
A new Bacillus cereus DNA-binding protein, HlyIIR, negatively regulates expression of B. cereus...
11
A new Bacillus cereus DNA-binding protein, HlyIIR, negatively regulates expression of B. cereus haemolysin II Zhanna I. Budarina, 1 Dmitri V. Nikitin, 1 Nikolay Zenkin, 2 Marina Zakharova, 1 Ekaterina Semenova, 2 Michael G. Shlyapnikov, 1 Ekaterina A. Rodikova, 3 Svetlana Masyukova, 1 Oleg Ogarkov, 2 Gleb E. Baida, 1 Alexander S. Solonin 1 and Konstantin Severinov 2 Correspondence Konstantin Severinov [email protected] Alexander S. Solonin [email protected] 1 The Institute of Biochemistry and Physiology of Micro-organisms, Nauki Avenue, 5, Pushchino, 142292 Russia 2 Waksman Institute for Microbiology, Rutgers, The State University of New Jersey, 190 Frelinghuysen Road, Piscataway, NJ 08854, USA 3 Pushchino State University, Nauki Avenue, 5, Pushchino, 142292 Russia Received 5 March 2004 Revised 23 June 2004 Accepted 4 August 2004 Haemolysin II, HlyII, is one of several cytotoxic proteins produced by Bacillus cereus, an opportunistic human pathogen that causes food poisoning. The hlyII gene confers haemolytic activity to Escherichia coli cells. Here a new B. cereus gene, hlyIIR, which is located immediately downstream of hlyII and regulates hlyII expression, is reported. The deduced amino acid sequence of HlyIIR is similar to prokaryotic DNA-binding transcriptional regulators of the TetR/AcrA family. Measurements of haemolytic activity levels and of hlyII promoter activity levels using gene fusions and primer-extension assays demonstrated that, in E. coli, hlyII transcription decreased in the presence of hlyIIR. Recombinant HlyIIR binds to a 22 bp inverted DNA repeat centred 48 bp upstream of the hlyII promoter transcription initiation point. In vitro transcription studies showed that HlyIIR inhibits transcription from the hlyII promoter by binding to the 22 bp repeat and RNA polymerase, and by decreasing the formation of the catalytically competent open promoter complex. INTRODUCTION Bacteria of the Bacillus cereus group, B. cereus, Bacillus anthracis, Bacillus mucoides and Bacillus thuringiensis, are important human and animal pathogens. While the pathogenic properties of bacteria of the B. cereus group are clearly distinct, analyses of genomic sequences, parti- cularly rRNA sequences, of member taxa reveal the very small amounts of sequence variation that would be expected for different strains of a single bacterial species (Ash et al., 1991). The differences in pathogenic properties of bac- teria of the B. cereus group are due to the production of different and non-overlapping sets of cytotoxic proteins, some of which are encoded by plasmid-borne genes. Haemolysin II, HlyII, is one of several cytotoxic proteins produced by B. cereus. B. cereus is a soil bacterium and an opportunistic human pathogen that causes sometimes- lethal food poisoning (Granum, 1997). Based on its deduced amino acid sequence, HlyII is a member of the family of oligomeric b-barrel toxins (Baida et al., 1999; Gouaux, 1998). This family also includes a number of Staphylococcus aureus cytolysins (a-toxin, leukocidins, c-haemolysin), Clostridium perfringens b-toxin (Gouaux, 1998) and B. cereus cytotoxin K (CytK) (Lund et al., 2000). These toxins are secreted in a soluble form and cause their cytotoxic effect by assembling into a transmembrane pore. The synthesis of b-barrel toxins is subject to genetic regula- tion. The synthesis of the prototypical member of the family, S. aureus a-toxin, occurs at the end of the exponential phase of growth and is regulated in a complex fashion by the agr (accessory gene regulator) (Recsei et al., 1986) and sar (Staphylococcus accessory regulator) loci. The products of agr genes are involved in positive regulation of a-toxin gene transcription. SarA, a pleiotropic regulator of S. aureus virulence genes, interacts directly with AT-rich sequences upstream of its target genes, which include the a-toxin gene; SarA also negatively controls expression of the agr genes (Chakrabarti & Misra, 2000). In the much less well- understood case of B. cereus CytK, putative binding sites for PlcR, a pleiotropic regulator of extracellular virulence factor gene expression in B. thuringiensis, have been identi- fied (Lund et al., 2000), suggesting that cytK expression is subject to positive regulation. Abbreviation: RNAP, RNA polymerase. 0002-7142 G 2004 SGM Printed in Great Britain 3691 Microbiology (2004), 150, 3691–3701 DOI 10.1099/mic.0.27142-0
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of A new Bacillus cereus DNA-binding protein, HlyIIR, negatively regulates expression of B. cereus...

A new Bacillus cereus DNA-binding protein,HlyIIR, negatively regulates expression ofB. cereus haemolysin II
Zhanna I. Budarina,1 Dmitri V. Nikitin,1 Nikolay Zenkin,2 Marina Zakharova,1
Ekaterina Semenova,2 Michael G. Shlyapnikov,1 Ekaterina A. Rodikova,3
Svetlana Masyukova,1 Oleg Ogarkov,2 Gleb E. Baida,1
Alexander S. Solonin1 and Konstantin Severinov2
Correspondence
Konstantin Severinov
Alexander S. Solonin
1The Institute of Biochemistry and Physiology of Micro-organisms, Nauki Avenue, 5, Pushchino,142292 Russia
2Waksman Institute for Microbiology, Rutgers, The State University of New Jersey,190 Frelinghuysen Road, Piscataway, NJ 08854, USA
3Pushchino State University, Nauki Avenue, 5, Pushchino, 142292 Russia
Received 5 March 2004
Revised 23 June 2004
Accepted 4 August 2004
Haemolysin II, HlyII, is one of several cytotoxic proteins produced by Bacillus cereus, an
opportunistic human pathogen that causes food poisoning. The hlyII gene confers haemolytic
activity to Escherichia coli cells. Here a new B. cereus gene, hlyIIR, which is located immediately
downstream of hlyII and regulates hlyII expression, is reported. The deduced amino acid
sequence of HlyIIR is similar to prokaryotic DNA-binding transcriptional regulators of the TetR/AcrA
family. Measurements of haemolytic activity levels and of hlyII promoter activity levels using
gene fusions and primer-extension assays demonstrated that, in E. coli, hlyII transcription
decreased in the presence of hlyIIR. Recombinant HlyIIR binds to a 22 bp inverted DNA repeat
centred 48 bp upstream of the hlyII promoter transcription initiation point. In vitro transcription
studies showed that HlyIIR inhibits transcription from the hlyII promoter by binding to the
22 bp repeat and RNA polymerase, and by decreasing the formation of the catalytically
competent open promoter complex.
INTRODUCTION
Bacteria of the Bacillus cereus group, B. cereus, Bacillusanthracis, Bacillus mucoides and Bacillus thuringiensis, areimportant human and animal pathogens. While thepathogenic properties of bacteria of the B. cereus groupare clearly distinct, analyses of genomic sequences, parti-cularly rRNA sequences, of member taxa reveal the verysmall amounts of sequence variation that would be expectedfor different strains of a single bacterial species (Ash et al.,1991). The differences in pathogenic properties of bac-teria of the B. cereus group are due to the production ofdifferent and non-overlapping sets of cytotoxic proteins,some of which are encoded by plasmid-borne genes.
Haemolysin II, HlyII, is one of several cytotoxic proteinsproduced by B. cereus. B. cereus is a soil bacterium and anopportunistic human pathogen that causes sometimes-lethal food poisoning (Granum, 1997). Based on its deducedamino acid sequence, HlyII is a member of the family ofoligomeric b-barrel toxins (Baida et al., 1999; Gouaux,1998). This family also includes a number of Staphylococcus
aureus cytolysins (a-toxin, leukocidins, c-haemolysin),Clostridium perfringens b-toxin (Gouaux, 1998) and B.cereus cytotoxin K (CytK) (Lund et al., 2000). These toxinsare secreted in a soluble form and cause their cytotoxiceffect by assembling into a transmembrane pore.
The synthesis of b-barrel toxins is subject to genetic regula-tion. The synthesis of the prototypical member of the family,S. aureus a-toxin, occurs at the end of the exponential phaseof growth and is regulated in a complex fashion by the agr(accessory gene regulator) (Recsei et al., 1986) and sar(Staphylococcus accessory regulator) loci. The products ofagr genes are involved in positive regulation of a-toxingene transcription. SarA, a pleiotropic regulator of S. aureusvirulence genes, interacts directly with AT-rich sequencesupstream of its target genes, which include the a-toxingene; SarA also negatively controls expression of the agrgenes (Chakrabarti & Misra, 2000). In the much less well-understood case of B. cereus CytK, putative binding sitesfor PlcR, a pleiotropic regulator of extracellular virulencefactor gene expression in B. thuringiensis, have been identi-fied (Lund et al., 2000), suggesting that cytK expression issubject to positive regulation.Abbreviation: RNAP, RNA polymerase.
0002-7142 G 2004 SGM Printed in Great Britain 3691
Microbiology (2004), 150, 3691–3701 DOI 10.1099/mic.0.27142-0

HlyII is most closely related to CytK of B. cereus and to thea-toxin of S. aureus. Up to now, no regulatory system forthe hlyII gene has been described. hlyII-like genes havebeen found in a number of B. cereus strains, in most of theB. thuringiensis strains tested (Budarina et al., 1994), andmost recently in B. anthracis (Read et al., 2002). The extentof haemolytic activity in these bacilli varies both betweenthese closely related species and between different strainsof the same species; however, the reasons for such variationare not known.
We have previously reported the cloning of a fragment ofB. cereus VKM-B771 genomic DNA containing the hlyIIgene and adjacent sequences (Sinev et al., 1993). Thisfragment, when introduced on a plasmid into E. coli, led toa haemolytic phenotype (Sinev et al., 1993; Baida et al.,1999). A perfect 22 bp inverted repeat has been found222 bp upstream of the hlyII ORF initiating ATG codon,and it was hypothesized that it could be required for trans-cription regulation of hlyII (Baida et al., 1999). However,no molecular mechanism for this regulation was proposed.In this paper, we describe a B. cereus ORF, hlyIIR, locateddownstream of hlyII. Sequence analysis revealed that HlyIIR(for HlyII Regulator) belongs to the Tet/AcrA family ofDNA binding transcription regulators. When hlyIIR wasintroduced into E. coli cells harbouring hlyII plasmids, thehaemolytic phenotype and hlyII transcription levels weredecreased. In vitro, recombinant HlyIIR specifically inter-acted with the 22 bp inverted repeat which is centred 48 bpupstream from the hlyII promoter transcription initiationpoint. HlyIIR binding to promoter DNA did not affectpromoter recognition by RNA polymerase (RNAP), butdecreased the number of catalytically active open promotercomplexes. Thus, our results indicate that HlyII synthesisis the subject of negative regulation by HlyIIR.
METHODS
Bacterial strains and growth conditions. The Escherichia coli
strain Z85 [thi, D(lac-proAB), D(srl-recA), hsdR, supE, Tn10(Tcr), (F9,
2traD, proAB, lacI, DM15)] was used in haemolytic phenotype
detection experiments and as a host for plasmid DNA. The hlyIIR
protein was expressed in E. coli strain M15pRep4 (Kmr, lac2, ara2,
gal2, mtl2, F2). Both strains were grown in LB liquid medium
(10 g bactotryptone, 5 g yeast extract, 5 g sodium chloride l21) at
37 uC. For growth on plates, the LB medium was supplemented with
15 g agar l21.
Haemolytic phenotype detection. The haemolytic phenotype of
recombinant cells was tested by the appearance of haemolytic zones
(clearance zones) around colonies grown on LB agar containing a
1% suspension of human erythrocytes. Cells were grown at 37 uCovernight and were then incubated for an additional 8–12 hours at
20 uC.
To measure the haemolytic phenotype quantitatively, cells were
grown in LB medium at 20 uC with vigorous aeration until the onset
of stationary phase. Haemolytic activity in cultured medium was
measured as described by Baida et al. (1999).
Plasmids and DNA manipulations. Molecular cloning was per-formed using standard protocols. Sequencing reactions were carriedout using the FemtoMol sequencing kit (Promega).
Previously described plasmids pUJ1 and pUJ2 (Sinev et al., 1993)contain the 2?9 kb EcoRI fragment of B. cereus VKM-B771 genomicDNA containing the hlyII gene cloned in opposing orientation in apUC19 vector. For construction of the pH2 plasmid, the hlyII gene wasPCR amplified using 59 primer ACGTTGTAAAACGACGGCCAGTG(anneals to the pUC19 vector part of pUJ1, upstream of the EcoRI siteused for the cloning of the 2?9 kb fragment of B. cereus DNA) and 39primer AATAAAGCTTTTATACTCATATTTG complementary toDNA 38–63 nt downstream of the hlyII termination codon andcarrying an engineered HindIII site (underlined). The amplifiedproduct was treated with EcoRI and HindIII and cloned into thepUC19 vector. For construction of the pRH2 plasmid, the hlyIIRgene was amplified with the same 59 primer as for the hlyII ampli-fication together with 39 primer GATGGATCCTTATTTCATCAA-AAT, which anneals to B. cereus DNA 24–44 nt downstream ofthe hlyII termination codon and carries an engineered BamHI site(underlined), using the pUJ2 plasmid as a template. The amplifiedproduct was treated with EcoRI and BamHI and cloned into thep15KS vector. For construction of the pFH1Lac plasmid, the 2420 bpBamHI–HindIII fragment of pUJ1 was replaced with the 3070 bpBamHI–HindIII lacZ fragment from plasmid pCL471 (Zabeau &Stanley, 1982). Plasmid pHR, expressing hexahistidine-tagged HlyIIR,was obtained by cloning the PCR-amplified hlyIIR ORF between theBamHI and HindIII sites of the pQE30 expression vector.
b-Galactosidase assays. The amounts of b-galactosidase in E. colicell extracts were determined with a quantitative colorimetric assayusing ONPG as a substrate, as described elsewhere (Nikolaev et al.,1992).
Primer extension reactions. Total RNA was extracted either fromE. coli Z85 cells transformed with appropriate hlyII plasmids andgrown in LB medium overnight or from cells scraped from thesurface of LB plates containing 1% human erythrocytes. RNA waspurified with the RNeasy RNA isolation mini kit (Qiagen), followingthe manufacturer’s instructions. RNA samples were then treatedwith RNase-free DNase and repurified using the Qiagen RNeasyprotocol. Total RNA (5 mg) was analysed by electrophoresis in 1%agarose gels to ascertain that the rRNA was intact. For primer exten-sion experiments, 10 mg of total RNA was reverse transcribed with5 U AMV Reverse Transcriptase (Roche) for 40 minutes at 42 uC in16AMV Reverse Transcriptase buffer, supplied by the manufacturer,in the presence of 1 pmol of the 32P-end-labelled primer CGGAC-GCTACGGCAACACTTTTAGCTATTCCC, which is complementaryto hlyII ORF positions 15–46. A side-by-side sequencing reactionwas performed with the same end-labelled primer and the pUJ1plasmid as a template, using the Cycle Sequencing system (Promega)according to manufacturer’s protocol. Primer extension reactionswere terminated by the addition of 50 ml RNase solution (100 U)(Qiagen), incubated for 15 minutes at room temperature andphenol/chloroform extracted, after which nucleic acids were pre-cipitated with ethanol. The pellet was dissolved in formamide-containing loading buffer, and products were resolved on a 6%sequencing gel and visualised using a PhosphoImager (MolecularDynamics).
Proteins. E. coli M15pREP4 transformed with the pHR plasmidwas inoculated into 200 ml LB medium containing 60 mg ampicillinl21 and 10 mg kanamycin l21. When the OD600 of the culturereached 0?5–0?7, cells were induced by the addition of 4 mM IPTG.After 3 h induction, cells were harvested and resuspended in 5 mlBuffer A (40 mM Tris/HCl, pH 7?5, 50 mM NaCl). Cells were lysedby sonication, cell debris was removed by centrifugation and the cellextract was loaded onto a Ni-NTA agarose column equilibrated with
3692 Microbiology 150
Z. I. Budarina and others

Buffer A. The column was washed and HlyIIR was eluted with250 mM imidazole in buffer A. HlyIIR-containing fractions werepooled and dialysed into buffer B (40 mM Tris/HCl, pH 7?5,50 mM NaCl, 1 mM EDTA, 1 mM b-mercaptoethanol). The proteinwas then loaded onto a 1 ml MonoQ column equilibrated in thesame buffer. The column was developed with a linear gradient ofNaCl concentration (50–1000 mM) in buffer B. Fractions containinghomogeneous HlyIIR were pooled, concentrated and stored under50% (v/v) glycerol at 220 uC.
E. coli RNAP core and s70 holoenzymes, and recombinant s70 ands565 were purified as described by Minakhin et al. (2003). B. cereusRNAP sA holoenzyme was partially purified from B. cereus VKM-B771 by performing polyethyleneimine P fractionation and heparinagarose affinity chromatography, using the standard procedure forE. coli RNAP purification. The resultant enzyme was ~75% pure.
DNA binding assays, footprinting and in vitro transcription.The standard HlyIIR–DNA binding reaction contained, in 20 mlbuffer (10 mM Tris/HCl, pH 7?5, 10 mM NaCl, 1 mM MgCl2),various amounts of HlyIIR, 200 ng radioactively labelled DNA or1 mg unlabelled DNA, and 1 mg BSA. Complexes were allowed toform for 20 min at 37 uC. Reactions were then loaded on 4% or12% polyacrylamide gels, or on 0?8% agarose gels. DNA complexesseparated on polyacrylamide gels were revealed by autoradiography;complexes resolved by agarose gel electrophoresis were stained byethidium bromide and visualized with UV irradiation. Whereindicated, 0?5, 1 and 2 mg of pUC19 competitor DNA was added tosamples prior to electrophoresis.
The hlyII promoter-containing DNA fragments used in in vitro trans-cription and footprinting reactions were created by PCR, using pUJ1 asa template. The standard, ‘wild-type’ fragment corresponded to posi-tions 294 to +214 relative to the hlyII transcription initiation startpoint. ‘Mutant’ fragments, lacking B. cereus sequences 27 and 47 basesupstream of the hlyII promoter initiation start point, were created byPCR.
To form open promoter complexes, 3 pmol E. coli RNAP s70
holoenzyme or B. cereus RNAP sA holoenzyme (purified as describedby Kashlev et al., 1996) was preincubated in 10 ml transcription buffer(20 mM Tris/HCl, pH 7?9, 40 mM KCl, 10 mM MgCl2) with 1 pmolof hlyII promoter DNA fragment for 10 min at 37 uC. HlyIIR(50 pmol) was added to the reaction either before or after RNAPaddition and open complex formation. After the HlyIIR addition, thereaction mixture was incubated for 10 min at 37 uC. Transcriptioncomplexes were next subjected to native gel analysis or footprinting, orwere supplemented with 500 mM GTP, 50 mM ATP and CTP, and[a-32P]UTP (30 Ci mmol21) to analyse in vitro transcription products.Transcription reactions were allowed to proceed for 10 min at 37 uC,and were terminated by the addition of an equal volume of formamide-containing gel loading buffer. Reaction products were separated on a20% polyacrylamide, 6 M urea gel and revealed by PhosphoImageranalysis. For native gel and footprinting analysis, the DNA fragmentcontaining the hlyII promoter was 39-end-labelled at the non-templatestrand with Klenow enzyme, using standard procedures. Samplescontaining hlyII promoter complexes were loaded directly on a 4%(37?5 : 1) polyacrylamide native gel, footprinted with DNase I orprobed with KMnO4, as described by Severinova et al. (1998). Thesamples were then analysed on a 6% sequencing gel and revealed on aPhosphoImager.
For primer extension analysis of in vitro transcribed RNA, promotercomplexes were formed during a 10 min 37 uC incubation of 10 pmolE. coli or B. cereus RNAP and 5 pmol hlyII promoter-containingfragment in 100 ml transcription buffer. NTP (500 mM) was added andthe reaction continued for 30 min at 37 uC. Reactions were extracted
with chloroform, and nucleic acids were precipitated with ethanol,dissolved in water and subjected to the primer extension assay. In vitrotranscribed RNA was reverse-transcribed with 100 U Super ScriptIII enzyme (Invitrogen) in the presence of 1 pmol 32P-end-labelledprimer complementary to nucleotide positions 158–182 of the hlyIIgene. Primer extension reactions were carried out for 50 min at 50 uCand terminated by a 5 min incubation at 85 uC. RNA was removedby RNase H treatment. Reaction products were extracted withchloroform, precipitated with ethanol and dissolved in formamide-containing loading buffer. As a reference, sequencing reactions wereperformed on a hlyII promoter-containing DNA fragment using thesame end-labelled primer as the one used in the primer extensionreaction. The fmol DNA Cycle Sequencing System (Promega) was usedfor sequencing, according to the manufacturer’s protocol. The exten-sion products were resolved on an 8% sequencing gel, together withthe sequencing reactions, and visualised using a PhosphorImager.
RESULTS
The hlyIIR gene negatively regulates B. cereushlyII expression in E. coli
Previously, we reported that E. coli plasmid pUJ1, contain-ing a ~2?9 1kb EcoRI fragment of B. cereus VKM-B771DNA cloned in pUC19, allowed E. coli cells to lyse erythro-cytes (Sinev et al., 1993; Baida et al., 1999). The haemolyticactivity was attributed to the presence of the hlyII (haemo-lysin II) gene, which was located in the left-hand side of the2?9 kb EcoRI fragment (Baida et al., 1999).
The hlyII gene (412 codons) is much shorter than theB. cereus DNA fragment of pUJ1. We determined thesequence of the entire 2?9 kb EcoRI fragment of pUJ1 andidentified an additional, 201-codon-long ORF downstreamof hlyII. The new ORF has an appropriately positionedShine–Dalgarno sequence. The new ORF is transcribed inthe same direction as hlyII; it is preceded by a putativepromoter and is followed by a putative r-independenttranscription terminator, suggesting that it is expressedindependently of hlyII.
To determine whether the product of the additional ORFis involved in haemolysis or not, E. coli expression plasmidscontaining either hlyII or the new gene were created(Fig. 1a), and their ability to impart haemolytic activityto E. coli was tested. As expected, the pUC19-based hlyIIplasmid pH2 imparted haemolytic activity to E. coli (datanot shown; see also Fig. 1b). In contrast, plasmid pRH2,which carried the downstream ORF with its putativeregulatory elements cloned in the p15KS vector, did notimpart haemolytic activity to E. coli, thus excluding thepossibility that the product of the second gene is involvedin haemolysis directly (data not shown; see also Fig. 1b).
Since replicons of the pUC19 and p15KS plasmids arecompatible, we tested the haemolytic activity of E. coli cellsharbouring both hlyII and the downstream ORF plasmids.The haemolytic activity of cells harbouring the pH2 (hlyII)plasmid and the p15KS vector plasmid was about 10 timeshigher than the haemolytic activity of cells carrying pH2and pRH2 (Fig. 1b). As expected, no haemolytic activity was
http://mic.sgmjournals.org 3693
Regulator of B. cereus haemolysin II expression
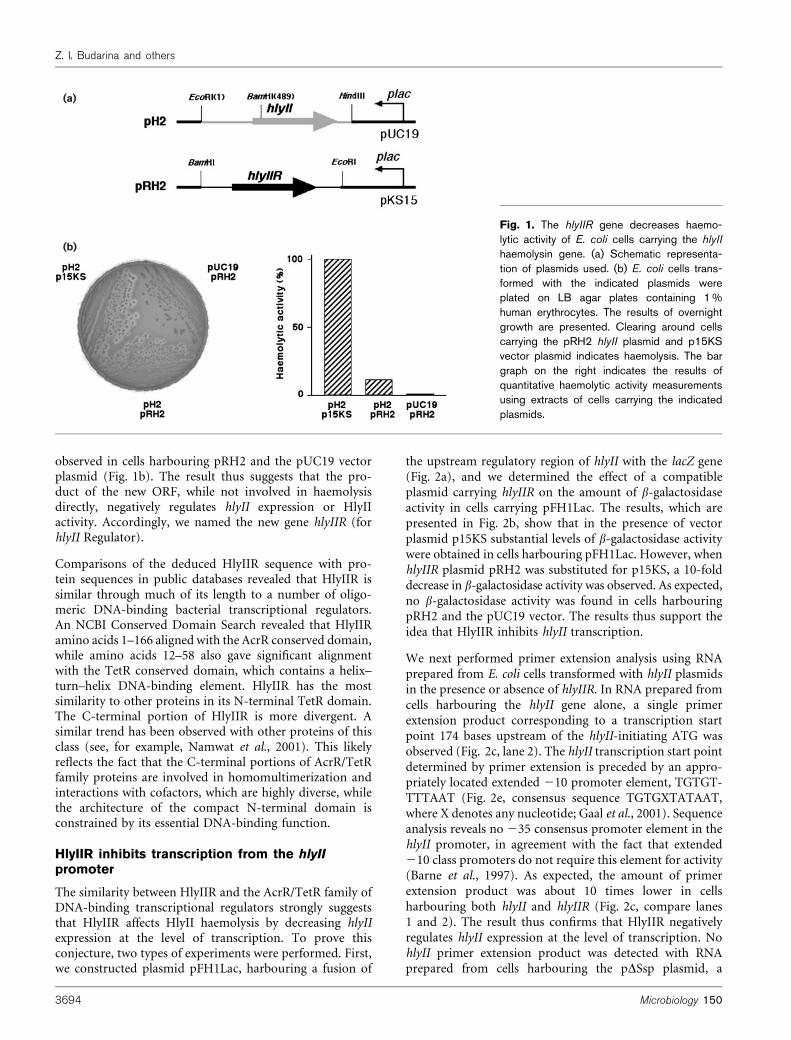
observed in cells harbouring pRH2 and the pUC19 vectorplasmid (Fig. 1b). The result thus suggests that the pro-duct of the new ORF, while not involved in haemolysisdirectly, negatively regulates hlyII expression or HlyIIactivity. Accordingly, we named the new gene hlyIIR (forhlyII Regulator).
Comparisons of the deduced HlyIIR sequence with pro-tein sequences in public databases revealed that HlyIIR issimilar through much of its length to a number of oligo-meric DNA-binding bacterial transcriptional regulators.An NCBI Conserved Domain Search revealed that HlyIIRamino acids 1–166 aligned with the AcrR conserved domain,while amino acids 12–58 also gave significant alignmentwith the TetR conserved domain, which contains a helix–turn–helix DNA-binding element. HlyIIR has the mostsimilarity to other proteins in its N-terminal TetR domain.The C-terminal portion of HlyIIR is more divergent. Asimilar trend has been observed with other proteins of thisclass (see, for example, Namwat et al., 2001). This likelyreflects the fact that the C-terminal portions of AcrR/TetRfamily proteins are involved in homomultimerization andinteractions with cofactors, which are highly diverse, whilethe architecture of the compact N-terminal domain isconstrained by its essential DNA-binding function.
HlyIIR inhibits transcription from the hlyIIpromoter
The similarity between HlyIIR and the AcrR/TetR family ofDNA-binding transcriptional regulators strongly suggeststhat HlyIIR affects HlyII haemolysis by decreasing hlyIIexpression at the level of transcription. To prove thisconjecture, two types of experiments were performed. First,we constructed plasmid pFH1Lac, harbouring a fusion of
the upstream regulatory region of hlyII with the lacZ gene(Fig. 2a), and we determined the effect of a compatibleplasmid carrying hlyIIR on the amount of b-galactosidaseactivity in cells carrying pFH1Lac. The results, which arepresented in Fig. 2b, show that in the presence of vectorplasmid p15KS substantial levels of b-galactosidase activitywere obtained in cells harbouring pFH1Lac. However, whenhlyIIR plasmid pRH2 was substituted for p15KS, a 10-folddecrease in b-galactosidase activity was observed. As expected,no b-galactosidase activity was found in cells harbouringpRH2 and the pUC19 vector. The results thus support theidea that HlyIIR inhibits hlyII transcription.
We next performed primer extension analysis using RNAprepared from E. coli cells transformed with hlyII plasmidsin the presence or absence of hlyIIR. In RNA prepared fromcells harbouring the hlyII gene alone, a single primerextension product corresponding to a transcription startpoint 174 bases upstream of the hlyII-initiating ATG wasobserved (Fig. 2c, lane 2). The hlyII transcription start pointdetermined by primer extension is preceded by an appro-priately located extended 210 promoter element, TGTGT-TTTAAT (Fig. 2e, consensus sequence TGTGXTATAAT,where X denotes any nucleotide; Gaal et al., 2001). Sequenceanalysis reveals no 235 consensus promoter element in thehlyII promoter, in agreement with the fact that extended210 class promoters do not require this element for activity(Barne et al., 1997). As expected, the amount of primerextension product was about 10 times lower in cellsharbouring both hlyII and hlyIIR (Fig. 2c, compare lanes1 and 2). The result thus confirms that HlyIIR negativelyregulates hlyII expression at the level of transcription. NohlyII primer extension product was detected with RNAprepared from cells harbouring the pDSsp plasmid, a
(a)
(b)
Fig. 1. The hlyIIR gene decreases haemo-lytic activity of E. coli cells carrying the hlyII
haemolysin gene. (a) Schematic representa-tion of plasmids used. (b) E. coli cells trans-formed with the indicated plasmids wereplated on LB agar plates containing 1%human erythrocytes. The results of overnightgrowth are presented. Clearing around cellscarrying the pRH2 hlyII plasmid and p15KSvector plasmid indicates haemolysis. The bargraph on the right indicates the results ofquantitative haemolytic activity measurementsusing extracts of cells carrying the indicatedplasmids.
3694 Microbiology 150
Z. I. Budarina and others
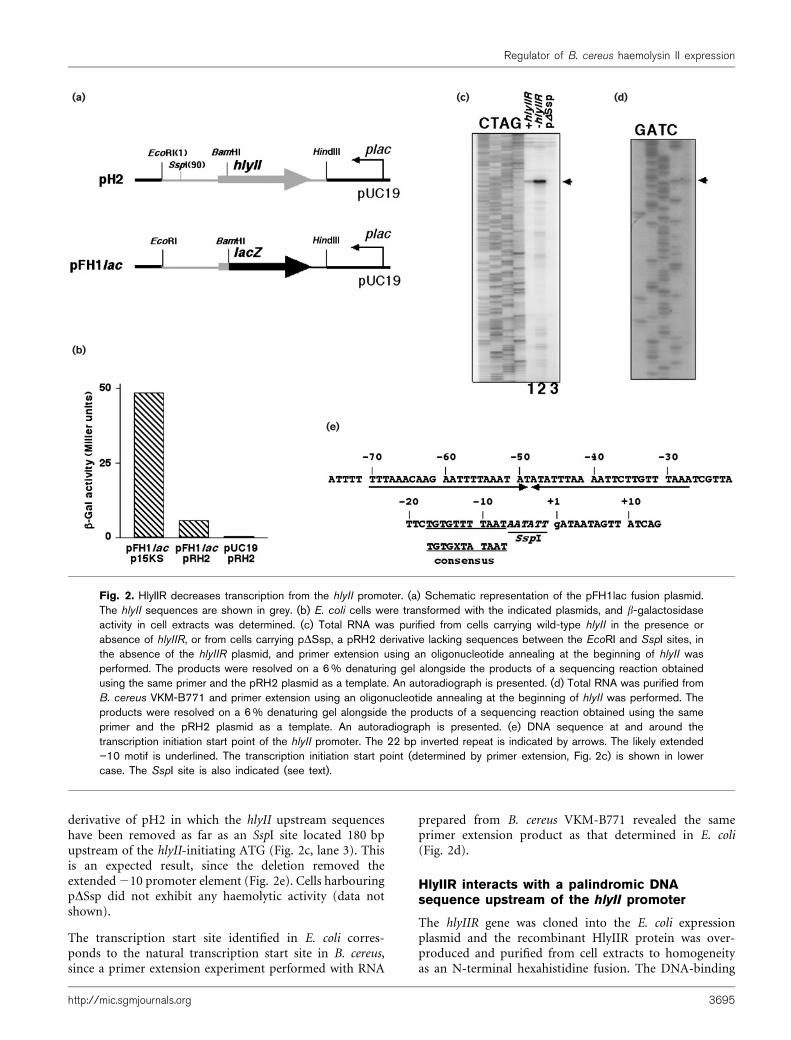
derivative of pH2 in which the hlyII upstream sequenceshave been removed as far as an SspI site located 180 bpupstream of the hlyII-initiating ATG (Fig. 2c, lane 3). Thisis an expected result, since the deletion removed theextended210 promoter element (Fig. 2e). Cells harbouringpDSsp did not exhibit any haemolytic activity (data notshown).
The transcription start site identified in E. coli corres-ponds to the natural transcription start site in B. cereus,since a primer extension experiment performed with RNA
prepared from B. cereus VKM-B771 revealed the sameprimer extension product as that determined in E. coli(Fig. 2d).
HlyIIR interacts with a palindromic DNAsequence upstream of the hlyII promoter
The hlyIIR gene was cloned into the E. coli expressionplasmid and the recombinant HlyIIR protein was over-produced and purified from cell extracts to homogeneityas an N-terminal hexahistidine fusion. The DNA-binding
(a)
(b)
(e)
(c) (d)
Fig. 2. HlyIIR decreases transcription from the hlyII promoter. (a) Schematic representation of the pFH1lac fusion plasmid.The hlyII sequences are shown in grey. (b) E. coli cells were transformed with the indicated plasmids, and b-galactosidaseactivity in cell extracts was determined. (c) Total RNA was purified from cells carrying wild-type hlyII in the presence orabsence of hlyIIR, or from cells carrying pDSsp, a pRH2 derivative lacking sequences between the EcoRI and SspI sites, inthe absence of the hlyIIR plasmid, and primer extension using an oligonucleotide annealing at the beginning of hlyII wasperformed. The products were resolved on a 6% denaturing gel alongside the products of a sequencing reaction obtainedusing the same primer and the pRH2 plasmid as a template. An autoradiograph is presented. (d) Total RNA was purified fromB. cereus VKM-B771 and primer extension using an oligonucleotide annealing at the beginning of hlyII was performed. Theproducts were resolved on a 6% denaturing gel alongside the products of a sequencing reaction obtained using the sameprimer and the pRH2 plasmid as a template. An autoradiograph is presented. (e) DNA sequence at and around thetranscription initiation start point of the hlyII promoter. The 22 bp inverted repeat is indicated by arrows. The likely extended”10 motif is underlined. The transcription initiation start point (determined by primer extension, Fig. 2c) is shown in lowercase. The SspI site is also indicated (see text).
http://mic.sgmjournals.org 3695
Regulator of B. cereus haemolysin II expression
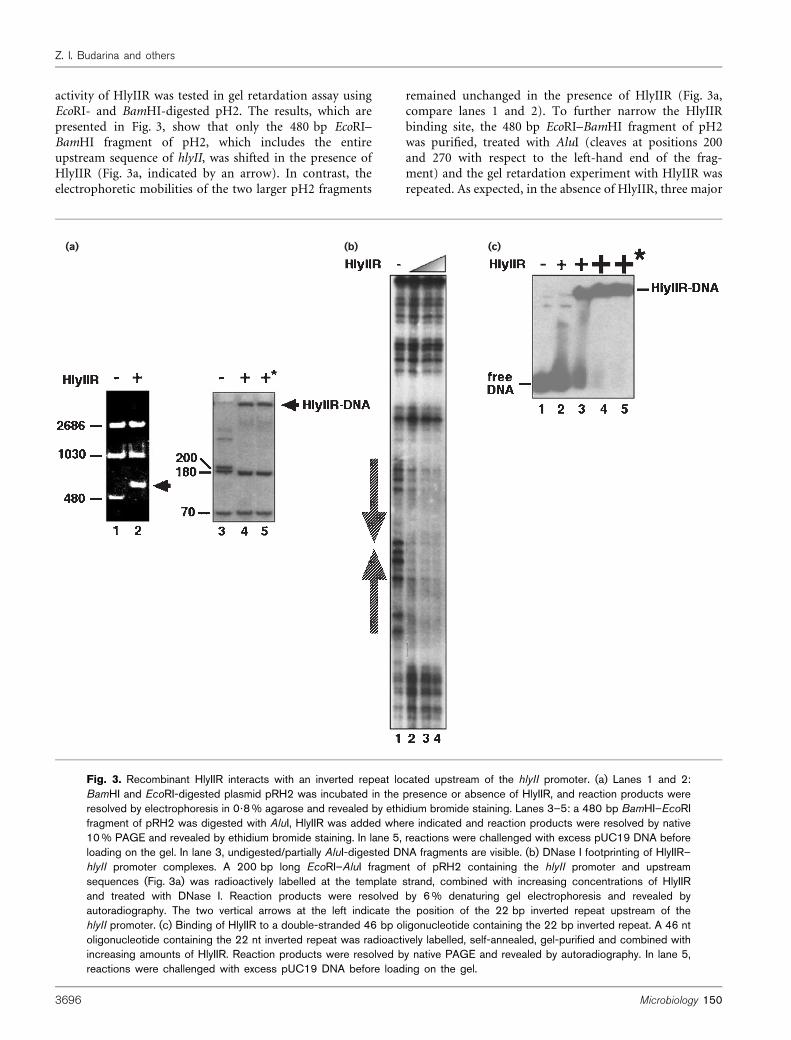
activity of HlyIIR was tested in gel retardation assay usingEcoRI- and BamHI-digested pH2. The results, which arepresented in Fig. 3, show that only the 480 bp EcoRI–BamHI fragment of pH2, which includes the entireupstream sequence of hlyII, was shifted in the presence ofHlyIIR (Fig. 3a, indicated by an arrow). In contrast, theelectrophoretic mobilities of the two larger pH2 fragments
remained unchanged in the presence of HlyIIR (Fig. 3a,compare lanes 1 and 2). To further narrow the HlyIIRbinding site, the 480 bp EcoRI–BamHI fragment of pH2was purified, treated with AluI (cleaves at positions 200and 270 with respect to the left-hand end of the frag-ment) and the gel retardation experiment with HlyIIR wasrepeated. As expected, in the absence of HlyIIR, three major
(a) (b) (c)
Fig. 3. Recombinant HlyIIR interacts with an inverted repeat located upstream of the hlyII promoter. (a) Lanes 1 and 2:BamHI and EcoRI-digested plasmid pRH2 was incubated in the presence or absence of HlyIIR, and reaction products wereresolved by electrophoresis in 0?8% agarose and revealed by ethidium bromide staining. Lanes 3–5: a 480 bp BamHI–EcoRIfragment of pRH2 was digested with AluI, HlyIIR was added where indicated and reaction products were resolved by native10% PAGE and revealed by ethidium bromide staining. In lane 5, reactions were challenged with excess pUC19 DNA beforeloading on the gel. In lane 3, undigested/partially AluI-digested DNA fragments are visible. (b) DNase I footprinting of HlyIIR–hlyII promoter complexes. A 200 bp long EcoRI–AluI fragment of pRH2 containing the hlyII promoter and upstreamsequences (Fig. 3a) was radioactively labelled at the template strand, combined with increasing concentrations of HlyIIRand treated with DNase I. Reaction products were resolved by 6% denaturing gel electrophoresis and revealed byautoradiography. The two vertical arrows at the left indicate the position of the 22 bp inverted repeat upstream of thehlyII promoter. (c) Binding of HlyIIR to a double-stranded 46 bp oligonucleotide containing the 22 bp inverted repeat. A 46 ntoligonucleotide containing the 22 nt inverted repeat was radioactively labelled, self-annealed, gel-purified and combined withincreasing amounts of HlyIIR. Reaction products were resolved by native PAGE and revealed by autoradiography. In lane 5,reactions were challenged with excess pUC19 DNA before loading on the gel.
3696 Microbiology 150
Z. I. Budarina and others
DNA fragments were observed (Fig. 3a, lane 3). In thepresence of recombinant HlyIIR, retardation of the largest,200 bp, fragment but not of the smaller, 180 and 70 bp,fragments was observed (Fig. 3a, lane 4). The interactionof HlyIIR with the 200 bp fragment was specific, since theaddition of a 10-fold excess of linearized pUC19 plasmidDNA did not change the retardation pattern (Fig. 3b, lane 5).
To locate the HlyIIR-binding site within the 200 bpfragment, we footprinted HlyIIR DNA complexes withDNase I. The results, presented in Fig. 3b, indicated thatHlyIIR specifically protected ~50 bp of DNA, betweenpositions 275 to 225 relative to the hlyII transcriptioninitiation start point, from DNase I digestion. The pro-tected region corresponds to the 22 bp inverted repeatcentred 48 bp upstream from the hlyII transcriptioninitiation start point (Fig. 2d).
We next proceeded to establish that the 22 bp invertedrepeat is sufficient for interaction with HlyIIR. We used asynthetic double-stranded DNA probe that contained theentire 22 bp inverted repeat. Gel-retardation experimentsrevealed that HlyIIR readily shifted this oligonucleotide(Fig. 3c, compare lanes 1 and 3) and that the HlyIIR–DNAcomplex was specific, as it withstood competition with alarge excess of pUC19 DNA (Fig. 3c, compare lanes 4and 5). We conclude that the 22 bp inverted repeat containsthe HlyIIR binding site.
Transcription inhibition by HlyIIR in vitro
We next studied the effect of recombinant HlyIIR ontranscription from the hlyII promoter in vitro. Both E. coli
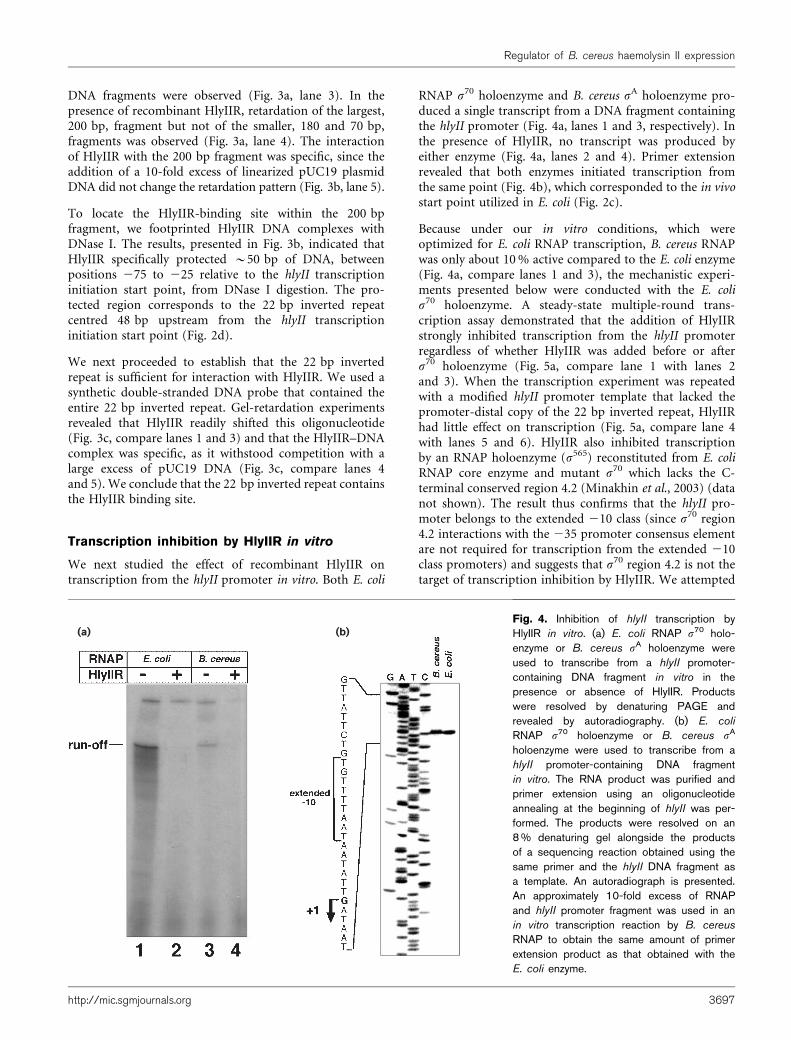
RNAP s70 holoenzyme and B. cereus sA holoenzyme pro-duced a single transcript from a DNA fragment containingthe hlyII promoter (Fig. 4a, lanes 1 and 3, respectively). Inthe presence of HlyIIR, no transcript was produced byeither enzyme (Fig. 4a, lanes 2 and 4). Primer extensionrevealed that both enzymes initiated transcription fromthe same point (Fig. 4b), which corresponded to the in vivostart point utilized in E. coli (Fig. 2c).
Because under our in vitro conditions, which wereoptimized for E. coli RNAP transcription, B. cereus RNAPwas only about 10% active compared to the E. coli enzyme(Fig. 4a, compare lanes 1 and 3), the mechanistic experi-ments presented below were conducted with the E. colis70 holoenzyme. A steady-state multiple-round trans-cription assay demonstrated that the addition of HlyIIRstrongly inhibited transcription from the hlyII promoterregardless of whether HlyIIR was added before or afters70 holoenzyme (Fig. 5a, compare lane 1 with lanes 2and 3). When the transcription experiment was repeatedwith a modified hlyII promoter template that lacked thepromoter-distal copy of the 22 bp inverted repeat, HlyIIRhad little effect on transcription (Fig. 5a, compare lane 4with lanes 5 and 6). HlyIIR also inhibited transcriptionby an RNAP holoenzyme (s565) reconstituted from E. coliRNAP core enzyme and mutant s70 which lacks the C-terminal conserved region 4.2 (Minakhin et al., 2003) (datanot shown). The result thus confirms that the hlyII pro-moter belongs to the extended 210 class (since s70 region4.2 interactions with the 235 promoter consensus elementare not required for transcription from the extended 210class promoters) and suggests that s70 region 4.2 is not thetarget of transcription inhibition by HlyIIR. We attempted
(a) (b)Fig. 4. Inhibition of hlyII transcription byHlyIIR in vitro. (a) E. coli RNAP s70 holo-enzyme or B. cereus sA holoenzyme wereused to transcribe from a hlyII promoter-containing DNA fragment in vitro in thepresence or absence of HlyIIR. Productswere resolved by denaturing PAGE andrevealed by autoradiography. (b) E. coli
RNAP s70 holoenzyme or B. cereus sA
holoenzyme were used to transcribe from ahlyII promoter-containing DNA fragmentin vitro. The RNA product was purified andprimer extension using an oligonucleotideannealing at the beginning of hlyII was per-formed. The products were resolved on an8% denaturing gel alongside the productsof a sequencing reaction obtained using thesame primer and the hlyII DNA fragment asa template. An autoradiograph is presented.An approximately 10-fold excess of RNAPand hlyII promoter fragment was used in anin vitro transcription reaction by B. cereus
RNAP to obtain the same amount of primerextension product as that obtained with theE. coli enzyme.
http://mic.sgmjournals.org 3697
Regulator of B. cereus haemolysin II expression
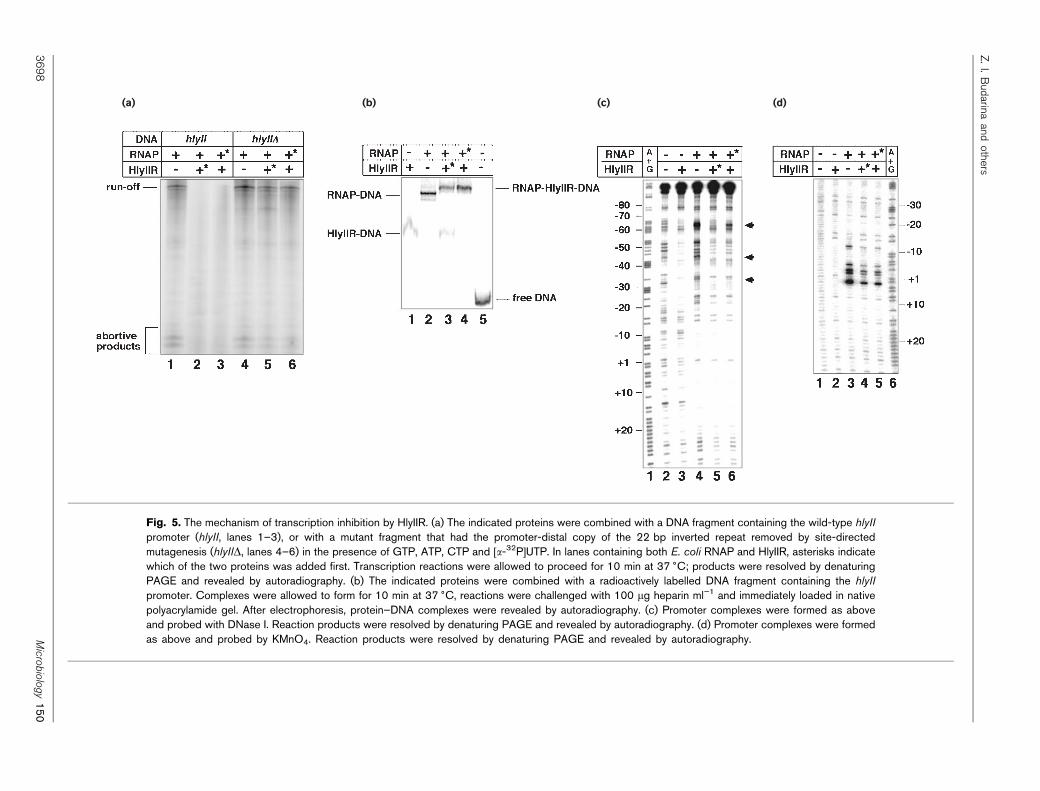
(a) (b) (c) (d)
Fig. 5. The mechanism of transcription inhibition by HlyIIR. (a) The indicated proteins were combined with a DNA fragment containing the wild-type hlyII
promoter (hlyII, lanes 1–3), or with a mutant fragment that had the promoter-distal copy of the 22 bp inverted repeat removed by site-directedmutagenesis (hlyIID, lanes 4–6) in the presence of GTP, ATP, CTP and [a-32P]UTP. In lanes containing both E. coli RNAP and HlyIIR, asterisks indicatewhich of the two proteins was added first. Transcription reactions were allowed to proceed for 10 min at 37 6C; products were resolved by denaturingPAGE and revealed by autoradiography. (b) The indicated proteins were combined with a radioactively labelled DNA fragment containing the hlyII
promoter. Complexes were allowed to form for 10 min at 37 6C, reactions were challenged with 100 mg heparin ml”1 and immediately loaded in nativepolyacrylamide gel. After electrophoresis, protein–DNA complexes were revealed by autoradiography. (c) Promoter complexes were formed as aboveand probed with DNase I. Reaction products were resolved by denaturing PAGE and revealed by autoradiography. (d) Promoter complexes were formedas above and probed by KMnO4. Reaction products were resolved by denaturing PAGE and revealed by autoradiography.
3698
Micro
biology
150
Z.I.Budarina
andothers

to determine the effect of HlyIIR on in vitro transcriptionfrom a hlyII promoter derivative that lacked both copies ofthe inverted repeat. However, such a DNA fragment hadno promoter activity in vitro, despite the fact that theextended 210 promoter element was retained, suggestingthat upstream elements of the hlyII promoter contribute toefficient promoter utilization. Be that as it may, we concludethat HlyIIR acts as a transcription inhibitor in vitro, andthat transcription inhibition is dependent on the presenceof an intact HlyIIR binding site, the 22 bp inverted repeat.
The inhibitory effect of HlyIIR could be due to simpleexclusion of RNAP from the promoter, or inactivation ofRNAP–hlyII promoter complexes. A gel-retardation experi-ment performed in the presence of the DNA competitorheparin revealed that, in the presence of HlyIIR and RNAPs70 holoenzyme, a supershifted complex with electrophore-tic mobility higher than that of either HlyIIR complex(Fig. 5b, lane 1) or RNAP open complex (Fig. 5b, lane 2)was formed (Fig. 5b, lanes 3 and 4). The formation of thesupershifted complex was independent of the order of addi-tion of RNAP or HlyIIR to the hlyII promoter fragment.The result thus indicated the presence of heparin-resistant,transcriptionally inactive ternary complexes containingpromoter DNA, HlyIIR and RNAP. Therefore, transcriptioninhibition by HlyIIR is not a simple consequence of stericexclusion of RNAP from the promoter by bound HlyIIR,since in this case no ternary complexes are expected. Closerinspection of the band-shift gel of Fig. 5b revealed that inthe presence of HlyIIR or RNAP alone, all of the promoterDNA fragment was complexed with proteins (Fig. 5b, lanes1 and 2, respectively). However, when RNAP was added toHlyIIR–DNA complexes, a mixture of supershifted com-plexes and HlyIIR-only complexes was observed (Fig. 5b,lane 3), suggesting that HlyIIR decreases the affinity ofRNAP for promoter DNA or, alternatively, that HlyIIRdestabilizes promoter complexes. We favour the first possi-bility, since when HlyIIR was added to preformed RNAPpromoter complexes, only the supershifted band corres-ponding to the ternary complex was observed (Fig. 5b, lane 4).
The hlyII promoter complexes formed by RNAP in thepresence or absence of HlyIIR were studied by DNase Ifootprinting. The results are presented in Fig. 5c. As canbe seen, HlyIIR and RNAP alone produced distinct DNaseI footprints (Fig. 5c, compare lane 2 with lanes 3 and 4).HlyIIR alone protected DNA from 217 to 260 (Fig. 5c,lane 3), while RNAP alone protected the DNA from posi-tions +20 to 213 (Fig. 5c, lane 4). In addition, RNAPchanged the pattern of promoter DNA digestion by DNaseI further upstream. DNA at around positions 224, 234,244, 254 and, most strikingly, 264 became hypersensitiveto DNase in the presence of RNAP. The extended protec-tion of promoter upstream sequences by RNAP is probablydue to wrapping of the very AT-rich upstream promoterDNA around RNAP (Burns et al., 1999). In the presence ofHlyIIR and RNAP, promoter DNA between positions +20and 213 remained fully protected (Fig. 5c, lanes 5 and 6).
The pattern of upstream promoter DNA protection waschanged and became distinct from those seen in the presenceof either HlyIIR or RNAP alone. The extent of hypersensi-tivity in the upstream promoter DNA decreased in thepresence of HlyIIR. However, in contrast to the situationseen in the presence of HlyIIR alone, no full protection ofupstream promoter DNA was seen in the presence of bothHlyIIR and RNAP. Several quantitative differences betweenthe HlyIIR/RNAP footprint and the RNAP-alone footprintsuggested that HlyIIR was present in these complexes(Fig. 5c, arrows). First, the striking hypersensitive band at264 was either missing (when RNAP was added to theHlyIIR–DNA complex, Fig. 5c, lane 5) or significantlyreduced (when HlyIIR was added to RNAP–promoter com-plexes, Fig. 5c, lane 6) in the presence of both HlyIIR andRNAP. Second, hypersensitive bands at positions 244 and233 were also missing when both proteins were present infootprinting reactions. We take these data, together with theband shift results of Fig. 5b, as an indication that promotercomplexes containing both HlyIIR and RNAP can form onthe hlyII promoter. It appears that, in the ternary complex,contacts made by either HlyIIR or RNAP are different fromcontacts made when either of these proteins is present alone.
The hlyII promoter complexes were also probed by KMnO4,a chemical agent specific for single-stranded thymines(Fig. 5d). As can be seen, and as expected, RNAP, but notHlyIIR, caused the appearance of KMnO4-sensitive bandsat positions +3, 21, 22, 25 and 212 (Fig. 5d, comparelanes 2 and 3). The degree of KMnO4 sensitivity decreased~3?5-fold in the presence of HlyIIR, but the pattern ofsensitive thymines remained the same (Fig. 5d, comparelane 3 with lanes 4 and 5). The result thus suggests thatHlyIIR negatively regulates hlyII transcription by interfer-ing with the formation of catalytically active RNAP openpromoter complexes.
The footprinting results presented above suggest thatHlyIIR may interact with the RNAP holoenzyme, at leastin the context of the hlyII promoter complex. To determinewhether HlyIIR can interact with free RNAP, a native gelanalysis of reactions containing pure HlyIIR, E. coli RNAPcore or s70 holoenzyme, and reactions containing HlyIIRwith either core or s70 holoenzyme was performed (Fig. 6a).As can be seen, HlyIIR caused a change in the electro-phoretic mobility of both the core and the holoenzyme(compare lanes 2 and 4 with lanes 2 and 5, respectively),suggesting an interaction. Analysis of the polypeptide com-position of bands from the native gels revealed that bandsshifted in the presence of HlyIIR contained, in addition tothe expected RNAP subunits, the HlyIIR protein (Fig. 6b,lanes 4 and 6). We therefore conclude that HlyIIR is ableto bind RNAP core and holoenzyme in solution, in theabsence of the hlyII promoter DNA.
DISCUSSION
The principal result of this work is the demonstration thatheterologous expression of B. cereus haemolysin II in E. coli
http://mic.sgmjournals.org 3699
Regulator of B. cereus haemolysin II expression

is negatively regulated by the product of the hlyIIR geneat the level of transcription. We demonstrate that HlyIIRinteracts with a palindromic sequence centred 48 bp up-stream of the hlyII transcription start point. The location ofthe HlyIIR binding site made it likely that HlyIIR wouldexert its negative effects on transcription by steric hindrance;however, this turned out not to be the case, since HlyIIR isable to interact with RNAP, and the resultant binarycomplex forms a ternary complex with hlyII promoterDNA. In vitro footprinting data indicate that the DNAcontacts made by RNAP and HlyIIR in the ternary complexare different from the contacts made when either proteinbinds hlyII promoter DNA on its own. Although ternarycomplexes are heparin resistant, the extent of promoteropening is significantly decreased compared to binaryRNAP–hlyII promoter complexes. Thus, it appears thatHlyIIR interferes with the process of isomerization of theRNAP closed promoter complex to the catalytically activeopen promoter complex. This interference must be due tospecific contacts between HlyIIR and the RNAP core, sinceHlyIIR binds with the same affinity to both core andholoenzymes, suggesting that the s subunit is not involvedin the interaction.
Although our data on hlyII promoter regulation wereobtained in a heterologous E. coli system, we believe thatthe hlyII promoter identified in this study is also active inB. cereus, since, in vitro, B. cereus RNAP initiates hlyIItranscription at the same site as E. coli RNAP, indicatingthat the relative positions of HlyIIR and RNAP bound to thehlyII promoter are the same in E. coli and B. cereus. Sinevet al. (1993) and Budarina et al. (1994) reported that in bothB. cereus and E. coli cells carrying the pUJ1 plasmid (con-tains both hlyII and hlyIIR), haemolysin II production wasactivated at the late logarithmic stage of growth. While thereason(s) for this is not known, it may have to do either
with negative regulation of hlyIIR expression or with theregulation of HlyIIR binding to DNA by low-molecular-massligands whose nature is yet to be determined.
Analyses of genomic sequences of B. anthracis demonstratethat sequences homologous to both hlyII and hlyIIR arepresent, as expected from the very close similarity betweenB. cereus and B. anthracis. However, unlike B. cereus, theanthrax bacterium is not generally known to producehaemolysins. Sequence analysis of B. anthracis DNA givessome clues to this apparent paradox. While the B. anthracisA2012 strain hlyII sequence is identical to the B. cereusVKM-B771 sequence (Read et al., 2002), an A correspond-ing to B. cereus hlyII position 232 is missing from theB. anthracis hlyII gene. In addition, a G at hlyII position1116 is substituted for an A. The frame shift resulting fromthe absence of an A at position 232 likely renders B. anthracishaemolysin II non-functional. In contrast, while the hlyIIgenes from B. cereus strains ATCC 14579 (Ivanova et al.,2003) and BGSC 6A5 (Miles et al., 2002) have accumulateda significant number of point substitutions compared tothe VKM-B771 sequence, they have maintained an intactreading frame which, at least in the case of BGSC 6A5, isknown to code for a functional haemolysin (Miles et al.,2002).
The relative position of the the hlyII and hlyIIR genes, andthe palindromic HlyIIR binding sites in front of hlyII isconserved. This allows some of the errors in annotationwhich arise due to sequence similarities between the proteinsequences of HlyII and CytK haemolysins to be corrected.Thus, in the recently published genome of B. cereus ATCC10987 (Rasko et al., 2004) a gene annotated as CytK isfollowed by a gene encoding a HlyIIR protein. The haemo-lysin gene is preceded by a palindromic sequence similar tothe HlyIIR binding site. Since CytK genes are not followedby genes encoding TetR-like regulators and are not precededby HlyIIR binding sites, we suggest that the B. cereus ATCC10987 cytotoxin K gene in fact encodes HlyII. As is also thecase with other B. cereus hlyII genes, the ATCC 10987 geneencodes a full-sized protein.
Interestingly, the ORF of the hlyII gene (annotated as ana-haemolysin precursor) is also interrupted in B. cereusG9241 (Hoffmaster et al., 2004), the only sequenced B.cereus strain which is associated with an illness resemblinginhalation anthrax and which carries a circular plasmid verysimilar to B. anthracis toxin-encoding plasmid pXO1. Theresult may indicate that some aspects of HlyII functionmay be incompatible with anthrax toxin function. In thisregard, the presence of frame-shift mutations in hlyIIgenes from B. anthracis, but not in those from B. cereus, isreminiscent of the previously described situation with theplcR gene, which encodes the major positive regulator ofpathogenicity factors, including haemolysins, in B. cereus.The ORF of the plcR gene is interrupted in B. anthracis,which explains why B. anthracis does not produce patho-genicity proteins characteristic of B. cereus. Inspection ofthe DNA sequence upstream of hlyII and hlyIIR reveals no
(a) (b)
Fig. 6. HlyIIR binds RNAP core and holoenzymes. The indi-cated proteins were combined and resolved by native 4–15%Phastgel. The gel was stained (a), bands were excised andpolypeptides were resolved by SDS-PAGE and revealed byCoomassie staining (b).
3700 Microbiology 150
Z. I. Budarina and others

PlcR-binding sites, suggesting that haemolysin II produc-tion is not subject to direct regulation by PlcR.
The sequence of the B. anthracis A2012 hlyIIR gene isidentical to that of B. cereus hlyIIR, except for a single G toA substitution that changes Gly2 in B. cereus HlyIIR to Gluin the B. anthracis protein. Further, the 22 bp palindromicHlyIIR operator is also present in front of B. anthracis hlyII.However, a single G to A change at the tenth position of thepromoter-distal copy of the inverted repeat has occurred. Acorresponding C to T change has occurred in the promoter-proximal copy of the B. anthracis repeat, thus preservingthe perfect 22 bp palindrome, and suggesting that this DNAsite continues to play a regulatory role. It is formally possiblethat a single Gly to Glu amino acid substitution changes theoperator specificity of B. anthracis HlyIIR. It is not clearwhy B. anthracis would keep a non-functional haemolysinII gene as well as a gene encoding an apparently functionalregulator protein. Reports on the isolation of haemolyticB. anthracis strains from several natural sources (Mikshiset al., 1999) indicate that haemolysis in B. anthracis may bemore widespread than previously anticipated. A systematiccomparison of hlyII sequences from various anthracoidbacilli strains and correlation with the haemolytic activityof these strains may thus be warranted.
ACKNOWLEDGEMENTS
This work was supported by NIH RO1 grant GM59295 and an NRC/DTRA exchange fellowship (K. S.) and the Russian Foundation forBasic Research (Project 03-04-48623) (A. S.). The work of D. V. N. wassupported by a Russian Science Support Foundation grant.
REFERENCES
Ash, C., Farrow, J. A., Dorsch, M., Stackebrandt, E. & Collins, M. D.(1991). Comparative analysis of Bacillus anthracis, Bacillus cereus,and related species on the basis of reverse transcriptase sequencing of16S rRNA. Int J Syst Bacteriol 41, 343–346.
Baida, G., Budarina, Z. I., Kuzmin, N. P. & Solonin, A. S. (1999).Complete nucleotide sequence and molecular characterization ofhemolysin II gene from Bacillus cereus. FEMS Microbiol Lett 180,7–14.
Barne, K. A., Bown, J. A., Busby, S. J. & Minchin, S. D. (1997).Region 2?5 of the Escherichia coli RNA polymerase s70 subunit isresponsible for the recognition of the ‘extended-10’ motif atpromoters. EMBO J 16, 4034–4040.
Budarina, Z. I., Sinev, M. A., Mayorov, S. G., Tomashevski, A. Y.,Shmelev, I. V. & Kuzmin, N. P. (1994). Hemolysin II is morecharacteristic of Bacillus thuringiensis than Bacillus cereus. ArchMicrobiol 161, 252–257.
Burns, H. D., Ishihama, A. & Minchin, S. D. (1999). Open complexformation during transcription initiation at the Escherichia coli galP1promoter: the role of the RNA polymerase a subunit at promoterslacking an UP-element. Nucleic Acids Res 27, 2051–2056.
Chakrabarti, S. K. & Misra, T. K. (2000). SarA represses agr operonexpression in a purified in vitro Staphylococcus aureus transcriptionsystem. J Bacteriol 182, 5893–5897.
Gaal, T., Ross, W., Estrem, S. T., Nguyen, L. H., Burgess, R. R. &Gourse, R. L. (2001). Promoter recognition and discrimination byEsS RNA polymerase. Mol Microbiol 42, 939–954.
Gouaux, E. (1998). a-Hemolysin from Staphylococcus aureus: an arche-
type of b-barrel, channel-forming toxins. J Struct Biol 121, 110–122.
Granum, P. E. (1997). Bacillus cereus. In Fundamentals in Food
Microbiology, pp. 327–336. Edited by M. Doyle, L. Beuchat &T. Montville. Washington, DC: American Society for Microbiology.
Hoffmaster, A. R., Ravel, J., Rasko, D. A. & 19 other authors (2004).Identification of anthrax toxin genes in a Bacillus cereus associatedwith an illness resembling inhalation anthrax. Proc Natl Acad Sci
U S A 101, 8449–8454.
Ivanova, N., Sorokin, A., Anderson, I. & 20 other authors (2003).Genome sequence of Bacillus cereus and comparative analysis withBacillus anthracis. Nature 423, 87–91.
Kashlev, M., Nudler, E., Severinov, K., Borukhov, S.,Komissarova, N. & Goldfarb, A. (1996). Histidine-tagged RNApolymerase of Escherichia coli and transcription in solid phase.
Methods Enzymol 274, 326–334.
Lund, T., De Buyser, M. L. & Granum, P. E. (2000). A new cytotoxin
from Bacillus cereus that may cause necrotic enteritis. Mol Microbiol38, 254–261.
Mikshis, N. I., Eremin, S. A. & Bolotnikova, M. F. (1999). Correlationof the virulence of Bacillus anthracis with expression of signs, codedfor by chromosomal genes. Molecular Genetics, Microbiology and
Virology (English translation of Molekulyarnaya Genetika Microbio-logija i Virusologija) 4, 25–28.
Miles, G., Bayley, H. & Cheley, S. (2002). Properties of Bacillus cereushemolysin II: a heptameric transmembrane pore. Protein Sci 11,
1813–1824.
Minakhin, L., Niedziela-Majka, A., Kuznedelov, K., Adelman, K.,Urbauer, J., Heyduk, T. & Severinov, K. (2003). Interaction of T4
AsiA with its target sites in the RNA polymerase s70 subunit leadsto distinct and opposite effects on transcription. J Mol Biol 326,
679–690.
Namwat, W., Lee, C. K., Kinoshita, H., Yamada, Y. & Nihira, T.(2001). Identification of the varR gene as a transcriptional regulatorof virginiamycin S resistance in Streptomyces virginiae. J Bacteriol
183, 2025–2031.
Nikolaev, I., Epishin, S., Zakharova, E., Kotenko, S. & Vinetskii, I.(1992). Molecular cloning of the gene for secreted beta-galactosidase
of the filamentous fungus Penicillium canescens. Molecular Biology(EnglishTranslation ofMolekulyarnayaBiologiya (Moscow)) 26, 869–875.
Rasko, D. A. J., Ravel, O. A., Okstad, E. & 12 other authors (2004).The genome sequence of Bacillus cereus ATCC 10987 reveals
metabolic adaptations and a large plasmid related to Bacillus
anthracis pXO1. Nucleic Acids Res 32, 977–988.
Read, T. D., Salzberg, S. L., Pop, M. & 10 other authors (2002).Comparative genome sequencing for discovery of novel polymor-phisms in Bacillus anthracis. Science 296, 2028–2033.
Recsei, P., Kreiswirth, B., O’Reilly, M., Schlievert, P., Gruss, A. &Novick, R. P. (1986). Regulation of exoprotein gene expression in
Staphylococcus aureus by agr. Mol Gen Genet 202, 58–61.
Severinova, E., Severinov, K. & Darst, S. A. (1998). Inhibition ofEscherichia coli RNA polymerase by T4 AsiA. J Mol Biol 279, 9–18.
Sinev, M. A., Budarina, Z. I., Gavrilenko, I. V., Tomashevskii, A. I. &Kuzmin, N. P. (1993). Evidence of the existence of hemolysin II from
Bacillus cereus: cloning the genetic determinant of hemolysin II. Mol
Biol (Russ) 27, 1218–1229.
Zabeau, M. & Stanley, K. K. (1982). Enhanced expression of cro-beta-
galactosidase fusion proteins under the control of the PR promoter ofbacteriophage lambda. EMBO J 1, 1217–1224.
http://mic.sgmjournals.org 3701
Regulator of B. cereus haemolysin II expression




















