
A noveltau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal...
17
Reactivation of Human Neurotropic JC Virus Expressing Oncogenic Protein in a Recurrent Glioblastoma Multiforme Luis Del Valle, MD,* S. Ausim Azizi, MD, PhD,² Barbara Krynska, MS, PhD,* Sahnila Enam,* Sidney E. Croul, MD,* and Kamel Khalili, PhD* Examination of the primary tumor of glioblastoma mul- tiforme and its recurrence for their association with JC virus revealed that, while the viral genome is present in both initial and recurrent tumors, expression of the viral oncoprotein T-antigen occurs only in the recurrent tu- mor cells. Accordingly, the level of inducible cellular transcription factors, including the p65 subunit of NF-kB and YB-1, which have the ability to stimulate JCV gene expression, was found to be higher in the recurrent tumor cells. These observations suggest that induction of the regulatory factors after resection of the primary tu- mor may have reactivated JC virus gene expression and led to redevelopment of the tumor in brain. Del Valle L, Azizi SA, Krynska B, Enam S, Croul SE, Khalili K. Reactivation of human neurotropic JC virus expressing oncogenic protein in a recurrent glioblastoma multiforme. Ann Neurol 2000;48:932–936 JC virus (JCV) is a human neurotropic polyomavirus infecting over 70% of the human population world- wide during early childhood. 1 Replication of JCV in the central nervous system (CNS) results in the fatal demyelinating disease progressive multifocal leukoen- cephalopathy (PML). 2,3 PML occurs primarily in adults with immunosuppressive conditions and fre- quently presents with rapidly progressing dementia and weakness. 4,5 The histological changes in PML occur primarily in glial cells and include destruction of oli- godendrocytes, the myelin-producing cells in the CNS leading to demyelinating plaques, and the appearance of bizarre astrocytes. These events rely on the product of the viral early gene T-antigen, which is preferentially expressed in glial cells. The specificity of the viral early gene expression is attributed to several cis-elements that are induced in response to extracellular stimuli. 6,7 The T-antigen of JCV is oncogenic, as its expression trans- forms glial cells in vitro 8 and induces the development of neural-origin tumors in experimental animals. 9 –11 Several clinical studies reported cases of concomitant PML and CNS neoplasm in patients with long- standing immunosuppressive disorders such as leuke- mia, lymphoma, and acquired immunodeficiency syn- drome (AIDS). 12 More recently, several studies have demonstrated the presence of JCV DNA sequence and expression of T-antigen in oligoastrocytoma and me- dulloblastomas from nonimmunosuppressed individu- als. 13–15 In this report, we describe detection of JCV in a glioblastoma multiforme (GBM) from an immuno- competent patient in whom resections were performed on the initial tumor and its recurrence several months later following chemotherapy and radiotherapy. Our results showed the presence of the JCV sequence in the primary and the recurrent tumor cells, and expression of T-antigen was found only in recurrent neoplastic cells. Interestingly, production of several inducible transcription factors that potentiate JCV early gene transcription was detected in the recurrent tumor, but not in the primary tumor, suggesting that alterations in the physiological condition of the patient after the ini- tial surgery and during the course of treatment may have reactivated JCV early gene transcription. Materials and Methods Immunohistochemical Analysis The left posterior parietal lobectomy specimen was prepared for hematoxylin and eosin and immunostaining using pAb416 (Oncogene Science, Boston, MA) for detection of T-antigen, DO-7 (Dako) for detection of p53, C-20 (Santa Cruz Biotechnology, Santa Cruz, CA) for detection of p65, and polyclonal anti-YB-1 antibody according to the proce- dure described previously. 14 Gene Amplification and Molecular Analysis DNA was extracted from the paraffin-embedded tissue using QIA amp tissue Kit (Qiagen, Valencia, CA) and polymerase chain reaction (PCR) amplification was performed using three sets of primers that amplified various regions of JCV as detailed previously. 14 Sequencing of the amplified DNA was carried out by automated fluorescent DNA cycle sequencing using the ABI Prism 377 DNA Sequencer-XL. Results and Discussion A 24-year-old immunocompetent caucasian man with no significant medical history experienced headaches behind the left eye that had begun to increase in in- tensity and that were associated with flashes in the From the *Center for Neurovirology and Cancer Biology, Labora- tories of Brain Tumor Biology and Neuropathology, College of Sci- ence and Technology, Temple University, and ²Department of Neurology, MCP Hahnemann University, Philadelphia, PA. Received Jun 8, 2000, and in revised form Jul 18. Accepted for publication Aug 8, 2000. Address correspondence to Dr Khalili, Center for Neurovirology and Cancer Biology, Laboratories of Brain Tumor Biology and Neuropathology, College of Science and Technology, Temple Uni- versity, 1900 North 12th Street, 015-96, Room 203, Philadelphia, PA 19122. BRIEF COMMUNICATIONS 932 Copyright © 2000 by the American Neurological Association
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of A noveltau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal...

Reactivation of HumanNeurotropic JC VirusExpressing OncogenicProtein in a RecurrentGlioblastoma MultiformeLuis Del Valle, MD,* S. Ausim Azizi, MD, PhD,†Barbara Krynska, MS, PhD,* Sahnila Enam,*Sidney E. Croul, MD,* and Kamel Khalili, PhD*
Examination of the primary tumor of glioblastoma mul-tiforme and its recurrence for their association with JCvirus revealed that, while the viral genome is present inboth initial and recurrent tumors, expression of the viraloncoprotein T-antigen occurs only in the recurrent tu-mor cells. Accordingly, the level of inducible cellulartranscription factors, including the p65 subunit ofNF-kB and YB-1, which have the ability to stimulate JCVgene expression, was found to be higher in the recurrenttumor cells. These observations suggest that induction ofthe regulatory factors after resection of the primary tu-mor may have reactivated JC virus gene expression andled to redevelopment of the tumor in brain.
Del Valle L, Azizi SA, Krynska B, Enam S,Croul SE, Khalili K. Reactivation of human
neurotropic JC virus expressing oncogenic proteinin a recurrent glioblastoma multiforme.
Ann Neurol 2000;48:932–936
JC virus (JCV) is a human neurotropic polyomavirusinfecting over 70% of the human population world-wide during early childhood.1 Replication of JCV inthe central nervous system (CNS) results in the fataldemyelinating disease progressive multifocal leukoen-cephalopathy (PML).2,3 PML occurs primarily inadults with immunosuppressive conditions and fre-quently presents with rapidly progressing dementia andweakness.4,5 The histological changes in PML occurprimarily in glial cells and include destruction of oli-godendrocytes, the myelin-producing cells in the CNS
leading to demyelinating plaques, and the appearanceof bizarre astrocytes. These events rely on the productof the viral early gene T-antigen, which is preferentiallyexpressed in glial cells. The specificity of the viral earlygene expression is attributed to several cis-elements thatare induced in response to extracellular stimuli.6,7 TheT-antigen of JCV is oncogenic, as its expression trans-forms glial cells in vitro 8 and induces the developmentof neural-origin tumors in experimental animals.9–11
Several clinical studies reported cases of concomitantPML and CNS neoplasm in patients with long-standing immunosuppressive disorders such as leuke-mia, lymphoma, and acquired immunodeficiency syn-drome (AIDS).12 More recently, several studies havedemonstrated the presence of JCV DNA sequence andexpression of T-antigen in oligoastrocytoma and me-dulloblastomas from nonimmunosuppressed individu-als.13–15 In this report, we describe detection of JCV ina glioblastoma multiforme (GBM) from an immuno-competent patient in whom resections were performedon the initial tumor and its recurrence several monthslater following chemotherapy and radiotherapy. Ourresults showed the presence of the JCV sequence in theprimary and the recurrent tumor cells, and expressionof T-antigen was found only in recurrent neoplasticcells. Interestingly, production of several inducibletranscription factors that potentiate JCV early genetranscription was detected in the recurrent tumor, butnot in the primary tumor, suggesting that alterations inthe physiological condition of the patient after the ini-tial surgery and during the course of treatment mayhave reactivated JCV early gene transcription.
Materials and MethodsImmunohistochemical AnalysisThe left posterior parietal lobectomy specimen was preparedfor hematoxylin and eosin and immunostaining usingpAb416 (Oncogene Science, Boston, MA) for detection ofT-antigen, DO-7 (Dako) for detection of p53, C-20 (SantaCruz Biotechnology, Santa Cruz, CA) for detection of p65,and polyclonal anti-YB-1 antibody according to the proce-dure described previously.14
Gene Amplification and Molecular AnalysisDNA was extracted from the paraffin-embedded tissue usingQIA amp tissue Kit (Qiagen, Valencia, CA) and polymerasechain reaction (PCR) amplification was performed usingthree sets of primers that amplified various regions of JCV asdetailed previously.14 Sequencing of the amplified DNA wascarried out by automated fluorescent DNA cycle sequencingusing the ABI Prism 377 DNA Sequencer-XL.
Results and DiscussionA 24-year-old immunocompetent caucasian man withno significant medical history experienced headachesbehind the left eye that had begun to increase in in-tensity and that were associated with flashes in the
From the *Center for Neurovirology and Cancer Biology, Labora-tories of Brain Tumor Biology and Neuropathology, College of Sci-ence and Technology, Temple University, and †Department ofNeurology, MCP Hahnemann University, Philadelphia, PA.
Received Jun 8, 2000, and in revised form Jul 18. Accepted forpublication Aug 8, 2000.
Address correspondence to Dr Khalili, Center for Neurovirologyand Cancer Biology, Laboratories of Brain Tumor Biology andNeuropathology, College of Science and Technology, Temple Uni-versity, 1900 North 12th Street, 015-96, Room 203, Philadelphia,PA 19122.
BRIEF COMMUNICATIONS
932 Copyright © 2000 by the American Neurological Association

temporal visual field of the right eye. Magnetic reso-nance imaging (MRI) of the brain revealed a large par-tially cystic mass measuring 7 cm located in the leftparietal lobe. A total resection of the mass was per-formed, and the patient received 6,210 cGy of postop-erative radiation in 34 fractions followed by BCNUchemotherapy. Histopathological study revealed a neo-plasm of neuroectodermal origin with differentiation ofglial elements. The tumor was classified as high-gradeglioma, that is, GBM. Histologically, the tumor wascomposed of numerous pleomorphic cells with atypi-cal, hyperchromatic nuclei and abundant eosinophyliccytoplasm.
After treatment, the patient had no symptoms for 15months except for right inferior quadrantanopsia anddifficulty reading. Follow-up MRI and thalium-singlephoton emission computed tomography (SPECT) re-vealed a recurrence of the tumor. Results from histo-
logical analysis of tissue from a left occipital craniot-omy and lobectomy showed a neoplasm of neuro-ectodermal origin with differentiation of glial elements.The recurrence showed identical characteristics to theoriginal tumor with the exception of increased mitoticfigures, cell pleomorphism, and areas of necrosis. Im-munohistochemistry against the intermediate filamentglial fibrillary acidic protein showed intense staining inthe cytoplasm of the neoplastic cells from both theoriginal tumor and the recurrence (data not shown).
Recent observations on the association of polyoma-viruses with human cancer12 prompted us to examinethe primary and recurrent tumor tissues for the pres-ence of the human polyomavirus, JCV, sequence. To-ward this end, total DNA was obtained from tissuesand analyzed by PCR technique using a set of primersthat amplify the N- and C-terminal regions of JCVT-antigen between nucleotides 4255 to 4427 and 2578
Fig 1. Detection of DNA sequence corresponding to JC virus ( JCV) in the original and recurrent tumors. (A) Structural organiza-tion of the JCV genome and the position of the DNA primers and probes used for polymerase chain reaction (PCR) amplificationand Southern blot analysis. The positions of the early genes (T/t-antigens) and late genes (Agno, VP1, VP2, and Vp3) are shown.The positions of the PCR primers used for amplification of the 173-, 230-, and 212-bp DNA fragments corresponding to theN-terminus and C-terminus of T-antigen, and the C-terminus of VP1, respectively, are shown. The location of the oligonucleotideDNA probes corresponding to the control region of the amplified sequences that are used for Southern blot hybridization is shown.(B) PCR amplification of JCV DNA sequence using primers from the N-terminus and C-terminus of T-antigen and the VP1 gene.PCR products from primary (I) and recurrence (II) were analyzed by Southern blot analysis by using specific DNA probes as shownin A. NC 5 negative control in which DNA from normal (without progressive multifocal leukoencephalopathy) samples was usedin PCR; PC 5 positive control in which DNA from JCV-transformed glial cells was examined.
Brief Communication: Del Valle at al: JC Virus Expressing Oncogenic Protein in GBM 933
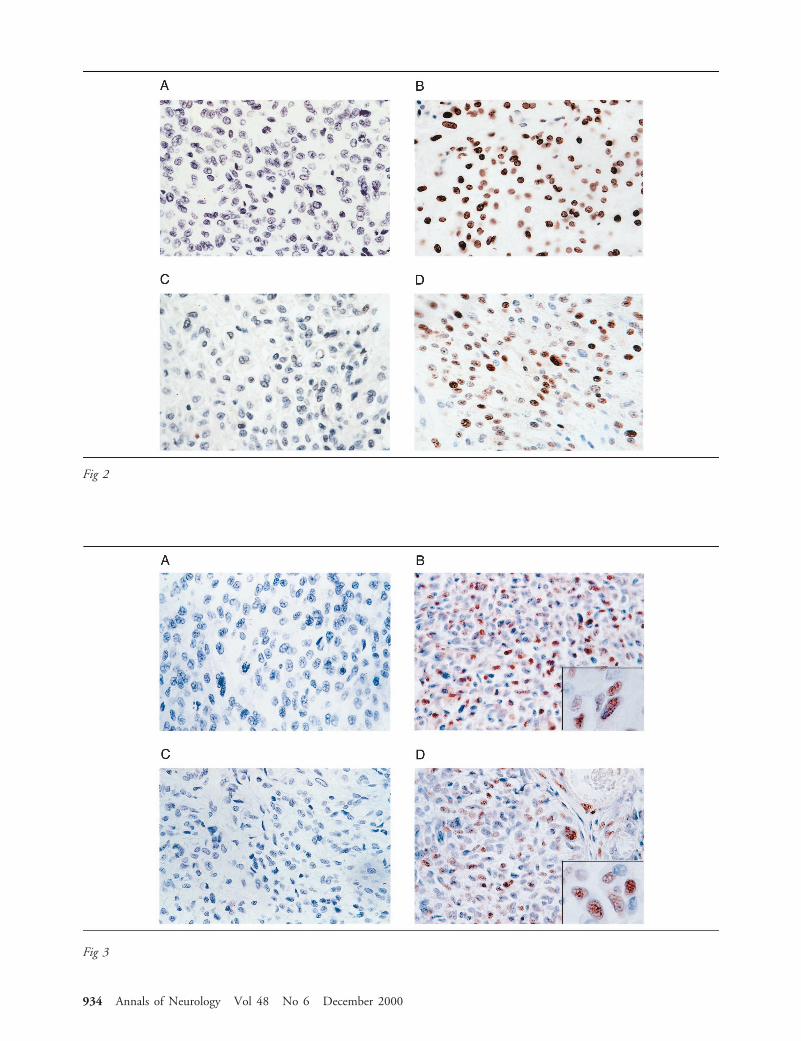
Fig 2
Fig 3
934 Annals of Neurology Vol 48 No 6 December 2000

to 2797, respectively. In addition, a pair of primerscorresponding to the viral late genome, between nucle-otides 1828 and 2039, was used (Fig 1A). All PCRproducts were analyzed by Southern blot using JCV-specific probes derived from the internal regions of theamplified fragments as shown in Figure 1A. Resultsshowed that both DNA samples obtained from the ini-tial and the recurrent tumors are positive for all threeregions of the JCV genome, indicative of the presence ofthe viral DNA in the tumor cells (see Fig 1B). The neg-ative control DNA was from a patient with no sign oftumor or PML. Sequence analysis of the amplified DNAverified the presence of JCV DNA in the tumor cells.
Next, expression of JCV T-antigen was determinedby immunohistochemistry. While the initial tumorcells were negative for T-antigen production (Fig 2A),cells from the recurrent tumor exhibited strongT-antigen positivity in the nuclei (see Fig 2B). Of thetotal tumor cells in a 403 microscopic field, over 90%of nuclei were immunopositive. We also examined theexpression of p53 in these samples. p53 is a tumor sup-pressor protein that, because of its short half-life, is notusually detectable in normal tissue. However, a muta-tion in the p53 or its association with several onco-proteins, including those from polyomaviruses, stabi-lizes this protein and maintains p53 in a nonfunctionalstate.16 As shown in Figure 2, the initial tumor tissue,which was negative for T-antigen, showed no strongstaining with anti-p53 antibody (see Fig 2C). However,a significant number of cells from the recurrent tumorshowed positive nuclear staining for p53 (see Fig 2D),implying that association of p53 with JCV T-antigen inthe recurrent tumor cells may increase its stability in anonfunctional state. These observations suggest the in-volvement of distinct pathways in the development ofGBM at the initial stage where T-antigen is absent andin the recurrent stages where T-antigen is expressed.
Reactivation of the JCV early promoter in the recur-rent tumor cells, which promotes the expression ofT-antigen, provided the rationale for the examinationof several inducible regulatory factors that participatein the control of viral gene transcription. In this re-spect, we focused our attention on the inducible tran-scription factor, NF-kB, since earlier studies revealedthe presence of functional kB in the control region ofthe JCV early regulatory sequences, which is responsive
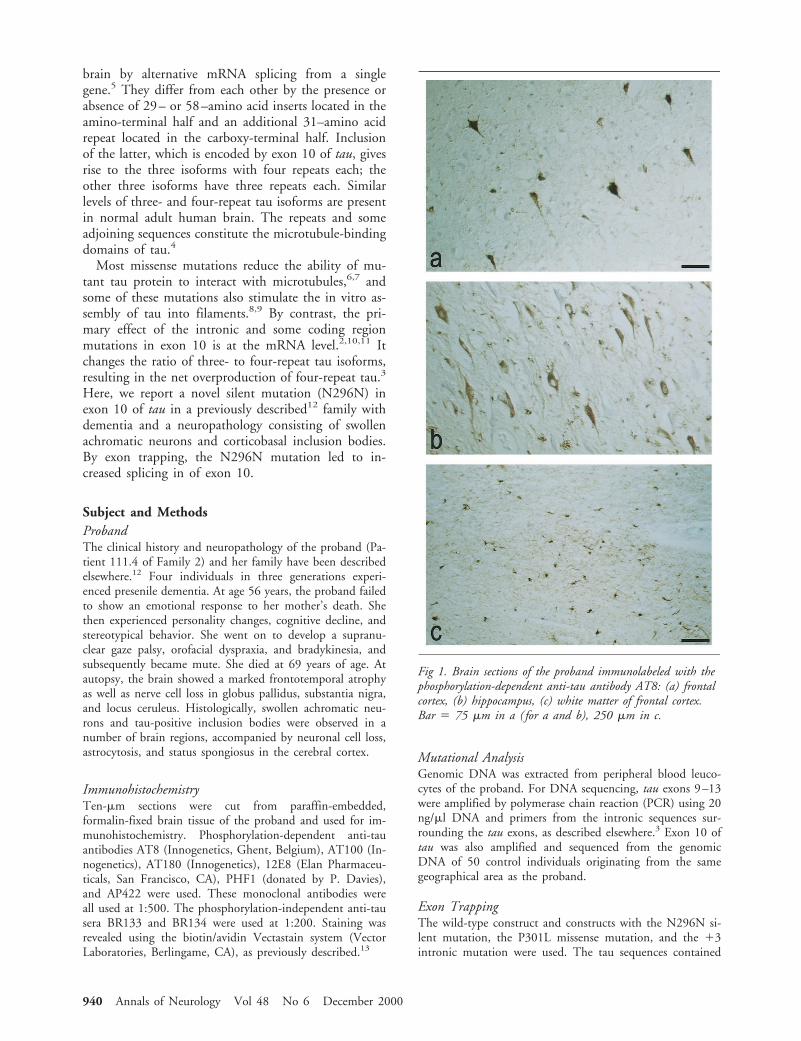
to NF-kB modulation.7 In addition, activation of theJCV promoter by YB-1,17,18 a transcription factor thatcan be induced under stress conditions,19 prompted usto examine the level of YB-1 gene expression in thesetumors. Immunohistochemical staining of the tumortissues revealed that, unlike the primary tumor cells,which showed no evidence for high level expression ofthe p65 subunit of NF-kB, the nuclei of the recurrenttumor cells indicates an increased level of p65 produc-tion (Fig 3, compare A and B). Similarly, the level ofYB-1 was significantly enhanced in the recurrent tumorcells in comparison to those seen in the primary tumor(compare C and D in Fig 3). This variation was not ob-served with a ubiquitous transcription factor such as Sp1(data not shown). These observations corroborate with thehypothesis that the enhancement of NFkB and YB-1 ac-tivities in the recurrent tumor results in the transcriptionalactivation of the JCV early gene in these tumor cells.
Reactivation of the viral early genome in the absenceof viral late gene expression and viral DNA replicationcan lead to accumulation of the early protein,T-antigen. This protein has the ability to interact withand functionally inactivate cellular proteins such asp53, which is involved in the control of cell prolifera-tion. Expression of JCV T-antigen in the recurrent tu-mor cells and detection of p53 suggests that the inter-action of T-antigen with p53 in the tumor cells mayinactivate this protein and lead to deregulated prolifer-ation of glioma in this patient. On the other hand, thelack of JCV T-antigen and p53 in the initial tumorsuggests that an alternative pathway may be involved inthe development of the initial tumor.
Expression of the JCV early gene, which is concur-rent with detection of p65 NF-kB and YB-1 in therecurrent tumor cells, suggests the involvement of theseregulators, at least in part, in the transcriptional acti-vation of the JCV promoter. Although the cause of thestimulation of the NF-kB pathway that occurs after theinitial tumor resection remains unclear, several events(eg, alteration of cytokine gene expression, exposure tochemicals, and radiation) are among the known NF-kBinducers.20 Furthermore, it has been shown that thestress-inducing agents can cause enhanced expression ofYB-1 in tumor cells.19 Experiments are currently inprogress to determine the effect of chemotherapy andradiation therapy on reactivation of JCV early gene
Fig 2. Immunohistological analysis of the primary tumor and recurrence. Immunostaining of the primary tumor samples (A) andrecurrent (B) with anti-T-antigen antibody. C and D represent nuclear immunostaining of the primary tumor and recurrence, re-spectively, with anti-p53 antibody. (Original magnification, 3400.)
Š
Fig 3. Immunohistological evaluation of primary tumor and recurrent tumors for expression of NF-kB p65 and YB-1. Immunohis-tochemistry against NF-kB p65 is negative in the primary tumor samples (A) and shows strong nuclear reactivity in the recurrence(B). (C) Staining of primary tumor with anti-YB-1 antibody. (D) Nuclear staining in the recurrent tumor was observed withYB-1 polyclonal antibody. (Original magnification, 3400; insets, 31,000.)
Š
Brief Communication: Del Valle at al: JC Virus Expressing Oncogenic Protein in GBM 935

transcription in an in vitro cell culture system and inexperimental animals.
This work was made possible by grants (PO1 NS3466) awarded bythe National Institutes of Health to Dr Khalili.
We thank past and present members of the Center for Neurovirol-ogy and Cancer Biology for their insightful discussion, and sharingof ideas and reagents. We also wish to thank C. Schriver for edito-rial assistance.
References1. Berger JR, Concha M. Progressive multifocal leukoencephalop-
athy: the evolution of a disease once considered rare. J Neuro-virol 1995;1:5–18
2. Padgett BL, Walker DL, ZuRhein GM, et al. Cultivation of apapova-like virus from human brain with progressive multifocalleukoencephalopathy. Lancet 1971;1:1257–1260
3. ZuRhein GM, Chow SM. Particles resembling papovavirions inhuman cerebral demyelinating disease. Science 1965;148:1477–1479
4. Astrom KE, Mancall EL, Richardson EP Jr. Progressive multi-focal leukoencephalopathy: a hitherto unrecognized complica-tion of chronic lymphocytic leukemia and lymphoma. Brain1958;81:93–111
5. Richardson EP Jr. Progressive multifocal leukoencephalopathy.N Engl J Med 1961;265:815–823
6. Raj GV, Khalili K. Transcriptional regulation: lessons from thehuman neurotropic polyomavirus, JCV. Virology 1995;213:283–291
7. Ranganathan PN, Khalili K. The transcriptional enhancer ele-ment, kB, regulates promoter activity of the human neurotropicvirus, JCV, in cells derived from the CNS. Nucleic Acids Res1993;21:959–964
8. Mandl CW, Frisque RJ. Characterization of cells transformed bythe human polyomavirus JC virus. J Gen Virol 1986;67:1733–1739
9. London WT, Houff SA, Madden DL, et al. Brain tumors inowl monkeys inoculated with a human polyomavirus (JCV).Science 1978;201:1246–1249
10. Ohsumi S, Ikehara I, Motoi M, et al. Induction of undifferen-tiated brain tumors in rats by a human polyomavirus (JCV).Japan J Cancer Res 1985;76:429–431
11. ZuRhein GM. Studies of JCV-induced nervous system tumorsin the Syrian hamster: a review. In: Polyomaviruses and humanneurological diseases. New York: Alan R Liss, 1983:205–221
12. Gallia GL, Gordon J, Khalili K. Tumor pathogenesis of humanneurotropic JC virus in the CNS. J Neurovirol 1998;4:175–181
13. Khalili K, Krynska B, Del Valle L, et al. Medulloblastomas andthe human neurotropic polyomavirus JC virus. Lancet 1999;353:1152–1153
14. Krynska B, Del Valle L, Croul S, et al. Detection of humanneurotropic JC virus DNA sequence and expression of the viraloncogenic protein in pediatric medulloblastomas. Proc NatlAcad Sci USA 1999;96:519–524
15. Rencic A, Gordon J, Otte J, et al. Detection of JCV DNAsequence and expression of the viral oncogene, T-antigen inbrain of immunocompetent patient with oligoastrocytoma. ProcNatl Acad Sci USA 1996;93:7352–7357
16. Oren M. Regulation of the p53 tumor suppressor protein.J Biol Chem 1999;274:36031–36034
17. Safak M, Gallia G, Khalili K. Reciprocal interaction betweentwo cellular proteins, Pura and YB-1, modulates transcriptionalactivity of human JCVCY in glial cells. Mol Cell Biol 1999;19:2712–2723
18. Safak M, Gallia GL, Ansari SA, Khalili K. Physical and func-tional interaction between the Y-box binding protein YB-1 andhuman polyomavirus JC virus large T-antigen. J Virol 1999;73:10146–10157
19. Koike KT, Uchiumi T, Ohga T, et al. Nuclear translocation ofthe Y-box binding protein by ultraviolet irradiation. FEBS Lett1997;417:390–394
20. Heike PL. Activators and targets of genes of Rel/NFkB tran-scription factors. Oncogene 1999;18:6853–6866
Herpes Simplex Virus Type1 (HSV-1)–Induced Retinitisfollowing Herpes SimplexEncephalitis: Indications forBrain-to-Eye Transmissionof HSV-1Jeroen Maertzdorf, MSc,* Allegonda Van der Lelij, PhD,†G. Seerp Baarsma, MD,‡Albert D. M. E. Osterhaus PhD,*and Georges M. G. M. Verjans, PhD*‡
Herpes simplex encephalitis is a severe neurological dis-ease with high mortality and morbidity rates. Reactivatedherpes simplex virus type 1 (HSV-1) can cause relapsesand might even spread to the retina, where it can inducea potentially blinding eye disease, known as acute retinalnecrosis. In the present study, the HSV-1 strains in thebrain and eye of 2 patients with acute retinal necrosisfollowing an episode of herpes simplex encephalitis weregenotyped. The HSV-1 strains in both the brain and eyewere identical in each patient, but they differed interin-dividually. The data suggest brain-to-eye transmission ofHSV-1 in these patients.
Maertzdorf J, Van der Lelij A, Baarsma GS,Osterhaus ADME, Verjans GMGM. Herpes
simplex virus type 1 (HSV-1)–induced retinitisfollowing herpes simplex encephalitis: indications
for brain-to-eye transmission of HSV-1.Ann Neurol 2000;48:936–939
From the *Department of Virology, Erasmus University, and ‡Rot-terdam Eye Hospital, Rotterdam; and †Department of Ophthalmol-ogy, Leiden University Medical Center, Leiden, The Netherlands.
Received Jun 12, 2000, and in revised form Jul 31. Accepted forpublication Aug 7, 2000.
Address correspondence to Dr Verjans, Department of Virology,Erasmus University, PO Box 1738, 3000 DR Rotterdam, TheNetherlands.
936 Copyright © 2000 by the American Neurological Association

Herpes simplex encephalitis (HSE), caused by an infec-tion of the brain by herpes simplex virus (HSV) is asevere disease with high mortality and morbidity rates.1
Reactivation and neuronal translocation of HSV canresult in relapses of HSE or new infections at anatomi-cally different sites, such as the eye. Clinical data suggestthat HSE may be a risk factor for the development ofacute retinal necrosis (ARN), a rapidly progressing andpotentially blinding eye disease induced by HSV.2–4
Two patients with HSE in whom ARN developedlater in life were included in this study. The HSV-1strains involved in both disease manifestations of eachpatient were genotyped using a newly developed poly-merase chaine reaction (PCR) method5 and subsequentnucleotide sequence analyses. The data indicate that inboth patients HSE and ARN were caused by a singleHSV-1 strain, suggesting transneuronal spread of thevirus from brain to eye.
Patients and MethodsPatientsPatient 1 was a 68-year-old man who had been admitted tothe hospital in a somnolent state. A viral encephalitis wassuspected, and computed tomographic scans showed a hypo-density in the right temporal region. A cerebrospinal fluid(CSF) sample showed leukocyte counts of 73 3 106/L. Di-agnosis of HSE was confirmed by detection of HSV-1 DNA,determined by PCR using virus-specific primers as described6
and HSV-specific antibodies in the CSF. Intravenous treat-ment with 10 mg/kg acyclovir three times daily for 2 weeksresulted in slow recovery. However, 9 months after dischargefrom the hospital, he experienced a unilateral decrease of vi-sual acuity (0.2). The diagnosis of ARN was made on clinicalgrounds and confirmed by detection of HSV-1 DNA andlocal HSV-specific antibody production in the aqueous hu-mor as described previously.6 Again the patient was treatedwith acyclovir intravenously, and maintenance therapy withvalacyclovir orally resulted in a slight improvement, with aremaining visual acuity of 0.5.
Patient 2 was a 64-year-old woman hospitalized because ofprogressive headache with vomiting and aphasia. Scansshowed a hypodense and space-occupying process in the lefttemporal region. A CSF sample showed a leukocyte count of44 3 106/L, and the diagnosis of HSE was confirmed bydetection of HSV-1 DNA6 and HSV-specific antibodies inthe CSF. A slow recovery was achieved after intravenoustreatment with 10 mg/kg acyclovir three times daily for 2weeks. Only 10 days after being discharged from the hospi-tal, this patient experienced unilaterally decreased visual acu-ity of 0.1. ARN was diagnosed 2 weeks later. An aqueoushumor sample contained HSV-1 DNA as determined byPCR, whereas no local HSV-specific antibody productioncould be detected.6 Again, this patient was given antiviraltreatment with acyclovir. However, despite maintenancetherapy, the remaining visual acuity was only finger countingat 3 meters.
HSV-1 Strain DifferentiationIsolation of DNA from the CSF and aqueous humor samplesfrom both patients, taken for diagnostic purposes, was per-formed as described previously.6 The HSV-1 strains in thesesamples were genotyped with a recently developed PCR-based DNA fingerprint assay that allows the rapid and accu-rate discrimination of up to 92% of unrelated HSV-1strains.5 The assay is based on the amplification of hyper-variable regions within the HSV-1 genes US1 and US12.These regions contain strain-to-strain differences in the num-ber of DNA repeats, termed reiteration IV (ReIV),7 resultingin variable amplicon lengths between HSV-1 strains. Sizeand specificity of the PCR products were determined on anagarose gel and Southern blotting with ReIV-specific probes.Nucleotide sequence analysis of gel-purified HSV-1 US12gene amplicons was performed with both PCR primers on aPerkin Elmer (Foster City, CA) automated sequencer using acommercially available kit according to the manufacturer’sinstructions (DYEnamic ET Terminator; Amersham Phar-macia, Cleveland, OH).
ResultsThe CSF- and aqueous humor–derived HSV-1 strainsfrom both patients were genotyped using a recentlydeveloped PCR assay.5 Although they were differentbetween the patients, the HSV-1 US1 and US12 am-plicons amplified from both CSF- and aqueous hu-mor–derived DNA samples from each patient were ofsimilar size (Fig 1). The nucleotide sequences of theUS12 amplicons were determined and aligned with thecorresponding sequence of HSV-1 strain 17 (HS1US;GenBank accession number 291490) (Fig 2). TheDNA sequence analyses revealed identical nucleotidesequences in CSF and aqueous humor samples fromeach patient. Comparison between the patients re-vealed, next to a difference in the number of ReIV el-ements (two and three times for Patients 1 and 2, re-spectively), four separate and unique point mutations(see Fig 2). These data suggest that in each patient thesame HSV-1 strain was involved in the pathogenesis ofboth HSE and ARN. Interestingly, next to the 22-bp-long repeating elements (ReIV), a new 45-bp-long re-peating element was identified in the US12 sequences.This 45-bp element (designated here as ReVIII) wasrepeated two and three times in the HSV-1 strains ob-tained from Patients 1 and 2, respectively. In the US12gene sequence of HSV-1 strain 17, the number ofReIV and ReVIII repeats are 5 and 1, respectively.
DiscussionSeveral studies have reported on the development ofHSV-induced ARN following an episode of HSE.2-4 Ithas been hypothesized that the induction of ARN inthese patients was due to reactivation of latent HSVwithin the brain and subsequent infection of the retina.Studies on the experimental ARN mouse model haveprovided evidence for this assumption. Herein, intraoc-
Brief Communication: Maertzdorf et al: Brain-to-Eye Transmission of HSV-1 in Humans 937

ular inoculation of mice with HSV-1 resulted in infec-tion of the brain and subsequent ARN in the contralat-eral eye. The virus was shown to reach the retina of thecontralateral eye by transaxonal spread through the opticnerve.8
Here, 2 ARN patients with a previous episode ofHSE were studied to determine whether a similarmode of brain-to-eye transmission of HSV-1 had oc-curred. Detailed genotypic analyses of the HSV-1strains located in the brain and eye samples from thesepatients strongly suggest that the viruses found in bothanatomical sites of each patient were identical but dif-
fered interindividually. To our knowledge, this is thefirst study to provide molecular evidence that a singleHSV-1 strain can cause HSE and subsequently ARN ina single individual. Analogous to the ARN mousemodel, this suggests that the virus may have spread fromthe brain to the eye, probably through the optic nerve.
The potential of HSV-1 to establish latency in thebrain9 and reactivate from neural cells poses a lifetimethreat of recurrent infections. Our findings should alertneurologists to the possibility that HSE may be fol-lowed by ARN, since only prompt and specializedmedical care may prevent the loss of sight in such pa-
Fig 1. Polymerase chain reaction–mediated genotypic analyses of HSV-1 strains located in cerebrospinal fluid (CSF) and aqueoushumor (AH) samples of two acute retinal necrosis patients with a history of herpes simplex encephalitis. Amplification and detectionof hypervariable regions in the herpes simplex virus type 1 genes US1 and US12 were performed as described previously.5 Left lane:100-bp molecular size marker.
Fig 2. DNA nucleotide sequences of the herpes simplex virus type 1 (HSV-1) US12 gene amplicons obtained from the aqueous hu-mor and cerebrospinal fluid samples of 2 acute retinal necrosis patients with a history of herpes simplex encephalitis. Comparison ofthe sequences between the patients and the homologous sequence of HSV-1 strain 17 revealed a high degree of variety within theUS12 region. The HSV-1 strain 17 US12 sequence was obtained from the GenBank database (HS1US; accession GI 291490:nucleotides 6683–6391). Reiterations (Re) IV and VIII are boxed, and unique point mutations are shaded. The start codon andthe encoded US12 amino acid sequence are indicated. The DNA sequences obtained have been submitted to the Genbank database(accession AF 290017 and AF290018 for Patients 1 and 2, respectively).
938 Annals of Neurology Vol 48 No 6 December 2000

tients. Patients recovering from HSV brain infectionsshould be closely monitored for viral eye infections,probably for the rest of their lives.
This study was funded in part by the Dr F. P. Fischer Stichting(J.M.) and SWOO, Rotterdamse Vereniging Blindenbelangen, andstichting HOF (G.M.G.M.V.).
References1. Whitley RJ. Herpes simplex virus infections of the central ner-
vous system: a review. Am J Med 1988;85:61–672. Pavesio CE, Conrad DK, Mc Cluskey PJ, et al. Delayed acute
retinal necrosis after herpetic encephalitis. Br J Ophthalmol1997;81:415–420
3. Levinson RD, Reidy R, Chiu MT. Acute retinal necrosis afterneonatal herpes encephalitis. Br J Ophthalmol 1999;83:123–124
4. Ganatra JB, Chandler D, Santos C, et al. Viral causes of theacute retinal necrosis syndrome. Am J Ophthalmol 2000;129:166–172
5. Maertzdorf J, Remeijer L, Van der Lelij A, et al. Amplification ofreiterated sequences of herpes simplex virus type 1 (HSV-1) ge-nome to discriminate between clinical HSV-1 isolates. J ClinMicrobiol 1999;37:3518–3523
6. Doornenbal P, Baarsma GS, Quint WGV, et al. Diagnostic as-says in cytomegalovirus retinitis: detection of herpesvirus by si-multaneous application of the polymerase chain reaction and lo-cal antibody analysis on ocular fluid. Br J Ophthalmol 1996;80:235–240
7. Umene K, Yoshida M. Reiterated sequences of herpes simplexvirus type 1 (HSV-1) genome can serve as physical markers forthe differentiation of HSV-1 strains. Arch Virol 1989;106:281–299
8. Matsubara A, Atherton SS. Spread of HSV-1 to the suprachias-matic nuclei and retina in T cell depleted BALB/c mice. J Neu-roimmunol 1997;80:165–171
9. Nicoll JAR, Love S, Kinrade E. Distribution of herpes simplexvirus DNA in the brains of human long-term survivors of en-cephalitis. Neuroscience Letters 1993;157:215–218
A Novel tau Mutation(N296N) in FamilialDementia with SwollenAchromatic Neuronsand CorticobasalInclusion BodiesMaria Grazia Spillantini, PhD,*† Hirotaka Yoshida, PhD,‡Claudia Rizzini, PhD,*† Peter L. Lantos, MD,§Nadeem Khan, MSc,§ Martin N. Rossor, MD,i
Michel Goedert, MD, PhD,‡ and Jeremy Brown, MD†
Familial dementia with swollen achromatic neurons andcorticobasal inclusion bodies is a neurodegenerative dis-ease that resembles corticobasal degeneration. It is char-acterized by the presence of abundant neuronal and glialtau protein deposits. Here we describe a novel silent mu-tation in exon 10 of tau (N296N) in this familial demen-tia. By exon trapping, the mutation produced an increasein the splicing in of exon 10, indicating that it probablycauses disease through an overproduction of four-repeattau.
Spillantini MG, Yoshida H, Rizzini C,Lantos PL, Khan N, Rossor MN, Goedert M,
Brown J. A novel tau mutation (N296N) infamilial dementia with swollen achromaticneurons and corticobasal inclusion bodies
Ann Neurol 2000;48:939–943
The discovery of mutations in the gene formicrotubule-associated protein tau in frontotemporaldementia and parkinsonism linked to chromosome 17(FTDP-17) has shown that dysfunction of tau proteinis sufficient to cause neurodegeneration and demen-tia.1–4 Known tau mutations are missense, deletion, orsilent mutations in the coding region and intronic mu-tations located close to the splice-donor site of the in-tron following exon 10. A filamentous pathology madeof hyperphosphorylated tau protein is a major neuro-pathological characteristic of FTDP-17.4
Six tau isoforms are produced in the adult human
From the *Brain Repair Centre and †Department of Neurology,University of Cambridge, and ‡MRC Laboratory of Molecular Bi-ology, Cambridge; and §Department of Neuropathology, Instituteof Psychiatry, and iDementia Research Group, National Hospitalfor Neurology and Neurosurgery, London, UK.
Received Jun 1, 2000, and in revised form Jul 20. Accepted forpublication Aug 7, 2000.
Address correspondence to Dr Spillantini, Brain Repair Centre, Uni-versity of Cambridge, Robinson Way, Cambridge CB2 2PY, UK.
Copyright © 2000 by the American Neurological Association 939
brain by alternative mRNA splicing from a singlegene.5 They differ from each other by the presence orabsence of 29– or 58–amino acid inserts located in theamino-terminal half and an additional 31–amino acidrepeat located in the carboxy-terminal half. Inclusionof the latter, which is encoded by exon 10 of tau, givesrise to the three isoforms with four repeats each; theother three isoforms have three repeats each. Similarlevels of three- and four-repeat tau isoforms are presentin normal adult human brain. The repeats and someadjoining sequences constitute the microtubule-bindingdomains of tau.4
Most missense mutations reduce the ability of mu-tant tau protein to interact with microtubules,6,7 andsome of these mutations also stimulate the in vitro as-sembly of tau into filaments.8,9 By contrast, the pri-mary effect of the intronic and some coding regionmutations in exon 10 is at the mRNA level.2,10,11 Itchanges the ratio of three- to four-repeat tau isoforms,resulting in the net overproduction of four-repeat tau.3
Here, we report a novel silent mutation (N296N) inexon 10 of tau in a previously described12 family withdementia and a neuropathology consisting of swollenachromatic neurons and corticobasal inclusion bodies.By exon trapping, the N296N mutation led to in-creased splicing in of exon 10.
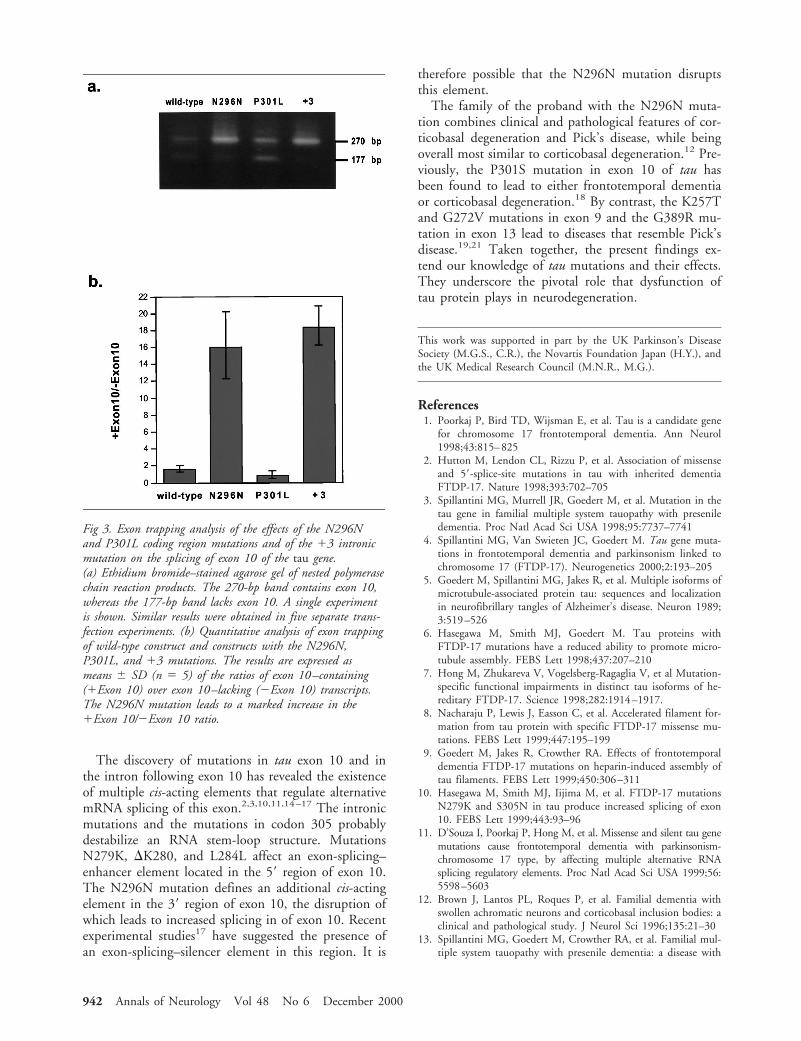
Subject and MethodsProbandThe clinical history and neuropathology of the proband (Pa-tient 111.4 of Family 2) and her family have been describedelsewhere.12 Four individuals in three generations experi-enced presenile dementia. At age 56 years, the proband failedto show an emotional response to her mother’s death. Shethen experienced personality changes, cognitive decline, andstereotypical behavior. She went on to develop a supranu-clear gaze palsy, orofacial dyspraxia, and bradykinesia, andsubsequently became mute. She died at 69 years of age. Atautopsy, the brain showed a marked frontotemporal atrophyas well as nerve cell loss in globus pallidus, substantia nigra,and locus ceruleus. Histologically, swollen achromatic neu-rons and tau-positive inclusion bodies were observed in anumber of brain regions, accompanied by neuronal cell loss,astrocytosis, and status spongiosus in the cerebral cortex.
ImmunohistochemistryTen-mm sections were cut from paraffin-embedded,formalin-fixed brain tissue of the proband and used for im-munohistochemistry. Phosphorylation-dependent anti-tauantibodies AT8 (Innogenetics, Ghent, Belgium), AT100 (In-nogenetics), AT180 (Innogenetics), 12E8 (Elan Pharmaceu-ticals, San Francisco, CA), PHF1 (donated by P. Davies),and AP422 were used. These monoclonal antibodies wereall used at 1:500. The phosphorylation-independent anti-tausera BR133 and BR134 were used at 1:200. Staining wasrevealed using the biotin/avidin Vectastain system (VectorLaboratories, Berlingame, CA), as previously described.13
Mutational AnalysisGenomic DNA was extracted from peripheral blood leuco-cytes of the proband. For DNA sequencing, tau exons 9–13were amplified by polymerase chain reaction (PCR) using 20ng/ml DNA and primers from the intronic sequences sur-rounding the tau exons, as described elsewhere.3 Exon 10 oftau was also amplified and sequenced from the genomicDNA of 50 control individuals originating from the samegeographical area as the proband.
Exon TrappingThe wild-type construct and constructs with the N296N si-lent mutation, the P301L missense mutation, and the 13intronic mutation were used. The tau sequences contained
Fig 1. Brain sections of the proband immunolabeled with thephosphorylation-dependent anti-tau antibody AT8: (a) frontalcortex, (b) hippocampus, (c) white matter of frontal cortex.Bar 5 75 mm in a ( for a and b), 250 mm in c.
940 Annals of Neurology Vol 48 No 6 December 2000

exon 10, as well as 34 nucleotides of upstream intronic se-quence and 85 nucleotides of downstream intronic sequence.PCR products were subcloned into the splicing vectorpSPL3, and exon trapping was done and the results quanti-fied as described elsewhere.10 PCR products were verified byDNA sequencing.
ResultsImmunohistochemical staining of frontal cortex, tem-poral cortex, and hippocampal formation of the pro-band showed extensive tau pathology in both nervecells and glial cells, as revealed by the phosphorylation-dependent anti-tau antibody AT8 (Fig 1). Similar re-sults were obtained with antibodies AT100, AT180,12E8, PHF1, and AP422, as well as with anti-tau seraBR133 and BR134 (data not shown). Sequencing ofthe proband’s genomic DNA showed wild-type se-quence in exons 9–13 of tau, with the exception ofexon 10, where a T to C transition was present in thethird base position at codon 296 (AAT to AAC) (Fig2). This change is silent at the amino acid level(N296N). It was not seen in DNA from 50 individualsoriginating from the same geographical region as theproband. Exon trapping was used to investigate the ef-fect of the N296N mutation on the splicing of exon10–containing transcripts, with the P301L and the 13intronic mutation being used as controls. The resultswere expressed as the ratios of exon 10–containingover exon 10–lacking transcripts (Fig 3). The P301Lmutation produced no significant change, whereas the13 intronic mutation produced a more than 10-foldincrease in the ratio, in confirmation of previous find-
ings (see Fig 3).2,14 The N296N mutation producedan approximately 10-fold increase in the ratio of exon10–containing over exon 10–lacking transcripts.
DiscussionThis study describes a novel mutation in tau in a fam-ily with dementia, frontotemporal atrophy, and degen-eration of subcortical brain regions, such as substantianigra and locus ceruleus. The N296N mutation is theeighth mutation to be described in the 31–amino acid–long exon 10 of tau. With the exception of P301L,P301S, and, possibly, DK280, these mutations(N279K, L284L, S305N and S305S) exert their pri-mary effect at the RNA level, where they lead to in-creased splicing in of exon 10. The same is true of theN296N mutation. By exon trapping, it produced amarked increase in the ratio of exon 10-containingover exon 10–lacking transcripts. In brain, this isknown to lead to a changed ratio of three- to four-repeat tau isoforms, resulting in an increase in solublefour-repeat tau.3 It is followed by the assembly of four-repeat tau into wide twisted ribbons in nerve cells andglial cells.13 The lack of frozen tissue precluded analysisof soluble and filamentous tau in the brain of the pro-band. By immunohistochemistry, an abundant neuro-nal and glial tau pathology was observed, consistentwith a primary effect of the N296N mutation at theRNA level. The extent of glial tau pathology was sim-ilar to that previously described for mutations in theintron following exon 10.13 These mutations also leadto increased splicing in of exon 10.2,14
Fig 2. (a) DNA sequence of exon 10 of tau from the proband. The arrow points to a T and a C nucleotide, indicative of theAAT to AAC transition at codon 296. (b) Nucleotide sequence of part of exon 10 of tau and of the intron following exon 10. Thesequence shown extends from codon 296 to nucleotide 125 of the intron (with the first nucleotide of the splice-donor site taken as11). Exonic sequences are shown in uppercase letters and intronic sequences in lowercase letters. The AAU to AAC transition atcodon 296 (N296N) is indicated.
Brief Communication: Spilantini et al: tau Mutation (N296N) in Familial Dementia 941
The discovery of mutations in tau exon 10 and inthe intron following exon 10 has revealed the existenceof multiple cis-acting elements that regulate alternativemRNA splicing of this exon.2,3,10,11,14–17 The intronicmutations and the mutations in codon 305 probablydestabilize an RNA stem-loop structure. MutationsN279K, DK280, and L284L affect an exon-splicing–enhancer element located in the 59 region of exon 10.The N296N mutation defines an additional cis-actingelement in the 39 region of exon 10, the disruption ofwhich leads to increased splicing in of exon 10. Recentexperimental studies17 have suggested the presence ofan exon-splicing–silencer element in this region. It is
therefore possible that the N296N mutation disruptsthis element.
The family of the proband with the N296N muta-tion combines clinical and pathological features of cor-ticobasal degeneration and Pick’s disease, while beingoverall most similar to corticobasal degeneration.12 Pre-viously, the P301S mutation in exon 10 of tau hasbeen found to lead to either frontotemporal dementiaor corticobasal degeneration.18 By contrast, the K257Tand G272V mutations in exon 9 and the G389R mu-tation in exon 13 lead to diseases that resemble Pick’sdisease.19,21 Taken together, the present findings ex-tend our knowledge of tau mutations and their effects.They underscore the pivotal role that dysfunction oftau protein plays in neurodegeneration.
This work was supported in part by the UK Parkinson’s DiseaseSociety (M.G.S., C.R.), the Novartis Foundation Japan (H.Y.), andthe UK Medical Research Council (M.N.R., M.G.).
References1. Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene
for chromosome 17 frontotemporal dementia. Ann Neurol1998;43:815–825
2. Hutton M, Lendon CL, Rizzu P, et al. Association of missenseand 59-splice-site mutations in tau with inherited dementiaFTDP-17. Nature 1998;393:702–705
3. Spillantini MG, Murrell JR, Goedert M, et al. Mutation in thetau gene in familial multiple system tauopathy with preseniledementia. Proc Natl Acad Sci USA 1998;95:7737–7741
4. Spillantini MG, Van Swieten JC, Goedert M. Tau gene muta-tions in frontotemporal dementia and parkinsonism linked tochromosome 17 (FTDP-17). Neurogenetics 2000;2:193–205
5. Goedert M, Spillantini MG, Jakes R, et al. Multiple isoforms ofmicrotubule-associated protein tau: sequences and localizationin neurofibrillary tangles of Alzheimer’s disease. Neuron 1989;3:519–526
6. Hasegawa M, Smith MJ, Goedert M. Tau proteins withFTDP-17 mutations have a reduced ability to promote micro-tubule assembly. FEBS Lett 1998;437:207–210
7. Hong M, Zhukareva V, Vogelsberg-Ragaglia V, et al Mutation-specific functional impairments in distinct tau isoforms of he-reditary FTDP-17. Science 1998;282:1914–1917.
8. Nacharaju P, Lewis J, Easson C, et al. Accelerated filament for-mation from tau protein with specific FTDP-17 missense mu-tations. FEBS Lett 1999;447:195–199
9. Goedert M, Jakes R, Crowther RA. Effects of frontotemporaldementia FTDP-17 mutations on heparin-induced assembly oftau filaments. FEBS Lett 1999;450:306–311
10. Hasegawa M, Smith MJ, Iijima M, et al. FTDP-17 mutationsN279K and S305N in tau produce increased splicing of exon10. FEBS Lett 1999;443:93–96
11. D’Souza I, Poorkaj P, Hong M, et al. Missense and silent tau genemutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNAsplicing regulatory elements. Proc Natl Acad Sci USA 1999;56:5598–5603
12. Brown J, Lantos PL, Roques P, et al. Familial dementia withswollen achromatic neurons and corticobasal inclusion bodies: aclinical and pathological study. J Neurol Sci 1996;135:21–30
13. Spillantini MG, Goedert M, Crowther RA, et al. Familial mul-tiple system tauopathy with presenile dementia: a disease with
Fig 3. Exon trapping analysis of the effects of the N296Nand P301L coding region mutations and of the 13 intronicmutation on the splicing of exon 10 of the tau gene.(a) Ethidium bromide–stained agarose gel of nested polymerasechain reaction products. The 270-bp band contains exon 10,whereas the 177-bp band lacks exon 10. A single experimentis shown. Similar results were obtained in five separate trans-fection experiments. (b) Quantitative analysis of exon trappingof wild-type construct and constructs with the N296N,P301L, and 13 mutations. The results are expressed asmeans 6 SD (n 5 5) of the ratios of exon 10–containing(1Exon 10) over exon 10–lacking (2Exon 10) transcripts.The N296N mutation leads to a marked increase in the1Exon 10/2Exon 10 ratio.
942 Annals of Neurology Vol 48 No 6 December 2000

abundant neuronal and glial tau filaments. Proc Natl Acad SciUSA 1997;94:4113–4118
14. Varani L, Hasegawa M, Spillantini MG, et al. Structure of tauexon 10 splicing regulatory element RNA and destabilization bymutations of frontotemporal dementia and parkinsonism linkedto chromosome 17. Proc Natl Acad Sci USA 1999;96:8229–8234
15. Clark LN, Poorkaj P, Wszolek Z, et al. Pathogenic implicationsof mutations in the tau gene in pallido-ponto-nigral degenera-tion and related neurodegenerative disorders linked to chromo-some 17. Proc Natl Acad Sci USA 1998;95:13103–13107
16. Grover A, Houlden H, Baker M, et al. 59 Splice site mutationsin tau associated with the inherited dementia FTDP-17 affect astem-loop structure that regulates alternative splicing of exon10. J Biol Chem 1999;274:15134–15143
17. D’Souza I, Schellenberg GD. Determinants of 4-repeat tauexpression: coordination between enhancing and inhibitorysplicing sequences for exon 10 inclusion. J Biol Chem 2000;275:17700–17709
18. Bugiani O, Murrell JR, Giaccone G, et al. Frontotemporal de-mentia and corticobasal degeneration in a family with a P301Smutation in tau. J Neuropathol Exp Neurol 1999;548:667–677
19. Spillantini MG, Crowther RA, Kamphorst W, et al. Tau pathol-ogy in two Dutch families with mutations in the microtubule-binding region of tau. Am J Pathol 1998;153:1359–1363
20. Murrell JR, Spillantini MG, Zolo P, et al. Tau gene mutationG389R causes a tauopathy with abundant Pick body-like inclu-sions and axonal deposits. J Neuropathol Exp Neurol 1999;58:1207–1216
21. Rizzini C, Goedert M, Hodges JR et al. Tau gene mutationK257T causes a tauopathy similar to Pick’s disease. J Neuro-pathol Exp Neurol 2000;59:990–1001
Therapeutic Benefit ofPolyamine-Modified Catalaseas a Scavenger of HydrogenPeroxide and Nitric Oxidein Familial AmyotrophicLateral Sclerosis TransgenicsJoseph F. Poduslo, PhD,* Shelly L. Whelan, BS,*Geoffry L. Curran, BS, BA, MBA,*and Thomas M. Wengenack, PhD*
Continuous subcutaneous administration of polyamine-modified catalase that has increased permeability at theblood-brain barrier showed both a highly significant de-lay in onset and an increase in survival in a transgenicmouse model of familial amyotrophic lateral sclerosishaving a point mutation in the gene encoding copper/zinc superoxide dismutase. These results suggest that hy-drogen peroxide–mediated oxidative stress with subse-quent free radical damage involving nitric oxide andpossibly hydroxyl radicals in motor neurons may be theculprit in familial amyotrophic lateral sclerosis.
Poduslo JF, Whelan SL, Curran GL,Wengenack TM. Therapeutic benefit of
polyamine-modified catalase as a scavenger ofhydrogen peroxide and nitric oxide in familial
amyotrophic lateral sclerosis transgenics.Ann Neurol 2000;48:943–947
Our laboratory has developed the technology to quan-tify the permeability of proteins at the blood-brain bar-rier (BBB),1 and we have demonstrated that the cova-lent attachment of the naturally occurring polyaminessignificantly increases their permeability at the BBBwithout substantially reducing their biological activity.2
This approach would, therefore, allow the systemic ad-ministration of therapeutic proteins for targets in thecentral or peripheral nervous system and eliminate theneed for invasive procedures, such as intraventricularcannulation or intraparenchymal injection. We haveextended this technology to the systemic administra-tion of polyamine-modified antioxidant enzymes such
From the *Molecular Neurobiology Laboratory, Departments ofNeurology and Biochemistry/Molecular Biology, Mayo Clinic andFoundation, Rochester, MN 55905.
Received Jan 27, 2000, and in revised form Jun 14. Accepted forpublication Aug 15, 2000.
Address correspondence to Dr Poduslo, Molecular NeurobiologyLaboratory, Departments of Neurology and Biochemistry/MolecularBiology, Mayo Clinic and Foundation, Rochester, MN 55905.
Copyright © 2000 by the American Neurological Association 943

as superoxide dismutase (SOD),3 which we have shownto be neuroprotective in a rat model of global cerebralischemia.4 We have also demonstrated that modifica-tion of catalase (CAT) with putrescine (PUT) increasesits permeability at the BBB two- to three-fold with pre-served enzymatic activity compared with that of nativeenzyme.5 More recently, we have evaluated the plasmapharmacokinetics, nervous system biodistribution andbiostability, and spinal cord BBB permeability of PUT-CAT.6 The delivery of exogenous SOD and CAT tothe nervous system may be effective, therefore, in treat-ing neurodegenerative diseases in which the underlyingmechanisms involve the action of free radicals and ox-idative damage.
The point mutations in the gene encoding copper/zinc SOD1 in some familial forms of amyotrophic lat-eral sclerosis (FALS)7 result in a neurotoxic gain offunction of the mutant SOD that causes selective de-generation of motor neurons.8–10 This gain of functionhas been hypothesized to involve (1) the increased per-oxidase activity as a result of the copper ions of mutantSOD catalyzing deleterious oxidation reactions of sub-strates by hydrogen peroxide (H2O2), (2) the increasedgeneration of hydroxyl radical upon reaction of copperin SOD with H2O2, (3) the increased nitration of ty-rosine by peroxynitrate, and (4) the increased loss ofcopper, zinc, or both from SOD with subsequent cel-lular metallotoxicity possibly involving copper chaper-ones. The first two hypotheses directly implicate H2O2
in playing a major role in these deleterious reactions.Since copper and ascorbate have been shown to be ef-fective inhibitors of catalase,11 the loss of copper frommutant SOD may also play a direct role in elevatingcytoplasmic H2O2 concentrations. Normally, the cell’sdefense mechanism to eliminate H2O2 involves catalaseor glutathione peroxidase. Very low levels of catalase,however, are found in the nervous system, whichmakes it particularly vulnerable to free radical-mediateddamage. The third hypothesis directly implicates nitricoxide (NO) as an important culprit. A consistent find-ing in ALS patients and mice is the increased concen-tration of nitrotyrosine12,13; hence, NO also likelyplays a prominent role in the free radical damage inALS. The nitration of tyrosine residues is catalyzed byperoxynitrite, which is formed by superoxide and NO,controlled by SOD,14,15 and elevated by mutant SOD.It has been demonstrated that catalase binds NO butdoes not significantly metabolize it in the absence ofH2O2.16 In the presence of H2O2, however, catalasecatalyzes NO breakdown.16 Catalase, therefore, can ef-fectively eliminate both H2O2 and NO and thus maybe beneficial in the treatment of ALS.
We have demonstrated the therapeutic benefit ofPUT-CAT in a transgenic mouse model of FALS withdramatic effects that delay both onset of clinical diseaseand progression of clinical weakness in a low- and a
high-expressor line of FALS mice.17 No significantchanges were observed in the survival of the PUT-CAT–treated high-expressor FALS mice, which sug-gests that the therapeutic window may be narrowerwith the more acute disease that develops in these an-imals. In contrast, a trend toward increased survivalwas seen, with the low-expressor mice having the morechronic form of the disease, which more closely mimicshuman ALS. Because the low-expressor mice weretreated only twice per week with intraperitoneal injec-tions of PUT-CAT, we rationalized that increasing thedosage by continuous subcutaneous osmotic pump de-livery might have a more dramatic effect on survival ofthe low-expressor animals. The following experimentswere designed to address this and to test the role ofH2O2 and NO removal by exogenous polyamine-modified catalase in this neurodegenerative animalmodel.
Material and MethodsAnimalsFounder male mice from a low-expressor line of FALS trans-genic mice, B6SJL-TgN (SOD1-G93A)1Gurdl (gene copy 8;our designation G1L/1)18 were obtained from Jackson Lab-oratories (Bar Harbor, ME) (stock #002300). The coloniesof the G1L/1 mice were maintained as heterozygotes bymating transgenic males with nontransgenic (1/1) B6SJLhybrid females. The transgenic progeny were identified byamplification of mouse tail DNA by the polymerase chainreaction technique specific for mutant human SOD1.
Polyamine Modification of CatalaseModification of CAT was performed using methods de-scribed previously.2–6,17
Treatment ProtocolThe G1L/1 transgenic mice were treated with 450 units/dayof PUT-CAT or sterile phosphate buffered saline (PBS; ve-hicle) by subcutaneously implanted osmotic pumps (AlzaCorp, Palo Alto, CA) starting between 35 and 42 days ofage. Initially, the Alzet pumps utilized were 2-week pumps(Model 1002, pumping rate 0.25 ml/hr with fill volume of100 ml) for the first 2 treatments. After the first month oftreatment, 4-week Alzet pumps were used for the duration ofthe experiment (Model 2004, pumping rate 0.25 ml/hr withfill volume of 200 ml).
Pump PlacementPumps were aseptically filled to capacity with filter-sterilizedPBS or PUT-CAT as indicated by the manufacturer to en-sure correct pump activation. Each mouse was anesthetizedby intraperitoneal injection of 0.3 to 0.5 ml of sodium phe-nobarbital diluted 1:10. A small, 2- to 3-cm incision wasmade on the back, and the pump was inserted into a subcu-taneous pocket. To reduce the risk of infection, the animals’water was replaced with a solution of amoxicillin (Biomoxoral suspension; Teva Pharm Co, Sellersville, PA) containingcherry flavoring and 5 g/L sucrose for 7 days.
944 Annals of Neurology Vol 48 No 6 December 2000

Clinical EvaluationThe clinical condition of the mice was monitored two tothree times per week according to procedures used by Gur-ney and co-workers,8 starting at 3 months of age and con-tinuing daily as the animals approached clinical onset. Theonset of clinical signs was scored by examining the mice forshaking limbs and/or the position of one or both back legs,hanging rather than splaying out, when the mice were sus-pended in the air by the tail. The age of clinical onset wasdetermined by the age (days) at which loss of splay or trem-ors of hindlimbs were observed in the mice. Examining themice for the loss of righting reflex determined the end stageof the disease. The mice were killed if they could not rightthemselves within 30 seconds when placed on either side ona flat surface. The treatment and clinical evaluation wereconducted by separate individuals, and thus the neurologicalsigns for determining onset and end stage of disease werescored in a blinded manner.
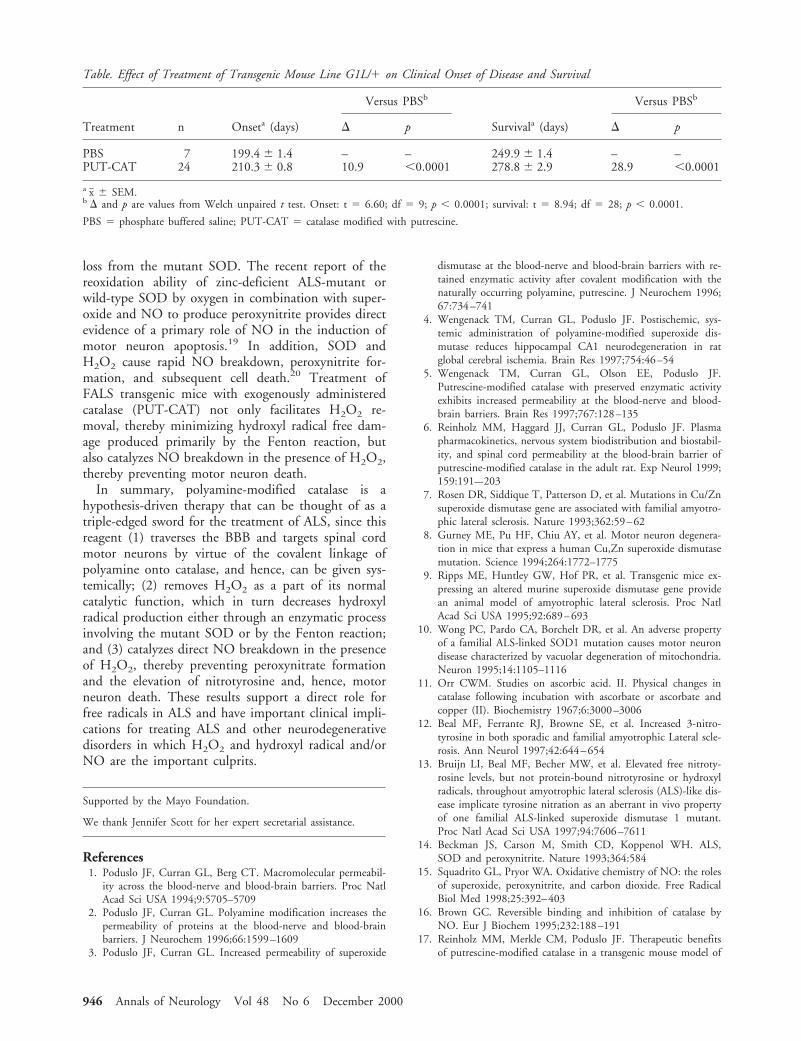
ResultsEffects of PUT-CAT on Onset of Clinical DiseasePart A of the Figure presents the probability curvesthat illustrate the age at which the clinical onset of dis-
ease occurred in each individual mouse. In our studies,the G1L/1 mice that were treated with PBS showedonset of clinical disease at 199.4 6 1.4 days (Table).This clinical onset was not significantly different fromthat observed in our previous study.17 PUT-CAT treat-ment of the G1L/1 mice significantly delayed the on-set of clinical disease by 10.9 days (p , 0.0001) com-pared with that observed in the PBS-treated mice (seeTable). Interestingly, in both studies, all PBS-treatedanimals had an onset in one hindlimb, which was fol-lowed by onset on the other side either at the sametime or within 4 days.
Effects of PUT-CAT on Survival TimesPart B of the Figure represents a probability curve thatdemonstrates the age at which each mouse was killed.In this study, PBS-treated G1L/1 mice exhibited clin-ical death at 249.9 6 1.4 days. This survival time alsowas not significantly different from what we reportedpreviously.17 PUT-CAT significantly increased survivalin the G1L/1 mice by 28.9 days (p , 0.0001) com-pared with PBS-treated mice (see Table). In 63% ofPUT-CAT treated mice, onset on one side was not im-mediately followed by onset in the other hindlimb.This led to asymmetrical progression of the disease onthe affected side, which would progress to complete pa-ralysis often before onset in the other limb could bedetected. Such asymmetrical progression of the diseaseis a common characteristic of both familial and spo-radic human ALS. Because many of the PUT-CATtreated mice were affected on only one side, they had aconsiderably easier time attaining food and water, andunder specialized conditions, could have reached evengreater survival times than reported here. Humanetreatment guidelines required that the asymmetricallyaffected mice be destroyed because, although they werenearly unaffected on one side, they did not have thebalance required to obtain food and water effectively inthe caging provided. The implications for the potentialfor human treatment are significant in that more indi-vidualized treatment of the mice could have easily in-creased their survival beyond that reported here.
DiscussionThese results demonstrate that the FALS clinical phe-notype (onset, survival) is significantly reduced whenthe animals are treated systemically with polyamine-modified catalase. This is the largest increase in survivalof any systemically delivered treatment in FALS trans-genic mice that has been published to date and hasimportant treatment implications for ALS patients.Our data suggest that H2O2-mediated oxidative stresswith subsequent free radical damage involving NO andpossibly hydroxyl radicals in motor neurons may wellbe the culprit in FALS. This is likely to be exacerbatedby the inhibition of catalase by copper as a result of its
Fig. The effects of phosphate buffered saline (PBS) and cata-lase modified with putrescine (PUT-CAT) on the probabilityof normal neurological signs (onset of clinical disease; A) andon the probability of survival (B) in the G1L/1 line of famil-ial amyotrophic lateral sclerosis transgenic mice.
Brief Communication: Poduslo et al: Polyamine-Modified Catalase in FALS 945
loss from the mutant SOD. The recent report of thereoxidation ability of zinc-deficient ALS-mutant orwild-type SOD by oxygen in combination with super-oxide and NO to produce peroxynitrite provides directevidence of a primary role of NO in the induction ofmotor neuron apoptosis.19 In addition, SOD andH2O2 cause rapid NO breakdown, peroxynitrite for-mation, and subsequent cell death.20 Treatment ofFALS transgenic mice with exogenously administeredcatalase (PUT-CAT) not only facilitates H2O2 re-moval, thereby minimizing hydroxyl radical free dam-age produced primarily by the Fenton reaction, butalso catalyzes NO breakdown in the presence of H2O2,thereby preventing motor neuron death.
In summary, polyamine-modified catalase is ahypothesis-driven therapy that can be thought of as atriple-edged sword for the treatment of ALS, since thisreagent (1) traverses the BBB and targets spinal cordmotor neurons by virtue of the covalent linkage ofpolyamine onto catalase, and hence, can be given sys-temically; (2) removes H2O2 as a part of its normalcatalytic function, which in turn decreases hydroxylradical production either through an enzymatic processinvolving the mutant SOD or by the Fenton reaction;and (3) catalyzes direct NO breakdown in the presenceof H2O2, thereby preventing peroxynitrate formationand the elevation of nitrotyrosine and, hence, motorneuron death. These results support a direct role forfree radicals in ALS and have important clinical impli-cations for treating ALS and other neurodegenerativedisorders in which H2O2 and hydroxyl radical and/orNO are the important culprits.
Supported by the Mayo Foundation.
We thank Jennifer Scott for her expert secretarial assistance.
References1. Poduslo JF, Curran GL, Berg CT. Macromolecular permeabil-
ity across the blood-nerve and blood-brain barriers. Proc NatlAcad Sci USA 1994;9:5705–5709
2. Poduslo JF, Curran GL. Polyamine modification increases thepermeability of proteins at the blood-nerve and blood-brainbarriers. J Neurochem 1996;66:1599–1609
3. Poduslo JF, Curran GL. Increased permeability of superoxide
dismutase at the blood-nerve and blood-brain barriers with re-tained enzymatic activity after covalent modification with thenaturally occurring polyamine, putrescine. J Neurochem 1996;67:734–741
4. Wengenack TM, Curran GL, Poduslo JF. Postischemic, sys-temic administration of polyamine-modified superoxide dis-mutase reduces hippocampal CA1 neurodegeneration in ratglobal cerebral ischemia. Brain Res 1997;754:46–54
5. Wengenack TM, Curran GL, Olson EE, Poduslo JF.Putrescine-modified catalase with preserved enzymatic activityexhibits increased permeability at the blood-nerve and blood-brain barriers. Brain Res 1997;767:128–135
6. Reinholz MM, Haggard JJ, Curran GL, Poduslo JF. Plasmapharmacokinetics, nervous system biodistribution and biostabil-ity, and spinal cord permeability at the blood-brain barrier ofputrescine-modified catalase in the adult rat. Exp Neurol 1999;159:191–-203
7. Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Znsuperoxide dismutase gene are associated with familial amyotro-phic lateral sclerosis. Nature 1993;362:59–62
8. Gurney ME, Pu HF, Chiu AY, et al. Motor neuron degenera-tion in mice that express a human Cu,Zn superoxide dismutasemutation. Science 1994;264:1772–1775
9. Ripps ME, Huntley GW, Hof PR, et al. Transgenic mice ex-pressing an altered murine superoxide dismutase gene providean animal model of amyotrophic lateral sclerosis. Proc NatlAcad Sci USA 1995;92:689–693
10. Wong PC, Pardo CA, Borchelt DR, et al. An adverse propertyof a familial ALS-linked SOD1 mutation causes motor neurondisease characterized by vacuolar degeneration of mitochondria.Neuron 1995;14:1105–1116
11. Orr CWM. Studies on ascorbic acid. II. Physical changes incatalase following incubation with ascorbate or ascorbate andcopper (II). Biochemistry 1967;6:3000–3006
12. Beal MF, Ferrante RJ, Browne SE, et al. Increased 3-nitro-tyrosine in both sporadic and familial amyotrophic Lateral scle-rosis. Ann Neurol 1997;42:644–654
13. Bruijn LI, Beal MF, Becher MW, et al. Elevated free nitroty-rosine levels, but not protein-bound nitrotyrosine or hydroxylradicals, throughout amyotrophic lateral sclerosis (ALS)-like dis-ease implicate tyrosine nitration as an aberrant in vivo propertyof one familial ALS-linked superoxide dismutase 1 mutant.Proc Natl Acad Sci USA 1997;94:7606–7611
14. Beckman JS, Carson M, Smith CD, Koppenol WH. ALS,SOD and peroxynitrite. Nature 1993;364:584
15. Squadrito GL, Pryor WA. Oxidative chemistry of NO: the rolesof superoxide, peroxynitrite, and carbon dioxide. Free RadicalBiol Med 1998;25:392–403
16. Brown GC. Reversible binding and inhibition of catalase byNO. Eur J Biochem 1995;232:188–191
17. Reinholz MM, Merkle CM, Poduslo JF. Therapeutic benefitsof putrescine-modified catalase in a transgenic mouse model of
Table. Effect of Treatment of Transgenic Mouse Line G1L/1 on Clinical Onset of Disease and Survival
Treatment n Onseta (days)
Versus PBSb
Survivala (days)
Versus PBSb
D p D p
PBS 7 199.4 6 1.4 – – 249.9 6 1.4 – –PUT-CAT 24 210.3 6 0.8 10.9 ,0.0001 278.8 6 2.9 28.9 ,0.0001
a x# 6 SEM.b D and p are values from Welch unpaired t test. Onset: t 5 6.60; df 5 9; p , 0.0001; survival: t 5 8.94; df 5 28; p , 0.0001.
PBS 5 phosphate buffered saline; PUT-CAT 5 catalase modified with putrescine.
946 Annals of Neurology Vol 48 No 6 December 2000

familial amyotrophic lateral sclerosis. Exp Neurol 1999;159:204–216
18. Gurney ME. Transgenic animal models of familial amyotrophiclateral sclerosis. J Neurol 1997;244:S15–S20
19. Estevez AG, Crow JP, Sampson JB, et al. Induction of NO-dependent apoptosis in motor neurons by zinc-deficient super-oxide dismutase. Science 1999;286:2498–2500
20. McBride AG, Borutaite V, Brown GC. Superoxide dismutaseand hydrogen peroxide cause rapid nitric oxide breakdown, per-oxynitrite production and subsequent cell death. Biochem Bio-phys Acta 1999;1454:275–288
CorrectionFor a Neurology Outcomes Research abstract in theSeptember issue (Ann Neurol 2000;48:497) and theAmerican Neurological Association meeting program(p 110), an author’s name was inadvertently left off.The corrected abstract follows:
3. Improving Quality of Care Using the GlobalHeadache InventoryDavid Hewitt, Ellen McKenzie, Chris O’Brien, andAnindya K. Do; Atlanta, GA
The Global Headache Inventory (GHI) was piloted in anacademic neurology clinic to assess patient satisfaction withheadache control. Thirty-seven patients evaluated for head-ache (mean age, 45 years; 73% female) completed baselineand follow-up surveys. Satisfaction with change in headachecontrol and attitudes toward completing the GHI were as-sessed. The scores reflecting satisfaction with headache con-trol were skewed, showing marked departure from normality(p , 0.05). The skewedness was positive (0.852) at time 1and negative (20.545) at time 2, demonstrating an improve-ment in satisfaction for this sample. Nonparametric tests forthe paired sample were used to test for improvement in sat-isfaction from time 1 to time 2. Wilcoxon’s Signed RankTest revealed significant improvement (p , 0.001). Most ofthe patients (n 5 27; 75%) indicated that the questionnairewas not a burden, and many (n 5 23; 66%) reported thatthe form was helpful. Almost all (n 5 33; 97%) believedthat the form should be used for headache patients in thefuture. Preliminary data indicate that the GHI may be valu-able in determining improvement in patient satisfaction withheadache control. The GHI improves communication andquality of care. Studies are needed to evaluate validity andreliability of this instrument.
Supported by Physicians’ Small Grants for AcceleratedPerformance Improvement 1999.
Brief Communication: Poduslo et al: Polyamine-Modified Catalase in FALS 947

STATEMENT OF OWNERSHIP MANAGEMENT AND CIRCULATION (Required by 39 U.S.C. 3685) 1. Publication Title: ANNALS OFNEUROLOGY 2. Publication no.: 0364–5134 Filing date: 10–01-00. 4. Frequency of issue: Monthly 5. No. of issues published annually: 12;6. Annual subscription price: $ 214.00. Complete mailing address of known office of publication: Lippincott Williams & Wilkins, 16522 HuntersGreen Parkway, Hagerstown, MD 21740-2116. Complete mailing address of the headquarters of general business offices of the publisher: 530Walnut Street, Philadelphia, PA 19106. Full names and complete mailing address of publisher, editor, and managing editor: Publisher: LippincottWilliams & Wilkins, Inc., 530 Walnut Street, Philadelphia, PA 19106; Editor: Richard T. Johnson, MD, 3103 N. Charles St., Second Floor,Baltimore, MD 21218: Sheila Rose Garrity, 3103 N. Charles St., Second Floor, Baltimore, MD 21218. Owner: American Neurological Associ-ation, 5841 Cedar Lake Road, Suite 108, Minneapolis, MN 55416. Known bondholders, mortgagees, and other security holders owning orholding 1% or more of total amount of bonds, mortgages, or other securities: None. 12. Purpose, function, and nonprofit status: Has not changedduring preceding 12 months. 13. Publication Name: ANNALS OF NEUROLOGY. 14. Issue Date for circulation data: Volume 48, Issue 2.Extent and nature of circulation: Average number of copies each issue during preceding 12 months: (a) Total No. copies (Net Press Run), 8,033.(b) Paid and/or requested circulation; (1) Paid/requested outside-county mail subscriptions stated on form 3541. (Include advertiser’s proof andexchange copies), 6,381,; (2) Paid in-county subscriptions (include advertiser’s proof and exchange copies), N/A; (3) Sales through dealers andcarriers, street vendors, counter sales, and other non-USPS paid distribution, N/A; (4) Other classes mailed through the USPS, N/A. (c) Total paidand/or requested circulation (sum of 15b (1), (2), (3), and (4), 6,381, (d) Free distribution by mail (samples, complimentary, and other free). (1)Outside-county as stated on form 3541, 361; (2) In country as stated on form 3541, N/A; (3) Other classes mailed through the USPS, N/A. (e)Free distribution outside the mail (carriers or other means), N/A. (f) Total free distribution (sum of 15d and 15e), 361. (g) Total distribution (Sumof 15c and 15f), 6,742. (h) Copies not distributed, 1,291. (i) Total (sum of 15g and h), 8,033. (j) Percent paid and/or requested circulation (15cdivided by 15g times 100). 97%. No. copies of single issue published nearest to filing date: (a) Total no. copies (Net Press Run), 7,700. (b) Paidand/or requested circulation; (1) Paid/requested outside-county mail subscriptions stated on form 3541, (Include advertiser’s proof and exchangecopies), 6,397; (2) Paid in-county subscriptions (include advertiser’s proof and exchange copies), N/A; (3) Sales through dealers and carriers, streetvendors, counter sales, and other non-USPS paid distribution, N/A; (4) Other classes mailed through the USPS, N/A. (c) Total paid and/orrequested circulation (sum of 15b. (1), (2), (3), and (4), 6,397. (d) Free distribution by mail (samples, complimentary, and other free), (1)Outside-county as stated on form 3541, 359; (2) In-county as stated on form 3541, N/A; (3) Other classes mailed through the USPS, N/A. (e)Free distribution outside the mail (carriers or other means), N/A. (f) Total free distribution (sum of 15d and 15e), 359. (g) Total distribution (Sumof 15c and 15f), 6,756. (h) Copies not distributed, 944. (i) Total (sum of 15g and h), 7,700. (j) Percent paid and/or requested circulation (15cdivided by 15g x 100), 95%. 16. This Statement of Ownership will be printed in volume 48, issue 6 of this publication. 17. I certify that thestatements made by me above are correct and complete. Michael Wells, Manager, Periodical Operations
948 Annals of Neurology Vol 48 No 6 December 2000




















