







Bahasa
Halaman
Hukum


Preparation and stabilization of gold nanoparticles formedby in situ reduction of aqueous chloroaurate ionswithin surface-modified mesoporous silica
Anirban Ghosh a, Chitta Ranjan Patra a, Priyabrata Mukherjee a,Murali Sastry b,*, Rajiv Kumar a,*
a Catalysis Division, National Chemical Laboratory, Pune 411008, Indiab Materials Chemistry Division, National Chemical Laboratory, Pune 411008, India
Received 1 July 2002; received in revised form 14 October 2002; accepted 18 November 2002
Abstract
A convenient method for the synthesis and simultaneous stabilization of gold nanoparticles in organo-functionalized
MCM-41 is described. The silanol groups present on the surface of the mesoporous matrix reduce aqueous chloroaurate
ions, resulting in the formation of gold nanoparticles, which are subsequently anchored to the host matrix via organic
functionality. The method, in which propylamine-MCM-41 or propylthiol-MCM-41 is treated with a dilute aqueous
solution of HAuCl4 at ambient conditions, is quite simple and environmentally benign as there is no need to add any
external reducing agent. The nano-Au-MCM-41 hybrid materials were characterized by XRD, UV–Vis, TEM, XPS,
EDX and N2 adsorption measurements.
� 2002 Elsevier Science Inc. All rights reserved.
Keywords: Auto-reduction; Gold nanoparticle; MCM-41; Organo-functionalized mesoporous silica; Organo-inorganic nanocomposite
1. Introduction
The discovery of mesoporous materials such asMCM-41 [1,2] with pore sizes ranging from 2 to 10
nm has opened up research not only in the field of
catalysis [3] but in materials science [4,5] as well.
Surface-modified MCM-41 materials [6], in which
the surface of MCM-41 contains organic func-
tionality, such as propylamine, propylthiol, vinyl,
etc., are emerging as important hosts for metallic
and semiconductor nanoparticles to form noveladvanced hybrid materials [7–9].
Generally, surfactant molecules containing
polar head groups like –SH, NH2, etc. are used
to stabilize nanoparticles and to prevent their ag-
glomeration to get the quantum dots. Propyl-
amine- and propylthiol-functionalized MCM-41
materials are particularly important in the sense
that they have the polar head group generally re-quired to stabilize the nanoparticles. Further, they
provide a solid support of well-defined pores to
the nanoclusters. Therefore, these nanocomposites
*Corresponding authors. Fax: +91-20-589-3952/3761.
E-mail addresses: [email protected] (M. Sastry), rajiv@
cata.ncl.res.in (R. Kumar).
1387-1811/02/$ - see front matter � 2002 Elsevier Science Inc. All rights reserved.doi:10.1016/S1387-1811(02)00626-1
Microporous and Mesoporous Materials 58 (2003) 201–211
www.elsevier.com/locate/micromeso

can be visualized as a three-layered entity, where
the outer sphere is the inorganic support, the
second sphere is the organic moiety attached to
the surface of MCM-41 and protruding inside the
pores, and the third sphere consists of the nano-
particles hooked to the polar head groups of theorganic moiety present inside the pores. These
three-layered nanocomposites may lead to some
interesting non-linear optical properties for future
opto-electronic applications [10], and they are
excellent materials for studying host–guest inter-
actions [11]. The stabilization of nanoparticles
inside the well-defined pores of MCM-41 materials
may lead to the formation of nanowires, which arethe building units for future nano-electronic cir-
cuits.
However, in most reports on the formation
and stabilization of metallic nanoparticles, except
those of Esumi et al. [12] using sugar balls and
Mukherjee et al. [13–15] using microorganisms, the
hosts are usually passive and do not actively par-
ticipate in the reduction of metal ions to formnanoparticles followed by their entrapment in the
host matrix. Some external methods, either physi-
cal (vapor deposition, lithography or radiolysis) or
chemical (reduction by borohydride, citrate etc.)
have to be employed for the formation of nano-
particles prior to their stabilization.
Realizing the immense importance of nano-
composites in various applications, we have re-cently developed a novel method for the synthesis
of gold nanoparticles by in situ reduction of
aqueous chloroaurate ions (AuCl�4 ) via silanol
groups present on the surface of amorphous silica
[16] or on the surfaces of organo-functionalized
MCM-41 [17]. Employing this method, we can
avoid the use of any external-reducing environ-
ment other than the host matrix. Now, we reportour detailed studies on the synthesis of gold
nanoparticles of controlled and uniform size
and their simultaneous stabilization using propyl-
amine- and propylthiol-functionalized MCM-41
materials in a single step. The organic functional
groups act as anchors for the nanoparticles
by binding them through covalent interactions,
thereby avoiding the requirement of external cap-ping agents like alkylamines or alkylthiols for
particle size control [16,18].
2. Experimental
Two different procedures were adopted for
the synthesis of siliceous MCM-41 (Si-MCM-41),
propylamine-modified MCM-41 (NH2-MCM-41)and propylthiol-modified MCM-41 (SH-MCM-
41) materials, as described in detail elsewhere [19,
20]. Tetraethyl orthosilicate (TEOS, Aldrich), 3-
aminopropyltrimethoxy silane (APTS, Aldrich), 3-
mercaptopropyltrimethoxy silane (MPTS, Aldrich)
and cetyltrimethylammomium bromide (CTABr,
SD Fine Chem, India) were used as received. In a
typical synthesis of organo-functionalized MCM-41 (NH2-MCM-41 or SH-MCM-41), the gel
composition was 1.0TEOS–yXPTS–0.21CTABr–0.32NaOH–75H2O–16MeOH [XPTS ¼ APTS orMPTS, y ¼ 0:17–0.4]. However, the samples pre-pared with y ¼ 0:4 were used in the majority of theexperiments. Methanol was used in the initial gel
mixture to reduce and control the fast hydrolysis
of XPTS. The reaction mixture was first stirred atroom temperature for 12 h, transferred to a 250
cm3 polypropylene bottle, and heated at 100 �C for36 h under static conditions and autogenous
pressure.
For the synthesis of siliceous MCM-41 (Si-
MCM-41) [19] two types of silica sources were
used, viz. sodium metasilicate (Na2SiO3, SD Fine
Chem, India) and fumed silica (SiO2, Aldrich),where the Na2SiO3:SiO2 ratio was 0.124:1. The
silica sources were suspended in a 25% aque-
ous solution of tetramethylammonium hydroxide
(TMAOH, Aldrich). This suspension was then
combined with an aqueous solution of CTABr.
The initial gel composition was 1.0SiO2–0.22
NaOH–0.1TMAOH–0.21CTABr–125H2O. The
synthesis gel was transferred to a 250 cm3 poly-propylene bottle and kept at 100 �C for 36 h. Allthe as-synthesized mesoporous samples were
washed with distilled water and acetone, and dried
at 95 �C for 4 h. The surfactant from the NH2-MCM-41 and SH-MCM-41 (1 g each) materials
was removed by solvent extraction with a mixture
of solvents containing 85 g of methanol and 2 g of
HCl (35.5%) under reflux conditions for 24 h [20].The as-prepared Si-MCM-41 material was calc-
ined in air at 540 �C for 12 h for complete removalof the template.
202 A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211

The Si-MCM-41, NH2-MCM-41 and SH-
MCM-41 materials were characterized by low-
angle X-ray diffraction (XRD) recorded on a
Rigaku D Max III VC instrument with Ni filtered
CuKa radiation (k ¼ 1:5404 �AA) in the 2h range of1.5–10� at a scan rate of 1�/min. The specific sur-face areas of the samples were determined by the
BET method through N2 adsorption at 77 K using
an Omnisorb CX-100 Coulter instrument. Prior to
the adsorption experiments, the samples were ac-
tivated at 150 �C for 6 h at 1:3� 10�2 Pa. The poresize distributions of the samples were computed by
the BJH model. Chemical analyses of the NH2-
MCM-41 and SH-MCM-41 materials were doneon a Carlo Erba EA1108 elemental analyzer.
Nano-Au-MCM-41 hybrid materials were
formed by in situ reduction of AuCl�4 ions as
follows: In a typical experiment, 1.0 g of the Si-
MCM-41, NH2-MCM-41 or SH-MCM-41 mate-
rial were separately treated with 100 cm3 of a 10�4
M HAuCl4 solution for 96 h. After the specified
duration, the samples were filtered, washed thor-oughly with copious amounts of water and ace-
tone and finally dried under vacuum at room
temperature. Thereafter, 0.5 g of the nano-Au–Si-
MCM-41, nano-Au–NH2-MCM-41 or nano-Au–
SH-MCM-41 material were separately stirred with
50 cm3 of distilled water for 12 h. The materials
thus obtained after aqueous treatment (designated
as Au–Si-MCM-41-w, Au–NH2-MCM-41-w andAu–SH-MCM-41-w, respectively) were filtered,
washed with water and dried under vacuum. All
the nano-Au-MCM-41 hybrid materials were
characterized by XRD, UV–Vis spectroscopy,
transmission electron microscopy (TEM), X-ray
photoelectron spectroscopy (XPS), energy-disper-
sive X-ray (EDX) analysis and N2 adsorption
measurements. Further, UV–Vis spectra of all thefiltrates obtained in the above experiments were
taken.
The UV–Vis spectra of the nano-Au-MCM-41
hybrid materials were recorded on a Shimadzu
UV-2101PC spectrophotometer operating in the
reflection mode at a resolution of 2 nm using bar-
ium sulphate as a standard for background cor-
rection. Further, UV–Vis spectra of all the filtratesobtained in the above experiments were taken on
the same instrument using distilled water as a
standard for background correction. In order to
estimate the size of the gold nanoparticles formed
by spontaneous reduction of chloroaurate ions,
XRD measurements of the nano-Au-MCM-41
hybrid materials were carried out on a Philips PW
1830 instrument operating at 40 kV voltage and acurrent of 30 mA with CuKa radiation betweenthe 2h range 30� and 45� at a scan rate of 1�/min.The nano-Au-MCM-41 samples were dispersed by
acetone on Holey carbon grids and TEM images
were scanned on a Jeol Model 1200 EX instrument
operated at an accelerating voltage of 100 kV. The
XPS measurements of the nano-Au-MCM-41 hy-
brid materials were carried out on a VG Micro-Tech ESCA 3000 instrument at a pressure better
than 1:3� 10�7 Pa. The general scan and C 1s,S 2p, N 1s, Si 2p and Au 4f core level spectra were
recorded with non-monochromatized MgKa ra-diation (photon energy ¼ 1253:6 eV) at a passenergy of 50 eV and an electron takeoff angle
(angle between electron emission direction and
surface plane) of 60�. The overall resolution of themeasurements was thus approximately 1 eV for the
XPS measurements. The core level spectra were
background-corrected using the Shirley algorithm
[21] and the chemically distinct species resolved
using a non-linear least squares procedure. The
core level binding energies (BE) were aligned with
respect to the Au 4f7=2 binding energy of 84 eV.
EDX analyses of the materials were carried out ona Kevex equipment attached to a Jeol JSM-5200
scanning microscope.
Catalytic hydrogenation reactions of different
olefinic substrates on the nano-Au-X-MCM-41
materials were performed in a 100 cm3 high-pres-
sure autoclave at a temperature of 100 �C and ahydrogen pressure of 4 MPa for 6 h at 400 rpm.
The reaction mixtures were analyzed by a Shima-dzu 17A series gas chromatograph containing a
capillary column (10% permethylated b-cyclodex-trin, 30 m� 0:32 mm� 0:25 lm film thickness)and a flame ionization detector.
3. Results and discussion
The porosity of all mesoporous samples was
evaluated via N2 adsorption. Fig. 1A and B show
A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211 203

typical N2 adsorption–desorption isotherms and
the corresponding pore size distribution curves
(insets) for the Si-MCM-41 and nano-Au–Si-
MCM-41 samples, respectively. All the samples
showed isotherms of type IV with inflection points
around P=P0 ¼ 0:3–0:45, which is characteristicof M41S type ordered mesoporous materials.
The samples exhibit complementary textural and
framework-confined mesoporosity, as evidenced
by the presence of two separate, well-defined hys-
teresis loops. One is in the P=P0 ¼ 0:3–0:45 regionand indicates framework-confined mesopores, and
the other one occurs at P=P0P 0:8 correspondingto capillary condensation in the inter-particle
pores [22]. The position of the inflection point inthe P=P0 ¼ 0:3–0:45 region depends on the dia-meter of the mesopores, and its sharpness indicates
the uniformity of the narrow pore size distribu-
tion. The specific BET surface areas, pore volumes
and average pore diameters calculated from the N2adsorption isotherms using the BJH model are
summarized in Table 1. After in situ reduction of
the AuCl�4 ions by the mesoporous samples, thenature of the N2 adsorption isotherms remained
the same, but a decrease of approximately 9%,
20% and 22% in the surface areas of nano-Au–Si-
MCM-41, nano-Au–NH2-MCM-41 and nano-
Au–SH-MCM-41, respectively, was observed. The
pore volumes of the materials also decreased by
approximately 8%, 18% and 22%, respectively,
after treatment with AuCl�4 . However, the averagepore diameters of the samples after entrapment of
Au nanoparticles did not change considerably,
indicating filling of part of the mesopores by gold
nanoparticles keeping the mesoporous structure
intact.
Results of elemental analyses of different NH2-
MCM-41 and SH-MCM-41 materials synthesized
with different TEOS: XPTS ratios, and EDX an-alyses of the corresponding nano-Au–NH2-MCM-
41 and nano-Au–SH-MCM-41 samples are given
in Table 2. From the table, it is evident that the
concentration of Au in the silicate matrices does
not significantly depend upon the population of
pendant –NH2 or –SH groups in the inner surfaces
of MCM-41.
After treatment of the Si-MCM-41, NH2-MCM-41 and SH-MCM-41 materials with AuCl�4ions for 96 h, it was observed that all materials had
attained a deep pink color, evidently due to the
presence of Au nanoparticles in the cavities of the
silicate matrix. Au nanoparticles, due to their
surface plasmon vibrations, have a characteristic
absorption band in the visible region of the elec-
tromagnetic spectrum at around 520–550 nm,which is responsible for the striking violet to pink
range of colors of the nanoparticles depending
Fig. 1. N2 adsorption–desorption isotherms and corresponding
pore size distribution curves (insets) for (A) Si-MCM-41 and
(B) nano-Au–Si-MCM-41 samples.
204 A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211

upon the particle size [23,24]. Fig. 2A shows theUV–Vis spectra recorded on the (a) parent Si-
MCM-41, (b) nano-Au–Si-MCM-41, and (c) Au–
Si-MCM-41-w materials. Fig. 2B presents the
UV–Vis spectra of (a) parent NH2-MCM-41,
(b) nano-Au–NH2-MCM-41, and (c) Au–NH2-
MCM-41-w. Fig. 2C shows the UV–Vis spectra of
the (a) parent SH-MCM-41, (b) nano-Au–SH-
MCM-41, and (c) Au–SH-MCM-41-w. A strongabsorption at approxmately 540 nm is observed
for all the mesoporous materials after treatment
with HAuCl4 solution and is a clear indication of
reduction of the AuCl�4 ions by all the three ma-
terials, viz. Si-MCM-41 (Fig. 2A, curve b), NH2-
MCM-41 (Fig. 2B, curve b), and SH-MCM-41
(Fig. 2C, curve b). This resonance is absent in the
parent Si-MCM-41 (Fig. 2A, curve a), NH2-MCM-41 (Fig. 2B, curve a) and SH-MCM-41
(Fig. 2C, curve a) materials as expected. An in-
teresting observation is the presence of an addi-
tional resonance at approximately 725 and 710 nm
in the case of the nano-Au–NH2-MCM-41 (Fig.
2B, curve b) and nano-Au–SH-MCM-41 (Fig. 2C,
curve b) materials, respectively. This feature arisesdue to excitation of longitudinal surface plasmon
vibrations due to close packing of the gold nano-
particles [25,26] within the cavities of these mate-
rials, which shows from anchoring of the gold
nanoparticles to the inner surface of the siliceous
matrices by the amine or thiol groups. It is known
that primary amines and thiols covalently bind to
gold nanoparticles [18], and may be the bindingmode in our study also.
To assess the stability of the gold nanoparticles
formed by in situ reduction within the MCM-41
matrix, the nano-Au–Si-MCM-41, nano-Au–NH2-
MCM-41 and nano-Au–SH-MCM-41 materials
were separately treated with distilled water for 12
h, filtered and dried. The UV–Vis spectra of the
samples after aqueous treatment, namely Au–Si-MCM-41-w (curve c), Au–NH2-MCM-41-w
(curve c) and Au–SH-MCM-41-w (curve c) are
shown in Fig. 2A–C, respectively. The character-
istic absorption at approximately 540 nm of gold
nanoparticles happens to be missing in Au–Si-
MCM-41-w (Fig. 2A, curve c), clearly indicating
Table 1
Pore diameters, pore volumes, BET surface areas, d1 0 0 spacings, unit cell parameters (a0) and framework thicknesses (FWT) of theMCM-41 samples
Sample Pore diameter (�AA) Pore volume (cm3 g�1) BET SA (m2 g�1) d1 0 0 (�AA) a0a (�AA) FWTb (�AA)
Si-MCM-41 36.61 1.14 1022 42.64 49.24 12.63
Nano-Au–Si-MCM-41 36.55 1.05 927 42.43 48.99 12.44
NH2-MCM-41 35.77 0.96 747 42.03 48.53 12.76
Nano-Au–NH2-MCM-41 35.69 0.79 596 41.83 48.30 12.61
SH-MCM-41 33.79 0.91 683 40.11 46.31 12.52
Nano-Au–SH-MCM-41 33.65 0.71 535 39.94 46.12 12.47
a a0 ¼ 2d1 0 0=ffiffiffi
3p.
b FWT ¼ a0 � pore diameter.
Table 2
Results of elemental analyses, and mean diameters of Au nanoparticles in different nano-Au-X-MCM-41 samples
Sample XPTSa:TEOS N (mmol g�1) S (mmol g�1) Au (wt.%) Mean diameter of Au nanoparticlesb (nm)
NH2-MCM-41 0.17 1.41 – 3.1 3.4� 0.50.25 2.06 – 3.2 3.4� 0.50.4 3.24 – 3.1 3.4� 0.5
SH-MCM-41 0.17 – 1.37 3.4 3.2� 0.50.25 – 2.01 3.5 3.2� 0.50.4 – 3.21 3.2 3.2� 0.5
aXPTS ¼ APTS or MPTS.bCalculated from the Debye–Scherrer equation.
A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211 205
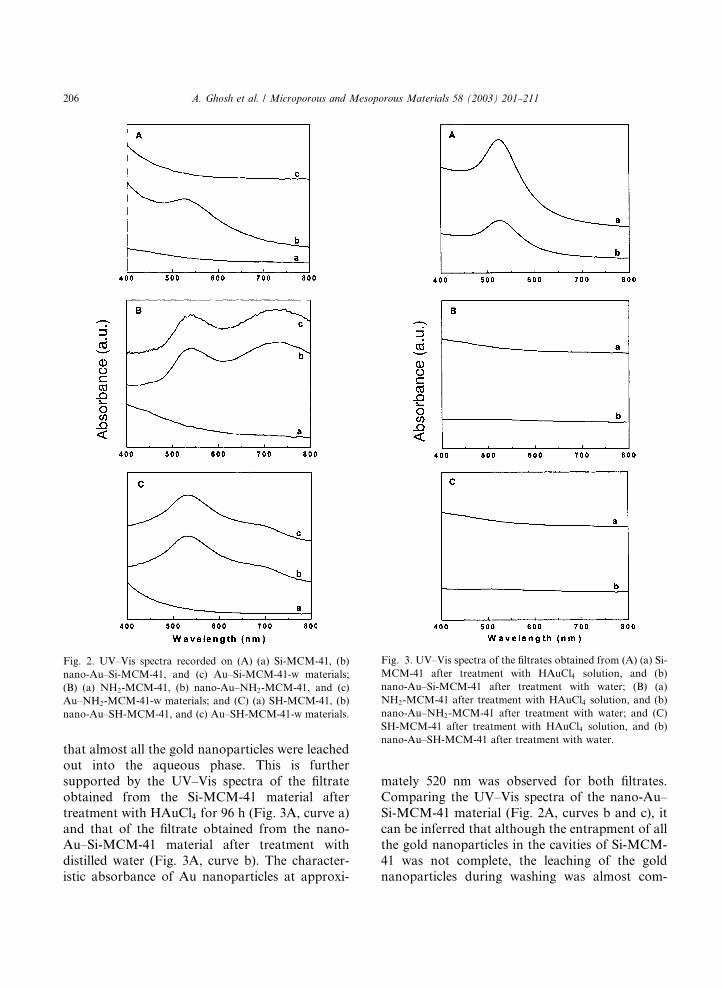
that almost all the gold nanoparticles were leached
out into the aqueous phase. This is further
supported by the UV–Vis spectra of the filtrate
obtained from the Si-MCM-41 material after
treatment with HAuCl4 for 96 h (Fig. 3A, curve a)
and that of the filtrate obtained from the nano-Au–Si-MCM-41 material after treatment with
distilled water (Fig. 3A, curve b). The character-
istic absorbance of Au nanoparticles at approxi-
mately 520 nm was observed for both filtrates.
Comparing the UV–Vis spectra of the nano-Au–
Si-MCM-41 material (Fig. 2A, curves b and c), it
can be inferred that although the entrapment of all
the gold nanoparticles in the cavities of Si-MCM-
41 was not complete, the leaching of the goldnanoparticles during washing was almost com-
Fig. 2. UV–Vis spectra recorded on (A) (a) Si-MCM-41, (b)
nano-Au–Si-MCM-41, and (c) Au–Si-MCM-41-w materials;
(B) (a) NH2-MCM-41, (b) nano-Au–NH2-MCM-41, and (c)
Au–NH2-MCM-41-w materials; and (C) (a) SH-MCM-41, (b)
nano-Au–SH-MCM-41, and (c) Au–SH-MCM-41-w materials.
Fig. 3. UV–Vis spectra of the filtrates obtained from (A) (a) Si-
MCM-41 after treatment with HAuCl4 solution, and (b)
nano-Au–Si-MCM-41 after treatment with water; (B) (a)
NH2-MCM-41 after treatment with HAuCl4 solution, and (b)
nano-Au–NH2-MCM-41 after treatment with water; and (C)
SH-MCM-41 after treatment with HAuCl4 solution, and (b)
nano-Au–SH-MCM-41 after treatment with water.
206 A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211

plete. Moreover, a weak absorbance at approxi-
mately 520 nm for the filtrate obtained after aque-
ous treatment of the nano-Au–Si-MCM-41material
(Fig. 3A, curve b) is further indicative of the
leaching of the gold nanoparticles in the aqueous
phase, which was previously inferred from the UV–Vis spectra of the Au–Si-MCM-41-w material (Fig.
2A, curve c). The UV–Vis spectra of the filtrates
obtained from the NH2-MCM-41 (Fig. 3B, curve a)
and SH-MCM-41 (Fig. 3C, curve a) materials after
treatment with HAuCl4, and those of the filtrates
obtained from the nano-Au–NH2-MCM-41 (Fig.
3B, curve b) and nano-Au–SH-MCM-41 (Fig. 3C,
curve b) materials after aqueous treatment did notshow any absorbance in this visible region, clearly
indicating that there is no leaching of Au nano-
particles in the case of NH2-MCM-41 and SH-
MCM-41 materials. Further, the UV–Vis spectra of
the Au–NH2-MCM-41-w (Fig. 2B, curve c) and
Au–SH-MCM-41-w (Fig. 2C, curve c) materials,
which are almost similar to those of the nano-
Au–NH2-MCM-41 (Fig. 2B, curve b) and nano-Au–SH-MCM-41 (Fig. 2C, curve b) materials,
respectively, including the additional absorbance at
approximately 725 and 710 nm, respectively, indi-
cate the stabilization of the close-packing of gold
nanoparticles in the open, string-like networks in-
side NH2-MCM-41 and SH-MCM-41.
Since all the organo-functionalized MCM-41
materials were synthesized by the ‘‘one-pot co-condensation’’ method, which leads to grafting of
all the organic functional groups on the inner
surfaces of the mesopores [27], quite obviously the
Au nanoparticles would be stabilized within the
mesopores only, and not on the outer surfaces, as
there is no binding moiety present on the outer
surfaces. So we can conclusively say that the Au
nanoparticles are present inside the pores of theorgano-functionalized MCM-41 materials, which
was previously inferred from the decrease in sur-
face area and pore volume of the materials.
Fig. 4 shows the low-angle XRD patterns of (a)
NH2-MCM-41, (b) nano-Au–NH2-MCM-41, (c)
SH-MCM-41, (d) nano-Au–SH-MCM-41, (e) Si-
MCM-41, and (f) nano-Au–Si-MCM-41. The
main (1 0 0) Bragg reflection is visible in all thematerials along with the weak (1 1 0), (2 0 0) and
(2 1 0) reflections, indicating a high degree of or-
dered hexagonal mesopores even after in situ for-
mation of gold nanoparticles within the cavities.
The d1 0 0 values along with the corresponding unitcell parameter (a0) of the different MCM-41 sam-ples are given in Table 1. The a0 values arecalculated by the equation a0 ¼ 2d1 0 0=
ffiffiffi
3p. To
evaluate the average size of the gold nanoparticles,
XRD measurements of the nano-Au–NH2-MCM-41 and nano-Au–SH-MCM-41 were carried out in
the 2h range from 30� to 45� with a scan rate of 1�/min. The diffraction patterns in the region of the
(1 1 1) Bragg reflection (2h ¼ 38:2�) are shown inthe inset of Fig. 4, the solid lines being the Lo-
rentzian fits to the respective (1 1 1) reflections.
Under the experimental conditions of the XRD
Fig. 4. XRD patterns of (a) NH2-MCM-41, (b) nano-Au–
NH2-MCM-41, (c) SH-MCM-41, (d) nano-Au–SH-MCM-41,
(e) Si-MCM-41, and (f) nano-Au–Si-MCM-41. Inset shows the
diffraction patterns of (a) nano-Au–SH-MCM-41 and (b) nano-
Au–NH2-MCM-41 in the region of the Au (1 1 1) Bragg re-
flection at 2h ¼ 38:2�, the solid lines being the Lorentzian fit tothe respective curves.
A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211 207

measurement (small slit width and monochroma-
tized CuKa radiation), it was felt that the XRDline-shape would be better approximated by a
Lorentzian than a Gaussian. Indeed, poorer chi-
square error values (which denote the goodness of
fit) were observed while fitting the (1 1 1) Braggreflection of gold to a Gaussian line-shape. A
similar pattern and peak width was obtained for
the nano-Au–Si-MCM-41 material (not shown).
The mean diameter of the gold nanoparticles was
estimated from the broadening of the (1 1 1) peak
using the Debye–Scherrer equation [28], which
yielded the values of approximately 3:4� 0:5 nmfor nano-Au–NH2-MCM-41 (inset of Fig. 4, curveb) and approximately 3:2� 0:5 nm for nano-Au–SH-MCM-41 (inset of Fig. 4, curve a), respec-
tively. These values are in good agreement with the
maximum possible particle size that can be ac-
commodated within the mesopores of the NH2-
MCM-41 and SH-MCM-41 materials, the mean
pore diameters of which, as we recollect, are 3.6
and 3.4 nm, respectively. The average diameter ofthe Au nanoparticles does not depend upon the
concentration of pendant –NH2 or –SH groups, as
evident from the samples prepared with different
TEOS: XPTS ratios (Table 2).
The TEM images of the nano-Au–NH2-MCM-
41 sample are given in Fig. 5A and B, the inset of
Fig. 5A showing a selected area electron diffrac-
tion pattern. The parallel fringes due to the side-onview of the long pores of MCM-41 are well visible
in both images. These equidistant parallel fringes
are characteristic features of separate layers, the
addition of which results in the formation of a
bunch of layers. The electron diffraction pattern
of the sample exhibits well-defined hexagonal
maxima, further confirming the periodicity of
the structure. The entrapped Au nanoparticles arevisible in Fig. 5B (highlighted by arrows in the
figure), which shows that the spatial distribution of
the nanoparticles in the perpendicular direction to
the channels is consistent with the pore spacing.
A chemical characterization of the nano-Au–
SH-MCM-41 and nano-Au–NH2-MCM-41 mate-
rials was carried out using XPS. Fig. 6 shows the
Au 4f and N1s core levels from the nano-Au–NH2-MCM-41 material (A and B) as well as the
Au 4f and S 2p core levels recorded from the nano-
Au–SH-MCM-41 sample (C and D). Strong sig-
nals from the gold nanoparticles entrapped in the
mesoporous silica matrix can be seen in both
samples (Fig. 6A and C). These core levels could
be fit to a single spin–orbit pair indicating no
chemical shifts arising due to complexation with–NH2 and –SH pendant groups in the MCM-41
matrix. The S 2p signal from the nano-Au–SH-
MCM-41 sample (Fig. 6D) could be satisfactorily
fit to a single spin–orbit pair separated by 1.14 eV.
It is interesting to note the absence of an addi-
tional high binding energy component in the S 2p
spectrum at approximately 168 eV that is often
observed in self-assembled monolayers of alkane-thiols on silver alloy surfaces [29]. The fact that
this component is not observed indicates that the
Fig. 5. (A,B) Representative TEM images of nano-Au–NH2-MCM-41. The arrows in (B) identify gold nanoparticles within the pores
of the NH2-MCM-41 material. The inset of (A) shows a selected area electron diffraction pattern recorded from the same sample.
208 A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211
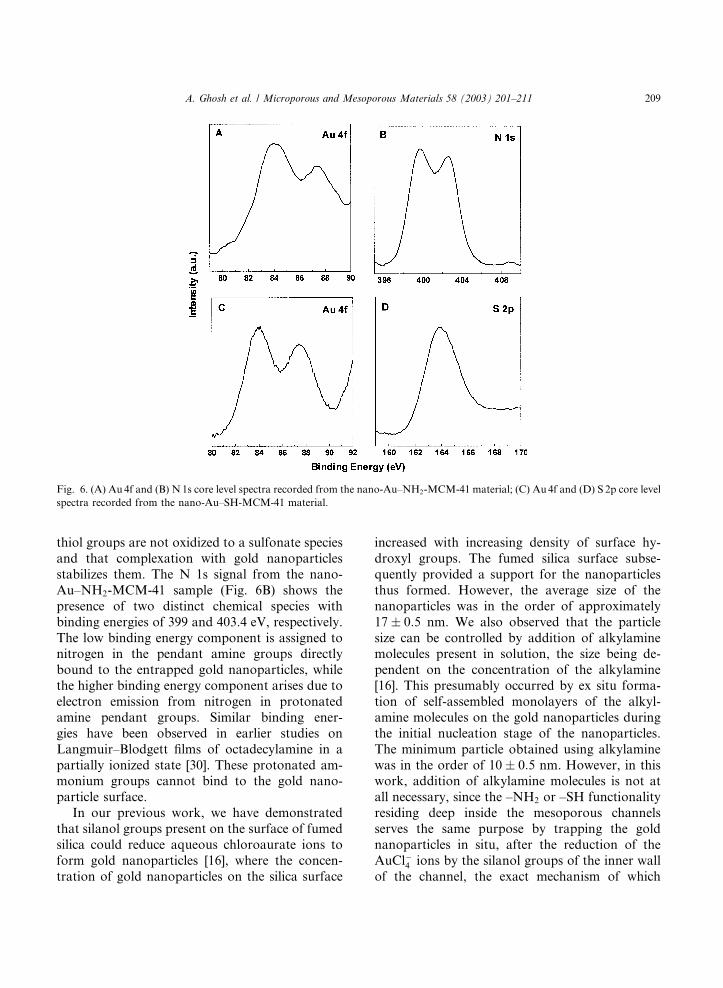
thiol groups are not oxidized to a sulfonate species
and that complexation with gold nanoparticles
stabilizes them. The N 1s signal from the nano-
Au–NH2-MCM-41 sample (Fig. 6B) shows the
presence of two distinct chemical species with
binding energies of 399 and 403.4 eV, respectively.
The low binding energy component is assigned tonitrogen in the pendant amine groups directly
bound to the entrapped gold nanoparticles, while
the higher binding energy component arises due to
electron emission from nitrogen in protonated
amine pendant groups. Similar binding ener-
gies have been observed in earlier studies on
Langmuir–Blodgett films of octadecylamine in a
partially ionized state [30]. These protonated am-monium groups cannot bind to the gold nano-
particle surface.
In our previous work, we have demonstrated
that silanol groups present on the surface of fumed
silica could reduce aqueous chloroaurate ions to
form gold nanoparticles [16], where the concen-
tration of gold nanoparticles on the silica surface
increased with increasing density of surface hy-
droxyl groups. The fumed silica surface subse-
quently provided a support for the nanoparticles
thus formed. However, the average size of the
nanoparticles was in the order of approximately
17� 0:5 nm. We also observed that the particlesize can be controlled by addition of alkylaminemolecules present in solution, the size being de-
pendent on the concentration of the alkylamine
[16]. This presumably occurred by ex situ forma-
tion of self-assembled monolayers of the alkyl-
amine molecules on the gold nanoparticles during
the initial nucleation stage of the nanoparticles.
The minimum particle obtained using alkylamine
was in the order of 10� 0:5 nm. However, in thiswork, addition of alkylamine molecules is not at
all necessary, since the –NH2 or –SH functionality
residing deep inside the mesoporous channels
serves the same purpose by trapping the gold
nanoparticles in situ, after the reduction of the
AuCl�4 ions by the silanol groups of the inner wall
of the channel, the exact mechanism of which
Fig. 6. (A) Au 4f and (B) N1s core level spectra recorded from the nano-Au–NH2-MCM-41 material; (C) Au4f and (D) S 2p core level
spectra recorded from the nano-Au–SH-MCM-41 material.
A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211 209

requires further work to be understood. The meanparticle sizes obtained here are far less than those
obtained earlier by fumed silica and alkylamines.
It is pertinent to mention here that the size of the
gold nanoparticles is of utmost importance in in-
fluencing their catalytic activity. Haruta [31] and
Bond and Thompson [32] have demonstrated in
recent years, that gold nanoparticles of an average
diameter ranging between 2 and 4 nm exhibit ex-cellent catalytic activity in a variety of reactions
including decomposition of trace amounts of am-
ines and conversion of CO to CO2. Hence, our
approach for the preparation and stabilization of
gold nanoparticles in the cavities of the ordered
MCM-41 type materials in the size range of ap-
proximately 3–4 nm offers distinct advantages in
exploiting the catalytic potential of these hybridmaterials, as has been briefly demonstrated in the
hydrogenation of styrene to ethylbenzene in our
recent short communication [17]. To evaluate the
catalytic potential of the nano-Au-MCM-41
hybrid materials, the hydrogenation of differ-
ent olefinic substrates was carried out in a high-
pressure autoclave. The results are summarized
in Table 3. From these results, it is evident thatthe nano-Au–NH2-MCM-41 and nano-Au–SH-
MCM-41 materials have fairly comparable cata-
lytic activities.
4. Conclusions
Gold nanoparticles can be synthesized and en-trapped within the pores of propylamine- and
propylthiol-functionalized MCM-41 materials by
auto-reduction of aqueous chloroaurate ions,which occurs via silanol groups present on the
inner surface of the mesopores. The Au-nano-X-
MCM-41 (where X ¼ –NH2 or –SH) materials are
examples for inorgano–organic nanocomposites
where the mesoporous matrix actively participates
in the reduction of the metal ions and acts as a
support for the nanoparticles through covalent
linkage by the pendant organic functionality,thereby perfectly accommodating them inside the
mesopores. The dimension of the pores in the
mesoporous material is in accordance with the size
range of the gold nanoparticles where they show
excellent catalytic activity. This makes the Au-
MCM-41 nano-sized hybrid materials quite excit-
ing for future applications in catalysis.
Acknowledgements
AG and CRP gratefully acknowledge the
Council of Scientific and Industrial Research,
Government of India for granting research fel-
lowships. The authors thank Drs. A.B. Mandale
and R. Parischa for their helpful co-operation incarrying out XPS and TEM experiments.
References
[1] C.T. Kresge, M.E. Leonowicz, W.J. Roth, J.C. Vartuli, J.S.
Beck, Nature 359 (1992) 710.
[2] J.S. Beck, J.C. Vartuli, W.J. Roth, M.E. Leonowicz, C.T.
Kresge, K.D. Schmitt, C.T.-W. Chu, D.H. Olson, E.W.
Sheppard, S.B. McCullen, J.B. Higgins, J.L. Schlenker, J.
Am. Chem. Soc. 114 (1992) 10834.
[3] A. Corma, Chem. Rev. 97 (1997) 2373.
Table 3
Results of catalytic hydrogenation of different olefinic substratesa by nano-Au-X-MCM-41 materials
Catalyst Substrate Conversion (mol%) Selectivityb (%)
Nano-Au–NH2-MCM-41 Cyclohexene 8.9 100
1-Hexene 18.5 100
1-Octene 13.5 100
Nano-Au–SH-MCM-41 Cyclohexene 9.7 100
1-Hexene 18.7 100
1-Octene 12.9 100
aReaction conditions: substrate ¼ 10 mmol; catalyst ¼ 200 mg, solvent ¼ toluene, 30 cm3; temperature ¼ 100 �C; H2 pressure ¼ 4MPa; reaction time ¼ 6 h.b Selectivity for cyclohexane, n-hexane and n-octane, respectively.
210 A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211

[4] Y. Sakamoto, M. Kaneda, O. Terasaki, D. Zhao, J.M.
Kim, G.D. Stucky, H.J. Shin, R. Ryoo, Nature 408 (2000)
449.
[5] Q. Huo, D.I. Margolese, G.D. Stucky, Chem. Mater. 8
(1996) 1147.
[6] X. Feng, G.E. Fryxell, L.-Q. Wang, A.Y. Kim, J. Liu,
K.M. Kemner, Science 276 (1997) 923.
[7] P. Mukherjee, M. Sastry, R. Kumar, Phys. Chem. Comm.
3 (2000) 15.
[8] Y. Xu, C.H. Langford, J. Phys. Chem. B 101 (1997) 3115.
[9] T. Hirai, H. Okubo, I. Komasawa, J. Phys. Chem. B 103
(1999) 4228.
[10] D.H. Gracias, J. Tien, T.L. Breen, C. Hsu, G.M. White-
sides, Science 289 (2000) 1170.
[11] K. Moller, T. Bein, Chem. Mater. 10 (1998) 2950.
[12] K. Esumi, T. Hosoya, A. Suzuki, K. Torigoe, Langmuir 16
(2000) 2978.
[13] P. Mukherjee, A. Ahmad, D. Mandal, S. Senapati, S.R.
Sainkar, M.I. Khan, R. Ramani, R. Parischa, P.V.
Ajayakumar, M. Alam, M. Sastry, R. Kumar, Angew.
Chem. Int., Ed. Engl. 40 (2001) 3585.
[14] P. Mukherjee, A. Ahmad, D. Mandal, S. Senapati, S.R.
Sainkar, M.I. Khan, R. Parischa, P.V. Ajayakumar, M.
Alam, R. Kumar, M. Sastry, Nano Lett. 1 (2001) 515.
[15] P. Mukherjee, S. Senapati, D. Mandal, A. Ahmad, M.I.
Khan, R. Kumar, M. Sastry, Chem. Bio. Chem. 3 (2002)
461.
[16] P. Mukherjee, C.R. Patra, A. Ghosh, M. Sastry, R.
Kumar, Chem. Mater. 14 (2002) 1678.
[17] P. Mukherjee, C.R. Patra, R. Kumar, M. Sastry, Phys.
Chem. Comm. 4 (2001) 24.
[18] D.V. Leff, L. Brandt, J.R. Heath, Langmuir 12 (1996)
4723.
[19] P. Mukherjee, R. Kumar, U. Schuchardt, in: L. Bonneviot,
F. B�eeland, C. Danumah, S. Giasson, S. Kaliaguine (Eds.),
Mesoporous Molecular Sieves, Studies in Surface Science
and Catalysis, vol. 117, Elsevier, Amsterdam, 1998, p. 351.
[20] P. Mukherjee, S. Laha, D. Mandal, R. Kumar, in: A.
Sayari, M. Jaroniec, T.J. Pinnavaia (Eds.), Nanoporous
Materials II, Studies in Surface Science and Catalysis, vol.
129, Elsevier, Amsterdam, 2000, p. 283.
[21] D.A. Shirley, Phys. Rev. B. 5 (1972) 4709.
[22] X. Chen, L. Huang, Q. Li, J. Phys. Chem. B. 101 (1997)
8460.
[23] S. Underwood, P. Mulvaney, Langmuir 10 (1994) 3427.
[24] S. Link, M.A. El-Sayed, J. Phys. Chem. B. 103 (1999)
4212.
[25] C.G. Blatchford, J.R. Campbell, J.A. Creighton, Surf. Sci.
120 (1982) 435.
[26] K.S. Mayya, V. Patil, M. Sastry, Langmuir 13 (1997) 3944.
[27] S.L. Burkett, S.D. Sims, S. Mann, Chem. Commun. (1996)
1367.
[28] R.C. Rau, Advances in X-ray Analysis, vol. 5, Plenum
Press, Inc., New York, 1962, p. 104.
[29] K. Bandyopadhyay, K.S. Mayya, K. Vijayamohanan, M.
Sastry, J. Electron. Spectrosc. Relat. Phenom. 87 (1997)
101.
[30] P. Ganguly, D.V. Paranjape, M. Sastry, J. Am. Chem. Soc.
115 (1993) 793.
[31] M. Haruta, Catal. Today 36 (1997) 153.
[32] G.C. Bond, D.T. Thompson, Catal. Rev. Sci. Eng. 41
(1999) 319.
A. Ghosh et al. / Microporous and Mesoporous Materials 58 (2003) 201–211 211
Top Related
Copyright © 2022 FDOKUMEN

