
Induction of Cardiogenesis in Embryonic Stem Cells via Downregulation of Notch1 Signaling
Upload
independentCategory
view
1download
0
doi:10.1182/blood-2005-06-2553Prepublished online September 15, 2005;
Frederick W Alt, Michelle Kelliher and A T LookJennifer O'Neil, Jennifer Calvo, Keith McKenna, Veena Krishnamoorthy, Jon C Aster, Craig H Bassing, Activating Notch1 mutations in mouse models of T-ALL
(1930 articles)Signal Transduction � (795 articles)Oncogenes and Tumor Suppressors �
(4217 articles)Neoplasia � (1653 articles)Brief Reports �
Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
digital object identifier (DOIs) and date of initial publication. theindexed by PubMed from initial publication. Citations to Advance online articles must include
final publication). Advance online articles are citable and establish publication priority; they areappeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet
Copyright 2011 by The American Society of Hematology; all rights reserved.20036.the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

1
Activating Notch1 Mutations in Mouse Models of T-ALL
Jennifer O’Neil1, Jennifer Calvo2, Keith McKenna1, Veena Krishnamoorthy2, Jon C.
Aster3, Craig H. Bassing4, Frederick W. Alt4, Michelle Kelliher2* and A. Thomas Look1*
1Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA 02115, USA. 2Department of Cancer Biology, University of Massachusetts Medical School, Worcester, MA 01605, USA. 3Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115. 4Howard Hughes Medical Institute, The Children’s Hospital, Department of Genetics, Harvard Medical School and The Center for Blood Research, Boston, MA 02115. *Both laboratories contributed equally to this work. Reprints: A. Thomas Look, Dana-Farber Cancer Institute, Mayer Bldg, Rm 630, 44 Binney Street, Boston, MA 02115, Fax 617-632-6989, Phone 617-632-5826 [email protected] abstract word count: 129 total text word count: 1477 Scientific Heading: Neoplasia
Blood First Edition Paper, prepublished online September 15, 2005; DOI 10.1182/blood-2005-06-2553
Copyright © 2005 American Society of Hematology
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

2
Abstract
Recent studies have demonstrated that the majority of T-ALL patients have
activating mutations in NOTCH1. We sought to determine whether these mutations are
also acquired in mouse models of T-ALL. We have sequenced the heterodimerization
domain and PEST domain of notch1 in our mouse model of TAL1-induced leukemia and
have found that 74% of the tumors harbor activating mutations in notch1. Cell lines
derived from these tumors undergo G0/G1 arrest and apoptosis when treated with a γ-
secretase inhibitor. In addition, we found activating notch1 mutations in 31% of thymic
lymphomas that occur in mice deficient for various combinations of the H2AX, p53 and
Rag2 genes. Thus, notch1 mutations are often acquired as a part of the molecular
pathogenesis of T-ALLs that develop in mice with known predisposing genetic
alterations.
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

3
Introduction
TAL1 is a basic helix-loop-helix (bHLH) transcription factor that is normally
expressed in hematopoietic cells, endothelial cells and cells of the central nervous system.
Through chromosomal translocation, interstitial deletion or biallelic activation, TAL1 is
misexpressed in thymocytes of 60 and 45% of pediatric and adult T-ALL patients
respectively1,2. Improved results in the treatment of pediatric T-ALL have been achieved
in recent years by intensifying chemotherapy regimens, leading to a five-year event free
survival rate approaching 80%3,4, however, patients whose lymphoblasts overexpress the
TAL1 oncogene have a less favorable prognosis than patients with activation of other
oncogenes5,6.
Double-strand breaks occur in mammalian cells as a result of exposure to DNA-
damaging agents such as ionizing radiation or during V(D)J recombination in
lymphocytes. Many T-ALL tumors harbor chromosomal translocations or
rearrangements that activate oncogenes, or create oncogenic fusion genes7. These
translocations and rearrangements likely occur as a result of errors in the repair of
double-strand breaks. The histone H2A variant, H2AX, plays a role in the cellular
response to IR-induced double-strand breaks8,9. H2AX deficiency alone causes only a
modest predisposition to cancer; however, mice deficient for both H2AX and p53 rapidly
develop T and B cell lymphomas and solid tumors demonstrating that H2AX acts as a
tumor suppressor in mice10,11. The fact that human H2AX (H2AFX) maps to 11q23, a
region that is frequently altered in human cancer, suggests that the human gene may also
function as a tumor suppressor10.
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

4
The NOTCH genes encode single pass transmembrane receptors that regulate
apoptosis, proliferation and cell fate determination in multicellular organisms. Binding
of NOTCH ligands initiates a series of proteolytic cleavages in NOTCH1. The last of
these cleavages, which is catalyzed by γ-secretase, results in the release of the
intracellular domain of NOTCH1 (ICN), permitting it to translocate to the nucleus and
form part of a multiprotein complex that regulates gene transcription12. Recent work
from our laboratories has revealed that activating mutations in NOTCH1 occur in over
50% of cases of human T-ALL13. Previous studies have demonstrated that the notch1
gene is a frequent site of retroviral insertional mutagenesis in mouse models of T-ALL14-
17 (see also http://RTCGD.ncifcrf.gov). In order to determine whether notch
mutations are acquired in mouse models of T-ALL, we sequenced the heterodimerization
domain and PEST domain of all four notch genes in tumors from our previously
established models of T-ALL.
Study Design
Mice
FVB/N lck-tal1 transgenic mice and tal1/+HEB+/- mice have been previously
described18,19. Ink4a/Arf+/- mice were obtained from the MMHCC mouse repository20
and mated to tal1 transgenic mice to obtain tal1/+Ink4a/Arf+/- mice (Calvo and
Kelliher, unpublished data) . 129Sv/ev p53-/-, H2AX-/-, H2AX-/-p53-/-, and
H2AX+/-p53-/- mice were described previously10. RAG2 deficient mice21 were
mated to the above mice to obtain H2AX-/-p53+/-RAG-/- and H2AX+/-p53-/-
RAG-/- mice.
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

5
Mutation Detection
Exon 26 and 27 of notch1 were amplified using the following primers: exon 26 forward
5’-ACGGGAGGACCTAACCAAAC-3’, exon 26 reverse 5’-
CAGCTTGGTCTCCAACACCT-3’, exon 27 forward 5’-
CGCTGAGTGCTAAACACTGG-3’, and exon 27 reverse 5’-
GTTTTGCCTGCATGTACGTC-3’. Exon 34 was amplified in two fragments using the
following primers: forward 1 5’-GCTCCCTCATGTACCTCCTG-3’, reverse 1 5’-
TAGTGGCCCCATCATGCTAT-3’, forward 2 5’-ATAGCATGATGGGGCCACTA-3’,
reverse 2 5’-CTTCACCCTGACCAGGAAAA-3’. The products were direct sequenced
at Agencourt Bioscience Corporation (Beverly, MA) and the results were analyzed using
Mutation Surveyor.
Gamma-Secretase Inhibitor Treatment
Murine tal1 tumor cell lines were either treated with 1μΜ DAPT [N–N-(3,5-
Difluorophenacetyl)-L-alanyl-(S)-phenylglycine t-butyl ester] (Calbiochem San Diego,
CA Cat#565770) or mock treated with DMSO for 6 days. The cells were fixed with
70% ethanol, stained with propidium iodide and analyzed by flow cytometry.
Western Blotting
Tal1 tumor cell lines were either untreated or treated with 1μM DAPT. Cell lysates were
fractionated on a SDS-PAGE gel and then transferred to PVDF. Intracellular Notch1 was
detected by probing the blot with the V1744 antibody (Cell Signaling Technology
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

6
Beverly, MA). The blot was then stripped and reprobed with an anti-β actin antibody
(Sigma St. Louis, MO) to insure equal loading.
RT-PCR
RNA was extracted from tal1 tumor cells that were untreated or treated with DAPT using
Trizol (Invitrogen Carlsbad, CA). cDNA was prepared with Superscript II reverse
transcriptase (Invitrogen). RT-PCR was then performed using primers specific for
Deltex, Hes1 and GAPDH22.
Results and Discussion
We sequenced the heterodimerization domain and PEST domain of all four notch
genes in tumors from tal1/+, tal1/+HEB+/-, and tal1/+Ink4a/Arf+/- mice. Of the 27
tumors that we analyzed, we found activating mutations in the notch1 gene in 20 samples
(74%) (Table 1). One tumor (9205) had a point mutation in the heterodimerization
domain leading to a leucine to proline change at residue 1668 (1679 in human NOTCH1).
Point mutations causing identical L to P substitutions have been observed in four human
T-ALL patients13. The majority of mutations that we detected in the mouse tumors were
in the PEST domain. Here, as in the human cell lines and samples, we found insertions,
deletions and point mutations resulting in premature stop codons and loss of the notch1
PEST domain. In contrast, we found no mutations in notch2, notch3, or notch4.
In addition to the notch1 mutations found in tal1 transgenic mice, we also found
notch1 mutations in 9 out of 29 (31%) of T cell tumors that developed in H2AX-/-, p53-/-,
H2AX-/-p53-/-, H2AX+/-p53-/-, p53-/-RAG-/-, H2AX-/-p53-/-RAG-/-, and H2AX-/-
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

7
p53+/-RAG-/- mice. One tumor (455) had an alanine to proline missense mutation in the
heterodimerization domain of notch1. This same mutation at the homologous residue in
human NOTCH1 (1702) was also observed in one primary sample from a T-ALL
patient13. However, as in the tal1 transgenic mice most of the mutations were in the
PEST domain. This data indicates that notch1 mutations are not specific to leukemias
arising in tal1 transgenic mice, but arise in diverse T-ALL-prone backgrounds. Of note,
notch1 mutations are significantly more common in tal1 transgenic mice compared to
mice that are heterozygous or deficient for p53 (p=0.0009), H2AX (p=0.006), or RAG
(p=0.0006) using a two-tailed Fisher’s exact test. Because of the complex genotypes of
the mice analyzed in this study, further experiments will be necessary to determine the
individual contributions of p53, H2AX or RAG deficiency to susceptibility to notch1
mutations.
The mutations we have found affecting full-length notch proteins in murine T-
ALL are predicted to activate notch pathway signaling in way that is dependent on
cellular γ-secretase activity. Therefore, to determine whether the tumor cells depend on
notch signaling, we treated tumor cell lines derived from these mice with the γ-secretase
inhibitor DAPT (Figure 1 and Supplemental Table 1). After treatment of the cell lines
with the inhibitor for six days, we found that the majority of cell lines exhibited a G0/G1
arrest and/or an increase in apoptosis, as indicated by cells with 45-70% sub-G0/G1 DNA
content (similar results were also seen after 3 days). In addition, we demonstrate that in
both sensitive and resistant cell lines, DAPT treatment inhibits the production of
activated notch (Figure 1E) and the transcription of the notch target genes deltex and hes1
(Figure 1F). Some tal1 tumor cell lines were resistant to the γ-secretase inhibitor
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

8
treatment. Two out of four γ-secretase resistant cell lines did not have mutations in
notch1. Resistant cell lines with mutations in notch1 have likely incurred additional
mutations rendering them independent of notch pathway signaling for growth and
survival since DAPT treatment does decrease notch1 signaling in these cells. In fact, our
previous studies have demonstrated that tumor 5146 displays constitutive NFκB
activation, therefore it may be dependent on NFκB signaling rather than notch signaling
for its growth/survival23. One cell line was sensitive to the γ-secretase inhibitor but did
not have a mutation in notch1. In this case, we hypothesize that there may be activating
mutations in one or more other components of the notch signaling pathway. However,
we cannot rule out the possibility that this cell line has a mutation in another substrate of
γ-secretase. This work provides further evidence that notch1 activation plays a key role
in the pathogenesis of T-ALL in both humans and in murine models and provides model
systems incorporating clinically relevant oncogenes and tumor suppressors for testing
therapeutics that target the NOTCH signaling pathway.
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

9
Figure Legend
Figure 1. Cell lines derived from tal1 tumors are sensitive to a γ-secretase inhibitor.
Tal1 tumor cell lines were treated with 1μΜ DAPT or DMSO (control) for six days and
the DNA content of propidium-iodide-stained cell populations was determined by flow
cytometry. The numbers over the cell populations indicate the percentage of cells in sub-
G0/G1, G0/ G1, S and G2/M (A-D). Tal1 tumor cells were untreated or treated with DAPT
for 40 hours. Western blot analysis using an antibody that specifically recognizes the
activated form of notch1 (E). A tal1 T-ALL cell line was either untreated or treated with
DAPT for 24 or 48 hours. RT-PCR analysis was performed with primers specific for
deltex, hes1 and GAPDH (F).
References
1. Ferrando AA, Look AT. Gene expression profiling in T-cell acute lymphoblastic leukemia. Semin Hematol. 2003;40:274-280. 2. Ferrando AA, Herblot S, Palomero T, et al. Biallelic transcriptional activation of oncogenic transcription factors in T-cell acute lymphoblastic leukemia. Blood. 2003. 3. Schrappe M, Reiter A, Ludwig WD, et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood. 2000;95:3310-3322. 4. Silverman LB, Gelber RD, Dalton VK, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood. 2001;97:1211-1218. 5. Ferrando AA, Neuberg DS, Dodge RK, et al. Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet. 2004;363:535-536. 6. Ferrando AA, Neuberg DS, Staunton J, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75-87. 7. Ferrando AA, Look AT. Clinical implications of recurring chromosomal and associated molecular abnormalities in acute lymphoblastic leukemia. Semin Hematol. 2000;37:381-395.
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

10
8. Bassing CH, Chua KF, Sekiguchi J, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A. 2002;99:8173-8178. 9. Celeste A, Petersen S, Romanienko PJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922-927. 10. Bassing CH, Suh H, Ferguson DO, et al. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359-370. 11. Celeste A, Difilippantonio S, Difilippantonio MJ, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371-383. 12. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770-776. 13. Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269-271. 14. Feldman BJ, Hampton T, Cleary ML. A carboxy-terminal deletion mutant of Notch1 accelerates lymphoid oncogenesis in E2A-PBX1 transgenic mice. Blood. 2000;96:1906-1913. 15. Hoemann CD, Beaulieu N, Girard L, Rebai N, Jolicoeur P. Two distinct Notch1 mutant alleles are involved in the induction of T-cell leukemia in c-myc transgenic mice. Mol Cell Biol. 2000;20:3831-3842. 16. Girard L, Jolicoeur P. A full-length Notch1 allele is dispensable for transformation associated with a provirally activated truncated Notch1 allele in Moloney MuLV-infected MMTV(D)/myc transgenic mice. Oncogene. 1998;16:517-522. 17. Yanagawa S, Lee JS, Kakimi K, Matsuda Y, Honjo T, Ishimoto A. Identification of Notch1 as a frequent target for provirus insertional mutagenesis in T-cell lymphomas induced by leukemogenic mutants of mouse mammary tumor virus. J Virol. 2000;74:9786-9791. 18. Kelliher MA, Seldin DC, Leder P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIalpha. Embo J. 1996;15:5160-5166. 19. O'Neil J, Shank J, Cusson N, Murre C, Kelliher M. TAL1/SCL induces leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell. 2004;5:587-596. 20. Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27-37. 21. Shinkai Y, Rathbun G, Lam KP, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855-867. 22. Deftos ML, Huang E, Ojala EW, Forbush KA, Bevan MJ. Notch1 signaling promotes the maturation of CD4 and CD8 SP thymocytes. Immunity. 2000;13:73-84. 23. O'Neil J, Ventura JJ, Cusson N, Kelliher M. NF-{kappa}B activation in premalignant mouse tal-1/scl thymocytes and tumors. Blood. 2003;102:2593-2596.
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

11
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

12
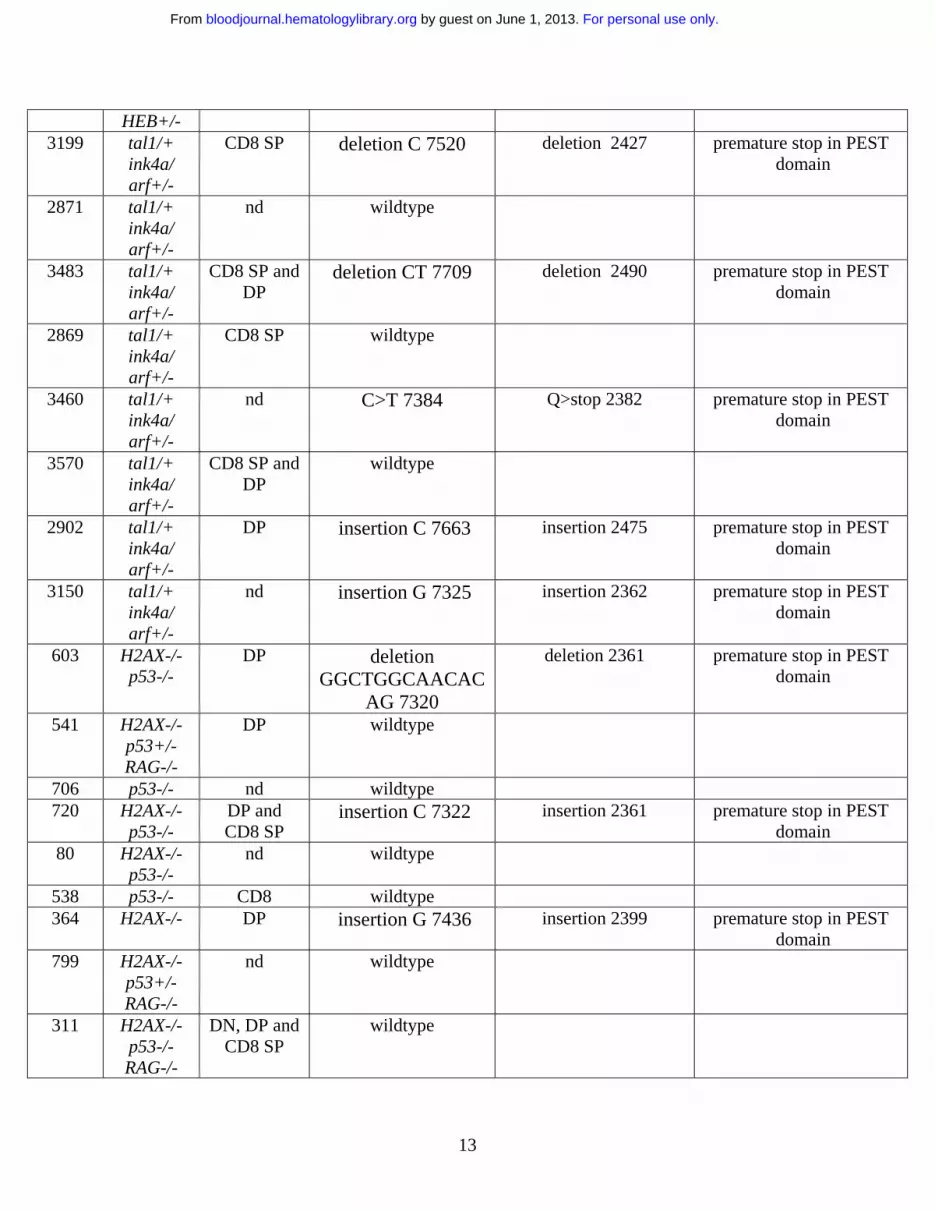
Table 1. Activating Notch 1 mutations occur at a high frequency in mouse T-ALL
Tumor Genotype Phenotype* Nucleotide Change┼
Amino Acid Change‡ Mutational Consequence
5046 tal1/+ nd insertion G 7501 insertion 2421 premature stop in PEST domain
5151 tal1/+ nd deletion CCCTGACCAGTG
GT 7716
deletion 2492 premature stop in PEST domain
5146 tal1/+ nd deletion CT 7709 deletion 2490 premature stop in PEST domain
1444 tal1/+ DP deletion AC 7320 deletion 2360 premature stop in PEST domain
5015 tal1/+ nd insertion CGTGG 7322
insertion 2361 premature stop in PEST domain
1330 tal1/+ CD8 SP wildtype 5148 tal1/+ nd wildtype 5188 tal1/+ nd insertion GG 7322 deletion 2361 premature stop in PEST
domain 1161 tal1/+ nd C>T 7495 Q>stop 2419 premature stop in PEST
domain 5145 tal1/+ nd insertion C 7322 insertion 2361 premature stop in PEST
domain 4862 tal1/+ DP deletion CT 7709 deletion 2490 premature stop in PEST
domain 1469 tal1/+ CD4 SP insertion
GGGGGGGG 7324 insertion 2361 premature stop in PEST
domain 1011 tal1/+ CD8 SP C>T 7495 Q>stop 2419 premature stop in PEST
domain 8998 tal1/+
HEB+/- nd C>T 7129 Q>stop 2296 premature stop in PEST
domain 9450 tal1/+
HEB+/- nd insertion C 7510 insertion 2420 premature stop in PEST
domain 9205 tal1/+
HEB+/- DP C>T 5243 L>P 1668 missense in
heterodimerization domain
9306 tal1/+ HEB+/-
nd wildtype
6839 tal1/+ HEB+/-
nd insertion CCTC 7321
insertion 2361 premature stop in PEST domain
8283 tal1/+ nd wildtype
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom
13
HEB+/- 3199 tal1/+
ink4a/ arf+/-
CD8 SP deletion C 7520 deletion 2427 premature stop in PEST domain
2871 tal1/+ ink4a/ arf+/-
nd wildtype
3483 tal1/+ ink4a/ arf+/-
CD8 SP and DP
deletion CT 7709 deletion 2490 premature stop in PEST domain
2869 tal1/+ ink4a/ arf+/-
CD8 SP wildtype
3460 tal1/+ ink4a/ arf+/-
nd C>T 7384 Q>stop 2382 premature stop in PEST domain
3570 tal1/+ ink4a/ arf+/-
CD8 SP and DP
wildtype
2902 tal1/+ ink4a/ arf+/-
DP insertion C 7663
insertion 2475 premature stop in PEST domain
3150 tal1/+ ink4a/ arf+/-
nd insertion G 7325 insertion 2362 premature stop in PEST domain
603 H2AX-/-p53-/-
DP deletion GGCTGGCAACAC
AG 7320
deletion 2361 premature stop in PEST domain
541 H2AX-/-p53+/-RAG-/-
DP wildtype
706 p53-/- nd wildtype 720 H2AX-/-
p53-/- DP and CD8 SP
insertion C 7322 insertion 2361 premature stop in PEST domain
80 H2AX-/-p53-/-
nd wildtype
538 p53-/- CD8 wildtype 364 H2AX-/- DP insertion G 7436 insertion 2399 premature stop in PEST
domain 799 H2AX-/-
p53+/-RAG-/-
nd wildtype
311 H2AX-/-p53-/-RAG-/-
DN, DP and CD8 SP
wildtype
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

14
859 H2AX+/-p53-/-
DP wildtype
618 H2AX-/-p53-/-
DN, DP and CD8 SP
deletion GCCACAGAACTTACAGCTCCAGCC
TCAGA 7365
deletion 2374 premature stop in PEST domain
455 H2AX-/- DP G>C 5308 A>P 1690 missense in heterodimerization
domain 727 H2AX-/-
p53-/- DP wildtype
613 H2AX+/-p53-/-
DP deletion CT 7709 deletion 2490 premature stop in PEST domain
84 H2AX-/-p53-/-
nd wildtype
310 H2AX+/-p53-/-
nd deletion ATGTACAACCGCTGGGCCCCAGCA
G 7490
deletion 2417 premature stop in PEST domain
308 H2AX-/-p53-/-RAG-/-
DN and CD8 SP
wildtype
761 H2AX-/-p53-/-RAG-/-
DP wildtype
728 H2AX-/-p53-/-
DP wildtype
274 H2AX+/-p53-/-
DN and CD8 SP
wildtype
814 H2AX+/-p53-/-
nd wildtype
634 H2AX-/-p53-/-
DP insertion T 7518 insertion 2426 premature stop in PEST domain
540 p53-/-RAG-/-
CD8 wildtype
696 H2AX-/-p53-/-
DN and CD8 SP
wildtype
24 H2AX-/-p53+/-RAG-/-
DP wildtype
414 H2AX-/-p53+/-RAG-/-
DP and CD8 SP
wildtype
814 H2AX-/-p53+/-
DN and CD8 SP
deletion AGTCTGCCTGTG
deletion 2425
premature stop in PEST domain
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom

15
RAG-/- CACACCATTCTGCCCCAGGAAAGCCAGGCCCTGCCCACATCACTGCCATCCTCCATGGTCCCACCCATGACCACTACCCAGTTC
CTGAC 7513 deletion
CTTCCCAGCACAGTTACTCCTCCTCCCCTGTGGACAACACCCCCAGCCACCAGCTGCAGGTGCCAGAGCACCCCTTCCTCACCCCATCCCCTGAGTCCCCTGACCAGTGGTCCAGCTCCTCCCCGCATTCCAACATCTCTGATTGGTCCGAGGGCATCTCCAGCCCGCCCACCACCATGCCGTCCCAGATCACCCACATTCCAGAGGCATTTAAAT
7619
deletion 2460
399 H2AX-/-p53+/-RAG-/-
DP and CD8 SP
wildtype
531 H2AX-/-p53+/-RAG-/-
DN wildtype
*nd-not determined ┼Numbers correspond to nucleotide position in notch1 cDNA. ‡Numbers indicate amino acid residue in notch1 at which mutation occurs.
For personal use only. by guest on June 1, 2013. bloodjournal.hematologylibrary.orgFrom
Copyright © 2022 FDOKUMEN




![Quantitative analysis of [11C]-erlotinib PET demonstrates specific binding for activating mutations of the EGFR kinase domain](https://static.fdokumen.com/doc/165x107/6345ca446cfb3d406409d7f9/quantitative-analysis-of-11c-erlotinib-pet-demonstrates-specific-binding-for-activating.jpg)















