







Bahasa
Halaman
Hukum


Preparation of Different Dendritic-Layered SilicateNanocomposites
Amal Amin,1 Rajarshi Sarkar,2 Charles N. Moorefield,3 George R. Newkome2,3,4
1Department of Polymers and Pigments, National Research Center, Dokki, Giza, Egypt
2Department of Chemistry, The University of Akron, Akron, Ohio 44325-4717
3The Maurice Morton Institute for Polymer Science, The University of Akron, Akron, Ohio 44325-4717
4Department of Polymer Science, The University of Akron, Akron, Ohio 44325-4717
Several dendrimer–clay nanocomposites have beenprepared. Firstly, the dendrimer (DE1)/clay nanocompo-site was obtained via an in situ free radical polymeriza-tion of a double bond-ended dendrimer (DE1), derivedfrom Behera’s amine by using 2,20-azobisisobutyroni-trile (AIBN), as initiator, and Cloisite 30 B, as nanofiller.Further free radical in situ copolymerization processeswere conducted between DE1, methyl methacrylate(MMA), and styrene (St). Two other dendrimer/claynanocomposites were prepared by the reaction ofsecond generation (G2)–36-acid dendrimer (DE2) andN,N0,N0,N0-tetrakis[2-hydroxy-1,1-bis(hydroxylmethyl)ethyl]-a,a,x,x-alkane-tetracarboxamide [6]-10-[6] Arbor-ols (DE3) with montmorillonite clay (MMT). POLYM. ENG.SCI., 53:2166–2174, 2013. ª 2013 Society of Plastics Engineers
INTRODUCTION
Since Toyota researchers first published [1–3] their
work on nylon 6-clay hybrid materials, many types of
polymer–clay nanocomposites (PCNs) [4] such as polyi-
mide [5], epoxy resin [6, 7], polystyrene [8–10], polycap-
rolactone [11], polypropylene [12], polyvinylalcohol [13],
and polyurethane [14] have been reported. The three main
categories of PCN that have been described include inter-
calated, flocculated, and exfoliated–delaminated nanocom-
posites. PCNs are generally prepared using a low-load
percentage of clay nanofiller that is homogeneously
dispersed within the polymer matrix. PCNs are currently
prepared by in situ polymerization, solution exfoliation,
and melt intercalation. PCN incorporation has been shown
to impart superior mechanical, thermal, and barrier prop-
erties when compared to materials strictly composed of
conventional polymers or composites [15–23].
Herein, it was thought to incorporate dendrimers as the
vital polymeric category in such nanocomposites. Den-
drimers are considered as an ubiquitous type of precisely
defined polymers [24, 25]. Dendrimers are a class of
regularly branched macromolecules with unique structural
and topological features whose properties are attracting
considerable interest from both scientists and technolo-
gists [26–28]. Dendrimers are different from traditional
polymers in that they possess a multibranched, three-
dimensional architecture with very low polydispersity and
high polyfunctionality. The dendrimers are potentially
usable in numerous applications such as catalysts, photo-
active and electronic materials, medicinal and functional
materials, biomedical materials, and specifically as drug
carriers [29–39]. Dendrimers are synthesized via a fully
controllable stepwise series of reactions. Two major
synthetic strategies are used to construct dendritic struc-
tures, namely, either a divergent or convergent approach.
Herein, three different dendritic materials with three
different functionalities were chosen and prepared such as
DE1, DE2, and DE3. The three dendritic architectures
were involved in forming nanocomposites with layered
silicates or clay to invest the impact of organic dendritic
moieties/inorganic clay hybrids in different potential
applications and mostly in biomedical ones. DE1 was dou-
ble bond-ended Behera’s amine-based 1 ? 3 C-branched
construct where Behera’s amine is considered as impor-
tant dendritic brick because of several reasons including
facile acylation of the amino moiety and quantitative
removal of the carboxylic acid protecting group and hence
transformation to versatile chemical active structures [40,
41]. Also, possible polymerization reactions can be
carried out on the double bond end of DE1 with other
Additional Supporting Information may be found in the online version of
this article.
Correspondence to: Amal Amin; e-mail: [email protected]
Contract grant sponsor: US-Egypt Grant.
DOI 10.1002/pen.23485
Published online in Wiley Online Library (wileyonlinelibrary.com).
VVC 2013 Society of Plastics Engineers
POLYMER ENGINEERING AND SCIENCE—-2013

vinyl monomers as was done actually in the current com-
munication with methyl methacrylate (MMA) and styrene
(St) to obtain several new functionalities and hence new
dendritic architectures. Two other water-soluble dendritic
architectures were prepared such as DE2 (3-oxo-6-oxa-2-
azaheptylidyne): (3-oxo-2- aza-pentylidyne): propionic
acid which ends with COOH [42] and DE3 (N,N0,N0,N00-tetrakis [2-hydroxy -1,1-bis(hydroxyl-methyl)ethyl]-a,a,x,
x-alkanetetracarboxamide or better a [6]-10-[6] Arborols)
which ends with OH [43].
EXPERIMENTAL
Materials
Cloisite (30 B) and montmorillonite (MMT) were
kindly provided by Southern clay products (Texas, USA).
All other chemicals were purchased from Sigma-Aldrich
and were used as received, unless further modified as
noted below [40–43]. Methyl methacrylate (MMA) and
styrene (St) monomers were purified by passing through
alumina columns, and then stored under argon.
Instrumentation
Molecular weights (�MnGPC) of the prepared polymers
were determined by gel permeation chromatography
(GPC) using an Agilent GPC-1100 with a refractive index
detector with 100, 104, and 105 A ultrastyragel columns
connected in series. Tetrahydrofuran (THF) and N,N-
dimethylformamide (DMF) were used, as the eluents, with
flow rates ca. 1 mL/min. Commercially available linear
polystyrene standards were used to calibrate the columns.
Proton nuclear magnetic resonance (1H NMR) spectra
were recorded using a Varian Mercury 300 MHz NMR;
tetramethylsilane (TMS) was used as the internal stand-
ard, and deuterated chloroform (CDCl3) and dimethyl
sulfoxide [(CD3)2SO)] were used as solvents. FT-IR spec-
tra were recorded using KBr pellets on a Digilab Excali-
bur FTS 3000.
X-ray diffraction (XRD), for measurement of the basal
spaces (d) between the layers of clay, was obtained on a
1D WAXD (wide angle X-ray diffraction) powder diffrac-
tion pattern using a Rigaku MultiFlex 2kW X-ray genera-
tor coupled to the diffractometer, which had a hot stage
for studing the phase transitions, as a function of tempera-
ture. The hot stage was calibrated with a deviation range
of 658C and the samples were scanned across a 2y range
of 3–358 at a scanning rate of 1 deg/min. Peak positions
were calibrated using silicon powder in the high-angle
region (\158).Transmission electron microscopy (TEM) measure-
ments were acquired with JEM 1200XII electron micro-
scope operating at 60 kV. Samples were prepared by drop
casting a polymer suspension onto carbon-coated copper
grids, followed by solvent evaporation in air.
Thermal gravimetric analysis (TGA) and differential
scanning calorimetry (DSC) data were obtained under a
nitrogen atmosphere using the TA Instruments Q50 Ther-
mogravimetric Analyzer and Q200 Differential Scanning
Calorimeter, respectively.
SYNTHETIC PROCEDURES
Dendritic DE1
Behera’s Amine. The nitrotriester (50 g, 112 mmol)
was added to an EtOH solution of Raney nickel, as previ-
ously described elsewhere (Scheme 1) [40]. The hydro-
genation was maintained for 45–75 min, while external
cooling was used so that the temperature does not exceed
558C. The catalyst was removed by filtration through a
Celite pad; the desired amine is soluble in EtOH. Then,
the solvent was removed in vacuo maintaining the
temperature below 558C to give an oil, which solidified
in vacuo to give (89%) pure Behera’s amine, as white crys-
tals: 1H NMR (CDCl3) [40]: d 1.44 (s, CH3, 27 H), 1.78
(m, CH2, 12 H); 13C NMR: d 27.8 (CH3), 29.8 (CH2CO),
34.2 (CCH2), 52.2 (CNH2), 80 (CMe3), 172.8 (CO2).
Synthesis of Di-tert-butyl 4-acryloylamino-4-(2-tert-
butoxycarbonylethyl)-heptanedioate (DE1). Acryloyl
chloride (1 g, 11 mmol) dissolved in CH2Cl2 (20 mL)
was added dropwise over a period of 15 min to a stirred
solution of Behera’s amine (4.6 g, 11 mmol) and Et3N
(3.1 mL, 22 mmol) in dry CH2Cl2 (100 mL) at 08C [41].
The resulting mixture was stirred for 2 h at 258C. Then, it
was washed with water and saturated brine, dried
(MgSO4), filtered, and concentrated in vacuo to give a
crude solid, which was chromatographed (SiO2) eluting
with an EtOAc/CHCl3 (1:9 v/v) solvent mixture. DE1 was
obtained (96%) as a white solid: m.p. 144–1458C; 1H
NMR (CDCl3) [41]: d 1.44 (27 H, s), 2.04 (6 H, t), 2.26
(6 H, t), 5.59 (1 H, dd), 6.03 (1 H, dd), 6.20 (1 H, s),
6.22 (1 H, dd); 13C NMR (CDCl3): d 27.9, 29.6, 29.9,
57.4, 80.3, 125.5, 131.7, 164.7, 172.7; IR 3290, 1710,
1654, 1622 cm21. Elemental analyses: calcd for
C25H43NO7: C, 63.94; H, 9.23; N, 2.98. Found: C, 63.68;
H, 9.30; N, 2.84.
Synthesis of Dendrimer DE2. (3-Oxo-6-oxa-2-azahep-
tylidyne): (3-oxo-2-azapentylidyne): propionic acid (DE2)
was prepared by hydrolysis of the corresponding ester
[(3-oxo-6-oxa-2-azaheptylidyne): (3-oxo-2-azapentyli-
dyne): tert-butyl propanoate] (E) via the described proce-
dure [42] by stirring (13.55 g, 2.22 mmol) of E in formic
acid (95%, 100 mL) for 12 h at 258C (Scheme 2). After-
ward, toluene (50 mL) was added to the concentrated
solution. The solution was evaporated in vacuo to give
a crude solid that was dissolved in a water (200 mL)/
acetone (10 mL) mixture and subsequently washed with
CH2Cl2 (50 mL) and EtOAc (50 mL). The aqueous phase
was boiled with charcoal (50 mg), filtered through Celite
and then concentrated in vacuo to give DE2, as white
solid: m.p. 132–1348C; 1H NMR (5% NaOD/p-dioxane/
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2013 2167

3.54 ppm) [42]: d 1.75, 1.99 (br, CH2CH2CO, 192 H),
2.32 (br, OCH2CH2CO, 8 H), 3.19 (br, CH2O, 8H), 3.48
(br, OCH2, 8 H); 13CNMR (5% NaOD/p-dioxane/66.4
ppm): d 29.7, 30.1 (CH2CH2CO), 37.0 (O CH2CH2CO),
67.7 (CH2O), 69.9 (OCH2), 173.1 (CONH), 179.7 (CO2);
IR 3363 (br, acid OH), 1718 (acid C¼¼O), 1648 (amide
C¼¼O) cm21. Elemental Analysis: (C177H268N16O92),
calcd: C, 51.95; H, 6.60, N, 5.48. Found: C, 51.74; H,
6.71; N, 5.30.
Synthesis of N,N0,N0,N0-Tetrakis[2-hydroxy-1,1–bis
(hydroxylmethyl)ethyl]-a,a,x,x-alkanetetracarboxamide
[6]-10-[6] Arborols (DE3). A mixture of tris (hydroxy-
methyl)aminomethane (8.0 mmol), tetraester (T, 2.0
mmol), and anhydrous K2CO3 (8.64 mmol) in Me2SO (5
mL) was stirred at 258C for 24 h (Scheme 3) [43]. The
solution was filtered and the solid was washed with
Me2SO (10 mL). The combined filtrate was concentrated
in vacuo. The residue was dissolved in water, precipitated
by the addition of acetone, and then filtered to give the
desired tetraamide (DE3), as a white solid, which was
washed with anhydrous EtOH. The arborol was obtained
in purity 97%. 13C NMR d 26.7 (b-CH2), 53.8 (a-CH),
60.5 (CH2OH), 171.3 (C¼¼O).
Preparation of Dendrimer/Clay Nanocomposites
p-DE1/Cloisite 30 B Nanocomposites. To a glass vial,
Cloisite 30 B (250 mg) and xylene (10 mL) were inserted
with continuous stirring, then DE1 (1 g, 22 3 1024 mol)
was added. The vial was closed with added argon. The
reaction mixture was placed at thermostated oil bath
adjusted at 708C. Afterward, AIBN (100 mg, 61 3 1025
SCHEME 1. Synthesis of DE1, p-DE1/MMA, and p-DE1/St nanocomposites.
2168 POLYMER ENGINEERING AND SCIENCE—-2013 DOI 10.1002/pen

mol) was dissolved in low amount of xylene (�2 mL)
and injected into the reaction. The polymerization proce-
dure was left for 44 h, then the vial was opened and the
nanocomposite was isolated, dried, and characterized (see
Supporting Information).
p-DE1/MMA or St Copolymers/Cloisite 30 B Nanocom-
posites. The previous sequence for addition of ingre-
dients was followed but DE1 and MMA or St were added
in 50/50 wt%. Also, MMA or St was added after closing
the vial under argon before adding AIBN solution. The
copolymerization was maintained in case of MMA and St
for 48 h to obtain �60% conversion. The amount of reac-
tants used were Cloisite 30 B (250 mg), xylene (10 mL),
DE1 (1.17 g, 25 3 1024 mol), MMA or St (25 3 1024
mol), and AIBN (100 mg, 61 3 1025 mol). To character-
ize the structure of the p-DE1 inside the formed polymer/
clay nanocomposite with 1H NMR and GPC, the nano-
composite was dissolved in THF to separate the polymer
solution from clay with centrifugation and filtration (see
Supporting Information).
DE2 or DE3/Montmorillonite/Nanocomposites. Clay
(250 mg, MMT) was dispersed in H2O (60 mL) with
continuous stirring for 24 h at 608C. At the same time, DE2
(500 mg, 12 3 1025 mol) or DE3 (66 3 1025 mol) was
separately stirred in H2O (40 mL) for 3 h at 608C. After-
ward, the dendrimer suspension was added to the clay, then
the total mixture was maintained for 24 h. The resulting
nanocomposites were separated, dried, and analyzed.
RESULTS AND DISCUSSION
The field of PCNs is a rapidly growing field of
research extending to comprise different types of
polymers [5–14]. Recently, efforts were exerted to form
nanocomposites by using new polymeric architectures,
such as hyperbranched polymers [44, 45]. Accordingly,
some dendritic compounds DE1, DE2, and DE3 were
involved in forming new nanocomposites with two types
of clay such as Cloisite 30 B and montmorillonite clay
(MMT) according to the functionalities of the dendritic
structures where Cloisite 30 B was chosen for DE1 and
its derivatives because it is organically modified nanoclay
[46] which fits with the hydrophobic nature of DE1 and
its derivatives with MMA and St. On the other hand,
montmorillonite clay with its hydrophilic OH ends was
suitable for DE2 and DE3 with COOH and OH polar end
groups, respectively.
Firstly, DE1 with double bond end was synthesized
from the reaction of Behera’s amine (Scheme 1) with
acryloyl chloride at 08C in dry CH2Cl2 [41]. Poly-DE1
(p-DE1)/clay nanocomposite was obtained by in situ free
radical polymerization of DE1 using AIBN and Cloisite
30 B at 708C. The same procedure was followed in case
SCHEME 2. Synthesis of DE2.
SCHEME 3. Synthesis of DE3.
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2013 2169

of p-DE1/MMA and p-DE1/St nanocomposites by an
in situ polymerization of DE1 with MMA or St under the
same reaction conditions using AIBN in the presence of
Cloisite 30 B at 708C. The polymerizations were left for
about 48 h and an almost 60% conversion was obtained
in each case. p-DE1 and the copolymers p-DE1/MMA and
p-DE1/St were separated from their nanocomposites and
identified with GPC and 1H NMR to confirm their struc-
tures as in situ formed polymers in the presence of clay.
GPC indicated �MnGPC of p-DE1, p-DE1/MMA, and
p-DE1/St as �2740, 4180, and 6320 g/mol, respectively.
The 1H NMR of p-DE1 (CDCl3) showed the chemical
shifts of (CH3)3 and CH3CHCO appeared at 1.3–1.4 and
1.8 ppm, respectively. The chemical shifts of
CH2CH2COOC and CH2CH2COOC ranged from 2 to 2.2
ppm. CH and NH bands were observed at 2.3 and 6.20
ppm, respectively. 1H NMR of p-DE1/MMA (CDCl3)
revealed some similar chemical shifts as in the parent
polymer p-DE1 for some functionalities such as in case of
CH3, CH2, CH, and NH which were observed at 1.3–2.2
and 5.4 ppm, respectively. Distinguishing characteristic
band for MMA unit appeared at 3.5 ppm referring to
OCH3. The 1H NMR of p-DE1/St (CDCl3) showed, in
addition to the similar bands of the parent p-DE1, 2.3
(CHph) and 7.2 ppm (ph).
On the other hand, the G2–DE2 was prepared from the
36-acid dendrimer by the hydrolysis of the corresponding
ester (E) as previously shown (Scheme 2) [42]. The third
dendritic compound DE3 was a 6-10-6 arborol, which was
prepared as shown in Scheme (3) by the reaction of tris
FIG. 1. XRD of p-DE1, p-DE1/MMA, and p-DE1/St nanocomposites.
FIG. 2. XRD of DE2 and DE3/MMT nanocomposites.
2170 POLYMER ENGINEERING AND SCIENCE—-2013 DOI 10.1002/pen

with tetraester (T) [43]. Both of DE2 and DE3/clay nano-
composites were prepared, as previously formed using
montmorillonite (MMT). Because of the hydroxyl and
carboxyl functionalities of both of DE2 and DE3, respec-
tively, polar–polar interactions were expected and hence
successful formation of nanocomposites with no need for
pretreatment of clay surface. The prepared nanocompo-
sites were characterized via XRD, TGA, DSC, and TEM.
Analyses of the Nanocomposites
XRD exhibited broadened peaks for p-DE1, p-DE1-
MMA, and p-DE1-St/nanocomposites, as shown in Fig. 1.
The practically recorded interlayer spacing (d) ¼ 18.8,
19.7, and 19 nm for p-DE1, p-DE1/MMA, and p-DE1-St/
nanocomposites, respectively, at lower 2y value with
respect to the pristine clay, whose d ¼ 1.85 nm at higher
2y [23]. Thereby, the interlayer distance between clay pla-
telets enlarged than the pristine clay by the effect of
inclusion of the new polymeric materials inside the clay
gallery. The previous fact revealed the disturbance of clay
ordering led to intercalated to semi-exfoliated structure of
the resulting nanocomposites.
In case of DE3-MMT nanocomposites, as shown in
Fig. 2, a sharp band appeared with interlayer spacing (d) ¼1.5 nm, which was more than that of the neat clay (MMT)
whose d spacing ¼ 1.25 nm [45]. This behavior was
expected to lead to intercalated morphology of the resulting
nanocomposite. On the other hand, XRD of DE2-MMT
nanocomposites recorded no peaks where exfoliation struc-
FIG. 3. TGA of p-DE1 parent, p-DE1, p-DE1/MMA, and p-DE1/St cloisite 30 B nanocomposites.
FIG. 4. TGA of DE2 and DE2/MMT nanocomposites.
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2013 2171

FIG. 5. TGA of DE3 and DE3/MMT nanocomposites.
FIG. 6. TEM of (a) p-DE1, (b) p-DE1/MMA, and (c) p-DE1/St cloisite nanocomposites.
ture was estimated. For the DE2 and DE3-MMT nanocom-
posites, it was expected that polar–polar interactions would
predominate in the formation of nanocomposites; however,
some repulsion forces, to some extent, were expected
between the polar ends whether on the clay surface or that
of the dendrimers DE2 (COOH) and DE3 (OH) themselves.
Therefore, that repulsion effect might be responsible for
widening of the interlayer spaces between the clay platelets
as in case of DE3/MMT nanocomposites or might cause
destruction of internal ordering of clay stacks, as in case of
DE2/MMT nanocomposites and hence led to exfoliation
structure of the resulting nanocomposite.
In the case of TGA, as shown in Fig. 3, the parent p-
DE1 was the least stable where 2.5% weight loss was
recorded at 1758C, then a gradual decomposition was
observed until 90% weight loss was observed at 4508C. p-
DE1/Cloisite nanocomposite lost about 40% of its weight
sample up to 7508C. With respect to p-DE1/MMA and p-
DE1/St–MMT nanocomposites, almost 2.5% loss happened
until 1758C, then gradual decomposition occurred on two
stages in case of p-DE1-MMA/Cloisite nanocomposite re-
cording 21% and 70% weight loss at 2408C and 4758C,
respectively. On the other hand, p-DE1/St/Cloisite nano-
composite gradually decomposed at 1758C up to almost
75% weight loss up to 8008C. It was clear that p-DE1-St/
Cloisite nanocomposite was more stable than p-DE1, p-
DE1/Cloisite, and p-DE1-MMA/Cloisite nanocomposites.
The DE2-MMT nanocomposite Fig. 4 demonstrated
higher thermal stability than its parent DE2 where the
nanocomposite lost 12.5% of its weight up to 5158C,
where a sharp decomposition began to reach 72.5%
weight loss, then another stage of decomposition was
indicated until 80% consumption of the total weight of
sample at 6758C. DE2 itself began its actual decomposi-
tion at 1968C, where it lost about 11% of its weight up to
2508C, then the sample completely degraded or 100% of
its weight at 7008C. In case of DE3 and its nanocomposite
(i.e., DE3/MMT, Fig. 5), DE3 started gradual decomposi-
tion at 1758C and continued up to 85% weight loss at
5008C, but in case of DE3-MMT nanocomposite, only
20% weight loss was recorded up to 8008C.
The DSC measurements were conducted to give the Tg
values of the polymers in the prepared nanocomposites.
The recorded Tg values of p-DE1, p-DE1/MMA, and
p-DE1-St nanocomposites were 131, 75, and 798C,
respectively. The Tg values of DE2/MMT and DE3-MMT
nanocomposites were 99 and 758C, respectively.
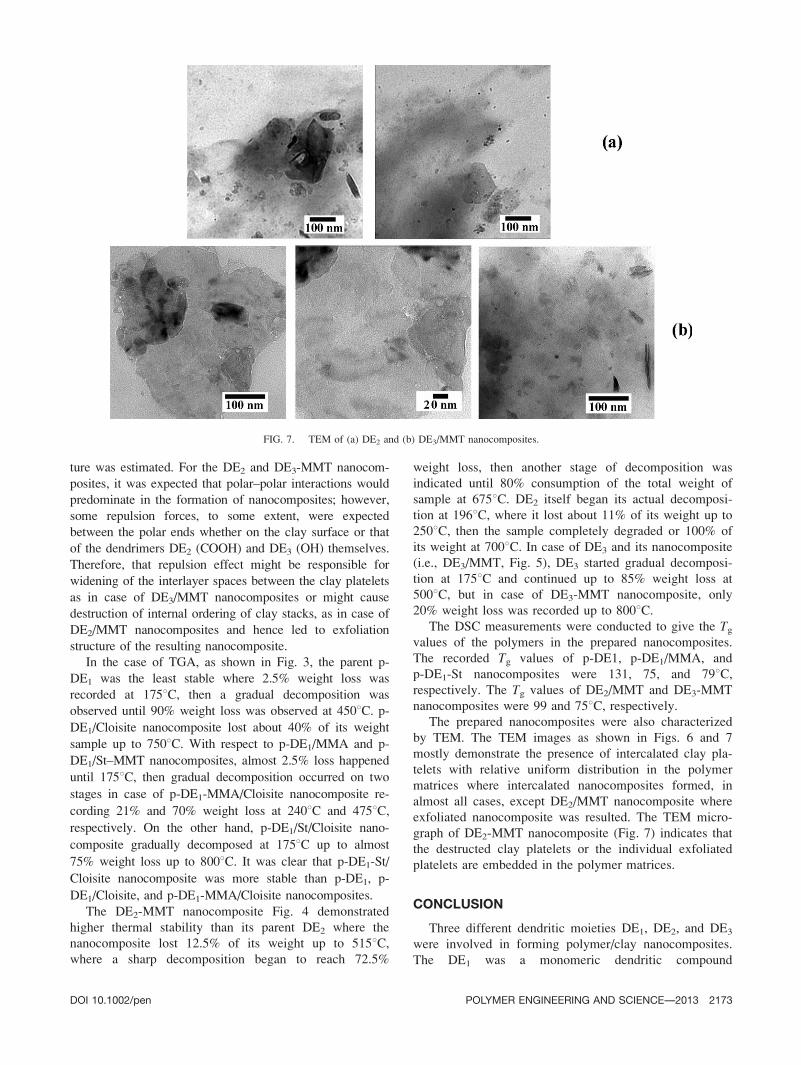
The prepared nanocomposites were also characterized
by TEM. The TEM images as shown in Figs. 6 and 7
mostly demonstrate the presence of intercalated clay pla-
telets with relative uniform distribution in the polymer
matrices where intercalated nanocomposites formed, in
almost all cases, except DE2/MMT nanocomposite where
exfoliated nanocomposite was resulted. The TEM micro-
graph of DE2-MMT nanocomposite (Fig. 7) indicates that
the destructed clay platelets or the individual exfoliated
platelets are embedded in the polymer matrices.
CONCLUSION
Three different dendritic moieties DE1, DE2, and DE3
were involved in forming polymer/clay nanocomposites.
The DE1 was a monomeric dendritic compound
FIG. 7. TEM of (a) DE2 and (b) DE3/MMT nanocomposites.
DOI 10.1002/pen POLYMER ENGINEERING AND SCIENCE—-2013 2173

terminated with a double bond, where it was subjected to
in situ free radical homopolymerization in the presence of
Cloisite 30 B. However, the other dendritic molecules
DE2 and DE3 formed nanocomposites with MMT without
further treatment as their skeletons include ¼¼COOH
and ��OH functional polar groups. Generally, the result-
ing nanocomposites demonstrated higher thermal stability
than the original polymers. Intercalated structures were
observed in almost all nanocomposites, except in case of
DE2/MMT nanocomposite, where exfoliation was
observed. The resulting dendrimers/clay nanocomposites
can be actively applied in several applications such as
drug delivery systems, which can be further developed in
future research.
ACKNOWLEDGMENT
The authors thank all workers at Department of Polymer
Science at the University of Akron, Akron, Ohio, USA.
REFERENCES
1. A. Usuki, Y. Kojima, M. Kawasumi, A. Okada, T. Kurau-
chi, and O. Kamigaito, Polym. Prepr., 31, 651 (1990).
2. A. Okada, M. Kawasumi, T. Kurauchi, and O. Kamigaito,
Polym. Prepr., 28, 447 (1987).
3. A. Okada and A. Usuki, Macromol. Mater. Eng., 291, 1449
(2006).
4. Y.P. Wang, X.W. Pei, X.J. Liu, K. Yuan, D.X. Zhang, Q.L.
Li, and Y.F. Wang, Polym. Compos., 26, 465 (2005).
5. M. Kakiage and S. Ando, J. Appl. Polym. Sci., 119, 3010
(2011).
6. X. Li, Z.-J. Zhan, G.-R. Peng, and W.-K. Wang, Appl. ClaySci., 55, 168 (2012).
7. M.M. Shokrieh, A.R. Kefayati, and M. Chitsazzadeh, Mater.Des., 40, 443 (2012).
8. M.H. Kim and O. Park, J. Appl. Polym. Sci., 125, E630
(2012).
9. N. Greesh, R. Sanderson, and P. Hartmann, Polym. Int., 61,
834 (2012).
10. M.W. Weimer, H. Chen, E.P. Giannelis, and D.Y. Sogah,
J. Am. Chem. Soc., 121, 1615 (1999).
11. H.F. Naguib, M.S. Abdel Aziz, S.M. Sherif, and G.R. Saad,
Appl. Clay Sci., 57, 55 (2012).
12. D.D.J. Rousseaux, M. Sclavons, P. Godard, and J.M.
Brynaert, React. Funct. Polym., 72, 17 (2012).
13. W. Zhen and C. Lu, Appl. Surf. Sci., 258, 6969 (2012).
14. M. Deka, A. Kumar, H. Deka, and N. Karak, Ionics, 18,
181 (2012).
15. M. Okamoto, Advance in Polymeric Nanocomposites, CMCPublishers, Tokyo (2004).
16. T.J. Pinavaia and G. Beall, Polymeric Clay Nanocomposites,
Wiley, New York (2000).
17. A. Arora, V. Choudhary, and D.K. Sharma, J. Polym. Res.,18, 843 (2011).
18. S.R. Razin, M.S. Kalajahi, V.H. Asl, and H.R. Mamaqani,
J. Polym. Res., 19, 9954 (2012).
19. K. Khezri, V.H. Asl, H.R. Mamaqani, and M.S. Kalajahi,
Polym. Compos., 33, 990 (2012).
20. H. Zhao, S.D. Argoti, B.P. Farrell, and D.A. Shipp, J. Appl.Polym. Sci. Part A: Polym. Chem., 42, 916 (2004).
21. B. Sahu and G. Pugazhenthi, J. Appl. Polym. Sci., 120, 2485
(2011).
22. R.M. Boumbimba, M. Bouquey, R. Muller, L. Jourdainne, B.
Triki, P. Hebraud, and P. Pfeiffer, Polym. Test., 31, 800 (2012).
23. H. Datta, A.K. Bhowmick, and N.K. Singha, J. Polym. Sci.Part A: Polym. Chem., 46, 5014 (2008).
24. G. Spataro, F. Malecaze, C.O. Turrin, V. Soler, C. Duhayon,
P.P. Elenad, J.P. Majoral, and A.M. Caminade, Eur. J. Med.Chem., 45, 326 (2010).
25. G.R. Newkome and C.D. Shreiner, Polymer, 49, 1 (2008).
26. W.D. Janga, C.H. Lee, and I.K. Kang, Prog. Polym. Sci.,34, 1 (2009).
27. D.A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, and
S. Martin, Polym. J., 17, 117 (1985).
28. D.A. Tomalia, Prog. Polym. Sci., 30, 294 (2005).
29. D.L. Jianga and T. Aida, Prog. Polym. Sci., 30, 403 (2005).
30. J.M.J. Frechet and D. Tomalia, Dendrimers and Other Den-
dritic Polymers, Wiley-VCH, Weinheim (2001).
31. T. Aida and D.L. Jiang, ‘‘Dendrimer Porphyrins and Metal-
loporphyrins: Syntheses, Structure, and Functions,’’ in ThePorphyrin Handbook: Inorganic, Organometallic, and Coor-dination Chemistry, Vol. 3, K.M. Kadish, K.M. Smith, and
R. Guilard, Eds., Academic Press, New York, 369 (2000).
32. U. Boas and P.M.H. Heegaard, Chem. Soc. Rev., 33, 43 (2004).
33. C.C. Lee, J.A. MacKay, J.M.J. Frechet, and F.C. Szoka,
Nat. Biotechnol., 23, 1517 (2005).
34. S. Svenson and D.A. Tomalia, Adv. Drug Delivery Rev., 57,
2106 (2005).
35. M. Najlah and A.D. Emanuele, Curr. Opin. Pharmacol., 6,
522 (2006).
36. A.M. Caminade, C.O. Turrin, and J.P. Majoral, Chem. Eur.J., 4, 7422 (2008).
37. E.R. Gillies and J.M.J. Frechet, Drug Discov. Today, 10, 35
(2005).
38. H.L. Crampton and E.E. Simanek, Polym. Int., 56, 489
(2007).
39. A. Myc, I.J. Majoros, T.P. Thomas, and J.R. Baker, Bioma-cromolecules, 8, 13 (2007).
40. G.R. Newkome and C.D. Weiss, OPPI, 28, 495 (1996).
41. G.R. Newkome, K.K. Kotta, and C.N. Moorefield, J. Org.Chem., 70, 4893 (2005).
42. J.M. Young, G.R. Baker, G.R. Newkome, K.F. Morris, and
C.S. Johnson, Macromolecules, 27, 3464 (1994).
43. G.R. Newkome, G.R. Baker, S. Arai, M.J. Saunders, P.S.
Russo, K.J. Theriot, C.N. Moorefield, E. Rogers, J.E. Miller,
T.R. Lieux, M.E. Murray, B. Phillips, and L. Pascal, J. Am.Chem. Soc., 112, 8458 (1990).
44. C.J.G. Plummer, M. Rodlert, and J.A.E. Manson, Chem.Mater., 14, 486 (2002).
45. A. Amin, A.S. Taha, and M.A. Abd El-Ghaffar, J. Appl.Polym. Sci., 118, 525 (2010).
46. H. Datta, A.K. Bhowmick, and N.K. Singha, J. Polym. Sci.Part A: Polym. Chem., 46, 5014 (2008).
2174 POLYMER ENGINEERING AND SCIENCE—-2013 DOI 10.1002/pen
Top Related
Copyright © 2022 FDOKUMEN

