
POTENSI METFORMIN -...
83
Transcript of POTENSI METFORMIN -...


POTENSI METFORMINSEBAGAI ANTI KANKER PAYUDARA
Irma Yanti RangkutiPoppy Anjelisa Zaitun Hasibuan
Tri WidyawatiYahwardiah Siregar
Yuandani
2019

POTENSI METFORMINSEBAGAI ANTI KANKER PAYUDARA
----------------------------------------------------Hak Cipta © 2019 pada penulis. dilarang keras mengutip,menjiplak, memphoto copy baik sebagian atau keseluruhandari isi buku ini tanpa mendapat izin tertulis danpengarang dan penerbit.
--------------------------------------Penulis:Irma Yanti Rangkuti
Poppy Anjelisa Zaitun HasibuanTri Widyawati
Yahwardiah SiregarYuandaniISBN : 978-623-7297-03-1---------------------------------------------------------Editor:
dr. Saiful Batubara, M.PdPenyunting:Drs. Saiful Anwar Matondang, M.A., Ph.DDesain Sampul Dan Tata Letak :
Syahlan, M.Pd-----------------------------------------------------------Penerbit:UISU PressRedaksi :JL. Sisingamangaraja – Teladan, Medan 20217Telp. (061) 7869790 email : [email protected] Pertama Juni 2019© HAK CIPTA DILINDUNGI OLEH UNDANG-UNDANG

KATA PENGANTAR
Puji syukur kehadirat Tuhan Yang Maha Esa
yang telah melimpahkan rahmad-Nya sehingga penulis
dapat menyelesaikan buku yang berjudul “Potensi
Metformin sebagai Anti Kanker Payudara”.
Kanker payudara merupakan salah satu masalah
kesehatan dunia, selain insidensinya semakin tahun
semakin meningkat tetapi juga merupakan penyakit
dengan angka kematian yang cukup tinggi. Berbagai
kendala mulai dari diagnosa, akses pengobatan serta
terapi terjadi terutama di Negara berkembang yang
menyebabkan keterlambatan diagnosa serta penangan
yang menyebabkan tingginya angka kematian akibat
kanker payudara. Terapi yang masih substandard salah
satu momok besar dalam penangan kasus ini. Sejumlah
penelitian telah dikembangkan untuk menemukan terapi
terbaik untuk kanker payudara. Berbagai obat serta
tanaman obat diuji coba untuk menemukan obat anti
kanker payudara dengan efektifitas yang tinggi untuk sel

kanker tanpa menimbulkan efek toksik untuk sel yang
normal.
Metformin merupakan obat pilihan untuk
diabetes melitus (DM) tipe 2. Berawal adanya penurunan
kasus kanker pada pasien DM tipe 2 yang menggunakan
metformin, memberikan inspirasi peneliti untuk meneliti
lebih jauh kemampuan anti kanker metformin.
Buku ini berisi hasil-hasil penelitian yang
menggunakan metformin sebagai anti kanker untuk
beberapa model sel kanker serta hasil penelitian terbaru
dari penulis sendiri yang ingin membuka secara jelas
efek anti kanker metformin terkhusus untuk kanker
payudara yang dilaksanakan di Laboratorium
Parasitologi Fakultas Kedokteran Universitas Gadjah
Mada Yogyakarta pada tahun 2018.
Buku ini dapat dijadikan salah satu rujukan bagi
peneliti yang berkecimpung dalam bidang kanker untuk
mengembangkan penelitian dalam bidang ini, mahasiswa
serta sejawat dokter yang langsung berhadapan dengan
pasien-pasien kanker payudara.
Ucapan terimakasih kami sampaikan sebesar-
besarnya kepada PT Dexa Medica Palembang yang telah

berkontribusi dalam penyediaan bahan baku metformin
HCl dan seluruh tim, DR. Poppy Anjelisa Zaitun
Hasibuan, S.Si, M.Si, Apt; dr. Tri Widyawati, M.Si,
Ph.D; Dr. med. dr. Yahwardiah Siregar dan Yuandani,
S.Si, M.Si, Ph.D; yang telah memberi semangat,
masukan serta dukungan hingga akhirnya penelitian ini
selesai. Semoga buku ini memberi manfaat bagi
kemajuan pendidikan dan penelitian kesehatan.

- 0 -
DAFTAR ISI
Kata Pengantar iDaftar Isi iv
BAB 1. PENDAHULUAN.................................................................. 1BAB 2. KANKER ............................................................................... 5
2.1 Tinjauan Umum Kanker..................................................... 52.2 Karakter Sel Kanker ........................................................... 52.3 Karsinogenesis ................................................................... 62.4 Siklus Sel............................................................................ 72.5 Mekanisme Apoptosis ..................................................- 10 -
BAB 3. KANKER PAYUDARA ..................................................- 15 -3.1 Epidemiologi ................................................................- 15 -3.2 Etiologi dan faktor risiko..............................................- 16 -3.3 Terapi ...........................................................................- 17 -3.4 Obat Anti Kanker .........................................................- 20 -
BAB 4. PENGUJIAN AKTIVITAS OBAT ANTI KANKER......- 26 -4.1 Uji Sitotoksisitas ..........................................................- 26 -4.1 Uji Siklus Sel dan Apoptosis Menggunakan
Flowcytometry ..............................................................- 27 -4.2 Uji Ekspresi Protein Menggunakan Imunositokimia........ 29
BAB 5. METFORMIN SEBAGAI ANTI KANKER....................- 31 -5.1 Mekanisme anti kanker metformin...............................- 32 -5.2 AMPK (Adenosine Monophosphate Kinase) ...............- 35 -5.3 Penelitian metformin sebagai anti kanker payudara.....- 40 -
KESIMPULAN .............................................................................- 62 -REFERENSI..................................................................................- 63 -

- 1 -
BAB 1 PENDAHULUAN
Kanker suatu kelainan genetik akibat mutasiDeoxyribonucleic Acid (DNA) sebagian besar terjadi spontanatau diinduksi dari lingkungan. Kelainan genetikmenyebabkan perubahan ekspresi atau fungsi gen kunci yangmengatur proses mendasar sel seperti pertumbuhan,pertahanan dan penuaan (senescence)1. Terdapat enam tandakhas kanker, yaitu: menghindari apoptosis, mencukupi sendirisinyal pertumbuhan, tidak peka terhadap sinyal antipertumbuhan, potensi replikasi tanpa batas, angiogenesisterus-menerus dan invasi ke jaringan dan metastasis2.
Kanker merupakan masalah kesehatan dunia, di tahun2008 terdapat 12,7 juta kasus kanker baru dan 7,6 jutameninggal akibat kanker. Kanker yang paling banyakterdiagnosa di seluruh dunia adalah kanker paru (1,6 juta,12,7%), kanker payudara (1.38 juta, 10.9%) dan kankerkolorektal (1.23 juta, 9.7%)3. Data Globocan tahun 2012menunjukkan 14.067.894 kasus baru kanker dan 8.201.575kematian akibat kanker di seluruh dunia dengan jenis kankerterbanyak, yaitu kanker payudara, kanker prostat dan kankerparu. Data kanker terbaru tahun 2018 menyebutkan kasuskanker tahun 2018 tercatat 18,1 juta dengan angka kematianakibat kanker sekitar 9,6 juta. Kanker paru menempati urutanpertama untuk kasus baru (11,6%, 2,094 juta) dengan angkakematian tertinggi sekitar 18,4% (1,8 juta). Kasus kankerterbanyak lainnya adalah kanker payudara disusul kankerkolorektal, prostat dan lambung5.
Kanker payudara merupakan jenis kanker terbanyakpada wanita, setiap tahun ditemukan peningkatan kasusdengan angka kematian yang cukup tinggi. Tahun 2002ditemukan 1,15 juta kasus baru dengan angka kematian akibatkanker payudara sekitar 411 ribu setiap tahun di seluruhdunia6. Tahun 2012 tercatat 1,7 juta kasus baru kankerpayudara dengan angka kematian 522 ribu7. Tahun 2018

- 2 -
sebanyak 2,089 juta kasus baru kanker payudara denganangka kematiannya sekitar 6,6% (627 ribu)5. Tahun 2050diperkirakan insiden kanker payudara seluruh dunia sekitar3,2 juta6. Untuk Indonesia, insiden kanker pada perempuansekitar 134 per 100.000 penduduk dengan kasus terbanyakkanker payudara sebesar 40 per 100.000 perempuan. EstimasiGlobocan, kematian di Indonesia akibat kanker payudarasekitar 16,6 kematian per 100.000 penduduk4. Data statistikWHO ini menunjukkan bahwa kanker payudara merupakanpenyakit yang banyak diderita dan penyebab kematiantertinggi di dunia dan Indonesia merupakan salah satu negaradengan kasus kanker payudara terbanyak.
Kanker payudara merupakan suatu penyakitterjadinya pertumbuhan yang berlebihan atau perkembangantidak terkontrol dari sel-sel jaringan payudara. Penyebabkanker payudara belum seluruhnya dapat difahami, namunada tiga hal yang mempengaruhi terjadi kanker payudara,yaitu perubahan genetik, pengaruh hormonal dan variabellingkungan2. Faktor risiko yang erat kaitannya denganpeningkatan insiden kanker payudara antara lain jenis kelaminwanita, usia diatas 50 tahun, riwayat keluarga dan genetik(Pembawa mutasi gen BRCA1, BRCA2, ATM atau TP53(p53)), riwayat penyakit payudara sebelumnya (DCIS padapayudara yang sama, LCIS, densitas tinggi pada mamografi),riwayat menstruasi dini (< 12 tahun) atau menopause lambat(>55 tahun), riwayat reproduksi (tidak memiliki anak dantidak menyusui), hormonal, obesitas, konsumsi alkohol,riwayat radiasi dinding dada, faktor lingkungan4. Hal inimenunjukkan bahwa risiko terjadinya kanker payudara padawanita akan meningkat apabila wanita tersebut memilikifaktor-faktor risiko seperti yang disebutkan. Faktor-faktorrisiko tersebut ada yang bersifat tidak dapat diubah (sepertijenis kelamin, usia dan faktor genetik) dan juga terdapatfaktor risiko yang dapat diubah (seperti obesitas, konsumsi

- 3 -
alkohol, radiasi dinding ada) yang dapat dimodifikasi denganmerubah gaya hidup sehat.
Kanker payudara secara klinis diklasifikasikanberdasarkan karakteristik morfologik meliputi infiltratingductal carcinoma, infiltrating lobular carcinoma, tubular,mucinous, medullary, dan adenoid cystic carcinoma.Klasifikasi lain berdasarkan ekspresi reseptor estrogen (ER)dan progesterone (PR) dan Her2 oncogene7 .
Terapi kanker payudara berdasarkan stadium danekspresi molekular. Terapi terdiri dari operatif, radioterapi,terapi hormonal, kemoterapi, transtuzumab dan terapisuportif8. Kemoterapi berperan penting dalam terapi kankerpayudara9,10,11, namun kemoterapi memiliki beberapapermasalahan, diantaranya efektifitas terapi yang bervariasidiantara individu, efek samping obat12 sampai resistensi13.
Resistensi terapi pada kanker payudara diantaranyaterjadi pada kasus kanker payudara dengan mutasi pada p53,diantaranya terhadap golongan antrasiklin14,15, dan nonantrasiklin, CMF16 dan juga terhadap 5-fluorouracil danmytomicin C14.
Metformin 1,1-dimetylbiguanida berasal dari alkaloidgalegine atau isoamylen guanidine, substansi aktif dariGallega officinales yang juga dikenal sebagai Goat’es Rue,telah digunakan sejak tahun 1958 sebagai obat diabetesmelitus (DM) tipe 2 di United Kingdom dan tahun 1995 diUnited States dan direkomendasikan sebagai first line therapyuntuk seluruh pasien yang baru didagnosa dengan DM tipe 2oleh American Diabetes Association 201417.
Penelusuran aktivitas anti kanker metforminberdasarkan data sebelumnyamenunjukkan penurunan risikoterjadinya kanker pada pasien DM tipe 2 yang menggunakanmetformin dibandingkan pasien yang tidak menggunakanmetformin seperti insulin, sulfonilurea, sitagliptin dan

- 4 -
pioglitazone18,19. Keseluruhan obat yang digunakan tersebutmerupakan obat anti diabetes melitus, dan ternyata setelahdibandingkan terjadinya kasus kanker setelah beberapa tahunpenggunaannya menunjukkan kasus kanker pada pasien DMtipe 2 yang menggunakan metformin lebih rendah dibandingobat anti diabetes yang lain.
Penelitian secara invitro telah dilakukan diantaranyake beberapa jenis sel kanker seperti sel WiDr20, sel Hep G-221,sel MCF-722 , MDA-MB-23123 dan MDA-MB-435 seluruhnyamenunjukkan efek penekanan pertumbuhan sel kanker.Aktifitas metformin terhadap sel WiDr dilaksanakan tahun2015 di Laboratorium Parasitologi Fakultas KedokteranUniversitas Gadjah Mada oleh Tim dari Fakultas KedokteranUniversitas Sebelas Maret Indonesia. Penelitian terhadap selHep G-2 dilaksanakan oleh Tim Rumah Sakit UniversitasShantou Guangdong China tahun 2013. Penelitian jugadilakukan terhadap varian sel kanker payudara lain yaituMCF-7 yang dilaksanakan tahun 2014 oleh Tim dari FakultasBiomedik dari Kanada dan Brazil, serta pengujian terhadapsel kanker payudara MDA-MBA-231 tahun 2015 oleh Timdari Departemen Biologi Kampolis USA bersama denganstaff dari Departemen onkologi Institut Kanker China.

- 5 -
BAB 2. KANKER
2.1 Tinjauan Umum Kanker
Kanker adalah pertumbuhan sel tidak beraturan yangmuncul dari satu sel. Kanker merupakan pertumbuhanjaringan secara otonom dan tidak mengikuti aturan danregulasi sel yang tumbuh normal. Tumor adalah istilah umumyang menunjukkan massa dari pertumbuhan jaringanabnormal. Pertumbuhan yang tidak terkendali tersebutdisebabkan kerusakan DNA, menyebabkan mutasi di gen vitalyang mengontrol pembelahan sel. Beberapa mutasi mungkindibutuhkan untuk mengubah sel normal menjadi sel kanker.Mutasi-mutasi tersebut sering diakibatkan agen kimia maupunfisik yang disebut karsinogen, mutasi dapat terjadi secaraspontan (diperoleh) ataupun diwariskan (mutasi germline)24.
2.2 Karakter Sel Kanker
Sel kanker memiliki perbedaan yang sangat signifikandengan sel normal antara lain:a. Sel kanker kehilangan kemampuan apoptosis sehingga sel
tersebut terus bertambah25.b. Sel kanker tidak mengenal komunikasi ekstraseluler atau
asosial. Komunikasi ekstraseluler ini diperlukan untukmenjalin koordinasi antar sel sehingga dapat salingmenunjang fungsi masing-masing. Sel kanker bersifatasosial berarti sel kanker dapat bertindak sendiri tanpaperduli apa yang dibutuhkan oleh lingkungannya. Hal iniditunjukkan dari kemampuan sel kanker memproduksigrowth factor sendiri, tidak tergantung rangsangan sinyalpertumbuhan luar untuk berproliferasi2.
c. Sel kanker tidak sensitif terhadap anti faktor pertumbuhanyang menyebabkan siklus sel tidak terhenti25.
d. Sel kanker bersifat invasif (menyerang jaringan sekitar),merusak jaingan tersebut dan bermetastasis (menyerangjaringan lain yang jauh)26.

- 6 -
e. Bersifat immortal (berpotensi melakukan replikasi tidakterbatas)25.
f. Sel kanker mampu membentuk pembuluh darah baru(neoangiogenesis)2.
2.3 Karsinogenesis
Karsinogenesis adalah suatu proses perubahanstruktur DNA yang bersifat irreversible sehingga terjadikanker26. Salah satu faktor terbentuknya kanker adalahdisebabkan adanya sel epitel yang terus berkembang(berproliferasi). Saat berproliferasi, genetik sel bisa berubahakibat adanya pengaruh agen karsinogen yang menyebabkanhilangnya penekanan terhadap proses proliferasi sel.Perubahan sel menjadi ganas juga melibatkan gen-gen yangmengatur pertumbuhan sel akibatnya sel berkembang tidakterkendali. Kanker terjadi melalui beberapa tingkat yaitu27:
a. Fase inisiasi: DNA dirusak akibat radiasi atau zatkarsinogen (radikal bebas).
b. Fase promosi: zat karsinogen tambahan (co-carcinogens)diperlukan sebagai promotor untuk mencetuskanproliferasi sel.
c. Fase progresi: gen-gen pertumbuhan yang diaktivasi olehkerusakan DNA mengakibatkan mitosis dipercepat danpertumbuhan liar dari sel-sel ganas.
Tahap inisiasi adalah tahap pertama padakarsinogenesis ketika sel normal mulai mengalami mutasioleh karsinogen27. Tahap karsinogenesis selanjutnya adalahpromosi merupakan tingkat lanjutan dari tahap inisiasi. Padatahap ini, sel mulai mengalami hiperplastik pada inti sel. Sel-sel akan memperoleh beberapa keuntungan selektif untuktumbuh sehingga pertumbuhannya menjadi cepat danberubah menjadi tumor jinak. Tahap promosi tidak melibatkanperubahan struktural dari genom secara langsung, tetapi

- 7 -
biasanya terjadi perubahan ekspresi gen yang terinisiasi28.
Pada tahap progresi, kemampuan pembelahan yangtinggi menuntun terbentuknya koloni sel yang lebih besarmelalui perubahan genetik lebih lanjut dan peningkatanmobilitas dan angiogenesis.Pada tahap ini, sel-sel tumordikatakan sebagai sel malignan. Pada fase ini juga akan terjadikarsinoma dan metastasis melalui aktivasi onkogen danmalfungsi dari enzim topoisomerase. Tahap metastasismerupakan tahap akhir dalam karsinogenesis.Pada tahap inisel kanker melakukan invasi ke jaringan-jaringan lain didalam tubuh melalui pembuluh darah, pembuluh limpa ataurongga tubuh.Sel malignan yang bermetastasis ini masukmelalui membran menuju saluran limfoid. Sel tersebut akanberinteraksi dengan sel limfoid yang digunakan sebagaiinangnya. Selanjutnya, sel kanker akan masuk ke jaringanlainnya membentuk tumor sekunder dengan didukungkemampuan neoangiogenesis yang dimilikinya2.
Tahap metastasis dapat berlangsung karenamelemahnya ikatan antarsel yang disebabkan olehterdegradasinya CAMs (Cell-cell Adhesion Molecules) dan E-cadherin sebagai molekul yang menjaga pertautan antarsel.Molekul-molekul tersebut diketahui sudah sangat sedikitbahkan tidak ditemukan lagi pada sel kanker sehingga prosesmetastasis dapat terus terjadi2. Adanya mutasi pada satu seltunggal normal sebagai akibat terpapar oleh karsinogen(inisiasi) akan menyebabkan progresi sel menjadi hiperplasia(promosi), displasia (progresi) dan pada akhirnya memilikikemampuan invasi ke jaringan sekitarnya (metastasis)28.
2.4 Siklus Sel
Siklus sel merupakan proses perkembangbiakan selyang memperantarai pertumbuhan dan perkembanganmakhluk hidup. Setiap sel baik normal maupun kankermengalami siklus sel. Siklus sel memiliki dua fase utama,

- 8 -
yakni fase S (sintesis) dan fase M (mitosis). Fase Smerupakan fase terjadinya replikasi DNA kromosom dalamsel sedangkan pada fase M terjadi pemisahan 2 set DNAkromosom tersebut menjadi 2 sel29.
Selain itu, terdapat fase yang membatasi kedua faseutama tersebut yang dinamakan Gap.G1 (Gap-1) terdapatsebelum fase S dan setelah fase S dinamakan G2 (Gap-2).Pada fase G1, sel melakukan persiapan untuk sintesis DNA.Fase ini merupakan fase awal cell cycle progression yangdiatur oleh faktor ekstraselular seperti mitogen dan molekuladhesi. Penanda fase ini adalah adanya ekspresi dan sintesisprotein sebagai persiapan memasuki fase S. Pada fase G2, selmelakukan sintesis lebih lanjut yang memadai untuk prosespembelahan sehingga sel siap melakukan pembelahan padafase M24.
Siklus sel dikontrol oleh beberapa protein danbertindak sebagai regulator positif dan negatif. Kelompoksiklin khususnya siklin D, E, A dan B merupakan protein yanglevelnya fluktuatif selama proses siklus sel. Siklin bersamadengan kelompok siklin dependent kinase (CDK) khususnyaCDK 4, 6 dan 2, bertindak sebagai regulator positif yangmemacu terjadinya siklus sel. Pada mamalia ekspresi kinase(CDK4, CDK2 dan CDC2/CDK1) terjadi bersamaan denganekspresi siklin (D, E, A dan B) secara berurutan seiringdengan jalannya siklus sel (G1-S-G2-M)29.
Aktivasi CDK dihambat oleh regulator negatif siklussel, yakni CDK inhibitor (CKI), yang terdiri dari Cip/Kipprotein (meliputi p21, p27, p57) dan keluarga INK4 (meliputip16, p18, p19). Selain itu, tumor suppressor protein yaitu p53dan pRb juga bertindak sebagai protein regulator negatif 30.
Aktivasi CDK memerlukan ekspresi siklin (cyc).Kompleks siklin-CDK dengan protein CKI dan adanyafosforilasi oleh Wee1 (tyrosin15)/ Myt1 (threonin14) dapat

- 9 -
menyebabkan inaktivasi CDK.Aktivasi kompleks siklin-CDKdiawali dengan proteolisis CKI oleh ubiquitin, kemudianfosforilasi CDK oleh CDK-activating kinase (CAK) padathreonin 161 dan penghilangan fosfat (defosforilasi) olehCdc25 fosfatase pada target fosforilasi Wee1(tyrosin15)/Myt1 (threonin14). CDK bekerja pada awal G1untuk mengaktifkan E2F-dependent transcription gen yangdiperlukan untuk fase S (di akhir G1 untuk menginisiasi faseS) dan juga di akhir G2 untuk menginisiasi mitosis (M)29.
Checkpoint pada G2 terjadi ketika ada kerusakanDNA yang akan mengaktivasi beberapa kinase termasukataxia telangiectasia mutated (ATM) kinase. Hal tersebutmenginisiasi dua kaskade untuk menginaktivasi Cdc25-CycBbaik dengan jalan memutuskan kompleks Cdc25-CycBmaupun mengeluarkan kompleks Cdc25-CycB dari nukleusatau aktivasi p21.Checkpoint pada fase G1 akan dapat dilaluijika (1) ukuran sel memadai; (2) ketersediaan nutrienmencukupi; dan (3) adanya faktor pertumbuhan (sinyal darisel yang lain). Checkpoint pada fase G2 dapat dilewati jikaukuran sel memadai dan replikasi kromosom terselesaikandengan sempurna26.
Checkpoint pada fase M (mitosis) terjadi jika benangspindle tidak terbentuk atau jika semua kromosom tidakdalam posisi yang benar dan tidak menempel dengansempurna pada spindle.Checkpoint tersebut bekerja denganmemonitor apakah kinetokor dan mikrotubul terhubung secarabenar. Jika tidak, kohesi kromatid akan tetap berlangsung danmikrotubul gagal untuk memendek sehingga kromatid tidakbergerak menjauh ke kutub yang berlawanan. Kontrolcheckpoint sangat penting untuk menjaga stabilitasgenomik.Kesalahan pada checkpoint akan meloloskan seluntuk berkembang biak meskipun terdapat kerusakan DNAatau replikasi yang tidak lengkap atau kromosom tidakterpisah sempurna sehingga akan menghasilkan kerusakan

- 10 -
genetik. Oleh karena itu, proses regulasi siklus sel mampuberperan dalam pencegahan kanker24.
2.5 Mekanisme Apoptosis
Apoptosis merupakan proses kematian sel terprogrampada sel tunggal, dengan gambaran morfologi dan biokimiawikhas tanpa menimbulkan reaksi radang. Gambaran histologisapoptosis berupa kondensasi kromatin, jisim apoptotik danfagositosis jisim apoptotik tanpa reaksi radang. Gambaranmorfologi apoptosis berupa pengecilan sel, kondensasikromatin dan fragmentasi inti31.
Apoptosis adalah mekanisme kematian sel yangterprogram yang dianggap sebagai komponen penting dariberbagai proses termasuk pergantian sel normal32. Apoptosisterjadi secara normal selama perkembangan dan penuaan seljuga sebagai mekanisme homeostasis untuk mempertahankanpopulasi sel dalam jaringan. Apoptosis juga terjadi sebagaimekanisme pertahanan seperti pada reaksi imun atau ketikasel-sel mengalami kerusakan oleh penyakit atau agenberbahaya33. Pada tingkat molekuler apoptosis dibagi menjadi3 fase yaitu fase inisiasi, fase eksekusi dan fase terminasi.Pada fase inisiasi apoptosis distimulasi berbagai macam faktorseperti rendahnya konsentrasi faktor pertumbuhan, radiasisinar gamma, obat-obatan kemoterapi dan sinyal dari deathreceptor.Fase eksekusi ditandai dengan penggelembunganmembran sel (blebbing), fragmentasi inti, kondensasikromatin dan degradasi DNA. Pada fase terminasi jisimapoptotik akan difagositosis oleh sel-sel fagosit34.
Apoptosis terjadi melalui 2 jalur yang dipicu olehbermacam-macam faktor baik internal (jalur intrinsik)maupun eksternal (jalur ekstrinsik)35. Jalur ekstrinsik dimulaidari adanya pelepasan molekul signal yang disebut ligan olehsel lain tetapi bukan berasal dari sel yang akan mengalamiapoptosis, seperti TNF-α, Fas-Ligand (Fas-L), TNF-related

- 11 -
apoptosis including ligand (TRAIL), dan Apo-3 ligand (Apo-3L). Ligan tersebut berikatan dengan death receptor yangterletak pada transmembran sel target yang menginduksiapoptosis. Death receptor yang terletak di permukaan seladalah famili reseptor TNF (Tumor Necrosis Factor), yangmeliputi CD 95, TNF-α receptor 1 (TNF-R1), TNF-α receptor2 (TNF-R2), Fas, death receptor-3 (DR-3)31.
Setelah berikatan dengan ligan yang sama, deathreceptor membentuk kompleks homotrimerik yangmenyebabkan protein adaptor intraseluler tertarik ke membransel seperti TNF-R1 dan DR-3 yang disebut TNFR-associateddeath domain protein (TRADD). Sedangkan Fas dan DR-4berinteraksi dengan Fas-associated death domain protein(FADD). FADD dan TRAAD tidak berinteraksi dengan DR-5sehingga diduga ada protein lain yang terlibat. Sinyal yangdiaktivasi oleh TNF-R1 atau DR- 3 terpecah pada tingkatTRADD.Translokasi inti faktor transkripsi nuclear factor-қB(NF-қB), dan aktivasi c-Jun N-terminal Kinase (JNK) dimulai.Sinyal TNF-α akan berikatan dengan sinyal jalur Fasmenyebabkan interaksi antara TRADD dengan FADD36.Ikatan-ikatan tersebut mengirimkan sinyal ke sitoplasmauntuk mengaktivasi caspase-8 selanjutnya terjadi kaskadecaspase untuk apoptosis37.
Jalur intrinsik
Radiasi ionisasi, kerusakan karena oksidasi radikalbebas, dan gangguan pada siklus sel dapat menyebabkanstress mitokondria yang menginduksi apoptosis jalur intrinsikdisebabkan oleh senyawa kimia atau kehilangan faktorpertumbuhan, sehingga menyebabkan gangguan padamitokondria dan terjadi pelepasan dua kelompok proteinproapoptosis dari intermembran mitokondria ke sitosol.Kelompok pertama seperti sitokrom c, Samc/ Diablo,Apoptosis Inducing Factor (AIF), dan omi/Htr2. Protein inimengaktifkan caspase yang bergantung jalur mitokondria.

- 12 -
Sitokrom c mengikat dan mengaktifkan Apaf-1 sertaprocaspase-9, membentuk apoptosome yang akanmengaktivasi kaskade caspase. Smac/DIABLO danHtrA2/Omi menginduksi apoptosis dengan menghambataktivitas IAP (inhibitors of apoptosis proteins) 32. Kontrol danregulasi peristiwa apoptosis mitokondria juga dapat terjadimelalui anggota dari famili protein Bcl-2.
Famili protein Bcl-2 mengatur permeabilitasmembran mitokondria dan dapat menjadi protein proapoptosisatau antiapoptosis. Protein penekan tumor p53 memiliki peranpenting dalam regulasi famili protein Bcl-2, dengan caraberikatan langsung dengan famili protein Bcl-2 antiapoptosisdan mengaktifkan famili protein Bcl-2 proapoptosis lalukemudian mengatur permeabilitas membran mitokondria.Sampai saat ini, total 25 gen telah diidentifikasi dalam familiprotein Bcl-2. Beberapa protein antiapoptosis termasuk Bcl-2,Bcl-x, Bcl-XL, Bcl-XS, Bcl-w, TAS, dan beberapa proteinproapoptosis termasuk Bcl-10, Bax, Bak, Bid, Bad, Bim, Bik,dan Blk. Protein ini memiliki peran penting karena dapatmenentukan apakah sel melakukan apoptosis atau tidak32.Diperkirakan bahwa mekanisme utama tindakan dari familiprotein Bcl-2 adalah regulasi pelepasan sitokrom c darimitokondria melalui perubahan permeabilitas membranmitokondria38.
Jalur ekstrinsik dan jalur intrinsik akan berakhir padatitik fase eksekusi yang dianggap jalur akhir apoptosis.Dimulai dengan aktivasi caspase yang mengaktifkanendonuklease sitoplasma, lalu mendegradasi badan inti, danmengaktivasi protease yang mendegradasi protein inti dansitoskeletal.Caspase adalah kelompok dari cysteine-asparticacid protease.Kaskade caspase berperan penting pada faseapoptosis sel. Caspase disintesis sebagai rantai tunggalzymogen tidak aktif yang terdiri dari N-terminal prodomaindiikuti oleh subunit besar berukuran 20 kDa dan subunit kecil

- 13 -
berukuran 10 kDa.Aktivasi dari zymogen didapatkan lewatpemecahan proteolitik pada tempat yang identik dengan motifcaspase yang dikenal. Mekanisme ini memungkinkan caspasedapat memproses dirinya sendiri atau caspase zymogen lain24.
Caspase dapat dibagi menjadi kelompok inisiatordan kelompok eksekutor. Caspase-3 merupakan salah satucontoh dari caspase kelompok eksekutor. Aktivasi caspase-3merupakan salah satu poin kunci dalam transmisi dari sinyalapoptosis karena aktifitas pemecahan caspase-3 menghasilkanberbagai morfologi dan berbagai jenis bahan biokimia dariapoptosis39. Caspase-3 diaktifkan dalam proses apoptosis baikoleh jalur ekstrinsik (death ligand) dan intrinsik (jalurmitokondria). Aktivasi dari caspase-3 diperlukan karena bilatidak maka aktivitas caspase akan membunuh sel-sel tanpapandang bulu. Sebagai caspase eksekutor, caspase-3 hampirtidak memiliki aktivitas sampai dibelah oleh caspase inisiatorsetelah peristiwa sinyal apoptosis telah terjadi40. Procaspase-3merupakan bentuk tidak aktif dari caspase-3 yang memilikiberat molekul 32 kDa yang bila diaktifkan akan menjadi subunit berukuran 20 kDa dan 12 kDa, seiring dengan perubahanmenjadi bentuk aktif maka sub unit 20 kDa secara otomatisberubah menjadi sub unit 17 kDa24.
Aktivasi dari procaspase-3 merupakan peran daricaspase-3, caspase-8, caspase-9, caspase-10, CPP32activating protease, serta granzyme B. Ketika caspase-3 aktifmaka akan menyebabkan pembelahan atau degradasi dariberbagai substrat yang bertinteraksi dengan caspase-3 sepertiprocaspase-3, procaspase-6, procaspase-9, DNA-PK, PKCγ,PARP (Poly ADP Ribose Polymerase), D4-GDI (D4 GDP-dissociation inhibitor), steroid response element-bindingprotein (SREBPs), U1-70kD, serta inhibitor of caspaseactivated deoxyribonuclease (ICAD). Ketika semua jenissubstrat telah diaktifkan, sel akan mengalami serangkaianperubahan, termasuk aktivasi gen terkait apoptosis, penurunan

- 14 -
kemampuan perbaikan DNA, aktivasi zymogen atau inaktivasienzim, pembongkaran sitoskeleton, dan fragmentasi kromatinyang pasti menyebabkan apoptosis24.
Caspase-3 bersama dengan caspase-6 dan caspase-7membelah berbagai substrat termasuk yang pada akhirnyamenyebabkan morfologi dan perubahan biokimia terlihat padasel apoptosis41. Fagositosis sel apoptosis adalah komponenterakhir dari apoptosis. Fagositosis sel-sel mati oleh selfagosit disebut efferocytosis42. Sel-sel sekarat yang menjalanitahap akhir apoptosis mengekspresikan molekul fagositikseperti phosphatidylserine di permukaan selnya.
Phosphatidylserine biasanya ditemukan pada bagiandalam permukaan membran plasma, tetapi didistribusikan dipermukaan esktraseluler selama apoptosis oleh protein yangdikenal sebagai scramblase. Molekul-molekul ini menandaisel untuk difagositosis oleh sel-sel yang memiliki reseptoryang sesuai, seperti makrofag43. Setelah pengenalan, selfagosit akan membentuk kembali sitoskeleton untukmelingkupi sel. Fagositosis sel yang sekarat oleh sel fagositterjadi secara tertib tanpa memunculkan respons peradangan44.

- 15 -
BAB 3. KANKER PAYUDARA
3.1 Epidemiologi
Kanker payudara merupakan jenis kanker terbanyakpada wanita dan merupakan jenis kanker terbanyak yangmenyebabkan kematian di seluruh dunia6. Tahun 2002terdiagnosa kanker payudara sebanyak 1,15 juta kasus barudengan angka mortalitas tertinggi pada wanita, 411 ribumenyebabkan kematian setiap tahunnya (sekitar 15%kematian) di seluruh dunia walaupun terdapat variasi yangbesar tentang insidensi, mortalitas dan angka bertahan hidup(survival rate) di Negara tertentu6.
Perlu dijelaskan bahwa angka insidensi merupakanangka yang menunjukkan jumlah kasus baru pada suatuperiode waktu tertentu di populasi tertentu juga, biasanyadituliskan dalam angka absolut dalam satu tahun untuk setiap100.000 penduduk. Sedangkan angka mortalitas merupakanangka yang menunjukkan jumlah kematian pada suatu periodetertentu pada suatu populasi tertentu juga, biasanya dituliskandalam angka absolut per tahun per 100.000 penduduk6. Datatahun 2008 menunjukkan dari 1,3 juta pasien yangterdiagnosa kanker payudara, sekitar 459 ribu meninggal.Data Globocan tahun 2012, sekitar 1,7 juta wanitaterdiagnosa kanker payudara dengan angka kematiannyasekitar 522 ribu7.
Tercatat di tahun 2018 sebanyak 2,089 juta kasusdengan angka kematiannya sekitar 6,6 % (627 ribu) di seluruhdunia 5. Tahun 2050 diperkirakan insiden kanker payudara diseluruh dunia sekitar 3,2 juta6. Untuk Indonesia, insidenkanker pada perempuan sekitar 134 per 100.000 pendudukdengan kasus terbanyak kanker payudara sebesar 40 per100.000 perempuan. Estimasi Globocan, kematian diIndonesia akibat kanker payudara sekitar 16,6 kematian per100.000 penduduk4. Terdapat perbedaan yang nyata ledakan

- 16 -
kasus kanker payudara antara Negara maju dan Negaraberkembang. Lebih setengah kasus baru kanker payudaraditemukan di Negara maju (seperti Amerika Utara kecualiMexiko dan Eropa Barat), namun lebih dari tiga per empatkematian akibat kanker payudara terjadi di Negaraberkembang. Hal ini berkaitan dengan masih rendahnyaskrining mammografi di Negara berkembang, diagnose yangterlambat dimana lebih dari 60% pasien dinyatakan kankerpayudara stadium lanjut III/IV), akses pengobatan yang burukserta rejimen pengobatan yang masih di bawah standar6.
3.2 Etiologi dan faktor risiko
Etiologi kanker payudara belum seluruhnya difahami,namun ada tiga hal yang mempengaruhi terjadi kankerpayudara yaitu perubahan genetik, pengaruh hormonal danvariabel lingkungan. Pada kanker payudara terdapat ekspresiproto onkogen HER2/Neu berlebihan, gen ini merupakankelompok reseptor faktor pertumbuhan epidermal,overekspresi pada sekitar 20% kanker payudara7. Perubahangenetik lainnya adalah amplifikasi gen RAS dan MYC, mutasigen supresor tumor RB dan TP531.
Perubahan genetik ini terjadi dalam beragamkombinasi dan didapat, sehingga menimbulkan berbagaisubtipe kanker payudara yang berbeda. Terdapat lima subtipekanker payudara berdasarkan ekspresi reseptor estrogen,progesteron dan Her-2 onkogen, dua klasifikasi jenis positifestrogen (luminal A dan luminal B), dan 3 jenis untukestrogen negatif (sub tipe Her2, basal like dan normal like).Luminal A (reseptor estrogen positif, HER2/Neu negatif);luminal B (reseptor estrogen positif, HER2/Neu terkspresiberlebihan).Subtipe Her2 (negative reseptor estrogen, positifHer2). Sub tipe basal like (negative reseptor estrogen,ekspresi mioepitel/sel basalmeningkat)7,1.

- 17 -
Survival rate dan prognosis buruk serta agresivitastinggi untuk kanker payudara jenis Her2 dan basal likejikadibandingkan jenis luminal7. Subtipe kanker payudara yangtidak mengekspresikan reseptor estrogen, progesteron danHer2 dinamakan triple negative breast cancer (TNBC), seringpada wanita muda Amerika-Afrika, mutasi BRCA-1, lebihagresif, lebih besar, survival rate jelek7. Subtipe lainnyaadalah inflammatory breast cancer (IBC)7.
Sepuluh persen kanker payudara berhubungan denganmutasi yang diwariskan dimana wanita yang mempunyai gen,cenderung menderita kanker bilateral, menderita kanker lain(seperti kanker ovarium), dan sepertinya mengalami mutasipada BRCA1 (pada lokus kromosom 17q21.3) atau BRCA2(pada pita kromosom 13q12-13). BRCA1 dan BRCA2merupakan gen supresor tumor, bila mengalami mutasi makakanker akan muncul. Berkaitan dengan hormonal, padakanker payudara terjadi kelebihan estrogen endogen.Estrogenmenstimulasi produksi faktor pertumbuhan (sepertitransforming growth factor α, platelet derived growth factordan fibroblast growth factor) memacu perkembangan tumorsecara parakrin dan autokrin 1.
3.3 Terapi
Terapi kanker payudara tergantung stadium penyakit.Stadium penyakit dibagi menjadi stadium 0, 1, IIa, IIb, IIIa,IIIb, IIIc dan IV.

- 18 -
Tabel 3.1 Pengelompokan Stadium Kanker Payudara
Dimana: (Tx) tumor primer tidak dapat dinilai. (T0) tidak ada tandatumor primer. (Tis) karsinoma in situ. (T1) diameter tumorterbesar 2 cm atau kurang. (T2) diameter tumor terbesarmelebihi 2 cm, teteapi kurang dari 5 cm. (T3) diameter tumorterbesar lebih dari 5 cm. (T4) tumor segala ukuran denganperluasan langsung ke (a) dinding dada atau (b) kulit.(Nx) kelenjar limfe regional tidak dapat dinilai. (N0) tidak adametastase ke kelenjar limfe regional. (N1) metastase kelenjarlimfe aksilaris ipsilateral yang mobile. (N2a) metastasekelenjar limfe yang terfiksir, baik satu dengan yang lain atauke struktur jaringan yang lain pada aksila ipsilateral. (N2b)metastase hanya pada kelenjar limfe mammaria iternaipsilateral yang secara klinis terlihat jelas. (N3a) metastase kekelenjar limfe infraklavikularis dan kelenjar limfe aksilarisipsilateral. (N3b) metastase ke kelenjar limfe mammariainterna dan kelenjar limfe aksilaris ipsilateral. (N3c) metastaseke kelenjar limfe supraklavikularis ipsilateral.
(Mx) metastase jauh tidak dapat dinilai. (M0) tidak adametastase jauh. (M1) didapatkan metastase jauh1.
3.3.1 Terapi Kanker Payudara Dini (Stadium I, IIa)
Terapi kanker payudara yang dini meliputi terapiuntuk tumor primer dan ajuvan.Untuk tumor primer dilakukanoperasi berupa mastektomi radikal yang dimodifikasi (MRM)atau operasi konservasi payudara (BCS). Terapi ajuvandilakukan untuk mengkontrol lokoregional tumor dengan

- 19 -
radioterapi dan untuk mengkontrol sistemik kankermenggunakan terapi hormonal atau kemoterapi.Terapihormonal hanya diberikan pada pasien dengan tumor yangmengekspresikan reseptor hormon steroid yaitu ER/PR,menggunakan obat lini pertama anti estrogen (tamoksifen,raloxifene), lini kedua berupa inhibitor aromatase(anastrozole, letrozole, exemestane, letrozole), lini ketigayaitu progesterone (mesestrol asetat) dan lini keempat berupaandrogen seperti fluoxymestrone.
Kemoterapi ajuvan yang digunakan untuk stadium dini, linipertama dengan CAF (cyclophosphamide, Adriamycin, 5-fluorouracil) atau adriamicin yang dapat diganti denganepirubicin karena adriamicin bersifat kardiotoksik, debganregimen FEC (5-fluorouracil, epirubicin, cyclophosphamide).Regimen ini diberikan setiap 1 hari x 6 siklus atau setiap 21hari x 4 siklus atau hanya Adriamycin dengncyclophosphamide (AC). Obat lini kedua berupa golongantaxane (paclitaxel dan docetaxel) dan lini ketiga berupagemcitabine. Kemoterapi ajuvan ini diberikan pada kasusdengan diameter tumor melebihi 1 cm atau diameter tumorkurang dari 1 cm dengan ER negatif, HER-2 positif, gradetinggi. Transtuzumab, suatu antibodi monoklonal memperkuatefek kemoterapi berbasis taxane,lima puluh persenmemperbaiki harapan hidup bebas penyakit45.
3.3.2 Terapi Kanker Payudara Lokal Lanjut
Terapi yang dilakukan untuk kanker payudara lokallanjut (T3 dan T4) bertujuan untuk mengkontrol lokoregionalyang baik dan mendapatkan kesembuhan dengankemoterapi.Untuk kanker stadium IIb dan IIIa (dengankeadaan ulserasi, edema kulit terbatas, terfiksir dengan ototpektoralis) reseksi dapat dilakukan, namun tidak untuk kankerstadium IIIb (dengan keadaan edema kulit yang luas, kelenjarlimfe interkostalis, edema lengan, atau kanker payudarainflamatorik). Kemoterapi yang diberikan untuk pasien kanker

- 20 -
payudara yang lanjut bertujuan menurunkan stadium tumor,meningkatkan kemungkinan dilakukannya konservasipayudara, tumor inoperable menjadi operabel, menghambatkecepatan kanker paska bedah.
Kemoterapi yang diberikan menggunakan regimenCT yaitu dengan pemberian golongan antrasiklin(doksorubisin, epirubisin) plus siklofosfamid (AC/EC) denganatau tanpa penambahan 5-fluorouracil, dan ditambahkangolongan taxan. Cara pemberiannya dengan doksorubisin 60mg/m2 secara intravena dan siklofosfamid 600 mg/m2 IVhari1, per siklus setiap 21 hari untuk 4 siklus yang diikutidengan paclitaxel 175 g/m2 IV setiap 3 jam per hari siklusuntuk setiap 21 hari untuk 4 siklus. Untuk stadium lanjut jugadiberikan terapi radiasi45.
3.3.3 Terapi Kanker Payudara Metastase
Kanker payudara metastase, pilihan terapi yang dapatdiberikan meliputi terapi hormonal, kemoterapi, transtuzumabdan terapi suportif.Tujuan terapi untuk stadium ini adalahuntuk memberikan efek paliatif untuk gejalaklinisnya.Kemoterapi diberikan pertama kali, diikuti eksisibenjolan/mastektomi simpleks untuk mengambil tumorfungata. Setelah kemoterapi tamoksifen diberikan selama limatahun, serial berulang obat kemoterapi lini kedua dan ketigajuga diberikan45.
3.4 Obat Anti Kanker
Obat anti kanker diklasifikasikan atas beberapagolongan besar, meliputi golongan alkilator, anti metabolit,produk alamiah, hormon dan antagonis, dan lain-lain.Golongan alkilator terbagi atas sub golongan mustarnitrogen (siklofosfamid, melfalan, mekloretamin, klorambusil,ifosfamid), etilenamin dan metilmelamin (trietilen-melaminTEM, thiotepa), metilhidrazin (prokarbazin), alkil sulfonate(busulfan), nitrosourea (karmustin, lomustin, semustin,

- 21 -
streptozotosin), dan platinum (cisplatin, karboplatin,oksaliplatin). Golongan metabolit terbagi atas sub golongananalog pirimidin (5-fluorourasil, sitarabin, 6-azauridin,floksuridin, gemsitabin), analog purin (6-merkaptopurin, 6-tioguanid, fludarabin, pentostatin), antagonis folat(metotreksat, pametreksed).
Produk alamiah terbagi atas sub golongan alkaloidvinka (vinblastin, vinkristin, vinorelbin), taxane (paklitaksel,dosetaksel), epidodofilotoksin (etoposide, teniposid),kamptotesin (irinotecan, topotekan), antibiotik (daktinomisin,antrasiklin atau daunorubisin, doksorubisin, mitramisn,antrasenedion atau itoksantron, mitomisin, bleomisin,mitomisin), enzim (L-asparaginase). Golongan hormon danantagonis terbagi atas sub golongan adrenokortikosteroid(prednison, hidrokortison), progestin (hidroksiprogesteronkaproat, medroksiprogesteron asetat, megestrol asetat),estrogen (dietilstilbesterol, etinil estradiol), anti estrogen(tamoksifen, toromifen), androgen (testosterone propionate,fluoksimesteron), anti androgen (flutamid), penghambatadrenokortikoid (mitotan, aminoglutetimid), analog GRH(leuprolid), penghambat aromatase (anastrozol, letrozol,eksemestan). Lain-lain termasuk sub golongan substitusi urea(hdroksiurea), derivate metilhidrazin (prokarbazin),differentiating agent (tretinoin, arsen trioksid), penghambattirosin kinase (imatinib, gefitinib), penghambat proteosom(bortezumib), modulator respon biologik (interferon alfa,interleukin 2), dan antibodi monoklonal46.
Doksorubisin merupakan antibiotik golonganantrasiklin yang banyak digunakan untuk terapi berbagaimacam jenis kanker seperti leukemia akut, kanker payudara,kanker tulang dan ovarium47. Senyawa ini diisolasi dariStreptomyces peucetius var caesius pada tahun 1960-an dandigunakan secara luas48. Doksorubisin dapat menyebabkankardiotoksisitas pada penggunaan jangka panjang, hal itu
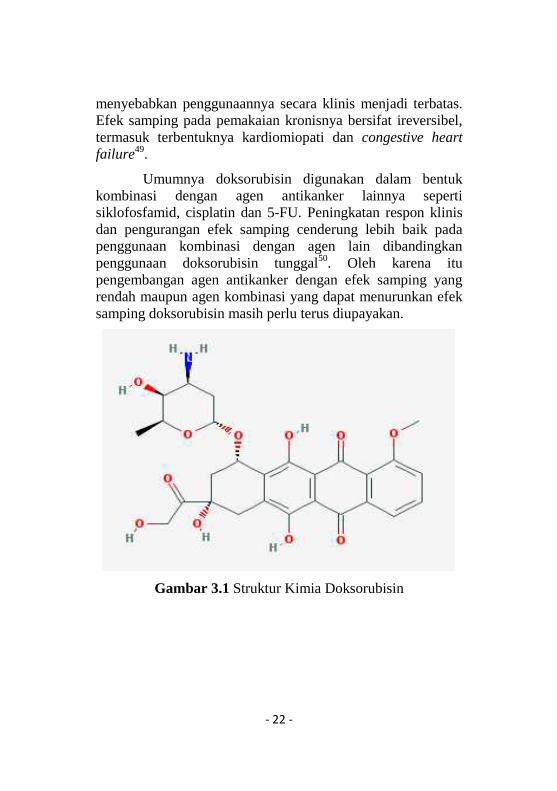
- 22 -
menyebabkan penggunaannya secara klinis menjadi terbatas.Efek samping pada pemakaian kronisnya bersifat ireversibel,termasuk terbentuknya kardiomiopati dan congestive heartfailure49.
Umumnya doksorubisin digunakan dalam bentukkombinasi dengan agen antikanker lainnya sepertisiklofosfamid, cisplatin dan 5-FU. Peningkatan respon klinisdan pengurangan efek samping cenderung lebih baik padapenggunaan kombinasi dengan agen lain dibandingkanpenggunaan doksorubisin tunggal50. Oleh karena itupengembangan agen antikanker dengan efek samping yangrendah maupun agen kombinasi yang dapat menurunkan efeksamping doksorubisin masih perlu terus diupayakan.
Gambar 3.1 Struktur Kimia Doksorubisin

- 23 -
Antibiotik antrasiklin seperti doksorubisin memilikimekanisme aksi sitotoksik melalui empat mekanisme yaitu:a. Penghambatan topoisomerase IIb. Interkalasi DNA sehingga mengakibatkan penghambatan
sintesis DNA dan RNA.c. Pengikatan membran sel yang menyebabkan aliran dan
transport iond. Pembentukan radikal bebas semiquinon dan radikal bebas
oksigen melalui proses yang tergantung besi dan prosesreduktif yang diperantarai enzim. Mekanisme radikalbebas ini telah diketahui bertanggungjawab padakardiotoksisitas akibat antibiotik antrasiklin50.Doksorubisin dapat berinterkalasi dengan DNA, secaralangsung akan mempengaruhi transkripsi dan replikasi.Doksorubisin mampu membentuk komplek tripartitdengan topoisomerase II dan DNA. Topoisomerase IIadalah suatu enzim tergantung ATP yang bekerjamengikat DNA dan menyebabkan double-strand breakpada ujung 3′fosfat sehingga memungkinkan penukaranstrand dan pelurusan DNA superkoil. Pelurusan strand inidiikuti dengan penyambungan strand DNA olehtopoisomerase II. Topoisomerase ini sangat pentingfungsinya dalam replikasi dan perbaikan DNA.Pembentukan kompleks tripartit tersebut akanmenghambat penyambungan kembali strand DNA,menyebabkan penghambatan daur sel terhenti di fase G1dan G2 serta memacu terjadinya apoptosis48,51.
Adanya gangguan pada sistem perbaikan DNA doublestrand akan memicu kerusakan sel, sedangkanoverekspresi transkripsi untuk perbaikan DNA mungkinterlibat dalam fenomena resistensi obat. Doksorubisindengan adanya gugus quinon yang dimilikinya jugamampu menghasilkan radikal bebas baik pada sel normalmaupun sel kanker51. Doksorubisin dapat membentukintermediate radikal semiquinon bersama dengan

- 24 -
MADPH melalui reaksinya dengan sitokrom P450reduktase, yang dapat bereaksi dengan oksigenmenghasilkan radikal anion superoksida, yang selanjutnyaakan menghasilkan hidrogen peroksida dan radikalhidroksil yang menyerang DNA46 dan mengoksidasi basapada DNA.Pembentukan radikal bebas ini secaraa signifikandistimulasi oleh interaksi antara doksorubisin dengan besi.Pertahanan enzimatik dalam sel seperti superoksiddismutase dan katalase merupakan hal penting untukmenjaga sel dari toksisitas doksorubisin50. Doksorubisindiberikan secara injeksi intravena dengan dosis padadewasa 60-75 mg/m2 sebagai suntikan tunggal setiap 3minggu sampai dosis total tidak lebih dari 550 mg/m2.Alternatif pemberiannya adalah 20 mg/m2 setiapminggu46.
Paclitaxel dan docetaxel merupakan obat kemoterapigolongan taxane, berasal dari tanaman pohon pinus pasifik(Taxus brevifolia) untuk paclitaxel dan dari pohon pinusEropa (Taxus baccata) untuk docetaxel.Obat ini telahdigunakan sejak dua dekade terakhir sebagai anti-tumor yangefektif untuk berbagai keganasan, misalnya kanker ovarium,paru, payudara, kepala dan leher, dan sebagainya. Kemoterapigolongan taxane bekerja dengan berikatan pada tubulinsubunit β, menginduksi polimerisasi tubulin dan menstabilkanmikrotubulus 1 Mikrotubulus yang dihasilkan dengankemoterapi golongan taxane resisten terhadap penguraian.
Hal ini mengakibatkan gangguan proses mitosis danakhirnya mengakibatkan apoptosis atau kematian sel.Kemoterapi golongan taxane bekerja pada siklus sel fase G2-M. Selain berikatan dengan tubulin, kemoterapi golongantaxane juga memiliki aktivitas fosforilasi onkoprotein yangmenghambat apoptosis yaitu bcl-2.Diperkirakan bahwa

- 25 -
fosforilasi bcl-2 menginaktivasi onkoprotein dan memicuterjadinya apoptosis.
Docetaxel tidak larut air, diformulasikan dalampolysorbate 80 (Tween 80).Docetaxel diberikan dengan dosis75-100 mg/m2 setiap 3 minggu atau 40 mg/m2 setiap minggu.Dengan pemberian setiap minggu, profil l keamanandocetaxel (toksisitas hematologi) lebih baik dibandingkansetiap 3 minggu. Kontak konsentrat docetaxel dengan plasticPVC tidak direkomendasikan dan untuk meminimalkanpajanan dengan plastic PVC, digunakan set infus non-PVC.
Berlawanan dengan paclitaxel, farmakokinetikdocetaxel bersifat linear. Perubahan AUC dan bersihanproporsional dengan perubahan dosis.Docetaxel berikatandengan protein plasma (> 90%), dimetabolisme oleh enzimcytochrome P450. Sebagian besar dosis diekskresikan di fesesdan sebagian kecil di urin (2-9%). Waktu paruh eliminasiterminal docetaxel sekitar 10-18 jam52. Sisplatin (cis-diammine dichloroplatinum) merupakan metal organikdengan mekanisme anti kanker dengan membunuh sel padasemua siklus pertumbuhannya, menghambat biosintesis DNAdan berikatan dengan DNA di N7 pada guanine membentukikatan silang (cross linking), juga membentuk ikatan kovalendengan adenin dan sitosin46.

- 26 -
BAB 4. PENGUJIAN AKTIVITAS OBAT ANTIKANKER
Aktivitas anti kanker suatu obat/tanaman obat dapatdilakukan melalui beberapa tahapan pengujian. Uji antikanker diawali dengan uji sitotoksisitas, setelahnya dapatdilakukan pengujian lain untuk mengetahui mekanismesitotoksisitas obat tersebut melalui uji siklus sel dan apoptosis,serta uji protein-protein yang berperan dalam pertumbuhankanker.
4.1 Uji Sitotoksisitas
Uji sitotoksisitas adalah suatu pengujian secara invitro menggunakan kultur sel yang bertujuan untukmempelajari potensi sitotoksik dari suatu senyawa yang akandikembangkan sebagai obat anti kanker atau memastikanbahwa suatu senyawa tidak bersifat toksik yang akandikembangkan sebagai kosmetik. Obat baru, kosmetik,makanan tambahan, dan sebagainya, harus melalui pengujiansitotoksisitas sebelum dirilis untuk digunakan oleh masyarakat53. Potensi sitotoksik dari suatu senyawa dapat merusakstruktur sel, dan umumnya digunakan untuk menghambat danmenghentikan pertumbuhan sel kanker54.
Pengujian ini idealnya dilakukan secara in vivo, akantetapi pada saat ini karena berbagai faktor, maka pengujian inilebih cenderung dilakukan secara in vitro. Metode ujisitotoksisitas secara in vitro merupakan langkah awalpengembangan obat, karena murah, mudah diukur, sertamudah untuk dikembangbiakkan53 .
Uji Sitotoksik Menggunakan Metode MTT
Metode MTT (microculture tetrazolium technique)adalah salah satu uji sitotoksisitas yang bersifat kuantitatif.Uji ini berdasarkan pengukuran intensitas warna (kolorimetri)

- 27 -
yang terjadi sebagai hasil metabolisme suatu substrat oleh selhidup menjadi produk berwarna55.
Pada uji ini digunakan garam MTT. Garam ini akanterlibat pada kerja enzim dehidrogenase. Prinsip uji MTTyaitu perubahan warna kuning garam tetrazolium yangtereduksi menjadi kristal formazan dalam mitokondria selhidup yang menghasilkan dehidrogenase. Sel yang matikarena efek sitotoksik tidak menghasilkan dehidrogensesehingga kristal formazan tidak terbentuk56.
Kristal formazan yang akan terlarut digunakansebagai indikator pengukuran. Setiap peningkatan ataupenurunan jumlah dari sel hidup dapat dideteksi denganmengukur konsentrasi formazan menggunakan plate readerpada panjang gelombang 595 nm (Kupcsik, 2011).Konsentrasi formazan berbanding lurus dengan jumlah selhidup56.
Formazan merupakan zat berwarna ungu yang tidaklarut dalam air sehingga dilarutkan menggunakan HCl 0,04 Ndalam isopropanol atau 10% SDS dalam HCl 0,01 N.Intensitas warna ungu yang terbentuk dapat ditetapkan denganspektrofotometri dan berkorelasi langsung dengan jumlah selyang aktif melakukan metabolisme, sehingga berkorelasidengan viabilitas sel. Persentase viabilitas dapat dihitungdengan persamaan sebagai berikut55.
4.1 Uji Siklus Sel dan Apoptosis MenggunakanFlowcytometry
Flowcytometry adalah teknik yang digunakan untukmenghitung dan menganalisis partikel mikroskopis yangtersuspensi dalam aliran fluida57. Prinsip dasar dari metode iniadalah berdasarkan fluoresensi. Suspensi sel atau partikel

- 28 -
yang hendak dianalisa disedot atau dialirkan. Alirandikelilingi oleh fluida yang sempit, sel akan melewati satudemi satu melalui sinar laser terfokus. Sinar laser akanmenyinari sel tersebut. Sel yang sesuai dengan cahaya laserdan panjang gelombang yang tepat dapat dipancarkan kembalisebagai fluoresensi jika sel mengandung zat alami fluorescentsatu atau lebih fluorochrome-labeled antibodi yang melekatpada permukaan atau struktur internal sel. Penyerapan cahayabergantung pada struktur internal sel serta ukuran danbentuknya.Cahaya fluoresensi terdeteksi oleh serangkaiandioda.
Filter optik berfungsi untuk memblokir cahaya yangtidak diinginkan. Hasil data disimpan melalui computer58.Dengan adanya fluorochrome yang memiliki kemampuanberinterkalasi dengan basa untai DNA seperti propidiumiodide (PI) maka setiap sel yang memiliki jumlah setkromosom yang berbeda akan memberikan intensitasfluoresensi yang berbeda. Semakin banyak set kromosommaka intensitas fluoresensi akan semakin besar. PenggunaanPI yang mampu mengikat dan melabel DNA memungkinkanuntuk mendapatkan hasil yang cepat.
Flowcytometry dapat digunakan untuk menganalisasiklus sel dengan pewarna PI59. Selain itu flowcytometry jugadapat menganalisa pemacuan apoptosis dengan pewarna PIdan penambahan antibodi Annexin V yang didasarkan padaprinsip bahwa sel apoptosis memiliki berbagai ciri khas, yangditandai dengan fragmentasi DNA dan hilangnya kandunganDNA inti serta redistribusi phosphatidylserine dari bagiandalam ke luar membran plasma60. Flowcytometer atau FACS(Fluorescence Activated Cell Sorting) digunakan untukmembaca intensitas fluoresensi tiap sel58.

- 29 -
4.2 Uji Ekspresi Protein Menggunakan Imunositokimia
Imunositokimia dimulai oleh penelitian yangdilakukan oleh Coons dan kawan-kawan pada awal 1940-anmenggunakan antibodi yang dilabeli agen fluoresen untukmendeteksi antigen suatu jaringan61. Immunositokimiamerupakan suatu teknik yang digunakan untuk mendeteksiantigen pada sel tertentu yang memungkinkan untukvisualisasi antigen dalam suatu sampel melalui aplikasi yangberurutan dari antibodi yang spesifik terhadap antigen(antibodi primer), antibodi sekunder terhadap antibodiprimer, sebuah kompleks enzim, dan substrat kromogenik.Aktivasi enzimatik dari substrat kromogenik akanmenghasilkan suatu reaksi perubahan warna yangmenunjukkan adanya antigen dan dapat diamati menggunakanmikroskop cahaya62.
Metode imunositokimia terbagi menjadi dua yaitumetode langsung dan metode tidak langsung.Antibodi yangmengikat fluoresen atau zat warna berikatan langsung denganantigen pada sel disebut metode langsung. Metode tidaklangsung yaitu apabila antigen diikat pada antibodi primersecara langsung, kemudian ditambahkan antibodi sekunderyang mengikat enzim seperti peroksidase, alkali fosfatase,atau glukosa oksidase. Antibodi sekunder berikatan denganantibodi primer, kemudian ditambahkan substrat kromogenyang diubah oleh enzim sehingga terjadi pembentukan warnayang mampu memberikan warna pada sel63.
Salah satu yang paling umum dan masih banyakdigunakan adalah pewarnaan sistem peroksidase denganmenggunakan 3,3' diaminobenzidine tetrahydrochloride(DAB). Endapan dari senyawa ini akan memberikan warnacoklat keemasan ketika beraksi dengan peroksidase danhidrogen peroksida. Warna coklat ini dapat diamati padabagian jaringan yang menunjukkan ekspresi dari suatu jenisprotein yang akan diperiksa64.

- 30 -
Imunositokimia dimanfaatkan untuk mengidentifikasiprotein dan makromolekul lain pada tingkat sel. Sampelkontrol diperlukan untuk menunjukan lokasi pelabelan yangbenar. Kontrol yang diperlukan untuk imunositokimia adalahkontrol antibodi primer yang menunjukan spesifitas antaraantibodi primer dan antigen, kontrol antibodi sekunder untukmenunjukan spesifitas ikatan antara antibodi sekunder danantibodi primer. Kontrol yang ketiga adalah kontrol labelyang menunjukan bahwa pelabelan yang terjadi merupakanhasil dari label yang ditambahkan dan bukan dari labelendogen65.

- 31 -
BAB 5. METFORMIN SEBAGAI ANTI KANKER
Metformin merupakan anti diabetik oral digunakanuntuk kasus diabetes melitus tipe 2, termasuk golonganbiguanida. Metformin suatu anti hiperglikemik, tidakmenyebabkan hipoglikemik dan tidak merangsang sekresiinsulin. Efek farmakodinamik metformin adalah terjadinyapenurunan produksi glukosa di hati dan meningkatkansensitifitas jaringan otot dan adiposa terhadap insulin, hal initerjadi akibat aktivasi metformin terhadap AMP-Kinase64.
Struktur metformin adalah sebagai berikut :
Gambar 5.1 Struktur Metformin
Metformin memasuki hepatosit melalui transporterkation organik 1 (organic cation ransporter, OCT), kemudianberakumulasi ke dalam matriks mitokondria menghasilkanmolekul ion positif yang dibawa membran mitokondria66.Interaksi terjadi antara rantai samping hidrokarbon apolar darimetformin dengan fosfolipid membran mitokondria67.
Penghambatan metformin terhadap kompleks Idimungkinkan metal-binding property obat ini. PeningkatanpH matriks mitokondria menyebabkan perubahan metforminmenjadi suatu bentuk deprotonated yang memiliki affinitastinggi terhadap ion tembaga68. Metformin menghambatreduksi ubikuinon secara non kompetitif melalui ikatan padapermukaan hidrofilik dan menangkap enzim dalam suatukonformasi deactive-like open loop69.

- 32 -
5.1 Mekanisme anti kanker metformin
Efek anti kanker metformin didasari dari dua mekanisme,secara langsung dan tidak langsung17.
a. Efek tidak langsung metformin.
Efek tidak langsung metformin dalam menimbulkan efekanti kanker terjadi melalui kemampuan metforminmenurunkan hiperinsulinemia70 melalui peningkatansensitisasi insulin terhadap glukosa, dan sensitifitas reseptorinsulin di otot skeletal71.
Diketahui bahwa hiperinsulinemia merupakan growthfactoryang mendukung perkembangan kanker 72 melalui duareseptor yaitu insulin receptor (IR) dan insulin-like growthfactor receptor (IGF-1R). Hiperinsulinemia menyebabkanpeningkatan insulin like growth factor (IGFs) akibatdownregulasi insulin-like growth factor binding protein(IGFBP), hal ini menyebabkan peningkatan IGFs bebas, IGF1dan aktivasi IGF-1R17. Aktivasi dua reseptor insulin tersebutmenyebabkan perangsangan kaskade transduksi intraseluleruntuk pertumbuhan dan pertahanan sel yaitu phosphoinositide3-kinase (PI3K) dan mitogen-activated protein-kinase(MAPK)73. Overekspresi IGF-1 terdapat pada kankerkolorektal, prostat dan payudara17. Pada pasien DM tipe 2,metformin potensial sebagai anti kanker dengan caramengurangi kadar glukosa, insulin dan IGF-117.
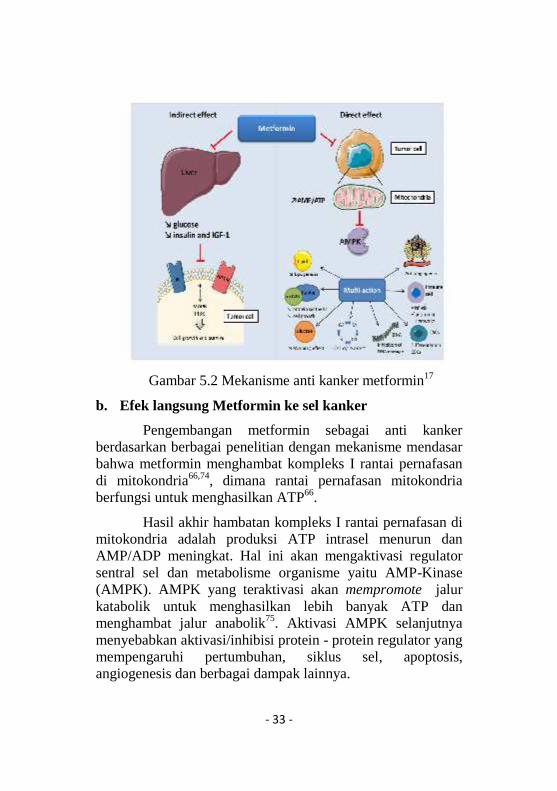
- 33 -
Gambar 5.2 Mekanisme anti kanker metformin17
b. Efek langsung Metformin ke sel kanker
Pengembangan metformin sebagai anti kankerberdasarkan berbagai penelitian dengan mekanisme mendasarbahwa metformin menghambat kompleks I rantai pernafasandi mitokondria66,74, dimana rantai pernafasan mitokondriaberfungsi untuk menghasilkan ATP66.
Hasil akhir hambatan kompleks I rantai pernafasan dimitokondria adalah produksi ATP intrasel menurun danAMP/ADP meningkat. Hal ini akan mengaktivasi regulatorsentral sel dan metabolisme organisme yaitu AMP-Kinase(AMPK). AMPK yang teraktivasi akan mempromote jalurkatabolik untuk menghasilkan lebih banyak ATP danmenghambat jalur anabolik75. Aktivasi AMPK selanjutnyamenyebabkan aktivasi/inhibisi protein - protein regulator yangmempengaruhi pertumbuhan, siklus sel, apoptosis,angiogenesis dan berbagai dampak lainnya.

- 34 -
Aktifitas anti kanker metformin secara langsungberdasarkan kepada aktivasi AMPK yang menyebabkanperubahan regulasi energi sel untuk metabolisme, kontrasdengan sel kanker yang membutuhkan energi dan nutrisitinggi bagi pertumbuhannya diantaranya untuk sintesisprotein, DNA dan produksi lipid76.
Gambar 5.3 Mekanisme kerja metformin72.
Setelah uptake metformin oleh transporter kation organic(OCT), metformin menyebabkan reduksi ATP via inhbisikompleks I rantai pernafasan mengawali aktivasi AMPK ,yang mengganggu ekspresi gen yang terlibat dalamglukoneogenesis, lipogenesis, sintesis protein danangiogenesis. ACC, acetylCoA carboxylase; F-1,6-biphosphatase; FAS, fatty acid synthase; IGF, insulin likegrowth factor; LKB-1, serine-threonine liver kinase B1;mTOR, mammalian target of rapamycin; PAI-1, plasminogen-activator inhibitor-1; PI3K, phosphoinositide 3-kinase;TORC2, transducer of regulated CREB-binding protein 2;

- 35 -
TSC2, tuberous sclerosis 2; VEGF, vascular endhothelialgrowth factor.
5.2 AMPK (Adenosine Monophosphate Kinase)
AMPK merupakan master sensor dan regulatorhomeostasis energi sel terutama dalam keadaan sel yangmengalami stress, pada keadaan ini pertumbuhan sel terhentiuntuk menghemat penggunaan energy di dalam sel77.
AMPK suatu heterotrimer serine/treonine kinase,terdiri dari satu sub unit katalitik α (terdiri dari residukatalitik treonin 172) dan dua sub unit regulasi β dan γ (terdiridari 4 tempat ikatan nukleotida AMP, ATP dan ADP), enzimini diekspresikan pada berbagai jaringan termasuk hati danotot78,79.
Unit α AMPK terdiri dari residu treonin (Thr172)difosforilasi proteinkinase upstream menghasilkan aktivasiAMPK. Kinase-kinase yang dapat mengaktifkan AMPKadalah liver kinase B1 (LKB1), calcium/calmodulin-dependent protein kinase (CaMKK) dan transforming growthfactor β (TGF- β)-activated kinase (TAK1)80. Unit β terdiridari suatu sentral CBM (carbohydrate-binding molecule) yangmemungkinkan AMPK berikatan dengan glikogen78.
AMPK dapat diaktifkan oleh perubahan ekstraselulerseperti deplesi ATP, glukosa rendah dan perubahan levelNADPH81. Obat-obatan seperti metformin dan beberapaNSAID82,83 dapat mengaktifkan AMPK, demikian jugadengan senyawa bahan alam seperti polifenol(resveratrol)84,85,86, flavonoid (quercetin) 87 serta herbal China(berberine)88,89,90.
Aktivasi AMPK selanjutnya akan mengaktifkanberbagai protein efektor diantaranya penghambatanmammalian target of rapamycin (mTOR)91, meregulasi p5392,

- 36 -
modulasi aktivitas faktor transkripsi dan koregulator–koregulator yang mengendalikan siklus sel93,94,95.
AMPK memfosforilasi tempat-tempat yang memilikisetidaknya satu rantai samping dasar (basic side chain) yaituR, K atau H dengan 3 atau 4 residu N-terminal (P-4 dan P-3)ke residu serine atau treonin. Adapun substrat AMPK adalahasetil-KoA karboksilase ACC1 (Ser-80), asetil-KoAkarboksilase ACC2 (Ser-221), HMG-KoA reduktase (Ser-872), hormone sensitive lipase (Ser-885), muscle glycogensynthase (Ser-8), 6-phosphofructo-2 kinase PFKFB3 (ser-461), 6-phosphofructo-2 kinase PFKFB2 (ser-469), TBC1D1(Ser-237), TSC2 (Ser-1345), Raptor (Ser-792), Raptor (Ser-722), SREBP-1c (Ser-396), p300 (Ser-89), CRTC2 (Ser-171),HDAC4 (Ser-246), HDAC5 (Ser-259), HDAC7 (Ser-194),ULK1 (Ser-467), ULK1 (Ser-556), ULK1 (Ser-575), ULK1(Ser-638)79.
AMPK berperan meregulasi metabolisme melaluihomeostasis energi dengan mengaktifkan jalur katabolikuntuk menghasilkan ATP. Jalur katabolik melalui aktivasiGLUT1 dan GLUT4 (akibat fosforilasi Rab-GAP TBC1D1),glikolisis melalui fosforilasi dan aktivasi 2 atau 4 isoformPFKFB dengan sintesis aktivator glikolitik fruktosa-2,6-bifosfat, oksidasi asam lemak melalui fosforilasi ACC2(malonil-KoA turun, suatu inhibitor ambilan asam lemak kemitokondria). Aktivasi AMPK juga menghambat beberapajalur anabolik seperti inhibisi asam lemak melalui fosforilasiACC1, sintesis kolesterol melalui fosforilasi HMG-KoAreduktase,sintesis glikogen melalui fosforilasi glikogensintase, sintesis RNA ribosomal melalui fosforilasi faktortranskripsi RNA polymerase I TIF-1A RRN3.
AMPK mendown regulasi ekspresi enzim sintesisasam lemak, glukoneogenesis (glukosa 6 fosfatase danfosfoenolpiruvat karboksikinase) pada level transkripsimelalui fosforilasi SREBP-1c. Fosforilasi CRTC2, suatu

- 37 -
koaktivator transkripsi CREB (cyclic AMP respond element-binding protein) menyebabkan eksklusi CRTC2 dari nukleus.Fosforilasi HDAC (histon deasetilase) menyebabkan ekslusiHDAC dari nukleus. Fosforilasi TSC2 dan raptormenyebabkan inhibisi kompleks m-TOR1 (TORC1) denganakibat hambatan inisiasi dan elongasi translasi pada sintesisprotein79.
Mekanisme hambatan pertumbuhan sel akibat aktivasiAMPK adalah melalui hambatan sintesis protein, lipid danRNA ribosomal serta hambatan pada siklus sel79,96. ReplikasiDNA terjadi di fase S siklus sel fase M mitosis. Kedua prosesini membutuhkan ATP. Efek penghambatan siklus sel terjadiakibat fosforilasi AMPK pada p53 di Ser15 (p21 targettranskripsi p53) dan p27 di Thr198 (Hardie, 2011). Fosforilasiini menyebabkan stabilisasi dan akumulasi p53 di nukleusmelalui transkripsi, lalu mengup-regulasi salah satu siklindependent kinase inhibitor p21 melalui mekanismetranskripsi. Fosforilasi p53 oleh AMPK diakibatkan karenatempat fosforilasi p53 di serine 15 motifnya serupa denganmotif ACC yang juga difosforilasi AMPK96.
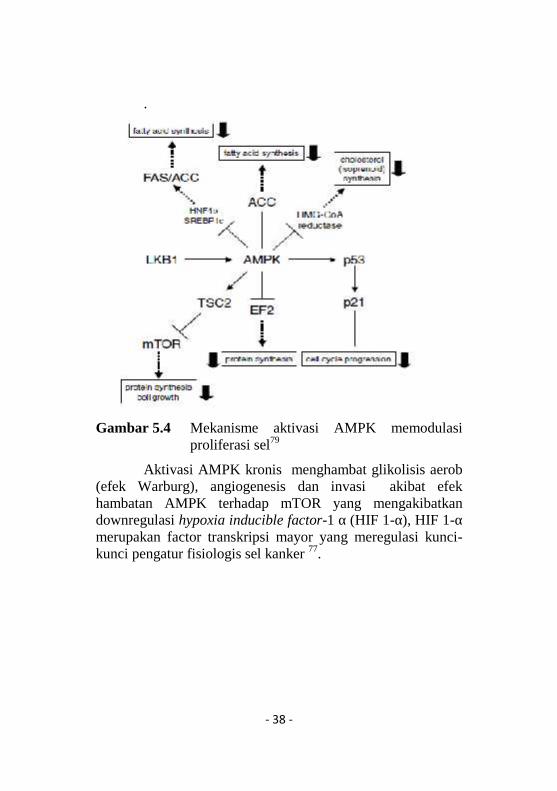
- 38 -
.
Gambar 5.4 Mekanisme aktivasi AMPK memodulasiproliferasi sel79
Aktivasi AMPK kronis menghambat glikolisis aerob(efek Warburg), angiogenesis dan invasi akibat efekhambatan AMPK terhadap mTOR yang mengakibatkandownregulasi hypoxia inducible factor-1 α (HIF 1-α), HIF 1-αmerupakan factor transkripsi mayor yang meregulasi kunci-kunci pengatur fisiologis sel kanker 77.

- 39 -
Gambar 5.5 Interaksi AMPK dengan metabolism sel 77
Peranan aktivasi AMPK terhadap apoptosis terjadimelalui beberapa mekanisme yaitu down modulate Akt (suatuprotein kinase B), BMF, aktivasi p53 dan interaksi AMPKdengan sinyalisasi MAPK (mitogen-activated protein kinase).Down modulate Akt menyebabkan inaktivasi FOXO3A (suatufaktor transkripsi) sehingga terjadi transaktivasi gen promoterBim. Pada keadaan reduksi ATP yang sangat parah AMPKmengaktivasi transkripsi protein proapoptotik BH3-onlyprotein BMF.Sinyalisasi MAPK terjadi melalui aktivasi p38MAPK (MAPK14). Fosforilasi p53 dapat terjadi langsungoleh AMPK atau melalui p38 MAPK. Aktivasi Bim, BMF,dan p53 selanjutnya akan menyebabkan pembentukan poripada mitokondria selanjutnya terjadi apoptosis secara internal.P53 menginduksi up-regulasi gen pro apoptotik seperti BAXdan PUMA78.

- 40 -
Gambar 5.6 Keterlibatan AMPK didalam apoptosis78
5.3 Penelitian metformin sebagai anti kanker payudara
Metformin bukan obat kanker, bahkan untuk kankerpayudara. Metformin merupakan anti hiperglikemik, obatpilihan utama untu penderita diabetes mellitus (DM) tipe 2yang direkomendasikan American Diabetes Association201417. Penelitian sebelumnya menyebutkan bahwa terdapatpenurunan risiko terjadinya kanker untuk penderita DM yangmenggunakan metformin18,19. Penelitian-penelitian invitrotelah pernah dilakukan ke beberapa jenis sel kanker seperti selWiDr model sel kanker kolorektal20, sel Hep G-2 model selkanker hepar21, dan sel MCF-722 , MDA-MB-23123 dan MDA-MB-435 model sel kanker payudara, seluruhnya menunjukkanefek penekanan pertumbuhan sel kanker.
Kanker payudara memiliki banyak jenis, diantaranyaadalah jenis duktal dengan model sel T47D. Sel T47Dmerupakan suatu continous cell line diisolasi dari jaringantumor duktal payudara seorang wanita berusia 54 tahun,mengekspresikan protein p53 yang termutasi (missencemutation) pada residu 194 (dalam zinc-binding domain,L2)97,98.

- 41 -
Pengujian aktifitas antikanker metformin terhadap selT47D mengikuti beberapa alur pemeriksaan, dimulai dari ujisitotoksisitas, selektifitas, siklus sel, apoptosis dan protein-protein pendukung. Uji sitotoksisitas, langkah awalpengembangan obat untuk menilai aktifitas sitotoksik obatbaru Kemampuan sitotoksik obat/zat dinyatakan dengan IC50
yaitu konsentrasi yang menyebabkan kematian pada 50%populasi sel.
Menurut US National Cancer Institute senyawasitotoksik dibagi atas empat kategori yaitu sangat toksik jikanilai IC50 ≤20 µg/mL, sitotoksik moderat atau cukup aktif nilaiIC50 21-200 µg/mL, kategori sitotoksik lemah jika IC50 201-500 µg/mL, dan tidak toksik jika IC50 ≥500 µg/mL. Semakinkecil nilai IC50 maka semakin besar aktivitas sitotoksiknya99.
Pengujian sitotoksik menggunakan metode MTTassay, suatu metode kuantitatif dengan mengacu pada nilaiabsorbansi yang didapat melalui pembacaan sel di bawahELISA reader dengan panjang gelombang 595 nm (Nugrohoet al, 2012). Prinsip MTT assay adalah kolorimetri(pengukuran intensitas warna) berdasarkan denganterbentuknya kristal formazan (berwarna ungu dan berserabut)pada sel hidup, menembus membran dan terakumulasi didalamnya. Pembentukan warna pada sel hidup ini sebagaihasil metabolisme suatu substrat oleh sel hidup menjadiproduk berwarna.
Sistem reduktase suksinat tetrazolium yang terdapatdalam mitokondria sel hidup yang termasuk dalam rantairespirasi akan mereduksi warna kuning MTT membentukkristal formazan yang berwarna ungu (Merloo et al, 2011).Sel mati tidak dapat membentuk kristal formazan karena selmati tidak mampu berespirasi sehingga enzim suksinattetrazolium yang dapat mereduksi garam MTT menjadiproduk formazan tidak dihasilkan, sehingga warna sel matitidak ungu namun akan tetap berwarna kuning. Semakin

- 42 -
banyak sel yang hidup maka warna ungu akan semakinpekat95.
Paparan metformin HCl terhadap sel T47D selama 24jam dengan serial konsentrasi metformin masing-masing5000; 2500; 1250; 312,5; 156,25 µM didapatkan IC50
metformin 1738,1875 ± 141,63 µg/mL100. Nilai ini sangat jauhsekali lebih besar bila dibandingkan dengan IC50 doxorubicin(kemoterapi yang telah digunakan untuk kanker payudaraselama ini) terhadap sel T47D yaitu 0,1 ± 0,002 µg/mL100
(Gambar 5.7, Gambar 5.8).
Gambar 5.7 Grafik Pengaruh Perbedaan KonsentrasiMetformin Terhadap Viabilitas Sel T47D.

- 43 -
Gambar 5.8 Grafik pengaruh Konsentrasi DoksorubisinTerhadap Viablitas Sel T47D
Kemampuan sitotoksik metformin yang sangat kecil terhadapsel T47D ini didukung kemampuan hambatan metforminterhadap siklus sel T47D yang sangat minim dan terjadi difase G0-G1. Uji siklus sel menggunakan metodeflowsitometri100 (Gambar 5.9 - 5.13, Tabel 1).
Gambar 5.9 Histogram Siklus Sel T47D Pada Sel Kontrol

- 44 -
Gambar 5.10 Histogram Siklus Sel T47D yangDipaparkan Metformin Konsentrasi 1738,2µg/mL
Gambar 5.11 Histogram Siklus Sel T47D yang DipaparkanMetformin Konsentrasi 3476,4 µg/mL

- 45 -
Gambar 5.12 Histogram Siklus Sel T47D yang DipaparkanDoksorubisin Konsentrasi 0,1 µg/mL
Gambar 5.13 Histogram Siklus Sel T47D yang DipaparkanDoksorubisin Konsentrasi 0,2 µg/mL

- 46 -
Tabel 1 Persentase Akumulasi Pada Tiap Fase dalam SiklusSel T47D
Pemanjangan fase G0-G1 juga terjadi terhadapsel MCF-7 yang dipapar dengan metformin konsentrasi2,5; 5; 10 dan 20 mM, dan semakin memanjang padapaparan metformin dua dan tiga hari berikutnya. Padahari pertama, pemanjangan fase G0-G1 akibat paparanmetformin 42,53 ± 0,35% dibanding kontrol 38,89 ±0,98. Fase G0-G1 pada sel yang dipapar metforminselama 48 jam menjadi 66,54 ± 0,04% dibandingkontrol 47,55±0,48%, dan setelah paparan metformin 72jam fase G0-G1 menjadi 69,14±0,24% dibanding kontrol58,93 ± 0,40%. Paparan metformin selama 48 jammeningkatkan sel yang mengalami apoptosis (fase subG1) dua kali lebih tinggi dibanding kontrol22.
Siklus sel merupakan jalan untuk sel tumbuh danberkembang, dan proses ini terjadi pada sel normal dan selkanker29. Akumulasi sel pada siklus sel merupakan salah satutarget utama agen antikanker. Siklus sel memiliki dua faseutama, yakni fase S (sintesis) dan fase M (mitosis)29. Selainitu, terdapat fase yang membatasi kedua fase utama tersebutyang dinamakan Gap.G1 (Gap-1) terdapat sebelum fase S dansetelah fase S dinamakan G2 (Gap-2). Checkpoint pada faseG1 akan dapat dilalui jika (1) ukuran sel memadai; (2)

- 47 -
ketersediaan nutrien mencukupi; dan (3) adanya faktorpertumbuhan (sinyal dari sel yang lain. Checkpoint pada faseG2 dapat dilewati jika ukuran sel memadai dan replikasikromosom terselesaikan dengan sempurna sedangkancheckpoint pada mitosis (M) terpenuhi bila semua kromosomdapat menempel pada gelendong (spindle) mitotik101.
Jika ada kekurangan protein esensial atau ditemukankelainan pada saat checkpoint, maka siklus tidak akanberlanjut.Pada fase G1, sel melakukan persiapan untuksintesis DNA. Fase ini merupakan fase awal cell cycleprogression yang diatur oleh faktor ekstraselular sepertimitogen dan molekul adhesi. Penanda fase ini adalah adanyaekspresi dan sintesis protein sebagai persiapan memasuki faseS102. Hambatan siklus sel T47D akibat paparan metforminpada G0-G1 diakibatkan sel kekurangan nutrisi akibat efekmetformin terhadap kompleks I rantai pernafasan mitokondriasehingga sel kekurangan ATP, selanjutmya aktivasi AMP-kinase terhadap berbagai substrat yang berperan dalammetabolisme sel mengakibatkan anabolisme sel terhambat,pertumbuhan sel terganggu22.
Berbeda halnya dengan doksorubisin yangmenunjukkan hambatan siklus sel pada fase S dan G2-M. Halini sesuai dengan kerja doksorubisin melalui hambatantopoisomerase II, interkalasi DNA, pengikatan membran selserta pembentukan radikal bebas semiquinon dan radikalbebas oksigen103. Checkpoint pada fase G2 dapat dilewati jikaukuran sel memadai dan replikasi kromosom terselesaikandengan sempurna 101. Doksorubisin dapat berinterkalasidengan DNA, secara langsung akan mempengaruhi transkripsidan replikasi. Doksorubisin mampu membentuk komplektripartit dengan topoisomerase II dan DNA.
Topoisomerase II adalah suatu enzim tergantung ATPyang bekerja mengikat DNA dan menyebabkan double-strandbreak pada ujung 3′fosfat sehingga memungkinkan penukaran

- 48 -
strand dan pelurusan DNA superkoil. Pelurusan strand inidiikuti dengan penyambungan strand DNA olehtopoisomerase II. Topoisomerase ini sangat penting fungsinyadalam replikasi dan perbaikan DNA. Pembentukan komplekstripartit tersebut akan menghambat penyambungan kembalistrand DNA, menyebabkan penghambatan daur sel terhenti difase G1 dan G2 serta memacu terjadinya apoptosis48.
Tak cukup uji siklus sel saja yang hanya mampumenghambat siklus sel T47D di fase G0-G1, kemampuanapoptosis metformin pun sangat minim bila dibandingkandoksorubisin (Gambar 5.14-5.18, Tabel 5.2)100.
Gambar 5.14 Uji Apoptosis Sel T47D Pada Sel KontrolDimana: R1 adalah sel hidup; R2 adalah sel mengalami apoptosis awal;
R3 dimana sel mengalami apoptosis akhir; R4 adalah selmengalami nekrosis

- 49 -
Gambar 5.15 Uji Apoptosis Sel T47D yang DipaparkanMetformin Konsentrasi 1738,2 µg/mL
dimana: R1 adalah sel hidup; R2 adalah sel mengalami apoptosis awal; R3adalah sel mengalami apoptosis akhir; R4 adalah sel mengalaminekrosis
Gambar 5.16 Uji Apoptosis sel T47D yang DipaparkanMetformin Konsentrasi 3476,4µg/mL
dimana: R1 adalah sel hidup; R2 adalah sel mengalami apoptosis awal; R3adalah sel mengalami apoptosis akhir; R4 adalah sel mengalaminekrosis
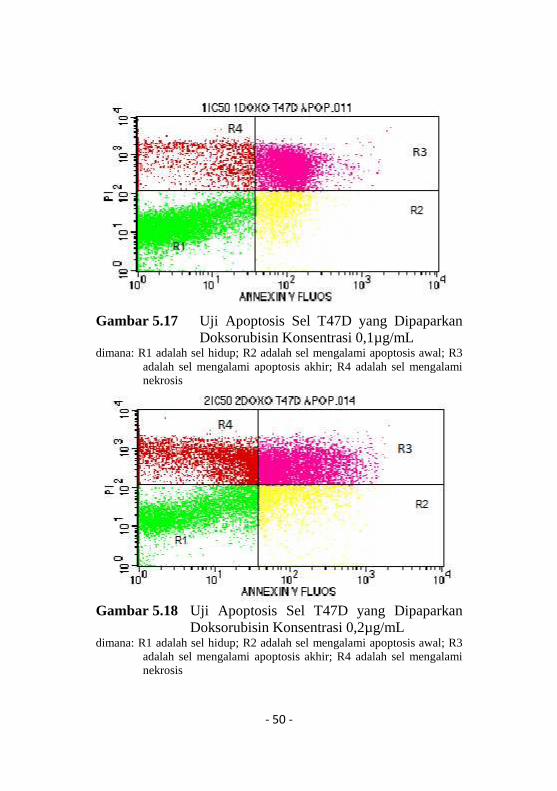
- 50 -
Gambar 5.17 Uji Apoptosis Sel T47D yang DipaparkanDoksorubisin Konsentrasi 0,1µg/mL
dimana: R1 adalah sel hidup; R2 adalah sel mengalami apoptosis awal; R3adalah sel mengalami apoptosis akhir; R4 adalah sel mengalaminekrosis
Gambar 5.18 Uji Apoptosis Sel T47D yang DipaparkanDoksorubisin Konsentrasi 0,2µg/mL
dimana: R1 adalah sel hidup; R2 adalah sel mengalami apoptosis awal; R3adalah sel mengalami apoptosis akhir; R4 adalah sel mengalaminekrosis

- 51 -
Selanjutnya, persentase sel di tiap kwadranditunjukkan pada Tabel 5.2. yang menunjukkan terjadinyapeningkatan apoptosis pada sel T47D yang dipaparkanmetformin 1738,2 µg/mL sebesar 3,72% dibanding kontrol2,37% dan apoptosis semakin meningkat pada sel yangdipaparkan metformin 3476,4 µg/mLsebesar 5,78%. Selainapoptosis, metformin meningkatkan nekrosis sel sebanyak1,42% dan meningkat menjadi 2,90% pada peningkatankonsentrasi metformin. Doksorubisin sebagai kontrol positifmenunjukkan efek apoptosis yang sangat besar (36,41%)dibandingkan sel kontrol dan metformin dan semakinmeningkat pada peningkatan konsentrasi (42,44%)100.
Tabel 5.2. Hasil Pengujian Apoptosis Sel T47D
Penelitian metformin terhadap sel T47D sebelumnyamenyatakan bahwa metformin menghambat pertumbuhan selT47D melalui aktivasi AMPK dan inhibisi histo H2Bmonoubikuitinasi 104 dan metformin menyebabkan apoptosisterhadap sel T47D dan meningkatkan ekspresi kas pase 8 danpase 9105. Penelitian tersebut menggunakan serial konsentrasi5, 10 dan 50 µM dan dievaluasi selama 24, 48 dan 72 jam,efek maksimum terjadi setelah paparan metformin 50 µMselama 72 jam105.
Penelitian lainnya terhadap sel MDA-MB-231 danMDA-MB-435 jenis sel kanker payudara dengan

- 52 -
menggunakan serial konsentrasi 1,25; 2,5; 5; 10 dan 20mmol/l selama 24, 48 dan 72 jam. Sel yang mengalamiapoptosis meningkat dari 7% dengan paparan metformin 5%menjadi 39,6% dengan paparan metformin 20 mM selama 24jam.
Hambatan metformin terhadap siklus sel danapoptosis T47D dikaitkan dengan menurunya ekspresi proteinp53 sel (5.19-5.23, Tabel 5.3)100.Pengujian ekspresi p53menggunakan metode imunositokimia, Sel yang positifmengekspresikan p53 secara visual terlihatsitoplasmanya mempunyai warna coklat/gelap,sedangkan sel yang tidak mengekspresikan p53berwarna transparan kebiruan 100.
Gambar 5.19. Ekspresi Protein p53 Pada Sel Kontrol T47DKet: lingkaran merah menunjukkan sel yang mengekspresikan p53
- 53 -
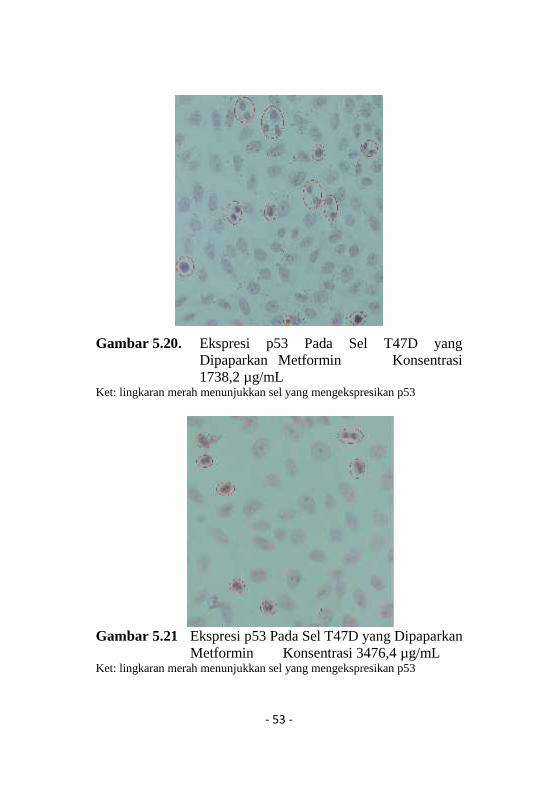
Gambar 5.20. Ekspresi p53 Pada Sel T47D yangDipaparkan Metformin Konsentrasi1738,2 µg/mL
Ket: lingkaran merah menunjukkan sel yang mengekspresikan p53
Gambar 5.21 Ekspresi p53 Pada Sel T47D yang DipaparkanMetformin Konsentrasi 3476,4 µg/mL
Ket: lingkaran merah menunjukkan sel yang mengekspresikan p53

- 54 -
Gambar 5.22 Ekspresi p53 Pada Sel T47D yangDipaparkan Doksorubisin Konsetrasi0,1 µg/mL
Ket: lingkaran merah menunjukkan sel yang mengekspresikan p53
Tabel 5.3 Analisa Ekspresi Protein p53 Pada Sel KankerT47D Menggunakan Uji Anova
Keterangan: Data dianalisis dengan one way Anova dengan uji lanjutBonferroni; * p<0,05 vs. kontrol
Metformin dan doksorubisin memilikikemampuan menurunkan ekspresi p53 sel T47D, namunberbeda dalam konsentrasi. Metformin membutuhkankonsentrasi yang sangat besar dibandingkan

- 55 -
doksorubisin untuk mendapatkan efek yang hampir samadengan doksorubisin dalam menurunkan p53. p53merupakan tumor suppressor yang mempertahankanstabilitas gen, berperan dalam siklus sel, apoptosis,senescence dan differensiasi. Mutasi p53 akanmenyebabkan kehilangan fungsi penjagaan kestabilangenom, bahkan memicu kanker106.
Penurunan ekspresi protein p53 yang mengalamimutasi pada sel T47D akan menurunkan pertumbuhansel kanker, P53 berperan sebagai mediator arrest siklussel107. Hal ini sesuai dengan penelitian terhadap selT47D dan sel MDA-MB-468 dengan membungkam p53mutan endogen melalui lentiviral shRNA yangmenunjukkan terjadinya kematian sel yang massifmelalui apoptosis dengan gambaran nuclear blebbing,PARP-1 cleavage, tetapi tidak terjadi pada MCF-7 atauMCF- 10A dengan keadaan p53 yang normal108.
Disebutkan juga bahwa p53 mutan merupakansuatu aktivator sejumlah gen seperti c-Myc,topoisomerase I dan MDR-1109 yang menyebabkanketidakstabilan genom serta terjadinya gangguan kontrolcheckpoint spindel110. P53 juga berefek pada protein bcl-2.Kontrol dan regulasi peristiwa apoptosis mitokondriajuga dapat terjadi melalui anggota dari famili proteinBcl-2.
Famili protein Bcl-2 mengatur permeabilitasmembran mitokondria dan dapat menjadi proteinproapoptosis atau antiapoptosis. Protein penekan tumorp53 memiliki peran penting dalam regulasi famili proteinBcl-2, dengan cara berikatan langsung dengan familiprotein Bcl-2 antiapoptosis dan mengaktifkan famili

- 56 -
protein Bcl-2 proapoptosis lalu kemudian mengaturpermeabilitas membran mitokondria. Koneksi p53 denganbcl2 terlihat dari gambar berikut, yang menerangkan bahwaregulasi p53 dapat terjadi langsung dengan mengikat bcl2,atau melalui PUMA, NOXA dan bim111.
Gambar 5.23 Koneksi p53 dengan bcl2111
Paparan metformin HCl selama 24 jam terhadap selT47D menyebabkan penuruan ekspresi protein bcl2. Hal inimendukung efek sitotoksik metformin (Gambar 5.24-5.27,Tabel 5.4).

- 57 -
Gambar 5.24 Ekspresi protein bcl-2 pada sel kontrol T47DKet: lingkaran merah menunjukkan sel yang mengekspresikan bcl-2
Gambar 5.25 Ekspresi bcl-2 Pada Sel T47D yangDipaparkan Metformin Konsentrasi 1738,2µg/mL
Ket: lingkaran merah menunjukkan sel yang mengekspresikan bcl-2

- 58 -
Gambar 5.26 Ekspresi bcl-2 pada sel T47D yangdipaparkan Metformin konsentrasi 3476,4µg/mL
Ket: lingkaran merah menunjukkan sel yang mengekspresikan bcl-2
Gambar 5.27 Ekspresi bcl-2 pada sel T47D yangdipaparkan Doksorubisin konsentrasi 0,1µg/mL
Ket: lingkaran merah menunjukkan sel yang mengekspresikan bcl-2

- 59 -
Tabel 5.4 Analisa Ekspresi Protein bcl-2 Pada Sel T47DMenggunakan Uji Anova
Ket: Data dianalisis dengan one way Anova dengan uji lanjut Bonferroni; *p < 0,05 vs. control; ** p < 0,01 vs. control; *** p < 0,001 vs. kontrol
Berdasarkan uji Anova terdapat perbedaan bermaknaekspresi bcl-2 pada sel T47D yang dipaparkan metforminkonsentrasi 3476,4 µg/mL dibandingkan doksorubisin,dimana nilai p sebesar 0,03 (p < 0,05)100.
Replikasi dan pembelahan DNA sel terjadi melaluisiklus sel, dimana siklus sel merupakan rangkaian sekuensyang meliputi fase G1 (pre sintesa), S (sintesa DNA), G2(premitotik) dan M (mitotik). Setiap fase dari siklus sel akanterjadi bila siklus sel sebelumnya telah lengkap dan akanberhenti jika terjadi defisiensi fungsi gen yang esensial. Tiapfase siklus sel akan diatur oleh siklin dan jenis siklin yangmengatur berbeda untuk tiap fase112.
Paparan metformin selama 24 jam terhadap sel T47Dmenimbulkan arrest siklus sel di fase G0-G1100, demikianjuga dengan penelitian Qu dan Cai21,113,21. Pada siklus selterjadi saat kritis berupa pengecekan (checkpoint) padabeberapa tempat diantaranya pada fase G1-S dan G2-M.Untuk checkpoint di fase G1-S diregulasi siklin dan siklindependent kinase inhibitor (CDKI) tertentu termasuk siklinD1, siklin E, p21 dan p27. Overekspresi siklin D1 dan siklin Emendukung perkembangan kanker114.

- 60 -
Sel T47D yang dipaparkan metformin selama 24 jammenunjukkan penurunan ekspresi siklin D1 (Gambar 5.28-5.30, Tabel 5.5). Penurunan ekspresi siklin D1 dapatmenjelaskan terjadinya arrest pada siklus sel fase G0-G1 dansesuai dengan penelitian Cai et al (2013) terhadap sel HepG2.
Gambar 5.28 Ekspresi Siklin D1 Pada Sel kontrol T47DKet: lingkaran merah menunjukkan sel yang mengekspresikan bcl-2
Gambar 5.29 Ekspresi Siklin D1 Pada Sel T47D yangDipaparkan Metformin Konsentrasi 1738,2µg/mL
Ket: lingkaran merah menunjukkan sel yang mengekspresikan Siklin D1

- 61 -
Gambar 5.30 Ekspresi Siklin D1 Pada Sel T47D yangDipaparkan Metformin Konsetrasi 3476,4µg/mL
Ket: lingkaran merah menunjukkan sel yang mengekspresikan Siklin D1
Tabel 5.5 Analisa Ekspresi Protein Siklin D1 Pada SelKanker T47D Menggunakan Uji Anova
Ket: Data dianalisis dengan one way Anova dengan uji lanjut Bonferroni;* p< 0,05 vs. kontrol
Berdasarkan Uji Anova didapatkan nilai p sebesar0,014 (p < 0,05), terdapat perbedaan bermakna ekspresi siklinD1 pada ketiga kelompok. Hasil sesuai Tabel 4.5menunjukkan adanya perbedaan bermakna ekspresi siklin D1pada kelompok sel yang dipaparkan metformin 3476,4 µg/mL(4,40 ± 3,89 %) dengan sel kontrol (14,61 ± 2,49 % %)dengan nilai p < 0,05.

- 62 -
KESIMPULAN
Metformin merupakan anti hiperglikemik yangmemiliki aktifitas anti kanker, bekerja langsung ke sel kankermelalui hambatan komlpeks I rantai pernafasan dengan hasilutama aktivasi AMPK sel yang menimbulkan hambatansintesis seluruh metabolisme sel dan bekerja tidak langasungmelalui penurunan hiperinsulinemia.
Pengembangan metformin sebagai anti kanker telahdiuji coba secara invitro dan invivo untuk beberapa jenis selkanker. Pengujian invitro untuk sel kanker payudara telahdiuji coba dengan hasil bahwa aktivitas anti kanker metforminterhadap sel kanker payudara jenis duktal (T47D) sangat kecilbila dibandingkan dengan efek sitotoksik doksorubisin, hal inijuga didukung dengan efek metformin terhadap siklus sel,apoptosis dan protein-protein pendukung . Uji invitro terhadapsel kanker payudara jenis duktal ini murni sel yangmengalami kanker payudara, tanpa mengalami diabetesmelitus, tanpa hiperinsulinemia. Padahal efek insulin sendirimemiliki peran penting dalam pembentukan kanker, dan halini yang tidak disentuh melalui uji invitro.
Selain terhadap jenis sel kanker payudara jenis duktal,uji invitro metformin terhadap sel kanker payudara jenis laintelah diuji coba dengan hasil bahwa memang metforminmampu menghambat pertumbuhan sel kanker payudaranamun dengan konsentrasi yang sangat besar. Berdasarkanhasil penelitian yang telah dilaksanakan dapat disimpulkanbahwa kemampuan metformin untuk menghambatpertumbuhan sel kanker payudara sangat kecil, dengankonsentrasi yang sangat besar. Sebagai monoterapi,metformin tidak mampu membunuh sel kanker payudara.

- 63 -
REFERENSI
1. Kumar, V., Abbas, A.K., Aster, J. . . Neoplasia. inBuku ajar patologi Robbins 155 (Elsevier Saunders,2013).
2. Kumar, V., Abas, A. K, and Foustro, N. PathologyBasic of Disease. (2005).
3. Ferlay, J. et al. Estimates of worldwide burden ofcancer in 2008: GLOBOCAN 2008. Int. J. Cancer127, 2893–2917 (2010).
4. Kementerian Kesehatan Republik Indonesia. StopKanker. (Pusat Data dan Informasi KesehatanRepublik Indonesia., 2016).
5. WHO. New Global Cancer Data : GLOBOCAN 2018.(2018).
6. Hortobagyi, G. N. et al. The global breast cancerburden: Variations in epidemiology and survival. Clin.Breast Cancer 6, 391–401 (2005).
7. Tao, Z. Q. et al. Breast Cancer: Epidemiology andEtiology. Cell Biochem. Biophys. 72, 333–338 (2015).
8. Shenoy, K. R dan Nileshwar, A. Kanker Payudara. inBuku ajar Ilmu Bedah 421–434 (Karisma, 2014).
9. Du XL, Jones DV, Z. D. Effectiveness of adjuvantchemotherapy for node-positive operable breast cancerin older woman. J Gerontol A Biol Sci Med Sci 60,1137–1144 (2005).
10. Colleoni, M. et al. Chemotherapy is more effective inpatients with breast cancer not expressing steroidhormone receptors: A study of preoperative treatment.Clin. Cancer Res. 10, 6622–6628 (2004).

- 64 -
11. Fujii, T. et al. Effectiveness of an adjuvantchemotherapy regimen for early-stage breast cancer.JAMA Oncol. 1, 1311–1318 (2015).
12. Kayl, A. E. & Meyers, C. A. Side-effects ofchemotherapy and quality of life in ovarian and breastcancer patients. Curr. Opin. Obstet. Gynecol. 18, 24–28 (2006).
13. Moiseenko, F., Volkov, N., Bogdanov, A., Dubina, M.& Moiseyenko, V. Resistance mechanisms to drugtherapy in breast cancer and other solid tumors: Anopinion. F1000Research 6, 288 (2017).
14. Geisler, S. et al. TP53 Gene Mutations Predict theResponse to Neoadjuvant Treatment with 5-Fluorouracil and Mitomycin in Locally AdvancedBreast Cancer. Clin. Cancer Res. 9, 5582–5588(2003).
15. Di Leo, A. et al. p-53 gene mutations as a predictivemarker in a population of advanced breast cancerpatients randomly treated with doxorubicin ordocetaxel in the context of a phase III clinical trial.Ann. Oncol. 18, 997–1003 (2007).
16. Andersson, J; Larsson, L; Klaar, S; Holmberg, L;Nilsson, J. et al. Worse survival for TP53 (p53)-mutated breast cancer patients receiving adjuvantCMF. Annal 16, 743–748 (2005).
17. Daugan, M., Dufaÿ Wojcicki, A., d’Hayer, B. &Boudy, V. Metformin: An anti-diabetic drug to fightcancer. Pharmacol. Res. 113, 675–685 (2016).
18. Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC,Thiru P, Yucel B, Wang T, Chen WW, Clish CB, S. D.metabolic determinants of cancer cell sensitivity toglucose limitation and biguanides. Nature 508, 108–

- 65 -
112 (2014).
19. Lin, C. M. et al. Association betweengastroenterological malignancy and diabetes mellitusand anti-diabetic therapy: A nationwide, population-based cohort study. PLoS One 10, 1–11 (2015).
20. Wibowo, Y. C., Mahanani, M. R., Budiani, D. R. &Mudigdo, A. P0101 Metformin inhibits cyclin D1expression in a p53-deficient colon cancer cell line invitro. Eur. J. Cancer 51, e20 (2015).
21. Cai, X. et al. Metformin suppresses hepatocellularcarcinoma cell growth through induction of cell cycleG1/G0 phase arrest and p21CIPand p27KIPexpressionand downregulation of cyclin D1 in vitro and in vivo.Oncol. Rep. 30, 2449–2457 (2013).
22. Queiroz, E. A. I. F. et al. Metformin induces apoptosisand cell cycle arrest mediated by oxidative stress,AMPK and FOXO3a in MCF-7 breast cancer cells.PLoS One 9, (2014).
23. Li, P., Zhao, M., Parris, A. B., Feng, X. & Yang, X.P53 is required for metformin-induced growthinhibition, senescence and apoptosis in breast cancercells. Biochem. Biophys. Res. Commun. 464, 1267–1274 (2015).
24. Ruddon, R. W. Cancer Biology. (Oxford UniversityPress., 2007).
25. Hanahan, D and Weinberg, R. Hallmarks of cancer :The next generation. Cell 144, 646–674 (2011).
26. Pecorino L. Molecular biology of cancer. Simplex(Oxfort University Press, 2012).doi:10.1017/CBO9781107415324.004
27. Diananda R. Mengenal seluk beluk kanker. (Kata hati,

- 66 -
2009).
28. Tsao AS; Kim ES; Hong WK. Chemoprevention ofCancer. Cancer J Clin 54, 150–180 (2004).
29. Nurse, P. A long twentieth century of the cell cycleand beyond. Cell 100, 71–78 (2000).
30. Foster, J. S., Henley, D. C., Ahamed, S. &Wimalasena, J. Estrogens and cell-cycle regulation inbreast cancer. Trends Endocrinol. Metab. 12, 320–327(2001).
31. Hadi, R. Mekanisme Apoptosis Pada Regresi SelLuteal. Maj. Kesehat. PharmaMedika 3, 246–254(2011).
32. Elmore, S. Apoptosis: A Review of Programmed CellDeath. Toxicol. Pathol. 35, 495–516 (2007).
33. Norbury, C. J. & Hickson, I. D. Cellular response toDNA damage. Annu Rev.Pharmacol.Toxicol 41, 367–401 (2001).
34. Krueger, A., Baumann, S., Krammer, P. H. &Kirchhoff, S. FLICE-Inhibitory Proteins : Regulatorsof Death Receptor-Mediated Apoptosis Downloadedfrom http://mcb.asm.org/ on October 21 , 2015 byUniversitaetsbibliothek Frankfurt am Main. Mol. Cell.Biol. 21, 8247–8254 (2001).
35. Igney, F. H. & Krammer, P. H. Death and Anti-Death:Tumour Resistance To Apoptosis. Nat. Rev. Cancer 2,277–288 (2002).
36. Chen G and Goeddel DV. TNF-R1 Signaling: Abeautiful pathway. Science (80-. ). 296, 1635–1636(2002).
37. Thorburn, A. Death receptor-induced cell killing. Cell.

- 67 -
Signal. 16, 139–144 (2004).
38. Scorrano, L. & Korsmeyer, S. J. Mechanisms ofcytochrome c release by proapoptotic BCL-2 familymembers. Biochem. Biophys. Res. Commun. 304, 437–444 (2003).
39. Chowdhury, I., Tharakan, B. & Bhat, G. K. Caspases -An update. Comp. Biochem. Physiol. - B Biochem.Mol. Biol. 151, 10–27 (2008).
40. Walters, J. et al. A constitutively active anduninhibitable caspase-3 zymogen efficiently inducesapoptosis. Biochem. J. 424, 335–345 (2009).
41. Slee, E. A., Adrain, C. & Martin, S. J. ExecutionerCaspase-3, -6, and -7 Perform Distinct, Non-redundantRoles during the Demolition Phase of Apoptosis. J.Biol. Chem. 276, 7320–7326 (2001).
42. Vandivier, R. W., Henson, P. M. & Douglas, I. S.Burying the dead: The impact of failed apoptotic cellremoval (efferocytosis) on chronic inflammatory lungdisease. Chest 129, 1673–1682 (2006).
43. Savill, J., Gregory, C. & Haslett, C. Eat Me or Die.Science (80-. ). 302, 1516–1517 (2003).
44. Hawkins LA and Devitt A. Current understanding ofthe mechanisms for clerance of apoptotic cells-A finebalance. J. Cell Death 6, 57–68 (2013).
45. Shenoy, K. R dan Nileshwar, A. Payudara. in Bukuajar Ilmu Bedah 421–434 (Karisma, 2014).
46. Nafrialdi dan Gan, S. Antikanker. in Farmakologi danterapi Departemen Farmakologi dan TerapeutikFakultas Kedokteran Universitas Indonesia 737–761(2016).

- 68 -
47. Childs, A. C., Phaneuf, S. L., Dirks, A. J., Phillips, T.& Leeuwenburgh, C. Doxorubicin Treatment in VivoCauses Cytochrome c Release and CardiomyocyteApoptosis , As Well As Increased MitochondrialEfficiency , Superoxide Dismutase Activity , and Bcl-2 : Bax Ratio 1. Cancer Res. 4, 4592–4598 (2002).
48. Minotti, G., Menna, P., Salvatorelli, E., Cairo, G. &Gianni, L. Anthracyclines : Molecular Advances andPharmacologic Developments in Antitumor Activityand Cardiotoxicity. Pharmacol Rev 56, 185–229(2004).
49. Han, X. et al. Protective effect of naringenin-7- O -glucoside against oxidative stress induced bydoxorubicin in H9c2 cardiomyocytes. Biosci. Trends 6,19–25 (2012).
50. Bruton, L., Lazo, J. S., and Parker, K. L. Goodman &Gilman’s The Pharmacological Basis of Therapeutics.(McGrawHill. Lange., 2005).
51. Gewirtz, D. A. A Critical Evaluation of theMechanisms of Action Proposed for the AntitumorEffects of the Anthracycline Antibiotics Adriamycinand Daunorubicin. Biochem. Pfarmacology 57, 727–741 (1999).
52. Lawrenti H. Kemoterapi Golongan Taxane. CDK-20940, 790–794 (2013).
53. Freshney, R. . Culture of animal cells: A manual ofbasic technique and specialized applications. (Wiley,2010).
54. Zuhud, E. A. Bukti Kedahsyatan: Sirsak MenumpasKanker. (AgroMedia Pustaka, 2011).
55. Kupcsik, L. Estimation of Cell Number Based on

- 69 -
Metabolic Activity: The MTT Reduction Assay.InStoddart, J.M. (ed.) Mammalian Cell Viability:Methods and Protocols. (Totowa, NJ: Humana Press,2011).
56. Nurhayati, E. SITOTOKSISITAS EKSTRAK DANSENYAWA FLAVONOID DAUN SUKUN (Artocarpus altilis ) TERHADAP SEL T47DMELALUI INDUKSI APOPTOSIS. (2012).
57. Sayed, D. M., El-attar, M. M. & Hussein, A. A. R. M.Evaluation of Flow Cytometric Immunophenotypingand DNA Analysis for Detection of Malignant Cells inSerosal Cavity Fluids. Diagn. Cytopathol. 37, (2009).
58. Shapiro, H. M. Practical Flow Cytometry. (Wiley-Liss., 2003).
59. Riccardi, C. & Nicoletti, I. Analysis of apoptosis bypropidium iodide staining and flow cytometry. Nat.Protoc. 1, 1458–1461 (2006).
60. Brumatti, G., Sheridan, C. & Martin, S. J. Expressionand purification of recombinant annexin V for thedetection of membrane alterations on apoptotic cells.Method 44, 235–240 (2008).
61. Merighi, A. and Lossi, L. The Evolution ofImmunocytochemistry in the Dissection of NeuralComplexity. in Immunocytochemistry and RelatedTechniques 1–35 (Springer, 2015).
62. Fetsch, P. A. Immunocytochemistry. in MedicalBiomethods Handbook 555–571 (Humana Press,2005).
63. Mao, S.-Y. and Mullins, J. M. Conjugation ofFluorochromes to Antibodies. in ImmunocytochemicalMethods and Protocols 43–48 (Humana Press, 2010).

- 70 -
64. Bratthauer, G. L. Overview of Antigen DetectionThrough Enzymatic Activity. in ImmunocytochemicalMethods and Protocols. . 231-241 (Humana Press,2010).
65. Burry, R. W. Controls for immunocytochemistry: Anupdate. J. Histochem. Cytochem. 59, 6–12 (2011).
66. Owen, M. R., Doran, E. & Halestrap, A. P. Evidencethat metformin exerts its anti-diabetic effects troughinhibition of complex 1 of the mitochondrialrespiratory chain. Biochem J 614, 607–614 (2000).
67. Viollet, B. et al. Cellular and molecular mechanisms ofmetformin : an overview . To cite this version : HALId : inserm-00658070. (2012).
68. Repiscak, P., Erhardt, S., Rena, G. & Paterson, M. J.Biomolecular Mode of Action of Metformin inRelation to Its Copper Binding Properties.Biochemistry 53, 787–795 (2014).
69. Bridges, H. R., Jones, A., Pollak, M. & Hirst, J. Effectsof metformin and other biguanides on oxidativephosphorylation in mitochondria. Biochem J 462, 475–487 (2014).
70. Pollak, M. Repurposing biguanides to target energymetabolism for cancer treatment. Nat. Med. 20, 591–593 (2014).
71. Barco, S. Del et al. Metformin : Multi-facetedprotection against cancer ABSTRACT : DIABETESAND CANCER : FROM. Oncotarget 2, 896–917(2011).
72. Morales, D. R. & Morris, A. D. Metformin in CancerTreatment and Prevention. Annu. Rev. Med. 66, 17–29(2015).

- 71 -
73. Weinberg SE, C. N. Targeting Mitochondria forCancer Therapy. Nat. Chem. Biol. 11, 9–15 (2015).
74. Wheaton, W. W. et al. Metformin inhibitsmitochondrial complex I of cancer cells to reducetumorigenesis. Elife 2014, 1–18 (2014).
75. Mihaylova, M. M. & Shaw, R. J. The AMPKsignalling pathway coordinates cell growth, autophagyand metabolism. Nat. Cell Biol. 13, 1016–1023 (2011).
76. Brown, K. A., Samarajeewa, N. U. & Simpson, E. R.Endocrine-related cancers and the role of AMPK. Mol.Cell. Endocrinol. 366, 170–179 (2012).
77. Choi, Y. K. & Park, K. G. Metabolic roles of AMPKand metformin in cancer cells. Mol. Cells 36, 279–287(2013).
78. Cardaci, S., Filomeni, G. & Ciriolo, M. R. Redoximplications of AMPK-mediated signal transductionbeyond energetic clues. J. Cell Sci. 125, 2115–2125(2012).
79. Hardie, D. G. AMP-activated protein kinase-an energysensor that regulates all aspects of cell function. GenesDev. 1–14 (2011). doi:10.1101/gad.17420111.crease
80. Chen, Z. et al. TAK1 activates AMPK-dependent celldeath pathway in hydrogen peroxide-treatedcardiomyocytes, inhibited by heat shock protein-70.Mol. Cell. Biochem. 377, 35–44 (2013).
81. Jeon, S. M., Chandel, N. S. & Hay, N. AMPKregulates NADPH homeostasis to promote tumour cellsurvival during energy stress. Nature 485, 661–665(2012).
82. Li, W., Saud, S. M., Young, M. R., Chen, G. & Hua,B. Targeting AMPK for cancer prevention and

- 72 -
treatment. Oncotarget 6, 7365–7378 (2015).
83. Abdelmonsif, D. A., Sultan, A. S., El-Hadidy, W. F. &Abdallah, D. M. Targeting AMPK, mTOR and β-Catenin by Combined Metformin and Aspirin Therapyin HCC: An Appraisal in Egyptian HCC Patients. Mol.Diagnosis Ther. 22, 115–127 (2018).
84. Hwang, J. T. et al. Resveratrol induces apoptosis inchemoresistant cancer cells via modulation of AMPKsignaling pathway. Ann. N. Y. Acad. Sci. 1095, 441–448 (2007).
85. Lin, J. N. et al. Resveratrol modulates tumor cellproliferation and protein translation via SIRT1 -dependent AMPK activation. J. Agric. Food Chem. 58,1584–1592 (2010).
86. Rashid, A. et al. Resveratrol enhances prostate cancercell response to ionizing radiation. Modulation of theAMPK, Akt and mTOR pathways. Radiat. Oncol. 6,1–12 (2011).
87. Shen, Y. et al. Quercetin and its metabolites improvevessel function by inducing eNOS activity viaphosphorylation of AMPK. Biochem. Pharmacol. 84,1036–1044 (2012).
88. Kim, H. S. et al. Berberine-induced AMPK activationinhibits the metastatic potential of melanoma cells viareduction of ERK activity and COX-2 proteinexpression. Biochem. Pharmacol. 83, 385–394 (2012).
89. Li, Y. et al. Activation of AMPK by berberinepromotes adiponectin multimerization in 3T3-L1adipocytes. FEBS Lett. 585, 1735–1740 (2011).
90. Jeong, H. W. et al. Berberine suppressesproinflammatory responses through AMPK activation

- 73 -
in macrophages. AJP Endocrinol. Metab. 296, E955–E964 (2009).
91. Wullschleger, S., Loewith, R. & Hall, M. N. TORsignaling in growth and metabolism. Cell 124, 471–484 (2006).
92. Jones, R. G. et al. AMP-activated protein kinaseinduces a p53-dependent metabolic checkpoint. Mol.Cell 18, 283–293 (2005).
93. Leclerc, I., Lenzner, C., Gourdon, L., Vaulont, S. &Kahn, A. Hepatocyte Nuclear Factor-4 a Involved inType 1 Maturity-Onset Diabetes of the Young Is aNovel Target of AMP-Activated Protein Kinase.Animals 50, 1515–1521 (2001).
94. Kawaguchi, T., Osatomi, K., Yamashita, H.,Kabashima, T. & Uyeda, K. Mechanism for fatty acid‘sparing’ effect on glucose-induced transcription:Regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J.Biol. Chem. 277, 3829–3835 (2002).
95. Liang, J. et al. The energy sensing LKB1-AMPKpathway regulates p27kip1 phosphorylation mediatingthe decision to enter autophagy or apoptosis. Nat. CellBiol. 9, 218–224 (2007).
96. Motoshima, H., Goldstein, B. J., Igata, M. & Araki, E.AMPK and cell proliferation - AMPK as a therapeutictarget for atherosclerosis and cancer. J. Physiol. 574,63–71 (2006).
97. Dai, X., Cheng, H., Bai, Z. & Li, J. Breast cancer cellline classification and Its relevance with breast tumorsubtyping. J. Cancer 8, 3131–3141 (2017).
98. Abcam. T47D (Human ductal breast epithelial tumor

- 74 -
cell line). Whole Cell Lysate (ab14899) datasheet(2007).
99. R, A. AKTIVITAS SITOTOKSIK EKSTRAKETANOL DAN FRAKSI - FRAKSI DAUNSelaginella willdenowii. (2018).
100. Rangkuti, I Y; Hasibuan, P A Z; Widyawati, T. TheCytotoxic Test of Metformin Hydrochloride to T47DBreast Cancer Cell. IJSRM 10, 1–7 (2018).
101. Pecorino, L. Molecular Biology of Cancer:Mechanisms, Targets, and Therapeutics. (OxfordUniversity Press., 2012).
102. Ruddon, R. W. Cancer Biology. (Oxford UniversityPress., 2007).
103. Bruton, L., Lazo, J. S., and Parker, K. L. E. M. L.Goodman & Gilman’s The Pharmacological Basis ofTherapeutics. (2005).
104. Du, Y. et al. Metformin inhibits histone H2Bmonoubiquitination and downstream genetranscription in human breast cancer cells. Oncol. Lett.8, 809–812 (2014).
105. Haji HA, Sheibak H, Khosraavi M, A. J. The effect ofmetformin on the expression of caspase 3, 8, 9 andPARP-1 in human breast cancer cell line T47D. IJHS3, 147–153 (2016).
106. Liu D, Song H, X. Y. A common gain of function ofp53 cancer mutants in inducing genetic instability.Oncogene 29, 949–956 (2010).
107. Kastan, M. B. et al. A mammalian cell cyclecheckpoint pathway utilizing p53 and GADD45 isdefective in ataxia-telangiectasia - ScienceDirect. Cell71, 587–597 (2017).

- 75 -
108. Lim, L. Y., Vidnovic, N., Ellisen, L. W. & Leong, C.O. Mutant p53 mediates survival of breast cancer cells.Br. J. Cancer 101, 1606–1612 (2009).
109. Murphy, K. L., Dennis, A. P. & Rosen, J. M. A gain offunction p53 mutant promotes both genomic instabilityand cell survival in a novel p53-null mammaryepithelial cell model. FASEB J. 14, 2291–2302 (2000).
110. Matas, D. et al. Integrity of the N-terminaltranscription domain of p53 is required for mutant p53interference with drug-induced apoptosis. EMBO J. 20,4163–4172 (2001).
111. Hemann, M. T. & Lowe, S. W. The p53-Bcl-2connection. Cell Death Differ. 13, 1256–1259 (2006).
112. Kumar, V., Abbas, A.K., Aster, J. . . Buku ajarpatologi Robbins. in Neoplasia 155 (ElsevierSaunders, 2013).
113. Tsai, H.-H. et al. Metformin promotes apoptosis inhepatocellular carcinoma through the CEBPD-inducedautophagy pathway. Oncotarget 8, 13832–13845(2017).
114. Biliran HJ, Wang Y, Banerjee S, et al. Overexpressionof cyclin D1 promotes tumor cell growth and confersresistance to cisplatin-mediated apoptosis in anelastase-myc transgene expressing pancreatic tumorcell line. Clin Cancer Res 11, 6075–6086. (2005).





















