Epidermolisis Bulosa Simpleks Dowling Meara
32
PRESENTASI KASUS Kepada Yth: Dipresentasikan pada : Hari/Tanggal : Jam : 10:00 WITA Epidermolisis bulosa simpleks yang diduga suatu varian Dowling Meara Oleh: Azhar Ramadan Nonci Pembimbing: dr. Luh Mas Rusyati, Sp.KK.
-
Upload
rama-nonci -
Category
Documents
-
view
113 -
download
2
description
case report
Transcript of Epidermolisis Bulosa Simpleks Dowling Meara

PRESENTASI KASUS Kepada Yth:
Dipresentasikan pada :
Hari/Tanggal :
Jam : 10:00 WITA
Epidermolisis bulosa simpleks yang diduga suatu varian Dowling Meara
Oleh:
Azhar Ramadan Nonci
Pembimbing:
dr. Luh Mas Rusyati, Sp.KK.
PROGRAM PENDIDIKAN DOKTER SPESIALIS I
BAGIAN/SMF ILMU KESEHATAN KULIT DAN KELAMIN
FAKULTAS KEDOKTERAN UNUD/RS SANGLAH DENPASAR

2013

16
PENDAHULUAN
Epidermolisis bulosa (EB) merupakan kelainan genetik berupa gangguan atau
ketidakmampuan kulit dan epitel lain melekat pada jaringan konektif di bawahnya
dengan manifestasi tendensi terbentuknya bula dan vesikel setelah terkena trauma
atau gesekan ringan.1 Beberapa penulis mendefinisikan EB sebagai suatu
kelompok penyakit herediter yang ditandai dengan terbentuknya bula pada kulit
dan mukosa terutama mukosa mulut dan esofagus. Bula dapat terbentuk karena
gesekan, trauma mekanik ringan maupun terjadi secara spontan. Penyakit ini
sering disebut mechanobullous disorders.1
Penyakit ini pertama kali dikemukakan oleh Koebner pada tahun 1886
sebagai epidermolisis bulosa herediter.2 Berdasarkan atas letaknya bula, terjadi
jaringan parut atau tidak, serta diturunkan secara genetik, maka EB dibagi menjadi
3 kelompok mayor yaitu EB simpleks, EB junctional dan EB distrofik serta
bentuk EB yang didapat.1,2 Masing-masing kelompok mayor tersebut mempunyai
beberapa varian.1-3
Penyakit ini jarang ditemukan, insidennya diperkirakan 1:50,000 kelahiran
per tahun.1-2 Dari tahun 1986-1990 insidens EB herediter di Amerika serikat
berkisar 19,6 kelahiran hidup per satu juta kelahiran terdiri atas EB simpleks 10,8,
EB junctional 2,0 dan EB distrofik dominan 2,0 serta EB distrofik resesif 2,0.
Diperkirakan prevalensi EB pada tahun 1990 di Amerika serikat 8,2 per satu juta
kelahiran. Prevalensi seluruh EB simpleks di Norwegia berkisar 1-14 per satu
juta,2 sedangkan di Inggris prevalensi EB simpleks tipe Weber-Cockayne (lokasi
tangan dan kaki) diperkirakan 10-20 per satu juta, tipe Koebner (generalisata)
hanya sekitar 2 per satu juta kelahiran.2,3 Selanjutnya penelitian Horn dan Tidman
pada tahun 1999, di Inggris didapatkan dari 130 pasien EB simpleks yang
terbanyak adalah tipe Koebner 53% diikuti tipe Weber-Cockayne sebanyak 42%
serta terdapat 5% penderita EB simpleks tipe Dowling-Meara.2
Berikut dilaporkan suatu kasus epidermolisis bulosa simpleks yang diduga
varian Dowling Meara pada seorang anak laki-laki berumur 13 tahun. Kasus ini
dilaporkan karena penyakit ini jarang ditemukan dan walaupun pengobatannya
hanya suportif dan paliatif, diharapkan dapat menambah pengetahuan kita dalam
hal penegakkan diagnosis mengingat banyaknya varian dari penyakit ini.

16
KASUS
Seorang anak laki-laki, usia 13 tahun, suku Bali, nomor rekam medis 01.59.13.28
datang ke poliklinik RSUP Sanglah Denpasar dengan membawa surat rujukan dari
dokter spesialis kulit dan kelamin dengan diagnosis suspek epidermolisis bulosa
herediter, saran untuk dilakukan biopsik kulit dan pemeriksaan histopatologi.
Penderita datang dengan keluhan utama luka pada seluruh tubuh yang
diawali dengan gelembung berair. Gelembung berair muncul sejak 5 hari yang
lalu, gelembung terdapat di daerah siku dan punggung belakang. Selain itu
penderita juga mengeluh sedikit nyeri pada daerah luka dan mengeluh sedikit
gatal pada beberapa tempat. Saat ini penderita tidak mengeluh demam. Selama
hidupnya penderita telah berulang kali mengalami keluhan yang sama, terutama
setelah melakukan aktivitas seperti mengangkat benda, berolahraga, memakai
sepatu, terjatuh dan sebagainya. Gelembung timbul di berbagai tempat seperti
tangan, kaki, lengan dan tungkai yang kemudian pecah spontan dengan
mengeluarkan cairan jernih. Keluhan seperti ini muncul pertama kali sejak umur
10 hari setelah lahir saat tali pusat lepas setelah itu timbul gelembung berisi air
pada lokasi pusar. Saat umur 12 hari penderita dirawat di rumah sakit karena
diare, dilokasi infus dipasang timbul luka yang sebelumnya diawali gelembung
kecil berisi air. Sejak saat itu timbul luka-luka diseluruh tubuh terutama setelah
terkena gesekan atau benturan. Gelembung berair kemudian menyebar sampai ke
kaki dan punggung atas. Penderita juga kadang-kadang mengeluh sedikit gatal
dan nyeri dirasa pada daerah luka dan bertambah berat terutama saat terkena
sedikit benturan atau saat cuaca panas. Selain itu pada tubuh dan ekstremitas
penderita juga terdapat bercak-bercak berwarna putih.
Gelembung berair dan luka biasanya dapat sembuh sendiri secara spontan
dan dalam beberapa minggu bekas luka dapat hilang. Penderita juga mengeluh
sering sariawan terutama apabila setelah mengkonsumsi makanan yang hangat
atau panas dan lebih kasar. Selain itu sejak 5 tahun yang lalu, penderita juga
mengeluh memiliki mata juling dan penglihatan ganda. Sejak bayi hingga
sekarang kondisi kulit cenderung semakin membaik walaupun tidak pernah
sembuh total. Penderita telah beberapa kali berobat ke puskesmas dan dokter
umum untuk penyakit ini, namun tidak pernah memperoleh kesembuhan.

16
Penderita adalah anak tunggal, kedua orang tua penderita tidak memiliki
keluhan yang sama seperti penderita, kedua orang tua penderita memiliki
hubungan keluarga (sepupu) yaitu memiliki satu kakek dan nenek yang sama.
Penderita tidak memiliki riwayat sakit asma, namun ibu penderita memiliki
keluhan sakit asma. Riwayat penyakit sistemik lainnya pada kedua orang tua
penderita seperti tekanan darah tinggi dan kencing manis disangkal. Dari riwayat
kelahiran, penderita lahir secara spontan ditolong oleh dokter spesialis kandungan
di rumah sakit dengan berat lahir 3,2 kg, panjang badan 51 cm. Penderita
mendapatkan air susu ibu hingga berusia 7 bulan, dan selanjutnya mengkonsumsi
susu formula. Nafsu makan penderita baik walaupun apabila mengkonsumsi
makanan yang sedikit keras dan kasar, agak sulit menelan.
Riwayat sosial, penderita seorang pelajar kelas 1 SMP dan tidak memiliki
masalah dalam perkembangan akademis. Penderita kadang-kadang merasa
berbeda dengan orang lain karena penyakitnya yang tak kunjung sembuh, namun
penderita tidak memiliki masalah dalam pergaulan dengan teman-temannya.
Pemeriksaan fisik ditemukan keadaan umum penderita baik, kesadaran
kompos mentis, berat badan 35 kg, tinggi badan 148 cm, tekanan darah 120/80
mmHg, denyut nadi 82x/menit, frekuensi pernafasan 20x/menit, suhu aksila
36,80C. Pada status generalis didapatkan kepala normosefali, kedua mata tidak
tampak anemia, ikterus maupun hiperemia, pupil isokor, reflek cahaya positif,
strabismus eksotropia. Pemeriksaan telinga, hidung dan tenggorokan didapatkan
kesan tenang dan pada leher tidak ditemukan pembesaran kelenjar getah bening.
Pada pemeriksaan toraks didapatkan suara jantung (S1 dan S2) tunggal, reguler,
tidak terdapat murmur. Suara nafas paru-paru vesikuler, tidak ditemukan adanya
rhonki ataupun wheezing. Pada pemeriksaan abdomen, hepar dan lien tidak teraba,
bising usus dalam batas normal, tidak terdapat distensi abdomen. Ekstremitas atas
dan bawah teraba hangat, terdapat edema pada kedua tungkai bawah.
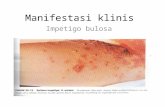
Status dermatologi lokasi pada kulit kepala, wajah, dada, perut, punggung,
bokong, ekstremitas atas dan bawah, tampak efloresensi vesikel dan bula multipel,
bentuk bulat, ukuran bula bervariasi Ø 1cm-3cm, dinding kendor berisi cairan
serous, diatas kulit normal. Di beberapa tempat terdapat makula hipopigmentasi
multipel, bentuk geografika, batas tegas. Di lokasi lain terdapat erosi multipel,

16
bentuk geografika, ukuran 0,5x1cm sampai 3x4cm beberapa ditutupi krusta tipis
coklat kehitaman. Lokasi kuku jari tangan tampak distrofi dan pada jari kaki
terdapat onikolisis.
Gambar 1 Gambar 2 Gambar 3
Gambar 4 Gambar 5 Gambar 6
Gambar 7 Gambar 8

16
Pemeriksaan penunjang darah lengkap didapatkan hasil leukosit 8,68 K/µL
(4,10-11,00); neutrofil 4,56 K/µL (2,50-7,50); limfosit 1,25 K/µL (1,00-4,00),
monosit 1,76 K/µL (0,10-1,20); eosinofil 0,20 K/µL (0,00-0,50); hemoglobin
12,45 g/dl (13,50-17,50); hematokrit 41,40 % (41,00-53,00); trombosit 398,00
K/µL (150,00-440,00). Pada pemeriksaan kimia klinik didapatkan albumin 3,45
g/dL (3,40-4,80); BUN 14,00 mg/dL (8,00-23,00); kreatinin 0,90 mg/dL (0,70-
1,20); natrium 138,00 mmol/L (136,00-145,00); kalium 4,60 mmol/L (3,5-5,1);
glukosa darah sewaktu 95,10 mg/dL (70,00-140,00).
Berdasarkan anamnesis tentang riwayat penyakit, pemeriksaan fisik dan
pemeriksaan penunjang didapatkan diagnosis kerja suspek epidermolisis bulosa
simpleks, dengan diagnosis banding epidermolisis bulosa tipe junctional, distrofik
dan epidermolisis akuisata. Penatalaksanaan yang diberikan adalah Gentasmisin
krim 2x sehari pada lesi erosi dan krusta, kompres terbuka NaCl 0,9% 3x sehari
lama setiap kompres 20menit. Direncanakan biopsi kulit pada lesi bula dan konsul
ke bagian Penyakit Mata serta konsul ke bagian Gizi untuk kebutuhan gizi
penderita, KIE tentang penyakit penderita.
Konsul ke bagian Penyakit Mata didiagnosis dengan Eksotropia intermiten
+ ambliopia Diberikan terapi Eyefresh tetes mata 4 x 1 tetes pada mata kanan dan
kiri. Rencana kontrol ke poliklinik mata dua minggu kemudian.
PENGAMATAN LANJUTAN
Pengamatan hari ke empat belas di poliklinik kulit dan kelamin RS Sanglah, dari
anamnesis didapatkan keluhan luka pada seluruh badan masih tetap, beberapa
sudah mengering, namun timbul gelembung berisi air yang baru di bebrapa tempat
antara lain di tangan, kaki, punggung dan bokong karena disebabkan akibat
benturan. Keluhan kulit yang tebal pada telapak kaki penderita juga masih ada.
Pemeriksaan fisik didapatkan keadaan umum baik, kesadaran kompos
mentis. Tekanan darah 120/80 mmHg, denyut nadi 85x/menit, frekuensi
pernafasan 18x/menit, suhu aksila 36,70C. Status generalis dalam batas normal.
Status dermatologi, lokasi pada kulit kepala, wajah, dada, perut, punggung,
ekstremitas atas dan bawah, masih sama seperti sebelumnya, tampak efloresensi
bula multipel, bentuk bulat, ukuran bula bervariasi Ø 1cm-3cm, dinding kendor

16
berisi cairan serus, diatas kulit normal. Di beberapa tempat terdapat makula
hipopigmentasi multipel, bentuk geografika, batas tegas. Di lokasi lain terdapat
erosi multipel, bentuk geografika, ukuran 0,5x1cm sampai 3x4cm beberapa
ditutupi krusta tipis coklat kehitaman. Lokasi kuku jari tangan tampak distrofi dn
pada jari kaki terdapat onikolisis.
Gambar 2 Gambar 3
Gambar 4 Gambar 5 Gambar 6
Gambar 7 Gambar 8

16
Hasil biopsi kulit multipel di dua lokasi bula pada regio antebrachii dan
regio kruris, sediaan kulit dari antebrachii terdiri dari epidermis, dermis, adneksa
kulit dan lemak subkutan. Tampak cleft di subepidermal dengan atap cleft
menunjukan epidermis yang sebagian mulai tampak nekrotik serta mengandung
beberapa sebaran apoptotic keratinocytes. Epidermis ini ditutupi oleh basket
weave keratin. Dermis atas menunjukan “preserved dermal papillae”. Pembuluh
darah yang melebar dengan infiltrate limfosit ringan perivaskuler serta fokus-
fokus pigmen incontinence.
Sediaan kulit berasal dari regio kruris terdiri dari epidermis dan dermis –
lemak subkutan yang terpisah (junctional separation?). Dermis atas menunjukan
gambaran “papilla dermis” yang terpreservasi, proliferasi pembuluh darah kecil
yang sebagian tampak melebar, serta infiltrat limfosit ringan perivaskuler.
Tampak pula sedikit sebaran netrofil di interstetiil dermis. Epidermis yang
terlepas tampak nekrotik (long standing erosion effect?). Pada kedua sediaan ini
lapisan dermis tidak menunjukan gambaran epidermolitik/akantolitik. Simpulan:
Subepidermal cleft accompanied by poor cell infiltrates. Gambaran klinis dan
morfologik sesuai untuk inherited epidermolysis bullosa. (Dowling Meara?)
Gambar 1. Gambar 2.
Gambar 3. Gambar 4.

16
Penderita didiagnosis kerja dengan follow-up epidermolisis bulosa
simpleks yang diduga suatu varian Dowling Meara. Penatalaksanaan Gentamisin
krim 2 kali sehari pada lesi erosi yang agak mengering, kompres terbuka dengan
NaCl 0,9% pada lesi erosi yang berkrusta sebelum aplikasi krim. KIE tentang
perawatan lesi bula dan erosi serta pencegahan dari trauma, menghindari cuaca
panas berlebihan, mengkonsumsi makanan tinggi kalori tinggi protein dan
menghindari makanan yang kasar dan panas.
PEMBAHASAN
Epidermolisis bulosa (EB) merupakan suatu spektrum genodermatosis yang
ditandai dengan adanya pembentukan bula secara spontan maupun trauma ringan.
EB adalah kelainan mekanobulosa (mechano-bullous disorders) yang terdiri atas
lebih dari 20 varian.2,3 Manifestasi penyakit bervariasi dari ringan sampai berat,
bahkan dapat mengakibatkan kematian.
Pertama-tama klasifikasi hanya didasarkan pada adanya jaringan parut
yang terbentuk kemudian, tetapi dengan makin canggihnya peralatan diagnostik
yang ada, maka terdapat berbagai variasi klasifikasi yang didasarkan kepada
penurunan genetik, gambaran klinis maupun pemeriksaan histologi.4,5 Dengan
menggunakan mikroskop biasanya hanya dapat dibedakan letak bula pada dermis
atau epidermis, tetapi mikroskop imunofluoresensi dapat menentukan letak bula di
daerah perbatasan dermis-epidermis dengan memperhatikan letak antigen
pemfigoid, proteoglikan dan jaringan kolagen di lamina basalis.5,6 Sedangkan
mikroskop elektron dapat melihat letak bula intra-epidermal. EB diklasifikasikan
dalam 3 kategori utama: (1) EB simpleks (EBS), (2) EB Junctional (EBJ) dan EB
distrofik (EBD).1,5 Diantara seluruh kasus EB, EBS adalah yang terbanyak.
EBS atau disebut juga EB epidermolitik ditandai dengan terbentuknya
bula intraepidermal, akibat defek pada gen yang mengkode keratin 5 dan 14.
Insidensinya 1 dalam 500.000 kelahiran.1,5 EBS sebagian besar diturunkan secara
autosomal dominan, akan tetapi terdapat pula kasus dengan penurunan autosomal
resesif.5-8 EBS memiliki beberapa varian, yaitu varian yang sering dijumpai antara
lain EBS lokalisata pada tangan dan kaki (Weber-Cockayne), EBS generalisata
(Koebner) dan EBS herpetiformis (Dowling Meara).5 Varian yang jarang dijumpai

16
antara lain varian EBS distrofi otot, EBS pigmentasi Mottled, EBS atresia pilori,
EBS superfisial, EBS Ogna dan EBS migrasi circinate. Varian yang bersifat auto
resesif antara lain EBS akantolitik letal, EBS defisiensi plakophilin dan EBS
defisiensi antigen pemfigoid bulosa-1 (varian yang terbaru ditemukan).1,7,8
EBS diduga terjadi akibat pembentukan enzim sitolitik dan pembentukan
protein abnormal yang sensitif terhadap perubahan suhu. Diduga defisiensi enzim
golactosylhidroxylysyl-glocosyltrans dan gelatinase (enzim degradase kolagen)
menyebabkan EBS.9 Selain diturunkan secara genetika autosom, diperkirakan
50% terjadi akibat mutasi pada gen pembentukan keratin terutama keratin 5 (K5)
dan kertin 14 (K14) yang terdapat di lapisan epidermis. Mutasi juga dapat terjadi
gen plectin (plektin). Plektin adalah protein yang terdapat di membran basal pada
attachment plague/hemidesmosom yang berfungsi sebagai penghubung filamen
intermedia ke membran plasma.10
Hampir semua tipe EB simpleks diturunkan secara autosomal dominan
kecuali pada EBS dengan distrofi otot, EBS autosomal resesif letal dan juga telah
dilaporkan EBS herpetiformis (Dowling Meara) serta EBS lokalisata yang
diturunkan secara resesif.3 Etiologi penyakit ini terjadi karena adanya mutasi gen

16
keratin.2,8 Mutasi terjadi kurang lebih 50% pada kode genetik keratin 5 atau 14
yang merupakan struktur utama pada lapisan keratin kulit.3,4,7
Beberapa peneliti menyatakan bahwa terjadi point mutations gen keratin
K5 dan K14 pada kromosom 12 dan 17. Lebih jelas lagi terjadi mis-sense mutasi
pada rangkaian asam amino pada keratin K5 dan K14. Perubahan asam amino ini
dapat menyebabkan perubahan struktur keratin. Keadaan ini dapat mengakibatkan
gangguan pembentukan jaringan filamen intermedia interseluler yang meluas dari
inti ke membran plasma yang menghubungkan struktur hemidesmosom dan
desmosome dengan keratinosit basal.11-14 Hal ini dibuktikan dalam penelitian tikus
transgenik yang mengalami mutasi keratin 14, didapatkan bula-bula di kulit tikus
tersebut seperti pada pasien EBS.15 Pada penelitian tersebut dibuktikan adanya
substitusi asam amino dapat menyebabkan rusaknya struktur jaringan filamen
keratin interseluler yang menyebabkan kertinosit basal rapuh sehingga mudah
terjadi bula intradermal karena trauma. Tidak semua pasien EBS mengalami
mutasi pada keratin 5 atau 14 namun dapat saja terjadi pada keratin 15 dan 17
yang terdapat juga di basal keratin.12 Dengan adanya mutasi pada gen keratin
menyebabkan terbentuknya struktur filamen keratin interseluler yang tidak stabil
yang mudah rusak karena truma ringan pada kulit. Sitolisis keratinosit dan bula
intradermal terjadi karena abnormalitas keratin.
Pada bentuk Dowling Meara yang berat, penggumpalan dari tonofilamen
dan pemindahan dari nukleus tampak ultrastruktural. Risiko lisis dan trauma sel
dibutuhkan untuk memicu timbulnya bula tergantung pada daerah mutasi dan
seberapa penting gen itu menghasilkan fungsi keratin. Daerah mutasi pada
sebagian besar Dowling Meara bentuk yang berat merupakan daerah yang paling
penting untuk fungsi keratin.10-11
Epidermolisis bulosa simpleks Dowling Meara (EBS herpetiformis)
merupakan bentuk paling berat dari EBS yng berasal dari mutasi gen keratin.1
Dowling dan Meara melaporkan pertama kali penyakit ini tahun 1945 yang
diderita oleh 4 anak berusia antara 3-7 tahun dengan gambaran klinis bula yang
timbul berhubungan dengan trauma dan menyerupai dermatitis herpetiformis
juvenilis. Sekitar tahun 1970, dengan pemeriksaan mikroskop elektron ditemukan

16
adanya abnormalitas pada keratinosit basal, yaitu adanya sitolisis sel basal dan
menyatunya tonofilamen.9
Tipe ini jarang terjadi namun sering cukup berat dan sering menimbulkan
kematian oleh karena luasnya daerah erosi pada masa neonatus. Awitan tipe ini
pada saat lahir sampai awal masa anak-anak. Mc-Grath dan kawan-kawan tahun
1991 pada penelitiannya terhadap 22 orang pasien EBS Dowling Meara,
didapatkan 12 orang penderita penyakit ini untuk pertama kali pada saat lahir dan
sisanya antara umur 1-5 hari. Sedangkan pada 7 orang pasien EBS Dowling
Meara yang dilaporkan oleh Hom dan Tidman pada tahun 1999, terdapat 4 orang
yang mempunyai awitan penyakit saat lahir dan sisanya antara 1-7 hari.2,8
Predileksi EBS Dowling Meara terutama pada tangan, kaki, muka dan
dada, ditandai dengan bula yang kecil, berkelompok (herpetiformis) sering
ditemui pada neonatus, tapi juga sering dilaporkan pada masa bayi dan anak-anak.
Bula tampak berkurang saat masa kanak-kanak dan dewasa. Dimasa dewasa, bula
jarang terjadi secara spontan sebagian besar terjadi karena trauma. Vesikel dan
bula hemoragik berkelompok lebih sedikit dan lebih cepat sembuh.5-9 Bula yang
yang pecah menimbulkan daerah erosi yang luas dan dapat terjadi infeksi
sekunder. Lesi kulit yang menyembuh biasanya meninggalkan makula hipo atau
hiperpigmentasi, jarang menimbulkan jaringan parut dan milia. Hiperkeratosis
pada telapak tangan dan kaki bisa terjadi pada usia 6-7 tahun, khususnya pada
anak yang lebih muda yang menunjukan bula palmoplantar.1 Hiperkeratosis ini
menimbulkan rasa nyeri bila disertai bula pada daerah tersebut.
Keterlibatan kuku yang rapuh sering terjadi pada varian Dowling Meara
disertai penebalan kuku ireguler, yang dapat tumbuh normal kembali tanpa
distrofi. Bula juga bisa terjadi pada daerah oral dan mukosa esofagus sehingga
sering menyebabkan terjadinya aspirasi makanan dan refluks gastro-esofagus dan
menyebabkan masalah dalam hal pemberian makanan untuk memenuhi kebutuhan
nutrisi. Terbentuknya gigi pada saat lahir telah dijelaskan berhubungan dengan
varian Dowling Meara.1 Pada gambaran histopatologi varian ini memiliki
perbedaan ultra-struktur yaitu terdapat clumping of tonofilmen.16
Pada kasus, penderita adalah seorang anak laki-laki berusia 13 tahun
memiliki keluhan luka diseluruh tubuh yang sebelumnya didahului oleh

16
munculnya bula pada kulit yang dicetuskan oleh trauma, gesekan atau benturan.
Distribusi bula berkelompok-kelompok kecil yang diikuti dengan pecahnya bula
menjadi erosi yang dapat sembuh tanpa meninggalkan jaringan parut atau milia.
Keluhan ini dialami penderita sejak umur 10 hari setelah kelahiran dan tetap ada
sampai sekarang walaupun cenderung ada perbaikan klinis dengan bertambahnya
usia. Penderita juga mengalami adanya penebalan pada kuku jari tangan sampai
hilangnya kuku jari kaki.
Pemeriksaan penunjang sangat dibutuhkan untuk mendiagnosis dan
memantau kemungkinan komplikasi yang dapat terjadi. Pemeriksaan laboratorium
darah pada EBS biasanya normal dan bila didapatkan anemia biasanya
berhubungan dengan adanya gangguan petumbuhan dan mal-absorbsi. Pada
anemia berat sering disertai penurunan kadar seng dalam serum ringan sampai
sedang.8 Teknik biopsi jaringan penderita EBS sangat penting. Biopsi sebaiknya
diambil dari tepi bula yang baru. Jika biopsi diambil dari bula yang lama maka
kemungkinan letak bula telah berubah karena regenerasi keratinosit pada dasar
bula atau karena degenerasi keratinosit di atas bula.10 Bula baru dapat diinduksi
dengan cara menggesek-gesek kulit dengan jari atau karet beberapa menit
sebelum biopsi. Lebih baik digunakan teknik biopsy shave atau elips.12
Pemeriksaan histopatologi dilakukan dengan mikroskop cahaya,
mikroskop elektron serta pemeriksaan imunohistokimia. Pemeriksaan rutin
dengan mikroskop cahaya tidak direkomendasikan untuk diagnostik. Sebagi baku
emas diagnostik EB digunakan mikroskop elektron. Selain dengan pewarnaan
hematoksolin eosin (HE) dapat juga dilakukan pewarnaan sediaan dengan PAS
(periodic acid Schiff) untuk melihat membran basalis. Diagnosis lain dapat
ditegakkan dengan monoklonal dan poliklonal antibodi LH7:2 atau AF1/AF2 juga
analisis DNA menggunakan metode PCR.9,12
Pada kasus, penderita diperiksa darah di laboratorium dan dilakukan biopsi
untuk pemeriksaan histopatologi. Dari hasil pemeriksaan darah didapatkan hasil
dalam batas normal, sedangkan dari histopatologi didapatkan suatu simpulan:
Subepidermal cleft accompanied by poor cell infiltrates. Gambaran klinis dan
morfologik sesuai untuk inherited epidermolysis bullosa. (Dowling Meara?)

16
Diagnosis banding dari EBS adalah EB tipe lainnya tergantung dari berat
ringannya gejala. Beberapa diagnosis banding antara lain: inkontinensia pigmenti
yaitu suatu kelainan multisistem terutama banyak diderita oleh wanita, diturunkan
secara X-linked dominant. Gambaran klinisnya pada stadium vesikuler sangat
mirip pada lesi awal EBS.3 Pemfigus neonatorum yaitu pemfigus pada masa
neonatal yang terjadi karena adanya substansi auto antibodi interseluler dari ibu
melalui plasenta. Pada pemeriksaan histologi dengan imunofleresensi langsung
ditemukan deposit interselulr IgG dan C3 pada kulit.10 Pemfigoid gestasional
memiliki gejala klinis terjadi saat lahir atau usia 3 hari, gambaran histopatologi
tampak bula subepidermal disertai serbukan sel eosinophil dan pada pemeriksaan
imunoflouresensi langsung didapatkan deposit IgG dan C3 pada membran
basalis.10,12
Pada kasus, penderita didiagnosis banding dengan EB tipe lainya yaitu EB
junctional, EB distrofik dan EB akuisata. Diagnosis banding epidermolisis bulosa
junctional, distrofik dapat disingkirkan dari anamnesis, pemeriksaan fisik,
predileksi dan tingkat keparahan. Selain itu diagnosis banding juga telah
disingkirkan dari hasil pemeriksaan histopatologi dimana dari hasil dapat
diketahui letak dari bulanya, walaupun belum dapat untuk memastikan sub tipe
nya secara pasti.
Seperti pada kelainan herediter lainnya, merupakan suatu tanggung jawab
dokter untuk memberikan informasi kepada orang tua tentang risiko terjadinya
abnormalitas yang diturunkan dari orang tua. Ketika kondisi dipastikan oleh suatu
gen dominan dan orang tua menderita, risiko terjadinya kelainan pada anak-
anaknya sebesar 50%. Pada suatu keluarga dimana seorang anak memiliki
abnormalitas akibat suatu gen resesif, risiko orang tua memiliki kemungkinan
terjadi abnormalitas pada keturunan selanjutnya di setiap kehamilan sebesar 25%.
Memberikan konseling genetik yang sesuai berdasarkan diagnosis yang akurat.
Karena perjalanan klinis dari bermacam bentuk epidermolisis bulosa sangat
beragam, terutama selama masa neonatus dan bayi, direkomendasikan bahwa
pasien sebaiknya dievaluasi sedini mungkin.1,3,13
Efek psikososial pada epidermolisis bulosa terutama bentuk yang berat,
pada individu dan keluarga yang terkena, memiliki efek paling dramatis diantara

16
penyakit kulit lainnya. Anak-anak yang menderita EB merasa memiliki kulit yang
gatal, nyeri, merasa tidak percaya diri, kesulitan berpartisipasi, sulit untuk
memahami individu yang lain dan merasa berbeda. Masalah-masalah ini
sebaiknya dibicarakan dan diberikan bantuan psikologis untuk pasien dan
keluarganya sebagai bagian dari perawatan optimal.13
Pada kasus, dari anamnesis tidak terlalu tampak adanya gangguan secara
psikologis dan mental dari penderita. Begitu pula dalam hal akademis penderita
memiliki kemampuan yang lebih secara akademis dibanding teman-temannya dan
tidak memiliki masalah dalam bersosialisasi.
Terapi epidermolisis bulosa adalah suportif dan paliatif, dengan
melindungi diri dari gesekan atau panas yang berlebihan, mencegah abrasi dan
konstriksi, penanganan infeksi sekunder, suplementasi dan penanganan nyeri.1,3
Untuk perawatan kulit, memberikan penjelasan dan edukasi pada keluarga pasien
atau perawat. Perawatan memerlukan kesabaran dan ketelitian, hindari trauma dan
gesekan. Dalam memilih pakaian maupun mainan harus yang ringan dan lembut.
Hindari penggunaan plester sehingga mencegah terjadinya fusi jari-jari. Pada
anak-anak hindari sepatu yang sempit atau yang terbuat dari kulit yang keras.
Kaos kaki dari bahan katun yang menyerap keringat untuk menghindari trauma
gesekan. Suhu lingkungan diusahakan agar cukup dingin, tempat tidur yang lunak
dan sprei yang halus. Ketika bula timbul, perluasan dapat dicegah dengan aspirasi
cairan bula secara aseptik. Apabila masih memungkinkan atap bula sebaiknya
dibiarkan tetap intak untuk melindungi kulit dasarnya. Bagian yang erosi diolesi
krim atau salep antibiotik.3,5
Pada kasus, penderita selain diberikan konseling dan penjelasan mengenai
jenis penyakit penderita, juga diberikan informasi mengenai perawatan kulit pada
lesi bula dan erosi juga cara-cara pencegahan dari trauma, gesekan atau benturan.
Selain itu penderita juga mendapatkan terapi antibiotik topikal untuk mencegah
terjadinya infeksi.
Dressing pelindung yang tidak melekat pada luka sebaiknya diaplikasikan
pada area erosi untuk membantu penyembuhan, namun harus mencegah
pengelupasan selanjutnya ketika penggantian dressing. Penggantian dressing
harus dilakukan secara steril untuk mencegah risiko terjadinya infeksi oleh

16
bakteri.1,3,5 Dressing pada lesi dapat menggunakan kasa gulung dengan plester
yang dilekatkan hanya pada dressing itu sendiri. Dressing dengan menggunakan
silver sulfadiazine dilaporkan telah memperbaiki keadaan pasien dari lesi
berulang. Pemberian steroid sistemik dan topikal umumnya tidak diperlukan pada
pasien dengan EB dan sebaiknya dihindari karena dapat memicu terjadi infeksi
dan efek samping yang lain.
Penanganan nyeri merupakan suatu hal yang penting pada perawatan EB,
terutama bayi. Pemberian suplemen nutrisi penting untuk pasien EB dengan
varian yang lebih parah untuk mencegah terjadinya gagal tumbuh kembang.
Kekurangan protein, zat besi dan darah melalui kulit yang terbuka menyebabkan
hipoalbumin defisiensi besi dan kekurangan mineral. Konsultasi ke ahli gizi
penting untuk memaksimalkan asupan kalori dan protein serta pemberian nutrisi
dan vitamin khusus seperti zat besi, zinc dan vitamin D3.13
Risiko terjadinya karsinoma sel basal tampak meningkat pada usia dewasa
dengan EBS Dowling Meara. Intervensi dini merupakan langkah tepat untuk
melakukan eksisi full-thickness dengan margin luas. Cetuximab (EGFR antagonis)
merupakan terapi terkini yang telah dilaporkan pada satu pasien dapat mengontrol
metastase pada karsinoma sel skuamus.14 Transplantasi gene-corrected cultured
epidermal stemscells menunjukan hasil kulit yang tampak nomal dalam satu
tahun. Studi terkini dilaporkan beberapa pasien telah menunjukan perbaikan
secara gradual setelah transplantasi stem cell.10
Secara umum prognosis EBS Dowling Meara baik, walaupun perjalanan
penyakitnya kronis. Diawali masa neonatus yang sering menimbulkan kematian
oleh karena luasnya daerah erosi yang dapat menyebabkan sepsis, namun dengan
pertambahan usia keadaan umum semakin membaik dengan lesi yang cepat
sembuh tanpa meninggalkan jaringan parut dan milia.3,5,13 Pada kasus prognosis
nya dubius karena kondisi penderita semakin membaik dan terdapat peningkatan
aktivitas yang signifikan.

16
SIMPULAN
Telah dilaporkan kasus epidermolisis bulosa simpleks yang diduga suatu varian
Dowling Meara (EBS herpetiformis) pada seorang anak laki-laki berusia 13 tahun.
Diagnosis ditegakkan berdasarkan anamnesis perjalanan penyakit penderita,
penelusuran riwayat penyakit dalam keluarga, pemeriksaan fisik dan pemeriksaan
penunjang laboratorium dan histopatologi. Epidermolisis bulosa simpleks
Dowling Meara ini diduga diturunkan secara autosomal resesif dari kedua orang
tua penderita dalam sebuah pernikahan keluarga yang keduanya membawa gen
karier. Penderita dan keluarga penderita diberikan konseling genetik dan
penjelasan mengenai penyakit, perawatan kulit, pencegahan terjadinya bula,
penanganan dan pencegahan infeksi, konseling gizi dengan tujuan meningkatkan
kulitas hidup penderita. Prognosis pada kasus dubius.

16
DAFTAR PUSTAKA
1. Paller AS, Mancini AJ. Bullous disorders of childhood. In: Hurwitz S. Clinical pediatric dermatology, a textbook of skin disorders of childhood and adolescence; fourth ed. Chicago, Illinois: Elsevier saunders; 2011. P.303-313.
2. Fine JD, Johnson LB, Suchindran C. The epidemiology of inherited epidermolysis bullosa: finding in the Us, Canadian and European study populations. In: Fine JD, Bauer EA, McGuire J, Moshel A, eds Clinical, Epidemologic and laboratory Advances and the Finding of the National Epidermolysis Bullosa Registry. Baltimore: John Hopkins University Press,1999:101-13
3. Marinkovich MP. Inherited Epidermolysis Bullosa. In: Goldsmith LA, Katz SI. Gilchrest BA, Paller AS, Leffell DJ, Wolf K. Eds Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York: McGraw-Hill; 2012. p.649-664
4. Coulombe PA, Kerns ML, Fuchs E: Epidermolysis bullosa simplex: a paradigm for disorders of tissue fragility. J Clin Invest 119:1784-1793, 2009
5. Fine JD: The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 58:931-50, 2008
6. LeBleu VS, Macdonald B, Kalluri R: Structure and function of basement membranes. Exp Biol Med (Maywood) 232:1121-1129, 2007
7. Natsuga K: Plectin deficiency leads to both muscular dystrophy and pyloric atresia in epidermolysis bullosa simplex. Hum Mutat 31(10): E1687-E1698, 2010
8. Fine JD & Burge SM. Genetic Blistering Diseases In: Burns T, Breathnach S, Cox N, Griffiths C, Eds Rook’s Textbook of Dermatology. 8th ed Massachusetts. Blackwell Science Ltd. 2010. P.39.1-40-2
9. Smith LT. Ultrasructural findings in Epidermolysis Bullosa. Arch Dermatol 1993; 129:1578-84
10. Hamada T, South AP, Mitsuhashi Y. Genotype-phenotype correlation in skin fragility-ctodermal dysplasia syndrome resulting from mutations in plakophin 1. Exp Dermatol 2002; 11: 107-14
11. Lin AN, Carter DM, eds Epidermolysis bullosa simplex: a clinical overview. In: Epidermolysis Bullosa: Basic and Clinical Aspect. New York: Springer, 1992: 89-117
12. Pfendner EG: Basic science of Epidermolysis Bullosa and diagnostic and molecular characterization: proceedings of the IInd International Symposium on Epidermolyis Bullosa, Santiago, Chile. Int J Dermatol 46:781-794, 2007
13. Tabolli S: Quality of life in patients with Epidermolysis Bullosa. Br J Dermatol 161:869-877, 2009
14. Arnold AW: Cetuximab therapy of metastizing cutaneous squamous cell carcinoma in patients with severe recessive dystrophyic epidermolysis bullosa. Dermatology 219(1): 80-83,2009
15. Bruckner-Tuderman L: Animal models of epidermolysis bullosa: update 2010. J Invest Dermatol 130:1485-1488, 2010

16
16. Weedon D. The vesiculobullous reaction patern: subepidermal blisters with little inflammation. In: Skin pathology. 2nd ed. Philadelphia: Churchill Livingstone.2002. p.144-152