
2010BOR14135.pdf - Thèses
253
N° d’ordre : 4135 THÈSE PRÉSENTÉE A L’UNIVERSITÉ BORDEAUX 1 ÉCOLE DOCTORALE DES SCIENCES CHIMIQUES Par Julien Pinaud POUR OBTENIR LE GRADE DE DOCTEUR SPÉCIALITÉ : POLYMÈRES Catalyse organique par les carbènes N-hétérocycliques (NHCs) et leur version supportée sur polymères à des fins de recyclage. Date de Soutenance le : 14 décembre 2010 Devant la commission d’examen formée de : M. H. Cramail Professeur, LCPO, Université Bordeaux1 Examinateur M. C. Detrembleur Chercheur FNRS, CERM, Université de Liège Examinateur M. Y. Gnanou Directeur de recherche, LCPO, Université Bordeaux 1 Examinateur M. D. Mecerreyes Head of Nanotechnology Unit, CIDETEC Rapporteur M. A. Mortreux Professeur, UCCS, Université de Lille 1 Rapporteur M. D. Taton Professeur, LCPO, Université Bordeaux 1 Examinateur
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of 2010BOR14135.pdf - Thèses

N° d’ordre : 4135
THÈSE
PRÉSENTÉE A
L’UNIVERSITÉ BORDEAUX 1
ÉCOLE DOCTORALE DES SCIENCES CHIMIQUES
Par Julien Pinaud
POUR OBTENIR LE GRADE DE
DOCTEUR
SPÉCIALITÉ : POLYMÈRES
Catalyse organique par les carbènes N-hétérocycliques (NHCs)
et
leur version supportée sur polymères à des fins de recyclage.
Date de Soutenance le : 14 décembre 2010
Devant la commission d’examen formée de :
M. H. Cramail Professeur, LCPO, Université Bordeaux1 Examinateur
M. C. Detrembleur Chercheur FNRS, CERM, Université de Liège Examinateur
M. Y. Gnanou Directeur de recherche, LCPO, Université Bordeaux 1 Examinateur
M. D. Mecerreyes Head of Nanotechnology Unit, CIDETEC Rapporteur
M. A. Mortreux Professeur, UCCS, Université de Lille 1 Rapporteur
M. D. Taton Professeur, LCPO, Université Bordeaux 1 Examinateur

1

2
À mes parents
et mes amis.

3

4
« N'essayez pas de devenir un homme qui a du succès.
Essayez de devenir un homme qui a de la valeur. »
Albert Einstein
«La joie de contempler et de comprendre,
voilà le langage que me porte la nature.»
Albert Einstein
« La science doit s’accommoder à la nature.
La nature ne peut s’accommoder à la science.»
Ferdinand Brunot

5

6
Remerciements

7

8
Je tiens tout d’abord à remercier M. Yves Gnanou et M. Henri Cramail, directeurs
successifs du LCPO pour m’avoir accueilli dans leur laboratoire, me permettant ainsi de
réaliser ma thèse dans un lieu où règnent la bonne humeur et la rigueur scientifique.
Merci aux membres du jury, Messieurs David Mecerreyes, André Mortreux,
Christophe Detrembleur et Henri Cramail pour avoir pris le temps d’examiner et de juger ce
travail.
Je remercie à nouveau M. Yves Gnanou, mon co-directeur de thèse, pour son soutien
et son aide d’un point de vue scientifique. Malgré la distance, il a su être présent au bon
moment, et a permis d’apporter une part d’originalité à mes travaux.
De simples remerciements ne suffisent pas pour exprimer ma profonde gratitude à
mon autre directeur de thèse, Daniel Taton. Au cours de ces quatre dernières années (avec
le stage de Master), il a su comprendre ma personnalité et ainsi me soutenir lorsque cela
était nécessaire. Sa porte était toujours ouverte, que ce soit pour des discutions scientifiques
ou personnelles. Son souci du détail ainsi que sa connaissance de la chimie sont pour moi un
modèle à suivre. Je lui souhaite un avenir scientifique prolifique ainsi que du courage dans le
management de ses autres étudiants ;).
Au cours de ces quatre années passées au LCPO, j’ai également eu la chance de
travailler avec une « équipe NHCs » remarquable. Xiaoshuan Feng, Jean Raynaud, Maréva
Fèvre, Joan Vignolle et Na Liu ont été des collègues précieux sans qui une bonne partie de ce
travail n’aurait pu se faire. Jonathan Arguillos a également contribué à ce travail lors de son
stage de Licence, je tiens donc à le remercier pour sa contribution mais également pour sa
sympathie au cours de ces deux mois.
Je tiens aussi à remercier Jean (Le chimiste fou !!) pour m’avoir si bien accueilli et
aidé en tant que voisin de paillasse. Il a été d’une grande aide pour ce qui est de la synthèse
et de la compréhension des propriétés des NHCs. Nous avons également passé de très bon
moments ensemble en dehors du labo, et je pense que ce sera encore le cas quand je serais
arrivé sur le « nouveau continent ».
Même si il n’est arrivé qu’en cours de route, Joan est vite devenu un membre
indispensable de l’équipe « NHCs » de par l’expérience qu’il avait dans ce domaine. Il a été
d’une aide précieuse pour ces travaux, même si parfois le monde des polymères lui est
apparût étrange (il est encore plus tatillon que Daniel, mais on ne lui en veut pas, c’est ça
formation d’organicien qui veut ça).
L’homme aux doigts d’or qu’est Feng m’a quasiment tout appris pour ce qui est de la
manipulation en chimie des polymères. Il était toujours présent lorsque j’avais une question
de chimie, et surtout avait toujours une réponse (il est pour moi la Bible de la chimie). Il a
bien tenté de m’apprendre le chinois, comme j’ai tenté de lui apprendre le français, mais
nous avons tous les deux échoué dans ce travail. Cela ne nous a pas empêchés d’avoir de
nombreuses discussions sur nos cultures différentes et sur un tas d’autres sujets. Pour tous

9
ces moments, je tiens particulièrement à le remercier, et j’espère que les autres personnes
du laboratoire seront un jour le reconnaître à sa juste valeur.
Na m’a également appris un bon nombre de choses sur la culture asiatique, mais
surtout elle m’a fait goûter des mets succulents. Je lui souhaite toute la réussite possible
pour la fin de sa thèse.
Enfin, j’aimerais remercier chaleureusement la petite danseuse du laboratoire, celle
qui mettait une ambiance de folie à toute heure de la journée, je veux bien sûre parler de
Maréva. Ce fût une voisine de paillasse excellente, et une amie très appréciée (j’espère bien
qu’elle le restera d’ailleurs). Je lui dirais juste de faire attention à « Papi » et de ne pas trop
le casser. Surtout, je lui dis à bientôt par Skype ou en chair et en os (pense d’ailleurs à
prendre les moumoutes que tu avais pour ma soutenance ;))
J’aimerais ensuite remercier les personnes que j’ai côtoyé au cours de ces trois
dernières années au laboratoire et dont certains sont devenus des amis.
Il y a tout d’abord Bertrand, mon acolyte au cours de ces 7 dernières années. Nous
avons effectué la grande majorité de notre vie d’étudiant ensemble, et c’est pourquoi je ne
pourrais, ici, m’étendre de trop. Il a été présent dans les bons moments comme dans les
mauvais, et a surtout été d’une grande aide pour l’informatique. J’espère que ces 7
dernières années ne sont que le début d’une amitié sincère qui durera encore longtemps.
Samira a également énormément compté (et comptera encore) lors de mon séjour
au LCPO. Elle est l’autre boute-en-train du laboratoire et va bien sûre de pair avec Maréva.
Même si ce n’était pas le cas quelques mois après son arrivée, elle est aujourd’hui une amie
proche et très importante à mes yeux. Si j’avais été « hallal » je ne sais pas ce qui aurait pu
se passer… Elle aussi a su être présente lorsque ça n’allait pas, mais également lorsque tout
allait pour le mieux (puis au sport aussi, d’ailleurs, continue de faire des abdos ;)).
Je voudrais aussi remercier Célia pour tous les bons moments qu’on a passé
ensemble au labo ou en soirée, Anne-Laure pour sa gaieté et m’avoir permis de reprendre le
jogging (dommage que ça n’ait pas été pareil vers la fin), Aurélie pour son agréable
compagnie et ses conseils , Stéphane pour sa bonne humeur et sa simplicité, Babeth pour
m’avoir aidé à organiser ma soutenance et surtout pour avoir « subit » mes blagues à propos
de sa cuisine, BuiBui rappelles-toi, tout ce qui s’est passé au JEPO reste au JEPO, Marc-Ellias
pour ses blagues vaseuses et ses conseils en drague, Aurélien pour nous avoir tant fait
rigoler à « trasher » les écrans des autres, Nico pour toutes les petites blagues qu’il a pu
faire, Chantal pour ces conseils avisés en habillement, mais également Charles pour les
bonnes soirées (en partant sans payer) au titi-twister, Stéphanie pour son rire et ses
« paroles d’hommes ». Il y a aussi ceux qui sont partis ou qui ne vont pas tarder: Cédric,
Floraine, Julie, Vincent, Flu, Jérome, Romain, Platoche, Anne-Claire, Willy, Bouk, Marie-
Hélène, Matthieu, Benjamin, Géraldine et j’en oublie surement. Ils ont notamment permis
de m’intégrer au laboratoire lors de mon arrivée en Master, mais nous avons également

10
passé de très bon moments ensemble comme à Saint-Céré. Et puis il y les petits nouveaux
(plus ou moins) avec qui j’ai déjà passé de très bons moments : Antoine, Jules, Jun,
Katherina, Lise, Thomas, Chrystilla, Charlotte, Clémence, Camille, Dan, Carine, Maïté…
Les permanents du laboratoire ont également étés d’une aide précieuse pour la
réalisation de cette thèse, que ce soit par leur conseils scientifiques ou plus personnels. Je
tiens ainsi à remercier particulièrement les membres de l’équipe enseignante de la chimie et
physico-chimie des polymères à l’université, messieurs Daniel Taton, Sébastien
Lecommandoux, Eric Papon, Henri Cramail, Stéphane Carlotti, Christophe Shatz, Jean-
François Le meins, Alain Soum… qui m’ont donné l’envie de continuer dans le monde
passionnant des polymères. Je voudrais aussi remercier Frédéric Peruch pour les discussions
que nous avons pu avoir, ce qui a notamment permis de confirmer mon choix de vie
professionnelle. Merci également à Emmanuel Ibarboure et Michel Schappacher pour leur
aide technique au cours de ce travail de thèse et pour leur bonne humeur au quotidien.
Gilles Pécastaing a tout d’abord été un collègue de bureau sympathique et je suis heureux
pour lui qu’il fasse aujourd’hui parti du LCPO. Les drôles de dames qu’on ne remercie assez
souvent ont aussi été d’une grande aide au cours de ces quatres dernières années, je veux
bien sur parler de Catherine, Corinne, Bernadette, Mimiet Nicole.
Au cours de ces trois années de thèse, j’ai également eu la chance d’enseigner à
l’université Bordeaux 1. Ce fût une expérience très enrichissante qui va (je pense) largement
influencer mon parcours professionnel. Pour m’avoir accepté dans son équipe et m’avoir
transmis son savoir-faire et sa passion de l’enseignement, je tiens ainsi à remercier
particulièrement Etienne Duguet. Cette expérience a également été l’occasion de travailler
avec d’autres moniteurs très sympathiques, je pense notamment à Jérémy, Laetitia, Cédric,
Florence, Aude, Laurianne…
Enfin j’aimerai remercier ceux qui me supportent depuis 27 ans, mes parents, qui
m’ont toujours soutenu et qui ont surtout fait tout ce qu’il fallait pour que j’en arrive là. Je
suis fier d’eux et de l’éducation qu’ils ont su me donner. Ils sont pour moi un modèle à suivre
car ils ont su gérer au mieux leur vie de famille et leur carrière professionnelle sans que l’une
n’interfère sur l’autre. Ils m’ont également permis d’avoir un frère sur qui je peux compter et
qui sait parfaitement me divertir que ce soit par ses spectacles ou lors des sorties que l’on
fait ensembles.
Merci à tous !!!!
(désolé pour ceux que j’oublis).

11

12
Sommaire

13

14
Liste des abréviations employées ......................................... 20
Introduction générale ........................................................... 26
Chapitre I : N-Heterocyclic carbenes in polymer synthesis ... 32
I.1) Essential features of carbenes ................................................ 34
I.1.1) Electronic structure ....................................................................... 35
I.1.2) Synthesis ........................................................................................ 36
I.1.3) Stabilization of carbenes ................................................................ 37
I.1.3.1) Electronic stabilization .............................................................................. 37
I.1.3.2) Steric stabilization ..................................................................................... 38
I.2) N-Heterocyclic carbenes (NHCs) ............................................. 39
I.2.1) Synthesis of N-Heterocyclic carbenes ............................................ 40
I.2.1.1) Deprotonation of azolium precursors ........................................................ 40
I.2.1.2) From masked NHCs ................................................................................... 42
I.2.2) Properties ...................................................................................... 46
I.2.3) Reactivity ....................................................................................... 47
I.2.3.1) Insertion reactions .................................................................................... 48
I.2.3.2) Dimerization and addition to double bonds ............................................... 48
I.2.3.3) 1,2-Migrations .......................................................................................... 49
I.3) NHCs as organocatalysts ........................................................ 51
I.3.1) In molecular chemistry .................................................................. 51
I.3.1.1) Benzoin condensation ............................................................................... 53
I.3.1.2) Stetter reaction ......................................................................................... 53
I.3.1.3) 1,2-Additions ............................................................................................ 54
I.3.1.4) Mukaiyama-Aldol...................................................................................... 55
I.3.1.5) Transesterification .................................................................................... 56
I.3.1.6) Ring opening reactions of three-membered rings ...................................... 57
I.3.2) In macromolecular synthesis ......................................................... 59

15
I.3.2.1) Step-growth polymerizations .................................................................... 59
I.3.2.2) Ring-opening polymerizations (ROP) ......................................................... 63
I.3.2.3) Group transfer polymerization of (Meth)Acrylates .................................... 74
I.3.2.4) NHCs as building blocks for polymer synthesis ........................................... 76
Chapitre II : Polymérisation par étapes du téréphtaldehyde
catalysée par les carbènes N-hétérocycliques : synthèse de
polybenzoïnes ....................................................................... 90
II.1) Contexte et objectifs ............................................................ 92
II.2) Influence des conditions expérimentales ............................ 97
II.2.1) Choix des catalyseurs NHCs ........................................................... 97
II.2.2) Effet du solvant de réaction ......................................................... 102
II.2.3) Influence du temps de réaction ................................................... 103
II.3) Détermination de la quantité de polybenzoïnes cycliques 107
II.3.1) Principe d’étalonnage de la SEC pour la caractérisation des
polybenzoïnes. ......................................................................................... 107
II.3.2) Détermination du pourcentage de polymères cycliques. ............. 110
II.3.3) Influence des conditions expérimentales sur le pourcentage de
polymères cycliques ................................................................................. 112
II.4) Conclusion et perspectives ................................................ 117
II.5) Références ......................................................................... 119
Chapitre III : Poly(Carbènes N-hétérocycliques), poly(NHC)s,
et leurs adduits de CO2, poly(NHC-CO2)s: nouveaux
organocatalyseurs recyclables de transestérification et de
condensation de la benzoïne .............................................. 120
III.1) Introduction ....................................................................... 122
III.2) Généralités sur les polymères liquides ioniques ................ 123

16
III.2.1) Les liquides ioniques ................................................................. 123
III.2.1.1) Des combinaisons d’anions et de cations variées .................................... 124
III.2.1.2) Synthèse des liquides ioniques ............................................................... 124
III.2.1.3) Propriétés et applications des liquides ioniques ..................................... 126
III.2.2) Les polymères liquides ioniques ............................................... 127
III.2.2.1) Monomères liquides ioniques portant un groupe imidazolium................ 128
III.2.2.2) Polymérisation radicalaire des MILs ....................................................... 129
III.2.2.3) Applications des PILs .............................................................................. 131
III.3) Objectifs............................................................................. 131
III.4) Synthèse des polymères liquides ioniques précurseurs des
poly(NHC)s .................................................................................... 133
III.4.1) Synthèse des monomères de type sels de 1-vinyl-3-
alkylimidazolium ...................................................................................... 134
III.4.2) Polymérisation et changement de contre-anion des sels de
poly(vinylimidazolium) ............................................................................. 135
III.4.2.1) Polymérisation radicalaire des MILs ....................................................... 135
III.4.2.2) Polymérisation RAFT/MADIX des MILs ................................................... 138
III.4.2.3) Changement d’anion .............................................................................. 147
III.5) Synthèse et évaluation des poly(NHC)s en organocatalyse
150
III.5.1.) Choix de la base ........................................................................ 150
III.5.3) Evaluation des poly(NHC)s en organocatalyse .......................... 153
III.5.3.1) Catalyse de transestérification ............................................................... 153
III.5.3.2) Evaluation en catalyse de condensation de la benzoïne .......................... 157
III.6) Synthèse et évaluation des poly(NHCs-CO2) en organocatalyse
..................................................................................................... 161
III.6.1) Synthèse des poly(NHC-CO2)s ................................................... 162
III.6.2) Evaluation en organocatalyse ................................................... 170
III.6.2.1) Evaluation en catalyse de transestérification .......................................... 170
III.6.2.2) Evaluation en catalyse de condensation de la benzoïne .......................... 173

17
III.7) Conclusion ......................................................................... 174
III.8) Références ......................................................................... 176
Chapitre IV : Molécules et polymères dérivés des
hydrogénocarbonates d’imidazolium : nouveaux précurseurs
de carbènes N-hétérocycliques .......................................... 180
IV.1) Introduction et objectifs .................................................... 182
IV.2) Des hydrogénocarbonates d’imidazolium aux carbènes N-
hétérocycliques: une réaction réversible ..................................... 185
IV.2.1) Synthèse et caractérisation des hydrogénocarbonates
d’imidazolium .......................................................................................... 186
IV.2.1.1) Méthodes de synthèse reportées dans la littérature ............................... 186
IV.2.1.2) Synthèse d’hydrogénocarbonates d’imidazolium par changement d’anion
........................................................................................................................... 187
IV.2.2) Génération in-situ de NHCs à partir des hydrogénocarbonates
d’imidazolium .......................................................................................... 194
IV.2.2.1) Formation d’adduits NHC-CS2 à partir du iPrIm+HCO3- ............................. 194
IV.2.2.2) Synthèse du complexe Au(iPrIm)Cl à partir de Au(SMe2)Cl et de iPrIm+HCO3-
........................................................................................................................... 196
IV.2.2.3) Utilisation du iPrIm+HCO3- comme précurseurs de NHCs pour
l’organocatalyse .................................................................................................. 199
IV.3) Synthèse et évaluation en organocatalyse
d’hydrogénocarbonates de poly(vinylimidazolium)...................... 202
IV.3.1) Synthèse d’hydrogénocarbonates de poly(vinylimidazolium) ... 202
IV.3.1.1) Synthèse du ViPrIm+HCO3- ...................................................................... 203
IV.3.1.2) Polymérisation radicalaire du ViPrIm+HCO3- (Voie B) .............................. 205
IV.3.1.3) Synthèse du PVEtPhIm+HCO3- au moyen d’un changement d’anion du
PVEtPhIm+Br- (Voie C) .......................................................................................... 209
IV.3.2) Evaluation en organocatalyse des PVIm+HCO3-. ........................ 212
IV.5) Références ......................................................................... 217

18
Conclusion générale ............................................................ 218
V. Experimental part (Partie expérimentale) ...................... 224
V.1) Characterization ................................................................ 226
V.1.1) Nuclear Magnetic Resonance (NMR) ....................................... 226
V.1.2) Size Exclusion Chromatography (SEC) ...................................... 227
V.1.2.1) SEC in THF ............................................................................................... 227
V.1.2.2) SEC in water............................................................................................ 227
V.1.2.3) SEC in DMF ............................................................................................. 227
V.1.3) Thermogravimetric analysis (TGA) ........................................... 227
V.1.4) Differential scanning calorimetry (DSC) ................................... 227
V.1.5) Infra-red spectroscopy ............................................................. 228
V.2) Preparation of solvents and reactive compounds ............. 228
V.2.1) Origin and purification of chemical products ........................... 228
V.2.2) purification of solvents ............................................................ 228
V.3) Experimental protocols related to chapter II ..................... 229
V.3.1) Synthesis of NHCs .................................................................... 230
V.3.1.1) Synthesis of 1,3-diisopropylimidazol-2-ylidene (NHC-I) ............................ 230
V.3.1.2) Synthesis of 1,3-ditert-butylimidazol-2-ylidene (NHC-II) .......................... 230
V.3.2) Step-growth polymerizations of terephtaldehyde ................... 231
V.4) Experimental protocols related to chapter III .................... 231
V.4.1) Monomer Synthesis ................................................................. 231
V.4.1.1) 1-vinyl-3-isopropylimidazolium bromide (ViIPrIm+Br-) ............................. 231
V.4.1.2) 1-vinyl-3-butylimidazolium bromide (ViBuIm+Br-) .................................... 232
V.4.1.3) 1-vinyl-3-(2-ethylbenzene)imidazolium bromide (ViEtBIm+Br-) ................ 232
V.4.2) Polymerization of vinylimidazolium salts ................................. 232
V.4.2.1) Free radical polymerization ..................................................................... 232
V.4.2.2) Controlled radical polymerization (MADIX) ............................................. 233

19
V.4.5.1) Procedure for transesterification catalysed by poly(NHC)s....................... 237
V.4.5.2) Procedure for Benzoin condensation catalysed by poly(NHC)s ................. 237
V.4.6) Preparation of poly(NHC-CO2) adducts .................................... 237
V.4.7) Procedure for organocatalysis by poly(NHC-CO2)s ................... 238
V.5) Experimental protocol related to chapter IV ..................... 238
V.5.1) Preparation of imidazolium salts.............................................. 238
V.5.1.1) Synthesis of iPrIm+Br- .............................................................................. 238
V.5.1.2) Synthesis of iPrIm+HCO3- ......................................................................... 239
V.5.1.2) Synthesis of iPrImCO2 ............................................................................. 239
V.5.2) Reactions with iPrIm+HCO3- ...................................................... 239
V.5.2.1) Synthesis of iPrImCS2 from iPrIm+HCO3- ................................................... 239
V.5.2.2) Synthesis of Au(iPrIm)Cl from iPrIm+HCO3- and Au(SMe2)Cl ...................... 240
V.5.2.3) In-situ generation of NHCs from iPrIm+HCO3- to catalyze transesterification
........................................................................................................................... 240
V.5.2.4) In-situ generation of NHCs from iPrIm+HCO3- to catalyze Benzoin
condensation ....................................................................................................... 240
V.5.3) Synthesis of poly(1-vinyl-3-alkylimidazolium hydrogenocarbonate)
(PViPrIm+HCO3-) ................................................................................................... 240
V.5.3.1) Synthesis of 1-vinyl-3-isopropylimidazolium hydrogenocarbonate
(ViPrIm+HCO3-) ..................................................................................................... 240
V.6) References ......................................................................... 242
Annexes .............................................................................. 244

20
Liste des abréviations employées

21

22
AED (DSC) : Analyse enthalpique différentielle (« differential scanning calrimetry »)
AIBN : 4,4’-Azobis(isobutyronitrile)
ATRP : polymérisation radicalaire par transfert d’atome (« atom transfer radical
polymerization »)
BHET : bis(2-hydroxyéthyl)téréphthalate
SEC : Chromatographie d’exclusion stérique (« steric exclusion chromatography »)
ε-CL : ε-caprolactone
D : dispersité des masses molaires
D3 : hexaméthylcyclotrisiloxane
D4 : octaméthylcyclotetrasiloxane
DAC : diamidocarbène
DBU : 1,8-Diazabicyclo[5.4.0]undec-7-ene
DFT : théorie de la fonctionnelle de la densité (« Density Functional Theory »)
DMAPN : 3-(diméthylamino)propionitrile
DMF : diméthylformamide
DMSO : diméthylsulfoxide
: degré de polymérisation
ESI-MS : spectrométrie de masse par ionisation électrospray (« electrospray ionization mass
spectrometry»)
EO : oxyde d’éthylène (ethylene oxide)
Et2O : diéthyl éther
GTP : polymérisation par transfert de groupe (« group tranfer polymerization »)
IDIPP : 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidène
IL : liquide ionique (« ionic liquid »)
IMes : 1,3-dimésitylimidazol-2-ylidène
Im+X- : sel d’imidazolium
iPrIm+X- : sel de 1,3-diisopropylimidazolium
iPrIm : 1,3-diisopropylimidazol-2-ylidène
iPrImCO2 : carboxylate de 1,3-diisopropylimidazolium
iPrImCS2 : thiocarboxylate de 1,3-diisopropylimidazolium
KHMDS : bis(triméthylsilyl)amidure de potassium
KPS : persulfate de potassium

23
LA : lactide
LCST : température critique inférieure de démixtion (« low critical solution temperature »)
LUMO : orbitale moléculaire de plus basse énergie inoccupée (« lowest unoccupied
molecular orbital »)
MADIX: « macromolecular design by interchange of xanthate »
MALDI-TOF: « matrix-assisted laser desorption/ionisation- time of flight mass spectrometry »
Me2Ime: 1,3,4,5-tétraméthylimidazol-2-ylidène
Me2IPr: 1,3-diisopropyl-4,5-diméthylimidazol-2-ylidène
MeOH-MeOD : méthanol-méthanol deutéré
MIL : monomère liquide ionique (« ionic liquid monomer »)
: masse molaire moyenne en nombre
MTS : 1-méthoxy-2-méthyl-1-triméthylsiloxypropène
: masse molaire moyenne en masse
NCA : N-carboxylanhydride
NHC : carbène N-hétérocyclique (« N-heterocyclic carbene »)
NHC-CO2 : carboxylate d’azolium
NHC-CS2 : thiocarboxylate d’azolium
Nu-E : composé nucléophile-électrophile
NVP : N-vinylpyrrolidone
p : conversion du monomère
PB : polybenzoïne ou poly(1,4-phénylène-1-oxo-2-hydroxyéthylène)
PCL : poly(ε-caprolactone)
PDMS : polydiméthylsiloxane
PEO : poly(oxyde d’éthylène) (« poly(ethylene oxide»)
PET : poly(téréphtalate d’éthylène) (« poly(ethylene terephtalate) »)
PIL : polymère liquide ionique (polymeric ionic liquid)
pKa : constante d’acidité logarithmique
PLA : polylactide
PNVP : poly(N-vinylpyrrolidone)
Poly(NIPAAm) : poly(N-isopropylacrylamide)
PPO : poly(oxyde de propylène) (« poly(propylene oxide) »)
PPV : poly(1,4-phenylènevinylène)

24
PS : polystyrène
P2VP : poly(2-vinylpyridine)
PVIm : sel de poly(1-vinylimidazolium)
PViPrIm+X- : sel de poly(1-vinyl-3-alkylimidazolium)
r. : rendement
RAFT : transfert de chaine réversible par addition-fragmentation (« reversible addition-
fragmentation chain transfer »)
ROMP : polymérisation par métathèse d’ouverture de cycle (« ring-opening metathesis
polymerization »)
ROP : polymérisation par ouverture de cyle (« ring-opening polymerization »)
RTIL : liquide ionique à température ambiante (« room temperature ionic liquid »)
t-BuONa : tertiobutylate de sodium
t-BuOK : tertiobutylate de potassium
TEA : triéthyl amine
Tf : triflate
TGA : analyse thermogravimétrique (« thermogravimetric analysis »)
THF : tétrahydrofurane
TMS : triméthylsilyle
TPA : téréphtaldéhyde
Ts : tosylate
TSIL : liquide ionique à tâche spécifique (« task-specific ionic liquid »)
V-501 : acide 4,4'-azobis(4-cyanovalerique)
V-70 : 2,2'-azobis(valéronitrile-4-méthoxy-2,4-diméthyle)
VBuIm+X- : sel de 1-vinyl-3-butylimidazolium
VIm+X- : sel de 1-vinylimidazolium
ViPrIm+X- : sel de 1-vinyl-3-isopropylimidazolium
VPhEtIm+X- : sel de 1-vinyl-3-(2-phényléthyl)imidazolium
ZROP : polymérisation zwittérionique par ouverture de cycle (« zwitterionic ring-opening
polymerization »)

25

26
Introduction générale

27

28
La plupart des réactions de synthèse organique de la chimie moléculaire nécessitent
l’emploi d’un catalyseur, le plus souvent de nature organométallique. Dans la nature
également, les processus réactionnels sont catalysés par des enzymes autorisant des
cinétiques rapides et des sélectivités élevées. Il existe une extrème variété de catalyseurs
enzymatiques et organométalliques (créés de la main de l’homme). Cependant, les effets
néfastes de l’accumulation de certains métaux dans l’environnement ainsi que l’actuelle
inflation des ressources minières, laissent présager de la disparition progressive de ces
catalyseurs.
Au cours des vingt dernières années, des catalyseurs purement organiques,
présentant des performances comparables aux catalyseurs métalliques et enzymatiques, ont
été développés. Parmi ceux-ci, les carbènes N-hétérocycliques (NHCs)1, longtemps utilisés
comme ligands de métaux de transition pour la formation de complexes organométalliques,
ont montré d’excellentes performances comme catalyseurs en chimie moléculaire2. Plus
récemment, et notamment au LCPO (thèse de Jean Raynaud soutenue en janvier 2010) dans
notre équipe, les NHCs ont été employés avec succès pour catalyser/activer diverses
réactions de polymérisation, en particulier les polymérisations (en chaine) procédant par
transfert de groupe (GTP pour « group tranfer polymerization » en anglais) et par ouverture
de cycle (ROP pour « ring-opening polymerization » en anglais). Dans la continuité de ces
travaux, nous nous sommes intéressés ici à l’utilisation de ces catalyseurs organiques pour la
réaction de polymérisation par étapes, en l’occurrence celle du téréphtaldéhyde. Cette
partie du travail de thèse sera discutée dans le chapitre II.
Les NHCs sont des molécules neutres possédant un carbone à 6 électrons de valence.
Parmi ceux-ci, 4 sont impliqués dans deux liaisons ζ, dont une au moins avec un atome
d’azote. Les deux électrons restants, sont regroupés dans une orbitale non-liante, conférant
au carbène un état singulet. Cet état est stabilisé par le doublet de l’atome d’azote qui opère
un effet π-donneur sur l’orbitale vide du carbène.
Ainsi, malgré la présence d’une orbitale vide et
d’une orbitale pleine, les NHCs présentent un fort
caractère nucléophile, contrairement aux autres
carbènes singulets qui sont ambiphiles. Cette
nucléophilie a notamment été mise à profit par
les chimistes pour activer la fonction carbonyle d’un grand nombre de molécules. Des
réactions telles les transestérifications ou encore les réactions de type Umpolung (inversion
de réactivité de la fonction carbonyle : condensation de la benzoïne et réaction de Stetter)
ont ainsi été catalysées par les NHCs.
Une telle réactivité présente cependant l’inconvénient de rendre les NHCs très
réactifs et sensibles à l’air et à la moindre trace d’humidité provoquant leur dégradation.
Ainsi, les NHCs doivent-ils être manipulés avec précaution sous atmosphère inerte et en
milieu anhydre.

29
Un des principaux objectifs de cette thèse a été de proposer des méthodes
permettant à l’expérimentateur, non seulement de manipuler plus facilement les NHCs, mais
aussi de les recycler pour les réutiliser.
A cet effet, le chapitre III décrira comment nous sommes parvenus à fixer les
catalyseurs NHCs sur des supports polymères et ainsi générer des « poly(NHC)s » pour des
réactions d’organocatalyse en milieu homogène.
En complément, nous discuterons de l’interêt de manipuler les NHCs sous une forme
« masquée », ce qui les rend moins sensibles à l’air et aux impuretés. Diverses formes de
protection ont en effet déjà été décrites dans la littérature, notamment en utilisant le CO2
pour former des adduits de type carboxylate d’imidazolium (NHC-CO2)3. Ceux-ci sont en-
effet connus pour subir une réaction de décarboxylation sous l’action de la température,
(re)formant alors les NHCs « libres ». Ce principe
de masquage/démasquage des NHCs a été pour
la première fois appliqué dans ce travail de thèse
aux versions poly(NHC)s, de façon à induire la
formation d’adduits de type « poly(NHC-CO2) ».
Dans ce même chapitre (III), nous montrerons
que de tels adduits sont plus aisément manipulables, car moins sensibles que leurs
homologues de type poly(NHC)s, à des fins de réactions d’organocatalyse « modèles » telles
que la condensation de la benzoïne et la transestérification.
Les adduits « poly(NHC-CO2) » eux-mêmes sont susceptibles de s’hydrater, selon la
nature des substituants sur les atomes d’azote, en formant des sels d’imidazolium à contre-
anions hydrogénocarbonate (HCO3-). A notre connaissance, de tels composés n’ont jamais
été employés comme précurseurs de carbènes. Dans le chapitre IV, nous montrerons
comment obtenir de manière simple de tels hydrogénocarbonates d’imidazolium et
comment ces précurseurs peuvent servir à générer des NHCs par simple chauffage, en
éliminant de l’eau et du dioxyde de carbone !
Un attrait supplémentaire à l’utilisation de ces précurseurs tient à la réversibilité du
processus, puisqu’on peut les reformer par simple carboxylation en milieu humide (CO2 +
H2O). Des « versions moléculaires » et des homologues supportés sur chaine polymère
seront présentés dans ce chapitre. Là encore, les réactions de condensation de la benzoïne
et de transestérification serviront de terrains d’investigation, utilisant les
hydrogénocarbonates d’imidazolium comme précatalyseurs organiques.
Ainsi, le chapitre I de ce manuscrit a pour objet d’éclairer le lecteur sur les spécificités
des carbènes N-hétérocycliques (NHCs). Il décrit en premier lieu les propriétés de ces
espèces ainsi que leurs méthodes de synthèse. L’état de l’art en matière de catalyse
organique par les NHCs est également présenté, mais de manière succincte, l’accent étant
mis plus particulierement sur l’utilisation récente des NHCs en chimie des polymères.

30
Comme déjà indiqué, le chapitre II est consacré à la polymérisation par étapes du
téréphtaldéhyde catalysée par les NHCs. Il illustre notamment comment la condensation de
la benzoïne a pu être étendue a un monomère de type bis-aldéhyde pour former des
« polybenzoïnes » de structure linéaire et cyclique.
Le chapitre III décrit la synthèse des « poly(NHC)s » par le biais d’une réaction de
déprotonation de sels de poly(vinylimidazolium). Ces derniers font partie de la famille des
« polymères liquides ioniques » dont une description succincte est faite en première partie
de ce chapitre. L’application de ces NHCs supportés à l’organocatalyse, ainsi que leur
protection par du CO2 complètent cette étude.
Au cours du chapitre IV, une nouvelle méthode de synthèse des
hydrogénocarbonates d’imidazolium, en version moléculaire comme en version polymère,
sera présentée. Il y est également démontré que ces composés permettent la génération in
situ de NHCs à des fins d’organocatalyse et de formation de complexes organométalliques.
Le lecteur retrouvera tous les détails permettant de reproduire et d’analyser les
produits décrits dans ce manuscrit dans la partie expérimentale. Les figures qui n’ont pas pu
être intégrées à certains chapitres sont quant à elles montrées en annexes.
1. Bourissou, D.; Guerret, O.; Gabbai, F. P.; Bertrand, G. Chemical Reviews 2000, 100, (1), 39-91. 2. Moore, J. L.; Rovis, T., Carbene catalysts. In Topics in Current Chemistry, 2010; Vol. 291, pp 77-144. 3. Delaude, L. European Journal of Inorganic Chemistry 2009, (13 SPEC. ISS.), 1681-1699.

31

32
Chapitre I : N-Heterocyclic carbenes in polymer synthesis

33

34
This chapter is aimed to be published as a highlight article that will be submitted by
the end of this year. In the first part, we describe the essential features of carbenes,
including their electronic properties, with a special focus on N-heterocyclic carbenes (NHCs).
The methods of their synthesis as well as their use as organic catalysts in molecular
chemistry are also briefly covered. The main purpose of this review is to give an account of
the past eight years in the utilization in polymer chemistry of carbenes, more particularly of
NHCs, that have emerged as very powerful organocatalysts of polymerization reactions, or
as building blocks bringing specific properties in polymer synthesis. The field of
organometallic chemistry, where carbenes have been extensively employed as ligands of
transition metal-based complexes for a wide variety of reactions (mostly C-C bond forming),
will not be discussed here but can be found in appropriate reviews1-6.
I.1) Essential features of carbenes
Carbenes are divalent species of carbon, highly reactive with a lifetime typically
under 1s (0,1-1 ns)7. Despite of this high reactivity, particular carbenes could be trapped in
matrices at low temperatures (<77 K), which has allowed their characterization via transient
absorption spectroscopy7. Carbenes were first mentioned in 1835 by Dumas who tried to
generate methylene carbene (CH2:), 1, by dehydration of methanol (Scheme I.1)8. Since this
work, many efforts have been directed towards isolation and stabilization of free carbenes.
Specific carbenes could be finally synthesized, characterized and isolated in the late 1980’s9-
11. The first was the phosphinosilyl-carbene, that is part of the family of the acyclic
phosphinocarbenes (5, Figure I.1), synthesized by Bertrand et al. in 198811. This work was
just followed by the isolation of 1,3-diadamantylimidazol-2-ylidene (6, with R = Ad and H), as
the first NHC isolated by Arduengo et al.12 Other carbenes like triplet diaryl carbenes (2,
Figure I.1) and acyclic aminocarbenes (4, Figure I.1) have also been characterized, but could
never been isolated. For more detailed information, the reader can refer to the special issue
of Chemical Reviews published in 200913-17.
Scheme I.1 : Attempt to dehydrate methanol for the generation of methylene carbene

35
Figure I.1 : Structure of some carbenes (NHCs are shown in red)10, 15, 17.
I.1.1) Electronic structure
Carbenes are neutral compounds with a divalent carbon possessing six peripheral
electrons in its valence shell. Four of them are involved in two σ-bonds while the two non-
bonded electrons remain at the central carbon10.
In the singlet ground state, these 2 electrons are paired in a σ2-type orbital
which is generally more stable than the pπ2-type orbital (structures A and B in Figure I.2).
These carbenes thus exhibit an ambiphilic character (can react as an electrophile or a
nucleophile) as they have a filled and a vacant orbital. They generally show an acute carbene
bond angle and adopt a sp2-hybridization. Note that an excited singlet state with a
configuration σ1pπ1 can also be envisaged (Structure C Figure I.2).
When one electron is located in each orbitals, these species are referred to as
triplet carbenes (structure D, Figure I.2) and can be described as diradicals. These triplet
carbenes exhibit an almost linear geometry and, in this case, adopt a σ1pπ1 configuration.
The latter configuration is almost equivalent to a px1py
1 as the p orbitals are degenerated
due to the quasi sp hybridization at the carbene center.

36
Figure I.2 : Electronic configurations of carbenes and relationship between the carbon bond
angle and the frontier of orbitals.
I.1.2) Synthesis
A common synthetic strategy to free carbenes is by α-elimination of haloalcanes 10, 17-
19. This method can be achieved either by using a strong base or upon thermolysis of the
precursor. A typical example is the treatment of chloroform by a base to produce the
dichlorocarbene (3, Scheme I.2)15, 17.
Scheme I.2 : The α-elimination reaction of HCl from chloroform, generating dichlorocarbene.
As for triplet carbenes, they are generally obtained by decomposition of diazo
compounds10. Some singlet carbenes can also be prepared by the latter method and upon
thermolysis or photolysis as well. In these processes, the precursor loses a N2 molecule,
giving the corresponding carbene (reaction A, Scheme I.3)9, 10, 17. Likewise, ketene20 and
diazirine21 compounds can serve as precursors releasing a CO and a N2 molecule
respectively, thus producing the targeted carbene (reactions B and C, Scheme I.3).

37
Scheme I.3 : Generation of carbenes by decomposition of diazo-type (A), ketene-type (B)
and diazirine-type (C) compounds.
Different and complementary strategies to synthesize NHCs have been reported and
will be discussed in section I.B.2.
I.1.3) Stabilization of carbenes
Stabilization of carbenes can be achieved either through electronic stabilization or
through steric stabilization.
I.1.3.1) Electronic stabilization
Substituents on a given carbene dictate its ground state multiplicity. Both inductive
and mesomeric effects (mesomeric effects being generally more important than inductive
ones) determine the σ-pπ gap hence the ground state multiplicity10, 17.
Inductive effect generally occurs by increasing the electronegativity of the
substituent. The σ-electron withdrawing substituents (electronegative atoms like halogens
or nitrogen) favor the singlet state by stabilizing the σ non-bonding orbital, thus increasing
the σ-pπ gap (Figure I.2). On the other hand, σ-electron donating subtituents (like Li or H)
lead to a decrease of the σ-pπ gap.
In the case of mesomeric effects, interactions between orbitals of the carbon and
appropriate p-type orbitals of the subtituents have to be considered. For this purpose,
substituents can be classified into two categories10:
π-electron-donating groups, namely X (-F, -Cl, -Br, -I, -NR2, -PR2, -OR, -SR, …) which
interact with the vacant orbital of the carbene. For example, this situation is occurring in
diaminocarbenes and NHCs.
π-electron-withdrawing groups, namely Z (-COR, -CN, -CF3, -BR2, -SiR3…) which
interact with the filled orbital of the carbene, like in the case of diborylcarbenes (never been
isolated).

38
Therefore, singlet carbenes can be classified in (X,X)-, (Z,Z)- and (X,Z)-carbenes, depending
on their substituents (Figure I.3):
Figure I.3 : Representation of the different electronic effects occurring in the stabilization of
singlet carbenes.
In (X,X)-carbenes, interactions between the vacant pπ orbital and the substituent lone-pairs
increase both the pπ orbital energy and the σ-pπ gap. The singlet ground state is thus favored
and these carbenes are predicted to exhibit a bent conformation. This is the case for N-
heterocyclics carbenes.
In the case of (Z,Z)-carbenes, interaction between vacant orbitals of the substituent and the
filled py orbital of the carbene is predominant. Thus the px orbital is not affected and the
degeneracy (px,py) is broken. By this way, (Z,Z)-carbenes possess a singlet ground state (pπ2).
Such carbenes coulds be characterized but have never been isolated.
When (X,Z)-carbenes are considered, the two electronic interactions described above
prevail. The lone pair of the X substituent interacts with the py vacant orbital which is thus
destabilized, and the vacant orbital of Z stabilizes the filled px orbital. This leads to an
increase of the σ-pπ gap that stabilizes the carbene center, and favors the singlet state.
I.1.3.2) Steric stabilization
Bulky substituents allow for a kinetic stabilization of carbenes10. When electronic
effects are negligible, steric effects also affect their ground state multiplicity. As emphasized
above, a linear geometry degenerate the pxpy orbital and favors the triplet state. So, by
increasing the steric hindrance of the substituents, the carbene bond angle is broadened and
the triplet state is favored. A good illustration of this effect is provided with the three
following alkyl-carbenes: dimethylcarbene (1)22, di(tert-butyl)carbene (10)23 and
diadamantylcarbene(11)24 (Figure I.4). These three species present theoretical bond angles
of 111°, 143° and 152° respectively, and only 1 exhibit a singlet state.

39
Figure I.4 : Dimethylcarbene, di(tert-butyl)carbene and diadamandtylcarbene with their
bond angle, showing the subtituent effect on the triplet or singlet state.
I.2) N-Heterocyclic carbenes (NHCs)
NHCs can be well stabilized through the electronic effects of the substituents on the
nitrogen atoms. The σ-electron withdrawing effect of nitrogen character combined with the
π-electron-donating give a large singlet to triplet gap and thus a stable singlet ground state.
This has lead NHCs to be fully characterized.
Ronald Breslow first thought of these carbenes in 195825. In particular, he suggested
that the active site of the coenzyme Thiamine (vitamin B1) during the acyloin/benzoin
condensation (see further in section I.C) was a carbene arising from the deprotonation of the
thiazolium ring (Figure I.5). Since this ground breaking paper, much research has been
conducted with the aim at developing and isolating stable NHCs that would be able of
catalyzing the benzoin condensation stereospecifically26.
Figure I.5 : Structure of the coenzyme Thiamine (vitamin B1) and its active carbenic form.
Attempts at isolating NHCs date back to the early 1960s with the studies of Wanzlick
and coworkers10, 27. This group has specifically investigated thiazol-2-ylidenes, imidazol-2-
ylidenes and imidazolin-2-ylidenes (Figure I.6). However, only the dimeric forms of these
NHCs could be isolated. The authors thus assumed that free carbenes were necessarly
involved in the so-called “Wanzlick’s equilibrium”28 (see section I.B.3). After some debate29-
31, this equilibrium has been evidenced three decades later by Denk et al.32. Finally, by
playing on nature of the substituents of the nitrogen atoms, Arduengo et al. succeeded in
synthesizing, characterizing and isolating the first NHCs as crystalline compounds in the early
1990s 12, 33.

40
Figure I.6 : Chemical structure of typical NHCs (6-9, 12)10,26and recently described NHCs (13-
16)34-36.
I.2.1) Synthesis of N-Heterocyclic carbenes
Three main strategies exist to prepare NHCs and their supported analogues or to
generate them in a reaction media (in situ generation).
I.2.1.1) Deprotonation of azolium precursors
The first stable NHC synthesized by Arduengo et al. in 1991 was the 1,3-di-
adamantylimidazol-2-ylidene (6, with R= Ad., Figure I.6)12. It could be obtained by
deprotonation of the imidazolium salt precursor, using one equivalent of NaH and a catalytic
amount of either DMSO or t-BuOK in THF. This reaction can also be performed in other
solvents and with others bases in a way to favor deprotonation and isolation of the final
product (Scheme I.3)10, 18, 37. Deprotonation of various commercially available azolium salt
precursors in order to produce carbenes is today a common method. This strategy is often
used for the in-situ generation of NHCs, but in this case, one has to keep in mind that
byproducts (bases or metallic compounds) might interfere with further undertaken
reactions.
Scheme I.3 : Generation of imidazol-2-ylidenes by deprotonation of azolium salts.
Recently, new classes of NHCs have been derived following such a strategy. Alder et
al38. and Stahl et al.36 have reported the synthesis of six- and seven-membered-ring-NHCs
(14, Figure I.6). Diamidocarbenes (13, Figure I.6) have also been described by Bielawski and

41
coll.20, 39. Lately, ionic diamidocarbenes 15 have been synthesized by Lavigne et al34. On their
side, Bertrand et al. have reported an original synthesis of mesoionic NHCs 16, using “click
chemistry” to synthesize the salt precursor35.
Alternatively, implementation of biphasic system (ionic liquid/THF) allows for the
migration of the generated NHC from the ionic liquid phase to the organic phase (THF)40, 41.
Interestingly, benzimidazolium salts featuring N-substituted long alkyl chain recently proved
efficient organic pre-catalysts to perform benzoin condensation in water (17, Scheme I.4) 42.
It was proposed that micellization in water of the iminium precursor, allowed for a
confinement of the corresponding NHC in the core of the micelle.
Scheme I.4 : Benzoin condensation in water catalyzed by benzimidazolium salts in the
presence of TEA.
Imidazolium salts, supported on either inorganic silica or most often onto organic
polymeric supports, which are further deprotonated in-situ can also be used as precursors of
supported organometallic complexes3, 43. It is interesting to note that the latter technique
has been rarely applied for the purpose of organocatalysis44-47. However, only a few reports
have described the direct synthesis of polymeric-supported NHCs (Figure I.7) or their
imidazolium analogues used as polymeric-supported pre-catalysts (Figure I.8). In all cases,
insoluble cross-linked polymers i.e. supports were employed, allowing for an easy recovery
of the (pre)catalysts by simple filtration 45, 47.
Figure I.7 : Synthesis of polymeric supported NHCs for the purpose of organocatalysis.

42
Figure I.8 : Polymeric supported imidazolium salts used as pre-organocatalysts.
I.2.1.2) From masked NHCs
Alternatively, synthesis of NHCs can be achieved by thermolysis of NHC adducts
(Scheme I.5). Upon heating, such precursors can indeed be subjected to an α-elimination
reaction, allowing the production of NHCs without the need for a strong base. This method
was first developed by Wanzlick et al. and was also used by Enders et al. to obtain the first
commercially available NHC in the late 1990s48. Heating 18 (with R = Ph) at 80 °C under
vacuum (0,1 mbar) releases methanol, forming the corresponding 1,2,4-triazol-5-ylidene (9,
Figure I.6). This strategy can also be applied to produce imidazolin-2-ylidenes (7, Figure I.6)26,
40, 49.
Scheme I.5 : Generation of NHCs from cleavable adducts (“masked” carbenes).

43
The latter method was further exploited by the Hedrick (IBM Almaden Research Center) and Waymouth (Stanford university) group in a way to generate carbenes catalysts
for ring opening polymerization of cyclic esters (see further section I.C.2.2)40. Other adducts
such as 19, obtained from a 1:1 reaction between imidazolin-2-ylidene 7 and alcohols, can
also be cleaved to release the free NHC27, 40, 49. It has to be mentioned, however, that
“protection” of NHCs with alcohols is specific to saturated NHCs 7.
For instance, Hedrick, Waymouth et al. developed NHC adducts featuring
halogenated substituents in C-1 position (e.g 23 and 25)50. In the solid state, adducts 25
were found to cleave at temperatures between 80 °C and 165 °C, depending on the
fluorinated substituent and the carbene used.
Silver(I)-based NHC complexes such as 24 have also been used as precursors to
synthetic free carbenes. Upon heating, the complex is decomposed into free NHCs while
AgCl precipitating out of the solution. This method has been successfully applied to trigger
the ROP of lactides through a NHC catalysis40. In organometallic chemistry, such silver-based
complexes are commonly used as transmetallating agents (ii, Scheme I.6). They can be
readily obtained by reacting silver oxide and imidazolium salts, as shown in Scheme I.651, 52.
Scheme I.6 : Synthesis of NHC-silver complexes from imidazolium halides and silver oxide.
Among “masked” carbenes, betaines adducts53 are certainly the most commonly
employed. NHC adducts of carbon disulfide (e.g 20, Scheme I.5) are stable adducts that
began to decompose at 200 °C only. For this reason, such adducts have never been applied
for in-situ generation of NHCs. Nonetheless, NHC-CS2 adducts have often served as probes to
evidence the presence of free carbenes in a given media50, 53, 54. Indeed, CS2 can react
quantitatively react with free NHCs (Scheme I.5), forming characteristic red crystals.
Other important NHC-betaines adducts are compounds 21 resulting from the
addition of carbon dioxide (CO2) with virtually all types of carbenes53. These NHC-CO2
adducts have attracted an increasing interest in molecular chemistry53, 55-64, in particular due
to the fact that related carbenes can be released upon heating at 50 °C in solution64 and 80
°C in bulk59. These carboxylate iminium-type adducts also represent convenient precursors
to prepare NHC metal complexes55, 60. In addition, they can serve as precatalysts for the
preparation of cyclic carbonates (Scheme I.7)56, 64.

44
Scheme I.7 : Synthesis of carbonates catalyzed by NHC-CO2 adducts63, 64.
In complement, Buchmeiser et al. have taken benefit of the easy formation of such
betaine-type adducts to achieve polymeric-supported NHCs by ring-opening metathesis
polymerization of a norbornene-based monomer (Scheme I.8)65. The corresponding
crosslinked polynorbornene-type resin supported precatalysts were found to catalyze the
cyanosilylation reaction of carbonylated reagents, under heating at 65 °C for 1 h65. No
attempts to recover and reuse these polymeric-supported catalysts have been mentioned.
Scheme I.8 : Structure of the polynorbornene-based supported NHC-CO2 derived by
Buchmeiser et al.65.
In this PhD thesis work, we also made use of such a strategy (CO2 “protection”) not
only to better handle NHCs but also to recycle them for a purpose of organocatalysis. In our
case, we employed soluble polymeric-supported NHC, referred to as poly(NHC)s, as potential
recyclable polymeric catalysts for both the benzoin condensation and the transesterification
reactions. These aspects will be described in chapter III.
Finally, existence of weak interactions between NHC and silicon atoms of
polydimethylsiloxane (PDMS) was exploited to encapsulate/protect both chemically and
physically NHCs in a PDMS-type oil, offering a convenient mean to manipulate NHC the

45
catalysts66. Density functional theory (DFT) and ab-initio calculations of the binding between
NHC and PDMS oligomers have shown that these interactions are very weak, about 2.8
kcal.mol-1, and that the Si-CNHC distance are rather large, between 1.982 Å and 1.988 Å
(Figure I.9), compare to other Si-NHC bonds (1.911 Å with tetrachlorosilane). The NHC-
polymer mixture (22, Scheme I.5) could thus be readily handled in air, without any particular
precautions; after solubilization, the free carbene could be released.
Figure I.9 : Interaction between NHC and PDMS.
I.2.1.3) Chemical or electrochemical reduction of imidazolium salts
Reduction of imidazolium salts or imidazol-2(3H)-thiones also leads to free NHCs. This
was first implemented by reacting potassium with sulfur adducts 26 in boiling THF (Scheme
I.9), as demonstrated by Kuhn et al.67. Similarly, Clyburne et al. found that 27 could be
reduced both electrochemically or chemically by metallic potassium (Scheme I.10)68.
However, the latter strategy has not been generalized to other imidazolium salts.
Scheme I.9 : Synthesis of NHCs by chemical reduction of imidazol-2(3H)-thiones.
Scheme I.10 : Synthesis of the dimesitylimidazol-2-ylidene 28 by chemical or electrochemical
reduction of the corresponding imidazolium salt.

46
I.2.2) Properties
With a vacant (pπ) orbital and a filled (σ) one, NHCs are expected to exhibit both a
nucleophilic and an electrophilic character. However, because of the strong interaction
between the nitrogen atoms and the pπ orbital, their electrophilic character is dramatically
reduced. Thus, NHCs are categorized as strong nucleophiles and excellent Lewis bases69.
Their pKa have been determined both experimentally and theoretically and are in the
range 15-30. Obviously, the pKa values of NHCs can be correlated with the nature of the
substituents on the nitrogen atoms, but also with the carbene bond angle (NCN), as
illustrated in Figure I.10. Electron-withdrawing groups on the nitrogen atoms decrease the
pKa value of NHC (pKa29pKa30pKa31>pKa32). Five-membered NHCs are also
characterized by lower pKas than their six-membered counterparts (pka34>pKa33). This
difference can be explained by the broader angle of NHC 34 (116°) compared to that of NHC
33 (102°) which lead to an increase of the p character of the ζ orbital, and thus to an
increase of the carbene basicity.
Figure I.10 : Structures and theoretical pKa values of some NHCs in DMSO69.
The structure of NHCs has also been evidenced by X-ray diffraction10, which has
revealed that the nitrogen atoms are in a planar environment. NHCs also exhibit rather short
N-C bond lengths (1.32-1.37Å) compared to their azolium precursors (1.28-1.33Å). This
feature indicates that N-C bonds possess a double bond character, resulting from the
nitrogen lone pair donation to the vacant orbital of the carbene 10. This strong interaction
tends to increase the LUMO energy. The range value of 100-120° for N-C-N bond angles
determined by X-ray diffraction studies is in good agreement with theoretical calculations for
a sp2 hybridized carbene. Thereby, NHCs are best described by the resonance forms II and III,
which can be summarized by structure IV (Figure I.11).

47
Figure I.11 : Resonance forms of NHCs.
Another feature of NHCs is the low field chemical shift of the carbene center in 13C
NMR spectroscopy70. The resonance of the carbonic center indeed appears at lower field (δ
= 205-260 ppm) compared to the corresponding atom of the iminium precursors (δ = 130-
160 ppm). The 13C NMR signals corresponding to thiazol-2-ylidenes and ring-expanded NHCs
are the most deshielded (δ = 240-260 ppm). Imidazolin-2-ylidenes resonate in a range of
235-245 ppm while the carbene signal of imidazol-2-ylidenes appears at higher fields (δ =
205-220 ppm).
I.2.3) Reactivity
Most of NHCs are air and moisture sensitive. Even if they cannot react with triplet
dioxygene, the ring structure is disrupted by hydrolysis producing a formamide compound
36 (Scheme I.11)71. Nevertheless, Arduengo and coll. Succeeded in generating an air stable
NHC (37, Scheme I.12) by reacting 1,3-dimesitylimidazol-2-ylidene with carbon
tetrachloride72.
Scheme I.11: Hydrolysis of NHCs.
Scheme I.12 : Synthesis of the “air-stable” 1,3-dimesityl-4,5-dichloroimidazol-2-ylidene 3772.

48
I.2.3.1) Insertion reactions
Unlike transient carbenes, NHCs cannot undergo cyclopropanation or insertion
reactions. This difference in reactivity can be explained by their non-ambiphilic character. In
contrast, NHCs can insert into activated A-H bonds only (A = O,N)18, and acidic C-H bonds
(Scheme I.13), by occurring a protonation (i, Scheme I.13) and a nucleophilic addition (ii,
Scheme I.13) . Moreover, these insertion reactions can be achieved with NHCs 948 and 773
but not with unsaturated NHCs 6 and 8 (Figure I.6). For the latter NHCs indeed, protonation
of the carbene (i) increases the aromatic character of the ring, hence, disfavor the addition
of A-.
Scheme I.13 : Stepwise insertion of NHCs into A-H bonds.
I.2.3.2) Dimerization and addition to double bonds
As already mentioned, NHCs have a tendency to dimerize which is referred to as the
Wanzlick equilibrium. NHCs 7 and 8 (Figure I.6) are particularly prone to such a dimerization
reaction74-76. Evidences for such equilibrium has been reported by Lemal et al. who
performed “cross-over experiments”29. For this purpose, heating a mixture of adducts 38
and 39, none mixed dimer 40 was formed in the presence of potassium hydride (Scheme
I.14). Thus, success in performing such reaction by Denk et al.32 has been attributed to the
presence of traces of acids acting as catalysts. As a matter of fact, it is likely that the
mechanism proceeds via a nucleophilic attack of one carbene molecule upon its conjugate
acid, followed by proton elimination (Scheme I.15).75, 76
Scheme I.14 : “Cross-over experiments” performed by Lemal et al.29

49
Scheme I.15 : Wanzlick’s equilibrium of imidazolin-2-ylidenes (NHCs 7).
Advantage of the nucleophilic character of NHCs has been also used in reactions
involving electrophilic alkenes18. In contrast to transient singlet or triplet carbenes and stable
monoaminocarbenes which react by forming cyclopropane derivatives ([1+2]-
cycloaddition)10, NHCs lead to methylene(tri)diazoline derivative (41, Scheme I.16)18.
Scheme I.16 : Addition of 1,2,4-triazol-5-ylidenes (NHCs 9) to activated C-C double bonds.
I.2.3.3) 1,2-Migrations
NHCs can also react with themselves in a different manner than in the Wanzlick’s
equilibrium, that is, through a 1,2-migration reaction (Scheme I.17). The first step in this
reaction consists in a nucleophilic attack of the carbene center to the substituent of an
adjacent heteroatom (most often a nitrogen atom)10, leading to compound 43 in Scheme
I.17. Then a transfer of the substituent (E) between 43 and 44 occurs with aromatic NHCs (6,
8 and 9, Figure I.6) via an intermolecular process. This 1,2-migration has only been
demonstrated for precursors featuring a hydrogen atom or a trialkylsilyl groups as
substituents (E), leading to compound-types 45.

50
Scheme I.17: 1,2-migrations in NHCs.
Recently, Bielawski et al. have shown that special diamidocarbenes 46 (DACs) can
split amonia forming primary amine derivatives, as shown in Scheme I.1839. This new class of
NHCs thus exhibit a different reactivity compared to common NHCs. Their electrophilic
character is indeed enhanced by the amido substituents of the carbene center, hence they
can react like acyclic amino carbenes77. Note that cyclic alkylamino carbenes (CAACs)
developed by Bertrand et al. were also shown to activate NH3 and H277. It has also been
reported that such six-membered ring diamidocarbenes (DACs) can form
diamidoketenimines through addition with isocyanides78.
Scheme I.18 : Split of ammonia by diamidocarbenes (DACs).

51
I.3) NHCs as organocatalysts
Use of NHCs as organocatalysts has began with the work of Ukai et al. regarding the
thiamine-catalyzed benzoin condensation in 194379. Lates Breslow has reported that this
catalysis involved the formation of a carbenic center25, the catalytic, synthetic and
mechanistic aspects of this reaction have been extensively investigated19, 26, 80, 81.
Besides their role as ligands for various transition metals, NHCs are nowadays widely
employed as powerfull organocatalysts for a variety of chemical transformations (Scheme
I.19)19, 26, 78, 82-90.
In the next section, NHC-catalyzed reactions in molecular chemistry will be described
only when such background reactions could be extended to polymerization reactions. The
reader can found more information about other NHC catalyzed reactions in excellent
reviews by the groups of Enders26, Nolan19 or Rovis82. In the second part, NHC-catalyzed
polymerization reactions will be overviewed as well as the use of NHCs as building blocks for
polymer synthesis.
I.3.1) In molecular chemistry
The use of NHCs as organocatalysts for organic transformations has received an
exponentially growing attention in the past ten years. NHCs have been found effective in a
variety of reactions such as condensation reactions, 1,2 additions, transesterification and
ring-opening reactions, among others. In most cases, it is important to note that these
reactions relate to an activation of a carbonyl group by the carbenic center. Scheme I.19,
though non exhaustive, illustrates the wide range of elementary reactions where NHCs were
reported to play a role as organocatalysts.
With aldehydes as substrates, addition of the carbene to the carbonyl leads to the
formation of the intermediate 47 (Scheme I.20), referred to as the “Breslow intermediate”.
This transient specie features an inverted polarity with regards to the carbonyl group, since
the carbon of the C=O becomes nucleophilic. It can thus be involved in so-called “Umpolung”
(German translation of polarity inversion) reactions, like in the benzoin condensation and
the Stetter reactions19, 26.
Scheme I.20 : Formation of the “Breslow intermediate” induce in both the benzoin
condensation and the Stetter reactions.

52
Scheme I.19 : Typical of NHC-catalyzed reactions in molecular chemistry.

53
I.3.1.1) Benzoin condensation
The first investigations of the benzoin condensation date back to 1832 with the works
by Wöhler and Liebig who studied the cyanide-catalyzed homocoupling of benzaldehyde26.
The mechanism of benzoin reaction does not depend on the nature of the catalyst (cyanide
or carbene), but involves the “Breslow intermediate” mentioned above (47, Scheme I.20) 25,
91. These nucleophilic species can further react with the electrophilic carbon of a second
aldehyde to form an adduct intermediate (48, Scheme I.21). Finally, elimination of the keto-
alcohol 49 allows regenerating of the catalyst. As benzoin condensation creates a
stereogenic center, a wide variety of catalysts has been developed in a way to induce a
stereoselective reaction. High selectivities (> 90% ee) can be well achieved with various
chiral NHCs.
Benzoin condensation can also be performed through an intramolecular fashion
forming cyclic β-keto-alcohol derivatives (Scheme I.22)19, 26, 82.
Scheme I.21 : Mechanism of the benzoin condensation.
Scheme I.22 : Intramolecular benzoin condensation leading to cyclic β-keto-alcool.
During this PhD work, the principle of this reaction has been extended to the step-
growth polymerization of a bis-aldehyde, namely terephtaldehyde. This will be the topic of
chapter II of this manuscript.
I.3.1.2) Stetter reaction
The concept of the benzoin condensation can also be applied to substrates consisting
of Michael acceptors, as originally established in the early 1970’s by Stetter et al., to achieve
1,4-diketones (51, Scheme I.23). The 1,4-addition of an aldehyde to an acceptor bearing an
activated double bond is thus referred to as the “Stetter reaction”. As in the benzoin
condensation, a catalytic pathway involving a Breslow intermediate (47, scheme I.23) has

54
been suggested; Subsequent addition of this Breslow intermediate to the α,β-unsaturated
ketone 50 by a nucleophilic attack. The reaction can also be performed with α,β-unsaturated
nitriles (52, Scheme I.24) and esters (53, Scheme I.24) to obtain 4-ketonitriles 54 and 4-
ketoesters 55, respectively. More information about substrates and related products that
can be accessed by the Stetter reaction can be found elsewhere19, 26, 82.
Scheme I.23 : Mechanism of the Stetter reaction.
Scheme I.23 : Formation of 4-ketonitriles and 4-ketoesters via the Stetter reaction.
I.3.1.3) 1,2-Additions
Other NHC-catalyzed reactions involving activation of carbonyl group include 1,2-
additions of aldehydes or ketones with appropriate nucleophiles19, 26, 82. In these reactions,
the alcoholate-type specie (56) formed after addition of the carbene to the carbonyl is
subsequently trapped by an electrophilic group (Scheme I.25).
Such 1,2-addition reactions can be conducted by using a Nu-E-type reagent
(Nu=nucleophile, E=electrophile) e.g. trimethyl(trifluoromethyl)silane or
trimethylsilylcyanide and leads to siloxanes 57 and 58.
During such reactions, activation of the trimethylsilyl group through the formation of
a pentavalent silicon center (61, Scheme I.26) can also be envisaged92, 93. These reactions are
generally quenched by an acid to obtain the corresponding alcohols (59 and 60, Scheme
I.25).
Scheme I.24 : NHC-catalyzed 1,2-addition reactions, through carbonyl activation (TMS =
trimethylsilyl).

55
Scheme I.25 : NHC-catalyzed silylation of aldehydes or ketones, through a pentavalent
silicon center (Nu = CN or CF3)92, 93.
I.3.1.4) Mukaiyama-Aldol
Besides carbonyl groups, NHCs can also activate the Si-O bond, in an identical fashion
to Lewis bases in the so-called catalyzed Mukaiyama aldol-type reactions. NHCs have been
reported to catalyze this reaction, presumably via the activation of the trialkylsilyl moiety of
the substrate involved (Scheme I.27)94.
Generally speaking, the Mukaiyama aldol consists in a C-C bond formation through a
catalyzed reaction between a silyl ketene acetal (forming an enolate-type specie, through
the catalyst activation) and an acceptor (a ketone or an aldehyde). A wide range of co-
catalysts (including Lewis acids and Lewis bases) can be employed, as reviewed by
Mukaiyama et al.95. With NHCs as catalysts, the pentacoordinate silicon in compound 62 is
thought to split to an enolate 64 and a silylated carbene moiety 63. Thus, the nucleophilic
coumpound 64 is allowed to attack the carbonyl group of a ketone 65, leading to the
alkoxide intermediate 66. Finally, the latter specie recovers the trimethylsilyl moiety
regenerating the NHC catalyst. The reaction is generally quenched with hydrochloric acid,
yielding monoadduct 67. Avoiding the last step of this reaction, i.e hydrolysis of silyl group,
silyl-enol-ethers are finally obtained96.
Scheme I.26 : Mukaiyama aldol between a silyl ketene acetal and carbonyl compound,
catalyzed by NHC94.

56
I.3.1.5) Transesterification
NHCs have also been found to efficiently catalyze transesterification reactions
involving an ester-type substrate and an alcohol19, 26, 82, as reported by Hedrick, Waymouth
et al.97 and by Nolan et al.98. High yields with a low catalyst loading can be reached.
Two mechanisms have been proposed to account for the reaction pathway. The NHC
catalyst can activate either the carbonyl group of the ester substrate (Path A Scheme I.28) or
the alcohol (Path B in Scheme I.28). In path A, the nucleophilic attack of the carbene to the
carbonyl group gives an azolium salt intermediate, 68, with an alkoxide as counter anion.
After protonation of this anion in the presence of another alcohol molecule, compound 69 is
produced while the alcohol 70 is released. A subsequent nucleophilic attack of the alkoxide
to the carbonyl of 69 leads to the formation of the targeted ester, 73.
The putative mechanism in path B does not form intermediates, strictly speaking.
Here the carbene would rather behave as a deprotonating agent of the alcohol (71, Scheme
I.28), followed by addition onto the carbonyl of the ester. After rearrangement (72) and
release of the alcohol 70, the ester 73 is obtained. The latter mechanism has been evidenced
by NMR characterization and X-Ray diffraction of complexes between various alcohols and a
peculiar NHC, namely 28 (Scheme I.10).
This reaction was later applied not only to secondary alcohols99, but also to amino
alcohols100. In the latter case, the authors have suggested that an unusual O-N acyl transfer
takes place, which leads to amide-type compound (74, Scheme I.29).
It has to be noted that esters and amides can also be synthesized using NHCs as
catalysts, in an oxidation reaction between an aldehyde84 and an alcohol101 or a nitroso
compounds102, respectively.
Scheme I.27 : Two proposed mechanisms for the NHC-catalyzed transesterification reaction.

57
Scheme I.28 : Amidation of esters by amino alcohols100.
I.3.1.6) Ring opening reactions of three-membered rings
Epoxides, aziridines and cyclopropanes have been also transformed through NHC-
triggered ring-opening reactions 19, 26, 82, 103, 104. In the case of the ring opening alkylation of
meso-epoxides (75, Scheme I.30), the role of NHC remains unclear as four different catalysts
(76-79) were effective to promote this reaction, including the imidazolium salt precursor 77.
Moreover, the presence of triethylaluminium, which can promote the alkylation of the
epoxide, leads to NHC-Al complexes, such as 78. The latter complex was found to be the
most efficient catalyst for this ring-opening reaction.
Scheme I.29 : Ring opening alkylation of meso-epoxides104.
The ring-opening of activated aziridines (80, Scheme I.30) can also be catalyzed by
NHCs in the presence of nucleophiles. As proposed by the authors, the mechanism likely
proceeds through the formation of a pentavalent silicon intermediate (61, Scheme I.30)
when silylated nucleophiles105 are employed. Note that the ring-opening of aziridines has
been also performed employing anhydrides106, oxidized aldehydes107, or enals103 as
nucleophiles. In this reaction, the presence of an electron-withdrawing group (such as N-
Tosyl group) seems to be crucial for the reaction outcome, since non activated aziridines do
not show any activity.

58
Scheme I.30 : NHC catalyzed ring-opening of aziridines in the presence of silylated
nucleophiles (TMS = trimethylsilyl; Ts = Tosyl; Nu = nucleophile)103, 105-107.
A method for carbon-carbon bond cleavage of formyl cyclopropyl units (81, Scheme
I.31) has also been reported using NHCs as catalysts108. When alcohols or thiols are used as
nucleophiles, esters 82 or thioesters 83, respectively, could be generated. Interestingly, the
acid-containing product 84 could also been synthesized when water was employed instead
of alcohols or thiols. The authors have proposed that the mechanism of this reaction
operates via a nucleophilic attack of the catalyst 85 to the aldehyde function of the substrate
81 (Scheme I.31). The adduct 86 formed is then directly converted to the Breslow
intermediate, 87, followed by ring-opening so as to generate the enolate-type specie 88. A
rapid proton-transfer then generates the more stable catalyst-bound enolate, 89, which,
after protonation, gives the activated carboxylate 90. Finally, a last step of nucleophilic
attack to the carbonyl group allows for the formation of the product while the free NHC is
released.
Scheme I.31 : Proposed mechanism for the ring-opening reaction of formyl cyclopropanes
catalyzed by NHCs108.

59
As mentioned above, the potential of NHC catalysts to other molecular reactions has
been exploited19, 26, 56, 82, 83, 86, 87, 89, 109-122. With the growing interest in organocatalysis19, 26, 40,
81, 90, 123-147 which provides new opportunities to mimic rates and selectivities of enzymes and
metal-based catalysts, it is likely that NHCs will find further utilizations in organocatalyzed
reactions in a near future.
As described in the next section, the potential of NHCs has also been applied to
trigger polymerization reactions.
I.3.2) In macromolecular synthesis
The last few decades have witnessed advances in polymer chemistry dominated by
metal-based catalysis. Most of synthetic polymers indeed require metallic species to catalyze
or initiate elementary reactions that bring about their synthesis, with the noticeable
exception of polymers obtained by free radical polymerization148, 149. In most cases, the
metallic catalyst represents minute amounts and yet remains in the final polymer. To
prevent the hazards associated with the presence of toxic metallic species that can freely
migrate out of the polymeric material when in service, a tedious purification process is
required which adds to the cost of the polymer produced. In sensitive domains such as
biomedical, packaging, and microelectronics, metal-based catalysts are prohibited. Thus,
organic and enzymatic catalysis appear today as the most reliable alternatives to metal-
mediated catalysis for polymer synthesis90.
This section discusses how NHCs have been employed as organic nucleophiles to
trigger different polymerization reactions, including step-growth and chain-growth
polymerizations. On the other hand, recent utilizations of NHCs as precursors to be
incorporated as building blocks in polymeric materials with specific properties will also be
described.
I.3.2.1) Step-growth polymerizations
It is worth mentioning that the first attempt to produce polymeric materials via a
NHC organocatalysis was reported by Jones et al. in the late 1990’s150, 151. These authors
have elegantly applied the principle of the Stetter reaction described above to
bifunctionalized monomers such as diformylarenes (91 and 92, Scheme I.32) and bis-
Mannich bases (93 and 94) in the presence of a thiazolylidene catalyst. Polymeric 1-4
diketones 95, 96 were synthesized in this way. Further ring closure of these polymeric
precursors has allowed for the production of conjugated alternating copolymers bearing
pyrroles (97), furyls (98) or thienyl moieties (99), as illustrated in Scheme I.32.

60
Scheme I. 32 : Synthesis of polymeric 1,4-diketones by thiazolylidene catalyzed step-growth
polymerization and their further ring-closure by Paal-knorr reactions150, 151.
In the course of their investigations of NHC-catalyzed transesterification reactions,
Hedrick et al. have also reported the synthesis of poly(ethyleneterephtalate) known as PET97.
This could be achieved using a two-step procedure, with NHCs as single catalytic
components for both the preparation of bis(2-hydroxyethyl)terephthalate (BHET) followed
by its subsequent self-condensation (Scheme I.33). The reaction is reversible in the presence
of methanol and the same NHCs were also effective for PET depolymerization (Scheme I.33)
under mild conditions (typically at 80 °C or even less)152.
The authors have also described the synthesis of moderate molecular weight -like
polyesters- (typically Mn ranging from 8,000 to 20,000 g.mol-1) poly(caprolactone) (PCL) and
poly(glycolide) by self-condensations of ethyl 6-hydroxyhexanoate and ethyl glycolate,
respectively, in bulk at 60 °C under vacuum for 24 h (Scheme I.34). In addition, a copolymer
was also obtained from the condensation of the two monomer types.
Scheme I.33 : Synthesis of PET by step-growth polymerization. The depolymerization of PET
via transesterification by a NHC catalysis is shown in blue97.

61
Scheme I.34 : Synthetic route to aliphatic polyesters by NHC-catalyzed polycondensation
reactions97.
Another contribution to the NHC-catalyzed step-growth polymerization has
been given by Gnanou et al. who reported that NHC 17 could catalyze the step growth
polymerization of di-isocyanates in the presence of diols to yield PUs (Scheme I.35),153 under
relatively mild conditions (50°C) and with low catalyst loading (< 1 %). However, it should be
noted that the as-prepared PUs exhibit rather low molecular weight (3000 - 5000 g.mol-1)
and broad dispersity ( 3), maybe due to moderate conversion of reactive functions.
Scheme I.35 : Synthesis of polyurethanes by NHC-catalyzed polycondensation between diols
and diisocyanates153.
NHCs adducts 100-110 (Figure I.12) were also shown to be active precatalysts for the
preparation of crosslinked polyurethanes from polyols and triisocyanates, as reported by
Buchmeiser et al. (Scheme I.36)154, 155. Polymerization were conducted at 60-70°C in CH2Cl2
to allow the thermal activation of 100-110 and the in-situ generation of free NHCs, as
evidenced by the apparition of the peak attributed to the azolium proton at δ = 10.7 ppm
during 1H NMR experiments. Indeed, it was proposed that these NHCs could activate the OH
function of polyols, due to their high basicity, forming alcoolate species and the
corresponding salt.
In the case of NHC-metal adducts 105-110, the authors proposed that two
mechanisms occurred simultaneously as a consequence of the dissociation of these adducts
into free NHC and metal salt, upon heating. While the free NHCs activate the alcohol, the
Lewis acidic metal salt can activate the isocyanate via complexation, both activations

62
explaining the high catalytic activities observed. Thus, the authors suggested that alcohol
activation only occurs with precatalysts 100-104 while NHC adducts 105-110 allow for a
dual-activation of both monomers (simultaneous activation of the alcohol and the
isocyanate) by in-situ generation of free NHC catalyst and metal catalyst upon heating.
Figure I.12 : Precatalysts employed by Buchmeiser et al. to prepare crosslinked
polyurethanes154, 155.
Scheme I.36 : Synthesis of crosslinked polyurethanes from polyols and triisocyanates in the
presence of NHC-adducts 100-110. 154, 155
Recently, inspired by the mechanism operating in the benzoin condensation (see
section I.C.1.1), our group employed a commercially available bis-aldehyde, namely
terephtaldehyde, that we polymerized by NHC-catalyzed step-growth polymerization
(Scheme I.37)156. This led to the formation of poly(1,4-phenylene-1-oxo-2-hydroxyethylene)s
(“polybenzoins”) under relatively mild conditions (THF or DMSO at 40 °C).

63
This work has been performed at the beginning of my PhD thesis and will be the
topics of chapter II of this manuscript. From the four catalysts employed in this study, 1,3,4-
triphenyl-1,2,4-triazol-5-ylidene was found to be the most active. As described in chapter II,
formation of cyclic polymers during the polymerization has been also noticed. The content of
these cyclic species could be evaluated and has been found to vary depending on the
reaction media used and the monomer conversion reached.
Scheme I.37 : NHC catalyzed benzoin condensation in step-growth polymerization156.
I.3.2.2) Ring-opening polymerizations (ROP)
Besides their use to trigger step-growth polymerization reactions, NHCs have been
mainly evaluated as organic catalysts in chain-growth polymerization reactions. The first
report dates back to 2002, when Hedrick, Waymouth et al. have exploited the ability of the
1,3-dimesitylimidazol-2-ylidene (IMes) (28, Scheme I.10) to catalyze transesterification
reactions to trigger the living ROP of cyclic esters157. Poly(L-lactide) (PLA), poly(ε-
caprolactone), poly(β-butyrolactone) and poly(δ-valerolactone) with controlled molecular
weights and narrow distributions could be obtained40, 41, 157, 158. In the case of lactide, IMes
found to be the most active catalyst, with a turnover frequency (number of monomer units
incorporated per second) close to 18 s-1. This particular NHC proved quite active even with
low catalyst loading (0.5 mol%), and molecular weights could be adjusted by varying the
ratio of monomer to initiator (Dpn= [M]/[I]) without changes in dispersity (D < 1.2).
Since this first report, several other NHCs, in their pure or under their masked form,
have been tested for the ROP of cyclic esters. In most cases, alcohols were employed to play
the role of initiators (controlling the chain length of the final polymer)40.
Two mechanisms have been proposed to account for the living character of the ROP
of cyclic esters. Interestingly, these two mechanisms find analogies with those of the NHC-
catalyzed transesterification discussed above (see Scheme I.28).
Thus, the “monomer-activated mechanism” involves a nucleophilic attack of the NHC
to the carbonyl group (path A, Scheme I.38), while the “chain-end activated mechanism”
operates by deprotonation of the O-H group of the initiator by the NHC (Path B, Scheme
I.38).

64
Scheme I.38 : Initiation step in the ring-opening polymerization of lactide and
subsequent propagation to form PLA40.
Evidences for both mechanisms have been reported and depending upon the nature
of the NHC catalyst, findings were in agreement with those described in the case of NHC-
catalyzed transesterifications.
Firstly, Hedrick, Waymouth et al. have argued that NHCs were unable to deprotonate
alcohols to initiate an anionic polymerization from the corresponding iminium alkoxide157.
This proposition was based on the fact that IMes has a pKa of 24 in DMSO, whereas the pKa
of the initiating benzyl alcohol is close to 29.
A further evidence of the “monomer activated mechanism” was given when
polymerization reactions of lactide were accomplished in total absence of alcohol as
initiator159, 160. In the latter case, cyclic PLAs were obtained, as a result of the deactivation of
zwitterionic species (Scheme I.39); in this case, molecular weights were directly controlled by
the initial ratio between monomer and NHC. These polymerization reactions proved very
fast, yielding high molecular weight polymers with moderate dispersities (D < 1.32) in only a
few seconds (4 s - 300 s for polymers of 5,300 g.mol-1 - 31,4000 g.mol-1 respectively). When
this polymerization reaction was applied to optically pure L-lactide, isotactic crystalline cyclic
PLA was formed, indicating that the polymerization proceeded with retention of
stereochemistry159.

65
Scheme I.39 : Zwitterionic ring-opening of lactide catalyzed by IMes and synthesis of cyclic
PLAs159, 161(Note that no ROH-type molecule has been employed here as initiator).
In complement to this study, and to further assess the role of the zwitterionic
intermediates in these polymerizations, the reaction between the saturated NHC 1,3-
dimesitylimidazolin-2-ylidene (111, Scheme I.40) and β-butyrolactone was investigated.162
When the two species were reacted in an equimolar amounts, generation of the zwitterionic
species, 112, by nucleophilic attack of the carbene on the lactone was followed by ring-
closure, forming a stable but labile spirocycle, 113. Indeed, the latter spiro-type intermediate
could be cleaved, allowing further propagation steps in the presence of monomer excess. In
this case, the spirocycle 113 serves as initiator for the ring-opening polymerization of both β-
propiolactone and β-butyrolactone and exhibits similar polymerization behavior to that of
111.
Scheme I.40 : Formation of a spirocycle from β-butyrolactone and the saturated NHC 1,3-
dimesitylimidazolin-2-ylidene41, 161.

66
The above results suggest that the ROP of cyclic esters proceed via the “activated
monomer mechanism”. However, other experiments have shown that ROP could occur via
the chain-end activation mechanism. Instead of monomer activation, NHCs can activate the
initiator or the active chain-end by H-bonding. This mechanism have been also reported for
other bases90, that can increase the nucleophilicity of the initiating or propagating alcohol to
facilitate nucleophilic attack on the cyclic ester monomer.
Thus, in the presence of an alcohol, it is likely that the NHC-catalyzed ROP of esters
operates by both mechanisms as a function of the initial experimental conditions, in
particular, depending upon the type of NHC catalyst used.
Interestingly, the same group have also resorted to amino- or amido-type reagents
serving as initiators163. By making use of bisamino- or tetraamino-terminated
poly(ethyleneoxide) (PEO) macroinitiators, H-shaped or super-H-shaped PEO/PLA-based
block copolymers could be achieved (Scheme I.41).
Specifically designed adducts resulting from the 1:1 addition of saturated NHC on a
tris-alcohol can also be used as a latent initiating catalytic system (release of both the
trifunctional initiator and the NHC catalyst in the mean time upon heating). Three-armed
PLA stars were thus obtained in a controlled fashion (Scheme I.42)49.
Scheme I.41 : Synthesis of H-shaped PEO/PLA-based block copolymers from NHC catalyzed
ROP of Lactide163.
Scheme I.42 : Polymerization of LA employing a tri-NHC-alcohol adduct and leading to tri-
armed star PLAs49.

67
The Interactions involved in alcohol complexes of IMes have been studied by NMR
spectroscopy and by X-ray crystallography. It has been found that such adducts are greatly
sensitive to the solvent and to the structure/nature of the alcohol164. The more sterically
congested the alcohol is, the weaker the interaction with the carbene is. A more acidic
alcohol thus leads to greater involvement of imidazolium alkoxides in equilibrium with other
hydrogen bond complexes (see Scheme I.38).
The “activated chain-end mechanism” has also been put forward to account for the
production of isotactic PLAs from rac-lactide and heterotactic PLAs with meso-lactide165, 166.
To support this hypothesis, experiments have been carried out in a way to epimerize either
the lactide monomer or the corresponding polymer40. When lactide is placed with an excess
(10 eq.) of CH3OD, in the presence of 1 equivalent of IMes, methyl lactate is obtained within
a few minutes. Analysis by 1H NMR of the resulting methyl lactate did not provide evidence
for incorporation of deuterium at the alpha-carbon of methyl lactate. Thus, epimerization of
methyl lactate or LA is not competitive with ring-opening of the cyclic ester. However, after
3 days at room temperature, 50% of the alpha hydrogens are substituted with deuterium,
indicating that the carbene is capable of enolizing methyl lactate, but at a rate that is much
slower than ring opening (Scheme I.43).
Scheme I.43 : Epimerization of butyl lactate by NHCs.
Similarly, ε-caprolactone (ε-CL) can be polymerized from mono- or multifunctional
alcohols as initiators167. Although IMes (28, Figure I.13) and 1,3-bis(2,6-
diisopropylphenyl)imidazol-2-ylidene (IDIPP), 114, can polymerize neat ε-CL, these NHCs are
not effective catalysts for the ROP in solution, and more electron-rich carbenes such as the
1,3,4,5-tetramethylimidazol-2-ylidene (Me2IMe), 115, and 1,3-diisopropyl-4,5-
dimethylimidazol-2-ylidene (Me2IPr), 116, must be employed. Unsaturated and less crowded
carbenes such as 29 also allow for the preparation of block copolymers featuring LA and ε-CL
monomer units41.
Even if the NHC catalyzed ROP of cyclic esters present the features of living
polymerizations and allow the production of well defined homo and block polyesters, one

68
has to keep in mind that NHCs can also catalyze transesterification. Thus, prolonged reaction
times after monomer consumption often yield to polyesters with broadened polydispersities
because of adverse transesterification side reactions41.
Figure I.13 : NHCs used as catalysts in the ROP of ε-CL41, 167.
NHC-induced ROP has been further extended to other cyclic monomers such as
carbonates (Scheme I. 44)168, cyclosiloxanes (Scheme I. 45)66, 169, 170 and N-carboxyanhydrides
(Scheme I. 46)171 and both ethylene and propylene oxides (see further). For carbonates and
N-carboxyanhydrides, the two mechanisms discussed above have again been suggested to
explain the controlled character of these polymerizations.
Scheme I.44 : NHCs catalyzed ROP of cyclic carbonates168.
Scheme I.45 : NHC-catalyzed ROPs of A) D3 and D4 reported by Baceiredo et al. 63,164 and
B) carbosiloxanes reported by Hedrick, Waymouth et al.163.

69
Scheme I.46 : NHC-catalyzed ROP of carboxylanhydrides171.
For instance, Zhang et al. have reported the synthesis of poly(α-peptoid)s that are
structural mimics of poly(α-peptide)s, via the direct NHC-initiated ROP of N-substituted N-
carboxyanhydrides (NR-NCA) (Scheme I.46). In contrast to poly(α-peptide)s where well-
defined secondary structures are stabilized through hydrogen bonding interactions, poly(α-
peptoid)s are free of such hydrogen bondings.
As for regular NCA monomers, NR-NCA can be polymerized through a nucleophilic
chain growth pathway that entails regioselective insertion of a nucleophilic initiator (e.g.,
primary amine) into the anhydride carbonyl group. The subsequent elimination of CO2 then
reforms an amino propagating chain-end.
These NR-NCA monomers could be polymerized in the presence of various amounts
of NHC 114 (Figure I.13) used as direct initiator thus controlling the polymer chain length.
Note here that the activated functional group (i.e., anhydride) of the NR-NCA monomer is
different from that of the polymer backbone (i.e., amide). As NHCs have not been reported
to trigger transamidation reactions, it is likely that inter- and intra-chain transfer (side)
reactions, if existing, are de facto reduced. This is thus a different situation compared to the
case of aliphatic polyesters discussed above, which can be subjected to transesterification
reshuffling reactions when catalyzed by NHCs.
Similarly to the polymerization of β-butyrolactone described above, ESI-MS analysis
have revealed that the “carbene” moiety was retained in the spirocycle adduct 117 when it
was precipitated in hexane. Thus, after complete conversion of the initial monomer, chain-
extension could be performed upon addition of a second batch of NR-NCA monomer,
confirming the living nature of the polymerization.
On the other hand, analysis by MALDI-TOF mass spectroscopy has revealed that the
corresponding polymers mainly consist of cyclic poly(α-peptoid)s formed by zwiterionnic
ring-opening polymerization (ZROP), followed by intramolecular cyclization and release of
the NHC moiety (Scheme I.46). The cyclic architecture was confirmed by viscometric
measurements. Interestingly, control over the polymer molecular weight and dispersity
could be maintained even at relatively high concentrations in monomer, in contrast to
common synthetic strategies of derivation of cyclic polymers that generally require dilute

70
conditions. Cyclic diblock copoly(α-peptoid)s could even be achieved by sequential ZROP of NMe-NCA and NBu-NCA monomers.
D3, D466, 170 or 2,2,5,5-Tetramethyl-1-oxa-2,5-disilacyclopentane169 being free of
carbonyl group, it might be postulated that polymerization occurs through a chain-end
activated mechanism (Path B, Scheme I.47). Nevertheless, as observed by Baceiredo et al.170,
a faster polymerization occurs when the amount of NHC, as compared to the alcohol
initiator, is increased. This effect has been attributed to the affinity of carbenes toward
silicon66 and the propensity of Si to hypervalency, which allows for ring opening of these
monomers. Thus, an activated monomer mechanism seems to be involved in ROP of cyclic
siloxanes (Path A, Scheme I.47).
Scheme I.47 : Mechanisms for the ROP of D4 proposed by Baceiredo et al.170
At LCPO, Taton, Gnanou et al. have shown that NHCs could serve not only as direct
initiators (via a ZROP mechanism) but also as organocatalysts when used in conjunction with
chain regulators of NuE-type, where Nu and E are the nucleophilic and the electrophilic
parts, respectively, (e.g. PhCH2OH, HC≡CCH2OH, N3SiMe3 and PhCH2OSiMe3), to trigger the
ROP of ethylene oxide.172, 173 In this way, α,ω-heterodifunctionalized poly(ethylene oxide)s
(PEOs) could be synthesized.
For instance, the 1,3-bis-(diisopropyl)imidazol-2-ylidene (30, Figure I.14) was found to
directly initiate the metal-free ROP of ethylene oxide (EO) at 50 °C in dimethylsulfoxide, in
absence of any other reagents. In other words, the latter polymerization proceeds via ZROP.
Without any other reagent added at the beginning, the PEO chain length is thus strictly
controlled by the EO to the NHC molar ratio (typically, [EO] / [NHC 30] = 100 / 1).

71
The growing species, namely 1,3-bis-(diisopropyl)imidazol-2-ylidinium alkoxide, were
quenched at the completion of ZROP by a functionalizing terminator of NuH or NuSiMe3-
type, leading to α-Nu,ω-OH (or α-Nu,ω-OSiMe3)-difunctionalized PEOs. The quantitative
introduction of the Nu moiety in α- position and of OH (or OSiMe3) in ω-position of PEO
chains, respectively, occurred through the nucleophilic substitution of the imidazolium
moiety by Nuδ- and the concomitant reaction of the ω-growing alkoxide of PEO chain with
Hδ+ (or Me3Siδ+), as illustrated in Scheme I.48.
In particular, α,ω-bis(hydroxy)-telechelic, α-benzyl,ω-hydroxy and α-azido,ω-hydroxy
difunctionalized PEOs were synthesized by NHC-initiated ZROP, using H2O, PhCH2OH and
N3SiMe3 as terminating agent, respectively.
Scheme I.48 : Functionalizing terminators of the zwitterionic ring opening polymerization of
ethylene oxide.174
A well-defined poly(ethylene oxide)-b-poly(ε-caprolactone) block copolymer could
also be directly synthesized by sequential ZROP using the same NHC as organic initiator,
without isolation of the PEO block intermediate.
Instead of using NHCs as direct initiators, Taton, Gnanou et al. have also reported the
ROP of EO with NHCs as real organocatalysts in conjunction with hydroxylated or
trimethylsilylated reagents introduced at the beginning and playing the role of chain
regulators (Figure I.14). Typical amounts of NHC catalyst equal to 10% molar relative to NuE
have been employed.

72
Figure I.14 : Ring-opening polymerization of ethylene oxide catalyzed by N-heterocyclic
carbenes in presence of OH- or Me3Si-containing chain regulators (107 and 108 were used
for comparison) 166, 167.
Similarly to ROP of cyclic esters,175 two distinct mechanisms of initiation and chain
growth can be contemplated to coexist, depending on the interaction of the O-H or O-TMS
chain regulators with NHC catalysts against that of the latter with the monomer (Scheme
I.49). Indeed, NHCs are nucleophilic and silicophilic enough to activate the electrophilic part
(E = H or SiMe3) of NuE chain regulators.176 Thus, both trimethylsilylated (121 and 123) and
hydroxylated (120 and 122) chain regulators could be activated by NHCs, followed by
monomer insertion as shown in path B of Scheme I.49 (chain end activation mechanism).
However, a nucleophilic attack by the NHC catalyst to the monomer is also plausible
(path A). Unambiguous evidences have been provided to comfort that NHCs can directly add
onto EO in absence of any other electrophile (ZROP).174 Protonation (or silylation) of the
zwitterionic intermediate species by an alcohol (or a silyl ether), followed by addition of the
resultant alkoxide on the activated azolium, generates a new alcohol (or silyl ether) with
incorporation of a monomer unit. In the meantime, the NHC catalyst is released and can
activate another EO molecule (path A, Scheme I.49). The mono-adduct, Nu-CH2CH2O-E, can
serve for subsequent chain propagation (activated monomer mechanism).
At this stage of their investigations, the authors have acknowledged that it is unclear
whether the NHC directly attacks the monomer, forming a zwitterionic intermediate or
activates the E moiety of the chain regulator, then the ω-OE moiety of the PEO chain, and
that more investigations would be needed.

73
Scheme I.49 : Proposed mechanisms of initiation in ring opening polymerization of ethylene
oxide and propylene oxide catalyzed by N-heterocyclic carbenes: monomer activation (path
A) and chain end activation (path B) 172, 173,174.
Recently, propylene oxide has also been polymerized using this two strategies,
with NHC being employed either as direct initiator of a ZROP or as true organocatalyst in
the presence of a chain regulator.177 Both methods allowed the authors to derive α,ω-
difinunctionalized PPOs using NHC 30 and a metal-free and solvent-free procedure,
whereas PPO is generally obtained by anionic ROP of PO carried out in low polar media
from alkali metal-based initiators.178, 179
When used as a direct organic initiator, the zwitterionic growing species were simply
quenched by H2O, leading to dihydroxytelechelic PPOs (path A, Scheme I.49). With NHC 30
as catalyst, the same hydroxylated or trimethylsilylated compounds already shown (120-123)
were employed as chain regulators for the metal-free ROP of PO at 50 °C (path B, Scheme
I.49). Both routes have implied PO polymerization in bulk and incomplete monomer
conversion (≤ 40% in all experiments), allowing an easy removal -and possible recycling- of
the residual low boiling point monomer by simple evaporation.

74
I.3.2.3) Group transfer polymerization of (Meth)Acrylates
Hedrick, Waymouth et al.180 and Taton, Gnanou et al.181, 182 at LCPO have reported
that the group transfer polymerization (GTP) of both acrylic and methacrylic monomers can
be efficiently carried out using NHCs as nucleophilic organic catalysts (Figure I.15), whereas
non-NHC catalysts are able to polymerize one category of monomers only.183
Figure I.15 : NHCs used as catalysts for GTP of (meth)acrylates 180, 181, 182.
GTP has been proposed in the mid 1980s as a convenient method to control the
polymerization of (meth)acrylic monomers at ambient temperature and above184, 185. The so-
called associate mechanism is based on the repetition of the Mukaiyama-Michael reaction
(see section I.3.1.4)95 involving the addition of silyl ketene acetal (SKA) onto an incoming
(meth)acrylic monomer. GTP is generally catalyzed by a metal-free nucleophile (Lewis base)
or a metal-based Lewis acid, for methacrylic and acrylic monomers, respectively.183 The
absence of a unique catalytic system that could be generalized to both families of monomers
did not permit, however, the synthesis of block copolymers featuring both monomer units
by sequential GTP.
Webster et al. originally suggested to name this mechanism “group transfer
polymerization” to account for the fact that the trimethylsilyl group remained at the chain-
end throughout polymerization.184 In other words, an associative (concerted) mechanism of
GTP involving a transfer of the trialkylsilyl moiety from the polymer chain-end to the inserted
monomer was originally put forward (Scheme I.50). However, this pathway was further
questioned, in particular by Quirk who proposed that GTP occurred by a dissociative
mechanism forming minute amounts of propagating enolates.186 These anionic species
which are formed in the presence of strong nucleophiles are the real active centers and that
are temporarily trapped by the silyl ketene acetals, forming dormant bis(enolato)siliconates
(Scheme I.50). As a matter of, the GTP mechanism seems to strongly depend on the overall
polymerization conditions and, in particular, on the nature catalysts.184-189
Works on the NHC-catalyzed GTP have reopened this debate on the exact mechanism
that operates in GTP (Scheme I.50).180-182 While Hedrick et al. have postulated a dissociative
mechanism forming enolate-type species using NHC 124 as catalyst180, Taton, Gnanou et al.
have provided a series of experimental evidences suggesting that GTP occurs by an
associative (concerted) mechanism, when catalyzed by NHC 30 or 31 (Scheme I.50).181, 182

75
Scheme I.50 : Associative and dissociative mechanisms for NHC-mediated GTP 180, 181, 182.
This discrepancy may eventually be explained by the difference in
nucleophilicity/silicophilicity of the different NHCs used. NHC 124 is indeed slightly more
nucleophilic than NHCs 30 and 31, hence the occurrence of two different mechanisms
depending on the NHC used is plausible. Yet, though the two mechanisms lead to the same
polymer, the polymerization kinetics as well as the properties of the final polymers
associated with each of them are different.180-182 GTPs catalyzed by NHCs 30 and 31 were
further applied to the synthesis of all-acrylic block copolymers.180-182
NHC-catalyzed GTPs could be typically performed at room temperature using 1-
methoxy-2-methyl-1-trimethylsiloxypropene (MTS) as initiator in polar (THF) or apolar
(toluene) medium. In this way, polymethacrylates and polyacrylates with molar masses in
the range 10,000-300,000 g.mol-1, corresponding to the initial [monomer] / [MTS] ratio and
with polydispersities lower than 1.2, were obtained in quantitative yields.
Taton, Gnanou et al. have noted several observations that support the existence of
the associative mechanism, when NHCs 30 and 31 are used as GTP catalysts:
Though the first-order kinetic plot ln[M]0/[M] versus time deviated from linearity at high
monomer conversion, no inhibition period was noted at low monomer conversion.
Moreover, the polymerization rate dramatically increased as the concentration of initiator
increased, with first-order dependence in initiator.
When mixed in 1 / 1 molar ratio, MTS and NHC 31 did not reveal the formation of
enolate-type species by 29Si or 13C NMR spectroscopy.
The production of well-defined poly(methyl methacrylate)s using 1 / 1 adducts of 31 and
MTS as initiating system.

76
The controlled polymerization of n-butyl and tert-butylacryates in the presence of 30 or
31, which suggests that back-biting and internal isomerisation can be minimized,
presumably because no enolate is generated.
The preparation of block copolymers based on acrylate-type and methacrylate-type
monomer units, irrespective of the order of addition of the two monomers, which would
not be conceivable from pure enolates formed by a dissociative mechanism.
Thus, pentacoordinate siliconates are thought to be the key intermediates in the
associative mechanism of GTP catalyzed by NHC 30 or 31 (see Scheme I.50). It is very likely
that other NHCs such as 124 used by Hedrick and Waymouth that are more nucleophilic /
silicophilic catalysts than 30 or 31 could bring about the GTP of (meth)acrylics via a
dissociative pathway.
In the past decade, NHCs have provided efficient “metal-free” routes to aliphatic
polyesters and other well-defined synthetic polymers including PEO, PPO, poly(α-peptoid)s,
poly(meth)acrylates and PDMS. Rates and selectivities of these organocatalysts are
competitive with the most active and selective metal-based catalysts. Moreover, they have
allowed the production of biocompatible polymers without metal-residues, which could be
used in biomedical applications. Metallic impurities can also be an inconvenient in polymers
for optoelectronic applications.
As described in the next section, NHCs can however serve as monomers for the
obtention of conjugated polymers free of metal residues.
I.3.2.4) NHCs as building blocks for polymer synthesis
Besides their use as organocatalysts in both step-growth and chain-growth
polymerization reactions, NHCs have been employed as true building blocks (synthons) for
polymers synthesis. This has been mainly investigated by the group of Bielawski190-192. The
basic idea here is to take benefit of the capability of NHCs to readily form adducts with a
variety of reagents such as CS2, azides or to dimerize, as highlighted in section I.2.1.2.
Synthesis of RAFT agents from NHCs
As mentioned above, CS2 can quantitatively react with NHCs, forming zwitterrionic
adducts. Bielawski et al. have taken advantage of such adducts (125, Scheme I.51) to derive
efficient cationic RAFT1 agents, 126 and 127. These controlling agents were subsequently
1 RAFT stands for the reversible-addition fragmentation chain transfer: for a review on RAFT, see ref 187-189.
193. Moad, G.; Rizzardo, E.; Thang, S. H. Acc. Chem. Res. 2008, 41, (9), 1133-1142, 194. Moad, G.;
Rizzardo, E.; Thang, S. H. Polymer 2008, 49, (5), 1079-1131, 195. York, A. W.; Kirkland, S. E.; McCormick, C.
L. Adv. Drug Delivery Rev. 2008, 60, (9), 1018-1036.
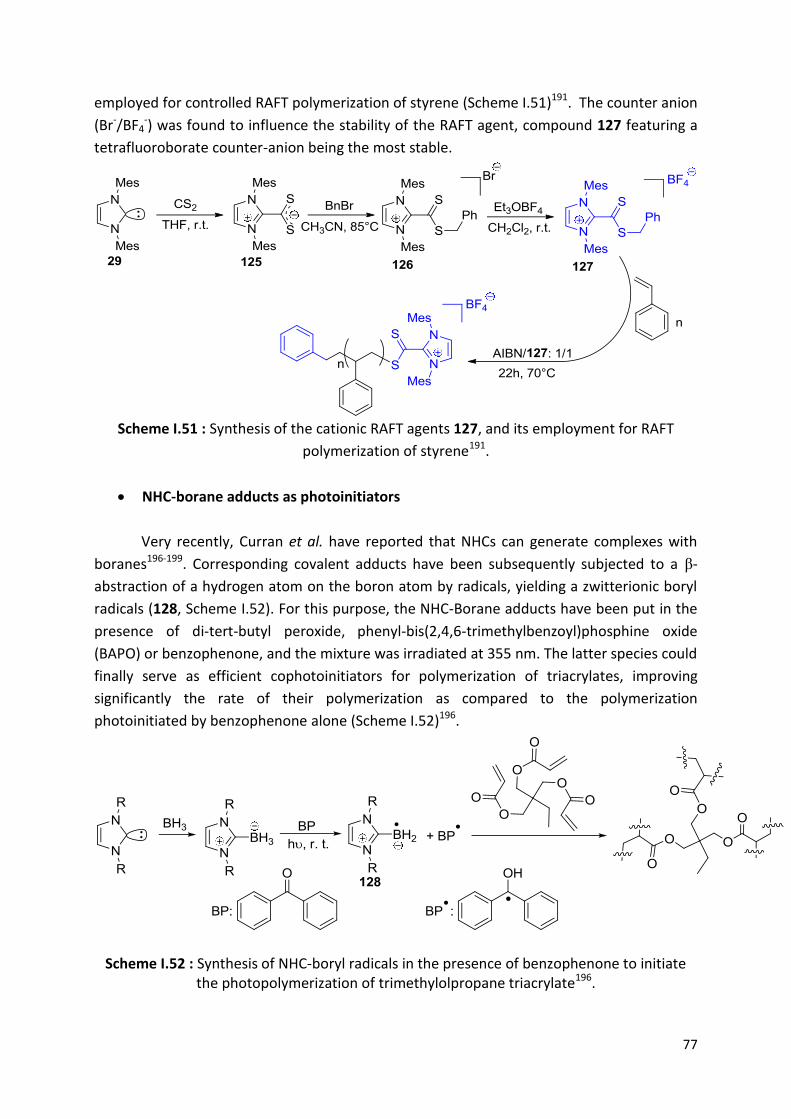
77
employed for controlled RAFT polymerization of styrene (Scheme I.51)191. The counter anion
(Br-/BF4-) was found to influence the stability of the RAFT agent, compound 127 featuring a
tetrafluoroborate counter-anion being the most stable.
Scheme I.51 : Synthesis of the cationic RAFT agents 127, and its employment for RAFT
polymerization of styrene191.
NHC-borane adducts as photoinitiators
Very recently, Curran et al. have reported that NHCs can generate complexes with
boranes196-199. Corresponding covalent adducts have been subsequently subjected to a β-
abstraction of a hydrogen atom on the boron atom by radicals, yielding a zwitterionic boryl
radicals (128, Scheme I.52). For this purpose, the NHC-Borane adducts have been put in the
presence of di-tert-butyl peroxide, phenyl-bis(2,4,6-trimethylbenzoyl)phosphine oxide
(BAPO) or benzophenone, and the mixture was irradiated at 355 nm. The latter species could
finally serve as efficient cophotoinitiators for polymerization of triacrylates, improving
significantly the rate of their polymerization as compared to the polymerization
photoinitiated by benzophenone alone (Scheme I.52)196.
Scheme I.52 : Synthesis of NHC-boryl radicals in the presence of benzophenone to initiate the photopolymerization of trimethylolpropane triacrylate196.

78
NHCs as reagents for post-modification polymerizations
By making the analogy with the Staudinger reaction between phosphines and
azides200, Bielawski et al. have found that NHCs can rapidly react with azides at ambient
temperature (Scheme I.53)190, 201-203. This reaction generally affords high yields and stable
1,3-disubstituted triazenes. Interestingly, no loss of N2 is observed at room temperature, in
contrast to the reaction between phosphines and azides where elimination of N2 occurs
readily.
Scheme I.53 : Formation of triazene compounds through the reaction between NHCs and
azides203.
This reaction has been exploited to introduce lateral functional groups onto
polystyrenics covering lateral azido groups, via a postpolymerization modification
procedure202. Coupling of NHCs with azides has been first used to functionalize poly(p-
azidomethylstyrene) (Scheme I.54). Upon heating at 135 °C, release of N2 finally takes place,
forming a guanidine functionalized polymer. As proposed by the authors, this Staudinger
reaction can be viewed as a complement to “click reaction”204 utilizing reactive azides to
generate functionalities and materials with unique properties204.
Scheme I.54 : Postpolymerization modification of poly(p-azidomethylstyrene) and release of
N2 upon eating of the triazeno-functionalized polymer.
NHCs as monomers
Based on the same latter idea consisting on the formation of an adduct between
NHCs and azides, Bielawski et al. have polymerized bis(NHC)s and bis(azide)s in a step
growth polymerization process (Scheme I.55)190. The obtained polytriazenes exhibited good
optical and electronic properties due to their conjugated structure, in addition to their high
solubility in common organic solvents. Considering the large number of methods known for
modifying the structures and electronic properties of NHCs10, as well as organic azides205, the
polymers reported herein may be well-adapted for a use in optoelectronic applications.

79
Scheme I.55 : Construction of new polymers based on bis(NHC)s and bis(azide)s190.
Conjugated polymers could also be obtained by self-coupling of bis(NHC)s involved in
the so-called Wanzlick equilibrium (see section I.2.3.2), which yields polyenetetramines
(Scheme I.56)206. Of particular interest, these polymerization reactions are reversible, the
bis(NHC)s being readily regenerated upon heating to 90 °C.
Scheme I.56 : Polymerization of Bis(NHC)s to produce polyenetetramines.
In addition, in the presence of PdCl2, these polymers formed main-chain
organometallic polymers consisting of NHC-Pd complexes (A, Scheme I.57)206. Oxidation of
the aforementionned polyenetetramines have been also performed in the presence of
excess iodine, which has resulted in the production of soluble conjugated polyelectrolytes
(B, Scheme I.57)207.
Scheme I.57 : Synthesis of main-chain organometallic Pd complexes polymers (A) and
soluble conjugated polyelectrolytes (B) from polyenetetramines206,207.
In a recent addition, the same group has implemented the step-growth
polymerization of these bis(NHC)s with bis(isothiocyanate)s, on the basis of the adduct
formation occuring between NHCs and isothiocyanates192,53. Purposely designed bis(NHC)s
and bis(isothiocyanate)s featuring long alkyl chains resulted in highly soluble aromatic
polymers (Scheme I.58). Here also, the polymers could be depolymerized upon heating while
adding one of the precursors. For this reason, they have been qualified as “structurally
dynamic polymers”.

80
Scheme I.58 : Synthetic methodology used to form structurally dynamic polymers derived
from bis(NHC)s and bis(isothiocyanate)s192.

81
I.4) Conclusion
Since Bertrand et al. and Arduengo et al. have described the first stable
carbenes in the 1990’s, the carbene chemistry has received a considerable attention in
molecular synthesis, and more recently, in macromolecular chemistry.
Thanks to their peculiar electronic properties, stable singlet carbenes, and in
particular NHCs, have not only become versatile ligands for transition metals, but also
powerful organocatalysts for a variety of organic transformations.
It is worth mentioning that many of these NHC-catalyzed reactions are based on the
activation of the carbonyl group (e.g. benzoin condensation, Stetter reaction,
transesterification, etc.), though other electrophilic groups such as trialkylsilyl can also be
activated, (e.g. cyanosilylation or trifluoromethylsilylation reactions).
Organocatalysis in its general sense can also offer new opportunities in polymer
synthesis, for instance for enhancing the rate of polymerization and improving the selectivity
to achieve polymeric structures that would be hard to access by a metal-mediated
polymerization. The field of organocatalyzed polymerization, however, is still in its infancy.
Since organic catalysts may operate differently as compared to metal-based ones, they offer
a diversity of mechanistic pathways to control the polymerization.
In this context, NHCs have already proven powerful in several organocatalyzed
polymerization reactions, though, up to date, their potential has been under-exploited. J.
Hedrick (IBM) and R. Waymouth (Stanford University) have first extensively employed NHCs
as organocatalysts for ring-opening polymerization (ROP) of cyclic esters, mainly ε-
caprolactone and lactide for the production of both cyclic and linear aliphatic polyesters.
Our group at LCPO has expanded the scope of NHCs to other polymerization reactions,
including ROP of both ethylene and propylene oxides and group transfer polymerization of
both acrylic and methacrylic monomers.
In some cases, it has not been clearly established whether the NHC directly activates
the monomer or the initiator/chain-ends or operates by cooperative dual activation. It turns
out that the polymerization mechanism dramatically depends on the NHC
structure/reactivity.
On the other hand, taking benefit of the reactivity of NHCs towards azides,
isothiocyanates or in a dimerization reaction, Bielawski et al. have reported the synthesis of
conjugated polymers through step-growth polymerizations involving bis(NHC)s. The work
described by this group provides a new aspect of the use of NHCs in macromolecular
synthesis, particularly for the production of optoelectronic materials.
Other opportunities such as enantio-asymmetric polymerizations using chiral NHCs
have yet to be investigated. Hence, challenges still remain concerning the use of carbenes in
macromolecular synthesis.

82
The main purpose of this PhD work is to further extend the possibilities offered by
NHCs. Two aspects of their use are covered: in one hand, exploiting their potential as
organocatalysts for step-growth polymerizations and, on the other hand, offering a better
manipulation and a possible recycling of such reactive species.
Chapter II of this manuscript presents how the benzoin condensation has been
applied to a bis-aldehyde, namely terephtaldehyde, in a step growth polymerization. The
two other chapters (III and IV) are dedicated to provide methodologies for an easier handling
of NHCs, by recycling them or generating them in-situ from more air-stable precursors.

83
I.5) References
1. Poyatos, M.; Mata, J. A.; Peris, E. Chem. Rev. 2009, 109, (8), 3677-3707. 2. Diez-Gonzalez, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, (8), 3612-3676. 3. Sommer, W. J.; Weck, M. Coord. Chem. Rev. 2007, 251, (5-6), 860-873. 4. Dröge, T.; Glorius, F. Angew. Chem., Int. Ed. 2010, 49, (39), 6940-6952. 5. Glorius, F., N-Heterocyclic Carbenes in Transition Metal Catalysis. In Topics in Organometallic Chemistry, Springer, Ed. 21: Berlin, Heidelberg, 2007. 6. Nolan, S. P., N-Heterocyclic Carbenes in Synthesis. In Wiley-VCH, Ed. 2006. 7. M. B. Smith; J. March, MARCH's advanced organic chemistry 6th Ed. In Wiley-Interscience, Ed. 2007; p 234. 8. Arduengo Iii, A. J.; Krafczyk, R. Chem. Unserer Zeit 1998, 32, (1), 6-14. 9. Tomioka, H. Acc. Chem. Res. 1997, 30, (8), 315-321. 10. Bourissou, D.; Guerret, O.; Gabbai, F. P.; Bertrand, G. Chem. Rev. 2000, 100, (1), 39-91. 11. Igau, A.; Grutzmacher, H.; Baceiredo, A.; Bertrand, G. J. Am. Chem. Soc. 1988, 110, (19), 6463-6466. 12. Arduengo Iii, A. J.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, (1), 361-363. 13. Arduengo, A. J.; Bertrand, G. Chem. Rev. 2009, 109, (8), 3209-3210. 14. Cadierno, V.; Gimeno, J. Chem. Rev. 2009, 109, (8), 3512-3560. 15. Hirai, K.; Itoh, T.; Tomioka, H. Chem. Rev. 2009, 109, (8), 3275-3332. 16. Mizuhata, Y.; Sasamori, T.; Tokitoh, N. Chem. Rev. 2009, 109, (8), 3479-3511. 17. Vignolle, J.; Cattoën, X.; Bourissou, D. Chem. Rev. 2009, 109, (8), 3333-3384. 18. Nair, V.; Bindu, S.; Sreekumar, V. Angew. Chem., Int. Ed. 2004, 43, (39), 5130-5135. 19. Marion, N.; Diez-Gonzalez, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, (17), 2988-3000. 20. Hudnall, T. W.; Bielawski, C. W. J. Am. Chem. Soc. 2009, 131, (44), 16039-16041. 21. Liu, M. T. H. Chem. Soc. Rev. 1982, 11, (2), 127-140. 22. Richards Jr, C. A.; Kim, S. J.; Yamaguchi, Y.; Schaefer Iii, H. F. J. Am. Chem. Soc. 1995, 117, (40), 10104-10107. 23. Gano, J. E.; Wettach, R. H.; Platz, M. S.; Senthilnathan, V. P. J. Am. Chem. Soc. 1982, 104, (8), 2326-2327. 24. Myers, D. R.; Senthilnathan, V. P.; Platz, M. S.; Jones Jr, M. J. Am. Chem. Soc. 1986, 108, (14), 4232-4233. 25. Breslow, R. J. Am. Chem. Soc. 1958, 80, (14), 3719-3726. 26. Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, (12), 5606-5655. 27. Wanzlick, H. W. Angew. Chem., Int. Ed. 1962, 1, (2), 75-80. 28. Alder, R. W.; Blake, M. E.; Chaker, L.; Harvey, J. N.; Paolini, F.; Schutz, J. Angew. Chem., Int. Ed. 2004, 43, (44), 5896-5911. 29. Lemal, D. M.; Lovald, R. A.; Kawano, K. I. J. Am. Chem. Soc. 1964, 86, (12), 2518-2519. 30. Winberg, H. E.; Carnahan, J. E.; Coffman, D. D.; Brown, M. J. Am. Chem. Soc. 1965, 87, (9), 2055-2056. 31. Wiberg, N. Angew. Chem., Int. Ed. 1968, 7, (10), 766-779. 32. Denk, M. K.; Hatano, K.; Ma, M. Tetrahedron Lett. 1999, 40, (11), 2057-2060. 33. Arduengo Iii, A. J.; Rasika Dias, H. V.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1992, 114, (14), 5530-5534. 34. Caesar, V.; Lugan, N.; Lavigne, G. J. Am. Chem. Soc. 2008, 130, (34), 11286-11287. 35. Gregorio, G.-B.; Jean, B.; Bruno, D.; Guy, B. Angew. Chem., Int. Ed. 2010, 49, (28), 4759-4762. 36. Scarborough, C. C.; Guzei, I. A.; Stahl, S. S. Dalton Trans. 2009, (13), 2284-2286. 37. de Fremont, P.; Marion, N.; Nolan, S. P. Coord. Chem. Rev. 2009, 253, (7-8), 862-892. 38. Alder, R. W.; Blake, M. E.; Bortolotti, C.; Bufali, S.; Butts, C. P.; Linehan, E.; Oliva, J. M.; Orpen, A. G.; Quayle, M. J. Chem. Commun. 1999, (3), 241-242. 39. Hudnall, T. W.; Moerdyk, J. P.; Bielawski, C. W. Chem. Commun. 2010, 46, (24), 4288-4290.

84
40. Kamber, N. E.; Jeong, W.; Waymouth, R. M.; Pratt, R. C.; Lohmeijer, B. G. G.; Hedrick, J. L. Chem. Rev. 2007, 107, (12), 5813-5840. 41. Nyce, G. W.; Glauser, T.; Connor, E. F.; Mick, A.; Waymouth, R. M.; Hedrick, J. L. J. Am. Chem. Soc. 2003, 125, (10), 3046-3056. 42. Iwamoto, K. i.; Hamaya, M.; Hashimoto, N.; Kimura, H.; Suzuki, Y.; Sato, M. Tetrahedron Lett. 2006, 47, (40), 7175-7177. 43. Cazin, C. S. J. C. R. Chim. 2009, 12, (10-11), 1173-1180. 44. Storey, J. M. D.; Williamson, C. Tetrahedron Lett. 2005, 46, (43), 7337-7339. 45. Zhang, Y.; Zhao, L.; Patra, P. K.; Hu, D.; Ying, J. Y. Nano Today 2009, 4, (1), 13-20. 46. Barrett, A. G. M.; Love, A. C.; Tedeschi, L. Org. Lett. 2004, 6, (19), 3377-3380. 47. MeiXuan, T.; Yugen, Z.; Jackie , Y. Y. Adv. Synth. Catal. 2009, 351, (9), 1390-1394. 48. Enders, D.; Breuer, K.; Raabe, G.; Runsink, J.; Teles, J. H.; Melder, J.-P.; Ebel, K.; Brode, S. Angew. Chem., Int. Ed. 1995, 34, (9), 1021-1023. 49. Csihony, S.; Culkin, D. A.; Sentman, A. C.; Dove, A. P.; Waymouth, R. M.; Hedrick, J. L. J. Am. Chem. Soc. 2005, 127, (25), 9079-9084. 50. Nyce, G. W.; Csihony, S.; Waymouth, R. M.; Hedrick, J. L. Chem.-Eur. J. 2004, 10, (16), 4073-4079. 51. Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B. Chem. Rev. 2009, 109, (8), 3561-3598. 52. Garrison, J. C.; Youngs, W. J. Chem. Rev. 2005, 105, (11), 3978-4008. 53. Delaude, L. Eur. J. Inorg. Chem. 2009, (13 ), 1681-1699. 54. Delaude, L.; Demonceau, A.; Wouters, J. Eur. J. Inorg. Chem. 2009, (13 ), 1882-1891. 55. Sauvage, X.; Demonceau, A.; Delaude, L. Adv. Synth. Catal. 2009, 351, (11-12), 2031-2038. 56. Kayaki, Y.; Yamamoto, M.; Ikariya, T. Angew. Chem., Int. Ed. 2009, 48, (23), 4194-4197. 57. Holbrey, J. D.; Reichert, W. M.; Tkatchenko, I.; Bouajila, E.; Walter, O.; Tommasi, I.; Rogers, R. D. Chem. Commun. 2003, 9, (1), 28-29. 58. Smiglak, M.; Holbrey, J. D.; Griffin, S. T.; Reichert, W. M.; Swatloski, R. P.; Katritzky, A. R.; Yang, H.; Zhang, D.; Kirichenko, K.; Rogers, R. D. Green Chem. 2007, 9, (1), 90-98. 59. Van Ausdall, B. R.; Glass, J. L.; Wiggins, K. M.; Aarif, A. M.; Louie, J. J. Org. Chem. 2009, 74, (20), 7935-7942. 60. Voutchkova, A. M.; Feliz, M.; Clot, E.; Eisenstein, O.; Crabtree, R. H. J. Am. Chem. Soc. 2007, 129, (42), 12834-12846. 61. Tommasi, I.; Sorrentino, F. Tetrahedron Lett. 2005, 46, (12), 2141-2145. 62. Tommasi, I.; Sorrentino, F. Tetrahedron Lett. 2006, 47, (36), 6453-6456. 63. Tommasi, I.; Sorrentino, F. Tetrahedron Lett. 2009, 50, (1), 104-107. 64. Zhou, H.; Zhang, W.-Z.; Liu, C.-H.; Qu, J.-P.; Lu, X.-B. J. Org. Chem. 2008, 73, (20), 8039-8044. 65. Pawar, G. M.; Buchmeiser, M. R. Adv. Synth. Catal. 2010, 352, (5), 917-928. 66. Fabien, B.; Tsuyoshi, K.; Mathias, D.; Gérard, M.; Fernando , P. C.; Antoine, B. Angew. Chem., Int. Ed. 2007, 46, (45), 8632-8635. 67. Kuhn, N.; Kratz, T. Synthesis 1993, (6), 561-562. 68. Gorodetsky, B.; Ramnial, T.; Branda, N. R.; Clyburne, J. A. C. Chem. Commun. 2004, 10, (17), 1972-1973. 69. Magill, A. M.; Cavell, K. J.; Yates, B. F. J. Am. Chem. Soc. 2004, 126, (28), 8717-8724. 70. Tapu, D.; Dixon, D. A.; Roe, C. Chem. Rev. 2009. 71. Denk, M. K.; Rodezno, J. M.; Gupta, S.; Lough, A. J. J. Organomet. Chem. 2001, 617-618, 242-253. 72. Arduengo, A. J.; Davidson, F.; Dias, H. V. R.; Goerlich, J. R.; Khasnis, D.; Marshall, W. J.; Prakasha, T. K. J. Am. Chem. Soc. 1997, 119, (52), 12742-12749. 73. Anthony , J. A., III; Calabrese, J. C.; Fredric, D.; Rasika Dias, H. V.; Jens , R. G.; Roland, K.; William , J. M.; Matthias, T.; Reinhard, S. Helv. Chim. Acta 1999, 82, (12), 2348-2364. 74. Wanzlick, H. W. Angew. Chem. Int. Ed. 1962, 1, (2), 75-80. 75. Chen, Y. T.; Jordan, F. J. Org. Chem. 1991, 56, (17), 5029-5038.

85
76. Graham, D. C.; Cavell, K. J.; Yates, B. F. J. Phys. Org. Chem. 2005, 18, (4), 298-309. 77. Frey, G. D.; Lavallo, V.; Donnadieu, B.; Schoeller, W. W.; Bertrand, G. Science 2007, 316, (5823), 439-441. 78. Hudnall, T. W.; Moorhead, E. J.; Gusev, D. G.; Bielawski, C. W. J. Org. Chem. 2010, 75, (8), 2763-2766. 79. Ukai, T. T., Ryuzo; Dokawa, Takashi. Yakugaku Zasshi 1943, 63, 296-300. 80. Jeffrey, S. J. Angew. Chem., Int. Ed. 2004, 43, (11), 1326-1328. 81. Enders, D.; Balensiefer, T. Acc. Chem. Res. 2004, 37, (8), 534-541. 82. Moore, J. L.; Rovis, T. Top. Curr. Chem. 2010, 291, 77-144. 83. Biju, A. T.; Wurz, N. E.; Glorius, F. J. Am. Chem. Soc. 2010, 132, (17), 5970-5971. 84. De Sarkar, S.; Studer, A. Org. Lett. 2010, 12, (9), 1992-1995. 85. Debray, J.; Le̕vêque, J.-M.; Philouze, C.; Draye, M.; Demeunynck, M. J. Org. Chem. 2010, 75, (6), 2092-2095. 86. Feroci, M.; Chiarotto, I.; Orsini, M.; Inesi, A. Chem. Commun. 2010, 46, (23), 4121-4123. 87. Nair, V.; Varghese, V.; Paul, R. R.; Jose, A.; Sinu, C. R.; Menon, R. S. Org. Lett. 2010, 12, (11), 2653-2655. 88. Sarkar, S. D.; Grimme, S.; Studer, A. J. Am. Chem. Soc. 2010, 132, (4), 1190-1191. 89. Vora, H. U.; Rovis, T. J. Am. Chem. Soc. 2010, 132, (9), 2860-2861. 90. Kiesewetter, M. K.; Shin, E. J.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2010, 43, (5), 2093-2107. 91. Lapworth, A. Journal of the Chemical Society, Transactions 1903, 83, 995-1005. 92. Kano, T.; Sasaki, K.; Konishi, T.; Mii, H.; Maruoka, K. Tetrahedron Lett. 2006, 47, (27), 4615-4618. 93. Suzuki, Y.; Bakar, M. D. A.; Muramatsu, K.; Sato, M. Tetrahedron 2006, 62, (17), 4227-4231. 94. Song, J. J.; Tan, Z.; Reeves, J. T.; Yee, N. K.; Senanayake, C. H. Org. Lett. 2007, 9, (6), 1013-1016. 95. Mukaiyama, T. Angew. Chem., Int. Ed. 2004, 43, (42), 5590-5614. 96. Song, J. J.; Tan, Z.; Reeves, J. T.; Fandrick, D. R.; Yee, N. K.; Senanayake, C. H. Org. Lett. 2008, 10, (5), 877-880. 97. Nyce, G. W.; Lamboy, J. A.; Connor, E. F.; Waymouth, R. M.; Hedrick, J. L. Org. Lett. 2002, 4, (21), 3587-3590. 98. Grasa, G. A.; Kissling, R. M.; Nolan, S. P. Org. Lett. 2002, 4, (21), 3583-3586. 99. Singh, R.; Kissling, R. M.; Letellier, M.-A.; Nolan, S. P. J. Org. Chem. 2003, 69, (1), 209-212. 100. Movassaghi, M.; Schmidt, M. A. Org. Lett. 2005, 7, (12), 2453-2456. 101. Maki, B. E.; Scheidt, K. A. Org. Lett. 2008, 10, (19), 4331-4334. 102. Wong, F. T.; Patra, P. K.; Seayad, J.; Zhang, Y.; Ying, J. Y. Org. Lett. 2008, 10, (12), 2333-2336. 103. Yadav, L. D. S.; Rai, V. K.; Singh, S.; Singh, P. Tetrahedron Lett. 2010, 51, (13), 1657-1662. 104. Zhou, H.; Campbell, E. J.; Nguyen, S. T. Org. Lett. 2001, 3, (14), 2229-2231. 105. Wu, J.; Sun, X.; Ye, S.; Sun, W. Tetrahedron Lett. 2006, 47, (28), 4813-4816. 106. Sun, X.; Ye, S.; Wu, J. Eur. J. Org. Chem. 2006, (21), 4787-4790. 107. Liu, Y.-K.; Li, R.; Yue, L.; Li, B.-J.; Chen, Y.-C.; Wu, Y.; Ding, L.-S. Org. Lett. 2006, 8, (8), 1521-1524. 108. Stephanie, S. S.; Jeffrey, W. B. Angew. Chem., Int. Ed. 2006, 45, (36), 6021-6024. 109. Chan, A.; Scheidt, K. A. J. Am. Chem. Soc. 2008, 130, (9), 2740-2741. 110. Suzuki, Y.; Ota, S.; Fukuta, Y.; Ueda, Y.; Sato, M. J. Org. Chem. 2008, 73, (6), 2420-2423. 111. Carmen, C.; Nicolas, D.; Andrew , D. S. Adv. Synth. Catal. 2009, 351, (17), 3001-3009. 112. DiRocco, D. A.; Oberg, K. M.; Dalton, D. M.; Rovis, T. J. Am. Chem. Soc. 2009, 131, (31), 10872-10874. 113. Gu, L.; Zhang, Y. J. Am. Chem. Soc. 2009, 132, (3), 914-915. 114. Hui, L.; Lin, Y.; Song, Y. Adv. Synth. Catal. 2009, 351, (17), 2822-2826. 115. Kawanaka, Y.; Phillips, E. M.; Scheidt, K. A. J. Am. Chem. Soc. 2009, 131, (50), 18028-18029. 116. Li, Y.; Shi, F.-Q.; He, Q.-L.; You, S.-L. Org. Lett. 2009, 11, (15), 3182-3185.

86
117. Molina, M. a. T.; Navarro, C.; Moreno, A.; CsakyÌ, A. G. J. Org. Chem. 2009, 74, (24), 9573-9575. 118. Nair, V.; Sinu, C. R.; Babu, B. P.; Varghese, V.; Jose, A.; Suresh, E. Org. Lett. 2009, 11, (24), 5570-5573. 119. Siti Nurhanna, R.; Yugen, Z.; Jackie , Y. Y. Angew. Chem., Int. Ed. 2009, 48, (18), 3322-3325. 120. Sun, F.-G.; Huang, X.-L.; Ye, S. J. Org. Chem. 2009, 75, (1), 273-276. 121. Thomas, B.; Yoann, C.; Jean, R. Adv. Synth. Catal. 2009, 351, (11-12), 1744-1748. 122. Vedachalam, S.; Zeng, J.; Gorityala, B. K.; Antonio, M.; Liu, X.-W. Org. Lett. 2009, 12, (2), 352-355. 123. Xu, L. W.; Li, L.; Shi, Z. H. Adv. Synth. Catal. 2010, 352, (2-3), 243-279. 124. Uria, U.; Vicario, J. L.; Badea, D.; Carrillo, L.; Reyes, E.; Pesquera, A. Synthesis 2010, (4), 701-713. 125. Terrasson, V.; De Figueiredo, R. M.; Campagne, J. M. Eur. J. Org. Chem. 2010, (14), 2635-2655. 126. Pihko, P. M.; Majander, I.; Erkkilae, A. Top. Curr. Chem. 2010, 291, 29-75. 127. Nielsen, M.; Jacobsen, C. B.; Holub, N.; Paixeo, M. W.; Jargensen, K. A. Angew. Chem., Int. Ed. 2010, 49, (15), 2668-2679. 128. Moyano, A.; El-Hamdouni, N.; Atlamsani, A. Chem.-Eur. J. 2010, 16, (18), 5260-5273. 129. Merino, P.; Marques-Lopez, E.; Tejero, T.; Herrera, R. P. Synthesis 2010, (1), 1-26. 130. Marques-Lopez, E.; Herrera, R. P.; Christmann, M. Nat. Prod. Rep. 2010, 27, (8), 1138-1167. 131. Marinetti, A.; Voituriez, A. Synlett 2010, (2), 174-194. 132. Jautze, S.; Peters, R. Synthesis 2010, (3), 365-388. 133. Werner, T. Adv. Synth. Catal. 2009, 351, (10), 1469-1481. 134. Rios, R.; Cardova, A. Curr. Opin. Drug Discovery Dev. 2009, 12, (6), 824-847. 135. Raj, M.; Singh, V. K. Chem. Commun. 2009, (44), 6687-6703. 136. Palomo, C.; Oiarbide, M.; Lopez, R. Chem. Soc. Rev. 2009, 38, (2), 632-653. 137. Marques-Lopez, E.; Diez-Martinez, A.; Merino, P.; Herrera, R. P. Curr. Org. Chem. 2009, 13, (16), 1585-1609. 138. Enders, D.; Wang, C.; Liebich, J. X. Chem.-Eur. J. 2009, 15, (42), 11058-11076. 139. Coles, M. P. Chem. Commun. 2009, (25), 3659-3676. 140. Buckley, B. R. Annual Reports on the Progress of Chemistry - Section B 2009, 105, 113-128. 141. Bertelsen, S.; Jergensen, K. A. Chem. Soc. Rev. 2009, 38, (8), 2178-2189. 142. Alba, A. N.; Companya, X.; Viciano, M.; Rios, R. Curr. Org. Chem. 2009, 13, (14), 1432-1474. 143. Grondal, C.; Jeanty, M.; Enders, D. Nature Chem. 2010, 2, (3), 167-178. 144. Gioia, C.; Bernardi, L.; Ricci, A. Synthesis 2010, (1), 161-170. 145. Cahard, D.; Xu, X.; Couve-Bonnaire, S.; Pannecoucke, X. Chem. Soc. Rev. 2010, 39, (2), 558-568. 146. Brazier, J. B.; Tomkinson, N. C. O. Top. Curr. Chem. 2010, 291, 281-347. 147. Albrecht, L.; Albrecht, A.; Krawczyk, H.; Jargensen, K. A. Chem.-Eur. J. 2010, 16, (1), 28-48. 148. Gnanou, Y.; Fontanille, M., Organic and Physical Chemistry of Polymers. John Wiley & Sons, Inc.: 2007. 149. Matyjaszewski, K., Radical Polymerization. Wiley-VCH Verlag GmbH & Co. KGaA: 2010; p 103-166. 150. Jones, R. A.; Karatza, M.; Voro, T. N.; Civcir, P. U.; Franck, A.; Ozturk, O.; Seaman, J. P.; Whitmore, A. P.; Williamson, D. J. Tetrahedron 1996, 52, (26), 8707-8724. 151. Jones, R. A.; Civcir, P. U. Tetrahedron 1997, 53, (34), 11529-11540. 152. Hedrick, J. L., Nyce, G. W., Waymouth, R. M. Heteroatom-stabilized carbenes and their precursors as depolymerization catalysts. 2006. 153. Mignani, G.; Destarac, M.; Taton, D.; Gnanou, Y.; Baceiredo, A.; Kato, T.; Bonnette, F.; Sivasankarapillai, G. SYNTHESIS OF URETHANES AND OF POLYURETHANES CATALYSED BY CARBENES. WO2005113626, 2010.

87
154. Bantu, B.; Manohar Pawar, G.; Wurst, K.; Decker, U.; Schmidt, A. M.; Buchmeiser, M. R. Eur. J. Inorg. Chem. 2009, 2009, (13), 1970-1976. 155. Bantu, B.; Pawar, G. M.; Decker, U.; Wurst, K.; Schmidt, A. M.; Buchmeiser, M. R. Chem.-Eur. J. 2009, 15, (13), 3103-3109. 156. Pinaud, J.; Vijayakrishna, K.; Taton, D.; Gnanou, Y. Macromolecules 2009, 42, (14), 4932-4936. 157. Connor, E. F.; Nyce, G. W.; Myers, M.; Mack, A.; Hedrick, J. L. J. Am. Chem. Soc. 2002, 124, (6), 914-915. 158. Dove, A. P.; Pratt, R. C.; Lohmeijer, B. G. G.; Culkin, D. A.; Hagberg, E. C.; Nyce, G. W.; Waymouth, R. M.; Hedrick, J. L. Polymer 2006, 47, (11), 4018-4025. 159. Culkin, D. A.; Jeong, W.; Csihony, S.; Gomez, E. D.; Balsara, N. P.; Hedrick, J. L.; Waymouth, R. M. Angew. Chem., Int. Ed. 2007, 46, (15), 2627-2630. 160. Jeong, W.; Shin, E. J.; Culkin, D. A.; Hedrick, J. L.; Waymouth, R. M. J. Am. Chem. Soc. 2009, 131, (13), 4884-4891. 161. Jeong, W.; Hedrick, J. L.; Waymouth, R. M. J. Am. Chem. Soc. 2007, 129, (27), 8414-8415. 162. Jeong, W.; Hedrick, J. L.; Waymouth, R. M. J. Am. Chem. Soc. 2007, 129, (27), 8414-8415. 163. Olivier, C.; Matthew , K. K.; Andrew, M.; Philippe, D.; James , L. H.; Robert , M. W. Angew. Chem., Int. Ed. 2007, 46, (25), 4719-4721. 164. Schmidt, M. A.; Müller, P.; Movassaghi, M. Tetrahedron Lett. 2008, 49, (27), 4316-4318. 165. Dove, A. P.; Li, H.; Pratt, R. C.; Lohmeijer, B. G. G.; Culkin, D. A.; Waymouth, R. M.; Hedrick, J. L. Chem. Commun. 2006, (27), 2881-2883. 166. Jensen, T. R.; Breyfogle, L. E.; Hillmyer, M. A.; Tolman, W. B. Chem. Commun. 2004, (21). 167. Kamber, N. E.; Jeong, W.; Gonzalez, S.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2009, 42, (5), 1634-1639. 168. Nederberg, F.; Lohmeijer, B. G. G.; Leibfarth, F.; Pratt, R. C.; Choi, J.; Dove, A. P.; Waymouth, R. M.; Hedrick, J. L. Biomacromolecules 2007, 8, (1), 153-160. 169. Lohmeijer, B. G. G.; Dubois, G.; Leibfart, F.; Pratt, R. C.; Nederberg, F.; Nelson, A.; Waymouth, R. M.; Wade, C.; Hedrick, J. L. Org. Lett. 2006, 8, (21), 4683-4686. 170. Rodriguez, M.; Marrot, S.; Kato, T.; Stérin, S.; Fleury, E.; Baceiredo, A. J. Organomet. Chem. 2007, 692, (4), 705-708. 171. Guo, L.; Zhang, D. J. Am. Chem. Soc. 2009, 131, (50), 18072-18074. 172. Raynaud, J.; Absalon, C.; Gnanou, Y.; Taton, D. J. Am. Chem. Soc. 2009, 131, (9), 3201-3209. 173. Raynaud, J.; Absalon, C.; Gnanou, Y.; Taton, D. Macromolecules 2010, 43, (6), 2814-2823. 174. Raynaud, J.; Absalon, C.; Gnanou, Y.; Taton, D. J. Am. Chem. Soc. 2009, 131, (9), 3201-3209. 175. Kamber, N. E.; Jeong, W.; Waymouth, R. M.; Pratt, R. C.; Lohmeijer, B. G. G.; Hedrick, J. L. Chem. Rev. 2007, 107, (12), 5813-5840. 176. Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, (12), 5606-5655. 177. Raynaud, J.; Ottou, W. N.; Gnanou, Y.; Taton, D. Chem. Commun. 2010, 46, (18), 3203-3205. 178. Hsieh, H. L.; Quirk, R. P., Anionic Polymerization: Principles and Practical Applications. New York, 1996. 179. Penczek, S.; Cypryk, M.; Duda, A.; Kubisa, P.; Slomkowski, S. Prog. Polym. Sci. 2007, 32, (2), 247-282. 180. Scholten, M. D.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2008, 41, (20), 7399-7404. 181. Raynaud, J.; Ciolino, A.; Baceiredo, A.; Destarac, M.; Bonnette, F.; Kato, T.; Gnanou, Y.; Taton, D. Angew. Chem., Int. Ed. 2008, 47, (29), 5390-5393. 182. Raynaud, J.; Gnanou, Y.; Taton, D. Macromolecules 2009, 42, (16), 5996-6005. 183. Webster, O. W., Group Transfer Polymerization: A Critical Review of Its Mechanism and Comparison with Other Methods for Controlled Polymerization of Acrylic Monomers. In New Synthetic Methods, Springer Berlin / Heidelberg: 2004; Vol. 167, pp 257-266. 184. Webster, O. W.; Hertler, W. R.; Sogah, D. Y.; Farnham, W. B.; RajanBabu, T. V. J. Am. Chem. Soc. 1983, 105, (17), 5706-5708. 185. Sogah, D. Y.; Hertler, W. R.; Webster, O. W.; Cohen, G. M. Macromolecules 1987, 20, (7), 1473-1488.

88
186. Quirk, R. P.; Ren, J. Macromolecules 1992, 25, (24), 6612-6620. 187. Dicker, I. B.; Cohen, G. M.; Farnham, W. B.; Hertler, W. R.; Laganis, E. D.; Sogah, D. Y. Macromolecules 1990, 23, (18), 4034-4041. 188. Quirk, R. P.; Kim, J. S. J. Phys. Org. Chem. 1995, 8, (4), 242-248. 189. Webster, O. W. Journal of Polymer Science Part A: Polymer Chemistry 2000, 38, (16), 2855-2860. 190. Daniel , J. C.; Dimitri , M. K.; Brent , C. N.; Andrew , G. T.; Christopher , W. B. Angew. Chem., Int. Ed. 2009, 48, (28), 5187-5190. 191. Coady, D. J.; Norris, B. C.; Lynch, V. M.; Bielawski, C. W. Macromolecules 2008, 41, (11), 3775-3778. 192. Norris, B. C.; Bielawski, C. W. Macromolecules 2010, 43, (8), 3591-3593. 193. Moad, G.; Rizzardo, E.; Thang, S. H. Acc. Chem. Res. 2008, 41, (9), 1133-1142. 194. Moad, G.; Rizzardo, E.; Thang, S. H. Polymer 2008, 49, (5), 1079-1131. 195. York, A. W.; Kirkland, S. E.; McCormick, C. L. Adv. Drug Delivery Rev. 2008, 60, (9), 1018-1036. 196. Tehfe, M.-A.; Makhlouf Brahmi, M.; Fouassier, J.-P.; Curran, D. P.; Malacria, M.; Fensterbank, L.; Lacote, E.; Lalevele, J. Macromolecules 2010, 43, (5), 2261-2267. 197. Walton, J. C.; Brahmi, M. M.; Fensterbank, L.; LacoÌ‚te, E.; Malacria, M.; Chu, Q.; Ueng, S.-H.; Solovyev, A.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, (7), 2350-2358. 198. Solovyev, A.; Ueng, S. H.; Monot, J.; Fensterbank, L.; Malacria, M.; Lacôte, E.; Curran, D. P. Org. Lett. 2010, 12, (13), 2998-3001. 199. Ueng, S. H.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Curran, D. P. Org. Lett. 2010, 12, (13), 3002-3005. 200. Bebbington, M. W. P.; Bourissou, D. Coord. Chem. Rev. 2009, 253, (9-10), 1248-1261. 201. Khramov, D. M.; Bielawski, C. W. J. Org. Chem. 2007, 72, (25), 9407-9417. 202. Coady, D. J.; Bielawski, C. W. Macromolecules 2006, 39, (26), 8895-8897. 203. Khramov, D. M.; Bielawski, C. W. Chem. Commun. 2005, (39), 4958-4960. 204. Binder, W. H.; Sachsenhofer, R. Macromol. Rapid Commun. 2007, 28, (1), 15-54. 205. Stefan, B.; Carmen, G.; Kerstin, K.; Viktor, Z. Angew. Chem., Int. Ed. 2005, 44, (33), 5188-5240. 206. Kamplain, J. W.; Bielawski, C. W. Chem. Commun. 2006, (16), 1727-1729. 207. Tennyson, A. G.; Kamplain, J. W.; Bielawski, C. W. Chem. Commun. 2009, (16), 2124-2126.

89

90
Chapitre II : Polymérisation par étapes du téréphtaldehyde
catalysée par les carbènes N-hétérocycliques : synthèse de
polybenzoïnes

91

92
II.1) Contexte et objectifs
La « condensation de la benzoïne » est une réaction créant une liaison C-C à partir de
deux molécules portant une fonction aldéhyde. Avec le benzaldéhyde comme substrat, une
telle réaction conduit à la formation d’un céto-alcool en beta d’un composé aromatique,
appelé benzoïne (Schéma II.1). Le produit de réaction possède un centre asymétrique et,
avec certains catalyseurs, la réaction peut-être stéréosélective. Cette réaction a largement
été étudiée depuis le début du siècle dernier, que ce soit du point de vue des catalyseurs
employés que de la compréhension du mécanisme mis en jeu1-4.
Schéma II.1 : condensation de la benzoïne à partir du benzaldéhyde.
C’est en 1832, avec les travaux de Wöhler et Liebig, que la condensation de la
benzoïne catalysée par des anions cyanates a été décrite pour la première fois1, 4. En traitant
de l’huile d’amande, composée de benzaldéhyde et d’acide cyanhydrique, avec de la
potasse, on forme en effet la benzoïne5. D’après le mécanisme proposé par Lapworth en
1903, les ions cyanates ont pour rôle d’attaquer le carbone électrophile de la fonction
aldéhyde du benzaldéhyde pour former un carbanion (Schéma II.2). L’espèce intermédiaire
ainsi formée présente alors une réactivité inversée de l’atome de carbone électrophile du
groupe carbonyle. Ceci permet une attaque nucléophile ultérieure sur le carbonyle d’un
autre aldehyde. Ce phénomène a par la suite été formalisé dans le concept de réaction dite
« Umpolung » (traduction de réaction inversée en allemand) détaillée par Seeback6.
Schéma II.2: Condensation de la benzoïne catalysée par les ions cyanates4.

93
L’utilisation de sels de thiazolium en tant que catalyseurs de la condensation de la
benzoïne n’est apparue qu’en 1943, avec les travaux de Ukai et al.7. En se fondant sur les
travaux de Lapworth en 1958, Ronald Breslow a décrit le mécanisme de la condensation de
la benzoïne catalysée par la thiamine (2-[3-[(4-amino-2-méthyl-pyrimidin-5-yl)méthyl]-4-
méthyl-thiazol-5-yl]éthanol)8. Les groupes thyazolium ou imidazolium sont présents dans de
nombreuses enzymes et participent à des transformations chimiques variées, que ce soit en
tant que sites actifs ou bien en tant que sites liants9.
Dans le cas de la co-enzyme Thiamine (1) (vitamine B1), le site actif est un groupe
thiazolium qui peut être déprotoné pour former un thyazol-2-ylidene (2), c'est-à-dire un
carbène N-hétérocyclique (NHC) (Schéma II.3). Lors de la condensation de la benzoïne, le
NHC s’additionne sur le groupe carbonyle du benzaldéhyde pour former un adduit de
thiazolium 3. Une déprotonation/reprotonation de ce composé amène à l’ « aldéhyde
activé » sous une forme stable et conjuguée, aussi appelée « intermédiaire de Breslow » 4.
Un tel agent nucléophile est alors capable de s’additionner sur un substrat électrophile tel
que le groupement carbonyle d’une seconde molécule d’aldéhyde. L’intermédiaire 5
régénère le carbène 2 en formant la benzoïne 6.
Schéma II.3 : Mécanisme de la condensation de la benzoïne catalysée par la thiamine8.

94
La découverte de ce mécanisme a été à l’origine de très nombreux travaux de
recherche, notamment pour la mise au point de nouveaux catalyseurs de la condensation de
la benzoïne, mais aussi pour l’utilisation des sels de thyazolium pour catalyser d’autres
réactions1, 2, 10. Une impulsion à ce domaine a été donnée par Stetter et coll. qui ont montré
que ces sels de thiazolium, en présence d’une base, peuvent catalyser la condensation
d’aldéhydes aliphatiques, contrairement aux ions cyanates5. Cette réaction a aussi été
étendue au formaldéhyde11, mais également à divers autres composés portant un groupe
carbonyle (cétones, aldéhydes, …), comme nous avons pu le voir au cours du chapitre I, avec
la réaction de Stetter (Schéma II.4).
Schéma II.4: Réaction de Stetter catalysée par les sels de thyazolium en présence d’une
base.
Du fait que cette réaction permet la formation d’un centre stéréogénique, différents
carbènes N-hétérocycliques (NHCs) ont été développés, notamment des catalyseurs
optiquement actifs4, 10. La génération de tels centres chiraux est particulièrement
importante dans le cas de molécules d’intérêt biologique, celles-ci pouvant devenir
totalement inactives, voir toxiques, si elles ne présentent pas la bonne configuration
spatiale12.
La condensation intramoléculaire de molécules bifonctionnelles (bis-aldéhydes ou
céto-aldéhydes) organocatalysée a également été envisagée à de nombreuses reprises. Une
telle réaction conduit à des composés cycliques, comme illustré Schéma II.5.13
Schéma II.5: condensation de la benzoïne intramoléculaire catalysée par un NHC14
asymétrique.
La condensation de bis-aldéhydes, c’est à dire la polymérisation par étapes de tels
monomères bifonctionnels a été décrite deux fois dans la littérature15, 16, mais,
curieusement, aucune étude portant sur la polycondensation organocatalysée par les NHCs,
n’avait été décrite au moment de démarrer cette thèse. Dans chaque cas, les ions cyanates
ont été utilisés comme catalyseurs de la condensation du téréphtaldéhyde, pour former le
poly(1,4-phénylène-1-oxo-2-hydroxyéthylène) ou polybenzoïne (Schéma II.6). Ceux-ci ont

95
ultérieurement été modifiés dans le but d’obtenir des polymères à propriétés spécifiques.
Ainsi, l’oxydation de la polybenzoïne, suivie d’une réaction avec des diamines
aromatiques ont permis à Stickney et al. de synthétiser des polyquinoxalines, polymères
connus pour leurs performances mécaniques et adhésives (Voie 1)16. Ces réactions ont
également été appliquées sur des polybenzoïnes (PBs) préparés à partir de l’isophtaldéhyde,
monomère présentant les fonctions aldéhydes en ortho du cycle aromatique. De leur côté,
Fernandez et al. ont utilisé le PB pour générer des dérivés du poly(1,4-phenylènevinylène)
(PPV), polymère connu pour être un très bon conducteur électronique (Voie 2)15. Pour cela,
le PB a été traité au moyen d’une base forte afin de former un polydianion. Celui-ci a par la
suite été exploité comme nucléophile afin de réagir avec le sulfate de diméthyle ou le
chlorure d’acétylène pour former respectivement, du PPV diacétate et du PPV
diméthyléther.
Schéma II.6: Polymérisation par étapes du téréphtaldéhyde et modification de la
polybenzoïne pour former des polyquinoxalines et des dérivés du poly(1,4-
phenylènevinylène) (PPV) 15, 16.
Dans cette partie du travail, nous avons utilisé les NHCs comme catalyseurs de la
polyaddition du téréphtaldéhyde, d’une part, afin d’élargir l’étendue des réactions de
polymérisations catalysées par les NHCs, et d’autre part, afin de corréler les résultats de ces
polymérisations avec la réactivité et la nucléophilie des carbènes testés. Jusqu’à présent, la

96
polymérisation du téréphtaldéhyde bien que peu étudiée, a utilisé des ions cyanates comme
catalyseurs15, 16. Or ces ions sont potentiellement capables de former de l’acide
cyanhydrique, un gaz très nocif. La catalyse par les NHCs est donc un moyen de surmonter ce
problème.
Comme présenté dans le chapitre I, les NHCs ont récemment émergé comme
catalyseurs organiques efficaces de différentes réactions de polymérisation en chaines telles
la polymérisation par ouverture de cycle des oxydes d’éthylène et de propylène17-19, des
esters cycliques et autres hétérocycles20 ou bien encore la polymérisation par transfert de
groupe des acrylates et méthacrylates d’alkyle21-23. Leur utilisation en catalyse de
polymérisation par étapes a également été décrite, mais de manière très succincte24-27.
Parmi ces dernières, le téréphtaldéhyde a été utilisé comme co-monomère du 1,1’-(1,4-
phénylène)diprop-2-ene-1-one dans une réaction de Stetter, au cours de laquelle un
intermédiaire de Breslow est généré (Schéma II.7)26. Les poly(1,4-dicétones) ainsi obtenus
ont par la suite été modifiés afin de former des copolymères alternés phénylène-pyrrole,
furyle ou thiényle.
Schémas II.7 : Polymérisation par étapes du téréphtaldéhyde et du 1,1’-(1,4-
phénylène)diprop-2-ene-1-one catalysée par les thyazol-2-ylidenes26.
L’objectif dans ce chapitre est de montrer que les NHCs permettent la catalyse de la
réaction de polymérisation par étapes du téréphtaldéhyde seul, pour former la
polybenzoïne. Pour cela, la condensation de la benzoïne a été appliquée à cette molécule
bifonctionnelle au moyen de différents organocatalyseurs.

97
II.2) Influence des conditions expérimentales
II.2.1) Choix des catalyseurs NHCs
Comme indiqué dans la partie bibliographique, l’organocatalyse de polymérisations
par les NHCs est un domaine émergent en chimie des polymères. Au LCPO, les travaux de
thèse de Jean Raynaud ont montré que les NHCs sont capables de catalyser la
polymérisation par transfert de groupe des acrylates et des méthacrylates d’alkyles. Il a
également été montré que les NHCs sont de bons catalyseurs -ou même amorceurs directs-
de la polymérisation par ouverture de cycle des oxiranes (oxyde d’éthylène et de propylène).
Certains des NHCs utilisés par J. Raynaud étaient ainsi disponibles pour le début de
cette étude. Nous avons donc utilisé les imidazol-2-ylidènes I à III (Figure II.1) pour catalyser
les premières expériences de polymérisation par étapes du téréphtaldéhyde (TPA). Par la
suite, le 1,3,4-triphényl-1,2,4-triazol-5-ylidene (NHC IV), c'est-à-dire le carbène développé
par Enders et commercialement disponible, a également été testé afin de faire varier la
nature du catalyseur. Les structures des différents carbènes utilisés pour la polymérisation
du téréphtaldéhyde sont montrées sur la Figure II.1.
Figure II.1: Structures des NHCs utilisés pour la catalyse de la polymérisation du TPA.
Tous ces NHCs ont été utilisés sous leur forme « nue », c'est-à-dire sans résidus
métalliques. Pour cela, les imidazol-2-ylidènes ont été synthétisés par déprotonation de sels
d’imidazolium au moyen d’une base forte (Schéma I.3 du paragraphe I.2.1.1). En fonction de
la nature des substituants du carbène, le produit de cette réaction a par la suite été distillé,
sublimé ou recristallisé.
Les carbènes étant des espèces très réactives et sensibles à l’air et à l’humidité, il est
nécessaire de les manipuler sous atmosphère inerte (azote ou argon). Il faut notamment les
préparer et les stocker en boite à gants, et effectuer par la suite les réactions de

98
polymérisation sous atmosphère d’azote ou sous vide. De même, les solvants et autres
réactifs ne doivent pas présenter de trace d’eau.
Comme décrit dans la littérature pour la condensation de la benzoïne28, les premières
expériences de polymérisation du TPA (Schéma II.8) ont été effectuées à température
ambiante, en utilisant une quantité catalytique de NHC (1% molaire par rapport au
monomère), dans le THF comme solvant.
Schéma II.8: Polyaddition du téréphtaldéhyde (TPA) catalysée par les NHCs.
Toutes ces réactions ont été réalisées sous atmosphère inerte, à partir de TPA
lyophilisé, pour préserver l’activité des catalyseurs NHCs. Lors de l’ajout du solvant sur le
monomère et le catalyseur, la solution a tout de suite pris une teinte orangée-rouge avec les
catalyseurs I, II et III et bleue marine avec le NHC IV (figure II.2). Après 30 minutes, les
réactions ont été stoppées, car la formation d’un précipité empêchait l’agitation de la
solution. Pour pallier cette difficulté, les réactions ont par la suite été conduites à 40°C afin
de favoriser la solubilisation du polymère formé. Cette température de réaction a aussi
permis de poursuivre les réactions pendant 24 h ou plus.
Figure II.2: Milieu réactionnel des polymérisations du TPA catalysé par différents NHCs.
III
III IV

99
Un suivi des polymérisations a alors été réalisé en effectuant des prélèvements des
milieux réactionnels. La conversion (p) des fonctions aldéhydes a été déterminée au moyen
des spectres de RMN 1H dans le DMSO-d6 des prélèvements effectués. Un spectre
représentatif et permettant de calculer la valeur de p est montré figure II.3. Pour cela,
l’intégration du groupement de pics entre 7,3 et 8,3 ppm, correspondant aux protons
aromatiques du TPA et des polymères, a volontairement été calibrée à 2. D’autre part,
l’intégration des pics au alentour de 10 ppm, correspondant aux fonctions aldéhydes
résiduelles, a été déterminée par rapport à cette valeur. La conversion p correspond alors à
la soustraction de cette dernière intégration à 1 (Cf. Figure II.3).
Figure II.3 : Spectre de RMN 1H (δ en ppm) dans le DMSO-d6 (*) de polybenzoïnes obtenus
après polymérisations du TPA catalysées par le NHC IV. (# = pic correspondant au MeOH
ajouté pour terminer la réaction). A titre de comparaison, un spectre de RMN 1H (δ en ppm)
dans le DMSO-d6 de la benzoïne pure est présenté en encadré.
Comme indiqué dans le Tableau II.1, la nature du NHC a une grande influence sur la
conversion du monomère, ainsi que sur la valeur des masses molaires des polymères formés.
Parmi les quatre catalyseurs employés, le NHC IV s’avère le plus efficace, en permettant des
conversions relativement élevées et l’obtention de polymères de plus fortes masses
molaires.
Si on compare les différents NHCs, leur efficacité peut être classée dans l’ordre
suivant : NHC I ≈ NHC II < NHC III << NHC IV. Cette tendance ne peut malheureusement pas
être totalement corrélée à la basicité ou à la nucléophilie des différents carbènes puisque
seul les pKas des NHC I et II ont été reportés29. Cependant, la légère augmentation d’activité
pour le NHC III comparée au NHC I et II, peut être expliquée par la présence des
substituants encombrants adamantyle du NHC III qui pourraient faciliter la libération du
dérivé de la benzoïne en fin de cycle catalytique (Cf. Schéma II.8).
10 9 8 7 6 5 4 3 2 1 ppm
2.500
3.330
5.959
6.045
6.125
6.247
6.401
6.456
6.535
6.615
7.322
7.339
7.446
7.468
7.489
7.610
7.628
7.833
7.851
7.867
7.884
7.905
7.932
7.952
7.974
8.005
8.027
8.043
8.117
8.137
8.146
9.937
9.947
10.036
10.053
OHC
O
OHCHO
n
a
b
c
a b+c
aromaticprotons
*#
Protons Aromatiques (Iarom. = 2)
b
c
a
a
b+ca
P = 1-Ia
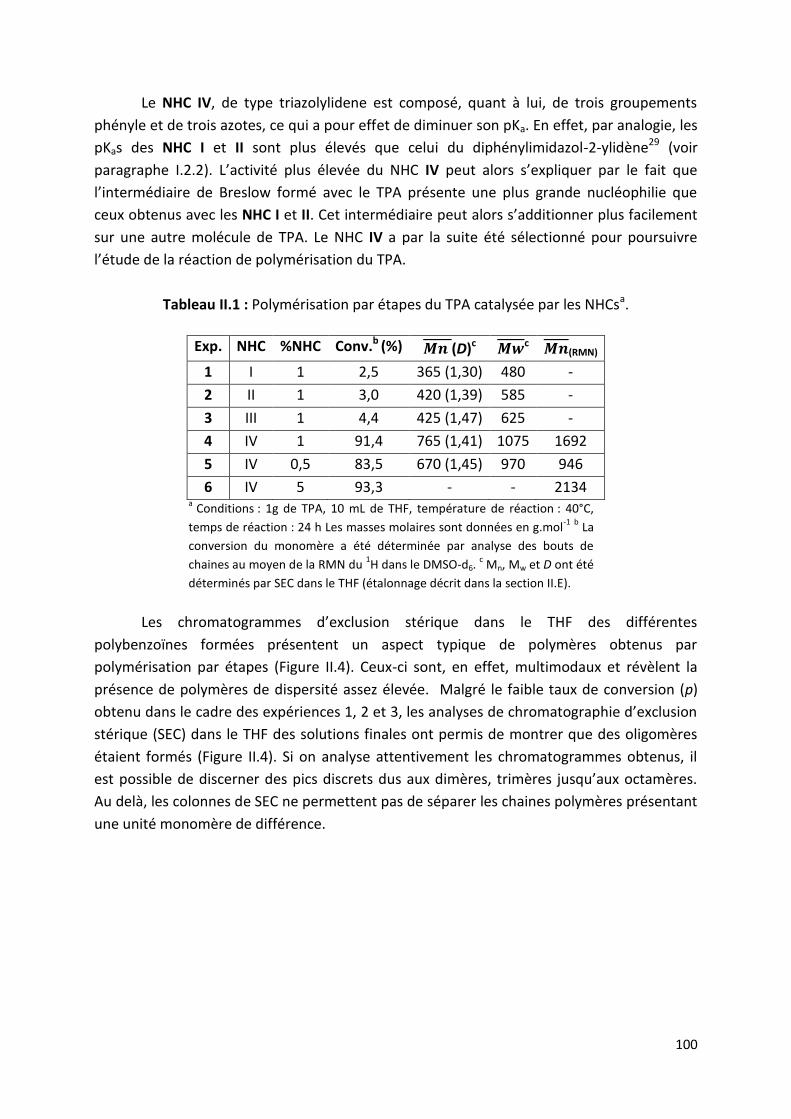
100
Le NHC IV, de type triazolylidene est composé, quant à lui, de trois groupements
phényle et de trois azotes, ce qui a pour effet de diminuer son pKa. En effet, par analogie, les
pKas des NHC I et II sont plus élevés que celui du diphénylimidazol-2-ylidène29 (voir
paragraphe I.2.2). L’activité plus élevée du NHC IV peut alors s’expliquer par le fait que
l’intermédiaire de Breslow formé avec le TPA présente une plus grande nucléophilie que
ceux obtenus avec les NHC I et II. Cet intermédiaire peut alors s’additionner plus facilement
sur une autre molécule de TPA. Le NHC IV a par la suite été sélectionné pour poursuivre
l’étude de la réaction de polymérisation du TPA.
Tableau II.1 : Polymérisation par étapes du TPA catalysée par les NHCsa.
Exp. NHC %NHC Conv.b (%) (D)c c (RMN)
1 I 1 2,5 365 (1,30) 480 -
2 II 1 3,0 420 (1,39) 585 -
3 III 1 4,4 425 (1,47) 625 -
4 IV 1 91,4 765 (1,41) 1075 1692
5 IV 0,5 83,5 670 (1,45) 970 946
6 IV 5 93,3 - - 2134 a
Conditions : 1g de TPA, 10 mL de THF, température de réaction : 40°C,
temps de réaction : 24 h Les masses molaires sont données en g.mol-1
b La
conversion du monomère a été déterminée par analyse des bouts de
chaines au moyen de la RMN du 1H dans le DMSO-d6.
c Mn, Mw et D ont été
déterminés par SEC dans le THF (étalonnage décrit dans la section II.E).
Les chromatogrammes d’exclusion stérique dans le THF des différentes
polybenzoïnes formées présentent un aspect typique de polymères obtenus par
polymérisation par étapes (Figure II.4). Ceux-ci sont, en effet, multimodaux et révèlent la
présence de polymères de dispersité assez élevée. Malgré le faible taux de conversion (p)
obtenu dans le cadre des expériences 1, 2 et 3, les analyses de chromatographie d’exclusion
stérique (SEC) dans le THF des solutions finales ont permis de montrer que des oligomères
étaient formés (Figure II.4). Si on analyse attentivement les chromatogrammes obtenus, il
est possible de discerner des pics discrets dus aux dimères, trimères jusqu’aux octamères.
Au delà, les colonnes de SEC ne permettent pas de séparer les chaines polymères présentant
une unité monomère de différence.

101
Figure II.4: Chromatogrammes obtenus par SEC dans le THF (détecteur RI) pour les
polymérisations par étapes du TPA catalysées par les NHCs (entrées 1 à 4 du tableau II.1).
La formation de ces oligomères n’est en revanche pas détectable par RMN du 1H dans
le DMSO-d6. En effet, pour déterminer le degré de polymérisation ( ̅̅ ̅̅ ̅̅ ) de ces
polymérisations par étapes, nous avons utilisé la relation de Carothers :
. Lorsque
p est inférieure à 0,5, le ̅̅ ̅̅ ̅̅ est alors inférieur à 2 et ne permet pas de déduire la formation
d’oligomères.
En appliquant la relation de Carothers aux valeurs obtenues pour les expériences 1, 2
et 3 des résultats proches de 1 sont alors obtenus, et ne sont pas vraiment significatifs. En
revanche lorsque celle-ci est utilisée pour l’expérience 4, on obtient :
Relation de Carothers :
avec p= 0,914
ce qui donne = 11,7 soit (RMN) = × 134 + 134 = 1692 g.mol-1.
(134 correspond à la fois à la masse molaire de l’unité répétitive et des bouts de chaines).
En utilisant 5% de NHC IV comme catalyseur (Tableau II.1, entrée 6), un polymère
insoluble dans le THF a été obtenu. Etant donné qu’aucune SEC dans le DMSO n’est
disponible au laboratoire, un moyen pratique permettant d’obtenir la masse molaire de ce
polymère est de le solubiliser dans le DMSO-d6, et de l’analyser par RMN 1H.

102
En comparant les valeurs de (RMN) des entrées 4 et 6, on note que l’utilisation
d’une quantité plus importante de catalyseur, afin de favoriser la croissance des chaines
n’est pas justifiée. De même, une quantité de 0,5% de NHC par rapport au monomère ne
permet pas d’atteindre des masses molaires aussi élevées qu’avec 1%. Cette valeur de 1%
semble donc être une valeur seuil pour obtenir des polymères de masses molaires de
quelques milliers de g/mol.
II.2.2) Effet du solvant de réaction
Afin de pallier les problèmes de solubilité du PB dans le THF, nous avons réalisé une
nouvelle série d’expériences de polymérisations dans d’autres solvants. Comme l’ont montré
Riffle et al.16, le PB est un polymère aromatique peu soluble dans de nombreux solvants,
notamment ceux apolaires. La présence des fonctions cétones et alcools du PB permettent
cependant sa solubilisation dans le DMSO. Nous avons donc utilisé ce solvant comme milieu
réactionnel des réactions de polymérisation du TPA (Tableau II.2).
Tableau II.2 : Polymérisation par étapes du TPA catalysée par les NHCs, conduite dans
différents solvants.
Exp. NHC %NHC Solvant Conv.b (%) (D)c c Mn(RMN)
4 4 1 THF 91,4 765 (1,41) 1075 1692
7 4 1 DMSO 78,3 650 (1,43) 930 751
8 4 1 THF + DMSO 81,5 680 (1,38) 940 858
Conditions : 1 g de TPA, 10 mL de THF, température de réaction : 40°C, temps de réaction :
24h. Les masses molaires sont données en g.mol-1
(étalonnage décrit dans la section II.E). b
La conversion du monomère a été déterminée par analyse des bouts de chaines au moyen
de la RMN du 1H dans le DMSO-d6
c Mn, Mw et D ont été déterminés par SEC dans le THF.
Comme attendu, le DMSO a permis de maintenir un milieu réactionnel homogène au
détriment cependant de la cinétique de polymérisation qui s’est avérée plus lente. Ceci a
conduit aussi à des polymères de plus faibles masses molaires, comparativement aux
réactions réalisées en conditions similaires dans le THF.
Des réactions dans un mélange THF + DMSO ont également été réalisées, en espérant
combiner les avantages du DMSO, i.e une bonne solubilité, et ceux du THF, i.e une cinétique
plus rapide. Ces conditions ont, en effet, permis d’obtenir des résultats intermédiaires, que
ce soit en termes de cinétique ou bien de masses molaires des polymères finaux.
L’ensemble de ces résultats nous amène à penser que la cinétique de polymérisation,
plus rapide dans le THF, pourrait être due à l’établissement d’associations intermoléculaires
privilégiées des différentes molécules dans ce solvant. On peut en effet penser que des
interactions de type empilements π-π par les groupes aromatiques des polymères et des
molécules monomères (Figure II.5) peuvent induire un phénomène de concentration locale

103
en monomère et polymère plus élevée, augmentant ainsi la cinétique de polymérisation.
Dans le cas du DMSO, solvant plus dissociant que le THF, une telle organisation des
molécules monomères et oligomères serait moins probable, entrainant ainsi une diminution
de la vitesse de polymérisation.
Figure II.5: Représentation des interactions π-π se déroulant entre les groupements
aromatiques des molécules monomères TPA et des oligomères, dans le THF.
II.2.3) Influence du temps de réaction
Dans le but de mieux comprendre l’effet de ces différents solvants sur la cinétique de
polymérisation du TPA, les réactions ont été conduites à différents temps. Pour cela, des
prélèvements ont été effectués à des intervalles réguliers, entre 0 et 120 h de
polymérisation en utilisant le NHC IV comme catalyseur. Les résultats obtenus sont
regroupés dans le Tableau II.3.
Tableau II.3 : Suivi cinétique de la polymérisation du TPD catalyée par le NHC IV dans
différents solvants.
Exp.a Temps
de réaction (h) Solvant
Conv.b
(%) (D)c c (RMN)
Cycles
(%)d
9 3 THF 81,3 690 (1,40) 970 850 4
10 17 THF 91,0 750 (1,41) 1060 1623 49
4 24 THF 91,4 765 (1,41) 1075 1692 51
11 72 THF 92,2 850 (1,54) 1150 1852 51
12 120 THF 93 ,3 820 (1,32) 1086 2134 59
7 24 DMSO 78,3 650 (1,43) 930 751 0
13 72 DMSO 82,0 705 (1,36) 955 878 5
14 120 DMSO 85,6 650 (1,52) 990 1065 30
8 24 THF + DMSO 81,5 680 (1,38) 940 858 6
15 72 THF + DMSO 86,9 875 (1,46) 1275 1157 15
16 120 THF + DMSO 91,2 880 (1,45) 1270 1657 42 a
Conditions : 1g de TPD, 10mL de THF, température de réaction : 40°C. Les masses molaires sont données en
g.mol-1
. b La conversion du monomère a été déterminée par analyse des bouts de chaines au moyen de la RMN
du 1H dans le DMSO-d6.
c Mn, Mw et D ont été déterminées par SEC dans le THF.
d Le pourcentage de cycles a pu
être déterminé comme décrit dans la section II.E.

104
Comme nous pouvions le prévoir, des temps de réaction plus longs permettent
d’obtenir des conversions plus élevées et des polymères de plus fortes masses molaires. Les
polymères obtenus après 24, 72 et 120 h dans le THF (expériences 4, 11 et 12 du tableau II.3)
présentent, respectivement, des températures de transition vitreuse de 112, 137 et 148°C
(Figure II.6), confirmant l’augmentation de la masse molaire avec le temps.
Figure II.6 : Analyse enthalpique différentielle des polymères obtenus par les expériences 4,
11 et 12. La température de transition vitreuse des polymères est donnée par le point
d’inflexion de la courbe (2ème montée en température à 10°C/min).
D’après les entrées 1-5 du tableau II.3, on note que la polymérisation du TPA dans le
THF permet d’obtenir des taux de conversion relativement élevés dès le début de la réaction
(81,3% au bout de 3h). En revanche, la vitesse de la polymérisation ralentit nettement par la
suite, et une augmentation de seulement 1% du taux de conversion est observé entre 24h et
170 h. La diminution de la concentration en fonctions aldéhydes peut expliquer cette
diminution de vitesse de réaction, après 24 h. En revanche, lorsque le DMSO et le mélange
THF + DMSO sont utilisés comme solvant(s), un temps de réaction de 120 h est nécessaire
pour pouvoir atteindre des masses molaires comparables à celles obtenues dans le THF (
≈ 1700 g/mol).
On note aussi qu’entre 72 et 120 h de réaction dans le THF, les masses molaires ont
tendances à diminuer et ce, malgré le fait que le taux de conversion augmente. Ceci peut
être expliqué par la formation de polymères cycliques. En effet, les macrocyles présentent
112.1°C(I)
136.9°C(I)
148.4°C(I)
-800
-600
-400
-200
0
He
at
Flo
w (
mW
/g)
60 80 100 120 140 160 180Temperature (°C)
Exp.4 (24h)––––––– Exp.9 (72h)––––––– Exp.10 (120h)–––––––
overlay
------ Exp. 4 (24h) ------ Exp. 11 (72h) ------ Exp. 12 (120h)

105
un temps d’élution plus faible en SEC comparé à leurs homologues linéaires de même masse
molaire30. Une discussion concernant la présence potentielle d’oligomères cycliques dérivés
de polybenzoïne 31 est présentée ci-après.
La formation de polymères cycliques au cours des réactions de polymérisations par
étapes est un phénomène bien connu32. Les polymères cycliques peuvent aussi se former au
cours de certaines polymérisations par ouverture de cycles (Figure II.7). Enfin, des méthodes
de synthèse peuvent être spécifiquement mises en œuvre pour préparer des polymères
présentant une telle morphologie30, 32.
Figure II.7: Formation de polymères cycliques.
Ainsi, dans le cadre de certaines polymérisations en chaîne, un équilibre entre
chaînes linéaires et polymères cycliques peut s’établir (A, Figure II.7). On peut par exemple
citer le polymérisation par ouverture de cycle des cyclosiloxanes, conduisant à la formation

106
de polydiméthylsiloxanes (PDMS) linéaires et cycliques, ou encore la polymérisation
cationique de l’oxyde d’éthylène au cours de laquelle des réactions de transfert induisent la
formation d’oligomère cycliques33.
D’autre part, des polymères cycliques peuvent être obtenus à partir de chaînes
polymères linéaires α-ω-difonctionnelles (B, Figure II.7). Deux possibilités sont envisageables
dans ce cas là :
Une réaction de cyclisation intramoléculaire (i, B, Figure II.7), lorsque la chaine
présente des bouts de chaines capables de réagir entre eux (polymère linéaire α-ω-
hétérofonctionnel)34, 35. Au Laboratoire, cette méthode à été mise à profit afin de
former des copolymères à blocs cycliques s’auto-assemblant en tubes de manière
élégante36. C’est également cette méthode qui permet d’expliquer la formation
potentielle de PBs cycliques dans notre cas, les fonctions aldéhydes en bout de
chaine du PB pouvant réagir entre elles.
Une réaction de cyclisation intermoléculaire (ii, B, Figure II.7) entre un polymère
linéaire α-α’-difonctionnel et une molécule difonctionnelle présentant deux
groupements réactifs, complémentaires des fonctions terminales de la chaîne
polymère.
Dans la plupart des cas de cyclisation par les bouts chaines, la formation de
macrocycles s’obtient dans des conditions réactionnelles très diluées32. Ceci permet en effet
de limiter les réactions intermoléculaires dans le cadre de la voie i, B ou d’éviter le couplage
de plusieurs chaines polymères au cours de la voie ii, B. Pour ce qui est des polymérisations
par étapes, la cyclisation apparaît donc en fin de polymérisation, lorsque seule une faible
quantité de groupements réactionnels restent dans le milieu30, 32.
Les polymères cycliques présentent des propriétés physiques différentes des
polymères linéaires, ce qui permet notamment de les caractériser au moyen de différentes
techniques d’analyses32. Ainsi, il a été montré que pour des polymères de même masse
molaire, le rapport de la viscosité entre polymères cycliques et polymères linéaires
([η]cyclique/[η]linéaire) est proche de 0,66. De fait, les polymères cycliques ont un volume
hydrodynamique plus faible que les polymères linéaires de même masse molaire. Les
macrocycles se caractérisent donc par une viscosité intrinsèque plus faible, une température
critique de solubilisation plus élevée ou encore un temps d’élution plus long en SEC.
Comme détaillé ci-après, cette dernière particularité nous a notamment permis
d’évaluer la quantité de PB cycliques obtenus au cours des différentes expériences de
polymérisations par étapes du TPA.

107
II.3) Détermination de la quantité de polybenzoïnes cycliques
La caractérisation des polymères issus du TPA a été réalisée au moyen de deux
techniques complémentaires : RMN du 1H, et SEC dans le THF. Ces deux techniques
permettent d’obtenir la masse molaire moyenne en nombre des polymères, notamment via
la relation de Carothers (
) s’agissant de la RMN.
Dans le cas où les polymères obtenus présentent exclusivement une structure
linéaire, une calibration en SEC avec des échantillons standards de même nature que le
polymère analysé (polybenzoïne) doit fournir des valeurs de identiques à celles
déterminées par RMN 1H.
Comme l’indiquent les résultats du tableau II.3, on obtient des valeurs différentes
lorsque les polymères sont caractérisés par ces deux méthodes. Les données obtenues par
SEC sont donc « erronées », malgré un étalonnage dont le principe est expliqué dans les
lignes suivantes. L’hypothèse de la formation de polymères cycliques au cours de la
polymérisation, ayant pour conséquence une diminution des masses molaires, peut
expliquer cette différences dans les valeurs de déterminées par SEC et RMN 1H.
II.3.1) Principe d’étalonnage de la SEC pour la caractérisation des
polybenzoïnes.
Le PB n’ayant été décrit que deux fois dans la littérature15, 16, les données sur ce
polymère sont rares. La calibration des colonnes de SEC au moyen d’échantillons
« standards » de PBs n’est donc pas possible. Afin de déterminer les masses molaires
« vraies » de ce polymère, nous avons tout d’abord utilisé la SEC sur la base d’une calibration
des colonnes au moyen d’étalons de polystyrène, en utilisant le THF comme solvant. Les
premières valeurs de ainsi obtenues par ce moyen sont donc apparentes ; de fait, elles
ne peuvent pas être directement comparées à celles obtenues par RMN 1H.
Comme déjà indiqué, la polymérisation par étapes du TPA a montré la formation de
pics discrets en SEC (Figure II.8), que nous avons attribués aux différents oligomères linéaires
(dimère, trimères, etc.). Etant donné que l’on connait exactement la masse de chacun de ces
oligomères, on peut assigner un temps d’élution précis à une masse molaire.
Cependant, il faut souligner que cette méthode indirecte pour calibrer notre système
de SEC n’est valable que pour des polymères de faibles masses (inférieurs à = 7). Au-
delà de cette valeur, en effet, les colonnes utilisées ne permettent plus de différencier de
pics discrets.
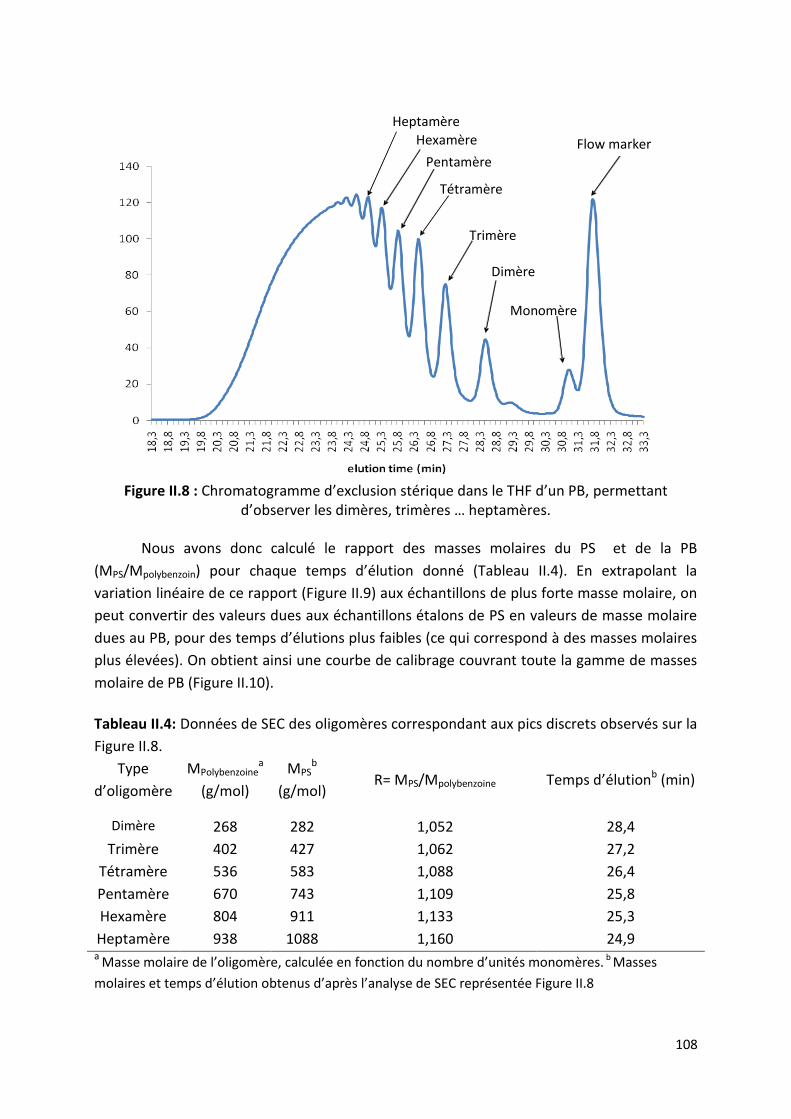
108
Figure II.8 : Chromatogramme d’exclusion stérique dans le THF d’un PB, permettant
d’observer les dimères, trimères … heptamères.
Nous avons donc calculé le rapport des masses molaires du PS et de la PB
(MPS/Mpolybenzoin) pour chaque temps d’élution donné (Tableau II.4). En extrapolant la
variation linéaire de ce rapport (Figure II.9) aux échantillons de plus forte masse molaire, on
peut convertir des valeurs dues aux échantillons étalons de PS en valeurs de masse molaire
dues au PB, pour des temps d’élutions plus faibles (ce qui correspond à des masses molaires
plus élevées). On obtient ainsi une courbe de calibrage couvrant toute la gamme de masses
molaire de PB (Figure II.10).
Tableau II.4: Données de SEC des oligomères correspondant aux pics discrets observés sur la
Figure II.8.
Type
d’oligomère
MPolybenzoinea
(g/mol)
MPSb
(g/mol) R= MPS/Mpolybenzoine Temps d’élutionb (min)
Dimère 268 282 1,052 28,4
Trimère 402 427 1,062 27,2
Tétramère 536 583 1,088 26,4
Pentamère 670 743 1,109 25,8
Hexamère 804 911 1,133 25,3
Heptamère 938 1088 1,160 24,9 a Masse molaire de l’oligomère, calculée en fonction du nombre d’unités monomères. b Masses
molaires et temps d’élution obtenus d’après l’analyse de SEC représentée Figure II.8
Flow marker
Dimère
Monomère
Trimère
Pentamère
Heptamère
Hexamère
Tétramère

109
Figure II.9 : Evolution du rapport MPS/ Mpolybenzoïne (rapport de la masse molaire du standard
PS sur la masse molaire du PB pour un temps d’élution donné) en fonction de la masse des
oligomères de PB.
Figure II.10 : Courbe de calibrage utilisée pour déterminer les masses des PBs synthétisés
par polymérisation par étapes du TPA catalysée par les NHCs.
A l’aide de cette courbe d’étalonnage, nous avons alors recalculé les masses molaires
obtenues par SEC et par RMN, et avons ainsi pu constater que les valeurs ne coïncidaient pas
exactement. Nous avons donc attribué cet écart à la présence de polybenzoïnes cycliques
y = 0,0002x + 1,0035 R² = 0,9907
1,04
1,06
1,08
1,1
1,12
1,14
1,16
1,18
1,2
0 200 400 600 800 1000 1200
R =
MP
S/M
po
lyb
enzo
in
Mpolybenzoin (g/mol)

110
dans nos échantillons. Dans une telle situation, les valeurs de déterminées par RMN 1H
sont faussées. En effet, les PBs cycliques ne présentent pas de fonctions aldéhydes,
comparativement à leurs équivalents linéaires ; l’intensité des pics correspondant aux
fonctions aldéhydes en RMN du 1H s’en trouve diminuée. Les valeurs de calculées à
partir de la relation de Carothers, pour les échantillons comportant une certaine proportion
de polymères cycliques sont donc surévaluées. Nous avons alors tenté de quantifier cette
proportion de polymères cycliques comme discuté ci-après.
II.3.2) Détermination du pourcentage de polymères cycliques.
En comparant les masses molaires obtenues par SEC, dont le calibrage indirect avec
les échantillons de PBs a été discuté précédemment, avec celles obtenues par RMN 1H, il est
possible de déterminer le pourcentage de polymères cycliques présents dans nos
échantillons.
Au cours de la polymérisation par étapes du TPA, les fonctions aldéhydes sont
réparties entre deux populations : le monomère résiduel et les polymères linéaires, à un
temps t donné.
En définitive, le milieu réactionnel contient trois types de populations : le monomère
résiduel, les polymères linéaires et les polymères cycliques. Le mécanisme de formation de
la population de cycles est illustré Schéma II.5 (les PBs cycliques ne présentent aucune
fonction aldéhyde).
Schéma II.9: Mécanisme de formation des PBs cycliques à partir de PBs linéaires via une
catalyse par le NHC IV.

111
D’après Flory, la proportion de chaque i-mère, pour un certain taux de conversion de
la réaction, est donnée par l’expression suivante37:
Ni/N0 = p(i-1)(i-p)2
Avec Ni : nombre de molecules de i-mères au temps t et N0 : nombre de molécules au début
de la réaction (i.e nombre de molécules de monomères à t = 0). Avec i = 1, on obtient la
fraction de molécules monomère résiduelles au temps t:
fmonomère = (1-p)2
Connaissant la valeur de p par RMN du 1H, on peut en déduire la concentration en fonctions
aldéhyde portées par le monomère restant au temps t:
[CHO]monomère = [CHO]0(1-p)2
Comme la concentration en fonctions aldéhydes dans le milieu réactionnel à un temps t
provient à la fois du monomère et des polymères linéaires, on peut écrire :
[CHO]t = [CHO]0 (1-p) = [CHO]monomère + [CHO]polym. lin.
où [CHO]0 est la concentration totale en fonctions aldéhydes au temps t = 0,
[CHO]monomère est la concentration en fonctions aldéhydes provenant du monomère résiduel
au temps t,
[CHO]polym. lin. est la concentration en fonctions aldéhydes provenant des polymères linéaires
formés au temps t.
soit
[CHO]polym. lin. = [CHO]0 [(1-p) - (1-p)2]
On sait également que, pour chaque fonction céto-alcool formée, deux fonctions aldéhydes
sont consommées. Ceci nous permet d’écrire :
([CHO]0/2) × p = [céto-alcool]polym. lin. + [céto-alcool]polym. cycl.
Dans le cas des polymères linéaires, leur structure chimique implique une relation simple
entre la concentration en fonctions céto-alcool et la concentration en fonctions aldéhydes :
[céto-alcool]polym. lin. = ( × [CHO]polym. lin.) / 2 = [CHO]0 [(1-p)- (1-p)2] / 2
Avec un déterminé à partir de la relation de Carothers, et en considérant qu’aucun
cycle n’est formé. Cette relation peut également s’écrire sous la forme :

112
[céto-alcool]polym. cycl. = [[CHO]0 × p – DPn × [CHO]0 × [(1-p)- (1-p)2]]/2
Cette expression devrait normalement donner une concentration nulle en fonctions céto-
alcools. Cependant, peut également être déterminé à partir des valeurs de
obtenues en SEC dans le THF.
Si l’on définit C comme étant le nombre de polymères cycliques à un instant t et T le nombre
total de molécules présentes dans le milieu réactionnel, on peut alors écrire :
C = T – N0(1-p)
Comme = M0N0/T, il vient
)1(10
pM
M
T
C n
Avec p obtenu à partir de la RMN du 1H dans le THF et déterminée par SEC dans le THF.
On obtient ainsi le pourcentage de polymères cycliques (C/T) présents dans le milieu
réactionnel. Quand les échantillons ont pu être analysés par SEC, cette dernière équation a
pu être appliquée. Les résultats du Tableau II.3 montrent ainsi que la population de
polymères cycliques varie en fonction du solvant et du temps de réaction, comme discuté ci-
dessous.
II.3.3) Influence des conditions expérimentales sur le pourcentage de
polymères cycliques
Le graphe montrant l’évolution de la fraction de polymères cycliques en fonction du
temps est montré Figure II.11.
Ce graphique indique clairement que la formation de polymères cycliques augmente
pour les derniers pourcentages de la conversion des fonctions aldéhydes. En effet, à ce
moment de la réaction de polymérisation, la concentration en fonctions aldéhydes dans le
milieu devient de plus en plus faible. La statistique de rencontre de deux fonctions aldéhydes
présentes au bout de deux chaines polymères différentes est donc très faible. Par
conséquent, la probabilité de rencontre de deux fonctions aldéhyde d’une même chaîne
(conduisant à des polymères cycliques) prévaut. Ainsi, pour un taux de conversion de plus en
plus élevé, la fraction de polymères cycliques aura tendance à augmenter.
Ce phénomène a notamment été discuté par Stepto et al. et Gordon et al.30. Un
diagramme théorique montrant l’évolution de la quantité de cycles en fonction de p dans le
cadre des polymérisations par étapes (Figure II.12) a d’ailleurs été établi par ces auteurs. On
peut remarquer que l’évolution de la fraction de polymères cycliques dans le cadre des

113
polymérisations par étapes du TPA, dans le THF et le mélange THF + DMSO (courbe rouge et
verte de la Figure II.11), est assez proche des prévisions de ces auteurs.
Figure II.11: Evolution de la fraction de PBs cycliques en fonction de p, dans différents solvants de réaction.
Figure II.12 : Evolution du pourcentage de polymères cycliques 5 en fonction de la conversion (p) dans le cadre de polymérisation par étapes, en faisant varier la concentration initiale en
monomère (∞ = dilution infinie). Diagramme de Gordon et Temple30.
0
10
20
30
40
50
60
70
78,3 81,3 81,5 82 85,6 86,9 91 91,2 91,4 92,2 93,3
THF DMSO THF + DMSO

114
Sur la Figure II.11, on peut également remarquer que l’utilisation d’un solvant peu
polaire, pour réaliser la polymérisation du téréphtaldéhyde, induit une augmentation du
pourcentage de polymères cycliques. En effet, pour un même taux de conversion, on
observe une fraction de polymères cycliques plus importante dans le THF, que dans le
mélange THF + DMSO et que dans le DMSO tout seul.
A nouveau, on peut relier cet effet à la solubilité du polymère dans le THF. Le
polymère étant peu soluble dans ce solvant, les chaines auront tendance à se replier sur elle-
même (Figure II.13). Cette conformation favorisera alors la rencontre des extrémités de la
même chaine polymère et, par là même, sa cyclisation.
Figure II.13: Conformation des chaines de polybenzoïne dans le THF ou le DMSO.
Comme l’ont décrit Endo et al.32, les polymères cycliques présentent des unités
monomères chimiquement et physiquement équivalentes, contrairement à leurs
homologues linéaires. Cette équivalence est due au fait que leurs propriétés ne sont pas
affectées par la nature des bouts de chaines.
Cependant, dans notre cas, une telle affirmation n’est pas pertinente. En-effet, les
unités monomères du polymère, peuvent s’enchainer de deux façons différentes : « tête-
tête » ou « tête-queue ». Ces deux types d’enchainement peuvent exister pour les
macrocycles comme pour les polymères linéaires.
Dans le schéma II.10 on note, par exemple, que si le monomère subit une attaque
nucléophile du catalyseur, et que par la suite un dimère est inséré, un enchainement « tête-
queue » est alors obtenu. Au contraire, si le dimère s’ajoute sur le catalyseur en premier
(Schéma II.11), par le même bout de chaîne qu’auparavant, l’insertion du monomère conduit
alors à un enchainement « tête-tête ». On peut donc légitimement s’interroger sur
l’influence de ces deux types d’enchainements sur la solubilité et sur la rigidité de la chaine
de polybenzoïne.
Pour rendre compte de la proportion de chaque type d’enchainement, il faudrait
couper sélectivement la chaine et analyser les produits réactionnels après clivage. Etant
donné la nature des liaisons C-C dans la chaine de polybenzoïne, cette réaction n’est pas
possible. Cependant, l’emploi d’un monomère bis-aldéhydique présentant une liaison
clivable en son sein, tel que l’ont rapporté Itsuno et al.38, permettrait, après qu’il ait été
polymérisé et segmenté, de déterminer le taux d’enchaînements « tête-tête » et « tête-
queue ».

115
Schéma II.10: Mécanisme de formation d’un enchaînement régulier « tête-queue » en présence d’un dimère et d’un monomère lors de la polymérisation par étapes du TPA.
Schéma II.11: Mécanisme de formation d’un enchaînement irrégulier « tête-tête » en présence d’un dimère et d’un monomère lors de la polymérisation par étapes du TPA.

116
De même, la condensation de la benzoïne conduit à la formation de centres stéréogéniques (Figure II.14). L’utilisation d’un catalyseur pour la polymérisation stéréospécifique du TPA, permettant de favoriser une configuration plutôt qu’une autre, permettrait sûrement l’obtention de polymères de tacticité définie. Leur morphologie et leur conformation rendrait alors la formation de macrocycles plus ou moins aisée.
Figure II.14 : les deux énantiomères de la benzoïne
Ces deux points mécanistiques indiquent notamment que les différents protons de la chaine polymère se placent dans des environnements différents. On comprend donc pourquoi les spectres RMN du 1H des polybenzoïnes synthétisés présentaient des groupements de pic relativement larges, quelques soient les protons observés.

117
II.4) Conclusion et perspectives
Ces travaux sont une adaptation de la catalyse par les NHCs de la réaction
de condensation de la benzoïne en chimie moléculaire. De par cette analogie, on peut
penser que chaque étape du processus de polymérisation implique la formation d’un
« intermédiaire de Breslow » par l’attaque initiale du carbène sur une fonction aldéhyde.
Nous avons montré que les NHCs catalysent efficacement la polymérisation par
étapes du téréphtaldéhyde pour conduire à des poly(1,4-phenylène-1-oxo-2-
hydroxyéthylène)s, encore appelés polybenzoïnes.
Quatre NHCs ont été testés, et parmi ceux-ci le 1,3,4-triphényl-1,2,4-triazol-5-ylidène
s’est avéré le plus efficace, les imidazolydènes ne permettant que la formation d’oligomères
de faible masse molaire ( max = 7). Ces polymérisations ont été conduites à 40°C, dans le
THF, le DMSO ou un mélange équivolumique de ces deux solvants.
La formation de polymères cycliques a été mise en évidence, grâce aux techniques
d’analyses complémentaires que sont la SEC et la spectroscopie RMN. A cet effet, une
méthode d’étalonnage particulière de la SEC dans le THF, a été mise au point. De même, des
calculs basés sur la théorie de Flory, concernant les polymérisations par étapes, ont permis
de quantifier la proportion de macrocycles au cours de nos expériences. L’augmentation de
cette proportion avec la conversion des fonctions aldéhydes a alors pu être démontrée. De
même, le pourcentage de macrocycles s’est avéré dépendant du solvant de polymérisation
utilisé.
Cette étude concernant un des tous premiers exemples de polymérisation par étapes
organocatalysée par les NHCs élargit ainsi le spectre des possibilités de catalyses de
polymérisations par les NHCs. Jusqu’à présent, l’organocatalyse de polymérisation par les
NHCs avait essentiellement concerné les polymérisations en chaine (par ouverture de cycle
et par transfert de groupe).
Ces résultats posent cependant un certain nombre de questions, notamment quant
au mécanisme exact de ces polymérisations. En particulier, deux éléments restent à éclaircir:
La nature des enchainements des unités monomères dans le polymère.
La formation de centres de chiralité au cours de la polymérisation.
De même, des conditions expérimentales (choix du solvant et du catalyseur
notamment) permettant d’obtenir des polymères de fortes masses molaires restent à
définir.

118
Etant donné l’efficacité des NHCs à catalyser d’autres réactions de condensation en
chimie moléculaire, cela laisse à penser que de telles réactions pourront seront bientôt être
transposées en chimie macromoléculaire. Parmi ces réactions, on peut par exemple citer la
condensation d’aldéhydes aliphatiques5 au moyen de la condensation de la benzoîne, qui
permettrait d’obtenir des polymères présentant une meilleure solubilité que la polybenzoïne
à partir de dialdéhyde aliphatiques. L’utilisation de l’aroylation des fluorobenzenes catalysée
par les NHCs39 (Schéma II.12) pourrait également être un moyen direct d’obtenir des
polymères conjugués.
Schéma II.12: Aroylation des fluorobenzenes catalysée par les NHCs.
La réactivité et l’instabilité des NHCs vis-à-vis de l’air et de l’eau, est cependant un
frein à leur utilisation en tant que catalyseurs de polymérisations. Les autres travaux réalisés
au cours de cette thèse se sont donc employés à faciliter l’emploi des NHCs dans de telles
conditions.

119
II.5) Références
1. Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, (12), 5606-5655. 2. Marion, N.; Diez-Gonzalez, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, (17), 2988-3000. 3. Jeffrey, S. J. Angew. Chem., Int. Ed. 2004, 43, (11), 1326-1328. 4. Enders, D.; Balensiefer, T. Acc. Chem. Res. 2004, 37, (8), 534-541. 5. Teles, J. H.; Johann-Peter, M.; Klaus, E.; Regina, S.; Eugen, G.; Wolfgang, H.; Stefan, B.; Dieter, E.; Klaus, B.; Gerhard, R. Helv. Chim. Acta 1996, 79, (1), 61-83. 6. Dieter, S. Angew. Chem., Int. Ed. 1979, 18, (4), 239-258. 7. Ukai, T. T., Ryuzo; Dokawa, Takashi. Yakugaku Zasshi 1943, 63, 296-300. 8. Breslow, R. J. Am. Chem. Soc. 1958, 80, (14), 3719-3726. 9. Zhao, H.; Foss, F. W.; Breslow, R. J. Am. Chem. Soc. 2008, 130, (38), 12590-12591. 10. Moore, J. L.; Rovis, T. Top. Curr. Chem. 2010, 291, 77-144. 11. Castells, J.; Geijo, F.; López-Calahorra, F. Tetrahedron Lett. 1980, 21, (47), 4517-4520. 12. LASZLO, P., CHIRALITÉ, chimie. In Encyclopaedia Universalys, 2008. 13. Sun, F.-G.; Huang, X.-L.; Ye, S. J. Org. Chem. 2009, 75, (1), 273-276. 14. Enders, D.; Niemeier, O.; Balensiefer, T. Angew. Chem., Int. Ed. 2006, 45, (9), 1463-1467. 15. Kaul, S. N.; Fernandez, J. E. Macromolecules 2002, 23, (11), 2875-2879. 16. Stickney, K. W.; Glass, T.; Riffle, J. S. Polymer Preprints 1994, 35, 531-532. 17. Raynaud, J.; Absalon, C.; Gnanou, Y.; Taton, D. J. Am. Chem. Soc. 2009, 131, (9), 3201-3209. 18. Raynaud, J.; Absalon, C.; Gnanou, Y.; Taton, D. Macromolecules 2010, 43, (6), 2814-2823. 19. Raynaud, J.; Ottou, W. N.; Gnanou, Y.; Taton, D. Chem. Commun. 2010, 46, (18), 3203-3205. 20. Kamber, N. E.; Jeong, W.; Waymouth, R. M.; Pratt, R. C.; Lohmeijer, B. G. G.; Hedrick, J. L. Chem. Rev. 2007, 107, (12), 5813-5840. 21. Raynaud, J.; Ciolino, A.; Baceiredo, A.; Destarac, M.; Bonnette, F.; Kato, T.; Gnanou, Y.; Taton, D. Angew. Chem., Int. Ed. 2008, 47, (29), 5390-5393. 22. Scholten, M. D.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2008, 41, (20), 7399-7404. 23. Raynaud, J.; Gnanou, Y.; Taton, D. Macromolecules 2009, 42, (16), 5996-6005. 24. Nyce, G. W.; Lamboy, J. A.; Connor, E. F.; Waymouth, R. M.; Hedrick, J. L. Org. Lett. 2002, 4, (21), 3587-3590. 25. Hedrick, J. L., Nyce, G. W., Waymouth, R. M. Heteroatom-stabilized carbenes and their precursors as depolymerization catalysts. 2006. 26. Jones, R. A.; Karatza, M.; Voro, T. N.; Civcir, P. U.; Franck, A.; Ozturk, O.; Seaman, J. P.; Whitmore, A. P.; Williamson, D. J. Tetrahedron 1996, 52, (26), 8707-8724. 27. Mignani, G.; Destarac, M.; Taton, D.; Gnanou, Y.; Baceiredo, A.; Kato, T.; Bonnette, F.; Sivasankarapillai, G. SYNTHESIS OF URETHANES AND OF POLYURETHANES CATALYSED BY CARBENES. WO2005113626, 2010. 28. Enders, D.; Kallfass, U. Angew. Chem., Int. Ed. 2002, 41, (10), 1743-1745. 29. Magill, A. M.; Cavell, K. J.; Yates, B. F. J. Am. Chem. Soc. 2004, 126, (28), 8717-8724. 30. Kricheldorf, H. R. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, (2), 251-284. 31. Zhou, H.; Campbell, E. J.; Nguyen, S. T. Org. Lett. 2001, 3, (14), 2229-2231. 32. Kobayashi, S.; Endo, K., Synthesis and Properties of Cyclic Polymers. In New Frontiers in Polymer Synthesis, Springer Berlin / Heidelberg: 2008; Vol. 217, pp 121-183. 33. Odian, G., Principles of polymerization 3rd ed. 1994. 34. Deffieux, A.; Schappacher, M. Cell. Mol. Life Sci. 2009, 66, (15), 2599-2602. 35. Laurent, B. A.; Grayson, S. M. Chem. Soc. Rev. 2009, 38, (8), 2202-2213. 36. Schappacher, M.; Deffieux, A. Science 2008, 319, (5869), 1512-1515. 37. Gnanou, Y.; Fontanille, M., Organic and Physical Chemistry of Polymers. John Wiley & Sons, Inc.: 2007. 38. Kumagai, T.; Itsuno, S. Macromolecules 2002, 35, (13), 5323-5325. 39. Suzuki, Y.; Ota, S.; Fukuta, Y.; Ueda, Y.; Sato, M. J. Org. Chem. 2008, 73, (6), 2420-2423.

120
Chapitre III : Poly(Carbènes N-hétérocycliques), poly(NHC)s,
et leurs adduits de CO2, poly(NHC-CO2)s: nouveaux
organocatalyseurs recyclables de transestérification et de
condensation de la benzoïne

121

122
III.1) Introduction
Comme discuté dans le chapitre bibliographique, la synthèse des NHCs est
généralement réalisée par déprotonation d’un sel d’azolium au moyen d’une base forte.
L’idée de base dans cette partie du travail est qu’une telle réaction pourrait être appliquée à
des précurseurs polymères portant ces unités de type azolium. C’est le cas des sels de
poly(vinylimidazolium), qui font partie de la famille des polymères liquides ioniques, qui
suscitent un intérêt croissant depuis un dizaine d’années. Ainsi, après une brève introduction
sur cette catégorie particulière de polyélectrolytes, nous décrirons comment nous avons
réussi à accéder à des supports polymères de NHCs, que nous désignerons dans ce travail
par « poly(NHC)s. Nous verrons également que ces derniers peuvent être protégés et ce, de
manière réversible à l’aide de dioxyde de carbone (Schémas III.1). Enfin, nous décrirons
comment ces supports catalytiques ont pu être utilisés et recyclés dans des réactions de
transestérification et de condensation de la benzoïne.
Schéma III.1 : Représentation succincte du travail décrit dans ce chapitre.

123
III.2) Généralités sur les polymères liquides ioniques
Les polymères liquides ioniques (abrégé PILs pour « polymeric ionic liquid »s en
anglais) sont des matériaux polymères obtenus à partir de liquides ioniques présentant une
fonction polymérisable1. Avant de définir plus exactement les PILs, il est nécessaire de
décrire les liquides ioniques dans leur acception classique et de présenter leurs propriétés1.
III.2.1) Les liquides ioniques
Du point de vue sémantique, le terme liquide ionique décrit un liquide composé
d’anions et de cations, tels les sels inorganiques et organiques fondus, que ce soit à
« basse » ou haute température2, 3. Aujourd’hui, ce terme est employé pour désigner les sels
présentant une température de fusion proche ou en dessous de 100 °C, et logiquement les
termes « liquide ionique à température ambiante » (RTIL, pour « room temperature ionic
liquid » en anglais) sont employés pour qualifier les sels présentant une température de
fusion proche ou inférieure à 25 °C. La Figure III.1 montre l’évolution du nombre de
publications et de brevets intégrant les mots « ionic liquid » au cours de ces dernières
années. Dans la suite de ce manuscrit, le terme liquide ionique, abrégé IL pour « ionic
liquid » en anglais, prendra le sens moderne du terme, et représentera donc les sels ayant
un point de fusion inférieur à 100 °C.
Figure III.1 : Evolution du nombre de publications et de brevets contenant le terme « liquide
ionique » de 1997 à 2009. Sources : SciFinder Scholar.

124
III.2.1.1) Des combinaisons d’anions et de cations variées
Si les ILs présentent autant d’intérêt, c’est en raison de leur composition chimique
qui peut être très variée, impliquant par là même diverses propriétés. Même si la nature du
cation, d’origine organique, ne se limite qu’à des composés issus de trois principaux
atomes4 (azote, soufre et phosphore), le nombre important d’anions, qu’ils soient
organiques ou inorganiques, offre maintes possibilités d’associations.
Comme le montre la Figure III.2, dans la plupart des cas, les cations utilisés sont à
base d’azote, tels les ammoniums, les imidazoliums, les pyridiniums, ou encore les
pyrrolidiniums. Les cations les plus utilisés sont souvent encombrés et dissymétriques. Cette
structure permet, en effet, d’obtenir des sels liquides en évitant leur cristallisation5. Les
cations de type sulfonium et phosphonium ne sont pas à négliger et contribuent également
à la diversité des ILs3. La Figure III.2 montre également quelques anions couramment utilisés,
qui comme nous verrons par la suite, influencent plus particulièrement les propriétés du IL
formé, notamment en terme d’hydrophobicité.
Figure III.2 : Cations et anions couramment utilisés dans les ILs. X désigne un halogène et Tf
le groupement CF3SO2.
III.2.1.2) Synthèse des liquides ioniques
Deux méthodes sont généralement utilisées pour préparer les liquides ioniques 6: par
neutralisation d’une solution contenant une amine au moyen d’un acide ou bien la
quaternérisation d’une amine ou d’un dérivé phosphoré par un halogénure d’alcane. Seule la
synthèse des ILs dérivés de cations imidazolium sera détaillée ici, car ces cations sont les
précurseurs de NHCs (Cf. chapitre bibliographique).

125
Le cycle imidazole (Figure III.3) qui les constitue présente la particularité d’être
amphotère : il est capable d’accepter et de donner des protons7. Cette propriété permet
notamment de réaliser des substitutions nucléophiles en position 3, notamment pour la
quaternérisation de l’amine par ionisation du cycle. Diverses réactions peuvent aussi être
effectuées sur la position 1, par exemple des substitutions nucléophiles, ce qui permet de
faire varier les structures des sels d’imidazolium. Enfin, en présence d’une base forte, le
proton en position 2 peut être arraché pour former un carbène N-hétérocyclique (NHC),
comme discuté plus loin7.
Figure III.3 : Structure du cycle imidazole.
Ainsi pour synthétiser des sels d’imidazolium à partir de l’imidazole (commercial), on
substitue généralement l’azote de la position 1 d’abord, au moyen d’une base et d’un
halogénure d’alcane (Schéma III.2, étape i)6. Une telle substitution nucléophile génère le 1-
alkylimidazole qui peut ensuite être quaternérisé en position 3, en présence d’un autre
halogénure d’alcane (Schéma III.2, étape ii)6. Cette dernière réaction se fait généralement
sous reflux mais nécessite souvent des temps de réaction importants (> 24h).
Schéma III.2 : Synthèse de sels d’imidazolium à partir de l’imidazole.
La réactivité des halogénures d’alcane croit dans l’ordre : Cl > Br > I >> F. Ainsi les sels
possédant un anion fluorure ne sont pas assez réactifs pour être préparés par le biais d’une
quaternérisation. De même, cette réaction peut être réalisée à température ambiante
lorsque les groupements halogènes sont remplacés par des groupements triflates ou
tosylates, connus pour être de bons groupes partants6. Les sels d’imidazolium ainsi formés
(Schéma III.2) sont en général purifiés par recristallisation ou décantation après un lavage
dans un non-solvant.

126
Une des particularités des sels d’imidazolium tient au fait que le contre-anion peut
être aisément changé, ce qui permet de moduler les propriétés physico-chimiques de ces
composés (solubilité, viscosité, point de fusion, stabilité thermique,…). Le changement
d’anion peut-être réalisé par métathèse entre deux sels (Schéma III.3)8. Celle-ci peut être
favorisée par la précipitation ou l’immiscibilité d’un des produits formés (généralement un
halogénure d’un métal alcalin). Le processus d’échange d’anion est réversible, mais nécessite
cependant des temps de réaction plus long (de l’ordre de la semaine) pour des rendements
non quantitatifs ( 70%).
Schéma III.3 : Echange d’anion sur un sel d’imidazolium.
III.2.1.3) Propriétés et applications des liquides ioniques
Les liquides ioniques sont connus pour présenter une faible tension de vapeur et une
stabilité thermique élevée3, 5. De plus, en raison d’une polarité variée (en fonction du cation
et de l’anion), ils sont souvent employés comme solvants de réaction pour solubiliser un
grand nombre de composés (organiques, inorganiques ou polymères) insolubles dans les
solvants organiques usuels4. Ils sont par exemple de très bons solvants de la cellulose3, 5. Ces
propriétés particulières les ont catalogués comme nouveaux solvants de la « chimie verte »
pour remplacer les composés organiques volatiles utilisés couramment2. Cependant, les
études récentes concernant la toxicité effective des ILs ont révélés que ceux-ci ne sont pas,
en réalité, inoffensifs9 pour la santé des êtres vivants, et des précautions sont aussi à
prendre quant à leur manipulation.
Une autre singularité des ILs est leur conductivité ionique élevée. Celle-ci peut
atteindre 10-2S.cm-1 dans une large fenêtre électrochimique de l’ordre de 6V3,5. Ceci
s’explique par le fait qu’à température ambiante, les liquides ioniques présentent une forte
concentration en ions, ainsi qu’une mobilité accrue. Cette propriété particulière, associée à
leur faible tension de vapeur, a permis de les utiliser dans un grand nombre d’applications
en tant qu’électrolytes, comme dans des batteries au lithium10, les dispositifs électroactifs7,
les piles à combustibles ou encore les cellules photovoltaïques.

127
D’autres utilisations des ILs peuvent être mentionnées : lubrifiants, catalyseurs en
synthèse organique, solvants pour l’élaboration de nanomatériaux ou de nouvelles
méthodes d’extractions, capture de CO2…2-6, 10-13.
En modifiant la structure du cation ou de l’anion, des liquides ioniques destinés à des
tâches spécifiques (« task specific ionic liquid » = TSIL en anglais) peuvent être élaborés, afin
de répondre à une utilisation donnée14.
Parmi les TSILs, ceux possédant une fonction polymérisable (MILs pour « monomeric
ionic liquids » en anglais) -généralement de type vinyle en position 1- conduisent aux
polymères liquides ioniques (PILs pour « polymeric ionic liquids »).
III.2.2) Les polymères liquides ioniques
L’intérêt des PILs est d’associer les propriétés des ILs discutées précédemment et les
propriétés spécifiques des polymères1. Par exemple, pour certaines applications comme
dans les batteries ou les piles à combustibles, l’état solide des PILs permet d’éviter certains
inconvénients des ILs, comme les fuites de liquide, la toxicité ou encore l’instabilité
dimensionnelle1. La polymérisation des MILs permet donc d’apporter des propriétés
avantageuses : stabilité renforcée, manipulation et mise en forme plus aisées (notamment
sous forme de films), en plus d’un contrôle de la structure au niveau meso- ou
nanoscopique.
La structure des PlLs peut être envisagée de différentes manières, selon la nature du
cation et de l’anion:
Chaîne polymère anionique avec des contre-cations libres1, 13 (A, Figure III.4).
Chaîne polymère cationique avec des contre-anions libres 1, 7, 13, 15(B, Figure III.4).
Il est également possible de combiner ces deux structures en formant des gels
physiques composés de chaines polymères anioniques et cationiques
interpénétrées1, 13 (C, Figure III.4).
Figure III.4 : Structures possibles des polymères liquides ioniques.

128
Les PILs ayant été les plus étudiés sont, là encore, ceux portant un cycle imidazolium.
Ces polymères sont généralement obtenus par polymérisation radicalaire conventionnelle
en solution, à l’aide d’un amorceur radicalaire se décomposant par voie thermique. D’autres
polymères cationiques et présentant des groupements ammoniums, pyridiniums ou
pyrolidiniums ont également été décrits15. Cependant, leur structure ne pouvant permettre
la génération d’un NHC, ils ne seront pas détaillés ici, tout comme les PILs de type A
représentés dans la Figure III.4. Pour plus d’informations concernant les PILs au sens large, le
lecteur peut se reporter à quelques revues récentes sur ce sujet1, 7, 15-17.
III.2.2.1) Monomères liquides ioniques portant un groupe imidazolium.
Deux familles de MILs ont principalement été étudiées : les monomères de type
vinylique et les monomères de type acrylate ou méthacrylate d’imidazolium.
De manière générale, les MILs de type (méth)acrylate sont obtenus en deux étapes1,
7, 18, 19 : réaction de couplage entre un chlorure de (méth)acryloyle et un halogénure d’alcool,
suivie d’une réaction de quaternérisation (Schéma III.4). Un avantage de cette méthode est
la possibilité de faire varier le groupe présent entre la fonction acrylate et le cycle
imidazolium. Ainsi, différents monomères/polymères ont pu être synthétisés avec des
longueurs et des natures de chaines pendantes variées1, 7, 18-21. De tels monomères ont
notamment été synthétisés au laboratoire, en collaboration avec David Mecerreyes, et ont
pu être polymérisés de manière contrôlée et séquentielle par la méthode RAFT (reversible
addition-fragmentation chain transfer)18, 19.
Schéma III.4 : Synthèse des monomères liquides ioniques de type acrylate.
Les MILs de type vinylique sont généralement plus faciles à obtenir, par
quaternérisation du noyau imidazole. Celle-ci peut être réalisée de deux manières
différentes, en fonction du monomère ciblé. Ainsi, en utilisant le p-chlorométhylstyrène et
un imidazole d’alkyle, des monomères de type D peuvent être obtenus (i, Schéma III.5). Les
monomères de type E sont, quant à eux, obtenus par quaternérisation du 1-vinylimidazole
avec un halogénure d’alcane (ii, Schéma III.5).
Comme dans le cas des ILs moléculaires, il est possible de moduler les
propriétés de ces PILs par changement d’anion.

129
Schéma III.5 : Synthèse des monomères liquides ioniques de type vinylique.
III.2.2.2) Polymérisation radicalaire des MILs
Les MILs de type acrylate et ceux de type N-vinyle sont généralement polymérisés par
voie radicalaire, amorcée thermiquement. Ces polymérisations sont relativement faciles à
mettre en oeuvre, mais conduisent à la formation de macromolécules relativement mal
définies. Quelques études récentes ont cependant montré qu’il est possible de contrôler la
polymérisation radicalaire de tels monomères18, 19, 22, 23.
Par exemple, la polymérisation par la méthode RAFT (Figure III.5) de MILs de type
méthacrylate ou méthacrylamide a permis d’obtenir des (co)polymères de structure bien
définie18, 19. Leur agrégation en solution a également été étudiée et a montré que ceux-ci
pouvaient s’assembler en micelles et en vésicules et ce, de manière réversible, par simple
changement d’anion19. Endo et al. ont décrit la polymérisation de monomères de type E
(Figure III.5) par la méthode RAFT/MADIX (macromolecular design via interchange of
xanthate)23. Cette polymérisation contrôlée/vivante a également permis l’extension de
macro-agents de transfert poly(NIPAAm) (poly(N-isopropylacrylamide)) afin d’aboutir à des
copolymères à blocs répondant à des stimuli thermiques.
La polymérisation radicalaire par transfert d’atomes (ATRP, « atome transfer radical
polymerization » en anglais) a également été décrite par Shen et al., avec des monomères de
type D présentant une chaine butyle comme substituant de l’imidazolium22.

130
Figure III.5 : Structures des monomères liquides ioniques et des agents de transferts
employés lors des polymérisations RAFT/MADIX décrites par Endo et al.23 et par Taton,
Gnanou et al.18, 19.
Les MILs peuvent aussi être polymérisés par d'autres méthodes selon le groupe
polymérisable porté par le cycle imidazolium17. Des PILs peuvent par exemple être obtenus
par polymérisation par étapes (Schéma III.6) ou bien par post-modification chimique (via une
réaction de quaternérisation) du poly(N-vinylimidazole) (Schéma III.7)1, 7, 17. Cette dernière
réaction n’étant pas quantitative, la polymérisation de sels de N-vinylimidazolium décrite
précédemment lui est généralement préférée.
Schéma III.6 : Synthèse de PILs au moyen d’une polymérisation par étapes.
Schéma III.7 : Modification de poly(N-vinylimidazole) par quaternérisation, et formation de
copolymères statistiques de PILs et de poly(N-vinylimidazole).

131
III.2.2.3) Applications des PILs
De par leur grande diversité structurale, les PILs ont été utilisés dans diverses
applications1, 7, 17. Ils ont notamment servi comme polyélectrolytes dans des batteries et des
piles à combustibles. Même si ils se sont avérés moins bons conducteurs que les ILs, avec
des conductivités de l’ordre de 10-4 S.cm-1, leur propriétés mécaniques restent très
intéressantes et ont permis de former des films conducteurs transparents. Tout comme les
ILs, ce sont également de très bons capteurs de CO2, pouvant parfois même adsorber deux
fois plus de gaz que leurs homologues moléculaires24. Les PILs en tant que polyélectrolytes
ont également été utilisés comme dispersants et stabilisants de nanoparticules métalliques
ou de polymères conducteurs en milieu aqueux. Enfin, l’utilisation de polymères portant des
fonctions imidazolium dans le domaine biomédical a montrée que ces PILs peuvent
s’associer à des molécules biologiques telles que des protéines ou des médicaments17.
Les PILs ont aussi été employés pour diverses applications comme l’ont notamment
montré Mecerreyes et al8, 25-28., Firestone et al.1, Ohno et al29, 30. ou encore Long et al.17. La
grande majorité d’entre eux ont été synthétisés par polymérisation radicalaire
conventionnelle. Peu d’études ont été consacrées à « l’ingénierie macromoléculaire des
PILs ». Récemment, Gnanou, Taton et al.18, 19, Endo et al.23 et Shen et al.22, ont décrit la
polymérisation radicalaire contrôlée de MILs ainsi que la synthèse de copolymères à blocs
dérivés.
Dans ce chapitre, nous montrerons comment les MILs et les PILs nous ont servis de
précurseurs pour élaborer des supports polymères de NHCs à des fins d’utilisation en
organocatalyse.
III.3) Objectifs
Comme indiqué dans la partie bibliographique, les NHCs représentent une classe de
catalyseurs organiques pour un grand nombre de réactions de la chimie moléculaire et, ont
plus récemment montré tout leur potentiel en chimie macromoléculaire. Les NHCs sont
cependant des molécules réactives et sensibles à l’air, nécessitant d’être manipulés avec
précaution dans des conditions inertes. Par ailleurs, leur synthèse et leur purification
peuvent s’avérer relativement complexes31-33. Pour rappel, les NHCs sont généralement
synthétisés par déprotonation d’un halogénure d’imidazolium à l’aide d’une base forte. En
fin de synthèse, une purification par distillation ou sublimation, conduit à un NHC dépourvu
de tout résidu métallique34.
Afin de faciliter l’utilisation des NHCs comme catalyseurs organiques, il est encore
nécessaire de développer des méthodes permettant d’abaisser leur coût ou de rendre leur
synthèse et leur manipulation plus aisées. Un autre objectif, important dans le domaine de la
catalyse, est de recycler le catalyseur après utilisation. Une technique souvent employée à

132
cet effet consiste à « fixer » le catalyseur sur un support polymère ou bien sur une particule
(organique ou inorganique) pouvant être filtrés et récupérés en fin de réaction.
Ces techniques de support/recyclage de catalyseurs, au sens très large, ont fait
l’objet d’une édition spéciale du journal « Chemical Reviews » en 200935-44. L’utilisation de
supports polymères pour des NHCs a d’ailleurs déjà été décrite dans la littérature, mais dans
la majorité des cas, les NHCs supportés ont été générés in-situ que ce soit pour leur
utilisation en tant qu’organocatalyseurs45-48 ou en tant que ligands de complexes
organométalliques 49.
Les exemples décrivant la présence de NHCs « nus » sur des supports restent donc
rares. Seuls Ying et coll. ont décrit, pendant ce travail de thèse, la synthèse et l’utilisation de
particules réticulées à base de poly(NHC)s pour l’organocatalyse (Schéma III.8)50, 51.
Schéma III.8 : Synthèse de particules de NHCs « nus » décrite par Ying et coll50, 51.
La synthèse de NHCs « nus » et supportés pour une utilisation en milieu homogène,
en d’autres termes des poly(NHC)s, reste donc une voie à développer. Dans ce chapitre,
nous montrerons d’une part, comment ces poly(NHC)s peuvent être générés à partir de PILs
porteurs de groupements imidazolium. D’autre part, nous discuterons de l’avantage
d’utiliser les poly(NHC)s, et les adduits qu’ils forment avec le CO2, comme catalyseurs
supportés à travers deux réactions modèles de la chimie moléculaire : la condensation de la
benzoïne et la transestérification.
Comme cela a été décrit plus haut, le laboratoire a déjà acquis une bonne expérience
pour préparer des copolymères à blocs PILs. Ces polymères présentant des groupes
imidazolium, il pourrait être envisagé de les utiliser comme précurseurs de NHCs supportés
par déprotonation sur le carbone en position 2. Cependant, ces polymères -de type
polyméthacrylate et polyméthacrylamido- n’ont pas été utilisés pour de travail de thèse.

133
En effet, les fonctions esters reliant le groupe imidazolium à la chaine polymère, pourraient
être dégradées dans les conditions basiques52 qui sont appliquées pour la génération des
NHCs. La stratégie présentée sur le Schéma III.9 a donc été adoptée.
Schéma III.9 : Stratégie de synthèse de poly(NHC)s décrite au cours de ce chapitre.
III.4) Synthèse des polymères liquides ioniques précurseurs des
poly(NHC)s
Pour disposer de supports polymères solubles, porteurs de NHCs « nus », nous nous
sommes tournés vers les sels de poly(1-vinyl-3-alkyl imidazolium) (PVIms) comme
précurseurs. Contrairement aux PILs de type (méth)acrylique, les PILs de type N-vinyle
présentent l’avantage de ne porter aucune fonction hydrolysable en milieu basique. La
présence du cycle imidazolium, peut alors permettre la formation de fonctions imidazol-2-
ylidene, par simple déprotonation du proton acide situé en position 2 (Schéma III.10).
Schéma III.10 : Formation de poly(NHC)s par déprotonation de sels de PVIms.

134
III.4.1) Synthèse des monomères de type sels de 1-vinyl-3-
alkylimidazolium
Trois MILs de type sels de 1-vinyl-3-alkylimidazolium ont été synthétisés par réaction
de quaternérisation du 1-vinylimidazole commercial, en présence d’un halogénure d’alkyle,
en l’occurrence les bromures de 1-vinyl-3-isopropylimidazolium (ViPrIm+Br-), de 1-vinyl-3-
butylimidazolium (VBuIm+Br-) et de 1-vinyl-3-(2-phényléthyl)imidazolium (VPhEtIm+Br-). Ces
synthèses sont illustrées par le Schéma III.11.
Schéma III.11 : Synthèse des monomères de type sels de 1-vinyl-3-alkylimidazolium.
Le choix des groupements alkyle a été fait de manière à moduler, non seulement la
solubilité des sels de PVIms précurseurs, mais aussi la nucléophilie des poly(NHC)s générés
ultérieurement par la modification de l’encombrement stérique autour du centre
carbénique.
En effet, il a été montré par Mecerreyes et coll. que la solubilité des PVIms ne varie
pas seulement en fonction du contre-anion, mais que celle-ci est également dépendante du
substituant présent sur l’atome d’azote du cycle imidazolium8. Par ailleurs, comme discuté
dans la partie bibliographique, la nucléophilie des NHCs de type imidazol-2-ylidènes diminue
lorsque l’encombrement stérique du NHC augmente. On peut donc supposer que cette
échelle de nucléophilie ne sera pas affectée pour la série des 3 poly(NHC)s homologues
ciblés.
La réaction de quaternérisation du 1-vinylimidazole avec les bromures d’isopropyle,
de n-butyle et de 2-phényléthyle permet d’obtenir les MILs correspondants, avec de bons
rendements. Les produits s’apparentent à une poudre blanche dans le cas du ViPrIm+Br- ou à
des liquides visqueux dans les cas du VBuIm+Br- et du VPhEtIm+Br-. Chacun de ces
monomères a pu être préparé à l’échelle de quelques grammes ( 10 grammes).
Les trois monomères ont été caractérisés par RMN du 1H dans le chloroforme
deutéré (CDCl3). Les spectres de RMN du 1H de ces MILs ont déjà été décrits par Mecerreyes
et coll.8 et Ritter et al.53.
Le spectre de RMN 1H du ViPrIm+Br- (Figure III.7) révèle ainsi la présence des pics
caractéristiques des protons du groupe vinyle à 7,45 ; 6,01 et 5,28 ppm, sous la forme de
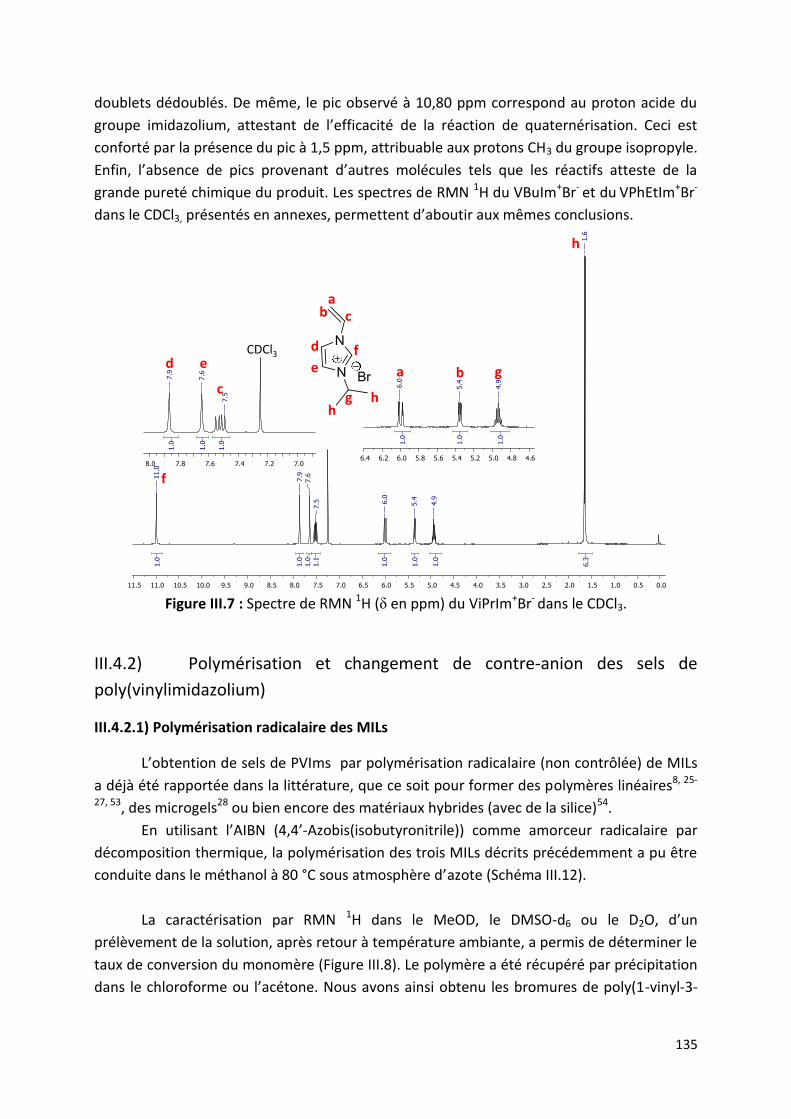
135
doublets dédoublés. De même, le pic observé à 10,80 ppm correspond au proton acide du
groupe imidazolium, attestant de l’efficacité de la réaction de quaternérisation. Ceci est
conforté par la présence du pic à 1,5 ppm, attribuable aux protons CH3 du groupe isopropyle.
Enfin, l’absence de pics provenant d’autres molécules tels que les réactifs atteste de la
grande pureté chimique du produit. Les spectres de RMN 1H du VBuIm+Br- et du VPhEtIm+Br-
dans le CDCl3, présentés en annexes, permettent d’aboutir aux mêmes conclusions.
Figure III.7 : Spectre de RMN 1H (δ en ppm) du ViPrIm+Br- dans le CDCl3.
III.4.2) Polymérisation et changement de contre-anion des sels de
poly(vinylimidazolium)
III.4.2.1) Polymérisation radicalaire des MILs
L’obtention de sels de PVIms par polymérisation radicalaire (non contrôlée) de MILs
a déjà été rapportée dans la littérature, que ce soit pour former des polymères linéaires8, 25-
27, 53, des microgels28 ou bien encore des matériaux hybrides (avec de la silice)54.
En utilisant l’AIBN (4,4’-Azobis(isobutyronitrile)) comme amorceur radicalaire par
décomposition thermique, la polymérisation des trois MILs décrits précédemment a pu être
conduite dans le méthanol à 80 °C sous atmosphère d’azote (Schéma III.12).
La caractérisation par RMN 1H dans le MeOD, le DMSO-d6 ou le D2O, d’un
prélèvement de la solution, après retour à température ambiante, a permis de déterminer le
taux de conversion du monomère (Figure III.8). Le polymère a été récupéré par précipitation
dans le chloroforme ou l’acétone. Nous avons ainsi obtenu les bromures de poly(1-vinyl-3-
ab c
d
ef
gh
h
d e
f
ca b g
h
CDCl3
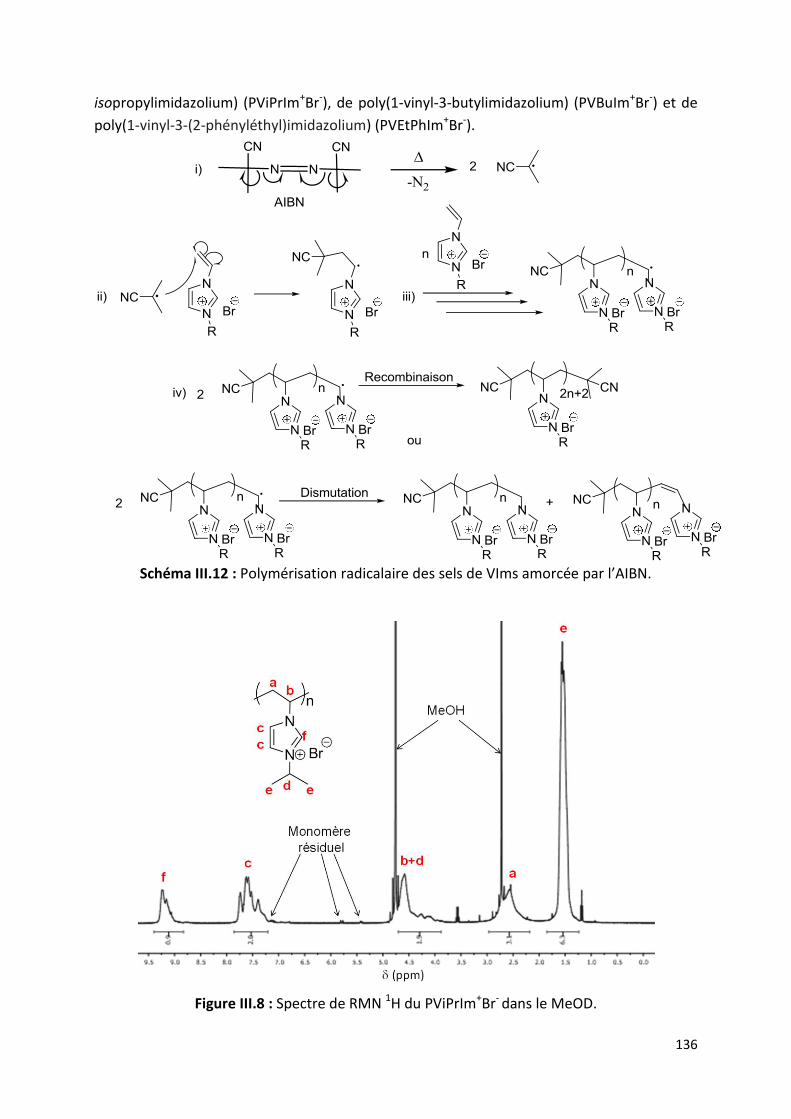
136
isopropylimidazolium) (PViPrIm+Br-), de poly(1-vinyl-3-butylimidazolium) (PVBuIm+Br-) et de
poly(1-vinyl-3-(2-phényléthyl)imidazolium) (PVEtPhIm+Br-).
Schéma III.12 : Polymérisation radicalaire des sels de VIms amorcée par l’AIBN.
Figure III.8 : Spectre de RMN 1H du PViPrIm+Br- dans le MeOD.

137
Comme attendu, les spectres de RMN 1H des polymères analysés présentent un
aspect très différent de ceux des monomères de départ. Par exemple, le spectre de RMN 1H
du PViPrIm+Br- révèle l’apparition d’un pic entre 2,2 et 3,0 ppm correspondant à la formation
des protons vinyliques des PILs. On note aussi la totale disparition des pics à 5,4 ; 5,8 et 7,2
ppm attribués à la fonction vinyle du monomère.
Les PILs solubles dans l’eau, à savoir le PViPrIm+Br- et le PVBuIm+Br-, ont aussi été
analysés par chromatographie d’exclusion stérique 35 en phase aqueuse. Malgré le fait que
ces polymères soient connus pour leur difficulté de caractérisation par cette méthode7, des
valeurs apparentes de Mn, Mw et de la dispersité (D) ont pu être ici obtenus. Pour cela, des
colonnes particulières, dédiées à l’élution de polymères cationiques, ont été utilisées en
présence d’acide formique à 0,3M dans l’eau comme solvant d’élution. Dans tous les cas,
des polymères ayant une distribution monomodale ont été obtenus (Figure III.9). On peut
noter que ces polymères possèdent des dispersités assez élevées, variant entre 2,3 et 3.
L’étalonnage des colonnes de SEC a été réalisé au moyen d’échantillons standards de poly(2-
vinylpyridine) (P2VP), des standards de PVIms n’existant pas. Cependant, la valeur des
masses molaires permet d’apprécier la taille des chaînes polymères de PVIms. Ainsi, les
PViPrIm+Br- obtenus présentent des masses molaires apparentes de l’ordre de 4600 g.mol-1
pour Mn et de 11400 pour Mw avec un indice de polydispersité de 2,5. Dans le cas des
PVBuIm+Br- des masses molaires apparentes plus élevées ont été obtenues (Mn = 11400
g.mol-1, Mw = 25260 g.mol-1 et D = 2,9) dans des conditions identiques de polymérisation.
On peut supposer que les interactions entre le polymère, les colonnes et le solvant sont
spécifiques dans chaque cas et toutes ces valeurs doivent être considérées comme relatives
et ne nous renseignent aucunement sur les masses molaires « vraies » de ces PILs.
Figure III.9 : Chromatogramme d’exclusion stérique dans une solution acide formique/ eau
(0,3M) d’un PViPrIm+Br- et d’un PVBuIm+Br- obtenus par polymérisation radicalaire
conventionnelle.
26 28 30 32 34 36 38 40 42 44
PViPrIm+Br- (Mn = 4590 g/mol;; D = 2,5)PVBuIm+Br- (Mn = 11400g/mol; D = 2,9)
Temps d’élution (min)

138
Des tentatives pour contrôler la polymérisation radicalaire des MILs de type N-
vinylimidazolium ont également été entreprises. Au delà de l’obtention de chaînes de PILs de
taille calibrée, il s’agissait en réalité de tirer profit du contrôle de ces polymèrisations pour
accéder à de nouveaux copolymères à blocs, par polymérisation séquentielle. A cet effet, des
agents de contrôle de type dithiocarbonate (appelés aussi xanthates) ont été employés pour
la polymérisation procédant par un mécanisme de type RAFT/MADIX. Cette partie de
l’étude, décrite dans le paragraphe suivant, a été menée en collaboration avec Jean Raynaud
et Xiaoshuang Feng, stagiaires post-doctoraux au LCPO, dans le cadre d’un projet européen
(acronyme : ORION) utilisant les ILs et les PILs à des fins d’applications dans le domaine de
l’énergie.
III.4.2.2) Polymérisation RAFT/MADIX des MILs
La possibilité de contrôler la polymérisation des sels de N-vinylimidazolium via la
méthode RAFT/MADIX a été évaluée. Dans le même temps, des travaux similaires ont été
publiés par le groupe de Endo en 200923. Ces travaux décrivent notamment la synthèse de
copolymères à blocs PVIm+Br--b-PNIPAAm, le PNIPAAm étant le poly(N-isopropylacrylamide),
incluant quelques résultats préliminaires de leur auto-assemblage en solution. Dans notre
cas, il s’agissait de tirer profit de structures auto-assemblées de copolymères à blocs dérivés
de monomères de type N-vinyle (N-vinylimidazolium et de N-vinylpyrolidone (NVP)) pour
induire la formation de poly(NHC)s dans les (nano)structures en vue de réactions catalysées
en milieu confiné.
Principe de la polymérisation RAFT/MADIX
La polymérisation RAFT/MADIX est une méthode de polymérisation fondée sur le
transfert de bouts de chaines polymères. Elle a été développée en 1998 par Rizzardo et
coll.55 56 et par Rhodia (MADIX)57, 58. Les polymérisations réalisées par ce moyen sont
considérées comme vivantes/contrôlées et permettent d’obtenir des polymères présentant
une faible dispersité (D) et des masses molaires contrôlées59-61.
Le mécanisme des polymérisations via la méthode RAFT/MADIX se distingue de celui
des polymérisations radicalaires conventionnelles par l’utilisation d’un agent de transfert de
type dithioester (Schéma III.13). Celui-ci induit des étapes de transfert réversible par des
étapes successives d’addition-fragmentation, en complément des étapes se produisant lors
de polymérisations radicalaires conventionnelles, ce qui permet le contrôle de la
polymérisation.

139
Schéma III.13 : Mécanisme des polymérisations RAFT/MADIX.
Quatre familles d’agents de transfert peuvent être employées (Figure III.10) ; ainsi,
les agents de transfert xanthates (brevetés par Rhodia) présentant un groupe Z, de type O-R,
permettent la polymérisation des esters vinyliques et des acrylates au cours des mécanismes
de type MADIX (MAcromolecular Design by Interchange of Xanthate), et les agents de
transfert tels que les dithioesters, trithiocarbonates et dithiocarbamates (brevetés par Du
Pont De Nemour) s’inscrivent dans les polymérisations de type RAFT, permettant la
polymérisation contrôlée des monomères styréniques ou méthacryliques.
Figure III.10 : Agents de transfert utilisés pour contrôler les polymérisations RAFT/MADIX.
Il a également été rapporté, au cours de ces dernières années, que les xanthates
permettent le contrôle de la polymérisation des monomères de type N-vinyle (NVP62, 63, N-
vinylcaprolactame64, N-vinylcarbazole65, N-vinylimidazole66). Il était donc logique de penser
que les sels de poly(1-vinyl-3-alkylimidazolium) puissent aussi être contrôlés au moyen de

140
polymérisations de type MADIX. Des développements récents, par C. Detrembleur et coll.,
utilisant des agents de contrôle métalliques à base de cobalt (au degré d’oxydation +II ou
+III) ont aussi montré que cette chimie permet de bien contrôler la polymérisation des
monomères de type N-vinyle67-69. La discussion qui suit a trait à nos premières tentatives de
contrôle de polymérisation radicalaire d’un sel d’imidazolium de type N-vinyle. Cette étude
n’a malheureusement pas pu être conduite à son terme.
Polymérisation MADIX des sels de vinylimidazoliums
Afin de déterminer les conditions expérimentales nécessaires à l’obtention de
polymères de taille contrôlée via polymérisation RAFT/MADIX, seul le VBuIm+Br- a été utilisé
comme monomère. De même, le xanthate X1, dont la structure est présentée Figure III.11, a
été utilisé en tant qu’agent de transfert. Il a notamment été décrit pour contrôler la
polymérisation du 1-vinylimidazole66 .
Figure III.11 : Structure de l’agent de transfert (xanthate) employé pour contrôler la
polymérisation des MILs.
Des polymérisations du VBuIm+Br- ont été conduites dans divers solvants de réaction,
tels que le méthanol, le chloroforme, le DMF ou le DMSO, en utilisant l’AIBN comme
amorceur. Différents taux d’AIBN, par rapport à X1, ont également été utilisés afin de
minimiser la formation de chaînes « mortes ».
La détermination des conditions expérimentales permettant d’obtenir les meilleurs
résultats ne fût pas aisée, en raison de la difficulté à caractériser les polymères obtenus. En
particulier, l’analyse par SEC dans l’eau des polymères obtenus, au moyen de colonnes
spécifiques pour les polymères cationiques, et d’un étalonnage avec des échantillons
standards de P2VP, a d’abord montré que la distribution des masses molaires des
PVBuIm+Br- était supérieure à 1,6. A titre d’exemple, un chromatogramme d’exclusion
stérique d’un de ces polymères est présenté Figure III.12. Par ailleurs, lorsque ceux-ci ont été
analysés par spectroscopie de RMN 1H, la présence de la fonction dithioester en bout de
chaîne polymère n’a pas pu être mise clairement en évidence. Ceci s’explique par le
recouvrement des pics attribués au PVBuIm+Br- avec les pics correspondants à la fonction
dithioester en bout de chaîne, comme le montre la Figure III.13. A noter que la synthèse de
polymères de même nature (poly(N-vinyl-3-alkylimidazolium)), et obtenus dans les mêmes
conditions, a été décrite par Endo et al.23 (c'est-à-dire dans le DMF, à 60 °C et avec un
rapport molaire AIBN/X1 de 0,5).

141
Figure III.12 : Chromatogramme SEC (détecteur RI) dans l’eau d’un PVBuIm+Br- préparé par
polymérisation MADIX en présence de X1 : = 7000 g/mol, D= 1,6.
Figure III.13 : Spectres de RMN 1H (δ en ppm) du VBuIm+Br-, de X1 et de PVBuIm+Br- obtenu
par polymérisation radicalaire en présence de X1.
769 869 969 1069 1169 1269 1369 1469
Temps d'élution (s)
+ Br
- PVBuIm

142
Synthèse de copolymères à blocs PVBuIm+Br--b-PNVP
Le contrôle de la polymérisation RAFT/MADIX du VBuIm+Br- n’ayant pu clairement
être établi au moyen des caractérisations décrites précédemment (RMN 1H et SEC), la
synthèse de copolymères à blocs à base de PNVP et de PVBuIm+Br- a d’abord été tentée par
polymérisation séquentielle de la NVP et du VBuIm+Br-, selon cet ordre d’addition des deux
monomères (Schéma III.14). Gnanou et al.62, Klumperman et al.63, 70 et d’autres encore71, 72
ont en effet déjà étudié dans le détail la polymérisation RAFT/MADIX de la NVP. De plus, la
PNVP est soluble dans le DMF, ce qui devrait permettre d’observer l’évolution des masses
molaires en SEC lors de la formation des copolymères à blocs visés. Ainsi, les polymérisations
MADIX du VBuIm+Br- ont tout d’abord été effectuées à partir de macroagents de transfert
PNVP.
Schéma III.14 : Synthèse de copolymères à blocs PNVP-b-PVBuIm+Br- par polymérisation
RAFT/MADIX séquentielle de la NVP puis du VBuIm+Br-.
Pour cela, La synthèse des blocs à base de PNVP a été réalisée par polymérisation
MADIX de la NVP, dans le dioxane, à une température 60 °C, au moyen de X1 comme agent
de transfert et d’AIBN comme source de radicaux. Après précipitation dans le diéthyl éther,
la caractérisation par RMN 1H dans le CD2Cl2 (Figure III.14) a révélé les massifs attendus, en
particulier du polymère obtenu, entre 1,2 et 4,2 ppm, mais également les pics attribués à
certains protons de la fonction dithioester en bout de chaîne, à 1,1 ppm et 4,6 ppm. Les
protons du CH2 de la chaîne vinylique étant attribués entre 3,0 et 3,4 ppm, il est alors
possible de déduire de ce spectre le du polymère, comme indiqué sur la Figure III.14.

143
Figure III.14 : Spectre de RMN 1H (δ en ppm) dans CD2Cl2 du PNVP obtenu par polymérisation
MADIX en présence de l’agent de transfert X1.
La caractérisation de ce PNVP par SEC dans le DMF (courbe orange Figure III.15),
atteste du bon contrôle de la polymérisation à travers la faible dispersité des masses
molaires (D = 1,07). Une masse molaire apparente de 18100 g/mol a ainsi été déterminée.
Celle-ci ne coïncide pas avec la masse molaire de 10000 g/mol calculée à partir du spectre de
RMN 1H, très probablement dû au fait que la calibration de la SEC (avec des étalons de PS)
n’est pas adaptée à la PNVP. De même, la polymérisation RAFT/MADIX d’acétate de vinyle
en présence de ces macroagents de transfert PNVP, a permis d’attester de la présence des
groupements dithiocarbonate en bout de chaîne. En effet, les copolymères à blocs obtenus
présentent un temps d’élution plus court en analyse par SEC dans le DMF et ce, pour une
grande majorité de la population (Cf. annexes).
Un tel précurseur a alors été employé comme macroagent de transfert RAFT/MADIX,
pour la polymérisation du VBuIm+Br-. Le chromatogramme de SEC du composé ainsi obtenu
a alors révélé que le macroagent de transfert PNVP ne permet pas l’obtention de
copolymères à blocs définis. On note, en effet, que la population du PNVP précurseur est
toujours présente, ainsi qu’une deuxième population de plus forte masse molaire (Figure
III.15). Ce résultat peut être attribué au fait que le bloc PNVP a extrémité xanthate ne joue
pas efficacement son rôle d’agent de transfert réversible via RAFT/MADIX. On peut supposer
que l’étape d’addition du VBuIm+Br- sur le groupement xanthate du PNVP induit la formation
d’un radical intermédiaire (voir Schéma III.15) dont la fragmentation n’est pas favorable à la
synthèse des composés visés. Nous ne pouvons, à ce stade de l’étude, conclure sur ce point
i
ad ch
b
e
f g
a , e, f, g
ib
c, h
d
DPn = Id/Ib

144
et une étude systématique serait nécessaire. Pour pallier cette difficulté, nous avons donc
choisi d’inverser l’ordre d’addition des deux monomères.
Figure III.15 : Chromatogrammes de SEC dans le DMF du macroagent de transfert PNVP
(courbe orange, RMN = 10000 g/mol, ces = 18100 g/mol, D = 1,07) et du polymère
obtenu par polymérisation du VBuIm+Br- en présence du macroagent de transfert PNVP
(courbe marron, deux populations).
Schéma III.15 : Equilibre proposé entre les différentes espèces radicalaires impliqués lors de
la polymérisation du VBuIm+Br- en présence du macroagent de transfert PNVP.
469 969
PNVP(10K)
temps d'élution (s)
PNVP(10K)-PVBuIm+Br
-(10K)?

145
Les PVBuIm+Br- obtenus par polymérisation RAFT/MADIX, décrits précédemment, ont
donc par la suite été employés comme macroagents de transfert pour la polymérisation de
la NVP (Schéma III.16).
Schéma III.16 : Synthèse des copolymères à blocs PVBuIm+Br--b-PNVP par polymérisation
RAFT/MADIX de la NVP en présence de macroagents de transfert PVBuIm+Br-.
La synthèse des copolymères à blocs PVBuIm+Br--b-PNVP a été réalisée en
polymérisant la NVP dans le DMF, à 60 °C et en utilisant 10% molaire d’AIBN par rapport aux
macroagents de transfert. La masse molaire de ces précurseurs a été évaluée en se basant
sur leur valeur théorique (équation I : rapport de la quantité de monomère par celle de
l’agent de transfert (nX1), en tenant compte du taux de conversion du monomère (x)).
équation I :
Ainsi, en faisant varier le taux de NVP par rapport à la quantité du macroagent de
transfert, trois copolymères à blocs PVBuIm+Br--b-PNVP on été obtenus. Ces copolymères
ont par la suite été injectés en SEC dans l’eau, dans les mêmes conditions que le PVBuIm+Br-
de départ. Cependant, les copolymères (présentant un bloc neutre associé à un bloc
cationique) n’ont pas pu être élués avec les colonnes de chromatographie utilisées en phase
aqueuse. D’autre part, le précurseur, quant à lui, n’a pas pu être élué par SEC dans le DMF,
contrairement aux copolymères dérivés (Figure III.16). On note néanmoins une
augmentation de la masse molaire avec l’augmentation du rapport *NVP+/*macroagent de
tranfert à base de PVBuIm+Br-], laissant penser à une certaine efficacité de la
copolymérisation séquentielle. Les distributions des masses molaires des composés sont
toutefois relativement larges (D 1,4).

146
Figure III.16 : Chromatogrammes de SEC dans le DMF de copolymères obtenus par
polymérisation MADIX séquentielle (Mnth : masse molaire théorique du macroamorceur,
déterminée d’après l’équation I ; r : rapport molaire entre les unités monomères VBuIm+Br-
et NVP ; Mn : masse molaire apparente du copolymère).
La caractérisation de ces copolymères par spectroscopie de RMN 1H dans le D2O
(Figure III.17), montre la présence des protons caractéristiques des deux types d’unités
monomères (NVP et VBuIm+Br-). En plus des pics du PVBuIm+Br- observés auparavant, il est
en effet possible d’attribuer le massif entre 3,0 et 3,6 ppm aux CH2 de la chaîne vinylique du
bloc PNVP. L’intégration de ce massif permet également de calculer le rapport (r) des unités
monomères NVP aux unités VBuIm+Br-, en comparant cette intégration à celle du massif
entre 7,2 et 7,8 ppm (r, Figure III.17).
Figure III.17 : Spectre de RMN 1H (δ en ppm) dans le D2O d’un copolymère PVBuIm+Br--b-
PNVP.
a b
cc
de
fg
h i
l k
j
c
g
r = m/n= Ih/Ic = 2,87
h
a, d, i
b, e, f, j, k, l

147
En conclusion, la polymérisation RAFT/MADIX permet l’obtention de copolymères à
blocs de type PVBuIm+Br--b-PNVP, de structure et de masse molaire bien contrôlée, même si
il faut reconnaitre que ces synthèses doivent être encore optimisés. En particulier, la
caractérisation de ces copolymères à doubles blocs hydrophiles reste compliquée. Au cours
de ce travail de thèse, Endo et al. ont d’ailleurs conclu dans le même sens en publiant la
polymérisation RAFT/MADIX de divers MILs de type N-vinylimidazolium23. Des études
complémentaires doivent donc être effectuées afin d’étendre la polymérisation à d’autres
sels de N-vinylimidazolium, mais aussi afin d’améliorer les techniques de caractérisation de
tels (co)polymères .
Etant donné l’influence du substituant alkyl sur la solubilité de ces polymères, on
peut penser que celui-ci influence également la polymérisabilité du monomère. Il doit en
être de même avec le contre-anion porté par le monomère. Il faudra donc déterminer les
conditions nécessaires à l’obtention de polymères contrôlés dans ces cas là : quel agent de
transfert xanthate pour quel monomère, et dans quel solvant ? De même, des études
cinétiques (Ln([M0]/[M])= f(t)) permettraient de vérifier le contrôle de ces polymérisations
radicalaires tout au long de la réaction.
III.4.2.3) Changement d’anion
La déprotonation de sels d’imidazolium pour former les NHCs a généralement lieu
dans des solvants aprotiques tels que le THF. Par analogie, les PVIms décrits précédemment
n’étant pas solubles dans le THF, nous avons procédé à un changement d’anion, en
remplaçant l’anion bromure par l’anion bis(triflurométhanesulfonyl)imide (NTf2-) (Figure
III.18).
Cette réaction de métathèse a été réalisée par ajout progressif d’une solution
aqueuse de LiNTf2 aux solutions de PVIms à chaine alkyle variable. Dans le cas du PViPrIm+Br-
et du PVBuIm+Br-, les polymères ont préalablement été solubilisés dans de l’eau déionisée.
En revanche le méthanol a été choisi comme solvant du PVEtPhIm+Br-, ce dernier n’étant pas
soluble dans l’eau. Après addition de quelques gouttes de la solution de LiNTf2, la formation
d’un précipité blanc est observée avec les trois polymères (Figure III.18), dû à la génération
des PVIms porteurs de NTf2- comme contre-anion. La précipitation du produit permet ici de
déplacer l’équilibre de la réaction vers la formation des polymères à contre-anion NTf2-,
conduisant ainsi à des rendements de réaction quantitatifs. Les précipités ainsi obtenus sont
alors filtrés et le PViPrIm+NTf2-, le PVBuIm+NTf2
- et le PVEtPhIm+NTf2- sont obtenus sous
forme d’une poudre blanche.

148
Figure III.18: Echange d’anion du Br- au NTf2
- réalisé sur les PVIms.
Les PILs à anion NTf2- ainsi formés sont insolubles dans l’eau et le méthanol, mais
solubles dans le THF. Leur caractérisation par RMN 13C dans le THF-d8 permet de vérifier
l’efficacité du changement d’anion. En effet, l’apparition d’un quadruplet bien défini à 120
ppm, avec une constante de couplage de l’ordre de 100 Hz, est caractéristique de la
présence du contre-anion NTF2-. En effet, le fluor ayant un spin ½, les 3 atomes de fluor
interagissent avec le carbone du contre-anion pour former un quadruplet. Ainsi, ce massif se
retrouve dans le spectre de RMN 13C du PViPrIm+NTf2- (Figure III.19), mais également dans
ceux du PVBuIm+ NTf2- et le PVEtPhIm+NTf2
- (Annexes).
Figure III. 19 : Spectre de RMN 13C de PViPrIm+NTf2
- dans le THF-d8.
a
N
N
NTf2
n
ab
c
cf
de
e
b+d
e
f
c
NTf2
THF
d (ppm)

149
Comme déjà indiqué, la solubilité des PILs précurseurs est un point clé en vue de la
synthèse des poly(NHC)s. En effet, la réaction de déprotonation des PVIms implique
l’utilisation de bases fortes qui ne doivent pas réagir avec le solvant.
Mecerreyes et coll. ont par ailleurs montré que, au-delà de la nature du contre-anion,
le groupement alkyl présent sur l’atome d’azote quaternaire a une grande influence sur la
solubilité des PILs. Les propriétés de solubilité des PVIms que nous avons obtenus, qu’ils
présentent un contre-anion bromure ou triflimide, sont rassemblées dans le Tableau III.1.
Tableau III.1: Solubilité des PVIms en fonction du contre-anion dans différents solvants (+:
soluble; -: insoluble; +-: polymère gonflé par le solvant).
PViPrIm+X - PVBuIm+X- PVPhEtIm+X-
X: Br X: NTf2 X: Br X: NTf2 X: Br X: NTf2
H2O + - + - - - MeOH + - + +- + + EtOH + - + - + +-
Acetone - + - + - + THF - +- - + - +
CHCl3 LCST + + + + +
Ainsi, la plupart des PVIms a contre-anion Br- sont solubles dans l’eau et les solvants
protiques. Seul le PVPhEtIm+Br- n’est pas soluble en milieu aqueux, ce qui n’a d’ailleurs pas
permis sa caractérisation par SEC. Nous avons également noté que les PViPrIm+Br- et
PVBuIm+Br- sont très hygroscopiques. Lorsque ceux-ci sont laissés à l’air, l’absorption d’eau
par ces polymères est telle, qu’ils passent d’une poudre blanche sèche à un liquide visqueux
en quelques minutes (15 min). Ils ont donc été conservés sous atmosphère sèche
(dessicateur sous vide) dès leur récupération. De même, les PVIm+Br- sont insolubles dans les
solvants moins polaires et aprotiques tels que l’acétone et le THF.
Lorsque le contre-anion Br- est remplacé par le NTf2-, la solubilité des polymères est
affectée. Ainsi, les PVIm+NTf2- sont solubles dans le THF, solvant idoine pour effectuer les
réactions de déprotonation. Seul le PViPrIm+NTf2- présente une assez faible solubilité dans ce
solvant, puisque lors de l’ajout de THF, le polymère est seulement gonflé par ce solvant, mais
ne se solubilise pas (représenté par +- dans le tableau).
On observe également que les PVIms sont solubles dans le chloroforme, quelque soit
le contre-anion utilisé. Toutefois, la température limite basse de solubilisation (Lower critical
solution temperature, LCST), de l’ordre de -20 °C, du PViPrIm+Br- ne permet pas sa
solubilisation à température ambiante. Celle-ci a été déterminée par analyse enthalpique
différentielle (AED) d’une goutte de la solution de polymère.

150
La synthèse et la polymérisation radicalaire des sels de N-vinylimidazolium sont donc
relativement faciles à mettre en œuvre. Celles-ci s’inspirent de travaux déjà décrits,
notamment par Mecerreyes et coll.8, 25, et ont été suivies d’une réaction de métathèse afin
de faire varier la solubilité des PILs générés. Ces derniers étant solubles dans le THF, il peut
maintenant être envisagé de les utiliser comme précurseurs de poly(NHC)s.
III.5) Synthèse et évaluation des poly(NHC)s en organocatalyse
L’étape suivante dans la synthèse des poly(NHC)s consiste à déprotoner des PILs à
contre-anion triflimide (voir Schéma III.9). Par analogie avec la synthèse des NHCs
moléculaires (voir chapitre bibliographique), la déprotonation des sels d’azoliums est
généralement effectuée avec un excès de base forte, à basse température31, 33, 73. Certains
NHCs moléculaires peuvent être obtenus purs, par distillation ou par sublimation. Pour
accéder aux poly(NHC)s, les PVIm+NTf2- décrits précédemment ont donc été déprotonés à
l’aide de différentes bases fortes dans le THF comme solvant.
III.5.1.) Choix de la base
La synthèse de poly(NHC)s à partir de PILs subissant une déprotonation au moyen
d’une base forte a déjà été décrite dans la littérature, mais à quelques reprises seulement45-
47, 50, 51 et, une seule fois sous une forme « nue » à des fins d’organocatalyse50, 51. Les autres
poly(NHC)s ont été générés in-situ et n’ont pas été obtenus sous une forme pure.
Contrairement aux PVIm+NTf2- que nous avons employés, les PILs décrits dans ces travaux
(Figure III.20) étaient pour la plupart insolubles dans le milieu réactionnel car issus de
polymères réticulés.
Figure III.20 : Précurseurs de poly(NHC)s générés in-situ reportés dans la littérature.

151
Afin d’obtenir des poly(NHC)s dénués de résidus métalliques issus de la base utilisée,
ce qui pourrait engendrer des réactions secondaires lors des tests en organocatalyse, deux
bases ont été sélectionnées : le KHMDS (bis(triméthylsilyl)amidure de potassium) et le NaH
(Figure III.21).
Figure III.21 : pKa des bases employées pour la déprotonation des PVIms.
Le NaH n’étant pas soluble dans le THF, cette base a été choisie comme agent
déprotonant en présence d’une quantité catalytique de t-BuOK. Il est ainsi possible de
séparer l’excès de base par filtration du milieu réactionnel, après réaction de déprotonation.
Le PViPrIm+NTf2- n’étant que gonflé par le THF, l’utilisation du NaH ne nous a pas paru
appropriée. Le KHMDS, soluble dans le THF, a donc été utilisé comme agent déprotonant
pour ce polymère. Afin d’éviter la présence de base résiduelle dans les solutions de
poly(NHC)s générées, celle-ci a été ajouté en quantité équivalente par rapport aux unités
monomères N-vinylimidazoliums.2
III.5.2) Synthèse des poly(NHC)s
La déprotonation des sels d’imidazoliums est une réaction exothermique,
généralement conduite à basse température, afin de ne pas dégrader les produits formés31,
33. Par ailleurs, les NHCs étant sensibles aux impuretés et à l’eau, la réaction doit être mise
en oeuvre sous atmosphère inerte et en milieu sec. Ces conditions ont été appliquées pour
la déprotonation des PVIm+NTf2- secs. Au cours de ces réactions (Schéma III.17), le milieu
réactionnel prend rapidement une teinte orangée (Figure III.22). En fin de réaction, les
solutions de poly(NHC) obtenues ont été filtrées sous atmosphère inerte de façon à éliminer
l’excés de base, avant leur utilisation pour les réactions d’organocatalyse.
2 Afin de déterminer la quantité de base pour déprotoner la totalité des sels d’imidazoliums présents sur les
chaînes polymères, les polymères ont été séchés puis pesés. En divisant cette masse par la masse molaire
d’une unité monomère du sel de PVIm, la quantité de matière de cycles imidazoliums a pu être déterminée, de
même que la quantité de matière de base à utiliser.

152
Schéma III.17 : Synthèse des poly(NHC)s par déprotonation des PVIm+NTf2- .
Figure III.22 : Photographie d’une solution de poly(NHC) obtenue après déprotonation de
PVIm+NTf2- puis filtration sous atmosphère inerte.
Les solutions de chacun de ces polymères 8 ont été caractérisées par spectroscopie de
RMN 1H et 13C dans le THF-d. La superposition des spectres de RMN 1H du PViPrIm+NTf2- et
du polyNHC-1 (Figure III.23) montre la disparition totale du pic correspondant au proton du
groupe imidazolium à 8,9 ppm, attestant de l’efficacité de la réaction de déprotonation. De
même, les spectres de RMN 1H dans le THF-d8 des polyNHC-2 et polyNHC-3 (voir annexes) ne
présentent pas de pics aux environs de 9 ppm, confirmant l’obtention de ces polymères. En
RMN 13C, aucun pic aux alentours de 220 ppm, caractéristique du carbène, n’a en revanche
pu être détecté malgré des temps d’accumulation importants. Par analogie avec les NHCs
moléculaires74, ceci peut être expliqué par la faible intensité du pic correspondant à ce
carbone quaternaire. De plus, les différents environnements, apportés par la chaîne
polymère, peuvent induire des massifs de pics relativement larges et de plus faible intensité.

153
Figure III.23 : Superposition des spectres de RMN 1H du PViPrIm+NTf2- et du PolyNHC-1 dans
le THF-d8.
III.5.3) Evaluation des poly(NHC)s en organocatalyse
Dans la partie bibliographique de ce manuscrit, il a été souligné que les NHCs
permettent de catalyser un grand nombre de réactions en chimie moléculaire comme en
chimie macromoléculaire. Ainsi, afin d’évaluer l’activité catalytique des NHCs supportés sur
polymères -poly(NHC)s- décrits précédemment, deux réactions modèles connues pour être
organocatalysées par les NHCs moléculaires et impliquant, dans les deux cas, l’activation de
la fonction carbonyle, ont été mises en oeuvre : la transestérification et la condensation de
la benzoïne. Ces deux réactions ne nécessitent pas la synthèse de réactifs particuliers et les
produits réactionnels sont facilement mis en évidence par spectroscopie de RMN. Comme
autres catalyseurs connus de la condensation de la benzoïne, on peut mentionner les
cyanures de métaux alcalins. L’obtention de la benzoïne, à partir du benzaldéhyde, est donc
un bon moyen d’attester de la présence et de l’efficacité de catalyseurs de type poly(NHC)s
dans le milieu réactionnel.
III.5.3.1) Catalyse de transestérification
Diverses réactions de transestérification peuvent être catalysées par les NHCs, de
manière quantitative, et avec de faibles quantités d’organocatalyseurs75, 76. L’équipe de
Nolan a notamment montré qu’il est possible de réaliser la transestérification entre l’acétate
a
a
d
b+d
b
c
c
e
e
f
a b
cc
d ee
ab
cc
de e
f
THF
THFH2O
HMDS

154
de vinyle et l’alcool benzylique, avec seulement 0,5% de dimésitylimidazol-2-ylidene
(Schéma III.18)77. De plus, cette réaction est caractérisée par une cinétique élevée : en
quelques minutes, des rendements quantitatifs sont obtenus. Nous avons donc choisi de
mettre en œuvre cette réaction afin d’évaluer le potentiel des poly(NHC)s comme
organocatalyseurs.
Schéma III.18 : Réaction de transestérification catalysée par les NHCs décrite par Nolan et
coll.77
La réaction de transestérification a donc été réalisée en milieu complètement
homogène au moyen des solutions de poly(NHC)s décrites auparavant (Schéma III.19). Au-
delà du pouvoir catalytique de ces NHCs supportés, il était intéressant d’essayer de recycler
ces catalyseurs et de les réutiliser. Pour des raisons de commodités, les conditions
expérimentales décrites par Nolan et al. ont été légèrement modifiées. Ainsi, 10% d’espèces
catalytiques de type poly(NHC)s par rapport à l’alcool benzylique ont été utilisées. Cette
quantité de 10% a été choisie afin de compenser la perte éventuelle d’activité catalytique
due à la mobilité restreinte du catalyseur supporté, comparée à son homologue moléculaire,
mais aussi afin d’avoir une quantité suffisante de polymère pour que celui-ci puisse être
filtré et recyclé.
Schéma III.19 : Réaction de transestérification catalysée par les poly(NHC)s
Pour récupérer les poly(NHC)s, un excès de diéthyl éther (Et2O) sec a été ajouté en fin
de réaction. Dans ce solvant, seul le catalyseur polymère précipite, ce qui permet de le
séparer du milieu réactionnel par simple filtration (Figure III.24). Etant donné que ce solvant
est en contact direct avec les poly(NHC)s, aucune trace d’eau ou autre impureté ne peut être
tolérée. Nous avons alors mis au point un montage expérimental permettant de filtrer le
catalyseur supporté à l’abri de l’air. Ce montage est montré en partie expérimentale.

155
Figure III.24 : Précipitation des poly(NHC)s dans le diéthyl éther.
Après évaporation des volatils du filtrat (THF, Et2O, acétate de vinyle et
acétaldéhyde), le rendement de la réaction a pu être déterminé par spectroscopie de RMN 1H. En effet, cette solution n’est plus composée que de l’acétate de benzyle (produit) et de
l’alcool benzylique (réactif) non converti.
Les pics caractéristiques des deux composés se retrouvent en effet dans le spectre de
RMN 1H obtenu (Figure III.25). Pour déterminer le rendement de la réaction de
transestérification, il faut alors simplement diviser l’intensité du pic correspondant au CH2 de
l’acétate de benzyle (présent à 5,13 ppm) par la somme des intensités des pics attribués aux
CH2 de l’alcool benzylique (4,7 ppm) et de l’acétate de benzyle (5,13 ppm).
Figure III.25 : Spectre de RMN 1H (δ en ppm) du filtrat obtenu après réaction de
transestérification. Le calcul du rendement de la réaction est indiqué dans l’encadré.
Diéthyléther
Photographie du milieu réactionnel de la transestérification (solvant THF)
Photographie du milieu réactionnel après ajout du diéthyl éther montrant la précipitation du
poly(NHC)
aa
aa
a
a’a’
a’a’
a’
bb’
c
a + a’
b
c
b’
rendement : Ib/(Ib+Ib’)

156
Cette méthodologie a été pratiquée pour les réactions impliquant les trois
poly(NHC)s. De cette manière, nous avons pu facilement récupérer les catalyseurs supportés
et ainsi les réutiliser avec des nombres de cycles allant de 2 à 4. Un récapitulatif des valeurs
de rendements obtenus pour ces réactions de transestérification est présenté dans le
Tableau III.2.
Tableau III.2 : Synthèse de l’acétate de benzyle à partir de l’alcool benzylique et de l’acétate
de vinyle par transestérification catalysée par les poly(NHC)s.
PolyNHC polyNHC-1
R= i-Pr
polyNHC-2
R=n-Bu
polyNHC-3
R= PhEt
Cycle r. (%) r. (%) r. (%)
1 100 85 100
2 14 27 57
3 13 - 10
4 - - 12 alcool benzylique (1 eq.), acetate de benzyle (1,2 eq.) et polyNHC (10% espèces actives) dans le THF à T. amb.
pendant 30 min. rendement (r.) en acétate de benzyle déterminé par RMN 1H.
On note d’emblée que les rendements obtenus lors du premier cycle de
transestérification sont très élevés quelque soit le poly(NHC) utilisé. En revanche, lors des
cycles suivants, l’activité catalytique des polymères diminue très nettement, notamment
pour les polymères présentant des groupements latéraux peu encombrants (polyNHC-1 et
polyNHC-2) ; on obtient en effet des rendements de 14% et 27%, respectivement. On peut
opérer un maximum de 3 cycles de transestérifications avec le polyNHC-1, alors qu’avec le
polyNHC-2, seuls 2 cycles sont possibles. Un rendement modéré de 57% est en revanche
obtenu lors du deuxième cycle catalytique avec le polyNHC-3. Avec ce catalyseur supporté,
on peut atteindre 4 cycles de réactions avec des rendements de l’ordre de 10%.
La perte d’activité catalytique des poly(NHC)s peut être attribuée à la désactivation
partielle des unités monomères de type NHC. En effet, malgré les précautions opératoires
utilisées lors des différentes manipulations (ajouts des solvants et réactifs), la probabilité
que quelques molécules d’eau pénètrent dans le milieu et hydrolysent les fonctions NHCs
(Schéma III.20) n’est pas négligeable.
Schéma III.20 : Hydrolyse des NHCs.
Afin d’expliquer la différence d’activité de nos poly(NHC)s avec les cycles
catalytiques, il est tentant d’analyser leur différence de structure. En effet, les polyNHC-1 et
polyNHC-2 présentent des groupements latéraux d’encombrement stérique plus faible que

157
dans le cas du polyNHC-3. De plus, comme on a pu l’observer sur les PVIm+Br- précurseurs,
seul le PVPhEtIm+Br- est hydrophobe, contrairement aux deux autres qui sont plus
hydrophiles et hygroscopiques. On peut donc supposer que le groupement phényléthyle
(PhEt) apporte une certaine protection au polyNHC-3, que ce soit par stabilisation stérique
du carbène que par son hydrophobicité.
Pour la première fois, des poly(NHC)s ont donc pu être utilisés et recyclés à des fins
d’organocatalyse en milieu homogène. A l’instar de leurs homologues moléculaires, ils
catalysent efficacement la réaction de transestérification au cours du premier cycle.
Cependant, on note une diminution très nette de l’activité catalytique lors des cycles
suivants, ce qui peut être corrélée à l’encombrement stérique du centre carbénique. Ainsi,
afin de confirmer cette tendance, ces même poly(NHC)s ont également été employés
comme organocatalyseurs de la condensation de la benzoïne dans plusieurs cycles de
catalyse.
III.5.3.2) Evaluation en catalyse de condensation de la benzoïne
La catalyse de la condensation de la benzoïne par les NHCs moléculaires a largement
été explorée et est encore l’objet de nombreuses études. Celle-ci permet la formation d’une
liaison C-C, générant un carbone asymétrique à partir de deux molécules d’aldéhyde
(typiquement le benzaldéhyde) (Schéma III.21). De ce fait, diverses recherches ont
notamment été menées dans le but de catalyser cette réaction de manière énantiosélective.
Pour ce faire, un grand nombre de catalyseurs asymétriques ont été développés75, 76, 78-86.
Schéma III.21 : Mécanisme de la condensation de la benzoïne catalysée par les NHCs.

158
Comme discuté dans le chapitre II, nous avons exploité cette réaction simple pour
synthétiser des « polybenzoïnes » par le biais de la polymérisation par étapes du
téréphtaldéhyde. Ici, nous nous proposons d’évaluer le potentiel des poly(NHC)s en tant
qu’organocatalyseurs. En particulier, nous avons tenté de recycler les poly(NHC)s pour
catalyser la condensation de la benzoïne en milieu homogène.
La possibilité de synthétiser la benzoïne au moyen de catalyseurs NHCs supportés ou
recyclables a déjà été évaluée46, 51, 81 (Figure III.26). Dans la plupart des cas, cependant, les
NHCs ont été générés in-situ46, 81. Quand ceux-ci ont été utilisés sous une forme « pure »51,
des tests de recyclage du catalyseur pour la condensation de la benzoïne n’ont pas été
rapportés.
Figure III.26 : NHCs supportés ou leurs précurseurs, dont la faculté à catalyser la
condensation de la benzoïne a été démontrée.
Pour démontrer l’aptitude des poly(NHC)s à catalyser la condensation de la benzoïne,
ces polymères ont été employés en appliquant le même protocole opératoire que pour la
transestérification. Le benzaldéhyde est utilisé ici en tant que réactif (Schéma III.22) et le
temps de réaction a été fixé à 24h conformément aux protocoles expérimentaux établis dans
la littérature46, 51, 81.
Schéma III.22 : Condensation de la benzoïne catalysée par les poly(NHC)s

159
En fin de réaction, le même protocole que celui décrit précédemment a été appliqué
afin de récupérer les catalyseurs organiques supportés. Les rendements en benzoïne ont là
aussi été déterminés à partir des spectres de RMN 1H dans le DMSO-d6 des solutions
récupérées.
En effet, les spectres de RMN 1H des produits dont un exemple représentatif est
montré sur la Figure III.27, permettent d’attribuer les signaux de la benzoïne, mais
également du benzaldéhyde n’ayant pas réagi. Ce dernier implique notamment la présence
d’un pic à 10,0 ppm, caractéristique du proton aldéhydique. Au cours de la réaction, cette
fonction réagit avec un autre aldéhyde pour former une liaison C-C de type céto-alcool.
Ainsi, le pic correspondant diminue en intensité avec l’avancement de la réaction, alors
qu’apparaît à 6,1 ppm le pic correspondant au CH formé. De même, la benzoïne et le
benzaldéhyde présentent tous les deux des protons aromatiques qui ne sont pas impliqués
au cours de la réaction. Leur déplacement chimique étant très peu affecté, il est possible de
calibrer l’intensité du massif observé entre 7,2 et 8,1 ppm à une valeur de 5. De plus, le
benzaldéhyde présente 5 protons aromatiques et un proton aldéhydique. On peut donc
déduire le rendement de la réaction par soustraction de l’intensité du pic à 10,0 ppm à 1.
Figure III.27 : Spectre de RMN1H dans le DMSO-d6 de la benzoïne, et calcul du rendement de
la condensation de la benzoïne à partir du spectre.
De façon similaire à l’étude de la transestérification, la condensation de la benzoïne a
été effectuée avec les différents catalyseurs supportés, après plusieurs recyclages. Les
rendements déterminés pour ces différentes réactions sont présentés dans le tableau III.3.
aaa
aa
b
a’a’a’
a’a’
b’
a’a’
a’
a’a’
b
a + a’ b’
r. (%) = (1- Ib) 100
Ia + Ia’ = 5
d (ppm)

160
Tableau III.3: Condensation de la benzoïne catalysée par les poly(NHC)s.
Cycle 1 2 3 4
r.(%) avec polyNHC-1 92 20 13 -
r.(%) avec polyNHC-2 28 23 - -
r.(%) avec polyNHC-3 85 72 46 35
benzaldéhyde (1 eq.), polyNHC (10% d’espèces actives) dans le THF à T. amb. pendant 24 h.
Les valeurs des rendements sont notablement plus faibles dès le premier cycle par
rapport aux valeurs observées dans le cas de la transestérification. Ces résultats nous
incitent d’abord à conclure que les poly(NHC)s présentent une plus faible activité catalytique
s’agissant de la condensation de la benzoïne. Cette tendance est d’autant plus marquée pour
le polyNHC-2 qui permet d’obtenir un rendement de 28% seulement au premier tour. De
même, malgré une baisse peu importante de ce rendement au deuxième tour, avec 23%, il
n’a pas été possible d’obtenir de benzoïne lors des cycles suivants à l’aide du polymère
recyclé. Comme pour la transestérification, on note cependant que le polyNHC-3 a permis
l’obtention de benzoïne sur le plus grand nombre de cycles. Le même comportement du
polymère a été observé, et les rendements obtenus diminuent avec le nombre de
recyclages, passant de 85% au premier tour à 72% et 46% pour le deuxième et troisième
cycle et se terminant par 35% lors du quatrième cycle.
Ainsi, les poly(NHC)s permettent de catalyser la condensation de la benzoïne, ce qui
implique la présence de groupements NHCs au sein du polymère. Cependant, l’activité
catalytique n’est pas maintenue lors des différents recyclages et ne permet plus l’obtention
de rendements quantitatifs en benzoïne. De même que pour la réaction de
transestérification, une corrélation entre la perte d’activité catalytique et l’encombrement
stérique du carbène, dû aux groupements latéraux des poly(NHC)s, peut être établie. Ainsi,
le groupement phényléthyl semble stabiliser plus efficacement le carbène, conférant au
polyNHC-3 une plus grande stabilité et une activité catalytique plus importante pour les
derniers cycles de catalyse de transestérification ou de condensation de la benzoïne.
Un avantage certain de fixer les NHCs sur un support polymère est de pouvoir
récupérer relativement facilement ces supports. De plus, l’utilisation de polymères linaires a
permis pour la première fois d’organocatalyser des réactions de chimie moléculaire en
milieu homogène, tout en autorisant le recyclage des NHCs.
Néanmoins, les poly(NHC)s eux-mêmes restent relativement sensibles à manipuler et
après quelques recyclages, on note une diminution très nette de l’activité catalytique. Celle-
ci est très probablement liée à la dégradation des unités carbéniques en présence
d’impuretés (H2O par exemple) apportées lors des différents ajouts de solvant et de réactifs.

161
Dans la continuité de nos efforts pour disposer d’organocatalyseurs efficaces, plus
facilement manipulables et potentiellement recyclables, nous avons alors utilisé le concept
de masquage/démasquage des NHCs (ici supportés) par réaction réversible avec le CO2
(carboxylation/décarboxylation).
III.6) Synthèse et évaluation des poly(NHCs-CO2) en organocatalyse
L’utilisation du CO2 pour masquer les NHCs est une méthode qui a largement été
décrite dans la littérature48, 87-96. En effet, en présence de CO2, à température ambiante et à
pression atmosphérique, le carbène peut s’additionner réversiblement sur le carbone du CO2
en formant des composés zwittérioniques stables à l’air (Schéma III.23). La réversibilité de
cette réaction, par libération du CO2 par simple chauffage aux environs de 50 °C selon la
nature du NHC de départ, a clairement été mise en évidence91, 95. Cette méthodologie a
notamment été mise à profit pour la synthèse de complexes organométalliques92, 96, mais
également pour générer des NHCs in-situ à des fins d’organocatalyse de réactions telles que
la cyanosylilation de composé carbonylés ou la formation de carbonates cycliques48, 91-94.
Schéma III.23 : Réaction réversible des NHCs avec le CO2.
Par analogie, nous avons donc tenté de générer des adduits entre les poly(NHC)s
décrits précédemment et le CO2, comme moyen de protection des organocatalyseurs
polymères. Dans la littérature, il a très récemment été décrit la synthèse d’adduits de
poly(NHC-CO2) mis à profit dans des réactions d’organocatalyse47, 48. Ces polymères ont été
synthétisés par des approches différentes de la nôtre, comme le montre le Schéma
III.24. Zhou et al. ont ainsi préparé des poly(NHC)s de type styrénique en présence de CO247,
tandis que Buchmeiser et al. ont décrit la synthèse par ROMP (« ring-opening metathesis
polymerization ») de particules réticulées, en copolymérisant un monomère zwittérionique
portant une fonction de type norbornène et un dérivé de type bis-norbornène48.

162
Schéma III.24 : Poly(NHC-CO2)s réalisés par Zhou et al., et polymérisation par ROMP d’un
monomère portant un adduit NHC-CO2 rapportée par Buchmeiser et al..
III.6.1) Synthèse des poly(NHC-CO2)s
La réaction de carboxylation des NHCs formant le zwittérion induit généralement sa
précipitation sous forme d’une poudre blanche en un milieu tel que le THF. Une simple
filtration permet ainsi la récupération du composé.
De manière avantageuse, le même phénomène a été observé lors de la réaction de
nos poly(NHC)s avec le CO2. Ainsi, afin de générer ces poly(zwittérion)s in-situ, le KHMDS a
été utilisé pour déprotoner les PVIm+NTf2- avant l’introduction d’un flux de CO2 (1 atm.)
(Schéma III.25). Le passage du CO2 au travers d’un dispositif permettant d’éliminer toute
trace d’eau dans le gaz, avant que celui-ci ne pénètre dans le milieu réactionnel, a également
permis d’éviter l’hydrolyse des groupes NHCs. La photographie du dispositif expérimental
mis en place pour cette réaction de carboxylation est montrée en partie expérimentale. La
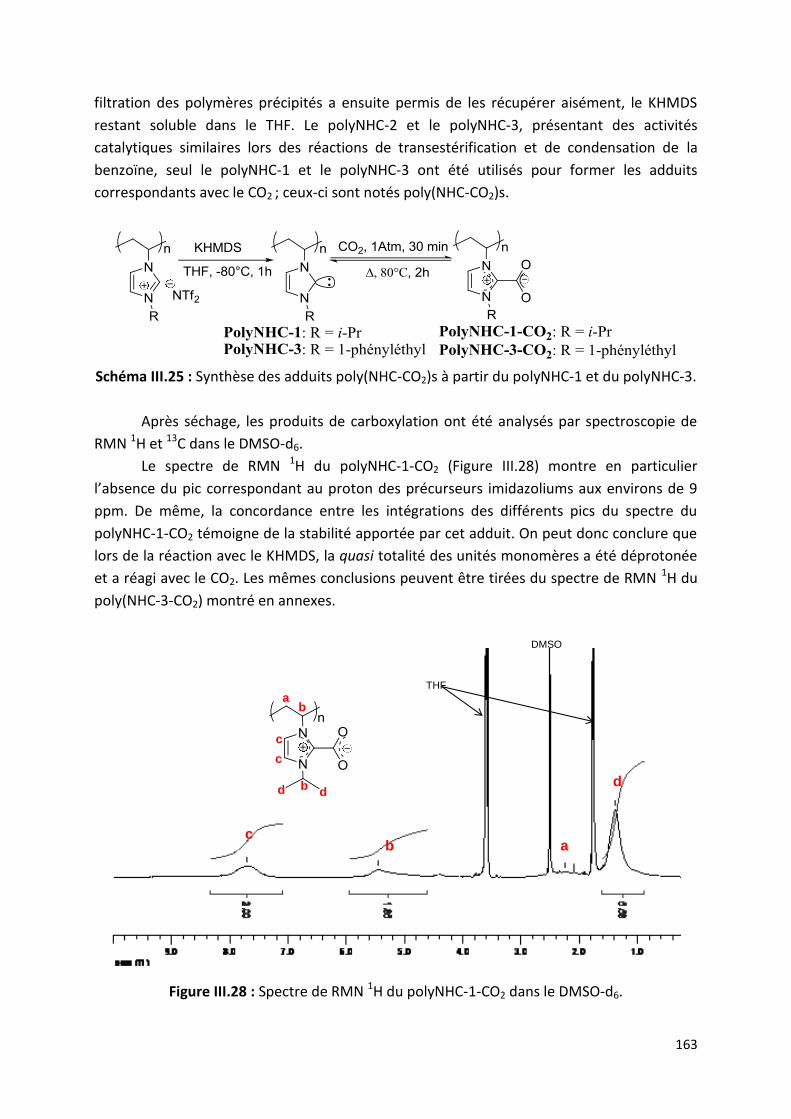
163
filtration des polymères précipités a ensuite permis de les récupérer aisément, le KHMDS
restant soluble dans le THF. Le polyNHC-2 et le polyNHC-3, présentant des activités
catalytiques similaires lors des réactions de transestérification et de condensation de la
benzoïne, seul le polyNHC-1 et le polyNHC-3 ont été utilisés pour former les adduits
correspondants avec le CO2 ; ceux-ci sont notés poly(NHC-CO2)s.
Schéma III.25 : Synthèse des adduits poly(NHC-CO2)s à partir du polyNHC-1 et du polyNHC-3.
Après séchage, les produits de carboxylation ont été analysés par spectroscopie de
RMN 1H et 13C dans le DMSO-d6.
Le spectre de RMN 1H du polyNHC-1-CO2 (Figure III.28) montre en particulier
l’absence du pic correspondant au proton des précurseurs imidazoliums aux environs de 9
ppm. De même, la concordance entre les intégrations des différents pics du spectre du
polyNHC-1-CO2 témoigne de la stabilité apportée par cet adduit. On peut donc conclure que
lors de la réaction avec le KHMDS, la quasi totalité des unités monomères a été déprotonée
et a réagi avec le CO2. Les mêmes conclusions peuvent être tirées du spectre de RMN 1H du
poly(NHC-3-CO2) montré en annexes.
Figure III.28 : Spectre de RMN 1H du polyNHC-1-CO2 dans le DMSO-d6.
dd
c
c
b
b
a
d
c
b a
THF
DMSO

164
Le spectre de RMN 13C du polyNHC-1-CO2 (Figure III.29) permet d’aboutir aux mêmes
conclusions. En effet, les pics observés à 143 ppm et 154 ppm indiquent que l’adduit
polyNHC-1-CO2 a bien été formé. Le déplacement de ces pics est caractéristique de tels
adduits95 et correspond aux carbones quaternaires e et f, issus de l’addition entre le carbène
et le carbone du CO2.
On note cependant que le quadruplet correspondant au carbone CF2 du groupement
triflate, présent dans le contre-anion du polymère précurseur (PViPrIm+NTf2-) est toujours
présent aux alentours de 120 ppm. On peut donc supposer que lors de la précipitation, le
polymère a emprisonné, en son sein, le sel de KNTf2 formé au moment de la déprotonation,
malgré le fait qu’il soit soluble dans le THF. Le spectre de RMN 13C du polyNHC-3-CO2 est
montré en annexe et présente également les pics attendus à 144 et 155 ppm.
Figure III.29 : Spectre de RMN 13C du polyNHC-1-CO2 dans le DMSO-d6.
Une autre preuve de la fonctionnalisation complète par le CO2 des poly(NHC)s
précurseurs est donnée par la perte de masse observée lors de l’analyse
thermogravimétrique (TGA) des adduits poly(NHC-CO2)s. En effet, l’analyse des
thermogrammes (Figure III.30) du polyNHC-1-CO2 et du polyNHC-3-CO2, montre des pertes
de masse de 25% et 19%, respectivement. Aux erreurs expérimentales près, ces valeurs
correspondent aux pertes de masses théoriques qui peuvent être déduites à partir de la
structure des unités monomères. Ainsi, si on divise la masse du CO2 (44 g.mol-1) par la masse
de l’unité monomère des polymères (180 g.mol-1 et 243 g.mol-1 respectivement), on trouve
des valeurs de 24,4% et 18,1%, respectivement.
b
c
c
bd d
ef
a
ppm (f1)50100150
0
1000
2000
3000
4000
15
3.9
55
14
1.4
98
12
4.2
48
12
1.0
50
11
8.5
17
11
7.7
98
11
4.6
54
50
.53
3
22
.27
0
c
d
efa+b
THFDMSO

165
Figure III.30 : Thermogrammes du polyNHC-1-CO2 et du polyNHC-3-CO2.
A partir de ces thermogrammes, il est également possible de déduire la température
moyenne de libération du CO2. La température de 140 °C, observée ici (pour une analyse en
masse), est en accord avec les valeurs de la littérature reportées dans le cas des adduits
moléculaires (de l’ordre de 150 °C) 95. On remarque aussi une perte de masse avant 100 °C
qui s’accentue à 95 °C. Celle-ci peut-être attribuée à l’évaporation de l’eau adsorbée sur les
polymères. La comparaison des thermogrammes des adduits moléculaires NHC-CO2 (tels que
décrits par Louie et coll.95) avec ceux des poly(NHC-CO2)s nous indique également que les
adduits supportés sur polymères sont bien plus stables que leur analogues moléculaires. En
effet, ces derniers présentent une perte totale de leur masse, c’est à dire une dégradation
complète des molécules, pour des températures comprises entre 180 °C et 200 °C. Les
polyNHC-1-CO2 et polyNHC-3-CO2 en revanche, sont stables jusqu’à une température d’au
moins 250 °C.
Il a également été démontré, qu’en solution, les adduits NHC-CO2 libèrent le CO2 à
plus basse température (50-80 °C)92, 95. Ainsi, afin de connaître la température de travail
pour régénérer les poly(NHC)s en solution, un suivi analytique par spectroscopie de RMN 13C
du PolyNHC-3-CO2 a été réalisé en fonction de la température. Pour diminuer le temps
d’acquisition de ces spectres (10 min au lieu de 4 h), le polyNHC-3 a été fonctionnalisé avec
du CO2 enrichi en 13C (13CO2 commercial). Ainsi, sur le spectre à 298K représenté Figure III.31,
seuls sont observés les pics correspondant aux carbones du solvant (40 ppm), aux carbones
aromatiques du polymère (127 ppm) et au carbone de la fonction carboxylate (154 ppm).
19%25%
50 100 150 200 25060
70
80
90
100m
asse (
%)
Temperature ( C)
PolyNHC-3-CO2PolyNHC-1-CO2

166
Le pic détecté à 159 ppm est attribuable à la forme hydratée du carboxylate
d’imidazolium (Schéma III.26), c'est-à-dire un hydrogénocarbonate d’imidazolium. Nous
reviendrons plus loin sur la présence de ce pic.
Lors des acquisitions des spectres à différentes températures, l’intensité du pic à 127
ppm a été prise comme référence afin de suivre l’évolution du groupement carboxylate. Une
superposition des spectres, réduite à un intervalle compris entre 150 et 165 ppm est incluse
dans la Figure III.31. Celle-ci montre qu’une température de 343 K (70 °C) doit être atteinte
avant d’observer une légère diminution de l’intensité du pic à 154 ppm, correspondant à la
libération des premières molécules de CO2. Lorsque la solution est placée à 353 K (80 °C),
aucune évolution n’est notée au cours des premières minutes. Cependant, après une heure
à cette température, l’intensité du pic correspondant au groupement carboxylate diminue
fortement. On peut donc supposer que la réaction de décarboxylation n’est pas instantanée
et nécessite un certain temps de chauffage. Enfin, lorsqu’une température de 363 K (90 °C)
est atteinte, le pic a presque totalement disparu, indiquant que la grande majorité du CO2 a
été libérée.
Figure III.31 : Spectre de RMN 13C (δ en ppm) du polyNHC-3-13CO2 dans le DMSO-d6, avec en
inclusion la superposition des spectres de la même solution, de 150 ppm à 165 ppm, et
obtenus à différentes températures.
165 160 155 150
298K
333K
343K
353K
353K + 1h
363K
a
a
a
a
a
a
b
b

167
Schéma III.26 : Hydratation des poly(NHC-CO2)s
La caractérisation par RMN 13C du polyNHC-3-CO2 à différentes températures nous
amène donc à penser qu’une température de 80 °C parait suffisante pour générer des
polyNHCs in-situ, par décarboxylation.
Au passage, ces expériences témoignent aussi de la réversibilité du processus de
carboxylation des poly(NHC)s, comme avec les homologues moléculaires. La température de
80 °C permet en effet la libération d’une majorité du CO2 au bout d’une heure, tout en
conservant des conditions de réaction relativement douces en vue des réactions
organocatalysées. Cependant, le 13CO2 libéré dans le tube par chauffage ne semble pas être
en proportion suffisante pour re-carboxyler le poly(NHC) formé. La comparaison des
températures de décarboxylation, obtenues par les deux techniques d’analyses (TGA et
RMN), montre cependant que les poly(NHC-CO2)s se comportent de la même manière que
leurs homologues moléculaires. De même, la différence de température de décarboxylation
entre ces deux états (solide et en solution) peut-être attribuée à l’énergie de solubilisation
des composés qui contribue en partie à l’énergie nécessaire pour libérer le CO2, comparé à
un composé en masse.
Il en est de même pour leur propension à s’hydrater. Les adduits NHC-CO2 eux-
mêmes peuvent en effet s’hydrater, comme l’on rapporté Louie et al.95. En fonction des
substituants des atomes d’azotes dans le cycle imidazolium, deux types d’hydratation ont
été observées95. Ainsi, les adduits présentant des groupements aryle ne forment que des
liaisons hydrogène entre l’eau et le carboxylate (Schéma III.27). Cependant, lorsque les
atomes d’azotes présentent des groupements alkyls, une réaction d’addition de l’eau sur le
carboxylate se produit. Il s’ensuit la formation d’un hydrogénocarbonate d’imidazolium
(Schéma III.28).
Schéma III.27 : Formation de liaisons hydrogènes lors de l’hydratation des adduits NHC-CO2s
présentant des groupements aryle95.

168
Schéma III.28 : Réaction entre les carboxylates d’alkylimidazolium et l’eau, formant des
hydrogénocarbonates d’imidazolium95.
Les adduits poly(NHC-CO2)s pourraient donc évoluer en s’hydratant au même titre
que leurs homologues moléculaires. Une telle réaction a ainsi été observée sur les polyNHC-
1-CO2 et polyNHC-3-CO2, comme en témoigne la présence du pic à 159 ppm déjà indiquée
auparavant. Ces polymères ne présentant que des substituants alkyle, leur hydratation est
supposée se produire de la même manière que la réaction présentée Schéma III.28.
Ceci a été confirmé par l’analyse spectroscopique de RMN dans le DMSO-d6 du
polyNHC-1-CO2 après que ce polymère ait été laissé à l’air pendant une nuit. Comme on peut
le voir sur le spectre de RMN 1H représenté Figure III. 32, l’hydratation du polymère est mise
en évidence par l’apparition d’un pic à environ 10 ppm correspondant au proton acide du
cycle imidazolium. Comme indiqué, cette réaction est également confirmée en RMN du 13C
(Figure III.33) par le déplacement des pics correspondants aux carbones quaternaires liant la
fonction carboxylate à l’imidazolium. Le carbone de l’imidazolium retrouve le déplacement
chimique observé dans le cas des cycles hydrogénés, et passe ainsi de 142 ppm à 135 ppm.
De même le pic correspondant au carbone du groupe carboxylate se déplace de 154 ppm à
159 ppm.
Dans la littérature, cette réaction d’hydratation des adduits de type NHC-CO2 n’est
pas décrite comme étant réversible. On peut donc s’interroger sur la possibilité de régénérer
les poly(NHC)s à partir des adduits de poly(NHC-CO2) hydratés. Dans le chapitre suivant de ce
manuscrit, nous montrerons comment nous avons tiré profit de ces formes hydratées.
Dans un premier temps, afin de pouvoir utiliser les polyNHC-1-CO2 et polyNHC-3-CO2
en tant que précurseurs de polyNHCs par thermolyse, ceux-ci ont cependant été conservés
sous atmosphère sèche.

169
Figure III.32 : Spectre de RMN 1H du polyNHC-1-CO2 hydraté dans le DMSO-d6.
Figure III.33 : superposition des spectres de RMN 13C dans le DMSO-d6 du polyNHC-1-CO2 et
du polyNHC-1-CO2 hydraté.
ppm (f1)5.010.0
0
50
100
150
9.7
40
7.7
59
4.5
17
3.4
80
1.4
56
0.7
4
2.0
7
1.3
5
3.0
6
6.0
0
ab
cc
df f
e
f
a
b+dce
ppm (f1)50100150
-1000
0
1000
2000
3000
4000
5000
6000
7000
15
9.2
10
13
5.2
47
12
8.0
82
12
4.0
15
12
1.1
58
11
9.7
15
11
7.8
35
11
4.4
43
66
.31
2
52
.66
6
22
.04
1
ppm (f1)50100150
0
1000
2000
3000
4000
15
3.9
55
14
1.4
98
12
4.2
48
12
1.0
50
11
8.5
17
11
7.7
98
11
4.6
54
50
.53
3
22
.27
0
a b
cc
ed d
fg
a’ b’
c’
c’f’
e’d’ d’
g’
fg c ab+e
d
g’ f’
d’
c’b’+e’
a’

170
III.6.2) Evaluation en organocatalyse
Les adduits NHC-CO2 permettent la génération in-situ de NHCs en libérant le CO2. De
ce fait, ils ont déjà été employés comme précurseurs d’organocatalyseurs pour des réactions
telles que la formation de carbonates cycliques, la cyanosilylation des cétones et des
aldéhydes ou encore la trimérisation des isocyanates48, 91, 93, 94.
Leur utilisation en tant que précurseurs de NHCs pour la catalyse des réactions de
transestérification ou de condensation de la benzoïne n’a cependant jamais été décrite.
Dans les paragraphes suivants, les adduits poly(NHC-CO2)s décrits précédemment vont être
employés afin de générer in situ des poly(NHC)s capables de catalyser ces deux réactions.
Nous pourrons ainsi discuter de l’utilité de masquer/démasquer les NHCs avec le CO2 dans le
but de protéger ces entités catalytiques lors des différents recyclages, pour une
manipulation plus aisée.
III.6.2.1) Evaluation en catalyse de transestérification
Les analyses par spectroscopie de RMN 13C du polyNHC-3-CO2 ont révélé que ce
polymère doit être chauffé en solution jusqu’à une température de 80 °C, pour une durée
minimum d’1h, afin de libérer la majeure partie du CO2 et ainsi régénérer le poly(NHC)s. Il
s’agissait donc ici d’utiliser les poly(NHC-CO2)s comme précurseurs in-situ de poly(NHC)s, en
d’autres termes comme précatalyseurs supportés pour les réactions de transestérification.
Ainsi, les réactions de transestérification ont été conduites à 80 °C pendant 2 h en présence
de poly(NHC-CO2)s (Schéma III.29).
Le même montage expérimental et le même protocole que ceux précédemment
décrits ont été utilisés. Toutefois, la précipitation du précatalyseur de type poly(NHC-CO2)
n’a pas été induite ici par l’ajout de diéthyl éther, mais simplement en appliquant une légère
pression de CO2, après retour à température ambiante. On peut en effet rappeler que les
poly(NHC-CO2)s ne sont pas solubles dans le THF à température ambiante, contrairement à
leurs homologues poly(NHC)s. Ainsi, après précipitation, les polymères ont pu être
directement recyclés par simple filtration. De la même manière, le filtrat a ensuite été
analysé par spectroscopie de RMN 1H afin de déterminer les rendements de
transestérification, comme discuté précédemment.

171
Schéma III.29 : Réaction de transestérification en présence des poly(NHC-CO2)s.
Les réactions de transestérification entre l’acétate de vinyle et l’alcool benzylique ont
donc été réalisées en utilisant le polyNHC-1-CO2 et le polyNHC-3-CO2 comme précurseurs de
catalyseurs NHCs supportés sur polymère, en milieu homogène. Les rendements en acétate
de benzyle, après différents recyclages des poly(NHC-CO2)s, sont donnés dans le tableau
III.4.
Tableau III.4 : Synthèse de l’acétate de benzyle à partir de l’alcool benzylique et de l’acétate
de vinyle par transestérification réalisée en présence de poly(NHC-CO2)s à 80 °C dans le THF.
Cycle 1 2 3 4 5 6 7 8
r.(%) avec polyNHC-1-CO2 94 51 23 - - - - -
r.(%) avec polyNHC-3-CO2 83 77 74 70 77 76 77 70 alcool benzylique (1 eq.), acetate de benzyle (1,2 eq.) et polyNHC (10%
espèces actives) dans le THF à 80 °C pendant 2 h. rendement (r.) en acétate de
benzyle déterminé par RMN 1H.
Lors du premier cycle de transestérification en présence des poly(NHC-CO2)s, des
rendements du même ordre de grandeur (94% pour le polyNHC-1-CO2 et 83% pour le
polyNHC-3-CO2) que ceux observés avec les poly(NHC)s parents ont été obtenus. On peut
donc conclure que la réaction de décarboxylation des précurseurs a bien eu lieu dans ces
conditions de réactions, permettant de régénérer les poly(NHC)s correspondants.
Qui plus est, après ajout de CO2 en fin de réaction, les poly(NHC-CO2)s peuvent être
aisément récupérés, recyclés et réutilisés dans un second cycle de transestérification. Les
rendements obtenus lors des deuxièmes cycles (de 77 et 51%) sont plus élevés que ceux
atteints lors du deuxième cycle avec les poly(NHC)s. La fonctionnalisation des poly(NHC)s
avec le CO2 permet donc de protéger plus efficacement ces NHCs supportés vis-à-vis de l’eau
et autres impuretés éventuelles. Ceci est d’autant plus probant avec le polyNHC-3-CO2, qui
permet d’atteindre des rendements de l’ordre de 75% jusqu’à 8 cycles d’organocatalyse.
Dans le cas du polyNHC-1-CO2, une baisse d’activité catalytique a toutefois été notée. Lors
du troisième cycle, en effet, on obtient un rendement de 23% seulement, et le quatrième
cycle s’est révélé infructueux (rendement nul). Cette perte progressive d’activité catalytique

172
peut en partie être expliquée par le fait que, contrairement au polyNHC-3-CO2, le polyNHC-
1-CO2 ne se dissout pas totalement dans le THF à 80 °C. Ainsi, seules quelques chaines
polymères dénuées de CO2 joueraient leur rôle de catalyseurs.
Des analyses par thermogravimétrie d’échantillons du polyNHC-1-CO2, avant et après
les cycles de transestérification (Figure III.34), ont par ailleurs montré que ce polymère ne
réincorpore pas totalement le CO2. En effet, comme il a été montré précédemment, celui-ci
libère la totalité du CO2 incorporé à environ 150 °C, ce qui correspond à une perte de masse
de 25%. Le même phénomène a été observé avant d’engager ce polymère dans une réaction
de transestérification. Cependant, après 1 cycle de transestérification, celui-ci subit une
perte de masse de 23%, ce qui indique que la totalité des unités monomères n’ont pas été
fonctionnalisées par le CO2. Ceci s’accentue après le 2ème cycle, avec une perte de masse de
seulement 13%.
Il apparaît donc que le polyNHC-1-CO2 se dégrade au fur et à mesure de son
utilisation, empêchant ainsi la réincorporation de CO2 sur toutes les unités monomères.
Figure III.34 : Thermogrammes d’échantillons de polyNHC-1-CO2 avant et après des cycles
de transestérification.
En conclusion, les poly(NHC-CO2)s, utilisés comme pré-catalyseurs de type NHC
supportés sur polymère, s’avèrent plus efficaces pour l’organocatalyse de réaction de
transestérification que les poly(NHC)s. De surcroit, ces adduits peuvent être recyclés
facilement et leur activité catalytique reste élevée sur un assez grand nombre de cycles. La
carboxylation des poly(NHC)s est donc un bon moyen de protéger/manipuler ces
organocatalyseurs supportés.
Masse (
%)
PolyNHC-1-CO2
PolyNHC-1-CO2 après 1 cyclePolyNHC-1-CO2 après 2 cycles
140 160 180 200 220
70
80
90
100
Temperature ( C)

173
Ces mêmes polymères ont été testés dans une autre réaction, en l’occurrence la
condensation de la benzoïne. Cette partie est discutée ci-après.
III.6.2.2) Evaluation en catalyse de condensation de la benzoïne
Comme précisé auparavant, la condensation de la benzoïne ne peut être catalysée
que par les NHCs ou les ions cyanates. La formation de benzoïne à partir de benzaldéhyde,
en présence de poly(NHC-CO2)s, est donc un bon moyen d’attester de la décarboxylation de
ces derniers et de la formation de poly(NHC)s.
Ainsi, les poly(NHC-CO2)s ont été employés pour la condensation de la benzoïne, en
appliquant le même protocole expérimental que celui décrit précédemment pour la réaction
de transestérification (Schéma III.30). Le benzaldéhyde a été utilisé en tant que réactif et un
temps de réaction de 24 h, au lieu de 2 h, a été choisi afin de concorder avec les protocoles
expérimentaux décrits auparavant.
Schéma III.30 : Condensation de la benzoïne catalysée par les poly(NHC-CO2)s.
Les pré-catalyseurs ont aussi été filtrés et recyclés, après qu’une pression de CO2 (1
atm.) ait été appliquée dans le montage expérimental. Les poly(NHC-CO2)s récupérés ont
ensuite été impliqués dans de nouveaux cycles de catalyse. Les rendements en benzoïne,
déterminés à partir des spectres de RMN 1H dans le DMSO-d6 des filtrats récupérés, sont
présentés dans le tableau III.5.
Tableau III.5 : Condensation de la benzoïne catalysée en présence des poly(NHC-CO2)s.
Cycle 1 2 3
r.(%) avec polyNHC-1-CO2 71 27 8
r.(%) avec polyNHC-3-CO2 65 35 21
benzaldéhyde (1 eq.), poly(NHC-CO2) (10% d’espèces actives) dans le THF à 80 °C pendant 24 h.

174
Au cours du premier cycle de catalyse avec les poly(NHC-CO2)s, les valeurs des
rendements sont légèrement plus faibles par rapports aux valeurs observées lors de
l’utilisation des poly(NHC)s (71 et 65% au lieu de 92 et 85%). Lors des cycles suivants, les
valeurs diminuent progressivement comme observé avec les poly(NHC)s. Après le deuxième
et le troisième cycle, le poly(NHC-3-CO2) présente même des activité catalytiques plus faibles
que son homologue non protégé (35% et 21% pour le polyNHC-3-CO2 et 72% et 46% pour le
polyNHC-3).
Etant donné l’aptitude des poly(NHC-CO2)s à catalyser la transestérification sur
plusieurs cycles, on aurait pu penser que ceux-ci se comporteraient de la même manière lors
de la catalyse de la condensation de la benzoïne. Le polyNHC-1-CO2 et le polyNHC-3-CO2, ne
sont actifs que sur 3 cycles impliquant par là même, la formation de groupements NHCs
après la réaction de décarboxylation. Cependant, cette activité catalytique n’est pas
conservée lors des différents recyclages et ne permet plus l’obtention de rendements
quantitatifs en benzoïne. Dans le cadre de cette réaction, le masquage/démasquage des
poly(NHC)s au moyen de CO2 ne paraît donc pas améliorer de façon significative la réaction
d’organocatalyse par rapport aux poly(NHC)s non masquées. Cette différence d’activité en
condensation de la benzoïne, comparée à la réaction de transestérification, pourrait
s’expliquer par la durée et la température à laquelle se déroule cette réaction. Il peut en
effet être envisagé qu’à une température de 80 °C, pendant 24 h, certains groupements
NHCs supportés se dégradent, et ne peuvent plus agir en tant que catalyseurs pour les
autres cycles de condensation de la benzoïne.
III.7) Conclusion
Les NHCs sont des molécules sensibles, par la même difficiles à manipuler et à
synthétiser. Il est donc nécessaire, comme cela a été réalisé pour un très grand nombre de
catalyseurs, de les supporter sur différents substrats, afin de pouvoir les recycler et de les
utiliser à nouveau. Le peu d’articles accessibles dans la littérature décrivant une telle
méthodologie nous a conduit à développer des supports de type poly(NHC). Ceux-ci ont été
employés en catalyse de transestérification et de condensation de la benzoïne, mais ont
également été protégés au moyen d’une réaction réversible avec le CO2.
Pour accéder à ces poly(NHC)s et à leurs adduits de CO2, nous avons tout d’abord
synthétisé des polymères liquides ioniques de type bromure de poly(1-vinyl-3-
alkylimidazolium). Leur propriétés de solubilité ne permettant pas de générer directement
des poly(NHC)s, un changement d’anion portés par les polymères a été effectué, en
remplaçant les ions bromures par les ions bis(trifluorométhanesulfonyl)amidure. D’autre
part, la technique de polymérisation RAFT/MADIX a également permis la synthèse de
PVBuIm+Br- de structure contrôlée. Ceux-ci ont par la suite été utilisés en tant que

175
macroagents de transfert pour la synthèse de copolymères à blocs de type PVBuIm+Br--b-
PNVP.
Un des objectifs visé en préparant de tels copolymères à blocs était de tirer profit de
leur auto-assemblage en solution, pour induire la formation de poly(NHC)s dans des
(nano)structures et ainsi catalyser des réactions en milieu confiné. Une telle méthodologie
n’a cependant pas été expérimentée, de par la difficulté à caractériser ces copolymères. La
formation de micelles ou de vésicules à partir des PVBuIm+Br--b-PNVP, au moyen de stimuli
tels qu’un changement d’anion reste donc a démontrer.
La déprotonation des PVIm+NTf2- au moyen des bases fortes (KHMDS et NaH), a
ensuite été appliquée afin de générer des poly(NHC)s. L’utilisation de CO2 pour protéger ces
derniers au moyen d’un masquage/démasquage a ensuite permis de protéger les fonctions
NHCs supportées sur polymères. Ces adduits ont été caractérisés par spectroscopie de RMN 1H et 13C, ainsi que par analyse thermogravimétrique. Ces deux techniques d’analyses ont
notamment montré que les poly(NHC-CO2)s libèrent le CO2 à des températures de l’ordre de
150 °C en masse et de 80 °C en solution.
Les NHCs supportés, qu’ils soient sous une forme protégée ou non, ont également
été évalués en catalyse organique moléculaire. Pour cela, des réactions de condensation de
la benzoïne et de transestérification ont été réalisées en utilisant les poly(NHC)s et leur
adduits de CO2 comme (pré)catalyseurs. Ceux-ci ont permis d’obtenir de bons rendements
pour les deux réactions. Ils ont également pu être recyclés aisément afin d’être à nouveau
mis en jeu en tant qu’organocatalyseurs. Dans la plupart des cas, l’activité catalytique des
poly(NHC)s, très élevée lors du premier cycle, n’a cependant pas été maintenue lors des
cycles suivants. Seul le polyNHC-3-CO2, s’est avéré être un bon catalyseur de la
transestérification et a conservé son activité catalytique sur près de 8 cycles.
La fonctionnalisation par du CO2 des NHCs supportés est apparue ici comme un
moyen de protection efficace de ces espèces sensibles. Elle a permis de conserver l’activité
catalytique des NHCs, tout en autorisant leur utilisation sur un grand nombre de cycles.
Ces polymères ne peuvent cependant pas être manipulés à l’air, du fait de leur
hydratation progressive dans une atmosphère humide. En effet, de même que pour leurs
analogues moléculaires, les poly(NHC-CO2)s présentant des substituants alkyls réagissent
avec l’eau pour former des hydrogénocarbonates d’imidazolium. Les adduits NHC-CO2
présentant des substituants aryle ne permettent pas une telle réaction d’hydratation, on
peut supposer qu’une fois supportés, ils seraient plus faciles à manipuler que les poly(NHC-
CO2)s développés par nos soins. Il reste cependant à démontrer que la réaction
d’hydratation de carboxylates d’alkylimidazolium est réversible, ce qui n’a jamais été décrit
dans la littérature.

176
III.8) Références
1. Green, O.; Grubjesic, S.; Lee, S.; Firestone, M. A. Polym. Rev. 2009, 49, (4), 339 - 360. 2. Seddon, K. R. J. Chem. Technol. Biotechnol. 1997, 68, (4), 351-356. 3. Tsukasa, T.; Tetsuya, T.; Ken-ichi, O.; Susumu, K. Adv. Mater. 2009, 22, (11), 1196-1221. 4. Chiappe, C.; Pieraccini, D. J. Phys. Org. Chem. 2005, 18, (4), 275-297. 5. Plechkova, N. V.; Seddon, K. R. Chem. Soc. Rev. 2008, 37, (1), 123-150. 6. Welton, T. Chem. Rev. 1999, 99, (8), 2071-2083. 7. Green, M. D.; Long, T. E. Polym. Rev. 2009, 49, (4), 291 - 314. 8. Marcilla, R.; Blazquez, J. A.; Rodriguez, J.; Pomposo, J. A.; Mecerreyes, D. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, (1), 208-212. 9. Wells, A. S.; Coombe, V. T. Org. Process Res. Dev. 2006, 10, (4), 794-798. 10. Lewandowski, A.; Aswiderska-Mocek, A. J. Power Sources 2009, 194, (2), 601-609. 11. Dominguez de Maria, P. Angew. Chem., Int. Ed. 2008, 47, (37), 6960-6968. 12. Ueki, T.; Watanabe, M. Macromolecules 2008, 41, (11), 3739-3749. 13. Lu, J.; Yan, F.; Texter, J. Prog. Polym. Sci. 2009, 34, (5), 431-448. 14. Ralf, G. Angew. Chem., Int. Ed. 49, (16), 2834-2839. 15. Jaeger, W.; Bohrisch, J.; Laschewsky, A. Prog. Polym. Sci. 2010, 35, (5), 511-577. 16. Gibson, H. W. Polym. Rev. 2009, 49, (4), 289 - 290. 17. Anderson, E. B.; Long, T. E. Polymer 2010, 51, (12), 2447-2454. 18. Vijayakrishna, K.; Jewrajka, S. K.; Ruiz, A.; Marcilla, R.; Pomposo, J. A.; Mecerreyes, D.; Taton, D.; Gnanou, Y. Macromolecules 2008, 41, (17), 6299-6308. 19. Vijayakrishna, K.; Mecerreyes, D.; Gnanou, Y.; Taton, D. Macromolecules 2009, 42, (14), 5167-5174. 20. Alexander, S. S.; Laurent, G.; Frédéric, V.; Elena, I. L.; Franck, M.; Inna, A. M.; Claude, C.; Dominique, T.; Irina, L. O.; Yakov, S. V. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, (17), 4245-4266. 21. Chen, H.; Choi, J.-H.; Salas-de la Cruz, D.; Winey, K. I.; Elabd, Y. A. Macromolecules 2009, 42, (13), 4809-4816. 22. Tang, H.; Tang, J.; Ding, S.; Radosz, M.; Shen, Y. J. Polym. Sci., Part A: Polym. Chem. 2005, 43, (7), 1432-1443. 23. Mori, H.; Yanagi, M.; Endo, T. Macromolecules 2009, 42, (21), 8082-8092. 24. Hasib-ur-Rahman, M.; Siaj, M.; Larachi, F. Chem. Eng. Process. 2010, 49, (4), 313-322. 25. Marcilla, R.; Blazquez, J. A.; Fernandez, R.; Grande, H.; Pomposo, J. A.; Mecerreyes, D. Macromol. Chem. Phys. 2005, 206, (2), 299-304. 26. Marcilla, R.; Ochoteco, E.; Pozo-Gonzalo, C.; Grande, H.; Pomposo, J. A.; Mecerreyes, D. Macromol. Rapid Commun. 2005, 26, (14), 1122-1126. 27. Marcilla, R.; Pozo-Gonzalo, C.; Rodri?guez, J.; Alduncin, J. A.; Pomposo, J. A.; Mecerreyes, D. Synth. Met. 2006, 156, (16-17), 1133-1138. 28. Marcilla, R.; Sanchez-Paniagua, M.; Lopez-Ruiz, B.; Lopez-Cabarcos, E.; Ochoteco, E.; Grande, H.; Mecerreyes, D. J. Polym. Sci., Part A: Polym. Chem. 2006, 44, (13), 3958-3965. 29. Ohno, H. Macromol. Symp. 2007, 249-250, 551-556. 30. Armand, M.; Endres, F.; MacFarlane, D. R.; Ohno, H.; Scrosati, B. Nat. Mater. 2009, 8, (8), 621-629. 31. Arduengo Iii, A. J.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, (1), 361-363. 32. Enders, D.; Breuer, K.; Raabe, G.; Runsink, J.; Teles, J. H.; Melder, J.-P.; Ebel, K.; Brode, S. Angew. Chem., Int. Ed. 1995, 34, (9), 1021-1023. 33. Bourissou, D.; Guerret, O.; Gabbai, F. P.; Bertrand, G. Chem. Rev. 2000, 100, (1), 39-91. 34. Arduengo Iii, A. J.; Rasika Dias, H. V.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1992, 114, (14), 5530-5534. 35. Barbaro, P.; Liguori, F. Chem. Rev. 2008, 109, (2), 515-529. 36. Buchmeiser, M. R. Chem. Rev. 2008, 109, (2), 303-321.

177
37. Fraile, J. M.; GarciÌ•a, J. I.; Mayoral, J. A. Chem. Rev. 2008, 109, (2), 360-417. 38. Minakata, S.; Komatsu, M. Chem. Rev. 2008, 109, (2), 711-724. 39. Wang, Z.; Chen, G.; Ding, K. Chem. Rev. 2008, 109, (2), 322-359. 40. Akiyama, R.; Kobayashi, S. Chem. Rev. 2009, 109, (2), 594-642. 41. Bergbreiter, D. E.; Tian, J.; Hongfa, C. Chem. Rev. 2009, 109, (2), 530-582. 42. Ikegami, S.; Hamamoto, H. Chem. Rev. 2009, 109, (2), 583-593. 43. Lu, J.; Toy, P. H. Chem. Rev. 2009, 109, (2), 815-838. 44. Trindade, A. F.; Gois, P. M. P.; Afonso, C. A. M. Chem. Rev. 2009, 109, (2), 418-514. 45. Barrett, A. G. M.; Love, A. C.; Tedeschi, L. Org. Lett. 2004, 6, (19), 3377-3380. 46. Storey, J. M. D.; Williamson, C. Tetrahedron Lett. 2005, 46, (43), 7337-7339. 47. Zhou, H.; Zhang, W.-Z.; Wang, Y.-M.; Qu, J.-P.; Lu, X.-B. Macromolecules 2009, 42, (15), 5419-5421. 48. Pawar, G. M.; Buchmeiser, M. R. Adv. Synth. Catal. 2010, 352, (5), 917-928. 49. Sommer, W. J.; Weck, M. Coord. Chem. Rev. 2007, 251, (5-6), 860-873. 50. MeiXuan, T.; Yugen, Z.; Jackie , Y. Y. Adv. Synth. Catal. 2009, 351, (9), 1390-1394. 51. Zhang, Y.; Zhao, L.; Patra, P. K.; Hu, D.; Ying, J. Y. Nano Today 2009, 4, (1), 13-20. 52. Murry, J. M., Fundamentals of organic chemistry 5th Ed. 1999. 53. Marcus, D.; Helmut, R. Macromol. Chem. Phys. 2009, 210, (9), 776-782. 54. Zhao, F.; Meng, Y.; Anderson, J. L. J. Chromatogr., A 2008, 1208, (1-2), 1-9. 55. Chiefari, J.; Chong, Y. K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T. P. T.; Mayadunne, R. T. A.; Meijs, G. F.; Moad, C. L.; Moad, G.; Rizzardo, E.; Thang, S. H. Macromolecules 1998, 31, (16), 5559-5562. 56. Gung, B. W.; Bailey, L. N.; Craft, D. T.; Barnes, C. L.; Kirschbaum, K. Organometallics 29, (15), 3450-3456. 57. Corpart, P.; Charmot, D.; Biadatti, T.; Zard, S.; Michelet, D. WO9858974, 1998. 58. Destarac, M.; Bzducha, W.; Taton, D.; Gauthier-Gillaizeau, I.; Zard, S. Z. Macromol. Rapid Commun. 2002, 23, (17), 1049-1054. 59. Moad, G.; Rizzardo, E.; Thang, S. H. Acc. Chem. Res. 2008, 41, (9), 1133-1142. 60. Moad, G.; Rizzardo, E.; Thang, S. H. Polymer 2008, 49, (5), 1079-1131. 61. York, A. W.; Kirkland, S. E.; McCormick, C. L. Adv. Drug Delivery Rev. 2008, 60, (9), 1018-1036. 62. Devasia, R.; Bindu, R. L.; Borsali, R.; Mougin, N.; Gnanou, Y. Macromol. Symp. 2005, 229, (1), 8-17. 63. Pound, G.; McKenzie, J. M.; Lange, R. F. M.; Klumperman, B. Chem. Commun. 2008, (27), 3193-3195. 64. Wan, D.; Zhou, Q.; Pu, H.; Yang, G. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, (11), 3756-3765. 65. Mori, H.; Ookuma, H.; Endo, T. Macromol. Symp. 2007, 249-250, 406-411. 66. Ge, Z.; Xie, D.; Chen, D.; Jiang, X.; Zhang, Y.; Liu, H.; Liu, S. Macromolecules 2007, 40, (10), 3538-3546. 67. Debuigne, A.; Poli, R.; Jérôme, C.; Jérôme, R.; Detrembleur, C. Progress in Polymer Science (Oxford) 2009, 34, (3), 211-239. 68. Debuigne, A.; Poli, R.; De Winter, J.; Laurent, P.; Gerbaux, P.; Wathelet, J.-P.; Jérôme, C.; Detrembleur, C. Macromolecules 2010, 43, (6), 2801-2813. 69. Debuigne, A.; Willet, N.; Jérôme, R.; Detrembleur, C. Macromolecules 2007, 40, (20), 7111-7118. 70. Pound, G.; Eksteen, Z.; Pfukwa, R.; McKenzie, J. M.; Lange, R. F. M.; Klumperman, B. Journal of Polymer Science Part A: Polymer Chemistry 2008, 46, (19), 6575-6593. 71. Fandrich, N.; Falkenhagen, J.; Weidner, S. M.; Pfeifer, D.; Staal, B.; Thünemann, A. F.; Laschewsky, A. Macromol. Chem. Phys. 2010, 211, (8), 869-878. 72. Wan, D.; Satoh, K.; Kamigaito, M.; Okamoto, Y. Macromolecules 2005, 38, (25), 10397-10405. 73. de Fremont, P.; Marion, N.; Nolan, S. P. Coord. Chem. Rev. 2009, 253, (7-8), 862-892. 74. Sun, F.-G.; Huang, X.-L.; Ye, S. J. Org. Chem. 2009, 75, (1), 273-276.

178
75. Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, (12), 5606-5655. 76. Marion, N.; Diez-Gonzalez, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, (17), 2988-3000. 77. Grasa, G. A.; Kissling, R. M.; Nolan, S. P. Org. Lett. 2002, 4, (21), 3583-3586. 78. Teles, J. H.; Johann-Peter, M.; Klaus, E.; Regina, S.; Eugen, G.; Wolfgang, H.; Stefan, B.; Dieter, E.; Klaus, B.; Gerhard, R. Helv. Chim. Acta 1996, 79, (1), 61-83. 79. Enders, D.; Kallfass, U. Angew. Chem., Int. Ed. 2002, 41, (10), 1743-1745. 80. Enders, D.; Balensiefer, T. Acc. Chem. Res. 2004, 37, (8), 534-541. 81. Iwamoto, K. i.; Hamaya, M.; Hashimoto, N.; Kimura, H.; Suzuki, Y.; Sato, M. Tetrahedron Lett. 2006, 47, (40), 7175-7177. 82. Enders, D.; Han, J. Tetrahedron Asymmetry 2008, 19, (11), 1367-1371. 83. Zhao, H.; Foss, F. W.; Breslow, R. J. Am. Chem. Soc. 2008, 130, (38), 12590-12591. 84. Enders, D.; Henseler, A. Adv. Synth. Catal. 2009, 351, (11-12), 1749-1752. 85. Pinaud, J.; Vijayakrishna, K.; Taton, D.; Gnanou, Y. Macromolecules 2009, 42, (14), 4932-4936. 86. Moore, J. L.; Rovis, T. Top. Curr. Chem. 2010, 291, 77-144. 87. Holbrey, J. D.; Reichert, W. M.; Tkatchenko, I.; Bouajila, E.; Walter, O.; Tommasi, I.; Rogers, R. D. Chem. Commun. 2003, 9, (1), 28-29. 88. Tommasi, I.; Sorrentino, F. Tetrahedron Lett. 2005, 46, (12), 2141-2145. 89. Tommasi, I.; Sorrentino, F. Tetrahedron Lett. 2006, 47, (36), 6453-6456. 90. Voutchkova, A. M.; Feliz, M.; Clot, E.; Eisenstein, O.; Crabtree, R. H. J. Am. Chem. Soc. 2007, 129, (42), 12834-12846. 91. Zhou, H.; Zhang, W.-Z.; Liu, C.-H.; Qu, J.-P.; Lu, X.-B. J. Org. Chem. 2008, 73, (20), 8039-8044. 92. Delaude, L. Eur. J. Inorg. Chem. 2009, (13 ), 1681-1699. 93. Kayaki, Y.; Yamamoto, M.; Ikariya, T. Angew. Chem., Int. Ed. 2009, 48, (23), 4194-4197. 94. Tommasi, I.; Sorrentino, F. Tetrahedron Lett. 2009, 50, (1), 104-107. 95. Van Ausdall, B. R.; Glass, J. L.; Wiggins, K. M.; Aarif, A. M.; Louie, J. J. Org. Chem. 2009, 74, (20), 7935-7942. 96. Sauvage, X.; Demonceau, A.; Delaude, L. Adv. Synth. Catal. 2009, 351, (11-12), 2031-2038.

179

180
Chapitre IV : Molécules et polymères dérivés des
hydrogénocarbonates d’imidazolium : nouveaux précurseurs
de carbènes N-hétérocycliques

181

182
IV.1) Introduction et objectifs
Comme indiqué et exploité dans le chapitre précédent, la formation de carboxylates
d’imidazolium par addition de CO2, sur les carbènes en général et sur les NHCs en particulier,
a largement été utilisée dans la littérature en synthèse organométallique1-3 et, plus
rarement, en catalyse organique4-8. Du fait de la réversibilité de cette réaction par
stimulation thermique9, divers complexes organométalliques peuvent en effet être
synthétisés par génération in-situ des NHCs1-3.
On peut cependant s’interroger quant à la stabilité à l’air de ces adduits
zwittérioniques et, plus généralement, à leur manipulation comme carbènes masqués. En
réalité, en présence d’eau, les carboxylates d’alkylimidazolium s’hydratent pour former des
hydrogénocarbonates d’alkylimidazolium (Schéma IV.1). A notre connaissance, la
réversibilité d’une telle réaction d’hydratation n’a jamais été discutée dans la littérature, si
bien que les hydrogénocarbonates d’imidazolium n’ont jamais été utilisés dans le but de
générer des carbènes.
Schéma IV.1 : Hydratation des carboxylates d’alkylimidazoliums, formant des
hydrogénocarbonates d’imidazoliums.
Dans ce chapitre, nous allons montrer que les adduits NHC-CO2-H2O peuvent être
facilement préparés et manipulés pour générer des NHCs par activation thermique. Outre
leur formation par hydratation d’adduits précurseurs NHC-CO2, nous montrerons que ces
hydrogénocarbonates d’imidazolium peuvent être très facilement obtenus par échange
d’anion à partir de sels d’azolium commerciaux (à contre-anion chlorure ou bromure). La
synthèse de ces composés ne nécessitera donc pas de passer par des intermédiaires NHC-
CO2, évitant ainsi la préparation préalable des carbènes et leur masquage subséquent par le
CO2. En d’autres termes, nous proposons ici une solution commode pour générer des NHCs
par activation thermique d’hydrogénocarbonates d’azolium (Schéma IV.2), avec comme
uniques sous produits de l’eau et du dioxide de carbone.
Nous démontrerons que, là encore, le processus de démasquage/masquage des
NHCs par un mélange H2O + CO2 est réversible, permettant le recyclage et une manipulation
facile des hydrogénocarbonates d’azolium, précurseurs de NHCs.
Schéma IV.2 : Activation thermique des hydrogénocarbonates d’azolium conduisant à la
formation de NHCs.

183
Enfin, en s’inspirant des travaux présentés dans le chapitre précédent, concernant la
formation de poly(NHC)s, nous proposerons une « version polymère » des
hydrogénocarbonates d’imidazolium comme supports précurseurs de NHCs exploités en
catalyse organique.
Dans la littérature, l’équipe de Jessop (Queens university, Canada) a montré que
l’utilisation de CO2 en présence d’amines permet d’inverser la polarité de solvants ou de
molécules organiques.
Schéma IV.3 : Inversion de la polarité de certaines amines par réaction avec H2O et CO2.
Ainsi, en présence de CO2 et d’eau, il a tout d’abord été montré que la DBU (1,8-
Diazabicyclo[5.4.0]undec-7-ene) forme un sel présentant un anion hydrogénocarbonate10
(Schéma IV.4). En remplaçant l’eau par de l’hexanol, Jessop et al. ont également montré que
la DBU se protone, formant alors un alkylcarbonate d’ammonium11 (Schéma IV.4). Un tel
adduit liquide ionique, en présence d’un flux d’argon ou d’azote peut reformer les réactifs de
départ. La réversibilité de cette réaction a notamment été mise à profit pour inverser la
polarité du milieu et permettre ainsi une démixtion entre le décane, l’hexanol et la DBU
(Figure IV.1).
Schéma IV.4 : Réaction réversible entre la DBU, le CO2 et l’eau ou l’hexanol10,11.
Figure IV.1: Principe d’inversion de polarité du solvant en fonction de l’atmosphère
environnante10,11.
Cette réaction a été étendue à des subtrats de type amidine non cyclique (Schéma
IV.5). En sélectionnant des amidines présentant une longue chaine alkyle, Jessop et coll. ont
ainsi développé le concept de « tensioactifs inversables » (« switchable surfactants »)12. De
manière astucieuse, ce concept a été exploité pour réaliser la polymérisation radicalaire en

184
émulsion du styrène. Ainsi, sous atmosphère de CO2, ces tensioactifs permettent la
stabilisation d’une émulsion du styrène dans l’eau. Après polymérisation du styrène (Figure
IV.2, A), en changeant l’atmosphère de CO2 par de l’argon, l’émulsion est déstabilisée et
conduit à la précipitation du polymère qui peut alors être récupéré facilement par filtration
(Figure IV.2, B).
Schéma IV.5 : Réversibilité de la réaction entre les amidines (1a et 1b), l’eau et le CO2
d’après Jessop et al.12.
Figure IV.2: A) Emulsion de polystyrène dans l’eau et stabilisée par 2a sous atmosphère de
CO2. B) Déstabilisation de l’émulsion de A en présence d’une atmosphère d’argon.
En utilisant le même procédé, l’addition de CO2 a par la suite été généralisée à
d’autres amidines, pour l’élaboration de solvants présentant une hydrophilie réversible13, 14,
et de liquides zwittérioniques de types carbonates ou carbamates d’amidinium15. Pour ces
derniers, la réversibilité de la réaction est effective à une température minimum de 50 °C,
formant des amidines présentant une fonction alcool ou amine (Schéma IV.6).
Schéma IV.6 : Liquides zwittérioniques formés lors de la réaction entre le CO2 et des
amidines présentant une fonction alcool ou amine15.
Récemment, la même équipe a montré qu’une telle réaction est également possible
avec des amines aliphatiques tertiaires16 ((2), Figure IV.3). L’addition de CO2 dans l’eau à ces
amines, induit une augmentation de la force ionique du milieu réactionnel. Avec ces
molécules, suivant le flux de gaz utilisé et en fonction de la température, la force ionique de

185
la solution aqueuse varie, ce qui autorise par exemple la séparation de certains solutés
comme le THF.
Figure IV.3: Réaction réversible entre des amines tertiaires, l’eau et le CO2, et structure des
différentes amines testées par le groupe de Jessop16.
Ce concept d’addition réversible de CO2 et de H2O (ou ROH) n’a pas, à notre
connaissance, été décrit avec les carbènes comme substrats à la place des amines ou des
amidines. C’est cette idée que nous développerons dans ce chapitre, avec pour objectif, non
pas l’obtention de « tensioactif inversables » pour la stabilisation/déstabilisation
d’émulsions, mais le masquage/démasquage de « NHCs inversables » pour une utilisation en
organocatalyse. Une telle réaction permettrait alors la formation de NHCs à partir
d’Im+HCO3-, au moyen de stimuli tels qu’un changement d’atmosphère, ou une élévation en
température du milieu.
IV.2) Des hydrogénocarbonates d’imidazolium aux carbènes N-
hétérocycliques: une réaction réversible
Comme déjà mentionné, les hydrogénocarbonates d’imidazolium (Im+HCO3-) ont déjà
été décrits dans la littérature. Ils ont notamment été utilisés comme sources de CO2 pour la
carboxylation des phénols (Schéma IV.7)17. Ils sont distribués par la société Sigma-Aldrich©,
qui les commercialise sous la forme de solutions aqueuses contenant du méthanol.
Nous avons cependant choisi de synthétiser ces composés, de manière à les obtenir
sous une forme pure.
Schéma IV.7 : Carboxylation des phénols catalysée par les hydrogénocarbonates
d’imidazolium.

186
IV.2.1) Synthèse et caractérisation des hydrogénocarbonates
d’imidazolium
IV.2.1.1) Méthodes de synthèse reportées dans la littérature
Pour mémoire, les Im+HCO3- n’ont jamais été employés comme précurseurs de NHCs.
Dans tous les protocoles reportés dans la littérature pour accéder aux Im+HCO3-, le passage
par un carboxylate d’imidazolium semble être une étape obligatoire. Ces derniers, comme
discuté dans les chapitres précédents, sont le plus souvent obtenus par carboxylation des
NHCs et leur hydratation ultérieure aboutit aux Im+HCO3- (Schéma IV.8). Cette étape
d’hydratation a pu être considérée comme indésirable par la plupart des auteurs, dans la
mesure où les adduits NHC-CO2 sont « dégradés », et le retour à la formation des NHCs n’a
jamais été envisagé.
Schéma IV.8 : Formation des carboxylates d’alkylimidazoliums par carboxylation des
imidazol-2-ylidènes, suivie d’une hydratation.
Alternativement à l’addition de CO2 sur les NHCs, une méthode de synthèse consiste
à quaternériser le cycle imidazole au moyen de diméthylcarbonate2, 18 (Schéma IV.5). Une
autre méthode est fondée sur la réaction de type Kolbe-Smith entre le bicarbonate de
sodium et un chlorure d’imidazolium, en présence d’une forte pression de CO2 (Schéma
IV.6)9.
Schéma IV.9 : Synthèse de carboxylates d’imidazolium par quaternérisation de 1-
alkylimidazoles par le DMC2, 18.
Schéma IV.10 : Réaction de Kolbe-Smith sur les chlorures d’imidazolium, permettant
l’obtention de carboxylates d’imidazolium9.

187
Comme on peut le remarquer, ces différentes réactions impliquent des conditions de
réactions relativement dures (températures et/ou pressions relativement élevées, solvant
peu commode). Pour que l’utilisation des Im+HCO3- présente un réel intérêt comme
précurseurs de NHCs, il nous a paru essentiel de mettre au point une méthode de synthèse
plus aisée.
IV.2.1.2) Synthèse d’hydrogénocarbonates d’imidazolium par changement d’anion
Comme cela a été décrit dans le chapitre précédent, le changement d’anion par
métathèse de sels d’imidazolium (Schéma IV.11) a largement été exploité. Une telle réaction
est généralement mise en œuvre dans des conditions douces. Ainsi, l’obtention de Im+HCO3-
par cette méthode nous est apparue évidente.
Schéma IV.11 : Changement d’anion des sels d’imidazolium par méthathèse entre un
halogénure d’imidazolium et un sel alcalin.
Le changement d’anion des halogénures d’imidazolium est en général effectué au
moyen d’un sel de cation alcalin. Nous avons donc tout d’abord tenté de synthétiser les
Im+HCO3-, par métathèse, entre le bromure de diisopropylimidazolium (iPrIm+Br-) et
l’hydrogénocarbonate de sodium (Schéma IV.12). Le méthanol a été choisi comme solvant
de cette réaction de manière à favoriser la réaction par la précipitation du sel NaBr formé.
Cependant, l’hydrogénocarbonate de sodium n’est que très faiblement soluble dans le
MeOH. Dans ces conditions, la réaction n’a pas permis l’obtention du produit ciblé sous une
forme pure.
Schéma IV.12 : Synthèse du iPrIm+HCO3- par métathèse.
Une autre méthode a donc été envisagée. L’hydrogénocarbonate de sodium (ou de
potassium) peut en réalité être obtenu par addition de CO2 sur la soude (ou la potasse). En
présence du sel d’imidazolium précurseur, dans le méthanol sec et avec un équivalent de

188
potasse, on obtient un précipité blanc de KBr au bout de quelques instants. Un premier
changement d’anion a alors été effectué permettant l’obtention du iPrIm+OH- (i, Schéma
IV.13). Le milieu a ensuite été placé sous atmosphère de CO2 (1bar) et le iPrIm+HCO3- a pu
être ainsi obtenu (ii, Schéma IV.13).
Schéma IV.13 : Synthèse du iPrIm+HCO3- par changement d’anion dans le méthanol au moyen
de potasse et de CO2.
Après évaporation du solvant, le composé obtenu a été caractérisé par spectroscopie
de RMN dans le DMSO-d6. Afin d’attribuer plus précisément les pics observés, l’adduit de
CO2 avec le NHC correspondant, iPrImCO2, a été synthétisé. Ce zwittérion a ensuite été
hydraté par exposition à l’air pendant une nuit, et caractérisé de la même manière par RMN 1H et 13C.
Pour synthétiser le carboxylate de diisopropylimidazolium (iPrImCO2), le NHC issu du
précurseur iPrIm+Br- a d’abord été préparé par déprotonation au moyen de KHMDS dans le
THF (i, schéma IV.14). La solution limpide de diisopropylimidazol-2-ylidène (iPrIm) ainsi
obtenue a ensuite été mise en présence de CO2 (1 bar) conduisant à la formation d’un
précipité blanc correspondant à l’adduit iPrImCO2 (ii, Schéma IV.14).
Schéma IV.14 : Synthèse du iPrImCO2 par déprotonation du iPrIm+Br- (i) permettant la
formation du iPrIm suivi d’une addition de CO2 sur ce dernier (ii).
Le spectre de RMN 1H dans le DMSO-d6 du composé iPrImCO2 (Figure IV.4) montre
trois pics distincts, à 1,4 ppm, 5,3 ppm et 7,8 ppm. Ceux-ci correspondent, respectivement, à
la résonance des protons CH3 et CH des groupements isopropyle latéraux, ainsi qu’aux CH de
la double liaison présente dans le cycle. L’absence de pic entre 9 et 10 ppm, correspondant
au proton acide du précurseur iPrIm+Br-, confirme l’obtention de l’adduit de CO2.
Le spectre de RMN 13C du iPrImCO2 révèle, quant à lui, la présence de deux pics à 142
et 153 ppm attribués respectivement aux carbones quaternaires de la partie imidazolium et
du groupement carboxylate. De plus, les groupements isopropyle sont représentés par les
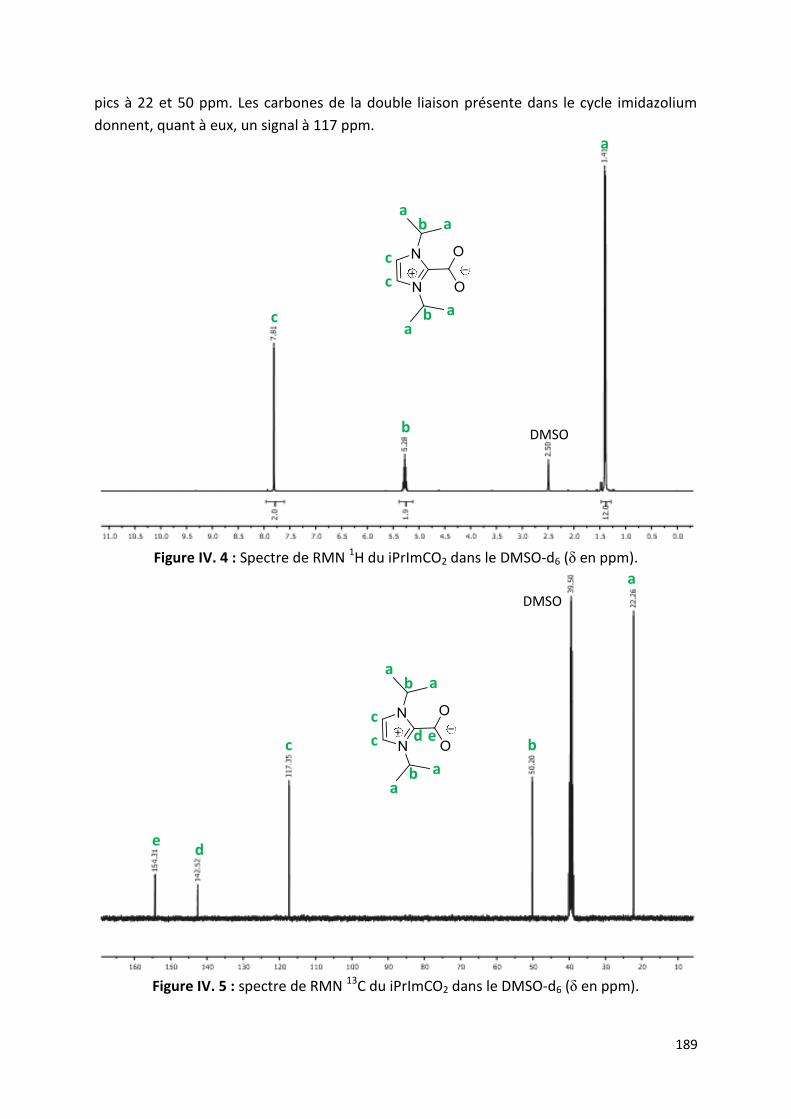
189
pics à 22 et 50 ppm. Les carbones de la double liaison présente dans le cycle imidazolium
donnent, quant à eux, un signal à 117 ppm.
Figure IV. 4 : Spectre de RMN 1H du iPrImCO2 dans le DMSO-d6 (δ en ppm).
Figure IV. 5 : spectre de RMN 13C du iPrImCO2 dans le DMSO-d6 (δ en ppm).
aa
aa
c
c
b
b
b
c
a
DMSO
aa
aa
c
c
b
b
d e
a
bc
de
DMSO

190
Par la suite, le composé iPrImCO2 a été placé à l’air pendant une nuit, et le produit
correspondant a été caractérisé par RMN 1 H et 13C dans le DMSO-d6. Dans ces conditions,
on s’attend à ce que l’humidité ambiante induise la formation du composé iPrIm+HCO3-.
On notera dans ce qui suit « iPrImCO2 hydraté» le produit résultant de l’hydratation
(volontaire ou subie) du carboxylate d’imidazolium (Schéma IV.14), tandis que le même
composé, mais obtenu par changement d’anion sera appelé « iPrIm+HCO3- » (Schéma IV.13).
Ainsi, le spectre de RMN 1H du iPrImCO2 hydraté (Figure IV. 6) révèle un pic à 8,8 ppm
correspondant au proton acide du cycle imidazolium, confirmant l’hydratation de l’adduit
NHC-CO2. Le pic à 4,4 ppm indique la présence d’une quantité relativement importante
d’eau dans le milieu, et ne permet pas d’observer le pic attribué au CH du groupe isopropyle.
Celui-ci donne en effet un signal à la même fréquence que les protons de l’eau.
On remarque également que le déplacement chimique du pic correspondant au CH
de la double liaison a varié de quelques dixièmes de ppm par rapport au spectre de l’adduit
non hydraté, en se déplaçant de 7,8 à 7,5 ppm. Cependant, les CH3 des deux groupes
isopropyle sont observés à 1,4 ppm, comme sur le spectre du iPrImCO2.
Figure IV.6 : Spectre de RMN 1H dans le DMSO-d6 du iPrImCO2 hydraté (δ en ppm).
aa
aa
c
cd
b
b
c
d
a
DMSO
H20
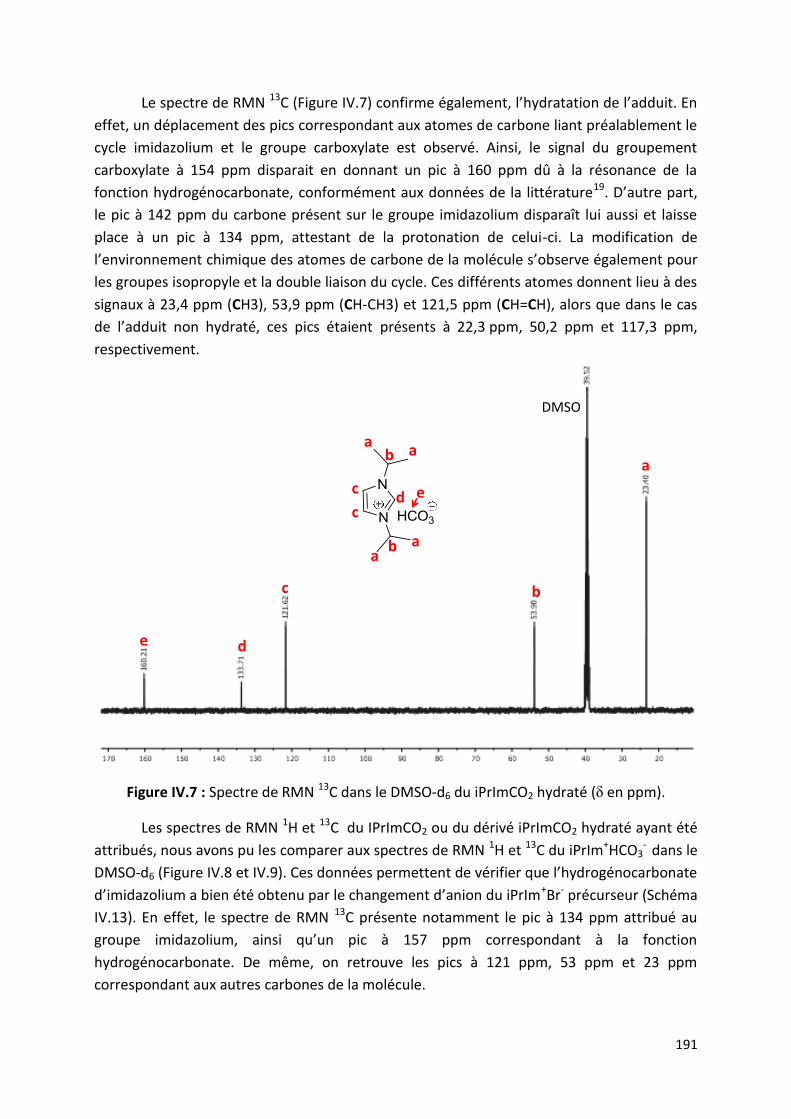
191
Le spectre de RMN 13C (Figure IV.7) confirme également, l’hydratation de l’adduit. En
effet, un déplacement des pics correspondant aux atomes de carbone liant préalablement le
cycle imidazolium et le groupe carboxylate est observé. Ainsi, le signal du groupement
carboxylate à 154 ppm disparait en donnant un pic à 160 ppm dû à la résonance de la
fonction hydrogénocarbonate, conformément aux données de la littérature19. D’autre part,
le pic à 142 ppm du carbone présent sur le groupe imidazolium disparaît lui aussi et laisse
place à un pic à 134 ppm, attestant de la protonation de celui-ci. La modification de
l’environnement chimique des atomes de carbone de la molécule s’observe également pour
les groupes isopropyle et la double liaison du cycle. Ces différents atomes donnent lieu à des
signaux à 23,4 ppm (CH3), 53,9 ppm (CH-CH3) et 121,5 ppm (CH=CH), alors que dans le cas
de l’adduit non hydraté, ces pics étaient présents à 22,3 ppm, 50,2 ppm et 117,3 ppm,
respectivement.
Figure IV.7 : Spectre de RMN 13C dans le DMSO-d6 du iPrImCO2 hydraté (δ en ppm).
Les spectres de RMN 1H et 13C du IPrImCO2 ou du dérivé iPrImCO2 hydraté ayant été
attribués, nous avons pu les comparer aux spectres de RMN 1H et 13C du iPrIm+HCO3- dans le
DMSO-d6 (Figure IV.8 et IV.9). Ces données permettent de vérifier que l’hydrogénocarbonate
d’imidazolium a bien été obtenu par le changement d’anion du iPrIm+Br- précurseur (Schéma
IV.13). En effet, le spectre de RMN 13C présente notamment le pic à 134 ppm attribué au
groupe imidazolium, ainsi qu’un pic à 157 ppm correspondant à la fonction
hydrogénocarbonate. De même, on retrouve les pics à 121 ppm, 53 ppm et 23 ppm
correspondant aux autres carbones de la molécule.
aa
aa
c
cd
b
b
e
a
bc
de
DMSO

192
Le spectre de RMN 1H n’est, quant à lui, pas tout à fait identique à celui observé pour
l’adduit hydraté ; les différents pics présentent tous un déplacement chimique légèrement
différent de ceux attendus. Ceci peut être expliqué par la quantité d’eau présente dans la
solution ayant servi à analyser le composé iPrImCO2 hydraté. Des interactions de type
liaisons hydrogène peuvent donc être envisagées entre l’eau et l’hydrogénocarbonate
d’imidazolium, ce qui induirait un environnement chimique différent pour les protons
observés. Le spectre de iPrIm+HCO3- ayant été réalisé dans des conditions anhydres, de telles
interactions ne peuvent avoir lieu. De même, le pic correspondant aux protons CH des
groupes isopropyle apparait à 4,7 ppm et n’est pas masqué ici par le pic de l’eau,
contrairement au spectre de l’adduit hydraté.
Figure IV.8 : Spectre de RMN 1H de iPrIm+HCO3- dans le DMSO-d6 (δ en ppm).
a
ad
b
b
b
aa
aa
c
cd
b
bc
c
b
aa
aa
c
c
b
b
c
c

193
Figure IV.9 : Spectre de RMN 13C de iPrIm+HCO3
- dans le DMSO-d6 (δ en ppm).
Les deux spectres de RMN du iPrIm+HCO3- révèlent également une seconde
population de pics de plus faible intensité correspondant exactement aux déplacements
chimiques des pics de iPrImCO2. On peut donc supposer que, dans des conditions anhydres,
une partie des molécules de iPrIm+HCO3- peut se déshydrater (Schéma IV.15). Ceci conduit
alors à la formation d’une faible quantité de iPrImCO2.
D’après l’intensité des deux populations de pics, environ 10% molaire de iPrImCO2
sont présents dans notre solution. On peut cependant supposer qu’en diluant à nouveau la
solution avec du DMSO anhydre, la quantité relative de iPrImCO2 augmentera. L’ajout de
tamis moléculaire dans la solution, servant à piéger l’eau formée, devrait permettre de
favoriser la formation de iPrImCO2. L’observation de ce composé dans la solution anhydre
est donc un premier signe de la formation potentielle de NHCs à partir des
hydrogénocarbonates d’imidazolium. D’autres expériences ont cependant été effectuées
pour mettre mieux en évidence la formation de NHCs à partir de ces composés.
Schéma IV.15 : Déshydratation du iPrIm+HCO3- en conditions anhydres, pouvant
substantiellement conduire à la formation de NHCs.
aa
aa
c
cd
b
b
e
aa
aa
c
c
b
b
d e
a
bc
d
e
ea
bcde
DMSO

194
IV.2.2) Génération in-situ de NHCs à partir des hydrogénocarbonates
d’imidazolium
La présence de NHCs au sein d’une solution peu être mise en évidence de plusieurs
manières. Ainsi, la formation d’adduits entre les NHCs et le disulfure de carbone (CS2) à
notamment été utilisée par les groupes de Hedrick et de Delaude pour diverses réactions6, 20,
21. Une telle réaction est en effet quantitative et quasi-instantanée, conduisant à la
formation d’adduits NHC-CS2 stables jusqu’à des températures de 200 °C5.
Par ailleurs, les NHCs sont aussi connus pour être de très bons ligands de divers
complexes organométalliques22, 23. La formation de complexes de NHCs stables permet donc
de mettre en évidence, de manière indirecte, la présence de NHCs en solution.
Enfin, la condensation de la benzoïne, réaction qui ne peut être catalysée que par les
NHCs ou les ions cyanates (voir chapitre II), peut également servir de réaction modèle et
argumenter en faveur de la présence de NHCs en solution.
Ces trois méthodes ont donc été mises en œuvre au cours de cette étude, afin
d’attester de la formation de NHCs à partir des hydrogénocarbonates d’imidazolium, en
l’absence totale de base.
IV.2.2.1) Formation d’adduits NHC-CS2 à partir du iPrIm+HCO3-
En présence de CS2, les NHCs forment des adduits zwitérioniques de couleur rouge
sang (Schéma IV.16). Lorsque ces adduits ne sont pas solubles dans le milieu, ils forment
alors des cristaux colorés qui peuvent être filtrés et analysés par diverses techniques tels que
la diffraction des rayons-X et la spectroscopie de RMN5, 20.
Schéma IV.16 : Réaction entre les NHCs et le CS2 formant des adduits NHC-CS2.
Afin de mettre en évidence la formation de NHCs à partir du iPrIm+HCO3-, une
solution de ce sel dans le THF anhydre a été préparée. Un excès de CS2 a ensuite été ajouté à
cette solution, avant qu’elle ne soit placée à une température de 80 °C pendant 24 h. Une
température de 80 °C a été choisie afin de permettre la libération du CO2 lors de la
formation de l’adduit iPrImCO2 observé précédemment. Ainsi, après une heure de réaction,
la solution prend une teinte rosée, indiquant la formation probable de l’adduit iPrImCS2.
Après extraction et lavage avec de l’acétone, le spectre de RMN 1H dans le DMSO-d6,
du composé rouge extrait, présente trois pics à 1,4 ppm, 4,6 ppm et 7,8 ppm et aucun pics à
9,6 ppm pouvant être attribué à iPrIm+HCO3- (Figure IV.10). Le spectre de RMN 13C montre,
quant à lui, un pic caractéristique à 226,2 ppm, qui est attribué au carbone du groupe
dithiocarboxylate (Figure IV.11). De même, le carbone en position 2 du groupe imidazolium

195
ne donne aucun signal aux environs de 135 ppm comme dans les sels bimoléculaires
d’imidazolium (iPrIm+Br- et iPrIm+HCO3-), mais présente un déplacement chimique de 147,6
ppm, caractéristique de ces zwitterions20.
De plus, la masse d’adduit NHC-CS2 récupéré a permis de déterminer un rendement
de réaction de 10%, en accord avec la quantité de iPrImCO2 déterminée à partir du spectre
de RMN 1H du iPrIm+HCO3-. Là aussi, on peut supposer que la présence de tamis moléculaire
dans le milieu réactionnel pourrait permettre d’accroître la formation de iPrImCO2 et donc
de iPrImCS2.
Figure IV.10 : Spectre de RMN 1H (δ en ppm) dans le DMSO-d6 de iPrImCS2.
aa
aa
a
b
b
c
c
b
c
DMSO

196
Figure IV.11 : Spectre de RMN 13C (δ en ppm) dans le DMSO-d6 de iPrImCS2.
Toutefois, les adduits NHC-CS2 peuvent aussi être formés directement par réaction
entre les adduits NHC-CO2 et CS25. Aussi, la formation de NHC-CS2 ne constitue pas une
preuve formelle de la présence de NHCs libres dans le milieu réactionnel. D’autres
investigations ont donc été entreprises pour apporter de nouveaux arguments quant à la
formation de NHCs à partir du iPrIm+HCO3-.
IV.2.2.2) Synthèse du complexe Au(iPrIm)Cl à partir de Au(SMe2)Cl et de iPrIm+HCO3-
Les NHCs sont particulièrement efficaces comme ligands pour se coordiner à des
métaux de transition et former toute une variété de complexes organométalliques grâce à
leurs propriétés de ζ-donation22. Ces complexes sont généralement obtenus par réaction de
substitution d’un ligand par un NHC « nu », ou bien par transmétalation à partir d’un
complexe de type Ag(I)-NHC24. Cependant, il a également été rapporté que les adduits NHC-
CO2 permettent la formation de complexes organométalliques, dans des conditions plus
faciles à mettre en œuvre, notamment à température ambiante2, 3.
Nous avons donc évalué l’aptitude des hydrogénocarbonates d’imidazolium, et
notamment du iPrIm+HCO3- décrit précédemment, à générer des complexes
organométalliques. Pour être innovant, un complexe n’ayant pas encore été décrit comme
possible à obtenir à partir d’adduits NHC-CO2, a été ciblé : Il s’agit du complexe Au(iPrIm)Cl.
aab
c
c
ba
a
d e
a
bc
de
DMSO

197
Ainsi avons-nous préparé ce composé à partir d’un mélange stœchiométrique de AuCl(SMe2)
et de iPrIm+HCO3- en présence de tamis moléculaire (Schéma IV.17). Pour cela, les deux
réactifs solides et le tamis moléculaire ont été suspendus dans du THF à température
ambiante et sous atmosphère inerte d’azote, la réaction ayant été suivie par RMN dans le
CDCl3.
Schéma IV.17 : Synthèse du complexe Au(iPrIm)Cl à partir de Au(SMe2)Cl et d’iPrIm+HCO3-.
Un prélèvement du milieu réactionnel au bout de 24 h a montré que la réaction
atteint un rendement de 57 %. Le spectre de RMN 1H dans le CDCl3 d’un tel prélèvement
révèle, en effet, la présence des pics correspondant au complexe Au(iPrIm)Cl à 7,0 ppm , 5,0
ppm et 1,4 ppm (Figure IV.12). De même, on retrouve les pics correspondants au composé
iPrIm+HCO3-, avec notamment le pic à 9,3 ppm attribué au proton acide du cycle
imidazolium. Le rendement de cette réaction a été déterminé à partir des valeurs des
intensité des pics : r. = IC/(IC+IC).
Figure IV.12 : Spectre de RMN 1H dans le CDCl3 (δ en ppm) d’un prélèvement de la réaction
entre le Au(SMe2)Cl et le iPrIm+HCO3- effectué au bout de 24 h.
bd
c
c
Au(SMe2)Cl
b
a
a
ab
c
cd
baa
aa
a
a
a
c c
b b
CD
Cl 3
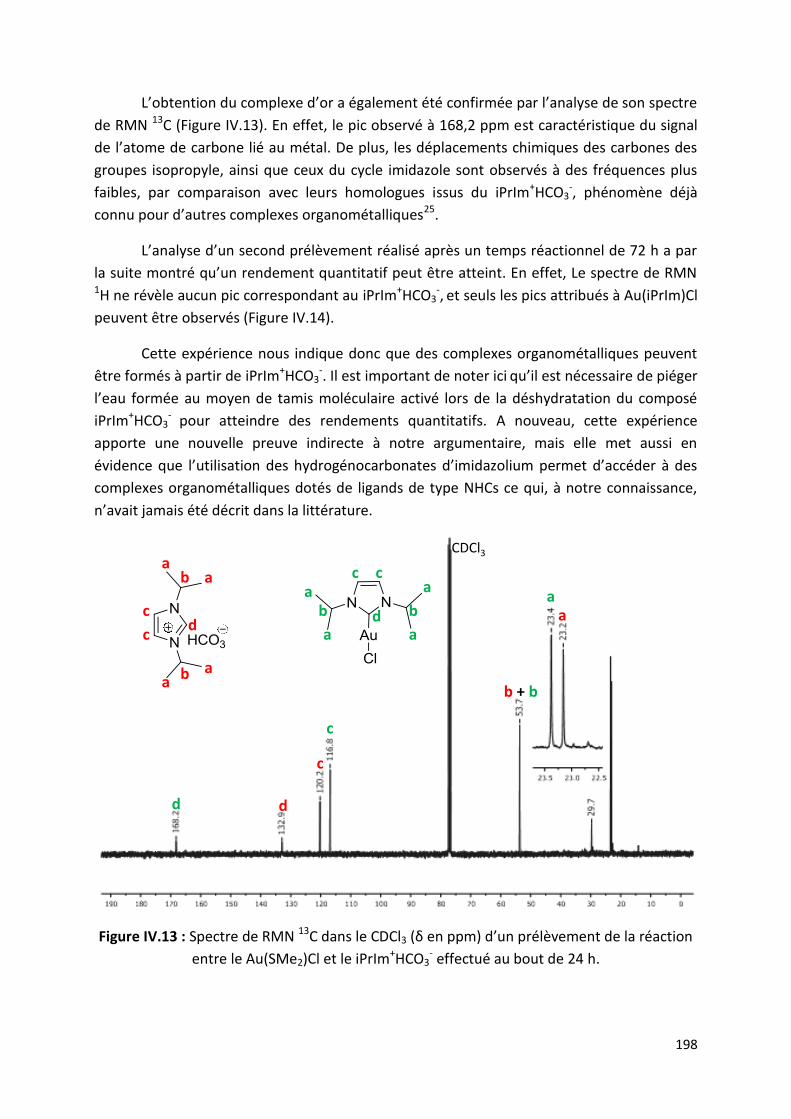
198
L’obtention du complexe d’or a également été confirmée par l’analyse de son spectre
de RMN 13C (Figure IV.13). En effet, le pic observé à 168,2 ppm est caractéristique du signal
de l’atome de carbone lié au métal. De plus, les déplacements chimiques des carbones des
groupes isopropyle, ainsi que ceux du cycle imidazole sont observés à des fréquences plus
faibles, par comparaison avec leurs homologues issus du iPrIm+HCO3-, phénomène déjà
connu pour d’autres complexes organométalliques25.
L’analyse d’un second prélèvement réalisé après un temps réactionnel de 72 h a par
la suite montré qu’un rendement quantitatif peut être atteint. En effet, Le spectre de RMN 1H ne révèle aucun pic correspondant au iPrIm+HCO3
-, et seuls les pics attribués à Au(iPrIm)Cl
peuvent être observés (Figure IV.14).
Cette expérience nous indique donc que des complexes organométalliques peuvent
être formés à partir de iPrIm+HCO3-. Il est important de noter ici qu’il est nécessaire de piéger
l’eau formée au moyen de tamis moléculaire activé lors de la déshydratation du composé
iPrIm+HCO3- pour atteindre des rendements quantitatifs. A nouveau, cette expérience
apporte une nouvelle preuve indirecte à notre argumentaire, mais elle met aussi en
évidence que l’utilisation des hydrogénocarbonates d’imidazolium permet d’accéder à des
complexes organométalliques dotés de ligands de type NHCs ce qui, à notre connaissance,
n’avait jamais été décrit dans la littérature.
Figure IV.13 : Spectre de RMN 13C dans le CDCl3 (δ en ppm) d’un prélèvement de la réaction
entre le Au(SMe2)Cl et le iPrIm+HCO3- effectué au bout de 24 h.
ab
c
cd
baa
a
a
a
a
a
c c
b bd
d
c
a
b + b
d
c
a
CDCl3
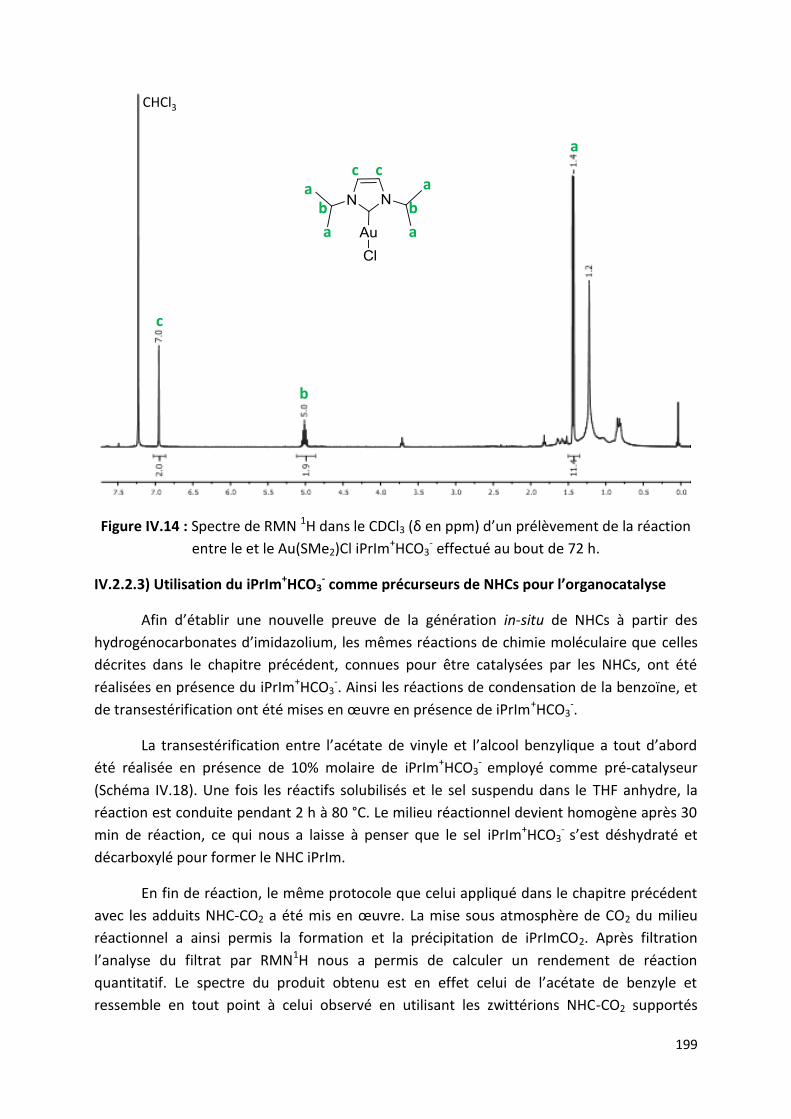
199
Figure IV.14 : Spectre de RMN 1H dans le CDCl3 (δ en ppm) d’un prélèvement de la réaction
entre le et le Au(SMe2)Cl iPrIm+HCO3- effectué au bout de 72 h.
IV.2.2.3) Utilisation du iPrIm+HCO3- comme précurseurs de NHCs pour l’organocatalyse
Afin d’établir une nouvelle preuve de la génération in-situ de NHCs à partir des
hydrogénocarbonates d’imidazolium, les mêmes réactions de chimie moléculaire que celles
décrites dans le chapitre précédent, connues pour être catalysées par les NHCs, ont été
réalisées en présence du iPrIm+HCO3-. Ainsi les réactions de condensation de la benzoïne, et
de transestérification ont été mises en œuvre en présence de iPrIm+HCO3-.
La transestérification entre l’acétate de vinyle et l’alcool benzylique a tout d’abord
été réalisée en présence de 10% molaire de iPrIm+HCO3- employé comme pré-catalyseur
(Schéma IV.18). Une fois les réactifs solubilisés et le sel suspendu dans le THF anhydre, la
réaction est conduite pendant 2 h à 80 °C. Le milieu réactionnel devient homogène après 30
min de réaction, ce qui nous a laisse à penser que le sel iPrIm+HCO3- s’est déshydraté et
décarboxylé pour former le NHC iPrIm.
En fin de réaction, le même protocole que celui appliqué dans le chapitre précédent
avec les adduits NHC-CO2 a été mis en œuvre. La mise sous atmosphère de CO2 du milieu
réactionnel a ainsi permis la formation et la précipitation de iPrImCO2. Après filtration
l’analyse du filtrat par RMN1H nous a permis de calculer un rendement de réaction
quantitatif. Le spectre du produit obtenu est en effet celui de l’acétate de benzyle et
ressemble en tout point à celui observé en utilisant les zwittérions NHC-CO2 supportés
a
a
a
a
c c
b b
b
a
c
CHCl3

200
comme catalyseurs de transestérification (Figure III.26). En d’autres termes, on peut former
l’acétate de benzyle à partir de l’alcool benzylique et de l’acétate de vinyle, en catalysant
simplement la réaction par un hydrogénocarbonate d’imidazolium comme générateur de
NHC, facile à synthétiser.
Schéma IV.18 : Organocatalyse de transestérification en présence de iPrIm+HCO3-.
Ces mêmes conditions expérimentales ont alors été appliquées à une solution de
benzaldéhyde en vue de sa réaction de condensation de la benzoïne (Schéma IV.19).
Cependant, ces conditions n’ont pas permis d’obtenir de bons rendements (r < 20%), ce que
nous avons attribué à un faible pourcentage de NHC formé, malgré un temps de réaction de
24 h.
D’après la littérature, l’obtention de rendements élevés en condensation de la
benzoïne nécessite des quantités catalytiques de NHCs plus importantes (10-30%)26 que
dans le cas des réactions de transestérification ; dans ce dernier cas, des taux de catalyseurs
de 0,5% sont suffisants pour atteindre de bons rendements27.
Schéma IV.19 : Condensation de la benzoïne en présence iPrIm+HCO3-.
Pour pallier cette difficulté, la réaction de condensation de benzoïne a été effectuée
en présence de tamis moléculaire activé (Schémas IV.20). La présence de cet additif a alors
conduit à des rendements quantitatifs et ce, dans des conditions réactionnelles plus douces
(T = 60 °C) et pour une plus faible quantité de catalyseur (2%). L’utilisation de tamis
moléculaire s’avère donc très pertinente. On peut bien sur attribuer cet effet au fait que le
tamis moléculaire piège les molécules d’eau issues du précatalyseur iPrIm+HCO3-.
Schéma IV.20 : Condensation de la benzoïne en présence iPrIm+HCO3- et de tamis
moléculaire.

201
Ce simple résultat obtenu en condensation de la benzoïne nous éclaire donc de
manière décisive quant au mécanisme de formation de NHCs à partir du iPrIm+HCO3-. En
effet, lors de la synthèse du iPrIm+HCO3-, nous avions remarqué qu’en milieu anhydre, une
partie de la molécule se déshydrate pour former l’adduit iPrImCO2 (voir partie IV.B.1.2).
Comme déjà discuté à plusieurs reprises, ces derniers sont connus pour subir une réaction
de décarboxylation en solution aux environs de 80 °C, autorisant alors la génération de NHCs
libres. Un mécanisme présenté Schéma IV.21 peut donc être proposé.
Schéma IV.21: Mécanisme supposé de la formation de NHCs à partir de iPrIm+HCO3-.
Nous avons ainsi montré que les sels d’hydrogénocarbonates d’imidazolium peuvent
être obtenus directement à partir de sels commerciaux (bromures ou chlorures
d’imidazolium) par un simple changement d’anion, sans qu’il soit nécessaire de passer par la
synthèse de NHCs. La caractérisation par RMN 1H et 13C des composés obtenus atteste de la
structure attendue, et montre la formation d’adduits NHC-CO2 lorsque les sels iPrIm+HCO3-
sont placés en milieu anhydre.
Ces sels peuvent également servir de précatalyseurs pour deux réactions standards
de la chimie moléculaire : la condensation de la benzoïne et les transestérifications,
prouvant ainsi que le véritable catalyseur est une entité carbénique, en l’occurrence un NHC.
La formation de ces espèces carbéniques à partir des hydrogénocarbonates d’imidazolium
est étayée par la possibilité de préparer, d’une part, des complexes organométalliques d’or
(Au(iPrIm)Cl) par substitution de ligand, et d’autre part, des adduits dithiocarboxylates
d’imidazolium (NHC-CS2) rouges sang en présence de CS2.
Le mécanisme proposé Schéma IV.17 illustre que seuls des composés non toxiques et
sans danger pour l’environnement (H2O et CO2) sont éliminés lors de la formation de NHCs à
partir des hydrogénocarbonates d’imidazolium.
D’après les différentes observations, on propose ici une réaction se déroulant en
deux étapes : une réaction de déshydratation, suivie d’une réaction de décarboxylation. On
peut cependant se demander si la réaction s’effectue bien dans cet ordre ou bien si les
composés sont éliminés en même temps. Dans le but d’éclaircir ce point, il pourrait être
intéressant de modéliser une telle réaction en effectuant des calculs théoriques rendant
compte des énergies mises en jeu lors des différentes étapes.

202
De même, ces modèles pourraient nous éclairer quant à la réversibilité de la
formation des NHCs à partir des hydrogénocarbonates d’imidazolium, observée lors de
l’ajout de CO2 en fin des réactions de transestérification et de condensation de la benzoïne.
Les hydrogénocarbonates d’imidazolium pouvant être facilement préparés, on peut
dès lors envisager un emploi à plus grande échelle de ces précatalyseurs dans l’optique
d’une manipulation encore plus aisée, mais aussi d’un recyclage des NHCs. Pour ce faire,
nous avons envisagé de supporter les hydrogénocarbonates d’imidazolium sur des chaînes
polymères, comme cela va être décrit dans la suite de ce chapitre.
IV.3) Synthèse et évaluation en organocatalyse
d’hydrogénocarbonates de poly(vinylimidazolium)
Même si le iPrIm+HCO3- permet l’obtention de NHCs de manière plus aisée, son
aspect moléculaire et son utilisation en quantité catalytique ne permettent pas un recyclage
par simple filtration. En effet, ce sel, insoluble dans le THF, a pu être reformé par addition de
CO2 en fin des réactions organocatalysées. Cependant, la faible quantité de produit présent
au fond du fritté, lors de la filtration, n’a permis de récupérer qu’une faible proportion du
précatalyseur de départ. Ainsi, comme dans le cas des NHCs et de leur adduits avec le CO2,
tels que décrits dans le chapitre III, nous avons choisi de supporter les hydrogénocarbonates
d’imidazolium sur un tronc polymère dérivé de polymère liquide ionique.
IV.3.1) Synthèse d’hydrogénocarbonates de poly(vinylimidazolium)
Dans le chapitre précédent, il a été mentionné que les carboxylates de
poly(vinylimidazolium)s, poly(NHC-CO2)s, peuvent progressivement s’hydrater pour former
des hydrogénocarbonates de poly(vinylimidazolium), Poly(VIm+HCO3-).
Une telle réaction d’hydratation constitue donc une première méthode permettant
d’obtenir les Im+HCO3- supportés (Voie A, Schéma IV.22), à l’instar de ce que nous avons
réalisé pour les « versions moléculaires ». Toutefois, nous avons également montré que ces
mêmes sels peuvent être obtenus par simple changement d’anion, à partir d’un halogénure
d’imidazolium correspondant, avec de la potasse suivie de l’addition de CO2. Ainsi, cette
réaction nous a permis de synthétiser divers Poly(VIm+HCO3-) via deux méthodes
différentes :
changement d’anion d’un halogénure de 1-vinyl-3-alkylimidazolium (VIm+X-) suivi de
la polymérisation de ces monomères par voie radicalaire (Voie B, Schéma IV.22).

203
polymérisation d’un monomère de type VIm+X- suivie du changement d’anion sur le
polymère liquide ionique correspondant. (Voie C, Schéma IV.22)
La voie A a en réalité déjà été décrite dans le chapitre précédent (Cf. III.F.1), elle ne
sera donc pas développée ici. Ainsi, seule la voie B et la voie C seront discutées, et plus
particulièrement la voie B. En effet, la voie C ne représente qu’une extension du changement
d’anion des VIm+Br- décrits précédemment, appliqué ici aux PVIm+Br-.
Schéma IV.22 : Différentes méthodes d’accès aux Poly(VIm+HCO3-).
IV.3.1.1) Synthèse du ViPrIm+HCO3-
Le changement d’anion effectué à partir du monomère liquide ionique de type
chlorure de N-vinyl-3-alkylimidazolium (voie B) n’a pas posé de difficultés particulières. Le
même protocole que pour l’obtention de la version « iPrIm+HCO3-» a été utilisé et conduit
dans ce cas à l’hydrogénocarbonate de 1-vinyl-3-isopropylimidazolium (ViPrIm+HCO3-).
Le spectre de RMN 1H dans le MeOD du ViPrIm+HCO3- révèle notamment les pics du
groupe vinyle à 5,5 ppm, 6,0 ppm et 7,4 ppm, sous la forme caractéristique de doublets
dédoublés (Figure IV.15). Cette analyse ayant été réalisée dans le MeOD, il n’est pas possible
d’observer le pic du proton acide du groupe imidazolium à environ 9 ppm. Cependant, le
changement d’anion a pu être confirmé par l’analyse du spectre de RMN 13C dans lequel les
pics correspondant au contre-anion hydrogénocarbonate et au carbone en position 2 du
cycle imidazole apparaissent, respectivement, à 135,2 ppm et 161,3 ppm (Figure IV.16).

204
Figure IV.15 : Spectre de RMN 1H (δ en ppm) dans le MeOD du ViPrIm+HCO3-.
Figure IV.16 : Spectre de RMN 13C (δ en ppm) dans le MeOD du ViPrIm+HCO3-.
ab
c
d
e
fg
g
bac
d ef
g
MeOH
ab
c
d
ef
f
g h
gh
b d c
a
e
f
MeOH

205
IV.3.1.2) Polymérisation radicalaire du ViPrIm+HCO3- (Voie B)
La polymérisation radicalaire (conventionelle) du ViPrIm+HCO3- a été effectuée en
utilisant différentes conditions (Tableau IV.1). Contrairement aux monomères porteurs de
l’anion halogénure (voir chapitre III), l’AIBN utilisé comme source de radicaux à 80 °C n’a pas
permis d’amorcer la polymérisation de ce monomère à contre-anion hydrogénocarbonate,
que ce soit dans le DMSO ou le MeOH comme solvants.
D’autres amorceurs permettant la formation de radicaux par décomposition
thermique (Figure IV.17) ont alors été testés, mais là encore, sans succès. Nous avons alors
choisi de polymériser le ViPrIm+CO3- à plus basse température, à l’aide d’autres amorceurs,
notamment le V-70 qui se décompose avec un temps de demi-vie de 10 h à 40 °C. Dans ces
conditions, on obtient finalement un polymère mais avec un rendement assez faible (40%)
après 72 h de réaction.
L’utilisation d’un système d’amorçage de type redox à température ambiante a
finalement permis l’obtention du PViPrIm+HCO3- avec de bien meilleurs rendements, en
particulier avec le système persulfate de potassium (KPS) en présence de
diméthylaminopropionitrile (DMAPN) pour un temps de réaction de 18 h (Schéma IV.23).
Tableau IV.1 : Conditions réactionnelles et résultats des polymérisations radicalaires du
ViPrIm+HCO3-.
entrée Amorceur Température
de
polymérisation
Solvant Temps
de
réaction
%
conv.a
Mn
(g/mol)b
Mw
(g/mol)b
Db
1 AIBN 80°C DMSO 144 h 0 - - -
2 AIBN 80°C MeOH 144 h 0 - - -
3 V-501 80°C DMSO 144 h 0 - - -
4 V-501 80°C MeOH 144 h 0 - - -
5 V-70 40°C DMSO 72 h 40 - - -
6 KPS +
DMAPN
T. ambiante DMSO 18 h 95 11400 30800 2,7
7 KPS +
DMAPN
T. ambiante MeOH 18 h 70 12400 45900 3,7
a. Déterminé à partir des intégrations des spectres de RMN
1H dans le MeOD.
b Valeurs obtenues par SEC dans
l’eau au moyen d’une calibration avec des standards de P2VP.

206
Figure IV.17 : Structures et températures de décomposition pour un temps de demi-vie de
10 h, des amorceurs azoïques utilisés pour la polymérisation du ViPrIm+HCO3-.
Schéma IV.23 : Réaction d’oxydoréduction entre le KPS et la DMAPN, conduisant à la
formation de radicaux hydroxyles à température ambiante.
Comme on peut le remarquer dans le Tableau IV.1, la température a donc une grande
importance sur l’efficacité de la polymérisation radicalaire du ViPrIm+HCO3-. En particulier,
on notera qu’il semble impossible de former le PViPrIm+HCO3- pour des températures
proches de la température de décarboxylation du iPrImCO2. Pour expliquer ce phénomène,
on peut proposer que les radicaux formés par décomposition thermique de l’amorceur
peuvent être piégés par des molécules de NHCs formées par activation thermique du
ViPrIm+HCO3-.
Il a en effet été rapporté par Apeloig et coll.28 et par West et al.29, 30 que les radicaux
peuvent s’additionner sur les NHCs pour former des radicaux centrés sur le cycle imidazole
(Schéma IV.24). On peut donc imaginer le même type de réaction dans notre milieu
réactionnel, les espèces radicalaires ainsi formées étant alors incapables d’amorcer la
polymérisation (Schéma IV.25).

207
Schéma IV. 24 : Addition d’un radical sur un NHC telle que rapportée par Apeloig et coll.28
Schéma IV.25 : Suggestion de mécanisme expliquant l’impossibilité de polymériser le
ViPrIm+HCO3- par décomposition thermique d’amorceurs azoïques.
La caractérisation des polymères obtenus par amorçage d’oxydo-réduction a été
réalisée par spectroscopie de RMN et par SEC dans l’eau.
Les spectres de RMN 1H et 13C (Figure IV.18 et IV.19) indiquent que la polymérisation
a bien eu lieu, car les différents signaux ne s’observent plus sous la forme de pics bien
définis, mais sous la forme de massifs larges. L’analyse spectroscopique ayant été réalisée
sur un prélèvement du milieu réactionnel de polymérisation, on peut également observer
des singulets dépassant de ces massifs et correspondant au monomère résiduel. Le spectre
de RMN 13C révèle également un pic à 160 ppm, de faible intensité, correspondant au
contre-anion hydrogénocarbonate, tout comme pour le spectre de RMN 13C du ViPrIm+HCO3-
(Cf. Figure IV.16).
Figure IV.18 : Spectre de RMN 1H (δ en ppm) dans le MeOD d’un prélèvement de la
polymérisation radicalaire correspondant à l’entrée 6 du tableau IV.1.
ab
c
d
e
fg
g
h
h d ec b a
f
g
ab
c
d
ef
f
g
e+bg
c + d
f
a

208
Figure IV.19 : Spectre de RMN 13C (δ en ppm) dans le MeOD d’un prélèvement de la
polymérisation radicalaire correspondant à l’entrée 6 du tableau IV.1.
L’analyse des polymères par SEC dans l’eau (Figure IV.20) au moyen de
colonnes spécifiques aux polymères cationiques a, quant à elle, permit d’évaluer les masses
molaires apparentes de ces polymères, ainsi que la dispersité des masses molaires (entrées 6
et 7 du Tableau IV.1). Comme discuté dans le chapitre III, l’analyse par SEC de tels polymères
cationiques est relativement difficile à mettre en place, en raison des interactions se
produisant entre le polymère, le solvant et les colonnes.
Les PViPrIm+ HCO3- ainsi obtenus ont par la suite été évalués en tant que précurseurs
de NHCs supportés pour la catalyse de transestérification et de condensation de la benzoïne.
Cependant, afin de comparer l’activité catalytique de tels supports avec celle des poly(NHC)s
et de leur adduits avec le CO2 (voir chapitre III), un autre dérivé à contre-anion
hydrogénocarbonate et porteur d’un substituant phényléthyle, en l’occurrence le
PVEtPhIm+HCO3- a également été synthétisé.
a
b
c
d
ef
f
g
ab
c
d
ef
f
g hh
ab d c
e
f + f
gh + h
abe
c + d
g
MeOH

209
Figure IV.20 : Chromatogrammes de SEC dans l’eau des PViPrIm+HCO3
- correspondants aux
entrées 6 et 7 du tableau IV.1.
IV.3.1.3) Synthèse du PVEtPhIm+HCO3- au moyen d’un changement d’anion du
PVEtPhIm+Br- (Voie C)
Nous avons montré précédemment que les hydrogénocarbonates d’imidazolium
moléculaires peuvent être facilement obtenus par simple changement d’anion à partir
d’halogénures d’imidazolium. Par extension, une telle réaction doit pouvoir être appliquée
aux PVIm+X- et ainsi permettre la synthèse de PVIM+HCO3- (voie C, Schéma IV.22).
Le PVEtPhIm+HCO3- a en effet pu être préparé en changeant l’anion bromure du
PVEtPhIm+Br- par l’anion hydrogénocarbonate, comme décrit ci-après (Schéma IV.26).
Le PVEtPhIm+Br- a d’abord été synthétisé par polymérisation radicalaire
conventionnelle amorcée par décomposition thermique de l’AIBN, comme décrit dans le
chapitre précédent. Le changement d’anion sur le polymère a ensuite été réalisé dans le
MeOH, en présence de potasse, et après filtration, en plaçant le polymère correspondant
sous atmosphère de CO2 pendant 30 minutes.
Schéma IV.26 : Synthèse du PVEtPhIm+HCO3
- par changement d’anion du PVEtPhIm+Br-.
2000 2500
Temps d'élution (s)
PViPrIm+HCO
3
- obtenu par amorçage de type rédox dans le MeOH
PViPrIm+HCO
3
- obtenu par amorçage de type rédox dans le DMSO

210
De même que pour les autres hydrogénocarbonates d’imidazolium, le spectre de
RMN 13C du PVEtPhIm+HCO3- révèle un pic à 161 ppm, correspondant au carbone de l’anion
hydrogénocarbonate (Figure IV.21). Deux pics à 129 et 131 ppm, correspondant aux
carbones aromatiques du groupement phényléthyle, sont également observés en
complément des autres pics du groupe imidazolium et de la chaine vinylique. Le groupement
aromatique se retrouve aussi dans le spectre de RMN 1H du PVEtPhIm+HCO3- aux environs de
7,5 ppm (Figure IV.22). Même si les rapports d’intégrations correspondent à la structure du
polymère, il n’est cependant pas possible d’observer le proton acide du groupe imidazolium,
en raison de l’échange de ce proton avec le solvant d’analyse (MeOD).
Figure IV.21 : Spectre de RMN 13C (δ en ppm) dans le DMSO-d6 du PVEtPhIm+HCO3- .
ab + dc
a b
cc
g
f
h
de
e
e e
e
e
e
fg
gh
MeOH

211
Figure IV.22 : Spectre de RMN 1H (δ en ppm) dans le DMSO-d6 du PVEtPhIm+HCO3- .
En conclusion, le PViPrIm+HCO3- et le PVEtPhIm+HCO3
- peuvent être préparés par trois
méthodes différentes. La voie A reste la plus difficile à mettre en oeuvre car elle nécessite
quatre étapes : polymérisation d’un halogénure d’alkyle, changement d’anion du polymère,
déprotonation suivie d’une carboxylation et enfin réaction d’hydratation. De plus, la réaction
de déprotonation doit se dérouler dans des conditions anhydres et nécessite donc la
purification des solvants et le séchage du polymère. Cependant, c’est la seule voie
permettant d’obtenir des PVim+HCO3- lorsque ceux-ci ne sont pas solubles dans le méthanol
(par exemple avec des substituants très hydrophobes). Il convient donc à l’utilisateur de
déterminer la méthode la plus adaptée à l’obtention des hydrogénocarbonates
d’imidazolium supportés désirés.
Les méthodes B et C sont, quant à elles, plus aisées à mettre en œuvre. Elles ne
nécessitent que des composés peu coûteux, potasse et CO2, mais doivent être réalisées dans
un solvant permettant à la fois la solubilisation des réactifs (monomère ou polymère et
potasse) et la précipitation du sel KBr. Dans le cas de monomères et de polymères liquides
ioniques non solubles dans le méthanol, on peut cependant envisager de réaliser le
changement d’anion au moyen de sels solubles dans des solvants organiques (THF, CH2Cl2…)
tels que les hydroxydes d’ammoniums, tout en prenant garde à ce que le sel d’halogénure
formé par la suite précipite dans le milieu réactionnel. Il convient donc à l’expérimentateur
de définir le bon triplet MIL ou PIL/solvant/sel d’échange.
a b
cc
fde
e
e e
e
abd
e
f
c
MeOH

212
IV.3.2) Evaluation en organocatalyse des PVIm+HCO3-.
Dans le chapitre précédent, nous avons montré que les poly(NHC-CO2)s sont capables
de catalyser la condensation de la benzoïne ainsi que la réaction de transestérification. Pour
cela, les réactions ont été effectuées à 80 °C, ce qui permet la libération du CO2 des adduits
et la formation de NHCs supportés sur les polymères vinyliques. Par ailleurs, il est bon de
rappeler que les poly(NHC-CO2)s nécessitent d’être stockés dans des conditions anhydres
pour ne pas subir de reaction d’hydratation conduisant à leur transformation progressive en
PVIm+HCO3-.
Les PVIm+HCO3- obtenus précédemment ont donc également été évalués comme
catalyseurs supportés de ces deux mêmes réactions modèles. Rappelons qu’en dehors de
nos travaux, aucune étude, à notre connaissance, a décrit l’utilisation
d’hydrogénocarbonates d’azolium en tant que précurseurs de NHCs. A fortiori, des
équivalents polymères n’ont pas non plus servi à cette fin. Dans un premier temps, les
mêmes conditions expérimentales que pour la catalyse par les poly(NHC-CO2)s ont donc été
appliquées.
Comme pour le iPrIm+HCO3-, qui permet aussi la catalyse de ces deux réactions,
l’efficacité de ces polymères a ensuite été déterminée en présence de tamis moléculaire
dans le milieu réactionnel.
Le PViPrIm+HCO3- ainsi que le PVEtPhIm+HCO3
- ont été évalués en catalyse de
transestérification. Ceci a été réalisé de façon à pouvoir comparer les rendements obtenus
avec ceux relevés pour la catalyse par les poly(NHC-CO2)s. Les transestérifications entre
l’acétate de vinyle et l’alcool benzylique ont donc été conduites à 80 °C pendant 2 h et avec
10 % de catalyseur (Schéma IV.23). En fin de réaction, le milieu réactionnel a été placé sous
atmosphère de CO2, après avoir été refroidi à température ambiante. Le polymère ainsi
régénéré et précipité, a ensuite été filtré et conservé à l’air pour un nouveau cycle de
transestérification. De la même manière que précédemment, le filtrat a été analysé par
spectroscopie de RMN 1H, ce qui a permis de calculer les rendements de réactions. Les
résultats obtenus pour la catalyse de transestérification par le PViPrIm+HCO3- et le
PVEtPhIm+HCO3- sont présentés dans le tableau IV.2.
Schéma IV.27 : Réaction de transestérification catalysée par les PVIm+HCO3-.

213
Tableau IV.2 : Rendement des réactions de transestérification catalysées par le
PViPrIm+HCO3- ou le PVEtPhIm+HCO3
-. Les rendements obtenus pour la catalyse par les
poly(NHC-CO2)s correspondants sont également rappelés en vert.
Cycle 1 2 3 4 5 6
Rendement (%) avec PViPrIm+HCO3-
(avec PolyNHC-1-CO2)
52
(94)
48
(51)
36
(23)
35
-
46
-
35
-
Rendement (%) avec PVEtPhIm+HCO3-
(avec PolyNHC-3-CO2)
25
(83)
37
(77)
5
(74)
-
(70)
-
(77)
-
(76)
Ainsi, les rendements obtenus montrent que les PVIm+HCO3- permettent la catalyse
de la réaction de transestérification sur plusieurs cycles, et ce, en absence de tamis
moléculaire. Cependant, les rendements sont plus faibles que lorsque cette réaction est
catalysée par les poly(NHC-CO2)s (voir chapitre III) ou bien par le iPrIm+HCO3- sous forme
moléculaire. La reformation de NHCs supportés en l’absence de tamis moléculaire paraît
donc être défavorisée, comme nous l’avons déjà noté avec le iPrIm+HCO3-. Afin de confirmer
cette tendance, ces polymères ont donc également été testés en catalyse de condensation
de la benzoïne (Schéma IV.24), et dans les mêmes conditions expérimentales. De la même
manière, ils ont été régénérés par la mise sous atmosphère de CO2 du milieu réactionnel et
ont par la suite été recyclés. Des rendements de 3% ont ainsi été obtenus sur 3 cycles avec le
PViPrIm+HCO3-, alors que le PVEtPhIm+HCO3
- n’a pas permis la catalyse de la condensation de
la benzoïne (Tableau IV.3). Ces résultats confortent donc l’hypothèse que seule une faible
quantité de NHCs peut être générée, en l’absence de tamis moléculaire servant à piéger
l’eau libérée par chauffage.
Schéma IV.28 : Condensation de la benzoïne catalysée par les PVIm+HCO3-.

214
Tableau IV.3 : Rendement des réactions de condensation de la benzoïne catalysées par le
PViPrIm+HCO3- ou le PVEtPhIm+HCO3
-, en absence ou en présence de tamis moléculaire. Les
rendements obtenus pour la catalyse par les poly(NHC-CO2)s correspondants sont également
rappelés en vert.
Cycle 1 2 3
Rendement (%) avec PViPrIm+HCO3-
(avec PolyNHC-1-CO2)
3
(71)
3
(27)
3
(8)
Rendement (%) avec PVEtPhIm+HCO3-
(avec PolyNHC-3-CO2)
0
(65)
0
(35)
-
(21)
Rendement (%) avec PViPrIm+HCO3-
en présence de tamis moléculaire 99 86
-
En présence de tamis moléculaire cette fois (Tableau IV.3, ligne 3), un rendement
quantitatif (>99%) a été obtenu lors du premier cycle de catalyse dans le cas de la
condensation de la benzoïne. Le polymère a également pu être recyclé et impliqué dans un
deuxième cycle de catalyse qui a alors conduit à un rendement de 86% en benzoïne.
La formation in-situ de NHCs supportés à partir de PVim+HCO3- (Schéma IV.29) est
donc réalisable en présence de tamis moléculaire. De même, il est possible de régénérer ces
précurseurs en présence de CO2 et d’eau (Schéma IV.29), ainsi que de les recycler par une
simple filtration du milieu réactionnel. D’après les différentes expériences menées ici, le
mécanisme de formation des NHCs, à partir de ces précurseurs, implique en premier lieu une
déshydratation qui conduit à la formation de carboxylates d’imidazolium. Pour une
température de 80 °C, ceux-ci subissent une décarboxylation permettant de générer les
NHCs. Ces différentes étapes sont réversibles, et il est ainsi possible de former à nouveau les
hydrogénocarbonates d’imidazolium par simple addition de CO2, et de H2O.

215
Schéma IV.29 : Mécanisme de formation et de régénération des NHCs supportés à partir des
PVIm+HCO3-.
IV.4) Conclusion
La génération in-situ de NHCs est une méthode bien connue, qui a largement été
utilisée dans le but de synthétiser des complexes organométalliques, ou bien
d’organocatalyser des réactions de chimie moléculaire et même, récemment des réactions
de polymérisation. Généralement, les NHCs sont obtenus à partir d’adduits de type azolium
qui se décomposent thermiquement dans le milieu réactionnel. Même si certains adduits
(ceux issus de CO2 et CS2) sont faciles à synthétiser et capables de générer tout type de
NHCs, d’autres (issus de CHCl3, ROH par exemple) nécessitent un assez grand nombre
d’étapes pour être obtenus purs, cette dernière méthode étant restreinte à la préparation
de quelques NHCs seulement.
Au cours de cette étude, nous avons montré que les hydrogénocarbonates
d’imidazolium et leurs homologues polymères peuvent servir de précurseurs très efficaces
de NHCs et de NHCs supportés. Ils peuvent être facilement synthétisés par un simple
changement d’anion, en utilisant des composés peu coûteux et inoffensifs pour
l’environnement, en l’occurrence des halogénures d’imidazolium, de la potasse et du CO2. La
formation de NHCs à partir de tels précurseurs se déroule en deux étapes : déshydratation
suivie d’une réaction de décarboxylation. Chacune peut être favorisée par des actions
simples telles que l’élimination d’eau (par emploi de tamis moléculaire) ou du CO2 (énergie
thermique). De surcroît, les précurseurs à contre-anion hydrogénocarbonate peuvent être
régénérés par simple addition de CO2 et de H2O. Il est alors possible de les recycler par
simple filtration, d’autant plus lorsque ceux-ci sont supportés sur une chaine polymère. Ces
différentes étapes restent cependant à être investiguées plus en détail, en effectuant par
exemple des calculs théoriques permettant de valider l’ordre dans lequel ces étapes ont lieu.

216
Ces sels peuvent également servir de précatalyseurs pour deux réactions standards
de la chimie moléculaire : la condensation de la benzoïne et les transestérifications, mais
également pour préparer, d’une part des complexes organométalliques d’or (Au(iPrIm)Cl)
par substitution de ligand, et d’autre part des adduits dithiocarboxylates d’imidazolium
(NHC-CS2) en présence de CS2. Les NHCs sont des organocatalyseurs efficaces de
nombreuses réactions de chimie moléculaire, mais également macromoléculaires. On peut
donc envisager que les hydrogénocarbonates d’azolium permettent de générer des NHCs
pour la catalyse de réactions telles que la cyanosilylation des cétones, ou bien encore pour la
polymérisation par transfert des groupes des méthacrylates d’alkyle.
Le changement d’anion pouvant être effectué sur la plupart des liquides ioniques, et
les adduits NHC-CO2s étant capables de générer tout type de NHCs, on peut supposer que
cette méthode de génération in-situ puisse être appliquée à la préparation d‘un grand
nombre de NHCs. Des expériences complémentaires sont donc nécessaires pour étayer cette
dernière hypothèse, par exemple, en synthétisant des hydrogénocarbonates de triazolium
ou de thiazolium puis en évaluant leur capacité à générer des NHCs in-situ. L’analyse par
diffraction des rayons X de ces sels permettrait également de connaître la structure des
différents composés, mais rendrait aussi compte des interactions pouvant s’établir entre le
cation azolium et le contre-anion hydrogénocarbonate, autorisant alors une explication
quant à la formation de NHCs à partir de ces composés. A partir de là, on pourrait aussi
envisager de supporter ces précatalyseurs comme nous avons pu le faire au cours de ce
travail.
De même, nous avons mis en évidence que la réaction de déshydratation est l’étape
clé permettant d’aboutir au NHCs. Elle peut être favorisée par l’élimination de l’eau formée,
que ce soit au moyen de tamis moléculaire ou bien par évaporation à des températures
supérieures à 100 °C. Ainsi, on peut supposer que la présence d’un contre-anion capable
d’éliminer un composé de même nature que l’eau, pour une température de vaporisation
plus faible, autoriserait la formation de NHCs de manière plus aisée. Par exemple, la
présence d’un anion mono-méthylcarbonate, c'est-à-dire issu de CO2 et de méthanol (en lieu
et place de H2O), permettrait la formation d’adduits de type NHC-CO2 pour des
températures plus basses et, en l’absence de tamis moléculaire.

217
IV.5) Références
1. Voutchkova, A. M.; Appelhans, L. N.; Chianese, A. R.; Crabtree, R. H. J. Am. Chem. Soc. 2005, 127, (50), 17624-17625. 2. Voutchkova, A. M.; Feliz, M.; Clot, E.; Eisenstein, O.; Crabtree, R. H. J. Am. Chem. Soc. 2007, 129, (42), 12834-12846. 3. Sauvage, X.; Demonceau, A.; Delaude, L. Adv. Synth. Catal. 2009, 351, (11-12), 2031-2038. 4. Zhou, H.; Zhang, W.-Z.; Liu, C.-H.; Qu, J.-P.; Lu, X.-B. J. Org. Chem. 2008, 73, (20), 8039-8044. 5. Delaude, L. Eur. J. Inorg. Chem. 2009, (13 ), 1681-1699. 6. Kayaki, Y.; Yamamoto, M.; Ikariya, T. Angew. Chem., Int. Ed. 2009, 48, (23), 4194-4197. 7. Tommasi, I.; Sorrentino, F. Tetrahedron Lett. 2009, 50, (1), 104-107. 8. Pawar, G. M.; Buchmeiser, M. R. Adv. Synth. Catal. 2010, 352, (5), 917-928. 9. Van Ausdall, B. R.; Glass, J. L.; Wiggins, K. M.; Aarif, A. M.; Louie, J. J. Org. Chem. 2009, 74, (20), 7935-7942. 10. Heldebrant, D. J.; Jessop, P. G.; Thomas, C. A.; Eckert, C. A.; Liotta, C. L. J. Org. Chem. 2005, 70, (13), 5335-5338. 11. Jessop, P. G.; Heldebrant, D. J.; Li, X.; Eckertt, C. A.; Liotta, C. L. Nature 2005, 436, (7054), 1102. 12. Liu, Y.; Jessop, P. G.; Cunningham, M.; Eckert, C. A.; Liotta, C. L. Science 2006, 313, (5789), 958-960. 13. Phan, L.; Jessop, P. G. Green Chem. 2009, 11, (3), 307-308. 14. Jessop, P. G.; Phan, L.; Carrier, A.; Robinson, S.; Darr, C. J.; Harjani, J. R. Green Chem. 2010, 12, (5), 809-814. 15. Heldebrant, D. J.; Koech, P. K.; Ang, M. T. C.; Liang, C.; Rainbolt, J. E.; Yonker, C. R.; Jessop, P. G. Green Chem. 2010, 12, (4), 713-721. 16. Mercer, S. M.; Jessop, P. G. ChemSusChem 2010, 3, (4), 467-470. 17. Krtschil, U.; Hessel, V.; Reinhard, D.; Stark, A. Chem. Eng. Technol. 2009, 32, (11), 1774-1789. 18. Holbrey, J. D.; Reichert, W. M.; Tkatchenko, I.; Bouajila, E.; Walter, O.; Tommasi, I.; Rogers, R. D. Chem. Commun. 2003, 9, (1), 28-29. 19. Bridges, N. J.; Hines, C. C.; Smiglak, M.; Rogers, R. D. Chem.-Eur. J. 2007, 13, (18), 5207-5212. 20. Delaude, L.; Demonceau, A.; Wouters, J. Eur. J. Inorg. Chem. 2009, (13 ), 1882-1891. 21. Nyce, G. W.; Csihony, S.; Waymouth, R. M.; Hedrick, J. L. Chem.-Eur. J. 2004, 10, (16), 4073-4079. 22. Diez-Gonzalez, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, (8), 3612-3676. 23. Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B. Chem. Rev. 2009, 109, (8), 3561-3598. 24. Garrison, J. C.; Youngs, W. J. Chem. Rev. 2005, 105, (11), 3978-4008. 25. De Fremont, P.; Scott, N. M.; Stevens, E. D.; Nolan, S. P. Organometallics 2005, 24, (10), 2411-2418. 26. Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, (12), 5606-5655. 27. Grasa, G. A.; Kissling, R. M.; Nolan, S. P. Org. Lett. 2002, 4, (21), 3583-3586. 28. Tumanskii, B.; Sheberla, D.; Molev, G.; Apeloig, Y. Angew. Chem., Int. Ed. 2007, 46, (39), 7408-7411. 29. Tumanskii, B.; Pine, P.; Apeloig, Y.; Hill, N. J.; West, R. J. Am. Chem. Soc. 2004, 126, (25), 7786-7787. 30. Tumanskii, B.; Pine, P.; Apeloig, Y.; Hill, N. J.; West, R. J. Am. Chem. Soc. 2005, 127, (23), 8248-8249.

218
Conclusion générale

219

220
L’objectif principal de cette thèse était d’apporter des solutions quant à la facilité de
manipulation des organocatalyseurs de la chimie moléculaire et, plus récemment de la
chimie des polymères, que sont les carbènes N-hétérocycliques (NHCs). Un autre objectif
était d’étendre l’utilisation des NHCs en tant que catalyseurs organiques de réactions de
polymérisation par étapes.
Si les NHCs sont énormément utilisés comme organocatalyseurs sélectifs d’un assez
grand nombre de réactions de la chimie moléculaire, leur potentiel vis à vis des réactions de
polymérisation n’a été qu’assez peu exploité. La plupart des exemples décrits depuis 2002 a
concerné les réactions de polymérisation en chaine, notamment celles procédant par
ouverture de cycle ou par transfer de groupe.
L’activité catalytique avérée des NHCs vis-à-vis de la réaction de condensation de la
benzoïne utilisant un mono-aldéhyde comme substrat, nous a incité à évaluer leur potentiel
pour la polymérisation par étapes d’un monomère de type bis-aldéhyde analogue, en
l’occurrence le téréphtaldéhyde.
S’agissant de la synthèse de « polybenzoïnes », quatre NHCs ont été testés en tant
que catalyseurs pour des réactions de polymérisation conduites dans des conditions
relativement douces à 40 °C dans le THF ou le DMSO. La caractérisation de ces polymères a
mis en évidence la présence de polybenzoïnes cycliques. Nous avons alors montré que la
proportion de ces macrocycles, déterminée au moyen de calculs basés sur la théorie de
Flory, augmente avec la conversion du monomère et dépend de la nature du solvant de
polymérisation. La polymérisation par étapes du téréphtaldéhyde étend ainsi le panel de
réactions de polymérisation catalysées par les NHCs. Les récentes avancées en catalyse
moléculaire utilisant les NHCs laissent présager que ces composés pourront catalyser
d’autres réactions de polymérisations par étapes.
Dans un deuxième temps, des méthodes originales permettant la
protection/déprotection ainsi que le recyclage des NHCs, après leur utilisation en
organocatalyse, ont été développées. En-effet, les NHCs sont des espèces sensibles,
susceptibles de se dégrader à l’air et/ou en présence d’humidité. Comme d’autres et nous-
même l’avons démontré, un moyen de mieux les manipuler consiste à les fixer sur un
support polymère linéaire.
Sur la base des méthodes de synthèse des NHCs impliquant la déprotonation de sels
d’azolium au moyen d’une base forte, nous avons ainsi élaboré des « poly(NHC)s » en
utilisant des polymères liquides ioniques de type poly(N-vinyl-3-alkylimidazolium) comme
précurseurs. Ceux-ci ont d’abord été préparés par polymérisation radicalaire de monomères
liquides ioniques, suivie d’un changement de l’anion halogénure par l’anion
bis(trifluorométhansulfonyl)amidure. Un travail préliminaire concernant la polymérisation
radicalaire contrôlée de type RAFT/MADIX de ces halogénures de N-vinyl-3-alkylimidazolium
a également été réalisé. Des copolymères à blocs liquides ioniques relativement définis de

221
type PVBuIm+Br--b-PNVP ont alors pu être synthétisés. De tels copolymères n’ont toutefois
pas pu être employés comme précurseurs de NHCs supportés.
Les poly(NHC)s s’avèrent des catalyseurs efficaces vis-à-vis de réactions modèles
telles que la condensation de la benzoïne et la réaction de transesterification. La
précipitation/récupération des poly(NHC)s, par ajout de non-solvant en fin de réaction,
permet le recyclage de ces catalyseurs supportés par simple filtration. Les polymères ainsi
récupérés ont été à nouveau impliqués dans des réactions organocatalysées, avec
cependant des activités de plus en plus faibles, au fur et à mesure du recyclage, en raison
très probablement de la dégradation progressive des entités carbéniques du support
polymère.
Pour pallier cette difficulté, les poly(NHC)s ont été « masqués » par addition de CO2.
En chauffant en solution les adduits correspondants, de type poly(NHC-CO2), la libération de
CO2 permet de reformer les NHCs supportés pour la catalyse des deux réactions sus-citées.
En appliquant des cycles de masquage/démasquage, les NHCs supportés peuvent ainsi être
recyclés et impliqués dans près de 8 cycles de catalyse, tout en conservant de très bonnes
activités catalytiques. Le recyclage de ces adduits poly(NHC-CO2)s peut être mis en oeuvre
par simple filtration du milieu réactionnel. Ainsi, les NHCs ont été supportés et recyclés au
moyen de simples filtrations, autorisant une manipulation plus aisée de ces entités
catalytiques. La mise à l’air des adduits poly(NHC-CO2)s a cependant révélée que ces derniers
s’hydratent sous la forme d’hydrogénocarbonate de poly(N-vinyl-3-alkylimidazolium). Avant
ce travail, de tels composés n’avaient jamais été utilisés comme précurseurs de NHCs.
La dernière partie de ce travail, apporte la preuve que les hydrogénocarbonates
d’imidazolium et leurs homologues polymères peuvent servir de précurseurs très efficaces
de NHCs et de NHCs supportés. Pour cela, nous avons d’abord mis au point une nouvelle
méthode de synthèse de ces précurseurs, en version moléculaire comme en version
supportée sur un tronc polymère, par simple changement d’anion (HCO3- en place de Br-) à
partir de sels précurseurs commerciaux de type bromure d’azolium. Cette réaction de
métathèse pouvant être appliquée à un grand nombre de liquides ioniques, on peut
supposer que d’autres hydrogénocarbonates d’azolium (triazolium, thyazolium…) pourraient
être préparés suivant cette méthode.
Après leur synthèse, nous avons alors démontré que l’activation par chauffage des
hydrogénocarbonates d’imidazolium génère des NHCs, du CO2 et de l’eau,
vraisemblablement en deux étapes : une déshydration suivie d’une décarboxylation. Qui plus
est, ce processus est réversible, les hydrogénocarbonates d’imidazolium pouvant être
régénérés par simple addition de CO2 en milieu humide sur les (poly)NHCs générés « libres ».

222
Le mécanisme de formation des NHCs, à partir des hydrogénocarbonates
d’imidazolium, n’ayant pas clairement été établi, des études, notamment théoriques,
doivent pouvoir attester l’ordre des différents évènements mis en jeu ainsi que l’ordre de
grandeur des énergies associées.
Nous avons ensuite utilisé ces sels comme précatalyseurs des deux mêmes réactions
mentionnées : condensation de la benzoïne et transestérification. Ces précurseurs nous ont
aussi servis à préparer, d’une part, des complexes organométalliques d’or (Au(iPrIm)Cl) par
substitution de ligand, et d’autre part, des adduits dithiocarboxylates d’imidazolium (NHC-
CS2) en présence de CS2. Dans le cas des hydrogénocarbonates d’imidazolium supportés,
ceux-ci ont été impliqués et recyclés dans plusieurs cycles de catalyse des réactions de
transestérification et de condensation de la benzoïne.
On peut dés lors envisager que les hydrogénocarbonates d’azolium pourraient être
employés comme pré-catalyseurs d’autres réactions en chimie moléculaire (cyanosilylation
des cétones, réaction de Stetter, etc.) comme en organocatalyse de réactions de
polymérisation (polymérisation par transfert de groupe des groupes des (méth)acrylates
d’alkyle, polymérisation par ouverture de cycle d’hétérocycles, polymérisation par étapes de
substrats monomères à fonctions carbonyle, etc…).
Ainsi, d’après ces différentes études, nous pouvons légitimement nous attendre à
l’essor des NHCs en synthèse macromoléculaire. Les différentes méthodes proposées dans
ce travail pour faciliter leur manipulation et leur utilisation, ainsi que leur recyclage,
devraient également contribuer au développement de ces nouveaux catalyseurs organiques.

223

224
V. Experimental part (Partie expérimentale)

225

226
This chapter is dedicated to the synthesis of compounds that were used in the course
of this PhD. It is divided in five main parts: description of material and techniques used for
characterization, preparation of solvents and reactive compounds, and the experimental
techniques and conditions used for the preparation of products described in each chapter
(Chapter II, III and IV).
V.1) Characterization
During the course of this PhD, several techniques were used to characterize the
chemical or physical properties of polymers and product obtained. The nuclear magnetic
resonance has been certainly the most employed technique and will be first described. Then,
the size exclusion chromatography, the thermogravimetric analysis and the differential
scanning calorimetry techniques for characterization will be detailed as well.
V.1.1) Nuclear Magnetic Resonance (NMR)
1H NMR and 13C NMR spectra were recorded on Bruker AC-400 spectrometer in
appropriate deuterated solvents. Resonance fields (δ) are explained in part per million
(ppm).
When analysis were performed in deuterated water (D2O > 99%), reference
used to calibrate 1H NMR spectrum was the residual peak of water (δ =4,79
ppm at 298K).
For analysis performed in CDCl3, references were the peak of chloroform
(CHCl3), 7,26 ppm at 298 K for 1H NMR and 77,0 ppm for 13C NMR analysis.
Spectrum recorded in DMSO-d6 were calibrated with the peak of DMSO at
2,54 ppm in 1H NMR analysis and 40 ppm in 13C NMR analysis.
When MeOD or THF-d8 were used as solvent for NMR spectroscopy analysis,
the 1H NMR spectrums were calibrated with the solvent residual peak at 3,31
ppm for MeOH and 1,85 and 3,76 ppm for THF; and the 13C NMR spectrums
with the peak at 49 ppm for MeOH and 25,6 and 68,0 ppm for THF.
During this last chapter, notation employed to described NMR spectrum will be
annotated as follow: s (singlet), d (doublet), t (triplet) and q (quadruplet) and m (multiplet).

227
V.1.2) Size Exclusion Chromatography (SEC)
Depending on polymer solubility and nature, molar masses and dispersities of the
different materials prepared during this PhD have been determinated employing the
following instrumentation techniques.
V.1.2.1) SEC in THF
Molar masses were determined by size exclusion chromatography (SEC) that was
performed using a 3-column set of TSK gel TOSOH (G4000, G3000, G2000 with pore sizes of
20, 75 and 200 Å respectively, connected in series) calibrated with polystyrene standards
with THF as eluent (1 mL/min) and trichlorobenzene as a flow marker at 25 0C, using both
refractometric and UV detectors (Varian).
V.1.2.2) SEC in water
Molar masses were determined by gel permeation chromatography (GPC) using a PL-
GPC50 plus Integrated GPC System equipped with PSS SUPREMA Max columns with pour
sizes of 30 and 1000 Å respectively, (connected in series) fitted with dual detectors
(refractometry and UV) and a water/ formic acid (0,3M) as the mobile phase at a flow rate of
0,6 mL/min. Pyridine was used as a flow-marker. Calibration curve was done using Poly(2-
vinylpyridine) as polymer standards.
V.1.2.3) SEC in DMF
Molar masses were determined by gel permeation chromatography (GPC) using a PL-
GPC50 plus Integrated GPC System equipped with PL gel 5μm mixed-D columns, (connected
in series) fitted with dual detectors (refractometry and UV) and DMF/LiBr (1g/L) as the
mobile phase at a flow rate of 0,8 mL/min at 80°C. Toluene was used as a flow-marker.
Calibration curve was done using Polystyrene as polymer standards.
V.1.3) Thermogravimetric analysis (TGA)
TGA analyses were performed on a TA instruments TGA-Q500, under Ar atmosphere
at a heating rate of 5°C/min.
V.1.4) Differential scanning calorimetry (DSC)
DSC measurements were performed on a DSC Q100 apparatus from TA Instruments.
Data were recorded during the second run for temperatures ranging from 20 to 200 °C at a
heating rate of 10 °C min-1. The cooling rate between the first and second runs was also
equal to 10 °C min-1. The glass transition temperature (Tg) was given by the inflection point
of the transition.

228
V.1.5) Infra-red spectroscopy
A Bruker Tensor 27 spectrometer was used for ATR-FTIR analysis.
V.2) Preparation of solvents and reactive compounds
V.2.1) Origin and purification of chemical products
For chapter II
Terephtaldehyde was purchased from Alfa Aesar and was dried azotopically with
dioxane.
For chapter III
1-vinylimidazole (99%), 1-Bromobutane (99%), 2-bromopropane and 2-
bromoethylbenzene were obtained from Alfa Aesar and used as received. Azobis(2-
methylpropionitrile) (AIBN, 99%) was received from Aldrich and was purified by
recrystallization from methanol. NaH (Aldrich, 60% in mineral oil), t-BuOK (Aldrich),
Potassium bis(trimethylsilyl)amide (KHMDS, Aldrich), Lithium
Bis(trifluoromethanesulfonyl)imide (LiNTf2, 98%, TCI) were used as received. Benzyl Alcolhol
(Aldrich) and Benzaldehyde (Aldrich) were distilled prior to use. Vinyl acetate (Aldrich) was
dried over CaH2 and distilled prior to use.
For chapter IV
Imidazole, chloro(dimethylsulfide)gold(I) (Au(SMe2)Cl,99%), Molecular sieves 3A and
potassium persulfate (KPS, 99%) were obtained from Alrich and used as received. KOH
(pellets, 85%), sodium bicarbonate (99%) and N,N’-dimethylaminopropionitrile (DMAPN,
98%) were purchased from Acros and used as received. CS2 (99,9%, Aldrich) was distilled
over Na metallic prior use. 4,4'-azobis(4-cyanovaleric acid) (V-501, 98%, Aldrich) and 2,2'-
azobis(valeronitrile-4-méthoxy-2,4-diméthyl) (V-70, 95%, Wako) were recrystallized from
methanol.
V.2.2) purification of solvents
When necessary, solvents were purified as follow:
- THF was distilled over Na/benzophenone.
- Diethyl Ether was distilled over polystyryllithium (PS-Li).
- DMSO was refluxed over CaH2 and was distilled under vacuum prior to use.

229
- Dioxanne was refluxed over CaH2 and was distilled under vacuum prior to use
- MeOH was distilled carefully over Na metallic before use.
- CO2 (N-45, Air Liquide) was purified by passing threw a Click-on inline Super Clean
Purifier (SGT) before use, as shown on the following picture of the experimental
equipment (Figure V.1).
Figure V.1 : Experimental set-up used to purify CO2.
V.3) Experimental protocols related to chapter II
All the experiments were performed under an inert atmosphere using standard
Schlenk techniques. Dry, oxygen-free solvents and monomers were employed. N-
heterocyclic carbenes NHC-III and NHC-IV were purchased from Sigma and STREM chemicals
and were used as such without further purification.
CO2 inflowfrom gas tube
Set-up for CO2
purificationClick-on inline Super Clean
Purifier
CO2 exit to reaction flask

230
V.3.1) Synthesis of NHCs
NHCs catalysts were prepared by slightly modifying already reported procedures1,
using the experimental setup shown Figure V.2.
Figure V.2 : Experimental set-up allowing the synthesis of « bare » NHCs
V.3.1.1) Synthesis of 1,3-diisopropylimidazol-2-ylidene (NHC-I)
A 150-mL round-bottom flask equipped with a stir bar was charged with 2.0 g (10.6
mmol) of 1,3-diisopropyl imidazolium chloride, solid potassium tert-butoxide (90 mg, 0.8
mmol) and sodium hydride (600 mg, corresponding to 15 mmol of pure NaH) as a 60%wt
dispersion in silicon oil. The flask was kept under vacuum. Then, 20 mL of freshly distilled
THF were added dropwise to the suspension at room temperature. A dark gray solution was
obtained immediately. The mixture was stirred for 5 h, then filtrated under vacuum (fritted
glass filter, porosity G5) and volatiles were removed under vacuum. The obtained pale
viscous liquid was subsequently distilled under vacuum to provide NHC-I as a clear liquid (1.3
g, 81% yield): 1H NMR (THF-d8) : 1.45 (s, CH3, 12 H), 4.45 (m, CH(CH3)2, 2 H), 6.95 (s, NCHC, 2
H); 13C NMR : 24.8 (s, CH3), 52.9 (s, CH-(CH3)2), 116.5 (s, NCHC), 212.9 (s, NCN).
V.3.1.2) Synthesis of 1,3-ditert-butylimidazol-2-ylidene (NHC-II)
A 150-mL round-bottom flask equipped with a stir bar was charged with 2.0 g (9.2
mmol) of 1,3-ditert-butyl imidazolium chloride, and 20 mL of freshly distilled THF. The
dispersion was stirred for 0.5h. Then, 7 mL of a 2M solution of nBu-Li in cyclohexane (14
mmol) was added dropwise at -78°C. A turbid solution was obtained immediately. The
mixture was allowed to warm up and stirred for 2 h, then filtrated under vacuum (fritted
glass filter, porosity G5) and volatiles were removed under vacuum. The obtained pale solid
was subsequently sublimed under vacuum to provide NHC-II as a white crystalline solid
(Figure V.3), (1.3 g, 78% yield): 1H NMR (THF-d8) : 1.6 (s, CH3, 18 H), 7.15 (s, NCHC, 2 H); 13C
NMR : 31.8 (s, -CH3), 56.3 (s, C(CH3)3), 115.7 (s, NCHC), 213.4 (s, NCN).

231
Figure V.3 : Crystallization of the NHC 1 after the fritted filter
V.3.2) Step-growth polymerizations of terephtaldehyde
All polymerization reactions were carried out under a dry and inert atmosphere using
vacuumed flame-dried special Schlenk apparatus equipped with a withdrawal vial on the
side of the main flask. In a typical procedure, a 50 mL flame dried special Schlenk apparatus
was charged with 1 g of terephtaldehyde (7.45 mmol), 22 mg of NHC-4 (0.074 x 10-3 mol) and
10 mL of THF (or DMSO). The Schlenk tube was left under stirring in an oil bath at 40 °C. At
precise time intervals, aliquots were withdrawn through vacuum flame-dried withdrawal vial
attached to the flask. A droplet of degassed MeOH was then introduced and the aliquot was
removed from the withdrawal vial. At the end, the reaction was quenched by adding few
drops of methanol to the reaction mixture. The polybenzoin was precipitated in water and
dried under vacuum.
V.4) Experimental protocols related to chapter III
V.4.1) Monomer Synthesis
V.4.1.1) 1-vinyl-3-isopropylimidazolium bromide (ViIPrIm+Br-) The product, 1-vinyl-3-isopropylimidazolium bromide, was prepared following the
procedures described in the literature2. Under vigorous stirring, 9.3 g (0.075 mol; 7.1mL) of
2-bromopropane were added dropwise to 5 g (0.053 mol, 4.8mL) of 1-vinylimidazole in a
100-mL, one-necked, roundbottom flask. The mixture was refluxed for 16h. The resulting
white-yellow powder was allowed to cool to room temperature, and then it was washed
several times with ethyl acetate. Before use, the product was dried under dynamic vacuum
until constant weight, and kept under vacuum (11.5 g, 100% yield).

232
1H NMR (CDCl3): δ 11.00 (s, N-CH-N, 1H), 7.88 (s, N-CH=CH-N, 1H), 7.66 (s, -CH=CH-N, 1H),
7.48 (dd, CH2=CH-N, 1H), 6.03 (dd, HCH=CH-N, 1H), 5.39 (dd, HCH=CH-N, 1H), 4.95 (h,
(CH3)2CH- 1H), 1.66 (d, (CH3)2CH-, 6H).
V.4.1.2) 1-vinyl-3-butylimidazolium bromide (ViBuIm+Br-)
The product, 1-vinyl-3-butylimidazolium bromide, was prepared following the
procedures described in the literature2. Under vigorous stirring, 13.7 g (0.1 mol, 9.5mL) of
bromobutane were added dropwise to 5 g (0.053 mol, 4.8mL) of 1-vinylimidazole in a 100-
mL, one-necked, roundbottom flask. The mixture was refluxed for 24h. The resulting brown
viscous liquid was allowed to cool to room temperature, and then it was washed several
times with ethyl acetate. Before use, the product was dried under dynamic vacuum until
constant weight (10.6g of ViBuIm+Br-, 87% yield), and keep under vacuum. 1H NMR (CDCl3): δ 10.64 (s, N-CH-N, 1H), 7.98 (s, N-CH=CH-N, 1H), 7,65 (s, N-CH=CH-N, 1H),
7.37 (dd, CH2=CH-N, 1H), 5.97 (dd, HCH=CH-N, 1H), 5.24 (dd, HCH=CH-N, 1H), 4.29 (t, CH3-
CH2-CH2-CH2-, 2H), 1.82 (m, CH3-CH2-CH2-CH2-, 2H), 1.24 (m, CH3-CH2-CH2-CH2-, 2H), 0.84 (t,
CH3-CH2-CH2-CH2-, 3H).
V.4.1.3) 1-vinyl-3-(2-ethylbenzene)imidazolium bromide (ViEtBIm+Br-)
The product, 1-vinyl-3-(2-ethylbenzene)imidazolium bromide, was prepared following
the same procedure used to synthesize ViBuIm+Br-. Under vigorous stirring, 2.8 g (0,015 mol;
2mL) of bromobutane were added dropwise to 1.04 g (0.011 mol, 1mL) of 1-vinylimidazole in
a 100-mL, one-necked roundbottom flask. The mixture was refluxed for 20h. The resulting
brown viscous liquid was allowed to cool to room temperature, and then it was washed
several times with ethyl acetate and then with THF. Before use, the product was dried under
dynamic vacuum until constant weight (2.6g, 85% yield), and keep under vacuum. 1H NMR (MeOD): δ 9.60 (s, N-CH-N, 1H), 8.04 (s, N-CH=CH-N, 1H), 7.75 (s, N-CH=CH-N, 1H),
7.3-7.5 (m, benzylics, 5H), 7.20 (dd, CH2=CH-N, 1H), 5.96 (dd, HCH=CH-N, 1H), 5.83 (q,
HCH=CH-N, 1H), 5.42(dd,-CH-(CH3)-Ph, 1H), 2.00 (d, -CH-(CH3)-Ph, 3H).
V.4.2) Polymerization of vinylimidazolium salts
V.4.2.1) Free radical polymerization
In a typical experiment, a 10 mL Schlenk tube was flame dried and charged with 1 g
of monomer, desirous amount of initiator (M/30) AIBN and 2 mL of methanol. The Schlenk
tube was subjected to five freeze thaw cycles and placed in a thermostated oil bath
previously maintained at 80 °C. Polymerization reaction was quenched after 3h by sudden

233
cooling. The obtained polymer was precipitated in Chloroform or Acetone to remove
residual monomer, filtrated and dried in vacuum. All polyvinylimidazolium salts were
recovered as a yellowish powder.
Poly(ViIPrIm+Br -) : ViIPrIm+Br – (1g; 4.7mmol); AIBN (25mg; 0.15mmol). Precipitated
in CHCl3. (1g, Yield 99%); Mn = 4600 g.mol-1 by SEC; D = 2.48. 1H NMR (MeOD): δ 9.3-10.0 (N-
CH-N, 0.9H due to partial exchange with MeOD), 7.5-8.3 (CH=CH, 2H), 4.5-5.2 (N-CH-CH2 and
N-CH-(CH3)2, integration no possible due to recovery with solvent residual peak), 2.5-3.3 (N-
CH-CH2, 2H), 1.4-1.9 (N-CH-(CH3)2, 6H).
Poly(ViBuIm+Br-) : ViBuIm+Br– (1g; 4.3mmol); AIBN(24mg; 0.14mmol). Precipitated in
acetone. (1g, Yield 99%); Mn = 8722g.mol-1 by SEC, D = 2.9. 1H NMR (D2O): δ 8.9-9.3 (N-CH-N,
0.55H due to partial exchange with D2O), 7.3-7.8 (CH=CH, 2H), 3.9-4.5 (N-CH2-CH2-CH2-CH3,
2H), 4.4-4.8 (N-CH-CH2, 1H), 2.4-3.0 (N-CH-CH2, 2H), 1.7-2.1 (CH3-CH2-CH2-CH2-, 2H), 1.3-1.6
(CH3-CH2-CH2-CH2-, 2H), 0.9-1.2 (CH3-CH2-CH2-CH2-, 3H).
Poly(ViEtBIm+Br -) : ViEtBIm+Br – (1g; 3.6mmol); AIBN(19mg; 0.12mmol). Precipitated
in Acetone. Not soluble in water. (1g, 99% yield). 1H NMR (DMSO-d6) δ 8.9-9.8 (N-CH-N, 1H),
6.8-8.0 (CH=CH and benzylics, 7H), 5.1-5.9 (N-CH-CH2 and N-CH-(CH3)-ph, 2H), 3.6-4.0 (N-CH-
CH2, 2H), 1.6-2.0 (N-CH-(CH3)-Benzyl, 3H).
V.4.2.2) Controlled radical polymerization (MADIX)
[1-(O-Ethylxanthyl)ethyl]benzene (X1) : X1 was prepared according reported
procedure3. The synthesis was conducted by stirring ethanol (40 mL) and KOH (5.6 g, 0.1
mol) until a clear solution was formed. CS2 (20 mL) was added into the solution slowly, and
the mixture was stirred for 10 h at room temperature before excessive CS2 was distilled off
at 70 °C. (1-bromoethyl)benzene (9.5 mL) in 20 mL of ethanol was added to the residual
solution, and the mixture was further stirred at 55-65 °C for 5 h. After removal of the
inorganic salt and most of the ethanol, 50 mL of water was added and the solution was
extracted with diethyl ether (3 × 40 mL); the combined organic layer was dried over MgSO4.
Removal of the inorganic salt and evaporation of the solvent afforded a yellow oil product.
Yield yellow oil: 60%. 1H NMR (ppm, CDCl3), δ : 1.26 (t, 3H, CH3), 1.58 (d, 3H, CH3), 4.49 (q,
2H, CH2), 4.77 (q, 1H, CHPh), 7.1-7.4 (m, 5H,Ph).
MADIX polymerization of VBuIm+Br- (macrotransfer agent) : In a typical experiment,
a 10 mL Schlenk tube was flame dried and charged with 4 g (17.2 mmol) of VBuIm+Br-, 86 mg
(0.38 mmol, DPn 45) of X1 and 12.5 mg of AIBN (X1/5 : 0.076 mmol) and 6 mL of DMF. The
Schlenk tube was subjected to five freeze thaw cycles and placed in a thermostated oil bath

234
previously maintained at 60 °C. Polymerization reaction was quenched after 21h by sudden
cooling. According 1H NMR spectra in D2O, monomer conversion was 60%. The obtained
polymer was precipitated in (1/1) Diethyl ether/acetone mixture and washed with acetone
to remove residual monomer. Final dried polymer was recovered as a white powder.(2.12 g,
53% yield). Mn = 9560 g.mol-1 by SEC in water, D = 1.64. 1H NMR (D2O): δ 8.9-9.3 (N-CH-N,
0.55H due to partial exchange with D2O), 7.3-7.8 (m, CH=CH, 2H), 3.9-4.5 (m, N-CH2-CH2-CH2-
CH3, 2H), 4.4-4.8 (m, N-CH-CH2, 1H), 2.4-3.0 (m, N-CH-CH2, 2H), 1.7-2.1 (m, CH3-CH2-CH2-CH2-
, 2H), 1.3-1.6 (m, CH3-CH2-CH2-CH2-, 2H), 0.9-1.2 (m, CH3-CH2-CH2-CH2-, 3H).
Synthesis of PVBuIm+Br--b-PNVP : In a typical experiment, a 10 mL Schlenk tube was
flame dried and charged with 1 g (0.161 mmol) of macrotransfer agent PVBuIm+Br- prepared
as mentioned above, 5.3 mg of AIBN (0.032 mmol), 2g (18 mmol, DPn 110) of NVP freshly
distilled and 3 mL of DMF. The Schlenk tube was subjected to five freeze thaw cycles and
placed in a thermostated oil bath previously maintained at 60 °C. Polymerization reaction
was quenched after 14h by sudden cooling. The obtained polymer was precipitated twice in
Diethyl ether. Final dried polymer was recovered as a white powder.(2.5 g, 83% yield). Mn =
60 kg.mol-1 by SEC in DMF, D = 1.19. 1H NMR (D2O): δ 7.3-7.8 (m, CH=CH, 2H), 3.5-4.7 (m, 5,8
H), 3.0-3.5 (m N-CH-CH2PNVP, 5,7H), 1.2-2.8 (m, 23.2H), 0.8-1.1 (m, CH3-CH2-CH2-CH2-, 3H).
PNVP macrotransfer agent (MADIX polymerization of NVP) : In a typical experiment,
a 20 mL Schlenk tube was flame dried and charged with 10 g (90 mmol) of NVP freshly
distilled, 226 mg (1 mmol, DPn 90) of X1 and 16,4 mg of AIBN (X1/10 : 0,1 mmol) and 5 mL
of dioxanne. The Schlenk tube was subjected to five freeze thaw cycles and placed in a
thermostated oil bath previously maintained at 60 °C. Polymerization reaction was quenched
after 14h by sudden cooling. According 1H NMR spectra in D2O, monomer conversion was
90%. The obtained polymer was precipitated in several times in Diethyl ether to remove
residual monomer. Final dried polymer was recovered as a white powder.(6.6 g, 65% yield).
Mn = 18100 g.mol-1 by SEC in DMF, D = 1.07. 1H NMR (CD2Cl2): δ 4.5-4.7 (m, CH3-CH2-O-, 2H),
3.5-4.2 (m, N-CH-CH2-, 91.4H), 2.9-3.5 (N-CH-CH2-, 178.8H), 2.4-3.0 (N-CH-CH2, 2H), 1.3-2.5
(m, protons from pyrrolidone cycle, 548.6H), 1.1-1.2 (m, CH3-CH-(Ph)-PNVP, 3H).
Synthesis of PNVP-b-PVBuIm+Br : In a typical experiment, a 10 mL Schlenk tube was
flame dried and charged with 2.5 g (0.26 mmol) of macrotransfer agent PNVP prepared as
mentioned above, 8.0 mg of AIBN (0,05 mmol), 1.5g (6.5 mmol, DPn 25) of VBuIm+Br- and 3
mL of DMF. The Schlenk tube was subjected to five freeze thaw cycles and placed in a
thermostated oil bath previously maintained at 60 °C. Polymerization reaction was quenched
after 21h by sudden cooling. The obtained polymer was analyzed by SEC in DMF, and as two
populations appeared, was not kept.

235
V.4.3) Anion exchange
Poly(1-vinyl-3-alkylimidazolium) bromide were dissolved in water or methanol and
mixed with an excess of a solution of (CF3SO3)2NLi in water or methanol. Immediately after
mixing, a new polymer precipitated as a result of the anion-exchange reaction. Poly(1-vinyl-
3-alkylimidazolium) N-triflate was recovered by filtration and dry under vacuum for at least
one night.
Poly(ViPrIm+NTf2-) : Anion exchange was carried out in water, and 1.5 eq. of LiNTf2
were used. Polymer was recovered as white powder. (92% yield). 1H NMR (THF-d8, see Figure
1): δ 1.3-1.6 (N-CH-(CH3)2, 6H ), 2.2-2.9 (N-CH-CH2, integration no possible due to recovery
with solvent residual peak), 4.0-4.6 (N-CH-CH2 and N-CH-(CH3)2, 2H), 7.0-8.0 (CH=CH, 2H),
8.5-9.0 (N-CH-N, 1H). 13C NMR (THF-d8, see Figure S04): δ 20-22 (N-CH-(CH3)2), 39-42 (N-CH-
CH2), 52-57 (N-CH-CH2 and N-CH-(CH3)2), 112-125 (CH=CH), 114, 118, 121 and 124 (NTf2-),
132-135 (N-CH-N).
Poly(VBuIm+NTf2-) : Anion exchange was carried out in water, and 1.5 eq. of LiNTf2
were used. Polymer was recovered as white powder. (86% yield). 1H NMR (THF-d8): δ 8.6-8.9
(N-CH-N, 1H), 7.2-8.0 (CH=CH, 2H), 4.0-4.5 (N-CH2-CH2-CH2-CH3, and N-CH-CH2, 3H), 2.4-3.0
(N-CH-CH2, 2H), 1.8-2.1 (N-CH2-CH2-CH2-CH3, 2H), 1.4-1.6 (N-CH2-CH2-CH2-CH3, 2H), 1.0-1.2
(N-CH2-CH2-CH2-CH3, 3H). 13C NMR (THF-d8): δ 134-136 (N-CH-N), 115-122 (CH=CH), 114, 118,
121 and 124 (NTf2-), 47-49 (N-CH2-CH2-CH2-CH3, and N-CH-CH2), 30-33 (N-CH-CH2), 28-30 (N-
CH2-CH2-CH2-CH3),18-20 (N-CH2-CH2-CH2-CH3, 2H), 15-17 (N-CH2-CH2-CH2-CH3, 3H).
Poly(ViEtPhIm+NTf2-) : Anion exchange was carried out in methanol, and 1.5 eq. of
LiNTf2 were used. After 2h of stirring, water was added in the media. Polymer was recovered
as white powder. (70% yield). 1H NMR (THF-d8, see figure S05): δ 8.8-9.7 (N-CH-N, 1H), 7.0-
8.0 (CH=CH and aromatics, 7H), 5.3-6.0 (N-CH-CH2, 1H), 4.2-5.1 (N-CH-(CH3)-Ph, 1H), 2.3-3.3
(N-CH-CH2, 2H), 1.8-2.1 (N-CH-(CH3)-Ph, 3H). 13C NMR (THF-d8, see figure S06): δ 133-140 (N-
CH-N), 126-130 (Ph), 114, 118, 121, 124 (NTf2-), 118-125 (CH=CH), 60-62 (N-CH-CH2), 52-59
(N-CH-(CH3)-Ph), 38-42 (N-CH-CH2, 2H), 18-21 (N-CH-(CH3)-Ph, 3H).
V.4.4) Preparation of Poly(NHC)s: All reactions were carried under an inert atmosphere, using schlenks technique. Before deprotonation, polymers were dried azotropically with dioxane when they were soluble.

236
PolyNHC-1 : A schlenk tube was charged with 479 mg (1.00 mmol) of
Poly(ViPrIm+NTf2 -) which were solubilised in 10 mL of THF after drying. Schlenk was placed in
a -80°C ethanol bath and KHMDS in THF (10mL) was added to the solution. KHMDS was
preferred to NaH because of the low solubility of the polymer, even if polyNHC-1 could not
be isolated as pure compound. Then reaction was left to go back to room temperature under
vigorous stirring. Obtained solution was then used directly for catalysis experiments. 1H NMR
(THF-d8, see Figure 2): δ 1.2-1.5 (N-CH-(CH3)2, 6H ), 1.9-2.4 (N-CH-CH2,2H), 3.4-3.8 (N-CH-CH2,
1H), 4.3-4.6 (N-CH-(CH3)2, 1H), 6.2-6.9 (CH=CH, 2H).
PolyNHC-2 and PolyNHC-3 : A schlenk tube was charged with 1mmol (422mg of
Poly(VBuIm+NTf2 -) or 479 mg of Poly(VEtPhIm+NTf2
-) ) of desired Polymer which was further
dried azotropically with dioxane. Polymer was solubilised in 10 mL of THF and the solution
was added to a suspension of NaH (2mmol, 80mg) and t-BuOK (0.2mmol, 22mg) in 10 mL of
THF previously cooled to -80°C. Reaction was left to go back to room temperature under
vigorous stirring before filtration. Obtained orange solution was used directly for catalysis
experiments. 1H NMR of polyNHC-2 (THF-d8, see figure S07): δ 7.2-8.0 (aromatics, 7H), 6.5-
7.1 (CH=CH, 2H) 5.2-5.7 (N-CH-CH2 and N-CH-(CH3)-Ph, 2H), 1.4-1.9 (N-CH-CH2, 2H), 1.0-1.3
(N-CH-(CH3)-Ph, 3H).
V.4.5) Procedure for organocatalysis by poly(NHC)s
As mentioned in Chapter III, the special flask employed to perform organocatalyzed
reactions by supported NHCs or their CO2-adduct is shown in Figure V.4.
Figure V.4 : Experimental setup employed for organocatalysis by poly(NHC)s.
Fritted glass filter
Introduction of solvents and
reagents
Round-bottom flask equipped with a stir bar
Exit of filtratedsolution

237
V.4.5.1) Procedure for transesterification catalysed by poly(NHC)s
Transesterification with polyNHC-1.
In a schlenk tube charged with a solution of polyNHC-1 (1 mmol in 10 mL of THF),
benzyl alcohol (10 mmol, 1mL) and vinyl acetate (12 mmol, 1,2 mL) were added. The
reaction mixture was left under stirring for half an hour before filtration under vacuum. THF
and remaining vinyl acetate were evaporated from the filtrate, and the obtained yellowish
solution was analysed by 1H NMR in CDCl3. Efficiency of the transesterification was
determinated by comparing pick of benzyl acetate (CH2, 5ppm) and pick of benzyl alcohol
(CH2, 4.5 ppm) . Recovered polymer was then used for further transesterification.
Transesterification with polyNHC-2 and polyNHC-3.
In a schlenk tube charged with a solution of polyNHC-2 or polyNHC-3 (1 mmol in 10
mL of THF), were added benzyl alcohol (10 mmol, 1mL) and vinyl acetate (12 mmol, 1.2 mL).
The reaction mixture was left under stirring for half an hour and polyNHC was precipated
adding dry diethyl ether. Reaction mixture was filtrated under vacuum, THF and remaining
vinyl acetate were evaporated from the filtrate, and the obtained yellowish solution was
analysed by 1H NMR in CDCl3. Recovered polymer was then dissolved in 10 mL of THF and
used for further transesterification.
V.4.5.2) Procedure for Benzoin condensation catalysed by poly(NHC)s
A similar procedure was used for benzoin condensation. Benzaldehyde (1mL,
10mmol) was used instead of Benzyl alcohol and vinyl acetate. After polymer recovery, THF
was evaporated from filtrate and the remaining mixture was analyzed by 1H NMR in DMSO-
d6.
V.4.6) Preparation of poly(NHC-CO2) adducts
A schlenk tube was charge with 1mmol of Poly(ViPrIm+NTf2-) (417mg) or
Poly(ViEtPhIm+NTf2-) (479mg) and polymer was dried azotropically with dioxane before
addition of 10mL of THF. In another Schlenk tube, a solution of 2mmol (600mg) of KHMDS in
THF was prepared. Solutions were mixed together in an ethanol bath at -80°C for 15 min and
then let to go back at room temperature under stirring. After 45 minutes, 1 Atm of CO2 was
flushed to the schlenk and PolyNHC-CO2 formed, directly precipitated in the media. CO2
atmosphere was maintained for 1 hour before polymer was filtrated under vacuum and
recovered as an orange powder. Polymer was directly used for catalysis experiments or kept
in a glove-box to avoid reaction with water.

238
PolyNHC-1-CO2: 1H (DMSO-d6): δ 1.0-1.6 (N-CH-(CH3)2, 6H), 2.0-2.8 (N-CH-CH2, 2H),
4.6-5.9 (N-CH-CH2 and N-CH-(CH3)2, 2H), 7.2-8.3 (CH=CH, 2H). 13C NMR (DMSO-d6, see Figure
3): δ 21-24 (N-CH-(CH3)2), 50-54 (N-CH-CH2, N-CH-CH2 and N-CH-(CH3)2), 116-121 (CH=CH),
114, 118, 121 and 124 (NTf2-), 140-143 ((N)2-C-C), 153-156 (C-CO2). IR (ATR) : νCO 1670cm-1.
TGA : loose of 25% weight is observed, as compared to 25% theoretical planned for
complete functionalization. Polymer was soluble in polar solvents (DMSO, Acetone) and
protic solvents and insoluble in THF, CH2Cl2.
PolyNHC-3-CO2: 13C NMR (DMSO-d6): δ 161-163 (C-CO2), 136-141 ((N)2-C-C), 128-132
(aromatics), 114, 118, 121 and 124 (NTf2-), 120-128 (CH=CH), 61-63 (N-CH-CH2), 53-55 (N-CH-
(CH3)-Ph), 40-47 (N-CH-CH2), 21-24 (N-CH-(CH3)-Benzyl). IR (ATR) : νCO 1670cm-1. TGA : loose
of 20% weight is observed, as compared to 18% planned for complete functionalization.
Polymer was soluble in polar solvents (DMSO, Acetone) and protic solvents and insoluble in
THF, CH2Cl2.
V.4.7) Procedure for organocatalysis by poly(NHC-CO2)s
Procedure for transesterification and benzoin condensation by polyNHC-CO2
adducts:
In a typical experiment, a schlenk tube was charged with 1mmol of polymer, and 10
equivalents of reactants. 5 mL of THF where then added, and the flask was placed in an oil
bath at 80°C for 2 (or 24) hours under vigorous stirring and vacuum. After this period,
schlenk tube was left to cool down to room temperature and 1 Atm of CO2 was added for 15
minutes under stirring. Reaction mixture was then filtered under vacuum, filtrate was
analyzed by 1H NMR spectroscopy before and recovered polymer was used for another run
of catalysis.
V.5) Experimental protocol related to chapter IV
V.5.1) Preparation of imidazolium salts
Imidazolium salts prepared by the following protocols are hydroscopic compounds,
thus, to avoid their hydratation they were kept in dry atmosphere after their synthesis and
purification.
V.5.1.1) Synthesis of iPrIm+Br-
The product, 1,3-diisopropylimidazolium bromide (iPrIm+Br-), was prepared following
the procedures described in the literature4. Potassium hydroxide 0.84 g (0.015 mol), was
added to a solution of 0.63 g (0.01 mol) of imidazole in 20 mL of DMSO, the mixture was

239
stirred for 30 min at room temperature, and 1.23 g (0.01 mol) of 2-bromopropane were
added dropwise under vigorous stirring and cooling with a water bath. After 2h, the mixture
was diluted with 200 mL of water and extracted with chloroform (6 × 25 mL), the combined
extracts were washed with water and dried over MgSO4 and the solvent was distilled off. A
solution of 1.0 g (6 mmol) of 1-isopropylimidazole and 2.3 g (18 mmol) of 2-bromopropane
in THF (20 mL) was then prepared. The mixture was stirred for 24h at 80°C, the precipitate
was filtered off and dried under reduce pressure. Product was recovered as a white powder.
Yield 70%. 1H NMR (DMSO-d6): δ 1.49 (d, 4J = 1.6 Hz, N-CH-(CH3)2, 12H), 4.63 (m, N-CH-(CH3)2,
2H), 7.94 (d, 4J = 1.7 Hz CH=CH, 2H), 9.35 (s, N-CH=N, 1H). 13C NMR: δ 22.4 (N-CH-(CH3)2),
52.3 (N-CH-(CH3)2), 120.7 (CH=CH), 133.6 (N-CH=N).
V.5.1.2) Synthesis of iPrIm+HCO3-
A schlenk tube was charge with 1.17g (5 mmol) of dried iPrIm+Br- and 286 mg (5.1
mmol) of KOH. MeOH (5 mL) dried over sodium was then distilled in the schlenk and the
mixture was stirred for 30 min. The write precipitate was filtrated off, before the filtrate was
placed under a CO2 atmosphere (1 atm.) for 30 min. Solvent was distilled off and white
powder recovered kept under dry atmosphere. Yield 90%. 1H NMR (DMSO-d6): δ 1.49 (d, 4J =
1.6 Hz, N-CH-(CH3)2, 12H), 4.67 (m, N-CH-(CH3)2, 2H), 8.02 (d, 4J = 1.7 Hz CH=CH, 2H), 9.64 (s,
N-CH=N, 1H). 13C NMR: δ 23.2 (N-CH-(CH3)2), 53.1 (N-CH-(CH3)2), 121.6 (CH=CH), 134.8 (N-
CH=N), 156.8 (HCO3-).
V.5.1.2) Synthesis of iPrImCO2
The reaction was carried under an inert atmosphere. In schlenk tube, 200mg (0.86
mmol) of iPrIm+Br- were suspended in 5 mL of dried THF. The suspension was cooled to -80°C
in an ethanol bath and a solution of 200 mg (1 mmol) of KHMDS in 5 mL of dried THF was
added. The mixture was stirred for 2h before filtration under inert atmosphere. The filtrate
was then placed under a CO2 atmosphere for 30 min, the white precipitate was filtrated and
kept under dry atmosphere or allowed for hydratation at air to yield iPrIm+HCO3-. Yield 90%.
1H NMR (DMSO-d6): δ 1.41 (d, 4J = 1.6 Hz, N-CH-(CH3)v2, 12H), 5.28 (m, N-CH-(CH3)2, 2H), 7.81
(d, 4J = 1.7 Hz CH=CH, 2H. 13C NMR: δ 22.3 (N-CH-(CH3)2), 50.2 (N-CH-(CH3)2), 117.3 (CH=CH),
142.5 (N-C-(CO2)=N), 154.3 (N-C-(CO2)=N).
V.5.2) Reactions with iPrIm+HCO3-
The following protocols describe the reactions performed to prove that iPrIm+HCO3-
can generate NHCs in-situ.
V.5.2.1) Synthesis of iPrImCS2 from iPrIm+HCO3-
The reaction was carried under an inert atmosphere. In schlenk tube, 100 mg (0.47
mmol) of iPrIm+HCO3- were suspended in dry THF. 1 mL (5 mmol) of CS2 were added

240
dropwise and the reaction mixture was stirred for 15 h at 80°C. THF and CS2 were distilled off
and the product was extracted twice with acetone. Acetone was distilled and product
(iPrImCS2) was recovered as red crystals or powder. Yield 10%. 1H NMR (DMSO-d6): δ 1.41 (d, 4J = 1.6 Hz, N-CH-(CH3)2, 12H), 4.61 (m, N-CH-(CH3)2, 2H), 7.76 (d, 4J = 1.7 Hz CH=CH, 2H. 13C
NMR: δ 21.8 (N-CH-(CH3)2), 49.7 (N-CH-(CH3)2), 116.0 (CH=CH), 147.6 (N-C-(CS2)=N), 226.2 (N-
C-(CS2)=N).
V.5.2.2) Synthesis of Au(iPrIm)Cl from iPrIm+HCO3- and Au(SMe2)Cl
The reaction was carried under an inert atmosphere. In schlenk tube, 100 mg (0.47
mmol) of iPrIm+HCO3-, 138 mg (0.47 mmol) of Au(SMe2)Cl and 10 beads of molecular sieve
3A were suspended in 5 mL of dry THF. The reaction mixture was stirred for 72h at room
temperature, filtered and solvent was distilled off. Yield >95%. 1H NMR (CDCl3): δ 1.41 (d, 4J =
1.6 Hz, N-CH-(CH3)2, 12H), 5.01 (m, N-CH-(CH3)a2, 2H), 7.02 (d, 4J = 1.7 Hz CH=CH, 2H). 13C
NMR: δ 23.4 (N-CH-(CH3)2), 53.7 (N-CH-(CH3)2), 116.8 (CH=CH), 168.2 (N-C-(Au)-N).
V.5.2.3) In-situ generation of NHCs from iPrIm+HCO3- to catalyze transesterification
In a round bottom flask, 106 mg (1 mmol) of iPrIm+HCO3-, 1 mL (10 mmol) of vinyl
acetate, 1 mL (10 mmol) of benzyl alcohol and 5 mL of THF were added. The reaction
mixture was stirred for 2h at 80°C and the solvent was distilled off. The remaining solution
was analyzed by 1H NMR spectroscopy in CDCl3. Yield in benzyl acetate : > 95%.
V.5.2.4) In-situ generation of NHCs from iPrIm+HCO3- to catalyze Benzoin condensation
In a round bottom flask, 106 mg (1 mmol) of iPrIm+HCO3-, 1 mL (10 mmol) of
benzaldhyde, 10 beads of molecular sieves 3A and 5 mL of THF were added. The reaction
mixture was stirred for 48h at 60°C and the solvent was distilled off. The remaining powder
was analyzed by 1H NMR spectroscopy in DMSO-d6. Yield in benzoin : > 95%.
V.5.3) Synthesis of poly(1-vinyl-3-alkylimidazolium hydrogenocarbonate)
(PViPrIm+HCO3-)
V.5.3.1) Synthesis of 1-vinyl-3-isopropylimidazolium hydrogenocarbonate (ViPrIm+HCO3-)
A schlenk tube was charge with 500g (2.3 mmol) of dried ViPrIm+Br- and 92 mg (2.3
mmol) of KOH. MeOH (2 mL) dried over sodium was then distilled in the schlenk and the
mixture was stirred for 30 min. The write precipitate was filtrated off, and the filtrate was
placed under a CO2 atmosphere (1 atm.) for 30 min. Solvent was distilled off and yellowish
powder recovered kept under dry atmosphere. Yield 95%. 1H NMR (MeOD): δ 8.11 (s, N-
CH=CH-N, 1H), 7.96 (s, -CH=CH-N, 1H), 7.38 (dd, CH2=CH-N, 1H), 6.04 (dd, HCH=CH-N, 1H),
5.48 (dd, HCH=CH-N, 1H), 4.81 (m, (CH3)2CH- 1H), 1.66 (d, (CH3)2CH-, 6H). 13C NMR : δ 23.1 (-

241
N-CH-(CH3)2), 55.1 (-N-CH-(CH3)2), 110.0 (CH2=CH-N-), 120.9 (-CH=CH-), 122.8 (-CH=CH-),
130.0 (CH2=CH-N-), 135.2 (-N-CH=N-), 161.3 (HCO3-).
V.5.3.2) Free radical polymerization of ViPrIm+HCO3-
In a typical experiment, a 10 mL Schlenk tube was flame dried and charged with 100
mg (0.5 mmol) of ViPrIm+HCO3-, 5.9 mg (0.02 mmol) KPS and 2 mL of DMSO or MeOH. The
Schlenk tube was subjected to five freeze thaw cycles and let to rise up at room temperature
before a drop of DMAPN was added under nitrogene flow. The reaction mixture was then
stirred for 18h at room temperature. Polymerization reaction was quenched by sudden
cooling. The obtained polymer was precipitated in Acetone to remove residual monomer,
filtrated and dried in vacuum. PViPrIm+HCO3- was recovered as a yellowish powder. Yield 95
%. Mn = 11400 g.mol-1 by SEC in H20, D = 2.7. 1H NMR (MeOD): δ 9.5-10.0 (m, N-CH=N-, 1H)
7.5-8.2 (m, N-CH=CH-N, 2H), 4.1-4.7 (m, (CH3)2CH- and -CH2-CH-(N)-, 2H), 1.3-1.6 (m,
(CH3)2CH-, 6H). 13C NMR : δ 21-24 (-N-CH-(CH3)2), 30-32 (-CH2-CH-(N)-), 40-44 ((-CH2-CH-(N)-),
55-58 (-N-CH-(CH3)2), 120-125 (-CH=CH-), 134-136 (-N-CH=N-), 160-162 (HCO3-).
V.5.3.3) Synthesis of PViPrIm+HCO3- (and PVEtPhIm+HCO3
-) by anion exchange of
PViPrIm+Br- (and PVEtPhIm+Br-)
A schlenk tube was charge with 1g (4.6 mmol of monomer units) of dried PViPrIm+Br-
(synthesized by free radical polymerization as described above) and 184 mg (4.6 mmol) of
KOH. MeOH (5 mL) dried over sodium was then distilled in the schlenk and the mixture was
stirred for 30 min. The write precipitate was filtrated off, and the filtrate was placed under a
CO2 atmosphere (1 atm.) for 30 min. Solvent was distilled off and yellowish powder
recovered kept under dry atmosphere. Yield 95%.
The same procedure was used for the preparation of PVEtPhIm+HCO3-.
V.5.3.3) Synthesis of PViPrIm+HCO3- by hydratation of PolyNHC-1-CO2
The polyNHC-1-CO2, synthesized as reported before, was exposed to air for one night
to be hydrated and was then dried under reduced pressure. The yellowish powder recovered
was kept under dry atmosphere. Yield: 100%.
V.5.4) Organocatalysis in the presence of PViPrIm+HCO3-.
Transesterification and benzoin condensation reactions were conducted using the
same protocol described in part V.D.7. Few beads of molecular sieves 3A were added in the
case of benzoin condensation. After recovery, polymer was kept at air without precaution.

242
V.6) References 1. Arduengo Iii, A. J.; Rasika Dias, H. V.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1992, 114, (14), 5530-5534. 2. Marcilla, R.; Blazquez, J. A.; Rodriguez, J.; Pomposo, J. A.; Mecerreyes, D. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, (1), 208-212. 3. Wan, D.; Satoh, K.; Kamigaito, M.; Okamoto, Y. Macromolecules 2005, 38, (25), 10397-10405. 4. Starikova, O. V.; Dolgushin, G. V.; Larina, L. I.; Ushakov, P. E.; Komarova, T. N.; Lopyrev, V. A. Russ. J. Org. Chem. 2003, 39, (10), 1467-1470.

243

244
Annexes

245

246
Annexe 1 : Spectre de RMN 1H (δ en ppm) dans le CDCl3 de VBuIm+Br-.
Annexe 2 : Spectre de RMN 1H (δ en ppm) dans l’acétone-d6 de VPhEtIm+Br-.
ab
c
d
ej
fg
h
i
bc a
dejf
gh
i
CDCl3
ab
c
d
ef
hgi
i
i i
i
ab
c
d efg
h
i
D2O Acetone

247
Annexe 3 : Spectre de RMN 1H (δ en ppm)dans le D2O du PVBuIm+Br-.
Annexe 4 : Spectre de RMN 1H (δ en ppm) dans le D2O du PVPhEtIm+Br-.
ab
c
ch
de
f
g
h a
b+dc
e
f
g
Acetone
H2O
g
ab
c
cg
d e
ff
f
ff
f+c
b
d
ae
MeOH
Acetone
DMSO

248
Annexe 5 : Spectre de RMN 13C (δ en ppm) dans le THF-d8 de PVBuIm+NTf2-
Annexe 6 : Spectre de RMN 13C dans le THF-d8 (δ en ppm) du PVPhEtIm+NTf2-.
ab
c
de
fg
h
i
e
i
h
g
f
c + db a
NTf2
THF
ab
c
cg
de
ff f
f
ff
ab
d
c
e
f
g
NTf2

249
Annexe7 : Spectre de RMN 1H (δ en ppm) du polyNHC-3 dans le THF-d8.
Annexe 8 : Spectre de RMN 1H (δ en ppm) dans le THF-d8 du PolyNHC-3.
ab
c
d
ef
g
h
h
g
f
e
c + d
b
a
THF
DMSO
ab
c
c
de
ff
ff f
f
c b+da
e
THF

250
Annexe 9 : Spectre de RMN 13C (δ en ppm) de polyNHC-3-CO2 dans le DMSO-d6
Annexe 10 : Chromatogrammes d’exclusion stérique dans le DMF du macroagent de
transfert PNVP et du copolymère à bloc PNVP-b-PVAc obtenus par polymérisation MADIX.
a
ab
c
c
gh
e df
f
f
ff
f
c
db e
f
h
g
KNTf2
THF
469 569 669 769 869 969 1069 1169 1269
PNVP(5K)
elution time (S.)
PNVP(5K)-PVAc

251
Résumé : Des carbènes N-hétérocycliques (NHC)s ont été employés comme catalyseurs
organiques de la polymérisation par étapes du téréphtaldéhyde. Cette partie est une
application en chimie des polymères de la réaction de « condensation de la benzoïne »
catalysée par les NHCs impliquant un mono-aldéhyde analogue. Des poly(1,4-phénylène-1-
oxo-2-hydroxyéthylène)s ou « polybenzoïnes » ont ainsi été obtenus par polymérisation en
solution dans le DMSO ou le THF comme solvant à une température inférieure à 40 °C. La
présence de chaines de polybenzoïne cycliques a pu être mise en évidence. La proportion de
ces cycles dépend de la nature du catalyseur carbénique et de la polarité du milieu
réactionnel.
Dans la deuxième partie du travail, des solutions simples ont été proposées pour
manipuler les NHCs de manière plus aisée, en évitant leur dégradation prématurée. Pour ce
faire, des supports polymères porteurs de sites carbéniques, c’est à dire des « poly(NHC)s »,
ont été développés et employés à des fins d’organocatalyse. Bien que recyclables, ces
« poly(NHC)s » restent assez sensibles aux traces d’impuretés. Un moyen de les protéger est
de les faire réagir avec le CO2. Les adduits ainsi formés, « poly(NHC-CO2)s », peuvent alors
être employés comme précurseurs pour générer les « poly(NHC)s » in situ, par simple
activation thermique, le retour aux « poly(NHC-CO2)s » pouvant être effectué par
carboxylation des « poly(NHC)s ». Enfin, des méthodes de synthèse de composés de type
imidazolium (version moléculaire) et polyimidazolium (version polymère supportée) à contre
anion hydrogénocarbonate (HCO3-) ont été développées. De tels précurseurs peuvent eux-
mêmes servir de pré-catalyseurs (moléculaires ou polymères) pour générer, par chauffage,
des NHCs et poly(NHC)s, offrant un moyen très pratique de mener des réactions
d’organocatalyse et de recycler les catalyseurs.
Abstract : N-Heterocyclic carbenes (NHCs) have been employed as organic catalysts for the
step-growth polymerization of terephtaldehyde. This part is an application to polymer
chemistry of the so-called “benzoin condensation”, reaction catalyzed by NHCs involving a
mono-aldehyde substrate. Poly(1,4-phenylene-1-oxo-2-hydroxyethylene)s or
« polybenzoins » have thus been obtained by polymerization reactions conducted in DMSO
or THF at temperatures below 40°C. Presence of cyclic polybenzoins has been put forward.
The content of such cyclic species was found to vary as a function of the NHC catalyst
employed and of the reaction media used.
In a second part, simple solutions have been proposed to easily handle NHCs, by
avoiding their degradation. For this purpose, polymer supports bearing NHCs moities, i.e
“poly(NHC)s”, have been developed and employed for the purpose of organocatalysis. Even
if “poly(NHC)s” were found to be recyclable, they still remain sensitive to impurities. Another
way to protect the carbenic centers is to react “poly(NHC)s” with CO2. The adducts thus
obtained, “poly(NHC-CO2)s”, can then be employed as precursors for the in situ generation
of “poly(NHC)s”, by a simple thermal activation. A further carboxylation of such generated
species allow for the recovering of “poly(NHC-CO2)s”. Finally, synthetic methods for the

252
preparation of imidazolium (molecular version) and polyimidazolium (supported polymer
version) salts with hydrogenocarbonate (HCO3-) as counter-anion have been developed.
Such precursors can serve as precatalysts (molecular or supported) to generate, by heating,
NHCs and poly(NHC)s, giving a practice way to conduct organocalysed reactions and recycle
the catalysts.




















