
All-Metal Aromaticity: Revisiting the Ring Current Model among Transition Metal Clusters
8
All-Metal Aromaticity: Revisiting the Ring Current Model among Transition Metal Clusters Zahra Badri, †,‡ Shubhrodeep Pathak, § Heike Fliegl, ∥ Parviz Rashidi-Ranjbar,* ,‡ Radovan Bast, ⊥,# Radek Marek, †,▽ Cina Foroutan-Nejad,* ,▽ and Kenneth Ruud* ,○ † CEITEC − Central European Institute of Technology, Masaryk University, Kamenice 5/A4, CZ−62500 Brno, Czech Republic ‡ School of Chemistry, College of Science, University of Tehran, Tehran, Iran § Raman Center for Atomic, Molecular and Optical Sciences, Indian Association for the Cultivation of Science, Kolkata 700 032, India ∥ Centre for Theoretical and Computational Chemistry, Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, N−0315 Oslo, Norway ⊥ Laboratoire de Chimie et Physique Quantiques (UMR 5626), CNRS/Universite ́ de Toulouse 3 (Paul Sabatier), 118 route de Narbonne, F−31062 Toulouse, France # Theoretical Chemistry and Biology, School of Biotechnology, Royal Institute of Technology, AlbaNova University Center, S-10691 Stockholm, Sweden ▽ National Center for Biomolecular Research, Faculty of Science, Masaryk University, Kamenice 5/A4, CZ−62500 Brno, Czech Republic ○ Centre for Theoretical and Computational Chemistry, Department of Chemistry, University of TromsøThe Arctic University of Norway, N-9037 Tromsø, Norway * S Supporting Information ABSTRACT: We present new insight into the nature of aromaticity in metal clusters. We give computational argu- ments in favor of using the ring-current model over local indices, such as nucleus independent chemical shifts, for the determination of the magnetic aromaticity. Two approaches for estimating magnetically induced ring currents are employed for this purpose, one based on the quantum theory of atoms in molecules (QTAIM) and the other where magnetically induced current densities (MICD) are explicitly calculated. We show that the two-zone aromaticity/antiaromaticity of a number of 3d metallic clusters (Sc 3 − , Cu 3 + , and Cu 4 2− ) can be explained using the QTAIM-based magnetizabilities. The reliability of the calculated atomic and bond magnetizabilities of the metallic clusters are verified by comparison with MICD computed at the multiconfiguration self-consistent field (MCSCF) and density functional levels of theory. Integrated MCSCF current strength susceptibilities as well as a visual analysis of the calculated current densities confirm the interpretations based on the QTAIM magnetizabilities. In view of the new findings, we suggest a simple explanation based on classical electromagnetic theory to explain the anomalous magnetic shielding in different transition metal clusters. Our results suggest that the nature of magnetic aromaticity/antiaromaticity in transition-metal clusters should be assessed more carefully based on global indices. 1. INTRODUCTION The term all-metal aromaticity was introduced by Wang et al. after the discovery of an Al 4 2− cluster in 2001, in order to explain its exceptional gas-phase stability. 1 After this important finding, numerous theoretical and experimental studies appeared, addressing different aspects of the chemistry of all- metal aromatic/antiaromatic clusters of both main-group (MG) and transition metals (TM). 2 The chemical community was quickly intrigued by the fascinating features of this new class of molecules, such as their multiple aromaticity, that is their simultaneous and even conflicting σ, π, δ, and φ aromaticity/ antiaromaticity. 2b,c,g,3,4 The aromatic nature of this new class of compounds has been exclusively studied using electronic and magnetic criteria. Applications of energetic or structural criteria are limited because it is difficult to decide on proper reference systems. 1,2,5 However, some issues regarding the magnetic aromaticity of certain all-TM clusters have remained unclear. In particular, the patterns of nucleus-independent chemical shift (NICS) scans 6 that is, the magnitude of the NICS 7 values at different heights above the molecular ring planeare not consistent Received: July 1, 2013 Published: October 14, 2013 Article pubs.acs.org/JCTC © 2013 American Chemical Society 4789 dx.doi.org/10.1021/ct4007184 | J. Chem. Theory Comput. 2013, 9, 4789−4796
Transcript of All-Metal Aromaticity: Revisiting the Ring Current Model among Transition Metal Clusters

All-Metal Aromaticity: Revisiting the Ring Current Model amongTransition Metal ClustersZahra Badri,†,‡ Shubhrodeep Pathak,§ Heike Fliegl,∥ Parviz Rashidi-Ranjbar,*,‡ Radovan Bast,⊥,#
Radek Marek,†,▽ Cina Foroutan-Nejad,*,▽ and Kenneth Ruud*,○
†CEITEC − Central European Institute of Technology, Masaryk University, Kamenice 5/A4, CZ−62500 Brno, Czech Republic‡School of Chemistry, College of Science, University of Tehran, Tehran, Iran§Raman Center for Atomic, Molecular and Optical Sciences, Indian Association for the Cultivation of Science, Kolkata 700 032, India∥Centre for Theoretical and Computational Chemistry, Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern,N−0315 Oslo, Norway⊥Laboratoire de Chimie et Physique Quantiques (UMR 5626), CNRS/Universite de Toulouse 3 (Paul Sabatier), 118 route deNarbonne, F−31062 Toulouse, France#Theoretical Chemistry and Biology, School of Biotechnology, Royal Institute of Technology, AlbaNova University Center, S-10691Stockholm, Sweden▽National Center for Biomolecular Research, Faculty of Science, Masaryk University, Kamenice 5/A4, CZ−62500 Brno, CzechRepublic○Centre for Theoretical and Computational Chemistry, Department of Chemistry, University of TromsøThe Arctic University ofNorway, N-9037 Tromsø, Norway
*S Supporting Information
ABSTRACT: We present new insight into the nature ofaromaticity in metal clusters. We give computational argu-ments in favor of using the ring-current model over localindices, such as nucleus independent chemical shifts, for thedetermination of the magnetic aromaticity. Two approaches forestimating magnetically induced ring currents are employed forthis purpose, one based on the quantum theory of atoms inmolecules (QTAIM) and the other where magneticallyinduced current densities (MICD) are explicitly calculated.We show that the two-zone aromaticity/antiaromaticity of anumber of 3d metallic clusters (Sc3
−, Cu3+, and Cu4
2−) can beexplained using the QTAIM-based magnetizabilities. Thereliability of the calculated atomic and bond magnetizabilities of the metallic clusters are verified by comparison with MICDcomputed at the multiconfiguration self-consistent field (MCSCF) and density functional levels of theory. Integrated MCSCFcurrent strength susceptibilities as well as a visual analysis of the calculated current densities confirm the interpretations based onthe QTAIM magnetizabilities. In view of the new findings, we suggest a simple explanation based on classical electromagnetictheory to explain the anomalous magnetic shielding in different transition metal clusters. Our results suggest that the nature ofmagnetic aromaticity/antiaromaticity in transition-metal clusters should be assessed more carefully based on global indices.
1. INTRODUCTION
The term all-metal aromaticity was introduced by Wang et al.after the discovery of an Al4
2− cluster in 2001, in order toexplain its exceptional gas-phase stability.1 After this importantfinding, numerous theoretical and experimental studiesappeared, addressing different aspects of the chemistry of all-metal aromatic/antiaromatic clusters of both main-group (MG)and transition metals (TM).2 The chemical community wasquickly intrigued by the fascinating features of this new class ofmolecules, such as their multiple aromaticity, that is theirsimultaneous and even conflicting σ, π, δ, and φ aromaticity/antiaromaticity.2b,c,g,3,4 The aromatic nature of this new class of
compounds has been exclusively studied using electronic andmagnetic criteria. Applications of energetic or structural criteriaare limited because it is difficult to decide on proper referencesystems.1,2,5
However, some issues regarding the magnetic aromaticity ofcertain all-TM clusters have remained unclear. In particular, thepatterns of nucleus-independent chemical shift (NICS)scans6that is, the magnitude of the NICS7 values at differentheights above the molecular ring planeare not consistent
Received: July 1, 2013Published: October 14, 2013
Article
pubs.acs.org/JCTC
© 2013 American Chemical Society 4789 dx.doi.org/10.1021/ct4007184 | J. Chem. Theory Comput. 2013, 9, 4789−4796

with the proposed magnetic aromaticity for many TMclusters.5b,f In the present work, we explain the above-mentioned discrepancies in the light of recent advances inthe analysis of the current density using the bond magnet-izability approach.8 Systems containing transition metals areknown to exhibit significant nondynamical correlation effectsdue to the presence of closely spaced nd, (n + 1)s, and (n + 1)porbitals having variable occupancies. A recent study by Pathaket al.9 demonstrated that nondynamical electron correlationsubstantially affects magnetic properties of molecules withdegenerate or near-degenerate ground states. It was inparticular shown that the inclusion of nondynamical correlationeffects can even reverse the picture obtained from a single-reference theory with respect to diatropicity or paratropicity ofsuch systems. Accordingly, DFT results should not be blindlytrusted for systems with an inherent multireference character,unless a genuine multiconfigurational theory confirms theirlegitimacy. We corroborate our QTAIM findings with thecalculation of magnetically induced current densities (MICD)at the multiconfigurational self-consistent field (MCSCF) levelof theory.9 We reinvestigate the aromatic characters of threeTM clusters from the 3d block (Sc3
−, Cu3+, and Cu4
2−). Thesestate-of-the-art computations provide new insight into thearomatic nature of the investigated TM clusters that have untilnow not been accessible to reliable calculations.The manuscript is organized as follows. In section 2, the
computational methods used are described, and in section 3, abrief overview of previous findings as well as new results arepresented and discussed for each of the clusters. Finalconclusions are drawn in section 4.
2. COMPUTATIONAL METHODSAll geometries were first optimized at the density functionaltheory (DFT) level using the B3LYP10 hybrid GGA exchange-correlation functional and the 6-311+G(d) basis set, as this hasrecently been shown to be sufficient for NICS calculations forthis group of metal clusters.5f Frequency calculations wereperformed to confirm that the structures correspond to localminima. As has been reported previously,5f the Cu4
2− cluster isa saddle point at the B3LYP/6-311+G(d) computational level,and for this reason the def2-QZVP11 basis set was alsoemployed to optimize the Cu4
2− cluster since the D4h Cu42− is a
local minimum at the B3LYP/def2-QZVP level of theory. Incase of Cu4
2−, two sodium salts of the cluster (C4v NaCu4− and
D4h Na2Cu4) were also optimized at the same level of theorybecause it is a well-known fact that doubly charged moleculesare prone to spontaneous electron emission. Magnetic responseproperties of each cluster were computed at the samecomputational levels using gauge including atomic orbitals(GIAOs).12 Since canonical molecular orbital (CMO) NICS13
values for Sc3− and Cu3
+ have not been reported previously inthe literature, their CMO−NICS values were computed at theB3LYP/6-311+G(d) level in order to investigate if any valuableinformation can be derived from CMO−NICS values. TheGaussian 09 suite of programs was employed in thesecalculations.14
Intra- and interatomic magnetizabilities within the context ofquantum theory of atoms in molecules (QTAIM)15 werecomputed for the DFT-based electron densities. Thecontribution of each individual molecular orbital to the intra-and interatomic magnetizabilities were computed as describedin ref 8. The accuracy of the calculations was controlled bykeeping the atomic integral of the one-electron density
Laplacian below 10−4 a.u. and the total energy differencebetween the QTAIM and DFT calculations below 0.5 kcal/mol.All QTAIM computations were performed employing theProaim integration method, as implemented in the AIMAllsuite of programs.16
In order to evaluate the validity of the DFT computations,magnetically induced current densities (MICD) of the differentclusters were plotted at the Hartree−Fock self-consistent field(HF−SCF), DFT (B3LYP), and complete active space self-consistent field (CASSCF) levels of theory using a def2-TZVPbasis set and the preoptimized geometries obtained using theB3LYP/6-311+G(d) (for Cu4
2− at the B3LYP/def2-QZVP)level with a development version of the Dalton 2011 package.17
The calculated current density plots were visualized usingPyNGL.18
The choice of active space for any system involving transitionmetals is a nontrivial task and warrants careful analysis in orderto achieve convergence as well as include the right amount ofcorrelation. The obvious choice would be to include all 3d, 4s,and 4p orbitals and all the electrons therein. However, thischoice (10 electrons in 27 orbitals in case of Sc3
−) was out ofreach with the available MCSCF implementation. Thus, wechose a viable active space of 10 electrons in 11 orbitals after athorough analysis of the occupancies of natural orbitals from anMP2 calculation for Sc3
−. The choice of active space in the caseof Cu3
+ is simpler considering that Cu has a completely filled3d level. It can be safely concluded that the ideal choice in thecase of Cu3
+ would be two electrons in all the 4s and 4porbitals, 12 for three Cu atoms. We brought this down to fiveorbitals after analyzing MP2 natural orbital occupancies. Finally,for Cu4
2−, similar to Cu3+, the choice of active space was
restricted to 6 and 12 of the 16 4s and 4p orbitals and the sixelectrons contained by them.The flux of the current density was integrated on a 2D plane,
perpendicular to the molecular ring plane using a Gauss−Lobatto quadrature grid, as described in ref 9. The integratedring current strength susceptibility, referred to as currentstrength hereafter, provides quantitative information regardingthe degree of aromaticity of a molecule as shown in variousstudies.19 The sign and magnitude of the calculated ringcurrents provide quantitative information on the aromatic,antiaromatic, or nonaromatic character of a molecule resultingin diatropic, paratropic, or vanishing net ring currents.20 Thecurrent strength for benzene lies between 11.8 nA/T (B3LYP/def2-TZVP) and 13.5 nA/T (CASSCF/aug-cc-pVDZ), whichcan be used as a reference value.NICS can be calculated with any code providing magnetic
shielding constants. The DALTON 2011 package,16 with itsimplementation of GIAO−CASSCF shielding constants,provides an excellent platform for the determination of NICSvalues at the CASSCF−GIAO level. In this study, we report thefour most commonly reported NICS indices, namely,NICS(0)iso, NICS(0)zz, NICS(1)iso, and NICS(1)zz. It isimportant to note here the differences in convention whencomparing results from the NICS, bond magnetizability, andMICD values. Negative NICS and bond magnetizabilitiesdenote aromatic character, whereas positive ones denoteantiaromaticity. In contrast, a positive MICD value indicatesan aromatic, whereas a negative MICD deontes an antiaromaticcharacter. The plots of the current densities are interpreted asbeing aromatic if they are clockwise and antiaromatic if they areanticlockwise. Negative atomic magnetizabilities denote the
Journal of Chemical Theory and Computation Article
dx.doi.org/10.1021/ct4007184 | J. Chem. Theory Comput. 2013, 9, 4789−47964790
presence of net local diatropic currents, and positive valuespoint to paratropic currents.
3. RESULTS AND DISCUSSIONSSc3
−. Sc3− was proposed as a double (σ + π) aromatic system
based on the large negative NICSiso values both at the centerand 1 Å above the center of the molecule at the DFT (B3LYP/6-311+G(d)) computational level by Chi and Liu.21 Later, itwas suggested to be a double (σ + δ) aromatic species.5b
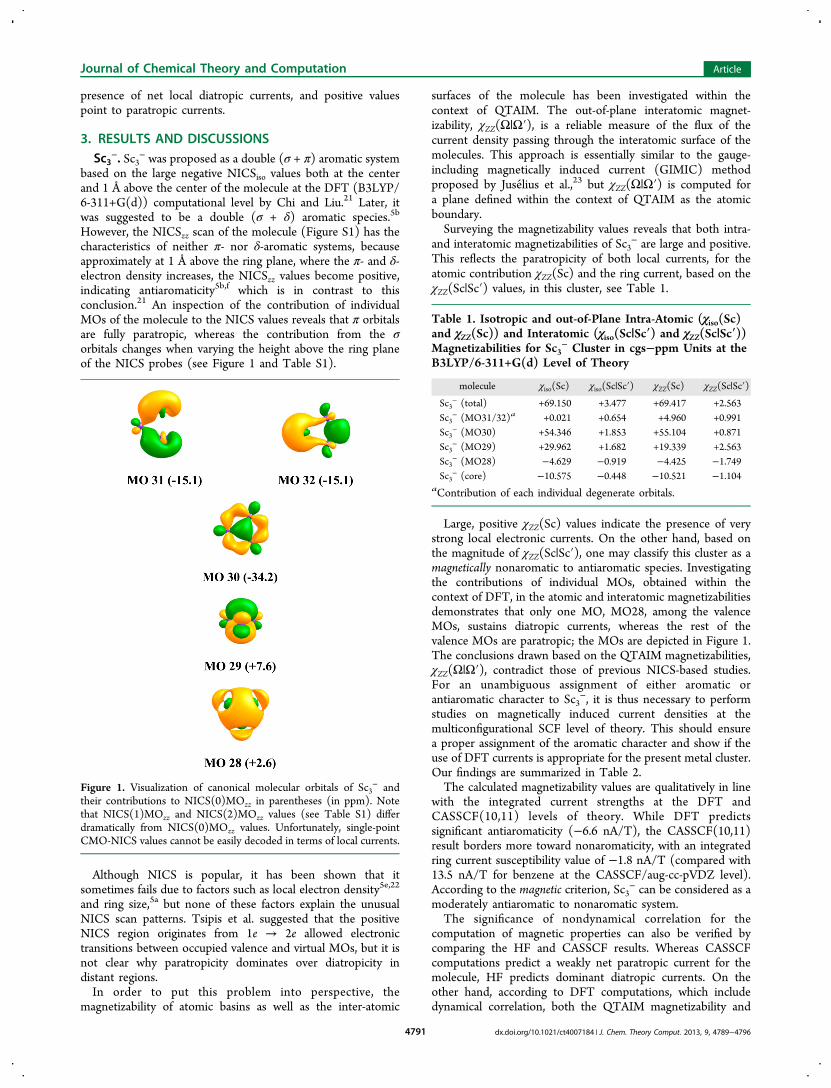
However, the NICSzz scan of the molecule (Figure S1) has thecharacteristics of neither π- nor δ-aromatic systems, becauseapproximately at 1 Å above the ring plane, where the π- and δ-electron density increases, the NICSzz values become positive,indicating antiaromaticity5b,f which is in contrast to thisconclusion.21 An inspection of the contribution of individualMOs of the molecule to the NICS values reveals that π orbitalsare fully paratropic, whereas the contribution from the σorbitals changes when varying the height above the ring planeof the NICS probes (see Figure 1 and Table S1).
Although NICS is popular, it has been shown that itsometimes fails due to factors such as local electron density5e,22
and ring size,5a but none of these factors explain the unusualNICS scan patterns. Tsipis et al. suggested that the positiveNICS region originates from 1e → 2e allowed electronictransitions between occupied valence and virtual MOs, but it isnot clear why paratropicity dominates over diatropicity indistant regions.In order to put this problem into perspective, the
magnetizability of atomic basins as well as the inter-atomic
surfaces of the molecule has been investigated within thecontext of QTAIM. The out-of-plane interatomic magnet-izability, χZZ(Ω|Ω′), is a reliable measure of the flux of thecurrent density passing through the interatomic surface of themolecules. This approach is essentially similar to the gauge-including magnetically induced current (GIMIC) methodproposed by Juselius et al.,23 but χZZ(Ω|Ω′) is computed fora plane defined within the context of QTAIM as the atomicboundary.Surveying the magnetizability values reveals that both intra-
and interatomic magnetizabilities of Sc3− are large and positive.
This reflects the paratropicity of both local currents, for theatomic contribution χZZ(Sc) and the ring current, based on theχZZ(Sc|Sc′) values, in this cluster, see Table 1.
Large, positive χZZ(Sc) values indicate the presence of verystrong local electronic currents. On the other hand, based onthe magnitude of χZZ(Sc|Sc′), one may classify this cluster as amagnetically nonaromatic to antiaromatic species. Investigatingthe contributions of individual MOs, obtained within thecontext of DFT, in the atomic and interatomic magnetizabilitiesdemonstrates that only one MO, MO28, among the valenceMOs, sustains diatropic currents, whereas the rest of thevalence MOs are paratropic; the MOs are depicted in Figure 1.The conclusions drawn based on the QTAIM magnetizabilities,χZZ(Ω|Ω′), contradict those of previous NICS-based studies.For an unambiguous assignment of either aromatic orantiaromatic character to Sc3
−, it is thus necessary to performstudies on magnetically induced current densities at themulticonfigurational SCF level of theory. This should ensurea proper assignment of the aromatic character and show if theuse of DFT currents is appropriate for the present metal cluster.Our findings are summarized in Table 2.The calculated magnetizability values are qualitatively in line
with the integrated current strengths at the DFT andCASSCF(10,11) levels of theory. While DFT predictssignificant antiaromaticity (−6.6 nA/T), the CASSCF(10,11)result borders more toward nonaromaticity, with an integratedring current susceptibility value of −1.8 nA/T (compared with13.5 nA/T for benzene at the CASSCF/aug-cc-pVDZ level).According to the magnetic criterion, Sc3
− can be considered as amoderately antiaromatic to nonaromatic system.The significance of nondynamical correlation for the
computation of magnetic properties can also be verified bycomparing the HF and CASSCF results. Whereas CASSCFcomputations predict a weakly net paratropic current for themolecule, HF predicts dominant diatropic currents. On theother hand, according to DFT computations, which includedynamical correlation, both the QTAIM magnetizability and
Figure 1. Visualization of canonical molecular orbitals of Sc3− and
their contributions to NICS(0)MOzz in parentheses (in ppm). Notethat NICS(1)MOzz and NICS(2)MOzz values (see Table S1) differdramatically from NICS(0)MOzz values. Unfortunately, single-pointCMO-NICS values cannot be easily decoded in terms of local currents.
Table 1. Isotropic and out-of-Plane Intra-Atomic (χiso(Sc)and χZZ(Sc)) and Interatomic (χiso(Sc|Sc′) and χZZ(Sc|Sc′))Magnetizabilities for Sc3
− Cluster in cgs−ppm Units at theB3LYP/6-311+G(d) Level of Theory
molecule χiso(Sc) χiso(Sc|Sc′) χZZ(Sc) χZZ(Sc|Sc′)Sc3
− (total) +69.150 +3.477 +69.417 +2.563Sc3
− (MO31/32)a +0.021 +0.654 +4.960 +0.991Sc3
− (MO30) +54.346 +1.853 +55.104 +0.871Sc3
− (MO29) +29.962 +1.682 +19.339 +2.563Sc3
− (MO28) −4.629 −0.919 −4.425 −1.749Sc3
− (core) −10.575 −0.448 −10.521 −1.104aContribution of each individual degenerate orbitals.
Journal of Chemical Theory and Computation Article
dx.doi.org/10.1021/ct4007184 | J. Chem. Theory Comput. 2013, 9, 4789−47964791

the magnetically induced current strength predict a moderateantiaromaticity for Sc3
−. Nevertheless, the aromaticity of thecluster is definitely ruled out.MICD plots for Sc3
− at the B3LYP and CASSCF(10,11)levels are shown in Figure 2. A notable difference between thecurrent density plot for Sc3
− and other molecules that havebeen studied in the literature so far is the presence of stronglocal paratropic currents around the Sc nuclei. These local
currents are also clearly reflected in large positive interatomicmagnetizability values (χ(Sc)).Although the MICD and the magnetizability calculated at the
DFT and CASSCF theoretical levels disprove the magneticaromaticity of Sc3
−, NICS(0) values are at all levels negativeand consistently suggest aromaticity, see Table 2. Theexplanation for the large negative NICS values near the ringplane and positive values 1 Å above the ring plane could be
Table 2. Magnetically Induced Current Densities (nA/T) and Various NICS Values in TM Clusters
clusters computational level MICD NICS(0)iso NICS(0)zz NICS(1)iso NICS(1)zz
Sc3− HF +2.5 −51.5 −9.5 −51.5 −2.1
DFT (B3LYP) −6.6 −17.5 −44.8 −21.8 +8.2CASSCF(10,11) −1.8 −32.0 −31.8 −27.7 +4.1
Cu3+ HF +6.3 −22.7 +1.2 −10.4 −15.0
DFT (B3LYP) +8.1 −28.4 −7.0 −12.2 −18.8CASSCF(2,5) +6.2 −22.4 +1.5 −10.2 −14.9
Cu42− HF +14.6 −12.4 −14.7 −9.5 −23.0
DFT (B3LYP) +15.0 −17.3 −19.7 −11.5 −25.1CASSCF(6,6) +14.4 −12.6 −14.6 −9.5 −22.8CASSCF(6,12) +13.8 −13.4 −14.9 −9.9 −22.9
Figure 2.MICD plots for Sc3− at (a) B3LYP and (b) CASSCF(10,11) levels on the ring plane of the molecule. Vertical and horizontal axes represent
dimensions of the space around the molecule in atomic units where the MICD is plotted. The vector arrow color intensity is proportional to thecurrent strength. Counterclockwise vectors represent paratropic currents. Both plots show the presence of strong local paratropic currents aroundthe scandium nuclei. HF and the intersection of DFT MICD plots are presented in Figure S2.
Figure 3. The influence of local electronic currents on the non-nuclear shielding values can be understood by this schematic representation. A stronglocal paratropic current as observed in Sc3
− (a) induces magnetic shielding around the center of the ring in a region similar to the blue egg-shapedregion in the picture. On the other hand, a strong local diatropic current as in the Cu clusters (b) induces a magnetic deshielding around the centerof the ring. The red egg-shaped zone in the figure represents this deshielded zone.
Journal of Chemical Theory and Computation Article
dx.doi.org/10.1021/ct4007184 | J. Chem. Theory Comput. 2013, 9, 4789−47964792
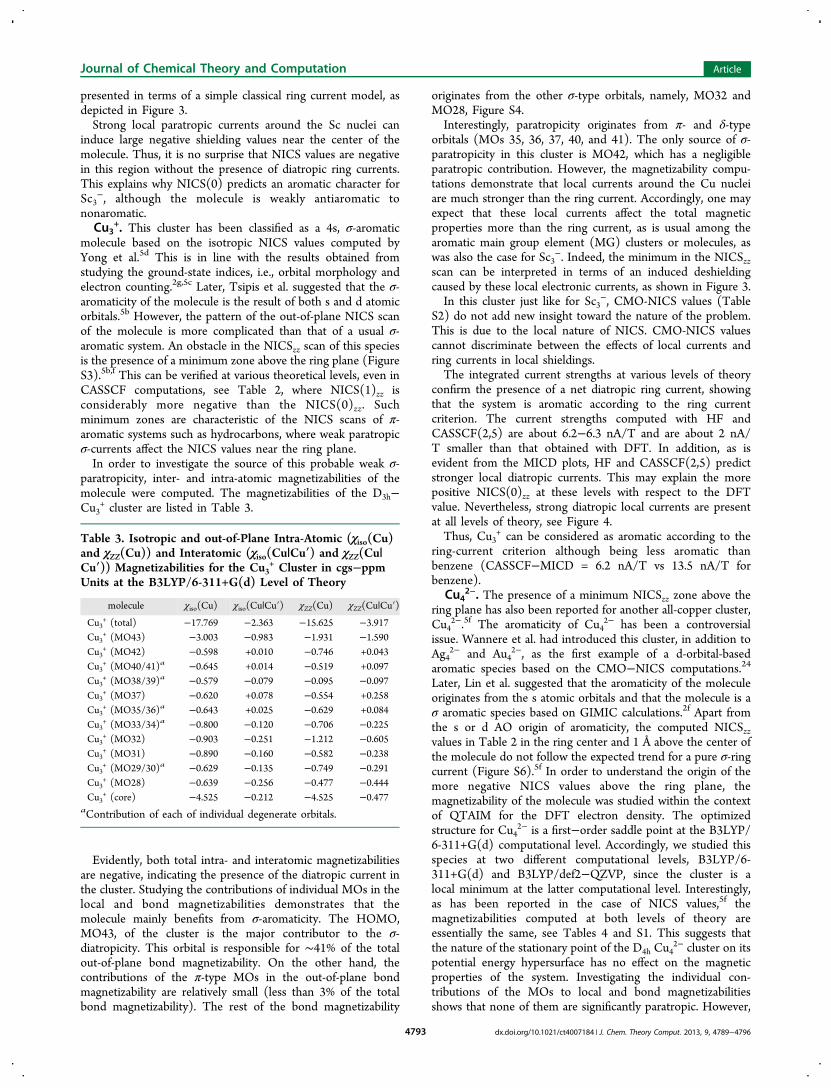
presented in terms of a simple classical ring current model, asdepicted in Figure 3.Strong local paratropic currents around the Sc nuclei can
induce large negative shielding values near the center of themolecule. Thus, it is no surprise that NICS values are negativein this region without the presence of diatropic ring currents.This explains why NICS(0) predicts an aromatic character forSc3
−, although the molecule is weakly antiaromatic tononaromatic.Cu3
+. This cluster has been classified as a 4s, σ-aromaticmolecule based on the isotropic NICS values computed byYong et al.5d This is in line with the results obtained fromstudying the ground-state indices, i.e., orbital morphology andelectron counting.2g,5c Later, Tsipis et al. suggested that the σ-aromaticity of the molecule is the result of both s and d atomicorbitals.5b However, the pattern of the out-of-plane NICS scanof the molecule is more complicated than that of a usual σ-aromatic system. An obstacle in the NICSzz scan of this speciesis the presence of a minimum zone above the ring plane (FigureS3).5b,f This can be verified at various theoretical levels, even inCASSCF computations, see Table 2, where NICS(1)zz isconsiderably more negative than the NICS(0)zz. Suchminimum zones are characteristic of the NICS scans of π-aromatic systems such as hydrocarbons, where weak paratropicσ-currents affect the NICS values near the ring plane.In order to investigate the source of this probable weak σ-
paratropicity, inter- and intra-atomic magnetizabilities of themolecule were computed. The magnetizabilities of the D3h−Cu3
+ cluster are listed in Table 3.
Evidently, both total intra- and interatomic magnetizabilitiesare negative, indicating the presence of the diatropic current inthe cluster. Studying the contributions of individual MOs in thelocal and bond magnetizabilities demonstrates that themolecule mainly benefits from σ-aromaticity. The HOMO,MO43, of the cluster is the major contributor to the σ-diatropicity. This orbital is responsible for ∼41% of the totalout-of-plane bond magnetizability. On the other hand, thecontributions of the π-type MOs in the out-of-plane bondmagnetizability are relatively small (less than 3% of the totalbond magnetizability). The rest of the bond magnetizability
originates from the other σ-type orbitals, namely, MO32 andMO28, Figure S4.Interestingly, paratropicity originates from π- and δ-type
orbitals (MOs 35, 36, 37, 40, and 41). The only source of σ-paratropicity in this cluster is MO42, which has a negligibleparatropic contribution. However, the magnetizability compu-tations demonstrate that local currents around the Cu nucleiare much stronger than the ring current. Accordingly, one mayexpect that these local currents affect the total magneticproperties more than the ring current, as is usual among thearomatic main group element (MG) clusters or molecules, aswas also the case for Sc3
−. Indeed, the minimum in the NICSzzscan can be interpreted in terms of an induced deshieldingcaused by these local electronic currents, as shown in Figure 3.In this cluster just like for Sc3
−, CMO-NICS values (TableS2) do not add new insight toward the nature of the problem.This is due to the local nature of NICS. CMO-NICS valuescannot discriminate between the effects of local currents andring currents in local shieldings.The integrated current strengths at various levels of theory
confirm the presence of a net diatropic ring current, showingthat the system is aromatic according to the ring currentcriterion. The current strengths computed with HF andCASSCF(2,5) are about 6.2−6.3 nA/T and are about 2 nA/T smaller than that obtained with DFT. In addition, as isevident from the MICD plots, HF and CASSCF(2,5) predictstronger local diatropic currents. This may explain the morepositive NICS(0)zz at these levels with respect to the DFTvalue. Nevertheless, strong diatropic local currents are presentat all levels of theory, see Figure 4.Thus, Cu3
+ can be considered as aromatic according to thering-current criterion although being less aromatic thanbenzene (CASSCF−MICD = 6.2 nA/T vs 13.5 nA/T forbenzene).
Cu42−. The presence of a minimum NICSzz zone above the
ring plane has also been reported for another all-copper cluster,Cu4
2−.5f The aromaticity of Cu42− has been a controversial
issue. Wannere et al. had introduced this cluster, in addition toAg4
2− and Au42−, as the first example of a d-orbital-based
aromatic species based on the CMO−NICS computations.24
Later, Lin et al. suggested that the aromaticity of the moleculeoriginates from the s atomic orbitals and that the molecule is aσ aromatic species based on GIMIC calculations.2f Apart fromthe s or d AO origin of aromaticity, the computed NICSzzvalues in Table 2 in the ring center and 1 Å above the center ofthe molecule do not follow the expected trend for a pure σ-ringcurrent (Figure S6).5f In order to understand the origin of themore negative NICS values above the ring plane, themagnetizability of the molecule was studied within the contextof QTAIM for the DFT electron density. The optimizedstructure for Cu4
2− is a first−order saddle point at the B3LYP/6-311+G(d) computational level. Accordingly, we studied thisspecies at two different computational levels, B3LYP/6-311+G(d) and B3LYP/def2−QZVP, since the cluster is alocal minimum at the latter computational level. Interestingly,as has been reported in the case of NICS values,5f themagnetizabilities computed at both levels of theory areessentially the same, see Tables 4 and S1. This suggests thatthe nature of the stationary point of the D4h Cu4
2− cluster on itspotential energy hypersurface has no effect on the magneticproperties of the system. Investigating the individual con-tributions of the MOs to local and bond magnetizabilitiesshows that none of them are significantly paratropic. However,
Table 3. Isotropic and out-of-Plane Intra-Atomic (χiso(Cu)and χZZ(Cu)) and Interatomic (χiso(Cu|Cu′) and χZZ(Cu|Cu′)) Magnetizabilities for the Cu3
+ Cluster in cgs−ppmUnits at the B3LYP/6-311+G(d) Level of Theory
molecule χiso(Cu) χiso(Cu|Cu′) χZZ(Cu) χZZ(Cu|Cu′)Cu3
+ (total) −17.769 −2.363 −15.625 −3.917Cu3
+ (MO43) −3.003 −0.983 −1.931 −1.590Cu3
+ (MO42) −0.598 +0.010 −0.746 +0.043Cu3
+ (MO40/41)a −0.645 +0.014 −0.519 +0.097Cu3
+ (MO38/39)a −0.579 −0.079 −0.095 −0.097Cu3
+ (MO37) −0.620 +0.078 −0.554 +0.258Cu3
+ (MO35/36)a −0.643 +0.025 −0.629 +0.084Cu3
+ (MO33/34)a −0.800 −0.120 −0.706 −0.225Cu3
+ (MO32) −0.903 −0.251 −1.212 −0.605Cu3
+ (MO31) −0.890 −0.160 −0.582 −0.238Cu3
+ (MO29/30)a −0.629 −0.135 −0.749 −0.291Cu3
+ (MO28) −0.639 −0.256 −0.477 −0.444Cu3
+ (core) −4.525 −0.212 −4.525 −0.477aContribution of each of individual degenerate orbitals.
Journal of Chemical Theory and Computation Article
dx.doi.org/10.1021/ct4007184 | J. Chem. Theory Comput. 2013, 9, 4789−47964793
as was the case for the Cu3+ cluster, considerable local diatropic
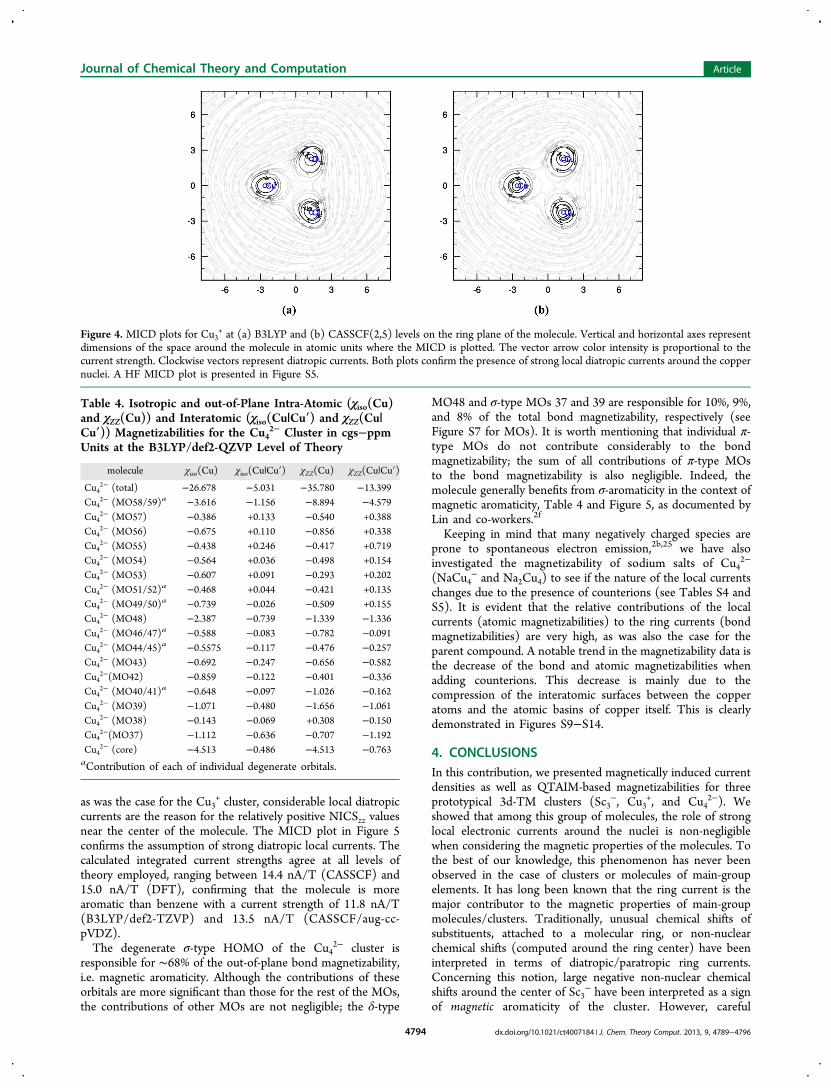
currents are the reason for the relatively positive NICSzz valuesnear the center of the molecule. The MICD plot in Figure 5confirms the assumption of strong diatropic local currents. Thecalculated integrated current strengths agree at all levels oftheory employed, ranging between 14.4 nA/T (CASSCF) and15.0 nA/T (DFT), confirming that the molecule is morearomatic than benzene with a current strength of 11.8 nA/T(B3LYP/def2-TZVP) and 13.5 nA/T (CASSCF/aug-cc-pVDZ).The degenerate σ-type HOMO of the Cu4
2− cluster isresponsible for ∼68% of the out-of-plane bond magnetizability,i.e. magnetic aromaticity. Although the contributions of theseorbitals are more significant than those for the rest of the MOs,the contributions of other MOs are not negligible; the δ-type
MO48 and σ-type MOs 37 and 39 are responsible for 10%, 9%,and 8% of the total bond magnetizability, respectively (seeFigure S7 for MOs). It is worth mentioning that individual π-type MOs do not contribute considerably to the bondmagnetizability; the sum of all contributions of π-type MOsto the bond magnetizability is also negligible. Indeed, themolecule generally benefits from σ-aromaticity in the context ofmagnetic aromaticity, Table 4 and Figure 5, as documented byLin and co-workers.2f
Keeping in mind that many negatively charged species areprone to spontaneous electron emission,2b,25 we have alsoinvestigated the magnetizability of sodium salts of Cu4
2−
(NaCu4− and Na2Cu4) to see if the nature of the local currents
changes due to the presence of counterions (see Tables S4 andS5). It is evident that the relative contributions of the localcurrents (atomic magnetizabilities) to the ring currents (bondmagnetizabilities) are very high, as was also the case for theparent compound. A notable trend in the magnetizability data isthe decrease of the bond and atomic magnetizabilities whenadding counterions. This decrease is mainly due to thecompression of the interatomic surfaces between the copperatoms and the atomic basins of copper itself. This is clearlydemonstrated in Figures S9−S14.
4. CONCLUSIONSIn this contribution, we presented magnetically induced currentdensities as well as QTAIM-based magnetizabilities for threeprototypical 3d-TM clusters (Sc3
−, Cu3+, and Cu4
2−). Weshowed that among this group of molecules, the role of stronglocal electronic currents around the nuclei is non-negligiblewhen considering the magnetic properties of the molecules. Tothe best of our knowledge, this phenomenon has never beenobserved in the case of clusters or molecules of main-groupelements. It has long been known that the ring current is themajor contributor to the magnetic properties of main-groupmolecules/clusters. Traditionally, unusual chemical shifts ofsubstituents, attached to a molecular ring, or non-nuclearchemical shifts (computed around the ring center) have beeninterpreted in terms of diatropic/paratropic ring currents.Concerning this notion, large negative non-nuclear chemicalshifts around the center of Sc3
− have been interpreted as a signof magnetic aromaticity of the cluster. However, careful
Figure 4. MICD plots for Cu3+ at (a) B3LYP and (b) CASSCF(2,5) levels on the ring plane of the molecule. Vertical and horizontal axes represent
dimensions of the space around the molecule in atomic units where the MICD is plotted. The vector arrow color intensity is proportional to thecurrent strength. Clockwise vectors represent diatropic currents. Both plots confirm the presence of strong local diatropic currents around the coppernuclei. A HF MICD plot is presented in Figure S5.
Table 4. Isotropic and out-of-Plane Intra-Atomic (χiso(Cu)and χZZ(Cu)) and Interatomic (χiso(Cu|Cu′) and χZZ(Cu|Cu′)) Magnetizabilities for the Cu4
2− Cluster in cgs−ppmUnits at the B3LYP/def2-QZVP Level of Theory
molecule χiso(Cu) χiso(Cu|Cu′) χZZ(Cu) χZZ(Cu|Cu′)Cu4
2− (total) −26.678 −5.031 −35.780 −13.399Cu4
2− (MO58/59)a −3.616 −1.156 −8.894 −4.579Cu4
2− (MO57) −0.386 +0.133 −0.540 +0.388Cu4
2− (MO56) −0.675 +0.110 −0.856 +0.338Cu4
2− (MO55) −0.438 +0.246 −0.417 +0.719Cu4
2− (MO54) −0.564 +0.036 −0.498 +0.154Cu4
2− (MO53) −0.607 +0.091 −0.293 +0.202Cu4
2− (MO51/52)a −0.468 +0.044 −0.421 +0.135Cu4
2− (MO49/50)a −0.739 −0.026 −0.509 +0.155Cu4
2− (MO48) −2.387 −0.739 −1.339 −1.336Cu4
2− (MO46/47)a −0.588 −0.083 −0.782 −0.091Cu4
2− (MO44/45)a −0.5575 −0.117 −0.476 −0.257Cu4
2− (MO43) −0.692 −0.247 −0.656 −0.582Cu4
2−(MO42) −0.859 −0.122 −0.401 −0.336Cu4
2− (MO40/41)a −0.648 −0.097 −1.026 −0.162Cu4
2− (MO39) −1.071 −0.480 −1.656 −1.061Cu4
2− (MO38) −0.143 −0.069 +0.308 −0.150Cu4
2−(MO37) −1.112 −0.636 −0.707 −1.192Cu4
2− (core) −4.513 −0.486 −4.513 −0.763aContribution of each of individual degenerate orbitals.
Journal of Chemical Theory and Computation Article
dx.doi.org/10.1021/ct4007184 | J. Chem. Theory Comput. 2013, 9, 4789−47964794

examination of the current density by means of integrated ringcurrent strengths using CASSCF and DFT levels of theory, aswell as bond magnetizabilities, unveils the role of strong localparatropic currents. Our study shows that Sc3
− has to beregarded as weakly antiaromatic to nonaromatic, in contrast toearlier work that predicted this molecule to be magneticallyaromatic. Studying molecules with NICSzz as well as CMO-NICS scans similar to that of Sc3
− may reveal additionalmolecular systems with strong local paratropic or diatropiccurrents. Of course, our study does not necessarily disprovearomaticity of all metallic clusters with patterns similar to thatfor Sc3
−. However, these types of systems should be carefullyre-examined, using QTAIM magnetizabilities as well as DFTand MCSCF current densities, when possible.On the other side of the periodic table, exceptionally strong
local diatropic currents around the copper nuclei deshield theinner space of Cu3
+ and Cu42− like for a typical weak paratropic
ring current. The magnetic response properties of Sc3−, Cu3
+,and Cu4
2− in addition to a number of f-block cyclic complexesthat were recently examined by Ramirez-Tagle et al.26 highlightthe significant role of local (atomic) electronic currents in themagnetic properties of this group of compounds. Finally, wepoint out that for the investigated TM clusters, the magneticaromaticity does not strongly correlate with electronic,structural, or energetic criteria of aromaticity as one perhapswould expect based on the conventional notion in organicchemistry. In organic compounds, aromaticity is associated withlarge band gaps so that the role of nondynamic correlationeffects in organic-aromatic systems is negligible.27 In contrast,nondynamic correlation plays a crucial role among TM clusters.It is shown that the magnetic properties of Cu4
2− do not changewith the changing nature of this cluster on its potential energyhypersurface at different DFT levels. In addition, Sc3
− is theglobal minimum on its PES,21 and ground-state propertiespredict it to be aromatic; however, the magnetic criterionsuggests an antiaromatic character for this species. These pointsindicate that the theory of aromaticity for TM clusters is stillnot fully understood.The above-mentioned findings highlight a deficiency of the
NICS approach. Lazzeretti discussed28 already in 2004 thatNICS is an index that estimates ring currents solely based on
the information available at one point in the molecular space. Ifthe ring current pattern in a molecule differs from the classicalring current pattern as seen for example for aromatichydrocarbons,29 then NICS cannot provide any reliableinformation regarding the true nature of the current density.On the basis of the above-mentioned discussion, it is highlyadvisible to employ current density-based approaches forstudying the magnetic aromaticity of TM clusters.
■ ASSOCIATED CONTENT*S Supporting InformationThe morphology of MOs and additional MICD plots. Thismaterial is available free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Authors*E-mail: [email protected].*E-mail: [email protected].*E-mail: [email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSZ.B., P.R.-R., and C.F-N. thank Patrick Bultinck and ShantShahbazian for their helpful discussions on an early version ofthis paper and Ramin Sakhtemani for his help in preparinggraphics for the manuscript. This work was supported by theProgram of “Emlpoyment of Newly Graduated Doctors ofScience for Scientific Excellence” (grant number CZ.1.07/2.3.00/30.009) cofinanced from European Social Fund and thestate budget of the Czech Republic (C.F.-N.), the project“CEITECCentral European Institute of Technology”(CZ.1.05/1.1.00/02.0068 to R.M.) from the European Region-al Development Fund, and by the Czech Science Foundation(P206/12/0539 to Z.B.). H.F. and K.R. thank the NorwegianResearch Council through the CoE Centre for Theoretical andComputational Chemistry (CTCC) Grant No. 179568/V30and the Norwegian Supercomputing Program (NOTUR)through a grant of computer time (Grant No. NN4654K).S.P. thanks the Department of Science and Technology, Govt.
Figure 5. MICD plots for Cu42− at (a) B3LYP and (b) CASSCF(6,12) levels in the ring plane of molecule. Vertical and horizontal axes represent
dimensions of the space around the molecule in atomic units where the MICD is plotted. The vector arrow color intensity is proportional to thecurrent strength. Clockwise vectors represent diatropic currents. Both plots confirm the presence of strong local diatropic currents around the coppernuclei. HF and CASSCF(6,6) MICD plots are presented in Figure S8.
Journal of Chemical Theory and Computation Article
dx.doi.org/10.1021/ct4007184 | J. Chem. Theory Comput. 2013, 9, 4789−47964795

of India, for his fellowship. P.R.-R. and Z.B. are grateful to theResearch Council of University of Tehran for financial support.Access to the computing and storage facilities owned by partiesand projects contributing to the National Grid InfrastructureMetaCentrum (Czech Republic) provided under the program“Projects of Large Infrastructure for Research, Development,and Innovations” (LM2010005) and the CERIT−SC comput-ing and storage facilities provided under the program CenterCERIT Scientific Cloud, part of the Operational ProgramResearch and Development for Innovations, reg. no. CZ. 1.05/3.2.00/08.0144, is appreciated.
■ REFERENCES(1) Li, X.; Kuznetsov, A. E.; Zhang, H. F.; Boldyrev, A. I.; Wang, L. S.Science 2001, 291, 859.(2) (a) Kuznetsov, A. E.; Birch, K. A.; Boldyrev, A. I.; Li, X.; Zhai, H.-J.; Wang, L. S. Science 2003, 300, 622. (b) Boldyrev, A. I.; Wang, L.-S.Chem. Rev. 2005, 105, 3716. (c) Tsipis, C. A. Coord. Chem. Rev. 2005,249, 2740. (d) Fliegl, H.; Lehtonen, O.; Lin, Y.-C.; Patzschke, M.;Sundholm, D. Theor. Chem. Acc. 2011, 129, 701. (e) Foroutan-Nejad,C.; Shahbazian, S.; Rashidi-Ranjbar, P. Phys. Chem. Chem. Phys. 2011,13, 4576. (f) Lin, Y.-C.; Sundholm, D.; Juselius, J.; Cui, L.-F.; Li, X.;Zhai, H. J.; Wang, L. S. J. Phys. A 2006, 110, 4244. (g) Zubarev, D. Y.;Averkiev, B. B.; Zhai, H. J.; Wang, L. S.; Boldyrev, A. I. Phys. Chem.Chem. Phys. 2008, 10, 257. (h) Foroutan-Nejad, C. Phys. Chem. Chem.Phys. 2012, 14, 9738. (i) Fowler, P. W.; Havenith, R. W. A.; Steiner, E.Chem. Phys. Lett. 2001, 343, 85. (j) Mandado, M.; Krishtal, A.; vanAlsenoy, C.; Bultinck, P.; Hermida-Ramon, J. M. J. Phys. Chem. A2007, 111, 11885. (k) Aromaticity and Metal Clusters; Chattaraj, P. K.,Ed.; CRC Press Taylor and Francis Group: Boca Raton, FL, 2011.(3) (a) Alexandrova, A. N.; Boldyrev, A. I.; Zhai, H.-J.; Wang, L.-S.Coord. Chem. Rev. 2006, 250, 2811. (b) Zubarev, D. Yu.; Boldyrev, A. I.J. Comput. Chem. 2007, 28, 251. (c) Galeev, T. R.; Boldyrev, A. I.Annu. Rep. Prog. Chem., Sect. C 2011, 107, 124.(4) Tsipis, A. C.; Kefalidis, C. E.; Tsipis, C. A. J. Am. Chem. Soc. 2008,130, 9144.(5) (a) Foroutan-Nejad, C.; Shahbazian, S.; Feixas, F.; Rashidi-Ranjbar, P.; Sola, M. J. Comput. Chem. 2011, 32, 2422. (b) Tsipis, A.C.; Depastas, I. G.; Karagiannis, E. E.; Tsipis, C. A. J. Comput. Chem.2010, 31, 431. (c) Feixas, F.; Matito, E.; Duran, M.; Poater, J.; Sola, M.Theor. Chem. Acc. 2011, 128, 419. (d) Yong, L.; Wu, S. D.; Chi, X. X.Int. J. Quantum Chem. 2007, 107, 722. (e) Foroutan-Nejad, C.; Badri,Z.; Shahbazian, S.; Rashidi-Ranjbar, P. J. Phys. Chem. A 2011, 115,12708. (f) Badri, Z.; Foroutan-Nejad, C.; Rashidi-Ranjbar, P. Phys.Chem. Chem. Phys. 2012, 14, 3471.(6) (a) Stanger, A. J. Org. Chem. 2006, 71, 883. (b) Jimenez-Halla, J.O. C.; Matitio, E.; Robles, J.; Sola, M. J. Organomet. Chem. 2006, 691,4359.(7) (a) Buhl, M.; van Wullen, C. Chem. Phys. Lett. 1995, 247, 63.(b) Chen, Z.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; Schleyer,P. v. R. Chem. Rev. 2005, 105, 3842.(8) Foroutan-Najad, C. J. Phys. Chem. A 2011, 115, 12555.(9) Pathak, S.; Bast, R.; Ruud, K. J. Chem. Theory Comput. 2013, 9,2189.(10) (a) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.(b) Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J.Phys. Chem. 1994, 98, 11623. (c) Becke, A. D. Phys. Rev. A 1988, 38,3098.(11) Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297.(12) (a) London, F. J. Phys. Radium 1937, 8, 397. (b) McWeeny, R.Phys. Rev. 1962, 126, 1028. (c) Ditchfield, R. Mol. Phys. 1974, 27, 789.(d) Dodds, J. L.; McWeeny, R.; Sadlej, A. J. Mol. Phys. 1980, 41, 1419.(e) Wolinski, K.; Hilton, J. F.; Pulay, P. J. Am. Chem. Soc. 1990, 112,8251. (f) Ruud, K.; Helgaker, T.; Kobayashi, R.; Jørgensen, P.; Bak, K.L.; Jensen, H. J. Aa. J. Chem. Phys. 1994, 100, 8178.(13) Fallah-Bagher-Shaidaei, H.; Wannere, C. S.; Corminboeuf, C.;Puchta, R.; Schleyer, P. v. R. Org. Lett. 2006, 8, 863.
(14) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci,B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.;Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima,T.; Honda, Y.; Kitao, O.;Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.;Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin,K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.;Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega,N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.;Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.;Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.;Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.;Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.;Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09,revision A.02; Gaussian, Inc: Wallingford CT, 2009.(15) (a) Bader, R. F. W. In Atoms in Molecules: A Quantum Theory;Oxford University Press: Oxford, 1990. (b) Keith, T. A.; Bader, R. F.W. J. Chem. Phys. 1993, 99, 3669. (c) Keith, T. A.; Bader, R. F. W.Chem. Phys. Lett. 1993, 210, 223. (d) Keith, T. A. In The QuantumTheory of Atoms in Molecules: From Solid State to DNA and DrugDesign; Matta, C. F., Boyd, R. J., Eds.; Wiley−VCH: Weinheim,Germany, 2007; Chapter 3.(16) Keith, T. A. AIMAll (version 11.02.27). http://aim.tkgristmill.com.(17) DALTON, a molecular electronic structure program. http://www.daltonprogram.org.(18) PyNGL, developed at the National Center for AtmosphericResearch. http://www.pyngl.ucar.edu.(19) (a) Fliegl, H.; Taubert, S.; Lehtonen, O.; Sundholm, D. Phys.Chem. Chem. Phys. 2011, 13, 20500. (b) Fliegl, H.; Lehtonen, O.;Patzschke, M.; Sundholm, D.; Lin, Y. C. Theor. Chem. Acc. 2011, 129,701.(20) Fliegl, H.; Sundholm, D.; Taubert, S.; Juselius, J.; Klopper, W. J.Phys. Chem. A 2009, 113, 8668.(21) Chi, X. X.; Liu, Y. Int. J. Quantum Chem. 2007, 107, 1886.(22) (a) Foroutan-Nejad, C.; Shahbazian, S.; Rashidi-Ranjbar, P.Phys. Chem. Chem. Phys. 2010, 12, 12630. (b) Foroutan-Nejad, C.;Shahbazian, S.; Rashidi-Ranjbar, P. Phys. Chem. Chem. Phys. 2011, 13,12655.(23) Juselius, J.; Sundholm, D.; Gauss, J. J. Chem. Phys. 2004, 121,3952.(24) Wannere, C. S.; Corminboeuf, C.; Wang, Z.-X.; Wodrich, M. D.;King, R. B.; Schleyer, P. v. R. J. Am. Chem. Soc. 2005, 127, 5701.(25) (a) Pyykko, P.; Zhao, Y.Mol. Phys. 1990, 70, 701. (b) Pyykko, P.Phys. Scr. 1990, T33, 52. (c) Pyykko, P.; Zhao, Y. J. Phys. Chem. 1990,94, 7753. (d) Zubarev, D. Y.; Boldyrev, A. I. J. Phys. Chem. A 2008,112, 7984.(26) Ramírez-Tagle, R.; Alvarado-Soto, L.; Arratia-Perez, R.; Bast, R.;Alvarez-Thon, L. J. Chem. Phys. 2011, 135, 104506.(27) (a) Zhou, Z.; Parr, R. G. J. Am. Chem. Soc. 1989, 111, 7371.(b) Zhou, Z.; Parr, R. G.; Garst, J. F. Tetrahedron Lett. 1988, 29, 4843.(28) Lazzeretti, P. Phys. Chem. Chem. Phys. 2004, 6, 217.(29) (a) Pelloni, S.; Lazzeretti, P. J. Phys. Chem. A 2011, 115, 4553.(b) Pelloni, S.; Monaco, G.; Lazzeretti, P.; Zanasi, R. Phys. Chem.Chem. Phys. 2011, 13, 20666. (c) Fliegl, H.; Sundholm, D. J. Org.Chem. 2012, 77, 3408. (d) Kaipio, M.; Patzschke, M.; Fliegl, H.;Pichierri, F.; Sundholm, D. J. Phys. Chem. A 2012, 116, 10257.(e) Fliegl, H.; Oczan, N.; Mera-Adasme, R.; Pichierri, F.; Juselius, J.;Sundholm, D. Mol. Phys. 2013, 111, 1364. (f) Pelloni, S.; Lazzeretti, P.J. Phys. Chem. A 2013, 117, 9083.
Journal of Chemical Theory and Computation Article
dx.doi.org/10.1021/ct4007184 | J. Chem. Theory Comput. 2013, 9, 4789−47964796




















