







Bahasa
Halaman
Hukum


www.elsevier.nl/locate/jorganchem
Journal of Organometallic Chemistry 613 (2000) 208–219
Synthesis, structure and spectroscopic properties of branchedoligosilanes
Silke Chtchian a, Rhett Kempe b, Clemens Krempner a,*a Fachbereich Chemie der Uni6ersitat Rostock, Buchbinderstrasse 9, D-18051 Rostock, Germany
b Institut fur Organische Katalyseforschung an der Uni6ersitat Rostock e. V., D-18055 Rostock, Germany
Received 3 April 2000; received in revised form 17 July 2000
Abstract
A number of various branched heptasilanes [(Me3Si)2MeSi]2MeSiX (2: X=Ph; 3: X=Me; 6: X=Br; 7: X=I; 8: X=Cl; 9:X=F; 10: X=OH) bearing two (Me3Si)2MeSi groups as well as branched decasilanes [(Me3Si)2MeSi]3Si�X (12: X=H; 13:X=Br; 14: X=Cl; 15: X=F; 16: X=OH) bearing three (Me3Si)2MeSi groups at one silicon centre were synthesized. Thestructures of the compounds prepared were elucidated on the basis of comprehensive NMR and MS studies. Additionally, themolecular structures of the decasilanes 12 and 14–16 were obtained from X-ray diffraction data, which verify a ca. spherical shapewith the core silicon atom at the centre of the sphere. The spatial demand of three (Me3Si)2MeSi groups forces a widening of theSi�Si�Si angles of the XSiSi3 tetrahedra toward the group X (X=H, Cl, F, OH) and a remarkable elongation of the central Si�Xbonds. The analysis of the Si�Si�Si�Si dihedral angles of the tetrasilane subunits in the decasilanes 12 and 14–16 indicates theexistence of three conformations denoted as anti, ortho and gauche and four different arrangements of pentasilane subunits,denoted as anti–gauche, anti–ortho, ortho–gauche and ortho–ortho. The absorption spectra of the heptasilanes 2–10 exhibitbroadened absorption maxima, shifted strongly to the red relative to those of the decasilanes 11–16. As a result of strongly limitedconformational flexibility due to steric overcrowding, the singlet excitation in 11–16 is at much higher energy than in linearpentasilanes. A comparison of NMR chemical shifts of 2–16 with those of branched silanes of the type (Me3Si)3SiX and(Me3Si)2MeSiX reveals that the replacement of methyl groups by additional SiMe3 groups in the b-position leads to a stronglow-field shift of the signal of the central nucleus. © 2000 Elsevier Science B.V. All rights reserved.
Keywords: Silicon; Silanes; Branched oligosilanes; Structure elucidation; UV–vis-spectroscopy
1. Introduction
Compounds containing branched oligosilyl sub-stituents of type (Me3Si)2RSi — (R=oligosilyl,trimethylsilyl, aryl, alkyl, hydrogen) have been shownto display unusual chemical behaviour as well as novelelectronic properties [1]. For example, Bock et al.demonstrated that radical anions as well as radicalcations of tris(trimethylsilyl)silyl derivatives are stabi-lized by delocalizing the negative or positive charge,respectively, into the oligosilyl groups [2]. The ability ofbranched oligosilyl substituents to act as a p-acceptorwas discussed by Pitt [3] in the course of studies of the
electronic spectra of heptamethyltrisil-2-yl derivativessubstituted by electronegative donor groups and byWest et al. [4] in the course of ESR studies of thephenyl-tris(trimethylsilyl)silane radical anion. Further-more, the remarkable steric requirements of branchedoligosilyl groups, especially of the space-filling(Me3Si)3Si group, made these substituents versatile lig-ands in main group [5] and in transition metal chem-istry [6].
Of special interest in the search for sterically moredemanding ligands for the kinetic stabilization of low-valent intermediates are compounds in which two orthree bulky (Me3Si)2RSi substituents are fixed at thesame central atom. Some of those derivatives, mainly ofheavier main Group IV elements, have been preparedand for a few of those compounds the X-ray crystalstructures were reported [7]. Recently, Apeloig, [8]
* Corresponding author. Tel.: +49-381-4981841; fax: +49-381-4981763.
E-mail address: [email protected] (C.Krempner).
0022-328X/00/$ - see front matter © 2000 Elsevier Science B.V. All rights reserved.PII: S 0022 -328X(00 )00518 -0

S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219 209
Marschner [9] and Oehme [10] succeeded in synthesiz-ing compounds containing two hypersilyl [11] groups atcarbon or silicon atoms, respectively, starting with(Me3Si)3SiLi×3THF [12]. Moreover, Klinkhammmer[13] reported the preparation and X-ray structure of atris(hypersilyl)stannyl anion, whereas the silicon andcarbon analogues of the compound had not been syn-thesized before. Reducing the bulkiness of the oligosilylsubstituents by going from the (Me3Si)3Si group to the(Me3Si)2MeSi group, Lambert succeeded in the fixationof three of those units at one central silicon atom insynthesizing a highly branched decasilane [14].
These recent developments prompted us to designbranched oligosilane structures by Si�Si linkage ofmethyl-bis(trimethylsilyl)silylmetal-derivatives [15] withsubstituted dichlorosilanes and trichlorosilanes via saltelimination reactions. This way it should be possible toget stable, hemispherical and space-filling moleculesthat might be used as ligands with exceptional stericproperties [16]. In this paper, we report the synthesis ofvarious halosilanes and hydroxysilanes substituted onthe central silicon atom by two or three (Me3Si)2MeSigroups. In addition, we discuss the results of the X-raystructure analysis of some selected compounds andreport the spectroscopic properties of the synthesizedoligosilanes.
2. Results and discussion
2.1. Synthesis of heptasilanes 2–10
As mentioned above, our strategy for preparingbranched oligosilanes involved the selective formationof Si�Si bonds by salt elimination reactions of methyl-bis(trimethylsilyl)silyllithium (1) with easily availabledichlorosilanes and trichlorosilanes. Normally, the re-quired starting material 1 can be prepared by a cleavageof methyl-tris(trimethylsilyl)silane with methyllithiumin tetrahydrofuran and can be used in situ for couplingreactions as described previously [16]. But the outcomeof those coupling reactions strongly depends on thetemperature and the solvent applied. Particularly, themetal–halogen exchange and the Si�Si bond cleavagewere observed in the case of polar solvents such astetrahydrofuran. Therefore, after changing the solventfrom tetrahydrofuran to pentane, the silanide 1 wasallowed to react with the appropriate chlorosilanes atlow temperatures. In fact, the reaction of two equiva-lents of 1 with dichloromethylphenylsilane, dichloro-dimethylsilane and dichloromethylsilane at −78°Cgave the heptasilanes 2, 3 and 4, respectively, in excel-lent yields (Scheme 1).
Starting with the hydridosilane 4 [17], bromosilane 6and iodosilane 7 were easily prepared by reaction withtribromomethane or triiodomethane at 120°C, whereasthe chlorosilane 8 could not be synthesized in this way.Nevertheless, compound 8 was obtained by selectivecleavage of the Si�Ph bond of phenylsilane 2 withtrifluoromethanesulfonic acid giving the silyl triflate 5.Without isolation of 5, the triflate group was replacedby chloride in the presence of LiCl in THF to give pure8. The corresponding fluorosilane 9 was synthesized byfluorination of the bromosilane 6 with TASF [18] intoluene. On the other hand, hydrolysis of 6 led to theformation of hydroxysilane 10. It should be emphasisedthat the isolated heptasilanes 2, 3 and 6–10 are easilyavailable, air-stable compounds, which could bepurified by Kugelrohr distillation in vacuum and wereobtained in high yields. The structures proposed forcompounds 2, 3 and 6–10 are in full agreement withthe straightforward NMR spectra and the MS data.
2.2. Synthesis of decasilanes 12–16
As mentioned above, Lambert [14] showed that thetreatment of three equivalents of 1 with methyl-trichlorosilane led to the formation of the highlybranched decasilane 11 (Scheme 2). In view of therelative bulkiness of the three (Me3Si)2MeSi groupsbonded at one silicon centre, the ease of formation of11 is really surprising. Therefore, it should be possibleto synthesize comparable structures, substituted bygroups such as hydrido, chloro or phenyl at the centralsilicon atom.Scheme 1. Synthesis of heptasilanes 2–10.
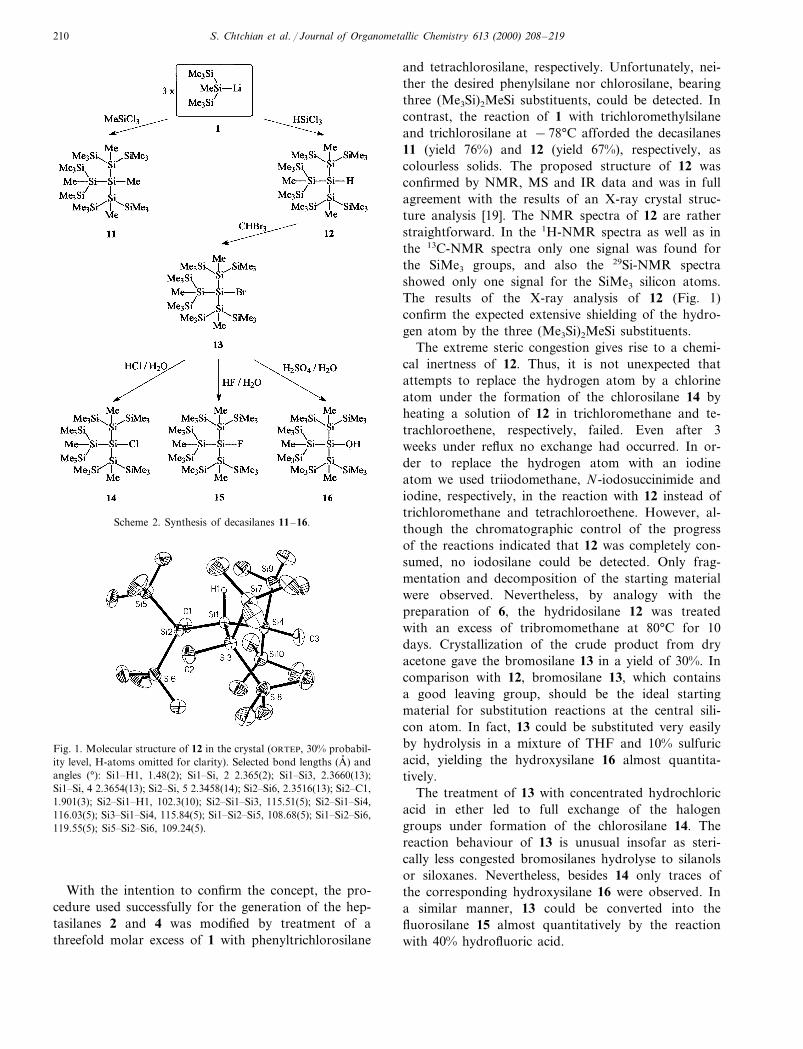
S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219210
Scheme 2. Synthesis of decasilanes 11–16.
and tetrachlorosilane, respectively. Unfortunately, nei-ther the desired phenylsilane nor chlorosilane, bearingthree (Me3Si)2MeSi substituents, could be detected. Incontrast, the reaction of 1 with trichloromethylsilaneand trichlorosilane at −78°C afforded the decasilanes11 (yield 76%) and 12 (yield 67%), respectively, ascolourless solids. The proposed structure of 12 wasconfirmed by NMR, MS and IR data and was in fullagreement with the results of an X-ray crystal struc-ture analysis [19]. The NMR spectra of 12 are ratherstraightforward. In the 1H-NMR spectra as well as inthe 13C-NMR spectra only one signal was found forthe SiMe3 groups, and also the 29Si-NMR spectrashowed only one signal for the SiMe3 silicon atoms.The results of the X-ray analysis of 12 (Fig. 1)confirm the expected extensive shielding of the hydro-gen atom by the three (Me3Si)2MeSi substituents.
The extreme steric congestion gives rise to a chemi-cal inertness of 12. Thus, it is not unexpected thatattempts to replace the hydrogen atom by a chlorineatom under the formation of the chlorosilane 14 byheating a solution of 12 in trichloromethane and te-trachloroethene, respectively, failed. Even after 3weeks under reflux no exchange had occurred. In or-der to replace the hydrogen atom with an iodineatom we used triiodomethane, N-iodosuccinimide andiodine, respectively, in the reaction with 12 instead oftrichloromethane and tetrachloroethene. However, al-though the chromatographic control of the progressof the reactions indicated that 12 was completely con-sumed, no iodosilane could be detected. Only frag-mentation and decomposition of the starting materialwere observed. Nevertheless, by analogy with thepreparation of 6, the hydridosilane 12 was treatedwith an excess of tribromomethane at 80°C for 10days. Crystallization of the crude product from dryacetone gave the bromosilane 13 in a yield of 30%. Incomparison with 12, bromosilane 13, which containsa good leaving group, should be the ideal startingmaterial for substitution reactions at the central sili-con atom. In fact, 13 could be substituted very easilyby hydrolysis in a mixture of THF and 10% sulfuricacid, yielding the hydroxysilane 16 almost quantita-tively.
The treatment of 13 with concentrated hydrochloricacid in ether led to full exchange of the halogengroups under formation of the chlorosilane 14. Thereaction behaviour of 13 is unusual insofar as steri-cally less congested bromosilanes hydrolyse to silanolsor siloxanes. Nevertheless, besides 14 only traces ofthe corresponding hydroxysilane 16 were observed. Ina similar manner, 13 could be converted into thefluorosilane 15 almost quantitatively by the reactionwith 40% hydrofluoric acid.
Fig. 1. Molecular structure of 12 in the crystal (ORTEP, 30% probabil-ity level, H-atoms omitted for clarity). Selected bond lengths (A, ) andangles (°): Si1�H1, 1.48(2); Si1�Si, 2 2.365(2); Si1�Si3, 2.3660(13);Si1�Si, 4 2.3654(13); Si2�Si, 5 2.3458(14); Si2�Si6, 2.3516(13); Si2�C1,1.901(3); Si2�Si1�H1, 102.3(10); Si2�Si1�Si3, 115.51(5); Si2�Si1�Si4,116.03(5); Si3�Si1�Si4, 115.84(5); Si1�Si2�Si5, 108.68(5); Si1�Si2�Si6,119.55(5); Si5�Si2�Si6, 109.24(5).
With the intention to confirm the concept, the pro-cedure used successfully for the generation of the hep-tasilanes 2 and 4 was modified by treatment of athreefold molar excess of 1 with phenyltrichlorosilane

S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219 211
2.3. Molecular structures and conformations of thedecasilanes 12 and 14–16
The synthesized branched decasilanes 12–16 (Scheme2) are all solid, air-stable compounds, which werepurified by crystallization and characterized on thebasis of NMR, MS and IR data (see Section 3). Inaddition, the molecular structures of the decasilanes 12[19] and 14–16 have been derived from X-ray diffrac-tion data. The results of the X-ray analyses give a goodidea of the steric congestion in the molecules. As we seein Figs. 1–4 the presented molecules have a threefoldaxis along the central Si�X bond (X=H, Cl, F, OH)and adopt a ca. spherical shape, with the core siliconatom at the centre of the sphere. The functional groupX is fully enclosed by the three (Me3Si)2MeSi sub-stituents, filling the hole between these groups.
Fig. 4. Molecular structure of 16 in the crystal (ORTEP, 30% probabil-ity level, H-atoms omitted for clarity). Selected bond lengths (A, ) andangles (°): Si1�O1, 1.689(4); Si1�Si2, 2.3773(15); Si1�Si3, 2.3814(11);Si1�Si4, 2.3770(11); Si2�Si5, 2.3603(13); Si2�Si6, 2.3569(14); Si2�C1,1.905(3); O1�Si1�Si2, 102.84(14); Si2�Si1�Si3, 114.40(4); Si2�Si1�Si4,114.88(4); Si3�Si1�Si4, 114.76(4); Si1�Si2�Si5, 108.11(5); Si1�Si2�Si6,120.65(5); Si5�Si2�Si6, 108.59(5).
Fig. 2. Molecular structure of 14 in the crystal (ORTEP, 30% probabil-ity level, H-atoms omitted for clarity). Selected bond lengths (A, ) andangles (°): Si1�Cl1, 2.1370(11); Si1�Si2, 2.3799(12); Si1�Si3,2.3771(12); Si1�Si4, 2.3829(12); Si2�Si5, 2.3663(13); Si2�Si6,2.3705(13); Si2�C1, 1.900(3); Cl1�Si1�Si2, 101.59(5); Si2�Si1�Si3,115.24(4); Si2�Si1�Si4, 117.35(4); Si3�Si1�Si4, 115.55(4); Si1�Si2�Si5,110.43(5); Si1�Si2�Si6, 118.78(5); Si5�Si2�Si6, 108.43(5).
The intramolecular distances between the group X(F, Cl, OH) and the three next neighbouring Me3Sicarbon atoms are relatively short. For example, theaverage C�X distances of 15 (C�F1, 3.45 A, ) and 16(C�O1, 3.50 A, ) correspond ca. with the sum of the vander Waals radii [20] of a methyl group (2.0 A, ) and afluorine (1.47 A, ) or a hydroxy group (1.52 A, ), respec-tively. Only the average distance C�Cl1 (3.58 A, ) in 14 isslightly smaller than the sum of the van der Waals radiiof a methyl group and chlorine atom amounting toabout 3.76 A, . The central Si�X bonds are remarkablyelongated (Si1�H1 1.482; Si1�F1 1.637; Si1�Cl1 2.137;Si1�O1 1.689 A, ). The spatial demand of the three(Me3Si)2MeSi groups forces a widening of the Si�Si�Siangles of the XSiSi3 tetrahedra toward the group Xwithin the range of 115–117°. The sum of the threeSi�Si�Si bond angles around the central silicon atomsare 347.55 (12), 346.26 (14), 348.14 (15) and 344.04°(16). Most of the Si�Si bond lengths lie unremarkablywithin the range of 2.360–2.370 A, . Only the threebonds emanating from the central silicon are slightlyelongated within the range 2.375–2.385 A, . At the posi-tions where the trimethylsilyl groups of the three(Me3Si)2Me Si substituents contact each other, the an-gles Si1�Si2�Si6 are significantly widened within therange 118.8–120.7°, reducing the steric interactions.Thus, the average distances of neighboring methylgroups are 4.0 (12, 14, 16) and 3.9 A, (15) and arecomparable with the sum of the van der Waals radii oftwo methyl groups amounting to about 4.0 A, . Theseresults are in good agreement with the structural dataobtained for the corresponding decasilane 11 reportedby Lambert et al. [14].
Very instructive is a view along the Si2�Si1 axis ofthe molecules, shown for the decasilane 14 (Fig. 5),which reveals that the conformation is predominantlydetermined by the steric repulsion of the three
Fig. 3. Molecular structure of 15 in the crystal (ORTEP, 30% probabil-ity level, H-atoms omitted for clarity). Selected bond lengths (A, ) andangles (°): Si1�F1, 1.637(2); Si1�Si2, 2.375(2); Si1�Si3, 2.377(2);Si1�Si4, 2.368(2); Si2�Si5, 2.357(2); Si2�Si6, 2.361(2); Si2�C1,1.890(4); F1�Si1�Si2, 102.39(10); Si2�Si�Si3, 115.04(6); Si2�Si1�Si4,115.70(6); Si3�Si1�Si4, 115.52(6); Si1�Si2�Si5, 107.97(6); Si1�Si2�Si6,119.58(7); Si5�Si2�Si6, 109.21(7).

S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219212
Fig. 5. Molecular structure of 14 in the crystal (ORTEP, 30% probabil-ity level, H-atoms and C-atoms omitted for clarity). Selected dihedralangles (°): Cl1�Si1�Si2�Si5, 21.71(6); Cl1�Si1�Si2�Si6, 147.84(5);Si3�Si1�Si2�Si5, 87.65(6); Si4�Si1�Si2�Si5, 131.03(5); Si3�Si1�Si2�Si6, 38.48(7); Si4�Si1�Si2�Si6, 102.84(6).
91 (ortho) and 162° (anti ). Furthermore, Michl pointedout that these values are nearly the same as in theconformers of linear dodecamethylpentasilane and thatthe all-anti forms seem to be the most stable of allconformers, as it was shown in n-Si4Me10 [21]. Theresults of our conformational analyses of the decasi-lanes 12 and 14–16 reveal four tetrasilane subunits withdifferent dihedral angles (Fig. 5) and 12 possible pen-tasilane subunits, each defined by two dihedral angles.Following the denomination of conformers by Michl etal. [21], we termed the dihedral angles obtained for 12and 14–16 as gauche (30–38), ortho (88–107) and anti(127–134°). In addition, Table 1 lists the dihedral an-gles for the 12 pentasilane subunits in the decasilanes 12and 14–16. We found a total of 12 ortho, six anti andsix gauche arrangements of the tetrasilane subunits andfour combinations of pentasilane subunits, denoted asanti–gauche, anti–ortho, ortho–gauche and ortho–ortho[14].
In conclusion, it should be stated that the arrange-ment of the tetrasilane as well as of the pentasilanechains is strongly determined by the space filling(Me3Si)2MeSi groups. Thus, it is not unexpected that inthe pentasilane subunits of the compounds 12 and14–16 no all-anti conformers were observed, which hasbeen noted to be optimal for delocalization or s-conju-gation [22]. Moreover, the conformational parametersof 14 as well as of 11 reported by Lambert et al. [14] arecomparable to those for 12, 15 and 16, indicating thatin these classes of compounds the space demand of thecentral group X (X=H, Me, Cl, F, OH) is ca. thesame.
2.4. UV spectra — conformational effects
The absorption spectra of typical linear permethy-loligosilanes strongly depend on the degree of catena-tion as well as on the conformation of the silicon-backbone [22]. Thus, the absorption maxima of oligo-silane chains move to longer wavelengths as the number
(Me3Si)2MeSi groups, while the Si�Cl1 group is playinga minor role. For example, neither is the trimethylsilylgroup (Si5) in a gauche conformation nor is thetrimethylsilyl group (Si6) in an anti conformation withCl1, since this would lead to an extreme approach ofthe neighbouring SiMe3 groups. Therefore, bothtrimethylsilyl groups evade the steric strain by rotatingthe (Me3Si)2MeSi group around the Si2�Si1 axis, de-creasing the dihedral angles Si5�Si2�Si1�Cl1 (21.71)and Si6�Si2�Si1�Cl1(147.84°). Obviously due to thesteric stress in the molecule the conformation of thetetrasilane subunits also differ strongly from the usualgauche or anti conformation of linear permethyloligosi-lanes. Thus, the dihedral angles in 14 were found to be38.48 (Si6�Si2�Si1�Si3), 102.84 (Si6�Si2�Si1�Si4), 87.65(Si5�Si2�Si1�Si3) and 131.03° (Si5�Si2�Si1�Si4).
Interestingly, calculations by Michl and coworkers[21] have shown that the relatively unstrained lineardecamethyltetrasilane has three conformational min-ima, whose energies lie within 1 kcal mol−1 of eachother, and whose dihedral angles are near 53 (gauche)
Table 1Tetrasilane dihedral angles (°) for compounds 12 and 14–16
Pentasilane subunits 14 1512 16 Conformers
88–128 89–12888–127Si5�Si2�Si1�Si3�Si7 90–129 ortho–anti89–107 ortho–ortho88–10688–108Si5�Si2�Si1�Si3�Si8 90–106
132–93 131–90Si5�Si2�Si1�Si4�Si9 133–94 134–95 anti–orthoanti–gauche134–31133–32131–36132–33Si5�Si2�Si1�Si4�Si10
Si6�Si2�Si1�Si3�Si7 39–127 38–128 37–128 35–129 gauche–anti35–106 gauche–ortho38–106Si6�Si2�Si1�Si3�Si8 37–10739–108
101–95 ortho–ortho103–90Si6�Si2�Si1�Si4�Si9 102–94102–93ortho–gauche101–31102–32Si6�Si2�Si1�Si4�Si10 103–36102–33ortho–anti93–127 90–129 93–128 95–129Si7�Si3�Si1�Si4�Si9
93–107 90–105Si7�Si3�Si1�Si4�Si10 93–107 95–105 ortho–orthogauche–anti30–12931–12836–129Si8�Si3�Si1�Si4�Si9 33–127
33–107 36–105 31–107 30–105 gauche–orthoSi8�Si3�Si1�Si4�Si10
S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219 213
Table 2UV absorption spectral data for compounds 3, 4, 6 and 8–16
Heptasilanes olmax (nm) Decasilanes lmax (nm) o
2.27×104 113 239.5254.8 3.40×104
253.44 1.72×104 12 238.5 a 3.42×104
2.10×104 14247.5 230.68 5.70×104
2.40×104 136 233.8245.5 4.08×104
1.55×104 15259.0 219.59 5.38×104
1.49×104 16 �225 a10 4.25×104262.2
a Shoulder.
of silicon atoms increases, with a limiting wavelengthnear 300 nm. Oligosilanes with more than three siliconatoms exhibit broad absorption maxima with multiplepeaks attributable to the presence of conformationalisomers [23]. Consequently, the degree of s-conjugationin the oligosilane chain is a very sensitive function ofthe chain conformation.
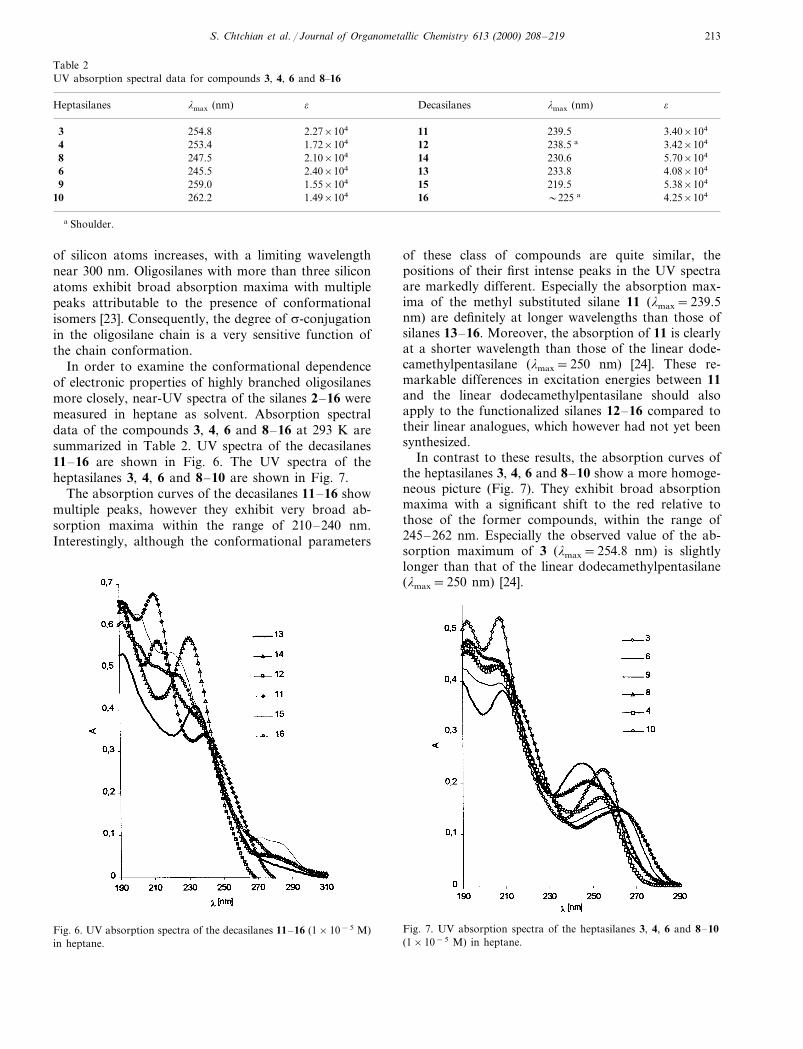
In order to examine the conformational dependenceof electronic properties of highly branched oligosilanesmore closely, near-UV spectra of the silanes 2–16 weremeasured in heptane as solvent. Absorption spectraldata of the compounds 3, 4, 6 and 8–16 at 293 K aresummarized in Table 2. UV spectra of the decasilanes11–16 are shown in Fig. 6. The UV spectra of theheptasilanes 3, 4, 6 and 8–10 are shown in Fig. 7.
The absorption curves of the decasilanes 11–16 showmultiple peaks, however they exhibit very broad ab-sorption maxima within the range of 210–240 nm.Interestingly, although the conformational parameters
of these class of compounds are quite similar, thepositions of their first intense peaks in the UV spectraare markedly different. Especially the absorption max-ima of the methyl substituted silane 11 (lmax=239.5nm) are definitely at longer wavelengths than those ofsilanes 13–16. Moreover, the absorption of 11 is clearlyat a shorter wavelength than those of the linear dode-camethylpentasilane (lmax=250 nm) [24]. These re-markable differences in excitation energies between 11and the linear dodecamethylpentasilane should alsoapply to the functionalized silanes 12–16 compared totheir linear analogues, which however had not yet beensynthesized.
In contrast to these results, the absorption curves ofthe heptasilanes 3, 4, 6 and 8–10 show a more homoge-neous picture (Fig. 7). They exhibit broad absorptionmaxima with a significant shift to the red relative tothose of the former compounds, within the range of245–262 nm. Especially the observed value of the ab-sorption maximum of 3 (lmax=254.8 nm) is slightlylonger than that of the linear dodecamethylpentasilane(lmax=250 nm) [24].
Fig. 6. UV absorption spectra of the decasilanes 11–16 (1×10−5 M)in heptane.
Fig. 7. UV absorption spectra of the heptasilanes 3, 4, 6 and 8–10(1×10−5 M) in heptane.

S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219214
As mentioned above the space demand of the(Me3Si)2MeSi group in the branched decasilanes 11–16leads to strong limitations in the conformational flexi-bility in the silicon backbones, i.e. interconversion ofconformers through rotations around Si�Si singlebonds are restricted. As a result, only four combina-tions of conformers anti–gauche, anti–ortho, ortho–gauche and ortho–ortho, but no anti–anti conformerswere observed in the pentasilane subunits, for which itis commonly believed that the anti–anti-form has thelowest singlet excitation energy of all conformers [23].Furthermore, it is well known, that the excitation en-ergy rapidly increases upon twisting from trans to orthoor gauche conformation. Thus, the obtained broadenedabsorption maxima of 11–16 shifted strongly to theblue, can be explained as a statistical distribution ofvarious conformers and, hence, absorb at a higherenergy that depends on twisting in the pentasilanechains. In contrast, the corresponding broadened ab-sorption maxima of the heptasilanes 3, 4, 6 and 8–10are significantly shifted to the red. This suggests, due tothe minor steric stress in the molecules, a considerablyhigher conformational flexibility in the silicon back-bones. Consequently, it is reasonable to assume that inthe four possible pentasilane pathways of 3, 4, 6 and8–10 besides a statistical distribution of other conform-ers also anti–anti conformers exist, causing a noticeablylower singlet excitation energy.
Interestingly, we found that the fluorosilane 9 andhydroxysilane 10 show a significant bathochromic shiftof the absorption, coupled with some reduction in theintensity relative to the methyl substituted silane 3. Thiscan be attributed to a weak interaction of the nonbond-ing electrons with the heptasilane skeleton [3]. Theseresults are completely different from those obtained forthe bulkier analogues 12–16, in which such a significantinteraction is not evident. Although sharp maxima ofabsorptions could not be determined in all cases, the
maximum moves clearly to shorter wavelengths relativeto the methyl substituted silane 11, suggesting that theelongated Si�X bonds render the non-bonding electroninteractions with the silicon backbone negligible.
2.5. IR and NMR spectra — trends of chemical shifts
The 29Si-NMR chemical shift data for the centralsilicon atom of the oligosilanes 2–16 are summarised inTable 3 together with the data for selected silanes,trisilanes and tetrasilanes reported in the literature. Asalready reported by Radeglia et al. [25] in the case ofsubstituted siloxanes and phenylsilanes, it can be seen,that the low-field shift value of the central silicon atomincreases with the electronegativity of the group X.Furthermore, the replacement of methyl groups inMe3SiX by one or more trimethylsilyl groups in thea-position leads to a strong high-field shift, which canbe attributed mainly to the electronegativity differencebetween these groups. For example, the observed valuefor the central silicon nucleus of tetrakis(trimethylsi-lyl)silane (d= −135.9), a compound in which fourtrimethylsilyl groups are fixed at the same silicon atom,is the lowest so far described for comparable oligosi-lanes. Contrary, the additional replacement of methylgroups of compounds (Me3Si)2MeSiX and (Me3Si)3SiXby trimethylsilyl groups in the b-position leads to alow-field shift of the signal for the central silicon nu-cleus. The observed influences on the chemical shift arein full agreement with the data for a series of branchedas well as linear permethylated oligosilanes, reported byIshikawa and West [26]. 1H-, 13C- and 19F-NMR chem-ical shifts (Table 4) observed for the central group Xindicate the same effect, a strong high-field shift bytrimethylsilyl substitution in the a-position and a stronglow-field shift as a result of b-substitution of additionalSiMe3 groups.
Table 3Comparison of 29Si-NMR shift values (ppm) of the heptasilanes and decasilanes 2–16 with the corresponding trisilanes and tetrasilanes measuredin benzene-d6
dSi*X=CH3 dSi*X=H dSi*X=F dSi*X=Cl dSi*X=Br dSi*X=I dSi*X=OH dSi*X=Ph
0.0 −17.5 a 32.0Me3Si*X 30.2 b −4.616.0 c, d8.7 b26.4 b
−72.7 f −46.2 f−48.5 e(Me3Si)2MeSi* X 8.8−22.8 f2.4 f9.9 f36.3 f
−88.3 −115.6 34.2 −12.6(Me3Si)3Si*X −24.8 g −56.8 g −9.7 −77.0[(Me3Si)2MeSi]2MeSi*X −33.0 −67.1 54.3 26.7 17.2 −14.2 26.1 −34.4
37.6[(Me3Si)2MeSi]3Si*X −55.4 −111.4 74.8 27.9 12.3
a Ref. [27].b Ref. [28].c Ref. [29].d Measured in CDCl3.e Ref. [26a].f Ref. [30].g Ref. [31].

S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219 215
Table 4Selected spectroscopic data of the silanes 4, 9, 11, 12 and 15 compared with those reported in the literature
dF (ppm) X=F dC (ppm) X=CH31JSi�H (Hz)Compounds nSi�H (cm−1)dH (ppm) X=H
−158.0 b 0.0Me3SiX 182 c4.04 a 2121 d
(Me3Si)2MeSiX 3.37 e −208.9 −6.9 f 163 g 2075 g
3.95[(Me3Si)2MeSi]2MeSiX −182.2 0.5 160 2058−255.1 −13.32.52 154 h(Me3Si)3SiX 2051
3.53[(Me3Si)2MeSi]3SiX −204.4 0.4 i 147 2017
a Ref. [34].b Ref. [35].c Ref. [36].d Ref. [37].e Ref. [38].f Ref. [26b].g Ref. [39].h Ref. [33].i Ref. [14].
Interestingly, the values of coupling constants1J(Si�H) of the selected hydridosilanes (Table 4) in-crease as substituents on the central silicon atoms be-come bulkier. It is well known that in substitutedhydridosilanes the s-character of the Si hybrid orbital iscorrelated with spin–spin coupling constants [32]. Thetrend of 1J(Si�H) indicates that the s-character of theSi�H orbitals becomes smaller as the substituents be-come bulkier. Especially the value of 1J(Si�H) of [(i-Pr)3Si]3SiH [33], a molecule with a nearly planarstructure of the central SiSi3 skeleton, is the lowestamong those of hydridosilanes so far reported.
Moreover, the values of the Si�H stretching frequen-cies, obtained for the hydridosilanes summarised inTable 4, indicate that the Si�H bonds are dramaticallyweakened by successive substitution of trimethylsilylgroups in the a-position and the b-positions. Thus, theobserved value for the hydridosilane 12 (2017 cm−1) isthe lowest so far described for comparable oligosilanes.Although the origin of the effect is unclear, the assump-tion of steric repulsion alone leading to a flattening ofthe central SiSi3 skeleton of 12 and making the Si�Hbond quite weak is not valid, since the stretching fre-quency of [(i-Pr)3Si]3SiH was found to be 2050 cm−1
[33]. However, it is expected that mainly s-conjugationinto the pentasilane chains together with inductive ef-fects of the trimethylsilyl groups decrease the Si�Hbond strength.
2.6. Conclusions
We have prepared various branched halosilanes andhydroxysilanes substituted at the central silicon atomby two or three (Me3Si)2MeSi groups. X-ray analysesrevealed the branched decasilanes 12 and 14–16 to behemispherical and space-filling molecules, in which thegroup X (X=H, Cl, F, OH) is strongly shielded andthe Si�Si�Si�Si dihedral angles are remarkably twisted.
Absorption spectra of the heptasilanes 2–10 exhibitbroadened absorption maxima, shifted strongly to thered relative to those of the highly branched decasilanes11–16. These results support the suggestion that if theconformational flexibility is strongly limited by stericovercrowding, the singlet excitation in higher oligosi-lane dendrimers tends to be localized at a relativelysmall number of silicon atoms in a way dictated bychain conformation. Finally, spectroscopic investiga-tions have shown that the properties of the Si�X func-tion are strongly influenced by electronic effects of thepentasilanes chains as well as by the space demand oftwo or three (Me3Si)2MeSi groups. Despite the exten-sive shielding of the Si�X function, the oligosilanes2–15 are proven to be reactive, and their chemicalbehaviour is the subject of current studies.
3. Experimental
3.1. General procedures and material
All reactions involving organometallic reagents werecarried out under an atmosphere of argon using stan-dard Schlenk techniques. Methyl-tris(trimethylsi-lyl)silane [31], methyl-bis(trimethylsilyl)silyllithium (1)[15] and 1,1,1,2,3,4,5,5,5-nonamethyl-2,4-bis(trimethyl-silyl)pentasilane (4) [17] were prepared as previouslydescribed. NMR: Bruker AC 250, Bruker ARX 300,Bruker ARX 400 (250 MHz, 62.9 MHz, 79.5 MHz and235.3 MHz, for 1H, 13C, 29Si and 19F, respectively). For1H-, 13C- and 29Si-NMR, benzene-d6 as solvent, TMS asinternal standard; for 19F-NMR, benzene-d6 as solvent,CFCl3 as internal standard. IR: Nicolet 205 FT-IR.MS: Intectra AMD 402, chemical ionization withisobutane as the reactant gas. UV–vis: Perkin–ElmerLambda 2, quartz cells of 1.0 cm path length andspectral grade n-heptane.

S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219216
3.2. 1,1,1,2,3,4,5,5,5-Nonamethyl-3-phenyl-2,4-bis(trimethylsilyl)pentasilane (2)
Dichloromethylphenylsilane (2.44 ml, 15 mmol) wasadded at −78°C to a solution of methyl-bis(trimethylsilyl)silyllithium (30 mmol) in pentane (100ml). Stirring was continued for 1 h and the mixture wasallowed to warm to room temperature (r.t.) within 2 h.After filtration and removal of the solvent under re-duced pressure the residue was distilled (170°C, 0.03mbar) to give 2 (5.99 g, 80%). M.p. 50°C. 1H-NMR: d
7.49–7.53, 7.06–7.2 (2m, phenyl, 5H), 0.71 (s, SiMePh,3H), 0.39 (s, SiMe, 6H), 0.11, 0.18 (2s, SiMe3, 2×18H).13C-NMR: d 140.2, 135.0, 128.3, 128.1 (C-aromat.),−2.9 (SiMePh), −10.1 (SiMe), 0.8, 1.1 (SiMe3). 29Si-NMR: d −34.4 (SiMePh), −80.5 (SiMe), −11.6,−11.8 (SiMe3). MS: (70 eV) m/z (%)=498 (5) [M+],483 (3) [M+−Me], 309 (100) [M+−SiMe(SiMe3)2].HRMS: Calc. for C21H50Si7 498.22387. Found:498.22974. Anal. Calc. for C21H50Si7 (499.235): C,50.52; H, 10.10. Found: C, 48.56; H, 9.99%.
3.3. 1,1,1,2,3,3,4,5,5,5-Decamethyl-2,4-bis(trimethylsilyl)pentasilane (3)
Dichlorodimethylsilane (1.82 ml, 15 mmol) wasadded at −78°C to a solution of methyl-bis(trimethylsilyl)silyllithium (30 mmol) in pentane (100ml). Stirring was continued for 1 h and the mixture wasallowed to warm to r.t. within 2 h. After filtration andremoval of the solvent under reduced pressure theresidue was distilled (120°C, 0.01 mbar) to give 3 (5.82g, 87%). M.p. 41°C. 1H-NMR: d 0.44 (s, SiMe2, 6H),0.28 (s, SiMe, 6H), 0.25 (s, SiMe3, 36H). 13C-NMR: d
0.5 (SiMe2), −10.7 (SiMe), 1.1 (SiMe3). 29Si-NMR: d
−33.0 (SiMe2), −81.6 (SiMe), −12.0 (SiMe3). MS:(70 eV) m/z (%)=436 (14) [M+], 421 (6) [M+−Me].Anal. Calc. for C16H48Si7 (437.162): C, 43.96; H, 11.07.Found: C, 43.97; H, 10.92%.
3.4. 3-Bromo-1,1,1,2,3,4,5,5,5-nonamethyl-2,4-bis(trimethylsilyl)pentasilane (6)
A mixture of 1.1.1.2.3.4.5.5.5-nonamethyl-2.4-bis(trimethylsilyl)-pentasilane (4) (5.3 g, 12 mmol) andCHBr3 (2.1 ml, 23 mmol) was heated to 120°C for 15 h.After removal of the volatiles, the residue was distilled(145°C, 0.005 mbar) to give 6 (5.76 g, 92%). M.p. 62°C.1H-NMR: d 0.99 (s, SiMeBr, 3H), 0.31 (s, SiMe, 6H),0.33, 0.24 (2s, SiMe3, 2×18H). 13C-NMR: d 5.2(SiMeBr), −10.4 (SiMe), 0.9, 0.87 (SiMe3). 29Si-NMR:d 17.2 (SiMeBr), −75.0 (SiMe), −12.0, −10.9(SiMe3). MS: (70 eV) m/z (%)=502 (0.4) [M+], 487 (9)[M+−Me], 429 (15) [M+−SiMe3], 348 (65) [M+−SiMe3−HBr]. HRMS: Calc. for C15H45
79BrSi7500.10923. Found: 500.10895. Anal. Calc. for
C15H45BrSi7 (502.03): C, 35.89; H, 9.04; Br, 15.92.Found: C, 34.30; H, 8.85; Br, 13.56%.
3.5. 3-Iodo-1,1,1,2,3,4,5,5,5-nonamethyl-2,4-bis-(trimethylsilyl)pentasilane (7)
A mixture of 1.1.1.2.3.4.5.5.5-nonamethyl-2.4-bis(trimethylsilyl)-pentasilane (4) (1.6 g, 3.8 mmol),CHI3 (1.6 g, 4 mmol) and toluene (20 ml) was heated to110°C for 20 h. After removal of the volatiles, theresidue was distilled (175°C, 0.03 mbar) to give 7 (1.41g, 68%). 1H-NMR: d 1.20 (s, SiMeI, 3H), 0.30 (s, SiMe,6H), 0.33, 0.26 (2s, SiMe3, 2×18H). 13C-NMR: d 2.9(SiMeI), −9.2 (SiMe), 1.0, 1.1 (SiMe3). 29Si-NMR: d
−14.2 (SiMeI), −75.0 (SiMe), −11.5, −10.6(SiMe3). MS: (70 eV) m/z (%)=548 (1) [M+], 475 (51)[M+−SiMe3]. Anal. Calc. for C15H45ISi7 (549.03): C,32.82; H, 8.26. Found: C, 32.50; H, 8.17%.
3.6. 3-Chloro-1,1,1,2,3,4,5,5,5-nonamethyl-2,4-bis-(trimethylsilyl)pentasilane (8)
Trifluoromethansulfonic acid (0.1 ml, 1.1 mmol) wasadded at r.t. to a stirred solution of compound 2 (0.5 g,1.0 mmol) in CH2Cl2. After removal of the solventunder reduced pressure, the solid residue was dissolvedin 10 ml THF and 50 mg (1.2 mmol) of anhydrous LiClwere added. Stirring was continued for 1 day. Afterfiltration and removal of the solvent under reducedpressure, the residue was distilled (165°C, 0.02 mbar) togive 8 (0.41 g, 90%). M.p. 47–48°C. 1H-NMR: d 0.84(s, SiMeCl, 3H), 0.30 (s, SiMe, 6H), 0.32, 0.23 (2s,SiMe3, 2×18H). 13C-NMR: d 6.3 (SiMeCl), −11.0(SiMe), 0.8, 0.83 (SiMe3). 29Si-NMR: d 26.7(SiMeCl),−75.4 (SiMe), −11.3, −11.2 (SiMe3). MS:(CI) m/z (%)=457 (4) [M++H], 441 (30) [M+−Me],421 (40) [M+−Cl], 348 (100) [M+−ClSiMe3]. Anal.Calc. for C15H45ClSi7 (457.57): C, 39.37; H, 9.91; Cl,7.75. Found: C, 39.30; H, 9.83; Cl, 7.54%.
3.7. 3-Fluoro-1,1,1,2,3,4,5,5,5-nonamethyl-2,4-bis-(trimethylsilyl)pentasilane (9)
A mixture of compound 6 (0.3 g, 0.6 mmol), tris(-dimethylamino)-sulfonium-difluorotrimethylsilicate(TASF) (0.181 g, 0.65 mmol) and toluene (10 ml) wasstirred at r.t. for 1 day. After filtration and removal ofthe volatiles, the residue was distilled (115°C, 0.02mbar) to give 9 (0.195 g, 74%). 1H-NMR: d 0.75 (d,3J=9.5 Hz, SiMeF, 3H), 0.31 (s, SiMe, 6H), 0.31, 0.22(2s, SiMe3, 2×18H). 13C-NMR: d 5.8 (d, 2J=12.4 Hz,SiMeF), −11.9 (SiMe), 0.7, 0.5 (SiMe3). 29Si-NMR: d
54.3 (d, 1J=331.5 Hz, SiMeF), −80.9 (SiMe), −11.5,−12.8 (SiMe3). 19F-NMR: d −182.2 (SiMeF). MS:(CI) m/z (%)=441 (10) [M++1], 425 (50) [M+−Me].HRMS: Calc. for C15H45FSi7 440.18428. Found:

S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219 217
440.18903. Anal. Calc. for C15H45FSi7 (441.120): C,40.84; H, 10.28. Found: C, 40.26; H, 10.39%.
3.8. 3-Hydroxy-1,1,1,2,3,4,5,5,5-nonamethyl-2,4-bis(trimethylsilyl)pentasilane (10)
Sulfuric acid (15 ml of 10%) was added to a solutionof compound 6 (0.3 g, 0.6 mmol) in tetrahydrofuran (30ml) and the mixture was vigorously stirred for 1 day atr.t. After addition of diethylether the organic phase wasseparated, dried with MgSO4, and evaporated. The oilyresidue was distilled (120°C, 0.005 mbar) to give 10(0.241 g, 92%). IR (Nujol) n=3672 cm−1 (OH). 1H-NMR: d 0.65 (s, SiMeOH, 3H), 0.53 (s, OH, 1H), 0.28(s, SiMe, 6H), 0.29, 0.24 (2s, SiMe3, 2×18H). 13C-NMR: d 6.7 (SiMeOH), −11.6 (SiMe), 0.9 (SiMe3).29Si-NMR: d 26.1 (SiMeOH), −81.5 (SiMe), −11.9,−12.8 (SiMe3). MS: (70 eV) m/z (%)=438 (2) [M+],365 (10) [M+−SiMe3], 348 (100) [M+−HOSiMe3].Anal. Calc. for C15H46OSi7 (439.134): C, 41.03; H,10.56. Found: C, 40.73; H, 10.38%.
3.9. 1,1,1,2,3,4,5,5,5-Nonamethyl-3-[1 %,2 %,2 %,2 %-tetramethyl-1 %-(trimethylsilyl)disilanyl]-2,4-bis(trimethylsilyl)pentasilane (11)
Trichloromethylsilane (1.18 ml, 10 mmol) was addedat −78°C to a solution of methyl-bis(trimethylsi-lyl)silyllithium (33 mmol) in pentane (100 ml). Stirringwas continued for 1 h, and the mixture was allowed towarm to r.t. within 2 h. After filtration and removal ofthe solvent under reduced pressure, the solid residuewas recrystallized from acetone to give 11 (4.64 g, 76%).The spectroscopic data obtained were in full agreementwith those reported [14].
3.10. 1,1,1,2,4,5,5,5-Octamethyl-3-[1 %,2 %,2 %,2 %-tetramethyl-1 %-(trimethylsilyl)disilanyl]-2,4-bis(trimethylsilyl)pentasilane (12)
Trichlorosilane (1.01 ml, 10 mmol) was added at−78°C to a solution of methyl-bis(trimethylsi-lyl)silyllithium (33 mmol) in pentane (100 ml). Stirringwas continued for 1 h, and the mixture was allowed towarm to r.t. within 2 h. After filtration and removal ofthe solvent under reduced pressure at 20°C, the solidresidue was suspended with cold acetone and filteredoff, yield 4.01 g (67%). M.p. 165–167°C. IR (Nujol)n=2016.7 cm−1 (SiH). 1H-NMR: d 3.53 (s, SiH, 1H),0.39 (s, SiMe, 9H), 0.31 (s, SiMe3, 54H). 13C-NMR: d
−6.1 (SiMe), −0.7 (SiMe3). 29Si-NMR: d −11.7(SiMe3), −80.8 (SiMe),−111.4 (d, 1J=147 Hz, SiH).MS: (CI) m/z (%)=597 (15) [M++1], 523 (30) [M+−SiMe3], 507 (55) [M+−Me−HSiMe3], 406 (100) [M+
−HMeSi(SiMe3)2]. Anal. Calc. for C21H64Si10 (597.60):C, 42.21; H, 10.79. Found: C, 41.89; H, 10.59%.
3.11. 3-Bromo-1,1,1,2,4,5,5,5-octamethyl-3-[1 %,2 %,2 %,2 %-tetramethyl-1 %-(trimethylsilyl)disilanyl]-2,4-bis(trimethylsilyl)pentasilane (13)
A mixture of compound 12 (3 g, 5.02 mmol) andCHBr3 (20 ml) was heated to 80°C for 10 days. Afterremoval of the volatiles, the solid residue was recrystal-lized twice from acetone to give 13 (1.02 g, 30%). M.p.164°C. 1H-NMR: d 0.51 (s, SiMe, 9H), 0.36 (s, SiMe3,54H). 13C-NMR: d 1.9 (SiMe3), −5.9 (SiMe). 29Si-NMR: d 12.3 (SiBr), −9.8 (SiMe3), −68.7 (SiMe).MS: (CI) m/z (%)=660 (30) [M+−Me], 595 (100)[M+−Br], 406 (95) [M+−BrMeSi(SiMe3)2]. HRMS:Calc. for C20H60
79BrSi10 659.1466. 659.1571. Anal. Calc.for C21H63BrSi10 (676.49): C, 37.29; H, 9.38. Found: C,35.64; H, 9.05%.
3.12. 3-Chloro-1,1,1,2,4,5,5,5-octamethyl-3-[1 %,2 %,2 %,2 %-tetramethyl-1 %-(trimethylsilyl)disilanyl]-2,4-bis(trimethylsilyl)pentasilane (14)
Concentrated HCl (20 ml) was added to a solution ofcompound 13 (0.1 g, 0.16 mmol) in diethylether (30 ml).The mixture was vigorously stirred for 7 days at r.t.The organic phase was separated, dried with MgSO4,and evaporated. The solid residue was recrystallizedfrom acetone to give 14 (0.085 g, 90%). M.p. 189°C.1H-NMR: d 0.5 (s, SiMe, 9H), 0.35 (s, SiMe3, 54H).13C-NMR: d 1.6 (SiMe3), −6.6 (SiMe). 29Si-NMR: d
27.9 (SiCl), −10.6 (SiMe3), −67.1 (SiMe). MS: (CI)m/z (%)=629 (5) [M+−H], 615 (30) [M+−Me], 595(54) [M+−Cl], 406 (100) [M+−ClMeSi(SiMe3)2].HRMS: Calc. for C20H60
35ClSi10 615.1972. Found:615.20764. Anal. Calc. for C21H63ClSi10 (632.04): C,39.91; H, 10.05. Found: C, 37.13; H, 9.85%.
3.13. 3-Fluoro-1,1,1,2,4,5,5,5-octamethyl-3-[1 %,2 %,2 %,2 %-tetramethyl-1 %-(trimethylsilyl)disilanyl]-2,4-bis(trimethylsilyl)pentasilane (15)
HF (20 ml of 40%) was added to a solution ofcompound 13 (0.1 g, 0.16 mmol) in ether (30 ml). Themixture was vigorously stirred for 4 days at r.t. Afteraddition of 100 ml of water the organic phase wasseparated, dried with MgSO4, and evaporated. Thesolid residue was recrystallized from acetone to give 14(0.082, g 90%). M.p. 184–185°C. 1H-NMR: d 0.50 (s,SiMe, 9H), 0.32 (s, SiMe3, 54H). 13C-NMR: d 1.2(SiMe3), −7.3 (SiMe). 29Si-NMR: d 74.8 (d, 1J=339.4Hz, SiF), −11.4 (SiMe3), −73.2 (SiMe). 19F-NMR: d
−204.4 (SiMeF). MS: (CI) m/z (%)=613 (4) [M+−H], 599 (55) [M+−Me], 425 (100) [M+−MeSi(SiMe3)2], 406 (40) [M+−MeFSi(SiMe3)2].HRMS: Calc. for C20H60FSi10 599.24451. Found:599.23718. Anal. Calc. for C21H63FSi10 (615.59): C,40.97; H, 10.32. Found: C, 37.76; H, 10.14%.

S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219218
3.14. 3-Hydroxy-1,1,1,2,4,5,5,5-octamethyl-3-[1 %,2 %,2 %,2 %-tetramethyl-1 %-(trimethylsilyl)disilanyl]-2,4-bis(trimethylsilyl)pentasilane (16)
Sulfuric acid (15 ml of 10%) was added to a solutionof compound 13 (0.1 g, 0.15 mmol) in tetrahydrofuran(30 ml) and the mixture was vigorously stirred for 1 dayat r.t. After the usual workup, the solid residue wasrecrystallized from acetone to give 16 (0.083 g, 92%).M.p.180°C. IR (Nujol) n=3654 cm−1 (OH). 1H-NMR:d 0.49 (s, SiMe, 9H), 0.33 (s, SiMe3, 54H). 13C-NMR: d
1.5 (SiMe3), −7.1 (SiMe). 29Si-NMR: d 37.6 (SiOH),−11.7 (SiMe3), −73.2 (SiMe), MS: (CI) m/z (%)=612(6) [M+], 595 (99) [M+−OH], 423 (100) [M+−MeSi(SiMe3)2]. HRMS: Calc. for C21H64OSi10 612.2666.Found: 612.26501. Anal. Calc. for C21H64OSi10
(613.60): C, 41.11; H, 10.51. Found: C, 39.47; H,10.38%.
3.15. Crystal structure determination of 12, 14, 15 and16
The crystal structure determinations were performedon a STOE-IPDS diffractometer with graphitemonochromated Mo–Ka radiation. The structures weresolved by direct methods (SHELXS-86) [40] and refinedby full matrix least-squares techniques against F2
(SHELXL-93) [41]. The position of the central hydrogenatom (H1) in compound 12 could be elucidated from
the difference map. XP (Siemens Analytical X-ray In-struments) was used for structure representations (forfull crystallographic data see Table 5).
4. Supplementary material
Crystallographic data (excluding structure factors)for the structures reported in this paper have beendeposited with the Cambridge Crystallographic DataCentre, CCDC no. 134653 for compound 12, no.134651 for compound 14, no. 134650 for compound 15and no. 134652 for 16. Copies of this information maybe obtained free of charge from: The Director, CCDC,12 Union Road, Cambridge, CB2 1EZ, UK (Fax:+44-1223-336-033; e-mail: [email protected] orwww: http://www.ccdc.cam.ac.uk).
Acknowledgements
We thank Professor Dr H. Oehme for his generoussupport and T. Groß for carrying out the X-ray diffrac-tion analyses. We gratefully acknowledge the supportof our research by the Deutsche Forschungsgemein-schaft and the Fonds der Chemischen Industrie. Wethank Dr M. Michalik, Dr W. Baumann and ProfessorN. Stoll for recording the NMR and MS spectra,respectively.
Table 5Summary of crystal and structure solution data of 12, 14, 15 and 16
12 14 15 16
C21H64Si10 C21H63FSi10Formula C21H63ClSi10 C21H64OSi10
632.065597.62Formula weight (g mol−1) 613.62615.6120.236(4) 18.154(4)a (A, ) 20.283(4) 20.339(4)
12.053(2)12.015(2)12.101(2)12.012(2)b (A, )19.061(4)19.174(4)c (A, ) 19.064(4)19.139(4)
a (°) 90.00 90.00 90.00 90.00117.32(3) 117.31(3)b (°) 90.42(3)117.43(3)
g (°) 90.00 90.00 90.00 90.00V (A, 3) 4152.5(14)4143.9(14)4187.2(14)4136.7(14)
0.9871.003 0.9820.960D (g cm−3)4 4Z 44
Monoclinic MonoclinicCrystal system Monoclinic MonoclinicSpace group P2(1)/cP2(1)/cP2(1)/cP2(1)/c
0.3310.388 0.3290.327m (Mo–Ka) (mm−1)Crystal size (mm) 0.5×0.3×0.2 0.3×0.3×0.2 0.2×0.3×0.4 0.3×0.3×0.2
293(2)293(2) 293(2) 293(2)Temperature (°C)2.04–24.38 1.99–21.99Scan range (2u) (°) 2.20–20.00 2.03-24.26
hkl Range −23/0, −13/13,−19/21 −20/20, −13/13, 0/21 0/23, −13/13,−22/19 −23/0, −13/13,−18/20Measured reflections 12167 9504 11964 12078
6555 5131Unique reflections 6567 6253359523523568Observed reflections 3556
284 289Refined parameters 289 293R1 [I\2s(I)] 0.044 0.036 0.043 0.039
0.105R2 for all 0.060 0.155 0.0790.855/1.024 0.859/0.983 0.601/0.864 0.794/0.985Goodness-of-fit

S. Chtchian et al. / Journal of Organometallic Chemistry 613 (2000) 208–219 219
References
[1] (a) M. Ballestri, C. Chatgillialoglu, K.B. Clark, D. Griller, B.Giese, B. Kopping, J. Org. Chem. 56 (1991) 678. (b) M. Aggar-wal, M.A. Ghuman, R.A. Geanangel, Main Group Met. Chem.14 (1991) 263. (c) R. West, in: S. Patai, Z. Rappoport (Eds.), TheChemistry of Organic Silicon Compounds, Part 2, Wiley,Chichester, 1989, pp. 1207.
[2] (a) H. Bock, J. Meuret, K. Ruppert, Angew. Chem. Int. Ed.Engl. 105 (1993) 413.(b) H. Bock, J. Meuret, K. Ruppert,Angew. Chem. Int. Ed. Engl. 32 (1993) 414. (c) H. Bock, J.Meuret, H. Schodel, Chem. Ber. 126 (1993) 2227. (d) H. Bock, J.Meuret, R. Baur, K. Ruppert, J. Organomet. Chem. 446 (1993)113.
[3] C.G. Pitt, J. Am. Chem. Soc. 91 (1969) 6613.[4] R.J. Sipe, R. West, J. Organomet. Chem. 70 (1974) 353.[5] (a) M. Haase, U. Klingebiel, R. Boese, M. Polk, Chem. Ber. 119
(1986) 369. (b) F. Elsner, H.-G. Woo, T.D. Tilley, J. Am. Chem.Soc. 110 (1988) 314. (c) A. Heine, D. Stalke, Angew. Chem. Int.Ed. Engl. 105 (1993) 90. (d) A. Heine, D. Stalke, Angew. Chem.Int. Ed. Engl. 32 (1993) 121. (e) G. Linti, W. Koestler, Angew.Chem. Int. Ed. Engl. 108 (1996) 593. (f) G. Linti, W. Koestler,Angew. Chem. Int. Ed. Engl. 35 (1996) 550. (g) H. Siegl, W.Krumlacher, K. Hassler, Monatsh. Chem. 130 (1999) 139.
[6] T.D. Tilley, in: S. Patai, Z. Rappoport (Eds.), The Chemistry ofOrganic Silicon Compounds, Part 2, Wiley, Chichester, 1989, p.1415.
[7] (a) K.W. Klinkhammer, W. Schwarz, Angew. Chem. Int. Ed.Engl. 107 (1995) 1448. (b) K.W. Klinkhammer, W. Schwarz,Angew. Chem. Int. Ed. Engl. 34 (1995) 1334. (c) K.W.Klinkhammer, S. Henkel, W. Schwarz, Angew. Chem. Int. Ed.Engl. 106 (1994) 712. (d) K.W. Klinkhammer, S. Henkel, W.Schwarz, Angew. Chem. Int. Ed. Engl. 32 (1994) 681. (e) S.P.Mallela, R.A. Geanangel, Inorg. Chem. 29 (1990) 3525. (f) A.M.Arif, A.H. Cowley, T.M. Elkins, J. Organomet. Chem. 325(1987) C11. (g) A.M. Arif, A.H. Cowley, T.M. Elkins R.A.Jones, J. Chem. Soc. Chem. Commun. (1986) 1776.
[8] Y. Apeloig, M. Yuzefovich, M. Bendikov, D. Bravo-Zhivo-tovskii, K. Klinkhammer, Organometallics 16 (1997) 1265.
[9] C. Marschner, Eur. J. Inorg. Chem. (1998) 221.[10] (a) T. Gross, R. Kempe, H. Oehme, Inorg. Chem. Commun. 1
(1998) 128. (b) T. Gross, R. Kempe, H. Oehme, Chem. Ber./Re-cueil 130 (1997) 1709.
[11] Using the term ‘hypersilyl’ we follow the proposal of N. Wiberg,made at the Xth International Symposium on OrganosiliconChemistry in Poznan, Poland, 1993.
[12] A.G. Brook, G. Gutekunst, J. Organomet. Chem. 225 (1982) 1.[13] K.W. Klinkhammer, Chem. Eur. J. 3 (1997) 1418.[14] J.B. Lambert, J.L. Pflug, J.M. Denari, Organometallics 15 (1996)
615.[15] K.M. Baines, A.G. Brook, R.R. Ford, P.D. Lickiss, A.K. Sax-
ena, W.J. Sawyer, B.A. Behnam, Organometallics 8 (1989) 693.
[16] (a) S. Chtchian, C. Krempner, in: N. Auner, J. Weis (Eds.),Organosilicon Chemistry IV, VCH, Weinheim, 2000, p. 352. (b)S. Chtchian, C. Krempner, XIIth Organosilicon Symposium,Sendai, Japan, May 1999, Abstract P087.
[17] J.B. Lambert, H. Wu, Organometallics 17 (1998) 4904.[18] TASF: tris(dimethylamino)sulfonium-difluorotrimethylsilicate.[19] Also, the structure of 10 has been reported by: D. Bravo-Zhivo-
tovskii, M. Yuzefovich, A. Shames, Y. Apeloig, K. Klinkham-mer at XIIth Organosilicon Symposium, Sendai, Japan, May1999, Abstract 4B13.
[20] A. Bondi, J. Phys. Chem. 68 (1964) 441.[21] (a) B. Albinsson, H. Teramae, J.W. Downing, J. Michl, Chem.
Eur. J. 2 (1996) 529. (b) B. Albinsson, D. Antic, F. Neumann, J.Michl, J. Phys. Chem. 103 (1999) 2184.
[22] R.D. Miller, J. Michl, Chem. Rev. 89 (1989) 1359.[23] (a) H.S. Plitt, V. Balaji, J. Michl, Chem. Phys. Lett. 213 (1993)
158. (b) H.S. Plitt, J.W. Downing, H. Teramae, M.K. Raymond,M. Michl, ACS Symp. Ser. 579 (1994) 376.
[24] H. Gilman, W.H. Atwell, G.L. Schwebke, J. Organomet. Chem.2 (1964) 369.
[25] (a) R. Radeglia, G. Engelhardt, J. Organomet. Chem. 67 (1974)C45. (b) G. Engelhardt, H. Jancke, R. Radeglia, H. Kriegsmann,M.F. Larin, V.A. Pestunovich, E.I. Dubinskaja, M.G.Voronkov, Z. Chem. 17 (1977) 376. (c) J. Schraml, R. Ponec, V.Chvalovsky, G. Engelhardt, H. Kriegsmann, M.F. Larin, V.A.Pestunovich, M.G. Voronkov, J. Organomet. Chem. 178 (1979)55.
[26] (a) M. Ishikawa, J. Iyoda, H. Ikeda, K. Kotake, T. Hashimoto,M. Kumada, J. Am. Chem. Soc. 103 (1981) 4845. (b) D.A.Stanislawski, R. West, J. Organomet. Chem. 204 (1981) 295.
[27] J.B. Lambert, S. Zhang, J. Chem. Soc. Chem. Commun. 4 (1993)383.
[28] E.V. Vanden Berghe, G.D. Vander Kelen, J. Organomet. Chem.59 (1973) 175.
[29] D.H. Barton, S.I. Parekh, J. Am. Chem. Soc. 115 (1993) 948.[30] K. Schenzel, K. Hassler, in: N. Auner, J. Weis (Eds.), Organosil-
icon Chemistry, vol. II, VCH, Weinheim, 1996.[31] H.C. Marsmann, W. Raml, E. Hengge, Z. Naturforsch. Teil B
35 (1980) 1541.[32] A. Rastelli, S.A. Pozzoli, J. Mol. Struct. 18 (1973) 469.[33] S. Kyushin, H. Sakurai, H. Matsumoto, Chem. Lett. (1998) 107.[34] B. Buerglen, E. Uhlig, Z. Chem. 28 (1988) 408.[35] D.C. England, F.J. Weigert, J.C. Calabrese, J. Org. Chem. 49
(1984) 4816.[36] R.K. Harris, B.J. Kimber, J. Organomet. Chem. 43 (1974) C70.[37] (a) A. Marchand, P. Gerval, F. Duboudin, M.-H. Gaufryau, M.
Joanny, P. Mazerolles, J. Organomet. Chem. 267 (1984) 93. (b)D.C. McKean, A.R. Morrisson, I. Torto, M.I. Kelly, Spec-trochim. Acta Part A 41 (1985) 25.
[38] M.E. Lee, M.A. Nath, P.P. Gaspar, Phosphorus, Sulfur, SiliconRelat. Elem. 56 (1991) 203.
[39] C. Chatgilialoglu, A. Guerrini, M. Lucarini, J. Org. Chem. 57(1992) 3405.
[40] G.M. Sheldrick, Acta Crystallogr., Sect. A 46 (1990) 467.[41] G.M. Sheldrick, University of Gottingen, Germany, 1993.
.
Top Related
Copyright © 2022 FDOKUMEN

