







Bahasa
Halaman
Hukum


Polyperoxidic Surfactants for Interface Modification andCompatibilization of Polymer Colloidal Systems. II.Design of Compatibilizing Layers
S. VORONOV,1 V. TOKAREV,1 V. DATSYUK,1 V. SEREDYUK,1 O. BEDNARSKA,1 K. ODUOLA,1 H. ADLER,2
C. PUSCHKE,2 A. PICH,2 U. WAGENKNECHT3
1 State University “Lvivska Polytechnica,” S. Bandera 12, Lviv 290646, Ukraine
2 Dresden University of Technology, Mommsenstr. 4, Dresden D-01062, Germany
3 Institute for Polymer Research, Hohe 6, Dresden D-01069, Germany
Received 13 October 1998; accepted 6 June 1999
ABSTRACT: The polymer peroxide surfactants obtained by copolymerization of a perox-ide monomer with maleic anhydride were either physically or chemically sorbed on thedispersed-phase surfaces, for example, on mineral fillers and latex particles. Subse-quent initiation of graft copolymerization from the surface resulted in the formation ofinterfacial compatibilizing polymer layers in water emulsions and dispersed-filledpolyethylene. The morphology of the resulting filled polymer was characterized byscanning electron microscopy. © 2000 John Wiley & Sons, Inc. J Appl Polym Sci 76: 1228–1239, 2000
Key words: polyperoxide surfactant; adsorption; graft copolymerization; core–shelllatex; filled polyethylene
INTRODUCTION
Filled polymers, aqueous polymer dispersions, aswell as polymer blends are typical colloidal sys-tems with a highly developed interface. The prop-erties of the end products—polymer compositesand latexes—are a function of the structure andproperties of the interfacial layers. It follows thatfurther advances in the chemistry of polymercomposites is dependent on the possibilities ofdesigning interfacial layers.
Compatibilization of solid polymers in melts isconsidered to be very similar to the stabilizationof polymer colloidal particles in the aqueousphase with the use of polymeric surfactants.1
These substances have a tendency to localize atthe interface, thereby lowering surface and inter-facial tensions. A similar function is performed bya compatibilizer in immiscible polymer blends.Since the late 1980s, compatibilization problemshave been progressively elucidated through reac-tive blending, wherein along with the blendingprocesses physical or chemical interaction mayoccur between the molecules of the diverse com-ponents. In particular, these processes effectivelyproceed when coupling agents and peroxides (rad-ical generators) are introduced into the polymerblends.2,3 As proposed in the literature,4 the rad-ical generator and the coupling agent should beideally distributed at the interfacial boundary ofthe polymers being blended in order to maximizecoupling reactions between the reactive centers.In a number of works, attempts to locate thegraft-copolymerization initiating sites at the
Correspondence to: S. Voronov.Journal of Applied Polymer Science, Vol. 76, 1228–1239 (2000)© 2000 John Wiley & Sons, Inc.
1228

phase boundary of the emulsion system5,6 and atthe interface of polymer blends4,7,8 using low mo-lecular weight peroxides and coupling agentsproved to be successful. To realize these objec-tives, peroxide groups were immobilized on thefiller surface via the chemisorption of a dicarboxy-lic peroxide9 or a functionalized dialkyl perox-ide.10
For the introduction of peroxide groups ontothe polymer surface, gamma irradiation,11 ultra-violet light,12 glow,13 and corona14 dischargetreatment, ozone oxidation,15 as well as plasmatreatment16 were employed. Polymer peroxidesurfactants (PPS) have been synthesized by em-ploying polymer ozonization with successivegrafting of vinyl monomers onto these polymersfor use as compatibilizers in polymer blends.17
Application of polymeric surfactants containingperoxide groups will obviously be promising forsolving the problems involved in compatibiliza-tion. These substances are capable of lowering thesurface and interfacial tensions, localizing thespecific amount of radical generating sites at theinterface, and initiating graft copolymerization.18
Adsorption of PPS on the surface of the dispersedphase results in the immobilization of peroxidegroups at the interface.19 Polyperoxide macromol-ecules are capable of acting as anchor polymers;the PPS macrochains are attached to the surfaceeither by physical adsorption or chemisorptionwith the surface groups. On the other hand, theycan either be chemically bonded to the polymermatrix macromolecules or form grafted polymerchains capable of strong interaction with the dis-persion medium.
This method allows an appropriately engi-neered design of interfacial layers with a definitestructure and nature,20 thereby permitting toseparately control the interactions of the result-ing polymer layer with the dispersed-phase sur-face and dispersion medium. In addition, it facil-
itates the optimization of the extent of these in-teractions.21 The process consists of the followingstages: (i) synthesis of the PPS, (ii) its sorption onthe dispersed-phase surface, and (iii) formation ofcompatibilizing polymer layers either by graft co-polymerization of the monomers initiated fromthe surface or by grafting the preformed polymermacromolecules through chain-transfer reac-tions.20
Synthesis of polymer surfactants includingthose with reactive groups and their applicationshave been well documented in the litera-ture.1,22–24 At the present time, research is inprogress on the quest for a universal compatibi-lizer.25 We propose that this problem could besolved to some extent by the appropriate synthe-sis of the specific PPS for each particular compati-bilization of colloidal system ingredients. Thissynthesis can be carried out by copolymerizationof peroxide monomers with monomers containingother functional groups. Being that there is awide range of both peroxide monomers, for exam-ple, hydroperoxides, peroxyesters, and dialkylperoxides18,26 and vinyl monomers, the synthesisof versatile surface-active polyperoxides havingvarious natures and reactivities becomes a tech-nically feasible process, making it a quite prom-ising route to the creation of compatibilizing lay-ers.18
In a previous article,27 we reported on thesynthesis and notable properties of polymer sur-factants with peroxide and anhydride functional-ities, which are obtained through the radical co-polymerization of the peroxide monomer 52tert-butylperoxy252methyl212hexen232yne (PM)with maleic anhydride (MA) or their copolymer-ization with styrene (S). Their structures areshown in Scheme 1.
In aqueous solutions, these polyperoxidesreadily transform into the form of PPS with car-boxylic acid groups (PM–MAc, PM–MAc–S) or
Scheme 1 Structure of polymer peroxide surfactants used.
POLYPEROXIDIC SURFACTANTS. II 1229

salts of the latter (PM–MS, PM–MS–S) in aque-ous alkaline solutions. This work was performedwith the aim to investigate the applicability ofPPS for interface modification and compatibiliza-tion of polymeric colloidal systems.
EXPERIMENTAL
Materials
Polymeric peroxide surfactants PM–MA and PM–MA–S were synthesized by the method describedearlier,27 and the characteristics of the productsused are presented in Table I. The monomersused—butyl acrylate (BA) and S—were indus-trial products and were conventionally purifiedbefore use.
Synthesis of Polystyrene (PS) Core Latex
Emulsion polymerization of styrene was per-formed in a 2% w/w aqueous solution of sodiumdodecyl sulfonate (SDS) supplied by Merck (Darm-stadt, Germany) at a 1 : 4 monomer-to-aqueousphase w/w ratio by a method described else-where.28 Ammonium persulfate (0.1% w/w withrespect to the monomer) was used as the initiatorand the process was carried out until 99% conver-sion, which was controlled gravimetrically by an-alyzing the solid residual after evaporation. Theresidual monomer content was estimated usinggas chromatography with a LXM 80 equipment(Russia) in accordance to the technique reportedelsewhere.29 The pH values of the latexes werecontrolled with the aid of a pH meter I-130 (Be-larus).
Surface-tension (s) measurements were per-formed with a K12 KRUESS tensometer (Ham-burg, Germany) by the Wilhelmy method with aplatinum/iridium plate at 293 K, taking into con-sideration the time necessary to attain adsorptionequilibrium. The saturation degree of the latex-adsorption layers was estimated by means of ad-
sorption titration30 of the latex samples with SDSusing surface tension measurements. Latex par-ticle-size measurements were performed with aMalvern Zeta-Sizer 3 equipment.
Grafting of PPS onto PS Core Latex
PPS grafting onto the PS latex particle surfacewas performed in a glassy reactor equipped witha reflux condenser under agitation at 300 rpmand at three temperatures: 343, 353, and 363 K,for a period of 10 h. The polyperoxide was em-ployed in the form of potassium salt (10% solutionin water at pH 9) at a 4% w/w concentrationrelative to the PS. The grafting kinetics was si-multaneously followed by two methods: adsorp-tion titration of the latex samples and potentio-metric titration of frozen serum samples.
By the first method, samples of the reactionmixture were taken and adsorption titrated everyhour. The amount of grafted polyperoxide on thelatex particle surface was estimated with respectto the concentration of the low molecular weightemulsifier displaced from the surface. It was es-tablished that the amount of the SDS emulsifierdisplaced from the polymer particle surface is di-rectly proportional to the grafted polyperoxideamount.
For the potentiometric titration, after every 2 hof synthesis, 6 mL of the latex samples weretaken, frozen in a cryostat at 261 K, and then keptin this state for a period of 20 h. Serum wasremoved by filtration through a blue ribbon filterpaper and a (1.0 . . . 1.2) 6 0.0002 g weight wassampled and added to 2 mL of a 0.1N NaOHsolution. The resulting mixture was then dis-solved in 50 mL of distilled water and titratedwith a 0.1N HCl solution up to a pH value of 2.5.The potentiometric data were differentiated andthe volume of HCl consumed on the polyperoxidecarboxylic groups’ titration was evaluated fromthe turning points. The amount of the ungrafted
Table I. Characteristics of the PPS Used
PPSComposition(mol fraction)
AnhydrideGroup Content
(wt %)Peroxide GroupContent (wt %)
MolecularWeight
PM–MA 0.54 : 0.46 14.26 6.02 7240PM–MA–S 0.25 : 0.49 : 0.26 18.23 3.32 23,600
1230 VORONOV ET AL.

polyperoxidic emulsifier was then estimated fromthe carboxylic group content in the serum. Thestatistical measurement errors were very close forthese two methods of analysis, and the relativeerror did not exceed 10%.
Core–Shell Latex Synthesis
Core–shell latexes were synthesized in glass re-actors equipped with a mechanical agitator, acapillary-type inert gas inlet, and a monomerfeeder. Polymerization of the shell monomer (BA)on the surface of core latex was initiated due toeither thermal decomposition of peroxide groupsof PPS immobilized at a temperature of 363 K ortheir Redox decomposition at 313 K using an Fe1/disodium salt of an ethylenediamine tetraaceticacid/formaldehyde sodium bisulfide (ITF) systemas the reducing agent prepared as earlier re-ported.31 The syntheses were carried out up to99% conversion of the shell monomer. Polymer-ization kinetics of the BA shell monomer wasstudied using chromatography as indicatedabove, during which 3 mL of the reaction sampleswere mixed with 5 mL of hexane and kept undermechanical mixing for 4 h until total extraction ofthe unreacted shell monomer was observed. Theprevious experiments showed that at the reportedconditions BA is extracted with hexane almostcompletely because of its very low solubility inwater (0.2% w/w at 293 K). After mixing, themixture was kept for 3 h for the purpose of sepa-ration and unreacted BA was estimated by chro-matography. Proton NMR spectra of the peroxidemonomer as well as PM–MA and PM–MAcpolyperoxides grafted onto the PS core surfaceand the core–shell polymers with a PS core andthe polyBA shell were taken with a DR 3500Bruker equipment (frequency: 500 MHz; solvent:CDCl3 1 CD3COOD).
Peroxide Copolymer Adsorption
Two different types of CaCO3 were used as re-ceived, namely, industrial chalk Saxolith 2 HEwith a specific surface (Ssp) of 1.35 m2/g and areagent-grade product with 6.00 m2/g. MgO, anindustrial product with 2.77 m2/g; Al(OH)3, anindustrial product with Ssp 5 0.68 m2/g; BaSO4,reagent grade with Ssp 5 0.25 m2/g; and reactive-grade solvents were also used as received.PM–MA copolymer adsorption on the MgO,BaSO4, CaCO3, and Al(OH)3 surfaces was carried
out by adding 10 mL of the copolymer solution inacetone to 2.5 g of the filler and the mixture waskept under mechanical stirring for 2 h. The sus-pension was separated by centrifugation and theresulting filler washed with acetone and, afterrepeated centrifugation, dried at room tempera-ture until a constant weight. The copolymer ad-sorption value on the surface was evaluated fromthe weight loss of the filler samples upon pyroly-sis at 823 K for a period of 1.5 h in the case ofAl(OH)3 and CaCO3 and at 973 K for a period of1 h in the case of MgO and BaSO4. For CaCO3, thecopolymer adsorption value was evaluated fromthe solid residual content after dissolving thefiller samples in a 5 % aqueous solution of HCl.
Chemisorption of PPS on a chalk surface wasperformed in the aqueous phase as follows: Acalculated amount of the PM–MA copolymer wasdissolved in a water/alkaline solution and the so-lution pH value was brought to 5.6 by adding HCl.Then, 20 g of CaCO3 was added to 200 mL of aPM–MAc aqueous solution under continuous me-chanical mixing. The chemisorption process oc-curred at a temperature of 293 K until the carbondioxide is totally evolved, usually in 5–6 h, afterwhich the suspension was centrifuged and peroxi-dized CaCO3 was separated, washed with water,and later dried, first at room temperature, then atelevated temperature (303 K), and, finally, undera vacuum at 313 K until a constant weight. Thecopolymer adsorption value was evaluated asmentioned above. In both cases of physical ad-sorption and chemisorption, adsorption valueswere determined from the composition of the de-composition products of PM units in the copoly-mer using chromatography.32
Polymer Filling
It is well known that low-density polyethylene iseasily crosslinked when free-radical reactions areperformed in the melt.7 Thus, polyethylene (Hos-talen, Germany, GC 7260) was used as the poly-mer matrix, while peroxidized chalk with achemisorbed PM–MAc content in the rangewithin 0.5–2% w/w served as the active filler. Thefiller content was varied between 0 and 70% w/w.
The filling process was carried out by reactiveextrusion in a twin-screw corotating extruder Mi-cro 27 (Leistritz, Nurnberg) with a screw diame-ter of 27 mm and length-to-diameter ratio of 36.The filler was introduced by gravimetric sidefeeding. The flow rate was near 10 kg/h, and the
POLYPEROXIDIC SURFACTANTS. II 1231
average residence time in the equipment was 2min. Processing occurred at a temperature rangeof 513–535 K at 200 rpm depending on the level ofthe filling. The samples for mechanical testingwere prepared with a Battenfeld BA 500/200press (injection-molding machine). For the mor-phology studies, the filled polymer fracture sam-ples were investigated using scanning electronmicroscopy (SEM, low-voltage DSM 982, CarlZeiss, Germany).
RESULTS AND DISCUSSION
Immobilization of Peroxide Groups on theDispersed-phase Particle Surface
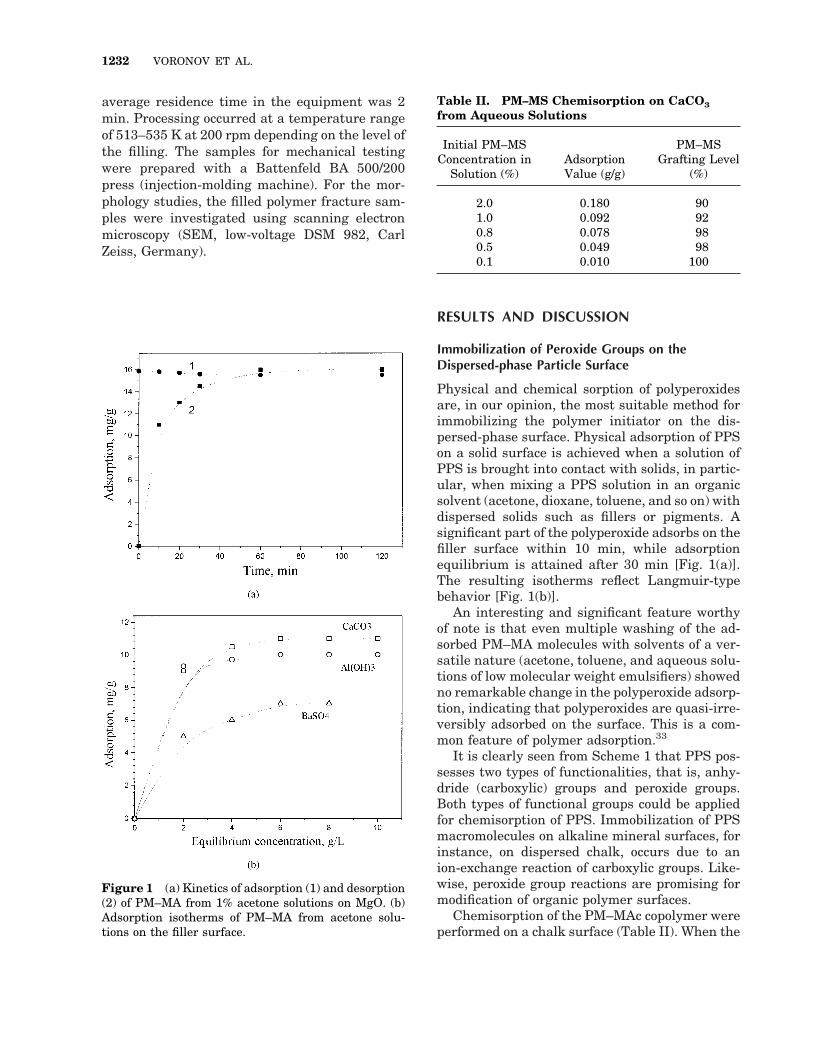
Physical and chemical sorption of polyperoxidesare, in our opinion, the most suitable method forimmobilizing the polymer initiator on the dis-persed-phase surface. Physical adsorption of PPSon a solid surface is achieved when a solution ofPPS is brought into contact with solids, in partic-ular, when mixing a PPS solution in an organicsolvent (acetone, dioxane, toluene, and so on) withdispersed solids such as fillers or pigments. Asignificant part of the polyperoxide adsorbs on thefiller surface within 10 min, while adsorptionequilibrium is attained after 30 min [Fig. 1(a)].The resulting isotherms reflect Langmuir-typebehavior [Fig. 1(b)].
An interesting and significant feature worthyof note is that even multiple washing of the ad-sorbed PM–MA molecules with solvents of a ver-satile nature (acetone, toluene, and aqueous solu-tions of low molecular weight emulsifiers) showedno remarkable change in the polyperoxide adsorp-tion, indicating that polyperoxides are quasi-irre-versibly adsorbed on the surface. This is a com-mon feature of polymer adsorption.33
It is clearly seen from Scheme 1 that PPS pos-sesses two types of functionalities, that is, anhy-dride (carboxylic) groups and peroxide groups.Both types of functional groups could be appliedfor chemisorption of PPS. Immobilization of PPSmacromolecules on alkaline mineral surfaces, forinstance, on dispersed chalk, occurs due to anion-exchange reaction of carboxylic groups. Like-wise, peroxide group reactions are promising formodification of organic polymer surfaces.
Chemisorption of the PM–MAc copolymer wereperformed on a chalk surface (Table II). When the
Figure 1 (a) Kinetics of adsorption (1) and desorption(2) of PM–MA from 1% acetone solutions on MgO. (b)Adsorption isotherms of PM–MA from acetone solu-tions on the filler surface.
Table II. PM–MS Chemisorption on CaCO3
from Aqueous Solutions
Initial PM–MSConcentration in
Solution (%)AdsorptionValue (g/g)
PM–MSGrafting Level
(%)
2.0 0.180 901.0 0.092 920.8 0.078 980.5 0.049 980.1 0.010 100
1232 VORONOV ET AL.
polyperoxide carboxylic group interacts withCaCO3 in aqueous solutions, an ion-exchange re-action takes place, which proceeds with the for-mation of carbonic acid, the latter being detectedby the evolution of carbon dioxide. Analysis of thepolymer present in the equilibrium solutionshowed that it contained bonded calcium, whoseconcentration increased as the chemisorption pro-cess proceeded. This is due to the displacement ofthe polyperoxide calcium salt from the surface bythe more active PM–MAc macromolecules. Theproduct of this reaction (calcium salt of the car-boxyl-containing polyperoxide) is slightly solublein water, thereby precipitating on the solid-phasesurface. Consequently, encapsulation of CaCO3particles is achieved both by polyperoxide chemi-sorption and precipitation of the polyperoxide cal-cium salt macromolecules on the particle surface.This method permits the immobilization of a sig-nificant concentration of peroxide groups (up to1023 mol/m2) on the surface, which is of 2–3 or-ders of magnitude higher than that obtained dur-ing physical adsorption or chemisorption of lowmolecular weight initiators. Hence, this is of im-mense value for modifying fillers with low specificsurfaces. Another very important result is thatthe peroxide groups are not consumed duringthese PPS adsorption or chemisorption processesand can be further used for the formation of com-patibilizing layers through graft copolymerizationof monomers or chain-transfer grafting of a pre-formed polymer.
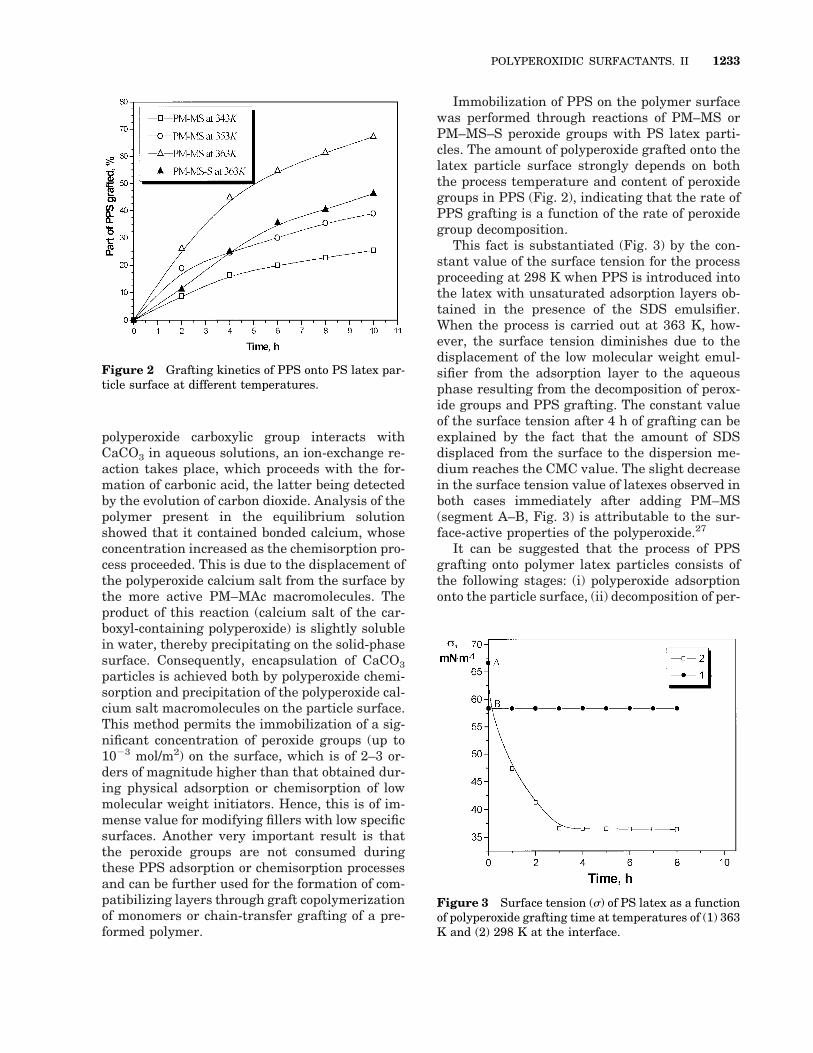
Immobilization of PPS on the polymer surfacewas performed through reactions of PM–MS orPM–MS–S peroxide groups with PS latex parti-cles. The amount of polyperoxide grafted onto thelatex particle surface strongly depends on boththe process temperature and content of peroxidegroups in PPS (Fig. 2), indicating that the rate ofPPS grafting is a function of the rate of peroxidegroup decomposition.
This fact is substantiated (Fig. 3) by the con-stant value of the surface tension for the processproceeding at 298 K when PPS is introduced intothe latex with unsaturated adsorption layers ob-tained in the presence of the SDS emulsifier.When the process is carried out at 363 K, how-ever, the surface tension diminishes due to thedisplacement of the low molecular weight emul-sifier from the adsorption layer to the aqueousphase resulting from the decomposition of perox-ide groups and PPS grafting. The constant valueof the surface tension after 4 h of grafting can beexplained by the fact that the amount of SDSdisplaced from the surface to the dispersion me-dium reaches the CMC value. The slight decreasein the surface tension value of latexes observed inboth cases immediately after adding PM–MS(segment A–B, Fig. 3) is attributable to the sur-face-active properties of the polyperoxide.27
It can be suggested that the process of PPSgrafting onto polymer latex particles consists ofthe following stages: (i) polyperoxide adsorptiononto the particle surface, (ii) decomposition of per-
Figure 2 Grafting kinetics of PPS onto PS latex par-ticle surface at different temperatures.
Figure 3 Surface tension (s) of PS latex as a functionof polyperoxide grafting time at temperatures of (1) 363K and (2) 298 K at the interface.
POLYPEROXIDIC SURFACTANTS. II 1233

oxide groups, and (iii) surface activation by achain-transfer reaction and grafting of thepolyperoxide macroradicals onto the latex particlesurface, leading to the formation of reactive inter-facial layers in accordance with Scheme 2.
In this process, contrary to that of PPS immo-bilization on mineral surfaces occurring at theexpense of anhydride (carboxylic) groups, the per-oxide groups of PM–MS are particularly decom-
posed in order to provide grafting of polyperoxideonto the latex particle surface. The estimatedamount of peroxide groups consumed was foundto be 24%. Therefore, the remaining peroxidegroups could be further utilized to initiate graftcopolymerization, for example, during the synthe-ses of core–shell latexes. The polyperoxides immo-bilized onto the dispersed-phase adsorption lay-ers (e.g., mineral fillers and latex particles) have
Scheme 2 Immobilization of PM–MS on the latex particles.
1234 VORONOV ET AL.
found potential applications for the formation ofcompatibilizing polymer layers, for the control ofthe electrosteric stability of latexes, as well as forthe creation of core–shell latexes.
Formation of Interfacial Polymer CompatibilizingLayers
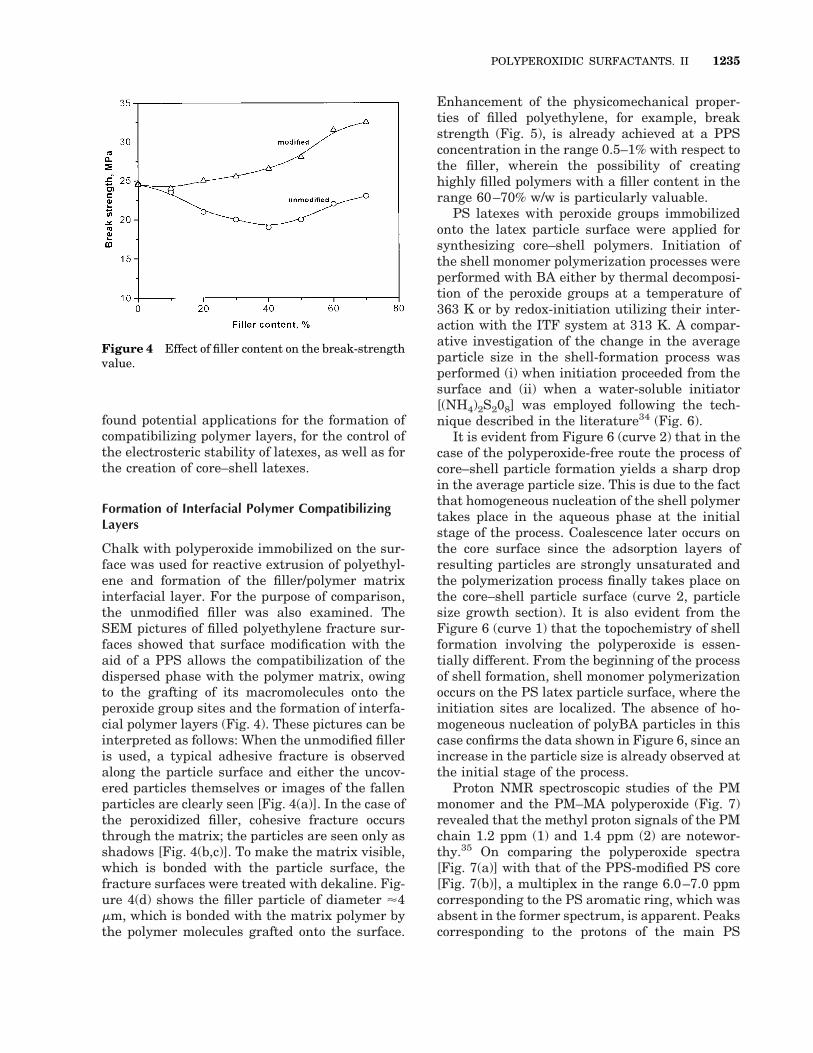
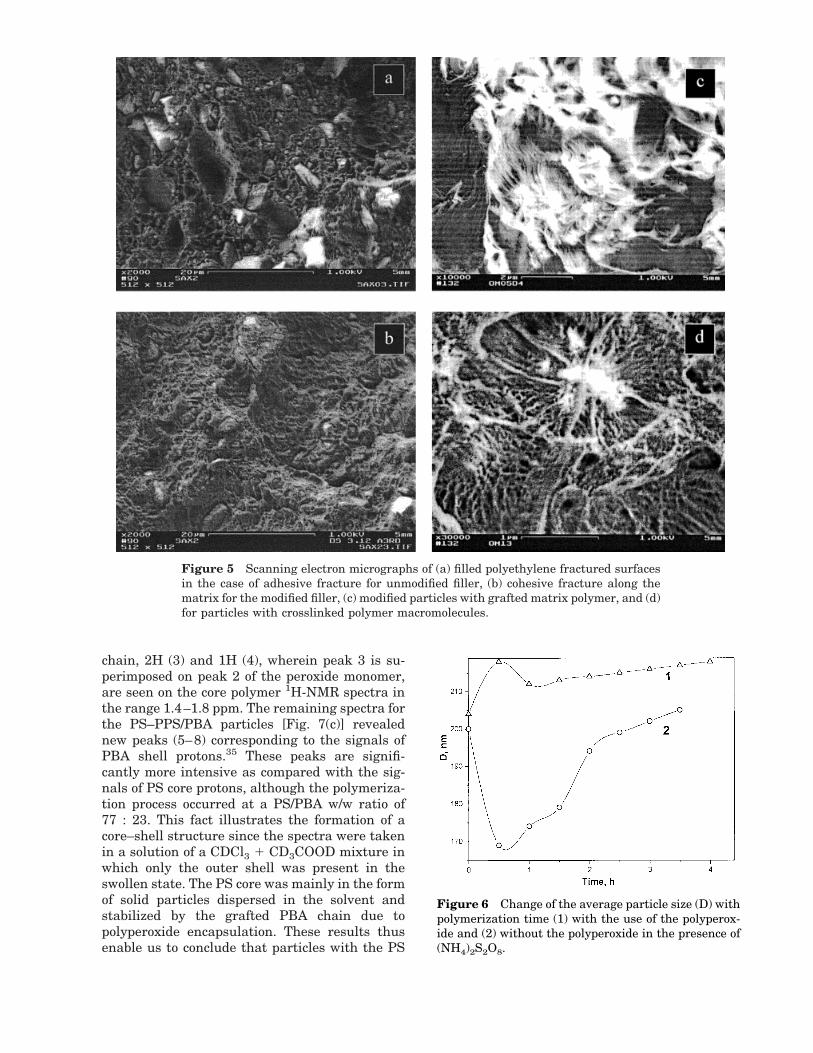
Chalk with polyperoxide immobilized on the sur-face was used for reactive extrusion of polyethyl-ene and formation of the filler/polymer matrixinterfacial layer. For the purpose of comparison,the unmodified filler was also examined. TheSEM pictures of filled polyethylene fracture sur-faces showed that surface modification with theaid of a PPS allows the compatibilization of thedispersed phase with the polymer matrix, owingto the grafting of its macromolecules onto theperoxide group sites and the formation of interfa-cial polymer layers (Fig. 4). These pictures can beinterpreted as follows: When the unmodified filleris used, a typical adhesive fracture is observedalong the particle surface and either the uncov-ered particles themselves or images of the fallenparticles are clearly seen [Fig. 4(a)]. In the case ofthe peroxidized filler, cohesive fracture occursthrough the matrix; the particles are seen only asshadows [Fig. 4(b,c)]. To make the matrix visible,which is bonded with the particle surface, thefracture surfaces were treated with dekaline. Fig-ure 4(d) shows the filler particle of diameter '4mm, which is bonded with the matrix polymer bythe polymer molecules grafted onto the surface.
Enhancement of the physicomechanical proper-ties of filled polyethylene, for example, breakstrength (Fig. 5), is already achieved at a PPSconcentration in the range 0.5–1% with respect tothe filler, wherein the possibility of creatinghighly filled polymers with a filler content in therange 60–70% w/w is particularly valuable.
PS latexes with peroxide groups immobilizedonto the latex particle surface were applied forsynthesizing core–shell polymers. Initiation ofthe shell monomer polymerization processes wereperformed with BA either by thermal decomposi-tion of the peroxide groups at a temperature of363 K or by redox-initiation utilizing their inter-action with the ITF system at 313 K. A compar-ative investigation of the change in the averageparticle size in the shell-formation process wasperformed (i) when initiation proceeded from thesurface and (ii) when a water-soluble initiator[(NH4)2S208] was employed following the tech-nique described in the literature34 (Fig. 6).
It is evident from Figure 6 (curve 2) that in thecase of the polyperoxide-free route the process ofcore–shell particle formation yields a sharp dropin the average particle size. This is due to the factthat homogeneous nucleation of the shell polymertakes place in the aqueous phase at the initialstage of the process. Coalescence later occurs onthe core surface since the adsorption layers ofresulting particles are strongly unsaturated andthe polymerization process finally takes place onthe core–shell particle surface (curve 2, particlesize growth section). It is also evident from theFigure 6 (curve 1) that the topochemistry of shellformation involving the polyperoxide is essen-tially different. From the beginning of the processof shell formation, shell monomer polymerizationoccurs on the PS latex particle surface, where theinitiation sites are localized. The absence of ho-mogeneous nucleation of polyBA particles in thiscase confirms the data shown in Figure 6, since anincrease in the particle size is already observed atthe initial stage of the process.
Proton NMR spectroscopic studies of the PMmonomer and the PM–MA polyperoxide (Fig. 7)revealed that the methyl proton signals of the PMchain 1.2 ppm (1) and 1.4 ppm (2) are notewor-thy.35 On comparing the polyperoxide spectra[Fig. 7(a)] with that of the PPS-modified PS core[Fig. 7(b)], a multiplex in the range 6.0–7.0 ppmcorresponding to the PS aromatic ring, which wasabsent in the former spectrum, is apparent. Peakscorresponding to the protons of the main PS
Figure 4 Effect of filler content on the break-strengthvalue.
POLYPEROXIDIC SURFACTANTS. II 1235
chain, 2H (3) and 1H (4), wherein peak 3 is su-perimposed on peak 2 of the peroxide monomer,are seen on the core polymer 1H-NMR spectra inthe range 1.4–1.8 ppm. The remaining spectra forthe PS–PPS/PBA particles [Fig. 7(c)] revealednew peaks (5–8) corresponding to the signals ofPBA shell protons.35 These peaks are signifi-cantly more intensive as compared with the sig-nals of PS core protons, although the polymeriza-tion process occurred at a PS/PBA w/w ratio of77 : 23. This fact illustrates the formation of acore–shell structure since the spectra were takenin a solution of a CDCl3 1 CD3COOD mixture inwhich only the outer shell was present in theswollen state. The PS core was mainly in the formof solid particles dispersed in the solvent andstabilized by the grafted PBA chain due topolyperoxide encapsulation. These results thusenable us to conclude that particles with the PS
Figure 6 Change of the average particle size (D) withpolymerization time (1) with the use of the polyperox-ide and (2) without the polyperoxide in the presence of(NH4)2S2O8.
Figure 5 Scanning electron micrographs of (a) filled polyethylene fractured surfacesin the case of adhesive fracture for unmodified filler, (b) cohesive fracture along thematrix for the modified filler, (c) modified particles with grafted matrix polymer, and (d)for particles with crosslinked polymer macromolecules.

core and the PBA shell are generally formed inthe process of BA polymerization initiated withpolyperoxide immobilized on PS latex particles.
CONCLUSIONS
In colloidal polymer systems (water dispersions ofpolymers and filled polymers), the disperse phaseand the dispersion medium could be compatibi-lized by introducing to the phase-boundarypolyperoxidic surfactants which are synthesizedthrough the copolymerization of peroxide mono-
mers like PM with other functional monomers, forexample, MA.
Adsorption of polyperoxidic surfactants on thedispersed-phase surface is obviously an especiallyappropriate method for immobilizing peroxidegroups on the interface. Furthermore, in dis-persed-filled polymers, there arises the possibilityof creating compatibilizing polymer layers result-ing from the grafting of the matrix polymer ontothe peroxidized filler surface. SEM observationsconfirmed the occurrence of graft copolymeriza-tion of the polymer matrix (polyethylene) on theperoxidized filler (chalk) surface. It was shownthat initiating graft copolymerization from the
Figure 7 Proton NMR spectra of (a) the peroxide monomer, (b) the PM–MAc polyper-oxide, (c) PS latex particles modified by the PM–MAc polyperoxide at 363 K in a PS-to-PM–MAc wt ratio of 100:5, (d) and the core–shell latex particles with PS/PM–MAccore and PBA shell at 363 K in a core/shell wt ratio of 100:30.
POLYPEROXIDIC SURFACTANTS. II 1237
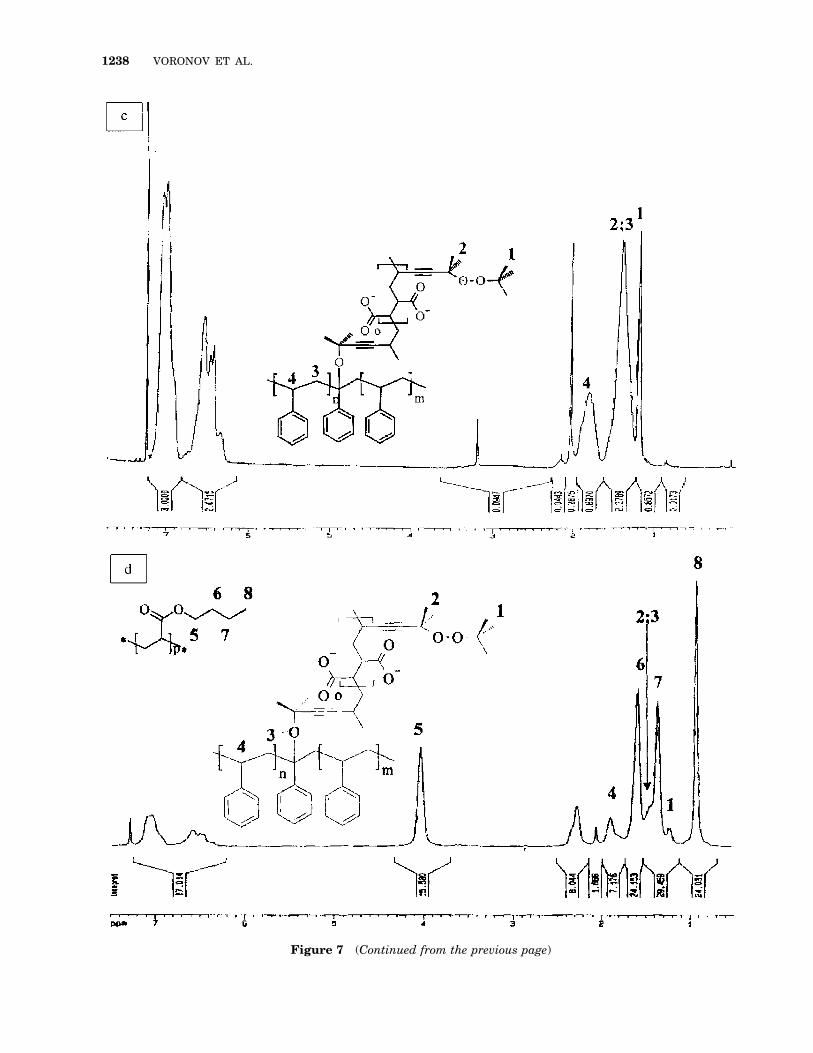
Figure 7 (Continued from the previous page)
1238 VORONOV ET AL.

dispersed-phase surface in water emulsions led tothe formation of core–shell polymer systems. Wepropose that an appropriately engineered synthe-sis of the particular PPS would greatly contributetoward solving the problems concerning the com-patibilization of the ingredients employed in wa-ter dispersions of polymers, filled polymer sys-tems, as well as polymer blends.
REFERENCES
1. Piirma, I. Polymeric Surfactants; Surfactant Sci-ence Series; Marcel Dekker: New York, 1992.
2. Xanthos, M. Polym Eng Sci 1991, 28, 1392.3. Xanthos, M.; Dagli, S. S. Polym Eng Sci 1991, 31,
929.4. Teh, J. W.; Rudin, A. Polym Eng Sci 1992, 32, 1678.5. Schneider, M.; Pitch, P.; Lambla, M. J Appl Polym
Sci 1996, 62, 273.6. Nuyken, O.; Bauer, R.; Bulliq-Peters, W.; Voit, B.;
Yang, D. Kautsch Gummi Kunstst 1994, 47, 903.7. Lambla, M.; Seadan, M. Polym Eng Sci 1992, 32,
1687.8. Xu, C.; Fang, Z.; Zhang, J. Polymer 1997, 38, 155.9. Popov, V. A.; Zvereva, Y. A.; Grishin, A. N.;
Palaeva, T. V.; Fomin, V. A. Vysokomol Soedin SerA 1982, 24, 1654 (in Russian).
10. Ivanchev, S. S.; Yenikolopyan, N. S.; Polozov, B. V.;Dmitrenko, A. V. ; Demidova, V. A.; Krupnik,A. M.; Litkovetz, A. K. Vysokomol Soedin Ser A1981, 23, 2064 (in Russian).
11. Suzuki, M.; Tamada, Y.; Iwata, H.; Ikada, Y. InPhysicochemical Aspects of Polymer Surfaces; Mit-tel, K.-L., Ed.; Plenum: New York, 1983; p 923.
12. Uchida, E.; Uyama, Y.; Ikada, Y. J Appl Polym Sci1993, 47, 417.
13. Chen, K. S.; Uyama, Y.; Ikada, Y. J Adhes SciTechnol 1992, 6, 1023.
14. Hoebergen, A.; Uyama, Y.; Okada, T.; Ikada, Y.J Appl Polym Sci 1993, 48, 1825.
15. Fujimoto, K.; Takebayashi, Y.; Inoue, H.; Ikada, Y.J Polym Sci Polym Chem Ed 1993, 31, 1035.
16. Kulik, E. A.; Ivanchenko, M. I.; Kato, K.; Sano, S.;Ikada, Y. J Polym Sci Polym Chem Ed 1995, 33,323.
17. Boutevin, B.; Pietrasanta, Y.; Robin, I. I.; Pabiot, I.;Leseur, B. Makromol Chem Makromol Symp 1992,57, 371.
18. Voronov, S.; Tokarev, V.; Petrovska, G. Heterofunc-tional Polyperoxides. Theoretical Basis of TheirSynthesis and Application in Compounds; LvivPolytechnic State University: Lviv, 1994.
19. Tokarev, V.; Kucher, R.; Voronov, S.; Ryabova, O.;Minko, S.; Kurganski, V. Dokl Akad Nauk SSSR1987, 293, 166 (in Russian); Chem Abstr 1987, 106,196838d.
20. Voronov, S.; Tokarev, V. In Proceedings of theEuro-Fillers’ 97 International Conference on FilledPolymers and Fillers, Manchester, 1997.
21. Voronov, S.; Tokarev, V.; Datzyuk, V.; Kozar, M.Prog Colloid Polym Sci 1996, 101, 189.
22. Guyot, A.; Tauer, K. Adv Polym Sci 1994, 111, 45.23. Tauer, K.; Kosmella, S. Polym Int 1993, 30, 253.24. Pichot, C.; Charleux, B.; Charreyre, M. T.; Revilla,
J. Makromol Chem Makromol Symp 1994, 88, 71.25. Vilgis, T. A.; Nooli, J. Macromolecules 1990, 23,
2941.26. Houben-Weyl; Methoden der organischen Chemie;
Bd E 13, Organische Peroxo-Verbindungen/hrsg(Von Heinz Kropp. Bearb. von A. Blaschette);Thime. Teilb: Stuttgart, New York; 1988; v1, p 367.
27. Voronov, S.; Tokarev, V.; Oduola, K.; Lastukhin,Yu. J Appl Polym Sci (in press).
28. Toroptzeva, A. M.; Belgorodskaya, K. V.; Boi-darenko, O. M. Laboratory Guide on Chemistry& Technology of Macromolecular Compounds;Khimiya: Leningrad, 1972 (in Russian).
29. Klesheva, M.; Zavyalov, Y.; Korpiva, I. Gas Chro-matographic Analysis in the Production ofPolymerization Plastic; Khimiya: Leningrad, 1978(in Russian).
30. Neyman, R. E. In Colloidal Chemistry of SyntheticLatexes; Voronej State University: Voronej, 1984(in Russian).
31. Reykhspheld, B.; Yerkova, L.; Ruban, V. In Labo-ratory Guide on Synthetic Rubber; Khimiya: Len-ingrad, 1967 (in Russian).
32. Vasilyev. V.; Puchin, V.; Tokarev, V.; Voronov, S.Izvest VUZov Khim Khim Technol 1983, 10, 1246(in Russian); Chem Abstr 1984, 100, 86215s.
33. Fleer, G. J.; Lyklema, T. In Adsorption from Solu-tion at the Solid/Liquid Interface; Rochester, C. H.,Ed.; Academic: New York, 1983; p 470.
34. Hidalgo, M.; Cavaille, J. Colloid Polym Sci 1992,270, 1208.
35. Bovey, F. A. In High Resolution NMR of Macromol-ecules; Academic: New York, London, 1972.
POLYPEROXIDIC SURFACTANTS. II 1239
Top Related
Copyright © 2022 FDOKUMEN

