







Bahasa
Halaman
Hukum


Speciation of adsorbed arsenic (V) on red mud
using a sequential extraction procedure
D. A. RUBINOS1,*, M. ARIAS
2, F. DIÂAZ-FIERROS
1AND M. T. BARRAL
1
1Departamento de EdafoloxõÂa e QuõÂmica AgrõÂcola, Facultade de Farmacia, Universidade de Santiago de
Compostela, 15782 Santiago de Compostela, Spain2AÂ rea de EdafoloxõÂa e QuõÂmica AgrõÂcola, Facultade de Ciencias de Ourense, Universidade de Vigo, 32004 Ourense,
Spain
ABSTRACT
The distribution of sorbed arsenic(V) among different geochemical fractions for arsenic(V)-loaded red
mud, an oxide-rich residue from bauxite refining that has been proposed as an adsorbent for arsenic,
was studied as a function of sorbed arsenic(V) concentration using a sequential extraction procedure.
The release of previously sorbed arsenic(V) was also studied as a function of pH and arsenic(V)
concentration. Most sorbed arsenic(V) (0.39ÿ7.86 mmol kgÿ1) was associated with amorphous and
crystalline Al and Fe oxides (24.1ÿ43.8% and 24.7ÿ59.0% of total sorbed arsenic, respectively).
Exchangeable arsenic was the smallest fraction (0.4ÿ5.2% of total sorbed arsenic). The distribution of
sorbed arsenic(V) was related to the arsenic surface coverage. For arsenic surface coverages >~30% the
percentage of arsenic(V) associated with the amorphous Al oxide fraction increased and that associated
with the crystalline oxide fraction decreased. The arsenic(V) exchangeable fraction increased from 1.4
to 756 mmol kgÿ1
as surface coverage increased from 388 to 7855 mmol kgÿ1. The release of sorbed
arsenic(V) from red mud was greater at alkaline pH values (maximum release of ~33% of previously
sorbed arsenic at pH = 12), but for high arsenic(V) initial concentration (0.2 mM arsenic) considerable
amounts of arsenic (6.5% of previously sorbed arsenic) were released at pH 4, in accordance with the
dissolution of amorphous Al oxides in the red mud. The results obtained suggest a greater mobility of
sorbed arsenic(V) as its surface concentration approaches saturation.
KEYWORDS: red mud, arsenic, sorption, speciation, sequential extraction.
Introduction
THE extremely toxic properties of inorganic
arsenic compounds are well-known. Arsenic is
classi®ed by the International Agency for
Research on Cancer (IARC) as a human
carcinogen (IARC, 1980). The occurrence of
arsenic in the environment can have natural
(weathering of rocks and volcanism) or anthro-
pogenic (petroleum re®ning, thermal power
plants, pesticides, ceramics, non ferrous smelting,
gold-mine tailings, ¯y ash leachates and fertilizer
production) origins (Prasad, 1994). The introduc-
tion of arsenic compounds to the food chain,
mainly through ingestion of contaminated
drinking water, is especially dangerous.
Drinking water is contaminated by arsenic in
many areas of the world; for example, excessive
concentrations of arsenic in groundwater have
affected large populations in West Bengal and
Bangladesh (Anawar et al., 2004; Nickson et al.,
1998). Recent studies on chronic exposure to
arsenic have shown that the consumption of water
containing concentrations of arsenic <50 mg lÿ1
can cause cancer over long exposure periods
(WHO, 2001). As a consequence, the World
Health Organisation has recently reduced their
recommended provisional guideline value for
arsenic in drinking water from 50 to 10 mg/l
(WHO, 1993).
In response, the search for and development of
new technologies for the removal of arsenic from
* E-mail: [email protected]
DOI: 10.1180/0026461056950273
Mineralogical Magazine, October 2005, Vol. 69(5), pp. 591±600
# 2005 The Mineralogical Society

aqueous systems have increased considerably.
Sorption methods are promising because they are
simple to perform, can be used in small-scale
treatment plants or household systems and are
easy to operate (GencË-Fuhrman et al., 2004a).
Several materials have already been proposed for
the removal of metals and metalloids from water,
including natural materials and specially designed
technical particles (Daus et al., 2004). Amongst
proposed sorbents for the removal of arsenic from
aqueous solutions are activated carbon (Daus et
al., 2004), activated bauxite (Gupta and Chen,
1978), activated alumina (Lin and Wu, 2001),
amorphous Al hydroxide (Anderson et al., 1976),
amorphous iron hydroxide (Pierce and Moore,
1980), hematite (Prasad, 1994), granular Fe
hydroxide (GIH) (Driehaus et al., 1998), zeolites
(Shevade and Ford, 2004) and clays (Frost and
Grif®n, 1977).
Red mud is an oxide-rich residue generated by
alumina production from bauxite using the Bayer
process. About two tons of red mud is generated
per ton of metallic aluminium. World production
of red mud is roughly 60 million tons per year
(Glenister, 1987). Consequently, efforts have
been made to use the red mud bene®cially, for
example for leveÂe construction material, road
embankments, land®ll cover, synthetic soils,
fertilizer ®llers, remediation of coastal erosion,
manufacture of bricks, production of ceramic
glazes, as a ®ller for polymer reinforcement and,
of course, as a cheap adsorbent for removal of
potentially toxic metals, phosphorous and dyes
(Apak et al., 1998; Arias et al., 1999; Gupta and
Sharma, 2002; LoÂpez et al., 1998), and inorganic
arsenic from water and wastewater (AltundogÆan
et al., 2000; Rubinos et al., 1998a), in view of its
high Fe, Al and Ti oxides concentrations.
Recently, several studies have tested a derivative
product of red mud, BauxsolTM
(trademark name
of seawater-neutralized red mud), as an uncon-
ventional sorbent for arsenic removal from
aqueous solutions (GencË et al., 2003; GencË-
Fuhrman et al., 2004a,b). The most recent
studies on the utilization of red mud for the
removal of arsenic have been directed towards the
search for treatments to improve removal
ef®ciency. However, to our knowledge, the
distribution of adsorbed arsenic on red mud and
its implications for arsenic removal have not been
previously investigated, bearing in mind the
heterogeneous composition of red mud. The
objectives of this study are: (1) to determine the
distribution of sorbed arsenic(V) among different
geochemical fractions in red mud; and (2) to
understand the desorption behaviour of arsenic(V)
in relation to the distribution of arsenic(V) in the
different phases of the sorbent.
Experimental methods
Red mud
Red mud used in the present study was obtained
from ALCOA Europe Aluminium factory, Lugo,
Spain. The red mud was air-dried, crushed and
sieved (0.250 mm sieve). The chemical composi-
tion, determined after acid digestion, shows that
main constituents of the red mud employed in this
study (%w/w) are: 37.22Ô0.33% Fe2O3,
20.10Ô0.59% TiO2, 12.40Ô1.07% Al2O3,
6 .30Ô0 .20% CaO, 4 .64Ô0 .41% Na2O,
3.81Ô0.16% SiO2, 0.51Ô0.02% P2O5, 0.36Ô0.02%
ZrO2, 0.30Ô0.01% Cr2O3, 0.14Ô0.00% MgO, loss
on ignition 11.34Ô0.02%. Red mud composition
depends on bauxite origin, although typically it is
rich in Fe oxides (25ÿ40%) and Al oxides
(15ÿ20%) (Lombi et al., 2002). The main
mineral components (as determined by X-ray
diffraction) are hematite, rutile, magnetite, boeh-
mite, ilmenite and zeolite-type minerals. The
zeolite content, determined by a cation exchange
capacity method (Rubinos et al., 1998b) is ~9%.
The surface area of the red mud, determined by the
N2 adsorption-BET method, is 23.7 m2gÿ1
and its
pH (as a 1:5 mixture in water) is 9.7Ô0.1. Particle-
size distribution analysis, performed by wet sieving
(for the fraction >50 mm) and the pipette method
(for the fraction <50 mm) showed a predominance
of silt and clay fractions (80%). The cation
exchange capacity (CEC) of the red mud, as
determined by the ammonium acetate (pH 7)
method, is 10.83Ô0.02 cmolc kgÿ1. It is important
to note that ~50% of the total CEC of red mud is
zeolite CEC (Rubinos et al., 1998b). Selective
dissolution analyses performed showed that the red
mud studied contains 196Ô7 mg gÿ1
of crystalline
and amorphous Fe, extracted by dithionite-citrate-
bicarbonate (DCB) (Mehra and Jackson, 1960).
Amorphous Fe and Al contents in red mud,
determined by extraction with 0.2 M ammonium
oxalate at pH 3 (Schwertmann, 1964), are
2.4Ô0.1 mg gÿ1
and 25Ô1.4 mg gÿ1, respectively.
Arsenic analysis
All arsenic analyses were performed by hydride
generation atomic absorption spectrophotometry
(HGAAS) using a Perkin-Elmer MHS-10 hydride
592
D. A. RUBINOS ET AL.

unit on an atomic absorption spectrophotometer
(Perkin-Elmer M2100). The system was cali-
brated using standard solutions of arsenic
prepared by dilution of a commercial standard
arsenic solution (1000 mg lÿ1
of arsenic). To
generate the AsH3, samples and standards were
reacted with a 3% NaBH4/1% NaOH solution (as
reducing agent) and 1.5% HCl solution. The
calculated detection limit for these conditions was
2 mg lÿ1
of arsenic. All solutions were prepared
with deionized MilliQPLUS
water.
Distribution of sorbed arsenic(V) on red mud
To study the distribution of sorbed arsenic(V) on
red mud, ®rst a series of samples of air-dried red
mud (1 g) were equilibrated in polypropylene
centrifuge tubes with 20 ml of solution containing
arsenic(V) (as Na2HAsO4´7H2O) and 0.1 M NaCl
(as background electrolyte). Initial arsenic(V)
concentrations employed ranged from 0.02 to
1 mM. The suspensions were shaken at room
temperature (20Ô1ëC) in a rotary shaker. The
arsenic(V) sorption stage was performed at natural
pH value of red mud (~9.2) without adjustment of
suspension pH during the equilibration step. After
24 h of shaking, suspensions were centrifuged
(14000 g, 10 min) and ®ltered through
WhatmanTM
40 ®lter papers (particle retention
rating at 98% ef®ciency = 8 mm). The concentra-
tion of arsenic in the ®ltered solution was analysed
by HGAAS and the concentration of sorbed
arsenic(V) on the red mud calculated as the
difference between the initial and ®nal concentra-
tion of arsenic in solution. Losses of arsenic by
adsorption to the centrifuge tubes or ®lters were
tested by preparing identical arsenic(V) solutions
and subjecting them to the same treatment as the
samples in the absence of red mud. The
concentration determined in these solutions was
assumed to be the arsenic(V) initial concentration.
Three arsenic(V)-loaded red mud samples were
prepared for each arsenic(V) concentration.
Immediately after the loading step, the red mud
samples containing arsenic were chemically
fractionated using the ®ve step sequential
extraction procedure for arsenic proposed by
Lombi et al. (1999) (Table 1). The extractant:red
mud ratio was 25:1. Sequential extractions
determine only operationally de®ned speciation
(Nirel and Morel, 1990), and depend on factors
including the concentration of the reagents,
duration of the extractions and the selectivity of
the reagents used to attack a given phase. Results
cannot simply be extrapolated to ®eld conditions
(Cappuyns et al., 2002). Despite their limitations,
sequential extraction procedures are a useful
approach to estimate partitioning of elements
between different geochemical fractions.
After the period of extraction, the ®ltered
extracts obtained from each step of the sequential
extraction scheme were analysed for arsenic by
HGAAS. The amounts of arsenic extracted in
each step were corrected (estimating the volume
of entrained solution after decanting the super-
natant by the difference in tube weight compared
to initial tube weight, accounting for the mass of
red mud) for arsenic contribution from previous
extraction step (Jackson and Miller, 2000).
Experiments were run in triplicate.
Arsenic(V) leaching at different pH
With the purpose of relating the distribution of
arsenic(V) between the different phases of the red
mud and the mobility of sorbed arsenic(V) on red
mud, we studied the remobilization of previously
sorbed arsenic(V) on red mud as a function of
solution pH. For this, arsenic(V)-loaded red mud
samples, prepared by reaction of 0.5 g of red mud
with 25 ml of 0.1 M NaCl solutions containing
0.02 mM or 0.2 mM As(V), were resuspended in
TABLE 1. Sequential extraction procedure for arsenic fractionation (Lombi et al., 1999).
Step Treatment Fraction
1 (NH4)2SO4 0.05 M ÿ 1 h shaking Exchangeable
2 NH4H2PO4 0.05 M ÿ 1 h shaking Specifically sorbed
3 NH4F 0.05M pH 7.0 ÿ 1 h shaking Al and organic matter-associated
4 NH4-oxalate 0.2 M pH 3.25 ÿ 4 h shaking
in the dark
Bound to amorphous oxides
5 NH4-oxalate 0.2 M + ascorbic acid 0.1 M ÿ
pH 3.25. 30 min shaking in water bath at 96ëC
Bound to crystalline oxides
SPECIATION OF ADSORBED AS(V)
593

arsenic-free 0.1 M NaCl solutions, the pH of which
was adjusted to the same pH value as that in the
arsenic(V)-loading step. pH adjustment was by
addition of small quantities of 0.1 M HCl or 0.1 M
NaOH using a Metrohm 702SM automatic titrator.
The pH values studied were moderate acidic (pH
4), neutral pH (pH 7), equilibrium pH of red mud
in the release solution without addition of base or
acid (pH 8.3) and an extremely alkaline value (pH
12). The suspensions were shaken for 1 h in a
rotary shaker at room temperature, then centrifuged
and ®ltered (WhatmanTM
40 ®lters). The same
process was repeated for the centrifuged solid
samples with another 25 ml of arsenic-free
solution and the pH of the suspensions was
readjusted again to the initial corresponding pH
value. After the shaking period, the suspensions
were centrifuged and ®ltered and the ®ltrates
obtained from the two desorption steps were
mixed and analyzed for arsenic by HGAAS. Al,
Fe, Si and Ca were also analyzed in the extracts by
atomic absorption spectrophotometry (AAS) to test
the solubility of the main components of red mud
at the pH values studied. The experiments were
conducted in triplicate.
Statistical analysis
Analyses of variance (ANOVA) and least
signi®cance difference tests (LSD) were
performed on the data using version 12.0 of the
SPSS for WindowsTM
program.
Results and discussion
Distribution of sorbed arsenic(V) on red mud
The arsenic loading procedure resulted in samples
of red mud with concentrations of sorbed
arsenic(V) of 0.39, 1.91, 3.58, 6.68 and 7.86
mmol of As kgÿ1. These values represent 4.9,
24.1, 45.3, 84.5 and 99.4% of the maximum
arsenic(V) sorption capacity for red mud,
calculated by ®tting the experimental sorption
data to the Langmuir equation (data not shown).
The red mud is a heterogeneous solid with a
number of components to which the arsenic(V)
species in solution may show different af®nities.
Therefore, arsenic species may sorb preferentially
onto some components of red mud to which they
show greater af®nity. As the arsenic(V) concen-
tration in solution increases and red mud
approaches saturation, arsenic will also sorb on
the `lower af®nity' components of red mud. It is
possible that this aspect can be re¯ected in the
distribution of sorbed arsenic(V) species between
the different components of red mud as a function
of surface coverage. Moreover, the nature and
strength of the bonds established between the
arsenic(V) species and the different phases of the
sorbent cannot be the same. Also, the components
of the red mud present different solubility
behaviour. This aspect would directly affect the
remobilization of associated arsenic(V), which in
turn would affect the net ef®ciency of the removal
process.
The amounts of arsenic extracted in each step
of the sequential fractionation for the arsenic-
loaded red mud samples are shown in Table 2.
The arsenic recovered with the sequential
extraction ranged between 74 and 99% of the
sorbed arsenic(V). The distribution of arsenic
among the different (operationally de®ned) phases
of the sorbent, expressed as percentage of
recovered arsenic, are shown in Fig. 1.
Two main aspects must to be noted. First, most
sorbed arsenic on red mud was extracted in steps 4
TABLE 2. Arsenic extracted in each step of the sequential fractionation procedure for the As(V)-loaded red
mud samples (data in mmol of As kgÿ1
of red mud). The ratio between arsenic extracted (% of total
extracted) in steps 5 and 4 is also shown (standard deviation in parenthesis, N = 3).
As(V)RM1
Step 1 Step 2 Step 3 Step 4 Step 5 AsASC/AsOX (%)2
388 1.4 (1.3) 46.7 (5.4) 17.2 (4.2) 92.7 (1.3) 227.3 (3.2) 2.45 (0.00)
1909 17.3 (4.5) 352.3 (50.4) 58.8 (4.3) 474.7 (25.8) 499.9 (19.2) 1.06 (0.10)
3580 55.1 (9.9) 556.8 (19.0) 119.4 (18.6) 1189.9 (129.3) 807.8 (55.7) 0.71 (0.17)
6680 168.2 (23.8) 1227.9 (11.4) 117.8 (30.9) 2102.5 (178.9) 1181.8 (102.7) 0.57 (0.10)
7855 755.5 (56.9) 1908.0 (28.9) 316.8 (139.1) 2739.7 (120.8) 1779.0 (145.1) 0.65 (0.02)
1As(V)RM is sorbed arsenic(V) on red mud
2AsASC and AsOX are the arsenic percentages extracted in steps 5 and 4, respectively.
594
D. A. RUBINOS ET AL.
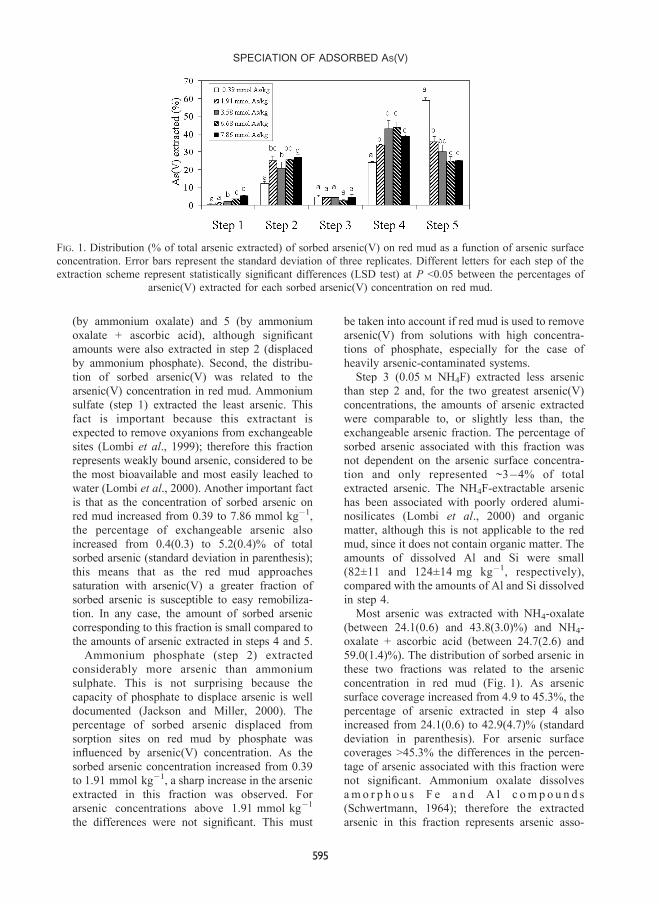
(by ammonium oxalate) and 5 (by ammonium
oxalate + ascorbic acid), although signi®cant
amounts were also extracted in step 2 (displaced
by ammonium phosphate). Second, the distribu-
tion of sorbed arsenic(V) was related to the
arsenic(V) concentration in red mud. Ammonium
sulfate (step 1) extracted the least arsenic. This
fact is important because this extractant is
expected to remove oxyanions from exchangeable
sites (Lombi et al., 1999); therefore this fraction
represents weakly bound arsenic, considered to be
the most bioavailable and most easily leached to
water (Lombi et al., 2000). Another important fact
is that as the concentration of sorbed arsenic on
red mud increased from 0.39 to 7.86 mmol kgÿ1,
the percentage of exchangeable arsenic also
increased from 0.4(0.3) to 5.2(0.4)% of total
sorbed arsenic (standard deviation in parenthesis);
this means that as the red mud approaches
saturation with arsenic(V) a greater fraction of
sorbed arsenic is susceptible to easy remobiliza-
tion. In any case, the amount of sorbed arsenic
corresponding to this fraction is small compared to
the amounts of arsenic extracted in steps 4 and 5.
Ammonium phosphate (step 2) extracted
considerably more arsenic than ammonium
sulphate. This is not surprising because the
capacity of phosphate to displace arsenic is well
documented (Jackson and Miller, 2000). The
percentage of sorbed arsenic displaced from
sorption sites on red mud by phosphate was
in¯uenced by arsenic(V) concentration. As the
sorbed arsenic concentration increased from 0.39
to 1.91 mmol kgÿ1, a sharp increase in the arsenic
extracted in this fraction was observed. For
arsenic concentrations above 1.91 mmol kgÿ1
the differences were not signi®cant. This must
be taken into account if red mud is used to remove
arsenic(V) from solutions with high concentra-
tions of phosphate, especially for the case of
heavily arsenic-contaminated systems.
Step 3 (0.05 M NH4F) extracted less arsenic
than step 2 and, for the two greatest arsenic(V)
concentrations, the amounts of arsenic extracted
were comparable to, or slightly less than, the
exchangeable arsenic fraction. The percentage of
sorbed arsenic associated with this fraction was
not dependent on the arsenic surface concentra-
tion and only represented ~3ÿ4% of total
extracted arsenic. The NH4F-extractable arsenic
has been associated with poorly ordered alumi-
nosilicates (Lombi et al., 2000) and organic
matter, although this is not applicable to the red
mud, since it does not contain organic matter. The
amounts of dissolved Al and Si were small
(82Ô11 and 124Ô14 mg kgÿ1, respectively),
compared with the amounts of Al and Si dissolved
in step 4.
Most arsenic was extracted with NH4-oxalate
(between 24.1(0.6) and 43.8(3.0)%) and NH4-
oxalate + ascorbic acid (between 24.7(2.6) and
59.0(1.4)%). The distribution of sorbed arsenic in
these two fractions was related to the arsenic
concentration in red mud (Fig. 1). As arsenic
surface coverage increased from 4.9 to 45.3%, the
percentage of arsenic extracted in step 4 also
increased from 24.1(0.6) to 42.9(4.7)% (standard
deviation in parenthesis). For arsenic surface
coverages >45.3% the differences in the percen-
tage of arsenic associated with this fraction were
not signi®cant. Ammonium oxalate dissolves
am o r p h o u s F e a n d A l c om p o u n d s
(Schwertmann, 1964); therefore the extracted
arsenic in this fraction represents arsenic asso-
FIG. 1. Distribution (% of total arsenic extracted) of sorbed arsenic(V) on red mud as a function of arsenic surface
concentration. Error bars represent the standard deviation of three replicates. Different letters for each step of the
extraction scheme represent statistically signi®cant differences (LSD test) at P <0.05 between the percentages of
arsenic(V) extracted for each sorbed arsenic(V) concentration on red mud.
SPECIATION OF ADSORBED AS(V)
595

ciated with amorphous oxides of the red mud. The
amounts of Al and Si extracted in step 4 were
26Ô1.4 and 2.5Ô0.4 mg gÿ1, respectively. The
ability of amorphous oxides to bind arsenic is well
established (Anderson et al., 1976) and several
works have found a strong correlation between
amorphous oxide content and arsenic(V) sorption
parameters for soils and sediments (Livesey and
Huang, 1981).
The percentage of arsenic extracted with
oxalate + ascorbic acid decreased from ~60 to
~25% of the total extracted arsenic as the
concentration of sorbed arsenic increased from
0.39 to 7.86 mmol kgÿ1, and, speci®cally, we
observed a sharp decrease in the arsenic extracted
as the arsenic surface coverage increased from
0.39 to 1.91 mmol kgÿ1
(Fig. 1).
When comparing the percentages of arsenic
extracted in step 5 with those extracted in step 4
we observed a reduction from 2.5 to ~0.6 as
surface coverage increased (Table 2). This shows
a preferential association of arsenic(V) with
crystalline oxides of red mud at low arsenic(V)
concentrations. It has been shown that arsenic
binds preferentially to Fe oxides in soils, and to a
lesser degree to Al oxides (Wauchope, 1975). At
surface coverages >~30%, and the sites in the
crystalline oxide surfaces of red mud become
occupied, arsenic(V) tends to sorb preferentially
onto amorphous oxides of red mud, probably a
consequence of their greater surface area and
porous structure. This suggests greater arsenic(V)
sorption capacity for amorphous oxides than for
crystalline oxides of red mud. Higher arsenic(V)
sorption capacities have been reported for
amorphous Al hydroxides than crystalline Fe
oxides (GarcõÂa-SaÂnchez et al., 2002).
Some sorbed arsenic(V) on red mud was not
extracted with the sequential extraction scheme
employed. This can be explained by the fact that
red mud contains 20% TiO2 as rutile. The ability
of rutile to sorb arsenate to a signi®cant degree
has been described (Fordham and Norris, 1979).
TiO2 in red mud is extremely stable, remained
undissolved (85%) even in strong acid (6 M HCl)
solutions.
It was observed that ~80% of sorbed arsenic(V)
was associated with the two oxalate fractions for
small arsenic concentrations, and this percentage
decreased to ~60% for high arsenic surface
concentrations. Arsenic associated with oxalate-
extractable fractions represents less mobile forms
of arsenic (Lombi et al., 1999), therefore the
reduction in the percentage of sorbed arsenic
associated with these fractions observed at high
arsenic(V) concentrations must be appreciated.
Selective dissolution analyses of red mud
resulted in calculated ratios Alox/Altotal = 0.38
and Feox/Fetotal = 0.007, i.e. the amorphous Fe
oxides content in red mud represent <1% of total
Fe, whereas amorphous Al oxides represent ~38%
of total Al content. Since at surface coverages
>~30%, arsenic is extracted predominantly in step
4, and considering the low amorphous Fe oxide
content of the red mud, the relative contribution
of amorphous Al oxides to the overall sorption
process increases above this surface coverage.
The predominant association of arsenic(V) with
amorphous Al oxide fraction at high surface
concentrations is very important, because some
amorphous aluminum compounds in red mud are
aluminosilicates integrated in the so-called
desilication product (DSP). The DSP is formed
during the Bayer process by reprecipitation of
dissolved silicates as sodium-alumino-silicates.
The DSP is not a single compound, but rather a
series of zeolites. Glenister and Thornber (1985)
observed that at pH values near 4 the Al and Si of
red mud show a sharp increase in solubility,
suggesting that at this pH the zeolite materials of
red mud could be dissolved. Therefore, if part of
the amorphous Al compounds are dissolved at
these pH values, the associated arsenic can
potentially be released. The results obtained
from the arsenic remobilization experiments at
different pH values seem to con®rm this
hypothesis.
Arsenic(V) leaching at different pH
Figure 2 shows the amounts of arsenic released at
pH 4, 7, 8.3 and 12 for two arsenic(V) initial
concentrations (0.02 and 0.2 mM). In general, the
amounts of arsenic released were small. It has
been suggested that the remarkably low reversi-
bility of arsenic(V) sorption on red mud indicates
that the mechanism governing the sorption
process involves chemisorption (GencË-Fuhrman
et al., 2004a). It has been shown that arsenate
forms inner-sphere surface complexes on both Al
and Fe oxides (Goldberg and Johnston, 2001; Sun
and Doner, 1996). Also, ligand exchange has been
proposed as the removal mechanism for adsorp-
tion of arsenate onto zeolites (Shevade and Ford,
2004). Rubinos et al. (2002) observed little ionic
strength dependence of arsenic(V) sorption on red
mud as a function of solution pH and shifts
towards higher pH values in titration curves of red
596
D. A. RUBINOS ET AL.
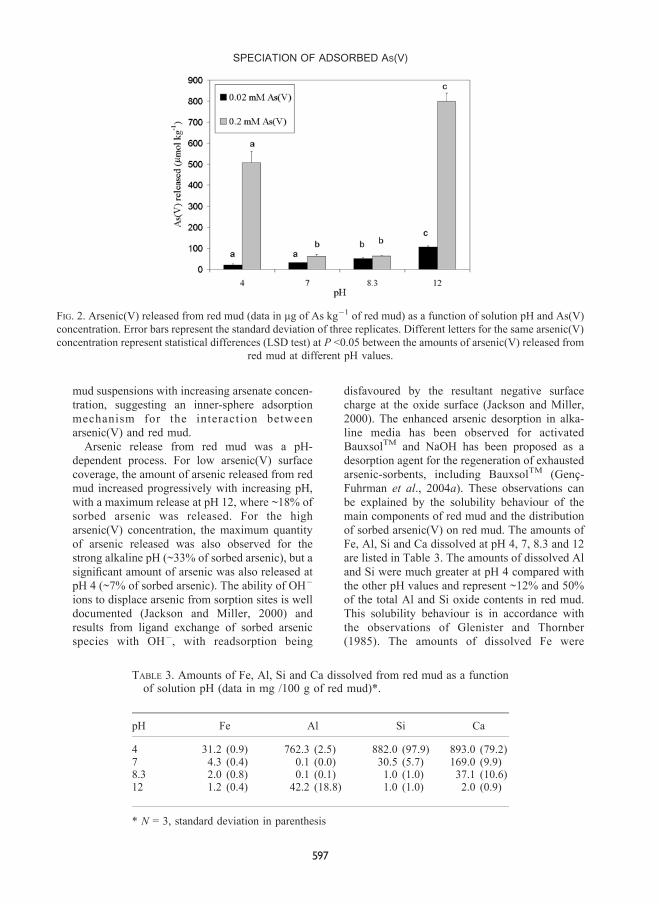
mud suspensions with increasing arsenate concen-
tration, suggesting an inner-sphere adsorption
mechanism for the interaction between
arsenic(V) and red mud.
Arsenic release from red mud was a pH-
dependent process. For low arsenic(V) surface
coverage, the amount of arsenic released from red
mud increased progressively with increasing pH,
with a maximum release at pH 12, where ~18% of
sorbed arsenic was released. For the high
arsenic(V) concentration, the maximum quantity
of arsenic released was also observed for the
strong alkaline pH (~33% of sorbed arsenic), but a
signi®cant amount of arsenic was also released at
pH 4 (~7% of sorbed arsenic). The ability of OHÿ
ions to displace arsenic from sorption sites is well
documented (Jackson and Miller, 2000) and
results from ligand exchange of sorbed arsenic
species with OHÿ
, with readsorption being
disfavoured by the resultant negative surface
charge at the oxide surface (Jackson and Miller,
2000). The enhanced arsenic desorption in alka-
line media has been observed for activated
BauxsolTM
and NaOH has been proposed as a
desorption agent for the regeneration of exhausted
arsenic-sorbents, including BauxsolTM
(GencË-
Fuhrman et al., 2004a). These observations can
be explained by the solubility behaviour of the
main components of red mud and the distribution
of sorbed arsenic(V) on red mud. The amounts of
Fe, Al, Si and Ca dissolved at pH 4, 7, 8.3 and 12
are listed in Table 3. The amounts of dissolved Al
and Si were much greater at pH 4 compared with
the other pH values and represent ~12% and 50%
of the total Al and Si oxide contents in red mud.
This solubility behaviour is in accordance with
the observations of Glenister and Thornber
(1985). The amounts of dissolved Fe were
FIG. 2. Arsenic(V) released from red mud (data in mg of As kgÿ1
of red mud) as a function of solution pH and As(V)
concentration. Error bars represent the standard deviation of three replicates. Different letters for the same arsenic(V)
concentration represent statistical differences (LSD test) at P <0.05 between the amounts of arsenic(V) released from
red mud at different pH values.
TABLE 3. Amounts of Fe, Al, Si and Ca dissolved from red mud as a function
of solution pH (data in mg /100 g of red mud)*.
pH Fe Al Si Ca
4 31.2 (0.9) 762.3 (2.5) 882.0 (97.9) 893.0 (79.2)
7 4.3 (0.4) 0.1 (0.0) 30.5 (5.7) 169.0 (9.9)
8.3 2.0 (0.8) 0.1 (0.1) 1.0 (1.0) 37.1 (10.6)
12 1.2 (0.4) 42.2 (18.8) 1.0 (1.0) 2.0 (0.9)
* N = 3, standard deviation in parenthesis
SPECIATION OF ADSORBED AS(V)
597

extremely small for all the pH values studied (a
maximum of only 0.12% of total Fe oxides
content at pH 4) re¯ecting the stability and poor
solubility of crystalline Fe oxides of red mud.
The increase in arsenic release observed at pH
4 for the high arsenic(V) concentration, but not
for the low arsenic(V) concentration, can be
explained considering the distribution of arsenic
in red mud. As we have seen before, at low
arsenic(V) concentrations, arsenic tends to bind
mainly to crystalline Fe oxides. Since these
compounds are not dissolved at pH 4, the
associated arsenic is not released, so the main
factor in¯uencing the arsenic release for low
concentrations will be the increasing concentra-
tion of OHÿ
ions in solution as the pH increases.
On the other hand, as arsenic(V) concentration
increases, the sorbed arsenic tends to associate
predominantly with amorphous Al compounds
(Fig. 2), that are in part soluble at pH 4 (Table 3),
thus releasing the associated arsenic. At alkaline
pH (12), the release of arsenic is again attributed
to the displacement of arsenic by OHÿ
ions.
Conclusions
The results obtained show that distribution of
sorbed arsenic(V) between the different geochem-
ical phases of red mud and the relative
contribution of the active components for
arsenic(V) sorption depend on arsenic(V)
surface concentration.
Most sorbed arsenic was associated with the
amorphous and crystalline oxide fraction as
observed from the amounts extracted in the two
acid ammonium oxalate steps, which show that
arsenic(V) is strongly sorbed by red mud.
However, it is important to note that as red mud
approaches its maximum arsenic(V)-sorption
capacity, the percentage of sorbed arsenic
associated with these two fractions diminish,
and the percentage of exchangeable (weakly
bound) arsenic increases. Moreover, for high
arsenic(V) concentrations, arsenic(V) associated
with the crystalline oxide fraction decreases and
arsenic(V) associated with the amorphous oxide
fraction increases, dominating at arsenic surface
concentrations above ~30% of maximum
arsenic(V) capacity for red mud. The dissolution
behaviour at different pH of the main components
of red mud involved in the arsenic(V) retention
process indicates that arsenic(V) associated with
the crystalline oxide fraction is less susceptible to
remobilization. Arsenic release from red mud is
in¯uenced by the pH of the release medium and
the arsenic concentration in the sorbent. Arsenic
release is enhanced in strongly alkaline media, but
for high arsenic surface concentrations arsenic
release occurs at acidic pH also, re¯ecting the
changes in the arsenic distribution in the red mud
as surface occupation proceeds. This must be
taken into account when using red mud for the
removal of arsenic(V) from heavily arsenic(V)-
contaminated systems.
References
AltundogÆan, H.S., AltundogÆan, S., TuÈmen, F. and
Bildik, M. (2000) Arsenic removal from aqueous
solutions by adsorption on red mud. Waste
Management, 20, 761ÿ767.
Anawar, H.M., Akai, J. and Sakugawa, H. (2004)
Mobilization of arsenic from subsurface sediments
by effect of bicarbonate ions in groundwater.
Chemosphere, 54, 753ÿ762.
Anderson, M.A., Ferguson, J.F. and Gavis, J. (1976)
Arsenate adsorption on amorphous aluminum hydro-
xide. Journal of Colloid and Interface Science, 54,
391ÿ399.
Apak, R., TuÈtem, E., Mehmet, H. and Hizal, J. (1998)
Heavy metal cation retention by unconventional
sorbents (red muds and ¯y ashes). Water Research,
32, 430ÿ440.
Arias, M., LoÂpez, E., NuÂnÄez, A., Rubinos, D., Soto, B.,
Barral, M.T. and DõÂaz-Fierros, F. (1999) Adsorption
of methylene blue by red mud, an oxide-rich
byproduct of bauxite re®ning. Pp. 361ÿ365 in:
Effect of Mineral-Organic-Microorganism
Interactions on Soil and Freshwater Environments
(J. Berthelin et al., editors). Kluwer Academic/
Plenum Publishers, New York.
Cappuyns, V., Van Herreweghe, S., Swennen, R.,
Ottenburgh, R. and Deckers, J. (2002) Arsenic
pollution at the industrial site of Reppel-Bocholt
(north Belgium). Science of the Total Environment,
295, 217ÿ240.
Daus, B., Wennrich, R. and Weiss, H. (2004) Sorption
materials for arsenic removal from water: a
comparative study.Water Research, 38, 2948ÿ2954.
Driehaus, W., Jekel, M. and Hildebrandt, J. (1998)
Granular ferric hydroxide ÿ a new adsorbent for the
removal of arsenic from natural water. Journal of
Water SRT ÿ Aqua, 47, 30ÿ35.
Fordham, A.W. and Norris, K. (1979) Arsenate-73
uptake by components of several acidic soils and its
implications for phosphate retention. Australian
Journal of Soil Research, 17, 307ÿ316.
Frost, R.R. and Grif®n, A. (1977) Effect of pH on
adsorption of arsenic and selenium from land®ll
leachate by clay minerals. Soil Science Society of
598
D. A. RUBINOS ET AL.

America Journal, 41, 53ÿ56.
GarcõÂa-SaÂnchez, A., AÂ lvarez-Ayuso, E. and RodrõÂguez-
MartõÂn, F. (2002) Sorption of As(V) by some
oxyhydroxides and clay minerals. Application to its
immobilization in two polluted mining soils. Clay
Minerals, 37, 187ÿ194.
GencË, H., Tjell, J.C., McConchie, D. and Schuiling, O.
(2003) Adsorption of arsenate from water using
neutralized red mud. Journal of Colloid and
Interface Science, 264, 327ÿ334.
GencË-Fuhrman, H., Tjell, J.C. and McConchie, D.
(2004a) Adsorption of arsenic from water using
activated neutralized red mud. Environmental
Science and Technology, 38, 2428ÿ2434.
GencË-Fuhrman, H., Tjell, J.C. and McConchie, D.
(2004b) Increasing the arsenate adsorption capacity
of neutralized red mud (Bauxsol). Journal of Colloid
and Interface Science, 271, 313ÿ320.
Glenister, D.J. (1987) Alcoa's experiences with alter-
native techniques for bauxite residue disposal and
the rehabilitation of old residue areas. Pp. 50ÿ70 in:
Tailings Disposal and Management Symposium (N.
Stockon editor). Murdoch University, Western
Australia.
Glenister, D.J. and Thornber, M.R. (1985) Alkalinity of
red mud and its application for the management of
acid wastes. Pp. 109ÿ113 in: Chemeca 85
Symposium Proceedings, Paper A8c.
Goldberg, S. and Johnston, C.T. (2001) Mechanisms of
arsenic adsorption on amorphous oxides evaluated
using macroscopic measurements, vibrational spectro-
scopy, and surface complexation modeling. Journal of
Colloid and Interface Science, 234, 204ÿ216.
Gupta, S.K. and Chen, K.Y. (1978) Arsenic removal by
adsorption. Journal of Water Pollution Control
Federation, 50, 493ÿ506.
Gupta, V.K. and Sharma, S. (2002) Removal of
cadmium and zinc from aqueous solution using red
mud. Environmental Science and Technology, 36,
3612ÿ3617.
International Agency for Research on Cancer (1980)
IARC Monographs on the Evaluation of
Carcinogenic Risks to Humans, Vol. 23, IARC,
Lyon, France.
Jackson, B.P. and Miller, W.P. (2000) Effectiveness of
phosphate and hydroxide for desorption of arsenic
and selenium species from iron oxides. Soil Science
Society of America Journal, 64, 1616ÿ1622.
Lin, T.F. and Wu, J.K. (2001) Adsorption of arsenite and
arsenate within activated alumina grains:
Equilibrium and kinetics. Water Research, 35,
2049ÿ2057.
Livesey, N.T. and Huang, P.M. (1981) Adsorption of
arsenate by soils and its relation to selected chemical
properties and anions. Soil Science, 131, 88ÿ94.
Lombi, E., Wenzel, W.W. and Sletten, R.S. (1999)
Arsenic adsorption by soils and iron-oxide-coated
sand: kinetics and reversibility. Journal of Plant
Nutrition and Soil Science, 162, 451ÿ456.
Lombi, E., Sletten, R.S. and Wenzel, W.W. (2000)
Sequentially extracted arsenic from different size
fractions of contaminated soils. Water, Air and Soil
Pollution, 124, 319ÿ332.
Lombi, E., Zhao, F.J., Zhang, G., Sun, B., Fitz, W.,
Zhang, H. and McGrath, S.P. (2002) In situ ®xation
of metals in soils using bauxite residue: chemical
assessment. Environmental Pollution , 118 ,
435ÿ443.
LoÂpez, E., Soto, B., Arias, M., NuÂnÄez, A., Rubinos, D.
and Barral, M.T. (1998) Adsorbent properties of red
mud and its use for wastewater treatment. Water
Research, 32, 1314ÿ1322.
Mehra, O.P. and Jackson, M.L. (1960) Iron oxide
removal from soils and clays by a dithionite-citrate
system buffered with sodium bicarbonate. Clays and
Clay Minerals, 7, 317ÿ327.
Nickson, R., McArthur, J., Burgess, W., Ahmed, K.M.,
Ravenscroft, P. and Rahman, M. (1998) Arsenic
poisoning of Bangladesh groundwater. Nature, 395,
338.
Nirel, P.M.V. and Morel, F.M.M. (1990) Pitfalls of
sequential extractions. Water Resources, 24,
1055ÿ1056.
Pierce, M.L. and Moore, C.B. (1980) Adsorption of
arsenite and arsenate on amorphous iron hydroxide
from dilute aqueous solutions. Environmental
Science and Technology, 14, 214ÿ216.
Prasad, G. (1994) Removal of arsenic(V) from aqueous
systems by adsorption onto some geological materi-
als. Pp. 133ÿ152 in: Arsenic in The Environment.
Part I: Cycling and Characterization (J.O. Nriagu,
editor). John Wiley & Sons, Inc., New York, USA.
Rubinos, D., Soto, B., Arias, M. and Barral, M.T.
(1998a) AdsorcioÂn de As(V) por lodos rojos de
bauxita (As(V) adsorption by bauxite red mud). V
Congreso Internacional de QuõÂmica de la ANQUE.
Tenerife, Spain. Abstract.
Rubinos, D., Vilaboy, K. and Barral, M.T. (1998b)
Quantitative determination of zeolites in red mud by
a cation-exchange capacity method. In: Analytical
methodology in the environmental ®eld (D. Prada et
al., editors). DiputacioÂn de La CorunÄa, La CorunÄa,
Spain.
Rubinos, D., Arias, M., Barral, M.T. and DõÂaz-Fierros,
F. (2002) Surface properties of red mud as in¯uenced
by arsenate adsorption. XIII Spanish-Italian
Congress on the thermodynamics of metal com-
plexes. Santiago de Compostela, Spain. Abstract.
Schwertmann, U. (1964) Differenzierug der Eisenoxide
des Bodens durch extraktion mit ammonium
oxalaLoÈsunh. Zeitscrift fuÈr P¯anzenernaÈhrung und
Bodenkunde, 105, 194ÿ202.
SPECIATION OF ADSORBED AS(V)
599

Shevade, S. and Ford, R.F. (2004) Use of synthetic
zeolites for arsenate removal from pollutant water.
Water Research, 38, 3197ÿ3204.
Sun, X. and Doner, H.E. (1996) An investigation of
arsenate and arsenite bonding structures on goethite
by FTIR. Soil Science, 161, 865ÿ872.
Wauchope, R.D. (1975) Fixation of arsenical herbicides,
phosphate and arsenate in alluvial soils. Journal of
Environmental Quality, 4, 355ÿ358.
World Health Organization, WHO (1993) WHO
Guidelines for drinking water quality, 2nd
edition.
Geneva, Switzerland.
World Health Organization, WHO (2001) Arsenic and
arsenic compounds. Environmental Health Criteria,
2nd
edition. Geneva, Switzerland.
[Manuscript received 4 November 2005:
revised 4 July 2005]
600
D. A. RUBINOS ET AL.
Top Related
Copyright © 2022 FDOKUMEN

