow fertility rate and high spontaneous abortion rate.Reproductive potential can, however, be evaluated inprimates by evaluation of hormones, menstrualcycles, semen parameters, and other indicators ofreproductive capability. In contrast, fertility mea-sured by pregnancy success is a well-establishedendpoint in rodents.
Embryofetal development studies can also beconducted in rodents using a surrogate molecule.There are, however, certain caveats concerningthe use of a surrogate molecule for embryofetaldevelopment studies that must be taken into con-sideration. The mechanisms that regulate largemolecular weight protein transport and diffusionacross the placenta are species dependent and
236
236 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

differ between rodent and primate placenta.28 Forantibodies that are known to be transported acrossthe placenta, the timing of placental transfer andthe efficiency of transfer differs between rodentsand primates.29,30 In rodents fetal exposure toantibodies occurs earlier in development than inprimates. Therefore, adverse effects may beobserved in the rodents that are not relevant tohumans and may therefore overpredict the poten-tial risk. For large molecular weight proteins thatdo not cross the placenta, embryofetal develop-ment studies are likely to be restricted to maternaleffects rather than direct teratogenic effects and astudy in rodents with the surrogate molecule maymodel these effects similarly as a study inprimates.
Pre- and postnatal development studies can alsobe conducted in rodents using the rodent surrogatemolecule. In this case, endpoints for establishingeffects of treatment on postnatal development arewell established in rodents. The rodent postnataldevelopment studies allow for an evaluation of sexualmaturity and second-generation reproductive perfor-mance that cannot be evaluated in nonhuman pri-mates because of the long period of time betweenbirth and sexual maturity. However, because thetoxicity of biopharmaceuticals is generally relatedto exaggerated pharmacology, unless there is ascientific reason why the molecule might affectsexual maturation, the absence of this information isnot considered to be critical to assessing human safety.
In addition to the discussed limitations of usingprimates for reproductive toxicity studies, the otherconsiderations for reproductive and developmentaltoxicity testing include the ethical use of primatesversus rodents, especially for fertility studies, and theduration of the studies. A primate embryofetaldevelopment study can take up to 1 year to completeand a primate pre- and postnatal development studycan take 2 years or more to complete. Reproductivetoxicity studies can be conducted in rodents using asurrogate molecule in a fraction of this time, andcould provide safety information earlier in the devel-opment process than is possible for primate studies.However, this advantage must be balanced with thepreviously mentioned disadvantages associated withdeveloping a surrogate molecule and the possibilitythat the surrogate molecule in the rodent may notbe pharmacologically identical to the clinical candi-date in the primate. Also the resources required todevelop, characterize, and test a surrogate molecule
can exceed that of conducting nonhuman primateDART studies, so there may not be any advantageto the sponsor in developing a surrogate molecule.The most relevant model to assess risk ofreproductive effects in humans should be utilizedand justified to the regulatory agencies. Therefore,because of their limitations, the surrogate moleculeapproach should be considered as an alternate tononhuman primate DART studies, not as an addi-tional species.
There are several examples of approved prod-ucts on the market in the United States for whichthe safety assessment included surrogate moleculesin the regulatory filing. These products includeActimmune (interferon-g; InterMune, Brisbane,CA), Remicade (infliximab; Centocor, Horsham, PA),Raptiva (efalizumab; Genentech, Inc, South SanFrancisco, CA, and Xoma Ltd, Berkeley, CA),Cimzia (Certolizumab pegol; UCB Inc, Smyrna,GA), and Solaris (eculizumab; Alexion Pharmaceu-ticals, Cheshire, CT). For example, to conduct athorough safety assessment of interferon (IFN)-g,which lacks activity in rodents, the sponsor electedto develop a recombinant murine IFN-g and usedthat product to conduct toxicology studies inmice.22,23 Efalizumab and infliximab, monoclonalantibodies recognizing human CD11a and tumornecrosis factor-a (TNF-a), respectively, are activein humans and chimpanzees only. For both prod-ucts, initial toxicology studies that supported thesafety of clinical trials, were conducted in chimpan-zees. To conduct a more thorough safety evaluation,which was necessary for product approval, thesponsors for these products elected to developantibodies that recognized mouse CD11a andTNF-a.21,24 Ecalizumab, an anti-C5 antibody,showed cross-reactivity only to humans. Therefore,a surrogate mouse antibody was developed andchronic and DART studies were conducted withthis surrogate molecule (Solaris approval informa-tion, http://www.fda.gov/). Certolizumab pegol isan anti-TNF antibody Fab0 fragment conjugatedwith PEG to extend the terminal plasma eliminationhalf-life. Inasmuch as certolizumab pegol does notcross-react with mouse or rat TNF-a, reproductionstudies were performed in rats using a rodentanti-murine TNF-a pegylated Fab fragment (cTNFPF), similar to certolizumab pegol. A few of theseexamples will be highlighted in more detail in thefollowing section, along with cases of molecules stillin development.
237
Alternative Strategies for Biopharmaceuticals / Bussiere et al 237
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

Case Study Examples Using AlternateApproaches to Safety Assessment
Approved Products
Efalizumab
Efalizumab is a recombinant humanized monoclonalIgG1 antibody specific for the a subunit (CD11a) ofleukocyte function associated antigen-1 (LFA-1)approved for treatment of chronic plaque psoriasis.Efalizumab binds specifically to human and chimpan-zee CD11a. As a result of the limited species binding,muM17, a murinized rat anti-mouse chimeric IgG2asurrogate antibody specific for murine CD11a wasdeveloped for preclinical safety evaluation. Themurine surrogate antibody was of the IgG2a isotypeto match the potential effector functions of an IgG1isotype in humans. The general preclinical safety pro-gram conducted with muM17 to support registrationof efalizumab included a tissue cross-reactivity study,1- and 6-month repeat dose toxicity studies, maleand female fertility, embryofetal development, andpre- and postnatal development studies. In addition,special immunotoxicology evaluations were con-ducted in a 1-month subcutaneous immunotoxicitystudy and as part of the pre-and post-developmentaltoxicity study to assess the effect of muM17 onimmune function.
Prior to initiation of the preclinical studies, thepharmacology of muM17 was characterized and theactivity of muM17 was demonstrated to be compara-ble with efalizumab in binding and pharmacodynamicassays. Characterization experiments included themeasurement of binding affinity of muM17 to murineCD11a, in vitro activity of muM17 using the murinemixed lymphocyte reaction, and in vivo activity in amouse model of delayed type hypersensitivity. Inaddition, muM17 was shown to have a comparablepharmacodynamic effect as efalizumab in inducingdownmodulation of CD11a on peripheral bloodT cells following ligand binding.
Results from preclinical studies using muM17were consistent with distribution of the target antigenand pharmacology of blocking CD11a/ICAM-1 (intra-cellular adhesion molecule-1) interactions. In thetissue cross-reactivity study, the pattern of stainingusing a panel of murine tissues was similar to resultsobserved with efalizumab on a panel of humantissues. Results from the male and female fertility, andembryofetal development studies, demonstrated no
effects on either fertility or fetal development fromadministration of muM17 to CD-1 mice. Consistentwith these findings, no toxicities were observed ineither the 1- or 6-month toxicity studies.
The results from the 1-month subcutaneousimmunotoxicology study demonstrated administra-tion of muM17 to young adult mice as 4 weekly dosesresulted in decreased IgM and IgG responses tosheep red blood cells (SRBC), a T-cell-dependentantigen, immediately following the dosing phase.This result is consistent with the biology of blockingLFA-1/ICAM-1 interactions. After serum levels ofmuM17 decreased below the level of detection fol-lowing a 4-week recovery period, humoral immuneresponses to SRBC were comparable with controls.Similar decreases in humoral immune responses toSRBC were demonstrated in F1 generation miceexposed to muM17 via the dam during gestation andlactation in the pre- and postnatal developmentstudy. However, in contrast to the normalization ofhumoral immune responses observed in the youngadult mice following clearance of muM17, humoralimmune responses to SRBC in the F1 mice remaineddecreased relative to age-matched vehicle controlanimals following a 22-week recovery period.
Additional data regarding placental transfer andantigenicity of the surrogate antibody were obtainedfrom the toxicology studies. Placental transfer ofmuM17 was confirmed in the reproductive toxicitystudies. Fetal and maternal serum concentrationswere approximately proportional whereas muM17concentrations in amniotic fluid were considerablylower than maternal serum. Antigenicity of muM17was shown to be low in all studies with an incidenceof less than 1% for the entire surrogate moleculeprogram.21 The adverse safety events or toxicitiesobserved in the clinic with efalizumab are reportedto include infection, malignancy, immune thrombo-cytopenia, and hemolytic anemia.31 Approximately2 years after marketing approval a Dear Health CareProfessional letter was authored notifying prescrib-ing physicians of potential toxicities.32 Most of theadverse events observed in the clinic were consistentwith the drug’s mechanism of action but were notpredicted based on the safety profile of the murinesurrogate antibody.
The surrogate antibody program utilized the con-sistent biology of LFA-1/ICAM-1 between species tobuild a robust preclinical program that was success-fully accepted by regulatory agencies in the UnitedStates, European Union, and Japan. It is important
238
238 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

to recognize that, because the use of chimpanzees isgreatly restricted and therefore of limited use forassessing safety, the mouse surrogate molecule effec-tively represented the primary safety evaluation; itwas not developed to allow evaluation of a second,rodent species. At first pass, use of the clinicalcandidate in chimpanzees may appear to be the morerelevant approach to safety assessment comparedwith the use of a surrogate in rodents. However, asaccess to chimpanzees becomes further restrictedin the future due to both supply (decreased breedingin United States33) and ethical considerations (sup-ported by proposal of the 2008 Great Ape ProtectionAct to the US Congress and adoption of similarlegislation in several EU countries), it will be vitalto provide sufficient scientific evidence regardingrelevance of the surrogate molecule in regulatorysubmissions, as well as complimentary data (ie, invitro assays) necessary to adequately bridge surrogatedata and relevance to humans.
Infliximab
Infliximab is an anti-human TNF-a (TNF) mAb thatwas first approved in the United States in 1998 forthe treatment of Crohn’s disease. Infliximab is a chi-meric IgG1 mAb that binds to human TNF and ishighly species specific (cross-reacts only with humanand chimpanzee TNF). Therefore, an analogousanti-TNF mAb (cV1q) that selectively inhibits thefunctional activity of mouse TNF was developed toassess chronic and reproductive toxicity.24 Similarto efalizumab, the murine surrogate antibody wasof the IgG2a isotype to match the potential effectorfunctions of an IgG1 isotype in humans. This surro-gate molecule inhibited disease activity in murinemodels of Crohn’s disease, and was thus demon-strated to be pharmacologically similar to infliximab.For registration, an embryofetal development toxicitystudy, a combined male and female fertility study,and a chronic, 6-month toxicity study in CD-1 micewas performed with the murine surrogate. Animmune response to the anti-murine mAb diddevelop in most animals, but this immune responsedid not affect exposure of the animals to the mAb(ie, no change in PK parameters). A nonterminalacute tolerability study was also conducted in chim-panzees with the anti-human TNF mAb.
Pre- and postnatal development studies in miceusing the murine surrogate antibody were also con-ducted postmarketing.34 In addition to all of the
standard evaluations of postnatal development, anevaluation of immune function was conducted in theF1 mice. When the results from the studies con-ducted with mice using the surrogate are comparedwith published information on TNF-deficientmice,35 the results are generally similar but not iden-tical. Although the genetically deficient mice andsurrogate-targeted mice show a general concordancewith regard to effects on fertility and embryofetaldevelopment, the genetically deficient mice show alack of splenic germinal centers and reduced func-tional immune responses that are not observed in thesurrogate molecule-treated mice. Therefore, thegenetically deficient mice are useful models forunderstanding the biology of TNF, but are imperfectmodels for evaluating the safety of mAb treatment.
This example illustrates an overall approach foran mAb that showed cross-reactivity only to humansand chimpanzees. This approach allowed for the safedosing of infliximab in clinical trials and was alsoacceptable to the regulatory agencies in the UnitedStates, European Union, and Japan. The adverseeffects of infliximab that have been observed in theclinic have been mostly related to selective downmo-dulation of immune responses leading to an increasein some opportunistic infections, which is directlyrelated to the pharmacology of the molecule. Asexpected, infections were not seen in the normalhealthy animals used in the toxicology studies, buthave been seen in TNF-a-deficient animals that arechallenged with specific pathogens. Therefore, tak-ing the entire weight of evidence into consideration,the human toxicities were predictable based uponthe nonclinical information and the pharmacologyof the molecule.
Although the toxicity studies conducted in micewith the surrogate molecule were acceptable to sup-port the clinical use of a human mAb with cross-reactivity only in humans and chimpanzees, themouse studies were not acceptable to the regulatoryagencies to support the clinical use of anotherhuman anti-TNF mAb that was pharmacologicallyactive in humans, chimpanzees, and macaques. Forthe latter mAb, regulatory agencies requiredembryofetal and postnatal development studies incynomolgus monkeys with the humanized mAb,36
despite DART studies previously conducted with asurrogate mAb, and significant chronic-use clinicalexperience with the original chimeric mAb, as wellas several other anti-TNF therapies. Therefore,although it has been proposed that surrogate
239
Alternative Strategies for Biopharmaceuticals / Bussiere et al 239
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

molecules could be used to reduce the use ofnonhuman primates,37 to ensure the regulatoryacceptance of a surrogate molecule-only approachfor a clinical candidate that shows cross-reactivityto nonhuman primates, a dialogue must be con-ducted between the company and the agencies toensure that all parties are in agreement with thescientific justifications for the approach.
Interferon-g
IFN-g is an immunomodulatory cytokine that ishighly species specific, with pharmacologic activityonly in humans and nonhuman primates. In sub-chronic toxicity studies in cynomolgus monkeys, theclinical side-effects that were observed (fever,lethargy, anorexia, and changes in hematology andchemistry parameters) were comparable with clinicaleffects seen in humans.23 However, a neutralizingantibody response was seen that attenuated theresponse in a 13-week study compared with the4-week monkey study. A murine version of IFN-g wasutilized in a 4-week mouse study, where the nature ofthe treatment-related findings and organ systemsaffected were similar to the observations in cynomol-gus monkeys treated with the human protein, and yetno neutralizing antibodies developed.38 Thus, in thiscase the murine surrogate was not developed to allowsafety evaluation in a second, rodent species, butrather to better characterize the response without theconfounding factor of neutralizing immunogenicity.
In developmental toxicity studies, both thehuman IFN-g and the murine surrogate moleculewere abortifacients in cynomolgus monkeys andmice, respectively. The murine surrogate moleculewas also used to evaluate potential developmentaland reproductive capacity of juvenile animals associ-ated with chronic treatment, because juvenilepatients with chronic granulomatous disease arethe main patient population (Actimmune; http://www.fda.gov/cder/foi/label/2007/103836s5098LBL.pdf). In this study, mice were treated with daily doses(0, 0.02, 0.2, or 2 mg/kg/d) from postnatal day 8through day 60 to determine the effects on matura-tion, behavioral/functional development, and repro-ductive capacity.22 Male mice in the high-dosegroup had delayed sexual maturation, reduced epidi-dymal and testes weights, reduced sperm count andconcentration, and sperm abnormalities, and showedreduced mating performance and fertility despite theabsence of altered histopathology of the testes.
Motor activity was also decreased in all mice in thehigh-dose group. Although it is unknown whetherthese findings would be found in humans treatedchronically with IFN-g, this information does appearin the label for Actimmune (along with the caveat ofunknown significance). Prior to conducting thesestudies with the surrogate molecule, a careful reviewand comparison of data regarding biochemical prop-erties, biological activity, and disposition profiles ofboth proteins in similar test systems was performed.The characterization of the mouse surrogate mole-cule in this case allowed for further exploration ofthe reproductive and behavioral effects of IFN-g thatwould have been difficult to evaluate in the nonhu-man primate.
Products in Development
Keliximab, Clenoliximab
Keliximab is a primatized IgG1 mAb directed towarddomain 1 of human CD4.15,39 Clenoliximab is anIgG4 version, developed to reduce Fc interactionswith Fc receptors and thus mitigate T-cell depletionand cytokine release. Keliximab and clenoliximabshow cross-reactivity only to human and chimpanzeeCD4. To evaluate the preclinical safety of thesemolecules, the sponsors developed a transgenicmouse that expressed human CD4 in place of mouseCD4. Both monoclonal antibodies were shown to bepharmacologically active in these KO/KI mice.15
With this KI mouse, the human therapeutic antibo-dies could be tested for nonclinical safety. The KImouse was characterized by demonstration of appro-priate expression of human CD4 and by evaluation ofimmune system function following challenge withinfection or tumor cells. The preclinical safety stud-ies conducted with keliximab in the KI mice includedsingle and repeated dose toxicology studies, male andfemale fertility, embryofetal development, pre- andpostnatal development studies with functionalimmune response evaluation in the F1 generation,and host defense assays.15,39 A nonterminal acutetolerability study was also conducted in chimpan-zees. Toxicity studies conducted in the mice showedthe expected reduction in CD4 cells. The mice diddevelop an immune response toward the human mAband in some instances anaphylactic reactionsoccurred. However, sufficient numbers of animalssurvived and exposure levels were sufficientlymaintained to adequately evaluate the toxicity.
240
240 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

This example demonstrates a novel approach inwhich KI mice expressing the human target antigenwere developed so that the human protein could betested in preclinical studies. These methods have thedisadvantage that human proteins can be highlyimmunogenic in animals and thereby limit the dura-tion of the studies, impact the exposure of the animalto the therapeutic, or lead to adverse effects. Alsothis approach requires development and extensivecharacterization of the animal model, including gen-otyping of all animals used for the studies, to ensureappropriate expression of human CD4. In addition,the model tests only specific inhibition of theintended target and may not expose any secondarypharmacology attributable to closely related ordownstream targets associated with administrationof the human protein. However, when the clinicalcandidate is active only in humans and chimpanzees,data in KI mice treated with the clinical candidatecan be useful in assessing safety.
Fully Human mAb (Anti-Cytokine Receptor)
A fully human mAb targeting a cytokine receptor wasdeveloped for treatment of allergic inflammatoryresponses (data on file, Amgen Inc). Because theactivity of the clinical molecule was limited tohumans and chimpanzees, a surrogate moleculeapproach was taken for the safety program to supportclinical development, utilizing 2 well-characterizedchimeric anti-mouse and anti-monkey surrogateantibodies. These molecules have similar respectiveactivity for murine and cynomolgus monkey cytokineinhibition as does the clinical molecule for humancytokine inhibition. The characterization of thesemolecules included binding and functional activityin cell-based assays in vitro (murine and monkey)and in vivo pharmacologic activity (murine only).The safety program along with various noteworthyrationales behind the necessity for many of thestudies is described below.
Prior to the first in human (FIH) clinical trial, themolecules that were available to conduct preclinicalstudies included the clinical molecule, which onlycross-reacted with the target in humans and chimpan-zees, and the anti-murine surrogate antibody. Asingle-dose chimpanzee study was conducted primar-ily to model the pharmacokinetics of the clinical mole-cule in a relevant species, and a 4-week mousetoxicology study with the anti-murine surrogate wasconducted as the definitive safety study to support the
FIH trial. Because the clinical candidate moleculewas not evaluated in the toxicology studies wherehistopathology evaluation was available, a rabbit localtolerance study was conducted with the clinicalmolecule to assess the irritation potential of theformulation to support the FIH trial.
Information from the literature indicated thatthe cytokine of interest is important in the mainte-nance of pregnancy. In a previous embryo-fetaldevelopment study conducted with a human solublecytokine receptor that had the same pharmacologicactivity (antagonism of the cytokine) in cynomolgusmonkeys, an increased frequency of spontaneousabortions and stillbirths was observed.40 To deter-mine if a better model could be established to under-stand the mechanism of this toxicity, a mouse studywas conducted with a murine surrogate molecule.The cynomolgus monkey reproductive findings werenot reproducible in mice with a murine surrogate ofthe soluble cytokine receptor or with an anti-murinecytokine receptor antibody, suggesting that themouse was not an appropriate species for evaluationof the reproductive effects following inhibition ofthis pathway. Thus, the cynomolgus monkey surro-gate molecule was developed primarily to evaluatereproductive toxicity. The murine surrogate antibodywas utilized for fertility evaluation, because an effecton fertility was not expected based on information inthe literature. The complete package for reproduc-tive toxicology evaluation thus consisted of a fertilitystudy with the murine surrogate molecule in miceand an embryofetal and prenatal development studywith the cynomolgus monkey surrogate molecule inthe cynomolgus monkey.
Results from the anti-monkey surrogate moleculefor the reproductive toxicology evaluations as well asavailability of results from a 1-month repeated-dosecynomolgus monkey toxicology study with the monkeysurrogate molecule were available at the time point forinitiation of the subchronic study. Although bothsurrogate molecules were similar to the clinicalcandidate, to limit resources originally dedicated to2 surrogate molecules, the cynomolgus monkeysurrogate molecule was the only one utilized toconduct the subchronic (3–month) and chronic(6-month) repeated-dose toxicology studies.
Fully Human mAb (Anti-Cytokine)
A fully human mAb targeting a cytokine was devel-oped for the treatment of inflammatory disease (data
241
Alternative Strategies for Biopharmaceuticals / Bussiere et al 241
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

on file, Amgen Inc). The clinical molecule bound torecombinant human cytokine with high affinity, buthad lower affinity (approximately 30-fold) for thecynomolgus monkey cytokine. In addition, the clini-cal molecule could neutralize the ability of thehuman cytokine to stimulate human cells in a cell-based assay, but was not able to efficiently neutralizethe cynomolgus monkey cytokine. This demonstratesthat binding alone may not be sufficient todemonstrate species relevance. A murine surrogatemolecule was not available at the time of initiationof the preclinical safety program. Thus, to enablepreclinical studies in cynomolgus monkeys, a surro-gate antibody was developed by fusing the F(ab) por-tion of a mouse anti-human cytokine, known toneutralize the cynomolgus monkey cytokine, withhuman Fc. The chimeric surrogate molecule hadnearly as high an affinity for the human cytokine asdid the clinical molecule, and higher binding affinityfor the cynomolgus monkey cytokine compared withthe clinical molecule. The surrogate molecule alsoneutralized the ability of cynomolgus monkey cyto-kine to stimulate cynomolgus monkey cells in a man-ner similar to the neutralization of human cytokineactivity on human cells by the clinical molecule.Thus, the surrogate molecule was used in the mon-key for the nonclinical safety program.
Toxicology studies were conducted in cynomol-gus monkeys with the surrogate molecule to supportthe early stage clinical development plan. Prior to theFIH clinical trial, studies conducted included arepeated-dose study of 1-month duration and a safetypharmacology study (cardiovascular, respiratory, andcentral nervous system). Results from the 1-monthstudy indicated a pharmacodynamic effect with thesurrogate molecule in the monkey that would be pre-dicted based on one of the known activities for thecytokine of interest, as noted in the literature. Inter-estingly, this pharmacodynamic effect was not evi-dent in the FIH trial, indicating that there may bea species difference in response to inhibition of thecytokine of interest or that there is a differencebetween the monkey surrogate molecule and theclinical candidate. This example highlights thechallenges when using a surrogate molecule and con-flicting data are generated. Further work is thenneeded to understand whether the difference is dueto the use of a different molecule, or whether theactivity of that target is different in the animalspecies compared with humans, or between differentanimal species (as seen in the previous example).
Fusion Protein
A soluble lymphotoxin b receptor consisting of theextracellular domain of human lymphotoxin b fusedto the Fc region of human IgG1 (LTbR-Fc) is currentlyin development for the treatment of rheumatoid arthri-tis. The pharmacologic activity is limited to humansand nonhuman primates. During development, ananti-murine lymphotoxin b receptor Fc IgG fusion sur-rogate molecule was generated to evaluate the pharma-cology of the soluble receptor in rodents. The Fc regionin the murine surrogate molecule was of the IgG2a iso-type to match the effector function of an IgG1 isotypein humans. Adult mice treated with the surrogateLTbR-Fc showed reduced immune responses andreduced disease activity in a number of murine diseasemodels.41 Mice that are genetically deficient in LTbhave also been generated and shown reduced immuneresponses and an absence of lymph nodes.42 Pregnantmice treatedwith the surrogateLTbR-Fcmolecule hadoffspring that lacked lymph nodes.43 However, admin-istration of the human LTbR-Fc to pregnant monkeys(a pharmacologically relevant species) was not associ-ated with an absence of lymph nodes.44 The differ-ences observed between the surrogate in the mouseand the human therapeutic in the monkeys may be dueto differences in the timing of lymph node develop-ment relative to placental transport of antibodies inmice versus primates, although monkey fetal exposureswere not reported in this study.
This example illustrates how results obtained with asurrogatemolecule inmiceorextrapolatedfromKOmicemay not necessarily be representative of the results thatare likely to occur in nonhuman primates or humans,particularly in developmental toxicity studies in whichspecies differences are known to exist in the stages ofembryofetal development. In this case, the surrogate andthe genetically deficient animals identified the potentialhazard, but may have overestimated the human risk.
Questions Regarding the Use of SurrogateMolecules
If a Surrogate Molecule Is Used to AssessAny Aspect of Safety, Is It a Requirementto Conduct All Toxicology Studies in ThatSpecies?
The nonclinical safety programs for small moleculedrugs routinely require assessment of general toxicity
242
242 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

in 2 species, including a rodent (usually the rat) anda nonrodent (usually the dog) (ICH M3 (R1)45). TheICH S6 guidance document for biopharmaceuticalsstates that ‘‘Safety evaluation programs shouldnormally include 2 relevant species. However, incertain justified cases, one relevant species maysuffice (e.g., when only one relevant species can beidentified or where the biological activity of thebiopharmaceutical is well understood).’’ Further-more, the guidance document goes on to say that‘‘even where two species may be necessary to charac-terize toxicity in short term studies, it may be possibleto justify the use of only one species for subsequentlong-term studies (e.g., if the toxicity profile in the2 species is comparable in the short term).’’ There-fore, for biopharmaceuticals that have a very specificmechanism of action that is well understood, such asmonoclonal antibodies and receptor fusion proteins,a 2-species assessment may not always be necessary.
For species-restricted biopharmaceuticals, themost common species used for nonclinical safetytesting is a nonhuman primate, usually a macaque.The toxicities that have been observed in macaquesfor biopharmaceuticals have generally been directlyrelated to the pharmacology of the molecule andoff-target toxicities are rarely observed. Because thesurrogate molecule has many limitations that havebeen mentioned previously, the surrogate moleculeprovides supportive information only in those situa-tions where there is no other option for nonclinicalsafety testing or where information can be obtainedin the rodent that cannot be adequately evaluatedin the nonhuman primate (eg, fertility). In cases inwhich a surrogate molecule is available and is consid-ered optimal for use in developmental and reproduc-tive toxicity evaluations, inclusion of standardtoxicity endpoints (ie, clinical pathology endpoints)in preliminary dose-ranging studies may complimentthe overall weight of evidence regarding drug safety.These may provide a useful comparison to thefindings seen in the general toxicology studies withthe clinical candidate.
Should a Surrogate Molecule BeDeveloped to Allow for Assessment ofCarcinogenicity?
The question of carcinogenicity is not so much oneof whether a surrogate molecule should be used forcarcinogenity testing, but whether 2-year bioassays
in rodents are relevant models for evaluatingcarcinogenic potential for biopharmaceuticals.46
Carcinogens generally fall into 3 major categories:genotoxic carcinogens, cellular proliferators, andimmune suppressants.47 Biopharmaceuticals have alarge molecular weight that precludes them from dif-fusing into cells and interacting with DNA. Therefore,biopharmaceuticals are unlikely to be genotoxiccarcinogens.
Biopharmaceuticals that induce cellular prolif-eration such as insulin-like growth factor and growthhormone are assumed to be associated with a greaterrisk of tumor development. Increased cellular prolif-eration can be detected with in vitro studies and maybe evident in repeated-dose toxicology studies ofsufficient duration. The absence of hyperplasia in arepeated-dose toxicity study may be indicative thatthe biopharmaceutical is unlikely to be carcinogenicbecause of increased proliferation, when consideredin context with the duration of dosing, level of anal-ysis, and known target biology. A recently publishedstudy has described the development of mouse- andrat-specific growth hormones for the evaluation ofcarcinogenic potential in 2-year bioassays assurrogate molecules for human growth hormone.48
The studies showed no increase in tumors in thetreated animals. However, it is not clear that thesenegative results in rodents will change either theperception or labeling of the product, even inthe context of 40 years of clinical experience usinghuman growth hormones.
Finally, immune suppressive agents areassumed to increase a patient’s susceptibility to cer-tain tumor types (especially lymphomas and skincancer) because of decreased host defense.49 Thishypothesis has been based upon clinical experience,not upon 2-year bioassays, which are frequentlynegative for small molecule nongenotoxic immuno-suppressive agents. Immunosuppressive drugs andimmunomodulating biopharmaceuticals carrywarnings on their product labels for a potentialincreased susceptibility to tumors. In a 2-year bioas-say, mice infected with both mouse leukemia virusand mammary tumor virus and then treated withabatacept, which inhibits T-cell activation, werefound to develop lymphomas and mammary tumors,respectively.50 Therefore, this study reinforces thehypothesis that immunosuppression can increasesusceptibility to oncogenic viruses but did notprovide new information and did not change thewarnings in the product label.
243
Alternative Strategies for Biopharmaceuticals / Bussiere et al 243
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

The carcinogenic risk assessment for biopharma-ceuticals can best be made based upon an under-standing of the biology of the molecule. Anexample of a rational, scientific-based assessmentof carcinogenicity potential in the absence of animalcarcinogenicity testing has been published forinterleukin-10.51 Overall, for the majority of bio-pharmaceuticals, there is little rationale for conduct-ing 2-year bioassays for biopharmaceuticals witheither the human therapeutic (if it shows rodentcross- reactivity) or with a surrogate molecule,because the assessment of carcinogenic potentialand communication of risk could be achieved byother means (ie, in vitro data, pharmacology data,appropriate wording in the product label, etc). Inthe end, assessment of carcinogenic potential shouldbe made on a case-by-case basis considering therelevance of carcinogenicity testing in the contextof target biology, to adequately communicate carci-nogenic risk to patients.
How Should the Relevance of theSurrogate Molecule to the ClinicalCandidate Be Determined? What Is theImportance of Understanding Target-Mediated Pharmacologic Effects VersusCompound-Related Off-Target Effects?
It is important that any compound selected for use asa surrogate molecule be shown to be pharmacologi-cally similar to the development compound, because,if not, it is possible that erroneous conclusionsabout safety could be reached. A few examples aredescribed below.
In the first example, a sponsor was developing anIgG1 Fc-mutated humanized mAb against a chemo-kine receptor present on T cells and monocytes thatwas believed to be important in leukocyte traffickingand activation in inflammatory and autoimmune dis-eases (C. Horvath, personal communication, 2009).The antibody had been tested extensively in vitrowith human cell systems and demonstrated to be aligand-binding antagonist with no signaling throughthe receptor and no target cell-depleting effects.These properties were confirmed in early clinicaltrials with the humanized mAb. To potentiallyaddress reproductive toxicology and host resistanceconcerns related to target blockade, an anti-rodentmAb was desired for use as a surrogate molecule fortoxicity testing. One such anti-rodent IgG2 mAb had
been published by an academic laboratory to beeffective in several animal models of autoimmunedisease and to induce no target cell depletion. ThismAb was subsequently in-licensed by the sponsor foruse as a development surrogate molecule and smallquantities were synthesized for evaluation. Theanti-rodent mAb was administered to normal miceand target cells were immunophenotyped and evalu-ated for receptor expression and blockade by flowcytometry. Unlike the development mAb, the surro-gate mAb resulted in depletion of all target cellswithin the circulating blood, as well as spleen tissue,within 15 min of dosing. T cells and monocytes didnot begin to return to the circulation until approxi-mately 3 days later. Upon reviewing the publicationsthat demonstrated efficacy with this rodent mAb, itwas found that the cell-depleting properties were notknown because early time points after dosing had notbeen evaluated. In light of this information (celldepletion with the surrogate molecule vs no targetcell depletion with the clinical molecule), the spon-sor questioned whether the results with the surrogatemAb in models of autoimmune disease would be reli-able indicators of the results that might be expectedfor the development mAb in human autoimmunediseases. An attempt to alter the cell-depletingproperties of the surrogate molecule by antibodyengineering was then undertaken. Despite almost2 years of dedicated efforts, the cell-depleting prop-erties of this mAb could not be eliminated by isotypeswitching or Fc mutations. The sponsor concludedthat the anti-rodent mAb could not be used as asurrogate molecule because either the surrogatemAb was not pharmacologically similar to the anti-human mAb (eg, different Fc-Fc receptor inter-actions) or the rodent species was not biologicallysimilar to humans (eg, different target expressionand/or function). In this example, a safety profilegenerated with the surrogate mAb may have over-estimated potential concerns (eg, extent of immuno-modulation) because the surrogate molecule resultedin destruction of the target cells, rather than block-ade of a specific cellular function.
The second example is a recent one in whichreliance on results of a surrogate mAb may have con-tributed to an underestimation of the potential safetyconcerns for the development mAb. TGN1412 was ahumanized IgG4 mAb directed against CD28 onT cells being developed for oncology and autoimmunediseases. In vitro, the TGN1412 ‘‘superagonist’’ mAbdirectly activated human T cells in the absence of a
244
244 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

second costimulatory signal and resulted in cytokinerelease and T-cell proliferation, with a bias toward aregulatory immunophenotype (Treg cells). However,when dosed to normal volunteers, TGN1412 resultedin massive cytokine release (a ‘‘cytokine storm’’),accompanied by rapid, severe T-cell depletion (within1 hour, the earliest time point at which T-cell countswere measured) associated with headache, rigors,myalgia, hypotension, tachycardia, fever, and multior-gan failure.52 This response of T-cell depletion andcytokine release had not been described in preclinicalstudies. The pharmacologic effect of CD28 stimula-tion had been evaluated extensively in rats with a sur-rogate anti-rat CD28 mAb, JJ316.53 This mAb wasshown to be effective in several animal models ofautoimmune disease, such as adjuvant-inducedarthritis (AA) and experimental allergic encephalo-myelitis (EAE), but not collagen-induced arthritis(CIA). In normal rats, the surrogate mAb was associ-ated with marked increases in blood and spleen T-cellcounts (up to *20-fold) and marked expansion (up to*6-fold) of lymphoid tissues, such as spleen andlymph nodes, within 3 days.54,55 No adverse effectsrelated to cytokine release were reported in this study.The results generated with the surrogate mAb there-fore suggested that administration of an anti-CD28mAb could be well tolerated.
Because TGN1412 (or an IgG1 version,TGN1112) recognized CD28 in rhesus and cynomol-gus monkeys, it was evaluated in these species andwas reported to be efficacious in a monkey modelof rheumatoid arthritis (CIA). In toxicology studiesin monkeys, TGN1412 was associated with minimalcytokine release and, at approximately 2 weeks afterdosing, with only mild increases in blood T-cellcounts (*2-fold) and minimal evidence of expansionof lymphoid tissues.56-58 These results for TGN1412in monkeys were in contrast to those obtained withJJ316 in rats, where profound lymphocytosisoccurred. Although a cytokine storm did not occurin monkeys, close examination of the reported lym-phocyte counts suggests that T cells may have beendepleted in monkeys after dosing (as they were inhumans). It is not possible to confirm this becauseearly postdose time points were not evaluated.T-cell depletion was, however, a prominent findingwhen TGN1412 was administered to another speciesexpressing human CD28þ T cells. When H2d Rag2–/–gc�/� KO mice are irradiated and their bonemarrow reconstituted with human CD34þ fetal liver(stem) cells, they develop human immune systems
(HIS) with all major human myeloid and lymphoidcellular compartments, including CD28þ human Tcells. When the original mouse precursor ofTGN1412 (mAb 5.11A1) was administered to thesemice, they developed rapid, profound T-cell deple-tion that persisted through 60 days.59 These resultsare in contrast to those obtained with the anti-rodent surrogate mAb. Thus, perhaps testing of thedevelopment compound in this ‘‘surrogate species’’expressing human T cells was more representativeof the potential safety concerns for the developmentmAb. These examples highlight some of the caveatsthat must be considered when electing to supportdevelopment products with surrogate compounds.Further, because the generation of surrogate mole-cules for toxicology studies is a time- and resource-consuming effort, use of a surrogate molecule shouldbe warranted only in special cases. Surrogates stud-ies should not be conducted based on ‘‘no effect’’ innonhuman primate studies if no toxicity was pre-dicted based on super pharmacology or the absenceof the target in a normal animal.
Discussion/Conclusions
This review illustrates that alternative approaches,including animal models of disease, KO/Tg or huma-nized mice, or surrogate molecules, can be appropri-ate to improve the predictive value of preclinicalsafety assessments for species-specific biopharma-ceuticals, although many caveats must be considered(Tables 3 and 4). Surrogate molecules may be partic-ularly useful for repeated-dose toxicity studies (incases where the molecule cross-reacts only with thehuman and chimpanzee target), or for specialty stud-ies such as fertility toxicity testing, immunotoxicitytesting, host resistance models, and so forth (in caseswhere the molecule cross-reacts with primates, butnot rodents). However, having a surrogate moleculeavailable does not obligate evaluation of a ‘‘secondspecies’’ or the conduct of carcinogenicity studies.Rather, alternative approaches should only be usedto evaluate safety when the clinical candidate cannotbe readily used to identify the hazard in an appropri-ate nonclinical species.
There are many issues to be considered whenpursuing alternative approaches with a surrogatemolecule. Indeed, it is critical to sufficiently under-stand the similarity and relevance of the surrogatemolecule to the clinical candidate (eg, based on
245
Alternative Strategies for Biopharmaceuticals / Bussiere et al 245
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

Table
3.
Spec
ific
Issu
esW
ith
Non
clin
ical
Eva
luat
ion
of
PK
/PD
/Toxi
city
of
Bio
ph
arm
aceu
tica
lsL
ackin
gC
ross
-Rea
ctiv
ity
inT
radit
ion
alS
pec
ies
(eg,
Rat
san
dD
ogs
)
Dev
elopm
ent
Str
ateg
y
Tes
tH
um
anT
estA
rtic
le(C
lin
ical
Can
did
ate)
in
NH
PS
pec
ies
Tes
tS
urr
oga
teR
oden
tT
est
Art
icle
(Hom
olo
gue)
inR
oden
tS
pec
ies
Tes
tK
nock
ou
tM
ice
Wit
hG
enet
icD
elet
ion
of
Tar
get
Pro
Con
Pro
Con
Pro
Con
Spec
ies
char
acte
rist
ics
Rel
ativ
eex
pen
seE
xpen
sive
Inex
pen
sive
Can
be
very
expen
sive
Nu
mber
sof
anim
als
and
sam
plin
g
Allow
sre
pea
ted
sam
plin
g
Gen
eral
lyre
qu
ires
low
n/g
rou
p;
gen
eral
lyn
ot
pow
ered
for
stat
isti
cal
sign
ific
ance
;
indiv
idu
als
may
pro
vide
toxi
city
sign
als
Allow
sh
igh
n/g
rou
p;
may
be
pow
ered
for
stat
isti
-ca
lsi
gnif
ican
ce
Usu
ally
does
not
allo
wre
pea
ted
sam
plin
g
Allow
sh
igh
n/g
rou
p;
may
be
pow
ered
for
stat
isti
cal
sign
ific
ance
Usu
ally
does
not
allo
w
repea
ted
sam
plin
g
Popu
lati
on
and
indiv
idu
al
anim
alch
arac
teri
zati
on
Het
eroge
neo
us
popu
la-
tion
(lik
eh
um
ans?
);
may
nee
dto
scre
enfo
r
sele
cted
attr
ibu
tes
Hom
oge
neo
us
popu
lati
on
Hom
oge
neo
us
popu
lati
on
May
hav
eto
ph
eno-
type
or
gen
oty
pe
indiv
idu
als
An
imal
avai
labilit
yG
ener
ally
good
Gen
eral
lygo
od
Gen
eral
lypoor;
lim
-
ited
nu
mber
of
ven
dors
;m
ayre
qu
ire
spec
ial
con
sider
atio
ns,
incl
udin
gsc
ale-
up
of
pro
du
ctio
nO
pport
un
isti
cor
bac
k-
grou
nd
infe
ctio
ns
Occ
asio
nal
lypre
sen
t;
sero
logy
test
ing
may
not
be
reliab
le
Gen
eral
lyn
ot
anis
sue;
bar
rier
der
ived
or
VA
F
stra
ins
avai
lable
Occ
asio
nal
lypre
sen
t;
espec
ially
iffr
om
acad
emia
;m
ay
nee
dto
be
reder
ived
Oth
eru
ses
for
spec
ies+
trea
tmen
tw
ith
test
arti
cle
Allow
ste
stin
gof
oth
ercl
inic
alin
dic
atio
ns
(lim
-
ited
nu
mber
of
dis
ease
model
s),
imm
un
oto
xici
ty,
chro
nic
toxi
city
and
repro
du
ctiv
e
and
dev
elopm
en-
tal
toxi
city
test
ing
Gen
eral
lyn
ot
use
dfo
r
host
resi
stan
ceas
says
,tu
mor
surv
eillan
ce
assa
ysor
carc
inoge
ni-
city
test
ing;
no
indu
s-
try
stan
dar
dfo
rN
HP
imm
un
oto
xici
tyor
imm
un
om
odu
lati
on
assa
ys
Allow
ste
stin
gof
oth
er
clin
ical
indic
atio
ns
(dis
ease
model
s),
imm
un
oto
xici
ty,
host
resi
stan
ceas
says
,
tum
or
surv
eillan
ce,
chro
nic
toxi
city
,re
pro
-
du
ctiv
ean
ddev
elop-
men
tal
toxi
city
and
carc
inoge
nic
ity
Allow
ste
stin
gof
oth
ercl
inic
alin
dic
atio
ns
(dis
-
ease
model
s),
imm
un
oto
xici
ty,
host
resi
stan
ceas
says
,tu
mor
surv
eillan
ce,
chro
nic
toxi
city
,
repro
du
ctiv
ean
ddev
elopm
enta
l
toxi
city
and
carc
i-
noge
nic
ity.
May
allo
wte
stin
gof
repla
cem
ent
ther
apie
s.
(con
tin
ued
)
246
246 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

Table
3.
(con
tin
ued
)
Dev
elopm
ent
Str
ateg
y
Tes
tH
um
anT
estA
rtic
le(C
lin
ical
Can
did
ate)
in
NH
PS
pec
ies
Tes
tS
urr
oga
teR
oden
tT
est
Art
icle
(Hom
olo
gue)
inR
oden
tS
pec
ies
Tes
tK
nock
ou
tM
ice
Wit
hG
enet
icD
elet
ion
of
Tar
get
Pro
Con
Pro
Con
Pro
Con
Rea
gen
ts,
assa
ys,
met
h-
ods,
con
trols
(eg,
clin
i-
cal
or
anat
om
icpat
holo
gy)
Gen
eral
lyav
aila
ble
,
oft
enad
apte
d
from
hu
man
Gen
eral
lyav
aila
ble
,oft
en
dif
fere
nt
from
hu
man
Gen
eral
lyav
aila
ble
,
may
be
dif
fere
nt
from
wild-t
ype
mic
e
CR
Oca
pac
ity
and
capab
ilit
ies
Gen
eral
lygo
od
Gen
eral
lygo
od
May
requ
ire
spec
ial
con
sider
atio
ns;
CR
Os
relu
ctan
tto
acce
pt
anim
als
from
non
-appro
ved
ven
dors
His
tori
cal
con
trols
Gen
eral
lyav
aila
ble
Gen
eral
lyav
aila
ble
May
not
be
avai
lable
;
may
nee
dto
gen
er-
ate;
oft
encr
eate
d
inn
on
trad
itio
nal
stra
ins;
no
indu
stry
con
sen
sus
for
‘‘ph
enoty
pin
g’’k/
o
Tar
get
bio
logy
inse
lect
edsp
ecie
sT
issu
eex
pre
ssio
nan
dfu
nct
ion
Tar
get
expre
ssio
nan
dfu
nct
ion
in
NH
Ps
may
be
com
par
able
toh
um
ans
(sh
ou
ld
dem
on
stra
tebio
-
logi
cal
acti
vity
of
clin
ical
can
did
ate)
Mu
stdem
on
stra
teth
atro
den
tsp
e-
cies
targ
etbio
logy
isre
leva
nt
for
hu
man
s
Roden
tsp
ecie
sk/
ota
rget
bio
logy
oft
endes
crib
ed
(val
idat
ed?)
inlite
ratu
re
Gen
etic
del
etio
nor
inse
rtio
nof
ata
rget
may
not
repre
sen
t
chro
nic
blo
ckad
eor
repla
cem
ent
by
test
arti
cle;
redu
n-
dan
tpat
hw
ays,
fals
ebio
logy
etc
Role
of
targ
etin
dis
ease
model
s
NH
Pm
odel
sof
dis
ease
are
gen
eral
lyle
ss
com
mon
than
roden
tm
odel
s
Role
of
roden
tta
rget
in
dis
ease
model
soft
en
des
crib
edin
lite
ratu
re
Gen
eral
lyn
ot
know
n
ifpre
dic
tive
of
role
inh
um
andis
ease
s
Role
of
roden
tta
rget
indis
ease
model
s
oft
endes
crib
edin
lite
ratu
re
Gen
eral
lyn
ot
know
n
ifpre
dic
tive
of
role
inh
um
andis
ease
s
Tes
tar
ticl
e
man
ufa
ctu
re
Su
pply
Allow
su
seofcl
inic
al
can
did
ate
Req
uir
esla
rge
amou
nt
of
clin
ical
can
did
ate
Req
uir
essm
all
amou
nt
of
hom
olo
gue
Req
uir
esdev
elop-
men
tof
hom
olo
-gu
ew
ith
GM
P-
like
man
ufa
ctu
re
Ch
arac
teri
zati
on
Rel
ies
on
exis
tin
g
bio
chem
ical
char
-ac
teri
zati
on
and
lot
rele
ase
spec
s
Req
uir
esdev
elop-
men
tof
new
bio
-ch
emic
alch
arac
-
teri
zati
on
and
lot
rele
ase
spec
s;m
ay
not
repre
sen
tcl
inic
alca
ndid
ate
(con
tin
ued
)
247
Alternative Strategies for Biopharmaceuticals / Bussiere et al 247
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

Table
3.
(con
tin
ued
)
Dev
elopm
ent
Str
ateg
y
Tes
tH
um
anT
estA
rtic
le(C
lin
ical
Can
did
ate)
in
NH
PS
pec
ies
Tes
tS
urr
oga
teR
oden
tT
est
Art
icle
(Hom
olo
gue)
inR
oden
tS
pec
ies
Tes
tK
nock
ou
tM
ice
Wit
hG
enet
icD
elet
ion
of
Tar
get
Pro
Con
Pro
Con
Pro
Con
Impu
riti
esor
con
tam
inan
ts
Allow
ste
stin
gof
pro
du
ct-r
elat
ed
impu
riti
es,
con
-
tam
inan
ts,
or
oth
erch
arac
teri
s-
tics
of
clin
ical
can
did
ate
(eg,
glyc
osy
lati
on
,h
ost
cell
pro
tein
s)
Hom
olo
gue
may
hav
epro
duct
-
rela
ted
impuri
ties
,
con
tam
inan
ts,
or
oth
erdif
fere
nce
s
from
hT
A(e
g,gl
y-
cosy
lati
on
,h
ost
cell
pro
tein
s);m
aynot
repre
sen
thT
A
Tes
tar
ticl
e
ph
arm
acolo
gy
Invi
tro
or
invi
vo
ph
arm
acolo
gy
Aff
init
y,sp
ecif
icit
y,
acti
vity
of
clin
ical
can
did
ate
in
NH
Ps
may
be
com
par
able
to
hu
man
s(m
ust
dem
on
stra
te)
Mu
stdem
on
stra
te
that
affi
nit
y,sp
e-ci
fici
ty,
acti
vity
of
hom
olo
gue
in
roden
tsp
ecie
sis
rele
van
tfo
rh
um
ans
Pote
nti
alse
con
dar
y
and
par
alle
lef
fect
sfr
om
gen
etic
man
ipu
lati
on
may
con
fou
nd
resu
lts
Rea
gen
ts,
assa
ys,
met
h-
ods,
con
trols
(eg,
to
det
ect
PK
,P
D,
imm
u-
noge
nic
ity
resp
on
ses)
Gen
eral
lyav
aila
ble
,
oft
enad
apte
d
from
clin
ical
can
-did
ate
met
hods
for
hu
man
s
Req
uir
esdev
elop-
men
tof
new
reag
ents
,as
says
,m
eth
ods
and
con
-
trols
spec
ific
for
hom
olo
gue
inro
den
tsp
ecie
s
PK
/PD
PK
/PD
of
clin
ical
can
did
ate
in
NH
Ps
may
be
com
par
able
to
hu
man
s
Mu
stdem
on
stra
te
that
hom
olo
gue
PK
/PD
inro
den
tsp
ecie
sis
rele
van
t
for
hu
man
s(c
om
-
par
able
tocl
inic
al
can
did
ate
inN
HP
s?)
Imm
un
oge
nic
ity
Clin
ical
can
did
ate
oft
en
imm
un
oge
nic
in
NH
Ps
Hom
olo
gue
may
be
imm
un
oge
nic
in
roden
tsp
ecie
sIm
mu
noto
xici
tyG
ener
alis
sues
No
indu
stry
stan
dar
dfo
r
NH
Pim
mu
noto
xor
imm
un
om
odu
lati
on
assa
ys;
lack
of
his
tori
-
cal
con
trols
,va
lidat
ed
assa
ys,
posi
tive
con
-
trols
,et
c;dif
ficu
ltie
sin
inte
rpre
tin
gre
sult
s
sin
cesm
all
n
May
be
able
toap
ply
met
hods
adap
ted
from
adu
ltst
udie
s(t
his
isth
etr
end)
Lac
kof
his
tori
cal
con
trols
,va
li-
dat
edas
says
,pos-
itiv
eco
ntr
ols
,et
c;
dif
ficu
ltie
sin
inte
rpre
tin
g
resu
lts
Gen
etic
del
etio
nor
inse
rtio
nof
ata
rget
may
not
repre
sen
tch
ron
icblo
ckad
e
or
repla
cem
ent
by
clin
ical
can
did
ate
CR
O,
Con
trac
tR
esea
rch
Org
aniz
atio
n;
GM
P,
Good
Man
ufa
ctu
rin
gP
ract
ice;
hT
A,
hu
man
test
arti
cle;
NH
P,
non
hu
man
pri
mat
e;P
D,
ph
arm
acodyn
amic
s;P
K,
ph
arm
acoki
net
ics;
VA
F,
Vir
us
An
tibody
Fre
e.
248
248 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from
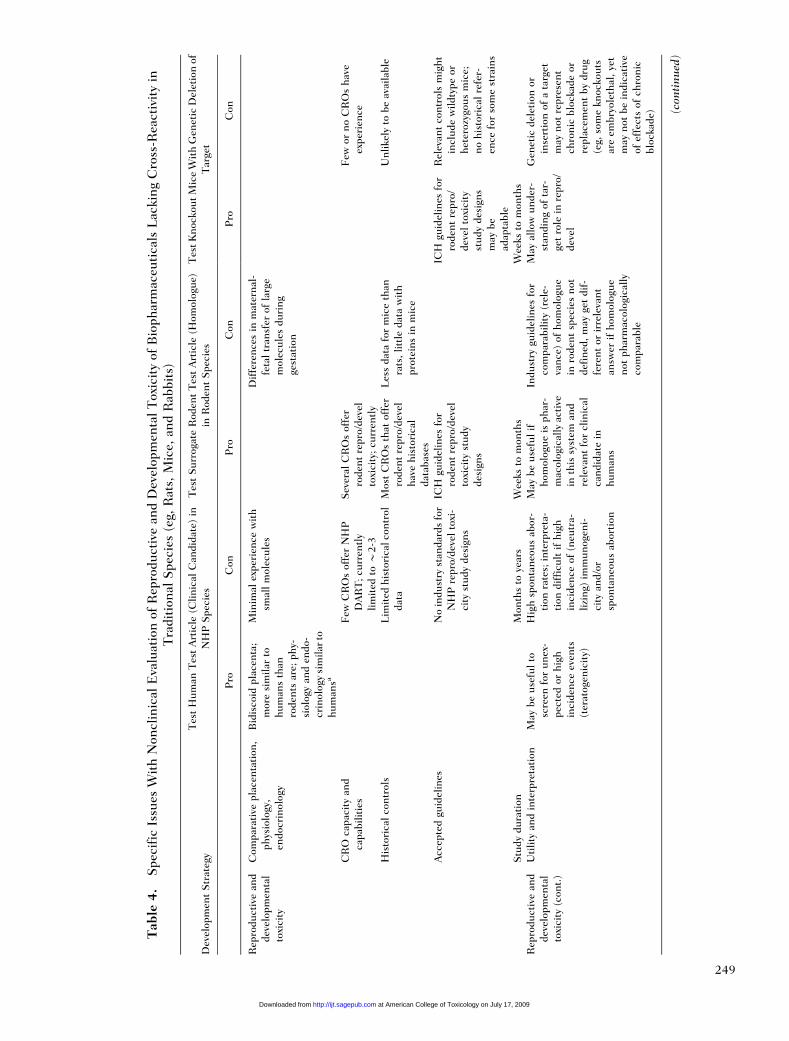
Table
4.
Spec
ific
Issu
esW
ith
Non
clin
ical
Eva
luat
ion
of
Rep
rodu
ctiv
ean
dD
evel
opm
enta
lT
oxi
city
of
Bio
ph
arm
aceu
tica
lsL
ackin
gC
ross
-Rea
ctiv
ity
inT
radit
ion
alS
pec
ies
(eg,
Rat
s,M
ice,
and
Rab
bit
s)
Dev
elopm
ent
Str
ateg
y
Tes
tH
um
anT
est
Art
icle
(Clin
ical
Can
did
ate)
in
NH
PS
pec
ies
Tes
tS
urr
oga
teR
oden
tT
est
Art
icle
(Hom
olo
gue)
inR
oden
tS
pec
ies
Tes
tK
nock
ou
tM
ice
Wit
hG
enet
icD
elet
ion
of
Tar
get
Pro
Con
Pro
Con
Pro
Con
Rep
rodu
ctiv
ean
ddev
elopm
enta
l
toxi
city
Com
par
ativ
epla
cen
tati
on
,ph
ysio
logy
,
endocr
inolo
gy
Bid
isco
idpla
cen
ta;
more
sim
ilar
to
hu
man
sth
an
roden
tsar
e;ph
y-
siolo
gyan
den
do-
crin
olo
gysi
milar
to
hu
man
sa
Min
imal
exper
ien
cew
ith
smal
lm
ole
cule
sD
iffe
ren
ces
inm
ater
nal
-fe
tal
tran
sfer
of
larg
e
mole
cule
sdu
rin
g
gest
atio
n
CR
Oca
pac
ity
and
capab
ilit
ies
Few
CR
Os
off
erN
HP
DA
RT
;cu
rren
tly
lim
ited
to*
2-3
Sev
eral
CR
Os
off
er
roden
tre
pro
/dev
elto
xici
ty;
curr
entl
y
Few
or
no
CR
Os
hav
e
exper
ien
ce
His
tori
cal
con
trols
Lim
ited
his
tori
calco
ntr
ol
dat
a
Most
CR
Os
that
off
er
roden
tre
pro
/dev
elh
ave
his
tori
cal
dat
abas
es
Les
sdat
afo
rm
ice
than
rats
,litt
ledat
aw
ith
pro
tein
sin
mic
e
Un
like
lyto
be
avai
lable
Acc
epte
dgu
idel
ines
No
indu
stry
stan
dar
ds
for
NH
Pre
pro
/dev
elto
xi-
city
stu
dy
des
ign
s
ICH
guid
elin
esfo
r
roden
tre
pro
/dev
elto
xici
tyst
udy
des
ign
s
ICH
guid
elin
esfo
r
roden
tre
pro
/dev
elto
xici
ty
stu
dy
des
ign
s
may
be
adap
table
Rel
evan
tco
ntr
ols
mig
ht
incl
ude
wildty
pe
or
het
erozy
gou
sm
ice;
no
his
tori
cal
refe
r-
ence
for
som
est
rain
s
Stu
dy
du
rati
on
Mon
ths
toye
ars
Wee
ksto
mon
ths
Wee
ksto
mon
ths
Rep
rodu
ctiv
ean
d
dev
elopm
enta
lto
xici
ty(c
on
t.)
Uti
lity
and
inte
rpre
tati
on
May
be
use
ful
to
scre
enfo
ru
nex
-pec
ted
or
hig
h
inci
den
ceev
ents
(ter
atoge
nic
ity)
Hig
hsp
on
tan
eou
sab
or-
tion
rate
s;in
terp
reta
-ti
on
dif
ficu
ltif
hig
h
inci
den
ceof
(neu
tra-
lizi
ng)
imm
un
oge
ni-
city
and/o
rsp
on
tan
eou
sab
ort
ion
May
be
use
ful
if
hom
olo
gue
isph
ar-
mac
olo
gica
lly
acti
ve
inth
issy
stem
and
rele
van
tfo
rcl
inic
al
can
did
ate
inh
um
ans
Indu
stry
guid
elin
esfo
r
com
par
abilit
y(r
ele-
van
ce)
of
hom
olo
gue
inro
den
tsp
ecie
sn
ot
def
ined
,m
ayge
tdif
-
fere
nt
or
irre
leva
nt
answ
erif
hom
olo
gue
not
ph
arm
acolo
gica
lly
com
par
able
May
allo
wu
nder
-
stan
din
gof
tar-
get
role
inre
pro
/
dev
el
Gen
etic
del
etio
nor
inse
rtio
nof
ata
rget
may
not
repre
sen
t
chro
nic
blo
ckad
eor
repla
cem
ent
by
dru
g
(eg,
som
ekn
ock
ou
tsar
eem
bry
ole
thal
,ye
t
may
not
be
indic
ativ
e
of
effe
cts
of
chro
nic
blo
ckad
e) (con
tin
ued
)
249
Alternative Strategies for Biopharmaceuticals / Bussiere et al 249
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

Table
4.
(con
tin
ued
)
Dev
elopm
ent
Str
ateg
y
Tes
tH
um
anT
est
Art
icle
(Clin
ical
Can
did
ate)
in
NH
PS
pec
ies
Tes
tS
urr
oga
teR
oden
tT
est
Art
icle
(Hom
olo
gue)
inR
oden
tS
pec
ies
Tes
tK
nock
ou
tM
ice
Wit
hG
enet
icD
elet
ion
of
Tar
get
Pro
Con
Pro
Con
Pro
Con
Oth
eru
ses
Eff
icac
ym
odel
sfo
r
test
ing
clin
ical
can
did
ate
effe
cts
on
repro
du
ctiv
ebio
logy
Dev
elopm
enta
l
(ped
iatr
ic)
imm
un
oto
xici
ty
Gen
eral
issu
esN
oin
du
stry
stan
dar
dfo
r
NH
Pim
mu
noto
xor
imm
un
om
odu
lati
on
assa
ys;
lack
of
his
tori
-
cal
con
trols
,va
lidat
ed
assa
ys,
posi
tive
con
-tr
ols
,et
c;dif
ficu
ltie
s
inin
terp
reti
ng
resu
lts
May
be
able
toap
ply
met
hods
adap
ted
from
adu
ltst
udie
s
(th
isis
the
tren
d)
Lac
kof
his
tori
cal
con
-
trols
,va
lidat
edas
says
,posi
tive
con
trols
,et
c;
dif
ficu
ltie
sin
inte
r-
pre
tin
gre
sult
s
Gen
etic
del
etio
nor
inse
rtio
nof
ata
rget
may
not
repre
sen
t
chro
nic
blo
ckad
eor
repla
cem
ent
by
dru
g
a.H
um
ans
hav
edis
coid
pla
cen
ta.
CR
O,
Con
trac
tR
esea
rch
Org
aniz
atio
n;
DA
RT
,dev
elopm
enta
lan
dre
pro
du
ctiv
eto
xico
logy
;IC
H,
Inte
rnat
ion
alC
on
fere
nce
on
Har
mon
izat
ion
;N
HP
,n
on
hu
man
pri
mat
e.
250
250 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

characteristics of the molecule, formulation, andpharmacologic activity). Examples have been givenin which surrogates have both under- or overpre-dicted the effects seen in humans, and produced dif-ferent results compared with the clinical candidatein nonhuman primates. However, the potential forover-and underprediction of human effects is a chal-lenge common to both traditional and alternativetoxicity assessments in any nonhuman species.When a difference between results or lack of predic-tivity occurs with a surrogate molecule, additionalquestions arise regarding whether the differencesbetween nonclinical and human studies are due topharmacological or molecular differences betweenthe surrogate and the biopharmaceutical, or simplydue to differences in species biology. Thus, it is crit-ical to fully understand and extensively characterizethe pharmacology of the surrogate molecule in theappropriate species both in vitro and in vivo, andensure that its pharmacology is as similar to that ofthe clinical candidate in humans as possible.Because the pharmacology is often poorly under-stood, the use of alternative approaches in the safetyassessment of biopharmaceuticals should be supple-mental and evaluated as part of a case-by-case,weight-of-evidence approach. However, any of thesealternatives can be valuable support for a biopharma-ceutical safety assessment program in which uniquechallenges of species specificity often make a stan-dard toxicology program inappropriate to conduct.
Acknowledgments
The authors appreciate the BioSafe leadership com-mittee, the PhRMA Biologics Technical Group, andthe FDA’s review of this manuscript and their criticalinput on these complex issues.
References
1. ICH S6. Preclinical Safety Evaluation of Biotechnology-
DerivedPharmaceuticals.1997. http://www.ich.org.Accessed
April 14, 2009.
2. Cavagnaro JA. Preclinical safety evaluation of
biotechnology-derived pharmaceuticals. Nat Rev Drug
Discov. 2002;1:469-475.
3. Haley PJ. Species differences in the structure and func-
tion of the immune system. Toxicology. 2003;188:49-71.
4. Mestas J, Hughes CCW. Of mice and not men:
differences between mouse and human immunology.
J Immunol. 2004;172:2731-2738.
5. Bolon B, Galbreath E, Sargent L, Weiss J. Genetic
engineering and molecular technology. In: Krinke G,
ed. The Laboratory Rat. London, England: Academic
Press; 2008:603.
6. Bolon B, Galbreath E. Use of genetically engineered
mice in drug discovery and development: wielding
Occam’s razor to prune the product portfolio. Int J
Toxicol. 2002;21:55-64.
7. Doetschman T. Interpretation of phenotype in
genetically engineered mice. Lab Anim Sci. 1999;49:
137-143.
8. Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2
deficiency: past lessons, current understanding and
future prospects. Oncogene. 2006;25:5885-5897.
9. Wellendorph P, Johansen L, Jensen A, et al. No evidence
for a bone phenotype in GPRC6A knockout mice under
normal physiological conditions. J Mol Endocrinol.
2009;42:215-223.
10. Lee GS. Phenotype of a calbindin-D9k gene knockout is
compensated for by the induction of other calcium trans-
porter genes in a mouse model. J Bone Min Res.
2007;22:1968-1978.
11. Koch PJ, de Viragh PA, Scharer E, et al. Lessons from
loricrin-deficient mice: compensatory mechanisms main-
taining skin barrier function in the absence of a major cor-
nified envelope protein. J Cell Biol. 2000;151:389-400.
12. Tanabe M, Matsumoto T, Shibuya K, et al. Compensa-
tory response of IL-1 gene knockout mice after pulmon-
ary infection with Klebsiella pneumoniae. J Med
Microbiol. 2005;54:7-13.
13. Hirano E, Knutsen RH, Sugitani H, et al. Functional res-
cue of elastin insufficiency in mice by the human elastin
gene: implications for mouse models of human disease.
Circ Res. 2007;101:523-531.
14. Inoue T. Concept of a ‘relevant animal model.’ In: D’Arcy
PF, Harron DWG, eds. In: Proceedings of The Fourth
International Conference on Harmonisation Brussels
1997. Belfast: The Queen’s University; 1998:209.
15. Bugelski PJ, Herzyk DJ, Rehm S, et al. Preclinical devel-
opment of keliximab, a Primatized anti-CD4 monoclonal
antibody, in human CD4 transgenic mice: characteriza-
tion of the model and safety studies. Hum Exp Toxicol.
2000;19:230-243.
16. Thoma-Uszynski S, Stenger S, Takeuchi O, et al. Induc-
tion of direct antimicrobial activity through mammalian
toll-like receptors. Science. 2001;291:1544-1547.
17. Veninga H, Becker S, Hoek RM, et al. Analysis of CD97
expression and manipulation: antibody treatment but
not gene targeting curtails granulocyte migration.
J Immunol. 2008;181:6574-6583.
18. Bussiere JL, Adler MW, Rogers TJ, Eisenstein TK.
Differential effects of morphine and naltrexone on the
251
Alternative Strategies for Biopharmaceuticals / Bussiere et al 251
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

antibody response in various mouse strains. Immuno-
pharmacol Immunotoxicol. 1992;14:657-673.
19. Bozic CR, Kolakowski LF Jr, Gerard NP, et al. Expres-
sion and biologic characterization of the murine chemo-
kine KC. J Immunol. 1995;154:6048-6057.
20. Krupa A, Walencka MJ, Shrivastava V, et al. Anti-KC
autoantibody:KC complexes cause severe lung inflam-
mation in mice via IgG receptors. Am J Resp Cell Mol
Biol. 2007;37:532-543.
21. Clarke J, Leach W, Pippig S, et al. Evaluation of a surro-
gate antibody for preclinical safety testing of an anti-
CD11a monoclonal antibody. Regul Toxicol Pharmacol.
2004;40:219-226.
22. Bussiere JL, Hardy LM, Hoberman AM, et al. Reproduc-
tive effects of chronic administration of murine inter-
feron-gamma. Reprod Toxicol. 1996;10:379-391.
23. Green JD, Terrell TG. Utilization of homologous pro-
teins to evaluate the safety of recombinant human
proteins—case study: recombinant human interferon-
gamma (rhIFN-gamma). Toxicol Lett. 1992;64-65 Spec
No: 321-327.
24. Treacy G. Using an analogous monoclonal antibody to
evaluate the reproductive and chronic toxicity potential
for a humanized anti-TNF-a monoclonal antibody. Hum
Exp Toxicol. 2000;19:226-228.
25. Hulett MD, Hogarth PM. Molecular basis of Fc receptor
function. Adv Immunol. 1994;57:1-127.
26. King DJ. Applications and Engineering of Monoclonal
Antibodies. Boca Raton, FL: CRC Press; 1998.
27. ICH S5(R2). Detection of Toxicity to Reproduction for
Medicinal Products & Toxicity to Male Fertility. http://
www.ich.org. Posted 1995. Accessed April 14, 2009.
28. Malassine A, Frendo JL, Evain-Brion D. A comparison of
placental development and endocrine functions between
the human and mouse model. Hum Reprod Update.
2003;9:531-539.
29. Fujimoto K, Terao K, Cho F, Honjo S. The placental
transfer of IgG in the cynomolgus monkey. Jpn J Med Sci
Biol. 1983;36:171-176.
30. Malek A, Sager R, Kuhn P, et al. Evolution of materno-
fetal transport of immunoglobulins during human preg-
nancy. Am J Reprod Immunol. 1996;36:248-255.
31. Haller CA, Cosenza ME, Sullivan JT. Safety issues spe-
cific to clinical development of protein therapeutics.
Clin Pharmacol Ther. 2008;84:624-627.
32. Geizen TJ, Mantel-Teeuwisse AK, Straus SMJM, et al.
Safety-related regulatory actions for biological approved
in the United States and the European Union. JAMA.
2008;300:1887-1896.
33. Knight A. The beginning of the end for chimpanzee
experiments? Philos Eth Hum Med. 2008;3:16.
34. Martin PL, Cornacoff JB, Treacy G. et al. Effects of
administration of a monoclonal antibody against
mouse tumor necrosis factor alpha during pregnancy
and lactation on the pre- and postnatal development
of the mouse immune system. Int J Toxicol. 2008;
27:341-347.
35. Pasparakis M, Alexopoulou L, Episkopou V, Kollias G.
Immune and inflammatory responses in TNF alpha-
deficient mice: a critical requirement for TNF alpha in
the formation of primary B cell follicles, follicular den-
dritic cell networks and germinal centers, and in the
maturation of the humoral immune response. J Exp Med.
1996;184:1397-1411.
36. Martin PL, Oneda S, Treacy G. Effects of an anti-TNFamonoclonal antibody, administered throughout preg-
nancy and lactation, on the development of the macaque
immune system. Am J Reprod Immunol. 2007;58:
136-149.
37. Chapman K. Opportunities for Reducing the Use of
Non-human Primates in the Development of Biologi-
cals—a Workshop Report. www.NC3Rs.org.UK. Posted
2006. Accessed April 14, 2009.
38. Terrell TG, Green JD. Comparative pathology of recom-
binant murine interferon-gamma in mice and recombi-
nant human interferon-gamma in cynomolgus monkeys.
Int Rev Exp Pathol. 1993;34 (Pt B):73-101.
39. Herzyk DJ, Bugelski PJ, Hart TK, Wier PJ. Practical
aspects of including functional endpoints in develop-
mental toxicity studies. Case study: immune function
in HuCD4 transgenic mice exposed to anti-CD4 MAb
in utero. Hum Exp Toxicol. 2002;21:507-512.
40. Carlock LL, Cowan LA, Oneda S, et al. A comparison of
effects on reproduction and neonatal development in
cynomolgus monkeys given human soluble IL-4R and
mice given murine soluble IL-4R. Reg Tox Pharmacol.
2009;53:226-234.
41. Gommerman JL, Browning JL. Lymphotoxin/light, lym-
phoid microenvironments and autoimmune disease. Nat
Rev Immunol. 2003;3:642-655.
42. Alimzhanov MB, Kuprash DV, Kosco-Vilbois MH, et al.
Abnormal development of secondary lymphoid tissues in
lymphotoxin beta-deficient mice. Proc Natl Acad Sci
USA. 1997;94:9302-9307.
43. Rennert PD, Browning JL, Mebius R, et al. Surface
lymphotoxin alpha/beta complex is required for the
development of peripheral lymphoid organs. J Exp Med.
1996;184:1999-2006.
44. Martin PL. Effects of human lymphotoxin-b receptor-
IgG1 (LTbR-Ig) fusion protein on lymph node develop-
ment in non human primates. In: Korte R, Vogel F,
Weinbauer GF, eds. Primate Models in Pharmaceutical
Drug Development. Muenster, Germany: Waxmann
Press; 2002:105.
45. ICH M3 (R1). Guidance on Nonclinical Safety Studies
for the Conduct of Human Clinical Trials and Marketing
Authorization for Pharmaceuticals. http://www.ich.org.
Accessed April 14, 2009.
46. Cavagnaro J. Preclinical evaluation of cancer hazard
and risk of biopharmaceuticals. In: Cavagnaro JA, ed.
252
252 International Journal of Toxicology / Vol. 28, No. 3, May/June 2009
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from

Preclinical Safety Evaluation of Biopharmaceuticals: A
Science-Based Approach to Facilitating Clinical Trials.
Hoboken, NJ: Wiley & Sons; 2008:399.
47. Cohen SM. Human carcinogenic risk evaluation: an
alternative approach to the two-year rodent bioassay.
Toxicol Sci. 2004;80:225-229.
48. Farris GM, Miller GK, Wollenberg GK, et al. Recombi-
nant rat and mouse growth hormones: risk assessment
of carcinogenic potential in 2-year bioassays in rats and
mice. Toxicol Sci. 2007;97:548-561.
49. Bugelski PJ, Herzyk DJ, Rehm S, et al. Immunomodula-
tory biopharmaceuticals and risk of neoplasia. In:
Cavagnaro JA, ed. Preclinical Safety Evaluation of Bio-
pharmaceuticals: A Science-Based Approach to Facilitating
Clinical Trials. Hoboken, NJ: Wiley & Sons; 2008:601.
50. Reilly TP, Abbott M, Golovkina T, et al. Role of immuno-
modulation by the selective costimulation modulator,
abatacept, in mouse mammary tumor virus (MMTV)-
initiated tumors. The Toxicologist. 2006;90:50.
51. Rosenblum IY, Dayan AD. Carcinogenicity testing of
IL-10: principles and practicalities. Hum Exp Toxicol.
2002;21:347-358.
52. Suntharalingam G. Perry MR, Ward S, et al. Cytokine
storm in a phase 1 trial of the anti-CD28 mono-
clonal antibody TGN1412. N Engl J Med. 2006;355:
1018-1028.
53. Tacke M, Hanke G, Hanke T, Hunig T. CD28-mediated
induction of proliferation in resting T cells in vitro and in
vivo without engagement of the T cell receptor: evidence
for functionally distinct forms of CD28. Eur J Immunol.
1997;27:239-247.
54. Lin CH, Hunig T. Efficient expansion of regulatory T
cells in vitro and in vivo with a CD28 superagonist. Eur
J Immunol. 2003;33:626-638.
55. Beyersdorf N, Hanke T, Kerkau T, Hunig T. CD28
superagonists put a break on autoimmunity by preferen-
tially activating CD4þCD25þ regulatory T cells. Auto-
immun Rev. 2006;5:40-45.
56. TeGenero AG. TGN1412 Investigational Medicinal
Product Dossier. Vol. 1. http://www.circare.org/foia5/
tgn1412dossier.pdf. Posted December 19, 2005. Accessed
May 11, 2009.
57. TeGenero AG. TGN1412 Investigator’s Brochure.
TGN1412 Humanized Agonistic Anti-CD28 Mono-
clonal Antibody. Vol. 1.1. http://www.circare.org/foia5/
tgn1412 investigatorbrochure.pdf. Posted December
19, 2005. Accessed May 11, 2009.
58. TeGenero AG. TGN1412 Consent Form/Patient Infor-
mation Sheet. A Phase-I, single-centre, double-blind,
randomised, placebo-controlled, single escalating-dose
study to assess the safety, pharmacokinetics, pharma-
codynamics and immunogenicity of TGN1412 adminis-
tered intravenously to healthy volunteers. Protocol
Number: TGN1412. Parexel Project Number: 68419.
Version: 02 Final. http://www.circare.org/foia5/tgn1412_
consentform.pdf. Posted February 9, 2006. Accessed May
11, 2009.
59. Legrand N, Cupedo T, van Lent AU, et al. Transient
accumulation of human mature thymocytes and regula-
tory T cells with CD28 superagonist in ‘‘human immune
system’’ Rag2(–/–)gammac(�/�) mice. Blood. 2006;
108:38-245.
For reprints and permissions queries, please visit SAGE’s Web site at http://www.sagepub.com/journalsPermissions.nav
253
Alternative Strategies for Biopharmaceuticals / Bussiere et al 253
at American College of Toxicology on July 17, 2009 http://ijt.sagepub.comDownloaded from
















![Testing Testing [Read-Only] - Czone - East Sussex](https://static.fdokumen.com/doc/165x107/6327b2a26d480576770d6757/testing-testing-read-only-czone-east-sussex.jpg)



