
Abnormal hepatobiliary and circulating lipid metabolism in the Long-Evans Cinnamon rat model of...
12
Abnormal hepatobiliary and circulating lipid metabolism in the Long-Evans Cinnamon rat model of Wilson's disease Emile Levy a, ⁎ , Sylvain Brunet a , Fernando Alvarez b , Ernest Seidman b , Guylaine Bouchard a , Enrique Escobar b , Steve Martin b a Department of Nutrition, CHU Sainte-Justine, Université de Montréal, 3175 Côte Sainte Catherine, Montréal, Québec, Canada H3T 1C5 b Department of Pediatrics, CHU Sainte-Justine, Université de Montréal, 3175 Côte Sainte Catherine, Montréal, Québec, Canada H3T 1C5 Received 9 June 2006; accepted 8 January 2007 Abstract Long-Evans Cinnamon (LEC) rats exhibit a genetic defect in Atp7b gene, which is homologous to the human Wilson's disease gene, resulting in an inability to mobilize copper from the liver. This study was undertaken to gain insight into the relationship between liver copper accumulation and plasma lipid profile, circulating lipoprotein composition, hepatic sterol metabolism and biliary lipid secretion rates in 12-week-old LEC rats compared to control Long-Evans rats. Concomitant with hepatic copper deposition, LEC rats displayed increased content of triglycerides (TGs), free cholesterol (FC) and cholesteryl ester (CE) in the liver. Hepatic concentrations of malondialdehyde (MDA), an index of lipid peroxidation were also significantly elevated in LEC rats (50%). This steatosis was associated with aberrant microsomal apolipoprotein (apo) B-100 and microsomal triglyceride transfer protein (MTP) content, hypotriglyceridemia, hypocholesterolemia and abnormalities in both circulating lipoprotein composition and size. Atypical hepatobiliary sterol metabolism was established by the assessment of the activity of key intracellular enzymes for cholesterol homeostasis, which demonstrated, with respect to controls, a 40% reduction in 3-hydroxy-3-methylglutaryl coenzyme A reductase, a 30% reduction in cholesterol 7α-hydroxylase, and a 54% reduction in acyl CoA:cholesterol acyltransferase. During a 6-h biliary drainage, a decline in the bile acid output was recorded and might be linked to the low protein expression of the bile salt export pump (BSEP or ABCB11). Our data emphasize the crucial role of copper balance in hepatic sterol homeostasis and lipoprotein metabolism in LEC rats. Additional studies are needed to delineate the mechanisms of these disorders. © 2007 Elsevier Inc. All rights reserved. Keywords: Lipid peroxidation; Sterol metabolism; Liver copper; MTP; Apo B Introduction Wilson's disease is an autosomal recessive disorder that is characterized by defective hepatic copper transport resulting in the accumulation of copper in the liver, kidney and central nervous system (Sternlieb, 2000). The excessive deposition of copper leads to hepatic, neuropsychiatric and other clinical manifestations. A wide variety of mutations in the copper- transporting P-type adenosine tri-phosphatase (ATP7B) gene are responsible for defective hepatic copper excretion in Wilson's disease (Bacon and Schilsky, 1999). The gene was cloned in 1993 (Bull et al., 1993), and its ATP7B product localizes to the trans-Golgi network or late endosomes under basal conditions (Hung et al., 1997; Harada et al., 2000), with a copper- dependent redistribution to an apical vesicular compartment (Schaefer et al., 1999; Roelofsen et al., 2000). This localization permits both copper incorporation into ceruloplasmin and may facilitate biliary copper excretion via lysosomes (Harada et al., 2000). The liver plays a central role in lipid and, particularly, cholesterol homeostasis (Turley and Dietschy, 1988). It constitutes the major site of lipogenesis and the production of very low density lipoprotein (VLDL), as well as a portion of high-density lipoproteins (HDL). In addition, the liver takes up and degrades various types of lipoproteins (Cooper, 1985; Wanon et al., 1998). Within hepatocytes, cholesterol is converted into bile acids; the biliary secretion of cholesterol, Life Sciences 80 (2007) 1472 – 1483 www.elsevier.com/locate/lifescie ⁎ Corresponding author. Centre de Recherche, CHU Sainte-Justine, 3175 Côte Ste-Catherine, Montréal, Québec, Canada H3T 1C5. Tel.: +1 514 345 7783; fax: +1 514 345 4999. E-mail address: [email protected] (E. Levy). 0024-3205/$ - see front matter © 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.lfs.2007.01.017
Transcript of Abnormal hepatobiliary and circulating lipid metabolism in the Long-Evans Cinnamon rat model of...

7) 1472–1483www.elsevier.com/locate/lifescie
Life Sciences 80 (200
Abnormal hepatobiliary and circulating lipid metabolism in the Long-EvansCinnamon rat model of Wilson's disease
Emile Levy a,⁎, Sylvain Brunet a, Fernando Alvarez b, Ernest Seidman b, Guylaine Bouchard a,Enrique Escobar b, Steve Martin b
a Department of Nutrition, CHU Sainte-Justine, Université de Montréal, 3175 Côte Sainte Catherine, Montréal, Québec, Canada H3T 1C5b Department of Pediatrics, CHU Sainte-Justine, Université de Montréal, 3175 Côte Sainte Catherine, Montréal, Québec, Canada H3T 1C5
Received 9 June 2006; accepted 8 January 2007
Abstract
Long-Evans Cinnamon (LEC) rats exhibit a genetic defect in Atp7b gene, which is homologous to the human Wilson's disease gene, resultingin an inability to mobilize copper from the liver. This study was undertaken to gain insight into the relationship between liver copper accumulationand plasma lipid profile, circulating lipoprotein composition, hepatic sterol metabolism and biliary lipid secretion rates in 12-week-old LEC ratscompared to control Long-Evans rats. Concomitant with hepatic copper deposition, LEC rats displayed increased content of triglycerides (TGs),free cholesterol (FC) and cholesteryl ester (CE) in the liver. Hepatic concentrations of malondialdehyde (MDA), an index of lipid peroxidationwere also significantly elevated in LEC rats (50%). This steatosis was associated with aberrant microsomal apolipoprotein (apo) B-100 andmicrosomal triglyceride transfer protein (MTP) content, hypotriglyceridemia, hypocholesterolemia and abnormalities in both circulatinglipoprotein composition and size. Atypical hepatobiliary sterol metabolism was established by the assessment of the activity of key intracellularenzymes for cholesterol homeostasis, which demonstrated, with respect to controls, a 40% reduction in 3-hydroxy-3-methylglutaryl coenzyme Areductase, a 30% reduction in cholesterol 7α-hydroxylase, and a 54% reduction in acyl CoA:cholesterol acyltransferase. During a 6-h biliarydrainage, a decline in the bile acid output was recorded and might be linked to the low protein expression of the bile salt export pump (BSEP orABCB11). Our data emphasize the crucial role of copper balance in hepatic sterol homeostasis and lipoprotein metabolism in LEC rats. Additionalstudies are needed to delineate the mechanisms of these disorders.© 2007 Elsevier Inc. All rights reserved.
Keywords: Lipid peroxidation; Sterol metabolism; Liver copper; MTP; Apo B
Introduction
Wilson's disease is an autosomal recessive disorder that ischaracterized by defective hepatic copper transport resulting inthe accumulation of copper in the liver, kidney and centralnervous system (Sternlieb, 2000). The excessive deposition ofcopper leads to hepatic, neuropsychiatric and other clinicalmanifestations. A wide variety of mutations in the copper-transporting P-type adenosine tri-phosphatase (ATP7B) gene areresponsible for defective hepatic copper excretion in Wilson'sdisease (Bacon and Schilsky, 1999). The gene was cloned in
⁎ Corresponding author. Centre de Recherche, CHU Sainte-Justine, 3175 CôteSte-Catherine, Montréal, Québec, Canada H3T 1C5. Tel.: +1 514 345 7783; fax:+1 514 345 4999.
E-mail address: [email protected] (E. Levy).
0024-3205/$ - see front matter © 2007 Elsevier Inc. All rights reserved.doi:10.1016/j.lfs.2007.01.017
1993 (Bull et al., 1993), and its ATP7B product localizes to thetrans-Golgi network or late endosomes under basal conditions(Hung et al., 1997; Harada et al., 2000), with a copper-dependent redistribution to an apical vesicular compartment(Schaefer et al., 1999; Roelofsen et al., 2000). This localizationpermits both copper incorporation into ceruloplasmin and mayfacilitate biliary copper excretion via lysosomes (Harada et al.,2000).
The liver plays a central role in lipid and, particularly,cholesterol homeostasis (Turley and Dietschy, 1988). Itconstitutes the major site of lipogenesis and the production ofvery low density lipoprotein (VLDL), as well as a portion ofhigh-density lipoproteins (HDL). In addition, the liver takes upand degrades various types of lipoproteins (Cooper, 1985;Wanon et al., 1998). Within hepatocytes, cholesterol isconverted into bile acids; the biliary secretion of cholesterol,

Table 1Characteristics of experimental groups
Gross characteristics Control LEC rat
Bilirubin (μmol/L) b1.0 19.8±6.5 a
ALT (UI/L) 45±4 756±69 b
Liver copper (μg/g of dry weight) 12.1±0.3 1943±78 b
Plasma Cu2+ (μmol/L) 19.9±0.5 15.8±1.6 c
Values are expressed as means±S.E.M. of n=6/group.a pb0.01 vs. control.b pb0.001 vs. control.c pb0.05 vs. control.
1473E. Levy et al. / Life Sciences 80 (2007) 1472–1483
either in unchanged form or as bile acids, represents the mainphysiological route of net cholesterol elimination from the body(Packard and Shepherd, 1982). Although it is well establishedthat liver injury can provoke profound abnormalities incirculating lipoproteins (Levy et al., 1995; Seidel, 1987), verylimited data are available on the detailed lipoprotein alterationsin Wilson's disease (Taniguchi et al., 1991). Moreover, littleattention has been paid to intrahepatic cholesterol metabolismand biliary sterol output despite the well-established liverdisturbances in this disorder.
Finally, copper-induced lipid peroxidation has not beenthoroughly examined in the blood circulation and hepatobiliarysystem in Wilson's disease. The purpose of the present studywas, therefore, to explore in detail the plasma lipid profile,circulating lipoprotein composition, hepatic cholesterol metab-olism, biliary sterol secretion and lipid peroxidation in theLong-Evans Cinnamon (LEC) rat. This animal model developsmany clinical and biochemical features of Wilson's disease,including liver copper accumulation, as a result of a deletion inthe rat Atp7b gene which shares 85% homology with the humanATP7B gene (Wu et al., 1994; Okayasu et al., 1992).
Materials and methods
Animals
The experiments were performed on male LEC and Long-Evans (LE) rats. The animal care and use was approved by theCommittee for Animal Care in Research Center of Sainte-Justine Hospital. The care and treatment of animals followed theguidelines outlined in the NIH “Guide for the Care and Use ofLaboratory Animals” (revised 1985) as prepared by the NationalAcademy of Sciences.
Confirmation of the nature of LEC rats was obtained byserum ceruloplasmin determinations using a modification of theo-dianisidine di-hydrochloride oxidation method (Schosinskyet al., 1974). Confirmation of the genetic defect was made byPCR amplification of a 300-nucleotide segment of exon 2(present in all rats) and a 200-nucleotide segment of exon 18(part of the region deleted in LEC rats and therefore onlydetected in normal rats). The rats, weighing ∼300–350 g, wereallowed free access to water and food and were maintained at22 °C with a 12-h light cycle. Rats were fasted overnight beforebeing used for the following studies, which included anequivalent number of males and females. The quantities of ratsemployed in the various experiments are mentioned in thelegends of the different figures.
Blood collection
Rats were anesthetized by the intraperitoneal injection ofpentobarbital (48 mg/kg body wt, i.p.) (Abbott Laboratories,Montreal). Blood was collected in an EDTA-containing tube(1 mg/mL) and separated immediately by low-speed centrifu-gation (2500 rpm, 24 min, 4 °C). For the determination ofmalondialdehyde (MDA), a fraction of plasma was immediatelyfrozen in liquid nitrogen and stored at −80 °C until analysis.
Lipoprotein isolation
Lipoproteins were isolated from fresh plasma by conven-tional discontinuous density gradient ultracentrifugation in aBeckman L5-65 preparative ultracentrifuge (Beckman Inc.,Montreal) by using a Ti-50 rotor as previously describedin detail (Levy et al., 1990). Briefly, after preliminarycentrifugation to remove chylomicrons, VLDL and low-densitylipoproteins (LDLs) were isolated at a density of 1.006 and1.063 g/mL, respectively, running at 40,000 rpm for 18 h at5 °C. The separation of HDL (1.21 g/mL) was performed at40,000 rpm for 48 h at 5 °C. Each fraction was dialyzedintensively against phosphate-buffered saline (PBS) with0.001 mol/L EDTA, pH 7.0 at 4 °C.
Plasma lipid and lipoprotein analysis
Plasma concentrations of total cholesterol (TC), freecholesterol (FC), and triglycerides (TGs) were measuredenzymatically using commercial kits (Boehringer Mannheim,Montreal, Canada) as previously described (Levy et al., 1989).Cholesteryl ester (CE) was calculated as the difference betweentotal and FC×1.7. The protein (PR) moiety of lipoproteins wasquantified according to Lowry et al. (1951) with bovine serumalbumin as a standard. Phospholipids (PLs) were determined byBartlett's (1959) method. HDL-cholesterol (HDL-C) wasmeasured after precipitating VLDL and LDL with phospho-tungstic acid (Levy et al.,1990, 1989). Electron microscopy oflipoprotein particles was performed using negative staining with1% phosphotungstic acid (pH 7.2) as described previously(Levy et al., 1989).
Hepatic lipid determination
To determine hepatic lipid content and lipid peroxidation, thelivers were promptly removed, rinsed with PBS containing3 mmol/L EDTA, blotted dry, and weighed. Special attentionwas paid to rapidly freeze aliquots in liquid nitrogen and keepthem at −80 °C until assayed. Then, a sample was homogenizedin 0.9% NaCl containing 3 mmol/L EDTA to prevent spuriouslipid peroxidation and to chelate any ionic iron that mighthave been released during tissue preparation. Aliquots ofhomogenates were lipid-extracted with chloroform–methanol(2:1, v/v), and their TG, FC, and CE were determined (Levyet al., 1992, 1996).
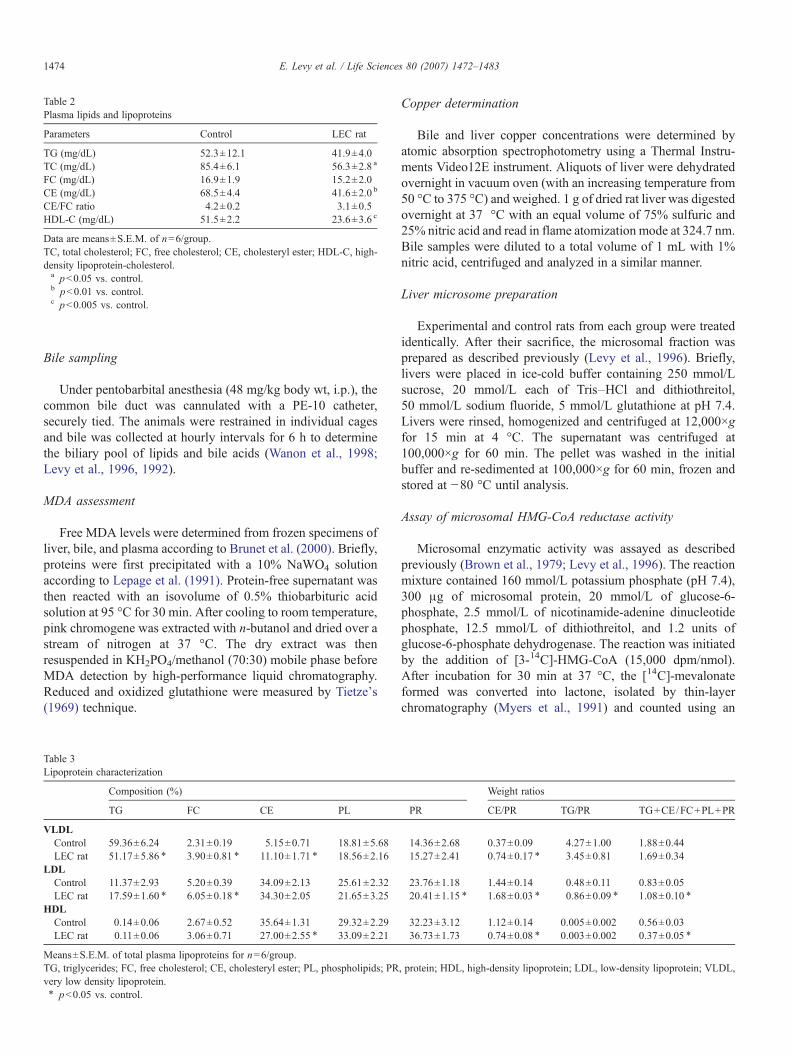
Table 2Plasma lipids and lipoproteins
Parameters Control LEC rat
TG (mg/dL) 52.3±12.1 41.9±4.0TC (mg/dL) 85.4±6.1 56.3±2.8 a
FC (mg/dL) 16.9±1.9 15.2±2.0CE (mg/dL) 68.5±4.4 41.6±2.0 b
CE/FC ratio 4.2±0.2 3.1±0.5HDL-C (mg/dL) 51.5±2.2 23.6±3.6 c
Data are means±S.E.M. of n=6/group.TC, total cholesterol; FC, free cholesterol; CE, cholesteryl ester; HDL-C, high-density lipoprotein-cholesterol.a pb0.05 vs. control.b pb0.01 vs. control.c pb0.005 vs. control.
1474 E. Levy et al. / Life Sciences 80 (2007) 1472–1483
Bile sampling
Under pentobarbital anesthesia (48 mg/kg body wt, i.p.), thecommon bile duct was cannulated with a PE-10 catheter,securely tied. The animals were restrained in individual cagesand bile was collected at hourly intervals for 6 h to determinethe biliary pool of lipids and bile acids (Wanon et al., 1998;Levy et al., 1996, 1992).
MDA assessment
Free MDA levels were determined from frozen specimens ofliver, bile, and plasma according to Brunet et al. (2000). Briefly,proteins were first precipitated with a 10% NaWO4 solutionaccording to Lepage et al. (1991). Protein-free supernatant wasthen reacted with an isovolume of 0.5% thiobarbituric acidsolution at 95 °C for 30 min. After cooling to room temperature,pink chromogene was extracted with n-butanol and dried over astream of nitrogen at 37 °C. The dry extract was thenresuspended in KH2PO4/methanol (70:30) mobile phase beforeMDA detection by high-performance liquid chromatography.Reduced and oxidized glutathione were measured by Tietze's(1969) technique.
Table 3Lipoprotein characterization
Composition (%)
TG FC CE PL
VLDLControl 59.36±6.24 2.31±0.19 5.15±0.71 18.81±5.68LEC rat 51.17±5.86 ⁎ 3.90±0.81 ⁎ 11.10±1.71 ⁎ 18.56±2.16
LDLControl 11.37±2.93 5.20±0.39 34.09±2.13 25.61±2.32LEC rat 17.59±1.60 ⁎ 6.05±0.18 ⁎ 34.30±2.05 21.65±3.25
HDLControl 0.14±0.06 2.67±0.52 35.64±1.31 29.32±2.29LEC rat 0.11±0.06 3.06±0.71 27.00±2.55 ⁎ 33.09±2.21
Means±S.E.M. of total plasma lipoproteins for n=6/group.TG, triglycerides; FC, free cholesterol; CE, cholesteryl ester; PL, phospholipids; PRvery low density lipoprotein.⁎ pb0.05 vs. control.
Copper determination
Bile and liver copper concentrations were determined byatomic absorption spectrophotometry using a Thermal Instru-ments Video12E instrument. Aliquots of liver were dehydratedovernight in vacuum oven (with an increasing temperature from50 °C to 375 °C) and weighed. 1 g of dried rat liver was digestedovernight at 37 °C with an equal volume of 75% sulfuric and25% nitric acid and read in flame atomization mode at 324.7 nm.Bile samples were diluted to a total volume of 1 mL with 1%nitric acid, centrifuged and analyzed in a similar manner.
Liver microsome preparation
Experimental and control rats from each group were treatedidentically. After their sacrifice, the microsomal fraction wasprepared as described previously (Levy et al., 1996). Briefly,livers were placed in ice-cold buffer containing 250 mmol/Lsucrose, 20 mmol/L each of Tris–HCl and dithiothreitol,50 mmol/L sodium fluoride, 5 mmol/L glutathione at pH 7.4.Livers were rinsed, homogenized and centrifuged at 12,000×gfor 15 min at 4 °C. The supernatant was centrifuged at100,000×g for 60 min. The pellet was washed in the initialbuffer and re-sedimented at 100,000×g for 60 min, frozen andstored at −80 °C until analysis.
Assay of microsomal HMG-CoA reductase activity
Microsomal enzymatic activity was assayed as describedpreviously (Brown et al., 1979; Levy et al., 1996). The reactionmixture contained 160 mmol/L potassium phosphate (pH 7.4),300 μg of microsomal protein, 20 mmol/L of glucose-6-phosphate, 2.5 mmol/L of nicotinamide-adenine dinucleotidephosphate, 12.5 mmol/L of dithiothreitol, and 1.2 units ofglucose-6-phosphate dehydrogenase. The reaction was initiatedby the addition of [3-14C]-HMG-CoA (15,000 dpm/nmol).After incubation for 30 min at 37 °C, the [14C]-mevalonateformed was converted into lactone, isolated by thin-layerchromatography (Myers et al., 1991) and counted using an
Weight ratios
PR CE/PR TG/PR TG+CE/FC+PL+PR
14.36±2.68 0.37±0.09 4.27±1.00 1.88±0.4415.27±2.41 0.74±0.17 ⁎ 3.45±0.81 1.69±0.34
23.76±1.18 1.44±0.14 0.48±0.11 0.83±0.0520.41±1.15 ⁎ 1.68±0.03 ⁎ 0.86±0.09 ⁎ 1.08±0.10 ⁎
32.23±3.12 1.12±0.14 0.005±0.002 0.56±0.0336.73±1.73 0.74±0.08 ⁎ 0.003±0.002 0.37±0.05 ⁎
, protein; HDL, high-density lipoprotein; LDL, low-density lipoprotein; VLDL,
1475E. Levy et al. / Life Sciences 80 (2007) 1472–1483
internal standard to correct for incomplete recovery. HMG-CoAreductase activity was expressed in nanomoles of mevalonatesynthesized per milligram of protein per minute.
Cholesterol 7α-hydroxylase activity
Microsomal cholesterol 7α-hydroxylase activity was mea-sured according to previous reports (Hylemon et al., 1989; Levyet al., 1996). Briefly, 1 mg of microsomal protein was diluted ina potassium phosphate buffer, pH 7.4, containing 50 mmol/LNaF, 5 mmol/L dithiothreitol, 1 mmol/L EDTA, 20% glycerol,
Fig. 1. Representative electron photomicrographs of negatively stained plasmalipoproteins. Lipoproteins were isolated by ultracentrifugation as described inMaterials and methods. Samples of VLDL, LDL and HDL were utilized forelectron microscopic size examination. The means of particle diameters in twoseparate samples of LEC and control rats were obtained after the evaluation of aminimum of 250 particles/samples of VLDL: 463±7 Å, and 458±9 Å; LDL:192±4 Å, and 233±7 Å; and HDL: 106±3 Å, and 84±6 Å, respectively.
0.015% CHAPS and 0.25 mmol/L desferroxamine. Reactionwas started by the addition of an NADPH regenerating systemcontaining 1 mmol/L NADP, 10 mmol/L glucose-6-phosphateand 0.15 UI glucose-6-phosphate dehydrogenase. Following a20-min reaction at 37 °C, the endogenous 7α-hydroxycholes-terol form was derivatized with a 0.1% cholesterol oxidasesolution. Dried petroleum ether extract was resuspended in 70%acetonitrile/30% methanol mobile phase, before its analysiswith high-performance liquid chromatography, with UVdetection at 240 nm, using 7β-hydroxycholesterol as an internalstandard. The 7α-hydroxycholesterol product was expressed aspmol/min/mg protein.
Acyl-coenzyme A:cholesterol acyltransferase (ACAT) activity
The determination of this enzyme was based on the assaydescribed previously (Levy et al., 1996). To the assay mixturecontaining 75 μg microsomal protein were added 5 μmol [14C]-oleoyl coenzyme A (specific activity∼10,000 dpm/μmol/L) toinitiate the reaction in a buffer solution (pH 7.5) consisting of0.1 mol/L of Tris–HCl, 0.25 mol/L of sucrose, and 1 mmol/L ofEDTA acid. After incubating for 10 min at 37 °C, the reactionwas stopped by adding chloroform/methanol (2:1, v/v) followedby [3H]-cholesteryl oleate as an internal standard to estimaterecovery.
Microsomal apo B-100 and microsomal triglyceride transferprotein (MTP) expression
To evaluate apolipoprotein (apo) B and MTP as well as ATP-binding cassette (ABCG8) and ABCB11 protein expression,tissue homogenate or microsome samples were adequatelyprepared for Western blotting as described previously (Levyet al., 2001a,b). Proteins were denatured in sample buffercontaining SDS and β-mercaptoethanol, separated on a 4–20%gradient SDS–PAGE, and electroblotted onto nitrocellulosemembranes. Nonspecific binding sites of the membranes wereblocked using defatted milk proteins followed by the addition ofspecific primary antibodies. The relative amount of primaryantibody was detected with species-specific horseradish perox-idase-conjugated secondary antibody. Blots were developed andthe protein mass was quantitated using HP Scanjet scannerequipped with a transparency adapter and software.
MTP activity assay
Liver microsomes used as the source of MTP activity wereisolated as described previously (Levy et al., 2001a,b, 2002).MTP activity was determined by the transfer of radiolabeledtriacylglycerol from donor small unilamellar vesicles (40 nmolegg phosphatidylcholine, 0.08 nmol [14C]-triacylglycerol,and 2 nmol cardiolipin) to acceptor small unilamellar ve-sicles (240 nmol egg phosphatidylcholine and 0.48 nmoltriacylglycerol) at 37 °C for 1 h. This assay was previouslydescribed in detail (Levy et al., 2001a,b, 2002). Lipid transferactivity is expressed as pmol/min/mg protein under verifiedlinear assay conditions (first-order kinetics).
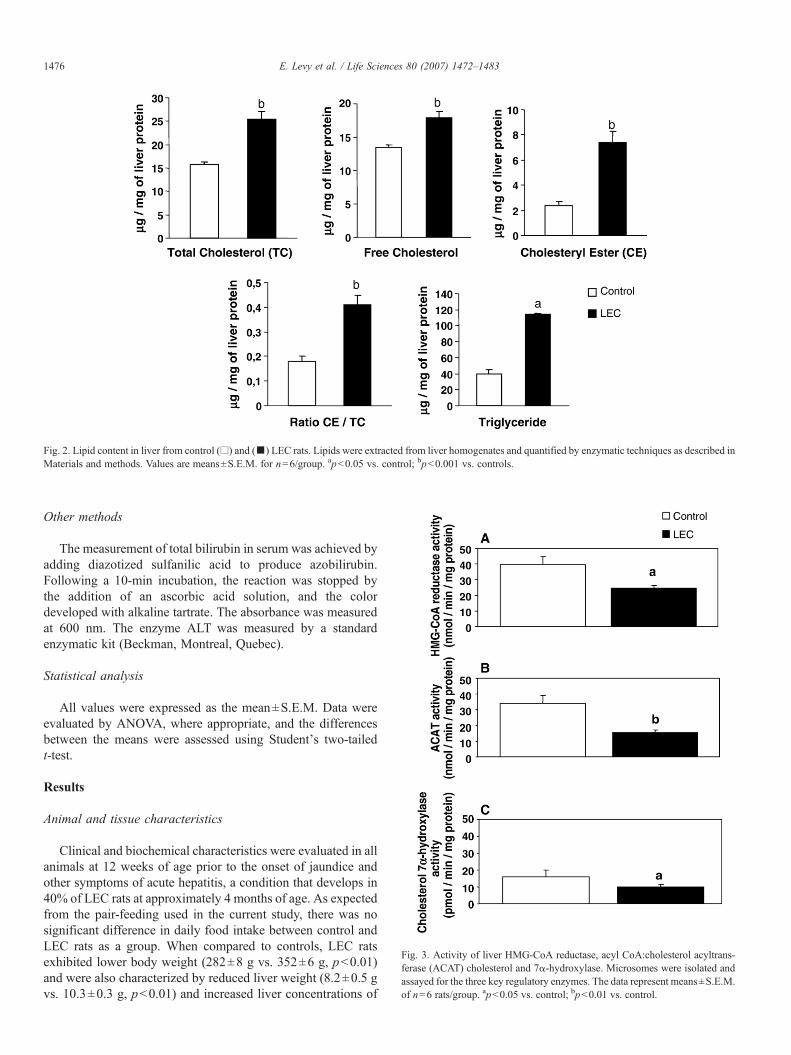
Fig. 2. Lipid content in liver from control (□) and (▪) LEC rats. Lipids were extracted from liver homogenates and quantified by enzymatic techniques as described inMaterials and methods. Values are means±S.E.M. for n=6/group. apb0.05 vs. control; bpb0.001 vs. controls.
Fig. 3. Activity of liver HMG-CoA reductase, acyl CoA:cholesterol acyltrans-ferase (ACAT) cholesterol and 7α-hydroxylase. Microsomes were isolated andassayed for the three key regulatory enzymes. The data represent means±S.E.M.of n=6 rats/group. apb0.05 vs. control; bpb0.01 vs. control.
1476 E. Levy et al. / Life Sciences 80 (2007) 1472–1483
Other methods
The measurement of total bilirubin in serum was achieved byadding diazotized sulfanilic acid to produce azobilirubin.Following a 10-min incubation, the reaction was stopped bythe addition of an ascorbic acid solution, and the colordeveloped with alkaline tartrate. The absorbance was measuredat 600 nm. The enzyme ALT was measured by a standardenzymatic kit (Beckman, Montreal, Quebec).
Statistical analysis
All values were expressed as the mean±S.E.M. Data wereevaluated by ANOVA, where appropriate, and the differencesbetween the means were assessed using Student's two-tailedt-test.
Results
Animal and tissue characteristics
Clinical and biochemical characteristics were evaluated in allanimals at 12 weeks of age prior to the onset of jaundice andother symptoms of acute hepatitis, a condition that develops in40% of LEC rats at approximately 4 months of age. As expectedfrom the pair-feeding used in the current study, there was nosignificant difference in daily food intake between control andLEC rats as a group. When compared to controls, LEC ratsexhibited lower body weight (282±8 g vs. 352±6 g, pb0.01)and were also characterized by reduced liver weight (8.2±0.5 gvs. 10.3±0.3 g, pb0.01) and increased liver concentrations of
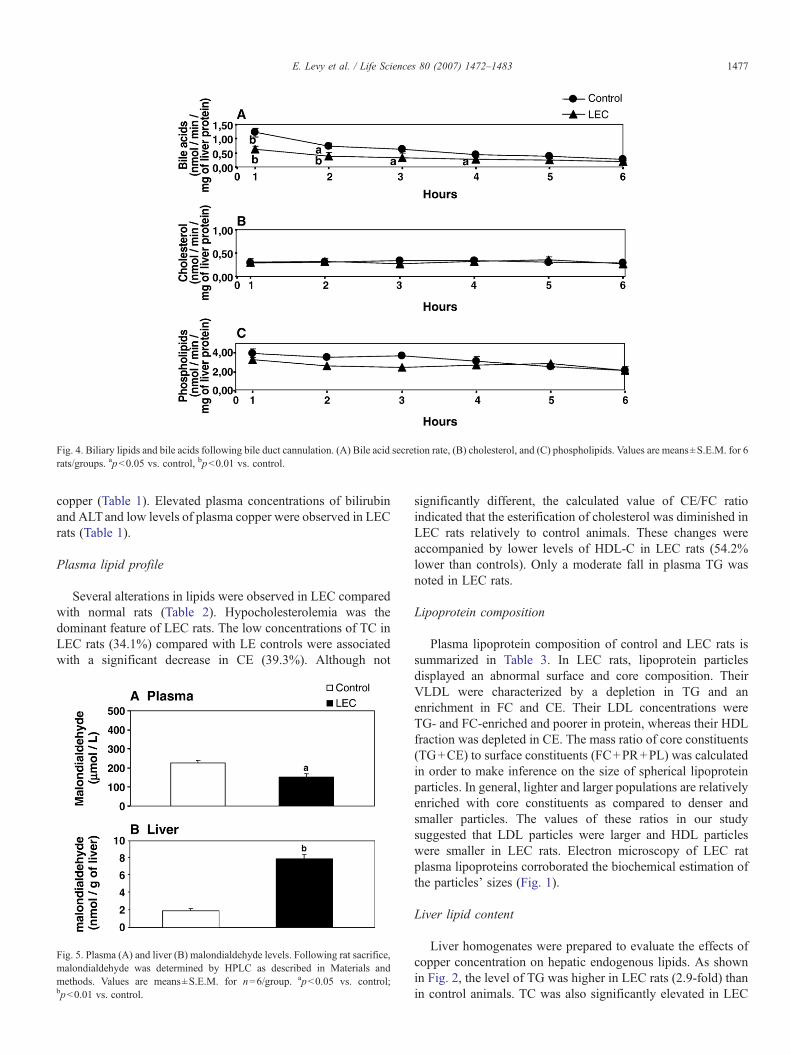
Fig. 4. Biliary lipids and bile acids following bile duct cannulation. (A) Bile acid secretion rate, (B) cholesterol, and (C) phospholipids. Values are means±S.E.M. for 6rats/groups. apb0.05 vs. control, bpb0.01 vs. control.
1477E. Levy et al. / Life Sciences 80 (2007) 1472–1483
copper (Table 1). Elevated plasma concentrations of bilirubinand ALTand low levels of plasma copper were observed in LECrats (Table 1).
Plasma lipid profile
Several alterations in lipids were observed in LEC comparedwith normal rats (Table 2). Hypocholesterolemia was thedominant feature of LEC rats. The low concentrations of TC inLEC rats (34.1%) compared with LE controls were associatedwith a significant decrease in CE (39.3%). Although not
Fig. 5. Plasma (A) and liver (B) malondialdehyde levels. Following rat sacrifice,malondialdehyde was determined by HPLC as described in Materials andmethods. Values are means±S.E.M. for n=6/group. apb0.05 vs. control;bpb0.01 vs. control.
significantly different, the calculated value of CE/FC ratioindicated that the esterification of cholesterol was diminished inLEC rats relatively to control animals. These changes wereaccompanied by lower levels of HDL-C in LEC rats (54.2%lower than controls). Only a moderate fall in plasma TG wasnoted in LEC rats.
Lipoprotein composition
Plasma lipoprotein composition of control and LEC rats issummarized in Table 3. In LEC rats, lipoprotein particlesdisplayed an abnormal surface and core composition. TheirVLDL were characterized by a depletion in TG and anenrichment in FC and CE. Their LDL concentrations wereTG- and FC-enriched and poorer in protein, whereas their HDLfraction was depleted in CE. The mass ratio of core constituents(TG+CE) to surface constituents (FC+PR+PL) was calculatedin order to make inference on the size of spherical lipoproteinparticles. In general, lighter and larger populations are relativelyenriched with core constituents as compared to denser andsmaller particles. The values of these ratios in our studysuggested that LDL particles were larger and HDL particleswere smaller in LEC rats. Electron microscopy of LEC ratplasma lipoproteins corroborated the biochemical estimation ofthe particles' sizes (Fig. 1).
Liver lipid content
Liver homogenates were prepared to evaluate the effects ofcopper concentration on hepatic endogenous lipids. As shownin Fig. 2, the level of TG was higher in LEC rats (2.9-fold) thanin control animals. TC was also significantly elevated in LEC
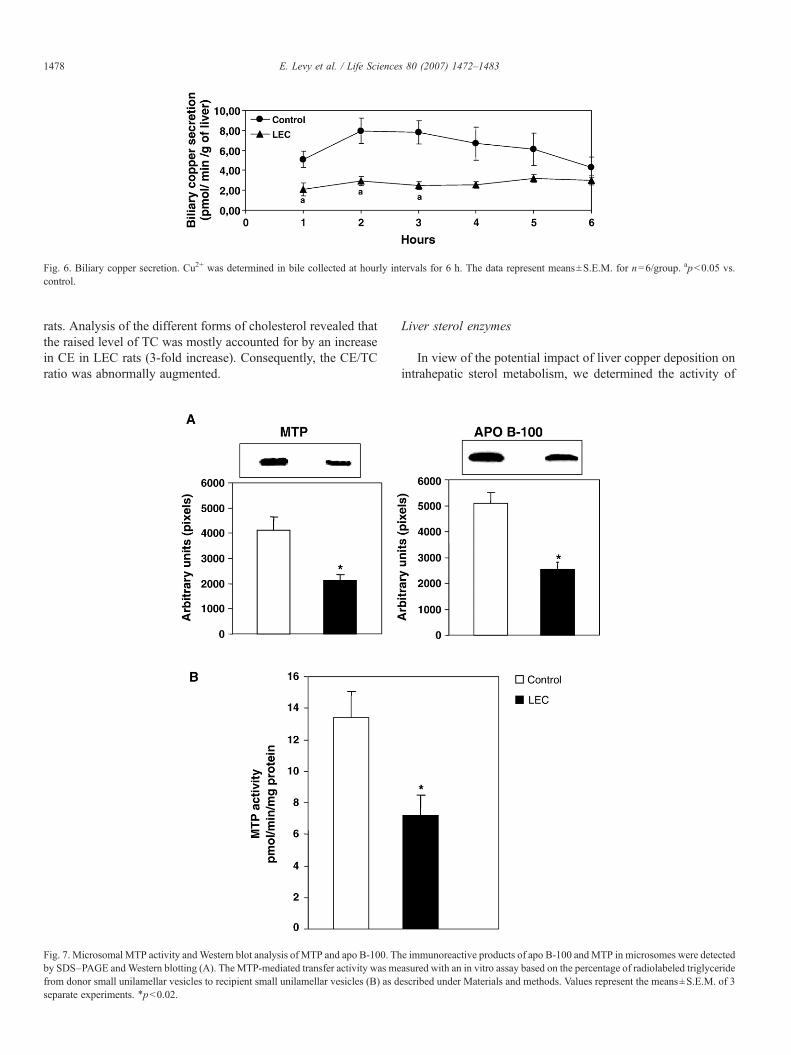
Fig. 6. Biliary copper secretion. Cu2+ was determined in bile collected at hourly intervals for 6 h. The data represent means±S.E.M. for n=6/group. apb0.05 vs.control.
1478 E. Levy et al. / Life Sciences 80 (2007) 1472–1483
rats. Analysis of the different forms of cholesterol revealed thatthe raised level of TC was mostly accounted for by an increasein CE in LEC rats (3-fold increase). Consequently, the CE/TCratio was abnormally augmented.
Fig. 7. Microsomal MTP activity andWestern blot analysis of MTP and apo B-100. Thby SDS–PAGE andWestern blotting (A). TheMTP-mediated transfer activity was mefrom donor small unilamellar vesicles to recipient small unilamellar vesicles (B) as dseparate experiments. ⁎pb0.02.
Liver sterol enzymes
In view of the potential impact of liver copper deposition onintrahepatic sterol metabolism, we determined the activity of
e immunoreactive products of apo B-100 andMTP in microsomes were detectedasured with an in vitro assay based on the percentage of radiolabeled triglycerideescribed under Materials and methods. Values represent the means±S.E.M. of 3
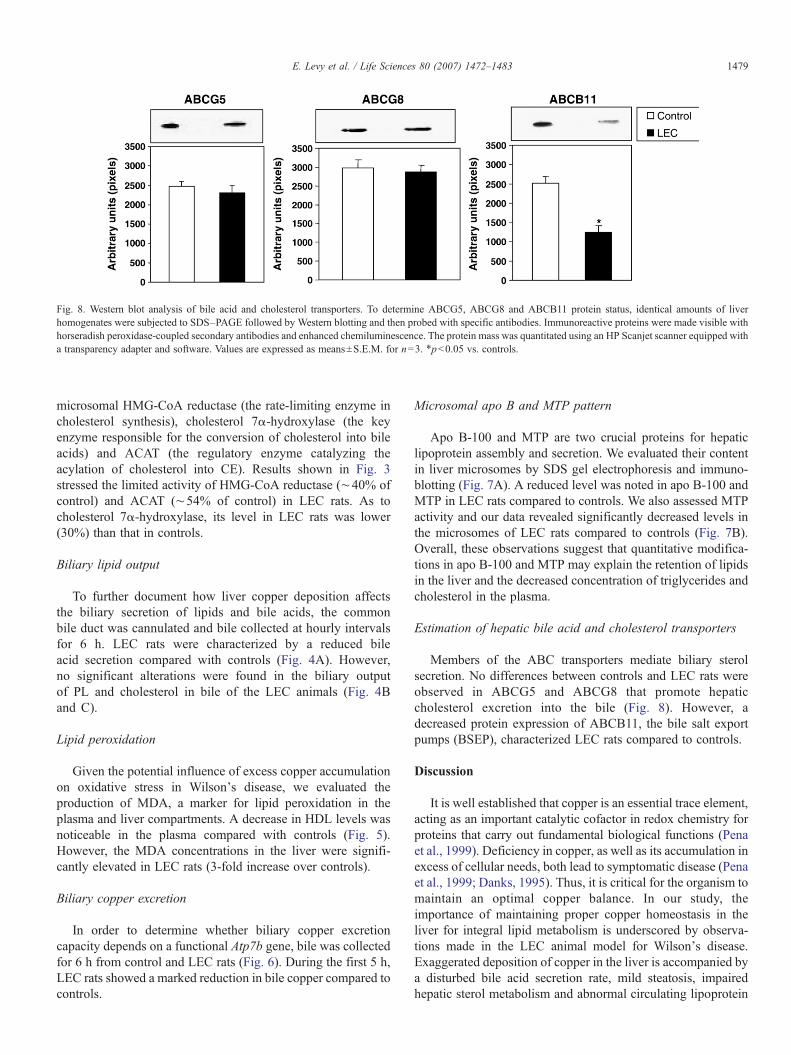
Fig. 8. Western blot analysis of bile acid and cholesterol transporters. To determine ABCG5, ABCG8 and ABCB11 protein status, identical amounts of liverhomogenates were subjected to SDS–PAGE followed by Western blotting and then probed with specific antibodies. Immunoreactive proteins were made visible withhorseradish peroxidase-coupled secondary antibodies and enhanced chemiluminescence. The protein mass was quantitated using an HP Scanjet scanner equipped witha transparency adapter and software. Values are expressed as means±S.E.M. for n=3. ⁎pb0.05 vs. controls.
1479E. Levy et al. / Life Sciences 80 (2007) 1472–1483
microsomal HMG-CoA reductase (the rate-limiting enzyme incholesterol synthesis), cholesterol 7α-hydroxylase (the keyenzyme responsible for the conversion of cholesterol into bileacids) and ACAT (the regulatory enzyme catalyzing theacylation of cholesterol into CE). Results shown in Fig. 3stressed the limited activity of HMG-CoA reductase (∼40% ofcontrol) and ACAT (∼54% of control) in LEC rats. As tocholesterol 7α-hydroxylase, its level in LEC rats was lower(30%) than that in controls.
Biliary lipid output
To further document how liver copper deposition affectsthe biliary secretion of lipids and bile acids, the commonbile duct was cannulated and bile collected at hourly intervalsfor 6 h. LEC rats were characterized by a reduced bileacid secretion compared with controls (Fig. 4A). However,no significant alterations were found in the biliary outputof PL and cholesterol in bile of the LEC animals (Fig. 4Band C).
Lipid peroxidation
Given the potential influence of excess copper accumulationon oxidative stress in Wilson's disease, we evaluated theproduction of MDA, a marker for lipid peroxidation in theplasma and liver compartments. A decrease in HDL levels wasnoticeable in the plasma compared with controls (Fig. 5).However, the MDA concentrations in the liver were signifi-cantly elevated in LEC rats (3-fold increase over controls).
Biliary copper excretion
In order to determine whether biliary copper excretioncapacity depends on a functional Atp7b gene, bile was collectedfor 6 h from control and LEC rats (Fig. 6). During the first 5 h,LEC rats showed a marked reduction in bile copper compared tocontrols.
Microsomal apo B and MTP pattern
Apo B-100 and MTP are two crucial proteins for hepaticlipoprotein assembly and secretion. We evaluated their contentin liver microsomes by SDS gel electrophoresis and immuno-blotting (Fig. 7A). A reduced level was noted in apo B-100 andMTP in LEC rats compared to controls. We also assessed MTPactivity and our data revealed significantly decreased levels inthe microsomes of LEC rats compared to controls (Fig. 7B).Overall, these observations suggest that quantitative modifica-tions in apo B-100 and MTP may explain the retention of lipidsin the liver and the decreased concentration of triglycerides andcholesterol in the plasma.
Estimation of hepatic bile acid and cholesterol transporters
Members of the ABC transporters mediate biliary sterolsecretion. No differences between controls and LEC rats wereobserved in ABCG5 and ABCG8 that promote hepaticcholesterol excretion into the bile (Fig. 8). However, adecreased protein expression of ABCB11, the bile salt exportpumps (BSEP), characterized LEC rats compared to controls.
Discussion
It is well established that copper is an essential trace element,acting as an important catalytic cofactor in redox chemistry forproteins that carry out fundamental biological functions (Penaet al., 1999). Deficiency in copper, as well as its accumulation inexcess of cellular needs, both lead to symptomatic disease (Penaet al., 1999; Danks, 1995). Thus, it is critical for the organism tomaintain an optimal copper balance. In our study, theimportance of maintaining proper copper homeostasis in theliver for integral lipid metabolism is underscored by observa-tions made in the LEC animal model for Wilson's disease.Exaggerated deposition of copper in the liver is accompanied bya disturbed bile acid secretion rate, mild steatosis, impairedhepatic sterol metabolism and abnormal circulating lipoprotein

1480 E. Levy et al. / Life Sciences 80 (2007) 1472–1483
composition. Additionally, our findings suggest induction ofoxidative stress along with copper accumulation.
Copper is important in a variety of physiological processes.It is an essential co-factor of numerous key metabolic enzymesand it enables reactions necessary for mitochondrial respiration,melanin biosynthesis, dopamine metabolism, iron homeostasis,connective tissue formation and peptide formation (Culotta andGitlin, 2001). However, excess copper must be rapidlyeliminated, thereby avoiding cellular toxicity. In our study,LEC rats exhibited a marked copper accumulation in the liverwith impaired biliary copper efflux. Therefore, the LEC rataffected by the ATP7Bmutation represents a valuable model fordissecting the disease mechanisms and the wide spectrum ofpathological changes manifested in Wilson's disease.
As observed in this study, the accumulated copper wasassociated with lipid peroxidation in liver cells, which maycontribute to both hepatocellular injury and to the developmentof hepatocarcinoma, a complication that is well described inLEC rats (Mori et al., 1994). Hussain et al. have recentlyreported that reactive oxygen, nitrogen and aldehyde species inthe non-tumorous liver of patients with Wilson's disease maycontribute to the mutation of the p53 tumor suppressor gene, themost prominent genetic alteration in the development of humancancer (Hussain et al., 2000). MDA formation followed thesame trend as copper deposition; the defective ATPase activityin LEC rats resulted in ∼160-fold copper liver accumulationand ∼4-fold liver MDA production more than controls. Thesechanges may, in turn, alter hepatocyte structure and function,including lipid transport.
The data presented here clearly indicate that sterolmetabolism is substantially altered in the liver of LEC rats.The activity of the principal rate-limiting enzymes catalyzingde-novo cholesterol synthesis, bile acid production andcholesterol esterification was markedly affected in the animalswith high liver copper deposition. Different mechanisms,including a direct copper effect or indirect lipid peroxidationcould have caused the enzymatic disturbances. Our previousstudies are noteworthy in this context, showing that theendoplasmic reticulum (ER) membrane to which HMG-CoAreductase, cholesterol 7α-hydroxylase and ACATare attached isvery sensitive to lipid peroxidation (Brunet et al., 2000). Indeed,free radical-mediated lipid peroxidation produced by ironresulted in a significant loss of polyunsaturated fatty acidsand a drop in membrane fluidity that, in turn, modified thefunctioning of the membrane-bound enzymes. Nevertheless,more studies are warranted to delineate the mechanismsresponsible for the marked enzyme inhibition noted in LECrats. As previously reported in LEC rats, copper cannot beincorporated into ceruloplasmin and the resulting absence ofperoxidase activity leads to iron accumulation in addition to thatof copper (Ma et al., 1997). It is likely that the combined effectof increased concentrations of copper and iron, both transitionmetals, detected in the liver of LEC rats, enhances the degree oflipid peroxidation (Ma et al., 1997). However, the observationson lipid processing in this study are quite different from thoseassociated with isolated hepatic iron overload (Brunet et al.,1999), suggesting that copper accumulation in itself, in the LEC
rat, may be responsible for the findings. One prior reportshowed that LEC rats developed increased hepatic TG, CE andcholesterol levels after the age of 8 weeks, while serum lipidschanged little, suggesting that the changes were related toprogressive copper accumulation (Taniguchi et al., 1991). InWilson's patients on-therapy, alterations in serum lipids similarto those observed in this study have been described (Rodo et al.,2000) and copper overload in calves leads to similar hepaticlipid changes (Jenkins and Kramer, 1989). It may be argued thatsimply the development of parenchymal inflammation was theprincipal factor affecting lipid metabolism. Clearly, the LECrats at 12 weeks, although asymptomatic, were alreadyexhibiting biochemical evidence of hepatitis including hyper-bilirubinemia, elevated ALT (15–20 times normal) and anincrease in plasma copper to normal levels. Liver histology wasalso abnormal in LEC rats (data not shown) showing typicalpleomorphic changes to hepatocytes, anisocytosis, mild focalsteatosis, and chronic inflammatory cell infiltration in the portalareas, composed mainly of macrophages. Apoptosis, cholan-giolar proliferation and fibrosis, which arise progressively overthe first 6 months of life, were not observed. These histologicalchanges correlate well with the biochemical abnormalities.Possibly, the study of control rats with chronic copperadministration and of LEC rats on iron-reduced diet or treatedwith copper chelators may help to clarify the issue of lipidprocessing.
Without lessening the impact of lipid peroxidation, theaccumulation of FC and bile acids in the liver may also hamperthe optimal activity of the specific enzymes (Taborsky, 1973;Samuni et al., 1981; Kim et al., 1988; Halliwell and Gutteridge,1985). Our experiments have documented low concentrationsof TG and cholesterol in the plasma together with increasedamounts of these lipids in the liver of LEC rats, suggestinglimited VLDL output. VLDL is the principal vehicle for thetransport of endogenous TG in the blood circulation. Themechanism of VLDL assembly is very complex and is regulatedat different levels by a variety of factors (Kang and Davis, 2000;Mason, 1998; Kendrick et al., 1998): a) apo B-100 formationand degradation (Kang and Davis, 2000; Mason, 1998;Kendrick et al., 1998); b) the availability of hepatic levels ofphosphatidylcholine (PC), CE and TG (Kang and Davis, 2000;Mason, 1998; Kendrick et al., 1998); c) adequate activity ofHMG-CoA reductase (Huff and Burnett, 1997), ACAT (Burnettet al., 1999), cholesterol 7α-hydroxylase (Wang et al., 1997)and MTP (Wetterau et al., 1992). Experiments of the presentstudy revealed a decreased protein expression of apo B-100 andMTP in LEC rats. MTP transfer activity was also diminished inthese animals. The apo B-100 decline may reflect inadequatelipidation of apo B by MTP, a critical player in the earlyposttranslational regulation of apo B-100 (Mitchell et al., 1998).Accordingly, genetic MTP deficiency has been shown to be thebasis for the lack of plasma apo B in patients withabetalipoproteinemia (Wetterau et al., 1992). Similarly, knock-out mice exhibited a marked reduction in plasma apo B and TGconcentrations because of the severely impaired hepaticsecretion of apo B-containing lipoproteins (Raabe et al.,1998). Thus, our results suggest that low MTP levels may

1481E. Levy et al. / Life Sciences 80 (2007) 1472–1483
confer sluggish assembly and export of TG-rich, apo B-containing lipoproteins in the liver of LEC rats. Theabnormalities in lipoprotein transport may also explain themagnitude of TG and cholesterol in the liver of these animals.
In the liver, the expression of ABCG5 and ABCG8 is seen incells lining the hepatobiliary tracts, both hepatocytes and ductalcells (Klett et al., 2004). They are necessary for the translocationof cholesterol into bile (Graf et al., 2003; Yu et al., 2005). Inorder to determine whether these ABC transporters are involvedin the accumulation of cholesterol in the liver, their protein masswas assessed by Western blotting. The absence of alterations intheir expression (Fig. 8) together with the normal biliarycholesterol output (Fig. 4) indicates that hepatic lipid retentionis mainly due to defects in lipoprotein assembly and secretion.
The bile salt export pump BSEP is the ABCB11 transporterrepresenting the predominant bile salt efflux system ofhepatocytes (Noe et al., 2002; Kullak-Ublick et al., 2004).Western blot analysis of liver homogenates detected lowABCB11 expression in LEC rats, which may explain theweak bile acid output recorded in these animals. We cantherefore hypothesize that the delay in bile acid export maycontribute to the decline of cholesterol 7α-hydroxylase activityby feedback inhibition.
Analysis of plasma lipids revealed a reduction in cholesterolesterification in LEC rats. This finding suggests limited activityin lecithin–cholesterol acyltransferase (LCAT), the majorenzyme responsible for the esterification of FC in circulatinglipoproteins.
Under normal circumstances, LCAT actively convertscholesterol and PC to CE and lyso-PC on the surface of HDL(Jonas, 2000). However, in conditions of oxidative stress, LCATactivity cannot optimally fulfill its function. Hydroperoxides ofPC and aldehydes were found to down-modulate LCAT activity(Bielicki and Forte, 1999; McCall et al., 1995). Similarly,oxidized HDL and products of LDL oxidation were noted toadversely affect LCAT functioning (Maziere et al., 1993;McCall et al., 2001). Therefore, it might be possible that thelipid peroxidation observed in LEC rats exerted deleteriouseffects on LCAT activity. Nevertheless, more studies arenecessary to establish the contribution of oxidative stress tothe abnormalities notes in the present work. This is particularlyimportant since no correlations were observed between copper-catalyzed lipid peroxidation and lipid disorders.
LEC rat model may differ in some respects from humanWilson's, a criticism that may be applied to many animal modelsof human disease. The LEC bears a single mutation resulting in alarge deletion of the C-terminal portion of the protein whereasthere are more than 200 different mutations now described inWilson's disease andmost patients are compound heterozygotes.The effect of such mutations on the function and localization ofthe ATPB7 protein is not well understood at present but almostcertainly will result in phenotypic heterogeneity in what weunderstand as Wilson's disease. Of note is the possibilityregarding the association of hepatocellular carcinoma andhepatitis with Wilson's disease. While it is true that hepatocel-lular carcinoma has been infrequently associated with Wilson'sdisease, it certainly has been reported in many publications over
the years. One should bear in mind that, in contrast to LEC rats,symptomatic WD patients are never simply observed but are allplaced on chelation or zinc therapy, which alters the naturalhistory of the illness. Hepatitis is in fact a well-knowncomponent of symptomatic Wilson's disease which can presentsilently, as an acute hepatitis and as an illness not infrequentlymistaken for autoimmune hepatitis because of similarities in thehistological findings of chronic active hepatitis. This point iswell articulated in the AASLD practice guidelines on Wilson'sdisease written by Roberts and Schilsky (2003).
In conclusion, our data confirm hepatic copper accumulationin LEC rats. Furthermore, our findings stressed the occurrenceof steatosis accompanied with disturbances in the threeregulatory enzymes of cholesterol homeostasis as well as thediminution of bile acid output via probably low expression ofBSEP. Significant changes were noticed in plasma lipid profileand lipoprotein composition through irregularities in apo B-100and MTP. Additional explorations are required to dissect thespecific contribution of copper accumulation and oxidativestress to these disorders in the LEC rat, a model for Wilson'sdisease.
Acknowledgements
This research was undertaken with grants from the HôpitalSainte-Justine Research Center, the Hospital for Sick Children'sFoundation and the Canadian Institutes of Health Research(CIHR).
The authors thank Mrs Schohraya Spahis for her experttechnical assistance.
References
Bacon, B.R., Schilsky, M.L., 1999. New knowledge of genetic pathogenesis ofhemochromatosis and Wilson's disease. Advances in Internal Medicine 44,91–116.
Bartlett, G.R., 1959. Phosphorus assay in column chromatography. Journal ofBiological Chemistry 234 (3), 466–468.
Bielicki, J.K., Forte, T.M., 1999. Evidence that lipid hydroperoxides inhibitplasma lecithin : cholesterol acyltransferase activity. Journal of LipidResearch 40 (5), 948–954.
Brown, M.S., Goldstein, J.L., Dietschy, J.M., 1979. Active and inactive forms of3-hydroxy-3-methylglutaryl coenzyme A reductase in the liver of the rat.Comparison with the rate of cholesterol synthesis in different physiologicalstates. Journal of Biological Chemistry 254 (12), 5144–5149.
Brunet, S., Thibault, L., Delvin, E., Yotov, W., Bendayan, M., Levy, E., 1999.Dietary iron overload and induced lipid peroxidation are associated withimpaired plasma lipid transport and hepatic sterol metabolism in rats.Hepatology 29 (6), 1809–1817.
Brunet, S., Thibault, L., Lepage, G., Seidman, E.G., Dube, N., Levy, E., 2000.Modulation of endoplasmic reticulum-bound cholesterol regulatoryenzymes by iron/ascorbate-mediated lipid peroxidation. Free RadicalBiology & Medicine 28 (1), 46–54.
Bull, P.C., Thomas, G.R., Rommens, J.M., Forbes, J.R., Cox, D.W., 1993. TheWilson disease gene is a putative copper transporting P-type ATPase similarto the Menkes gene. Nature Genetics 5 (4), 327–337.
Burnett, J.R., Wilcox, L.J., Huff, M.W., 1999. Acyl coenzyme A: cholesterolacyltransferase inhibition and hepatic apolipoprotein B secretion. ClinicaChimica Acta 286 (1–2), 231–242.
Cooper, A.D., 1985. Role of the liver in the degradation of lipoproteins.Gastroenterology 88 (1 Pt 1), 192–205.

1482 E. Levy et al. / Life Sciences 80 (2007) 1472–1483
Culotta, V.C., Gitlin, J.D., 2001. Disorders of copper transport. In: Scriver, C.R.,Beaudet, A.L., Sly, S.W., Valley, D. (Eds.), The Molecular and MetabolicBasis of Inherited Disease. McGraw-Hill, New York, pp. 3105–3126.
Danks, D.M., 1995. Disorders of copper transport. In: Scriver, C.R., Beaudet,A.L., Sly, S.W., Valley, D. (Eds.), The Metabolic and Molecular Basis ofInherited Disease. McGraw-Hill, New York, pp. 2211–2235.
Graf, G.A., Yu, L., Li, W.P., Gerard, R., Tuma, P.L., Cohen, J.C., Hobbs, H.H.,2003. ABCG5 and ABCG8 are obligate heterodimers for protein traffickingand biliary cholesterol excretion. Journal of Biological Chemistry 278 (48),48275–48282.
Halliwell, B., Gutteridge, J.M.C., 1985. Lipid peroxidation: a radical chainreaction. In: Halliwell, B. (Ed.), Free Radicals in Biology and Medicine.Clarendon Press, Oxford, pp. 139–189.
Harada, M., Sakisaka, S., Terada, K., Kimura, R., Kawaguchi, T., Koga, H.,Taniguchi, E., Sasatomi, K., Miura, N., Suganuma, T., Fujita, H., Furuta, K.,Tanikawa, K., Sugiyama, T., Sata, M., 2000. Role of ATP7B in biliarycopper excretion in a human hepatoma cell line and normal rat hepatocytes.Gastroenterology 118 (5), 921–928.
Huff, M.W., Burnett, J.R., 1997. 3-Hydroxy-3-methylglutaryl coenzyme Areductase inhibitors and hepatic apolipoprotein B secretion. Current Opinionin Lipidology 8 (3), 138–145.
Hung, I.H., Suzuki, M., Yamaguchi, Y., Yuan, D.S., Klausner, R.D., Gitlin, J.D.,1997. Biochemical characterization of the Wilson disease protein andfunctional expression in the yeast Saccharomyces cerevisiae. Journal ofBiological Chemistry 272 (34), 21461–21466.
Hussain, S.P., Raja, K., Amstad, P.A., Sawyer, M., Trudel, L.J., Wogan, G.N.,Hofseth, L.J., Shields, P.G., Billiar, T.R., Trautwein, C., Hohler, T., Galle,P.R., Phillips, D.H., Markin, R., Marrogi, A.J., Harris, C.C., 2000.Increased p53 mutation load in nontumorous human liver of Wilsondisease and hemochromatosis: oxyradical overload diseases. Proceedingsof the National Academy of Sciences of the United States of America 97(23), 12770–12775.
Hylemon, P.B., Studer, E.J., Pandak, W.M., Heuman, D.M., Vlahcevic, Z.R.,Chiang, J.Y., 1989. Simultaneous measurement of cholesterol 7 alpha-hydroxylase activity by reverse-phase high-performance liquid chromatog-raphy using both endogenous and exogenous [4-14C]cholesterol as substrate.Analytical Biochemistry 182 (2), 212–216.
Jenkins, K.J., Kramer, J.K., 1989. Influence of excess dietary copper on lipidcomposition of calf tissues. Journal of Dairy Science 72 (10), 2582–2591.
Jonas, A., 2000. Lecithin cholesterol acyltransferase. Biochimica et BiophysicaActa 1529 (1–3), 245–256.
Kang, S., Davis, R.A., 2000. Cholesterol and hepatic lipoprotein assembly andsecretion. Biochimica et Biophysica Acta 1529 (1–3), 223–230.
Kendrick, J.S., Wilkinson, J., Cartwright, I.J., Lawrence, S., Higgins, J.A., 1998.Regulation of the assembly and secretion of very low density lipoproteins bythe liver. Biological Chemistry 379 (8–9), 1033–1040.
Kim, K., Kim, I.H., Lee, K.Y., Rhee, S.G., Stadtman, E.R., 1988. The isolationand purification of a specific “protector” protein which inhibits enzymeinactivation by a thiol/Fe(III)/O2 mixed-function oxidation system. Journalof Biological Chemistry 263 (10), 4704–4711.
Klett, E.L., Lee, M.H., Adams, D.B., Chavin, K.D., Patel, S.B., 2004.Localization of ABCG5 and ABCG8 proteins in human liver, gall bladderand intestine. BMC. Gastroenterology 4 (1), 21.
Kullak-Ublick, G.A., Stieger, B., Meier, P.J., 2004. Enterohepatic bile salttransporters in normal physiology and liver disease. Gastroenterology 126(1), 322–342.
Lepage, G., Munoz, G., Champagne, J., Roy, C.C., 1991. Preparative stepsnecessary for the accurate measurement of malondialdehyde by high-performance liquid chromatography. Analytical Biochemistry 197 (2),277–283.
Levy, E., Lepage, G., Bendayan, M., Ronco, N., Thibault, L., Galeano, N.,Smith, L., Roy, C.C., 1989. Relationship of decreased hepatic lipase activityand lipoprotein abnormalities to essential fatty acid deficiency in cysticfibrosis patients. Journal of Lipid Research 30 (8), 1197–1209.
Levy, E., Thibault, L., Garofalo, C., Messier, M., Lepage, G., Ronco, N., Roy,C.C., 1990. Combined (n-3 and n-6) essential fatty deficiency is a potentmodulator of plasma lipids, lipoprotein composition, and lipolytic enzymes.Journal of Lipid Research 31 (11), 2009–2017.
Levy, E., Garofalo, C., Thibault, L., Dionne, S., Daoust, L., Lepage, G., Roy,C.C., 1992. Intraluminal and intracellular phases of fat absorption areimpaired in essential fatty acid deficiency. American Journal of Physiology262 (2 Pt 1), G319–G326.
Levy, E., Bendayan, M., Thibault, L., Lambert, M., Paradis, K., 1995.Lipoprotein abnormalities in two children with minimal biliary excretion.Journal of Pediatric Gastroenterology and Nutrition 20 (4), 432–439.
Levy, E., Garofalo, C., Rouleau, T., Gavino, V., Bendayan, M., 1996. Impact ofessential fatty acid deficiency on hepatic sterol metabolism in rats.Hepatology 23 (4), 848–857.
Levy, E., Menard, D., Delvin, E., Stan, S., Mitchell, G., Lambert, M., Ziv, E.,Feoli-Fonseca, J.C., Seidman, E., 2001a. The polymorphism at codon 54 ofthe FABP2 gene increases fat absorption in human intestinal explants.Journal of Biological Chemistry 276 (43), 39679–39684.
Levy E., Stan S., Garofalo C., Delvin E.E., Seidman E.G., Menard D., 2001a.Immunolocalization, ontogeny, and regulation of microsomal triglyceridetransfer protein in human fetal intestine. American Journal of Physiology.Gastrointestinal and Liver Physiology 280 (4), G563–G571.
Levy, E., Stan, S., Delvin, E., Menard, D., Shoulders, C., Garofalo, C., Slight, I.,Seidman, E., Mayer, G., Bendayan, M., 2002. Localization of microsomaltriglyceride transfer protein in the Golgi: possible role in the assembly ofchylomicrons. Journal of Biological Chemistry 277 (19), 16470–16477.
Lowry, O.H., Rosebrough, N.J., Farr, A.L., Randall, R.J., 1951. Proteinmeasurement with the Folin phenol reagent. Journal of Biological Chemistry193 (1), 265–275.
Ma, Y., Zhang, D., Kawabata, T., Kiriu, T., Toyokuni, S., Uchida, K., Okada, S.,1997. Copper and iron-induced oxidative damage in non-tumor bearing LECrats. Pathology International 47 (4), 203–208.
Mason, T.M., 1998. The role of factors that regulate the synthesis and secretionof very-low-density lipoprotein by hepatocytes. Critical Reviews in ClinicalLaboratory Sciences 35 (6), 461–487.
Maziere, J.C., Myara, I., Salmon, S., Auclair, M., Haigle, J., Santus, R., Maziere,C., 1993. Copper- and malondialdehyde-induced modification of highdensity lipoprotein and parallel loss of lecithin cholesterol acyltransferaseactivation. Atherosclerosis 104 (1–2), 213–219.
McCall, M.R., Tang, J.Y., Bielicki, J.K., Forte, T.M., 1995. Inhibition oflecithin–cholesterol acyltransferase and modification of HDL apolipopro-teins by aldehydes. Arteriosclerosis, Thrombosis, and Vascular Biology 15(10), 1599–1606.
McCall, M.R., Carr, A.C., Forte, T.M., Frei, B., 2001. LDL modified byhypochlorous acid is a potent inhibitor of lecithin–cholesterol acyltransfer-ase activity. Arteriosclerosis, Thrombosis, and Vascular Biology 21 (6),1040–1045.
Mitchell, D.M., Zhou, M., Pariyarath, R., Wang, H., Aitchison, J.D., Ginsberg,H.N., Fisher, E.A., 1998. Apoprotein B100 has a prolonged interaction withthe translocon during which its lipidation and translocation change fromdependence on the microsomal triglyceride transfer protein to independence.Proceedings of the National Academy of Sciences of the United States ofAmerica. 95 (25), 14733–14738.
Mori, M., Hattori, A., Sawaki, M., Tsuzuki, N., Sawada, N., Oyamada, M.,Sugawara, N., Enomoto, K., 1994. The LEC rat: a model for humanhepatitis, liver cancer, and much more. American Journal of Pathology 144(1), 200–204.
Myers, B.M., Prendergast, F.G., Holman, R., Kuntz, S.M., LaRusso, N.F., 1991.Alterations in the structure, physicochemical properties, and pH ofhepatocyte lysosomes in experimental iron overload. Journal of ClinicalInvestigation 88 (4), 1207–1215.
Noe, J., Stieger, B., Meier, P.J., 2002. Functional expression of the canalicularbile salt export pump of human liver. Gastroenterology 123 (5), 1659–1666.
Okayasu, T., Tochimaru, H., Hyuga, T., Takahashi, T., Takekoshi, Y., Li, Y.,Togashi, Y., Takeichi, N., Kasai, N., Arashima, S., 1992. Inherited coppertoxicity in Long-Evans cinnamon rats exhibiting spontaneous hepatitis: amodel of Wilson's disease. Pediatric Research 31 (3), 253–257.
Packard, C.J., Shepherd, J., 1982. The hepatobiliary axis and lipoproteinmetabolism: effects of bile acid sequestrants and ileal bypass surgery.Journal of Lipid Research 23 (8), 1081–1098.
Pena, M.M., Lee, J., Thiele, D.J., 1999. A delicate balance: homeostatic controlof copper uptake and distribution. Journal of Nutrition 129 (7), 1251–1260.

1483E. Levy et al. / Life Sciences 80 (2007) 1472–1483
Raabe, M., Flynn, L.M., Zlot, C.H., Wong, J.S., Veniant, M.M., Hamilton, R.L.,Young, S.G., 1998. Knockout of the abetalipoproteinemia gene in mice:reduced lipoprotein secretion in heterozygotes and embryonic lethality inhomozygotes. Proceedings of the National Academy of Sciences of theUnited States of America 95 (15), 8686–8691.
Roberts, E.A., Schilsky, M.L., 2003. A practice guideline on Wilson disease.Hepatology 37 (6), 1475–1492.
Rodo, M., Czonkowska, A., Pulawska, M., Swiderska, M., Tarnacka, B., Wehr,H., 2000. The level of serum lipids, vitamin E and low density lipoproteinoxidation in Wilson's disease patients. European Journal of Neurology 7 (5),491–494.
Roelofsen, H., Wolters, H., Van Luyn, M.J., Miura, N., Kuipers, F., Vonk, R.J.,2000. Copper-induced apical trafficking of ATP7B in polarized hepatomacells provides a mechanism for biliary copper excretion. Gastroenterology119 (3), 782–793.
Samuni, A., Chevion, M., Czapski, G., 1981. Unusual copper-inducedsensitization of the biological damage due to superoxide radicals. Journalof Biological Chemistry 256 (24), 12632–12635.
Schaefer, M., Hopkins, R.G., Failla, M.L., Gitlin, J.D., 1999. Hepatocyte-specific localization and copper-dependent trafficking of the Wilson'sdisease protein in the liver. American Journal of Physiology 276 (3 Pt 1),G639–G646.
Schosinsky, K.H., Lehmann, H.P., Beeler, M.F., 1974. Measurement ofceruloplasmin from its oxidase activity in serum by use of o-dianisidinedihydrochloride. Clinical Chemistry 20 (12), 1556–1563.
Seidel, D., 1987. Lipoproteins in liver disease. Journal of Clinical Chemistry andClinical Biochemistry 25 (9), 541–551.
Sternlieb, I., 2000. Wilson's disease. Clinics in Liver Disease 4 (1), 229–2ix.Taborsky, G., 1973. Oxidative modification of proteins in the presence of ferrous
ion and air. Effect of ionic constituents of the reaction medium on the natureof the oxidation products. Biochemistry 12 (7), 1341–1348.
Taniguchi, M., Sugiyama, T., Taniguchi, N., 1991. Abnormal lipid metabolismin LEC rats. In: Mori, M., Yoshida, M.C., Takeichi, N., Taniguchi, N. (Eds.),The LEC Rat, A New Model for Hepatitis and Liver Cancer. Springer-Verlag, Tokyo, pp. 169–174.
Tietze, F., 1969. Enzymic method for quantitative determination of nanogramamounts of total and oxidized glutathione: applications to mammalian bloodand other tissues. Analytical Biochemistry 27 (3), 502–522.
Turley, S.D., Dietschy, J.M., 1988. The metabolism and excretion ofcholesterol by the liver. In: Arias, I.M., Jacoby, H., Popper, H., Schachter,D., Schafritz, D.A. (Eds.), The Liver: Biology and Pathology. Raven, NewYork, pp. 617–641.
Wang, S.L., Du, E.Z., Martin, T.D., Davis, R.A., 1997. Coordinate regulation oflipogenesis, the assembly and secretion of apolipoprotein B-containinglipoproteins by sterol response element binding protein 1. Journal ofBiological Chemistry 272 (31), 19351–19358.
Wanon, J., Guertin, F., Brunet, S., Delvin, E., Gavino, V., Bouthillier, D., Lairon,D., Yotov, W., Levy, E., 1998. The effects of cholesterol uptake from high-density lipoprotein subfractions on biliary sterol secretion in rats withessential fatty-acid deficiency. Hepatology 27 (3), 779–786.
Wetterau, J.R., Aggerbeck, L.P., Bouma, M.E., Eisenberg, C., Munck, A.,Hermier, M., Schmitz, J., Gay, G., Rader, D.J., Gregg, R.E., 1992. Absenceof microsomal triglyceride transfer protein in individuals with abetalipo-proteinemia. Science 258 (5084), 999–1001.
Wu, J., Forbes, J.R., Chen, H.S., Cox, D.W., 1994. The LEC rat has a deletion inthe copper transporting ATPase gene homologous to the Wilson diseasegene. Nature Genetics 7 (4), 541–545.
Yu, L., Gupta, S., Xu, F., Liverman, A.D., Moschetta, A., Mangelsdorf, D.J.,Repa, J.J., Hobbs, H.H., Cohen, J.C., 2005. Expression of ABCG5 andABCG8 is required for regulation of biliary cholesterol secretion. Journal ofBiological Chemistry 280 (10), 8742–8747.




















