
A p56lck-independent Pathway of CD2 Signaling Involves Jun Kinase
10
A p56 lck -independent Pathway of CD2 Signaling Involves Jun Kinase* (Received for publication, March 5, 1998, and in revised form, June 5, 1998) Raute Sunder-Plassmann‡ and Ellis L. Reinherz§ From the Laboratory of Immunobiology, Dana-Farber Cancer Institute and Department of Medicine, Harvard Medical School, Boston, Massachusetts 02115 The p56 lck Src family non-receptor tyrosine kinase has been shown to be critical for T lymphocyte differentiation and activation. Hence in the absence of p56 lck , T cell re- ceptor triggered activation does not occur. We now pro- vide evidence for a CD2-based signaling pathway which, in contrast to that of the T cell receptor, is independent of p56 lck . CD2-mediated interleukin-2 production occurs via activation of Jun kinase in cell lines lacking p56 lck . Jun kinase then facilitates the binding of c-Jun/c-Fos het- erodimers to the AP-1 consensus site and the subsequent transcriptional activity of the interleukin-2 promoter. These data elucidate differences between TCR and CD2 signaling pathways in the same T cells. CD2 is a cell surface glycoprotein which is expressed on immature thymocytes and on mature T cells and NK cells (1–3). The ligand for human CD2 is CD58 (LFA-3), a ubiqui- tously expressed cell surface protein found on many cell types including antigen presenting cells (4, 5). Unlike integrins such as CD11a/CD18 (LFA-1), the adhesion between CD2 and CD58 is not dependent on TCR 1 triggering (6, 7). Rather, the CD2- CD58 interaction between T cells and their cognate partners facilitates the T cell recognition process and subsequent T cell activation (8 –10). Cross-linking of CD2 molecules by specific pairs of mAbs recognizing distinct epitopes on human CD2 initiates a signal- ing cascade which leads to T cell cytokine production, prolifer- ation, and cytolytic activity (11–13). For optimal mAb-mediated activation two antibody specificities are needed, one directed against conventional CD2 epitopes and the second against an activation-associated epitope of CD2, termed CD2R. The CD2R epitope has recently been mapped to the flexible linker region between the two extracellular domains of CD2 (14). Its appear- ance coincides with reorientation of the adhesion domain (D1) relative to the membrane proximal domain (D2) of the CD2 extracellular segment. Such domain reorientation occurs upon CD58 binding to CD2 and following T cell activation (14). Furthermore, the CD2 interaction with CD58 regulates the responsiveness of activated human T cells to IL-12 (15, 16). Hence, both IL-12-stimulated T cell proliferation and interfer- on-g production are markedly augmented by CD2 ligation. Finally, some studies have uncovered a unique role for CD2 in the regulation of anergy (17). Stimulation of anergized alloreac- tive T cells with a combination of specific alloantigen in con- junction with CD2-CD58 co-receptor ligation reverses the an- ergic state. In contrast, identical allostimulation but in the absence of CD2-CD58 co-ligation on T cells and allostimulators, respectively, fails to restore responsiveness (17). Signaling through CD2 is dependent on its cytoplasmic do- main (8, 18 –20). Comparison of CD2 from various species shows the highest homology occurring in this segment (21, 22). While the cytoplasmic domain has no intrinsic protein-tyrosine kinase activity and no tyrosine residues which might serve as docking sites for SH2 domains upon phosphorylation (reviewed in Refs. 23 and 24), stimulation via CD2 leads to the tyrosine phosphorylation of several intracellular proteins (25–27). For these activation events CD2 signaling requires the presence of the CD3z chain in T cells (27–31) or FceRIg in CD16 expressing NK cells (32). The CD2 tail contains several proline-rich regions (19). Two of these (PPPGHR) are known to be important in inducing IL-2 production (33). Related proline-rich sequences bind to SH3 domains of non-receptor protein-tyrosine kinases of the Src family (23). For example, the SH3 domain of p56 lck has been shown to bind to the rat CD2 cytoplasmic tail at least in vitro (34). Peptides corresponding to residues 269 –270 and 200 –310 (according to the human CD2 numbers) bind to p56 lck -GST fusion proteins (34). In vivo intracellular colocalization exper- iments, however, suggest, that p59 fyn rather than p56 lck has a predominant association with CD2 (35), perhaps by binding through the p59 fyn SH3 domain. The activation of protein tyrosine kinases is one of the prox- imal steps in signaling cascades which ultimately lead to the activation of T cell effector function. For p56 lck it has been shown that this kinase is involved in the activation of the Ras/Raf/MAPK pathway (36). The function of MAPK is mani- fold. Several studies reported that MAPK can phosphorylate and activate the transcription factors ELK, Jun, and Stat-1 and thereby participates in the regulation of gene expression (37). A different pathway leading to Jun activation is the Rac/JNK pathway (38). Recently, the Pyk2 tyrosine kinase has been described, shown to be involved in the activation of JNK (39), and has been linked to TCR signaling via p59 fyn (40). In this study, we have begun to dissect the differences in TCR- and CD2-based signaling pathways. Since p56 lck is thought to be most important at a proximal step in CD3 sig- naling, we investigated the role p56 lck plays in CD2 signaling. To this end, we have employed the Jurkat variant JCaM1.6s * This work was supported in part by National Institutes of Health Grants AI21226 and AI19807. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. ‡ Supported by Erwin Schro ¨dinger Auslandsstipendium J01036- MED from the Fonds zur Fo ¨ rderung der Wissenschaftlichen Forschung, Vienna, Austria. § To whom correspondence should be addressed: Laboratory of Im- munobiology, Dana-Farber Cancer Institute, 44 Binney St., Boston, MA 02115. Tel.: 617-632-3412; Fax: 617-632-3351; E-mail: ellis_reinherz @dfci.harvard.edu. 1 The abbreviations used are: TCR, T cell receptor; mAb, monoclonal antibody; IL, interleukin; MAPK, mitogen-activated protein kinase; PLC, phospholipase C; PMSF, phenylmethylsulfonyl fluoride; PAGE, polyacrylamide gel electrophoresis; PMA, phorbol 12-myristate 13-ace- tate; DTT, dithiothreitol; GST, glutathione S-transferase; EMSA, elec- trophoretic mobility shift assay. THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 273, No. 37, Issue of September 11, pp. 24249 –24257, 1998 © 1998 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. This paper is available on line at http://www.jbc.org 24249 by guest on December 3, 2015 http://www.jbc.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of A p56lck-independent Pathway of CD2 Signaling Involves Jun Kinase

A p56lck-independent Pathway of CD2 Signaling Involves JunKinase*
(Received for publication, March 5, 1998, and in revised form, June 5, 1998)
Raute Sunder-Plassmann‡ and Ellis L. Reinherz§
From the Laboratory of Immunobiology, Dana-Farber Cancer Institute and Department of Medicine, Harvard MedicalSchool, Boston, Massachusetts 02115
The p56lck Src family non-receptor tyrosine kinase hasbeen shown to be critical for T lymphocyte differentiationand activation. Hence in the absence of p56lck, T cell re-ceptor triggered activation does not occur. We now pro-vide evidence for a CD2-based signaling pathway which,in contrast to that of the T cell receptor, is independent ofp56lck. CD2-mediated interleukin-2 production occurs viaactivation of Jun kinase in cell lines lacking p56lck. Junkinase then facilitates the binding of c-Jun/c-Fos het-erodimers to the AP-1 consensus site and the subsequenttranscriptional activity of the interleukin-2 promoter.These data elucidate differences between TCR and CD2signaling pathways in the same T cells.
CD2 is a cell surface glycoprotein which is expressed onimmature thymocytes and on mature T cells and NK cells(1–3). The ligand for human CD2 is CD58 (LFA-3), a ubiqui-tously expressed cell surface protein found on many cell typesincluding antigen presenting cells (4, 5). Unlike integrins suchas CD11a/CD18 (LFA-1), the adhesion between CD2 and CD58is not dependent on TCR1 triggering (6, 7). Rather, the CD2-CD58 interaction between T cells and their cognate partnersfacilitates the T cell recognition process and subsequent T cellactivation (8–10).
Cross-linking of CD2 molecules by specific pairs of mAbsrecognizing distinct epitopes on human CD2 initiates a signal-ing cascade which leads to T cell cytokine production, prolifer-ation, and cytolytic activity (11–13). For optimal mAb-mediatedactivation two antibody specificities are needed, one directedagainst conventional CD2 epitopes and the second against anactivation-associated epitope of CD2, termed CD2R. The CD2Repitope has recently been mapped to the flexible linker regionbetween the two extracellular domains of CD2 (14). Its appear-ance coincides with reorientation of the adhesion domain (D1)relative to the membrane proximal domain (D2) of the CD2extracellular segment. Such domain reorientation occurs upon
CD58 binding to CD2 and following T cell activation (14).Furthermore, the CD2 interaction with CD58 regulates theresponsiveness of activated human T cells to IL-12 (15, 16).Hence, both IL-12-stimulated T cell proliferation and interfer-on-g production are markedly augmented by CD2 ligation.Finally, some studies have uncovered a unique role for CD2 inthe regulation of anergy (17). Stimulation of anergized alloreac-tive T cells with a combination of specific alloantigen in con-junction with CD2-CD58 co-receptor ligation reverses the an-ergic state. In contrast, identical allostimulation but in theabsence of CD2-CD58 co-ligation on T cells and allostimulators,respectively, fails to restore responsiveness (17).
Signaling through CD2 is dependent on its cytoplasmic do-main (8, 18–20). Comparison of CD2 from various speciesshows the highest homology occurring in this segment (21, 22).While the cytoplasmic domain has no intrinsic protein-tyrosinekinase activity and no tyrosine residues which might serve asdocking sites for SH2 domains upon phosphorylation (reviewedin Refs. 23 and 24), stimulation via CD2 leads to the tyrosinephosphorylation of several intracellular proteins (25–27). Forthese activation events CD2 signaling requires the presence ofthe CD3z chain in T cells (27–31) or FceRIg in CD16 expressingNK cells (32).
The CD2 tail contains several proline-rich regions (19). Twoof these (PPPGHR) are known to be important in inducing IL-2production (33). Related proline-rich sequences bind to SH3domains of non-receptor protein-tyrosine kinases of the Srcfamily (23). For example, the SH3 domain of p56lck has beenshown to bind to the rat CD2 cytoplasmic tail at least in vitro(34). Peptides corresponding to residues 269–270 and 200–310(according to the human CD2 numbers) bind to p56lck-GSTfusion proteins (34). In vivo intracellular colocalization exper-iments, however, suggest, that p59fyn rather than p56lck has apredominant association with CD2 (35), perhaps by bindingthrough the p59fyn SH3 domain.
The activation of protein tyrosine kinases is one of the prox-imal steps in signaling cascades which ultimately lead to theactivation of T cell effector function. For p56lck it has beenshown that this kinase is involved in the activation of theRas/Raf/MAPK pathway (36). The function of MAPK is mani-fold. Several studies reported that MAPK can phosphorylateand activate the transcription factors ELK, Jun, and Stat-1 andthereby participates in the regulation of gene expression (37). Adifferent pathway leading to Jun activation is the Rac/JNKpathway (38). Recently, the Pyk2 tyrosine kinase has beendescribed, shown to be involved in the activation of JNK (39),and has been linked to TCR signaling via p59fyn (40).
In this study, we have begun to dissect the differences inTCR- and CD2-based signaling pathways. Since p56lck isthought to be most important at a proximal step in CD3 sig-naling, we investigated the role p56lck plays in CD2 signaling.To this end, we have employed the Jurkat variant JCaM1.6s
* This work was supported in part by National Institutes of HealthGrants AI21226 and AI19807. The costs of publication of this articlewere defrayed in part by the payment of page charges. This article musttherefore be hereby marked “advertisement” in accordance with 18U.S.C. Section 1734 solely to indicate this fact.
‡ Supported by Erwin Schrodinger Auslandsstipendium J01036-MED from the Fonds zur Forderung der Wissenschaftlichen Forschung,Vienna, Austria.
§ To whom correspondence should be addressed: Laboratory of Im-munobiology, Dana-Farber Cancer Institute, 44 Binney St., Boston, MA02115. Tel.: 617-632-3412; Fax: 617-632-3351; E-mail: [email protected].
1 The abbreviations used are: TCR, T cell receptor; mAb, monoclonalantibody; IL, interleukin; MAPK, mitogen-activated protein kinase;PLC, phospholipase C; PMSF, phenylmethylsulfonyl fluoride; PAGE,polyacrylamide gel electrophoresis; PMA, phorbol 12-myristate 13-ace-tate; DTT, dithiothreitol; GST, glutathione S-transferase; EMSA, elec-trophoretic mobility shift assay.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 273, No. 37, Issue of September 11, pp. 24249–24257, 1998© 1998 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 24249
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from

which lacks functional p56lck and is unable to transmit TCRsignals leading to IL-2 production. We here report that al-though the TCR pathway is not functional in JCaM1.6s, thesecells can be induced to produce IL-2 by stimulation via CD2.Furthermore, we present evidence for a p56lck-independentCD2 signal transduction pathway which uses Pyk2 and JNKand leads to the binding of c-Jun/c-Fos heterodimers to theAP-1 consensus site, important for initiation of IL-2transcription.
EXPERIMENTAL PROCEDURES
Cell Lines and Antibodies
Monoclonal antibodies (mAbs): anti-T111, anti-T112, and anti-T113
recognizing different epitopes of human CD2 (11) and the anti-humanCD3e mAbs 2AD2A2 and RW28C8 (41) were developed in our labora-tory at Dana Farber Cancer Institute, Boston, MA. 4G10 (anti-phos-photyrosine, IgG2b) was kindly provided by Tom Roberts (Dana FarberCancer Institute). Rabbit antisera against ZAP70, p56lck, and Pyk2were kind gifts from J. Bolen (DNAX, Palo Alto, CA), A. Veillette(McGill Cancer Center, Montreal, Quebec, Canada), and J. Schlessinger(New York University Medical Center, New York), respectively. Anti-bodies against PLC-g1 were obtained from Upstate Biotechnology Inc.,Lake Placid, NY, and rabbit antisera against p59fyn, Erk1/MAPK, JNK,c-Jun, JunB, JunD, c-Fos, Fra1, Fra2, and FosB were purchased fromSanta Cruz Biotechnology Inc., Santa Cruz, CA. Rabbit antiserumagainst pMAPK was obtained from New England Biolabs, Beverly, MA.
Cell Lines-The Jurkat variant, JCaM1.6, that lacks p56lck (42) wasobtained from ATCC (Rockville, MD) and sorted for high CD2 and CD3expression (JCaM1.6s) and further sorted for those cells which show afast and high rise in intracellular free Ca21 levels following CD2 stim-ulation (JCaM1.6s.S3). Both JCaM1.6s and JCaM1.6s.S3 were ana-lyzed for p56lck by in vitro kinase activity and p56lck was found to be notenzymatically active. J77 is a CD2posCD3posCD8neg subclone of theJurkat cell line (29).
Ca21 Assay
Ca21 assay was performed as described (43). Increase in intracellularfree Ca21 in Indo-1 (Molecular Probes Inc., Eugene, OR) loaded cellswas induced by either a combination of the anti-CD2 mAbs anti-T112
and anti-T113 (1:100) or 20 mg/ml biotinylated RW28C8 alone or byfurther cross-linking with 100 mg/ml avidin (Sigma). Maximal Ca21
influx was induced by the addition of 10 mg/ml 4-bromo-calcium iono-phore A23187 (Sigma). The analysis was performed on an EPICS V cellsorter (Coulter, Hialeah, FL).
Analysis of Tyrosine Phosphorylation
Immunoprecipitation was performed as described previously (43). Inbrief, cells (1 3 107) were stimulated for 5 min at 37 °C with either acombination of the anti-CD2 mAbs anti-T112 and anti-T113 (1:100) orthe anti-CD3e mAb 2AD2A2 (1:100). Cells were then washed withice-cold TBS (50 mM Tris-HCl, pH 7.4, 150 mM NaCl), resuspended inlysis buffer (1 3 107 cells/ml; 1% Triton X-100 in TBS, 10 mg/ml leu-peptin (Sigma), 0.2 TIU/ml aprotinin (Sigma), 1 mM PMSF (Sigma), 5mM EDTA, 1 mM Na3VO4 (Fisher Scientific, Fair Lawn, NJ), 5 mM
Na2H2P2O7 (Sigma), and 5 mM sodium fluoride (Fisher) or 25 mM
b-glycerophosphate (Sigma)) and rotated for 30 min at 4 °C followed bycentrifugation at 13,000 3 g for 5 min. Proteins were immunoprecipi-tated from 1 ml of postnuclear lysate with 10 ml of GammaBind Plus-Sepharose (Pharmacia Biotech Inc., Uppsala, Sweden) preincubatedwith the respective Abs. Beads bound immune complexes were washedwith lysis buffer and TBS and were eluted by boiling in SDS (Laemmli)sample buffer. The samples were analyzed by SDS-PAGE and Westernblotted as described below.
In Vitro Kinase Assay—Lysis of cells (1 3 107 cells/ml) and immuno-precipitation was carried out as described above. After washing withlysis buffer and kinase buffer (100 mM NaCl, 5 mM MnCl2, 5 mM MgCl2,20 mM Hepes, pH 7.4) the beads bound immune complexes were resus-pended in 50 ml of kinase buffer containing 2 mM ATP and were incu-bated with 10 mCi of [g-32P]ATP for 15 min at room temperature.Kinase reaction was stopped by addition of ice-cold lysis buffer contain-ing 20 mM EDTA. After washing 3 times with lysis buffer/EDTA andonce with Tris-HCl, pH 7.4, the phosphorylated proteins were eluted byboiling in SDS sample buffer. The proteins were analyzed by 9% SDS-PAGE and after drying the gels were subjected to autoradiography.Some in vitro kinase assays were carried out in the presence of 10mg/sample poly(Glu-Tyr) (4:1) (Sigma). The kinase reaction was stopped
by boiling in SDS sample buffer and the samples were further processedas described above.
Western Blotting—Western blotting onto nitrocellulose membranesof proteins resolved by SDS-PAGE was performed according to stand-ard procedures. Protein detection was carried out by incubating themembranes with 1:1000 dilutions of specific mAbs (4G10, PLC-g1) orrabbit heteroantisera (ZAP70, p56lck, p59fyn, Pyk2, pMAPK, Erk1/MAPK, Raf) for 1 h at room temperature. The blots were washed withTBS/Triton X-100 (0.05%) and incubated with 1:2000 dilutions ofHRPO-labeled anti-mouse IgG2b, anti-mouse IgG, or anti-rabbit Abs(Caltag Laboratories, San Francisco, CA), respectively, for 1 h at roomtemperature. After washing with TBS/Triton X-100, the proteins werevisualized by enhanced chemiluminescence (ECL Western blotting de-tection reagents, Amersham International, Little Chalfont, Bucking-hamshire, United Kingdom) and exposing the membranes to films forvarious time intervals. Unspecific binding of Abs was inhibited bypreincubating the membranes with blocking buffer (TBS containingeither 5% fetal calf serum or 5% non-fat dry milk and 10 mM NaN3) forat least 2 h at room temperature.
Jun Kinase Assay
1 3 107 cells were stimulated for 30 min at 37 °C with either acombination of the anti-CD2 mAbs anti-T112 and anti-T113 (1:100), theanti-CD3e mAb 2AD2A2 (1:100), or 25 ng/ml phorbol ester (PMA). Cellswere then washed with ice-cold TBS and lysed for 30 min at 4 °C in 100ml of JNK-lysis buffer (25 mM HEPES pH 7.7, 300 mM NaCl, 1.5 mM
MgCl2, 0.1% Triton X-100, 0.5 mM DTT, 0.2 mM EDTA, 2 mg/ml leupep-tin (Sigma), 1 mM PMSF (Sigma), 0.1 mM Na3VO4 (Fisher Scientific), 20mM b-glycerophosphate (Sigma) followed by centrifugation at 13,000 3g for 5 min. The supernatants were then diluted 1:4 to a final concen-tration of 20 mM HEPES pH 7.7, 75 mM NaCl, 2.5 mM MgCl2, 0.05%Triton X-100, 0.5 mM DTT, 0.1 mM EDTA, 2 mg/ml leupeptin (Sigma), 1mM PMSF (Sigma), 0.1 mM Na3VO4 (Fisher Scientific), 20 mM b-glyc-erophosphate (Sigma) and rotated for 4 h at 4 °C with 10 ml of Gam-maBind Plus-Sepharose (Pharmacia) preincubated with anti-JNK Ab.Bead-bound immune complexes were washed with lysis buffer andkinase buffer (20 mM MgCl2, 20 mM b-glycerophosphate, 0.1 mM
Na3VO4, 2 mM DTT, 10 mM Hepes, pH 7.4) and subsequently resus-pended in 50 ml of kinase buffer containing 20 mM ATP and 2 mg/sampleGST-Jun fusion protein (kindly provided by M. Karin) and were incu-bated with 5 mCi of [g-32P]ATP for 20 min at 30 °C. The kinase reactionwas stopped by boiling in SDS (Laemmli) sample buffer and the sam-ples were analyzed by 10% SDS-PAGE and after drying the gels weresubjected to autoradiography.
IL-2 Production Assay
1 3 105 cells/well were stimulated in Immulon enzyme-linked immu-nosorbent assay plates (Dynatech Laboratories Inc., Chantilly, VA)with either PMA alone (25 ng/ml, Sigma), PMA plus a combination ofthe anti-CD2 mAbs anti-T112 and anti-T113 (1:100), PMA plus theanti-CD3e mAb RW28C8 (1 mg/well, precoated overnight onto the platesat 4 °C) or PMA plus calcium ionophore A23187 (1 mg/ml). After 48 hculture supernatants were harvested and examined by human IL-2specific enzyme-linked immunosorbent assays (Endogen Inc., Cam-bridge, MA).
Nuclear Extracts and EMSA
5 3 107 cells, unstimulated or stimulated for 4 or 6 h at 37 °C witheither a combination of stimulatory anti-CD2 (anti-T112 plus anti-T113,1:100) or with a 1:100 dilution of anti-CD3e mAb 2AD2A2, were incu-bated on ice for 15 min with 400 ml of ice-cold Buffer A (10 mM HEPESpH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.2 mM PMSF) andspun at 13,000 rpm for 10 s. The pellets were resuspended in 100 ml ofice-cold buffer C (10 mM HEPES pH 7.9, 420 mM NaCl, 1.5 mM MgCl2,0.2 mM EDTA, 25% (v/v) glycerol, 0.5 mM DTT, 0.2 mM PMSF) and leftfor 30 min on ice. After spinning for 5 min at full speed, the superna-tants were removed, aliquoted, and stored at 280 °C. The proteinconcentration in the supernatants was determined by BCA Assay(Pierce) and 10 mg/sample were used in the EMSA.
The double-stranded oligonucleotides (AP1 consensus, NF-kB con-sensus, NF-AT consensus, and Oct 1 consensus) and the Abs for thesupershifts (anti-c-Jun, JunB, JunD, c-Fos, FRA1, FRA2, Fos-B, NF-kBp50 and p65) used in these EMSA were purchased from Santa CruzBiotechnology.
10 mg of NE were incubated in binding buffer (10 mM HEPES pH 7.5,30 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 12% (v/v) glycerol, 0.5 mM DTT,0.2 mM PMSF) for 30 min on ice with 2 mg of poly(dI-dC) (Boehringer
p56lck-independent CD2 Signaling24250
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from

Mannheim) and either 2 ml of the respective Ab or a 100-fold excess ofunlabeled oligonucleotide as competitor. Subsequently, 0.5 ng of 32P-labeled oligo were added and incubated with the NE at room tempera-ture for an additional 20 min. The proteins were separated on 5.2(EMSA) or 4% (supershift) gels. The gels were dried and subjected toautoradiography.
RESULTS
The CD2posCD3pos JCaM1.6s Cells do Not Express p56lck—JCaM1.6 is a Jurkat cell line variant, which lacks functionalp56lck and is unresponsive to TCR stimulation such that it failsto generate IL-2 upon anti-CD3 cross-linking (42). To excludethe possibility that different TCR and/or CD2 surface expres-sion levels between Jurkat and JCaM1.6 might account for acomponent of the signaling defect, JCaM1.6 cells were sortedon a FACSVantage for high co-expression of both CD2 andCD3, equivalent to the surface expression number of thesereceptors found on the Jurkat line J77 (Fig. 1A). The sortedJCaM1.6 (referred to as JCaM1.6s) as well as JCaM1.6s cells,further sorted for maximal Ca21 mobilization upon CD2 cross-linking (termed JCaM1.6s.S3), were analyzed by in vitro ki-nase assay for p56lck autophosphorylation and poly(Glu-Tyr)substrate phosphorylation activity. As shown in Fig. 1B, and incontrast to J77, JCaM1.6s does not express detectable amountsof functional p56lck, as judged by in vitro autophosphorylationof unstimulated, anti-CD2-, or anti-CD3e-stimulated JCaM1.6scells. Similarly, by in vitro kinase assay, anti-p56lck antibodyimmunoprecipitates from JCaM1.6s.S3 cells do not reveal au-tophosphorylation activity or any enzymatic activity using thepoly(Glu-Tyr) substrate (data not shown).
CD2 Stimulation in the Absence of p56lck Leads to a Pro-longed and High Amplitude Rise in Intracellular Free Ca21—Although TCR cross-linking by anti-CD3e mAb or anticlono-typic mAb does not activate JCaM1.6 cells (42), the status ofthe CD2 pathway in these cells was not defined. To first ad-dress the integrity of CD2 mediated signaling, we analyzed
Ca21 mobilization in both JCaM1.6s, the Ca21 sorted JCaM1.6s.S3 and J77 following anti-CD2 or anti-CD3e stimulation. Incontrast to J77, in which both anti-CD2 and anti-CD3e mAbsinduced a long lasting and high rise in intracellular free Ca21,only anti-CD2 stimulation via the combination of anti-T112
plus anti-T113 mAbs resulted in a substantial Ca21 influx inJCaM1.6s (data not shown). After sorting, the Ca21 response ofJCaM1.6s.S3 to anti-CD2 stimulation was equal or even higherthan in J77 (Fig. 2A). Addition of biotinylated anti-CD3e mAbalone was not sufficient for Ca21 mobilization in JCaM1.6s(data not shown) or in JCaM1.6s.S3 (Fig. 2A). Rather extensivecross-linking of the biotinylated anti-CD3e mAb by avidin wasrequired to lead to a detectable Ca21 transient. This appears asa low magnitude rise in Ca21 of short duration and mostprobably is due to the initial release of Ca21 from intracellularCa21 stores (Fig. 2A) (41, 44, 45).
PLC-g1 Is Tyrosine Phosphorylated and Activated by CD2Stimulation in the Absence of p56lck—Given that phosphoryla-tion and activation of PLC-g1 leads to increased phosphatidyl-inositol turnover and the release of Ca21 from intracellularCa21 stores (45, 46), the phosphorylation status of PLC-g1 wasdetermined prior to and following stimulation in the J77 andJCaM1.6s cells. In J77, the stimulation via both CD2 and CD3eresults in a strong tyrosine phosphorylation of PLC-g1 asshown by anti-Tyr(P) Western blot analysis of anti-PLC-g1immunoprecipitations (Fig. 2B). In contrast, only activation viaCD2 leads to a clear tyrosine phosphorylation of PLC-g1 inJCaM1.6s (Fig. 2B), whereas anti-CD3e stimulation had a mi-nor effect (Fig. 2B). These differences were not a consequence ofgel loading of the anti-PLC-g1 immunoprecipitates as revealedby sequential Western blotting with anti-PLC-g1 antibody.
CD2 but Not CD3 Stimulation Results in Interleukin 2 Produc-
FIG. 1. The phenotype and p56lck in vitro kinase activity of J77and JCaM1.6s. A, J77 (parental Jurkat cells) and JCaM1.6s cells werestained with anti-T111 (anti-CD2) or RW28C8 (anti-CD3e) mAbs andanalyzed on a FACScan. The x axis indicates the log fluorescenceintensity. Equal expression of CD2 or CD3 is shown for both cell lines.B, J77 or JCaM1.6s cells were left unstimulated or were stimulated viaaCD2 (anti-T112 plus anti-T113 mAbs) or aCD3e (2AD2A2 mAb). Ly-sates were immunoprecipitated with a p56lck-specific antiserum, sub-jected to in vitro kinase assay and analyzed on SDS-PAGE followed byautoradiography. The arrows indicate the migration position of p56lck.
FIG. 2. Analysis of [Ca21]i in J77 and JCaM1.6s.S3. A, Ca21
mobilization in J77 and JCaM1.6s.S3. Cells were loaded with Indo-1.Calcium flux was then analyzed on an EPICS V flow cytometer. Whereindicated, a combination of the anti-CD2 mAbs anti-T112 plus anti-T113(CD2) or biotinylated anti-CD3e mAb RW28C8 (aCD3ebiot), avidin, andCa21 ionophore were added sequentially. B, tyrosine phosphorylation ofPLC-g1 in J77 and JCaM1.6s.S3. Cells were either unstimulated (2) orstimulated with a combination of anti-CD2 mAbs or the anti-CD3e mAb2AD2A2 and cell lysates were immunoprecipitated with anti-PLC-g1Ab. Immunoprecipitates were subjected to SDS-PAGE followed byWestern blotting with anti-phosphotyrosine mAb 4G10 (left panel). Thestripped blots were subsequently immunoblotted with PLC-g1 Ab (rightpanel). The migration position of PLC-g1 is indicated by the arrows (at;135 kDa).
p56lck-independent CD2 Signaling 24251
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from

tion in the Absence of p56lck—To next determine whether theCD2 pathway in JCaM1.6s could elicit IL-2 production, we de-termined the levels of IL-2 secretion in the supernatant of anti-CD2 stimulated JCaM1.6s cells in a human IL-2 specific enzyme-linked immunosorbent assay (Table I) and by intracellularstaining of IL-2 in these stimulated JCaM1.6s (data not shown).Parallel analysis was performed in the same cells following anti-CD3e mAb stimulation. As shown in Table I, stimulation via CD2in JCaM1.6s cells results in substantial IL-2 production, in fact toa level equivalent to 50% of the maximal induction measuredupon bypassing receptor triggering with the combination of cal-cium ionophore plus PMA. In contrast, and as expected, no IL-2production is induced by TCR cross-linking by anti-CD3e. Datafrom different subclones of JCaM1.6s obtained by sequentialsorting for Ca21 mobilization following CD2 stimulation areshown (JCaM1.6s.S1, JCaM1.6s.S2, and JCaM1.6s.S3). Theamount of IL-2 produced increases with enhanced Ca21 respon-siveness to CD2 stimulation.
Neither p59fyn nor ZAP70 Are Detectably Tyrosine Phospho-rylated following CD2 Stimulation in JCaM1.6s—Tyrosinephosphorylation and activation of kinases such as the Src ki-nases p56lck or p59fyn or the related tyrosine kinase ZAP70 aremajor events in TCR signaling and are also thought to beinvolved in signal transduction via CD2 (47). The Jurkat var-iant JCaM1.6s gives us the opportunity to investigate theseevents in the absence of p56lck. To this end, we performed aseries of Western blots with J77 and JCaM1.6s cells followinganti-CD2 or anti-CD3e stimulation. In J77, activation via bothCD2 and CD3 results in the tyrosine phosphorylation of p56lck
and the appearance of a second p60lck band due to the serine/threonine phosphorylation of p56lck (48) (Fig. 3A, upper panel).As expected, both lck bands are absent in the phosphotyrosineblot of p56lck immunoprecipitates from JCaM1.6s. Note thatthe p58 band appearing in the Tyr(P) blot of both J77 andJCaM1.6s is nonspecific, being unrelated to p56lck. Reanalysisof the stripped blot with anti-lck antiserum shows that thesep56- and p60-phosphorylated proteins in J77 indeed representp56lck (Fig. 3A, lower panel).
In J77, stimulation via the CD3 pathway leads to a strongtyrosine phosphorylation of ZAP70, whereas activation via CD2induces weaker but definite ZAP70 phosphorylation (Fig. 3B,upper panel). In contrast to this observation, in JCaM1.6sneither CD2 nor CD3 stimulation induce the phosphorylationof ZAP70 (Fig. 3B, upper panel). Nevertheless, similar amountsof ZAP70 were immunoprecipitated in each lane, as shown bythe results of reprobing the stripped Tyr(P) blot with an anti-ZAP70 rabbit antiserum (Fig. 3B, lower panel).
In both J77 and JCaM1.6s, the tyrosine phosphorylation ofp59fyn increases minimally if at all after either CD2 or CD3stimulation (Fig. 3C, upper panel). The corresponding p59fyn
blot probed with anti-p59fyn antibody is shown in Fig. 3C, lowerpanel. Note that the band below p59fyn is related to immuno-globulin heavy chain.
The Ras-MAPK Pathway Is Not Functional in the Absence ofp56lck—TCR stimulation of T cells leads to the activation of Rasresulting in the serine phosphorylation and activation of Raf,then subsequently the activation of the MAPK signaling cas-cade and finally to the initiation of IL-2 transcription (49). Weinvestigated the activation of Raf and MAPK by Western blot-ting of the total lysate of either unstimulated or anti-CD2 oranti-CD3 stimulated J77 or JCaM1.6s.S3 cells with rabbit an-tisera specific for Raf, pMAPK, or MAPK (Fig. 4, A-C). In J77,both CD2 and CD3 stimulation induced the phosphorylationand molecular weight shift of Raf (Fig. 4A) and the subsequentphosphorylation of MAPK as shown by Western blotting withantisera specific for the phospho-MAPK (Fig. 4B). In contrast,in JCaM1.6s.S3 stimulation via CD2 had no effect on eitherkinase (Fig. 4, A and B). Surprisingly, anti-CD3 triggering inJCaM1.6s.S3 led to the phosphorylation and molecular weightshift of Raf (Fig. 4A) but not to the functional activation of Rafas judged by the very low or absent phosphorylation of MAPK(Fig. 4B). As shown in Fig. 4C by sequential immunoblottingwith a MAPK specific antiserum, the described differenceswere not due to different amounts of MAPK in the total lysates.Similarly, in J77, but not in JCaM1.6s.S3, stimulation viaeither CD2 or CD3 enhanced the kinase activity of MAPKtoward myelin basic protein in an in vitro kinase assay (datanot shown). Additionally, CD2 stimulated IL-2 production inJCaM1.6s.S3 was not inhibited by coculture with the MEK1-inhibitor PD98059, which interferes with the MAPK pathwayupstream of MAPK (data not shown).
CD2 but Not CD3 Stimulation Activates Jun Kinase in theAbsence of p56lck—Initiation of IL-2 transcription requires thebinding of certain DNA-binding proteins to specific regionswithin the IL-2 promoter (50). One of these DNA-binding ele-ments is the AP-1 complex, which consists of homo- or het-erodimers of members of the Jun and Fos protein family (51).In order to bind to and activate the IL-2 promoter, these dimersmust assemble and the participating proteins become phospho-rylated (51). Jun kinase (JNK) is involved in the serine phos-phorylation and activation of Jun family members (52). Follow-ing CD2 stimulation in JCaM1.6s.S3 cells, activation of JNK isreadily detected as shown by in vitro kinase activity using aGST-Jun fusion protein substrate (Fig. 5A). CD3 stimulation
TABLE ICD2 triggering induces IL-2 production in the absence of p56lck
Cell culture supernatants were analyzed for IL-2 content after 48 hstimulation with A23187, a-CD3e mAb RW28C8, or a-CD2 mAbs anti-T112 plus anti-T113; or after no stimulation (none). PMA was added toall wells at 25 ng/ml. S.D. of triplicate samples ,5% for all entries.
StimulusIL-2 (ng/ml)
None A23187 a-CD3e a-CD2
J77 0.05 17.95 6.17 23.56JCaM1.6s 0.01 6.09 0.08 2.60JCaM1.6s.S1 0.01 5.77 0.09 2.77JCaM1.6s.S2 0.05 14.39 0.17 7.36JCaM1.6s.S3 0.03 43.76 0.16 21.13
FIG. 3. Analysis of protein-tyrosine kinase activation in J77and JCaM1.6s.S3. Cells were either unstimulated (2) or stimulatedwith either a combination of the anti-CD2 mAbs anti-T112 plus anti-T113 (aCD2) or the anti-CD3e mAb 2AD2A2 (aCD3) and cell lysateswere immunoprecipitated with antisera specific for p56lck, ZAP70, orp59fyn, respectively. Immunoprecipitates in panels A-C using the des-ignated Abs were subjected to SDS-PAGE followed by Western blottingwith anti-phosphotyrosine mAb 4G10 (upper row). The blots werestripped and subsequently immunoblotted with the respective Abs (low-er row). The migration positions of p56lck, ZAP70, or p59fyn are indicatedby arrows.
p56lck-independent CD2 Signaling24252
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from

induced JNK activation is significantly lower than CD2 in-duced JNK activation in JCaM1.6s.S3, whereas both pathwaysactivate JNK in J77 to a comparable level (Fig. 5A). The aver-age increase of JNK activity from three independent experi-ments following CD2 or CD3 stimulation compared with un-stimulated cells is as follows: JCaM1.6s.S3: CD2, 2.96 6 0.8;CD3, 1.44 6 0.4; J77: CD2, 1.54 6 0.2; CD3, 1.72 6 0.5-foldincrease (over background unstimulated controls as deter-mined by scanning the autoradiographs on a PhosphorImager).
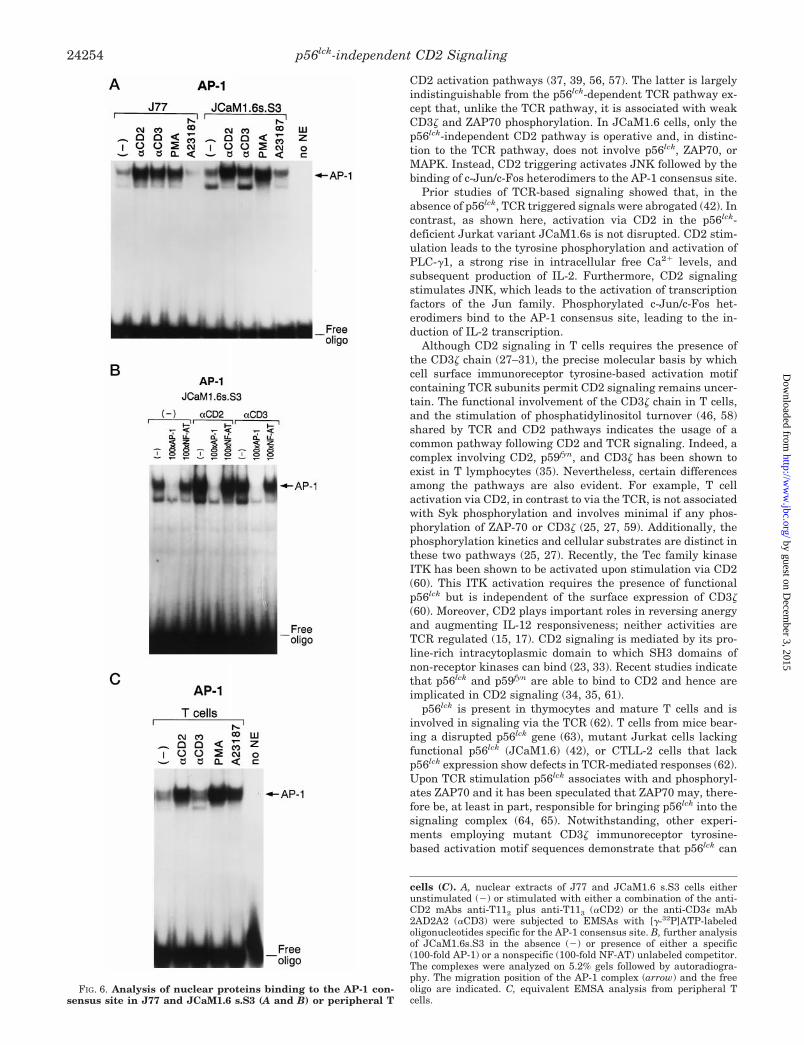
Following CD2 Stimulation There Are Predominantly c-Jun/c-Fos Heterodimers Binding to the AP-1 Consensus Site—TheIL-2 promoter contains several binding sites for transcriptionfactors which control IL-2 gene expression including AP-1, NF-kB, NF-AT, and the CD28RE (50, 53). In the case of the AP-1complex, the major kinase which serine phosphorylates Jun isJNK (52). Since JNK is activated in JCaM1.6s.S3 following CD2stimulation (Fig. 5A), it was important to determine how thisactivation would affect AP-1 binding. As shown by EMSA (elec-tromobility shift assay) in Fig. 6A, compared with CD3 stimula-tion, the CD2 induced Jun kinase activity in JCaM1.6s.S3 isaccompanied by enhanced expression and binding of AP-1 com-plexes to the AP-1 consensus site. This interaction of AP-1 pro-teins with the AP-1 consensus site double-stranded oligonucleo-tide is specific for AP-1, as shown by competition experimentsusing an excess of unlabeled AP-1 oligonucleotide (Fig. 6B). InJ77, on the other hand, AP-1 complex binding is similar between
CD2 and CD3 stimulation (Fig. 6A).The AP-1 complex consists of homo- or heterodimers of pro-
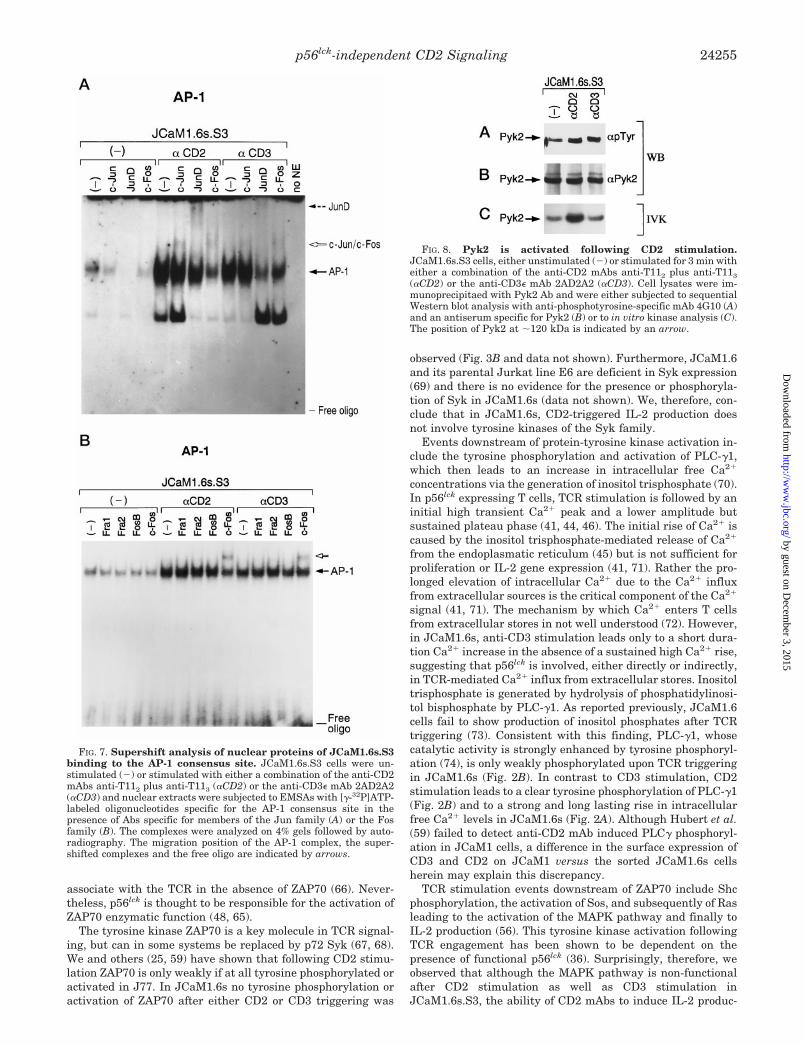
teins of the Jun and Fos family (54). It has been suggested thatJun/Fos heterodimers have a higher affinity for the AP-1 siteand are more efficient in activating IL-2 gene expression (55).The Jun family contains at least three members, c-Jun, JunB,and JunD, while the Fos family includes c-Fos, Fra1, Fra2, andFosB (51, 53). Following CD2 stimulation of JCaM1.6s.S3 bothhomodimers and heterodimers appear to bind to the AP-1 con-sensus site as shown by supershift of the AP-1 proteins withthe respective Abs specific for JunD, c-Jun, and c-Fos (Fig. 7, Aand B). These AP-1 complexes do not contain JunB (data notshown), Fra1, Fra2, or FosB (Fig. 7B). Given that the super-shift with anti-c-Jun and anti-c-Fos Abs results in a mobilityshift of their respective complexes to the same position in thegel (Fig. 7A), we infer that c-Jun/c-Fos heterodimers areformed. In contrast, the different mobility of the complex in thesupershift with anti-JunD Ab implies that JunD/JunD ho-modimers are present. CD3 stimulation is less efficient in AP-1induction in JCaM1.6s.S3 (Fig. 6A). Moreover, with anti-CD2stimulation the AP-1 complex is supershifted to the greatestextent by anti-c-Fos mAb. In contrast, with anti-CD3 mAbstimulation anti-JunD mAb effects the greatest shift. Theseresults imply that the AP-1 dimer is primarily a complex con-sisting of JunD/JunD homodimers following anti-CD3stimulation (Fig. 7A).
Although not shown, analysis of the other transcriptionalfactors involved in the regulation of IL-2 production includingNF-AT, NF-kB, and Oct-1 (50, 53) showed similar induciblenuclear protein binding profiles to the NF-AT, NF-kB, or Oct-1consensus site in JCaM1.6s.S3 following CD2 or CD3 trigger-ing; moreover, supershift analysis with Abs specific for c-Jun,JunD, or c-Fos revealed no difference in the composition of thecomplex binding to the NF-AT site upon either anti-CD2 oranti-CD3 stimulation. Similarly, Abs specific for the p50 or p65subunit of the NF-kB complex did not indicate differentialeffects of these stimulatory pathways as judged by supershiftanalysis of nuclear proteins binding to the NK-kB consensussite (data not shown).
To determine if resting T cells would respond to CD2 stim-ulation similarly in terms of JNK activation and binding ofAP-1 proteins, we performed corresponding experiments infreshly isolated peripheral T cells. JNK is activated via bothCD2 and to a lesser extent, via CD3 (Fig. 5B), suggesting, asdiscussed below, an important role for the CD2 signaling path-way in T cell co-stimulation. In addition, CD2 but not CD3stimulation also efficiently leads to the activation and bindingof AP-1 proteins to the AP-1 consensus site (Fig. 6C).
The Tyrosine Kinase Pyk2 Is Activated following Stimulationvia CD2 in the Absence of p56lck—Recently, Pyk2 a tyrosinekinase homologous to the focal adhesion kinase (FAK) has beenidentified and linked to the JNK pathway, (39). Pyk2 activa-tion, like JNK activation, requires a strong Ca21 signal such asthat provided by CD2 stimulation in JCaM1.6s.S3. We inves-tigated the activation of Pyk2 in JcaM1.6s.S3 following anti-CD2 or anti-CD3 mAb triggering by Western blot analysis andin vitro kinase assay. Pyk2 is phosphorylated in response toboth CD2 and CD3 triggering (Fig. 8A), but activated only afterCD2 stimulation as judged by in vitro autophosphorylation(Fig. 8C). Equivalent amounts of Pyk2 were precipitated asshown by sequential immunoblotting of the phosphotyrosineblot (Fig. 8A) with a Pyk2-specific rabbit antiserum (Fig. 8B).
DISCUSSION
In this study, we identify a p56lck-independent CD2 signal-ing pathway capable of inducing IL-2 production. Fig. 9 offers aschematic view of p56lck-independent as well as -dependent
FIG. 4. Analysis of the MAPK pathway in J77 and JCaM1.6s.S3.Cells were either unstimulated (2) or stimulated with either a combi-nation of the anti-CD2 mAbs anti-T112 plus anti-T113 (aCD2) or theanti-CD3e mAb 2AD2A2 (aCD3) and total lysates were subjected toSDS-PAGE followed by Western blotting (WB) with antisera specific forRaf (A) or phosphorylated MAPK (pMAPK) (B). The pMAPK blot wasstripped and subsequently immunoblotted with an antiserum specificfor MAPK (C). The migration position of Raf (at ;74 kDa), pMAPK (at;42 and 44 kDa), and MAPK (at ;42 and 44 kDa) are indicated byarrows.
FIG. 5. JNK is activated in JCaM1.6s.S3 and peripheral T cellsfollowing CD2 stimulation. J77 and JCaM1.6s.S3 cells (A) or freshlyisolated peripheral T cells (B) were either unstimulated (2) or stimu-lated with either a combination of the anti-CD2 mAbs anti-T112 plusanti-T113 (aCD2), the anti-CD3e mAb 2AD2A2 (aCD3), or PMA and celllysates were immunoprecipitated with an antiserum specific for JNK.Subsequently, the immunoprecipitates were subjected to an in vitrokinase assay in the presence of a GST-Jun fusion protein as a substrateand analyzed on SDS-PAGE followed by autoradiography. The migra-tion position of the GST-Jun fusion protein is indicated by an arrow.
p56lck-independent CD2 Signaling 24253
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from
CD2 activation pathways (37, 39, 56, 57). The latter is largelyindistinguishable from the p56lck-dependent TCR pathway ex-cept that, unlike the TCR pathway, it is associated with weakCD3z and ZAP70 phosphorylation. In JCaM1.6 cells, only thep56lck-independent CD2 pathway is operative and, in distinc-tion to the TCR pathway, does not involve p56lck, ZAP70, orMAPK. Instead, CD2 triggering activates JNK followed by thebinding of c-Jun/c-Fos heterodimers to the AP-1 consensus site.
Prior studies of TCR-based signaling showed that, in theabsence of p56lck, TCR triggered signals were abrogated (42). Incontrast, as shown here, activation via CD2 in the p56lck-deficient Jurkat variant JCaM1.6s is not disrupted. CD2 stim-ulation leads to the tyrosine phosphorylation and activation ofPLC-g1, a strong rise in intracellular free Ca21 levels, andsubsequent production of IL-2. Furthermore, CD2 signalingstimulates JNK, which leads to the activation of transcriptionfactors of the Jun family. Phosphorylated c-Jun/c-Fos het-erodimers bind to the AP-1 consensus site, leading to the in-duction of IL-2 transcription.
Although CD2 signaling in T cells requires the presence ofthe CD3z chain (27–31), the precise molecular basis by whichcell surface immunoreceptor tyrosine-based activation motifcontaining TCR subunits permit CD2 signaling remains uncer-tain. The functional involvement of the CD3z chain in T cells,and the stimulation of phosphatidylinositol turnover (46, 58)shared by TCR and CD2 pathways indicates the usage of acommon pathway following CD2 and TCR signaling. Indeed, acomplex involving CD2, p59fyn, and CD3z has been shown toexist in T lymphocytes (35). Nevertheless, certain differencesamong the pathways are also evident. For example, T cellactivation via CD2, in contrast to via the TCR, is not associatedwith Syk phosphorylation and involves minimal if any phos-phorylation of ZAP-70 or CD3z (25, 27, 59). Additionally, thephosphorylation kinetics and cellular substrates are distinct inthese two pathways (25, 27). Recently, the Tec family kinaseITK has been shown to be activated upon stimulation via CD2(60). This ITK activation requires the presence of functionalp56lck but is independent of the surface expression of CD3z(60). Moreover, CD2 plays important roles in reversing anergyand augmenting IL-12 responsiveness; neither activities areTCR regulated (15, 17). CD2 signaling is mediated by its pro-line-rich intracytoplasmic domain to which SH3 domains ofnon-receptor kinases can bind (23, 33). Recent studies indicatethat p56lck and p59fyn are able to bind to CD2 and hence areimplicated in CD2 signaling (34, 35, 61).
p56lck is present in thymocytes and mature T cells and isinvolved in signaling via the TCR (62). T cells from mice bear-ing a disrupted p56lck gene (63), mutant Jurkat cells lackingfunctional p56lck (JCaM1.6) (42), or CTLL-2 cells that lackp56lck expression show defects in TCR-mediated responses (62).Upon TCR stimulation p56lck associates with and phosphoryl-ates ZAP70 and it has been speculated that ZAP70 may, there-fore be, at least in part, responsible for bringing p56lck into thesignaling complex (64, 65). Notwithstanding, other experi-ments employing mutant CD3z immunoreceptor tyrosine-based activation motif sequences demonstrate that p56lck can
FIG. 6. Analysis of nuclear proteins binding to the AP-1 con-sensus site in J77 and JCaM1.6 s.S3 (A and B) or peripheral T
cells (C). A, nuclear extracts of J77 and JCaM1.6 s.S3 cells eitherunstimulated (2) or stimulated with either a combination of the anti-CD2 mAbs anti-T112 plus anti-T113 (aCD2) or the anti-CD3e mAb2AD2A2 (aCD3) were subjected to EMSAs with [g-32P]ATP-labeledoligonucleotides specific for the AP-1 consensus site. B, further analysisof JCaM1.6s.S3 in the absence (2) or presence of either a specific(100-fold AP-1) or a nonspecific (100-fold NF-AT) unlabeled competitor.The complexes were analyzed on 5.2% gels followed by autoradiogra-phy. The migration position of the AP-1 complex (arrow) and the freeoligo are indicated. C, equivalent EMSA analysis from peripheral Tcells.
p56lck-independent CD2 Signaling24254
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from
associate with the TCR in the absence of ZAP70 (66). Never-theless, p56lck is thought to be responsible for the activation ofZAP70 enzymatic function (48, 65).
The tyrosine kinase ZAP70 is a key molecule in TCR signal-ing, but can in some systems be replaced by p72 Syk (67, 68).We and others (25, 59) have shown that following CD2 stimu-lation ZAP70 is only weakly if at all tyrosine phosphorylated oractivated in J77. In JCaM1.6s no tyrosine phosphorylation oractivation of ZAP70 after either CD2 or CD3 triggering was
observed (Fig. 3B and data not shown). Furthermore, JCaM1.6and its parental Jurkat line E6 are deficient in Syk expression(69) and there is no evidence for the presence or phosphoryla-tion of Syk in JCaM1.6s (data not shown). We, therefore, con-clude that in JCaM1.6s, CD2-triggered IL-2 production doesnot involve tyrosine kinases of the Syk family.
Events downstream of protein-tyrosine kinase activation in-clude the tyrosine phosphorylation and activation of PLC-g1,which then leads to an increase in intracellular free Ca21
concentrations via the generation of inositol trisphosphate (70).In p56lck expressing T cells, TCR stimulation is followed by aninitial high transient Ca21 peak and a lower amplitude butsustained plateau phase (41, 44, 46). The initial rise of Ca21 iscaused by the inositol trisphosphate-mediated release of Ca21
from the endoplasmatic reticulum (45) but is not sufficient forproliferation or IL-2 gene expression (41, 71). Rather the pro-longed elevation of intracellular Ca21 due to the Ca21 influxfrom extracellular sources is the critical component of the Ca21
signal (41, 71). The mechanism by which Ca21 enters T cellsfrom extracellular stores in not well understood (72). However,in JCaM1.6s, anti-CD3 stimulation leads only to a short dura-tion Ca21 increase in the absence of a sustained high Ca21 rise,suggesting that p56lck is involved, either directly or indirectly,in TCR-mediated Ca21 influx from extracellular stores. Inositoltrisphosphate is generated by hydrolysis of phosphatidylinosi-tol bisphosphate by PLC-g1. As reported previously, JCaM1.6cells fail to show production of inositol phosphates after TCRtriggering (73). Consistent with this finding, PLC-g1, whosecatalytic activity is strongly enhanced by tyrosine phosphoryl-ation (74), is only weakly phosphorylated upon TCR triggeringin JCaM1.6s (Fig. 2B). In contrast to CD3 stimulation, CD2stimulation leads to a clear tyrosine phosphorylation of PLC-g1(Fig. 2B) and to a strong and long lasting rise in intracellularfree Ca21 levels in JCaM1.6s (Fig. 2A). Although Hubert et al.(59) failed to detect anti-CD2 mAb induced PLCg phosphoryl-ation in JCaM1 cells, a difference in the surface expression ofCD3 and CD2 on JCaM1 versus the sorted JCaM1.6s cellsherein may explain this discrepancy.
TCR stimulation events downstream of ZAP70 include Shcphosphorylation, the activation of Sos, and subsequently of Rasleading to the activation of the MAPK pathway and finally toIL-2 production (56). This tyrosine kinase activation followingTCR engagement has been shown to be dependent on thepresence of functional p56lck (36). Surprisingly, therefore, weobserved that although the MAPK pathway is non-functionalafter CD2 stimulation as well as CD3 stimulation inJCaM1.6s.S3, the ability of CD2 mAbs to induce IL-2 produc-
FIG. 7. Supershift analysis of nuclear proteins of JCaM1.6s.S3binding to the AP-1 consensus site. JCaM1.6s.S3 cells were un-stimulated (2) or stimulated with either a combination of the anti-CD2mAbs anti-T112 plus anti-T113 (aCD2) or the anti-CD3e mAb 2AD2A2(aCD3) and nuclear extracts were subjected to EMSAs with [g-32P]ATP-labeled oligonucleotides specific for the AP-1 consensus site in thepresence of Abs specific for members of the Jun family (A) or the Fosfamily (B). The complexes were analyzed on 4% gels followed by auto-radiography. The migration position of the AP-1 complex, the super-shifted complexes and the free oligo are indicated by arrows.
FIG. 8. Pyk2 is activated following CD2 stimulation.JCaM1.6s.S3 cells, either unstimulated (2) or stimulated for 3 min witheither a combination of the anti-CD2 mAbs anti-T112 plus anti-T113(aCD2) or the anti-CD3e mAb 2AD2A2 (aCD3). Cell lysates were im-munoprecipitaed with Pyk2 Ab and were either subjected to sequentialWestern blot analysis with anti-phosphotyrosine-specific mAb 4G10 (A)and an antiserum specific for Pyk2 (B) or to in vitro kinase analysis (C).The position of Pyk2 at ;120 kDa is indicated by an arrow.
p56lck-independent CD2 Signaling 24255
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from

tion was not compromised. These observations strongly indi-cate that CD2 stimulation can activate signaling pathwaysdistinct from CD3 stimulation.
Pyk2 was recently identified as a tyrosine kinase, which isinvolved in Jun kinase activation and associates with theadapter protein Grb2 (39). By forming a complex containingPyk2 and the GDP-GTP exchange factor Vav (75), Grb2 canlink Pyk2 to the GTP-binding protein Rac, whose activationfinally leads to the activation of JNK (57). In our study, weshow that, following both CD2 and CD3 stimulation, Pyk2 istyrosine phosphorylated in JCaM1.6s.S3 cells, but only CD2triggering activates Pyk2 enzymatically, providing a possiblelink to a stimulation pathway which leads to JNK activation.
JNK is important in the regulation of the IL-2 promoter sinceJNK activation correlates with IL-2 production (38, 51, 52, 76).IL-2 promoter regulation involves several transcription factorssome of which are JNK sensitive (50, 51, 77–80). The AP-1transcription factor can bind to the IL-2 promoter either di-rectly at the AP-1 site (81, 82) or together with NF-AT or Oct attheir respective binding sites (83–86). The AP-1 complex iscomposed of proteins of the Jun and Fos family (51). JNK, aCa21-sensitive serine/threonine kinase of the MAP kinase fam-ily, is critically involved in the post-transcriptional stimulationof AP-1 activity by phosphorylating the activation domain ofc-Jun. Su et al. (38) reported that two signals are necessary toefficiently activate JNK, including different combinations ofA23187, TPA, anti-CD3 mAb, or anti-CD28 mAb. This dualactivation requirement and the reported Ca21 sensitivity dis-tinguishes JNK from other members of the MAPK/JNK family.
The involvement of CD2 stimulation in the activation of JNKhas not yet been previously investigated. In both J77 andJCaM1.6s.S3, JNK is activated following stimulation via CD2(Fig. 5A), suggesting that CD2 utilizes a p56lck independentpathway which leads to the activation of JNK. The strong andlong lasting Ca21 mobilization observed in JCaM1.6s.S3 follow-ing CD2 stimulation might sensitize JNK to p56lck independentsignals involved in JNK activation (38). Furthermore, in theabsence of p56lck, anti-CD2 stimulation is accompanied by theenhanced binding of the c-Jun/c-Fos heterodimers to the consen-sus AP-1 sequence leading to IL-2 production. A previous studyperformed by stimulating Jurkat cells with superantigen pulsedHLA-DR transfectants revealed differences in the composition ofthe NF-ATzAP-1 complexes following costimulation with either
LFA-3 or B7 (87). These differences were due to the dimerizationof JunD with different members of the Fos family (Fra1 andFra2), indicating a selective induction of certain nuclear factorsdepending on the costimulatory pathway. In our current study,we observed no significant difference in the binding of nuclearproteins to the NF-AT consensus site upon anti-CD2 versus anti-CD3 stimulated JCaM1.6s.S3. Since the MAPK pathway, knownto be involved in Fos transcription, is non-functional in JCaM1.6,the lack of Fra1 and Fra2 Fos family members complexed to theAP-1 site in JCaM1.6s.S3 is perhaps not unexpected. In addition,the coordinate action of TCR engagement and the B7 versusLFA-3 costimulatory signal could differentially induce Fos familyproteins. In the study by Parra et al. (87), AP-1 and NF-kBcomplexes binding to their respective site in the IL-2 promoterrevealed no differences after either type of costimulation. Bystimulation via CD2 alone, we found not only JunD but alsoc-Jun induced and heterodimerizing with c-Fos in JCaM1.6s.S3(Fig. 7A). Since the AP-1 site in the IL-2 promoter is a relativelylow affinity site (83), Fos-Jun heterodimers, which are moreeffective in DNA binding and transactivation (55), might be re-quired for optimal activity. Additionally, c-Jun and c-Fos in con-trast to JunB, Fra1, and Fra2 are more efficient transactivators(55, 88, 89). The function of JunD in IL-2 gene transcription is notdefined yet.
Anergy, a state of T cell unresponsiveness to antigenic chal-lenge, which is induced by TCR stimulation in the absence ofthe CD28 costimulatory signal (90), is accompanied by prefer-ential induction of the inhibitory p50-p50 NF-kB homodimerand a reduced binding of AP-1 to the IL-2 promoter (91, 92).Recently, it was shown in an alloreactive system that alloan-tigen stimulation induced T cell anergy can be reversed inthose cells after culture in IL-2 for 7 days only by costimulationwith CD58 (17). This ability of CD2 stimulation to reverseanergy is unique and distinct from costimulatory moleculessuch as CD28 (17). Our observation that CD2 stimulation canactivate JNK and leads to the induction and binding of c-Jun/c-Fos heterodimers to the AP-1 site might provide a basis forthe role CD2 plays in regulating anergy. This possibly remainsto be investigated in anergized T cells.
The data presented in this study provide evidence for a CD2signaling pathway distinct from that of the TCR and capable ofinducing IL-2 production in the absence of p56lck. This CD2stimulation induced signaling cascade does not involve either
FIG. 9. Signal transduction via CD2.p56lck-dependent and independent activa-tion pathways following CD2 stimulationare based on prior results (37, 39, 56, 57,75) and the current studies. In JCaM1.6,only the p56lck independent signal trans-duction pathway is functional leading viaJNK and subsequent c-Jun activation tothe binding of the AP-1 complex to theIL-2 promoter and the initiation of IL-2transcription. Both pathways apparentlyactivate PLC-g1 leading to the generationof a rise in intracellular free Ca21, result-ing in a calmodulin-dependent activationof calcineurin. The latter dephosphory-lates NF-AT resulting in its nucleartranslocation.
p56lck-independent CD2 Signaling24256
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from

p56lck or ZAP70, key molecules in T cell activation via the TCR,and can also be initiated by CD2 triggering in peripheral Tcells. CD2 activates Pyk2 and undoubtedly other kinases, andsubsequently stimulates JNK independent of p56lck. The pre-cise definition of the intermediate steps in this activation cas-cade will now be of interest to determine.
Acknowledgments—We thank Drs. A. Rao and T. Roberts for carefulreview of the manuscript.
REFERENCES
1. Howard, F. D., Ledbetter, J. A., Wong, J., Bieber, C. P., Stinson, E. B., andHerzenberg, L. A. (1981) J. Immunol. 126, 2117–2122
2. Kamoun, M., Martin, P. J., Hansen, J. A., Brown, M. A., Siadak, A. W., andNowinski, R. C. (1981) J. Exp. Med. 152, 207–212
3. Rodewald, H. R., Awad, K., Moingeon, P., D’Adamio, L., Rabinowitz, D.,Shinkai, Y., Alt, F. W., and Reinherz, E. L. (1993) J. Exp. Med. 177,1079–1092
4. Selvaraj, P., Plunkett, M. L., Dustin, M., Sanders, M. E., Shaw, S., andSpringer, T. A. (1987) Nature 326, 400–403
5. Krensky, A. M., Sanchez-Madrid, F., Robbins, E., Nagy, J. A., Springer, T. A.,and Burakoff, S. J. (1983) J. Immunol. 131, 611–616
6. Dustin, M. L., and Springer, T. A. (1989) Nature 341, 619–6247. Moingeon, P. E., Lucich, J. L., Stebbins, C. C., Recny, M. A., Wallner, B. P.,
Koyasu, S., and Reinherz, E. L. (1991) Eur. J. Immunol. 21, 605–6108. Moingeon, P., Chang, H. C., Sayre, P. H., Clayton, L. K., Alcover, A., Gardner,
P., and Reinherz, E. L. (1989a) Immunol. Rev. 111, 111–1449. Moingeon, P., Chang, H. C., Wallner, B. P., Stebbins, C., Frey, A. Z., and
Reinherz, E. L. (1989) Nature 339, 312–31410. Koyasu, S., Lawton, T., Novick, D., Recny, M. A., Siliciano, R. F., Wallner,
B. P., and Reinherz, E. L. (1990) Proc. Natl. Acad. Sci. U. S. A. 87,2603–2607
11. Meuer, S. C., Hussey, R. E., Fabbi, M., Fox, D., Acuto, O., Fitzgerald, K. A.,Hodgdon, J. C., Protentis, J. P., Schlossman, S. F., and Reinherz, E. L.(1984) Cell 36, 897–906
12. Brottier, P., Boumsell, L., Gelin, C., and Bernard, A. (1985) J. Immunol. 135,1624–1631
13. Siliciano, R. F., Pratt, J. C., Schmidt, R. E., Ritz, J., and Reinherz, E. L. (1985)Nature 317, 428–430
14. Li, J., Smolyar, A., Sunder-Plassmann, R., and Reinherz, E. L. (1996) J. Mol.Biol. 263, 209–226
15. Gollob, J. A., Li, J., Kawasaki, H., Daley, J. F., Groves, C., Reinherz, E. L., andRitz, J. (1996) J. Immunol. 157, 1886–1893
16. Gollob, J. A., Li, J., Reinherz, E. L., and Ritz, J. (1995) J. Exp. Med. 182,721–731
17. Boussiotis, V. A., Freeman, G. J., Griffin, J. D., Gray, G. S., Gribben, J. G., andNadler, L. M. (1994) J. Exp. Med. 180, 1665–1673
18. He, Q., Beyers, A. D., Barclay, A. N., and Williams, A. F. (1988) Cell 54,979–984
19. Chang, H. C., Moingeon, P., Lopez, P., Krasnow, H., Stebbins, C., and Rein-herz, E. L. (1989) J. Exp. Med. 169, 2073–2083
20. Bierer, B. E., Bogart, R. E., and Burakoff, S. J. (1990) J. Immunol. 144,785–789
21. Brown, M. H., Sewell, W. A., Mason, D. Y., Rothbard, J. B., and Crumpton,M. J. (1988) Eur. J. Immunol. 18, 1223–1227
22. Clayton, L. K., Ramachandran, H., Pravtcheva, D., Chen, Y. F., Diamond,D. J., Ruddle, F. H., and Reinherz, E. L. (1988) J. Immunol. 140, 3617–3621
23. Pawson, T., and Gish, G. D. (1992) Cell 71, 359–36224. Cohen, G. B., Ren, R., and Baltimore, D. (1995) Cell 80, 237–24825. Jin, Y. J., Kaplan, D. R., White, M., Spagnoli, G. C., Roberts, T. M., and
Reinherz, E. L. (1990) J. Immunol. 144, 647–65226. Ley, S. C., Davies, A. A., Druker, B., and Crumpton, M. J. (1991) Eur. J.
Immunol. 21, 2203–220927. Howard, F. D., Moingeon, P., Moebius, U., McConkey, D. J., Yandava, B.,
Gennert, T. E., and Reinherz, E. L. (1992) J. Exp. Med. 176, 139–14528. Breitmeyer, J. B., Daley, J. F., Levine, H. B., and Schlossman, S. F. (1987)
J. Immunol. 139, 2899–290529. Alcover, A., Alberini, C., Acuto, O., Clayton, L. K., Transy, C., Spagnoli, G. C.,
Moingeon, P., Lopez, P., and Reinherz, E. L. (1988) EMBO J. 7, 1973–197730. Bockenstedt, L. K., Goldsmith, M. A., Dustin, M., Olive, D., Springer, T. A.,
and Weiss, A. (1988) J. Immunol. 141, 1904–191131. Moingeon, P., Lucich, J. L., McConkey, D. J., Letourneur, F., Malissen, B.,
Kochan, J., Chang, H. C., Rodewald, H. R., and Reinherz, E. L. (1992) Proc.Natl. Acad. Sci. U. S. A. 89, 1492–1496
32. Arulanandam, A. R. N., Koyasu, S., and Reinherz, E. L. (1991) J. Exp. Med.173, 859–868
33. Chang, H. C., Moingeon, P., Pedersen, R., Lucich, J., Stebbins, C., andReinherz, E. L. (1990) J. Exp. Med. 172, 351–355
34. Bell, G. M., Fargnoli, J., Bolen, J. B., Kish, L., and Imboden, J. B. (1996)J. Exp. Med. 183, 169–178
35. Gassmann, M., Amrein, K. E., Flint, N. A., Schraven, B., and Burn, P. (1994)Eur. J. Immunol. 24, 139–144
36. Gupta, S., Weiss, A., Kumar, G., Wang, S., and Nel, A. (1994) J. Biol. Chem.269, 17349–17357
37. Hunter, T., and Karin, M. (1992) Cell 70, 375–37838. Su, B., Jacinto, E., Hibi, M., Kallunki, T., Karin, M., and Ben-Neriah, Y. (1994)
Cell 77, 727–73639. Tokiwa, G., Dikic, I., Lev, S., and Schlessinger, J. (1996) Science 273, 792–79440. Quian, D., Lev, S., van-Oers, N. S. C., Dikic, I., Schlessinger, J., and Weiss, A.
(1997) J. Exp. Med. 185, 1253–125941. Weiss, M. J., Daley, J. F., Hodgdon, J. C., and Reinherz, E. L. (1984) Proc. Natl.
Acad. Sci. U. S. A. 81, 6836–684042. Straus, D., and Weiss, A. (1992) Cell 70, 585–59343. Koyasu, S., Tse, A. G. D., Moingeon, P., Hussey, R. E., Mildonian, A.,
Hannisian, J., Clayton, L. K., and Reinherz, E. L. (1994) Proc. Natl. Acad.Sci. U. S. A. 91, 6693–6697
44. Alcover, A., Weiss, M. J., Daley, J. F., and Reinherz, E. L. (1986) Proc. Natl.Acad. Sci. U. S. A. 83, 2614–2618
45. Berridge, M. J., and Irvine, R. F. (1989) Nature 341, 197–32546. Imboden, J. B., and Stobo, J. D. (1985) J. Exp. Med. 161, 44647. Weiss, A., and Littman, D. R. (1994) Cell 76, 263–27448. Fischer, S., Marie-Cardine, A., Ramos-Morales, F., Bougeret, C., Soula, M.,
Maridonneau-Parini, I., and Benarous, R. (1994) Cell. Mol. Biol. 40,605–609
49. Izquierdo, P. M., Reif, K., and Cantrell, D. (1995) Immunol. Today 16, 159–16450. Crabtree, G. R. (1989) Science 243, 355–36151. Angel, P., and Karin, M. (1991) Biochim. Biophys. Acta 1072, 129–15752. Hibi, M., Lin, A., Smeal, T., Minden, A., and Karin, M. (1993) Genes Dev. 7,
2135–214853. Jain, J., Loh, C., and Rao, A. (1995) Curr. Opin. Immunol. 7, 333–34254. Jain, J., McCaffrey, P. G., Valge-Archer, V. E., and Rao, A. (1992) Nature 356,
801–80455. Ryseck, R.-P., and Bravo, R. (1991) Oncogene 6, 533–54256. Cantrell, D. (1996) Annu. Rev. Immunol. 14, 259–27457. Crespo, P., Bustelo, X. R., Aaronson, D. S., Coso, O. A., Lopez-Barahona, M.,
Barbacid, M., and Gutkind, J. S. (1996) Oncogene 13, 455–46058. Pantaleo, G., Olive, D., Poggi, A., Kozumbo, W. J., Moretta, L., and Moretta, A.
(1987) Eur. J. Immunol. 17, 55–6059. Hubert, P., Lang, V., Debre, P., and Bismuth, G. (1996) J. Immunol. 157,
4322–433260. King, P. D., Sadra, A., Han, A., Liu, X.-R., Sunder-Plassmann, R., Reinherz,
E. L., and Dupont, B. (1996) Int. Immunol. 8, 1707–171461. Bell, G. M., Bolen, J. B., and Imboden, J. B. (1992) Mol. Cell. Biol. 12,
5548–555462. Anderson, S. J., Levin, S. D., and Perlmutter, R. M. (1994) Adv. Immunol. 56,
151–17863. Molina, T. J., Kishira, K., Siderovski, D. P., van-Ewijk, W., Narendran, A.,
Timms, E., Wakeham, A., Paige, C. J., Hartmann, K.-U., Veillette, A.,Davidson, D., and Mak, T. W. (1992) Nature 357, 161–164
64. Iwashima, M., Irving, B. A., van-Oers, N. S. C., Chan, A. C., and Weiss, A.(1994) Science 263, 1136–1139
65. van-Oers, N. S. C., Killeen, N., and Weiss, A. (1996) J. Exp. Med. 183,1053–1062
66. Sunder-Plassmann, R., Lialios, F., Madsen, M., Koyasu, S., and Reinherz, E. L.(1997) Eur. J. Immunol. 27, 2001–2009
67. Burkhardt, A. L., Stealey, B., Rowley, R. B., Mahajan, S., Prendergast, M.,Fargnoli, J., and Bolen, J. B. (1994) J. Biol. Chem. 269, 23642–24647
68. Latour, S., Chow, L. M. L., and Veillette, A. (1996) J. Biol. Chem. 271,22782–22790
69. Fargnoli, J., Burkhardt, A. L., Laverty, M., Kut, S. A., van-Oers, N. S. C.,Weiss, A., and Bolen, J. B. (1995) J. Biol. Chem. 270, 26533–26537
70. Wange, R. L., and Samelson, L. E. (1996) Immunity 5, 197–20571. Gelfand, E. W., Cheung, R. K., Mills, G. B., and Grinstein, S. (1988) Eur.
J. Immunol. 18, 917–92272. Gardner, P., Alcover, A., Kuno, M., Moingeon, P., Weyand, C. M., Goronzy, J.,
and Reinherz, E. L. (1989) J. Biol. Chem. 264, 1068–107673. Goldsmith, M. A., and Weiss, A. (1987) Proc. Natl. Acad. Sci. U. S. A. 84,
6879–688374. Nishibe, S., Wahl, M. I., Hernandez-Sotomayor, S. M. T., Tonks, N. K., Rhee,
S. G., and Carpenter, G. (1990) Science 250, 1253–125675. Ramos-Morales, F., Druker, B. J., and Fischer, S. (1994) Oncogene 9,
1917–192376. Derijard, B., Hibi, M., Wu, I.-H., Barrett, T., Su, B., Deng, T., Karin, M., and
Davis, R. J. (1994) Cell 76, 1025–103777. Durand, D. B., Shaw, J. P., Bush, M. R., Replogle, R. E., Balgaje, R., and
Crabtree, G. R. (1988) Mol. Cell. Biol. 8, 1715–172478. Kamps, M. P., Corcoran, L., LeBowitz, J. H., and Baltimore, D. (1990) Mol.
Cell. Biol. 10, 5464–547279. Fraser, J. D., Irvine, B. A., Crabtree, G. R., and Weiss, A. (1991) Science 251,
313–31680. Granelli-Piperno, A., and Nolan, P. (1991) J. Immunol. 147, 2734–273981. Muegge, K., Williams, T. M., Kant, J., Karin, M., Chiu, R., Schmidt, A.,
Siebenlist, U., Young, H. A., and Durum, S. K. (1989) Science 246, 249–25182. Serfling, E., Barthelmaes, R., Pfeuffer, I., Schenk, B., Zarius, S., Swoboda, R.,
Mercurio, F., and Karin, M. (1989) EMBO J. 8, 465–47383. Jain, J., Valge-Archer, V. E., and Rao, A. (1992) J. Immunol. 148, 1240–125084. Ullman, K., Northrop, J., Admon, A., and Crabtree, G. (1993) Genes Dev. 7,
188–19685. DeGrazia, U., Felli, M. P., Vacca, A., Farina, A. R., Maroder, M., Cappabianca,
L., Meco, D., Farina, M., Screpanti, I., Frati, L., and Gulino, A. (1994)J. Exp. Med. 180, 1485–1497
86. Northrop, J. P., Ho, S. N., Chen, L., Thomas, D. J., Timmerman, L. A., Nolan,G. P., Admon, A., and Crabtree, G. R. (1994) Nature 369, 497–502
87. Parra, E., Varga, M., Hedlund, G., Kalland, T., and Dohlsten, M. (1997) Mol.Cell. Biol. 17, 1314–1323
88. Chiu, R., Angel, P., and Karin, M. (1989) Cell 59, 979–98689. Suzuki, T., Okuno, H., Yoshida, T., Endo, T., Nishina, H., and Iba, H. (1991)
Nucleic Acids Res. 19, 5537–554290. Schwartz, R. H. (1990) Science 248, 1349–135691. Kang, S. M., Bart, B., Tran, A. C., Brorson, K., Schwartz, R. H., and Lenardo,
M. J. (1992) Science 257, 1134–113892. Sundstedt, A., Sigvardsson, M., Leanderson, T., Hedlund, G., Kalland, T., and
Dohlsten, M. (1996) Proc. Natl. Acad. Sci. U. S. A. 93, 979–984
p56lck-independent CD2 Signaling 24257
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from

ReinherzRaute Sunder-Plassmann and Ellis L. Signaling Involves Jun Kinase
-independent Pathway of CD2 lck A p56CELL BIOLOGY AND METABOLISM:
doi: 10.1074/jbc.273.37.242491998, 273:24249-24257.J. Biol. Chem.
http://www.jbc.org/content/273/37/24249Access the most updated version of this article at
.JBC Affinity SitesFind articles, minireviews, Reflections and Classics on similar topics on the
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/273/37/24249.full.html#ref-list-1
This article cites 92 references, 52 of which can be accessed free at
by guest on Decem
ber 3, 2015http://w
ww
.jbc.org/D
ownloaded from
![arXiv:1702.05747v2 [cs.CV] 4 Jun 2017](https://static.fdokumen.com/doc/165x107/631c4ea1b8a98572c10cd804/arxiv170205747v2-cscv-4-jun-2017.jpg)
![arXiv:1510.02125v3 [cs.CL] 3 Jun 2016](https://static.fdokumen.com/doc/165x107/6313c62ec72bc2f2dd0423d4/arxiv151002125v3-cscl-3-jun-2016.jpg)






![arXiv:1912.04007v3 [math.NA] 14 Jun 2021](https://static.fdokumen.com/doc/165x107/63176943f68b807f88039fe2/arxiv191204007v3-mathna-14-jun-2021.jpg)





![arXiv:1606.04930v1 [cs.LG] 15 Jun 2016](https://static.fdokumen.com/doc/165x107/632157a3e9691360fe022f38/arxiv160604930v1-cslg-15-jun-2016.jpg)





