
extraction, purification and distribution of fish collagen ...

Vol. 39, No. 2, May 1996 BIOCHEMISTRY ond MOLECULAR BIOLOGY INTERNATIONAL Pages 403-413
A NOVEL METHOD FOR PURIFICATION OF CATALYTIC ANTIBODIES TOWARD DNA FROM SERA OF PAT1ENTS WITH
LYMPHOPROLIFERATIVE DISEASES
A.V. Kozyr
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 32 Vavilov Street, Moscow, 117984, Russia
Received February 16, 1996 Received after revision March 22, 1996
SUMMARY
A novel highly efficient method of purification of DNA-hydrolyzing antibodies from sera of patients with lymphoproliferative diseases was developed. The method suggests purification of serum sample using Cibacron Blue 3GA, ion exchange chromatography on DEAE-Sepharose FF, PrG affinity chromatography and selection of the DNA-binding fraction on DNA-Sephacryl. The application of the method allows to obtain the stable and active polyclonal preparation with increased content of DNA-hydrolyzing antibodies characterized by specific binding constant determined using heteropolymeric oligonucleotide substrate. The unusual properties of anti-DNA catalytic antibodies in respect to their interactions with DEAE ion exchanger are discussed.
Key words: DNA-hydrolyzing antibodies, lymphoproliferative diseases, Cibacron Blue 3GA, DEAE, DNA-Sephacryl
INTRODUCTION
Recent years have made a great contribution to the studies of catalytic antibodies (abzymes). All catalytic antibodies described to the time can be divided into three specific groups, i.e. antibodies raised against transition state analogs of different chemical transformations, antiidiotypic antibodies mounted toward enzyme paratope and those occurring spontaneously in the organism [1, 2]. The studies of the latter
403
1039-9712/96/020403-11 $05.00/0 Copyright © 1996 by Academic Press Australia. All rights of reproduction in any form reserved.

Vol. 39, No. 2, 1996 BIOCHEMISTRYand MOLECULAR BIOLOGY INTERNATIONAL
type of catalytic antibodies (also called natural abzymes) are likely to contribute to the knowledge on enzymatic activity in organism. History of their discovery and latest investigations provide a firm basis for the conclusion, that their occurrence in the organism is strongly associated with certain pathologies: VIP-hydrolyzing antibodies were found in sera of patients with asthma [3], DNA-hydrolyzing antibodies were detected in the sera of patients with autoimmune and lymphoproliferative disorders and AIDS [4-6], thyroglobulin-hydrolyzing antibodies were found in the sera of patients with Hashimoto thyroiditis [7]. Further investigations of distribution of natural catalytic antibodies in sera of patients with various diseases could afford their diagnostic application in clinical practice, emphasize their role in the pathological process and develop new strategies for therapeutics.
The goal of the present work was to develop fast and reliable technique for experimental work with polyclonal DNA-hydrolyzing antibodies from sera of patients with lymphoproliferative diseases and suggest a novel method of their isolation and purification.
MATERIALS AND METHODS
All chemicals were from Sigma and Merck. All purification steps were performed on Waters 470 HPLC unit (Waters-Millipore) with peak detection using Waters 994 Photodiode Array detector and peak area calculation using the Waters MAXIMA 820 software. Prepacked or home-packed Pharmacia HR columns were used throughout the work. Sera of patients with lymphoproliferative diseases were kindly provided by physicians from Department of hematology and intensive therapy of Russian Hematological Center, Russian Academy of Medical Sciences. Blood samples were taken from patients using vein puncture and in accordance with the rules of Declaration on work with human subjects under the supervision of the Institutional Review Board.
All steps of antibody purification were conducted at 0 ° C. The serum sample clarified from residual blood cells and fibrin clots by centrifugation was diluted twofold with buffer containing 40mMTris-HC1 pH 7.5 and 100mM NaCI. Crude antibodies were subsequently precipitated from the sample by addition of equal volume of saturated ammonium sulphate. The precipitate was separated by centrifugation, dissolved in buffer A (20 mM Tris-HCl pH 8.8) containing 300 mM NaCI, and applied to the Pharmacia Fast Desalting
404

Vol. 39, No. 2, 1996 BIOCHEMISTRY and MOLECULAR BIOLOGY INTERNATIONAL
column packed with Sephadex G-25 in order to remove salt and lipid contaminants.
Affinity chromatography on Cibacron Blue 3GA (HR 16/10 column packed with 10ml of Cibacron Blue 3GA-SepharoseFF, Pharmacia) was performed in buffer containing 20mMTris-HCl pH 8.8, 300 mM NaCI at the flow rate 0.2 ml/min; the bound fraction was eluted by the same buffer contained 3.3 M NaC1.
Anion exchange chromatography on DEAE-Sepharose FF (10 ml of the ion exchanger packed into the HR 10/10 column) was conducted in buffer A containing 35 mM NaCI and the bound fraction was eluted by the same buffer with 1M NaCl.
Eluted fraction was applied to Protein G affinity column in buffer containing Tris-HC1 pH 7.5, 50 mM NaC1, the column was washed with 20 column volumes of the same buffer, and antibodies were eluted by 100 mM GIy-HC1 pH 2.6.
Separation of the DNA-binding antibody fraction was performed on DNA-Sephacryl in buffer, containing 20mMTris-HC1 pH 7.5, 10 mMNaC1 and the bound fraction was eluted by 1MNaCI in the same buffer. DNA-hydrolyzing activity of antibodies was measured as described in [4]. One unit of DNA-abzyme was determined as amount of purified serum IgG capable to catalyse the conversion of 1 mkg of supercoiled plasmid DNA into circular form under specified conditions (Tris-HCl pH7.5, 50 mM NaC1, 10 mM MgC12, 37°C, 10 hours). A full convertion of supercoiled plasmid DNA into circular form was considered as 1 unit of DNA hydrolyzing activity. Purity of the antibody preparation was checked by SDS-PAGE according to Laemmli [8] and immunoblotting using anti-~: and anti-)~ light chain antibodies (Sigma) according to [9].
RESULTS AND DISCUSSION
The difficulties facing investigators on the path of isolation and purification of natural abzymes are provided by two main reasons. Polyclonal antibodies from human sera require several steps of fractionation to minimize the presence of non-catalytic antibodies in the final preparation. Antibodies are generally stable proteins and thereby harsh isolation conditions, i.e. strong ion exchangers, low and high pH cause little damage to their structure. The information about higher- order structure of naturally occurring abzymes and mechanism of catalysis is not sufficient to the date. The structure of the enzyme active
405

Vol. 39, No. 2, 1996 BIOCHEMISTRYand MOLECULAR BIOLOGY INTERNATIONAL
center is frequently very unstable in vitro and proned to denaturation under non-physiological conditions. Thus it is advisable to consider abzymes as enzymes with possible presence of fragile structures and not as conformationally stable antibodies. The traditional approach, described in [10] has proved itself in isolation of natural abzymes from patients with autoimmune pathologies (systemic lupus erythematosus, rheumatoid arthritis, scleroderma etc.). Anti-DNA antibodies spontaneously occurring in autoimmune pathologies were described to be highly cationic due to the increased ratio of Lys, Arg, and His aminoacid residues, as it was determined by isoelectric focusing [11, 12]. However, our data on chromatofocusing of the DNA-hydrolyzing antibodies isolated from sera of patients with lymphoproliferative diseases indicate, that DNA-hydrolyzing antibody fraction has pI near pH6-6.5. This fact suggests leukemic DNA-abzymes to contain the lower ratio of positively charged aminoacid residues, or alternatively their total charge could be close to the neutral due to the increased presence of alkaline and acidic residues as well.
The attempts of isolation and purification of leukemic DNA-abzymes using the traditional method [10] had often resulted in loss of enzymatic activity. The stability of the leukemic DNA-hydrolyzing antibodies highly depends on quantity and duration of purification steps and the rigidity of treatment. Thus, the labile nature of these antibodies dictated to develop more gentle purification technique than it was permissible for lupus-derived anti-DNA abzymes. The scheme of purification is presented in Fig. 1.
At the first step, it was necessary to remove the serum lipids contaminating the sample and hindering the chromatography procedures. We used desalting column (Pharmacia Fast Desalting HR 10/10) packed with Sephadex G-25 for delipidation. The advantage of this step was that simultaneous desalting, delipidation and buffer exchange was achieved. At the next step, the preparation was applied to the column with Cibacron Blue 3GA immobilized on Sepharose (Pharmacia). This step was aimed at serum albumin and proteases removal, on the basis of ' the method earlier described in [13]. Unfortunately, it was hard to avoid adsorption of certain antibodies on this column, possibly due to the high diversity in properties of polyclonal antibodies.
406

Vol. 39, No. 2, 1996 BIOCHEMISTRYand MOLECULAR BIOLOGY INTERNATIONAL
IgG fraction of serum sample-----~Sephadex G-25~Cibacron 3GA-~Protein G precipitated with 1 50% of saturated (NH4)2804
DEAE
Pr~ein G activity
IgG fra!tion with DN~ -~Y:il;~ialgrylCtivity
DNA-binding IgG fraction with increased level of DNA-hydrolyzing activity
Figure 1. The scheme of leukemic DNA-hydrolyzing antibodies purification.
IgG without
DNA-hydrolyzing
As it comes from chromatofocusing experiments, the leukemic DNA-hydrolyzing antibodies reveal a great difference in the net charge in buffers with different pH values. However, a number of experiments outlined the decrease in DNA-hydrolyzing activity after purification on MonoQHR5/5co lumn (Pharmacia) using the buffer containing 20 mM Tris-HC1 pH 9. The loss of activity could be provided by violent denaturating influence of the active groups of the column (strongly charged quarternary amines). Due to the fact, that leukemic DNA-hydrolyzing antibodies are expected to have lower positive charge, than earlier investigated antibodies from sera of patients with autoimmune diseases, it was interesting to try a strong cation exchanger for their purification. The total antibody preparation purified on the Protein G column was further applied to the MonoS HR 5/5 column (Pharmacia) in 100 mM GIy-HCI pH 4. The abzymes purified on the MonoS displayed higher level of DNA-hydrolyzing activity in comparison with that obtained on MonoQ, but nevertheless, the activity was observed in the non-retented fraction. So we chose DEAE-Sepharose Fast Flow (Pharmacia) for anion-exchange chromatography (step 3 in Fig. 1), since the interaction of the antibody charged groups with DEAE is weaker and helps to maintain the starting
407

Vol. 39, No. 2, 1996 BIOCHEMISTRY and MOLECULAR BIOLOGY INTERNATIONAL
level of activity. The flow-through fraction from the Cibacron Blue 3GA sorbent was dialyzed overnight against 20 volumes of 20 mM Tris-HC1 buffer pH 8.8 containing 35 mM NaC1 and applied to the HR 10/10 column packed with DEAE-Sepharose FF (Pharmacia). Antibodies of the IgG fraction bound to the column made up about 10% of the total IgG contents in the sample as juged by Protein G chromatography. Surprisingly, the same conditions recommended by the manufacturer for purification of antibodies using DEAE sorbents, were destined to eliminate IgG fraction binding to the DEAE column. This suggests the unusual properties of antibodies bound to the DEAE column.
It is known that at least a part of the anti-DNA autoantibodies family displays high degree of cross-reactivity with negatively charged polyanions (i.g. heparin, proteins, and phospholipids) [14-16]. The promiscous nature of binding capability of these antibodies strongly suggests the possibility of more pronounced interactions of such antibodies comparing to those from conventional antibody repertoire. In addition, antibodies isolated from healthy organisms also display variability in interactions with DEAE support that results in broad asymmetric elution peak of these molecules from the anion exchanger [17]. Retention of DNA-abzymes on the DEAE support thus appears to be the extension of known effect and not the absolutely new phenomenon.
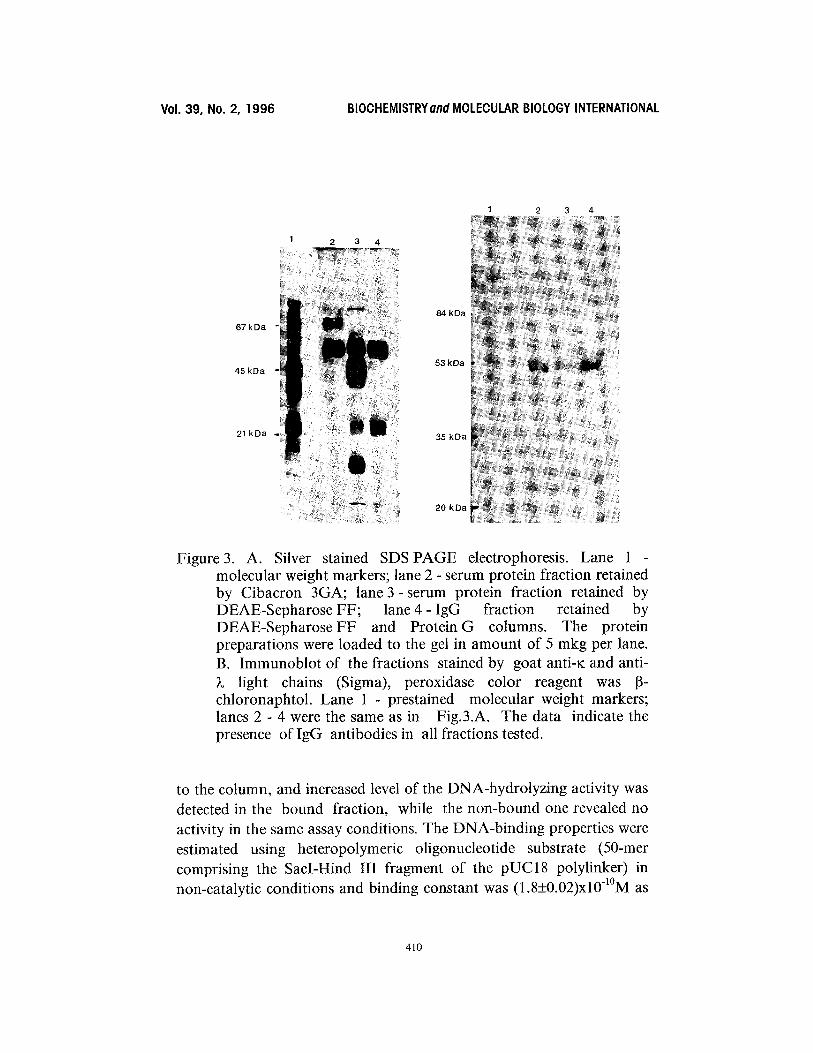
The final step of purification was performed on HiTrap Protein G column (Pharmacia) in order to obtain pure IgG fraction. The Protein G chromatography was also used to calculate the IgG content in all the fractions arised from the purification procedure. The results on DNA-hydrolyzing activity of IgG fractions purified thus far from different stages of purification are depicted in Table 1. DNA-hydrolyzing activity of antibodies was measured in the fractions retained by Cibacron Blue 3GA, DEAE-Sepharose FF and in the flow- through from both columns. In all cases significant level of activity was observed in the IgG fraction adsorbed by DEAE but passed through the Cibacron (Fig.2). The homogenity of purified catalytic antibody fraction was checked by silver staining of the Protein G eluate resolved by SDS-PAGE (Fig.3A, lane 4). The fraction lacks enzymatic contamination, as judged by the inhibition assay [4]. The protein spectra of the fractions yielded by different steps of purification are presented in Fig.3A. The results of immunoblotting (Fig.3B) indicate the presence of
408

Vol. 39, No. 2, 1996 BIOCHEMISTRYand MOLECULAR BIOLOGY INTERNATIONAL
1 2 3 4 5
Figure 2. DNA-hydrolyzing activity of antibodies from sera of leukemic patients from different steps of purification determined as described in [4] in equal reaction conditions. Supercoiled plasmid pUC19 was used as a substrate. Lane 1 - marker plasmid in three forms (supercoiled, linear and circular); lane 2 - control plasmid incubated in the same conditions without antibodies; lane 3- the plasmid incubated with IgG fraction retained by Cibacron 3GA; lane 4- the plasmid incubated with IgG fraction retained by DEAE-Sepharose FF; lane 5 - the plasmid incubated with IgG fraction passed through both columns.
IgG in bound fractions at all steps of purification and confirm, that two high molecular bands in lane 4, Fig.3A are presumably products of incomplete cleavage of S-S bonds by the reducing agent.
It is obvious, that the level of activity and purity of antibody fraction capable to hydrolyse DNA can be increased by affinity adsorption on the support with attached DNA. Earlier this selection was conducted using DNA-cellulose [4]. DNA affinity sorbents frequently have a number of drawbacks, such as non-specific binding, ligand leakage and poor flow characteristics. We used DNA-Sephacryl sorbent, prepared as described in [18] (step 5 in Fig. 1). No sorbent leakage was observed in 20 succsesive experiments and the column capacity has decreased very slightly. The sorbent permitted higher flow rate to be
applied than DNA-cellulose. About 1/20 of the total IgG fraction bound
409
Vol. 39, No. 2, 1996 BIOCHEMISTRYand MOLECULAR BIOLOGY INTERNATIONAL
67 k Da
45 kDa
21kDa
:3 4
84 kDa
53 kDa
35 kDa
20 kDa
1 2 3 4
Figure 3. A. Silver stained SDS PAGE electrophoresis. Lane 1 molecular weight markers; lane 2 - serum protein fraction retained by Cibacron 3GA; lane 3- serum protein fraction retained by DEAE-Sepharose FF; lane 4 - IgG fraction retained by DEAE-SepharoseFF and Protein G columns. The protein preparations were loaded to the gel in amount of 5 mkg per lane. B. Immunoblot of the fractions stained by goat anti-K and anti- )~ light chains (Sigma), peroxidase color reagent was 13- chloronaphtol. Lane 1 - prestained molecular weight markers; lanes 2 - 4 were the same as in Fig.3.A. The data indicate the presence of IgG antibodies in all fractions tested.
to the column, and increased level of the DNA-hydrolyzing activity was detected in the bound fraction, while the non-bound one revealed no activity in the same assay conditions. The DNA-binding properties were estimated using heteropolymeric oligonucleotide substrate (50-mer comprising the SacI-Hind III fragment of the pUC18 polylinker) in non-catalytic conditions and binding constant was (1.8_+0.02)xl 0-~°M as
410
Vol. 39, No. 2, 1996 BIOCHEMISTRYand MOLECULAR BIOLOGY INTERNATIONAL
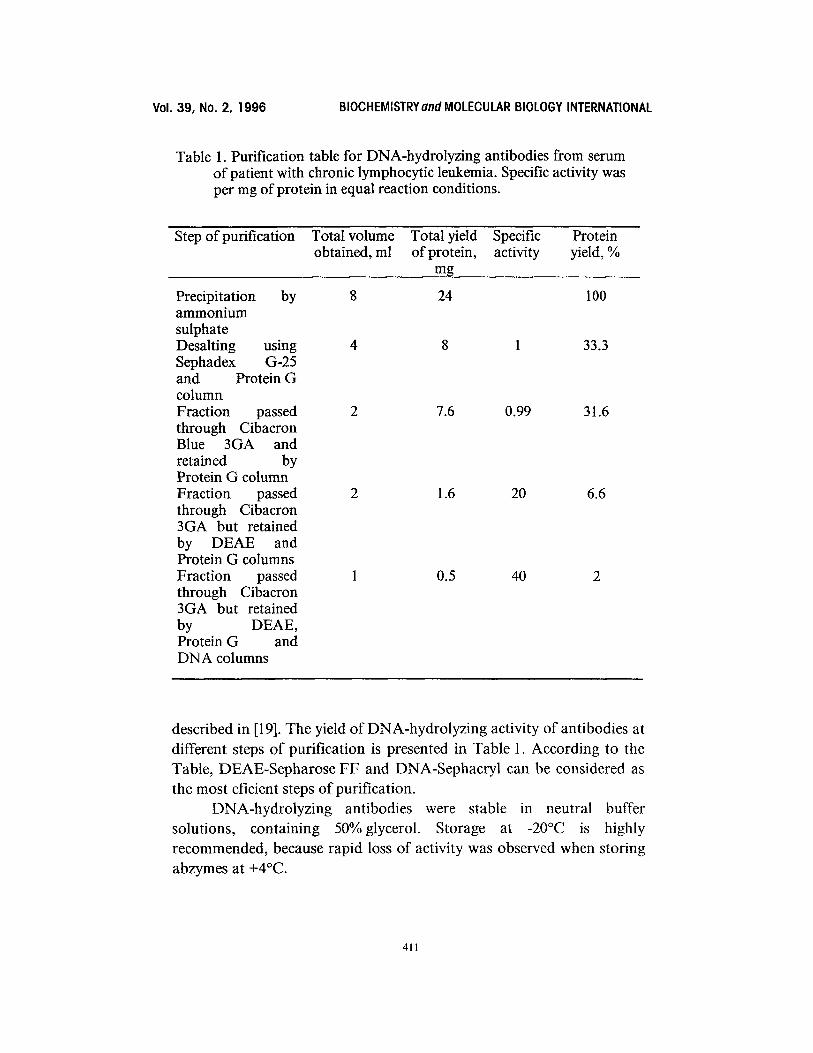
Table 1. Purification table for DNA-hydrolyzing antibodies from serum of patient with chronic lymphocytic leukemia. Specific activity was per mg of protein in equal reaction conditions.
Step of purification Total volume Total yield Specific Protein obtained, ml of protein, activity yield, %
mg
Precipitation by 8 24 100 ammonium sulphate Desalting using 4 8 1 33.3 Sephadex G-25 and Protein G column Fraction passed 2 7.6 0.99 31.6 through Cibacron Blue 3GA and retained by Protein G column Fraction passed 2 1.6 20 6.6 through Cibacron 3GA but retained by DEAE and Protein G columns Fraction passed 1 0.5 40 2 through Cibacron 3GA but retained by DEAE, Protein G and DNA columns
described in [19]. The yield of DNA-hydrolyzing activity of antibodies at different steps of purification is presented in Table 1. According to the Table, DEAE-Sepharose FF and DNA-Sephacryl can be considered as the most eficient steps of purification.
DNA-hydrolyzing antibodies were stable in neutral buffer solutions, containing 50% glycerol. Storage at -20°C is highly recommended, because rapid loss of activity was observed when storing abzymes at +4°C.
411

Vol. 39, No. 2, 1996 BIOCHEMISTRY and MOLECULAR BIOLOGY INTERNATIONAL
CONCLUSIONS
The DNA-hydrolyzing antibodies from sera of patients with lymphoproliferative diseases were found to reveal some features other than earlier described antibodies from sera of patients with autoimmune pathologies. In general, leukemic DNA-abzymes can be considered as substantially more labile than those found in systemic lupus erythematosus. Strong ion exchangers and temperatures above 0°C cause rapid loss of DNA-hydrolizing activity of the antibodies. These observations strongly suggest that the nature of DNA-cleaving activity of abzymes from the sera of patients with leukemia differs both from that of SLE-derived abzymes and blood DNAses.
The adequate purification technique for the leukemic DNA abzymes implies the use of dye affinity column to remove residual albumin and some proteases followed by weak anion exchanger. All purification steps must be done on ice. The unusual behaviour of anti-DNA abzymes from the sera of patients with leukemia on DEAE column permitted to separate them from the majority of other antibodies and prompts to examine whether some other autoantibodies exist with similar separation profiles on the DEAE column.
The method developed allows to reach a 40 fold purification of DNA-abzymes from total IgG fraction avoiding the loss of activity j udging by the specific activity of antibody fraction (Table 1).
ACKNOWLEDGEMENTS
The author thanks Prof. A.G. Gabibov and Dr. A.V. Kolesnikov for valuable help and consultations, E.S. Alexandrova for technical assistance and S. Dubiley for help with preparation of this manuscript. The work was supported by International Science Foundation (grant number M77300).
REFERENCES
1. Suzuki, H. (1994) J. Biochem. 115, 623-628. 2. Izdayar, L., Friboulet, A., Remy, M.H., Roseto, A.,
(1993) Proc. Natl. Acad. Sci. USA 90, 8876-8880. 3. Paul, S., Voile, D.J., Beach, C.M., Johnson, D.R.,
Massey, R.J. (1989) Science 244, 1158-1161.
& Thomas, D.
Powell, M.J., &
412

Vol. 39, No. 2, 1996 BIOCHEMISTRYand MOLECULAR BIOLOGY INTERNATIONAL
4. Schuster, A.M., Gololobov, G.V., Kvashuk, O.A., Bogomolova, A.E., Smirnov, I.V., & Gabibov, A.G. (1992) Science 256, 665-667.
5. Kozyr, A.V., Kolesnikov, A.V., Yakhnina, E.I., Astsaturov, I.A., Varlamova, E.Yu., Kirillov, E.V., & Gabibov, A.G. (1996) Bulletin of Exp. Biol. Med., in press (in Russian).
6. Gololobov, G.V., Mikhalap, S.V., Starov, A.V., Kolesnikov, A.V., & Gabibov, A.G. (1994) Appl. Biochem. Biotechnol. 47, 305-315.
7. Li, L., Paul, S., Tyutyulkova, S., Kazatchkine, M.D., Kaveri, S., (1995) J. Immunol. 154, 3328-3332.
8. Laemmli, E.K., (1970) Nature 227, 680-685. 9. Towbin, H., Staehelin, T., & Gordon, J. (1979)
Proc. Natl. Acad. Sci. USA 76, 4350-4354. 10. Gololobov, G.V., Chernova, E.A., Schourov, D.V., Smirnov, I.V.,
Kudelina, I.A., and Gabibov A.G. (1995) Proc. Natl. Acad. Sci. USA 92, 254-257.
11. Datta, S.K., Patel, H., & Berry, D. (1987) J. Exp. Med., 165, 1252-1268. 12. Herron, J.N., He, X.M., Ballard, D.W., Blier, P.R., Pace, P.E.,
Bothwell, A.L.M., Voss, E.W., Edmundson, Jr., & Edmundson, A.B. (1991 ) Proteins: structure, function and genetics, 11, 159-175.
13. Ashton, A.R. & Polya, G.M. (1978) Biochem. J., 175, 507. 14. Lymberi, P., Dighiero, G., Ternynck, T, & Avrameas, S.
(1985) Eur. J. Immunol. 15, 702-705. 15. Kofler, R., Noonars, D.I., Levy, D.E., Wilson, M.C., Moller, N.P.,
Dixon, F.J., & Theofilopoulos, A.N. (1985) J. Exp. Med. 161,805-815. 16. Dighiero, G., (1991) Autoreactive B-cell repertoire, in: Molecular
Immunobiology of self reactivity. C.A. Bona & A. Kaushik, eds., M. Dekker Inc., New York, 39-69.
17. Immunochemistry in practice (1987) A. Johnstone & R. Thorpe, eds., R. Clay Ltd., Chichester, Sussex, 51-52.
18. Bunemann, H., Westhoff, P., & Herrmann, R.C. (1982) Nucl. Acids Res. 10, 7163-7180. 19. Aleksandrova, E.S., (1996) Mol. Biol. (in Russian, in press).
413
Copyright © 2022 FDOKUMEN




















