
Ab initio study of electron transport in 4-(3-nitro-4-tetrafluorophenylthiolate-ethynyl,...
11
ORIGINAL PAPER Ab initio study of electron transport in 4-(3-nitro-4-tetrafluorophenylthiolate-ethynyl, phenylethynyl) benzenethiolate Lilia Serrato-Villegas & Marco Gallo & Marcos Delgado-Ríos & Maria Teresa Romero & Daniel Glossman-Mitnik Received: 28 February 2011 /Accepted: 20 April 2011 /Published online: 11 May 2011 # Springer-Verlag 2011 Abstract The electron transport of the 4-(3-nitro-4- tetrafluorophenylthiolate-ethynyl, phenylethynyl) benzene- thiolate (S-FNPPB-o) molecule assembled in two Au (111) electrodes, was studied using two approaches: in the first approximate approach an electric field was applied to the pure molecule attached to two thiolate ends fixed, and in the second approach we used the nonequilibrium Green´s function formalism (NEGF) coupled to DFT to calculate the I-V curve and the voltage dependence of the transmission function in the extended system, molecule plus electrodes. By applying an electric field to the pure molecule plus thiolate ends fixed, and visualizing the changes in the spatial distribution of the frontier molecular orbitals, we can expect based on the continuity of the conduction pathway in electron transport, that if electron transport occurs through the frontier orbitals, only the LUMO orbital would create an open channel for electron transport due to its delocalized nature and large orbital density at the thiolate groups. The NEGF calculations indicate that at applied voltages lower than ±0.8 V, the current is related to transmission values through the tails of the broad LUMO orbital, and since this orbital is the one closer to the Fermi energy, and we observed very low current values in this region, higher current values at positive bias than at negative bias. As the voltage exceeds ±0.8 V the current increases from the contribution of more states from the broadened part of the transmission function from the LUMO orbital, and when the voltage approaches ±2 V, the LUMO+1 orbital enters into the bias window and the current increases again. Keywords Ab initio calculations . Electron transport . Frontier orbitals . Molecular electronics . Nonequilibrium Green´s functions Introduction The semiconductor industry today is facing a rapid path to the development of small circuits, portable cell phones, small supercomputers, faster internet communications, etc. Without computers, scientists today could not develop new pharmaceuticals, track climate changes, develop nanosen- sors, or image the human brain. In the 1970s Gordon Moore, one of the founders of Intel, predicted that the number of components crammed into integrated circuits L. Serrato-Villegas Facultad de Ciencias Químicas, Universidad Autónoma de Coahuila, Blvd. Venustiano Carranza s/n. Saltillo, Coahuila, Mexico, C.P. 25200 M. Gallo (*) Facultad de Ciencias Químicas, Universidad Autónoma de San Luis Potosí, Av. Manuel Nava No. 6 Zona Universitaria, San Luis Potosí S.L.P., C.P. 78210, Mexico e-mail: [email protected] M. Delgado-Ríos Universidad Autónoma de Ciudad Juárez, Henri Dunant 4016, Zona Pronaf, Cd. Juárez, Chihuahua, Mexico, C.P. 32310 M. T. Romero Facultad de Ciencias Físico-Matemáticas, Universidad Autónoma de Coahuila, Conjunto Universitario Camporredondo Edificio “D”, C.P.( 25000 Saltillo Coahuila, Mexico D. Glossman-Mitnik Centro de Investigación en Materiales Avanzados, S.C., Miguel de Cervantes 120, Complejo Industrial Chihuahua, Chihuahua, Mexico 31109 J Mol Model (2012) 18:611–621 DOI 10.1007/s00894-011-1106-4
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Ab initio study of electron transport in 4-(3-nitro-4-tetrafluorophenylthiolate-ethynyl,...

ORIGINAL PAPER
Ab initio study of electron transportin 4-(3-nitro-4-tetrafluorophenylthiolate-ethynyl,phenylethynyl) benzenethiolate
Lilia Serrato-Villegas & Marco Gallo &
Marcos Delgado-Ríos & Maria Teresa Romero &
Daniel Glossman-Mitnik
Received: 28 February 2011 /Accepted: 20 April 2011 /Published online: 11 May 2011# Springer-Verlag 2011
Abstract The electron transport of the 4-(3-nitro-4-tetrafluorophenylthiolate-ethynyl, phenylethynyl) benzene-thiolate (S-FNPPB-o) molecule assembled in two Au (111)electrodes, was studied using two approaches: in the firstapproximate approach an electric field was applied to the puremolecule attached to two thiolate ends fixed, and in the secondapproach we used the nonequilibrium Green´s functionformalism (NEGF) coupled to DFT to calculate the I-V curveand the voltage dependence of the transmission function in theextended system, molecule plus electrodes. By applying an
electric field to the pure molecule plus thiolate ends fixed, andvisualizing the changes in the spatial distribution of thefrontier molecular orbitals, we can expect based on thecontinuity of the conduction pathway in electron transport,that if electron transport occurs through the frontier orbitals,only the LUMO orbital would create an open channel forelectron transport due to its delocalized nature and largeorbital density at the thiolate groups. The NEGF calculationsindicate that at applied voltages lower than ±0.8 V, the currentis related to transmission values through the tails of the broadLUMO orbital, and since this orbital is the one closer to theFermi energy, and we observed very low current values in thisregion, higher current values at positive bias than at negativebias. As the voltage exceeds ±0.8 V the current increases fromthe contribution of more states from the broadened part of thetransmission function from the LUMO orbital, and when thevoltage approaches ±2 V, the LUMO+1 orbital enters into thebias window and the current increases again.
Keywords Ab initio calculations . Electron transport .
Frontier orbitals .Molecular electronics . NonequilibriumGreen´s functions
Introduction
The semiconductor industry today is facing a rapid path tothe development of small circuits, portable cell phones,small supercomputers, faster internet communications, etc.Without computers, scientists today could not develop newpharmaceuticals, track climate changes, develop nanosen-sors, or image the human brain. In the 1970s GordonMoore, one of the founders of Intel, predicted that thenumber of components crammed into integrated circuits
L. Serrato-VillegasFacultad de Ciencias Químicas,Universidad Autónoma de Coahuila,Blvd. Venustiano Carranza s/n. Saltillo,Coahuila, Mexico, C.P. 25200
M. Gallo (*)Facultad de Ciencias Químicas,Universidad Autónoma de San Luis Potosí,Av. Manuel Nava No. 6 Zona Universitaria,San Luis Potosí S.L.P., C.P. 78210, Mexicoe-mail: [email protected]
M. Delgado-RíosUniversidad Autónoma de Ciudad Juárez, Henri Dunant 4016,Zona Pronaf, Cd. Juárez,Chihuahua, Mexico, C.P. 32310
M. T. RomeroFacultad de Ciencias Físico-Matemáticas,Universidad Autónoma de Coahuila,Conjunto Universitario Camporredondo Edificio “D”,C.P.( 25000 Saltillo Coahuila, Mexico
D. Glossman-MitnikCentro de Investigación en Materiales Avanzados,S.C., Miguel de Cervantes 120, Complejo Industrial Chihuahua,Chihuahua, Mexico 31109
J Mol Model (2012) 18:611–621DOI 10.1007/s00894-011-1106-4

would double every year, and “Moore’s Law” has held truefor the past 20 years [1].
However, the technology to place billions of transistorsin small electronic circuits is currently facing fundamentallimits [2]. The continued thinning of silicon for miniatur-izing circuits will destroy the band structure for themovement of electrons. These limitations can be surpassedby engineering single molecules to serve as transistors,diodes, and molecular wires. However at the nanoscalequantum effects must be considered in the design ofelectronic components.
The study of electron transport using organic moleculesanchored in electrodes will allow the design of newelectronic devices with various functionalities such as:memories, transistors, diodes, molecular switches, conduc-tors, integrated circuits and organic photovoltaic cellsdevices. Organic molecules can be potential candidates forelectronic devices if they present switching, conduction,rectifying behavior, and also if they have complexnonlinear current-voltage (I-V) characteristics such asnegative differential resistance (NDR) [3]. Molecularinterruption and NDR behavior can take place in an organicmolecule attached to electrodes due the changes in theshape, occupation and energy of molecular orbitals (MOs)[3]. To achieve efficient electron transport and highelectronic transmission values, strong electronic couplingamong the molecule and electrodes is necessary. Thiscoupling is provided by the energetic alignment of themolecular states with the electrode Fermi energy, signifi-cant orbital overlap between the structural units within themolecule and at the molecule-electrodes interfaces (spectraldensity) [4].
Kim et al. [5] pointed out that new electronic devicescan be constructed by using external fields such aselectrical and magnetic fields. Since an electric field is apowerful mean to change the energy and shape ofmolecular orbitals, the study of the effect of electricalfields on molecular orbitals would permit the creation ofnew electronic devices that can function as molecularswitches, diodes, rectifiers, etc.
In the last decade there have been many theoreticalstudies that have used the NEGF-DFT formalism to studythe electron transport of molecules attached to electrodes,since this method has been able to reproduce experimentaldata [6]. Seminario et al. [7] studied theoretically thecurrent-voltage characteristics of the Aun-S-(p-C6H4)-S-Aun (n=1–5) molecular junction connected to bulk gold inexcellent agreement with a break junction experiment andwith other ab initio calculations. Seminario et al. [8] studiedthe current–voltage characteristics of benzene, naphthalene,and anthracene attached to different types of nanoelectrodeconformations using gold atoms as electrodes. They foundthat conductances were high and tunneling barriers were
low when compared to saturated alkanes and unsaturatedoligophenylene vinylenes.
Kim et al. [9] studied the electrode effect on a molecularelectronic device. They studied different electrodes com-posed of Au, Ru, and carbon nanotubes (CNT) type (5,5)connected by thiolate groups and amide groups to an alkynemolecule using NEGF combined with DFT. Their studyshowed that the effect of different electrodes and differentlinkers was noticed by displaying different broadeningextent for the given molecular energy levels and differentalignment of the energy levels with respect to the Fermienergy of the electrodes.
They obtained nonlinear current-voltage characteristicsby using carbon nanotubes as electrodes. Their “CNT-amide” case showed the effect of the zero transmission gap,where the current is almost zero until it reaches 1.5 V,where the broadened LUMO orbital appears in the biasenergy windows and the current surges thereafter. It is theLUMO orbital of the alkyne which contributes to thecurrent surge since it is the orbital closer to the Fermienergy of the electrodes.
Lee et al. [10] studied the molecular level alignmentsand electron transport characteristics based on the NEGFapproach combined with DFT for six conjugated molecules(p-terphenyl PTP, and its derivatives, CPTP, NiPTP,CoPTP4, CoPTP5, and FePTP) containing different typesof conjugated frameworks. They observe that by combiningnon and antiaromatic components there was an increase inthe conductance since the HOMO level lay closer to theFermi level, concluding that the manipulation of theconjugated framework is an important factor for controllingthe electron transport characteristics.
Car et al. [11] studied the electronic properties of metal–molecule–metal systems at zero bias for benzene–, diben-zene–, and xylyl–dithiol molecules using ab initio calcu-lations. Their study focused in understanding how themolecular orbitals change upon adsorption on the electrodeand the orbitals closer to the Fermi level, which areimportant for the electron transport process. For thedithiol-benzene molecular junction, they noticed that theLUMO state is the one closer to the Fermi energy of theelectrodes and it is located at 0.16 eV below the Fermienergy. By studying the local density of states (LDOS),they found the existence of states available for conductionaround the Fermi energy which are delocalized throughoutthe molecule. They found that the insertion of methyl groupbetween the sulfur and the carbon ring decouples the twosubsystems and suppresses the LDOS at the Fermi energy.
Dilabio et al. [12] carried density functional theorycalculations combined with NEGF techniques to model theelectron transport through disubstituted benzenedithiolmolecules with electron acceptor and electron withdrawinggroups. The molecules were bonded by thiol groups to
612 J Mol Model (2012) 18:611–621

leads composed of 3x3 gold, 5x5 gold and 3x3 aluminumelectrodes. For the disubstituted 3x3 Au-benzenedithiol-Ausystems, the absence of transmission states from −0.4 to−0.2 eV, resulted in negative differential resistance in the I-V curve. This NDR feature is not observed in the 5x5 goldelectrodes due to the existence of states in the HOMO-LUMO transmission gap. The effect of electron donatingand withdrawing groups attached to the benzene was anincrease and a decrease in the current, respectively.
Xia et al. [13] used the NEGF formalism combined withdensity functional theory, to study the electronic transportproperties of the butadienimine-based optical molecularswitch with and without substituents. The two forms,namely the oxo-amine(keto) and hydroxyl-imine(enol)form, can reversibly switch from each other, making themgood candidates for light-driven molecular switches. Fromthe calculated I-V curves they found that the current fromthe enol form is higher than for the keto form, making itpossible to create a molecular switch. By studying thespatial distribution of the frontier orbitals, they found thatboth the HOMO and the LUMO are delocalized transportchannels for the enol, while the HOMO and LUMO for theketo form are both localized orbitals obstructing theelectron transport, because the electrons that enter themolecule at the energy of these orbitals have lowprobability of reaching the other end of the molecule. Theyalso found that the switching performance can be improvedby using appropriate electron donors or electron acceptorsubstituents.
Yang et al. [14] showed the effect that size of the basisset plays in determining the position of the Fermi energy ofthe lead in the molecular HOMO-LUMO gap, since thisposition is determined by the molecule-electrode chargetransfer. For example: for the Au(111)-OPE3-Au(111)molecular junction, the use of single zeta polarized basissets leads to the incorrect situation of the Fermi energybeing closer to the LUMO state, while using double andtriple zeta polarized basis sets gives the Fermi energy closerto the HOMO orbital.
Chelikowsky et al. [15] recently performed first-principles calculations on the electron transport in nanotubejunctions decorated with small Au nanoparticles. Theircalculations showed that the conductance of nanotubejunctions was increased only by the introduction of odd-numbered Au nanoparticles.
Goddard et al. [16] performed NEGF calculationsshowing that molecular junctions of cyclic and acyclic1,2-dithiolanes sandwiched between two gold electrodesexhibited essentially the same insulating current–voltagecharacteristics at moderate bias voltages, despite thedifference in their adsorption states.
Zhao et al. [17] performed DFT theoretical calculationabout the substituent effect on the molecular wire, oligo
(phenylene ethynylene) (OPE), using the electron-donating NH2 group, and the electron-withdrawing NO2
group. They considered in the DFT calculations theinfluence from an external electric field (EF), without themolecules being connected to electrodes and withoutterminal groups such as thiols, amides, etc. They statedthat the electrical properties of molecular electronicdevices are related to the electronic structure of the bridgemolecule.
Their study focused on the spatial distributions ofmolecular orbitals, especially frontier orbitals: HOMOand LUMO, as good indicators of electron transport ofthe molecular wire, only in the absence of quantuminterference [18], since frontier orbitals generally domi-nate the coupling to the electrode and the electrontransmission tunnel. Energy levels such as frontier orbitalscan be transmission channels [19], stating that a conduct-ing channel is a molecular orbital that is fully delocalizedalong the molecular backbone and a non-conductingchannel is a localized molecular orbital, that cannotconnect both ends of the molecule to the metallic electro-des. The important conclusion from their work was thedevelopment of a static treatment at the DFT level forpredicting the current–voltage behavior, by analyzing onlythe spatial distribution and energy levels under theinfluence of an external EF without having to resort tothe complex NEGF formalism. Later Zhao et al. [20]investigated molecular rectification in the porphyrin-basedmolecular junction originating from asymmetricalelectrode-molecule contacts using density functional the-ory combined with the NEGF method. They employedtwo thiol groups on the left side of the gold surface andone thiol group on the right side to increase the electroniccoupling. The Au(111) surface was simulated using a 4×4cell with periodic boundary conditions, and the supercellconsisted of two layers of Au atoms to the left and threelayers to the right with 16 atoms for each layer in thescattering region. Their transportation calculations in theseasymmetrical junctions showed obvious rectification, animportant effect in the design of molecular diodes.
Stokbro et al. [4] by calculating theoretically usingNEGF-DFT the conductance of junctions containing mol-ecules connected by thiol end groups, with different levelsof conjugation and of different lengths, found that theavailability of orbital density on the molecule end contactatoms is an important factor for obtaining high conductancevalues and can dominate both the conjugation of themolecule and its alignment with the electrode Fermi energy.When the conductive frontier molecular orbital lackssubstantial orbital density on the end group, the conductionwill be reduced significantly, even in there exists good thealignment between the molecular orbitals and the electrodeFermi energy.
J Mol Model (2012) 18:611–621 613

In this work we study the electronic transport of the S-FNPPB-o molecule connected with thiolate groups at theends as shown in Fig. 1, using two approaches. The first ofthese is the statical approach proposed by Zhao et al., [17,20], in which we study the molecular geometric andelectronic properties of the molecule under an appliedelectric field with the thiolate ends fixed. From this studywe hope to predict junction properties using the pathcontinuity approach for the electron transport, and a seconddynamical approach in which we employ the NEGF-DFTformalism to obtain the current–voltage characteristics at abias range form −2 to 2 V and the influence of the bias onthe transmission spectrum. The objective of this study is toelucidate the electronic transport of S-FNPPB-o to deter-mine if NDR is present in this molecule or note othernonlinear current voltage characteristics that can help in thedesign of new nanoelectronic devices.
We focus on the S-FNPPB-o fluorinated oligo(phenyl-ene ethynylene)s (OPE) molecule, since this moleculehas been previously synthesized by Tour et al. [21] withthe aim of using fluorination to alter the thermal, chemical,and electronic properties of OPE molecules and makethem suitable for integration with solid state devices. The4-(3-nitro-4-pentafluorophenylethynyl,phenylethynyl)ben-zenethiol (FNPPB-o) molecule can form stable self-assembled monolayers (SAMs) in gold and platinumsurfaces. Tour et al. also observed that the use offluorocarbons produced materials with increased thermalstability and chemical resistance, necessary characteristicsfor high-temperature production processes of SAMs onmetal surfaces, such as gas-phase physical vapor deposi-tion (PVD). Tour et al. also stated that molecularelectronic devices based on this fluorinated moleculemay lower the HOMO energy and the correspondingSchottky energy barrier, increasing the electron transmis-sion from a bulk contact through the frontier orbitals of achemisorbed organic film.
Tour et al. [22] also observed that FNPPB-o moleculesembedded in amide-containing alkanethiol SAMs on Au{111), presented bias-dependent switching as a function ofthe interaction between the dipole moment of the OPEs andthe electric field applied with the scanning tunnelingmicroscope.
Computational methodology
Figure 2a shows a schematic representation of the S-FNPPB-o molecular junction and Fig. 2b shows the S-FNPPB-o molecular junction, replacing the fluor atomswith hydrogen atoms named OPE-NO2 molecular junction.Before the introduction of an electrical field EF, the S-FNPPB-o molecule was fully optimized at the GGA-PBE/DNP level of theory using DMOL3 [23–25]. Then, theterminal sulfur atoms were fixed in space to simulate theconnection to the electrodes. Then we carry geometricaloptimizations at the same level of theory in the presence ofan external EF in the neutral state for this molecule sincethe remaining bonds in the sulfur atoms in the diradical aresaturated by gold atoms. Based on experimental resultsfrom SAMs attached to gold surfaces, it is generallyaccepted that hydrogen atoms of the thiol terminal groupdesorbs, while the sulfur atom in the radical binds stronglyto the gold surface [4, 11, 26]. The EF ranging from zero to0.257 V/Å is defined as uniform and aligned along the twoterminal carbon–sulfur inter-atomic vector, comparablewith most experimental measurements [17]. Both positiveand negative EF where applied to the molecule.
The disadvantage of this static computational methodol-ogy based in applying an EF to the S-FNPPB-o molecule isthat it does not take into consideration the effect of theelectrodes interface in the electronic transport. A morerigorous dynamic study was carried using the NEGF-DFTformalism to obtain the current–voltage characteristics ofthe molecular junction as well as the transmission spectrumof the molecular junction at different voltages (S-FNPPB-oattached to two Au(111) electrodes).
An overview of the NEGF-DFT formalism has beengiven by Stokbro et al. [27–29]. The conduction pathwayfor coherent motion of charge is comprised of threesegments: the intramolecular segment and the twomolecule-electrode interfaces. To achieve efficient conduc-tance, strong electronic coupling among the three segmentsshould be achieved. Such coupling is provided by closeenergetic lineup of the molecular states with the electrodeFermi energy (small injection gap) and by significantorbital overlap between the structural units within themolecule and at the molecule-electrodes interfaces (spectraldensity). We used the Atomistix Toolkit (ATK) softwarepackage [30] to study the electronic transport properties ofa molecule assembled in two Au (111) electrodes. Thisprogram combines DFT with NEGF to simulate theelectronic transport in single molecule devices undernonequilibrium conditions.
For the simulation of the molecular junction, thepreviously optimized S-FNPPB-o molecule at the DFTGGA-PBE/DNP level was translated into the Au junctionwith the Au(111) surface as a common feature in theFig. 1 Schematic representation of the S-FNPPB-o molecule
614 J Mol Model (2012) 18:611–621

software. (DNP) stands for double numerical plus polari-zation basis set. The Au(111) surface was simulated using a4×4 cell with periodic boundary conditions, and thesupercell consisted of two layers of gold atoms to the leftand three layers to the right with 16 atoms for each layer inthe scattering region. The complete molecular junction wasrelaxed using DNP basis set for all atoms and the GGA-PBE functional in the calculation, and the introduction ofrelativistic effects for some core electrons. All atoms exceptthe gold atoms, which were kept fixed at their bulkpositions, were relaxed until the force on each was lessthan 0.05 eV/Å. With this procedure we obtained endsulfur-surface distances of 1.95, and 1.97Å in the localenergy minimum, that is, the hollow site on the Au surface.This Au-S distance is in agreement with values obtained inthe literature [4, 10, 11, 13, 26, 31].
A bias voltage from −2.0 to 2.0 V was applied betweenthe two metal leads, and the electric current was generatedusing the Landauer-Buttiker formula [32–35].
The current through a single molecule connected by twometal electrodes is given by the following expression:
I ¼ 2e
h
ZmL
mR
T E;Vbð Þ fR E;Vbð Þ � fL E;Vbð Þ½ �dE ð1Þ
where fR(E, Vb), and fL(E, Vb) are the Fermi-Dirac functionsfor the left and right electrodes at energy E under the biasvoltage, Vb; T(E, Vb) is the sum of the transmissionprobabilities of all channels available at energy E underbias Vb [36], μL and μR are the chemical potentials of theleft and right electrodes; and [μL(Vb), μR(Vb)] is the energyregion for the current referred as the bias energy window.The bias energy window is given by μL=EF - eVb/2 and
μR=EF+eVb/2, and EF is the Fermi energy which is theaverage value of the chemical potential of the left and rightelectrodes, is usually set to zero [20].
From this equation 1, it can be observed that onlyelectrons with energies close to the Fermi level and withinthe bias energy windows add to the current. Since thecurrent is the integral of the transmission coefficient in thebias energy window, analysis of the transmission canprovide a comprehensible understanding of the electrontransport behavior.
Results and discussions
Table 1 shows the spatial distribution of the HOMO andLUMO for the S-FNPPB-o molecule at different electricalfields, frontier orbitals HOMO and LUMO, can becomegood indicators of electron transport of the molecular wirein the absence of quantum interference [18] because theydominate the coupling to the electrode and form theelectron transmission tunnel, and in the majority of thecases these orbitals align closer with the Fermi energy ofthe electrodes, becoming important channels for electrontransport. In the coherent tunneling regime, the electrontransport of a molecular junction can be studied in terms ofthe continuity of the conduction pathway.
In order to study and be able to modulate the electricalproperties of molecular wires, it is important to visualizehow the frontiers molecular orbital spatiate in response toan external EF. Taking into consideration that a conductingchannel is a molecular orbital that is fully delocalized alongthe molecular backbone; conversely, a non-conductingchannel is a localized molecular orbital, which cannotconnect both ends of the molecule to the metallic contacts
Fig. 2 (a)Molecular junctionstudied in this work with fluoratoms (b) OPE-NO2 molecularjunction, with hydrogen atomsinstead of fluor atoms
J Mol Model (2012) 18:611–621 615

Table 1 Spatial distribution of the HOMO and LUMO orbitals under an electric field for the S-FNPPB-o molecule
0.001 0.051
0.002 0.103
0.003 0.154
0.004 0.206
0.005 0.257
Electric Field homo lumo
ua V/Å
-0.005 -0.257
-0.004 -0.206
-0.003 -0.154
-0.002 -0.103
-0.001 -0.051
0 0.000
616 J Mol Model (2012) 18:611–621

and the electrons that enter at the energy of these localizedorbital have very low probabilities of transmission to theother end. [13, 17, 19].
From Table 1, we can observe that under the EF applied,the HOMO spatial distribution remains localized aroundonly one of the sulfur end atom at most EF, and the HOMOorbital delocalizes only at a negative EF value equal to−0.005 au; on the contrary, we observe that the LUMOorbital delocalizes in the molecular backbone at all positiveEF with a large orbital density at the sulfur atoms. Atnegative EF we observe scarce orbital density at the sulfur
left end side in the EF range from −0.001 to −0.002 au,signaling low electron transport at these EF. From theresults from Table 1 we can perceive that if the frontierorbitals align closer to the Fermi energy, the electrontransport would go mostly through the LUMO orbital,being the transport larger at positive EF, than at negative EFdue to the small orbital density at the left end side on thesulfur atom in the LUMO orbital.
Figure 3a shows the transmission spectrum and densitystates at 0 V bias for the S-FNPPB-o molecular junction, thezero energy corresponds to the Fermi energy of the electrodes,
Fig. 3 (a) Transmission spec-trum and DOS at 0 V for OPE-NO2 and S-FNPPB-o molecularjunctions (b) Transmissionspectrum and DOS at 2 V forOPE-NO2 and S-FNPPB-ojunctions (c) Transmissionspectrum and DOS at −2 V forOPE-NO2 and S-FNPPB-o mo-lecular junctions
J Mol Model (2012) 18:611–621 617

we observe that the LUMO orbital (−0.93 eV) lies muchcloser to the Fermi level than the HOMO orbital (−1.46 eV),and the electron transmission goes through the unoccupiedmolecular states, also the LUMO+1 orbital lies at 1.13 eV.From this figure we can see how the LUMO orbital is broader
with respect to the HOMO and LUMO+1 orbitals. FromFig. 3a at 0 V we can see a region from 0 to 0.8 eV showingnear zero transmission values. This behavior might be due tothe lack of states in the electrodes to couple with themolecular orbitals from the S-FNPPB-o bridge.
From the density of states at 0 V in Fig. 3a we canobserve a marked peak at the LUMO+1 orbital. In the sameFig. 3a, we compare the DOS and the transmissionspectrum at 0 V, for the S-FNPPB-o molecule replacingthe fluor atoms with hydrogen atoms named OPE-NO2
molecular junction, in order to assess the effect of thefluorination on the electron transport. We observe onlyminor differences in the transmission spectrum by compar-ing both molecular junctions. With respect to the DOS, weobserve in the OPE-NO2 molecular junction the appearanceof a marked peak near the HOMO orbital, absent in the S-FNPPB-o molecular junction.
The bias dependence of the transmission spectrum andDOS at 2 V and at -2 V is shown in Figs. 3b and c. Fromthese figures we can see that the LUMO+1 orbital becomescloser to the Fermi energy at 2 V than at -2 V. We can alsoobserve that the broad LUMO peak at 0 V center at −0.9 eVdoes not appears at this energy at -2 V and 2 V, and at thesevoltages there are strong peaks now centered at −1.33 eVand −1.79 eV.
Fig. 5 I-V curves for the OPE-NO2 and S-FNPPB-o molecularjunctions
Fig. 4 Complete dependence ofthe transmission spectrums as afunction of the applied voltagerange [−2 V, 2 V] for the S-FNPPB-o molecular junction
618 J Mol Model (2012) 18:611–621
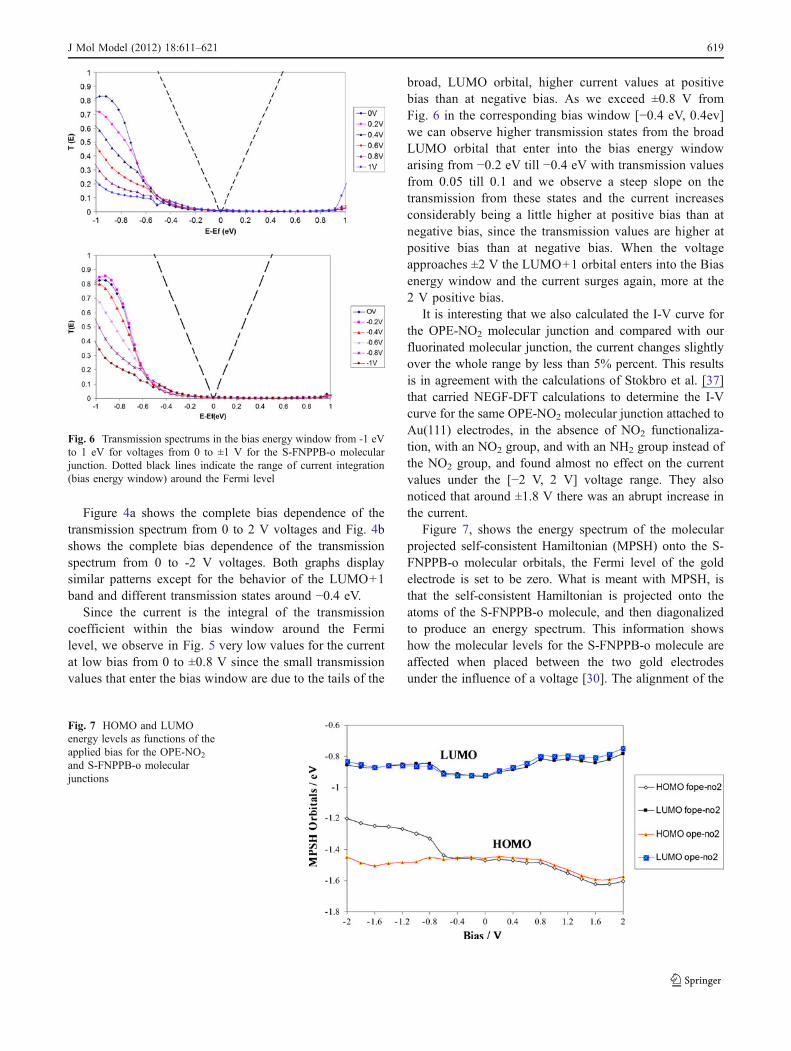
Figure 4a shows the complete bias dependence of thetransmission spectrum from 0 to 2 V voltages and Fig. 4bshows the complete bias dependence of the transmissionspectrum from 0 to -2 V voltages. Both graphs displaysimilar patterns except for the behavior of the LUMO+1band and different transmission states around −0.4 eV.
Since the current is the integral of the transmissioncoefficient within the bias window around the Fermilevel, we observe in Fig. 5 very low values for the currentat low bias from 0 to ±0.8 V since the small transmissionvalues that enter the bias window are due to the tails of the
broad, LUMO orbital, higher current values at positivebias than at negative bias. As we exceed ±0.8 V fromFig. 6 in the corresponding bias window [−0.4 eV, 0.4ev]we can observe higher transmission states from the broadLUMO orbital that enter into the bias energy windowarising from −0.2 eV till −0.4 eV with transmission valuesfrom 0.05 till 0.1 and we observe a steep slope on thetransmission from these states and the current increasesconsiderably being a little higher at positive bias than atnegative bias, since the transmission values are higher atpositive bias than at negative bias. When the voltageapproaches ±2 V the LUMO+1 orbital enters into the Biasenergy window and the current surges again, more at the2 V positive bias.
It is interesting that we also calculated the I-V curve forthe OPE-NO2 molecular junction and compared with ourfluorinated molecular junction, the current changes slightlyover the whole range by less than 5% percent. This resultsis in agreement with the calculations of Stokbro et al. [37]that carried NEGF-DFT calculations to determine the I-Vcurve for the same OPE-NO2 molecular junction attached toAu(111) electrodes, in the absence of NO2 functionaliza-tion, with an NO2 group, and with an NH2 group instead ofthe NO2 group, and found almost no effect on the currentvalues under the [−2 V, 2 V] voltage range. They alsonoticed that around ±1.8 V there was an abrupt increase inthe current.
Figure 7, shows the energy spectrum of the molecularprojected self-consistent Hamiltonian (MPSH) onto the S-FNPPB-o molecular orbitals, the Fermi level of the goldelectrode is set to be zero. What is meant with MPSH, isthat the self-consistent Hamiltonian is projected onto theatoms of the S-FNPPB-o molecule, and then diagonalizedto produce an energy spectrum. This information showshow the molecular levels for the S-FNPPB-o molecule areaffected when placed between the two gold electrodesunder the influence of a voltage [30]. The alignment of the
Fig. 6 Transmission spectrums in the bias energy window from -1 eVto 1 eV for voltages from 0 to ±1 V for the S-FNPPB-o molecularjunction. Dotted black lines indicate the range of current integration(bias energy window) around the Fermi level
Fig. 7 HOMO and LUMOenergy levels as functions of theapplied bias for the OPE-NO2
and S-FNPPB-o molecularjunctions
J Mol Model (2012) 18:611–621 619

molecular orbitals in relation to the Fermi level is veryimportant, since states near the Fermi level are the maincontributors to electron transport [36].
From Fig. 7 we can observe that the HOMO for the S-FNPPB-o molecular junction decreases under positive biasand increases at negative bias, while the LUMO for the S-FNPPB-o molecular junction, remains more or less at thesame level, leading to an asymmetrical HOMO-LUMO gap(HLG). Compared with the OPE-NO2 molecular junction,the effect of the fluorination is to increase the HOMOorbital from −0.8 to −2 V. At other voltages the HOMOlevels for both junctions do not differ significantly. Also,when comparing the LUMO orbitals for both junctions wedo not observe any significant difference.
Conclusions
We have presented theoretical calculation where an electricfield was applied to the S-FNPPB-o molecule without goldelectrodes, ‘static treatment’. From the visualization of thespatial distribution of the frontier orbitals, we noticed thatonly the LUMO orbital is delocalized at the molecularbackbone at most EF ranges, signaling and importantelectron transport channel. We perceive qualitatively thatthe electron transport would be larger at positive EFs thanat negative EFs, due to the small orbital density at the leftend side on the sulfur atom at some negative EFs values inthe LUMO orbital.
By carrying NEGF-DFTcalculations we calculated the I-Vcurve for the S-FNPPB-o molecular junction and the biasdependence of the transmission spectrum. The transmissionspectrum at 0 Volts has three peaks closer to the Fermi energyof the electrodes. However, only one transmission peakcorresponding to the LUMO orbital is closer to the Fermienergy. That means that there are three electron transportchannels, but only one of the channels, namely the LUMO,makes its contribution to the current in the [−0.9 eV, 0.9 eV]energy bias window. Only when the voltage approaches ±2 Vdoes the LUMO+1 orbital enter into the [−1 eV, 1 eV] biasenergy window and we observe an increase in the current, butat the voltage range studied the HOMO orbital never entersinto the bias energy window.
From our calculations we observed that the I-V curve forthe OPE-NO2 molecular junction compared with our S-FNPPB-o fluorinated molecular junction changes by lessthan 5% percent, so the fluorination does not change the I-V characteristics in the OPE-NO2 molecular junction in thevoltage range studied.
It is interesting to note that both first principlecalculations, the static EF treatment and the NEGF-DFTformalism, coincide in that the electronic transport has to bethrough the unoccupied frontier orbitals.
Acknowledgments This work was supported by Programa deMejoramiento del Profesorado (PROMEP) under grant No. 103.5/08/3301. L. Serrato-Villegas gratefully acknowledges a doctoral fellow-ship from the National Science and Technology Council in Mexico.M.T. R, M. D-R, D. G-M and Marco Gallo are Researchers from theNational Science and Technology Council in Mexico.
References
1. Moore GE (1965) Cramming more components onto integratedcircuits. Electronics 8:114–117
2. Service RF (2001) Molecules get wired. Science 294:2442–2443.doi:10.1126/science.294.5551.2442
3. Seminario JM, Araujo RA, Yan LJ (2004) Negative differentialresistance in metallic and semiconducting clusters. J Phys Chem B108:6915–6918. doi:10.1021/jp037781l
4. Revital C, Stokbro K, Martin JML, Ratner MA (2007) Chargetransport in conjugated aromatic molecular junctions: Molecularconjugation and molecule − electrode Coupling. J Phys Chem C111:14893–14902. doi:10.1021/jp0795309
5. Kim WY, Kim KS (2010) Tuning molecular orbitals in molecularelectronics and spintronics. Acc Chem Res 43:111–120.doi:10.1021/ar900156u
6. Lindsay SM, Ratner MA (2007) Molecular transport junctions:Clearing mists. Adv Mater 19:23–31. doi:10.1002/adma.200601140
7. Derosa PA, Seminario JM (2001) Electron transport throughsingle molecules: scattering treatment using density functional andgreen function theories. J Phys Chem B 105:471–481.doi:10.1021/jp003033+
8. Yan L, Bautista EJ, Seminario JM (2007) Ab Initio analysis ofelectron currents through benzene, naphthalene, and anthracenenanojunctions. Nanotechnology 18:485701(1–8). doi:10.1088/0957-4484/18/48/485701
9. Cho Y, Kim WY, Kim KS (2009) Effect of electrodes onelectronic transport of molecular electronic devices. J Phys ChemA 113:4100–4104. doi:0.1021/jp810467q
10. Lee SU, Belosludov RV, Mizuseki H, Kawazoe Y (2007) Controlof electron transport by manipulating the conjugated framework. JPhys Chem C 111:15397–15403. doi:10.1021/jp074294n
11. Piccinin S, Selloni A, Scandolo S, Car R, Scoles G (2003)Electronic properties of metal-molecule-metal systems at zerobias: A periodic density functional study. J Chem Phys 119:6729–6735. doi:10.1063/1.1602057
12. Smeu M, Wolkow RA, DiLabio GA (2008) Theoretical investi-gation of electron transport modulation through benzenedithiol bysubstituent groups. J Chem Phys 129:034707(1–8). doi:10.1063/1.2955463
13. Xia C, Liu D, Fang C, Zhao P (2010) The I-V characteristics ofthe butadienimine-based optical molecular switch: An ab initio.Physica E 42:1763–1768. doi:10.1016/j.physe.2010.01.044
14. Ke S, Baranger HU, Yang W (2007) Electron transport throughsingle conjugated organic molecules: Basis set effects in ab initiocalculations. J Chem Phys 127:144107-1-144107-6. doi: 10.1063/1.2770718
15. Khoo KH, Chelikowsky JR (2009) Electron transport acrosscarbon nanotube junctions decorated with Au nanoparticles:Density functional calculations. Physical review B 79:205422(1–6). doi:10.1103/PhysRevB.79.205422
16. Jang YH, Goddard WA III (2008) Electron transport throughcyclic disulfide molecular junctions with two different adsorptionstates at the contact: A density functional theory study. J PhysChem C 112:8715–8720. doi:10.1021/jp800201z
17. Li Y, Zhao J, Yin G (2007) Theoretical investigations of oligo(phenylene ethylene) molecular wire: Effects from substituents
620 J Mol Model (2012) 18:611–621

and external electric field. Comput Mater Sci 39:775–781.doi:10.1016/j.commatsci.2006.09.010
18. Solomon GC, Andrews DQ, van Duyne RP, Ratner MA (2008)When things are not as they seem: Quantum interference turnsmolecular electron transfer “rules” upside down. J Am Chem Soc130:7788–7789. doi:10.1021/ja801379b
19. Nitzan A, Ratner MA (2003) Electron transport in molecular wirejunctions. Science 30:1384–1389. doi:10.1126/science.1081572
20. Zhao J, Yu C, Wang N, Liu H (2010) Molecular rectificationbased on asymmetrical molecule-electrode contact. J Phys ChemC 114:4135–4141. doi:10.1021/jp905713a
21. Maya F, Chanteau SH, Cheng L, Stewart MP, Tour JM (2005)Synthesis of fluorinated oligomers toward physical vapor deposi-tion molecular electronics candidates. Chem Mater 17:1331–1345.doi:10.1021/cm0486161
22. Lewis PA, Inman CE, Maya F, Tour JM, Hutchison JE, Weiss PS(2005) Molecular engineering of the polarity and interactions ofmolecular electronic switches. J Am Chem Soc 127:17421–17426.doi:10.1021/ja055787d
23. a) www.accelrys.com24. Delley B (1990) An all-electron numerical method for solving the
local density functional for polyatomic molecules. J Chem Phys92:508–518. doi:10.1063/1.458452
25. Delley B (2000) From molecules to solids with the DMol3approach. J Chem Phys 113:7756–7764. doi:10.1063/1.1316015
26. Xia CJ, Fang CF, Zhao P, Liu HC (2010) Effects of contact atomicstructure on the electron transport of pyridine-substituted dithie-nylethene optical molecular switch. Eur Phys J D 59:375–378.doi:10.1140/epjd/e2010-00196-2
27. Stokbro K, Taylor J, Brandbyge M, Mozos JL, Ordejon P (2003)Theoretical study of the nonlinear conductance of Di-thiol benzenecoupled to Au(1 1 1) surfaces via thiol and thiolate bonds. ComputMater Sci 27:151–160. doi:10.1016/S0927-0256(02)00439-1
28. Stokbro K (2008) First-principles modeling of electron transport. JPhys Condens Mater 20:064216(1–6). doi:10.1088/0953-8984/20/6/064216
29. Datta S (2005) Quantum Transport: Atom to transistor. CambridgeUniversity Press, New York
30. www.quantumwise.com31. Miao L, Seminario JM (2007) Electronic and structural properties
of oligophenylene ethynylenes on Au(111) surfaces. J Chem Phys126:184706(1–7). doi:10.1063/1.2734545
32. Datta S (1996) Electronic Transport in the Mesoscopic Systems.Cambridge University Press, New York
33. Meir Y, Wingreen NS (1992) Landauer formula for the currentthrough an interacting electron region. Phys Rev Lett 68:2512–2515. doi:10.1103/PhysRevLett.68.2512
34. Landauer R (1957) Spatial variation of currents and fields due tolocalized scatterers in metallic conduction. IBM J Res Dev 1:223–231. doi:10.1147/rd.13.0223
35. Buttiker M (1988) Coherent and sequential tunneling in seriesbarriers. IBM J Res Dev 32:63–75. doi:10.1147/rd.321.0063
36. Bai P, Li E, Neerja CP (2005) Theoretical investigation of metal-molecule interface with terminal group. IEEE Trans Nanotechnol4:422–429. doi:10.1109/TNANO.2005.851282
37. Stokbro K, Taylor J, Brandbyge M, Ordejón P (2003) TranSI-ESTA: A spice for molecular electronics. Ann NY Acad Sci1006:212–226. doi:10.1196/annals.1292.014
J Mol Model (2012) 18:611–621 621




















