
Activated carbon supported Ni–Ca: Influence of reaction parameters on activity and stability of...
8
Activated carbon supported Ni–Ca: Influence of reaction parameters on activity and stability of catalyst on methane reformation Karina Dı ´az, Vı ´ctor Garcı ´a, Juan Matos * Laboratorio de Fisicoquı ´mica de Superficies, Centro de Quı ´mica, Instituto Venezolano de Investigaciones Cientı ´ficas, I.V.I.C., Apartado 21827, Caracas 1020-A, Venezuela Received 21 February 2006; received in revised form 9 May 2006; accepted 13 May 2006 Available online 12 June 2006 Abstract The objective of the present work was to study the influence of some reaction parameters (temperature, chemical nature of pre-treat- ment gas, mass catalyst) on the yield of methane reforming with carbon dioxide on Ni (5%)–Ca (1%)/AC at atmospheric pressure. The obtained results show that the best system was pre-treated under He flow at 650 °C and then following the reaction at the same temper- ature. It was also detected that Ca plays a co-support role inhibiting the deactivation of catalyst at long periods of reaction. It can be concluded that it is possible to employ activated carbon as support for methane dry reformation obtaining representative methane con- versions up to 40% at mild experimental conditions. Ó 2006 Elsevier Ltd. All rights reserved. Keywords: Methane reformation; Ni–Ca catalyst; Activated carbon 1. Introduction Venezuela has high quantities of natural gas stocks that by 2003 were estimated around 148,000 billions of cubic feet [1] being the first country in Latin-America and the ninth in the world with more natural gas stocks. Therefore, a clear potential in the production and conversion of natu- ral gas in the country is being taken into account for the future years. Particularly, it is important to convert the main component of natural gas methane, in up-grading compounds [2,3] by means, for example, of dry reforma- tion with carbon dioxide to yield syngas as it is indicated in reaction (1): CH 4 + CO 2 ! 2CO + 2H 2 H 2 /CO = 1 ð1Þ Different H 2 /CO proportions of syngas have been employed in different important industrial processes [4–8] as methanol, dimethyl ether or Fischer–Tropsch synthesis. Several catalysts have been studied [9–11] for up-grading natural gas. Among them, noble metals as Rh, Pt, Ru and Ir yield high activity and selectivity, but due to the low availability and high price of such metals, nickel has been explored and used as a possible substitute of noble metals [11,12]. However, some studies have shown that Ni has a higher tendency to be deactivated than noble met- als by the deposition of carbon [9,10]. Also, studies con- cerning the dry reformation of methane on supported nickel catalysts [12–15] have shown that support can play an important role on the activity and deactivation by cok- ing. Deactivation of catalysts can be attenuated or even suppressed when the metal is supported on a metal oxide with a strong Lewis basicity [13,16,17] as for example alka- line earth metal oxides, such as MgO, CaO, SrO or BaO which can act as supports [17] or structural promoters [18]. On the other hand, activated carbon (AC) as catalytic support has showed several desirable properties [19,20], e.g., low cost, high surface area, possibility to modify the pore size distribution and surface functionalities, reductive properties, as well as facile recovery of the active metals from the spent catalysts by burning off the support [21]. 0016-2361/$ - see front matter Ó 2006 Elsevier Ltd. All rights reserved. doi:10.1016/j.fuel.2006.05.011 * Corresponding author. Tel.: +58 212 5041922; fax: +58 212 5041350. E-mail address: [email protected] (J. Matos). www.fuelfirst.com Fuel 86 (2007) 1337–1344
-
Upload
independent -
Category
Documents
-
view
6 -
download
0
Transcript of Activated carbon supported Ni–Ca: Influence of reaction parameters on activity and stability of...

www.fuelfirst.com
Fuel 86 (2007) 1337–1344
Activated carbon supported Ni–Ca: Influence of reaction parameterson activity and stability of catalyst on methane reformation
Karina Dı́az, Vı́ctor Garcı́a, Juan Matos *
Laboratorio de Fisicoquı́mica de Superficies, Centro de Quı́mica, Instituto Venezolano de Investigaciones Cientı́ficas, I.V.I.C.,
Apartado 21827, Caracas 1020-A, Venezuela
Received 21 February 2006; received in revised form 9 May 2006; accepted 13 May 2006Available online 12 June 2006
Abstract
The objective of the present work was to study the influence of some reaction parameters (temperature, chemical nature of pre-treat-ment gas, mass catalyst) on the yield of methane reforming with carbon dioxide on Ni (5%)–Ca (1%)/AC at atmospheric pressure. Theobtained results show that the best system was pre-treated under He flow at 650 �C and then following the reaction at the same temper-ature. It was also detected that Ca plays a co-support role inhibiting the deactivation of catalyst at long periods of reaction. It can beconcluded that it is possible to employ activated carbon as support for methane dry reformation obtaining representative methane con-versions up to 40% at mild experimental conditions.� 2006 Elsevier Ltd. All rights reserved.
Keywords: Methane reformation; Ni–Ca catalyst; Activated carbon
1. Introduction
Venezuela has high quantities of natural gas stocks thatby 2003 were estimated around 148,000 billions of cubicfeet [1] being the first country in Latin-America and theninth in the world with more natural gas stocks. Therefore,a clear potential in the production and conversion of natu-ral gas in the country is being taken into account for thefuture years. Particularly, it is important to convert themain component of natural gas methane, in up-gradingcompounds [2,3] by means, for example, of dry reforma-tion with carbon dioxide to yield syngas as it is indicatedin reaction (1):
CH4 + CO2! 2CO + 2H2 H2/CO = 1 ð1Þ
Different H2/CO proportions of syngas have beenemployed in different important industrial processes [4–8]as methanol, dimethyl ether or Fischer–Tropsch synthesis.
0016-2361/$ - see front matter � 2006 Elsevier Ltd. All rights reserved.
doi:10.1016/j.fuel.2006.05.011
* Corresponding author. Tel.: +58 212 5041922; fax: +58 212 5041350.E-mail address: [email protected] (J. Matos).
Several catalysts have been studied [9–11] for up-gradingnatural gas. Among them, noble metals as Rh, Pt, Ruand Ir yield high activity and selectivity, but due to thelow availability and high price of such metals, nickel hasbeen explored and used as a possible substitute of noblemetals [11,12]. However, some studies have shown thatNi has a higher tendency to be deactivated than noble met-als by the deposition of carbon [9,10]. Also, studies con-cerning the dry reformation of methane on supportednickel catalysts [12–15] have shown that support can playan important role on the activity and deactivation by cok-ing. Deactivation of catalysts can be attenuated or evensuppressed when the metal is supported on a metal oxidewith a strong Lewis basicity [13,16,17] as for example alka-line earth metal oxides, such as MgO, CaO, SrO or BaOwhich can act as supports [17] or structural promoters [18].
On the other hand, activated carbon (AC) as catalyticsupport has showed several desirable properties [19,20],e.g., low cost, high surface area, possibility to modify thepore size distribution and surface functionalities, reductiveproperties, as well as facile recovery of the active metalsfrom the spent catalysts by burning off the support [21].

1338 K. Dı́az et al. / Fuel 86 (2007) 1337–1344
In some hydrotreatment reactions, as thiophene hydrodes-ulfuration [22] and ethylene hydrogenation [19,20], AC hasreceived some attention as it was earlier believed to renderhigher activities in such reactions than conventional formu-lations supported on alumina or other common supports[19–22]. However, it is very well known that active carbonhas seldom been reported as a catalytic support for meth-ane reforming reactions, principally because it can suffergasification with carbon dioxide by reverse Boudouardreaction or by steam (Eqs. (2) and (3), respectively), whereboth
C + CO2! 2CO ð2ÞC + H2O!CO + H2 ð3Þ
reactions occur spontaneously at temperature around700 �C or higher. This system has already been studied ata lower temperature such as 55 �C and at atmospheric pres-sure. It was found [23,24] that carbon supports practicallydo not suffer an important variation by means of reverseBoudouard reaction. In addition, activated carbon Ni–Casupported catalysts showed moderate but representativemethane conversion values in those mild experimental con-ditions. Having this in mind, the main objective of the pres-ent work was to study the influence of some reactionparameters (temperature, chemical nature of pre-treatmentgas, mass catalyst) on the activity and stability of an acti-vated carbon Ni–Ca supported catalyst in methane dry ref-ormation and compared it to the one obtained on othercatalytic support as c-alumina.
2. Experimental
2.1. Catalyst preparation
The catalyst employed in this study Ni (5%)–Ca (1%)/AC was synthesized by incipient wetness impregnation ofan activated carbon (AC) of commercial origin [(Merck),Langmuir surface area 907 m2 g�1, around 90% of micro-pore surface, ash content <1 wt%] with nickel and calciumnitrate solutions, heating on a stirring plate at 80 �C toapparent dryness and drying at 120 �C in static air ovenfor 3 h. The order of impregnation is given by the sequenceof elements, i.e., Ni followed of Ca. This catalyst was cho-sen because it has shown the best activity in previous stud-ies [23,24]. The catalyst was not calcined, but forconvenience in the comparison with conventional alu-mina-supported catalyst the composition is reported asthe calculated content of NiO (5%) and CaO (1%) and itwas labeled Ni–Ca/AC.
2.2. Catalyst characterization
Homogeneity in the preparation of catalyst was verifiedfrom gravimetric analysis of ashes obtained after submit-ting the catalyst to the pyrolysis of carbonaceous materialat 550 �C for 16 h in a ceramic oven (muffle) from Heraeus
following a standard test [23]. Catalyst composition waschecked from these ashes by inductively coupled plasma-atomic emission (ICP) in a Perkin–Elmer Optima 3000apparatus [23,24]. Characterization was performed by X-ray photoelectron spectroscopy (XPS) and by N2 adsorp-tion isotherms at 77 K. XPS was employed to determinethe nature of the chemical species on the surface of samplesun-pretreated, pre-treated and after the catalytic test. Thiswas carried out by means of an ESCALAB 220i-XL spec-trometer (VG scientific) equipped with a hemisphericalelectron analyzer and a double anode Mg–Al non-mono-chromatic X-ray source. The pressure in the analysischamber was kept below 10–9 Torr. Pre-treated and post-reaction samples were protected from exposition to theatmosphere by immersion into a hydrocarbon solvent(purified heptanes) while transferring from the reactor tothe preparation chamber of the spectrometer. Texturalcharacterization of catalysts was performed by means ofN2 adsorption at 77 K in order to obtain the Langmuir sur-face areas. The full isotherms in the range of 0.03 up to630 Torr were measured employing a MicromeriticsASAP-2010 apparatus. Other experimental conditions intextural characterization have already been reported bysome of us [19,20,25].
2.3. Catalyst pre-treatments and catalytic test
Samples were submitted to in situ pre-treatments beforeactivity test under helium or hydrogen flow (50 mL/min,14.5 psi pressure) starting at room temperature up to550–800 �C with a delta of 50 �C using a heating rate of10 �C/min. Time of pre-treatment (60 min) was countedfrom the moment the final temperature was reached. Activ-ity was followed in a continuous flow system (quartz reac-tor) flowing CH4 and CO2 at nearly atmospheric pressure(14.5 psi). Methane was purchased from Matheson(UHP) and CO2 from BOC (purity higher than 99.9%).Analysis of CH4 was carried out with gas chromatography(Varian 3700 GC apparatus) with FID detection employinga Porapak R packed column at 50 �C. The reaction condi-tions employed were: 100–200 g of catalyst sample; 80 mL/min flow of both CH4 and CO2, and the reaction tempera-ture was from 550 to 800 �C (similar than that of pre-treat-ments). In the present conditions CH4 could only bedetected in the reactor gaseous stream off; therefore, cata-lytic activity is reported as percent methane conversion asa function of time. Some activity tests were done by dupli-cates or triplicate, the reproducibility of results being betterthan 5%.
3. Results and discussion
3.1. Characterization of catalyst
Table 1 lists the gravimetric and ICP chemical analysisof catalyst. Ash values obtained from the pyrolysis of threesamples indicate a good homogeneity of catalyst prepared.
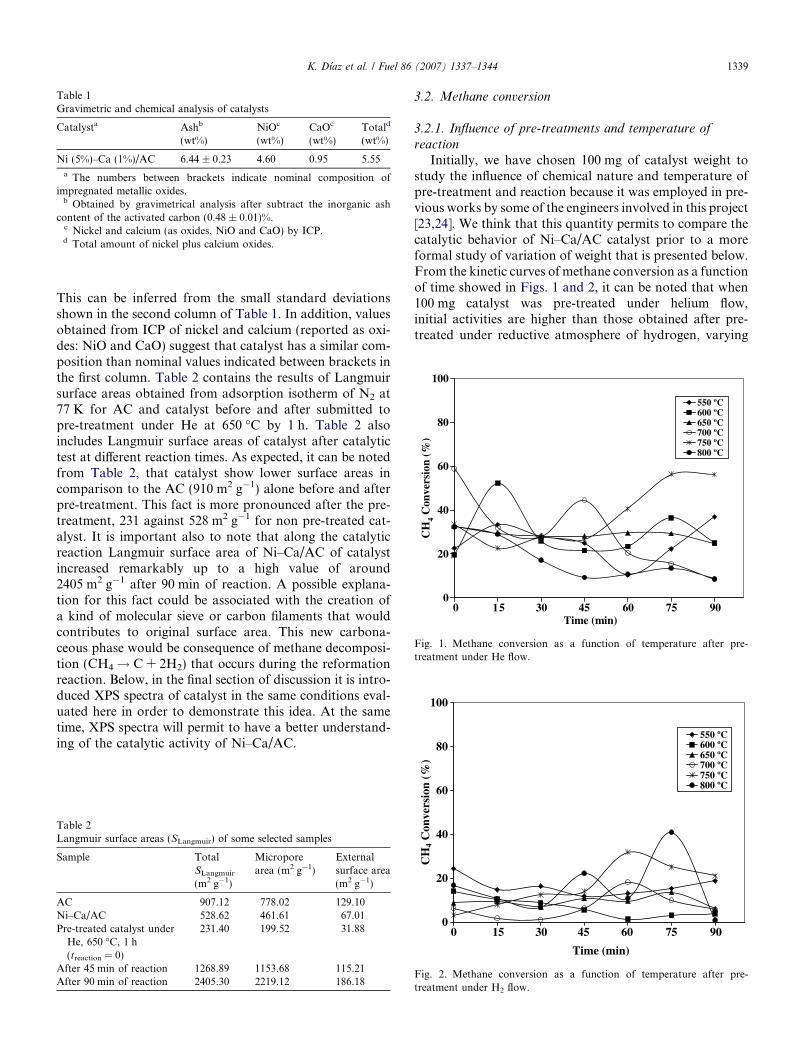
Table 1Gravimetric and chemical analysis of catalysts
Catalysta Ashb
(wt%)NiOc
(wt%)CaOc
(wt%)Totald
(wt%)
Ni (5%)–Ca (1%)/AC 6.44 ± 0.23 4.60 0.95 5.55
a The numbers between brackets indicate nominal composition ofimpregnated metallic oxides.
b Obtained by gravimetrical analysis after subtract the inorganic ashcontent of the activated carbon (0.48 ± 0.01)%.
c Nickel and calcium (as oxides, NiO and CaO) by ICP.d Total amount of nickel plus calcium oxides.
0
20
40
60
80
100
0 15 30 45 60 75 Time (min)
CH
4 C
onve
rsio
n (%
)
550 ºC600 ºC650 ºC700 ºC750 ºC800 ºC
90
Fig. 1. Methane conversion as a function of temperature after pre-treatment under He flow.
80
100
550 ºC600 ºC
K. Dı́az et al. / Fuel 86 (2007) 1337–1344 1339
This can be inferred from the small standard deviationsshown in the second column of Table 1. In addition, valuesobtained from ICP of nickel and calcium (reported as oxi-des: NiO and CaO) suggest that catalyst has a similar com-position than nominal values indicated between brackets inthe first column. Table 2 contains the results of Langmuirsurface areas obtained from adsorption isotherm of N2 at77 K for AC and catalyst before and after submitted topre-treatment under He at 650 �C by 1 h. Table 2 alsoincludes Langmuir surface areas of catalyst after catalytictest at different reaction times. As expected, it can be notedfrom Table 2, that catalyst show lower surface areas incomparison to the AC (910 m2 g�1) alone before and afterpre-treatment. This fact is more pronounced after the pre-treatment, 231 against 528 m2 g�1 for non pre-treated cat-alyst. It is important also to note that along the catalyticreaction Langmuir surface area of Ni–Ca/AC of catalystincreased remarkably up to a high value of around2405 m2 g�1 after 90 min of reaction. A possible explana-tion for this fact could be associated with the creation ofa kind of molecular sieve or carbon filaments that wouldcontributes to original surface area. This new carbona-ceous phase would be consequence of methane decomposi-tion (CH4! C + 2H2) that occurs during the reformationreaction. Below, in the final section of discussion it is intro-duced XPS spectra of catalyst in the same conditions eval-uated here in order to demonstrate this idea. At the sametime, XPS spectra will permit to have a better understand-ing of the catalytic activity of Ni–Ca/AC.
Table 2Langmuir surface areas (SLangmuir) of some selected samples
Sample TotalSLangmuir
(m2 g�1)
Microporearea (m2 g�1)
Externalsurface area(m2 g�1)
AC 907.12 778.02 129.10Ni–Ca/AC 528.62 461.61 67.01Pre-treated catalyst under
He, 650 �C, 1 h(treaction = 0)
231.40 199.52 31.88
After 45 min of reaction 1268.89 1153.68 115.21After 90 min of reaction 2405.30 2219.12 186.18
3.2. Methane conversion
3.2.1. Influence of pre-treatments and temperature of
reaction
Initially, we have chosen 100 mg of catalyst weight tostudy the influence of chemical nature and temperature ofpre-treatment and reaction because it was employed in pre-vious works by some of the engineers involved in this project[23,24]. We think that this quantity permits to compare thecatalytic behavior of Ni–Ca/AC catalyst prior to a moreformal study of variation of weight that is presented below.From the kinetic curves of methane conversion as a functionof time showed in Figs. 1 and 2, it can be noted that when100 mg catalyst was pre-treated under helium flow,initial activities are higher than those obtained after pre-treated under reductive atmosphere of hydrogen, varying
0
20
40
60
0 15 30 45 60 75 90
Time (min)
CH
4 C
onve
rsio
n (%
) 650 ºC700 ºC750 ºC800 ºC
Fig. 2. Methane conversion as a function of temperature after pre-treatment under H2 flow.
1340 K. Dı́az et al. / Fuel 86 (2007) 1337–1344
from 40% to 60% (Fig. 1) against 5% to 20% (Fig. 2).This is a general trend for most levels of temperature studiedalong the reaction time. This clear difference in catalyticactivity of Ni–Ca/AC can be attributed to the formationof nickel carbide [23,24] that will be discussed below in thegeneral discussion Section 3.3. In addition, Figs. 1 and 2clearly show that samples are not in steady-state conditionsand in most of the cases studied the catalytic trends lookvery unstable, excepting when catalyst was pre-treatedunder He flow at 650 �C. In this case, Ni–Ca/AC developeda great stability along the studied reaction time studied. Itis also important to note from Figs 1 and 2 an increasingtrend for methane conversion at 750 �C with reaction timefor catalyst pre-treated under He and H2, particularly inthe early case where after 90 min of reaction the catalystdevelops some stability. This increase in activity at 750 �Ccould be associated with the occurrence of a paralleltransformation as reverse Boudouard reaction (C +CO2! 2CO). It is logical to take into account this reactionbecause it is spontaneous at temperatures around 700 �C[26]. This reaction would help to increase methane catalytictransformation only if carbon deposits produced frommethane disproportion react preferentially with carbondioxide instead of the catalytic support. This is becausedecreases of surface area in activated carbon as a conse-quence of reverse Boudouard reaction or by hydrogen gas-ification of support (in the case of H2 pre-treatment ofcatalyst) would affect the activity of catalyst. The influenceof both reactions was verified testing the reactivity of CO2
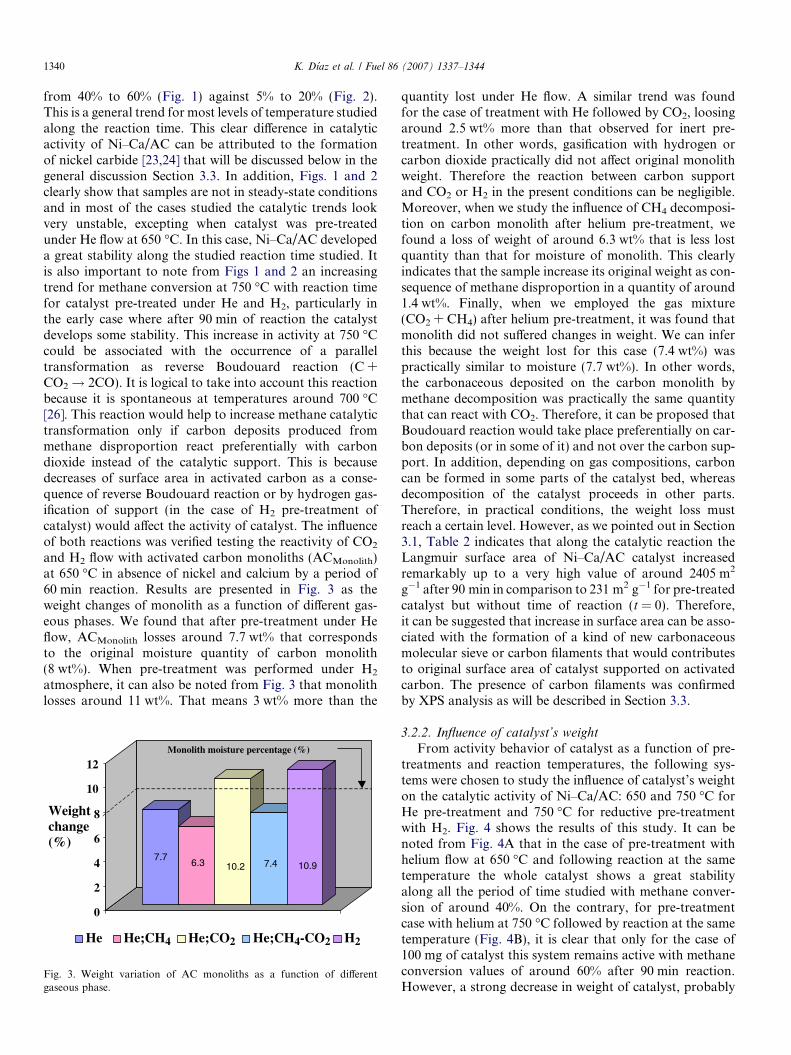
and H2 flow with activated carbon monoliths (ACMonolith)at 650 �C in absence of nickel and calcium by a period of60 min reaction. Results are presented in Fig. 3 as theweight changes of monolith as a function of different gas-eous phases. We found that after pre-treatment under Heflow, ACMonolith losses around 7.7 wt% that correspondsto the original moisture quantity of carbon monolith(8 wt%). When pre-treatment was performed under H2
atmosphere, it can also be noted from Fig. 3 that monolithlosses around 11 wt%. That means 3 wt% more than the
7.76.3 10.2 7.4 10.9
0
2
4
6
8
10
12
He He;CH4 He;CO2 He;CH4-CO2 H2
Monolith moisture percentage (%)
Weightchange(%)
Fig. 3. Weight variation of AC monoliths as a function of differentgaseous phase.
quantity lost under He flow. A similar trend was foundfor the case of treatment with He followed by CO2, loosingaround 2.5 wt% more than that observed for inert pre-treatment. In other words, gasification with hydrogen orcarbon dioxide practically did not affect original monolithweight. Therefore the reaction between carbon supportand CO2 or H2 in the present conditions can be negligible.Moreover, when we study the influence of CH4 decomposi-tion on carbon monolith after helium pre-treatment, wefound a loss of weight of around 6.3 wt% that is less lostquantity than that for moisture of monolith. This clearlyindicates that the sample increase its original weight as con-sequence of methane disproportion in a quantity of around1.4 wt%. Finally, when we employed the gas mixture(CO2 + CH4) after helium pre-treatment, it was found thatmonolith did not suffered changes in weight. We can inferthis because the weight lost for this case (7.4 wt%) waspractically similar to moisture (7.7 wt%). In other words,the carbonaceous deposited on the carbon monolith bymethane decomposition was practically the same quantitythat can react with CO2. Therefore, it can be proposed thatBoudouard reaction would take place preferentially on car-bon deposits (or in some of it) and not over the carbon sup-port. In addition, depending on gas compositions, carboncan be formed in some parts of the catalyst bed, whereasdecomposition of the catalyst proceeds in other parts.Therefore, in practical conditions, the weight loss mustreach a certain level. However, as we pointed out in Section3.1, Table 2 indicates that along the catalytic reaction theLangmuir surface area of Ni–Ca/AC catalyst increasedremarkably up to a very high value of around 2405 m2
g�1 after 90 min in comparison to 231 m2 g�1 for pre-treatedcatalyst but without time of reaction (t = 0). Therefore,it can be suggested that increase in surface area can be asso-ciated with the formation of a kind of new carbonaceousmolecular sieve or carbon filaments that would contributesto original surface area of catalyst supported on activatedcarbon. The presence of carbon filaments was confirmedby XPS analysis as will be described in Section 3.3.
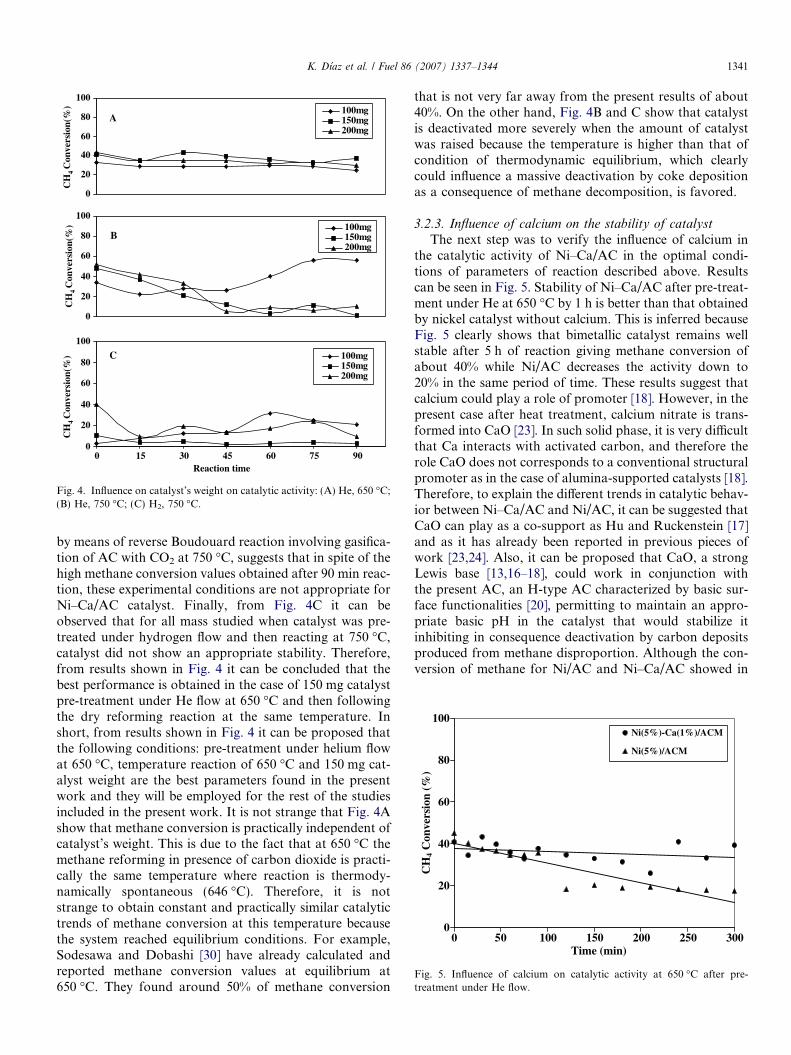
3.2.2. Influence of catalyst’s weight
From activity behavior of catalyst as a function of pre-treatments and reaction temperatures, the following sys-tems were chosen to study the influence of catalyst’s weighton the catalytic activity of Ni–Ca/AC: 650 and 750 �C forHe pre-treatment and 750 �C for reductive pre-treatmentwith H2. Fig. 4 shows the results of this study. It can benoted from Fig. 4A that in the case of pre-treatment withhelium flow at 650 �C and following reaction at the sametemperature the whole catalyst shows a great stabilityalong all the period of time studied with methane conver-sion of around 40%. On the contrary, for pre-treatmentcase with helium at 750 �C followed by reaction at the sametemperature (Fig. 4B), it is clear that only for the case of100 mg of catalyst this system remains active with methaneconversion values of around 60% after 90 min reaction.However, a strong decrease in weight of catalyst, probably
0
20
40
60
80
100
CH
4 C
onve
rsio
n(%
)C
H4
Con
vers
ion(
%)
100mg150mg200mg
0
20
40
60
80
100100mg150mg200mg
0
20
40
60
80
100
0 15 30 45 60 75 90
CH
4 C
onve
rsio
n(%
) 100mg150mg200mg
C
A
B
Reaction time
Fig. 4. Influence on catalyst’s weight on catalytic activity: (A) He, 650 �C;(B) He, 750 �C; (C) H2, 750 �C.
0
20
40
60
80
100
0 50 100 150 200 250 300Time (min)
CH
4 C
onve
rsio
n (%
)
Ni(5%)-Ca(1%)/ACM
Ni(5%)/ACM
Fig. 5. Influence of calcium on catalytic activity at 650 �C after pre-treatment under He flow.
K. Dı́az et al. / Fuel 86 (2007) 1337–1344 1341
by means of reverse Boudouard reaction involving gasifica-tion of AC with CO2 at 750 �C, suggests that in spite of thehigh methane conversion values obtained after 90 min reac-tion, these experimental conditions are not appropriate forNi–Ca/AC catalyst. Finally, from Fig. 4C it can beobserved that for all mass studied when catalyst was pre-treated under hydrogen flow and then reacting at 750 �C,catalyst did not show an appropriate stability. Therefore,from results shown in Fig. 4 it can be concluded that thebest performance is obtained in the case of 150 mg catalystpre-treatment under He flow at 650 �C and then followingthe dry reforming reaction at the same temperature. Inshort, from results shown in Fig. 4 it can be proposed thatthe following conditions: pre-treatment under helium flowat 650 �C, temperature reaction of 650 �C and 150 mg cat-alyst weight are the best parameters found in the presentwork and they will be employed for the rest of the studiesincluded in the present work. It is not strange that Fig. 4Ashow that methane conversion is practically independent ofcatalyst’s weight. This is due to the fact that at 650 �C themethane reforming in presence of carbon dioxide is practi-cally the same temperature where reaction is thermody-namically spontaneous (646 �C). Therefore, it is notstrange to obtain constant and practically similar catalytictrends of methane conversion at this temperature becausethe system reached equilibrium conditions. For example,Sodesawa and Dobashi [30] have already calculated andreported methane conversion values at equilibrium at650 �C. They found around 50% of methane conversion
that is not very far away from the present results of about40%. On the other hand, Fig. 4B and C show that catalystis deactivated more severely when the amount of catalystwas raised because the temperature is higher than that ofcondition of thermodynamic equilibrium, which clearlycould influence a massive deactivation by coke depositionas a consequence of methane decomposition, is favored.
3.2.3. Influence of calcium on the stability of catalyst
The next step was to verify the influence of calcium inthe catalytic activity of Ni–Ca/AC in the optimal condi-tions of parameters of reaction described above. Resultscan be seen in Fig. 5. Stability of Ni–Ca/AC after pre-treat-ment under He at 650 �C by 1 h is better than that obtainedby nickel catalyst without calcium. This is inferred becauseFig. 5 clearly shows that bimetallic catalyst remains wellstable after 5 h of reaction giving methane conversion ofabout 40% while Ni/AC decreases the activity down to20% in the same period of time. These results suggest thatcalcium could play a role of promoter [18]. However, in thepresent case after heat treatment, calcium nitrate is trans-formed into CaO [23]. In such solid phase, it is very difficultthat Ca interacts with activated carbon, and therefore therole CaO does not corresponds to a conventional structuralpromoter as in the case of alumina-supported catalysts [18].Therefore, to explain the different trends in catalytic behav-ior between Ni–Ca/AC and Ni/AC, it can be suggested thatCaO can play as a co-support as Hu and Ruckenstein [17]and as it has already been reported in previous pieces ofwork [23,24]. Also, it can be proposed that CaO, a strongLewis base [13,16–18], could work in conjunction withthe present AC, an H-type AC characterized by basic sur-face functionalities [20], permitting to maintain an appro-priate basic pH in the catalyst that would stabilize itinhibiting in consequence deactivation by carbon depositsproduced from methane disproportion. Although the con-version of methane for Ni/AC and Ni–Ca/AC showed in
0
20
40
60
80
100
0 15 30 45 60 75 90Time (min)
CH
4 C
onve
rsio
n (%
)
Ni(5%)/Al2O3 sin calcinar
Ni(5%)/Al2O3 calcinado
Ni(5%)/ACM
Fig. 6. Influence of support on methane conversion at 650 �C after heliumpre-treatment.
1342 K. Dı́az et al. / Fuel 86 (2007) 1337–1344
Fig. 5 did not change linearly, this figure clearly show thestability of the catalyst in presence of calcium. However,
0
20000
40000
60000
80000
100000
120000
140000
286288290Binding
286288290Binding
286288290Binding
Inte
nsit
y (u
.a)
0
20000
40000
60000
80000
100000
120000
140000
Inte
nsit
y (u
.a)
Ether286.40 eV
0
20000
40000
60000
80000
100000
120000
140000
Inte
nsit
y (u
.a)
Ether286.40 eV
B
C
A
Ether286.40
Fig. 7. XPS spectra in the C 1s region of Ni–Ca/AC catalyst: (A) After pre-treaafter 90 min of reaction at 650 �C.
in spite of catalyst in presence of calcium (Fig. 5) may seemstable in short period of time (5 h) a study about stability inlonger period of time must be performed as well as a com-parison of it with other kind of catalyst as for example alu-mina-supported nickel shown in Fig. 6 in the followingsection for much shorter period of time.
3.2.4. Influence of support
In order to verify the potential of the present system inthe dry reformation of methane, three different catalysts (inabsence of calcium) were studied at the optimal conditionsdescribed before: a non-calcined Ni (5%)/AC, Ni (5%) cat-alysts supported on a commercial c-Al2O3, denoted by Ni(5%)/c-Al2O3, and calcined or not at 550 �C by 16 h inan oven (muffle) under atmospheric static air. This compar-ison is shown in Fig. 6. It is remarkable that activated car-bon looks as the best support for nickel in the present mildconditions for dry reforming of methane (pre-treatment ofcatalysts under helium flow at 650 �C and reaction at thesame temperature of pre-treatment at atmospheric pres-sure). This can be inferred because along the reaction time
280282284 Energy (eV)
280282284 Energy (eV)
280282284 Energy (eV)
C-C284.65 eV
Ni3C283.82
CarbonFilaments282.80 eV
C-C284.78 eV
Ni3C283.90 eV
CarbonFilaments282.60 eV
C-C284.68
Ni3C283.9
ted under 650 �C He flow (t = 0); (B) after 45 min of reaction at 650 �C; (C)
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
850855860865870875880885890Binding Energy (eV)
Inte
nsit
y (u
.a.)
non pre-treatment P-T45minP-R
855.8
861.8
873.3
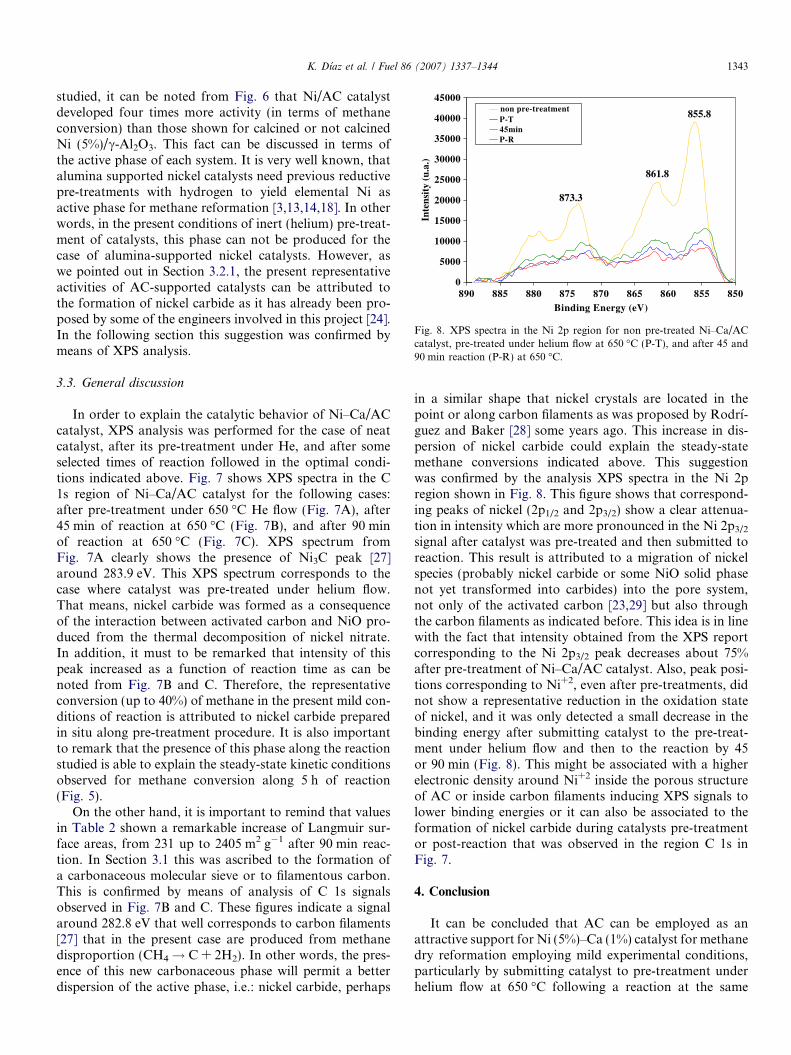
Fig. 8. XPS spectra in the Ni 2p region for non pre-treated Ni–Ca/ACcatalyst, pre-treated under helium flow at 650 �C (P-T), and after 45 and90 min reaction (P-R) at 650 �C.
K. Dı́az et al. / Fuel 86 (2007) 1337–1344 1343
studied, it can be noted from Fig. 6 that Ni/AC catalystdeveloped four times more activity (in terms of methaneconversion) than those shown for calcined or not calcinedNi (5%)/c-Al2O3. This fact can be discussed in terms ofthe active phase of each system. It is very well known, thatalumina supported nickel catalysts need previous reductivepre-treatments with hydrogen to yield elemental Ni asactive phase for methane reformation [3,13,14,18]. In otherwords, in the present conditions of inert (helium) pre-treat-ment of catalysts, this phase can not be produced for thecase of alumina-supported nickel catalysts. However, aswe pointed out in Section 3.2.1, the present representativeactivities of AC-supported catalysts can be attributed tothe formation of nickel carbide as it has already been pro-posed by some of the engineers involved in this project [24].In the following section this suggestion was confirmed bymeans of XPS analysis.
3.3. General discussion
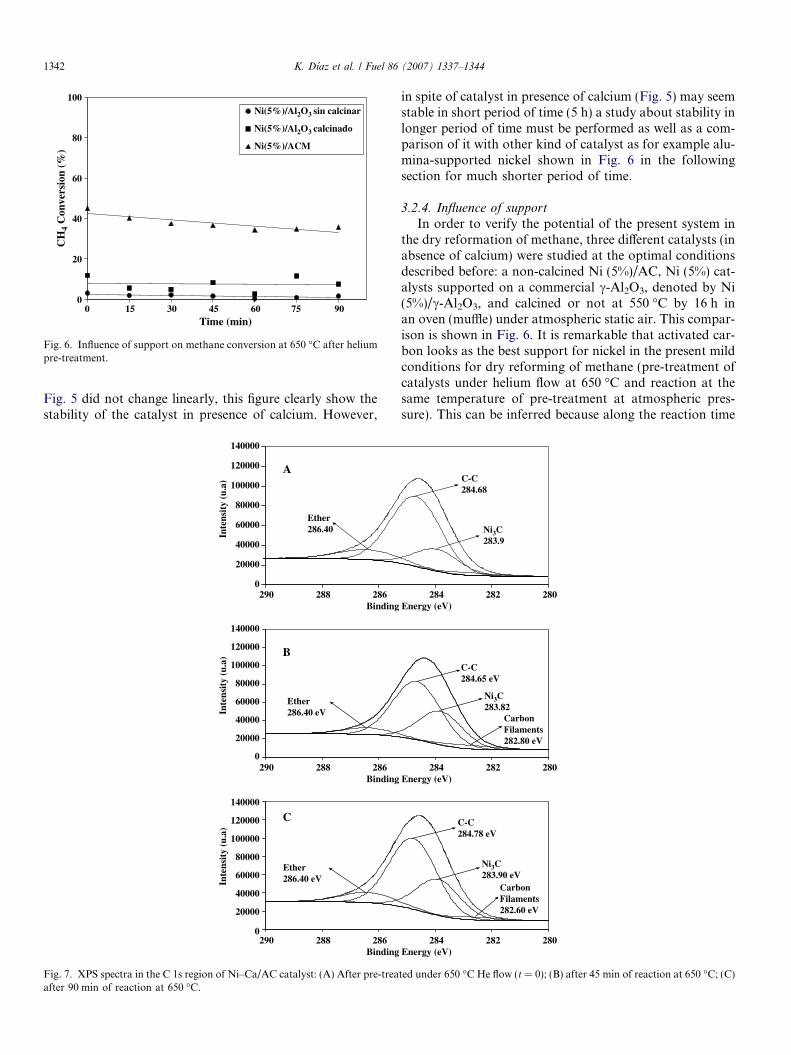
In order to explain the catalytic behavior of Ni–Ca/ACcatalyst, XPS analysis was performed for the case of neatcatalyst, after its pre-treatment under He, and after someselected times of reaction followed in the optimal condi-tions indicated above. Fig. 7 shows XPS spectra in the C1s region of Ni–Ca/AC catalyst for the following cases:after pre-treatment under 650 �C He flow (Fig. 7A), after45 min of reaction at 650 �C (Fig. 7B), and after 90 minof reaction at 650 �C (Fig. 7C). XPS spectrum fromFig. 7A clearly shows the presence of Ni3C peak [27]around 283.9 eV. This XPS spectrum corresponds to thecase where catalyst was pre-treated under helium flow.That means, nickel carbide was formed as a consequenceof the interaction between activated carbon and NiO pro-duced from the thermal decomposition of nickel nitrate.In addition, it must to be remarked that intensity of thispeak increased as a function of reaction time as can benoted from Fig. 7B and C. Therefore, the representativeconversion (up to 40%) of methane in the present mild con-ditions of reaction is attributed to nickel carbide preparedin situ along pre-treatment procedure. It is also importantto remark that the presence of this phase along the reactionstudied is able to explain the steady-state kinetic conditionsobserved for methane conversion along 5 h of reaction(Fig. 5).
On the other hand, it is important to remind that valuesin Table 2 shown a remarkable increase of Langmuir sur-face areas, from 231 up to 2405 m2 g�1 after 90 min reac-tion. In Section 3.1 this was ascribed to the formation ofa carbonaceous molecular sieve or to filamentous carbon.This is confirmed by means of analysis of C 1s signalsobserved in Fig. 7B and C. These figures indicate a signalaround 282.8 eV that well corresponds to carbon filaments[27] that in the present case are produced from methanedisproportion (CH4! C + 2H2). In other words, the pres-ence of this new carbonaceous phase will permit a betterdispersion of the active phase, i.e.: nickel carbide, perhaps
in a similar shape that nickel crystals are located in thepoint or along carbon filaments as was proposed by Rodrı́-guez and Baker [28] some years ago. This increase in dis-persion of nickel carbide could explain the steady-statemethane conversions indicated above. This suggestionwas confirmed by the analysis XPS spectra in the Ni 2pregion shown in Fig. 8. This figure shows that correspond-ing peaks of nickel (2p1/2 and 2p3/2) show a clear attenua-tion in intensity which are more pronounced in the Ni 2p3/2
signal after catalyst was pre-treated and then submitted toreaction. This result is attributed to a migration of nickelspecies (probably nickel carbide or some NiO solid phasenot yet transformed into carbides) into the pore system,not only of the activated carbon [23,29] but also throughthe carbon filaments as indicated before. This idea is in linewith the fact that intensity obtained from the XPS reportcorresponding to the Ni 2p3/2 peak decreases about 75%after pre-treatment of Ni–Ca/AC catalyst. Also, peak posi-tions corresponding to Ni+2, even after pre-treatments, didnot show a representative reduction in the oxidation stateof nickel, and it was only detected a small decrease in thebinding energy after submitting catalyst to the pre-treat-ment under helium flow and then to the reaction by 45or 90 min (Fig. 8). This might be associated with a higherelectronic density around Ni+2 inside the porous structureof AC or inside carbon filaments inducing XPS signals tolower binding energies or it can also be associated to theformation of nickel carbide during catalysts pre-treatmentor post-reaction that was observed in the region C 1s inFig. 7.
4. Conclusion
It can be concluded that AC can be employed as anattractive support for Ni (5%)–Ca (1%) catalyst for methanedry reformation employing mild experimental conditions,particularly by submitting catalyst to pre-treatment underhelium flow at 650 �C following a reaction at the same

1344 K. Dı́az et al. / Fuel 86 (2007) 1337–1344
temperature. In these optimal reaction conditions Ni–Ca/AC catalyst develops methane conversions as high as 40%and this yield for methane reforming was maintained insteady-state kinetic regimen at least by along 5 h of reaction.
Acknowledgements
The authors acknowledge Mary Labady and YraidaDı́az for technical support in Surface Area analysis andXPS spectra, respectively.
References
[1] Oil Gas J 2004;102:47.[2] Tomishige K. Catal Today 2004;89:405.[3] Bradford M, Vannice M. Catal Rev—Sci Eng 1999;41:1.[4] Bibby DM, Chang CD, Howe RF, Yurchak S. In: Delmon B, Yates
Jr JT, editors. Studies on surface science catalysis, vol. 36. Amster-dam: Elsevier; 1988.
[5] Ayers WM, editor. Proceedings of the ACS symposium series. Wash-ington (DC): ACS; 1988.
[6] Rostrup-Nielsen JR. Catal Today 1994;21:257.[7] Aasberg-Petersen K, Bak Hansen JH, Christensen TS, Dybkjaer I,
Seier-Christensen P, Stub-Nielsen C, Winter-Madsen SEL, Rostrup-Nielsen JR. Appl Catal A 2001;221:379.
[8] Fujimoto K. J Jpn Assoc Petrol Technol 2001;66:152.
[9] Richardson JT, Paripatyadar SA. Appl Catal 1990;61:293.[10] Ashcroft AT, Cheetham AK, Green MLH, Vernon PDF. Nature
1991;352:225.[11] Rostrup-Nielsen JR, Bak Hansen JH. J Catal 1993;144:38.[12] Zhang Z, Verykios XE. Appl Catal A 1996;138:109.[13] Bradford MCJ, Vannice MA. Appl Catal A 1996;142:73.[14] Gadalla AM, Sommer ME. J Am Ceram Soc 1989;72:683.[15] Chen Y, Ren J. Catal Lett 1994;29:39.[16] Chen TG, Yamazaki O, Tomoshige K, Fujimoto K. Catal Lett
1996;39:91.[17] Hu YH, Ruckenstein E. Catal Lett 1996;36:145.[18] Dias JAC, Assaf JM. Catal Today 2003;85:59.[19] Matos J, Brito JL, Laine J. Appl Catal A 1997;152:27.[20] Matos J, Laine J. Appl Catal A 2003;241:25.[21] Abotzi GMK, Scaroni AW. Fuel Process Technol 1989;22:107.[22] Duchet JC, van Oers EM, de Beer VHJ, Prins R. J Catal 1983;80:386.[23] Matos J, Dı́az K, Garcı́a V, Actas XIX SICAT, Mexico, 2004. p.
1391.[24] Matos J, Dı́az K, Garcı́a V, Cordero TC, Brito JL. Catal Lett, in
press.[25] Laine J, Labady M, Severino F, Yunes S. J Catal 1997;166:384.[26] Cheng Y, Yamazaki O, Tomishige K, Fujimoto K. Catal Lett
1996;39:91.[27] Kroll CH, Delichure P, Mirodatos C. Kinet Catal 1996;37:698.[28] Rodrı́guez NM, Baker RTK. J Catal 1993;140:16.[29] Dı́az K, Garcı́a V, BSc, Thesis, Venezuelan Central University,
Chemistry Engineering Faculty, October 2005.[30] Sodesawa T, Dobashi A. React Kinet Catal Lett 1979;12:107.



![A reformáció nyelve [The Language of the Reformation]](https://static.fdokumen.com/doc/165x107/63143f8a3ed465f0570b0e70/a-reformacio-nyelve-the-language-of-the-reformation.jpg)
















