







Bahasa
Halaman
Hukum


Protein Kinases C-g and -d Are Involved in Insulin-like GrowthFactor I–Induced Migration of Colonic Epithelial Cells
FREDERIC ANDRE, VERONIQUE RIGOT, MARYSE REMACLE–BONNET, JOSE LUIS, GILBERT POMMIER,and JACQUES MARVALDIESA CNRS 6032, Laboratoire de Biochimie Cellulaire, Universites d’Aix-Marseille I et II, Faculte de Pharmacie, Marseille, France
Background & Aims: The mechanisms by which epithe-lial cells migrate during the repair of damaged colonicmucosa are poorly understood. This study investigatedthe insulin-like growth factor I (IGF-I) signaling pathwayleading to HT29-D4 human colonic epithelial cell linemigration. Methods: IGF-stimulated cell migration wasdetermined using a wound model in the presence orabsence of kinase inhibitors. Activation of proteinkinase C (PKC) was determined by immunodetection.Results: IGF-I and insulin induce cell migration withoutaffecting cell proliferation through their cognate recep-tors. Des(1-3)-IGF-I, a truncated analogue of IGF-I, wasmore potent than IGF-I, suggesting that IGF-bindingproteins secreted in the medium modulated IGF-I–induced cell migration. Inhibition of phosphatidylinosi-tol 3-kinase, PKC, and mitogen-activated protein ki-nases eliminated cell restitution. Long-term exposureof cells to phorbol myristate acetate caused the deple-tion of PKC-d and -g and prevented also IGF-I–inducedcell motility. IGF-I also induced activation of PKC-dand -g only. Conclusions: IGF-I stimulates colonic restitu-tion through the activation of multiple signaling pathwaysincluding activation of phosphatidylinositol 3-kinase,PKC-d and -g, and mitogen-activated protein kinases.
The colonic epithelium forms a continuous physicaland functional barrier that protects the internal
environment of the body from the fluctuating externalmilieu. A variety of physiological and pathological factorsare known to disrupt this epithelial barrier, leading to theappearance of wounds due to shedding of surface epithe-lial cells.1 This epithelial barrier has a striking ability torelease superficial wounds to maintain critical intestinalbarrier function and homeostasis. As for other epithelia ofthe gastrointestinal tract, the repair of damaged colonicmucosa is a biphasic process. The first phase of repairinvolves the sloughing of damaged epithelial cells andmigration of remaining viable cells to restore epithelialcontinuity. This process, termed restitution,2 appears tobe independent of proliferation.3 The second phaserequires a proliferative response that restores a normalarchitecture.4 Proliferation of mucosal epithelial cells is
known to be regulated by several peptide growth factors,but the mechanisms involved in epithelial cell restitutionare less understood. Studies of cell restitution are limitedby the difficulty in distinguishing the effects of cellmigration from the secondary regenerative phase of cellproliferation.
To overcome the difficulties inherent to in vivo studies,many in vitro techniques have been developed to betterunderstand some of the fundamental aspects of epithelialrestitution.5 Although the detailed mechanisms of cellrestitution are not understood, it is clear that the dynamicand reciprocal interactions involving cell adhesion mol-ecules, extracellular matrix, and soluble factors are crucialfor this process. Recent studies have shown that extracel-lular matrix components are able to activate intestinalepithelial restitution.6,7 In addition, intestinal epithelialcell migration has been reported to be enhanced byvarious soluble factors including interleukin 2,8 epi-dermal growth factor (EGF),6,9,10 hepatocyte growthfactor/scatter factor,11,12 transforming growth factor b(TGF-b),6,13 and FGF.11
Insulin-like growth factors (IGF-I and IGF-II) aremultifunctional regulatory peptides that share structuralhomology with proinsulin but mediate primarily prolif-erative, differentiative, and survival effects depending onthe target cell and the presence of other hormones andgrowth factors.14 IGFs mediate these effects through cellsurface receptors, mainly the IGF-I receptor (IGF-IR), atyrosine kinase heterotetramer that is 70% homologousto the insulin receptor (IR).15 In addition, biological
Abbreviations used in the paper: BrdU, 5-bromo-28-deoxyuridine;BSA, bovine serum albumin; des(1-3)-IGF-I, NH2-terminally trun-cated insulin-like growth factor I analogue; DMEM, Dulbecco’smodified Eagle medium; EGF, epidermal growth factor; ERK, extracel-lular-regulated protein kinase; IGF, insulin-like growth factor; IGFBP,IGF-binding protein; IGF-IR, IGF-I receptor; IR, insulin receptor;IRS-1, insulin receptor substrate; MAb, monoclonal antibody; MAPK,mitogen-activated protein kinase; PI 3-K, phosphatidylinositol 3-ki-nase; PKC, protein kinase C; PMA, phorbol 12-myristate 13-acetate;TGF, transforming growth factor.
r 1999 by the American Gastroenterological Association0016-5085/99/$10.00
GASTROENTEROLOGY 1999;116:64–77

effects of IGFs are modulated by a family of IGF-bindingproteins (IGFBPs) that control the bioavailability of IGFsto IGF receptors.14 Accumulating evidence indicates thatIGFs are promoters of cell motility for a variety of normaland malignant cell types,16 suggesting a role in neovascu-larization, immune response, and metastatic spread ofmalignant tumor cells. IGF-I and -II have been shown toregulate the balance between proliferation, differentia-tion, and survival of colonic cancer cells in vitro17,18 andthe intestinal adaptability after surgical resection invivo,19,20 suggesting a role of IGFs on intestinal epithelialcell restitution.
To investigate the role of insulin and IGFs on HT29-D4cells, derived from a human colonic adenocarcinoma, weused a migration assay termed monolayer woundingassay. The findings suggest that IGF-I and, to a lesserextent, insulin induce intestinal epithelial cell restitutionthrough their respective receptors. IGF-I–induced cellmigration was independent of cell proliferation. Usingvarious kinase inhibitors we demonstrated that activationof tyrosine phosphorylation, phosphatidylinositol 3-ki-nase (PI 3-K), protein kinase C (PKC) isoforms, andmitogen-activated protein kinase (MAPK) cascade isrequired for IGF-I signaling pathway leading to epithe-lial cell migration. Interestingly, IGF-I caused subcellu-lar relocalization of PKC-g and -d specifically, suggestingtheir implication in the IGF-I–induced migratory pro-cess.
Materials and Methods
ReagentsFetal bovine serum was obtained from Sera-Lab (Craw-
ley Down, England); Dulbecco’s modified Eagle medium(DMEM) from GIBCO (Cergy-Pontoise, France); and Dulbec-co’s phosphate-buffered saline (PBS) from Oxoid (Basingstoke,England). Calphostin C, EGF, 4,5,7-trihydroxyisoflavone (genis-tein), wortmannin, bovine serum albumin (BSA), bovineinsulin, horseradish peroxidase–conjugated protein A, wheatgerm agglutinin, LY292004, tetramethyl rhodamine isothio-cyanate (TRITC)-conjugated wheat germ agglutinin, andTRITC-conjugated phalloidin were purchased from Sigma (LaVerpilliere, France). Bisindolylmaleimide GF109203X and5-bromo-28-deoxyuridine (BrdU) labeling and detection kitwere from Boehringer Mannheim (Le Meylan, France); phorbol12-myristate 13-acetate (PMA) and 4 a-phorbol 12-myristate13-acetate (4aPMA) from ICN Biomedical (Orsay, France);and des(1-3)-IGF-I and des(1-6)-IGF-II from Gropep (Ad-elaide, Australia). Recombinant IGF-I and IGF-II were fromBachem (Bubbendorf, Switzerland). 125I-IGF-II was purchasedfrom Amersham (Les Ulis, France). Rapamycin and 28-amino-38-methoxyflavone (PD98059) were from Alexis Biochemicals(San Diego, CA).
Antibodies
Rabbit polyclonal antibody against IR (B6) was ob-tained from Immunotech (Marseille, France) and mouse mono-clonal antibody (MAb) against IGF-IR (aIR3) from OncogeneScience (Uniondale, NY). Rabbit polyclonal antibodies againstIGFBP-2 and IGFBP-6 were from Upstate Biotechnology(Lake Placid, NY). Mouse isozyme-specific anti-PKC (-a,-bI/bII, -g, -e, -d, -µ, -l, -i, and -z) MAbs were fromTransduction Laboratories (Lexington, KY); rabbit anti–PKC-h from Santa Cruz Biotechnologies (Santa Cruz, CA); andrabbit anti-active MAPK from Promega (Madison, WI).Horseradish peroxidase–conjugated sheep anti-mouse immuno-globulin (Ig) was purchased from Amersham. Sheep anti-mouse and anti-rat fluorescein isothiocyanate–conjugated anti-bodies were obtained from Sigma.
Cell Migration
The human colonic adenocarcinoma HT29-D4 cell linewas routinely cultured in DMEM containing 10% fetal calfserum as described previously.21 Culture medium was renewedevery 2 days, and cells were used between passages 10 and 25.Wounding assays were essentially performed according toBurk22 with slight modifications. Briefly, cells were seeded into35-mm diameter 6-well plates at 1.5 3 105 cells/cm2 andallowed to grow to confluency during 4 days. Confluent cellmonolayers were wounded by pressing a sterilized razor bladedown onto the plate to cut the cell sheet and to mark on theplate a sharp visible demarcation at the wound edge. The bladewas then gently moved to one side to remove part of the cellmonolayer sheet. Two approximately 15–20-mm wounds sepa-rated by about 10 mm were made in the same well. Thewounded monolayers were then washed 4 times in DMEM toremove cell debris and incubated for 24 hours at 37°C inserum-free DMEM containing 0.1% BSA with or without testsubstances. At the end of incubation, cells were rinsed twicewith PBS, fixed in 3% paraformaldehyde in PBS for 20minutes at 4°C, and stained using the May–Grunwald/Giemsamethod. Migration of HT29-D4 cells was assessed using aninverted Zeiss MI microscope by numbering the cell nucleiobserved across the wound borders. To avoid observer bias, 10microscopic fields were analyzed for each wound in a blindedfashion. Migration results are expressed as the average 6 SD ofthe number of migrating cells per microscopic field (magnifica-tion, 3203). All data presented in Results are from at least 3independent experiments performed in duplicate. In someexperiments, kinase inhibitors were added 2 hours beforewounding and during the time of cell migration. None of thesecomponents affected cell viability, as verified by the trypan blueexclusion test. When required, antiinsulin or anti–type I IGF-Ireceptor MAbs were added to the cells 90 minutes before theaddition of des(1-3)-IGF-I or PMA. To distinguish betweenenhanced restitution by migration and proliferation, HT29-D4cells were treated by 5 µg/mL mitomycin C for 2 hours beforeperforming wounding assays and during the 24-hour incuba-tion with test substances. Preliminary experiments showed thatincubation of HT29-D4 cells with 5 µg/mL mitomycin C for
January 1999 IGF-I–INDUCED CELL MIGRATION REQUIRES PKCs 65

24 hours completely inhibited proliferation without affectingcell viability.
Cell Proliferation Assay
Confluent monolayers were wounded as describedabove, washed extensively, and incubated in DMEM/0.1% BSAsupplemented or not with 7 nmol/L des(1-3)-IGF-I. After anincubation period of 22 hours, 10 µmol/L BrdU was added andincubation continued for an additional 2-hour period. Afterwashing the wounded monolayers in PBS, cells were fixed in70% ethanol (in 50 mmol/L glycine buffer, pH 2.0) for 40minutes at 220°C. Cells that have incorporated BrdU intoDNA were detected using the BrdU labeling and detection kitaccording to the manufacturer’s instructions. Cell proliferationin wounded monolayers was visualized by light microscopyusing an inverted Zeiss MI microscope.
PKC Detection
Confluent HT29-D4 cells were incubated in DMEM/0.1% BSA for 24 hours with or without 100 nmol/L PMA or 7nmol/L des(1-3)-IGF-I. Cells were washed twice with PBS,then scrapped into boiling lysis buffer containing 63 mmol/LTris-HCl, pH 6.8, 10% glycerol, 4% sodium dodecyl sulfate(SDS), 1 mmol/L sodium orthovanadate, and a mixture ofprotease inhibitors (1 mmol/L iodoacetamide, 1 mmol/Lphenylmethyl sulfonyl fluoride, 500 U/mL aprotinin, 1 µg/mLleupeptin, 1 µmol/L pepstatin, and 1 mmol/L orthophenanthro-lin). After sonication to fragment DNA, lysates were cleared bycentrifugation at 15,000g for 15 minutes. Proteins fromsupernatants were mixed with Laemmli sample buffer contain-ing 1 mmol/L dithiothreitol and boiled for 5 minutes.
When required, soluble and cytoskeletal fractions wereprepared as described by Hinck et al.23 Cells treated for 90minutes with or without 7 nmol/L des(1-3)-IGF-I or 100nmol/L PMA were rinsed twice with PBS and homogenized for10 minutes at 4°C with gentle rocking in 50 mmol/L NaCl, 10mmol/L PIPES (pH 6.8), 3 mmol/L MgCl2, 0.5% TritonX-100, and 300 mmol/L sucrose supplemented with themixture of protease inhibitors. After centrifugation at 15,000gfor 10 minutes at 4°C, the supernatant was considered as theTriton-soluble fraction. The pellet was triturated in the samevolume of 20 mmol/L Tris (pH 7.5), 5 mmol/L EDTA, 2.5mmol/L EGTA, and 1% SDS and boiled at 100°C for 5minutes. After centrifugation for 10 minutes at 15,000g, thesupernatant was considered as the Triton-insoluble fraction.The Triton-soluble fractions usually contained 2-fold moreproteins than the Triton-insoluble ones, as determined with theMicro BCA protein assay (Pierce, Rockford, IL). This ratio wasnot significantly altered under any of the conditions studied.We therefore analyzed equivalent amounts of Triton-solubleand Triton-insoluble fractions.
Proteins were resolved by SDS–polyacrylamide gel electro-phoresis (PAGE), then transferred onto a nitrocellulose sheet(Hybond-C extra; Amersham) for 1 hour at 100 V in 20mmol/L Tris-HCl buffer (pH 8.0) containing 20% methanolplus 150 mmol/L glycine. Nonspecific protein binding sites
were blocked by an overnight incubation in PBS containing5% nonfat dried milk. Immunodetection was carried out byincubating the nitrocellulose sheet for 3 hours with isozyme-specific anti-PKC mouse MAbs (PKC-a, -bI/-bII, and -z, 0.5µg/mL; PKC-d, -g, -µ, -i, -l, and -u, 1 µg/mL) or with a 1/500dilution of rabbit anti–PKC-h polyclonal antibody. Blots werethen incubated with the appropriate horseradish peroxidase–conjugated secondary antibodies. Immunoreactive proteinswere visualized using an enhanced chemiluminescence system(Amersham). All dilutions and washing steps were performedin PBS containing 5% nonfat dried milk and 0.2% Tween 20.
Immunoblots were quantitated by scanning autoradiographswith a Sharp JX-325 laser densitometer, and absorbance curveswere integrated using the ImageMaster software (PharmaciaBiotech, Saint Quentin en Yvelines, France).
Detection of Activated MAPK
Confluent HT29-D4 cells were incubated for 2 hoursin DMEM/0.1% BSA alone or with 7 nmol/L des(1-3)-IGF-I,10 nmol/L EGF, or 100 nmol/L PMA; washed twice with PBS;and scrapped into boiling 63 mmol/L Tris-HCl buffer (pH 6.8)containing 10% glycerol, 4% SDS, 1 mmol/L orthovanadate,and the mixture of proteinase inhibitors. After sonication tofragment DNA, lysates were clarified by centrifugation at15,000g for 15 minutes. Extracted proteins (60 µg) wereseparated by 10% acrylamide SDS-PAGE and electrotrans-ferred onto a nitrocellulose sheet. After saturation for 2 hours atroom temperature with 50 mmol/L Tris-HCl (pH 7.4) plus 150mmol/L NaCl (TBS) containing 1% BSA, the nitrocellulosesheet was incubated overnight with anti-active MAPK poly-clonal antibodies in TBS containing 0.05% Tween 20 and0.1% BSA. Bound antibodies were detected by horseradishperoxidase–conjugated antibodies to rabbit Igs and revealedwith the light-based enhanced chemiluminescence system.
Immunocytochemistry
Confluent cell monolayers cultured on glass coverslipswere wounded with an asepticized plastic toothpick andwashed extensively with DMEM. Cells were then incubated inserum-free medium with or without des(1-3)-IGF-I (7 nmol/L)or PMA (100 nmol/L) for 2 hours. After 3 rinses with PBS, cellswere fixed in 3% formaldehyde for 20 minutes at 4°C, washed3 times in PBS, and incubated for 10 minutes in 50 mmol/LNH4Cl in PBS to remove fixative. Cells were permeabilizedwith acetone at 220°C for 5 minutes, air-dried, rehydrated inPBS, and incubated for 5 minutes in PBS containing 4% BSAto minimize nonspecific labeling. All following immunocyto-chemical reactions were performed in a moist chamber at roomtemperature in PBS containing 4% BSA. Cells were incubatedwith anti-PKC mouse MAbs (10 µg/mL) for 3 hours, washed 4times for 5 minutes, and incubated with fluorescein isothiocya-nate–conjugated sheep Igs directed against mouse Igs (1/50)for 45 minutes. After 4 washes, cells were mounted inglycerol/PBS, 9/1 (vol/vol), containing 1 mg/mL phenylenedi-amine and applied to a glass slide. Cells were examined with anOlympus BH-2 photomicroscope. The specificity of the label-
66 ANDRE ET AL. GASTROENTEROLOGY Vol. 116, No. 1

ing was verified by the absence of staining with the secondaryantibody alone.
IGFBP Analysis
Confluent cell monolayers were incubated for 24 hoursin serum-free medium containing 0.005% BSA in the absenceor presence of 14 nmol/L des(1-3)-IGF-I, 14 nmol/L IGF-I, or100 nmol/L PMA. Conditioned media were collected, centri-fuged to eliminate cellular debris, and stored at 220°C untiluse. IGFBPs in conditioned media were analyzed by Westernligand blotting24 using 125I-IGF-II as a probe and by Westernimmunoblotting using anti–IGFBP-2 and anti–IGFBP-6 anti-bodies and an enhanced chemiluminescence system to visualizeimmunoreactive proteins, as described previously.25
Results
IGF-I and Insulin Induce HT29-D4Cell Migration
Figure 1 shows photographs of HT29-D4 cellrestitution after monolayer wounding. When cell mono-layer was wounded and incubated for 24 hours inserum-free medium alone, only a few cells crossed thestarting line marked on the plate (average of 5 cells perfield), indicating that HT29-D4 cells did not migratewithout exogenous stimulus (Figure 1A). Similarly, noepithelial cell restitution was observed when 10% fetalcalf serum was added (data not shown). Because mol-ecules of the insulin family have been shown to influence
migration of various cell types, we investigated whethercell restitution could be induced by insulin, IGF-I, orIGF-II. Addition of IGF-I (7 nmol/L) and, to a lesserextent, IGF-II (7 nmol/L) or insulin (7 nmol/L) toserum-deprived medium induced significant migrationof HT29-D4 cells into the denuded area (Figure 1B, C,and D, respectively). The phenomenon was observed asearly as 5 hours after the injury in the presence of thepeptides. Cells that migrated into the wounded areaappeared perpendicularly oriented to the original woundedge and migrated as a linked sheet to resurface theepithelial defect.
To better characterize this inducible epithelial cellrestitution, we next quantified the cell migration process.Monolayers were wounded and incubated in serum-freemedium containing peptide concentrations ranging from0.01 to 100 nmol/L. Cells migrating across the woundborder were counted after incubation for 24 hours at37°C. As shown in Figure 2A, IGF-I induced HT29-D4cell migration in a dose-dependent manner with about a9-fold stimulation at 100 nmol/L compared with cellscultured in serum-free medium alone (45 vs. 5 migratingcells per field). Insulin also increased the rate of migrationof HT29-D4 cells, but was less potent than IGF-I at eachconcentration tested.
HT29-D4 cells synthesize and secrete several IGFBPs.26
Because IGFBPs modulate IGFs’ bioavailability and,
Figure 1. Cell migration in an in vitro model of epithelial restitution. Wounds were made with a razor blade on confluent monolayers of HT29-D4cells as described in Materials and Methods. After washing with DMEM, monolayers were cultured for 24 hours in serum-free medium in the (A)absence or (B) presence of 7 nmol/L IGF-I, (C) IGF-II, or (D) insulin. Arrowheads indicate the edges of the wounds (bar 5 20 µm).
January 1999 IGF-I–INDUCED CELL MIGRATION REQUIRES PKCs 67

hence, biological effects,14 we next studied the responseof HT29-D4 cells to des(1-3)-IGF-I, an IGF-I truncatedanalogue that binds with high affinity to the IGF-IR butnot to IGFBPs. As shown in Figure 2A, des(1-3)-IGF-Iinduced a dose-dependent migration of HT29-D4 cells.This effect could be observed for concentration as low as0.01 nmol/L and reached a plateau for a concentration of7 nmol/L. Induced des(1-3)-IGF-I cell migration wasalways higher than that observed using IGF-I, suggestingthat secreted IGFBPs partially inhibited IGF-I–stimu-lated cell migration. This result was further confirmed bycomparing the effect of IGF-II to its NH2-terminallytruncated analogue, des(1-6)-IGF-II (Figure 2B). Des(1-6)-IGF-II was also more potent than its natural counter-part in increasing the rate of HT29-D4 cell motility. Forsubsequent experiments, des(1-3)-IGF-I was thereforeused at a concentration of 7 nmol/L, i.e., the concentra-
tion inducing the maximal stimulation of HT29-D4 cellmigration.
To delineate whether enhancement of cell restitutionwas directly mediated through IR and/or IGF-IR, wepreincubated wounded monolayers for 90 minutes with30 µg/mL aIR3 or B6, two blocking antibodies raisedagainst the IGF-IR and IR, respectively. Cells were thenincubated for 24 hours with 7 nmol/L of either des(1-3)-IGF-I or insulin, a concentration that only stimulates thecognate receptor. As shown in Figure 3, aIR-3 antibodydramatically reduced the HT29-D4 cell migratory re-sponse to IGF-I, but had no effect on the migratoryresponse to insulin. Conversely, B6 antibody inhibitedthe migratory response to insulin only. These resultsclearly indicate that the effects of insulin and IGF-I oncell restitution were mediated by their respective recep-tors.
IGF-I–Induced Cell Migration IsIndependent of Cell Proliferation
IGF-I has been shown to induce proliferation ofseveral cell types. To determine whether restitutionobserved in response to IGF-I was caused by an increasein cell migration or cell proliferation, HT29-D4 cellswere treated with mitomycin C, both before performingwounding assays and during migration. As shown inFigure 4A, cell restitution was stimulated by des(1-3)-IGF-I in a dose-dependent manner with mitomycinC–treated or untreated cells. Further proof that enhance-ment of restitution is not caused by epithelial cellproliferation was obtained by detecting cells that have
Figure 2. Effects of insulin, IGFs, and their truncated analogues oncell restitution. (A) Monolayers were wounded and incubated for 24hours in serum-free medium in the presence of insulin (j), IGF-I (s), ordes(1-3)-IGF-I (h) in concentrations ranging from 0.01 to 100 nmol/L.Cells were fixed and stained, and migration of HT29-D4 cells wasassessed as described in Materials and Methods. Bars representmean 6 SD (n 5 4 duplicate assays). (B) Wounded monolayers wereincubated for 24 hours in serum-free medium containing 7 nmol/LIGF-I, IGF-II, or their truncated analogues des(1-3)-IGF-I and des(1-6)-IGF-II, respectively.
Figure 3. Insulin- and IGF-I–induced cell restitution is mediatedthrough their own receptors. Cell monolayers were wounded andtreated for 90 minutes with or without 30 µg/mL aIR3 or B6antibodies. Cells were then incubated for 24 hours with 7 nmol/Ldes(1-3)-IGF-I or 7 nmol/L insulin, fixed, and counted. Bars representmean 6 SD (n 5 2 duplicate assays).
68 ANDRE ET AL. GASTROENTEROLOGY Vol. 116, No. 1

incorporated BrdU into DNA. As shown in Figure 4B,few nuclei of migrating cells exhibited uptake of BrdU.Moreover, no significant difference in the labeling patternof monolayers, treated or untreated with des(1-3)-IGF-I,
was observed. Taken together, these findings suggest thatthe effect of IGF-I on HT29-D4 cell restitution isindependent of cell proliferation.
PKCs Are Involved indes(1-3)-IGF-I–Induced Cell Migration
In wounding assays, PMA was also able to inducecell migration (Figure 5A). We next tested the impact of2 PKC inhibitors, calphostin C and bisindolylmaleimide,
Figure 4. Des(1-3)-IGF-I–induced cell migration is independent of cellproliferation. (A) Cell monolayers were incubated in the absence (O)or presence (h) of 5 µg/mL mitomycin C for 2 hours. Monolayers werethen wounded and cultured for an additional 24-hour period with orwithout 5 µg/mL mitomycin C and various concentrations of des(1-3)-IGF-I. Cells were fixed and stained, and migrating cells were counted.Bars represent mean 6 SD (n 5 4 duplicate assays). (B and C)Confluent monolayers were wounded and incubated in (B) DMEM/0.1% BSA alone or (C) with 7 nmol/L des(1-3)-IGF-I. After 22 hours, 10µmol/L BrdU was added and incubation continued for an additional 2hours. Cells that have incorporated BrdU into DNA were detected asdescribed in Materials and Methods (bar 5 10 µm). Arrowheadsindicate the edge of the wounds.
Figure 5. PKCs are involved in des(1-3)-IGF-I–induced migration. (A)Monolayers were wounded and incubated for 90 minutes in theabsence or presence of 10 µmol/L calphostin C (lane 2) or 10 µmol/Lbisindolylmaleimide (lane 3). Wounded monolayers were incubated foran additional 24-hour period with or without PKC inhibitors and in theabsence (Ctrl) or presence of 100 nmol/L PMA or 7 nmol/L des(1-3)-IGF-I. Migrating cells were counted as described before. Bars repre-sent mean 6 SD (n 5 3 duplicate assays). (B) Effect of PKCdown-regulation on PMA- and des(1-3)-IGF-I–stimulated migration ofHT29-D4 cells. Cell monolayers were pretreated with 100 nmol/L PMAor its inactive isomere 4aPMA for 24 hours, then wounded andwashed extensively. Pretreated and untreated control cell monolayerswere further incubated for 24 hours with no addition (lane 1), 100nmol/L PMA (lane 2), or 7 nmol/L des(1-3)-IGF-I (lane 3). Migratingcells were counted as described before. Bars represent mean 6 SD(n 5 3 duplicate assays).
January 1999 IGF-I–INDUCED CELL MIGRATION REQUIRES PKCs 69

on des(1-3)-IGF-I– and PMA-induced cell migration.Wounded monolayers were pretreated for 90 minuteswith these 2 PKC inhibitors and exposed to 7 nmol/Ldes(1-3)-IGF-I or 100 nmol/L PMA for 24 hours in thepresence of PKC inhibitors. As expected, both inhibitorstotally inhibited PMA-induced cell migration. However,they also prevented des(1-3)-IGF-I–induced cell migra-tion (Figure 5A). These results suggested that PKCs areinvolved in the IGF-I pathway leading to HT29-D4 cellmigration. To provide additional evidence that IGF-I–induced cell motility occurred as a consequence of PKCactivation, we down-regulated PKC by preincubation ofHT29-D4 cells for 24 hours with 100 nmol/L PMA.Control or PKC down-regulated cells were wounded andsubsequently allowed to migrate for 24 hours in thepresence of 7 nmol/L des(1-3)-IGF-I or 100 nmol/LPMA. Figure 5B clearly indicates that on prolongedexposure to PMA, HT29-D4 cells were no more able tomigrate in the presence of PMA or IGF-I. Moreover,long-term exposure to 4aPMA, an inactive isomer ofPMA, did not influence IGF-I– and PMA-induced cellmigration.
IGF-I as Well as PMA InduceIGFBP-6 Secretion
The implication of PKCs in IGF-I–inducedHT29-D4 cell migration incited us to determine whetherthese kinases could be involved in other IGF-I–inducedbiological effects. Many studies in numerous cell modelshave reported that IGF-I is able to modulate IGFBPsecretion.14 We therefore determined whether IGF-I andPMA could modify the rate of IGFBPs’ secretion byHT29-D4 cells. Western ligand blot revealed the pres-ence of several molecular species of IGFBP in conditionedmedia from HT29-D4 cells (Figure 6A). The 24–26-kilodalton and 30-kilodalton molecular forms correspondto 3 IGFBP-4 isoforms, as reported previously by Culous-cou and Shoyab.27 Neither IGF-I, des(1-3)-IGF-I, norPMA seemed to alter IGFBP-4 level in conditionedmedia. Interestingly, all these compounds significantlyincreased the level of a broad diffuse 32–38-kilodaltonband corresponding to both IGFBP-2 (34 kilodaltons)and IGFBP-6 (32–38 kilodaltons) that superimposed onWestern ligand blot.25 To determine whether this in-crease reflected an increase in IGFBP-2, -6, or both, weperformed Western immunoblots. As shown in Figure 6Band C, IGF-I, des(1-3)-IGF-I, and PMA caused a potentincrease in secretion of IGFBP-6 but not of IGFBP-2.Taken together, these data suggest that PKCs are in-volved in the IGF-I signaling pathway.
PKC-d and -g Are Involved in theIGF-I–Induced Cell Migration
Because IGF-I–induced HT29-D4 cell migrationrequires activation of PKCs, we wanted to determinewhich PKC isozymes are responsible for inducing thisphenomenon. We first determined the PKC isozymesexpressed in HT29-D4 cells by Western blotting withisozyme-specific antibodies (Figure 7A). Using whole-cell lysate, we detected 6 PKC isozymes: PKC-a, -g, -d,-e, -i/l, and -µ. In contrast, PKC-bI/-bII, -u, -h, and -z(Figure 7A) did not appear to be expressed at a significantlevel. The molecular-weight values observed were consis-tent with reported data concerning the molecular weightsfor PKC isozymes. Interestingly, long-term incubation(24 hours) with 100 nmol/L PMA induced the loss of
Figure 6. (A) Western ligand blot and (B and C) Western immunoblotanalysis of IGFBPs secreted by HT29-D4 cells. Cell monolayers wereincubated for 24 hours in serum-free medium in the absence (lane 1)or presence (lane 2) of 100 nmol/L PMA, 14 nmol/L IGF-I (lane 3), or14 nmol/L des(1-3)-IGF-I (lane 4). Samples of conditioned media wereconcentrated 50-fold and submitted to electrophoresis on 12.5%SDS-polyacrylamide gels in the absence of reducing agents andtransferred to nitrocellulose sheets. IGFBPs were probed with either(A) 125I-IGF-II, (B) polyclonal antiserum to IGFBP-6, or (C) polyclonalantiserum to IGFBP-2, and IGFBPs were visualized by autoradiographyand enhanced chemiluminescence, respectively. The apparent molecu-lar weights (kilodaltons) of IGFBPs are indicated on the left.
70 ANDRE ET AL. GASTROENTEROLOGY Vol. 116, No. 1

both PKC-d and -g, but did not significantly affect theexpression of the other PKC isoforms in HT29-D4 celllysates (Figure 7B). This result indicates that bothPKC-d and -g isoforms were depleted on long-term PMAtreatment.
That long-term exposure of cells to PMA inhibitedIGF-I–induced migration and depleted PKC-g and -dprompted us to identify which of the expressed PKCswere involved in the IGF-I signaling pathway. A maincharacteristic of PKCs in many cell types is theirsubcellular redistribution within the cell on activation.To test such a possibility, Triton-soluble and Triton-insoluble (cytoskeletal) fractions of HT29-D4 cells treatedor untreated for 90 minutes with 7 nmol/L des(1-3)-IGF-I or 100 nmol/L PMA were prepared, and therelative distribution of PKC isozymes among the frac-tions was determined by Western blotting. As observedin Figure 8, in control cells PKC-d was mostly found inthe cytoskeletal fraction, whereas PKC-a, -g, and -e were
mainly detected in the Triton-soluble one. After incuba-tion with 7 nmol/L des(1-3)-IGF-I for 90 minutes,PKC-d level decreased in the cytoskeleton fraction andincreased in the Triton-soluble one. Scanning of theautoradiograms showed that 79% and 21% of PKC-dwere detected in the Triton-insoluble fraction and in theTriton-soluble fractions, respectively. Regarding PKC-g,des(1-3)-IGF-I also induced a significant reduction inTriton-soluble fraction with a corresponding increase inthe cytoskeletal fraction. Forty-seven percent of thePKC-g remained associated in the Triton-soluble frac-tion, whereas 53% was found in the cytoskeletal fraction.Interestingly, des(1-3)-IGF-I did not induce translocationto the cytoskeletal fraction of both PKC-a and -e. ForPKC-d and -g, PMA exerted the same effects as des(1-3)-IGF-I but also caused the translocation of PKC-a.
To further confirm the translocation of these PKCs, weused immunofluorescence labeling with PKC isozymeantisera after treatment of cells by des(1-3)-IGF-I orPMA. When control untreated cells were wounded,anti–PKC-d MAb stained cytoskeleton actin filaments(Figure 9A), because the same staining pattern was foundin cells labeled with TRITC-conjugated phalloidin (datanot shown). On the other hand, staining with anti–PKC-g MAb revealed that this enzyme was located incap-like perinuclear structures, becoming weaker towardthe peripheral cytoplasm (Figure 9B). A conformity ofstaining patterns was found in cells labeled with anti–
Figure 7. Expression of PKC isoforms in HT29-D4 cells. Cells wereincubated for 24 hours in DMEM/0.1% BSA (A) alone or (B) with 100nmol/L PMA. Total lysates extracted from HT29-D4 cells were submit-ted to electrophoresis on 10% SDS-polyacrylamide gel and transferredonto nitrocellulose sheets. PKCs were detected with isozyme-specificantibodies and visualized using enhanced chemiluminescence. cPKC,conventional PKC isoforms; nPKC, novel PKC isoforms; aPKC, atypicalPKC isoforms. Numbers on the left-hand side refer to the migrationposition of molecular-weight standards (kilodaltons).
Figure 8. Des(1-3)-IGF-I induces translocation of PKC-d and -g. HT29-D4cells were incubated for 90 minutes in DMEM/0.1% BSA alone or with7 nmol/L des(1-3)-IGF-I or 100 nmol/L PMA. Triton-soluble (S) andTriton-insoluble (I) fractions were prepared as described in Materialsand Methods. PKC-a, -g, -d, and -e were detected as described inFigure 7.
January 1999 IGF-I–INDUCED CELL MIGRATION REQUIRES PKCs 71
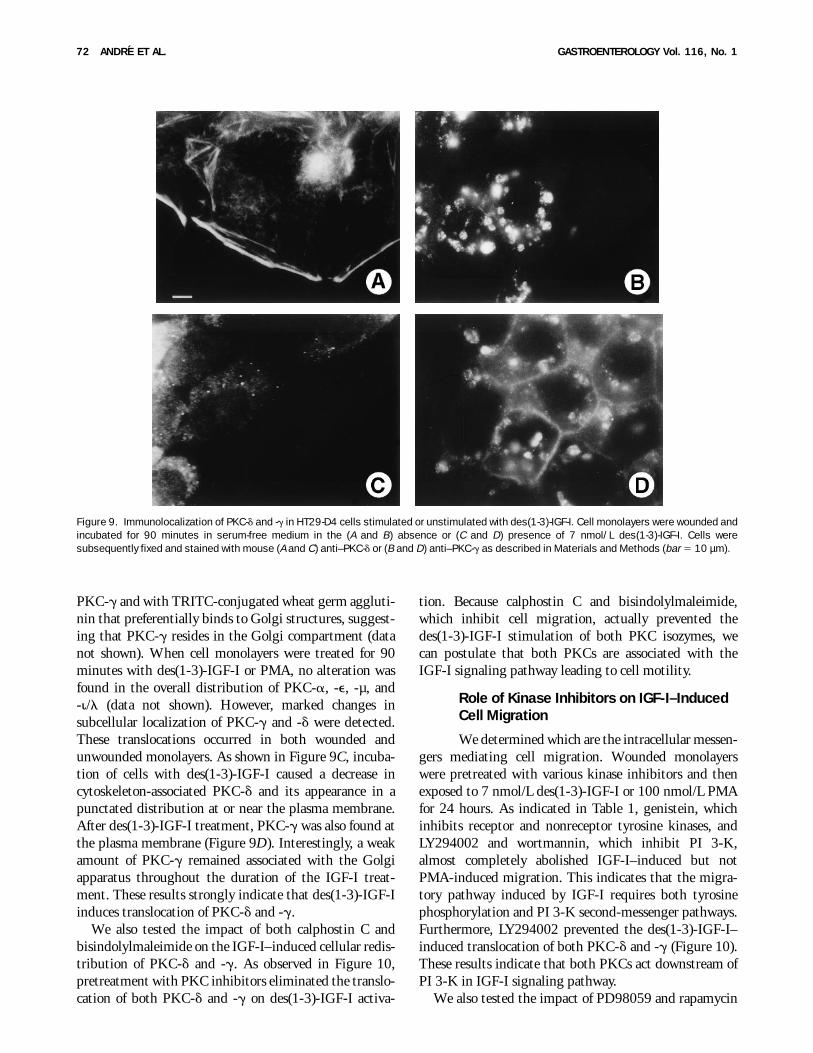
PKC-g and with TRITC-conjugated wheat germ aggluti-nin that preferentially binds to Golgi structures, suggest-ing that PKC-g resides in the Golgi compartment (datanot shown). When cell monolayers were treated for 90minutes with des(1-3)-IGF-I or PMA, no alteration wasfound in the overall distribution of PKC-a, -e, -µ, and-i/l (data not shown). However, marked changes insubcellular localization of PKC-g and -d were detected.These translocations occurred in both wounded andunwounded monolayers. As shown in Figure 9C, incuba-tion of cells with des(1-3)-IGF-I caused a decrease incytoskeleton-associated PKC-d and its appearance in apunctated distribution at or near the plasma membrane.After des(1-3)-IGF-I treatment, PKC-g was also found atthe plasma membrane (Figure 9D). Interestingly, a weakamount of PKC-g remained associated with the Golgiapparatus throughout the duration of the IGF-I treat-ment. These results strongly indicate that des(1-3)-IGF-Iinduces translocation of PKC-d and -g.
We also tested the impact of both calphostin C andbisindolylmaleimide on the IGF-I–induced cellular redis-tribution of PKC-d and -g. As observed in Figure 10,pretreatment with PKC inhibitors eliminated the translo-cation of both PKC-d and -g on des(1-3)-IGF-I activa-
tion. Because calphostin C and bisindolylmaleimide,which inhibit cell migration, actually prevented thedes(1-3)-IGF-I stimulation of both PKC isozymes, wecan postulate that both PKCs are associated with theIGF-I signaling pathway leading to cell motility.
Role of Kinase Inhibitors on IGF-I–InducedCell Migration
We determined which are the intracellular messen-gers mediating cell migration. Wounded monolayerswere pretreated with various kinase inhibitors and thenexposed to 7 nmol/L des(1-3)-IGF-I or 100 nmol/L PMAfor 24 hours. As indicated in Table 1, genistein, whichinhibits receptor and nonreceptor tyrosine kinases, andLY294002 and wortmannin, which inhibit PI 3-K,almost completely abolished IGF-I–induced but notPMA-induced migration. This indicates that the migra-tory pathway induced by IGF-I requires both tyrosinephosphorylation and PI 3-K second-messenger pathways.Furthermore, LY294002 prevented the des(1-3)-IGF-I–induced translocation of both PKC-d and -g (Figure 10).These results indicate that both PKCs act downstream ofPI 3-K in IGF-I signaling pathway.
We also tested the impact of PD98059 and rapamycin
Figure 9. Immunolocalization of PKC-d and -g in HT29-D4 cells stimulated or unstimulated with des(1-3)-IGF-I. Cell monolayers were wounded andincubated for 90 minutes in serum-free medium in the (A and B) absence or (C and D) presence of 7 nmol/L des(1-3)-IGF-I. Cells weresubsequently fixed and stained with mouse (A and C) anti–PKC-d or (B and D) anti–PKC-g as described in Materials and Methods (bar 5 10 µm).
72 ANDRE ET AL. GASTROENTEROLOGY Vol. 116, No. 1

on induced restitution. These drugs inhibit MAP/extracellular-regulated protein kinase (ERK) and pp70S6 kinase (pp70S6K), respectively. As shown in Table 1,rapamycin failed to prevent both des(1-3)-IGF-I– orPMA-induced cell restitution, suggesting that the effectof IGF-I on cell migration is not mediated by pp70S6K.On the other hand, PD98059 dramatically inhibitedboth PMA- and des(1-3)-IGF-I–induced cell restitution.However, PD98059 did not affect the des(1-3)-IGF-I–induced translocation of both PKC-d and -g (Figure 10),suggesting that MAPKs are activated downstream ofPKCs. To test such a possibility, we analyzed MAPKactivation in response to des(1-3)-IGF-I. Incubation with7 nmol/L des(1-3)-IGF-I, 10 nmol/L EGF, or 100 nmol/LPMA resulted in a rapid activation of ERK1 and ERK2(Figure 11; lanes 2, 7, and 10, respectively) which can becompletely blocked by incubating the cells with PD98059(lanes 6, 9, and 13). Interestingly, LY294002 preventedMAPK activation on IGF-I stimulation (lane 3). Thisconfirms that activation of PI 3-K is required in theIGF-I signaling pathway. Importantly, PKC inhibitorspotently inhibited (75%–80% by scanning densitom-etry) IGF-I–induced ERK1 activation (lanes 4 and 5 vs.lane 2). However, PKC inhibitors weakly affected IGF-I–induced ERK2 activation (20% by scanning densitom-etry). The same effects were observed when PMA wasused instead of IGF-I (lane 12 vs. lane 10). Together theseresults indicate that MAPK pathway, namely, the activa-tion of ERK1, was downstream of PKCs in the IGF-Isignaling pathway leading to cell motility. To excludethat this inhibition of MAPK was a nonspecific effect ofthe PKC inhibitors, we tested their impact on theEGF-induced MAPK activation. Reports have shownthat EGF activates MAPK through the ras/raf activation
pathways, via a PKC-independent process.28 As expected,PKC inhibitors did not prevent EGF-induced ERK1activation (lane 8).
Discussion
Cell restitution has been observed after a variety ofmucosal injuries in both upper and lower gastrointestinaltract. Although the detailed mechanisms of cell restitu-tion are not yet understood, studies using in vitro modelshave shown that soluble cytokines and growth factors arecritical for this process.1 IGFs and IGFBPs are producedby most cell types,14 including gastrointestinal cells,18
and function locally via autocrine/paracrine regulatoryloops. Because IGF-IR is widely distributed in thegastrointestinal tract, the IGF system is thought toconstitute an important modulator of the basic functionsof the cells lining the gastrointestinal tract.17 However,no information is currently available on the effects ofIGFs in intestinal epithelial cell restitution.
In the present study, we showed that IGF-I promotes
Figure 10. Effect of kinase inhibitors on PKC-g and -d activities.Subconfluent monolayers were incubated for 90 minutes with orwithout kinase inhibitors: 5 µmol/L LY294002, 10 µmol/L calphostinC (calph C), 10 µmol/L bisindolylmaleimide (BIM), or 10 µmol/LPD98059. Cells were then incubated for 90 minutes with or without 7nmol/L des(1-3)-IGF-I. Triton-soluble (S) and Triton-insoluble (I) frac-tions were prepared as described in Materials and Methods. (A) PKC-gand (B) PKC-d were detected as described in Figure 7.
Table 1. Effect of Kinase Inhibitors on Agonist-InducedMigration
InhibitorNo
treatment Des(1-3)-IGF-I PMA
No inhibitor 6 6 2 60 6 4 87 6 4Genistein (50 µmol/L) 8 6 2 16 6 2 83 6 4Wortmannin (50 nmol/L) 5 6 1 17 6 3 89 6 9LY294002 (1 µmol/L) 6 6 1 15 6 2 85 6 3PD98059 (10 µmol/L) 6 6 1 13 6 4 15 6 6Rapamycin (10 nmol/L) 5 6 1 59 6 2 80 6 8
NOTE. Cell monolayers were wounded and incubated for 90 minutes inthe presence or absence of the kinase inhibitors. Genistein, inhibitorof protein tyrosine kinases; wortmannin, inhibitor of phosphatidylinosi-tol 3-kinase; LY294002, inhibitor of MAPK; and rapamycin, inhibitor ofpp70 S6 kinase. Monolayers were cultured for an additional 24-hourperiod with or without kinase inhibitors and in the presence of 100nmol/L PMA or 7 nmol/L des(1-3)-IGF-I. Cells were then fixed andstained, and migrating cells were counted as described in Materialsand Methods. Values represent the mean 6 SD of the number of cellsmigrating per field from 3 replicate assays.
January 1999 IGF-I–INDUCED CELL MIGRATION REQUIRES PKCs 73

HT29-D4 cell restitution. Several lines of evidenceindicate that the IGF-I–induced restitution is indepen-dent of cell proliferation: (1) IGF-I-stimulated woundrepair was unaffected by treatment with DNA synthesisinhibitors such as mitomycin C; (2) only a few proliferat-ing cells were found in the wounded areas; and (3) wehave previously shown that des(1-3)-IGF-I did not alterHT29-D4 cell proliferation.29 The migratory effect wasblocked by aIR-3 MAb raised against the IGF-IR,indicating that IGF-I–induced cell restitution requiredIGF-IR ligation. Insulin and IGF-II were also able toinduce HT29-D4 cell restitution, although they were lesspotent than IGF-I. IGF-II was reported to stimulatemigration through IGF-IR30 or the IGF-IIR.31 BecauseHT29-D4 cells do not express the IGF-IIR at the cellsurface,32 it seems likely that IGF-II–induced cell restitu-tion also occurs through IGF-IR. The migratory effect ofinsulin appears to be directly mediated through IR: (1)insulin was able to induce cell restitution at a concentra-tion (7 nmol/L) that was not sufficient to stimulateIGF-IR14,33; and (2) B6, a blocking antibody raisedagainst IR, blocked the migratory response to insulin,whereas aIR-3 did not. However, we cannot rule out thatat higher insulin concentrations, cell restitution occurredvia cross reaction with the IGF-IR.
The data in this report also point out the role ofIGFBPs in the modulation of HT29-D4 cell restitution.In our migration assay, the truncated analogues des(1-3)-IGF-I and des(1-6)-IGF-II, which exhibit a reducedbinding capacity to IGFBPs,34 have a greater potency tostimulate HT29-D4 cell migration than their naturalcounterparts. This strongly suggests that IGFBPs par-tially inhibit IGF-induced cell migration by sequestrat-ing IGF-I and IGF-II and impairing with the interactionof IGFs with IGF-IR. HT29-D4 cells secrete IGF-II26
and simultaneously express IGF-IR.32 However, thesecreted IGF-II is totally sequestered in the extracellularmedium of these cells by IGFBP-2, -4, and -6, thuspreventing its autocrine interaction with IGF-IR at thecell surface.29 This lack of constitutive IGF-II–mediatedregulatory autocrine function could explain, at least inpart, why HT29-D4 cells do not spontaneously migrate.
The cellular mechanisms involved in the IGF-I–induced cell migration process are largely unknown.Recent investigations indicate that IGF-I–induced cellmigration occurs through a pathway involving IGF-IRtyrosine autophosphorylation, binding and phosphoryla-tion of insulin receptor substrate (IRS)-1, and associationof the PI 3-K with IRS-1 followed by its activation.35,36
In agreement with this, we previously reported thatIGF-IR autophosphorylation and insulin IRS-1 phosphor-ylation are required for IGF-I signal transduction inHT29-D4 cells.25 Furthermore, by using synthetic inhibi-tors, we show in this study that tyrosine kinases, PI 3-K,PKCs, MAPK cascade, but not pp70S6K, are critical forthe IGF-I–induced HT29-D4 cell migration.
Conflicting reports have been published on PKCrequirement in IGF-I–induced cell migration.37,38 In thisstudy we provide several lines of evidence that PKCsignaling can regulate IGF-I–induced cell restitution.First, we inhibited the enzymatic activity of the PKCs by2 compounds with different action sites on the kinase:calphostin C, which inhibits PKCs by competing at thebinding site with diacylglycerol and phorbol esters,39 andbisindolylmaleimide, which interacts with the adenosinetriphosphate–binding site.40 Both compounds preventedPMA- and IGF-I–induced cell restitution. Second, down-regulation of PKCs by long-term exposure of cells toPMA before induction of cell restitution dramaticallyinhibited both PMA- and IGF-I–induced HT29-D4 cellrestitution. In addition, the effect of optimal doses ofIGF-I on cell migration was not further enhanced byPMA (not shown). Interestingly, both PMA and IGF-Iwere found to increase IGFBP-6 secretion, suggestingthat activation of PKCs is also involved in IGF-I–inducedbiological effects other than cell migration in HT29-D4cells. According to this hypothesis, PMA41 as well asIGF-I29 have been shown to induce HT29-D4 celldifferentiation.
PKCs represent a multigene family of phospholipid-dependent serine/threonine tyrosine kinases consisting of11 members divided into groups on the basis of theirstructural and biochemical properties.42 ConventionalcPKCs (-a, -bI, -bII, and -g), novel nPKCs (-d, -e, -h,and -u), atypical aPKCs (-z and -i/l), and PKC-µ,although structurally related to the other PKCs, repre-sent a new PKC subgroup. HT29-D4 cells express 2
Figure 11. Effect of kinase inhibitors on MAPKs’ activation. Subconflu-ent monolayers were incubated for 90 minutes in serum-free mediumwith or without kinase inhibitors: 5 µmol/L LY294002 (lanes 3 and11), 10 µmol/L calphostin C (lanes 4, 8, and 12), 10 µmol/Lbisindolylmaleimide (lane 5), or 10 µmol/L PD98059 (lanes 6, 9, and13). Cells were then incubated for 60 minutes with DMEM/0.1% BSAalone (lane 1) or with 7 nmol/L des(1-3)-IGF-I (lanes 2–6), 10 nmol/LEGF (lanes 7–9), or 100 nmol/L PMA (lanes 10–13). After cell lysis,60 µg of extracted proteins was resolved on SDS-PAGE and blottedonto a nitrocellulose sheet. Active MAPKs (ERK 1 and ERK 2) wererevealed by specific antiactive MAPK antibody.
74 ANDRE ET AL. GASTROENTEROLOGY Vol. 116, No. 1

conventional PKCs (-a and -g), 2 novel PKCs (-d and -e),and 1 atypical PKC (-i/l) and PKC-µ. Given thebiochemical properties (i.e., sensibility to phorbol esters)of PKCs, the fact that long-term exposure of HT29-D4cells to PMA prevented the IGF-I–induced cell restitu-tion implicates that only cPKC (-a, -g) and/or nPKCs(-d, -e) could be involved in the IGF-I–induced migra-tion. Because only PKC-d and PKC-g were depletedduring long-term PMA treatment and because PKC-dand PKC-g depletion prevents des(1-3)-IGF-I–inducedmigration, these PKCs are probably involved in theIGF-I pathway that led to cell motility. However, thepersistent detection of PKC-a and -e after long-termexposure of cells to PMA could be explained by the factthat PKCs display differential sensitivities to down-regulation by PMA.43
Because PKCs convey intracellular signals by movingbetween cell compartments, we determined by 2 indepen-dent procedures which isoforms of PKC were involved inthe IGF signaling pathway. By immunoblotting andimmunocytochemical analysis, we showed that des(1-3)-IGF-I causes the translocation of PKC-d and -g only.Assuming that translocation is a prerequisite for activa-tion of conventional and novel PKCs, these resultsindicate that both PKC-g and -d are involved in the cellmigration–associated IGF-I signaling pathway. Becauseno chemical inhibitor discriminating between PKC-dand -g is yet available, to our knowledge, we cannotdetermine whether PKC-g or PKC-d, or both, areresponsible for IGF-I–induced HT29-D4 cell restitution.Interestingly, in addition to PKC-g and -d, PMA alsoinduced the translocation of PKC-a, indicating thatactivation of PKCs other than PKC-d and -g mayparticipate in the PMA-induced cell restitution.
The question of where PKC feeds into the IGF-Isignaling pathways remains to be elucidated. We showedthat PKCs act downstream of tyrosine kinases and PI 3-Kin IGF-I signaling pathway: (1) neither genistein,LY294002, nor wortmannin had an effect on PMA-induced restitution, whereas they inhibited IGF-I–induced cell restitution; and (2) PI 3-K inhibitorLY294002 prevented PKCs’ translocation on IGF-I acti-vation. Supporting this idea, several PKC isoforms havebeen identified as targets of PI 3-K.44 On the other hand,several lines of evidence indicate that PKCs may actupstream of MAPK activation. First, PD98059 abolishedIGF-I– as well as PMA-induced cell restitution. Second,PD98059 did not prevent IGF-I–induced PKC activa-tion. Third, inhibition of PKC activity prevented theactivation of ERK1 on IGF-I activation. Accordingly,MAPK has been shown to be a downstream target of PI3-K, and both enzymes are involved in the mesangial cell
migration induced by the platelet-derived growth fac-tor.45 Moreover, MacKenzie et al.46 and Kilgour et al.47
showed that PKC-d and PI 3-K are both required for thefull activation of MAPK by growth hormone. Our datasuggest that IGF-I induces epithelial colonic cells restitu-tion through activation of multiple signaling pathwaysincluding activation of PI 3-K and MAPK cascade. Inagreement with this conclusion, Parrizas et al.48 showedthat IGF-I is able to prevent PC-12 cells’ apoptosis byactivation of multiple signaling pathways includingactivation of the MAPK cascade and activation of PI 3-Kand pp70S6K. ERK1 and ERK2 display differentialsensitivities to PKC inhibitors in response to PKCinhibitors. PKC inhibitors essentially prevented ERK1activation, but ERK2 activation was only weakly af-fected. The biological significance of such a differentialeffect is unknown.
Several growth factors have been shown to functionthrough a TGF-b1–dependent pathway in entero-cytes.8,9,11 Thus, it will be important to determinewhether wounded HT29-D4 cells secrete active TGF-b1because this information could facilitate additional mecha-nistic studies on wound healing. TGF-b1 has also beenreported to induce secretion of IGFBP-3,49 supportingthe idea that both IGF-I and TGF-b1 systems may beintimately connected.
Studies have suggested a direct role of the extracellularmatrix molecules in intestinal epithelial cell restitu-tion.6,7 Cell interactions with the extracellular matrix aremediated in part by integrins, a family of cell surfaceadhesion receptors.50 Recently, it has become clear thatintegrins function as true receptors transmitting signalsto the cell interior on interaction with extracellularmatrix components.51 Connections between componentsof growth factor signaling pathways and integrin-mediated signaling pathways have begun to emerge.38,52–54
The mechanisms by which IGF-I receptors induce inte-grin-dependent cell migration are not understood. Thecell migration model used in this study therefore seems tobe appropriate for clarifying the mechanisms involved incross-talk between IGF-IR and integrin receptor signal-ing pathways.
References1. Wilson AJ, Gibson PR. Epithelial migration in the colon: filling the
gap. Clin Sci 1997;93:97–108.2. Silen W, Ito S. Mechanisms for rapid re-epithelialization of the
gastric mucosal surface. Annu Rev Physiol 1985;47:217–229.3. Feil W, Lacy ER, Wong YM, Burger D, Wenzl E, Starlinger M,
Schliessel R. Rapid epithelial restitution of human and rabbitcolonic mucosa. Gastroenterology 1989;97:685–701.
4. Graham M. Stricture formation: pathophysiological and therapeu-tic concepts. In: MacDermott RP, Stenson WF, eds. Inflammatorybowel disease. New York: Elsevier, 1992:323–335.
January 1999 IGF-I–INDUCED CELL MIGRATION REQUIRES PKCs 75

5. Manske M, Bade EG. Growth factor–induced cell migration:biology and methods of analysis. Int Rev Cytol 1994;155:49–93.
6. Basson M, Modlin I, Flynn S, Jena B, Madri J. Independentmodulation of enterocyte migration and proliferation by growthfactors, matrix proteins, and pharmacologic agents in an in vitromodel of mucosal healing. Surgery 1992;112:299–308.
7. Ohtaka K, Watanabe S, Ryozo I, Hirose M, Sato N. Role ofextracellular matrix on colonic cancer cell migration and prolifera-tion. Biochem Biophys Res Commun 1996;220:346–352.
8. Dignass AU, Podolsky DK. Interleukin 2 modulates intestinalepithelial cell function in vitro. Exp Cell Res 1996;225:422–429.
9. Dignass AU, Podolsky DK. Cytokine modulation of intestinalepithelial cell restitution: central role of transforming growthfactor b. Gastroenterology 1993;105:1323–1332.
10. Polk D. Epidermal growth factor receptor–stimulated intestinalepithelial cell migration requires phospholipase C activity. Gastro-enterology 1998;114:493–502.
11. Dignass AU, Lynch-Devaney K, Podolsky DK. Hepatocyte growthfactor/scatter factor modulates intestinal epithelial cell prolifera-tion and migration. Biochem Biophys Res Commun 1994;202:701–709.
12. Nustra A, Parkos CA, Bacarra AE, Godowski PJ, Delp-Archer C,Rosen EM, Madara JL. Hepatocyte growth factor/scatter factoreffects on epithelia. Regulation of intercellular junctions intransformed and nontransformed cell lines, basolateral polariza-tion of c-met receptor in transformed and natural intestinalepithelia, and induction of rapid wound repair in a transformedmodel epithelium. J Clin Invest 1994;93:2056–2065.
13. Ciacci C, Lind SE, Podolsky DK. Transforming growth factor b
regulation of migration in wounded rat intestinal epithelial mono-layers. Gastroenterology 1993;105:93–101.
14. Gockerman A, Prevette T, Jones JI, Clemmons DR. Insulin-likegrowth factor (IGF)-binding proteins inhibit the smooth muscle cellmigration responses to IGF-I and IGF-II. Endocrinology 1995;136:41168–41173.
15. LeRoith K, Werner H, Beitner-Johnson D, Robert CT. Molecularaspects of the insulin-like growth factor I receptor. Endocr Rev1995;16:143–163.
16. Leventhal PS, Feldman EL. Insulin-like growth factors of cellmotility. Trends Endocrinol Metab 1997;8:1–6.
17. Guo Y-S, Narayan S, Yallampalli C, Singh P. Characterization ofinsulin-like growth factor I receptors in human colon cancer.Gastroenterology 1992;102:1101–1108.
18. Singh P, Rubin N. Insulin-like growth factors and binding proteinsin colon cancer. Gastroenterology 1993;105:1218–1237.
19. Lemmey A, Martin A, Read L, Tomas F, Owens P, Ballard F. IGF-Iand the truncated analogue des-(1-3)IGF-I enhance growth in ratsafter gut resection. Am J Physiol 1991;260:E213–E219.
20. Lemmey A, Ballard F, Martin A, Tomas F, Howarth G, Read L.Treatment with IGF-I peptides improves functions of the remnantgut following small bowel resection in rats. Growth Factors1994;10:243–252.
21. Fantini J, Abadie B, Tirard A, Remy L, Ripert JP, El Battari A,Marvaldi J. Spontaneous and induced dome formation by twoclonal cell populations derived from a human adenocarcinomacell line. J Cell Sci 1986;83:235–249.
22. Burk RR. A factor from a transformed cell line that affects cellmigration. Proc Natl Acad Sci USA 1973;70:369–372.
23. Hinck L, Nathke IS, Papkoff J, Nelson WJ. Dynamics of cadherin/catenin complex formation: novel protein interactions and path-ways of complex assembly. J Cell Biol 1994;125:1327–1340.
24. Hossenlopp P, Seurin D, Segovia-Quinson B, Hardouin S, BinouxM. Analysis of serum insulin-like growth factor binding proteinsusing Western blotting: use of the method for titration of thebinding proteins and competitive binding studies. Anal Biochem1986;154:137–143.
25. Remacle-Bonnet M, Garrouste F, El Atiq F, Marvaldi J, Pommier G.Cell polarity of the insulin-like growth factor system in humanintestinal epithelial cells. Unique apical sorting of insulin-likegrowth factor binding protein-6 in differentiated human coloncancer cells. J Clin Invest 1995;96:192–200.
26. Culouscou JM, Remacle-Bonnet M, Garrouste F, Fantini J, Mar-valdi J, Pommier G. Production of insulin-like growth factor II(IGF-II) and different forms of IGF-binding proteins by HT29 humancolon carcinoma cell line. J Cell Physiol 1990;143:405–415.
27. Culouscou JM, Shoyab M. Purification of a colon cancer cellgrowth inbibitor and its identification as an insulin-like growthfactor binding protein. Cancer Res 1991;51:2813–2819.
28. Quintaje S, Rebsamen M, Church D, Vallotton M, Lang U. MAPkinase mediates epidermal growth factor– and phorbol ester–induced prostacyclin formation in cardiomyocytes. J Mol CellCardiol 1998;30:933–945.
29. Remacle-Bonnet M, Garrouste F, El Atiq F, Roccabianca M,Marvaldi J, Pommier G. Des-(1-3)-IGF-I, an insulin-like growthfactor analog used to mimic a potential IGF-II autocrine loop,promotes the differentiation of human colon carcinoma cells. IntJ Cancer 1992;52:910–917.
30. Stracke ML, Engel JD, Wilson LW, Rechler MM, Liotta LA,Schiffmann E. The type I insulin–like growth factor is a motilityreceptor in human melanoma cells. J Biol Chem 1989;264:21544–21549.
31. Minniti CP, Kohn EC, Grubb JH, Sly WS, Oh Y, Moller RG,Rosenfeld R, Helman LJ. The insulin-like growth factor II (IGF-II)/mannose 6-phosphate receptor mediates IGF-II–induced motilityin human rhabdomyosarcoma cells. J Biol Chem 1992;267:9000–9004.
32. Remacle-Bonnet M, Culouscou JM, Garrouste F, RabenandrasanaC, Marvaldi J, Pommier G. Expression of type I, but not type II,insulin-like growth factor receptor on both undifferentiated anddifferentiated HT29 human colonic carcinoma cell line. J ClinEndocrinol Metab 1992;75:609–616.
33. Bornfeldt KE, Raines EW, Nakano T, Graves LM, Krebs EG, RossR. Insulin-like growth factor-I and platelet-derived growth factor-BBinduce directed migration of human arterial smooth muscle cellsvia signaling pathways that are distinct from those of prolifera-tion. J Clin Invest 1994;93:1266–1274.
34. Forbes B, Szabo L, Baxter RC, Ballard FJ, Wallace JC. Classifica-tion of the insulin-like growth factor binding proteins into threedistinct categories according to their binding specificities. Bio-chem Biophys Res Commun 1988;157:196–202.
35. Kotani K, Yoneawa K, Hara K. Involvement of phosphoinositide3-kinase in insulin- or IGF-I–induced membrane ruffling. EMBO J1994;13:2313–2321.
36. Izumi T, Saeki Y, Akanuma Y, Takaku F, Kasuga M. Requirementfor receptor-intrinsic kinase activities during ligand-induced mem-brane ruffling in KB cells. J Biol Chem 1988;261:141–147.
37. Miyata Y, Nishida E, Koyasu S, Yahara I, Sakai H. Protein kinaseC–dependent and –independent pathways in the growth factor–induced cytoskeletal reorganization. J Biol Chem 1989;264:565–568.
38. Klemke RL, Yebra M, Bayna EM, Cheresh DA. Receptor tyrosinekinase signaling required for integrin avb5-directed cell motilitybut not adhesion on vitronectin. J Cell Biol 1994;3:859–866.
39. Kobayashi E, Nakano H, Morimoto M, Tamaoki T. Calphostin C(UCN-1028C), a novel microbial compound, is a highly potent andspecific inhibitor of protein kinase C. Biochem Biophys ResCommun 1989;159:548–553.
40. Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T,Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L,Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is apotent and selective inhibitor of protein kinase C. J Biol Chem1991;266:15771–15781.
76 ANDRE ET AL. GASTROENTEROLOGY Vol. 116, No. 1

41. Delezay O, Baghdiguian S, Fantini J. The development of Na1-dependent glucose transport during differentiation of an intesti-nal epithelial cell clone is regulated by protein kinase C. J BiolChem 1995;270:12536–12541.
42. Jaken S. Protein kinase C isozymes and substrates. Curr OpinCell Biol 1996;8:168–173.
43. MacKenzie S, Fleming I, Housley M, Anderson N, Kilgour E.Growth hormone and phorbol esters require specific proteinkinase C isoforms to activate mitogen-activated protein kinasesin 3T3-F442A cells. Biochem J 1997;324:159–165.
44. Shimizu Y, Hunt III SW. Regulating integrin-mediated adhesion:one more function for PI 3 kinase? Immunol Today 1996;17:565–573.
45. Choudhury GG, Karamitsos C, Hernandez J, Gentilini A, BardgetteJ, Abboud HE. PI-3-kinase and MAPK regulate mesangial cellproliferation and migration in response to PDGF. Am J Physiol1997;273:F931–F938.
46. MacKenzie S, Fleming I, Housley M, Anderson N, Kilgour E.Growth hormone and phorbol esters require specific proteinkinase C isoforms to activate mitogen-activated protein kinasesin 3T3-F442A cells. Biochem J 1997;324:159–165.
47. Kilgour E, Gout I, Anderson N. Requirement for phosphoinositide3-OH kinase in growth hormone signalling to the mitogen-activated protein kinase and p70s6k pathways. Biochem J1996;315:517–522.
48. Parrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor 1inhibits apoptosis using the phosphatidylinositol 38-kinase andmitogen-activated protein kinase pathways. J Biol Chem 1997;272:154–161.
49. Oh Y, Muller H, Neg L, Rosenfeld R. Transforming growth factor-beta induce cell growth inhibition in human breast cancer cells is
mediated through insulin-like growth factor-binding protein-3 ac-tion. J Biol Chem 1995;270:13589–13592.
50. Hynes RO. Integrins: versatility, modulation, and signaling in celladhesion. Cell 1992;69:11–25.
51. Juliano RL, Haskill S. Signal transduction from the extracellularmatrix. J Cell Biol 1993;120:577–585.
52. Vuori K, Ruoslahti E. Association of insulin receptor substrate-1with integrins. Science 1994;266:1576–1578.
53. Doerr ME, Jones JI. The roles of integrins and extracellular matrixproteins in the insulin-like growth factor I–stimulated chemotaxisof human breast cancer cells. J Biol Chem 1996;271:2443–2447.
54. Jones JI, Prevette T, Gockerman A, Clemmons DR. Ligandoccupancy of the avb3 integrin is necessary for smooth musclecells to migrate in response to insulin-like growth factor I. ProcNatl Acad Sci USA 1996;93:2482–2487.
Received April 7, 1998. Accepted October 13, 1998.Address requests for reprints to: Jacques Marvaldi, M.D., ESA
CNRS 6032, Laboratoire de Biochimie Cellulaire, Universited’Aix-Marseille I et II, Faculte de Pharmacie, 27 BoulevardJean Moulin, 13385 Marseille, France. e-mail: [email protected]; fax: (33) 49-183-5653.
Supported by the Association de la Recherche Contre le Cancer,the Ligue Nationale Contre le Cancer, and the Groupement desEntreprises Francaises dans la lutte contre le Cancer.
The authors thank Drs C. Montixi (CNRS UPRES-A 6032,Marseille) and H. Feracci (CNRS-UMR 144, Institut Curie, Paris) forcritical review of the manuscript and B. Khalil and H. Bouteille forthe preparation of illustrations.
January 1999 IGF-I–INDUCED CELL MIGRATION REQUIRES PKCs 77
Top Related
Copyright © 2022 FDOKUMEN

