
A Heteroleptic Ferrous Complex with Mesoionic Bis(1,2,3-triazol-5-ylidene) Ligands: Taming the MLCT...
12
& Iron Carbene Photochemistry | Very Important Paper| A Heteroleptic Ferrous Complex with Mesoionic Bis(1,2,3-triazol-5- ylidene) Ligands: Taming the MLCT Excited State of Iron(II) Yizhu Liu, [a, b] Kasper S. Kjær, [a, c] Lisa A. Fredin, [d] Pavel ChƁbera, [a] Tobias Harlang, [a] Sophie E. Canton, [e] Sven Lidin, [b] Jianxin Zhang, [b] Reiner Lomoth, [f] Karl-Erik Bergquist, [b] Petter Persson, [d] Kenneth WȨrnmark,* [b] and Villy Sundstrçm* [a] Abstract: Strongly s-donating N-heterocyclic carbenes (NHCs) have revived research interest in the catalytic chemis- try of iron, and are now also starting to bring the photo- chemistry and photophysics of this abundant element into a new era. In this work, a heteroleptic Fe II complex (1) was synthesized based on sequentially furnishing the Fe II center with the benchmark 2,2’-bipyridine (bpy) ligand and the more strongly s-donating mesoionic ligand, 4,4’-bis(1,2,3-tri- azol-5-ylidene) (btz). Complex 1 was comprehensively char- acterized by electrochemistry, static and ultrafast spectrosco- py, and quantum chemical calculations and compared to [Fe(bpy) 3 ](PF 6 ) 2 and (TBA) 2 [Fe(bpy)(CN) 4 ]. Heteroleptic com- plex 1 extends the absorption spectrum towards longer wavelengths compared to a previously synthesized homo- leptic Fe II NHC complex. The combination of the mesoionic nature of btz and the heteroleptic structure effectively desta- bilizes the metal-centered (MC) states relative to the triplet metal-to-ligand charge transfer ( 3 MLCT) state in 1, rendering it a lifetime of 13 ps, the longest to date of a photochemical- ly stable Fe II complex. Deactivation of the 3 MLCT state is pro- posed to proceed via the 3 MC state that strongly couples with the singlet ground state. Introduction Manipulation of the metal–ligand interaction is central to tran- sition-metal (TM) chemistry. It directly influences the electronic density on the metal center and the energy level ordering of different states of the complex, thus playing a critical role in determining a wide spectrum of fundamental properties. [1] A prominent example is the photophysics and photochemistry of TM complexes, in which the metal-to-ligand charge transfer (MLCT), metal-centered (MC), and ligand-centered (LC) states can be substantially manipulated relative to each other depending on the geometric configuration of the surrounding ligands and their s-donor/p-acceptor strengths. [2] The first-row TM elements are abundant and environmentally benign, but intrinsically possess much less ligand-field splitting compared to their second- and third-row congeners. For the former, the coordination environment is of special importance, as the above-mentioned states can be very sensitively tuned depend- ing on the variation of the ligand-field strength imposed by a specific type of ligation. This opens up possibilities to manipulate the way the molecules absorb and dissipate the photon energy. The octahedral polyimine complexes of d 6 Fe II , for example, have been most commonly characterized by the low-lying quintet MC state ( 5 MC), [3] which is a direct result of the smaller ligand-field splitting of Fe [4] compared to its second- and third- row congeners. [1] Under these circumstances, the d electrons overcome the energy gap between the t 2g and e g * orbitals to avoid the repulsion of pairing, yielding the high-spin electronic configuration that may be either the ground state (GS) or a state accessible from the singlet GS through thermal fluctu- ation or photophysical cascades following photoexcitation, the latter being termed as light-induced excited-state spin trapping (LIESST). [3a] While such properties have potential applications in displays or memory storage devices, [5] they generate a series of unfavorable features in the context of [a] Dr. Y. Liu, Dr. K.S. Kjær, Dr. P. ChƁbera, T. Harlang, Prof. V. Sundstrçm Department of Chemical Physics, Lund University P.O. Box 124, 22100 Lund (Sweden) E-mail: [email protected] [b] Dr. Y. Liu, Prof. S. Lidin, Dr. J. Zhang, + Dr. K.-E. Bergquist, Prof. K. WȨrnmark Centre for Analysis and Synthesis Department of Chemistry, Lund University P.O. Box 124, 22100 Lund (Sweden) E-mail : [email protected] [c] Dr. K. S. Kjær Department of Physics, Technical University of Denmark 2800 Kongens Lyngby (Denmark) [d] Dr. L. A. Fredin, Prof. P. Persson Theoretical Chemistry Division, Lund University P.O. Box 124, 22100 Lund (Sweden) [e] Dr. S. E. Canton Department of Synchrotron Instrumentation, Lund University P.O. Box 124, 22100 Lund (Sweden) [f] Prof. R. Lomoth Department of Chemistry—ĸngstrçm Laboratory, Uppsala University P.O. Box 523, 75120 Uppsala (Sweden) [ + ] Current address: School of Environment and Chemical Engineering Tianjin Polytechnic University Tianjin, 300087 (China) Supporting information for this article (including experimental details) is available on the WWW under http://dx.doi.org/10.1002/chem.201405184. Chem. Eur. J. 2015, 21, 3628 – 3639 # 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 3628 Full Paper DOI: 10.1002/chem.201405184
Transcript of A Heteroleptic Ferrous Complex with Mesoionic Bis(1,2,3-triazol-5-ylidene) Ligands: Taming the MLCT...

& Iron Carbene Photochemistry | Very Important Paper |
A Heteroleptic Ferrous Complex with Mesoionic Bis(1,2,3-triazol-5-ylidene) Ligands: Taming the MLCT Excited State of Iron(II)
Yizhu Liu,[a, b] Kasper S. Kjær,[a, c] Lisa A. Fredin,[d] Pavel Ch�bera,[a] Tobias Harlang,[a]
Sophie E. Canton,[e] Sven Lidin,[b] Jianxin Zhang,[b] Reiner Lomoth,[f] Karl-Erik Bergquist,[b]
Petter Persson,[d] Kenneth W�rnmark,*[b] and Villy Sundstrçm*[a]
Abstract: Strongly s-donating N-heterocyclic carbenes(NHCs) have revived research interest in the catalytic chemis-try of iron, and are now also starting to bring the photo-chemistry and photophysics of this abundant element intoa new era. In this work, a heteroleptic FeII complex (1) wassynthesized based on sequentially furnishing the FeII centerwith the benchmark 2,2’-bipyridine (bpy) ligand and themore strongly s-donating mesoionic ligand, 4,4’-bis(1,2,3-tri-azol-5-ylidene) (btz). Complex 1 was comprehensively char-acterized by electrochemistry, static and ultrafast spectrosco-py, and quantum chemical calculations and compared to
[Fe(bpy)3](PF6)2 and (TBA)2[Fe(bpy)(CN)4] . Heteroleptic com-plex 1 extends the absorption spectrum towards longerwavelengths compared to a previously synthesized homo-leptic FeII NHC complex. The combination of the mesoionicnature of btz and the heteroleptic structure effectively desta-bilizes the metal-centered (MC) states relative to the tripletmetal-to-ligand charge transfer (3MLCT) state in 1, renderingit a lifetime of 13 ps, the longest to date of a photochemical-ly stable FeII complex. Deactivation of the 3MLCT state is pro-posed to proceed via the 3MC state that strongly coupleswith the singlet ground state.
Introduction
Manipulation of the metal–ligand interaction is central to tran-sition-metal (TM) chemistry. It directly influences the electronicdensity on the metal center and the energy level ordering ofdifferent states of the complex, thus playing a critical role in
determining a wide spectrum of fundamental properties.[1] Aprominent example is the photophysics and photochemistry ofTM complexes, in which the metal-to-ligand charge transfer(MLCT), metal-centered (MC), and ligand-centered (LC) statescan be substantially manipulated relative to each otherdepending on the geometric configuration of the surroundingligands and their s-donor/p-acceptor strengths.[2] The first-rowTM elements are abundant and environmentally benign, butintrinsically possess much less ligand-field splitting comparedto their second- and third-row congeners. For the former, thecoordination environment is of special importance, as theabove-mentioned states can be very sensitively tuned depend-ing on the variation of the ligand-field strength imposed bya specific type of ligation. This opens up possibilities tomanipulate the way the molecules absorb and dissipate thephoton energy.
The octahedral polyimine complexes of d6 FeII, for example,have been most commonly characterized by the low-lyingquintet MC state (5MC),[3] which is a direct result of the smallerligand-field splitting of Fe[4] compared to its second- and third-row congeners.[1] Under these circumstances, the d electronsovercome the energy gap between the t2g and eg* orbitals toavoid the repulsion of pairing, yielding the high-spin electronicconfiguration that may be either the ground state (GS) ora state accessible from the singlet GS through thermal fluctu-ation or photophysical cascades following photoexcitation, thelatter being termed as light-induced excited-state spintrapping (LIESST).[3a] While such properties have potentialapplications in displays or memory storage devices,[5] theygenerate a series of unfavorable features in the context of
[a] Dr. Y. Liu, Dr. K. S. Kjær, Dr. P. Ch�bera, T. Harlang, Prof. V. SundstrçmDepartment of Chemical Physics, Lund UniversityP.O. Box 124, 22100 Lund (Sweden)E-mail : [email protected]
[b] Dr. Y. Liu, Prof. S. Lidin, Dr. J. Zhang,+ Dr. K.-E. Bergquist, Prof. K. W�rnmarkCentre for Analysis and SynthesisDepartment of Chemistry, Lund UniversityP.O. Box 124, 22100 Lund (Sweden)E-mail : [email protected]
[c] Dr. K. S. KjærDepartment of Physics, Technical University of Denmark2800 Kongens Lyngby (Denmark)
[d] Dr. L. A. Fredin, Prof. P. PerssonTheoretical Chemistry Division, Lund UniversityP.O. Box 124, 22100 Lund (Sweden)
[e] Dr. S. E. CantonDepartment of Synchrotron Instrumentation, Lund UniversityP.O. Box 124, 22100 Lund (Sweden)
[f] Prof. R. LomothDepartment of Chemistry—�ngstrçm Laboratory, Uppsala UniversityP.O. Box 523, 75120 Uppsala (Sweden)
[+] Current address :School of Environment and Chemical EngineeringTianjin Polytechnic UniversityTianjin, 300087 (China)
Supporting information for this article (including experimental details) isavailable on the WWW under http://dx.doi.org/10.1002/chem.201405184.
Chem. Eur. J. 2015, 21, 3628 – 3639 � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3628
Full PaperDOI: 10.1002/chem.201405184

solar energy harvesting, such as ligand lability and ultra-short-lived photoactive MLCT manifolds.[6] Recently, however, wehave demonstrated that by incorporating strongly s-donatingN-heterocyclic carbene (NHC) ligands, the lifetime of the3MLCT excited state of FeII complexes can be extended by twoorders of magnitude from about 100 fs to 9 ps in photochemi-cally stable FeII complexes.[7] This was rationalized by the intactbonding between the FeII center and the NHC carbon that ef-fectively destabilizes the 3MC and 5MC states.[8] This result wasrewarding as a long-lived 3MLCT state is the foundation ofmany photochemical applications, and a FeII complex with anextended lifetime would allow this earth-abundant, inexpen-sive, and environmentally benign metal to be utilized for thispurpose. Therefore, we are encouraged to further exploit thelimits of such interaction using strong-field ligands to achieveeven longer-lived 3MLCT states.
Iron NHC chemistry has been extensively developed in thefield of catalysis[9] since its first report in 2000.[10] Therein thesuperiority of NHC ligands has been manifested by increasingthe electron density on Fe and thus its reactivity towards or-ganic substrates.[9a] Interestingly, the electron density also dic-tates the excited-state properties of FeII complexes, making thelittle explored photophysics of Fe NHC complexes promising.[7]
While in our previous report, bis(tridentate) FeII NHC com-plexes were investigated in comparison to [Fe(tpy)2](PF6)2
(tpy = 2,2’:6’:2“-terpyridine),[7] herein we explore the other stan-dard octahedral ligand approach, namely the tris(bidentate)configuration. In general, bidentate ligands allow for morefreedom towards ideal octahedral geometry, and thereforea stronger ligand field could be expected than with tridentateligands, as demonstrated by [RuII(bpy)3](PF6)2 (bpy = 2,2’-bipyri-dine) versus [RuII(tpy)2](PF6)2.[11] Furthermore, by developingheteroleptic tris(bidentate) structures, one can selectivelychoose the number of s-donating or p-accepting ligation sites,thus allowing more fine-tuning of the electronic states andpromoting versatility and functionality. To realize such a strat-egy, bis(NHC) ligands that can be regarded as analogues of thebenchmark bpy[12] and 1,10-phenanthroline (phen)[13] ligandsare considered for the constructing of heteroleptic FeII com-plexes. In this study, we focus on the 4,4’-bis(1,2,3-triazol-5-yli-denes) (btz) ligand. The carbene moiety herein is mesoionic innature[14] and is believed to be even more strongly s-donatingcompared to normal NHC ligands owing to the formal nega-tive charge on the carbene carbon in the classical drawings ofthe resonance structure.[12b, 15] The 1,2,3-triazol-5-ylidene moietyhas been previously coupled with heterocycles to construct(C^N)[16] and (C^C)[17] heterobidentate complexes. However,there are few reports on homobidentate complexes incorpo-rating two triazolylidene moieties in one ligand (btz).[12b, d]
Therefore, we aimed for a heteroleptic complex 1 (Scheme 1)containing one bpy ligand and two btz ligands. The bpyligand ensures the presence of MLCT transitions in this com-plex while the btz ligands are intended to tune the electronicproperties compared to traditional FeII polyimine complexes.To the best of our knowledge, this is the first six-coordinate oc-tahedral TM complex based on the btz-type ligands. As a NHCanalogue of the benchmark complex [Fe(bpy)3](PF6)2, it is the
first heteroleptic FeII NHC complex bearing spatially separatedfour NHC sites and a bpy ligand. It is also the first heterolepticFeII NHC complex of which detailed excited-state photophysicsis comprehensively investigated and reported.
Interestingly, Gros et al.[18] also very recently reported hetero-leptic FeII NHC complexes comprised of a tpy derivative anda NHC ligand that is very close to our previous report.[7] How-ever, the work of Gros et al. was motivated by extending theabsorption spectrum of the series of complexes and there wasno investigation of the excited-state dynamics. The difficulty ofstudying the excited-state photophysics of heteroleptic com-plexes lies in the possible multiple isoenergetic MLCT states.[19]
In relation to complex 1, while the mesoionic nature shouldenhance the s-donor strength of the btz ligand, the increasednumber of N atoms may lower its p* energy compared tonormal NHC ligands, so that it could lie at a similar level as pol-ypyridines.[16e] However, owing to the lack of related complexesas stated above, there is no reference to whether the btz-typeligands are electrochemically active or photoredox-active. Toaddress this issue, comparison was made to (nBu4N)2[Fe(bpy)-(CN)4] , complex 2, which is also shown in Scheme 1. First of all,the ancillary cyanide ligands are also strong-field ligands in tra-ditional transition-metal chemistry.[1] More importantly, the3MLCT state can only be localized on bpy as it is the only sitewithin the molecule to accommodate the excited electron ina MLCT state.
Results and Discussion
Synthesis of complex 1
The synthesis of the heteroleptic complex 1 is shown inScheme 2 and described in detail in the Supporting Informa-tion. Generally speaking, the synthesis of heteroleptic FeII six-coordinate complexes is not as straightforward as for their RuII
congeners, especially when the ligand bears considerabledonor strength that may eventually induce the disproportiona-tion of the coordinatively unsaturated intermediates.[22] Thistendency can be remedied by kinetic barriers for the formationof homoleptic complexes,[23] or if the partially coordinated syn-thetic intermediates can be readily isolated due to solubilityissues.[24] In the recent work of Gros et al. on a heteroleptic FeII
NHC complex, Fe(tpy)Cl3 was employed as the intermediate.[18]
However, the employment of the NHC ligand generated in situas a reductant to bring FeIII back to FeII significantly limited theyield. In our approach, the heteroleptic FeII structure could be
Scheme 1. Chemical structures of complexes 1 and 2.
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3629
Full Paper

achieved by directly ligating the FeII center with first bpy andthen the btz ligand generated in situ, by the isolation ofFe(bpy)Cl2.[20, 25] This is also different from the not-executed op-posite approach, namely assembling the NHC first and theother ancillary ligand later, which would either have involvedthe air-sensitive intermediate FeCl2(PPh3)2
[26] or be limited bythe electron stoichiometry (two electrons per Fe for two NHCsites) based on an electrochemical method.[27] Before success-fully obtaining complex 1 bearing the p-tolyl-pendants, variousattempts were performed to utilize 1-alkyl-substituted btz-typeligands with both primary (n-octyl) and secondary (3-pentyl)alkyl substituents, which are sufficiently large to stabilize theazide intermediate in the click chemistry (Scheme 2) but steri-cally allow the realization of the six-coordinate structure. Un-fortunately, the resulting complexes were not stable enough.They existed transiently after isolation of the crude productand decomposed gradually during the workup and could onlybe characterized by mass spectrometry. In contrast, complex 1is robust. It precipitated from the reaction mixture as a hexa-fluorophosphate (PF6
�) salt and just filtration and washingwere sufficient to obtain the analytically pure compound. Sucha discrepancy could be due to the additional stabilization ofthe whole coordination sphere introduced by the rigid p-tolylgroup, which makes the two btz ligands like clamps, with thepossibility of holding each other as well as the third bpyligand through p–p interactions. It should be noted thatduring the characterization stage we observed temperature-and concentration-dependent peak broadening in the 1H NMRspectra of complex 1, which is not completely understood yet.However, the 1H NMR spectrum at a concentration below1 mm at room temperature iswell-defined. Therefore, NMRspectra (see the Supporting In-formation) were taken at 1 mm
and the concentration of the so-lution samples used in all of themeasurements described belowwere also restricted to be belowthis threshold.
Efforts to grow crystals suita-ble for X-ray diffraction analysis
failed by all means for complex 1. However, after changing thecounter ion from PF6
� to Br� , high-quality crystals could bereadily obtained by slow diffusion of diethyl ether into the cor-responding methanolic solution of the bromide complex (1 Br).Figure 1 shows the resulting X-ray structure of the coordina-tion cation (see the Supporting Information for details). Thecomplex adopts a distorted octahedral configuration as ex-pected. The p-tolyl N-substituents of the two btz ligands stayin close to perpendicular conformation relative to the btzplanes, so that they behave like intertwining clamps, holdingthe whole coordination sphere together. For each btz ligand,one p-tolyl group is almost parallel to and vertically overlap-ping the cis-pyridine rings of the bpy ligand. The interplanardistances fall in the range of 3.3–3.6 �, indeed suggesting p–p
interactions between the aromatic planes. The other p-tolylgroup is on top of the cis-triazolylidene ring of the other btzligand at a slightly tilted angle to avoid the steric hindrancecoming from the other p-tolyl group on the latter. This demon-strates that the p-tolyl group is of modest size so as not tocongest the coordination sphere.
The key structural parameters of [Fe(bpy)3](PF6)2[28] and com-
plexes 1 and 2[29] are listed in Table 1. The bonding betweenthe Fe center and bpy ligand is almost identical for 1 and 2 interms of the Fe�N bond distances and the N-Fe-N bite angle.Owing to the stronger trans influence of btz as a stronger-fieldligand compared to bpy, the bpy ligand in 1 is slightly pushedaway from the Fe center compared to in [Fe(bpy)3](PF6)2, sothat longer Fe�N bond distances and smaller N-Fe-N biteangles are seen. In 1, the Fe�C bonds that are trans to the pyr-idine rings are slightly shorter by 0.03–0.05 � than those transto the triazolylidene rings owing to the larger trans influenceof the later, giving rise to nonequivalent halves within each btzligand. A similar situation is seen in 2 as CN� is also a stronger-
Figure 1. X-ray crystal structure of complex 1 Br.[48] Ellipsoids are set at 30 %probability; solvent molecules, counterions, and hydrogen atoms areomitted for clarity.Scheme 2. Synthetic pathways for complex 1. a) Ethanol, 10 equiv FeCl2,
60 8C,[20] 82 % based on bpy; b) K2CO3, CuSO4, Na ascorbate, pyridine,tBuOH/H2O, RT,[21] 70 %; c) 1) MeOTf, ClCH2CH2Cl, �78 8C to RT to 100 8C,[12d]
2) NH4PF6(aq.), 85 %; d) tBuOK, THF, �78 8C to RT, 45 %.
Table 1. Selected structural parameters of [Fe(bpy)3](PF6)2, 1, and 2.
rFe–N
[�]rFe–Ccis
[�]rFe–Ctrans
[�]N-Fe-N[8]
Ccis-Fe-Ctrans
[8]N-Fe-Ctrans
[8]Ccis-Fe-Ccis
[8]
[Fe(bpy)3](PF6)2[a] 1.97 81.8
1 1.99, 2.0 1.99, 2.02 1,96, 1.97 80.5 79.3, 80.0 172.7, 178.1 172.62[b] 1.99, 2.0 1.93, 1.94 1.89, 1.91 80.6 88.4–91.1 174.2, 175.6 178.3
[a] The structural data of [Fe(bpy)3](PF6)2 are taken from reference [28] . [b] The structural data of 2 are takenfrom reference [29] . Potassium (K+) instead of tetra-n-butylammonium (TBA+) was used as the counter-iontherein.
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3630
Full Paper

field ligand than bpy. However, without the structural constrainimposed by the conjugated system as in the btz ligands in 1,the cyanide ligands in 2 can be freely arranged around the Fecenter. Therefore, the Fe�C bonds in the latter are generallyshorter by 0.06–0.08 � compared to those in 1, and the C-Fe-C“bite angles”, if termed likewise, are very close to 908 incomparison with those of about 808 in 1.
Electrochemistry and steady-state absorption spectroscopy
Cyclic voltammetry (CV; Figure 2) was performed in acetonitrile(MeCN) to evaluate the energy levels of the three compounds.The key parameters are listed in Table 2 for comparison. In thecathodic region, [Fe(bpy)3](PF6)2 shows a one-electron reversi-ble redox process at + 0.68 V vs. the ferrocenium/ferrocene(Fc+/Fc) redox couple and is assigned to the oxidation of theFeII center.[30] The potential of this process is drastically shifteddown to �0.35 V and �0.63 V vs. Fc+/Fc in 1 and 2, respective-ly, the latter in excellent agreement with a previous report.[31]
Considering the structural analogy among these complexes, itis evident that substitution of two bpy ligands for C-based li-gands significantly altered the electronic properties of the FeII
center. A similar cathodic shift was observed for RuIII/II withboth triazolylidene-containing[16c] and CN�[32] ligands. A directcomparison between the two is, however, not possible owingto the more intimate interaction between the Fe center andthe donating carbon atoms in 2 in terms of the shorter Fe�Cbond distances and the closer-to-perfect octahedral geometry.Although it is tempting to correlate the more energetic FeII
center with the superior donor strength of the C-based li-gands, the oxidation of the FeII center is nonetheless formallyremoving an electron from the dp (t2g) orbital, which in prin-
ciple do not interact with the ligand s-orbitals[33] and thusshould not necessarily be destabilized by the latter. In thisregard, the indeed widely observed cathodic shift of the metaloxidation potential upon coordination with NHC ligands,[16c, e, 34]
including our previous[7] and present studies, may point toa synergistic effect of the poor p-accepting ability of NHC anda non-perfect octahedricity that attenuates the dpM–p*L
overlap.[4]
In concert with the negatively shifted oxidation potential ofthe Fe center, the energy levels of the bpy ligands are alsodestabilized in the sequence [Fe(bpy)3](PF6)2 to 1 to 2. In theanodic region, [Fe(bpy)3](PF6)2 shows two closely separatedone-electron reversible redox processes at �1.75 and �1.94 Vvs. Fc+/Fc, which can be ascribed to the sequential reductionof two bpy ligands in the tris(bpy) complexes of d6 transitionmetals.[30, 32] In complex 1, ligand-based reduction happens ata much more negative potential. It is essentially irreversible,which has been similarly observed for other related NHC-con-taining complexes.[7, 34b] Differential pulse voltammetry (DPV)was carried out to help to resolve the potentials of the ligand-based reduction processes, and the result is shown in the Sup-porting Information, Figure S12. Three closely spaced reductionprocesses are found at �2.28, �2.42, and �2.64 V vs. Fc+/Fc,respectively. Although an anodic shift of more than 500 mV isseen for the lowest-energy ligand-based reduction, this is toa lesser degree than observed for its FeIII/II oxidation. The over-all result is therefore a decreased MLCT transition energy, con-sistent with the red-shift of the lowest-energy MLCT bandfrom 520 nm of [Fe(bpy)3](PF6)2 to 609 nm of 1. This improvesthe overlap with the solar spectrum compared to our first-gen-eration FeII NHC complexes (Figure 3 and Table 2).[7] In com-
Figure 2. Cyclic voltammograms of [Fe(bpy)3](PF6)2 (L.), 1 (c), and 2 (d)in deaerated MeCN.
Table 2. Redox potentials and spectroscopic parameters of [Fe(bpy)3](PF6)2, 1, and 2 in MeCN.
Eox[a] [V] Ered1
[a] [V] Ered2[a] [V] lmax [nm] (e [103 L mol�1 cm�1])
[Fe(bpy)3](PF6)2 + 0.68 �1.75 �1.94 248 (27.5), 298 (62.2), 349 (6.03), 486 (6.88, sh), 520 (7.98)1 �0.35 �2.28[b] �2.42[b] 243 (23.1, sh), 300 (29.5), 432 (5.08), 609 (3.26)2 �0.63 �2.47[c] – 244 (15.5), 298 (14.9), 438 (5.51), 678 (3.32[d))
[a] Potential values are reported vs. the Fc+/Fc redox couple. [b] Irreversible peaks. [c] Quasi-reversible peak. [d] From reference [35] .
Figure 3. a) The steady-state UV/Vis absorption spectra of [Fe(bpy)3](PF6)2
(L.), 1 (c), and 2 (d) in MeCN. The inset shows the normalized MLCTbands. b) The solvatochromism of complex 1.
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3631
Full Paper

plex 2, reduction did not happen until the potential was asnegative as �2.47 V vs. Fc+/Fc. As bpy is the only redox-activeligand in this complex, such a reduction is ascribed to beingcentered on bpy. Although 2 has been studied intermittentlyfor several decades, this bpy-based reduction has never beenreported. A somewhat astonishing point here is that such anenergetic reduction is quasi-reversible, whereas reductions ofthe ligands in NHC-containing complexes, not only of the com-plex 1 reported here, but also of our first-generation Fe NHCcomplexes,[7] are essentially irreversible. This in turn indicatesthe instability of the reduced NHC radical anion, and that theligand-based reduction processes in 1 are an intermingling ofthe two different types (btz as NHC and bpy, respectively) of li-gands. Another interesting point to note is that, in accordancewith the higher electron density on the bpy ligand in com-plexes 1 and 2 compared to in [Fe(bpy)3](PF6)2, the chemicalshifts of the protons on the bpy ligands are also positioned ata higher field (Supporting Information, Figure S10). However,with complex 2 holding a more energetic bpy ligand, the pro-tons are less-shielded compared to those of bpy in complex 1.This can be explained based on the crystal structure as a resultof the bpy ligand being sandwiched between two p-tolylgroups so that extra shielding is imposed.
Figure 3 a illustrates the steady-state UV/Vis absorption ofthe three complexes in MeCN. In the UV region, characteristicligand-centered bands have more or less identical peak posi-tions (Table 2). In the visible region, 1 and 2 show similar MLCTbands in terms of shape and extinction coefficient,[35] both red-shifted compared to [Fe(bpy)3](PF6)2 with less than half of theintensity. A further red-shift is observed from 1 to 2 which canbe understood by a roughly 300 mV anodic shift of the Fe-based oxidation potential but only 200 mV anodic shift of theligand-based reduction. Complex 2 is extremely solvatochromic(Supporting Information, Figure S13),[35] but different solventsshow very little effect on the absorption maxima of the MLCTbands of complex 1 (Figure 3 b, except for a significantly dis-tinct spectrum in H2O, which is probably related to solubilityissues). Although solvatochromism could be expected for com-
plex 1, owing to the dipolar nature of the MLCT state,[36] theabsence of a significant effect may lie in the planar btz frag-ments in 1 acting as an insulator between the FeII center andthe solvent molecules compared to the rather linear andmediating CN� in 2. As will be discussed in the next section,different solvents also exert drastically less influence on theexcited-state dynamics of 1 compared to that reported for2.[37] As a consequence, complex 1 is advantageous in termsof the versatility of applications.
Femtosecond transient absorption spectroscopy
To investigate the excited-state dynamics of 1, femtosecond(fs) transient absorption (TA) spectroscopic studies were carriedout. Considering the above-mentioned possibility of multipleMLCT states localized on different types of ligands in 1 as men-tioned above, 2 was studied for comparison as a complexwhere the MLCT states could only be localized on the bpyligand. In 1987, Winkler et al. reported that in acetone, com-plex 2 displayed an excited state that was MLCT in nature witha lifetime within the instrumental response (of 25 ps).[37]
Strangely, there was no follow-up since that study until recent-ly when Gaffney et al. unambiguously identified the excitedstate in DMSO as a 3MLCT state with a lifetime of 18 ps usingfs X-ray emission spectroscopy (XES).[38] With this result asa basis, it is much easier to understand the behavior ofcomplex 1 by comparing with 2.
Figure 4 shows the time-resolved differential absorptionspectra of complexes 1 and 2 excited at 615 and 705 nm, re-spectively, in MeCN. Both wavelengths were chosen as slightlylower in energy than their lowest-energy MLCT absorptionmaxima. The gaps seen in the spectra are due to the strongscattering at the pump wavelengths (615 nm for complex 1and 705 nm for complex 2) and the primary laser source(around 800 nm). Despite the speculation about multiple MLCTstates of complex 1, a striking similarity is observed at firstsight when comparing the two. Upon excitation, both com-plexes instantaneously show two well-defined ground-state
Figure 4. The transient absorption (TA) spectra of a) complex 1 excited at 615 nm and b) complex 2 excited at 705 nm in MeCN.
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3632
Full Paper
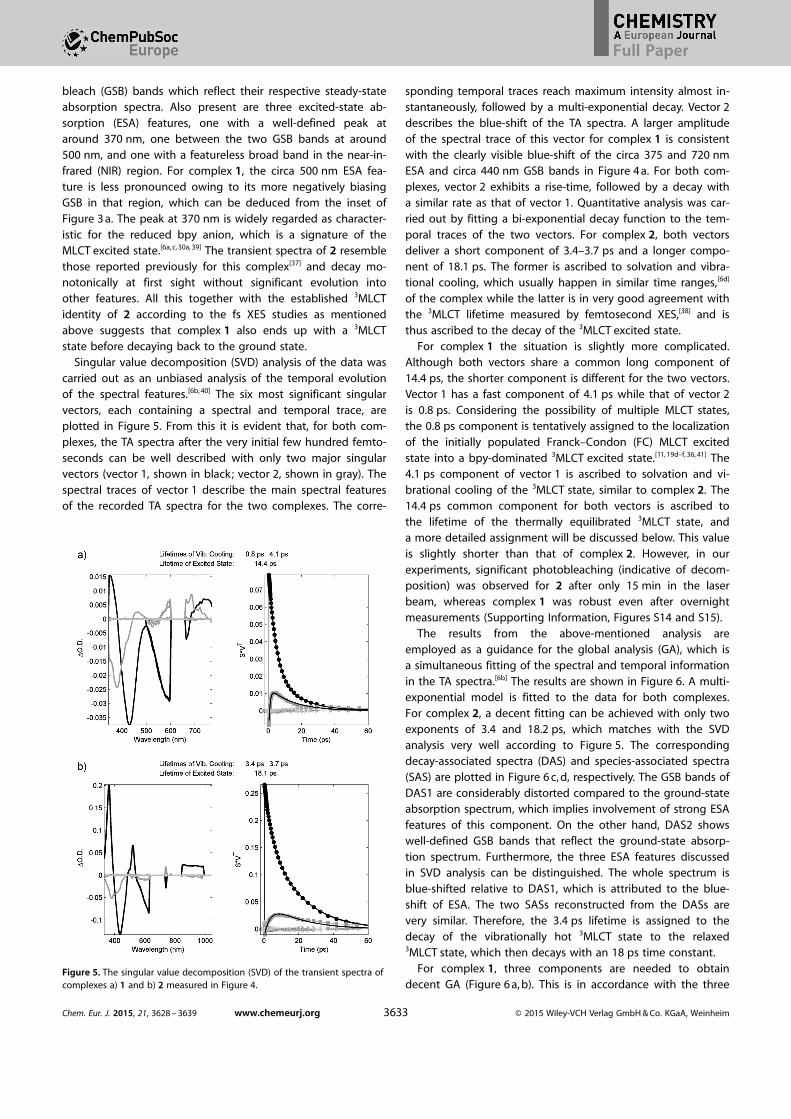
bleach (GSB) bands which reflect their respective steady-stateabsorption spectra. Also present are three excited-state ab-sorption (ESA) features, one with a well-defined peak ataround 370 nm, one between the two GSB bands at around500 nm, and one with a featureless broad band in the near-in-frared (NIR) region. For complex 1, the circa 500 nm ESA fea-ture is less pronounced owing to its more negatively biasingGSB in that region, which can be deduced from the inset ofFigure 3 a. The peak at 370 nm is widely regarded as character-istic for the reduced bpy anion, which is a signature of theMLCT excited state.[6a, c, 30a, 39] The transient spectra of 2 resemblethose reported previously for this complex[37] and decay mo-notonically at first sight without significant evolution intoother features. All this together with the established 3MLCTidentity of 2 according to the fs XES studies as mentionedabove suggests that complex 1 also ends up with a 3MLCTstate before decaying back to the ground state.
Singular value decomposition (SVD) analysis of the data wascarried out as an unbiased analysis of the temporal evolutionof the spectral features.[6b, 40] The six most significant singularvectors, each containing a spectral and temporal trace, areplotted in Figure 5. From this it is evident that, for both com-plexes, the TA spectra after the very initial few hundred femto-seconds can be well described with only two major singularvectors (vector 1, shown in black; vector 2, shown in gray). Thespectral traces of vector 1 describe the main spectral featuresof the recorded TA spectra for the two complexes. The corre-
sponding temporal traces reach maximum intensity almost in-stantaneously, followed by a multi-exponential decay. Vector 2describes the blue-shift of the TA spectra. A larger amplitudeof the spectral trace of this vector for complex 1 is consistentwith the clearly visible blue-shift of the circa 375 and 720 nmESA and circa 440 nm GSB bands in Figure 4 a. For both com-plexes, vector 2 exhibits a rise-time, followed by a decay witha similar rate as that of vector 1. Quantitative analysis was car-ried out by fitting a bi-exponential decay function to the tem-poral traces of the two vectors. For complex 2, both vectorsdeliver a short component of 3.4–3.7 ps and a longer compo-nent of 18.1 ps. The former is ascribed to solvation and vibra-tional cooling, which usually happen in similar time ranges,[6d]
of the complex while the latter is in very good agreement withthe 3MLCT lifetime measured by femtosecond XES,[38] and isthus ascribed to the decay of the 3MLCT excited state.
For complex 1 the situation is slightly more complicated.Although both vectors share a common long component of14.4 ps, the shorter component is different for the two vectors.Vector 1 has a fast component of 4.1 ps while that of vector 2is 0.8 ps. Considering the possibility of multiple MLCT states,the 0.8 ps component is tentatively assigned to the localizationof the initially populated Franck–Condon (FC) MLCT excitedstate into a bpy-dominated 3MLCT excited state.[11, 19d–f, 36, 41] The4.1 ps component of vector 1 is ascribed to solvation and vi-brational cooling of the 3MLCT state, similar to complex 2. The14.4 ps common component for both vectors is ascribed tothe lifetime of the thermally equilibrated 3MLCT state, anda more detailed assignment will be discussed below. This valueis slightly shorter than that of complex 2. However, in ourexperiments, significant photobleaching (indicative of decom-position) was observed for 2 after only 15 min in the laserbeam, whereas complex 1 was robust even after overnightmeasurements (Supporting Information, Figures S14 and S15).
The results from the above-mentioned analysis areemployed as a guidance for the global analysis (GA), which isa simultaneous fitting of the spectral and temporal informationin the TA spectra.[6b] The results are shown in Figure 6. A multi-exponential model is fitted to the data for both complexes.For complex 2, a decent fitting can be achieved with only twoexponents of 3.4 and 18.2 ps, which matches with the SVDanalysis very well according to Figure 5. The correspondingdecay-associated spectra (DAS) and species-associated spectra(SAS) are plotted in Figure 6 c, d, respectively. The GSB bands ofDAS1 are considerably distorted compared to the ground-stateabsorption spectrum, which implies involvement of strong ESAfeatures of this component. On the other hand, DAS2 showswell-defined GSB bands that reflect the ground-state absorp-tion spectrum. Furthermore, the three ESA features discussedin SVD analysis can be distinguished. The whole spectrum isblue-shifted relative to DAS1, which is attributed to the blue-shift of ESA. The two SASs reconstructed from the DASs arevery similar. Therefore, the 3.4 ps lifetime is assigned to thedecay of the vibrationally hot 3MLCT state to the relaxed3MLCT state, which then decays with an 18 ps time constant.
For complex 1, three components are needed to obtaindecent GA (Figure 6 a, b). This is in accordance with the three
Figure 5. The singular value decomposition (SVD) of the transient spectra ofcomplexes a) 1 and b) 2 measured in Figure 4.
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3633
Full Paper
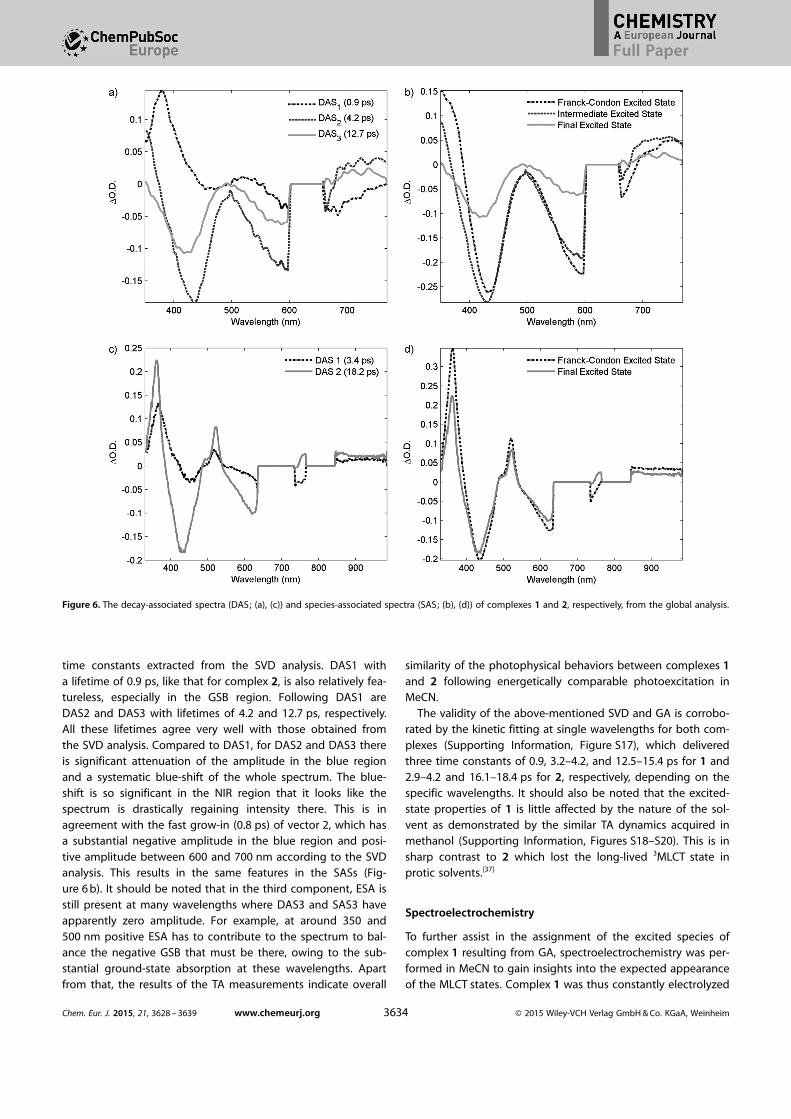
time constants extracted from the SVD analysis. DAS1 witha lifetime of 0.9 ps, like that for complex 2, is also relatively fea-tureless, especially in the GSB region. Following DAS1 areDAS2 and DAS3 with lifetimes of 4.2 and 12.7 ps, respectively.All these lifetimes agree very well with those obtained fromthe SVD analysis. Compared to DAS1, for DAS2 and DAS3 thereis significant attenuation of the amplitude in the blue regionand a systematic blue-shift of the whole spectrum. The blue-shift is so significant in the NIR region that it looks like thespectrum is drastically regaining intensity there. This is inagreement with the fast grow-in (0.8 ps) of vector 2, which hasa substantial negative amplitude in the blue region and posi-tive amplitude between 600 and 700 nm according to the SVDanalysis. This results in the same features in the SASs (Fig-ure 6 b). It should be noted that in the third component, ESA isstill present at many wavelengths where DAS3 and SAS3 haveapparently zero amplitude. For example, at around 350 and500 nm positive ESA has to contribute to the spectrum to bal-ance the negative GSB that must be there, owing to the sub-stantial ground-state absorption at these wavelengths. Apartfrom that, the results of the TA measurements indicate overall
similarity of the photophysical behaviors between complexes 1and 2 following energetically comparable photoexcitation inMeCN.
The validity of the above-mentioned SVD and GA is corrobo-rated by the kinetic fitting at single wavelengths for both com-plexes (Supporting Information, Figure S17), which deliveredthree time constants of 0.9, 3.2–4.2, and 12.5–15.4 ps for 1 and2.9–4.2 and 16.1–18.4 ps for 2, respectively, depending on thespecific wavelengths. It should also be noted that the excited-state properties of 1 is little affected by the nature of the sol-vent as demonstrated by the similar TA dynamics acquired inmethanol (Supporting Information, Figures S18–S20). This is insharp contrast to 2 which lost the long-lived 3MLCT state inprotic solvents.[37]
Spectroelectrochemistry
To further assist in the assignment of the excited species ofcomplex 1 resulting from GA, spectroelectrochemistry was per-formed in MeCN to gain insights into the expected appearanceof the MLCT states. Complex 1 was thus constantly electrolyzed
Figure 6. The decay-associated spectra (DAS; (a), (c)) and species-associated spectra (SAS; (b), (d)) of complexes 1 and 2, respectively, from the global analysis.
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3634
Full Paper
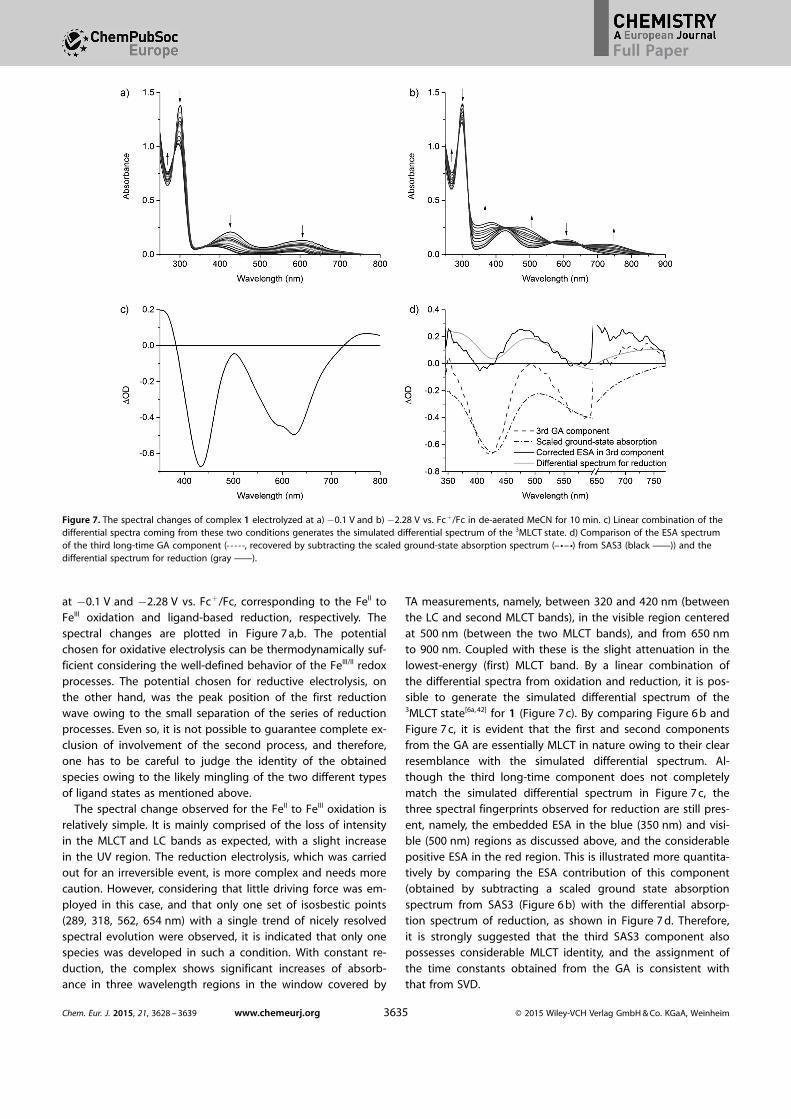
at �0.1 V and �2.28 V vs. Fc+/Fc, corresponding to the FeII toFeIII oxidation and ligand-based reduction, respectively. Thespectral changes are plotted in Figure 7 a,b. The potentialchosen for oxidative electrolysis can be thermodynamically suf-ficient considering the well-defined behavior of the FeIII/II redoxprocesses. The potential chosen for reductive electrolysis, onthe other hand, was the peak position of the first reductionwave owing to the small separation of the series of reductionprocesses. Even so, it is not possible to guarantee complete ex-clusion of involvement of the second process, and therefore,one has to be careful to judge the identity of the obtainedspecies owing to the likely mingling of the two different typesof ligand states as mentioned above.
The spectral change observed for the FeII to FeIII oxidation isrelatively simple. It is mainly comprised of the loss of intensityin the MLCT and LC bands as expected, with a slight increasein the UV region. The reduction electrolysis, which was carriedout for an irreversible event, is more complex and needs morecaution. However, considering that little driving force was em-ployed in this case, and that only one set of isosbestic points(289, 318, 562, 654 nm) with a single trend of nicely resolvedspectral evolution were observed, it is indicated that only onespecies was developed in such a condition. With constant re-duction, the complex shows significant increases of absorb-ance in three wavelength regions in the window covered by
TA measurements, namely, between 320 and 420 nm (betweenthe LC and second MLCT bands), in the visible region centeredat 500 nm (between the two MLCT bands), and from 650 nmto 900 nm. Coupled with these is the slight attenuation in thelowest-energy (first) MLCT band. By a linear combination ofthe differential spectra from oxidation and reduction, it is pos-sible to generate the simulated differential spectrum of the3MLCT state[6a, 42] for 1 (Figure 7 c). By comparing Figure 6 b andFigure 7 c, it is evident that the first and second componentsfrom the GA are essentially MLCT in nature owing to their clearresemblance with the simulated differential spectrum. Al-though the third long-time component does not completelymatch the simulated differential spectrum in Figure 7 c, thethree spectral fingerprints observed for reduction are still pres-ent, namely, the embedded ESA in the blue (350 nm) and visi-ble (500 nm) regions as discussed above, and the considerablepositive ESA in the red region. This is illustrated more quantita-tively by comparing the ESA contribution of this component(obtained by subtracting a scaled ground state absorptionspectrum from SAS3 (Figure 6 b) with the differential absorp-tion spectrum of reduction, as shown in Figure 7 d. Therefore,it is strongly suggested that the third SAS3 component alsopossesses considerable MLCT identity, and the assignment ofthe time constants obtained from the GA is consistent withthat from SVD.
Figure 7. The spectral changes of complex 1 electrolyzed at a) �0.1 V and b) �2.28 V vs. Fc+/Fc in de-aerated MeCN for 10 min. c) Linear combination of thedifferential spectra coming from these two conditions generates the simulated differential spectrum of the 3MLCT state. d) Comparison of the ESA spectrumof the third long-time GA component (a, recovered by subtracting the scaled ground-state absorption spectrum (d) from SAS3 (black c)) and thedifferential spectrum for reduction (gray c).
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3635
Full Paper
DFT and TD-DFT calculations
Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were carried out to assist in the understand-ing of the properties of complex 1. The ground-state structurewas optimized using Gaussian 09[43] with PBE0 j6-311G(d,p) andwith a complete acetonitrile polarizable continuum model(PCM). The structure is parameterized by the average distancesbetween Fe and either bpy (qbpy) or btz (qbtz), and the octahe-dricity (O-value), which is a measure of the mean absolute de-viation of the set of all metal–ligand bond angles from thevalues in an “ideal” octahedral structure, as listed in Table 3.
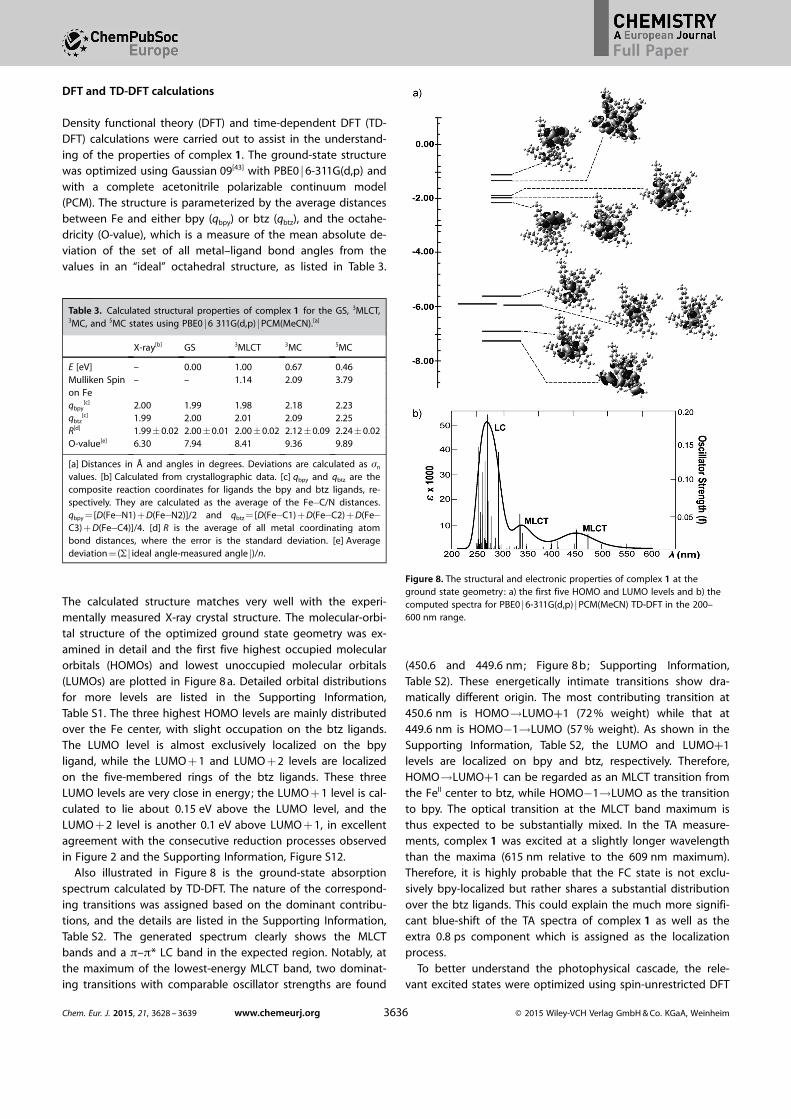
The calculated structure matches very well with the experi-mentally measured X-ray crystal structure. The molecular-orbi-tal structure of the optimized ground state geometry was ex-amined in detail and the first five highest occupied molecularorbitals (HOMOs) and lowest unoccupied molecular orbitals(LUMOs) are plotted in Figure 8 a. Detailed orbital distributionsfor more levels are listed in the Supporting Information,Table S1. The three highest HOMO levels are mainly distributedover the Fe center, with slight occupation on the btz ligands.The LUMO level is almost exclusively localized on the bpyligand, while the LUMO + 1 and LUMO + 2 levels are localizedon the five-membered rings of the btz ligands. These threeLUMO levels are very close in energy; the LUMO + 1 level is cal-culated to lie about 0.15 eV above the LUMO level, and theLUMO + 2 level is another 0.1 eV above LUMO + 1, in excellentagreement with the consecutive reduction processes observedin Figure 2 and the Supporting Information, Figure S12.
Also illustrated in Figure 8 is the ground-state absorptionspectrum calculated by TD-DFT. The nature of the correspond-ing transitions was assigned based on the dominant contribu-tions, and the details are listed in the Supporting Information,Table S2. The generated spectrum clearly shows the MLCTbands and a p–p* LC band in the expected region. Notably, atthe maximum of the lowest-energy MLCT band, two dominat-ing transitions with comparable oscillator strengths are found
(450.6 and 449.6 nm; Figure 8 b; Supporting Information,Table S2). These energetically intimate transitions show dra-matically different origin. The most contributing transition at450.6 nm is HOMO!LUMO+1 (72 % weight) while that at449.6 nm is HOMO�1!LUMO (57 % weight). As shown in theSupporting Information, Table S2, the LUMO and LUMO+1levels are localized on bpy and btz, respectively. Therefore,HOMO!LUMO+1 can be regarded as an MLCT transition fromthe FeII center to btz, while HOMO�1!LUMO as the transitionto bpy. The optical transition at the MLCT band maximum isthus expected to be substantially mixed. In the TA measure-ments, complex 1 was excited at a slightly longer wavelengththan the maxima (615 nm relative to the 609 nm maximum).Therefore, it is highly probable that the FC state is not exclu-sively bpy-localized but rather shares a substantial distributionover the btz ligands. This could explain the much more signifi-cant blue-shift of the TA spectra of complex 1 as well as theextra 0.8 ps component which is assigned as the localizationprocess.
To better understand the photophysical cascade, the rele-vant excited states were optimized using spin-unrestricted DFT
Table 3. Calculated structural properties of complex 1 for the GS, 3MLCT,3MC, and 5MC states using PBE0 j6 311G(d,p) jPCM(MeCN).[a]
X-ray[b] GS 3MLCT 3MC 5MC
E [eV] – 0.00 1.00 0.67 0.46Mulliken Spinon Fe
– – 1.14 2.09 3.79
qbpy[c] 2.00 1.99 1.98 2.18 2.23
qbtz[c] 1.99 2.00 2.01 2.09 2.25
R[d] 1.99�0.02 2.00�0.01 2.00�0.02 2.12�0.09 2.24�0.02O-value[e] 6.30 7.94 8.41 9.36 9.89
[a] Distances in � and angles in degrees. Deviations are calculated as sn
values. [b] Calculated from crystallographic data. [c] qbpy and qbtz are thecomposite reaction coordinates for ligands the bpy and btz ligands, re-spectively. They are calculated as the average of the Fe�C/N distances.qbpy = [D(Fe�N1) + D(Fe�N2)]/2 and qbtz = [D(Fe�C1) + D(Fe�C2) + D(Fe�C3) + D(Fe�C4)]/4. [d] R is the average of all metal coordinating atombond distances, where the error is the standard deviation. [e] Averagedeviation = (S j ideal angle-measured angle j)/n.
Figure 8. The structural and electronic properties of complex 1 at theground state geometry: a) the first five HOMO and LUMO levels and b) thecomputed spectra for PBE0 j6-311G(d,p) jPCM(MeCN) TD-DFT in the 200–600 nm range.
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3636
Full Paper

(uDFT) calculations. The lowest triplet (3MC, 3MLCT) and quintet(5MC, 5MLCT) states can be assigned as MLCT or MC based ontheir Fe spin density, which is shown in the Supporting Infor-mation, Table S3. The key structural and energetic parametersare listed in Table 3. Specifically, qbpy and qbtz of these states areplotted as the gray dots in the inset of Figure 9. From this, the
structural distortion of the excited states relative to theground state can be easily ascertained. With the projection ofthese gray dots along the diagonal direction of the inset (aver-age Fe–ligand distance q) as the horizontal coordinates andtheir corresponding energy as the vertical coordinates, a pro-jected potential energy surface (PPES) diagram is plotted inFigure 9. The 3MLCT state is structurally akin to the groundstate, lying only 1 eV above the latter, which is much smallerthan calculated for [Fe(bpy)3]2 + in previously reported stud-ies,[44] but consistent with the previous conclusion of reducedexcitation energy for complex 1. The 3MLCT state is largelylocalized on bpy (Supporting Information, Table S3), which isconsistently predicted to be lower in energy than btzaccording to the ordering of the LUMO levels. This supportsthe assignment of the longest component from both SVD andGA as a bpy-localized 3MLCT state.
The lowest-energy triplet MC state (3MC) shows a typicalstructural distortion owing to the population of the eg* anti-bonding orbitals. Compared to the ground and 3MLCT statesthe average Fe–bpy distance is elongated by 0.2 �, which isonly seen in the structural distortion of the quintet MC state(5T2) of [Fe(bpy)3]2 + , and is much larger than that of the tripletMC states (3T1, 3T2, and so on) therein.[44, 45] This can be under-stood as being due to the much larger electron density on theFe center. On the other hand, the Fe–btz bonds also stretch byaround 0.1 �, and the bite angles are smaller, as manifested bythe increased deviation from ideal octahedricity (higher O-
value). The 3MC minimum lies 0.67 eV above the ground state,which is less than the corresponding [Fe(bpy)3]2 + 3MC(1.24 eV).[44] However, the difference between the energyminima of the 3MLCT and 3MC is reduced to only 0.33 eV com-pared to 1.26 eV for [Fe(bpy)3]2+ .[44] In other words, the 3MCstate is significantly destabilized relative to the 3MLCT state incomplex 1, and its minimum is moved even further away com-pared to [Fe(bpy)3]2+ . A similar situation is seen for the lowest-energy quintet excited state (5MC). An increase of 0.25 � in theaverage Fe–ligand distances is observed, accompanied with aneven larger distortion from ideal octahedricity. The energy dif-ference between the 3MLCT and 5MC minimum is lowered to0.54 eV compared to 1.81 eV in [Fe(bpy)3]2+ .[44]
For common FeII polyimine complexes, the 5MC state isusually regarded as the main state for deactivating thephotochemically interesting 3MLCT state,[6d] while for most RuII
analogues, the thermally activated access to the triplet 3MCstate is the main deactivation mechanism.[11, 46] Such a discrep-ancy clearly arises from the weaker ligand interaction of FeII
compared to RuII. For this reason, in FeII complexes such as[Fe(bpy)3]2 + , the triplet 3T1 state is so short-lived that only anultrafast (femtosecond) X-ray emission technique[6f] and theo-retical calculations[44] can shed light on its dynamics. With in-creased ligand field strength, however, the 3T1 state may beginto dictate the deactivation cascade.[4] Recently, the role of thetriplet MC states in the mechanism of the 3MLCT deactivationcascade has received increased attention.[4, 6f, 44, 47] Indeed, in ourfirst-generation FeII NHC complex, the 3MC state is shown tobe critical to the major 3MLCT deactivation pathway. In thiswork,[8] the strong s-donation imposed by the NHC ligandswas shown to effectively destabilize the 5MC state and push itfar away along the molecular coordinate compared tocommon FeII polypyridine complexes. The consequence of thisis that the 3MLCT state is first intercepted by the 3MC surfacebefore the 5MC surface crosses the 3MLCT surface. In the pres-ent study, an even stronger s-donation than our first genera-tion FeII NHC complex is achieved in complex 1 thanks to thecombination of the mesoionic nature of btz and its heterolep-tic structure. Therefore, complex 1 features an even smaller3MLCT–3MC energy gap (0.33 eV) and an even larger structuraldistortion in its 3MC state, which contribute to its longer3MLCT lifetime measured by TA spectroscopy. Notably, accord-ing to Figure 9, the energy of the singlet ground state at theoptimized 3MC structure is very close to that of the 3MC state.Therefore, it is expected that once complex 1 is in the 3MCstate, it will quickly decay to the ground state.
Conclusions
We have successfully synthesized the first heteroleptic tetra-kis(NHC) FeII bpy complex (1) using the bis(1,2,3-triazol-5-yli-dene) ligand and the uncommon Fe(bpy)Cl2 intermediate. Thisis also the first octahedral TM complex based on this type ofbtz ligand. A strong ligand field is demonstrated to be im-posed in complex 1 without sacrificing stability and solventcompatibility, yielding a remarkably electron-rich Fe center.Electrochemistry, ultrafast spectroscopy, and computational
Figure 9. Projected potential energy surfaces (PPESs) versus q. Gray pointsare optimized minima, and black points are single-point energies calculatedat the minimum geometries (· S0, singlet state; � T1, triplet states ; + Q1,quintet states). The gray lines schematically show the PPESs. Inset : the struc-tural differences between calculated minima geometries in terms of qbpy andqbtz. The dashed diagonal is used as the horizontal axis (average iron–liganddistance q) of the main figure, to which the gray points in the inset areprojected.
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3637
Full Paper

studies revealed that in such a heteroleptic structure, the btzligand is also redox- and photoredox-active, like the bench-mark bpy ligand. Understanding of the photophysical behaviorof complex 1 is aided by the comparison with complex 2 and[Fe(bpy)3]2 + , as well as theoretical calculations. The employ-ment of the strongly s-donating btz ligand effectively destabil-izes both the triplet and quintet metal-centered states andmoves their minima far away from that of the photochemicallyvaluable 3MLCT state along the molecular coordinate (first co-ordination sphere bond distances). This results in a long-lived3MLCT state of 13 ps compared to the 130 fs of that for[Fe(bpy)3]2 + , and such a success achieved from a completelydifferent approach, namely the tris(bidentate) instead of thebis(tridentate) configuration, which was adopted in our previ-ous report,[7] demonstrates that Fe NHC chemistry is indeeda viable strategy for improving the photophysical properties ofFeII complexes. The MLCT lifetime is the longest for a photo-chemically stable FeII complex to date, raising hopes that re-placing expensive and rare RuII for inexpensive and earth-abun-dant FeII, for the purpose of solar energy harvesting, will bepossible. In this respect, it is important that the heterolepticcomplex 1 also increases the overlap with the solar spectrumto above 750 nm, a bathochromic shift of 200 nm compared toour previously synthesized homoleptic FeII NHC complex. Thequantum chemical calculations also suggest a 3MLCT deactiva-tion pathway that proceeds through the short-lived 3MC state.Thus, the photophysical mechanism of complex 1 resemblesthat of most RuII complexes, although the MLCT excited statelifetime is still significantly shorter. Therefore, the knowledgeand principles developed for RuII complexes may now be ap-plied to FeII complexes, turning research on the latter in a newdirection that is amenable to further improvement with theever enlarged library of photochemically interestingcompounds.
Acknowledgements
This work was supported by the Crafoord Foundation, theSwedish Research Council (VR), the Knut and Alice Wallenberg(KAW) Foundation, the European Research Council (ERC,226136-VISCHEM) and the Swedish Energy Agency. We thankDr. Erling Thyrhaug for the assistance in measuring high-concentration UV/Vis absorption spectroscopy and Sofia Ess�nfor measuring HR-MS. P.P. acknowledges support from the NSCand LUNARC supercomputing facilities.
Keywords: iron · femtosecond spectroscopy · N ligands ·metal-to-ligand charge transfer · N-heterocyclic carbenes
[1] B. N. Figgis, M. A. Hitchman, Ligand Field Theory and Its Applications,Wiley-VCH, New York, 2000.
[2] V. Balzani, G. Bergamini, S. Campagna, F. Puntoriero, Top. Curr. Chem.2007, 280, 1 – 36.
[3] a) A. Hauser, Top. Curr. Chem. 2004, 234, 155 – 198; b) M. A. Halcrow,Polyhedron 2007, 26, 3523 – 3576.
[4] L. L. Jamula, A. M. Brown, D. Guo, J. K. McCusker, Inorg. Chem. 2014, 53,15 – 17.
[5] O. Kahn, Science 1998, 279, 44 – 48.
[6] a) J. E. Monat, J. K. McCusker, J. Am. Chem. Soc. 2000, 122, 4092 – 4097;b) W. Gawelda, A. Cannizzo, V. T. Pham, F. van Mourik, C. Bressler, M.Chergui, J. Am. Chem. Soc. 2007, 129, 8199 – 8206; c) C. Consani, M. Pre-mont-Schwarz, A. Elnahhas, C. Bressler, F. van Mourik, A. Cannizzo, M.Chergui, Angew. Chem. 2009, 121, 7320 – 7323; Angew. Chem. Int. Ed.2009, 48, 7184 – 7187; d) A. Cannizzo, C. J. Milne, C. Consani, W. Gawel-da, C. Bressler, F. van Mourik, M. Chergui, Coord. Chem. Rev. 2010, 254,2677 – 2686; e) K. Haldrup, G. Vanko, W. Gawelda, A. Galler, G. Doumy,A. M. March, E. P. Kanter, A. Bordage, A. Dohn, T. B. van Driel, K. S. Kjaer,H. T. Lemke, S. E. Canton, J. Uhlig, V. Sundstrom, L. Young, S. H. South-worth, M. M. Nielsen, C. Bressler, J. Phys. Chem. A 2012, 116, 9878 – 9887;f) W. Zhang, R. Alonso-Mori, U. Bergmann, C. Bressler, M. Chollet, A.Galler, W. Gawelda, R. G. Hadt, R. W. Hartsock, T. Kroll, K. S. Kjaer, K. Kubi-cek, H. T. Lemke, H. W. Liang, D. A. Meyer, M. M. Nielsen, C. Purser, J. S.Robinson, E. I. Solomon, Z. Sun, D. Sokaras, T. B. van Driel, G. Vanko, T. C.Weng, D. Zhu, K. J. Gaffney, Nature 2014, 509, 345 – 348.
[7] Y. Liu, T. Harlang, S. E. Canton, P. Ch�bera, K. Su�rez-Alc�ntara, A. Fleck-haus, D. A. Vithanage, E. Gçransson, A. Corani, R. Lomoth, V. Sundstrçm,K. W�rnmark, Chem. Commun. 2013, 49, 6412 – 6414.
[8] L. A. Fredin, M. P�pai, E. Rozs�lyi, G. Vank�, K. W�rnmark, V. Sundstrçm,P. Persson, J. Phys. Chem. Lett. 2014, 5, 2066 – 2071.
[9] a) K. Riener, S. Haslinger, A. Raba, M. P. Hogerl, M. Cokoja, W. A. Herr-mann, F. E. Kuhn, Chem. Rev. 2014, 114, 5215 – 5272; b) M. J. Ingleson,R. A. Layfield, Chem. Commun. 2012, 48, 3579 – 3589.
[10] J. Louie, R. H. Grubbs, Chem. Commun. 2000, 1479 – 1480.[11] S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini, V. Balzani, Top. Curr.
Chem. 2007, 280, 117 – 214.[12] a) S. Schick, T. Pape, F. E. Hahn, Organometallics 2014, 33, 4035 – 4041;
b) S. Hohloch, L. Suntrup, B. Sarkar, Organometallics 2013, 32, 7376 –7385; c) J. M. Aizpurua, M. Sagartzazu-Aizpurua, Z. Monasterio, I.Azcune, C. Mendicute, J. I. Miranda, E. Garcia-Lecina, A. Altube, R. M.Fratila, Org. Lett. 2012, 14, 1866 – 1868; d) G. Guisado-Barrios, J. Bouf-fard, B. Donnadieu, G. Bertrand, Organometallics 2011, 30, 6017 – 6021;e) S. Sanz, A. Azua, E. Peris, Dalton Trans. 2010, 39, 6339 – 6343; f) M.Poyatos, W. McNamara, C. Incarvito, E. Clot, E. Peris, R. H. Crabtree, Or-ganometallics 2008, 27, 2128 – 2136; g) D. H. Jeong, W. J. Park, J. H.Jeong, D. G. Churchill, H. Lee, Inorg. Chem. Commun. 2008, 11, 1170 –1173.
[13] a) V. Gierz, A. Seyboldt, C. Maichle-Mçssmer, K. W. Tçrnroos, M. T. Spei-del, B. Speiser, K. Eichele, D. Kunz, Organometallics 2012, 31, 7893 –7901; b) V. Gierz, C. Maichle-Mçssmer, D. Kunz, Organometallics 2012,31, 739 – 747.
[14] a) B. Schulze, U. S. Schubert, Chem. Soc. Rev. 2014, 43, 2522 – 2571;b) K. F. Donnelly, A. Petronilho, M. Albrecht, Chem. Commun. 2013, 49,1145 – 1159; c) R. H. Crabtree, Coord. Chem. Rev. 2013, 257, 755 – 766;d) A. Krger, M. Albrecht, Aust. J. Chem. 2011, 64, 1113; e) J. D. Crowley,A.-L. Lee, K. J. Kilpin, Aust. J. Chem. 2011, 64, 1118 – 1132; f) G. Guisado-Barrios, J. Bouffard, B. Donnadieu, G. Bertrand, Angew. Chem. 2010, 122,4869 – 4872; Angew. Chem. Int. Ed. 2010, 49, 4759 – 4762; g) M. Hecken-roth, E. Kluser, A. Neels, M. Albrecht, Dalton Trans. 2008, 6242 – 6249.
[15] a) H. V. Huynh, G. Frison, J. Org. Chem. 2013, 78, 328 – 338; b) J. C. Bern-hammer, G. Frison, H. V. Huynh, Chem. Eur. J. 2013, 19, 12892 – 12905;c) D. Yuan, H. V. Huynh, Organometallics 2012, 31, 405 – 412; d) H. V.Huynh, Y. Han, R. Jothibasu, J. A. Yang, Organometallics 2009, 28, 5395 –5404; e) D. G. Gusev, Organometallics 2009, 28, 6458 – 6461; f) A. K.Phukan, A. K. Guha, S. Sarmah, R. D. Dewhurst, J. Org. Chem. 2013, 78,11032 – 11039.
[16] a) S. Sinn, B. Schulze, C. Friebe, D. G. Brown, M. Jager, E. Altuntas, J.Kubel, O. Guntner, C. P. Berlinguette, B. Dietzek, U. S. Schubert, Inorg.Chem. 2014, 53, 2083 – 2095; b) A. Bolje, S. Hohloch, D. Urankar, A.Pevec, M. Gazvoda, B. Sarkar, J. Kosmrlj, Organometallics 2014, 33,2588 – 2598; c) V. Leigh, W. Ghattas, R. Lalrempuia, H. Muller-Bunz, M. T.Pryce, M. Albrecht, Inorg. Chem. 2013, 52, 5395 – 5402; d) D. G. Brown,P. A. Schauer, J. Borau-Garcia, B. R. Fancy, C. P. Berlinguette, J. Am. Chem.Soc. 2013, 135, 1692 – 1695; e) D. G. Brown, N. Sanguantrakun, B.Schulze, U. S. Schubert, C. P. Berlinguette, J. Am. Chem. Soc. 2012, 134,12354 – 12357; f) B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Gorls,U. Kohn, E. Altuntas, A. Baumgaertel, M. D. Hager, A. Winter, B. Dietzek,J. Popp, L. Gonzalez, U. S. Schubert, Chem. Eur. J. 2011, 17, 5494 – 5498.
[17] a) A. Petronilho, J. A. Woods, S. Bernhard, M. Albrecht, Eur. J. Inorg.Chem. 2014, 708 – 714; b) R. Lalrempuia, H. Muller-Bunz, M. Albrecht,
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3638
Full Paper

Angew. Chem. 2011, 123, 10144 – 10148; Angew. Chem. Int. Ed. 2011, 50,9969 – 9972; c) D. B. Grotjahn, D. B. Brown, J. K. Martin, D. C. Marelius,M. C. Abadjian, H. N. Tran, G. Kalyuzhny, K. S. Vecchio, Z. G. Specht, S. A.Cortes-Llamas, V. Miranda-Soto, C. van Niekerk, C. E. Moore, A. L. Rhein-gold, J. Am. Chem. Soc. 2011, 133, 19024 – 19027; d) Y. Wei, A. Petronil-ho, H. Mller-Bunz, M. Albrecht, Organometallics 2014, 33, 5834 – 5844.
[18] T. Duchanois, T. Etienne, M. Beley, X. Assfeld, E. A. Perpte, A. Monari,P. C. Gros, Eur. J. Inorg. Chem. 2014, 3747 – 3753.
[19] a) R. Ghosh, D. K. Palit, Phys. Chem. Chem. Phys. 2014, 16, 219 – 226;b) J. T. Hewitt, P. J. Vallett, N. H. Damrauer, J. Phys. Chem. A 2012, 116,11536 – 11547; c) W. Henry, C. G. Coates, C. Brady, K. L. Ronayne, P. Ma-tousek, M. Towrie, S. W. Botchway, A. W. Parker, J. G. Vos, W. R. Browne,J. J. McGarvey, J. Phys. Chem. A 2008, 112, 4537 – 4544; d) S. Wallin, J.Davidsson, J. Modin, L. Hammarstrçm, J. Phys. Chem. A 2005, 109,4697 – 4704; e) G. B. Shaw, D. J. Styers-Barnett, E. Z. Gannon, J. C. Grang-er, J. M. Papanikolas, J. Phys. Chem. A 2004, 108, 4998 – 5006; f) G. B.Shaw, C. L. Brown, J. M. Papanikolas, J. Phys. Chem. A 2002, 106, 1483 –1495.
[20] F. F. Charron, W. M. Reiff, Inorg. Chem. 1986, 25, 2786 – 2790.[21] J. T. Fletcher, B. J. Bumgarner, N. D. Engels, D. A. Skoglund, Organometal-
lics 2008, 27, 5430 – 5433.[22] W. M. Reiff, B. Dockum, M. A. Weber, R. B. Frankel, Inorg. Chem. 1975, 14,
800 – 806.[23] R.-A. Fallahpour, M. Neuburger, M. Zehnder, New J. Chem. 1999, 23, 53 –
61.[24] a) H. V. Phan, P. Chakraborty, M. Chen, Y. M. Calm, K. Kovnir, L. K. Keni-
ley, Jr. , J. M. Hoyt, E. S. Knowles, C. Besnard, M. W. Meisel, A. Hauser, C.Achim, M. Shatruk, Chem. Eur. J. 2012, 18, 15805 – 15815; b) Z. Ni, A. M.McDaniel, M. P. Shores, Chem. Sci. 2010, 1, 615 – 621; c) D. L. Reger, C. A.Little, A. L. Rheingold, R. Sommer, G. J. Long, Inorg. Chim. Acta 2001,316, 65 – 70.
[25] G. Albertin, S. Antoniutti, M. Bortoluzzi, Inorg. Chem. 2004, 43, 1328 –1335.
[26] O. Kaufhold, F. E. Hahn, T. Pape, A. Hepp, J. Organomet. Chem. 2008,693, 3435 – 3440.
[27] B. Liu, Y. Zhang, D. Xu, W. Chen, Chem. Commun. 2011, 47, 2883 – 2885.[28] a) A. Hauser, C. Enachescu, M. L. Daku, A. Vargas, N. Amstutz, Coord.
Chem. Rev. 2006, 250, 1642 – 1652; b) S. Dick, Z. Kristallogr. New Cryst.Struct. 1998, 213, 356.
[29] M. Nieuwenhuyzen, B. Bertram, J. F. Gallagher, J. G. Vos, Acta Crystallogr.Sect. C 1998, 54, 603 – 606.
[30] a) P. S. Braterman, J.-I. Song, R. D. Peacock, Inorg. Chem. 1992, 31, 555 –559; b) C.-T. Lin, W. Bçttcher, M. Chou, C. Creutz, N. Sutin, J. Am. Chem.Soc. 1976, 98, 6536.
[31] M. Yang, D. W. Thompson, G. J. Meyer, Inorg. Chem. 2002, 41, 1254 –1262.
[32] A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser, A. von Zelew-sky, Coord. Chem. Rev. 1988, 84, 85 – 277.
[33] H. Jacobsen, A. Correa, A. Poater, C. Costabile, L. Cavallo, Coord. Chem.Rev. 2009, 253, 687 – 703.
[34] a) S. U. Son, K. H. Park, Y. S. Lee, B. Y. Kim, C. H. Choi, M. S. Lah, Y. H.Jang, D. J. Jang, Y. K. Chung, Inorg. Chem. 2004, 43, 6896 – 6898; b) L. H.Chung, K. S. Cho, J. England, S. C. Chan, K. Wieghardt, C. Y. Wong, Inorg.Chem. 2013, 52, 9885 – 9896.
[35] H. E. Toma, M. S. Takasugi, J. Solution Chem. 1983, 12, 547 – 561.[36] E. M. Kober, B. P. Sullivan, T. J. Meyer, Inorg. Chem. 1984, 23, 2098 – 2104.[37] J. R. Winkler, C. Creutz, N. Sutin, J. Am. Chem. Soc. 1987, 109, 3470 –
3471.[38] K. Gaffney, private communication.[39] C. Creutz, M. Chou, T. L. Netzel, M. Okumura, N. Sutin, J. Am. Chem. Soc.
1980, 102, 1309 – 1319.[40] I. H. van Stokkum, D. S. Larsen, R. van Grondelle, Biochim. Biophys. Acta
2004, 1657, 82 – 104.[41] a) A. T. Yeh, C. V. Shank, J. K. McCusker, Science 2000, 289, 935 – 938;
b) R. A. Malone, D. F. Kelley, The Journal of Chemical Physics 1991, 95,8970 – 8976; c) L. F. Cooley, P. Bergquist, D. F. Kelley, J. Am. Chem. Soc.1990, 112, 2612 – 2617.
[42] A. M. Brown, C. E. McCusker, J. K. McCusker, Dalton Trans. 2014, 43,17635 – 17646.
[43] Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G.E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Men-nucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K.Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O.Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr. , J. E. Peralta, F. Ogliaro,M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Ko-bayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyen-gar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B.Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann,O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin,K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg,S. Dapprich, A. D. Daniels, �. Farkas, J. B. Foresman, J. V. Ortiz, J. Cio-slowski, and D. J. Fox, Gaussian, Inc. , Wallingford CT, 2009.
[44] C. Sousa, C. de Graaf, A. Rudavskyi, R. Broer, J. Tatchen, M. Etinski, C. M.Marian, Chem. Eur. J. 2013, 19, 17541 – 17551.
[45] M. P�pai, G. Vank�, C. de Graaf, T. Rozgonyi, J. Chem. Theory Comput.2013, 9, 509 – 519.
[46] Q. Sun, S. Mosquera-Vazquez, L. M. Daku, L. Guenee, H. A. Goodwin, E.Vauthey, A. Hauser, J. Am. Chem. Soc. 2013, 135, 13660 – 13663.
[47] a) I. M. Dixon, F. Alary, M. Boggio-Pasqua, J. L. Heully, Inorg. Chem. 2013,52, 13369 – 13374; b) A. Marino, P. Chakraborty, M. Servol, M. Lorenc, E.Collet, A. Hauser, Angew. Chem. Int. Ed. 2014, 53, 3863 – 3867; Angew.Chem. 2014, 126, 3944 – 3948.
[48] CCDC 1018263 (1) contains the supplementary crystallographic data forthis paper. These data can be obtained free of charge from The Cam-bridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_re-quest/cif.
Received: September 8, 2014Published online on December 11, 2014
Chem. Eur. J. 2015, 21, 3628 – 3639 www.chemeurj.org � 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3639
Full Paper







![N-[(4Z )-1-(3-Methyl-5-oxo-1-phenyl-4,5-dihydro-1Hpyrazol- 4-ylidene)hexyl]benzenesulfonohydrazide](https://static.fdokumen.com/doc/165x107/631d41f1f26ecf94330a76af/n-4z-1-3-methyl-5-oxo-1-phenyl-45-dihydro-1hpyrazol-4-ylidenehexylbenzenesulfonohydrazide.jpg)



![N ′-[1-(2,4-Dioxo-3,4-dihydro-2 H -1-benzopyran-3-ylidene)ethyl]thiophene-2-carbohydrazide](https://static.fdokumen.com/doc/165x107/63252fe2c9c7f5721c01f37f/n-1-24-dioxo-34-dihydro-2-h-1-benzopyran-3-ylideneethylthiophene-2-carbohydrazide.jpg)








