







Bahasa
Halaman
Hukum


ORTom: a multi-species approach based on conservedco-expression to identify putative functionalrelationships among genes in tomato
Laura Miozzi • Paolo Provero • Gian Paolo Accotto
Received: 25 August 2009 / Accepted: 11 April 2010 / Published online: 22 April 2010
� Springer Science+Business Media B.V. 2010
Abstract Co-expressed genes are often expected to be
functionally related and many bioinformatics approaches
based on co-expression have been developed to infer their
biological role. However, such annotations may be unre-
liable, whereas the evolutionary conservation of gene
co-expression among species may form a basis for more
confident predictions. The huge amount of expression data
(microarrays, SAGE, ESTs) has already allowed functional
studies based on conserved co-expression in animals. Up to
now, the implementation of analogous tools for plants
has been strongly limited probably by the paucity and
heterogeneity of data. Here we present ORTom, a tomato-
centred EST data-mining approach based on conserved
co-expression in the Solanaceae family. ORTom can be
used to predict functional relationships among genes and
to prioritize candidate genes for targeted studies. The
method consists in ranking ESTs co-expressed with a gene
of interest according to the level of expression pattern
conservation in phylogenetically-related plants (potato,
tobacco and pepper) to obtain lists of putative functionally-
related genes. The lists are then analyzed for Gene
Ontology keyword enrichment. The web server ORTom
has been implemented to make the results publicly-
available and searchable. Few biological examples on how
the tool can be used are presented.
Keywords Solanaceae � Conserved co-expression �Functional genomics � Expressed sequence tag (EST) �Systems biology
Introduction
Technological advances in the post-genomic era have
allowed measurement of the expression level of thousand
of genes simultaneously. As a consequence, a huge amount
of transcriptomic data is becoming publicly available. This
huge amount of information can be useful to get new
insight on how living systems work. A number of bioin-
formatics approaches have been developed to help in
deciphering the biological role of genes; several are based
on gene co-expression and the so-called ‘guilt-by-associa-
tion’ (GBA) principle (Eisen et al. 1998). According to
this, genes sharing a common expression pattern are likely
to be involved in related functions. Thus a gene of
unknown function, co-expressed with them, can be sup-
posed to be involved in the same process.
The importance of co-expression in inference of gene
function has been highlighted by several studies indicating
that proteins associated in stable complexes generally
show similar expression profiles (Jansen et al. 2002).
Co-expression can be also correlated to neighbouring genes
(Fukuoka et al. 2004) or to enzymes belonging to the same
metabolic pathway (Gachon et al. 2005). A systematic
survey of five co-expression networks using microarray
data from three mammalian organisms has demonstrated
the broad applicability of the GBA approach in predicting
gene functions (Wolfe et al. 2005). Co-expression has
Electronic supplementary material The online version of thisarticle (doi:10.1007/s11103-010-9638-z) contains supplementarymaterial, which is available to authorized users.
L. Miozzi (&) � G. P. Accotto
Istituto di Virologia Vegetale, CNR, Strada delle Cacce 73,
10135 Turin, Italy
e-mail: [email protected]
P. Provero
Molecular Biotechnology Center and Dipartimento di Genetica,
Biologia e Biochimica, Universita di Torino, Via Nizza 52,
10100 Turin, Italy
123
Plant Mol Biol (2010) 73:519–532
DOI 10.1007/s11103-010-9638-z

already been useful to annotate genes in yeast and barley
(Wu et al. 2002; Faccioli et al. 2005) and several bioin-
formatic tools exploiting microarray-based co-expression
have been developed (for a review on co-expression tools
for plant biology see Usadel et al. 2009).
However, prediction reliability can be compromised by
noise inherent in microarray and other technologies
employed. A way to improve prediction is to exploit evolu-
tionary conservation of co-expression, integrating data from
other species. Considering Saccharomyces cerevisiae and
Caenorhabditis elegans, Van Noort et al. (2003) have shown
that conserved co-expression among orthologs or para-
logs can more accurately predict function than simple
co-expression. Of the conserved co-expressed gene pairs, for
which functional annotation was available between those
two organisms, 89% were part of the same protein complex
(Teichmann and Babu 2002). Moreover, according to
Bhardwaj and Lu (2005), co-expression of interacting pro-
tein pairs tends to be conserved among human, mouse, yeast
and Escherichia coli. A comparative study of expression
profiles from S. cerevisiae, C. elegans, E. coli, A. thaliana,
D. melanogaster, and H. sapiens showed that functionally-
related sets of genes frequently belong to conserved
co-expressed modules, even when the evolutionary distance
among the organisms is considerable (Bergmann et al. 2004).
Functional annotation using conserved co-expression has
been exploited for model organisms, mainly animals (Daub
and Sonnhammer 2008; Ramani et al. 2008; Obayashi et al.
2008; Pellegrino et al. 2004) and particular attention has
been paid to the prediction of human candidate disease genes
(Ala et al. 2008; Oti et al. 2008). In plant biology, only the
GeneCAT tool (Mutwil et al. 2008) has considered the
co-expression of genes in more than one species, particularly
Arabidopsis and barley. However, it does not search for
statistically significant conserved co-expression but, given
two orthologs, it allow the users to compare the lists of
co-expressed genes.
Most of the work described above has involved the use
of microarray data. However, other kinds of experimental
data are available. The integration of array and SAGE
datasets significantly improved the functional annotations
of human genes (Miozzi et al. 2008), while Wu and
co-workers (Wu et al. 2005) pointed out the importance of
the already available EST resources. The huge amount of
EST data available in public databanks can be a remark-
ably rich alternative source of information but, as far as we
know, the use of EST-based conserved co-expression to
find functional relationships among genes in plants has not
yet been exploited.
Tomato (Solanum lycopersicum L.) is a model organism
among the Solanaceae, a family comprising several other
economically important crops (potato, tobacco, pepper,
eggplant) as well as ornamental and medicinal plants
(petunia, deadly nightshade). The Solanaceae is a medium-
size family of about 90 genera and 3,000–4,000 species,
almost half of which are in the genus Solanum. In spite of
its importance, the sequencing of the euchromatin portion
of the tomato genome, considered the gene rich region
(Wang et al. 2006) has been just completed and, at the
moment, only a provisional assembly is available (http://
www.sgn.cornell.edu/about/tomato_sequencing.pl). Major
array databases, such as ArrayExpress (http://www.ebi.ac.
uk/microarray-as/ae/) and GEO (http://www.ncbi.nlm.nih.
gov/geo/), as well as dedicated repositories like the Tomato
Functional Genomics Database (http://ted.bti.cornell.edu/)
contain data for only a few tens of microarray experi-
ments, moreover performed on several different platforms.
However, some EST repositories for tomato are available
(http://www.sgn.cornell.edu/; http://biosrv.cab.unina.it/tom
atestdb/) and, among them, the DFCI Gene Index Project
(http://compbio.dfci.harvard.edu/tgi/) contains thousands
of tomato ESTs assembled in virtual transcripts named
Tentative Consensus (TC) together with collections of TCs
from other Solanaceae. Putative orthologous relationships
among TCs are available through the eukaryotic gene
orthologues (EGO) database (http://compbio.dfci.harvard.
edu/tgi/ego/).
In this study we describe ORTom, a tomato-centred
data-mining method, that uses publicly-available EST data
for functional prediction and candidate gene prioritization
on the basis of the level of EST presence/absence profile
conservation. The core of our approach is the comparison
of transcriptional presence/absence patterns derived from
the best studied members of the Solanaceae family, i.e.,
tomato, potato, tobacco and pepper. Because of the limited
availability of information on EST library construction
(i.e., normalized, not normalized, subtracted), we reduced
the gene expression level at only two states: presence or
absence in a given library. Form now on, the EST presence/
absence profiles will be indicated by the term ‘‘expression
profiles’’ and the term ‘‘co-expression’’ will be used to
indicate similar EST presence/absence patterns. To show
how ORTom can be used to improve functional annotation
and to infer biological relationships among genes, few
biological examples are presented.
Materials and methods
Computational method
Expression data and measure of co-expression
We considered EST expression data for four Solanaceae
species: tomato, potato, tobacco and pepper. For each of
these, publicly available tentative consensus (TC) data
520 Plant Mol Biol (2010) 73:519–532
123

belonging to release 11, 11, 3 and 2, respectively, and
originating from different kinds of tissues and/or devel-
oping stages were downloaded through DFCI Gene Index
Project (http://compbio.dfci.harvard.edu/tgi/). According to
the DFCI definition, TCs are virtual transcripts created by
assembling ESTs. The number of ESTs, TCs and libraries
available for each species is reported in Table 1.
As a first step, a presence/absence matrix was con-
structed for all TC sequences belonging to each organism;
the presence/absence profile of each TC was a string con-
stituted of as many bits as libraries, with a bit equal to 1
indicating that the TC is represented in the corresponding
library and 0 otherwise. The best way to treat ESTs data
would be to differentiate among normalized, not normal-
ized and subtracted libraries. Unfortunately, the available
information on library construction is very limited, not
homogeneous and not standardized and at the moment it’s
not possible to set up an automatic way to extract this
information from the DFCI database. To correctly mine the
ESTs data, without such information, we decided to con-
sider only the presence/absence of genes, as already done
by Faccioli et al. (2005).
To measure the similarity between the presence/absence
profiles, for all pairs (TC1, TC2) of Tentative Consensus
we calculated the binary asymmetric distance, defined as
the ratio of the number of bits that are 1 for only one of the
two TCs over the total number of bits that are 1 in at least
one of the two TCs. The choice of the optimal similarity
measure is critical (Usadel et al. 2009). Shmulevich and
Zhang (2002) pointed out that several distances assume to
operate on continuous-level expression values and con-
sidered the Hamming distance as a natural choice for
investigating the differential expression using binary
expression data. However, this distance is given simply by
the number of different bits between the two strings, while
the binary asymmetric distance can capture the fact that
having both genes expressed in the same library is bio-
logically more significant than having both genes non-
expressed. Indeed, Glazko et al. (2005) showed that the
binary asymmetric distance (to which they refer as Jaccard
distance) performed best among several dissimilarity defi-
nitions in analyzing genome-wide binary expression data.
For each Tentative Consensus in a given species we
defined a group of TCs with similar presence/absence
profiles, considering no more than the first 400 TCs with
the highest similarity value. Other cut-off values (100 and
300) were tested and gave essentially similar results (data
not shown).
TCs with similar EST presence/absence profiles were
defined ‘‘co-expressed’’.
Definition of orthologous TCs
The second step was to define orthologous TCs. Orthology
relationships among TCs belonging to different species were
obtained from the latest release (release 13) of the Eukary-
otic Gene Orthologues database (http://compbio.dfci.har
vard.edu/tgi/ego/). This database, organized in orthologous
clusters generated by pair-wise comparison between TC
sequences, allowed us to directly link orthology and
expression data. Out of 20,680 tomato TCs, 12,612 (about
61%) had an ortholog in potato, 6,240 (about 30%) in
tobacco and 3,950 (about 19%) in pepper; 2,553 tomato TCs
(about 12%) had at least one ortholog in all three species.
Identification of groups of TCs with conserved
co-expression
Co-expressed tomato TCs were searched according to the
level of conservation of co-expression in at least one of the
other species considered (potato, tobacco and pepper),
using Fisher’s exact test. Given a tomato TC (TCtom) and
its ortholog in one of the other species (TCorth), the group
of tomato TCs co-expressed with TCtom was deemed to
show conserved co-expression if the number of tomato TCs
belonging to this group and having an ortholog among the
TCs co-expressed with TCorth was statistically significant
(P-value \ 10-3).
For a given tomato TCtom, the list of tomato TCs
showing conserved co-expression was ranked according to
the number of species for which the co-expression was
conserved.
Functional characterization of groups of TCs
with conserved co-expression
The groups of tomato TCs showing conserved co-expres-
sion were searched for GO term enrichment, using the
Gene Ontology annotations provided by the DFCI Gene
Index Project (http://compbio.dfci.harvard.edu/tgi/). For
each group we used Fisher’s exact test to evaluate the
probability that the TCs annotated to a given GO keyword
were significantly overrepresented. A TC was consid-
ered to be annotated to a GO term if it was directly
annotated to it or to any of its descendants in the GO graph.
A P-value \ 10-6 was considered as statistically signifi-
cant for overrepresentation.
Table 1 Dataset information
Species No. of libraries No. of TCs No. of ESTs
Tomato 104 20,680 196,114
Potato 60 30,152 194,910
Tobacco 48 9,912 51,044
Pepper 22 4,163 21,322
Plant Mol Biol (2010) 73:519–532 521
123

The percentage of false positives (PFP) among the
putative gene annotations was estimated using randomized
TC lists: we randomized the TC names 100 times inde-
pendently and recorded the number of putative annotations
obtained from each set of randomized lists. The PFP was
then calculated as the ratio between the number of pre-
dicted annotations obtained from the randomized TC lists
and the number obtained from the true lists. The overall
PFP was calculated as the average over all the obtained
PFPs.
Bench-based methods
Biological material
S. lycopersicum cv. Moneymaker seeds were surface ster-
ilized (Ethanol 70% (v/v) for 3 min; dip in Tween 20,
sodium hypochlorite 5% (v/v) for 13 min, rinse with dis-
tilled water), germinated in petri dishes with 0.6% (w/v)
agar for 5 days in the dark (25�C) and 4 days in the light.
Seedlings were then transferred to pots containing sterile
quartz sand. Plants were maintained in a growth chamber
with 14 h of light (24�C) and 10 h of dark (20�C) and
watered twice per week: once with 125 ml of the Modified
Long-Ashton solution (Trotta et al. 1996) and once with
water. 28 days after potting plants were inoculated with
TSWV (strain T1012). Approximately 1 g of infected
tomato leaf tissue was homogenized in 10 ml of inocula-
tion buffer (10 mM DIECA, 5 mM EDTA, 20 mM
Na2SO3). Inoculum was applied on the upper side of leaves
by rubbing with carborundum. Mock-inoculated plants,
used as control, were inoculated with non-infected tomato
leaf tissue and kept under the same conditions.
RNA extraction
Fourteen days after inoculation, systemically infected
leaves and the corresponding leaves of mock inoculated
plants were harvested and frozen in liquid N2. Total RNA
was purified using Trizol (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer’s instructions. Quality and
quantity of total RNA were checked using the Experion
(Bio-Rad, Hercules, CA, USA). RNA extracted from three
to five plants were pooled to obtain three biological repli-
cates for each experimental condition.
Real-time quantitative RT–PCR analysis
Total RNA was treated with DNAse (Ambion, Foster City,
CA, USA) according to the manufacturer’s instructions and
RNA was subsequently quantified using a NanoDrop 1000
Spectrophotometer (Thermo Fisher Scientific, Waltham,
MA, USA).
For each sample, 4 lg of total RNA was used to
synthesize cDNA using Stratascript reverse transcriptase
(Stratagene, La Jolla, CA, USA) and SUPERase-In
RNase Inhibitor (Applied Biosystems/Ambion, Austin, TX,
USA) according to the manufacturer’s instructions. Primers
(Online Resource 1) were designed using Primer 3 software
(http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi),
on the basis of TC and the more recent corresponding SGN
sequences.
PCRs were carried out in 96-well plates with the
Applied Real-Time PCR Detection System (Applied Bio-
systems, Foster City, CA, USA), according to the following
cycling parameters: 95�C for 10 min (1 cycle), 15 s at
95�C 1 min at 60�C (40 cycles). Each reaction was con-
ducted in triplicate in a final volume of 10 ll containing
about 20 ng of template cDNA, 300 nM gene-specific
primers, 2X Power SYBR Green Master Mix.
A melting curve analysis (95–60�C with a cooling rate
of 0.5�C per 15 s and continuous fluorescence measure-
ment) was performed after each reaction to exclude the
generation of non-specific PCR products. PCR efficiency
was determined for each set of primers by using standard
curves on six standard tomato DNA serial dilutions.
The ubiquitin tomato gene Ubi3 (X58253), verified to be
not regulated in our experimental conditions (data not
shown), was used as a reference gene; the comparative
threshold cycle (Ct) method was used to calculate relative
expression levels (Rasmussen 2001).
Results and discussion
The aim of this work is to develop a data-mining approach
using publicly available EST expression data to identify
genes likely to be functionally related to a gene of interest
and provide a reliable method to prioritize candidate genes
for targeted experiments.
Our basic assumption is that genes belonging to the same
functional module are likely to be co-expressed and main-
tain their co-expression among closely related species. It
worth mentioning here the recently proposed neutral theory
of transcriptome evolution whereby divergence in transcript
abundance among taxa would be selectively neutral and
likely to be of little or no functional significance (Khaito-
vich et al. 2004; Broadley et al. 2008). In the neutral theory
of evolution, positive selection has little or no role in
determining divergence between species, but negative
(purifying) selection is assumed to be responsible of the
conservation of characters. Indeed the divergence of gene
expression profiles has been confirmed to be significantly
lower for orthologous genes than for randomly chosen ones
(Jordan et al. 2005). Therefore conserved co-expression
among orthologs can be used to achieve higher accuracy in
522 Plant Mol Biol (2010) 73:519–532
123
function prediction than simple co-expression. Such
co-expression, when phylogenetically conserved in more
than two species, may significantly increase the reliability
of predictions, allowing more accurate inferences of func-
tional relationships among genes. Being a condition-inde-
pendent approach (Usadel et al. 2009), ORTom consider
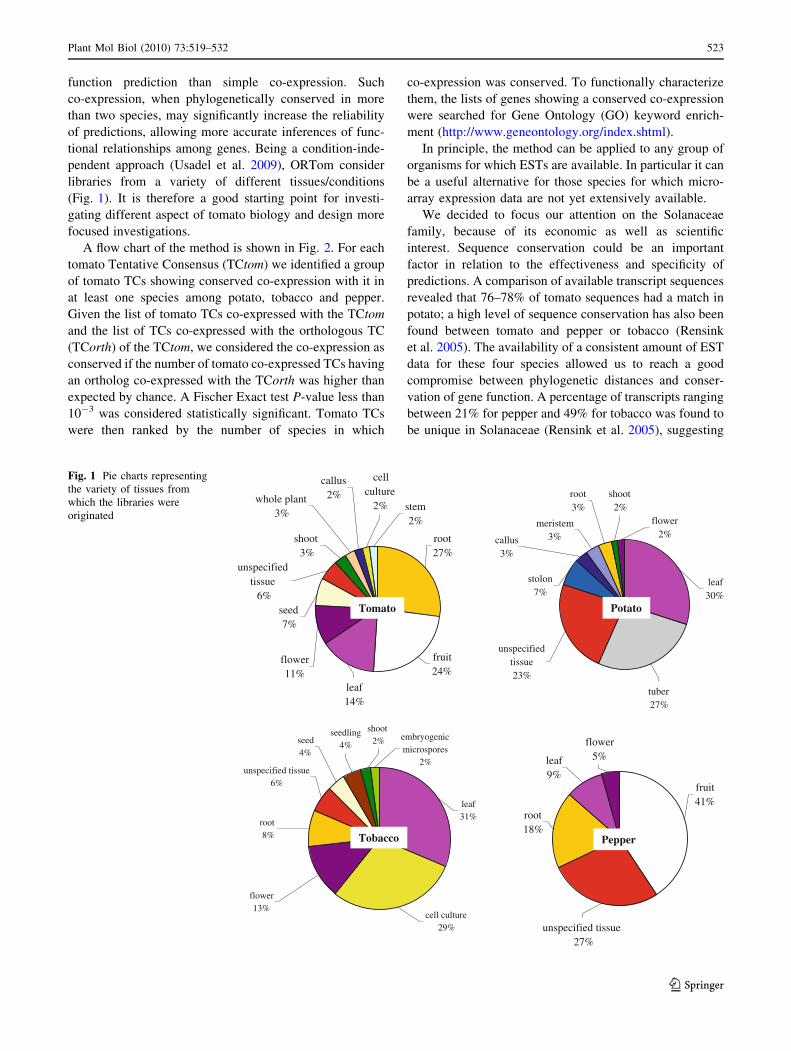
libraries from a variety of different tissues/conditions
(Fig. 1). It is therefore a good starting point for investi-
gating different aspect of tomato biology and design more
focused investigations.
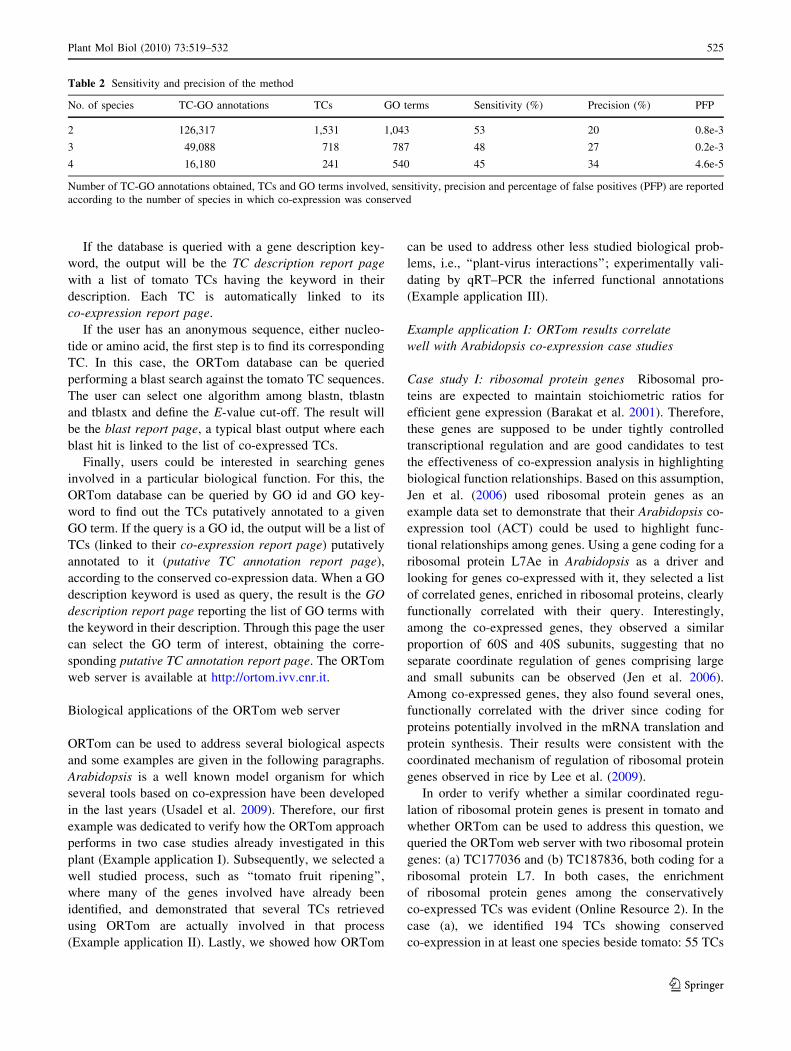
A flow chart of the method is shown in Fig. 2. For each
tomato Tentative Consensus (TCtom) we identified a group
of tomato TCs showing conserved co-expression with it in
at least one species among potato, tobacco and pepper.
Given the list of tomato TCs co-expressed with the TCtom
and the list of TCs co-expressed with the orthologous TC
(TCorth) of the TCtom, we considered the co-expression as
conserved if the number of tomato co-expressed TCs having
an ortholog co-expressed with the TCorth was higher than
expected by chance. A Fischer Exact test P-value less than
10-3 was considered statistically significant. Tomato TCs
were then ranked by the number of species in which
co-expression was conserved. To functionally characterize
them, the lists of genes showing a conserved co-expression
were searched for Gene Ontology (GO) keyword enrich-
ment (http://www.geneontology.org/index.shtml).
In principle, the method can be applied to any group of
organisms for which ESTs are available. In particular it can
be a useful alternative for those species for which micro-
array expression data are not yet extensively available.
We decided to focus our attention on the Solanaceae
family, because of its economic as well as scientific
interest. Sequence conservation could be an important
factor in relation to the effectiveness and specificity of
predictions. A comparison of available transcript sequences
revealed that 76–78% of tomato sequences had a match in
potato; a high level of sequence conservation has also been
found between tomato and pepper or tobacco (Rensink
et al. 2005). The availability of a consistent amount of EST
data for these four species allowed us to reach a good
compromise between phylogenetic distances and conser-
vation of gene function. A percentage of transcripts ranging
between 21% for pepper and 49% for tobacco was found to
be unique in Solanaceae (Rensink et al. 2005), suggesting
leaf14%
fruit24%
root27%
stem2%
cellculture
2%
callus2%whole plant
3%
shoot3%
unspecified tissue
6%seed7%
flower11%
tuber27%
unspecifiedtissue23%
stolon7%
callus3%
meristem3%
root3%
shoot2%
flower2%
leaf30%
leaf31%
embryogenic microspores
2%
shoot2%
seedling4%seed
4%
unspecified tissue6%
root8%
flower13%
cell culture29%
fruit41%
flower5%leaf
9%
root18%
unspecified tissue27%
Tomato Potato
Tobacco Pepper
Fig. 1 Pie charts representing
the variety of tissues from
which the libraries were
originated
Plant Mol Biol (2010) 73:519–532 523
123

the existence of genes characteristic of this family. In this
perspective, our method will provide a useful tool to
investigate Solanaceae-specific functional modules.
Conserved co-expression and functional annotation
According to release 13 of the EGO database, 13,032 TCs
(about 63%) belonging to the DFCI tomato release 11 have
an ortholog in at least one species among potato, tobacco
and pepper. By our method, we found that for 1,609 of
those TCs, corresponding to approximately 12.3%, it is
possible to define groups of TCs showing conserved
co-expression in at least one of the other three species
considered (P-value \ 10-3); 2,553 TCs have an ortholog
in all the Solanaceae considered. Among them we selected
262 TCs for which it was possible to identify a list of TCs
with conserved co-expression in tomato, potato, tobacco
and pepper. This collection of transcripts could be con-
sidered as a core of best candidates to be experimentally
tested for functional relationships.
Lists of co-expressed TCs, ranked according to the
number of species in which we observe conserved
co-expression, can be already used to prioritize candidate
genes for targeted experiments (see example applications).
Moreover, to improve their functional annotation, we
searched each group of co-expressed TCs for Gene
Ontology term enrichment. Statistically significant GO
keywords (exact Fisher test P-value \ 10-6) were con-
sidered as new putative functional annotations, subject to
experimental validation. The percentage of false positives
(PFP) was estimated according to the procedure described
in the method section.
We calculated the sensitivity as the proportion of true
GO annotations recalled by the method over the number of
true annotations available on the DFCI website. The pre-
cision was evaluated as the percentage of recalled anno-
tations over all the putative GO annotations obtained
(Table 2). As expected, our approach slightly decreases in
sensitivity when the number of species in which the
co-expression is conserved increases, due to the lower
number of transcripts for which the conserved co-expres-
sion is detected. On the other hand, in the same conditions
the precision of the predictions tends to improve, sup-
porting the hypothesis that if the co-expression is con-
served in several species, the functional inference is more
reliable (Bergmann et al. 2004). Both sensitivity and pre-
cision of the method will be improved as new expression
data becomes available.
Structure and use of the ORTom web server
The results obtained with the proposed method have been
stored in a MySQL database, and an easy to use web
interface has been provided to query it for further investi-
gations (Fig. 3). ORTom can be inspected using five dif-
ferent query forms on: (1) tomato TC id, (2) gene
description keyword, (3) sequence, (4) GO annotation id,
and (5) GO annotation keyword. When a tomato TC id
is used as a query, the user will obtain as output a
co-expression report page with the list of tomato TCs
showing conserved co-expression with the query. A link
allows the user to download a tab-delimited file with this
list. Each tomato TC is associated with its orthologs in the
other species and is linked to the graphical presence/
absence profile page, displaying a graphical representation
of presence/absence profiles. Each profile is linked to a
page reporting the list of libraries used. To facilitate the
selection of candidate genes for targeted experiments, the
list of TCs is sorted according to the number of species in
which co-expression is conserved. A link allows the user to
obtain the putative GO annotations related to the TC query
(putative GO report page). Putative annotations are sorted
according to the Fisher’s exact test P-value.
ESTs expression
data
GOenrichment
ORTom web
interface
Lists of genes showing conserved
co-expression
MySQL database
Orthologs from EGO database
Calculate co-expression of genes for each species
Lists of co-expressed genes ranked according to the number of species where the co-expression
is conserved
tomato
potato
tobacco
pepper
ESTs expression
data
EST data from DFCI
database
Putative GO annotations
Fig. 2 ORTom data-mining approach flow chart
524 Plant Mol Biol (2010) 73:519–532
123
If the database is queried with a gene description key-
word, the output will be the TC description report page
with a list of tomato TCs having the keyword in their
description. Each TC is automatically linked to its
co-expression report page.
If the user has an anonymous sequence, either nucleo-
tide or amino acid, the first step is to find its corresponding
TC. In this case, the ORTom database can be queried
performing a blast search against the tomato TC sequences.
The user can select one algorithm among blastn, tblastn
and tblastx and define the E-value cut-off. The result will
be the blast report page, a typical blast output where each
blast hit is linked to the list of co-expressed TCs.
Finally, users could be interested in searching genes
involved in a particular biological function. For this, the
ORTom database can be queried by GO id and GO key-
word to find out the TCs putatively annotated to a given
GO term. If the query is a GO id, the output will be a list of
TCs (linked to their co-expression report page) putatively
annotated to it (putative TC annotation report page),
according to the conserved co-expression data. When a GO
description keyword is used as query, the result is the GO
description report page reporting the list of GO terms with
the keyword in their description. Through this page the user
can select the GO term of interest, obtaining the corre-
sponding putative TC annotation report page. The ORTom
web server is available at http://ortom.ivv.cnr.it.
Biological applications of the ORTom web server
ORTom can be used to address several biological aspects
and some examples are given in the following paragraphs.
Arabidopsis is a well known model organism for which
several tools based on co-expression have been developed
in the last years (Usadel et al. 2009). Therefore, our first
example was dedicated to verify how the ORTom approach
performs in two case studies already investigated in this
plant (Example application I). Subsequently, we selected a
well studied process, such as ‘‘tomato fruit ripening’’,
where many of the genes involved have already been
identified, and demonstrated that several TCs retrieved
using ORTom are actually involved in that process
(Example application II). Lastly, we showed how ORTom
can be used to address other less studied biological prob-
lems, i.e., ‘‘plant-virus interactions’’; experimentally vali-
dating by qRT–PCR the inferred functional annotations
(Example application III).
Example application I: ORTom results correlate
well with Arabidopsis co-expression case studies
Case study I: ribosomal protein genes Ribosomal pro-
teins are expected to maintain stoichiometric ratios for
efficient gene expression (Barakat et al. 2001). Therefore,
these genes are supposed to be under tightly controlled
transcriptional regulation and are good candidates to test
the effectiveness of co-expression analysis in highlighting
biological function relationships. Based on this assumption,
Jen et al. (2006) used ribosomal protein genes as an
example data set to demonstrate that their Arabidopsis co-
expression tool (ACT) could be used to highlight func-
tional relationships among genes. Using a gene coding for a
ribosomal protein L7Ae in Arabidopsis as a driver and
looking for genes co-expressed with it, they selected a list
of correlated genes, enriched in ribosomal proteins, clearly
functionally correlated with their query. Interestingly,
among the co-expressed genes, they observed a similar
proportion of 60S and 40S subunits, suggesting that no
separate coordinate regulation of genes comprising large
and small subunits can be observed (Jen et al. 2006).
Among co-expressed genes, they also found several ones,
functionally correlated with the driver since coding for
proteins potentially involved in the mRNA translation and
protein synthesis. Their results were consistent with the
coordinated mechanism of regulation of ribosomal protein
genes observed in rice by Lee et al. (2009).
In order to verify whether a similar coordinated regu-
lation of ribosomal protein genes is present in tomato and
whether ORTom can be used to address this question, we
queried the ORTom web server with two ribosomal protein
genes: (a) TC177036 and (b) TC187836, both coding for a
ribosomal protein L7. In both cases, the enrichment
of ribosomal protein genes among the conservatively
co-expressed TCs was evident (Online Resource 2). In the
case (a), we identified 194 TCs showing conserved
co-expression in at least one species beside tomato: 55 TCs
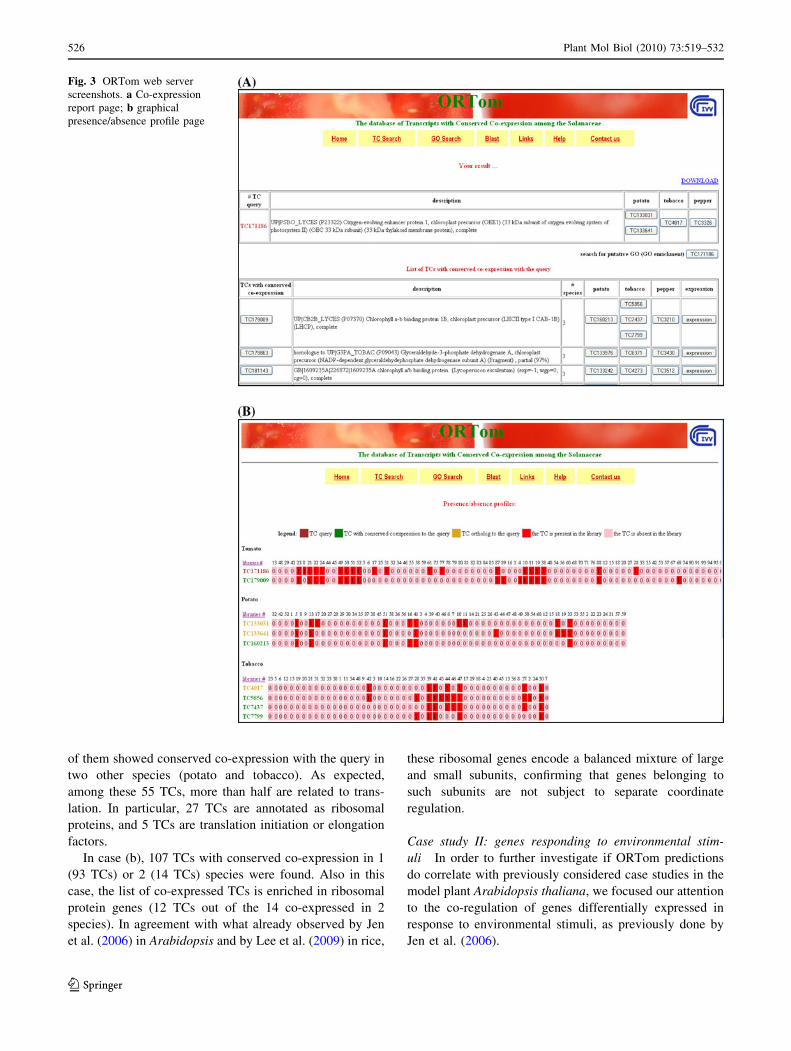
Table 2 Sensitivity and precision of the method
No. of species TC-GO annotations TCs GO terms Sensitivity (%) Precision (%) PFP
2 126,317 1,531 1,043 53 20 0.8e-3
3 49,088 718 787 48 27 0.2e-3
4 16,180 241 540 45 34 4.6e-5
Number of TC-GO annotations obtained, TCs and GO terms involved, sensitivity, precision and percentage of false positives (PFP) are reported
according to the number of species in which co-expression was conserved
Plant Mol Biol (2010) 73:519–532 525
123
of them showed conserved co-expression with the query in
two other species (potato and tobacco). As expected,
among these 55 TCs, more than half are related to trans-
lation. In particular, 27 TCs are annotated as ribosomal
proteins, and 5 TCs are translation initiation or elongation
factors.
In case (b), 107 TCs with conserved co-expression in 1
(93 TCs) or 2 (14 TCs) species were found. Also in this
case, the list of co-expressed TCs is enriched in ribosomal
protein genes (12 TCs out of the 14 co-expressed in 2
species). In agreement with what already observed by Jen
et al. (2006) in Arabidopsis and by Lee et al. (2009) in rice,
these ribosomal genes encode a balanced mixture of large
and small subunits, confirming that genes belonging to
such subunits are not subject to separate coordinate
regulation.
Case study II: genes responding to environmental stim-
uli In order to further investigate if ORTom predictions
do correlate with previously considered case studies in the
model plant Arabidopsis thaliana, we focused our attention
to the co-regulation of genes differentially expressed in
response to environmental stimuli, as previously done by
Jen et al. (2006).
Fig. 3 ORTom web server
screenshots. a Co-expression
report page; b graphical
presence/absence profile page
526 Plant Mol Biol (2010) 73:519–532
123

By using their co-expression based tool ACT, those
authors highlighted the functional correlation among genes
implicated in the heat shock response. Choosing as a
ORTom query the TC170016, coding for a heat shock
cognate 70 kDa protein 1, we selected 12 TCs with con-
served co-expression in 2 species (potato and pepper)
(Online Resource 2). Among them, 4 TCs were
clearly correlated to the query because of their involve-
ment in the response to environmental stimuli: TC175284
and TC179427 annotated as heat shock proteins (HSP),
TC169983 and TC170750 annotated as catalases, enzymes
involved in the response to oxidative stress. Since plant
HSPs are known to respond to a wide range of environ-
mental stresses, including heat, cold, drought, salinity and
oxidative stress (Wang et al. 2004b), the presence of
catalases among the co-expressed TCs is not surprising. A
fifth TC (TC186716) coding for an ATP-dependent Clp
protease ATP-binding subunit clpA, a chaperone involved
in the degradation of denatured proteins, was correlated to
the query by the fact that heat shock proteins are known to
function as chaperones, playing an important role in protein
folding, assembly, translocation and degradation (Wang
et al. 2004b). Such piece of evidence allows correlating our
query with three other co-expressed TCs (TC173852,
TC178671, and TC170633) involved in protein synthesis/
turnover. Eventually, more than 60% of the selected co-
expressed TCs were functionally related with the query.
These two case studies confirm that ORTom results
correlate well with those based on co-expression in Ara-
bidopsis, for which a wide amount of microarray data is
available.
Example application II: tomato fruit ripening
Tomato has been a model plant for the study of fleshy fruit
ripening and a lot of information is available in the liter-
ature on this process. Therefore, we decided to focus our
first example of ORTom web server application on this
biological process. As query we used the transcript
TC172177, annotated as a ripening regulated protein-like,
but lacking any other information from the literature. The
ORTom output consisted in 56 TCs showing conserved
co-expression with TC172177. Four of them showed con-
served co-expression in two other species (potato and
tobacco), and 52 only in one (potato or tobacco) (Online
Resource 3). According to the basic assumption of func-
tional correlation among conserved co-expressed genes,
several of the 56 selected TCs should be involved in the
ripening process and therefore functionally correlated with
the query. Alba et al. (2005), analyzing the transcriptional
changes during tomato fruit ripening, identified 869 genes
differentially expressed in developing tomato pericarp. Out
of 56 TCs putatively involved in ripening, according to
ORTom prediction, 13 have no homology (blastn
e-value B 10-10) among the sequences spotted in the
TOM1 array used in that study. Out of the remaining 43
TCs, 24 (more than 50%) have been found differentially
expressed during tomato ripening (Online Resource 3).
Interestingly, among them there are several genes indicated
by ORTom as involved in ripening but for which little or
no other evidence for such role is present in the literature.
In particular, among the TCs selected by ORTom, we
found two TCs (TC171077 and TC171403) clearly involved
in tomato ripening, as confirmed by their annotation and by
the data of Alba et al. (2005); they encode the fruit-ripening
protein E4 and the ripening-related mRNA ERT13,
respectively (Cordes et al. 1989; Picton et al. 1993). A third
TC (TC176732) shows similarity with the protein PM23
involved in seed maturation, fundamental in the ripening
process. Four are related to ethylene, a well known hormone
essential for fruit ripening (Alexander and Grierson 2002).
They are a 1-aminocyclopropane-1-carboxylate oxidase 4
(TC169966), a key enzyme in the ethylene biosynthesis; an
S-adenosylmethionine synthase 2 (TC170924), involved in
the Yang cycle, an early step in the biosynthesis of ethylene
(Wang et al. 2002); an S-adenosyl-L-homocysteine hydro-
lase (TC170042), involved in the S-adenosyl-L-methionine
cycle for the regeneration of methionine, the starting com-
pound in ethylene biosynthesis (Ravanel et al. 2004); and a
jasmonate and ethylene responsive factor 3 (TC170506), a
gene mainly induced by ethylene in tomato (Wang et al.
2004a). Three TCs (TC171281, TC177883, and TC174916)
are annotated as alcohol dehydrogenases (ADH); Longhurst
et al. (1990) showed that ADH activity decreases during the
early stages of ripening and then increases in the post-cli-
macteric period. The authors proposed that the increase
during ripening may contribute to flavour development.
TC171808 is annotated as a GDP-mannose phyrophos-
phorylase (GMP), an enzyme for the synthesis of the
ascorbic acid. Two studies on tomato (Zou et al. 2006) and
acerola (Badejo et al. 2007) indicate that GMP activity is
highest in fruits. TC170725 encodes an eukaryotic transla-
tion initiation factor 5A-4 (eIF-5A-4). Wang et al. (2005)
found that three members of tomato eIF-5A family were up-
regulated in parallel as the fruit begins to senesce and soften
and that plants where the activation of eIF-5A was inhibited
exhibited delayed fruit postharvest softening and senes-
cence. Interestingly, changes in transcriptional profiles of
ADH, GMP and eIF-5A were found associated with early
specialization of tomato fruit tissue (Lemaire-Chamley
et al. 2005). Finally, two TCs (TC 174083 and TC170496),
encoding a citrate synthase and an oxoglutarate/malate
translocator, respectively, can be related to the alteration of
citric and malic acid concentrations during tomato ripening
(Jeffery et al. 1984, 1986; Carrari et al. 2006).
Plant Mol Biol (2010) 73:519–532 527
123

Other TCs form a second class of transcripts, whose
relations with the ripening process are, according to the
limited literature available, still uncertain. The first one is
TC170007, encoding a 14-3-3 protein which was indicated
as an ethylene-dependent regulatory gene involved in rip-
ening (Alba et al. 2005) and found differentially expressed
particularly in exocarp during tissue specialization (Lem-
aire-Chamley et al. 2005); The 14-3-3 proteins, implicated
in the regulation of several physiological processes such as
regulation of primary metabolism, plant response to biotic
and abiotic stimuli (Finnie et al. 1999), have been already
isolated from ripening tomato fruits and their involvement
in fruit development was suggested (Laughner et al. 1994).
A second one is TC172410, annotated as a RAB7C protein.
It belongs to the RAB family, part of RAS superfamily of
small GTPases, regulators of membrane traffic pathways
(Stenmark and Olkkonen 2001). Other Rab mRNAs were
already observed to accumulate during tomato (Loraine
et al. 1996; Lu et al. 2001; Alba et al. 2005), mango (Zainal
et al. 1996), and apricot fruit ripening (Mbeguie-A-Mbeguie
et al. 1997). A third one is TC175454, which encodes a
disulfide isomerase protein, involved in the protein folding;
a precursor of this enzyme has been isolated in tomato fruits
in a survey of major protein variations during pericarp
development and ripening (Faurobert et al. 2007). The role
of these three TCs in the ripening process is therefore worth
investigating in depth by further experimental studies.
The 39 remaining TCs consist of ribosomal proteins (9
TCs), histones (2 TCs), unknown expressed proteins (4
TCs), TCs for which annotations are too generic to be
correlated with ripening in the literature (13 TCs), and TCs
with an annotation for which is difficult to suppose, at the
moment, an involvement in ripening on the basis of liter-
ature (11 TCs). Since these TCs were selected after a
search with ORTom, it is possible that some among them
are also involved in ripening, and they are therefore
potential candidates to be considered in studies on this
important biological process. The possible involvement of
17 of these genes is supported by the fact that they were
found differentially expressed during ripening in a tomato
microarray study (Alba et al. 2005). In our opinion, based
on these data, particular attention should be paid for
example to the nucleic acid binding protein (TC182771) or
to the Zinc finger transcription factor-like protein
(TC174072), as potential transcriptional regulators, or to
those TCs just annotated until now as expressed proteins
but for which no more information is available.
This example confirms that ORTom web server can
retrieve the functional annotation of those TCs whose the
function is already known from the literature and can be
useful to infer the biological role of TCs associated with
poor or no functional annotation.
Example application III: plant-virus interactions
To better elucidate how the ORTom web server can be used
to improve functional gene annotation, we used it to find new
candidate genes likely to be involved in plant-virus inter-
actions resulting in systemic infection. We chose as query a
tobacco gene, coding for an oxygen-evolving enhancer
protein 1, chloroplastic, OEE1 (Acc. No. X64349) known to
interact with Tobacco mosaic virus (TMV) replicase, and
tested our supposed functional annotation experimentally by
qRT-PCR.
Isolated in a yeast two-hybrid experiment by Abbink
et al. (2002), OEE1 was down-regulated in systemically
infected leaves, while its silencing resulted in a tenfold
increase of TMV accumulation. An analogous viral
increase was observed when silenced plants were infected
by two other RNA viruses, belonging to different genera,
Alfalfa mosaic virus (AMV) and Potato virus X (PVX),
suggesting this effect is not specific to a particular virus.
Focusing our attention on tomato as a model plant, we
speculated that: (1) a homologous tomato gene could be
involved in systemic virus infection; (2) genes showing
conserved co-expression with it are likely to be function-
ally correlated to this gene and therefore involved in the
systemic infection process.
To test the hypothesis (1), we first searched for
homologous genes in tomato and analysed their expression
in tomato leaves systemically infected by Tomato spotted
wilt virus (TSWV), an RNA virus, known causing huge
crop losses worldwide (Prins and Goldbach 1998).
Blasting the sequence X64349 reported in Abbink et al.
(2002) through the ORTom web server identified two TC
sequences (E-value = 0.0), TC171186 and TC170466,
both annotated as oxygen-evolving enhancer protein 1,
chloroplast precursor (OEE1), a 33 k subunit of the oxy-
gen-evolving system of photosystem II. Quantitative
RT-PCR experiments, with primers designed on TC171186
were performed on TSWV infected leaves, showing that
OEE1 is down-regulated. This indicates that OEE1 is
involved in the tomato-TSWV interaction, just as the
homologous gene is in systemic infection of N. benthami-
ana by TMV, AMV and PVX.
To test the hypothesis (2), we used as queries the two
TCs annotated as OEE1 and obtained two lists of TCs
showing conserved co-expression in potato, tobacco and
pepper. Considering that eight TCs were presents in both
lists and others showed high sequence similarity, the
dataset was reduced to a total of 11 transcripts (see Table 3
for gene description).
We hypothesized that these genes are involved in virus
infection, and investigated their regulation in TSWV-
infected leaves by qRT-PCR. For 7 out of 11 genes, down-
528 Plant Mol Biol (2010) 73:519–532
123
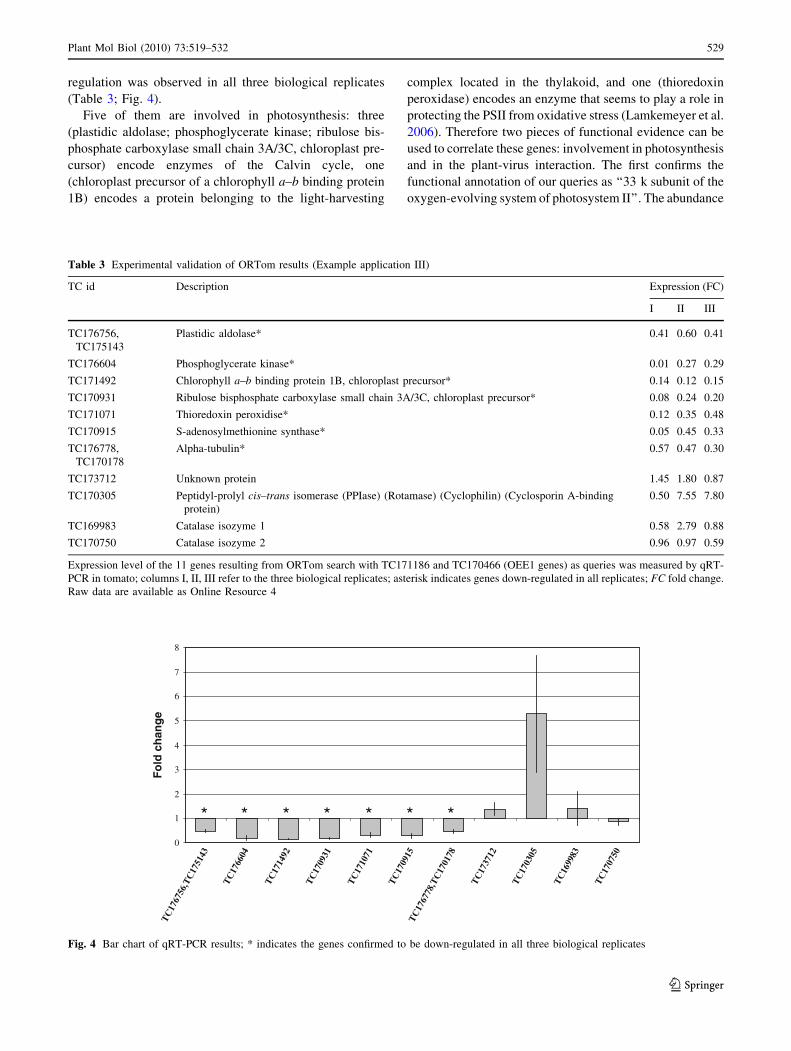
regulation was observed in all three biological replicates
(Table 3; Fig. 4).
Five of them are involved in photosynthesis: three
(plastidic aldolase; phosphoglycerate kinase; ribulose bis-
phosphate carboxylase small chain 3A/3C, chloroplast pre-
cursor) encode enzymes of the Calvin cycle, one
(chloroplast precursor of a chlorophyll a–b binding protein
1B) encodes a protein belonging to the light-harvesting
complex located in the thylakoid, and one (thioredoxin
peroxidase) encodes an enzyme that seems to play a role in
protecting the PSII from oxidative stress (Lamkemeyer et al.
2006). Therefore two pieces of functional evidence can be
used to correlate these genes: involvement in photosynthesis
and in the plant-virus interaction. The first confirms the
functional annotation of our queries as ‘‘33 k subunit of the
oxygen-evolving system of photosystem II’’. The abundance
Table 3 Experimental validation of ORTom results (Example application III)
TC id Description Expression (FC)
I II III
TC176756,
TC175143
Plastidic aldolase* 0.41 0.60 0.41
TC176604 Phosphoglycerate kinase* 0.01 0.27 0.29
TC171492 Chlorophyll a–b binding protein 1B, chloroplast precursor* 0.14 0.12 0.15
TC170931 Ribulose bisphosphate carboxylase small chain 3A/3C, chloroplast precursor* 0.08 0.24 0.20
TC171071 Thioredoxin peroxidise* 0.12 0.35 0.48
TC170915 S-adenosylmethionine synthase* 0.05 0.45 0.33
TC176778,
TC170178
Alpha-tubulin* 0.57 0.47 0.30
TC173712 Unknown protein 1.45 1.80 0.87
TC170305 Peptidyl-prolyl cis–trans isomerase (PPIase) (Rotamase) (Cyclophilin) (Cyclosporin A-binding
protein)
0.50 7.55 7.80
TC169983 Catalase isozyme 1 0.58 2.79 0.88
TC170750 Catalase isozyme 2 0.96 0.97 0.59
Expression level of the 11 genes resulting from ORTom search with TC171186 and TC170466 (OEE1 genes) as queries was measured by qRT-
PCR in tomato; columns I, II, III refer to the three biological replicates; asterisk indicates genes down-regulated in all replicates; FC fold change.
Raw data are available as Online Resource 4
0
1
2
3
4
5
6
7
8
TC
1767
56,T
C17
5143
TC
1766
04
TC
1714
92
TC
1709
31
TC
1710
71
TC
1709
15T
C17
6778
,TC
1701
78
TC
1737
12
TC
1703
05
TC
1699
83
TC
1707
50
Fo
ld c
han
ge
* * ** * * *
Fig. 4 Bar chart of qRT-PCR results; * indicates the genes confirmed to be down-regulated in all three biological replicates
Plant Mol Biol (2010) 73:519–532 529
123

of transcripts correlated with photosynthesis among those
co-expressed with the query in all the Solanaceae considered
makes this annotation more reliable.
Two more genes, not involved in photosynthesis, were
experimentally validated for coexpression with the query:
an alpha-tubulin and an S-adenosylmethionine synthase.
Previous proteomic studies highlighted the importance of
alpha-tubulin in viral infection, showing that TMV move-
ment protein (MP) (Heinlein et al. 1995) and viral RNA
(Mas and Beachy 1999) co-localized with microtubules
and endoplasmic reticulum. Microtubules directly interact
with the TMV MP during late stages of infection (Ashby
et al. 2006). Moreover, evidence of a role of TMV repli-
case in cell-to-cell movement (Hirashima and Watanabe
2003) suggests that virus replication and movement in
TMV are functionally linked.
S-adenosylmethionine synthetase (ADS) catalyzes the
formation of S-adenosylmethionine (AdoMet) from methio-
nine and ATP. AdoMet is the major methyl donor in plants
and is involved in the methylation of lipids, proteins and
nucleic acids (Fontecave et al. 2004). Moreover it is a
common precursor of polyamines and ethylene biosynthesis
(Walters 2000), two pathways known to be involved in
plant-virus interactions.
Several authors have observed an increase of conjugated
and free polyamines during the hypersensitive response
(HR) to TMV infection suggesting a role in virus resis-
tance, possibly inducing programmed cell death or affect-
ing virus multiplication (Walters 2003). Yamakawa et al.
(1998) showed that spermidine accumulates in tobacco
leaves reacting hypersensitively to TMV, and can induce
acidic pathogenesis-related proteins and resistance to TMV
via a salicylic acid-independent pathway. On the other
hand, ethylene is a phytohormone well known as principal
modulator in various mechanisms by which plants react to
pathogens (Broekaert et al. 2006). Genes encoding ADS
have been cloned from various plants, but until now no
specific regulation of them in response to pathogen attack
was reported, making it difficult to figure out the exact role
of ADS in the process; however, we obtained experimental
evidence that this enzyme is down-regulated under viral
infection.
Conclusions
We have shown that ORTom is a useful data-mining
method to extract information from publicly-available EST
data. Since this kind of data is still the most representative
for many organisms, including several plants, we believe
that ORTom is an effective approach to infer putative
functions for genes of interest and to prioritize candidate
genes for further experiments. The method, which was
applied to Solanaceae but could be extended to other
groups, has the potential to limit costly and time-consum-
ing non-targeted experiments and lead more rapidly to
improved gene annotations.
Acknowledgments This work was funded in part by the projects
‘‘GenoPom’’ (MIUR, Italy) and B74 (Ricerca Scientifica Applicata
2004, Regione Piemonte, Italy). P. P. gratefully acknowledges sup-
port from the Associazione Italiana per la Ricerca sul Cancro (AIRC).
The authors thank Christian Damasco, Stefano Ghignone and Matteo
Giaccone for their advice in developing the web site and Robert
G. Milne for revising the English.
References
Abbink TE, Peart JR, Mos TN, Baulcombe DC, Bol JF, Linthorst HJ
(2002) Silencing of a gene encoding a protein component of the
oxygen-evolving complex of photosystem II enhances virus
replication in plants. Virology 295:307–319. doi:10.1006/viro.
2002.1332
Ala U, Piro RM, Grassi E, Damasco C, Silengo L, Oti M, Provero P,
Di Cunto F (2008) Prediction of human disease genes by human-
mouse conserved coexpression analysis. PLoS Comput Biol
4:e1000043. doi:10.1371/journal.pcbi.1000043
Alba R, Payton P, Fei Z, McQuinn R, Debbie P, Martin GB, Tanksley
SD, Giovannoni JJ (2005) Transcriptome and selected metabo-
lite analyses reveal multiple points of ethylene control during
tomato fruit development. Plant Cell 17(11):2954–2965. doi:
10.1105/tpc.105.036053
Alexander L, Grierson D (2002) Ethylene biosynthesis and action in
tomato: a model for climacteric fruit ripening. J Exp Bot
53(377):2039–2055. doi:10.1093/jxb/erf072
Ashby J, Boutant E, Seemanpillai M, Groner A, Sambade A,
Ritzenthaler C, Heinlein M (2006) Tobacco mosaic virus
movement protein functions as a structural microtubule-associ-
ated protein. J Virol 80:8329–8344. doi:10.1128/JVI.00540-06
Badejo AA, Jeong ST, Goto-Yamamoto N, Esaka M (2007) Cloning
and expression of GDP-D-mannose pyrophosphorylase gene and
ascorbic acid content of acerola (Malpighia glabra L.) fruit at
ripening stages. Plant Physiol Biochem 45(9):665–672. doi:
10.1016/j.plaphy.2007.07.003
Barakat A, Szick-Miranda K, Chang IF, Guyot R, Blanc G, Cooke R,
Delseny M, Bailey-Serres J (2001) The organization of cyto-
plasmic ribosomal protein genes in the Arabidopsis genome.
Plant Physiol 127:398–415. doi:10.1104/pp.010265
Bergmann S, Ihmels J, Barkai N (2004) Similarities and differences in
genome-wide expression data of six organisms. PLoS Biol 2:E9.
doi:10.1371/journal.pbio.0020009
Bhardwaj N, Lu H (2005) Correlation between gene expression
profiles and protein–protein interactions within and across
genomes. Bioinformatics 21:2730–2738. doi:10.1093/bioinfor
matics/bti398
Broadley MR, White PJ, Hammond JP, Graham NS, Bowen HC,
Emmerson ZF, Fray RG, Iannetta PP, McNicol JW, May ST
(2008) Evidence of neutral transcriptome evolution in plants.
New Phytol 180(3):587–593. doi:10.1111/j.1469-8137.2008.
02640.x
Broekaert WF, Delaure SL, De Bolle MFC, Cammue BPA (2006)
The role of ethylene in host–pathogen interactions. Annu Rev
Phytopathol 44:393–416. doi:10.1146/annurev.phyto.44.070505.
143440
Carrari F, Baxter C, Usadel B, Urbanczyk-Wochniak E, Zanor MI,
Nunes-Nesi A, Nikiforova V, Centero D, Ratzka A, Pauly M,
530 Plant Mol Biol (2010) 73:519–532
123

Sweetlove LJ, Fernie AR (2006) Integrated analysis of metab-
olite and transcript levels reveals the metabolic shifts that
underlie tomato fruit development and highlight regulatory
aspects of metabolic network behavior. Plant Physiol 142(4):
1380–1396. doi:10.1104/pp.106.088534
Cordes S, Deikman J, Margossian LJ, Fischer RL (1989) lnteraction
of a developmentally regulated DNA-binding factor with sites
flanking two different fruit-ripening genes from tomato. Plant
Cell 1(10):1025–1034. doi:10.1105/tpc.1.10.1025
Daub CO, Sonnhammer EL (2008) Employing conservation of co-
expression to improve functional inference. BMC Syst Biol 2:81.
doi:10.1186/1752-0509-2-81
Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster
analysis and display of genome-wide expression patterns. Proc
Natl Acad Sci USA 95:14863–14868
Faccioli P, Provero P, Herrmann C, Stanca AM, Morcia C, Terzi V
(2005) From single genes to co-expression networks: extracting
knowledge from barley functional genomics. Plant Mol Biol
58:739–750. doi:10.1007/s11103-005-8159-7
Faurobert M, Mihr C, Bertin N, Pawlowski T, Negroni L, Sommerer
N, Causse M (2007) Major proteome variations associated with
cherry tomato pericarp development and ripening. Plant Physiol
143(3):1327–1346. doi:10.1104/pp.106.092817
Finnie C, Borch J, Collinge DB (1999) 14-3-3 proteins: eukaryotic
regulatory proteins with many functions. Plant Mol Biol 40(4):
545–554. doi:10.1023/A:1006211014713
Fontecave M, Atta M, Mulliez E (2004) S-adenosylmethionine:
nothing goes to waste. Trends Biochem Sci 29:243–249. doi:
10.1016/j.tibs.2004.03.007
Fukuoka Y, Inaoka H, Kohane IS (2004) Inter-species differences of
co-expression of neighboring genes in eukaryotic genomes.
BMC Genomics 5:4. doi:10.1186/1471-2164-5-4
Gachon CMM, Langlois-Meurinne M, Henry Y, Saindrenan P (2005)
Transcriptional co-regulation of secondary metabolism enzymes
in Arabidopsis: functional and evolutionary implications. Plant
Mol Biol 58:229–245. doi:10.1007/sl1103-005-5346-5
Glazko G, Gordon A, Mushegian A (2005) The choice of optimal
distance measure in genome-wide datasets. Bioinformatics
21(Suppl 3):iii3–iii11. doi:10.1093/bioinformatica/bti1201
Heinlein M, Epel BL, Padgett HS, Beachy RN (1995) Interaction of
tobamovirus movement proteins with the plant cytoskeleton.
Science 270:1983–1985. doi:10.1126/science.270.5244.1983
Hirashima K, Watanabe Y (2003) RNA helicase domain of tobamo-
virus replicase executes cell-to-cell movement possibly through
collaboration with its non conserved region. J Virol 77:12357–
12362. doi:10.1128/JVI.77.22.12357-12362.2003
Jansen R, Greenbaum D, Gerstein M (2002) Relating whole-genome
expression data with protein–protein interactions. Genome Res
12:37–46. doi:10.1101/gr.205602
Jeffery D, Smith C, Goodenough P, Prosser I, Grierson D (1984)
Ethylene-independent and ethylene-dependent biochemical
changes in ripening tomatoes. Plant Physiol 74(1):32–38
Jeffery D, Goodenough PW, Weitzman PDJ (1986) Enzyme activities
in mitochondria isolated from ripening tomato fruit. Planta
168(3):390–394. doi:10.1007/BF00392366
Jen CH, Manfield IW, Michalopoulos I, Pinney JW, Willats WG,
Gilmartin PM, Westhead DR (2006) The Arabidopsis co-
expression tool (ACT): a WWW-based tool and database for
microarray-based gene expression analysis. Plant J 46(2):336–
348. doi:10.1111/j.1365-313X.2006.02681.x
Jordan IK, Marino-Ramırez L, Koonin EV (2005) Evolutionary
significance of gene expression divergence. Gene 345(1):119–
126. doi:10.1016/j.gene.2004.11.034
Khaitovich P, Weiss G, Lachmann M, Hellmann I, Enard W, Muetzel
B, Wirkner U, Ansorge W, Paabo S (2004) A neutral model of
transcriptome evolution. PLoS Biol 2(5):E132. doi:10.1371/jour
nal.pbio.0020132
Lamkemeyer P, Laxa M, Collin V, Li W, Finkemeier I, Schottler MA,
Holtkamp V, Tognetti VB, Issakidis-Bourguet E, Kandlbinder A,
Weis E, Miginiac-Maslow M, Dietz KJ (2006) Peroxiredoxin Q
of Arabidopsis thaliana is attached to the thylakoids and
functions in context of photosynthesis. Plant J 45:968–981. doi:
10.1111/j.1365-313X.2006.02665.x
Laughner B, Lawrence SD, Ferl RJ (1994) Two tomato fruit
homologs of 14-3-3 mammalian brain proteins. Plant Physiol
105(4):1457–1458
Lee TH, Kim YK, Pham TT, Song SI, Kim JK, Kang KY, An G, Jung
KH, Galbraith DW, Kim M, Yoon UH, Nahm BH (2009)
RiceArrayNet: a database for correlating gene expression from
transcriptome profiling, and its application to the analysis of
coexpressed genes in rice. Plant Physiol 151(1):16–33. doi:
10.1104/pp.109.139030
Lemaire-Chamley M, Petit J, Garcia V, Just D, Baldet P, Germain V,
Fagard M, Mouassite M, Cheniclet C, Rothan C (2005) Changes
in transcriptional profiles are associated with early fruit tissue
specialization in tomato. Plant Physiol 139(2):750–769. doi:
10.1104/pp.105.063719
Longhurst TJ, Tung HF, Brady CJ (1990) Developmental regulation
of the expression of alcohol dehydrogenase in ripening tomato
fruits. J Food Biochem 14(6):421–433. doi:10.1111/j.1745-4514.
1990.tb00804.x
Loraine AE, Yalovsky S, Fabry S, Gruissem W (1996) Tomato
Rab1A homologs as molecular tools for studying Rab geranyl-
geranyl transferase in plant cells. Plant Physiol 110(4):1337–
1347
Lu C, Zainal Z, Tucker GA, Lycett GW (2001) Developmental
abnormalities and reduced fruit softening in tomato plants
expressing an antisense Rab11 GTPase gene. Plant Cell 13(8):
1819–1833. doi:10.1105/TPC.010069
Mas P, Beachy RN (1999) Replication of tobacco mosaic virus on
endoplasmic reticulum and role of the cytoskeleton and virus
movement protein in intracellular distribution of viral RNA.
J Cell Biol 147:945–958
Mbeguie-A-Mbeguie D, Gomez RM, Fils-Lycaon B (1997) Molecular
cloning and nucleotide sequence of a Rab7 small GTP-binding
protein from apricot fruit. Gene expression during fruit ripening
(PGR97-117). Plant Physiol 114:1569
Miozzi L, Piro RM, Rosa F, Ala U, Silengo L, Di Cunto F, Provero P
(2008) Functional annotation and identification of candidate
disease genes by computational analysis of normal tissue gene
expression data. PLoS ONE 3:e2439. doi:10.1371/journal.pone.
0002439
Mutwil M, Obro J, Willats WG, Persson S (2008) GeneCAT—novel
webtools that combine BLAST and co-expression analyses.
Nucleic Acids Res 36(Web Server issue):W320–W326. doi:
10.1093/nar/gkn292
Obayashi T, Hayashi S, Shibaoka M, Saeki M, Ohta H, Kinoshita K
(2008) COXPRESdb: a database of coexpressed gene networks
in mammals. Nucleic Acids Res 36:D77–D82. doi:10.1093/nar/
gkm840
Oti M, van Reeuwijk J, Huynen M, Brunner H (2008) Conserved co-
expression for candidate disease gene prioritization. BMC
Bioinformatics 9:208. doi:10.1186/1471-2105-9-208
Pellegrino M, Provero P, Silengo L, Di Cunto F (2004) CLOE:
identification of putative functional relationships among genes
by comparison of expression profiles between two species. BMC
Bioinformatics 5:179–189. doi:10.1186/1471-2105-5-179
Picton S, Gray J, Barton S, AbuBakar U, Lowe A, Grierson D (1993)
cDNA cloning and characterisation of novel ripening-related
mRNAs with altered patterns of accumulation in the ripening
Plant Mol Biol (2010) 73:519–532 531
123

inhibitor (rin) tomato ripening mutant. Plant Mol Biol 23(1):
193–207. doi:10.1007/BF00021431
Prins M, Goldbach R (1998) The emerging problem of tospovirus
infection and nonconventional methods of control. Trends
Microbiol 6:31–35. doi:10.1016/S0966-842X(97)01173-6
Ramani AK, Li Z, Hart TG, Carlson MW, Boutz DR, Marcotte EM
(2008) A map of human protein interactions derived from co-
expression of human mRNAs and their orthologs. Mol Syst Biol
4:180. doi:10.1038/msb.2008.19
Rasmussen R (2001) Quantification on the LightCycler. In: Meuer
SC, Wittwer C, Nakagawara K (eds) Rapid cycle real-time PCR
methods and applications. Springer, Heidelberg, pp 21–34
Ravanel S, Block MA, Rippert P, Jabrin S, Curien G, Rebeille F,
Douce R (2004) Methionine metabolism in plants: chloroplasts
are autonomous for de novo methionine synthesis and can import
S-adenosylmethionine from the cytosol. J Biol Chem 279(21):
22548–22557. doi:10.1074/jbc.M313250200
Rensink WA, Lee Y, Liu J, Iobst S, Ouyang S, Buell CR (2005)
Comparative analyses of six solanaceous transcriptomes reveal a
high degree of sequence conservation and species-specific
transcripts. BMC Genomics 6:124–138. doi:10.1186/1471-2164-
6-124
Shmulevich I, Zhang W (2002) Binary analysis and optimization-
based normalization of gene expression data. Bioinformatics
18(4):555–565. doi:10.1093/bioinformatics/18.4.555
Stenmark H, Olkkonen VM (2001) The Rab GTPase family. Genome
Biol 2(5):REVIEWS3007. doi: 10.1186/gb-2001-2-5-reviews
3007
Teichmann SA, Babu MM (2002) Conservation of gene co-regulation
in prokaryotes and eukaryotes. Trends Biotechnol 20:407–410.
doi:10.1016/S0167-7799(02)02032-2
Trotta A, Varese GC, Gnavi E, Fusconi A, Sampo S, Berta G (1996)
Interactions between the soil-borne root pathogen Phytophthoranicotianae var parasitica and arbuscular mycorrhizal fungus
Glomus mosseae in tomato plants. Plant Soil 185:199–209. doi:
10.1007/BF02257525
Usadel B, Obayashi T, Mutwil M, Giorgi FM, Bassel GW, Tanimoto
M, Chow A, Steinhauser D, Persson S, Provart NJ (2009) Co-
expression tools for plant biology: opportunities for hypothesis
generation and caveats. Plant Cell Environ 32:1633–1651. doi:
10.1111/j.1365-3040.2009.02040.x
Van Noort V, Snel B, Huynen MA (2003) Predicting gene function by
conserved co-expression. Trends Genet 19:238–242. doi:
10.1016/S0168-9525(03)00056-8
Walters DR (2000) Polyamines in plant–microbe interactions. Physiol
Mol Plant Pathol 57:137–146. doi:10.1006/pmpp.2000.0286
Walters D (2003) Resistance to plant pathogens: possible roles for
free polyamines and polyamine catabolism. New Phytol 159:
109–115. doi:10.1046/j.1469-8137.2003.00802.x
Wang KL, Li H, Ecker JR (2002) Ethylene biosynthesis and signaling
networks. Plant Cell 14(Suppl):S131–S151
Wang H, Huang Z, Chen Q, Zhang Z, Zhang H, Wu Y, Huang D,
Huang R (2004a) Ectopic overexpression of tomato JERF3 in
tobacco activates downstream gene expression and enhances salt
tolerance. Plant Mol Biol 55(2):183–192. doi:10.1007/s11103-
004-0113-6
Wang W, Vinocur B, Shoseyov O, Altman A (2004b) Role of plant
heat-shock proteins and molecular chaperones in the abiotic
stress response. Trends Plant Sci 9(5):244–252. doi:10.1016/j.
tplants.2004.03.006
Wang TW, Zhang CG, Wu W, Nowack LM, Madey E, Thompson JE
(2005) Antisense suppression of deoxyhypusine synthase in
tomato delays fruit softening and alters growth and development.
Plant Physiol 138(3):1372–1382. doi:10.1104/pp.105.060194
Wang Y, Tang X, Cheng Z, Mueller L, Giovannoni J, Tanksley SD
(2006) Euchromatin and pericentromeric heterochromatin: com-
parative composition in the tomato genome. Genetics 172:2529–
2540. doi:10.1534/genetics.106.055772
Wolfe CJ, Kohane IS, Butte AJ (2005) Systematic survey reveals
general applicability of ‘‘guilt-by-association’’ within gene
coexpression networks. BMC Bioinformatics 6:227–237. doi:
10.1186/1471-2105-6-227
Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R,
Altschuler SJ (2002) Large-scale prediction of Saccharomycescerevisiae gene function using overlapping transcriptional clus-
ters. Nat Genet 31:255–265. doi:10.1038/ng906
Wu X, Walker MG, Luo J, Wei L (2005) GBA server: EST-based
digital gene expression profiling. Nucleic Acids Res 33:W673–
W676. doi:10.1093/nar/gki480
Yamakawa H, Kamada H, Satoh M, Ohashi Y (1998) Spermine is a
salicylate-independent endogenous inducer for both tobacco
acidic pathogenesis-related proteins and resistance against
tobacco mosaic virus infection. Plant Physiol 118:1213–1222
Zainal Z, Tucker GA, Lycett GW (1996) A rab11-like gene is
developmentally regulated in ripening mango (Mangifera indicaL.) fruit. Biochim Biophys Acta 1314(3):187–190
Zou LP, Li HX, Ouyang B, Zhang JH, Ye ZB (2006) Cloning,
expression, and mapping of GDP-D-mannose pyrophosphorylase
cDNA from tomato (Lycopersicon esculentum). Acta Genet Sin
33(8):757–764. doi:10.1016/S0379-4172(06)60108-X
532 Plant Mol Biol (2010) 73:519–532
123
Top Related
Copyright © 2022 FDOKUMEN

