







Bahasa
Halaman
Hukum


FEMS MICROBIOLOGY LETTERS Volume 246, Issue 2, Pages 151-294 (15 May 2005)
1. Editorial board • EDITORIAL BOARD Pages iii-vi
2. Fluorescence in situ hybridisation (FISH) – the next generation • SHORT SURVEY Pages 151-158 Katrin Zwirglmaier
3. Biotin biosynthesis, transport and utilization in rhizobia • SHORT SURVEY Pages 159-165 Karina Guillén-Navarro, Sergio Encarnación and Michael F. Dunn
4. The Pseudomonas aeruginosa pirA gene encodes a second receptor for ferrienterobactin and synthetic catecholate analogues • SHORT COMMUNICATION Pages 167-174 Bart Ghysels, Urs Ochsner, Ute Möllman, Lothar Heinisch, Michael Vasil, Pierre Cornelis and Sandra Matthijs
5. Novel target genes of PsrA transcriptional regulator of Pseudomonas aeruginosa • SHORT COMMUNICATION Pages 175-181 Milan Kojic, Branko Jovcic, Alessandro Vindigni, Federico Odreman and Vittorio Venturi
6. Isolation and characterisation of the lipopolysaccharide from Acidiphilium strain GS18h/ATCC55963, a soil isolate of Indian copper mine • SHORT COMMUNICATION Pages 183-190 Rabindranath Bera, Abhijit Nayak, Asish Kumar Sen, Biswa Pronab Chowdhury and Ranjan Bhadra
7. Comprehensive analysis of classical and newly described staphylococcal superantigenic toxin genes in Staphylococcus aureus isolates • SHORT COMMUNICATION Pages 191-198 Katsuhiko Omoe, Dong-Liang Hu, Hiromi Takahashi-Omoe, Akio Nakane and Kunihiro Shinagawa

8. Passive immunisation of hamsters against Clostridium difficile infection using antibodies to surface layer proteins • SHORT COMMUNICATION Pages 199-205 Julie B. O’Brien, Matthew S. McCabe, Verónica Athié-Morales, George S.A. McDonald, Déirdre B. Ní Eidhin and Dermot P. Kelleher
9. Morphological and molecular taxonomy of Pythium longisporangium sp. nov. isolated from the Burgundian region of France • SHORT COMMUNICATION Pages 207-212 Bernard Paul, Kanak Bala, Sabine Gognies and Abdel Belarbi
10. Polymorphism and gene conversion of the 16S rRNA genes in the multiple rRNA operons of Vibrio parahaemolyticus • SHORT COMMUNICATION Pages 213-219 Narjol González-Escalona, Jaime Romero and Romilio T. Espejo
11. Induction of murine macrophage TNF-α synthesis by Mycobacterium avium is modulated through complement-dependent interaction via complement receptors 3 and 4 in relation to M. avium glycopeptidolipid • SHORT COMMUNICATION Pages 221-228 Vida R. Irani and Joel N. Maslow
12. Overexpression of a hydrogenase gene in Clostridium paraputrificum to enhance hydrogen gas production • SHORT COMMUNICATION Pages 229-234 Kenji Morimoto, Tetsuya Kimura, Kazuo Sakka and Kunio Ohmiya
13. RirA is the iron response regulator of the rhizobactin 1021 biosynthesis and transport genes in Sinorhizobium meliloti 2011 • SHORT COMMUNICATION Pages 235-242 Caroline Viguier, Páraic Ó Cuív, Paul Clarke and Michael O’Connell
14. Polymerase chain reaction for identification of aldoxime dehydratase in aldoxime- or nitrile-degrading microorganisms • SHORT COMMUNICATION Pages 243-249 Yasuo Kato, Satoshi Yoshida and Yasuhisa Asano

15. The gene encoding xylulose-5-phosphate/fructose-6-phosphate phosphoketolase (xfp) is conserved among Bifidobacterium species within a more variable region of the genome and both are useful for strain identification • SHORT COMMUNICATION Pages 251-257 Xianhua Yin, James R. Chambers, Kathleen Barlow, Aaron S. Park and Roger Wheatcroft
16. The stabilization of housekeeping transcripts in Trypanosoma cruzi epimastigotes evidences a global regulation of RNA decay during stationary phase • SHORT COMMUNICATION Pages 259-264 Ana María Cevallos, Mariana Pérez-Escobar, Norma Espinosa, Juliana Herrera, Imelda López-Villaseñor and Roberto Hernández
17. Genotypic and phenotypic characterization of a biofilm-forming Serratia plymuthica isolate from a raw vegetable processing line • SHORT COMMUNICATION Pages 265-272 Rob Van Houdt, Pieter Moons, An Jansen, Kristof Vanoirbeek and Chris W. Michiels
18. Chemotypes significance of lichenized fungi by structural characterization of heteropolysaccharides from the genera Parmotrema and Rimelia • SHORT COMMUNICATION Pages 273-278 Elaine Rosechrer Carbonero, Caroline Grassi Mellinger, Sionara Eliasaro, Philip Albert James Gorin and Marcello Iacomini
19. Isolation of genes differentially expressed during the fruit body development of Pleurotus ostreatus by differential display of RAPD • SHORT COMMUNICATION Pages 279-284 Masahide Sunagawa and Yumi Magae
20. Author index Volume 246 • INDEX Pages 285-287
21. Subject index Volume 246 • INDEX Pages 289-294
Copyright © 2005 Federation of European Microbiological Societies

Volume 246, 2005
Chief EditorJ.A. Cole, School of Biosciences, University of Birmingham, Edgbaston, B15 2TT Birmingham, United Kingdom. Tel: +44-121-414 5440; Fax: +44-121-414
5925; E-mail: [email protected]
MiniReviews EditorsR.I. Aminov, Gut Immunology and Microbiology, Rowett Research Institute, Greenburn Road, Bucksburn, AB21 9SB Aberdeen, Scotland, United Kingdom.
Tel: +44-1224-716 643; Fax: +44-1224-716 687; E-mail: [email protected]
Phylogeny; Molecular ecology; Antibiotic resistance; Bacterial genetics; Intestinal microbiology and microbial genomics
I. Henderson, Bacterial Pathogenesis and Genomics Unit, Division of Immunity and Infection, The Medical School, University of Birmingham, Edgbaston,
B15 2TT Birmingham, United Kingdom. Tel: +44-121-414 4368; Fax: +44-121-414 3599; E-mail: [email protected]
Microbial pathogenesis; Gram-negative bacteria; Infection; Cellular microbiology; Autotransporter proteins; Protein secretion
R.C. Staples, Boyce Thompson Institute, Cornell University, Tower Road, NY 14850 Ithaca, United States of America. Tel: +1-607-257 4889; Fax: +1-607-
254 1242; E-mail: [email protected]
Development, physiology, cell biology and molecular biology of filamentous fungi including fungal pathogens of plants and animals
Editors and their specialist fields
BIOTECHNOLOGY
S. Casella, Dipartimento di Biotecnologie Agrarie, Agripolis, Universita di Padova, Via dell’Universita 16, 35020 Legnaro Padova, Italy.
Tel: +39-049-827 2922; Fax: +39-049-827 2929; E-mail: [email protected]
Microbial physiology; Microbial biotechnology; Soil microbiology; Plant-bacteria interaction; Nitrogen metabolism
W. Kneifel, Department of Food Science and Technology, BOKU-University of Natural Resources and Applied Life Sciences, Muthgasse 18,
A-1190 Vienna, Austria. Tel: +43-1-36006-6290; Fax: +43-136006-6266; E-mail: [email protected]
Food fermentation; Lactic acid bacteria; Microbiological quality criteria of foods; Bacterial strain safety and virulence; product development and quality
assessment of functional foods (pro- and prebiotics); Food safety (hygiene issues)
D. Mattanovich, Institut fur Angewandte Mikrobiologie, Universitat fur Bodencultur Wien, Muthgasse 18, A-1190 Vienna, Austria. Tel: +43-1-360-066 569;
Fax: +43-1-369 7615; E-mail: [email protected]
Biotechnology, especially recombinant protein production with bacteria; Yeasts and filamentous fungi; Physiology of production strains; Metabolic
engineering
ENVIRONMENTAL MICROBIOLOGY; PLANT-MICROBE INTERACTIONS
E. Baggs, School of Biological Sciences, University of Aberdeen, Cruickshank Building, St Machar Drive, Aberdeen, AB24 3UU, United Kingdom. Tel: +44
(0)-122-427 2691; Fax: +44 (0)-122-427 2703 ; E-mail: [email protected]
Soil bacterial ecology, particularly in relation to nitrogen and carbon cycling; Functional genes involved in denitrification; Impacts of soil management,
pollution or climate change
C. Edwards, Division of Microbiology and Genomics, School of Biological Sciences, University of Liverpool, The Biosciences Building, L69 7ZB Liverpool,
United Kingdom. Tel: +44-151 795 4573; Fax: +44-151 795 4410; E-mail: [email protected]
Molecular ecology of micro-organisms; Novel methods for monitoring bacterial activity and biodiversity; Biogeochemical cycles (particularly methane cycling
bacteria); Bioremediation and environmental biotechnology; Extreme environments; Molecular methods
H-P.E. Kohler, Environmental Microbiology and Molecular Ecotoxicology, EAWAG, Ueberlandstrasse 133, CH-8600 Duebendorf, Switzerland. Tel: +41-1-
823 5521; Fax: +41-1-823 5547; E-mail: [email protected]
Microbial degradation and environmental fate of organic pollutants; Biochemistry of mono- and dioxygenases; Microbial transformation of chiral compounds
Y. Okon, Dept. of Plant Pathology & Microbiology, Faculty of Agricultural, Food & Environmental Quality Sciences, The Hebrew University of Jerusalem,
The Rehovot Campus, 76100 Rehovot, Israel. Tel: 972-8-948 9216; Fax: 972-8-946 6794; E-mail: [email protected]
Plant growth promoting bacterial-rhizosphere associations; Symbiotic and non-symbiotic biological nitrogen fixation; Physiology and ecology of Azospirillum
as a model system for rhizosphere studies
A. Oren, The Institute of Life Sciences, The Hebrew University of Jerusalem, 91904 Jerusalem, Israel. Tel: +972-2-658 4951; Fax: +972-2-652 8008;
E-mail: [email protected]
Microbial ecology and physiology; Halophilic micro-organisms; Photosynthetic prokaryotes
EUKARYOTIC CELLS
L.F. Bisson, Dept of Viticulture and Enology, 1311 Haring Hall, University of California at Davis, One Shields Avenue, CA 95616-8749 Davis, United States
of America. Tel: +1-530-752 1717; Fax: +1-530-752 0382; E-mail: [email protected]
Molecular biology, genetics, biochemistry, physiology, ecology and applications of yeasts
R. Fischer, Applied Microbiology, University of Karlsruhe, Hertzstrasse 16, D-76187 Karlsruhe, Germany. Tel: 49-721-608-4630; Fax: 49-721-608-8932;
E-mail: [email protected]
Cellular and molecular biology of filamentous fungi, especially polarized growth and development; Cytoskeleton, molecular motors and organelle movement;
Spore formation; Aspergillus nidulans
MICROBIOLOGY LETTERS

G.M. Gadd, Division of Environmental and Applied Biology, Biological Sciences Institute, School of Life Sciences, University of Dundee, DD1 4HN Dundee,
Scotland, United Kingdom. Tel: +44-1382-344 765; Fax: +44-1382-348 216; E-mail: [email protected]
Yeast and fungal physiology, ecology and differentiation; Metal-microbe interactions; Heavy metals and toxicology
N. Gunde-Cimerman, Department of Biology, Biotechnical Faculty, University of Ljubljana, Vecna Pot 111, 1000 Ljubljana, Slovenia. Tel: +386-1-423 3388;
Fax: +386-1-257 3390; E-mail: [email protected]
Physiology, ecology and biodiversity of fungi, especially in extreme (hypersaline and cold) environments; Biotechnologically important fungi and production of
extracellular enzymes and secondary metabolites; Pathogenic fungi and medicinal mushrooms; Culture collections and strain preservation.
M. Jacquet, Institut de Genetique et Microbiologie, UMR8621 CNRS, Universite Paris-Sud, Bat 400, 91405 Orsay Cedex, France. Tel: +33-16915 7963;
Fax: +33-16915 4629; E-mail: [email protected]
Yeast molecular and cell biology; Signal transduction in fungus
B. Paul, Laboratoire des Sciences de la Vigne, Institut Jules Guyot, Universite de Bourgogne, BP 27877, 21078 Dijon, France. Tel: +33-380-396326; Fax:
+33-380-396326; E-mail: [email protected]
Mycology, in particular biological control of plant diseases; The genera Botrytis and Pythium; Aquatic phycomycetes
B.A. Prior, Department of Microbiology, University of Stellenbosch, Private Bag XI, 7602 Matieland, South Africa. Tel: +27-21-808 5856; Fax: +27-21-808
5846; E-mail: [email protected]
Stress responses by yeast to the environment; Microbial solute channels; Fungal biotechnology; Hemicellulose biodegradation by fungi
C. Remacle, Genetics of Microorganisms, Department of Life Sciences B22, University of Liege, Bld du Rectorat 27, B-4000 Liege, Belgium.
Tel: +32-4366 3812; Fax: +32-4366 3840; E-mail: [email protected]
Genetics and molecular biology of lower eukaryotes with emphasis on cell organelles; The function and biogenesis of mitochondria and chloroplasts
P. Schaap, Division of Cell and Developmental Biology, University of Dundee, MSI/WTB Complex, Dow Street, Dundee DD1 5EH, UK. Tel: +44 1382 348
078; Fax: +44 1382 345 386; E-mail: [email protected]
Cellular and developmental biology of social amoebae; Signal transduction, especially the role of cyclic nucleotide signalling pathways in the regulation of
developmental decisions, sporulation and responses to stress; Evolutionary relationships between eukaryote cyclic nucleotide signalling proteins and their
prokaryote ancestors
D.P. Wakelin, High Street (Kirtlands), WR12 7AL Broadway, Worcestershire, United Kingdom. Tel: +44-1386-852 747; E-mail: [email protected]
Parasitology; Helminthology; Host immunity; Intestinal immunity; Intestinal inflammation; Immunoepidemiology; Genetics of resistance
GENETICS AND MOLECULAR BIOLOGY
R.S. Buxton, Division of Mycobacterial Research, National Institute for Medical Research, The Ridgeway, Mill Hill, NW7 1AA London,
United Kingdom. Tel: 020 8816 2225; Fax: 020 8906 4477; E-mail: [email protected]
Mycobacteria, especially pathogenesis; Microbial genetics and molecular biology; Gene regulation; Two-component signal transduction
K. Forchhammer, Institut fur Mikrobiologie und Molekularbiologie, Justus-Liebig-Universitat, Heinrich-Buff-Ring 26-32, D-35392 Giessen,
Germany. Tel: +49-641-9935 545; Fax: +49-641-9935 549; E-mail: [email protected]
Physiology and molecular genetics of cyanobacteria; Microbial nitrogen control; Bacterial signal transduction through serine/threonine phosphorylation/
dephosphorylation
R.P. Gunsalus, Department of Microbiology and Molecular Genetics, 1602 MSB, University of California (UCLA), CA 90095 Los Angeles, United States of
America. Tel: +1-310-206 8201; Fax: +1-310-206 5231; E-mail: [email protected]
Molecular genetics; Microbial physiology; Methanogenesis; Anaerobic cell function; Electron transport; Metabolism
D.J. Jamieson, School of Life Sciences, Heriot-Watt University, Riccarton, EH14 4AS Edinburgh, Scotland, United Kingdom. Tel: +44-131-451 3644; Fax:
+44-131-451 3009; E-mail: [email protected]
Molecular biology; Genetics and biochemistry of yeasts
A. Klier, Dept des Biotechnologies, Unite de Biochemie Microbienne, Institut Pasteur, 25 Rue du Docteur Roux, 75724 Paris, Cedex 15, France. Tel: +33-1-
44 27 6995; Fax: +33-1-44 27 6995; E-mail: [email protected]
Molecular biology, genetics, biochemistry and physiology of gram-positive bacteria
E. Ricca, Dipartimento di Fisiologia Generale ed Ambientale, Universita’ Federico II, Via Mezzocannone 16, 80134 Napoli, Italy. Tel: +39-81-253 4636; Fax:
+39-81-551 4437; E-mail: [email protected]
Bacterial differentiation; Sporulation; Gene expression in gram-positives; Bacteria as vaccine vehicles and as probiotics; Display of molecules on bacterial
surfaces
W. Schumann, Institute of Genetics, Universitat Bayreuth, D-95440 Bayreuth, Germany. Tel: +49-921-552 708; Fax: +49-921-552 710;
E-mail: [email protected]
Bacterial genetics, especially stress genes; Bacteriophages; Transposition
M.R. Soria, Professor of Biochemistry and Molecular Biology, Department of Experimental & Clinical Medicine ‘‘G. Salvatore’’, Magna Graecia University
School of Medicine, Via T.Campanella 115, 88100 Catanzaro, Italy. Tel: +39-961-770 880; Fax: +39-961-777 435; E-mail: [email protected]
Functional genomics of host-parasite interactions; Regulation of gene expression; Angiogenesis
GENOMICS AND BIOINFORMATICS
M.Y. Galperin, National Center for Biotechnology Information, National Library of Medicine, National Institute of Health, Building 38A, Room 507, Maryland
20894 Bethesda, United States of America. Tel: +1-301-435 5910; Fax: +1-301-435-7794; E-mail: [email protected]
Microbial genomics; Bio-informatics; Modelling of metabolic pathways; Evolution of metabolism
O.P. Kuipers, Dept. for Genetics, Rijksuniversiteit Groningen, Kerklaan 30, 9751 HN Laren, Netherlands. Tel: +31-50-3632093/2092;
Fax: +31-5-3632348; E-mail: [email protected]
Genetics and biotechnological applications of gram-positive bacteria (lactid acid bacteria, bacilli); Functional genomics; Bacteriocins;
Protein engineering

PATHOGENICITY INCLUDING VETERINARY MICROBIOLOGY
P.W. Andrew, Department of Infection, Immunity and Inflammation, University of Leicester, PO Box 138 (University Road), LE1 9HN Leicester, United
Kingdom. Tel: +44-116-252 2941; Fax: +44-116-252 5030; E-mail: [email protected]
Microbial pathogenicity; Intracellular parasites
M.J. Bidochka, Department of Biological Sciences, Brock University, Glenridge Ave 500, ON L2S 3A1 St. Catharines, Canada. Tel: +1-905-688 5550 ext
3392; Fax: +1-905-688 1855; E-mail: [email protected]
Microbial pathogenicity, especially pathogenic fungi; Microbial population genetics and phylogeography
T.H. Birkbeck, Division of Infection and Immunity, Institute of Biomedical & Life Sciences, Joseph Black Building, University of Glasgow,
G12 8QQ Glasgow, Scotland, United Kingdom. Tel: +44-141-330 5843; Fax: +44-141-330 4600; E-mail: [email protected]
Microbial toxins and pathogenicity in human and animal diseases; Immunochemistry; Fish disease
H.B. Deising, Faculty of Agriculture, Phytopathology and Plant Protection, Martin-Luther University Halle-Wittenberg, Ludwig-Wucherer Strasse 2, D-06099
Halle, Germany. Tel: +49-345-552 2660; Fax: +49-345-552 7120; E-mail: [email protected]
Fungal pathogenicity and virulence; Fungus-plant interactions, especially biochemistry and molecular biology of fungus-plant interactions; Fungal
morphogenesis, especially infection structure differentiation; Fungicide resistance
R. Delahay, Institute of Infection, Immunity & Inflammation, University of Nottingham, Floor C, West Block, Queen’s Medical Centre, Nottingham, NG7 2UH.
Tel: +44 115-924 9924 Ext 42449; Fax: +44 115-970 9923; E-mail: [email protected]
Microbial pathogenicity, especially enteric pathogens; enteropathogenic Escherichia coli; Helicobacter; Host-pathogen interaction; Bacterial virulence
secretion systems (Type III and IV in particular); Protein–protein interaction
M.C. Enright, Royal Society University Research Fellow, Department of Biology and Biochemistry, University of Bath, Bath BA2 7AY, United Kingdom. Tel:
+44 1225-386871; Fax: +44 1225-386779; E-mail: [email protected]
Staphylococcus aureus; Streptococci; MRSA; molecular epidemiology evolution; antibiotic resistance; virulence; biochemistry; physiology and genetics
streptococci
J.-I. Flock, Department of Laboratory Medicine, Division of Clinical Bacteriology, Karolinska Institutet, Huddinge University Hospital F82, SE-141 86
Stockholm, Sweden. Tel: +46-8-5858 1169; Fax: +46-8-711 3918; Email: [email protected]
Genetics and virulence factors of Staphylococci; Adherence of gram-positive bacteria; Experimental infection models in animals;
Function of antibodies against surface structures of gram-positive bacteria; Microbial immunity and vaccines against gram-positive bacteria
K. Hantke, Mikrobiologie/Membranphysiologie, Universitat Tubingen, Auf der Morgenstelle 28, D-72076 Tubingen, Germany. Tel: +49-7071-297 4645; Fax:
+49-7071-295 843; E-mail: [email protected]
Bacterial metal transport and regulation, especially iron, manganese and zinc; Functions of outer membrane and periplasmic proteins of gram-negative
bacteria; Colicins and microcins; Pathogenicity and iron
J.M. Ketley, Department of Genetics, University of Leicester, University Road, LE1 7RH Leicester, United Kingdom. Tel: +44-116-252 3434; Fax: +44-116-
252 3378; E-mail: [email protected]
Vibrio cholerae; Campylobacters; Pathogenic enteric bacteria; Pathogenesis; Microbial genomics; Gene regulation
R.Y.C. Lo, Department of Microbiology, University of Guelph, ON, N1G 2W1 Guelph, Canada. Tel: +1-519-824 4120; Fax: +1-519-837 1802;
E-mail: [email protected]
Microbial pathogenicity; Bacterial genetics; Physiology and biochemistry
T. Mitchell, Division of Infection and Immunity, Institute of Biomedical and Life Sciences, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ,
Scotland, United Kingdom, Tel: +44 141-330 4642; Fax: +44 141-330 3727; E-mail: [email protected]
Microbial pathogenicity, especially in Gram-positive bacteria and mainly streptococci; Bacterial protein toxins; Vaccine development; Bacterial virulence
gene expression; Genomic variation in bacterial pathogens; Use of bacterial microarrays
M. Mitsuyama, Department of Microbiology, Graduate School of Medicine (Rm203, Bldg D), Kyoto University, Yoshida-Konoe-cho, Sakyo-ku, 606-8501
Kyoto, Japan. Tel: +81-75-753 4441; Fax: +81-75-753 4446; E-mail: [email protected]
Bacterial pathogenicity; Medical bacteriology; Intracellular bacteria; Immune response to infection
M. Schembri, School of Molecular and Microbial Sciences, University of Queensland, Building 76, QLD 4072 Brisbane, Australia. Tel: +61-7-3365 3306;
Fax: +61-7-3365 4699; E-mail: [email protected]
Microbial pathogenicity, especially gram-negative bacteria; Bacterial adhesins; Biofilms; Bacterial gene regulation and DNA microarrays; Bacterial display
systems; Vaccine development
S. Schwarz, Molecular Microbiology and Diagnostics, Institute for Animal Breeding, Federal Agricultural Research Centre (FAL), Holtystr. 10, D-31535
Neustadt-Mariensee, Germany. Tel: +49-5034-871-241; Fax: +49-5034-871-246; E-mail: [email protected]
Molecular biology of Staphylococci; Antibiotic resistance mechanisms; Mobile genetic elements and horizontal gene transfer; Pathogenicity; Molecular
epidemiology; Gram-positive cocci; Pasteurellaceae (Pasteurella, Mannheimia, Actinobacillus) and Enterobacteriaceae (Salmonella, Escherichia),
Bordetella
S. Smith, Department of Microbiology, Moyne Institute, Trinity College, 2 Dublin, Ireland. Tel: +353-1-6083713; Fax: +353-1-6799294;
E-mail: [email protected]
Microbial pathogenicity; Gram-negative bacteria; Bacterial adhesion and invasion; Outer membrane proteins; Fimbriae and pili; Proteomics and genomics;
Bacterial gene regulation
A.H.M. van Vliet, Department of Gastroenterology and Hepatology (L-459), Erasmus MC, Dr. Molewaterplein 40, 3015 GD Rotterdam, Netherlands. Tel:
+31-10-463 5944; Fax: +31-10-463 2793; E-mail: [email protected]
Microbial pathogenesis and genetics, especially of Helicobacter and Campylobacter; Bacterial gene regulation; Microbial metal metabolism
W. Wade, Department of Microbiology, Dental Institute, King’s College London, Guy’s Hospital, Floor 28, Guy’s Tower, SE1 9RT London,
United Kingdom. Tel: +44-20-7188 3872; Fax: +44-20-7188 3871; E-mail: [email protected]
Clinical microbiology; Oral microbiology; Molecular microbial ecology; Molecular diagnostics; Bacterial systematics; Anaerobic bacteria

P.H. Williams, Department of Genetics, University of Leicester, University Road, LE1 7RH Leicester, United Kingdom. Tel: +44-116-252 3436; Fax: +44-
116-252 3378; E-mail: [email protected]
Molecular genetic and cell biological analysis of the pathogenesis of infectious diseases, especially the role of microbial iron uptake, both in infection and in
the survival, persistence and resuscitation of severely stressed micro-organisms; Virulence mechanisms of enteric pathogens
C. Winstanley, Division of Medical Microbiology, University of Liverpool, Duncan Building, Daulby Street, Liverpool L69 3GA, United Kingdom. Tel: +44-
151-706 4388; Fax: +44-151-706 5805; E-mail: [email protected]
Pathogenicity of and genetic variation amongst Gram-negative bacteria; Pseudomonas and Burkholderia; Pathogenicity islands
PHYSIOLOGY AND BIOCHEMISTRY
J.R. Andreesen, Institut fur Mikrobiologie, Martin-Luther-Universitat Halle-Wittenberg, Kurt-Mothes-Straße 3, D-06120 Halle, Germany. Tel: +49-345-552
6350; Fax: +49-345-552 7010; E-mail: [email protected]
Physiology and biochemistry of anaerobic bacteria; Metal(oids) involved in biochemical reactions (Mo, W, Se, Te, Zn) but not transport
R.A. Bonomo, Infectious Diseases Section, Louis Stokes Cleveland Veterans Affairs Medical Center, East Blvd 10701, Ohio 44106 Cleveland, United
States of America. Tel: +1-216-791-3800x4399; Fax: +1-216-231-3482; E-mail: [email protected]
Beta-lactamases; Resistance to beta-lactams; Mechanisms of antimicrobial resistance
R.A. Burne, Department of Oral Biology, College of Dentistry (Room D5-18), University of Florida, 1600 S.W. Archer Road, FL 32610 Gainesville, United
States of America. Tel: +1-352-392 4370; Fax: +1-352-392 7357; E-mail: [email protected]
Oral microbiology; Environmental regulation of bacterial gene expression; Stress tolerance; Biofilms; Streptococci
J.A. Cole, School of Biosciences, University of Birmingham, Edgbaston, B15 2TT Birmingham, United Kingdom. Tel: +44-121-414 5440; Fax: +44-121-414
5925; E-mail: [email protected]
Microbial physiology, especially the regulation of anaerobic metabolism of enteric bacteria; Nitrate and nitrite reduction by bacteria; Microbial pathogenicity
of gonococci; Bacterial cytochrome biosynthesis and electronic transfer pathways
C. Dahl, Institut fur Mikrobiologie und Biotechnologie, Rheinische Friedrich-Wilhelms Universitat Bonn, Meckenheimer Allee 168, vD-53115 Bonn,
Germany. Tel: +49-228-732 119; Fax: +49-228-737 576; E-mail: [email protected]
Physiology, biochemistry, molecular biology and genetics of anoxygenic phototropic bacteria; Microbial sulfur metabolism; Electron transport
A.M. George, Dept of Cell and Molecular Biology, University of Technology, Sydney, PO Box 123 (Broadway), NSW 2007 Sydney, Australia.
Tel: +61-2-9514 4158; Fax: +61-2-9514 4003; E-mail: [email protected]
Molecular biology and biochemistry of multidrug resistance in bacteria and higher organisms; Bacterial resistance to antibiotics; Membrane transport; ABC
transporters
J.A. Gil, Dept de Microbiologıa, Facultad de Biologıa, Universidad de Leon, 24071 Leon, Spain. Tel: +34-987-291 503; Fax: +34-987-291 479;
E-mail: [email protected]
Antibiotic biosynthesis and resistance; Actinomycetes and corynebacteria
D. Jahn, Institute for Microbiology, Technical University of Braunschweig, Spielmannstr. 7, 38106 Braunschweig, Germany. Tel: +49-531-391 5804; Fax:
+49-531-391 5854; E-mail: [email protected]
Bacterial biochemistry and bioenergetics; Enzyme mechanisms; Tetrapyrroles; Control of bacterial gene expression
W.J. Mitchell, School of Life Sciences, Heriot-Watt University, Riccarton, EH14 4AS Edinburgh, Scotland, United Kingdom. Tel: +44-131-451 3459; Fax:
+44-131-451 3009; E-mail: [email protected]
Regulation of bacterial gene expression; Solute transport, particularly the bacterial phosphotransferase system
S. Mongkolsuk, Laboratory of Biotechnology, Chulabhorn Research Institute, Lak Si, 10210 Bangkok, Thailand. Tel: (662) 574-0623 ext. 3816; Fax: (662)
574-2027; E-mail: [email protected]
Bacterial biochemistry, physiology and genetics of stresses and metals; Regulation of gene expression; Environmental microbiology;
Plantmicrobe interactions
M. Moracci, Institute of Protein Biochemistry -CNR, Via P. Castellino 111, 80131 Naples, Italy. Tel: +39 081 613 2271; Fax: +39 081 613 2277;
E-mail: [email protected]
Physiology and biochemistry of hyperthermophilic Bacteria and Archaea; Control of gene expression in thermophilic Archaea; Biotechnological applications
of enzymes from extremophiles
S. Rimsky, Enzymologie et Cinetique Structurale, LBPA, UMR 8113, Ecole Normale Superieure de Cachan/CNRS, Universite Paris XI, Avenue du
President Wilson 61, 94235 Cachan Cedex, France. Tel: +33-1-4740 7676; Fax: +33-1-4740 7684; E-mail: [email protected]
Protein-DNA interaction; Bacterial chromatin organisation; Protein-protein interaction (non-membrane); DNA chemical/enzymatic reactivity
S. Silver, Department of Microbiology and Immunology, Room E-704, University of Illinois, S. Wolcott Avenue 835, IL-60612-7344 Chicago, United States
of America. Tel: +1-312-996 9608; Fax: +1-312-996 6415; E-mail: [email protected]
Bacterial membrane transport; Molecular genetics and biochemistry; Metal-resistance mechanisms; Gram-positive bacteria and pseudomonads
J. Simon, School of Biological Sciences, University of East Anglia, NR4 7TJ Norwich, United Kingdom. Tel: +44-1603-593 250; Fax: +44-1603-592 250;
E-mail: [email protected]
Bacterial metabolism and bioenergetics, especially anaerobic respiration; Maturation of electron transport enzymes
A. Steinbuchel, Institut fur Molekulare Mikrobiologie und Biotechnologie, Westfalische Wilhelms-Universitat, Correnstraße 3, D-48149 Munster, Germany.
Tel: +49-251-833 9821; Fax: +49-251-833 8388; E-mail: [email protected]
Metabolism and biotechnological production of biopolymers (Polyesters, Cyanophycin and other poly(amino acids)); Microbial degradation of rubber
B. Ward, ICMB, Darwin Building, Kings Buildings, University of Edinburgh, EH9 3JR Edinburgh, Scotland, United Kingdom. Tel: +44-131 650 5370;
E-mail: [email protected]
Microbial physiology of gram-negative bacteria important in medicine or food; Campylobacter jejuni
A. Yokota, Laboratory of Microbial Resources and Ecology, Graduate School of Agriculture, Hokkaido University, 060-8589 Sapporo, Japan.
Tel: +81-11-706 2501; Fax: +81-11-706 4961; E-mail: [email protected]
Energy metabolism in gram-positive bacteria and Escherichia coli; Lactic acid bacteria and bifidobacteria as probiotics

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 151–158
MiniReview
Fluorescence in situ hybridisation (FISH) – the next generation
Katrin Zwirglmaier *
Department of Biological Sciences, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK
Received 25 February 2005; received in revised form 8 April 2005; accepted 13 April 2005
First published online 27 April 2005
Edited by R.I. Aminov
Abstract
Fluorescence in situ hybridisation (FISH) has become one of the major techniques in environmental microbiology. The original
version of this technique often suffered from limited sensitivity due to low target copy number or target inaccessibility. In recent
years there have been several developments to amend this problem by increasing signal intensity. This review summarises various
approaches for signal amplification, focussing especially on two widely recognised varieties, tyramide signal amplification and mul-
tiply labelled polynucleotide probes. Furthermore, new applications for FISH are discussed, which arise from the increased sensi-
tivity of the method.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Fluorescence in situ hybridisation; Tyramide signal amplification; Polynucleotide probes
1. Introduction
Fluorescence in situ hybridisation (FISH) allows the
visualisation of prokaryotic cells in their natural envi-
ronment. In short, cells are fixed (i.e., they are not viable
anymore and the status quo of their DNA and RNA is
preserved), permeabilised to facilitate access of the
probe to the target site and then hybridised with nucleicacid probes. The probes are either directly labelled with
a fluorochrome or the dye is introduced in a secondary
detection step. The samples can then be analysed by epi-
fluorescence or laser scanning microscopy or flow
cytometry. The classic FISH technique relied solely on
(usually 16S) rRNA as probe target. The rRNA imme-
diately suggests itself as the ideal target because it is
present in all living cells in relatively high copy numbers.Furthermore, since it is traditionally used as phyloge-
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.015
* Tel.: +44 24765 22572; fax: +44 24765 23701.
E-mail address: [email protected].
netic marker a lot of sequence data is available for probe
design.
Since its origins some 20 years ago [1] this technique
has become an invaluable tool for environmental micro-
biologists and has spawned numerous variations. The
reasons for this popularity are obvious: (1) FISH allows
the detection of cells regardless of their culturability.
With as little as 0.3% of bacteria in soil and <0.1% inmarine water being culturable [2] FISH offers a glimpse
at the full bacterial biodiversity. (2) The possibility to
detect cells in situ allows an insight into the structure
of microbial communities and may help to unveil their
ecological function.
Despite these promising features the classic FISH
protocol suffers from some limitations. A major draw-
back is the often very low signal intensity. This canpartly be attributed to insufficient cell permeability
preventing access of the probes into the cells and to
the target site. The introduction of various chemical
and enzymological pre-treatments (reviewed in [2]) can
. Published by Elsevier B.V. All rights reserved.

152 K. Zwirglmaier / FEMS Microbiology Letters 246 (2005) 151–158
alleviate this problem to some extent. However, cell per-
meability always has to be carefully balanced against
cell integrity to avoid cell loss.
Apart from permeability issues, the main reason for
weak signals with the classic rRNA targeted FISH is
the low ribosome content found in slowly growing ormetabolically inactive cells in environmental samples.
Problems of this nature have led to a number of different
approaches for signal amplification being developed in
recent years. This improved sensitivity of the method
ultimately negated the dogma that FISH requires high
copy numbers of the target molecule and paved the
way for new applications, allowing microbiologists to
move away from 16S rRNA and instead target other nu-cleic acids present in lower copy numbers, such as
mRNA, plasmids or even single copy genes. This trend
is further aided by the increasing amount of available
sequence data gathered from multiple genome projects.
This review summarises recent developments in FISH
technology, focussing on different approaches for signal
amplification and discussing possible new applications
for FISH arising from these developments.
2. Different ways for increasing FISH signals
2.1. Multiple probes for one target organism
The number of probes binding to their target is limited
by the number of available targets (i.e., ribosomes).Therefore, one obvious solution to get better signals is
to use several probes targeting different regions of the
16S rRNA. In various studies [3–5] combinations of up
to five probes were used. Although this led to stronger
signals, the effect was not as pronounced as anticipated
and generally did not exceed a 2–3-fold amplification
compared to hybridisations with single probes. A further
complication is that it is usually difficult to design severalprobes with the same specificity.
The inverse strategy (using one probe that carries
multiple labels) has been shown to be unsuitable for sig-
nal amplification, since it leads to increased non-specific
binding and a poor signal to noise ratio [6].
2.2. Helper oligos
As well as low ribosome content the signal intensity
is also strongly affected by the accessibility of the target
sequence. The ribosomal RNA has a well-described
complex secondary and tertiary structure with many
loops and helices and embedded ribosomal proteins
leaving some stretches of the rRNA more accessible to
probes than others. Studies by Fuchs et al. [7,8] and
Behrens [9,10] resulted in an ‘‘accessibility map’’ ofthe rRNA, defining regions with strong or weak hybrid-
isation signals, thus allowing a more directed probe
design. Unfortunately, some of the highly variable
(and therefore valuable for specific probe design) re-
gions turned out to be rather inaccessible. This problem
was addressed with the introduction of so-called helper-
oligos [11]. These are unlabelled oligonucleotides which
bind in close vicinity to the target site of the labelledprobe and open the secondary structure of the rRNA,
which facilitates the binding of the labelled probe. A
combination of one (mono)labelled probe and up to
four unlabelled helper oligonucleotides resulted in up
to 25-fold signal amplification compared to the labelled
probe alone. However, it is essential that the helper oli-
gonucleotides have the same specificity as the probe,
which again can cause problems with probe design.
2.3. PNA probes
Another option to address the problem of target
accessibility is to use peptide nucleic acid (PNA) probes
[12–14]. Due to their uncharged peptide backbone these
probes have a much higher affinity to their hybridisation
target than DNA-oligonucleotides do. This allows thehybridisation parameters to be changed to very high
temperatures and low salt concentrations, which weak-
ens the secondary structure of the rRNA and therefore
increases target accessibility. PNA-FISH yields up to
5-fold stronger signals than the classic FISH using
DNA oligonucleotides. A practical drawback of this ap-
proach is the currently still rather high price for PNA
oligos.
2.4. Treatment with chloramphenicol to increase rRNA
content
Instead of amplifying the signal by introducing more
probes or multiple labels it is also possible to artificially
increase the number of target molecules to allow more
probes to bind. One way to do this is to incubate envi-ronmental samples with the antibiotic chloramphenicol
[15].
Chloramphenicol is an inhibitor of protein synthesis
and rRNA degradation. Cell division is also inhibited,
thus leading to an accumulation of rRNA in the cell.
In a study by Ouverney et al. [15] 93–99% of all
DAPI stained cells in a marine sample could be de-
tected by FISH after chloramphenicol treatment as op-posed to 75% in an untreated sample. This effect of
chloramphenicol is also interesting for a second reason,
as it indicates that rRNA is present initially, disproving
earlier assumptions that low fluorescent cell counts in
marine environments are due to high levels of dead
cells.
A possible limitation of this approach is a potential
shift in community composition after incubation withchloramphenicol, since the antibiotic does not affect
archaebacteria or eukaryotes.

K. Zwirglmaier / FEMS Microbiology Letters 246 (2005) 151–158 153
2.5. In situ PCR
Another way to increase the number of target mole-
cules and thereby the signal intensity is in situ PCR
[16]. Although strictly speaking not a FISH technique
it deserves to be mentioned in the context of signalamplification. PCR is carried out inside the cell using la-
belled nucleotides to allow later detection of the PCR
products.
While this method has the advantage to be applicable
not only to rRNA (rDNA), but any other gene or even
mRNA, it nevertheless still suffers from technical diffi-
culties. Vigorous enzymatic pre-treatment of the cells
is required to allow the Taq polymerase to access thecell, which subsequently can result in considerable efflux
of PCR product or cell loss.
2.6. Bacterial chromosomal painting (BCP)
This technique differs from the others in that it does
not rely on a single target gene but rather uses the whole
genome as a target. It is therefore closely related to solu-tion based genomic DNA:DNA reassociation [17] and
promises a higher resolution and possible differentiation
of closely related strains.
In the bacterial species concept strains are defined as
belonging to the same species if they show >97% identity
on rRNA level and 70% on the whole genome [18]. Con-
sequently, differentiation of strains with rRNA targeted
probes is at best difficult and often impossible, whereasit is still possible when the rest of the genome is consid-
ered [17].
In bacterial chromosomal painting (BCP), [19,20] the
whole genome of a target organism is used as a probe.
Fluorescently labelled probes are generated by nick
translation with the size of the probe fragments ranging
between 50 and 200 bp. Although promising, one of the
drawbacks of this technique is that hybridisation timesare unusually long (2 days). BCP has been shown to al-
low differentiation of Salmonella serotypes [19] and has
also been applied to marine samples [20].
2.7. Enzymatic signal amplification – TSA-FISH
A very dramatic increase of sensitivity can be
achieved by enzyme-mediated signal amplification.TSA-FISH (tyramide signal amplification) [21], also
known as CARD-FISH (catalysed reporter deposition)
[22] is based on the deposition of fluorescently labelled
tyramide by peroxidase activity. Horseradish peroxidase
(HRP) is introduced in the target cells either by using
HRP-labelled probes or via a secondary detection of
digoxigenin labelled probes and an HRP-coupled antidi-
goxigenin antibody. The enzyme then catalyses the oxi-disation of the fluorochrome labelled tyramide
substrate, leading to a deposition of the highly active
intermediates in the vicinity of the enzyme by covalent
binding to electron-rich protein moieties. The resulting
signals are up to 12 times stronger than in hybridisations
with traditional fluorescence labelled probes and 3–
4-fold stronger than in hybridisations with multiple
monolabelled fluorescent probes [23]. A drawback ofthis approach is the need for vigorous pre-treatment of
the cells using e.g., lysozyme and/or proteinase to enable
the relatively large HRP molecule to enter the cell. This
can lead to loss of some cell types in heterogenous envi-
ronmental samples. Cell integrity and cell wall perme-
ability thus have to be carefully balanced.
Despite these difficulties, this approach is becoming
increasingly popular and has recently been used to de-tect not only rRNA, but also plasmids [5], tmRNA
[24] and mRNA [25].
2.8. Polynucleotide probes and RING-FISH
Another approach to increase the sensitivity of FISH
is the use of polynucleotide probes. These probes can
range in length between 100 and several hundred basepairs. They are made of ssRNA [25–30] or dsDNA
[19,20,31], generated either by in vitro transcription
[25,28,29], nick translation [19,20] or PCR [31] and carry
multiple labels, either fluorescent dyes or digoxigenin/
biotin for a secondary detection. The signal amplifica-
tion achieved with these probes is based on both the
multiple labelling and the secondary structures formed
by the long probes. These secondary structures involvenot only intra-, but also intermolecular binding, which
results in a network of probes (see Fig. 1). The huge po-
tential of polynucleotide probes lies in the fact that a
network allows the incorporation of probe molecules,
which are not directly connected to the target site.
Therefore, the detectable fluorescence is no longer pro-
portional to the number of available targets. [32]. A
characteristic feature of in situ hybridisations with poly-nucleotide probes is the ring-shaped or halo-like appear-
ance of the fluorescence signal. This is especially true for
hybridisations with (intermediate–high concentrations
of) ssRNA [26–28] and dsDNA probes [31]. The halo,
which can be seen as an extracellular probe network an-
chored at the intracellular probe specific target site, is
usually not observed with very low probe concentrations
[25,29], hydrolysis of the probes to shorter fragmentsprior to the hybridisation [19,20] or excessive permeabil-
isation of the cell wall with lysozyme or other treatments
[27,33].
rRNA targeted polynucleotide probes have been ap-
plied in various studies in recent years [27,29,30,34].
However, due to the structure of the rRNA, with
patches of highly conserved sequence, they can only be
used at a group specific, rather than species specific level.Their special value therefore lies in the possibility to
target other, low copy nucleic acids.

Fig. 1. Schematic illustration of the formation of probe networks, which are the presumed origin of the ring-shaped hybridisation signal seen with
RING-FISH: (a) secondary structure of polynucleotide probe GAP-E targeting a 258 bp fragment of the E. coli glycerol aldehyde phosphate
dehydrogenase (GAPDH) gene, (b) simplified and schematised secondary structure, (c) denaturation step prior to hybridisation leads to linearised
probe molecules, (d) during hybridisation intra- and intermolecular renaturation of secondary structure leads to formation of probe network. The
network is anchored at the intracellular probe-specific target site, (e) interconnected probes accumulate mainly outside the target cell due to the
limited permeability of the cell wall, resulting in a ring-shaped hybridisation signal. The specificity of the signal is based on the intracellular anchor.
Probe networks not connected to an intracellular probe-specific target site will be washed away during post-hybridisation wash steps,
(f) epifluorescence micrograph of a RING-FISH signal using probe GAP-E targeting the GAPDH gene.
154 K. Zwirglmaier / FEMS Microbiology Letters 246 (2005) 151–158
This has been implemented in the development of
RING-FISH (recognition of individual genes), a FISH
variety, which relies on the use of high concentrations
of polynucleotide probes and is characterised by a
ring-shaped fluorescence signal. RING-FISH has been
shown to allow the detection of plasmid encoded and
even chromosomal single copy genes [28,33].
3. Moving away from rRNA as target
Most of the above methods were developed with the
intention of improving detection of rRNA in cells with
low ribosome content. Due to its high copy number
rRNA has long been regarded as the only suitable mol-
ecule for bacterial FISH. However, with the develop-ment of potent signal amplification techniques,
especially TSA and polynucleotide probes, it became
possible to move away from the high copy (104–105 in
an actively growing cell) rRNA as the sole target for
fluorescence in situ hybridisation and instead target a
variety of other nucleic acids such as mRNA, tmRNA,
plasmids and chromosomal DNA, thereby opening the
door for new applications for FISH.
3.1. tmRNA
tmRNA, also called 10SaRNA or SsrA is a small sta-
ble RNA (length in Escherichia coli: 365 nt), which has
been shown to be involved in the degradation of trun-cated proteins. With copy numbers of approximately
103 in metabolically active cells it is slightly less abundant
than rRNA but still easily detectable with TSA [24].
Compared to rRNA it has the advantage of being more
accessible, since it is not complexed with ribosomal pro-
teins. tmRNA has so far been detected in all completely
sequenced bacterial genomes (in 17 of 20 phyla) and in
certain phage, mitochondrial and plastidial genomes,but not (yet) in archaeal or eukaryotic genomes.
3.2. Plasmids
The copy number of plasmids ranges (depending on
the type of plasmid) from 101 to 103 per cell. This

K. Zwirglmaier / FEMS Microbiology Letters 246 (2005) 151–158 155
suggests them as a target for an improved sensitivity
FISH protocol. Also, as they are made of DNA, they
are more stable than mRNA, which, although present
in comparable copy numbers, requires special handling
to avoid degradation.
The polynucleotide probe based RING-FISH wasused to detect different types of plasmids with high,
medium and low copy numbers [28].
Various modifications of the protocol were necessary
to account for the decreased target copy number and
target type.
To allow hybridisation to the dsDNA, a denaturation
step prior to the hybridisation had to be introduced. The
RNA:DNA hybrid (polynucleotide RNA probe andDNA target) is thermodynamically weaker than the
RNA:RNA hybrid (RNA probe and rRNA target) in
conventional fluorescence in situ hybridisations, calling
for more relaxed hybridisation conditions. Finally, the
decreased target copy number decelerates the formation
of the signal amplifying probe network, requiring longer
hybridisation times.
The signal intensity was the same for high, mediumand low copy plasmids, but slightly weaker than for
rRNA targeted probes.
A TSA-FISH based approach was used for an indi-
rect detection of ColE1 related plasmids [5]. The RNA
II, a 555 nt transcript, which regulates plasmid replica-
tion by acting as primer for the DNA synthesis was
targeted with a combination of up to seven HRP la-
belled oligonucleotide probes resulting in strong sig-nals in cells containing the plasmid. This study
compared signal amplification gained from using mul-
tiple fluorescently monolabelled probes and tyramide
signal amplification. TSA was shown to be the supe-
rior technique.
3.3. mRNA
The detection of mRNA presents a special challenge
due to its inherent instability. However, in the context
of elucidating the role of individual species within an
ecosystem the prospect of being able to detect mRNA
in individual cells in situ is very attractive. In recent
years there have been various reports describing suc-
cessful detection of mRNA with FISH [25,35–37]. All
these studies employ digoxigenin labelled probes. Theywere detected via TSA or, in one early study [35], by a
colour reaction mediated by alkaline phosphatase.
Only one study used digoxigenin labelled oligonucleo-
tide probes [37], while the others were based on poly-
nucleotide transcript probes carrying multiple
digoxigenin labels. This approach combines the signal
amplification gained from introducing multiple labels
(via the multiply labelled polynucleotide probes) withthe amplification through enzyme mediated deposition
of fluorescent dye.
3.4. Genomic DNA
Detecting a chromosomal single copy gene in situ is
the ultimate challenge for signal amplification with
FISH. As with many other developments (such as
TSA, in situ PCR and mRNA detection) this has al-ready been a well-established technique for eukaryotic
cells, e.g. [38,39], before it was adapted for use in pro-
karyotes in the form of RING-FISH. While the
eukaryotic ‘‘chromosomal painting’’ uses probes with
a length of several kb, probe length for the prokaryotic
RING-FISH is only about 150–800 nt [28]. Character-
istic for RING-FISH is the use of multiply labelled
polynucleotide probes in a very high concentration(250 ng/ll), a denaturation step prior to the hybridisa-
tion and a rather long hybridisation period (up to
24 h).
It has been applied to detect the housekeeping gene
glycerol aldehyde 3-phosphate dehydrogenase (GAPDH)
inE. coli, a virulence factor in the plant pathogenXantho-
monas campestris, as well as a fragment of the RNA
polymerase gene rpoC1 in Synechococcus ([28] andZwirglmaier and Scanlan, unpublished).
3.5. Specificity of polynucleotide probes
As studies with plasmids, mRNA and genomic DNA
have shown, signal amplification with polynucleotide
probes, possibly further enhanced by TSA clearly has
the potential to detect any low copy nucleic acid target.One question that remains to be clarified is the specific-
ity of these probes. In contrast to oligonucleotide
probes, where a single mismatch can be discriminated,
polynucleotide probes clearly require more sequence dif-
ferences for a specific signal. Other factors, such as the
secondary structure of the probe and the fact that in
case of a network formation probably not the complete
probe binds to the target also influence specificity. Cur-rently there is only limited data about the threshold for
mismatch discrimination. Ludwig et al. [40] found the
cut-off point for positive/negative hybridisation signals
to be between 78% and 85% sequence identity using a
probe targeting the domain III of the 23S rRNA in
membrane based hybridisations. A more detailed study
of the same target region using FISH recently described
a cut-off point of 72–75% [33]. Similar conclusions canbe drawn from the study by Pernthaler [25], where a
probe targeting the mRNA of the pmoA gene (coding
for subunit A of the particulate methane monooxygen-
ase) was shown to detect methylotrophic symbionts of
Bathymodiolus azoricus with a similarity of around
80%, but not the more distantly related (64%) Methylo-
cystis echinoides.
Sequence alignment and conservativity profiles to de-tect highly variable regions in a target sequence should
therefore be an integral part of future probe design.

156 K. Zwirglmaier / FEMS Microbiology Letters 246 (2005) 151–158
4. Monitoring bacterial activity
Currently the major question in microbial ecology of
who is out there can reliably be answered with FISH
technology. The next big question as to what they are
doing out there has been addressed more recently bycombining FISH with various other techniques.
Bacterial activity is linked to DNA synthesis. The
detection of newly synthesised DNA by monitoring
the uptake and incorporation of tritiated thymidine
[41] or, more recently, bromodioxyuridine BrdU [42–
44] is therefore a way to differentiate active from inactive
cells. Combining this technique with FISH consequently
allows an assertion of which parts of a bacterial commu-nity are metabolically active [42].
Information beyond a general assessment of cellular
activity can be gained from microautoradiography
(MAR). Radioactively labelled substrate is added to
an environmental sample and its uptake is monitored.
A subsequent phylogenetic identification of the cells by
FISH then gives a picture about the substrate utilisation
of different cells in a bacterial community and may allowconclusions about the underlying food-network. FISH-
MAR, also known as STAR-FISH (substrate tracking
autoradiography) has recently been applied in various
studies [15,45–48].
An even more detailed account of the activity of a
single microbial cell can be gained by studying gene
expression. The combination of mRNA and rRNA tar-
geted FISH [25] offers a true insight into the ‘‘blackbox’’ of complex biocommunities. Although currently
not yet a standard technique, future optimisations and
improvements could turn it into one of the core methods
for environmental microbiologists.
5. Conclusions and outlook
Fluorescence in situ hybridisation has seen some ma-
jor improvements since its development some 20 years
ago and is nowadays one of the chief techniques in envi-
ronmental microbiology. With the increased sensitivity
of the method and the ability to detect genes and mRNA
the questions that can be addressed with FISH are
changing. While originally developed to describe the
composition of a bacterial community, it is now alsopossible to look at the activity of individual members
and their ecological function. The correlation of phylog-
eny and physiology is becoming an ever more important
topic in our attempts to understand ecological systems.
The feasibility of in situ gene expression studies presents
a quantum leap in this context. The rapidly growing
amount of sequence data, with new bacterial genomes
being published almost weekly, further supports thesedevelopments, allowing comparative sequence analysis
and directed probe design for any given gene.
Apart from gene expression, combined rRNA and
DNA targeted FISH could unveil cases of horizontal
gene transfer. This would be of interest not only for evo-
lutionists, but also in the context of genetically modified
organisms and their potential impact on ecosystems.
Another new application for ultrasensitive FISHmight be the detection of viral infections. As with many
other developments (including the original FISH tech-
nique), this has already been applied to eukaryotic cells
[49,50], but would probably require some modification
and optimisation for use in prokaryotes.
With the problem of sensitivity solved, the next desir-
able step in the future of FISH technology would be an
efficient automation to increase the amount of samplesthat can be analysed, maybe by optimising existing flow
cytometry techniques or even a microarray based
approach.
References
[1] DeLong, E.F., Wickham, G.S. and Pace, N.R. (1989) Phyloge-
netic stains: ribosomal RNA-based probes for the identification of
single cells. Science 243, 1360–1363.
[2] Amann, R.I., Ludwig, W. and Schleifer, K.H. (1995) Phylogenetic
identification and in situ detection of individual microbial cells
without cultivation. Microbiol. Rev. 59, 143–169.
[3] Amann, R.I., Binder, B.J., Olson, R.J., Chisholm, S.W., Deve-
reux, R. and Stahl, D.A. (1990) Combination of 16S rRNA-
targeted oligonucleotide probes with flow cytometry for analyzing
mixed microbial populations. Appl. Environ. Microbiol. 56,
1919–1925.
[4] Lee, S.H., Malone, C. and Kemp, P.F. (1993) Use of multiple 16S
rRNA targeted fluorescent probes to increase signal strength and
measure cellular RNA from natural planktonic bacteria. Mar.
Ecol. Prog. Ser. 101, 193–201.
[5] Juretschko, S., Schonhuber, W., Kulakauskas, S., Ehrlich, D.S.,
Schleifer, K.H. and Amann, R. (1999) In situ detection of
Escherichia coli cells containing ColE1-related plasmids by
hybridization to regulatory RNA II. Syst. Appl. Microbiol. 22,
1–8.
[6] Wallner, G., Amann, R. and Beisker, W. (1993) Optimizing
fluorescent in situ hybridization with rRNA-targeted oligonu-
cleotide probes for flow cytometric identification of microorgan-
isms. Cytometry 14, 136–143.
[7] Fuchs, B.M., Wallner, G., Beisker, W., Schwippl, I., Ludwig, W.
and Amann, R. (1998) Flow cytometric analysis of the in situ
accessibility of Escherichia coli 16S rRNA for fluorescently
labeled oligonucleotide probes. Appl. Environ. Microbiol. 64,
4973–4982.
[8] Fuchs, B.M., Syutsubo, K., Ludwig, W. and Amann, R. (2001) In
situ accessibility of Escherichia coli 23S rRNA to fluorescently
labeled oligonucleotide probes. Appl. Environ. Microbiol. 67,
961–968.
[9] Behrens, S., Ruhland, C., Inacio, J., Huber, H., Fonseca, A.,
Spencer-Martins, I., Fuchs, B.M. and Amann, R. (2003) In
situ accessibility of small-subunit rRNA of members of the
domains Bacteria, Archaea, and Eucarya to Cy3-labeled
oligonucleotide probes. Appl. Environ. Microbiol. 69, 1748–
1758.
[10] Behrens, S., Fuchs, B.M., Mueller, F. and Amann, R. (2003) Is the
in situ accessibility of the 16S rRNA of Escherichia coli for Cy3-
labeled oligonucleotide probes predicted by a three-dimensional

K. Zwirglmaier / FEMS Microbiology Letters 246 (2005) 151–158 157
structure model of the 30S ribosomal subunit? Appl. Environ.
Microbiol. 69, 4935–4941.
[11] Fuchs, B.M., Glockner, F.O., Wulf, J. and Amann, R. (2000)
Unlabeled helper oligonucleotides increase the in situ accessibility
to 16S rRNA of fluorescently labeled oligonucleotide probes.
Appl. Environ. Microbiol. 66, 3603–3607.
[12] Worden, A.Z., Chisholm, S.W. and Binder, B.J. (2000) In situ
hybridization of Prochlorococcus and Synechococcus (marine
cyanobacteria) spp. with RRNA-targeted peptide nucleic acid
probes. Appl. Environ. Microbiol. 66, 284–289.
[13] Perry-O�Keefe, H., Rigby, S., Oliveira, K., Sorensen, D., Stender,
H., Coull, J. and Hyldig-Nielsen, J.J. (2001) Identification of
indicator microorganisms using a standardized PNA FISH
method. J. Microbiol. Meth. 47, 281–292.
[14] Oliveira, K., Procop, G.W., Wilson, D., Coull, J. and Stender, H.
(2002) Rapid identification of Staphylococcus aureus directly from
blood cultures by fluorescence in situ hybridization with peptide
nucleic acid probes. J. Clin. Microbiol. 40, 247–251.
[15] Ouverney, C. and Fuhrman, J. (1997) Increase in fluorescence
intensity of 16S rRNA in situ hybridization in natural samples
treated with chloramphenicol. Appl. Environ. Microbiol. 63,
2735–2740.
[16] Hodson, R., Dustman, W., Garg, R. and Moran, M. (1995) In
situ PCR for visualization of microscale distribution of specific
genes and gene products in prokaryotic communities. Appl.
Environ. Microbiol. 61, 4074–4082.
[17] Grimont, P.A. (1988) Use of DNA reassociation in bacterial
classification. Can. J. Microbiol. 34, 541–546.
[18] Stackebrandt, E. and Goebel, B. (1994) Taxonomic note: a place
for DNA–DNA reassociation and 16S rRNA sequence analysis in
the present species definition in bacteriology. Int. J. Syst.
Bacteriol. 44, 846–849.
[19] Lanoil, B. and Giovannoni, S. (1997) Identification of bacterial
cells by chromosomal painting. Appl. Environ. Microbiol. 63,
1118–1123.
[20] Lanoil, B.D., Carlson, C.A. and Giovannoni, S.J. (2000) Bacterial
chromosomal painting for in situ monitoring of cultured marine
bacteria. Environ. Microbiol. 2, 654–665.
[21] Schonhuber, W., Fuchs, B., Juretschko, S. and Amann, R. (1997)
Improved sensitivity of whole-cell hybridization by the combina-
tion of horseradish peroxidase-labeled oligonucleotides and tyra-
mide signal amplification. Appl. Environ. Microbiol. 63, 3268–
3273.
[22] Pernthaler, A., Pernthaler, J. and Amann, R. (2002) Fluorescence
in situ hybridization and catalyzed reporter deposition for the
identification of marine bacteria. Appl. Environ. Microbiol. 68,
3094–3101.
[23] Lebaron, P., Catala, P., Fajon, C., Joux, F., Baudart, J. and
Bernard, L. (1997) A new sensitive, whole-cell hybridization
technique for detection of bacteria involving a biotinylated
oligonucleotide probe targeting rRNA and tyramide signal
amplification. Appl. Environ. Microbiol. 63, 3274–3278.
[24] Schonhuber, W., Le Bourhis, G., Tremblay, J., Amann, R. and
Kulakauskas, S. (2001) Utilization of tmRNA sequences for
bacterial identification. BMC Microbiol. 1, 20.
[25] Pernthaler, A. and Amann, R. (2004) Simultaneous fluorescence
in situ hybridization of mRNA and rRNA in environmental
bacteria. Appl. Environ. Microbiol. 70, 5426–5433.
[26] Stoffels, M., Ludwig, W. and Schleifer, K.H. (1999) rRNA probe-
based cell fishing of bacteria. Environ. Microbiol. 1, 259–271.
[27] Trebesius, K.H., Amann, R., Ludwig, W., Muhlegger, K. and
Schleifer, K.H. (1994) Identification of whole fixed bacterial cells
with nonradioactive 23S rRNA-targeted polynucleotide probes.
Appl. Environ. Microbiol. 60, 3228–3235.
[28] Zwirglmaier, K., Ludwig, W. and Schleifer, K.H. (2004) Recog-
nition of individual genes in a single bacterial cell by fluorescence
in situ hybridization – RING-FISH. Mol. Microbiol. 51, 89–96.
[29] DeLong, E.F., Taylor, L.T., Marsh, T.L. and Preston, C.M.
(1999) Visualization and enumeration of marine planktonic
archaea and bacteria by using polyribonucleotide probes and
fluorescent in situ hybridization. Appl. Environ. Microbiol. 65,
5554–5563.
[30] Pernthaler, A., Preston, C.M., Pernthaler, J., DeLong, E.F. and
Amann, R. (2002) Comparison of fluorescently labeled oligonu-
cleotide and polynucleotide probes for the detection of pelagic
marine bacteria and archaea. Appl. Environ. Microbiol. 68, 661–
667.
[31] Zimmermann, J., Ludwig, W. and Schleifer, K.H. (2001) DNA
polynucleotide probes generated from representatives of the genus
Acinetobacter and their application in fluorescence in situ
hybridization of environmental samples. Syst. Appl. Microbiol.
24, 238–244.
[32] Zwirglmaier, K., Ludwig, W. and Schleifer, K.H. (2003)
Improved fluorescence in situ hybridization of individual micro-
bial cells using polynucleotide probes: the network hypothesis.
Syst. Appl. Microbiol. 26, 327–337.
[33] Fichtl, K. (2005) Polynucleotide probe based enrichment of
bacterial cells: development of probes for species of clinical
relevance. PhD thesis, Technical University Munich. Available
from: http://tumb1.biblio.tu-muenchen.de/publ/diss/karin.php.
[34] Karner, M.B., DeLong, E.F. and Karl, D.M. (2001) Archaeal
dominance in the mesopelagic zone of the Pacific Ocean. Nature
409, 507–510.
[35] Hahn, D., Amann, R. and Zeyer, J. (1993) Detection of mRNA in
Streptomyces cells by whole-cell hybridization with digoxigenin-
labeled probes. Appl. Environ. Microbiol. 59, 2753–2757.
[36] Wagner, M., Schmid, M., Juretschko, S., Trebesius, K.H., Bubert,
A., Goebel, W. and Schleifer, K.H. (1998) In situ detection of a
virulence factor mRNA and 16S rRNA in Listeria monocytogenes.
FEMS Microbiol. Lett. 160, 159–168.
[37] Bakermans, C. and Madsen, E.L. (2002) Detection in coal tar
waste-contaminated groundwater of mRNA transcripts related to
naphthalene dioxygenase by fluorescent in situ hybridization with
tyramide signal amplification. J. Microbiol. Meth. 50, 75–84.
[38] Rogan, P.K., Cazcarro, P.M. and Knoll, J.H. (2001) Sequence-
based design of single-copy genomic DNA probes for fluorescence
in situ hybridization. Genome Res. 11, 1086–1094.
[39] Sharma, A.K. and Sharma, A. (2001) Chromosome painting –
principles, strategies and scope. Meth. Cell Sci. 23, 1–5.
[40] Ludwig, W., Dorn, S., Springer, N., Kirchhof, G. and Schleifer,
K.H. (1994) PCR-based preparation of 23S rRNA-targeted
group-specific polynucleotide probes. Appl. Environ. Microbiol.
60, 3236–3244.
[41] Fuhrman, J.A. and Azam, F. (1982) Thymidine incorporation as
a measure of heterotrophic bacterioplankton production in
marine surface waters: evaluation and field results. Marine Biol.
66, 109–120.
[42] Pernthaler, A., Pernthaler, J., Schattenhofer, M. and Amann, R.
(2002) Identification of DNA-synthesizing bacterial cells in
coastal North Sea plankton. Appl. Environ. Microbiol. 68,
5728–5736.
[43] Steward, G.F. and Azam, F. (1999) Bromodeoxyuridine as an
alternative to 3H-thymidine for measuring bacterial productivity
in aquatic samples. Aq. Microbial Ecol. 19, 57–66.
[44] Urbach, E., Vergin, K.L. and Giovannoni, S.J. (1999) Immuno-
chemical detection and isolation of DNA from metabolically
active bacteria. Appl. Environ. Microbiol. 65, 1207–1213.
[45] Lee, N., Nielsen, P.H., Andreasen, K.H., Juretschko, S., Nielsen,
J.L., Schleifer, K.-H. and Wagner, M. (1999) Combination of
fluorescent in situ hybridization and microautoradiography – a
new tool for structure–function analyses in microbial ecology.
Appl. Environ. Microbiol. 65, 1289–1297.
[46] Ouverney, C.C. and Fuhrman, J.A. (1999) Combined microau-
toradiography-16S rRNA probe technique for determination of

158 K. Zwirglmaier / FEMS Microbiology Letters 246 (2005) 151–158
radioisotope uptake by specific microbial cell types in situ. Appl.
Environ. Microbiol. 65, 1746–1752.
[47] Teira, E., Reinthaler, T., Pernthaler, A., Pernthaler, J. and
Herndl, G.J. (2004) Combining catalyzed reporter deposition-
fluorescence in situ hybridization and microautoradiography to
detect substrate utilization by bacteria and Archaea in the deep
ocean. Appl. Environ. Microbiol. 70, 4411–4414.
[48] Gray, N.D., Howarth, R., Pickup, R.W., Jones, J.G. and Head,
I.M. (2000) Use of combined microautoradiography and fluores-
cence in situ hybridization to determine carbon metabolism in
mixed natural communities of uncultured bacteria from the genus
Achromatium. Appl. Environ. Microbiol. 66, 4518–4522.
[49] Alonso, M.C., Cano, I., Castro, D., Perez-Prieto, S.I. and
Borrego, J.J. (2004) Development of an in situ hybridisation
procedure for the detection of sole aquabirnavirus in infected fish
cell cultures. J. Virol. Meth. 116, 133–138.
[50] Plummer, T.B., Sperry, A.C., Xu, H.S. and Lloyd, R.V. (1998) In
situ hybridization detection of low copy nucleic acid sequences
using catalyzed reporter deposition and its usefulness in clinical
human papillomavirus typing. Diagn. Mol. Pathol. 7, 76–84.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 159–165
MiniReview
Biotin biosynthesis, transport and utilization in rhizobia
Karina Guillen-Navarro, Sergio Encarnacion, Michael F. Dunn *
Programa de Ingenierıa Metabolica, Centro de Ciencias Genomicas, Universidad Nacional Autonoma de Mexico, A.P. 565-A, Cuernavaca,
Morelos, Mexico
Received 25 February 2005; received in revised form 12 April 2005; accepted 13 April 2005
First published online 27 April 2005
Edited by R.I. Aminov
Abstract
Biotin, a B-group vitamin, performs an essential metabolic function in all organisms. Rhizobia are a-proteobacteria with the
remarkable ability to form a nitrogen-fixing symbiosis in combination with a compatible legume host, a process in which the impor-
tance of biotin biosynthesis and/or transport has been demonstrated for some rhizobia–legume combinations. Rhizobia have also
been used to delimit the biosynthesis, metabolic effects and, more recently, transport of biotin. Molecular genetic analysis shows that
an orthodox biotin biosynthesis pathway occurs in some rhizobia while others appear to synthesize the vitamin using alternative
pathways. In addition to its well established function as a prosthetic group for biotin-dependent carboxylases, we are beginning
to delineate a role for biotin as a metabolic regulator in rhizobia.
� 2005 Published by Elsevier B.V. on behalf of the Federation of European Microbiological Societies.
Keywords: Biotin biosynthesis; Rhizobia; Rhizobia–legume symbiosis
1. Introduction
Root nodule bacteria, collectively known as rhizobia,
are a crucial component of the global nitrogen cycle be-
cause they reduce atmospheric nitrogen to ammonia in
symbiotic association with a compatible plant host and
thus reduce the need for synthetic nitrogen fertilizers.
Before establishing symbiosis, rhizobia must survive inthe soil awaiting the presence of a suitable host legume.
Infection of the host requires multiplication in the rhizo-
sphere as well as during early phases of the infection.
Mature, nitrogen-fixing intracellular rhizobia (bacter-
oids) require large amounts of energy and reductant de-
rived from the catabolism of plant-supplied organic
acids. Consequently, the metabolism and growth factor
0378-1097/$22.00 � 2005 Published by Elsevier B.V. on behalf of the Feder
doi:10.1016/j.femsle.2005.04.020
* Corresponding author. Tel.: +52 73 311 4662; fax: +52 73 317 5094.
E-mail address: [email protected] (M.F. Dunn).
requirements of rhizobia have long been studied (for re-
views, see [1–4]).
Biotin (vitamin H) has an essential metabolic func-
tion as the CO2-carrying prosthetic group of selected
carboxylases, decarboxylases and transcarboxylases
[5]. De novo biotin biosynthesis occurs in many pro-
karyotes while others are partly or totally dependent
on external sources. The purpose of this review is tosummarize what is known about biotin biosynthesis,
transport and utilization in rhizobia. Rhizobia are
the only prokaryotes in which novel regulatory roles
for biotin have been investigated. Biotin transport is
important for the establishment of symbiosis in some
rhizobia, and they are the only prokaryotes in which
genes encoding biotin transport proteins have been
identified. A new aspect of biotin biosynthesis in rhizo-bia is the probable presence of novel pathways in
some species.
ation of European Microbiological Societies.

160 K. Guillen-Navarro et al. / FEMS Microbiology Letters 246 (2005) 159–165
2. Biotin requirement of rhizobia
Early studies on biotin used Rhizobium leguminosa-
rum bv. trifolii to demonstrate that ‘‘heat-stable Rhizo-
bium growth factor’’ was identical to ‘‘coenzyme R’’
from Azotobacter and that both were, in fact, identicalto biotin [6]. Based on their growth response to biotin
in defined media, rhizobia may be grouped with
respect to their ability to biosynthesize the vitamin.
Biotin auxotrophs are incapable of biotin biosynthesis
and require external sources for growth [6–8]. An eco-
logically interesting example is provided by the non-
symbiotic Mesorhizobium loti strains isolated from
soils [9,10]. These isolates lack a 500 kb region of theirchromosome termed the ‘‘symbiosis island’’ which, in
addition to a variety of symbiosis-specific functions,
encodes the biosynthesis of thiamine, nicotinic acid
and biotin.
Biotin prototrophs synthesize biotin de novo and
show neither a growth nor a significant metabolic re-
sponse to exogenous biotin [6–8,11]. For example, Rhi-
zobium tropici CFN299 grows well in minimal mediumsubcultures in the absence of biotin and maintains a
high level of pyruvate carboylase activity and holo-en-
zyme protein regardless of biotin supplementation
[12,13].
Biotin bradytrophs synthesize biotin but either do
so inefficiently or only under certain growth conditions
[11,14]. A controversy exists as to whether Rhizobium
etli and Sinorhizobium meliloti fit into this class[11,12,15] or with the biotin auxotrophs [16]. It is
important to note that when S. meliloti Rm1021 was
grown in biotin-free medium, a concomitant several-
fold increase in biomass and extracellular biotin, de-
tected with an ELISA assay, were found, indicating
that this strain can produce the vitamin de novo
[15]. Growth studies show that S. meliloti strain
GR4B is a biotin bradytroph whose synthesis of bio-tin, detected with a standard bioassay, was dependent
upon growth conditions [11].
Wild-type R. etli strain CE3 behaves as a biotin aux-
otroph when serially subcultured in minimal medium,
where very low biotin-dependent carboxylase activities
and protein levels confirm the presence of a biotin
starved state. Growth is restored not only in the pres-
ence of exogenous biotin but also by supplementationwith thiamine, pimelic acid (a biotin precursor), fuma-
rate plus malate, cAMP, glutamate, proline, or oxygen
([12]; unpublished results). S. meliloti Rm1021 behaves
similarly to R. etli CE3 with respect to the ability of thi-
amine to prevent biotin auxotrophy [12]. Given that
both S. meliloti and R. etli lack genes homologous to
most or all of the orthodox biotin biosynthesis genes
(see Section 6), the challenge of providing an unequivo-cal demonstration of their ability to synthesize the vita-
min remains.
3. Biotin-dependent carboxylases and biotin–protein
ligase in rhizobia
Biotin and carbon dioxide are essential for the
growth of rhizobia [1,6,17]. Genome sequence and
biochemical analysis show that rhizobia contain thebiotin-dependent enzymes pyruvate carboxylase
(PYC), acetyl-CoA carboxylase (ACC), and two or
more acyl-CoA carboxylases, including propionyl-
CoA carboxylase (PCC) [18]. PYC is required for
growth on sugars or pyruvate and, although its inacti-
vation has no effect on nodulation and nitrogen fixa-
tion in S. meliloti, R. etli or R. tropici [13,19], it
would be interesting to determine whether it plays arole in rhizosphere competition, since sugars are ex-
creted to the rhizosphere by legume roots [20]. The
symbiotic phenotype of a rhizobial PCC mutant has
not been determined but inactivation of the S. meliloti
methylmalonyl-CoA mutase, which catalyzes the step
following that of PCC during propionyl-CoA degrada-
tion, does not affect symbiotic performance [21,22].
ACC has not been characterized but would be expectedto be essential for fatty acid synthesis [18] and thus
viability.
Apo-biotin-dependent carboxylases are converted to
their active holo-enzymes by biotin–protein ligase
(BPL) [5,23]. The BPLs of Bacillus and enteric bacteria
are called BirA�s (biotin regulators) because their N-
terminal helix-turn-helix motif binds to and represses
biotin operon transcription [23,24]. In these organisms,BirA catalyzes the conversion of biotin into biotinoyl-
AMP, which functions with BirA as a co-repressor:
when the intracellular concentration of biotin is elevated
(e.g., in biotin-supplemented cultures), more BirA-biot-
inoyl-AMP is formed and transcription is repressed.
The middle and C-terminal portion of BirA contain
the catalytic residues for ligating biotin to target
proteins [23].Genome sequence analysis of M. loti, B. japonicum,
S. meliloti (http://www.kazusa.or.jp/rhizobase) R. etli
(G. Davila, V. Gonzalez, R. Gomez and P. Bustos,
unpublished), and R. leguminosarum bv. viciae (http://
www.sanger.ac.uk/Projects/R_leguminosarum) shows
that their deduced BPL gene products lack the N-termi-
nus found in BirA�s and retain only the catalytic motifs
required for biotinylating apo-carboxylases. Thesemonofunctional BPLs are present in many prokaryotes
and all eukaryotes.
The fact that rhizobia contain multiple biotin-depen-
dent carboxylases raises the question of how biotin is
partitioned among them by a single BPL. Western blot-
ting experiments designed to follow the biotinylation of
the carboxylases in biotin-starved R. etli cells pulsed
with biotin suggest that the relative level of each apo-carboxylase determines the amount of holo-carboxylase
formed (M. Dunn, unpublished). It is not known if the

K. Guillen-Navarro et al. / FEMS Microbiology Letters 246 (2005) 159–165 161
BPL has the same affinity for each of the different apo-
carboxylases.
4. Effect of biotin on gene expression
Biotin affects gene expression in eukaryotes [25] but
little information exists on biotin as a BirA-independent
regulatory molecule in prokaryotes. Proteome analysis
shows that biotin markedly alters global protein expres-
sion in R. etli [26] and S. meliloti [27,28]. In R. etli, how-
ever, most of the changes in protein expression caused
by biotin were similar to those observed with thiamine
supplementation or growth in complex medium [26].Thus most of the changes observed with biotin reflect
the general metabolic state of the cells rather than a spe-
cific effect of the vitamin, and so without appropriate
controls claims of biotin-dependent gene expression
must be interpreted with caution.
Gene fusion assays show that S. meliloti bhdA
(encoding b-hydroxybutyrate dehydrogenase), bioS
(putative biotin-responsive regulatory gene), and copC
(possible copper resistance gene) are induced in response
to culture biotin supplementation [27–29]. In contrast,
pcm (encoding L-isoaspartyl protein repair methyltrans-
ferase), sinI (homoserine lactone autoinducer syntha-
tase) and sinR (homoserine lactone autoinducer
transcriptional regulator) are repressed under these con-
ditions [27]. Proteome analysis revealed that proteins
whose levels decreased under biotin limitation includedthe gene product of the down-regulated copC mentioned
above, 50S ribosomal protein L7/L12, RNA polymerase
x subunit, periplasmic transporter substrate binding
proteins (two for sugars, one for amino acids) and 2-
keto-3-deoxy-6-phosphogluconate aldolase (part of the
Entner-Doudoroff pathway). As Heinz and Streit [27]
discuss in detail, there is some correlation between these
results and the physiological response of S. meliloti tobiotin. For instance, the upregulated BdhA participates
in the degradation of the carbon storage polymer poly-
b-hydroxybutyrate (PHB), consistent with the finding
that biotin supplementation prevents PHB accumula-
tion in S. meliloti [12,28]. Regulation of PHB degrada-
tion by biotin could prevent its accumulation in
bacteroids [3] and promote its accumulation and grad-
ual utilization in oligotrophic environments like soil.
5. Biotin and the rhizobia–legume symbiosis
The effect of biotin on symbiosis depends on the rhi-
zobia–legume combination in question, and many natu-
rally-occuring, symbiotically proficient rhizobia are
biotin auxotrophs. A M. loti R7A biotin auxotroph(bioA::Tn5) was indistinguishable from the wild-type in
colonizing the Lotus corniculatus rhizosphere. However,
the biotin phenotype of this mutant is leaky [14] perhaps
due to the presence of a second copy of bioA, as occurs
in M. loti MAFF303099 [30]. Studies with S. meliloti
and R. etli bioN and bioM biotin uptake mutants indi-
cate that high affinity uptake is required for efficient
nodulation of their respective legume hosts ([5,15]; K.Guillen-Navarro, submitted). Interestingly, S. meliloti
bioN mutants engineered for biotin overproduction by
the introduction of the E. coli bio operon were also
found to compete poorly with the wild-type in the alfalfa
rhizosphere, perhaps due to the reduced viability ob-
served in the overproducing strains [29].
Determining the absolute symbiotic requirement for
biotin in rhizobia is complicated by the fact that thevitamin is excreted from the roots of host plants
[15,31]. The very low bacteroid activities of biotin-
dependent carboxylases in the bradytroph R. etli CE3
indicate that little biotin is synthesized by, or available
to, the microsymbiont. In contrast, bacteroids of the
biotin prototroph R. tropici CFN299 from nodules
formed on the same host (bean) have high activities,
indicating de novo synthesis of the vitamin ([32]; M.Dunn, unpublished).
6. Biotin biosynthesis
Escherichia coli and Bacillus species are the model
organisms to which we owe most of our understanding
of biotin biosynthesis (Fig. 1). Bacillus spp. are able totake up (apparently by passive diffusion) and use pimelic
acid as a precursor of biotin. Pimelic acid is derived
through an unknown pathway which may involve the
postulated fatty acid synthase-like activities of BioX
and BioI [33]. Pimelic acid is activated to its CoA deriv-
ative by pimeloyl-CoA synthetase, the product of bioW
[34]. E. coli is unable to utilize pimelic acid as a biotin
precursor and instead synthesizes pimeloyl-CoA, possi-bly from acetyl-CoA [35], using BioH, a probable car-
boxyl esterase [36] and the yet uncharacterized BioC
[33]. bioW homologs do not occur in the sequenced
genomes of M. loti, B. japonicum, S. meliloti or
A. tumefaciens.
The M. loti gene encoding BioZ is part of the bioBF-
DAZ operon (Fig. 2) and shows similarity to b-ketoacid-acyl synthases. BioZ can functionally complement E.
coli bioH, but not bioC, mutants. Based on this, Sullivan
et al. [14] proposed that BioZ catalyzes both the conden-
sation of a thioester with an odd number of carbon
atoms to produce pimeoyl-ACP and its subsequent
transacetylation to pimeloyl-CoA. This hypothesis is
consistent with recent enzymological data on the E. coli
BioH [36].
Four enzymes convert pimeloyl-CoA to biotin,namely BioF (7-keto-8-aminopelargonic acid synthase),
BioA (7,8-diaminopelergonic acid aminotransferase),

C
O
CH2(CH2)4COOH
C
O
CH2(CH2)4COOH
HO
S CoA
CH2(CH2)4COOH
O NH2
H3C
CH2(CH2)4COOH
NH2
H3C
NH2
CH2(CH2)4COOH
HN
H3C
NH
O
CH2(CH2)4COOH
HN NH
O
S
Bacillus spp.
precursor
BioX, BioI Pimelic acid
ATP, CoA BioW
L-alanine
pimeloyl-CoA E. coli
precursor
BioC, BioH BioF
7-keto-8-aminoperlargonic acid(KAPA)
SAM BioA
7,8-diaminopelargonicacid (DAPA)
ATP, CO 2 BioD
d-Dethiobiotin
NADPH, SAM, Flavodoxin BioB
d-Biotin
KAPA synthase
DAPA synthase
Dethiobiotin synthetase
Biotin synthetase
pimeloyl-CoA synthase
At, Bj, Ml, Rla, Smb
At, Bj, Ml, Re, Rl, Sm, NGRc
At, Bj, Ml
At, Bj, Ml
Fig. 1. The orthodox biotin biosynthetic pathway derived largely from studies utilizing Bacillus spp. and E. coli. Where gene homologs encoding the
main biosynthesis pathway enzymes exist in rhizobia, they are designated by the following abbreviations: At, A. tumefaciens; Bj, B. japonicum; Ml,M.
loti, NGR, Rhizobium sp. NGR234; Re, R. etli; Rl, R. leguminosarum bv. viciae; Sm, S. meliloti. aHomolog with low sequence identity to other BioFs.bHomolog present in genome but does not complement an E. coli bioF mutant. cData obtained from a partial genome sequence [42].
162 K. Guillen-Navarro et al. / FEMS Microbiology Letters 246 (2005) 159–165
BioD (dethiobiotin synthetase) and BioB (biotin synthe-tase) (Fig. 1). The physical arrangement of gene clusters
encoding these orthodox biotin biosynthetic enzymes
are presented in Fig. 2. In M. loti R7A, a functional bio-
BFDA operon was confirmed by complementation of E.
coli mutants inactivated in one of these genes [14]. Ent-
cheva and co-workers [5] used genome sequence analysis
and complementation tests with E. coli biotin mutants
to identify putative biotin biosynthesis genes in S. melil-
oti. bioF and bioA homologs, potentially encoding the
first two enzymes for the pimeloyl-CoA to biotin path-
way, were found, but only the bioF homolog could com-
plement the corresponding E. coli mutant. Homologs
for the last enzymes of the pathway (bioD and bioB)
were not found. Genes possibly encoding BioH and
BioZ, involved in pimeloyl-CoA synthesis, were alsofound but could not complement their respective E. coli
mutants (a bioI homolog was encountered but comple-
mentation was not tested since no homolog occurs in
E. coli). The genome sequence of R. etli CE3 contains
a bioA homolog on plasmid f, but no bioB, bioD or bioF
homologs (unpublished results), while that of R. legu-
minosarum bv. viciae contains bioA and bioF homologs
but lacks homologs for bioB and bioD.
7. Biotin transport
Active biotin uptake occurs in E. coli but the trans-
port system involved is not known [37]. An S. meliloti

B. subtilis
M. loti
A. tumefaciens
B. sphaericus
E. coli
B. japonicum
bioD bioA bioY bioB bioX bioW bioF
orf1 bioA bioF bioB bioC bio D bioH
bioB bioF bioD bioA panD bioA bioY dct A
bioB bioF bioD bioA bioA bioA bioY bioN bioM
panD bioYbioB bioF bioD bioA orf bioZ bioA
bioW bioA bioF bioD bioB bioI orf2
orf bioM bioN bioYR. etli / S. meliloti
Fig. 2. Biotin biosynthesis gene clusters in selected prokaryotes. Data were obtained from the literature cited in the text or by homology searches of
the following genome databases: A. tumefaciens, http://www.ncbi.nlm.hih.gov/genomes/MICROBES/Complete.html; B. japonicum, http://
www.kazusa.or.jp/rhizobase; R. etli, G. Davila, V. Gonzalez, R. Gomez and P. Bustos, unpublished. Contiguous arrows represent gene clusters
and spaces denote genes or clusters in other parts of the genome. The drawings are not to scale.
K. Guillen-Navarro et al. / FEMS Microbiology Letters 246 (2005) 159–165 163
mutant inactivated in bioS, the biotin-upregulated genementioned in Section 4, has a higher level of biotin up-
take than the wild-type [38]. bioS encodes a LysR type
protein and its role in biotin uptake would appear to
be regulatory [28]. Both S. meliloti and R. etli contain
operons (bioMNB) encoding products involved in biotin
uptake or retention which are identically organized and
share high sequence identity ([5], Guillen-Navarro et al.,
submitted). Very similar operon exists in R. leguminosa-
rum bv. viciae and A. tumefaciens but have not been
characterized experimentally. The gene originally desig-
nated bioB in S. meliloti [5] does not resemble a biotin
synthase (the classical bioB product) but instead has
similarity to bioY, first implicated in biotin biosynthesis
in Bacillus sphaericus because of its proximity to other
genes involved in biotin biosynthesis [39]. We refer here
to the S. meliloti and R. etli ‘‘bioB’’ homologs as bioY.Sequence analysis and experimental data [5,40] suggest
that bioM and bioN are ABC-type transporters for bio-
tin and encode the ATPase and permease components,
respectively. BioY has six probable transmembrane do-
mains like those of transport permeases but constitutes
its own family in the Pfam database [40]. In S. meliloti,
uptake experiments with a high concentration of exter-
nal biotin (40 nM) showed that a bioM mutant was defi-cient in biotin retention but not uptake [5]. We used low
external biotin concentrations (10–100 pM) to show that
a R. etli bioM mutant had significantly reduced uptake
of biotin but was not defective in retaining it (Guillen-
Navarro, submitted). Overexpression of bioY in wild-
type S. meliloti Rm1021 allows better than wild-type
growth in medias supplemented with dethiobiotin. It
was suggested that BioY might play a role in convertingdethiobiotin to biotin by a mechanism distinct from that
of a classical biotin synthase [5]. However, because com-
mercially available dethiobiotin contains biotin as a con-taminant [41], extra copies of bioY may promote growth
in dethiobiotin-supplemented cultures by allowing more
efficient uptake of the contaminating biotin.
BlastN analysis was used to determine the presence of
homologs of bioB, bioD, bioF and bioA (the orthodox
biotin biosynthesis genes) and bioY (the putative high
affinity transport component) in 159 sequenced genomes
(including 37 incomplete genomes) in the GenBank andKEGG databases. We found that (i) nearly 16% of the
genomes contained only bioY, (ii) 39% lacked bioY
and contained all of the orthodox biosynthetic genes,
(iii) nearly 18% contained bioY and all of the orthodox
biosynthetic genes and (iv) the remainer contained bioY
plus one or two of the classical genes. The genomes
encoding all of the genes included those of A. tumefac-
iens and M. loti, which could benefit from possessingboth the orthodox biosynthetic route and high affinity
uptake capability, since both species colonize plant tis-
sues but also survive as saprophytes in soil.
8. Perspectives
Rhizobia make enlightening subjects for the study ofbiotin metabolism and utilization owing to characteris-
tics which differ from the standard model organisms
including (i) their ability to enter into symbiosis, which
has been disected at the molecular level and for which
the importance of biotin is dependent on the symbiotic
combination; (ii) the presence of multiple biotin-depen-
dent carboxylases; (iii) absence of BirA regulatory func-
tions; (iv) preliminary data indicating a metabolicregulatory function for biotin and (v) the apparent pres-
ence of novel biosynthetic pathways. We need to persue

164 K. Guillen-Navarro et al. / FEMS Microbiology Letters 246 (2005) 159–165
the work on possible novel biotin biosynthesis pathways
with a rigorous biochemical and physiological charac-
terization, including the use purified precursors to dem-
onstrate actual substrate/product relationships. The
application of global methodologies such as proteomics
and transcriptomics in rhizobia will allow further identi-fication of genes and gene products regulated by biotin.
Our knowledge of biotin uptake and the regulation of its
utilization can also be greatly expanded with rhizobia as
experimental organisms.
Acknowledgments
We apologize to the authors of papers which were not
cited because of the publishers space limitations. K.
G-N. was supported by graduate student fellowships
138526 from CONACyT and 202327 and 202363 from
DGAPA-UNAM. We thank G. Davila, V. Gonzalez,
R. Gomez and P. Bustos for access to the R. etli genome
sequence prior to publication.
References
[1] Allen, E.K. and Allen, O.N. (1950) Biochemical and symbiotic
properties of the rhizobia. Bacteriol. Rev. 14, 273–330.
[2] Dunn, M.F. (1998) Tricarboxylic acid cycle and anaplerotic
enzymes in rhizobia. FEMS Microbiol. Rev. 22, 105–123.
[3] Lodwig, E. and Poole, P. (2003) Metabolism of Rhizobium
bacteroids. Crit. Rev. Plant Sci. 22, 37–78.
[4] Streit, W. and Entcheva, P. (2003) Biotin in microbes, the genes
involved in its biosynthesis, its biochemical role and perspectives
for biotechnological production. Appl. Microbiol. Biotechnol. 61,
21–31.
[5] Entcheva, P., Phillips, D. and Streit, W. (2002) Functional
analysis of Sinorhizobium meliloti genes involved in biotin
synthesis and transport. Appl. Environ. Microbiol. 68, 2843–
2848.
[6] West, P.M. and Wilson, P.W. (1940) Biotin as a growth stimulant
for the root nodule bacteria. Enzymologia 8, 152–162.
[7] Graham, P.H. (1963) Vitamin requirements of root nodule
bacteria. J. Gen. Microbiol. 30, 245–248.
[8] Bunn, C.R., McNeill, J.J. and Elkan, G.H. (1970) Effect of biotin
on fatty acids and phospholipids of biotin-sensitive strains of
Rhizobium japonicum. J. Bacteriol. 102, 24–29.
[9] Sullivan, J.T. and Ronson, C.W. (1998) Evolution of rhizobia by
the acquisition of a 500-kb symbiosis island that integrates into a
phe-tRNA gene. Proc. Natl. Acad. Sci. USA 95, 5145–5149.
[10] Sullivan, J.T., Eardly, B.D., van Berkum, P. and Ronson, C.W.
(1996) Four unnamed species of nonsymbiotic rhizobia isolated
from the rhizosphere of Lotus corniculatus. Appl. Environ.
Microbiol. 62, 2818–2825.
[11] Sierra, S., Rodelas, B., Martınez-Toledo, M.V., Pozo, C. and
Gonzalez-Lopez, J. (1999) Production of B-group vitamins by two
Rhizobium strains in chemically defined media. J. Appl. Micro-
biol. 86, 851–858.
[12] Encarnacion, S., Dunn, M., Willms, K. and Mora, J. (1995)
Fermentative and aerobic metabolism in Rhizobium etli. J.
Bacteriol. 177, 3058–3066.
[13] Dunn, M.F., Encarnacion, S., Araıza, G., Vargas, M.C., Davalos,
A., Peralta, H., Mora, Y. and Mora, J. (1996) Pyruvate
carboxylase from Rhizobium etli: Mutant characterization, nucle-
otide sequence, and physiological role. J. Bacteriol. 178, 5960–
5970.
[14] Sullivan, J., Brown, S., Yocum, R. and Ronson, C. (2001) The bio
operon on the acquired symbiosis island of Mesorhizobium sp.
strain R7A includes a novel gene involved in pimeloyl-CoA
synthesis. Microbiologica 147, 1315–1322.
[15] Streit, W.R., Joseph, C.M. and Phillips, D.A. (1996) Biotin and
other water-soluble vitamins are key growth factors for alfalfa
root colonization by Rhizobium meliloti 1021. Mol. Plant-Microbe
Interact. 9, 330–338.
[16] Watson, R.J., Heys, R., Martin, T. and Savard, M. (2001)
Sinorhizobium meliloti cells require biotin and either cobalt or
methionine for growth. Appl. Environ. Microbiol. 67, 3767–3770.
[17] Lowe, R.H. and Evans, H.J. (1962) Carbon dioxide requirement
for growth of legume nodule bacteria. Soil Sci. 94, 351–356.
[18] Dunn, M.F., Araıza, G., Encarnacion, S., Finan, T.M. and Mora,
J. (2002) Characteristics and metabolic roles of biotin-dependent
enzymes in rhizobia In: Nitrogen Fixation: Global Perspectives
(Finan, T., O�Brian, M.R., Layzell, D.B., Vessey, J.K. and
Newton, W., Eds.), pp. 158–162. CABI, Wallingford, Oxon, UK.
[19] Dunn, M.F., Araıza, G. and Finan, T.M. (2001) Cloning and
characterization of the pyruvate carboxylase from Sinorhizobium
meliloti Rm1021. Arch. Microbiol. 176, 355–363.
[20] Rovira, A.D. (1960) Plant root exudates. Bot. Rev. 35, 35–59.
[21] Dunn, M.F., Araıza, G. and Mora, J. (2004) Biochemical
characterization of a Rhizobium etli monovalent cation-stimulated
acyl-coenzyme A carboxylase with a high substrate specificity
constant for propionyl-coenzyme A. Microbiologica 150, 399–
406.
[22] Charles, T.C. and Aneja, P. (1999) Methylmalonyl-CoA mutase
encoding gene of Sinorhizobium meliloti. Gene 226, 121–127.
[23] Cronan Jr, J.E. (1989) The E. coli bio operon: transcriptional
repression by an essential protein modification enzyme. Cell 58,
427–429.
[24] Bower, S., Perkins, J., Yocum, R.R, Serror, P., Sorokin, A.,
Rahaim, P., Howitt, C.L., Prasad, N., Ehrlich, S.D. and Pero, J.
(1995) Cloning and characterization of the Bacillus subtilis birA
gene encoding a repressor of the biotin operon. J. Bacteriol. 178,
2572–2575.
[25] Rodriguez-Melendez, R. and Zempleni, J. (2003) Regulation of
gene expression by biotin. J. Nutr. Biochem. 14, 680–690.
[26] Encarnacion, S., Guzman, Y., Dunn, M.F., Hernandez, M.,
Vargas, M.C. and Mora, J. (2003) Proteome analysis of aerobic
and fermentative metabolism in Rhizobium etli CE3. Proteomics
3, 1077–1085.
[27] Heinz, E.B. and Streit, W.R. (2003) Biotin limitation in Sinorhi-
zobium meliloti strain 1021 alters transcription and translation.
Appl. Environ. Microbiol. 69, 1206–1213.
[28] Hofmann, K., Heinz, E.B., Charles, T.C., Hoppert, M., Liebl, W.
and Streit, W.R. (2000) Sinorhizobium meliloti strain 1021 bioS
and bdhA gene transcriptions are both affected by biotin available
in defined medium. FEMS Microbiol. Lett. 182, 41–44.
[29] Streit, W.R. and Phillips, D.A. (1996) Recombinant Rhizobium
meliloti strains with extra biotin synthesis capability. Appl.
Environ. Microbiol. 62, 3333–3338.
[30] Kaneko, T., Nakamura, Y., Sato, S., Asamizu, E., Kato, T.,
Sasamoto, S., Watanabe, A., Idesawa, K., Ishikawa, A., Kawa-
shima, K., Kimura, T., Kishida, Y., Kiyokawa, C., Kohara, M.,
Matsumoto, M., Matsuno, A., Mochizuki, Y., Nakayama, S.,
Nakazaki, N., Shimpo, S., Sugimoto, M., Takeuchi, C., Yamada,
M. and Tabata, S. (2000) Complete genome structure of the
nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA
Res. 7, 331–338.
[31] Rovira, A.D. and Harris, J.R. (1961) Plant root excretions in
relation to the rhizosphere effect. V. The exudation of B-group
vitamins. Plant Soil 14, 199–214.

K. Guillen-Navarro et al. / FEMS Microbiology Letters 246 (2005) 159–165 165
[32] Dunn, M.F., Araıza, G., Cevallos, M.A. and Mora, J. (1997)
Regulation of pyruvate carboxylase in Rhizobium etli. FEMS
Microbiol. Lett. 157, 301–306.
[33] Lemoine, Y., Wach, A. and Jeltsch, J.-M. (1996) To be free or
not: the fate of pimelate in Bacillus sphaericus and in Escherichia
coli. Mol. Microbiol. 19, 645–647.
[34] Ploux, O., Soularue, P., Marquet, A., Gloeckler, R. and Lemoine,
Y. (1992) Investigation of the first step of biotin biosynthesis in
Bacillus sphaericus: purification and characterization of the
pimeloyl-CoA synthase, and uptake of pimelate. Biochem. J.
287, 685–690.
[35] Ifuku, O., Miyaoka, H., Koga, N., Kishimoto, J., Haze, S.,
Wachi, Y. and Kajiwara, M. (1994) Origin of carbon atoms of
biotin: 13C-NMR studies on biotin biosynthesis in Escherichia
coli. Eur. J. Biochem. 220, 585–591.
[36] Sanishvili, R., Yakunin, A., Laskowski, R., Skarina, T., Evd-
okimova, E., Doherty-Kirby, A., Lajoie, G., Thornton, T.,
Arrowsmith, C., Savchenko, A., Joachimiak, A. and Edwards,
A. (2003) Integrating structure, bioinformatics and enzymology to
discover function. BioH, a new carboxyl esterase from Escherichia
coli. J. Biol. Chem. 278, 26039–26045.
[37] Piffeteau, A. and Gaudry, M. (1985) Biotin uptake: influx, efflux
and countertransport in Escherichia coli K12. Biochim. Biophys.
Acta 816, 77–82.
[38] Streit, W.R. and Phillips, D.A. (1997) A biotin-regulated locus,
bioS, in a possible survival operon of Rhizobium meliloti. Mol.
Plant-Microbe Interact. 10, 933–937.
[39] Gloeckler, R., Ohsawa, I., Speck, D., Ledoux, C., Bernard, S.,
Zinsius, M., Villeval, D., Kisou, T., Kamogawa, K. and Lemoine,
Y. (1990) Cloning and characterization of the Bacillus sphaericus
genes controlling the bioconversion of pimelate into dethiobiotin.
Gene 87, 63–70.
[40] Rodinov, D.A., Mironov, A. and Gelfand, M. (2002)
Conservation of the biotin regulon and the BirA regula-
tory signal in Eubacteria and Archaea. Genome Res. 12,
1507–1516.
[41] Bowman, W.C. and DeMoll, E. (1993) Biosynthesis of biotin
from dethiobiotin by the biotin auxotroph Lactobacillus planta-
rum. J. Bacteriol. 175, 7702–7704.
[42] Viprey, V., Rosenthal, A., Broughton, W.J. and Perret, X. (2000)
Genetic snapshots of the Rhizobium species NGR234 genome.
Genome Biol. 1, 0014.1–0014.17.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 167–174
The Pseudomonas aeruginosa pirA gene encodes a second receptorfor ferrienterobactin and synthetic catecholate analogues
Bart Ghysels a, Urs Ochsner b,1, Ute Mollman c, Lothar Heinisch c, Michael Vasil b,Pierre Cornelis a,*, Sandra Matthijs a
a Department of Molecular and Cellular Interactions, Laboratory of Microbial Interactions, Flanders Interuniversity Institute of Biotechnology (VIB6),
Vrije Universiteit Brussel, Brussels, Belgiumb University of Colorado Health Science Center, Denver, USA
c Hans Knoll Institut, Jena, Germany
Received 6 January 2005; received in revised form 1 April 2005; accepted 4 April 2005
First published online 20 April 2005
Edited by S. Silver
Abstract
Actively secreted iron chelating agents termed siderophores play an important role in the virulence and rhizosphere competence
of fluorescent pseudomonads, including Pseudomonas aeruginosa which secretes a high affinity siderophore, pyoverdine, and the low
affinity siderophore, pyochelin. Uptake of the iron–siderophore complexes is an active process that requires specific outer membrane
located receptors, which are dependent of the inner membrane-associated protein TonB and two other inner membrane proteins,
ExbB and ExbC. P. aeruginosa is also capable of using a remarkable variety of heterologous siderophores as sources of iron, appar-
ently by expressing their cognate receptors. Illustrative of this feature are the 32 (of which 28 putative) siderophore receptor genes
observed in the P. aeruginosa PAO1 genome. However, except for a few (pyoverdine, pyochelin, enterobactin), the vast majority of
P. aeruginosa siderophore receptor genes still remain to be characterized. Ten synthetic iron chelators of catecholate type stimulated
growth of a pyoverdine/pyochelin deficient P. aeruginosa PAO1 mutant under condition of severe iron limitation. Null mutants of
the 32 putative TonB-dependent siderophore receptor encoding genes engineered in the same genetic background were screened for
obvious deficiencies in uptake of the synthetic siderophores, but none showed decreased growth stimulation in the presence of the
different siderophores. However, a double knock-out mutant of ferrienterobactin receptor encoding gene pfeA (PA 2688) and pirA
(PA0931) failed to be stimulated by 4 of the tested synthetic catecholate siderophores whose chemical structures resemble enterob-
actin. Ferric-enterobactin also failed to stimulate growth of the double pfeA–pirA mutant although, like its synthetic analogues, it
stimulated growth of the corresponding single mutants. Hence, we confirmed that pirA represents a second P. aeruginosa ferric-
enterobactin receptor. The example of these two enterobactin receptors probably illustrates a more general phenomenon of sider-
ophore receptor redundancy in P. aeruginosa.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Pseudomonas aeruginosa; Enterobactin; Catecholate siderophores; pfeA; pirA; TonB-dependent receptors
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.010
* Corresponding author. Tel.: 02 6291906; fax: 02 6291902.
E-mail address: [email protected] (P. Cornelis).1 Present address: Replidyne Inc., 1450 Infinite Drive, Louisville, CO
80027, USA.
1. Introduction
Pseudomonas aeruginosa is a Gram-negative bacte-
rium endowed with an extremely versatile metabolism,
reflected in its ability to colonize a wide variety of
. Published by Elsevier B.V. All rights reserved.

168 B. Ghysels et al. / FEMS Microbiology Letters 246 (2005) 167–174
habitats, from mammalian host to the rhizosphere of
plants [1]. As for most organisms, iron is indispensable
for survival of P. aeruginosa. Unfortunately, iron, de-
spite being one of the most abundant elements in the
earth crust, is rarely freely accessible and its acquisi-
tion demands significant adaptations from micro-organisms. A common microbial strategy for iron
acquisition is the production of low-molecular mass
iron-chelating compounds named siderophores [2].
For Gram-negative bacteria, uptake of the ferrisidero-
phore complexes into the cell necessitates specialized
receptors acting as gated porin channels that recognize
and actively internalize the ferrisiderophore complexes
[2]. As a rule, ferrisiderophore receptors recognizeexclusively one iron–siderophore complex, but excep-
tions to this rule have been reported [3–5]. Since the
outer membrane is devoid of energy sources, all these
receptors rely on a conserved protein, called TonB,
and two other proteins, ExbB and ExbC, also located
in the inner membrane, to transduce energy generated
in the cytoplasmic membrane to the receptor protein
[2]. Although three TonB homologues have been de-scribed in P. aeruginosa, only TonB1 seems to be in-
volved in the uptake of ferrisiderophores [6]. Just
like outer-membrane porins, these receptor proteins
are shaped of a large C-terminal domain of 22 antipar-
allel b-strands, which form a membrane spanning
b-barrel [7]. What distinguishes TonB dependent
receptors from porins is an additional domain known
as �cork� or �plug� that blocks the b-barrel domainand by using energy transduced by TonB, allows selec-
tive uptake of siderophore/iron complexes [8].
P. aeruginosa secretes a high affinity siderophore
pyoverdine (PVD), and another siderophore, pyochelin
(PCH), which displays lower iron affinity compared to
PVD [9,10]. Besides producing endogenous sidero-
phores, P. aeruginosa has the capacity to take up and
utilize numerous siderophores secreted by other micro-organisms including those of other bacteria (aerobactin,
enterobactin and its precursor 2,3-dihydrobenzoic acid
and breakdown product N-(2,3-dihydrobenzoyl)-L-ser-
ine), pyoverdines/pseudobactin from other pseudomo-
nads, cepabactin, fungal siderophores (deferrioxamines,
dererrichrysin, deferrirubin, coprogen), synthetic chela-
tors, (e.g. nitrilotriacetic acid) and naturally occuring
chelators such as citrate and myo-inositol hexakisphos-phate [10]. Not surprisingly, the P. aeruginosa PAO1
genome [11] counts no less than 32 genes with ferrisider-
ophore receptor gene signature [9]. Only three of them
were previously matched with a siderophore ligand: fpvA
(ferri-PVD uptake) [12], fptA (ferric PCH uptake) [13]
and pfeA (ferrienterobactin uptake) [14]. Recently, we
described a gene, fpvB, which encodes a second receptor
for PVD [15]. The active transport of siderophore–ironcomplexes across the outer membrane of Gram-negative
bacteria has caught the interest of scientists exploring
possible novel concepts of anti-microbial drug delivery.
One possible approach to overcome the problem of resis-
tance in P. aeruginosa [16] lies in the synthesis of antibi-
otics conjugated with compounds active as siderophores
[17]. Two different carrier concepts are currently under
evaluation: making use of either a derivative of a naturalsiderophore or artificial synthetic siderophore entities
[17]. Tests on several Gram-negative species have dem-
onstrated the applicability of both type of conjugates,
exhibiting minimal inhibitory concentrations (MICs)
that are significantly lower (up to 100 times) compared
to the associated free drugs. However, drug conjugates
with synthetic siderophore analogues are often easier to
produce [17] and may be designed for application againsta broader range of species. Hence, while exploring the
properties of synthetic siderophore analogues for active
drug delivery it is important to map the receptors that
mediate their uptake. An ideal siderophore drug carrier
is taken up by several receptors in order to minimize
the chances of resistance development.
Many pathogenic bacteria, including P. aeruginosa,
have outer membrane receptors for heterologous trans-port of ferrienterobactin (FeEnt), a siderophore pro-
duced by enteric bacteria [14,18]. It has been
suggested that P. aeruginosa can take up enterobactin
via two distinct uptake systems, one of ‘‘high affinity’’
induced by enterobactin, the second of ‘‘low affinity’’
not induced by enterobactin [19]. The pfeA gene,
encoding the high affinity enterobactin receptor, has
been cloned and sequenced [14]. Synthetic analoguesthat mimic enterobactin, but change certain aspects
of its chemistry were previously used to determine the
structural feature of the siderophore that are important
to its transport. These studies have shown that the iron
binding centre contains the primary determinants of
the uptake reaction and that replacement of the natural
macrocyclic ring had little effect on ferrienterobactin
transport [20].We evaluated the siderophore properties of a number
of synthetic catecholate siderophore analogues on P.
aeruginosa and tried to map their receptors and con-
firmed earlier suggestions for the presence of two ferri-
enterobactin uptake systems in P. aeruginosa PAO1
and identified the second, low affinity ferric-enterobactin
uptake mediating receptor as the product of pirA. Since
both ferrienterobactin receptors are also involved in theuptake of several synthetic enterobactin analogues, they
represent good candidate drug carriers.
2. Materials and methods
2.1. Bacterial strains, media and growth conditions
The different P. aeruginosa mutations in putative fer-
risiderophore receptor genes used in this study are listed

Table 1
List of 36 P. aeruginosa PAO1 TonB-dependent receptors genes
Gene no. Gene name Identified ligand Gene no. Gene name
PA 2398 fpvA Ferripyoverdine PA 4675
PA 4168 fpvB Ferripyoverdine PA 1302
PA 4221 fptA Ferripyochelin PA 4837
PA 2688 pfeA Ferrienterobactin PA 2911
PA 0931* pirA Ferrienterobactin PA 1922 cirA
PA 4710 phuR Heme PA 2289
PA 3408 hasR Heme PA 0192
PA 1910 ufrA PA 0434
PA 0674* pigC PA 0781
PA 0470* fiuA PA 1365
PA 4514* piuA PA 1613
PA 1322* pfuA PA 2057
PA 3901 fecA PA 2089
PA 2466 optS PA 3268
PA 0151 PA 2335 optO
PA 4897 optI PA 4156
PA 4675 iutA PA 2590
PA 1302 hxuC PA 2070
List of 36 P. aeruginosa PAO1 TonB-dependent receptors genes for
which the corresponding knock-out mutants were engineered in an
unmarked allelic pvdD pchEF mutant.* Fur-regulated genes picked up by the SELEX technique [21].
B. Ghysels et al. / FEMS Microbiology Letters 246 (2005) 167–174 169
in Table 1. P. aeruginosa wild-type and mutants were
grown under conditions of good aeration at 37 �C either
in Casamino acid medium (CAA, low iron medium) or
LB medium. The ferrisiderophore growth stimulation
assays were performed in CAA supplemented with
10 lM of the iron chelator ethylenediamine dihydroxy-
phenylacetic acid (EDDHA) and 200 lM dipyridil (in
agar medium) or CAA with 5 lM EDDHA and100 lM dipyridil (in liquid medium) for iron-limiting
conditions. For ferrienterobactin stimulation assays, a
wild-type E. coli strain (MC4100) producing enterobac-
tin was grown in CAA medium plus 0.2% glucose during
48 h. The supernatant was collected after centrifugation
and filter-sterilized. Another E. coli strain, H6876, an
entC derivative of MC4100 was grown under the same
conditions. This strain is unable to produce enterobac-tin. The supernatants of both E. coli strains (15% V/V)
were added to LB-agar plates containing EDDHA and
dipyridil as described in 2.1.
2.2. Siderophores utilization assay
The catecholate synthetic siderophore analogues are
shown in Fig. 1. Petri dishes of CAA solid agar mediumcontaining 10 lM EDDHA + 200 lM dipyridil were
used. Two hundred microlitres of a 105 CFU/ml cell sus-
pension of the mutant was spread on the medium sur-
face. A sterile paper disc impregnated with 5 ll of
2 mM siderophore solution was placed on top of the
agar plate. Siderophore usage was detected, after 1 day
of incubation at 37 �C, as a halo of growth around the
filter disc. For the utilization of enterobactin, LB-ED-
DHA-dipyridil agar plates were used which contained
15% (V/V) filter-sterilized supernatants of E. coli
MC4100 (enterobactin producer) and H6876 (entC mu-
tant of MC4100, unable to produce enterobactin). On
these plates, 0.1 ml of dilutions of P. aeruginosa cells
(from 108 to 103 CFU per ml) was inoculated and theplates incubated overnight at 37 �C.
2.3. Liquid growth stimulation assays
For more accurate analysis, growth was assessed in
microtiter plates using a Bio-Screen C incubator (Life
Technologies�). Briefly, the following protocol was
used: pre-cultures (2–3 ml) were grown overnight inCAA medium. The next day the pre-cultures were used
to inoculate in a 1:100 ratio 3 ml cultures in CAA med-
ium, which were grown till OD600 = 0.5. Serial dilutions
in CAA were performed to reach a final 1:5000 dilution.
The following parameters were programmed to be exe-
cuted by the apparatus: each well contains: 295 ll of
(CAA + 5 lM EDDHA + 100 lM 2-2 0dipyridil) with
5 ll of 2 mM of the to be tested siderophores and 5 llof DMSO for control wells (solvent used to dissolve
the siderophores); shaking for 30 s every 3 min, absor-
bance measured every 20 min at 600 nm and tempera-
ture at 37 �C.
3. Results
3.1. Mutants in putative ferrisiderophore receptor genes in
P. aeruginosa PAO1
A siderophore-free background was created in P.
aeruginosa PAO1 by making unmarked deletions in
pvdD (pyoverdine biosynthesis) and pchEF (pyochelin
biosynthesis) [15]. Candidate siderophore receptor
genes of P. aeruginosa PAO1 were originally pickedup by a cycle selection procedure to identify iron re-
pressed genes that are directly regulated by the Ferric
Uptake Regulator (Fur) [21,22]. Five of these genes
were found to be similar to known siderophore recep-
tor genes (Table 1). More candidate siderophore recep-
tor genes were counted in the completed P. aeruginosa
PAO1 genome sequence (http://www.pseudomo-
nas.com) [9,11]. No less than 36 ORFs carry the signa-ture of TonB dependent receptor encoding genes. Four
of them are the previously identified ferrisiderophore
receptor genes for respectively ferri-pyoverdine (fpvA,
PA2398; fpvB, PA4168), ferri-pyochelin (fptA,
PA4221) and ferrienterobactin (pfeA, PA2688). Also
included are the TonB dependent receptors involved
in haem-uptake encoded by phuR (PA4710) and hasR
(PA3408) [23]. In the earlier created siderophore-freemutant (pvdD pchEF) of P. aeruginosa PAO1, �candi-date� siderophore receptor genes, 36 in total, were
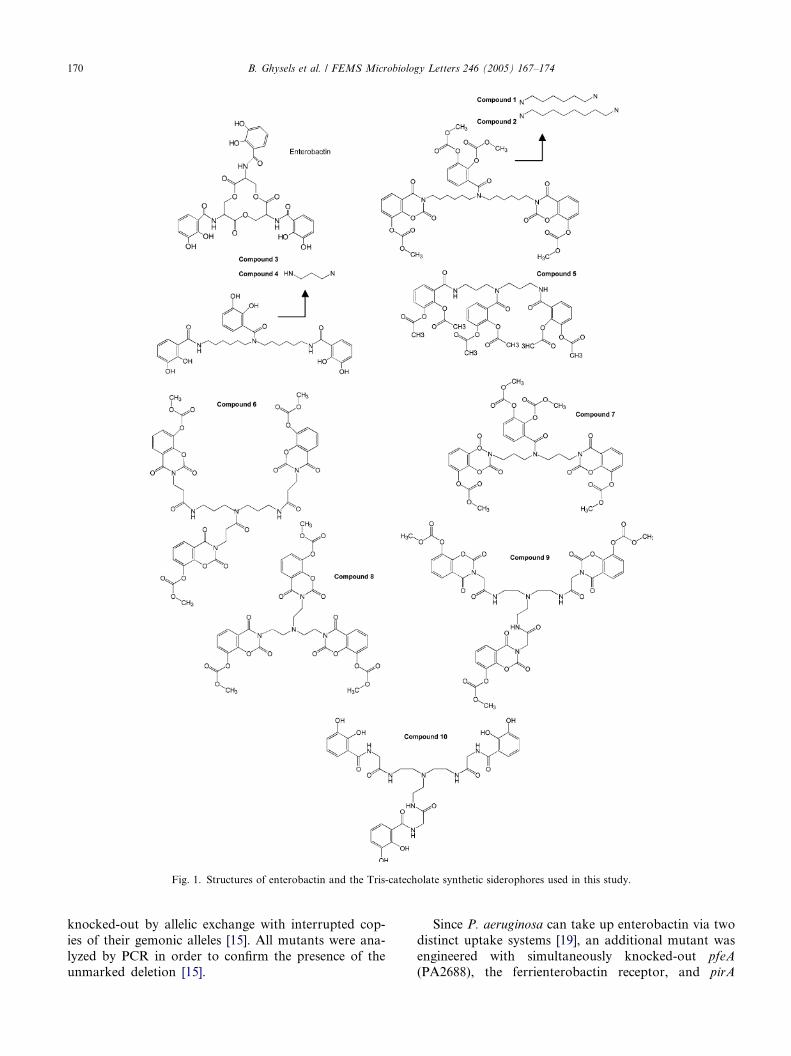
Fig. 1. Structures of enterobactin and the Tris-catecholate synthetic siderophores used in this study.
170 B. Ghysels et al. / FEMS Microbiology Letters 246 (2005) 167–174
knocked-out by allelic exchange with interrupted cop-
ies of their gemonic alleles [15]. All mutants were ana-
lyzed by PCR in order to confirm the presence of theunmarked deletion [15].
Since P. aeruginosa can take up enterobactin via two
distinct uptake systems [19], an additional mutant was
engineered with simultaneously knocked-out pfeA
(PA2688), the ferrienterobactin receptor, and pirA

B. Ghysels et al. / FEMS Microbiology Letters 246 (2005) 167–174 171
(PA0931), the candidate ferrisiderophore receptor gene
within the PAO1 genome with the highest similarity to
pfeA (72% similarity between their translation
products).
3.2. Utilization of synthetic catecholates by the different
TonB-dependent mutants
The P. aeruginosa PAO1 pvdD pchEF mutant car-
ries deletions in genes for PVD and PCH synthesis
and is therefore unable to grow in an iron restricted
situation created by the presence of both a ferric iron
chelator (10 lM EDDHA) and ferrous iron chelator
(200 lM 2-2 0-dipyridil). We aimed to select syntheticsiderophore analogues that are capable of stimulating
growth of this siderophore deficient mutant in presence
of EDDHA and dipyridil, which, in other words func-
tion as xeno-siderophores that can be assimilated by
P. aeruginosa. Ten ‘‘catecholate’’ compounds were
used [24] (see Table 2). The ‘‘catecholates’’ represent
either the free forms (compounds 3, 4 and 10) and
the protected forms (compounds 1, 2 and 5–9). Therewere 2 types of protected forms, form ‘‘a’’ as aliphatic
acyloxy group (compound 5), form ‘‘b’’ as heterocyclic
benzoxazine residue (compounds 6 and 8) and mixed
forms of both (compounds 1, 2 and 7). The basic
structures for the catecholates were either linear (com-
pounds 1–7) or tripodal (compounds 8–10). We as-
sume, that the protected forms can split off to the
free catecholates under physiological conditions sinceobviously only these structures can form iron com-
plexes. Additionally it should be mentioned that the
antibiotic conjugates of the protected catecholates are
active as antibacterials via uptake by ferrisiderophore
transport pathways [24–27].
Table 2
Summary of the results of growth stimulation tests
Catecholate
compound
Siderophore pvdD pchEF pvdD pchEF
pfeA pirA pfeA pirA
1 HKI 9824013 ++ ++ ++ ++
2 HKI 9824014 ++ ++ ++ ++
3 HKI 9824030 ++ ++ ++ ++
4 HKI 9824043 ++ ++ ++ ++
5 HKI 9824080 ++ ++ ++ ++
6 HKI 9924127 ++ ++ ++ ++
7 HKI 9824032 ++ ++ ++ �8 HKI 10024023 ++ ++ ++ �9 HKI 10024024 ++ ++ + �10 HKI 10024025 ++ ++ + �Summary of the results of growth stimulation tests peformed with the
synthetic siderophore analogues on the pvdD pchEF siderophore pro-
duction deficient background strain, and the mutants in pfeA, pirA and
the double pfeA pirA knock-out, all created in the pvdD pchEF back-
ground. Strong growth stimulation (++), weak/delayed growth stim-
ulation (+), no growth stimulation (�). The catecholate synthetic
siderophore structures are presented in Fig. 1.
All 10 catecholate compounds stimulated the
growth of the siderophore-deficient P. aeruginosa
PAO1 pvdD pchEF mutant under these conditions of
extreme iron limitation. Although each of the putative
TonB-dependent receptor genes of P. aeruginosa PAO1
had been inactivated, none of the 36 single mutantsfailed to be stimulated by any of the selected synthetic
siderophores, suggestive of a redundancy in sidero-
phore uptake systems in P. aeruginosa. Interestingly,
the synthetic enterobactin analogues 7–10 stimulated
single knock-out mutants of pfeA (PA2688), the high
affinity enterobactin receptor gene, and PA0931 (pirA)
its closest homologue within P. aeruginosa PAO1, but
failed to stimulate a mutant with both genes inacti-vated (Table 2).
3.3. Growth stimulation by enterobactin
Since we did not have purified enterobactin, we
looked at the growth stimulation conferred by the addi-
tion of cell-free supernatant from a culture of wild-type
E. coli MC4100 (enterobactin producer) and an entC
derivative from the same strain (unable to produce ente-
robactin) grown under iron-limiting conditions. As
shown in Fig. 2, the supernatant from MC4100 stimu-
lated the growth of the P. aeruginosa pvdD pchEF mu-
tant (Fig. 2(a)), pvdD pchEF pfeA (Fig. 2(b)) and pvdD
pchEF pirA (Fig. 2(c)), but not of the mutant pvdD
pchEF pfeA pirA (Fig. 2(d)). As could be expected, the
supernatant from the entC mutant could not stimulatethe growth of any of these P. aeruginosa strains (results
not shown). This observation confirms that PA0931
(pirA) serves as second ferrienterobactin receptor next
to pfeA.
3.4. Growth kinetics of pfeA and pirA in response to
stimulation by catecholates
The previous growth stimulation tests were per-
formed with siderophore impregnated filter-discs on
CAA-agar medium containing EDDHA and dipyridil.
In order to determine a hierarchical order between the
PfeA and PirA receptors in affinity for the different
enterobactin-like ligands, we kinetically measured
growth responses of the pfeA and pirA mutants to-
wards the different synthetic enterobactin analoguesin EDDHA- and dipyridil-containing liquid CAA cul-
tures. When compared to the pvdD pchEF strain and
the single pfeA mutant in the same genetic back-
ground, a delayed growth response of the pirA mutant
was observed towards compounds 9 (data not shown)
and 10 (Fig. 3) but not to 7 (Fig. 3) or 8 (result not
shown). In contrast to ferrienterobactin which is taken
up preferentially by PfeA [14,19] the iron complexeswith the synthetic siderophore analogues 9 and 10
are more efficiently taken up by PirA.
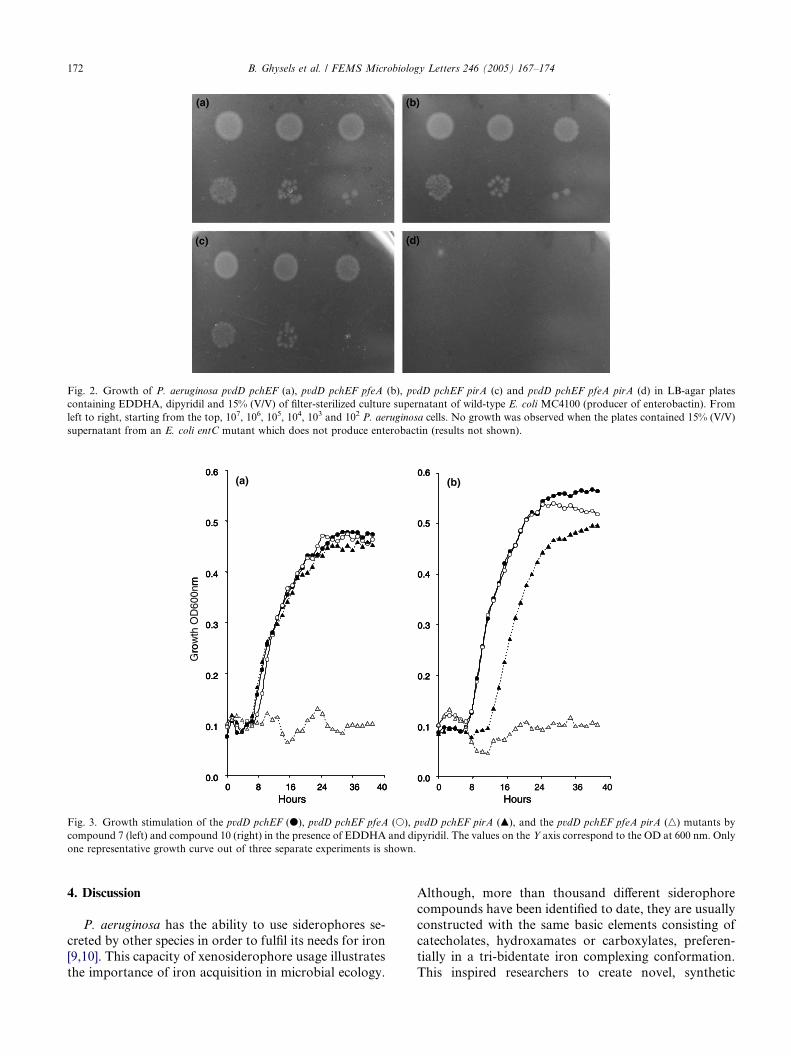
Gro
wth
OD
600n
m
(a) (b)
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0 8 16 24 36 40Hours
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0 8 16 24 36 40Hours
rom
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0 8 16 24 36 400 8 16 24 36 40Hours
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0 8 16 24 36 40Hours
Fig. 3. Growth stimulation of the pvdD pchEF (d), pvdD pchEF pfeA (s), pvdD pchEF pirA (m), and the pvdD pchEF pfeA pirA (n) mutants by
compound 7 (left) and compound 10 (right) in the presence of EDDHA and dipyridil. The values on the Y axis correspond to the OD at 600 nm. Only
one representative growth curve out of three separate experiments is shown.
Fig. 2. Growth of P. aeruginosa pvdD pchEF (a), pvdD pchEF pfeA (b), pvdD pchEF pirA (c) and pvdD pchEF pfeA pirA (d) in LB-agar plates
containing EDDHA, dipyridil and 15% (V/V) of filter-sterilized culture supernatant of wild-type E. coli MC4100 (producer of enterobactin). From
left to right, starting from the top, 107, 106, 105, 104, 103 and 102 P. aeruginosa cells. No growth was observed when the plates contained 15% (V/V)
supernatant from an E. coli entC mutant which does not produce enterobactin (results not shown).
172 B. Ghysels et al. / FEMS Microbiology Letters 246 (2005) 167–174
4. Discussion
P. aeruginosa has the ability to use siderophores se-
creted by other species in order to fulfil its needs for iron[9,10]. This capacity of xenosiderophore usage illustrates
the importance of iron acquisition in microbial ecology.
Although, more than thousand different siderophore
compounds have been identified to date, they are usually
constructed with the same basic elements consisting of
catecholates, hydroxamates or carboxylates, preferen-tially in a tri-bidentate iron complexing conformation.
This inspired researchers to create novel, synthetic

B. Ghysels et al. / FEMS Microbiology Letters 246 (2005) 167–174 173
siderophore compounds based on these naturally con-
served themes. Shared chemo-structural motifs between
synthetic and natural siderophores, allow their ferrisi-
derophore complexes to be taken up by the same cog-
nate receptors. Catecholate derivatives were generated
with high siderophore activities in strains of P. aerugin-osa and E. coli [25]. b-Lactam conjugates of these sidero-
phores showed enhanced antibacterial activities which
could be attributed to the active iron uptake routes used
by the conjugates to penetrate the bacterial cells [24–27].
The 10 synthetic catecholate siderophores used in this
study stimulated the growth of a siderophore-negative
P. aeruginosa under conditions of strong iron limitation,
indicating that these siderophores had sufficient ironbinding affinity to displace iron from EDDHA and
dipyridil and could be assimilated by the cell. The fact
that the growth of all siderophore-negative mutants with
a single receptor gene inactivation could be stimulated
by the 10 compounds suggests the presence of at least
two receptors for a given ferrisiderophore. Such receptor
redundancy has interesting implications for the use of
synthetic xenosiderophore analogues as drug carriers.Indeed, when a siderophore-drug conjugate can pene-
trate the cell via several independent receptors, the risk
of resistance development is significantly reduced.
Receptor redundancy, on the other hand, complicates
the mapping of receptor genes by a knock-out approach
since the dysfunctional receptor phenotype can be
masked by another receptor recognizing the same li-
gand. We recently demonstrated the applicability ofthe idea that receptor pairs with high sequence similarity
mediate the uptake of the same ligand [15], providing a
rational base for engineering multiple receptors knock-
out mutants. The pfeA gene, encoding the high affinity
enterobactin receptor, has been cloned and sequenced
[14]. Nonetheless, PfeA-deficient mutants display
growth, albeit reduced, in an enterobactin supple-
mented, iron-restricted minimal medium [19,28]. Thebest candidate for a second ferrienterobactin receptor
is the product of PA0931, dubbed pirA, which displays
substantial similarity with pfeA. With the receptor mu-
tants engineered in a pyoverdine and pyochelin-free
background, we unambiguously confirmed that P. aeru-
ginosa indeed counts two ferrienterobactin transporting
receptors, PfeA and PirA. Therefore the situation in P.
aeruginosa is similar to the situation in Salmonella enter-
ica where two receptors, FepA and IroN mediate the
transport of ferrienterobactin [29]. Interestingly, we
could not detect any difference in growth stimulation
by ferrienterobactin of the pfeA or pirA mutant (Fig.
2), which seems to be in contradiction with the results
obtained before [19,28]. This could be due to the differ-
ence of genetic background since we used a mutant of P.
aeruginosa which is unable to produce either pyoverdineof pyochelin. Growth kinetics of the mutants suggested
that two of the synthetic enterobactin analogues tested
are preferentially transported by the PirA receptor in
contrast to enterobactin for which PfeA acts as the high
affinity receptor. It is therefore likely that PirA trans-
ports another yet unknown natural siderophore different
from enterobactin as its primary substrate. Another
enterobactin-like siderophore, bacillibactin, is producedby the Gram-positive Bacillus subtilis [30] and it would
be interesting to look at the transport of this ferrisider-
ophore using the same set of mutants described in this
study. Another interesting question for the future is to
understand why only compounds 7–10 are taken by
fepA and pirA like enterobactin. It has to be mentioned
that compounds 8–10 are tripodal while compound 7 is
the only linear catecholate analogue which is taken upby these two receptors. Also, it would be interesting to
discover which TonB-dependent receptors mediate the
transport of compounds 1–6 since their growth stimula-
tion properties are not affected by the fepA pirA
mutations.
Acknowledgements
Support for these studies was provided in part by a
grant from the National Institutes of Allergy and Infec-
tious Diseases (AI15940) to Michael Vasil and of the
VUB OZR to Bart Ghysels. We thank Dr. Klaus Han-
tke for providing the E. coli MC4100 and H6876 strains.
References
[1] Goldberg, J. (1999) Pseudomonas: global bacteria. Trends Micro-
biol. 8, 55–57.
[2] Andrews, S.C., Robinson, A.K. and Rodrıguez-Quinones, F.
(2001) Bacterial iron homeostasis. FEMS Microbiol. Rev. 27,
215–237.
[3] Meyer, J.M., Geoffroy, V.A., Baysse, C., Cornelis, P., Barelmann,
I., Taraz, K. and Budzikiewicz, H. (2002) Siderophore-mediated
iron uptake in fluorescent Pseudomonas: characterization of the
pyoverdine-receptor binding site of three cross-reacting pyover-
dines. Arch. Biochem. Biophys. 397, 179–183.
[4] Meyer, J.M., Stintzi, A. and Poole, K. (1999) The ferripyoverdine
receptor FpvA of Pseudomonas aeruginosa PAO1 recognizes the
ferripyoverdines of P. aeruginosa PAO1 and P. fluorescens ATCC
13525. FEMS Microbiol. Lett. 170, 145–150.
[5] De Chial, M., Ghysels, B., Beatson, S.A., Geoffroy, V., Meyer,
J.M., Pattery, T., Baysse, C., Chablain, P., Parsons, Y.N.,
Winstanley, C., Cordwell, S.J. and Cornelis, P. (2003) Identifica-
tion of type II and type III pyoverdine receptors from Pseudo-
monas aeruginosa. Microbiology 149, 821–831.
[6] Zhao, Q. and Poole, K. (2002) Mutational analysis of the TonB1
energy coupler of Pseudomonas aeruginosa. J. Bacteriol. 184,
1503–1513.
[7] Koebnik, R., Locher, K.P. and Van Gelder, P. (2000) Structure
and function of bacterial outer membrane proteins: barrels in a
nutshell. Mol. Microbiol. 37, 239–253.
[8] Ferguson, A.D. and Deisenhofer, J. (2002) TonB-dependent
receptors-structural perspectives. Biochim. Biophys. Acta 1565,
318–332.

174 B. Ghysels et al. / FEMS Microbiology Letters 246 (2005) 167–174
[9] Cornelis, P. and Mathhijs, S. (2002) Diversity of siderophore-
mediated iron uptake systems in fluorescent pseudomonads: Not
only pyoverdines. Environ. Microbiol. 4, 787–798.
[10] Poole, K. and McKay, G.A. (2003) Iron acquisition and its
control in Pseudomonas aeruginosa: many roads lead to Rome.
Front. Biosci. 8, D661–D686.
[11] Stover, C.K. et al. (2000) Complete genome sequence of Pseudo-
monas aeruginosa PA01, an opportunistic pathogen. Nature 406,
959–964.
[12] Poole, K., Neshat, S., Krebes, K. and Heinrichs, D.E. (1993)
Cloning and nucleotide sequence analysis of the ferripyoverdine
receptor gene fpvA of Pseudomonas aeruginosa. J. Bacteriol. 175,
4597–4604.
[13] Ankenbauer, R.G. and Quan, H.N. (1994) FptA, the Fe(III)-
pyochelin receptor of Pseudomonas aeruginosa: a phenolate
siderophore receptor homologous to hydroxamate siderophore
receptors. J. Bacteriol. 176, 307–319.
[14] Dean, C.R. and Poole, K. (1993) Cloning and characterization of
the ferrienterobactin receptor gene (pfeA) of Pseudomonas aeru-
ginosa. J. Bacteriol. 175, 317–324.
[15] Ghysels, B., Dieu, B.T., Beatson, S.A., Pirnay, J.P., Ochsner,
U.A., Vasil, M.L. and Cornelis, P. (2004) FpvB, an alternative
type I ferripyoverdine receptor of Pseudomonas aeruginosa.
Microbiology 150, 1671–1680.
[16] Poole, K. and Srikumar, R. (2001) Multidrug efflux in Pseudo-
monas aeruginosa: components, mechanisms and clinical signifi-
cance. Curr. Top. Med. Chem. 1, 59–71.
[17] Budzikiewicz, H. (2001) Siderophore-antibiotic conjugates used as
trojan horses against Pseudomonas aeruginosa. Curr. Top. Med.
Chem. 1, 73–82.
[18] Raymond, K.N., Dertz, E.A. and Kim, S.S. (2003) Enterobactin,
an archetype for microbial iron transport. Proc. Natl. Acad. Sci.
USA 100, 3584–3588.
[19] Poole, K., Young, L. and Neshat, S. (1990) Enterobactin-
mediated iron transport in Pseudomonas aeruginosa. J. Bacteriol.
172, 6991–6996.
[20] Ecker, D.J., Matzanke, B.F. and Raymond, K.N. (1986) Recog-
nition and transport of ferrienterobactin in Escherichia coli. J.
Bacteriol. 167, 666–673.
[21] Ochsner, U.A. and Vasil, M.L. (1996) Gene repression by the ferric
uptake regulator in Pseudomonas aeruginosa: cycle selection of
iron-regulated genes. Proc. Natl. Acad. Sci. USA 93, 4409–4414.
[22] Vasil, M.L. and Ochsner, U.A. (1999) The response of Pseudo-
monas aeruginosa to iron: genetics, biochemistry and virulence.
Mol. Microbiol. 34, 399–413.
[23] Ochsner, U.A., Johnson, Z. and Vasil, M.L. (2000) Genetics and
regulation of two distinct haem-uptake systems, phu and has, in
Pseudomonas aeruginosa. Microbiology 146, 185–198.
[24] Heinisch, L., Gebhardt, P., Heidersbach, R., Reissbrodt, R. and
Mollmann, U. (2002) New synthetic catecholate-type sidero-
phores with triamine backbone. BioMetals 15, 133–144.
[25] Heinisch, L., Wittmann, S., Stoiber, T., Scherlitz-Hofmann, I.,
Ankel-Fuchs, D. and Mollmann, U. (2003) Synthesis and
biological activity of tris- and tetrakiscatecholate siderophores
based on poly-aza alkanoic acids or alkylbenzoic acids and their
conjugates with b-lactam antibiotics. Arzneimittelforschung 53,
188–195.
[26] Heinisch, L., Wittmann, S., Stoiber, T., Berg, A., Ankel-Fuchs, D.
and Mollmann, U. (2002) Highly antibacterial active aminoacyl-
penicillin conjugates with bis-catecholate siderophores based on
secondary diamino acids and related compounds. J. Med. Chem.
45, 3032–3040.
[27] Wittmann, S., Scherlitz-Hofmann, I., Mollmann, U., Ankel-
Fuchs, D. and Heinisch, L. (2000) 8-acyloxy-1,3-benzoxazine-2,4-
diones as siderophore components for antibiotics. Arzneimittelf-
orschung/Drug Research 50, 752–757.
[28] Dean, C.R., Neshat, S. and Poole, K. (1996) PfeR, an enterob-
actin-responsive activator of ferrienterobactin receptor gene
expression in Pseudomonas aeruginosa. J. Bacteriol. 178, 5361–
5369.
[29] Rabsch, W., Methner, U., Voigt, W., Tschape, H., Reissbrodt, R.
and Williams, P.H. (2003) Role of receptor proteins for enterob-
actin and 2,3-dihydroxybenzoylserine in virulence of Salmonella
enterica. Infect. Immun. 71, 6953–6961.
[30] May, J.J., Wendrich, T.M. and Marahiel, M.A. (2001) The dhb
operon of Bacillus subtilis encodes the biosynthetic template for
the catecholic siderophore 2,3-dihydroxybenzoate-glycine-threo-
nine trimeric ester bacillibactin. J. Biol. Chem. 276, 7209–7217.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 175–181
Novel target genes of PsrA transcriptional regulatorof Pseudomonas aeruginosa
Milan Kojic a,b,*, Branko Jovcic a, Alessandro Vindigni b,Federico Odreman b, Vittorio Venturi b
a Laboratory for Molecular Genetics of Industrial Microorganisms, Institute of Molecular Genetics and Genetic Engineering, Vojvode Stepe 444a,
11010 Belgrade, Serbia and Montenegrob International Centre for Genetic Engineering and Biotechnology, Area Science Park, Padriciano 99, 34012 Trieste, Italy
Received 23 February 2005; received in revised form 31 March 2005; accepted 4 April 2005
First published online 15 April 2005
Edited by S. Silver
Abstract
The PsrA transcriptional regulator is involved in stationary phase induced transcriptional regulation of rpoS and in negative
auto-regulation in Pseudomonas aeruginosa. This study was designed to determine whether other loci were regulated by PsrA in
P. aeruginosa. Computer search was performed of the PsrA binding motif (G/CAAAC N2–4 GTTTG/C) against the P. aeruginosa
genome sequence. Four of 14 analysed promoters responded to and bound PsrA; (i) divergent promoters controlling PA2952/
PA2951 and PA2953, (ii) promoter of PA0506 and (iii) upstream region of PA3571. Promoters PA0506 and PA2952–PA2951 were
regulated negatively whereas promoters of PA2953 and PA3571 were regulated positively by PsrA. Two dimensional sodium dode-
cyl sulphate polyacrylamide gel electrophoresis (2D SDS-PAGE) analysis on total proteins from P. aeruginosa PAO1 and psrA
knock-out derivative was also performed resulting in the identification of 11 protein spots which were differentially regulated. These
studies have indicated PsrA as a global regulator.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: PsrA regulon; Stationary phase
1. Introduction
In their natural environment, bacteria are often chal-
lenged by constantly changing nutrient availability and
by exposure to various forms of physical stress, including
osmotic, oxidative and temperature shock. Exposure to
starvation and stresses leads to reduction or cessation
of growth, known as stationary phase, resulting in a ma-jor switch of gene expression that allows the cell to cope
with the new conditions [1]. A very simple and effective
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.003
* Corresponding author. Tel.: +381 11 3975 960; fax: +381 11 3975
808.
E-mail address: [email protected] (M. Kojic).
mechanism employed by bacteria to bring about such a
major switch in gene expression is the use of alternative
sigma factors that alter RNA polymerase core specificity
[2]. The central regulator during stationary phase in
Pseudomonas spp., as in other Gram-negative bacteria,
is the stationary phase RpoS alternative sigma factor
[1,3,4]. In Escherichia coli, RpoS regulates more than
100 genes involved in cell survival, cross-protectionagainst various stresses and in virulence [2]. Similarly
in Pseudomonas aeruginosa RpoS regulates a large set
of genes as recently demonstrated using a microarray
transcriptome analysis [5]. The levels of RpoS within
a bacterial cell are carefully controlled and increase
considerably at the onset of stationary phase. The
. Published by Elsevier B.V. All rights reserved.

176 M. Kojic et al. / FEMS Microbiology Letters 246 (2005) 175–181
regulatory mechanisms governing this control have been
extensively studied in E. coli revealing that regulation
takes place at the level of transcription, translation and
post-translational level all responding to various envi-
ronmental stimuli [1]. Regulation has also been studied
to a lesser extent in the fluorescent pseudomonads high-lighting that unlike in E. coli, transcriptional regulation
plays a major role [6]. The global two-component
GacA/GacS system and the N-acyl homoserine lactone
dependent quorum sensing systems are involved in the
regulation of rpoS; the precise mechanisms of these reg-
ulatory controls are unknown and their effect is rather
marginal only mildly affecting rpoS transcription. We
have shown that a TetR family regulator, designatedPsrA, plays a major role in positively regulating rpoS
transcription at the entry of P. aeruginosa into stationary
phase [7,8]. psrA knock-out mutants displayed 90%
reduction in rpoS promoter activity and 50% in protein
levels. DNA-binding studies showed that PsrA binds
specifically to the rpoS promoter at a sequence �35 to
�59 which contains a palindromic motif C/GAAAC
N2–4 GTTTG/C. In addition, PsrA negatively autoregu-lates its own expression through binding to a similar
sequence in its own promoter [8].
In this study, we identified four new genes, involved
in response to stationary phase, regulated by PsrA tran-
scriptional regulator.
2. Materials and methods
2.1. Strains, plasmids, media and chemicals
The strains used in this study included E. coli DH5a[9], E. coli pRK2013 [10] and P. aeruginosa PAO1
(Holloway collection). P. aeruginosa PAO1 and its
psrA and rpoS knock-out mutants have been described
previously [7,11]. E. coli and P. aeruginosa strains weregrown in LB medium [12] at 37 �C. The following anti-
biotic concentrations were used: ampicillin, 100 lg/ml
(E. coli); kanamycin, 100 lg/ml (E. coli) and 300
lg/ml (PAO1); tetracycline, 15 lg/ml (E. coli) and
500 lg/ml (PAO1); gentamicin, 100 lg/ml (PAO1).
The plasmids used in this study are listed in Table A
(Supplementary data). The plasmid transcriptional fu-
sions were constructed as follows. Primers (Table B,Supplementary data) were designed in a way to ampl-
ify promoter regions starting from ATG and ending up
to 700 bp upstream. Amplified DNA fragments from
total genomic PAO1 DNA were treated with BamHI
and KpnI restriction enzymes and cloned in pBlue-
scriptKS (or SK) digested with the same restriction en-
zymes or directly cloned into pBluescriptKS digested
with SmaI, resulting in pBPA constructs (Table A,Supplementary data). pBPA constructs were sequenced
and fragments were then transferred into promoter
probe vector pMP220 using different restriction en-
zymes (BamHI/BglII and KpnI, XbaI and KpnI, EcoRI
and KpnI) to yield pMPA constructs (Table A, Supple-
mentary data).
2.2. Computer analysis
The genome of P. aeruginosa PAO1 ([13] http://
www.pseudomonas.com) was searched with the FIND-
PATTERNS (GCG) program using the PsrA binding
motif (G/CAAAC N2–4 GTTTG/C) derived from align-
ment of PsrA binding sequence in two promoters known
to be regulated by PsrA. However, to investigate the
importance of the spacing between the two motifs andto identify a maximum number of candidate genes, we
purposely expanded the spacing range to 2–4 nt of the
palindromic sequence.
2.3. Preparation of total cell proteins and 2-D gel
electrophoresis
Total cell proteins were prepared from overnightcultures of P. aeruginosa PAO1 and PAO1 psrA::Tn5.
Briefly, 6 mg of wet weight pellet was resuspended in
500 ll of solution [7 M urea, 2 M thiourea, 40 mM
dithiothreitol (DTT), 2% w/v 3-[(3-Cholamidopropyl)-
dimethylammonio]-1-propanesulfonate (CHAPS)] and
sonicated 4 · 15 s on ice. For the 2-D analysis, 20 ll(240 lg of total proteins) of sonicated sample was
mixed with 230 ll of the same solution to which1.5 ll of IPG buffer was added before loading. Two-
dimensional gel electrophoresis was performed using
immobilized pH gradient (pH 3–10 NL, IPG buffers)
13 cm long strips (Amersham Pharmacia Biotech).
Strips were rehydrated with entire protein sample for
2 h at room temperature under dry strip-cover fluid
(Amersham Pharmacia Biotech). Isoelectric focusing
was conducted using IPGphor Isoelectric Focusing Sys-tem (Amersham Pharmacia Biotech) at 20 �C. Proteinswere focused for 2 h at 1 kV, 5 h at 5 kV, 3 h at 1 kV,
for a total of 30 kV. IPG strips were equilibrated in
50 mM Tris–HCl pH 8.8, 6 M urea, 30% v/v glycerol,
2% w/v SDS and 15 mM DTT for 20 min at room tem-
perature. Strips were embedded on top of 15 · 15 cm,
12.5% SDS–PAGE gel for the second dimension.
Broad Range Prestained Protein Marker (6–175 kDa)purchased from BioLabs was loaded on the same gel
at one end of the strip. Protein spots were visualized
by Coomassie staining.
2.4. Mass spectrometric sequencing and protein
identification
The selected protein spots were cut out from the Coo-massie Brilliant blue-stained gels and placed in a silicon-
ized microcentrifuge tubes that had been rinsed with

M. Kojic et al. / FEMS Microbiology Letters 246 (2005) 175–181 177
ethanol, water and ethanol. An internal sequence analy-
sis of the protein spots was performed by using an elec-
tronspray ionization mass spectrometer (LCQ DECA
XP, ThermoFinnigam). The bands were digested with
trypsin, and the resulting peptides were extracted with
water and 60% acetonitrile–1% trifluoroacetic acid.The fragments were then analyzed by mass spectros-
copy, and the proteins were identified by analysis of
the peptides and by using the annotated P. aeruginosa
genome (www.pseudomonas.com).
2.5. Electrophoretic mobility shift
DNA mobility shift assays with purified His6-PsrAwere performed with modified previously described pro-
cedure [8]. Fragments carrying the promoters of genes
coding for acyl CoA dehydrogenase, electron transfer
protein, MmsR and MFS transporter were purified from
the plasmid constructs with BamHI–KpnI restriction en-
zymes. Purified DNA (0.1 pmol) was labelled at its Bam-
HI site with the Klenow fragment of DNA polymerase
and [a-32P]dCTP. Radiolebeled fragments (1000 cpm)and various quantities of purified His6-PsrA (PsrA pro-
tein with six histidine residues at N terminus) (from 0 to
150 ng) were incubated for 30 min at room temperature
in 10 ll reaction mixtures containing 1 · binding buffer
(20 mM HEPES-KOH pH 7.9, 20% v/v glycerol,
0.2 mM ethylendiaminetetraacetic acid disodium salt
(EDTA); 0.1 M KCl, 0.5 mM phenylmethanesulphonyl
fluoride (PMSF), 1 mM DTT), 10 lg of bovine serumalbumin (carrier protein), 400 ng of salmon sperm
(non-specific competitor) DNA and 1.5 mM MgCl2.
Supershifting was performed by incubating the reaction
mixtures with anti-PsrA antibodies for an additional
15 min at room temperature. Samples were than loaded
onto a non-denaturing 4.5% polyacrylamide 0.5 · TBE
(44.5 mM Tris, 44.5 mM boric acid, 0.5 mM EDTA)
�3% v/v glycerol gel, which was prerun for 1 h at110 V at room temperature, the samples were also run
at 110 V.
2.6. Recombinant DNA techniques
Digestion with restriction enzymes, agarose gel elec-
trophoresis, purification of DNA fragments, ligation
with T4 DNA ligase, end filling with the Klenow frag-ment of DNA polymerase, transformation of E. coli
and SDS–PAGE analysis were performed as previously
described [14]. Analytical amounts of plasmids were iso-
lated by procedure described by Birnboim [15], whereas
preparative amounts were purified with Qiagen columns
(Qiagen, Hilden, D). Total DNA from Pseudomonas was
isolated by Sarkosyl-pronase lysis [16]. Triparenatal
matings from E. coli to Pseudomonas were performedwith an E. coli (pRK2013) helper strain as previously
described [10].
2.7. Reporter gene fusion assays
Transcriptional fusions of all promoters possibly reg-
ulated by PsrA were made using pMP220 which harbors
a promoterless b-galactosidase lacZ gene. b-galactosi-dase activity was determined by method essentially de-scribed by Miller [12] with the modifications of Stachel
[17]. Miller units were defined as OD420 · 1000/
OD600 · T(min) · V(ml). All measurements were done
in triplicate and the mean value is given.
3. Results
3.1. Genomic analysis of the P. aeruginosa PAO1
chromosome for sequences representing potential PsrA
binding promoters
Having previously established that the TetR family
regulator PsrA was an important positive transcrip-
tional regulator of rpoS and a negative auto-regulator
via specific DNA-binding to a region of rpoS and psrA
promoters [6–8], it was of interest to determine whether
PsrA was transcriptionally regulating other loci in the P.
aeruginosa genome. The P. aeruginosa PAO1 genome
was therefore subjected to a degenerate pattern search
using the PsrA binding consensus sequences. The subse-
quence used to search the P. aeruginosa genome was
SAAAC N2–4 GTTTS where S was C or G and 2–4
was the spacing between the two palindromic motifs.This search resulted in the identification of the previ-
ously reported psrA and rpoS binding sites and in 16
new possible PsrA-binding sites distributed randomly
on the chromosome of P. aeruginosa PAO1 (Table 1).
Regions including 600 bp downstream of the potential
PsrA binding sites were examined for the presence of
open reading frames (ORFs) (Fig. 1 and Table 1). Fig.
1 illustrates the specific region in the chromosome wherethese putative binding sites were located with respect to
which ORF and Table 1 shows the precise location of
the putative binding site, the putative DNA-binding se-
quence and the possible downstream ORF that this
PsrA-site might be regulating.
3.2. Gene expression analysis of putative PsrA regulated
promoters identified by comparative genome analysis
In order to determine whether the putative PsrA
binding sites identified using a comparative genome
analysis search (see above) represented transcriptionally
regulated PsrA-dependent promoters, we tested 14 of
them by cloning with adjacent DNA into the lacZ
wide-host range pMP220 promoter probe vector. These
14 putative binding sites were located in or near inter-genic regions and what was believed to be a complete
promoter of a putative ORF. Of these, four putative

Table 1
Predicted binding sites for PsrA in the Pseudomonas aeruginosa PAO1 genome and b-galactosidase activity of these promoters
Binding site PA number of
downstream
genes
Position on PAO1
chromosome
Gene/protein Distance from
ATG
b-galactosidase activity (MU)
WT psrA
mutant
rpoS
mutant
Fold
change
GAAAC CC GTTTC PA0413 453497–453508 pilL 618 1282 1368 1315 NS
CAAAC GCCT GTTTG PA0506 564778–564791 Acyl CoA dehydrogenase 123 4305 13,545 3912 3.15
GAAAC TGAA GTTTC PA0806 883112–883125 Hypothetical 652 2965 2830 3201 NS
GAAAC GTAT GTTTC PA1394 1515759–1515772 Hypothetical 661 2130 2270 2050 NS
GAAAC CG GTTTC PA2258 2487773–2487784 ptxR 491 1069 1171 1120 NS
GAAAC CG GTTTC PA2259 2487784–2487773 ptxS 72 1030 1213 1153 NS
GAAAC CG GTTTC PA2260 2488926–2488937 Hypothetical 13 1224 1228 1280 NS
CAAAC TCC GTTTG PA2673 3021019–3021031 hplV 91 1256 1320 1304 NS
CAAAC AAAC GTTTG PA2952 3312671–3312684 etfB 202 4800 9940 4950 2.1
CAAAC GTTT GTTTG PA2953 3312684–3312671 Electron transfer
flavoprotein–ubiquinone
oxidoreductase
106 6650 5520 6470 0.83
GAAAC GTAT GTTTC PA3006 3367686–3367699 psrA 16 3890 24,378 4015 6.3
CAAAC ACTT GTTTG 3367699–3367712
GAAAC CGGG GTTTC PA3571 4003410–4003423 mmsR 309 3230 2357 3206 0.72
GAAAC CAGC GTTTC PA3595 4029672–4029685 MFS transporter 73 1268 1231 1259 NS
CAAAC TTCC GTTTG PA3622 4059323–4059336 rpoS 411 28,789 4190 27,630 0.15
GAAAC GCCC GTTTC PA4420 4955753–4955766 Hypothetical 183 2465 2716 2640 NS
GAAAC CG GTTTC PA4963 5572071–5572082 Hypothetical 143 3933 3753 3345 NS
NS – not significant, fold change – ratio of promoter activities (MU) in psrA mutant versus WT.
PA0505
PA0506 PA0507
PsrA-BS
123bp
PA0506
PsrA-BS
mmsRmmsA
309bp
mmsB
PA2952PA2953
PA3006PA3007
PA3571
PA3595
PA3622
PsrA-BS73bp
PA3594 PA3595 PA3596
PsrA-BS
rpoS
411bp
nlpD
fdxA
pcm
lexA
PsrA-BS
psrA
16bp 191bp
PA2953
PsrA-BS
etfB
106bp202bp
etfA
Fig. 1. Location of the putative PsrA binding sites for six promoters
regulated by PsrA and for MFS transporter in the P. aeruginosa PAO1
genome (for more details see Table 1). These sites were found using a
degenerate pattern search against the PAO1 genome. The position of
the putative binding site is given as well as its distance to the nearest
translation start codon of an annotated ORF. The PA number refers
to the possible ORF that PsrA might be transcriptionally regulating
(see text for further details). PsrA-BS – PsrA binding site.
178 M. Kojic et al. / FEMS Microbiology Letters 246 (2005) 175–181
binding sites were not tested (PA0099, PA1318, PA5372,
PA5451) since they were very distant from the annotated
translational start codons and were not in an intergenic
region and thus were most probably not located in puta-
tive gene promoters. Of the 14 tested putative gene pro-
moters, 4 were shown to be regulated by PsrA since
transcriptional fusions were behaving in a PsrA depen-
dent manner (Table 1). These were the promoter ofgenes PA0506 encoding a probable acyl-CoA dehydro-
genase, of operon PA2952–PA2951 encoding an elec-
tron transfer flavoprotein b-subunit and a subunit
respectively, of PA2953 encoding an electron transfer
flavoprotein–ubiquinone oxidoreductase and of
PA3571 encoding the transcriptional regulator MmsR.
Two of the promoters (PA0506 and PA2952–PA2951)
were regulated negatively whereas promoters ofPA2953 and PA3571 were regulated positively by PsrA.
3.3. Identification of PsrA regulated proteins
In order to characterise PsrA regulated genes more
fully in P. aeruginosa PAO1 we compared the protein
expression pattern in stationary phase of the wild type
strain PAO1 versus the PAO1psrA::Tn5 mutant bytwo dimensional (2D) SDS–PAGE gel electrophoresis.
Total protein extracts and analysis was performed in
triplicate as described in Section 2. The 2D, analysis, re-
vealed differences in protein levels between PAO1 and
PAO1 psrA::Tn5 mutant in all three experiments in 11
protein spots (Fig. 2). These 11 protein spots were se-
lected for further analysis; proteins present in spots 1,
2, 3, 5, 6, 7, 8, 9 and 10 (electron transfer flavoprotein

Fig. 2. Comparative 2-D gel analysis of total proteins of P. aeruginosa PAO1 (panel A) and P. aeruginosa PAO1 psrA::Tn5 (panel B). Numbers of
encircled protein spots refer to those represented in Table 2. kDa, kilo Daltons.
M. Kojic et al. / FEMS Microbiology Letters 246 (2005) 175–181 179
b-subunit; acyl-CoA-dehydrogenase; neomycin-kana-
mycin phosphotransferase from transposon Tn5; fatty
acid oxidation complex b-subunit; acyl-CoA-dehydroge-
nase; isocitrate dehydrogenase and elongation factor Tu;
DnaK protein; fatty acid oxidation complex a-subunitand GroEL, respectively) were over-expressed in
PAO1 psrA::Tn5 mutant, in contrast spots 4 (caraba-
mate kinase) and 11 (conserved hypothetical protein)
were more expressed in P. aeruginosa PAO1. Peptide
mass fingerprinting of tryptic digested fragments was
performed on all the 11 protein spots. Each protein spot
resulted in the identification of one protein with the only
exception of spot 7 which represented two proteins (iso-citrate dehydrogenase and elongation factor Tu). Pro-
tein spots numbered 2 and 6 contained the same
protein, annotated as PA0506, an acyl-CoA dehydroge-
nase of the same nominal mass of 66 kDa, but different
pI value, 5.62 (which correspond to calculated pI value
from aminoacid sequence) for spot 2 and about 4 for
spot 6. The difference in pI value could be the result of
post-translational modifications. The encoding genefor PA0506 contained a functional PsrA binding site
in its gene promoter as previously demonstrated (see
above). Spot number 3, present only in the PAO1
psrA::Tn5 mutant, was the neomycin–kanamycin phos-
photransferase from transposon Tn5. Protein spot num-
ber 1 represented protein PA2952 encoding an electron
Fig. 3. Retardation of the movement of a DNA fragment containing
dehydrogenase (BamHI-KpnI fragment of 429 bp) [A] electron transfer fl
oxidoreductase (BamHI-KpnI fragment of 377 bp) [B], mmsR BamHI-Kpn
fragment of 251 bp) [D] by purified PsrA protein. The amounts of PsrA prot
was used with anti-PsrA antibodies (lane 5). A 100-fold excess amount of the
added, except for mmsR promoter (100-fold excess amount of the same un
DNA, lane 6 and 7, respectively).
transfer flavoprotein b-subunit of which the gene, etfB,
contained a functional PsrA binding site and was shown
to be regulated by PsrA (see above). Interestingly, spots
5 and 9 were proteins PA3013 and PA3014 encoded by
faoA and faoB which are organized in an operon in-volved in fatty acid metabolism. The promoters of all
the genes encoding for the identified proteins in this
analysis were cloned in the lacZ promoter probe vector
pMP220 (as described in Section 2) and the expression
was determined in strain PAO1, PAO1psrA::Tn5 and
PAO1rpoS::Tn5. The b-galactosidase activities as ex-
pected for the two promoters previously identified using
a comparative genome search for PsrA binding sites (seeabove) display PsrA dependent expression. All other
gene promoter activities were comparable when ob-
tained in strain PAO1 and the psrA knock-out mutant.
The promoter activities were also tested in PAO1-
rpoS::Tn5 as PsrA is a positive transcriptional regulator
of rpoS; all promoters displayed comparable activities in
PAO1rpoS::Tn5 when compared to wild type PAO1.
3.4. Protein–DNA binding studies of PsrA regulated
promoters
In order to establish whether the identified PsrA-reg-
ulated promoters could bind PsrA, mobility shift assays
with the (i) etfBA promoter, PA2592/2591 (ii) the pro-
promoters (composed of complete intergenic region) of acyl CoA
avoprotein b-subunit and electron transfer flavoprotein-ubiquinone
I fragment of 417 bp) [C] and MFS transporter gene (BamHI-KpnI
ein used were 0, 50, 100 and 150 ng (lines 1–4, respectively) and 150 ng
same (lane 6) and psrA promoter (lane 7) unlabeled DNA fragment was
labeled DNA and 10-fold excess amount of unlabeled psrA promoter

180 M. Kojic et al. / FEMS Microbiology Letters 246 (2005) 175–181
moter of PA0506 (a probable acyl-CoA dehydrogenase),
and (iii) the mmsR promoter (PA3571), were performed.
As a control experiment the promoter of PA3595
(encoding a probable major facilitator superfamily,
MFS, transporter) was also used since it contained a
putative PsrA binding region however transcriptionalstudies showed that PsrA had no effect on its transcrip-
tion (Table 1). The three promoters which displayed
PsrA dependent expression were retarded and thus
shown to bind PsrA, and the shift was not observed in
the presence of excess unlabeled fragment (Fig. 3). A
supershift was detected in the presence of anti-PsrA
antibodies (Fig. 3). These results confirmed that these
gene promoters are regulated by PsrA. The promoterof PA3595 showed no retardation (Fig. 3) confirming
the transcriptional fusion data that PsrA was not in-
volved in its regulation.
4. Discussion
In this study, several new loci have been found whichare regulated at the transcriptional level by the TetR
family regulator PsrA of P. aeruginosa. PsrA has been
originally identified as a positive transcriptional regula-
tor of the stationary phase rpoS sigma factor, activating
transcription at the onset of stationary phase [7]. In
addition it was demonstrated that PsrA acts as a strong
negative autoregulator and the binding site in rpoS and
psrA promoters has been determined and was shown tobe well conserved [8]. Searching for the PsrA binding
motif in the P. aeruginosa genome revealed 18 putative
binding sites (Table 1 and Fig. 1), however only 4 of
the 14 tested were responding and could bind to PsrA
as determined with transcriptional fusions and pro-
tein–DNA gel retardation assays (Fig. 1, Fig. 3 and Ta-
ble 1). The search for the PsrA binding site was
performed using the consensus, SAAAC N2–4 GTTTS,it cannot be excluded that PsrA can bind to variants
of this sequence and therefore using this genome search
we did not find other functional PsrA binding sites.
Alternatively, of the 10 gene promoters tested which
contained a putative PsrA binding site but did not dis-
play any PsrA dependence, it cannot be excluded that
in some other environmental/growth condition these
promoters could become PsrA-dependent. ComparingPsrA binding motifs of promoters confirmed to be regu-
lated with PsrA indicate that the functional binding site
was C/GAAAC N4 GTTTG/C and that spacing of four
nucleotides was important between the two conserved
palindromic motifs. Interestingly, the promoter of
PA3595, which encodes a major facilitator superfamily
(MFS) transporter, contained a perfect GAAAC N4
GTTTC consensus, however we found that it was notregulated and does not bind PsrA in vitro (Table 1,
Fig. 3). It could be possible that other sequences are re-
quired outside this palindrome or possibly other factors
are required for PsrA recognition in certain gene pro-
moters. In summary, we have identified four new loci
which are directly regulated by PsrA in addition to the
already known rpoS and psrA promoters. All these pro-
moters have been shown to be able to bind PsrA andhave a very well conserved palindromic DNA sequence.
In order to identify other PsrA regulated loci, we also
performed total 2-D protein analysis of P. aeruginosa
versus P. aeruginosa psrA::Tn5 and could identify 11
protein spots, out of approximately 300, which were dif-
ferentially regulated in psrA::Tn5 mutant; two spots
were more expressed, eight were less and one was not
detectable in PAO1 wild type comparing to PAO1psrA::Tn5 (Fig. 2). The fact that RpoS and PsrA were
not identified using this approach indicates that there
are probably more proteins which are differentially ex-
pressed and were not detected here under these experi-
mental conditions. Interestingly however, three spots
represented proteins of which the encoding gene had a
PsrA-binding site as found in the comparative genome
search and as demonstrated with transcriptional fusionstudies and DNA-binding assays (see above). One of
these, PA0506 encoding an acyl-CoA dehydrogenase,
was detected twice probably due to having different pI
values possibly because of post-translational modifica-
tions. Of the remaining proteins which were differen-
tially expressed, the gene promoter was tested for PsrA
dependent transcriptional expression. Surprisingly all
promoters, with the exception of PA0506 and PA2592which contained a PsrA binding site, did not display
PsrA dependent transcription in stationary phase in P.
aeruginosa. The reason for this is not known, however
the fact that these protein spots were observed to be dif-
ferentially regulated in three independent experiments.
It could be that PsrA affected the levels of some of these
proteins through post-transcriptional and/or post-trans-
lational levels of control either directly and/or indirectly.PsrA has been shown to regulate rpoS expression in re-
sponse to stationary phase [6]. A stress encountered by
bacteria in stationary phase is starvation for energy-
yielding carbon source resulting in the induction of the
starvation-stress response [18,19]. Upon induction of
this response, numerous structural and physiological
changes in the cellular envelope occur in starved cells
of Gram-negative enteric bacteria. These include in-creased lipopolysaccharide in the outer membrane, a
shift from phosphatidylglycerol to diphosphatidylglyc-
erol in the inner membrane, decrease in the relative
amounts of long-chain monounsaturated fatty acid
and increased thickness and cross-linking of the peptido-
glycan as well as expanded attachment of the murein
layer to the outer membrane [20]. Degradation of these
fatty acids through b-oxidation, mediated by acyl-CoA-dehydrogenases, would generate acetyl-CoA to feed the
tricarboxylic acid (TCA) cycle, yielding C-compound

M. Kojic et al. / FEMS Microbiology Letters 246 (2005) 175–181 181
intermediates and electron/H+ ion donors for energy
production. This enzyme also catalyses a,b-dehydroge-nation of acyl-CoA esters and transfers electrons to
an electron transfer flavoprotein via the same mecha-
nism. The acyl-CoA-dehydrogenases (PA0506), the elec-
tron transfer flavoprotein (PA2951/PA2952) andelectron transfer flavoprotein-ubiquinone oxidoreduc-
tase (PA2953) were shown here to be all regulated by
PsrA in response to stationary phase and could there-
fore be part of the same cascade in this process in P.
aeruginosa linking up these gene products for the first
time.
In summary, we have identified new loci regulated by
the TetR family regulator PsrA, 4 of which have a func-tional PsrA box in their gene promoter. PsrA could
therefore play an important role in the adaptation to
stationary phase.
Acknowledgements
We are grateful to Rodolfo Garcia Carlos for help inpreparing 2-D SDS–PAGE electrophoresis and for
offering us the use of his laboratory facilities. We would
also like to thank Kristian Vlahovicek for computer
assistance. This work was funded by the ICGEB Collab-
orative Research Program, grant CRP/YUG02-01, and
partially supported by Ministry for Science and Envi-
ronmental Protection of Serbia, grant No. 1442.
Appendix A. Supplementary data
Supplementary data associated with this article can
be found, in the online version at doi:10.1016/j.fem-
sle.2005.04.003 [21].
References
[1] Hengge-Aronis, R. (2002) Signal transduction and regulatory
mechanisms involved in control of the sigma(S) (RpoS)
subunit of RNA polymerase. Microbiol. Mol. Biol. Rev. 66,
373–395.
[2] Ishihama, A. (2000) Functional modulation of Escherichia coli
RNA polymerase. Annu. Rev. Microbiol. 54, 499–518.
[3] Jorgensen, F., Bally, M., Chapon-Herve, V., Michel, G., Laz-
dunski, A., Williams, P. and Stewart, G.S. (1999) RpoS-depen-
dent stress tolerance in Pseudomonas aeruginosa. Microbiology
145, 835–844.
[4] Suh, S.J., Silo-Suh, L., Woods, D.E., Hassett, D.J., West, S.E.
and Ohman, D.E. (1999) Effect of rpoS mutation on the stress
response and expression of virulence factors in Pseudomonas
aeruginosa. J. Bacteriol. 181, 3890–3897.
[5] Schuster, M., Hawkins, A.C., Harwood, C.S. and Greenberg,
E.P. (2004) The Pseudomonas aeruginosa RpoS regulon and its
relationship to quorum sensing. Mol. Microbiol. 51, 973–985.
[6] Venturi, V. (2003) Control of rpoS transcription in Escherichia
coli and Pseudomonas: why so different?. Mol. Microbiol. 49, 1–9.
[7] Kojic, M. and Venturi, V. (2001) Regulation of rpoS gene
expression in Pseudomonas: involvement of a TetR family
regulator. J. Bacteriol. 183, 3712–3720.
[8] Kojic, M., Aguilar, C. and Venturi, V. (2002) TetR family
member psrA directly binds the Pseudomonas rpoS and psrA
promoters. J. Bacteriol. 184, 2324–2330.
[9] Hanahan, D. (1983) Studies on transformation of Escherichia coli
with plasmids. J. Mol. Biol. 166, 557–580.
[10] Figurski, D.H. and Helinski, D.R. (1979) Replication of an
origin-containing derivative of plasmid RK2 dependent on a
plasmid function provided in trans. Proc. Natl. Acad. Sci. USA
76, 1648–1652.
[11] Whiteley, M., Parsek, M.R. and Greenberg, E.P. (2000) Regula-
tion of quorum sensing by RpoS in Pseudomonas aeruginosa. J.
Bacteriol. 182, 4356–4360.
[12] Miller, J.H. (1972) Experiments in Molecular Genetics. Cold
Spring Harbor Laboratory, Cold Spring Harbor, NY.
[13] Stover, C.K., Pham, X.Q., Erwin, A.L., Mizoguchi, S.D.,
Warrener, P., Hickey, M.J., Brinkman, F.S., Hufnagle, W.O.,
Kowalik, D.J., Lagrou, M., Garber, R.L., Goltry, L., Tolentino,
E., Westbrock-Wadman, S., Yuan, Y., Brody, L.L., Coulter,
S.N., Folger, K.R., Kas, A., Larbig, K., Lim, R., Smith, K.,
Spencer, D., Wong, G.K., Wu, Z., Paulsen, I.T., Reizer, J., Saier,
M.H., Hancock, R.E., Lory, S. and Olson, M.V. (2000) Complete
genome sequence of Pseudomonas aeruginosa PA01, an opportu-
nistic pathogen. Nature 406, 959–964.
[14] Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular
Cloning: A Laboratory Manual, 2nd ed. Cold Spring Harbor
Laboratory, Cold Spring Harbor, NY.
[15] Birnboim, H.C. (1983) A rapid alkaline extraction method for the
isolation of plasmid DNA. Meth. Enzymol. 100, 243–255.
[16] Better, M., Lewis, B., Corbin, D., Ditta, G. and Helinski, D.R.
(1983) Structural relationships among Rhizobium meliloti symbi-
otic promoters. Cell 35, 479–485.
[17] Stachel, S.E., An, G., Flores, C. and Nester, E.W. (1985) A Tn3
lacZ transposon for the random generation of b-galactosidasegene fusions: application to the analysis of gene expression in
Agrobacterium. EMBO J. 4, 891–898.
[18] Spector, M.P., DiRusso, C.C., Pallen, M.J., Garcia del Portillo,
F., Dougan, G. and Finlay, B.B. (1999) The medium-/long-chain
fatty acyl-CoA dehydrogenase (fadF) gene of Salmonella typhimu-
rium is a phase 1 starvation-stress response (SSR) locus. Micro-
biology 145 (Pt 1), 15–31.
[19] Spector, M.P., Garcia del Portillo, F., Bearson, S.M., Mahmud,
A., Magut, M., Finlay, B.B., Dougan, G., Foster, J.W. and
Pallen, M.J. (1999) The rpoS-dependent starvation-stress response
locus stiA encodes a nitrate reductase (narZYWV) required for
carbon-starvation-inducible thermotolerance and acid tolerance
in Salmonella typhimurium. Microbiology 145 (Pt 11), 3035–3045.
[20] Huisman, G.W., Siegele, D.A., Zambrano, M.M. and Kolter, R.
(1996)Morphological and physiological changes during stationary
phase in Escherichia coli and Salmonella. In: Cellular and
Molecular Biology (Neidhart, F.C., et al., Eds.), 2nd ed., pp.
1672–1682. American Society for Microbiology, Washington, DC.
[21] Spaink, H.P., Okker, R.J.H., Wijffelmann, C.A., Pees, E. and
Lugtemberg, B.J.J. (1987) Promoter in the nodulation region of
the Rhizobium leguminosarum Sym plasmid pRL1JI. Plant. Mol.
Biol. 9, 27–39.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 183–190
Isolation and characterisation of the lipopolysaccharide fromAcidiphilium strain GS18h/ATCC55963, a soil isolate
of Indian copper mine
Rabindranath Bera a, Abhijit Nayak c, Asish Kumar Sen b,Biswa Pronab Chowdhury b, Ranjan Bhadra a,*
a Department of Cellular Biochemistry, Indian Institute of Chemical Biology, 4, Raja S.C. Mullick Road, Kolkata 700 032, Indiab Department of Organic Chemistry, Indian Institute of Chemical Biology, 4, Raja S.C. Mullick Road, Kolkata 700 032, India
c Division of Health and Education for Disabled, Pandit Sunderlal Sharma Central Institute of Vocational Education, Bhopal 46201, India
Received 28 February 2005; accepted 4 April 2005
First published online 15 April 2005
Edited by K. Hantke
Abstract
The lipopolysaccharide (LPS) of the Gram-negative Acidiphilium strain GS18h/ATCC55963, a new soil isolate, exhibited
very low endotoxic activity as determined by Limulus gelation activity, lethal toxicity in galactosamine (GalN) sensitised mice,
and level of tumor necrosis factor alpha (TNFa) in the blood serum of BALB/c mice. Analysis of the LPS, specially of lipid
A which usually accounts for the toxicity, revealed the latter to contain glucosamine and phosphate besides fatty acids, of
which 14:0(3-OH), 18:0(3-OH), 18:1 and 19:0(cyclo) are the major components, while 12:0, 16:0, 19:1, 20:0(3-OH) and
20:1(3-OH) are present in small amounts. The 14:0(3-OH) and 18:0(3-OH) fatty acids are amide-linked, whereas the rest
are ester bound. Glucose, galactose, mannose, rhamnose, heptose, galacturonic acid and 3-deoxy-D-manno-oct-2-ulosonic acid
(Kdo) were present in the polysaccharide part of this LPS. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–
PAGE) of the LPS showed a macromolecular heterogeneity distinctly different from those of Escherichia coli or Salmonella.
The toxicity of this LPS being extremely low attributed to fatty acid composition of its lipid A, promises potential therapeutic
application.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Acidiphilium ATCC55963; Lipopolysaccharide; Lipid A; Lethal toxicity
1. Introduction
As a major constituent of the outer leaflet of the cell
wall lipid bilayer [1], Gram-negative bacterial LPS vary
widely in composition as reflected in their multiple sero-
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.001
* Corresponding author. Fax: +91 33 2473 5197/0284.
E-mail address: [email protected] (R. Bhadra).
type [2,3]. The hydrophilic polysaccharide and lipophiliclipid A moieties are the components of the LPS mole-
cule. Lipid A expresses numerous biological activities
such as antitumor activity as also protection against
X-irradiation and bacterial infection in higher verte-
brates [4], but it is not acceptable clinically since it acts
as an agent for endotoxic shock induced death, a human
health problem yet to be solved. The toxicity of lipid A
depends on the type of hexosamine present, the degree
. Published by Elsevier B.V. All rights reserved.

184 R. Bera et al. / FEMS Microbiology Letters 246 (2005) 183–190
of phosphorylation, the presence of phosphate substitu-
ents and, most notably, the chemical structure, chain
length as well as the number and location of fatty acyl
groups [5,6]. Purified lipid A reported from few bacterial
sources [7,8] has very little toxicity and has notably dif-
ferent fatty acid composition (in respect of chain length,type of linkage, unsaturation and fatty acid hydroxyl-
ation) as also the degree of amino sugar phosphoryla-
tion. So lipid A may exist as a toxic or a non-toxic
entity depending on the constituents present.
Studies on the compositional analysis of the LPS
from Acidiphilium species [9–11] are limited. These spe-
cies have been suggested to be related to the acidophilic
Thiobacillus, the LPS of which is known to be non-toxicor weakly toxic [12]. The biological activity of Acidiphi-
lium LPS therefore may have interesting feature and va-
lue like the LPS of Thiobacillus. It is significant that the
LPS of the photobacteria Rhodobacter capsulatus and
Rhodopseudomonas sphaeroides were found to be non-
toxic and antagonistic to toxic LPS [12], which led to
the development of a preventive for endotoxemia [13].
We were interested by the finding that a new acidophilicGram-negative isolate, placed phylogenetically along
with Acidiphilium cryptum and Acidiphilium symbioticum
[14], has been characterised from the copper mine area
of Ghatshila, India, and submitted to the patent depos-
itory of ATCC and also to the national depository at
IMTECH, Chandigarh, (India) (designated as Acidiphi-
lium ATCC55963 and Acidiphilium strain GS18h,
respectively). It was therefore important to investigatethe LPS from this strain, described as Acidiphilium
ATCC55963 throughout this study. This LPS was found
to be nearly devoid of toxicity as described in a patent
[15] giving some preliminary observations. In this study
we have tested this LPS in vitro for its endotoxic activity
and studied for its biological activity in murine model,
which may be indicative of its clinical usefulness. To find
an explanation for its very low toxicity, the detailedcompositional analysis of this LPS was made by deter-
mining the constituent sugars and characterising the
chemical nature and linkage of the fatty acids present.
2. Materials and methods
2.1. Bacterial strain and culture condition
In a typical culture, Acidiphilium ATCC55963 was
grown in a complex medium containing (NH4)2SO4
(15 mM), MgSO4 (2 mM), K2HPO4 (1.4 mM), KCl
(1.3 mM), glucose (5.5 mM) and yeast extract (0.01%),
under aerobic condition at pH 3 (adjusted with 1 N
H2SO4) and at 30 �C under constant stirring for 72 h.
The bacteria were then harvested by centrifugation at6000g (4 �C, 30 min) and washed thrice with the same
culture medium without glucose and yeast extract.
2.2. Extraction of the lipopolysaccharide and preparation
of lipid A
The LPS was extracted from wet cells instead of dry
cells by a conventional hot phenol–water method
[16,17]. The lyophilised crude LPS was suspended inpyrogen free water (2% w/v) to form a fine suspension
of LPS, and subjected to repeated (thrice) ultra centrifu-
gation at 105,000g (at 4 �C for 4 h) for the removal of
RNA and DNA. Finally the pellet was suspended in
pyrogen free water and treated with chilled 90% ethanol
(4 times the volume of the suspension) and kept at 4 �Cfor 24 h to get the LPS as a precipitate, which was col-
lected by centrifugation (5000g, at 4 �C for 10 min). Thiswas further purified to remove the endotoxin protein by
the ‘‘phenol re-extracted LPS’’ procedure by treating
with triethylamine, phenol, and deoxycholate (DOC)
as per a reported method [18]. The absorbance of the
LPS solution was measured at 260 nm to determine
the presence of any contaminating nucleic acid. To re-
move the phospholipids, the LPS was washed thrice
with chloroform:methanol:water (16:8:1).Lipid A was released from the pure LPS by hydroly-
sis in aqueous 1% acetic acid at 100 �C for 90 min [19].
The precipitated lipid A was recovered by centrifugation
(5000g, 30 min) and washed thrice with hot water. The
supernatant of the hydrolysate (degraded oligosaccha-
ride) was concentrated and freeze-dried. The precipi-
tated material was then lyophilised and further
purified by an established procedure [20]. Briefly, thecrude lipid was suspended in a two-phase solvent chlo-
roform/methanol/water (10:5:6) and centrifuged. The
lower organic layer was recovered, filtered using ultra-
free-filter vessels with Durapore membrane (pore diam-
eter 0.45 lm, Millipore), and evaporated to dryness.
This material was used for fatty acid analysis.
2.3. Sodium dodecyl sulfate polyacrylamide gel
electrophoresis
Sodium dodecyl sulfate polyacrylamide gel electro-
phoresis (SDS–PAGE) was performed by incorporating
4 M urea in a 13% separating gel, containing 0.1% (w/v)
SDS with the buffer system of Laemmli [21]. Electropho-
resis was conducted with a constant current of 35 mA
for 5 h. After SDS–PAGE, the gel was fixed and thenoxidised with periodate; LPS bands were visualised by
silver staining method as described previously [22].
2.4. Lethal toxicity of LPS in galactosamine-treated mice
Lethal toxicity test was performed in galactosamine-
sensitised mice as described previously [23]. Briefly, a
group of six BALB/c mice, 8–12 weeks old and weighing20 ± 2 g, were injected intraperitoneally with a mixture
of D-galactosamine (GalN; 18 mg/mouse; Sigma) and

R. Bera et al. / FEMS Microbiology Letters 246 (2005) 183–190 185
various concentrations of LPS in 0.2 ml pyrogen free
saline. Another group containing six naıve mice, which
received only 0.2 ml pyrogen free saline without any
LPS, was used as control. The survivability of the mice
was recorded up to a period of 72 h after the administra-
tion of LPS, and the lethal toxicity was expressed as100% loss of survivability.
2.5. Limulus amebocyte lysate test
The pyrogenicity of Acidiphilium ATCC55963 LPS
was examined by studying its gel forming activity with
Limulus amebocyte lysate (Endosafen Charleston,
USA) and the test was performed as previously de-scribed [24]. Briefly, the LPS solution and the reference
positive standard (E. coli serotype 0111:B4, Sigma) were
diluted serially with pyrogen free water; heparin (Sigma)
was used as negative control. The test solution was then
added to an equal volume of the lysate and the mixture
incubated at 37 �C for 1 h according to the manufac-
turer�s instructions. Formation of a gel or flocculation
in the tube indicated a positive result.
2.6. Measurement of TNFa levels in serum of LPS treated
mice
Female BALB/c mice (8–12 weeks old, weighing
20 ± 2 g) were subjected to i.p. injection with different
doses of E. coli (serotype 0111:B4) and Acidiphilium
ATCC55963 LPS in a total of 0.2 ml solution in pyrogenfree saline, taking a group of 5 mice for each dose. An-
other group of 5 naıve mice, which received only 0.2 ml
pyrogen free saline without any LPS, was used as con-
trol. All the mice from each group were sacrificed and
bled by cardiac puncture 90 min after treatment. The
blood samples pooled from each group were allowed
to clot for collecting the serum. The serum was centri-
fuged (5000g for 5 min) and collected to store at�70 �C until use. The immunoactive TNFa in serum
samples were analysed with a commercial ELISA kit
(Quantikine, R&D Systems, Minneapolis, MN, USA).
2.7. Compositional analysis
Neutral sugars were liberated from purified LPS and
polysaccharide by hydrolysis with 2 M trifluroaceticacid at 120 �C for 2 h. The hydrolysate was evaporated
to dryness, then reduced with NaBH4 and acetylated
with acetic anhydride:pyridine (1:1, v/v) at room tem-
perature for 12 h or by heating on a boiling water bath
for 45 min. The resulting alditol acetate derivatives [25]
were analysed by gas liquid chromatography (GLC)
and GLC–mass spectrometry, using a Hewlett–Packard
gas chromatograph (model 6890 plus, equipped with aflame ionisation detector) and a Jeol mass spectrometer
(Model JMS-AX500). Glucosamine (GlcN) was released
by hydrolysis in 4 N HCl at 100 �C for 10 h and deter-
mined by the same method as used for neutral sugars.
3-Deoxy-D-manno-oct-2-ulosonic acid (Kdo) was deter-
mined by the thiobarbituric acid method [26]. Phospho-
rus was determined colorimetrically by ashing procedure
as described previously [27]. Galacturonic acid wasdetermined by a colorimetric method [28] and by
GLC–mass spectrometry after derivatisation of the car-
boxylic group to methyl ester.
Fatty acids were determined as methyl ester deriva-
tives by GLC comparison with authentic standards
and also by the analysis of both electron impact (EI)
and chemical ionisation (CI) mass spectra. GC–MS-EI
was done on Hewlett–Packard 5890 Series II gas chro-matograph equipped with a DB-1 column
(30 m · 0.25 mm · 0.25 lm) and connected to a mass
selective detector (MSD model HP 5970) using the tem-
perature program as follows: 80 �C! 2 min ! 20 �C/min ! 160 �C! 2 min ! 2 �C/min ! 200 �C ! 10 �C/min ! 250 �C! 11 min, injector temp: 250 �C and
detector temp: 280 �C. Chemical ionisation mass spec-
trometry was done on an instrument from Agilent Tech-nologies (6890N Network GC system followed by
detection on 5973 Network MSD). The column and
temperature program were the same as mentioned be-
fore. Ammonia was used as the reagent gas for chemical
ionisation mass spectrometry. For quantitative fatty
acid analysis, methyl ester of tetradecanoic acid was
used as an internal standard as the fatty acid was absent
in this LPS. Total fatty acids were liberated from LPS bytrans-esterification with 2 N HCl in methanol at 85 �Cfor 16 h in nitrogen filled sealed glass tubes [29]. Ester-
bound fatty acids were liberated by treatment of LPS
or lipid A with sodium methylate (0.25 N CH3ONa) at
37 �C for 16 h [30]. After cleavage of the ester-bound
fatty acids, amide linked fatty acids were determined
by the method described above for total fatty acids
[29]. Identification of hydroxylated fatty acids was con-firmed by GC–MS analysis of the trimethylsilylated
(TMS) derivatives.
3. Results
3.1. Isolation and purification of the LPS
The yield of wet bacterial cells was 2 g per liter for
Acidiphilium ATCC55963 grown in mineral-glucose-
yeast extract medium. As acetone dried cells of acido-
philic species could not be dispersed homogeneously in
water, the wet cells were subjected to hot phenol–water
extraction for preparing the LPS effectively. The yield of
the purified LPS was 90 mg from 20 g wet cells, about
2.2% of the total cell mass on dry weight basis. SDS–PAGE analysis of the purified LPS did not show any
band which could be lighted up with Coomassie blue,

186 R. Bera et al. / FEMS Microbiology Letters 246 (2005) 183–190
indicating it to be free of associated proteins (sometimes
termed as endotoxin proteins). Absorption at 260 nm
was found to be insignificant, suggesting the absence
of nucleic acids in the purified LPS.
3.2. SDS–polyacrylamide gel electrophoresis analysis
SDS–PAGE analysis was performed to determine the
structural characteristic of the Acidiphilium ATCC55963
LPS and to compare it with those of E. coli serotype
0111:B4 and Salmonella typhimurium (Sigma, USA).
As shown in Fig. 1, both E. coli and Salmonella LPS
were resolved into a large number of bands in stepladder
pattern, corresponding to various polysaccharide chainlengths anchored to the core lipid-A and characteristic
of smooth type LPS. The Acidiphilium ATCC55963
LPS was different from the above two LPS. It showed
condensed laddering bands starting from the middle of
the ladder of the other two LPS and extending upto
the low molecular weight region. At the bottom portion,
the laddering was not very distinct for Acidiphilium
ATCC55963 LPS. Thus the results clearly indicated thatit is of S- or SR-type, but not truly R-type as proposed
for other Acidiphilium species where the stepladder pat-
tern of resolution was absent there [9].
Fig. 1. Silver stained SDS–PAGE patterns with lipopolysaccharides
(LPS) isolated from E. coli serotype 0111:B4 (lane 1), Salmonella
typhimurium (lane 2) Acidiphilium ATCC55963 (lane 3).
3.3. Endotoxic properties of the LPS from Acidiphilium
ATCC55963
3.3.1. Lethal toxicity and pyrogenicity
The lethal toxicity of the new LPS was tested using
standard D-galactosamine-sensitised BALB/c mouse asthe test animal and compared with the reference toxic
LPS from E. coli serotype 0111:B4. The survival of the
mouse was recorded at 24, 48, and 72 h after the admin-
istration of the LPS and the results are shown in Table 1.
In this study, E. coli LPS exhibited 100% lethality
(LD100) and 50% (LD50) lethality at 80 and 50 ng/
mouse, respectively. On the other hand, the lethality of
Acidiphilium ATCC55963 LPS was not observed below500 lg per mouse (Table 1) and there was no loss of sur-
vivability up to a dose of 400 lg per mouse. A dose of
1000 g per mouse had to be administered to obtain
100% lethality. This indicated that the lethal toxicity
of Acidiphilium ATCC55963 LPS to BALB/c mice is ex-
tremely low, even negligible, and the LPS may be con-
sidered as non-toxic compared to E. coli LPS.
Similarly, the results obtained in the LAL assay showedthat the lowest concentration of LPS that produced a
positive test was 0.01 ng/ml for E. coli and >10 ng/ml
for Acidiphilium ATCC55963. So the toxic potency of
the new LPS was more than 1000 times less than that
of the E. coli LPS.
3.3.2. TNFa production after LPS administration in naive
BALB/c mice
Since serum TNFa levels peaked at 1–2 h after inject-
ing the toxic LPS [31] into mice, immunoreactive TNFawas measured at 90 min after administering Acidiphilium
ATCC55963 LPS. In mice treated with LPS, the circula-
tory level of TNFa, the primary mediator of LPS toxic-
ity, increased in a dose dependent manner (Table 2). In
saline treated control serum, the level of TNFa detected
Table 1
Lethal toxicity of Acidiphilium ATCC55963 and reference E. coli LPS
in galactosamine-sensitised BALB/c mice
Dose of LPS (lg/mouse) No. of dead mice/
No. of tested mice
% of lethality
after 72 h
Acidiphilium ATCC55963 24 h 48 h 72 h
10 0/6 0/6 0/6 0
100 0/6 0/6 0/6 0
200 0/6 0/6 0/6 0
400 0/6 0/6 0/6 0
500 1/6 2/6 2/6 33.33
750 2/6 4/6 4/6 66.66
1000 4/6 6/6 6/6 100
E.coli 0111:B4
0.01 0/6 0/6 0/6 0
0.02 1/6 1/6 1/6 16.66
0.04 2/6 2/6 2/6 33.33
0.05 2/6 3/6 3/6 50
0.08 4/6 6/6 6/6 100

Table 2
Serum TNFa and lethality induced by E. coli and Acidiphilium
ATCC55963 LPS in BALB/c mousea
Inducer Dose
(lg/mouse)
TNFa(ng/ml)
Survivors/
Total (%)b
E. coli 0111:B4 LPS 10 8.30 ± 1.10 20/20 (100)
25 18.16 ± 1.75 20/20 (100)
50 26.65 ± 3.25 15/20 (75)
100 42.25 ± 2.54 8/20 (40)
200 48.56 ± 4.25 0/20 (0)
Acidiphilium
ATCC55963 LPS
10 3.42 ± 0.75 20/20 (100)
100 10.33 ± 1.15 20/20 (100)
1000 26.85 ± 2.40 20/20 (100)
1500 27.75 ± 1.30 20/20 (100)
Control saline 0.65 ± 0.25 10/10 (100)
a Blood samples were collected from each group at 90 min after
treatment. Tabulated values represent means of four experiments
(n = 5 for each group).b Cumulative mortality followed over 72 h after both type LPS
administration.
Table 3
Chemical composition of LPS, lipid A and polysaccharide part of
Acidiphilium ATCC55963
Component Amount (nmol mg�1)
LPS Polysaccharide moiety Lipid A moiety
Glc 275 745 –a
Gal 280 772 –
Man 398 1095 –
Rha 136 355 –
Hep 67 186 –
Kdo 356 995 –
GalA 325 890 –
GlcN 545 – 984
Phosphorus 775 568 332
Ester-linked fa
12:0 25 – 42
16:0 32 – 56
18:1 234 – 372
19:1 42 – 65
19:1(cyclo) 145 – 218
20:0(3-OH) 28 – 56
20:1(3-OH) 62 – 90
Amide-linked fa
14:0(3-OH) 145 – 240
18:0(3-OH) 592 – 1056
Abbreviations are used as: Glc, glucose; Gal, galactose; Man, man-
nose; Rha, rhamnose; GlcN, glucosamine; GalA, galacturonic acid;
Kdo, 3-deoxy-D-manno-oct-2-ulosonic acid; Hep, heptose.a None. fa, fatty acids. The position of OH group is indicated.
R. Bera et al. / FEMS Microbiology Letters 246 (2005) 183–190 187
was negligible. The results showed that the level of
TNFa induced by E. coli LPS was always higher than
that of the newly identified pure LPS.
3.4. Compositional analysis of the LPS
The constituent carbohydrates and the fatty acids
along with their linkage pattern present in the LPS ofAcidiphilium ATCC55963 have been included in Table
3. As determined by GLC and GLC–mass spectrometry
as alditol acetate derivatives, the constituent sugars in-
clude glucose (Glc), galactose (Gal), mannose (Man),
rhamnose (Rha), glucosamine (GlcN) and a minor
amount of heptose. Glucosamine is present only in lipid
A and usually is the main carbohydrate backbone of this
lipid in most of the Gram-negative bacterial LPS,though galactosamine is present in place of glucosamine
in some Acidiphilium species [9]. Analysis of lipid A, ob-
tained after treating the LPS with 1% aqueous acetic
acid, indicated that the O-specific polysaccharide core
moiety was linked to lipid A via a Kdo molecule. Kdo
and heptose are characteristic components of core oligo-
saccharides in LPS and are useful markers of Gram-neg-
ative bacterial cell wall constituents. Glucose, galactose,mannose and rhamnose are common monosaccharides
of other Acidiphilium species like A. cryptum and A.
symbioticum [9]. Based on the GLC profile of fatty acids
(Fig. 2), the major fatty acids identified were 18:0(3-
OH), 18:1, 14:0(3-OH) and 19:0(cyclo). Other fatty acids
present in small amounts were 12:0, 16:0, 19:1, 20:0(3-
OH) and 20:1(3-OH). Though the mass fragmentation
patterns of 19:0(cyclo) and 19:1 are indistinguishable,the presence of both in this LPS (Fig. 2) could be ascer-
tained from the difference in their retention times. The
ester-linked and amide-bound fatty acids in lipid A are
present in almost the same molar ratio as in the LPS
(Table 3).
4. Discussion
The analysis of LPS establishes that the architecturalelements of the molecule are: (i) the lipophilic lipid A
consisting of a phosphorylated disaccharide linked with
hydroxylated or non-hydroxylated fatty acids, (ii) the
core oligosaccharide short in length and with Kdo,
and (iii) the outer-most O-specific side chain having re-
peated units of specific oligosaccharides displaying the
chemical and serological determinants for the identifica-
tion of the species and strains [2,3,32]. The LPS of Acid-iphilium ATCC55963 contains all the usual constituents
of Gram-negative bacterial LPS, but is distinctly differ-
ent from the fatty acid profile of the earlier reported li-
pid A of A. crypticum and A. symbioticum [9]. Thus,
except for 12:0 and 14:0(3-OH), all the remaining fatty
acids in Acidiphilium ATCC55963 LPS were different.
Again, instead of 14:0(3-OH) as major constituent as
in LPS of A. crypticum and A. symbioticum, 18:0(3-OH) was the highest (about 40% of total fatty acid) con-
tributor in Acidiphilium ATCC55963 LPS. The cellular
fatty acid 18:1, reported by other investigators from
other Acidiphilium species [10,11], was also present in

Fig. 2. Fatty acids profile of Acidiphilium ATCC55963 LPS. Fatty acid methyl esters were obtained by methanolysis as procedure described in
Section 2 and converted to TMS derivative. Separation was performed on a DB-1 column connected to HP 5890 Series II gas chromatograph
equipped with mass selective detector HP5970 using temperature program as follows: 80 �C! 2 min! 20 �C/min! 160 �C! 2 min! 2 �C/min! 200 �C! 10 �C/min! 250 �C! 11 min, injector temp: 250 �C and detector temp: 280 �C. Unidentified peaks and peaks of substances not
belonging to fatty acid methyl esters or artifacts are not marked.
188 R. Bera et al. / FEMS Microbiology Letters 246 (2005) 183–190
this LPS. Acidiphilium ATCC55963 LPS or lipid A con-tains two long chain b-hydroxy fatty acids, 20:0(3-OH)
and 20:1(3-OH), besides 18:0(3-OH) and 3-hydroxy tet-
radecanoic acid. Two other fatty acids, 19:0(cyclo) and
19:1, which are not common as the constituents of lipid
A, were also detected. It should be mentioned here that
fatty acids 19:1 and 19:0(cyclo) were recently found in
Mesorhizobium huakuii LPS [33].
The compositional diversity of any LPS is related toits biological activity, and endotoxicity is imparted to
LPS by its lipid A moiety [5,6]. The types of fatty acids
present, their linkages, and the state of sugar phosphor-
ylation in lipid A determine its toxicity. However the
presence of low amounts of LPS in the body fluid pro-
tects the host by enhancing resistance to infection and
malignancy through the release of immunomodulators
[34]. An appropriate control of the LPS response istherefore a central element in preserving the fine balance
between its harmful effect and toxicity. In order to ex-
plore the potentiality of the natural LPS as immunolog-
ical adjuvant, the type and amount of LPS present in the
host body fluid are of critical importance. The composi-
tion of Acidiphilium ATCC55963 LPS, specially the type
of fatty acids present, is strikingly different from those of
E. coli or Salmonella LPS. The lower activity of the lipid
A possessing relatively longer fatty acids was evidencedin Salmonella minnesota type lipid A using chemically
synthesised material [35,36], as well as in lipid A from
Porphyromonas gingivalis [30]. Indeed, the Acidiphilium
ATCC55963 LPS was found to contain mostly long
chain fatty acids that are either b-hydroxylated or unsat-
urated. In galactosamine-sensitised mouse, it was about
10,000 times less toxic than E. coli LPS. The chemical
nature, unsaturation and chain length of the fatty acidsin lipid A of Acidiphilium ATCC55963 LPS are com-
mensurate with such low or non-toxic activity. The fatty
acid compositions of two extensively studied non-toxic
LPS from R. sphaeroides and R. capsulatus are however
considerably different, since 3-oxotetradecanoic acid, 3-
hydroxytetradecanoic acid, 3-hydroxydecanoic acid and
7-tetradecenoic acid were reported as their major con-
tributory fatty acids [12].The host response to LPS is not induced directly but
mediated by immunomodulators, and TNFa is consid-
ered the principal mediator of LPS toxicity. In an in vivo
system, production of TNFa by E. coli and Acidiphilium
ATCC55963 LPS was compared. Dose dependent re-
lease was noticed in both the cases, but at each dose
the extent of release was lower for Acidiphilium
ATCC55963 LPS. Despite being able to induce compa-

R. Bera et al. / FEMS Microbiology Letters 246 (2005) 183–190 189
rable levels of TNFa, no endotoxic shock was induced
by Acidiphilium ATCC55963 LPS in BALB/c mice under
comparable conditions. In all experiments, cumulative
mortality was followed over 72 h after treatment,
although 75% of the deaths occurred within 24 h. So it
was difficult to correlate the lethality with the level ofserum TNFa, since no loss of survivability was observed
in the case of Acidiphilium ATCC55963 LPS treated
mouse. The composition of lipid A or LPS is crucial
for the toxicity as evidenced by Salmonella LPS. Salmo-
nella monophosphoryl lipid A (MPL) induced high level
of TNFa comparable to Salmonella LPS and diphos-
phoryl lipid A, but endotoxic shock and subsequent
death was induced only by the last two [31]. Thereforethe level of serum of TNFa is not the only determinant
for lethal toxicity, as Acidiphilium ATCC55963 LPS re-
mained non-lethal (inspite of producing comparable le-
vel of serum TNFa). If the induction of serum TNFawithout harming the host is of any therapeutic signifi-
cance then Acidiphilium ATCC55963 LPS may be con-
sidered as a candidate. Further investigation on this
LPS for detailed structure determination to elucidatethe structure–activity relationship in the mammalian
host will undoubtedly establish its novel biological prop-
erties as a non-toxic LPS and work on this aspect is cur-
rently going on.
Acknowledgements
We thank our Director Prof. Siddhartha Roy for his
patronization of this study. We are indebted to Dr. Co-
lin Goding, editor Pigment Cell Research and Dr. Basu-
dev Achari of our institute for critically reviewing the
manuscript. We also acknowledge Dr. P.C. Banerjee of
our institute for his kind cooperation in maintaining the
strain. Thanks are due to DBT, Government of India
for financial assistance (R. Bera).
References
[1] Nikaido, H. and Vaara, M. (1985) Molecular basis of bacterial
outer membrane permeability. Microbiol. Rev. 49, 1–32.
[2] Luderitz, O., Jann, K. and Wheat, R. (1968) Somatic and capsular
antigens of Gram-negative bacteria In: Comprehensive Biochem-
istry: Extracellular and Supporting Structures (Florkin, M. and
Stotz, E.H., Eds.), Vol. 26A, pp. 105–227. Elsevier Publishing
Company, Amsterdam.
[3] Galanos, C., Freudenberg, M.A., Luderitz, O., Rietschel, E.T.
and Westphal, O. (1979) Chemical, physiological and biological
properties of bacterial lipopolysaccharides. Prog. Clin. Biol. Res.
29, 321–332.
[4] Nowotny, A. (1983) Beneficial Effects of Endotoxins. Plenum
Publishing Corp., New York.
[5] Qureshi, N., Takayama, K. and Ribi, E. (1982) Purification and
Structural determination of nontoxic lipid A obtained from the
lipoplysaccharide of Salmonella typhimurium. J. Biol. Chem. 257,
11808–11815.
[6] Takayama, K., Qureshi, N., Ribi, E. and Cantrell, J.L. (1984)
Separation and Characterization of toxic and nontoxic formed of
lipid A. Rev. Infect. Dis. 6, 439–443.
[7] Golenbock, D.T., Hampton, R.Y., Qureshi, N., Takayama, K.
and Raetz, C.R.H. (1991) Lipid A-like molecules that antagonize
the effects of endotoxins on human monocytes. J. Biol. Chem.
266, 19490–19498.
[8] Henricson, B.E., Perera, P.Y., Qureshi, N., Takayama, K. and
Vogel, S.N. (1992) Rhodopseudomonas sphaeroides lipid A derived
block in vitro induction of tumor necrosis factor and endotoxin
Tolerance by smooth lipopolysaccharide and monophosphoryl
lipid A. Infect. Immun. 60, 4285–4290.
[9] Basu, S., Das, S. and Banerjee, P.C. (1994) Lipopolysaccharides
of the acidophilic heterotrophic bacteria Acidiphilium cryptum
and Acidiphilium symbioticum. FEMS Microbiol. Lett. 118, 65–
70.
[10] Kishimoto, N., Kosako, Y. and Tano, T. (1991) Acidobacterium
capsulatum gen. Nov. sp. Nov.: an acidophilic chemorgano-
trophic bacterium containing menaquinone from acidic mineral
environment. Curr. Microbiol. 22, 1–7.
[11] Urakami, T., Tamaoka, J., Suzuki, K-I. and Komagata, K. (1989)
Acidomonas gen. Nov., incorporating Acetocacter methanolicus
as Acidomonas methanolica comb. Nov.. Int. J. Syst. Bacteriol.
39, 50–55.
[12] Mayer, H., Bhat, R., Masoud, H., Radziejewska-Lebrecht, J.,
Widemann, C. and Kraus, J.H. (1989) Bacterial lipopolysaccha-
rides. Pure Appl. Chem. 61, 1271–1282.
[13] Christ, W.J., Asano, O., Robidoux, A.L., Perez, M., Wang, Y.,
Dubuc, G.R., Gavin, W.E., Hawkins, L.D., McGuinness, P.D.
and Mullarkey, M.A., et al. (1995) E5531, a pure endotoxin
antagonist of high potency. Science 268, 80–83.
[14] Banerjee, P.C., Ray, M.K., Koch, C., Bhattacharyya, S., Shivaji,
S. and Stackebrandt, E. (1996) Molecular characterization of two
Acidophilic heterotrophic bacteria isolated from a copper mine of
India. Sys. Appl. Microbiol. 19, 78–82.
[15] Bhadra, R., Nayak, A., Bandyopadhyay, P.C. and Basu, S.
(1998) Process for preparing nontoxic lipopolysaccharides from
Acidiphilium species. United States Patent. Patent number:
5,846,789.
[16] Westphal, O., Luderitz, O. and Bister, F. (1952) Uber die
extraction von bacterien mit phenol wasser. Naturforsh. Z. 7b,
148–155.
[17] Westphal, O. and Jann, K. (1965) Bacterial lipopolysaccharides –
extraction with phenol–water and further applications of the
procedure. Meth. Carbohydr. Chem. 5, 83–91.
[18] Hirschefeld, M., Ying, M., Weis, J.H., Vogel, S.N. and Weis, J.J.
(2000) Cutting edge: repurification of lipoplysaccharide eliminates
signaling through both human and murine toll like receptor 2. J.
Immunol. 165, 618–622.
[19] Luderitz, O., Westphal, O., Staub, A.M. and Nikaido, H. (1971)
In: Microbiol Toxins (Weinbaum, G., Kadis, S. and Ajl, S.J.,
Eds.), Vol. 4, p. 145. Academic Press, New York.
[20] Qureshi, N., Kattashov, I., Walker, K., Doroshenko, V., Cotter,
R.J., Takayama, K., Sievert, T.R., Rice, P.A., Lin, J.S.L. and
Golenbock, D.T. (1997) Structure of the monophosphoryl lipid A
moiety obtained from the lipopolysaccharide of Chlamydia
trachomatis. J. Biol. Chem. 272, 10594–10600.
[21] Laemmli, U.K. (1970) Cleavage of structural proteins during the
assembly of the head of bacteriophage T4. Nature 227, 680–685.
[22] Tsai, C.M. and Frasch, C.E. (1982) A sensitive silver stain for
detecting lipopolysaccharide in polyacrylamide gels. Anal. Bio-
chem. 119, 115–119.
[23] Galanos, C., Freudenberg, M.A. and Reutler, W. (1979) Galac-
tosamine-induced sensitization to the lethal effects of endotoxin.
Proc. Natl. Acad. Sci. USA 76, 5939–5943.

190 R. Bera et al. / FEMS Microbiology Letters 246 (2005) 183–190
[24] Ogawa, T. (1994) Immunobiological properties of chemically
defined lipid A from lipopolysaccharide of Porphyromonas
(Bacteroides) gingivalis. Eur. J. Biochem. 219, 737–742.
[25] Albershein, P., Nevins, D.J., English, P.D. and Karr, A. (1967) A
method for the analysis of sugars in plant cell-wall polysaccha-
rides by gas–liquid chromatography. Carbohydr. Res. 5, 340–345.
[26] Brade, H., Galanos, C. and Luderitz, O. (1983) Differential
determination of the 3-deoxy-D-manno-octulosonic acid residues
in lipopolysaccharides of Salmonella Minnesota rough mutants.
Eur. J. Biochem. 131, 195–200.
[27] Ames, B.N. (1966) Assay of inorganic phosphate, total phosphate
and phosphatases. Meth. Enzymol. 8, 115–118.
[28] Blumenkrantz, N. and Asboe-Hansen, G. (1973) New method for
quantitative determination of uronic acids. Anal. Biochem. 54,
484–489.
[29] Rietschel, E.T., Hase, S., King, M., Redmond, J. and Lehmann,
V. (1977) Chemical structure of lipid A. Microbiology 177, 262–
268.
[30] Wollenweber, H.-W. and Rietschel, E.T. (1990) Analysis of
lipopolysaccharide (lipid A) fatty acids. J. Microbiol. Meth. 11,
195–211.
[31] Kiener, P.A., Marek, F., Rodgers, G., Lin, P-F., Warr, G. and
Desiderio, J. (1988) Induction of tumor necrosis factor, IFN-c,and acute lethality in mice by toxic and non-toxic forms of lipid
A. J. Immunol. 141, 870–874.
[32] Lugtenberg, B. and van Alphen, L. (1983) Molecular arachitec-
ture and functioning of the outer membrane of Escherichia coli
and other Gram-negative bacteria. Biochem. Biophys. Acta 737,
51–115.
[33] Choma, A. (1999) Fatty acid composition of Mesorhizobium
huakuii lipopolysaccharides. Identification of 27-oxooctocosanoic
acid. FEMS Microbiol. Lett. 177, 257–262.
[34] Ulmer, A.J., Rietschel, E.Th., Zahringer, U. and Heine, H. (2002)
Lipopolysaccharide; structure, bioactivity, receptors, and signal
transduction. Trends Glycosci. Glycotechnol. 14, 53–68.
[35] Galanos, C., Luderitz, O., Freudenberg,M., Brade, L., Schade, U.,
Rietschel, E.Th., Kusumoto, S. and Shiba, T. (1986) Biological
activity of synthetic heptaacyl lipid A representing a component of
Salmonella minnesota R595 lipid A. Eur. J. Biochem. 160, 55–59.
[36] Tanamoto, K., Lida, T., Haishima, Y. and Azumi, S. (2001)
Endotoxic properties of lipid A from Comamonas testosteroni.
Microbiology 147, 1087–1094.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 191–198
Comprehensive analysis of classical and newlydescribed staphylococcal superantigenic toxin genes
in Staphylococcus aureus isolates
Katsuhiko Omoe a,*, Dong-Liang Hu b, Hiromi Takahashi-Omoe c,Akio Nakane b, Kunihiro Shinagawa a
a Department of Veterinary Medicine, Faculty of Agriculture, Iwate University, Ueda 3-18-8, Morioka, Iwate 020-8550, Japanb Department of Bacteriology, Hirosaki University School of Medicine, 5 Zaifu-cho, Hirosaki, Aomori 036-8562, Japan
c Chemical Management Center, National Institute of Technology and Evaluation, 2-49-10 Nishihara, Shibuya-ku, Tokyo 151-0066, Japan
Received 27 January 2005; received in revised form 6 April 2005; accepted 6 April 2005
First published online 19 April 2005
Edited by S. Schwarz
Abstract
We describe a comprehensive detection system for 18 kinds of classical and newly described staphylococcal superantigenic toxin
genes using four sets of multiplex PCR. Superantigenic toxin genotyping of Staphylococcus aureus for 69 food poisoning isolates and
97 healthy human nasal swab isolates revealed 32 superantigenic toxin genotypes and showed that many S. aureus isolates harbored
multiple toxin genes. Analysis of the relationship between toxin genotypes and toxin genes encoding profiles of mobile genetic ele-
ments suggests its possible role in determining superantigenic toxin genotypes in S. aureus as combinations of toxin gene-encoding
mobile genetic elements.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Staphylococcus aureus; Enterotoxin; Multiplex PCR; Genotyping; Mobile genetic elements
1. Introduction
Staphylococcal enterotoxins (SEs) are emetic toxins,
and staphylococcal food poisoning resulting from the
consumption of food contaminated with SEs is one of
the most common food-borne illnesses [1]. In addition,
SEs and the SE-related toxin, toxic shock syndrome tox-
in-1 (TSST-1), are members of the superantigenic toxin
family and have the ability to stimulate large popula-tions of T cells having a particular Vb element in their
T-cell receptors (TCR). This stimulation subsequently
leads to a massive proliferation of T cells and the uncon-
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.007
* Corresponding author. Tel.: +81 19 621 6221; fax: +81 19 621 6223.
E-mail address: [email protected] (K. Omoe).
trolled release of proinflammatory cytokines, whichcause life-threatening TSS [2–4]. SEs have been divided
into five serological types (SEA though to SEE) based
on their antigenicity [1]. In recent years, new types of
SEs (SEG, SEH, SEI, SEJ, SEK, SEL, SEM, SEN,
SEO, SEP, SEQ, SElR and SEU) have been reported
[2,5–11]. Several attempts to detect superantigenic toxin,
SE and TSST-1 genes in S. aureus isolates have been
made; these studies have shown that multiple superanti-genic toxin genes are commonly found among S. aureus
isolates [12–17]. These newly described SEs have been
designated as members of the SE family based on their
sequence similarity with classical SEs. The International
Nomenclature Committee for Staphylococcal Superanti-
gen Nomenclature (INCSSN) has recommended that
. Published by Elsevier B.V. All rights reserved.

192 K. Omoe et al. / FEMS Microbiology Letters 246 (2005) 191–198
only staphylococcal superantigens that induce emesis
following oral administration in a monkey model should
be designated as SE while other related toxins that either
lack emetic properties in this model or have not been
tested should be designated staphylococcal entero-
toxin-like (SEl) superantigens [18]. Based on this recom-mendation from INCSSN, the toxins SEJ, SEK, SEL,
SEM, SEN, SEO, SEP, SEQ, and SEU should be re-
named SElJ, SElK, SElL, SElM, SElN, SElO, SElP,
SElQ, and SElU, respectively.
On the other hand, it has been known that certain SE
(SEl) and TSST-1 genes are associated with mobile ge-
netic elements such as pathogenicity islands, prophages,
and plasmids [6,7,9,19–23]. These facts imply that super-antigenic toxin genes are transferred horizontally be-
tween staphylococcal strains. There is a possibility that
these mobile genetic elements have played an important
role in the evolution of S. aureus as a pathogen. To date,
there is a need for a method to comprehensively detect
and identify the large family of superantigenic toxin
genes; such a method would be a powerful tool for evo-
lutionary analysis of the pathogenicity of S. aureus, aswell as for diagnostic and epidemiological purposes.
Here, we report fine superantigenic toxin genotyping
of S. aureus isolates using a multiplex PCR system that
is capable of detecting 18 kinds of staphylococcal super-
antigenic toxin genes. The analysis between superanti-
genic toxin genotypes and toxin genes encoding
profiles of mobile genetic elements provides a hypothesis
on possible role for determination of superantigenic tox-in genotypes in S. aureus.
2. Materials and methods
2.1. Bacterial strains and culture media
A total of 177 S. aureus samples were used in thisstudy. Of these, 11 strains were reference strains, includ-
ing full genome sequencing strains (N315; DDBJ/Gen-
Bank/EMBL BA000018, Mu50; BA000017, MW2;
BA000033) (Table 1). Sixty-nine isolates were obtained
Table 1
Staphylococcal superantigenic toxin genotypes of Staphylococcus aureus refe
Strain Superantigenic toxin genotype
N315 sec, seg, sei, sell, selm, seln, selo
Mu50 sea, sec, seg, sei, sell, selm, seln
MW2 sea, sec, seh, selk, sell, selq
RN4220 no SE gene
196E sea, sed, selj, selr
S6 sea, seb, selk, selq
FRI-361 sec2, sed, seg, sei, selj, sell, selm
FRI-326 see, selq
FRI-569 seh
834 sec, seg, sei, sell, selm, seln, selo
Saga 1 seg, sei, selm, seln, selo, selp
from 30 food poisoning outbreaks diagnosed by 10 local
government laboratories in Japan from 1990 to 2002; S.
aureus isolates were isolated from patient feces, patient
vomit, or the foods involved, and collected from the lab-
oratories. Among these 30 food poisoning outbreaks, 6
were diagnosed as SE-unidentified, meaning that all S.aureus isolates from each outbreak were negative for
production of SEA to SED by commercial SET-RPLA
kit (DENKA Seiken Co. Ltd., Tokyo, Japan). In this
study, we included 21 isolates obtained from these SE-
unidentified outbreaks. In addition to the 69 food poi-
soning isolates, 97 isolates were obtained from nasal
swabs of healthy humans in Japan from 2000 to 2004.
Bacterial cultures were grown in brain heart infusion(BHI) broth prior to purification of genomic DNA.
2.2. DNA purification
Total DNA of S. aureus was purified with the QIA-
amp DNA purification kit (Qiagen GmbH, Hilden, Ger-
many) according to the manufacturer�s instructions. Theconcentration of DNA solution was determined accord-ing to A260 values.
2.3. Primers
The nucleotide sequences of all PCR primers used in
this study and their respective amplified products are
listed in Table 2. The primer sets used to detect selj, selk,
sell, selm, seln, selo, selp, selq and selr genes were de-signed according to published nucleotide sequences
[6,7,9,20–23]. These primer sets were designed to anneal
to unique regions and generate amplicons that would al-
low identification of each se gene based on the molecular
weight of its PCR product (Table 2). The primer sets
used to detect tst-1 and sea to see were those described
by Becker et al. [12]. The primer sets used to detect
seg, seh and sei were described by Omoe et al. [17]. Asan internal positive control, we used primers that are
specific to S. aureus to amplify femA and femB genes
[16,24]. To construct a multiplex PCR system, four sets
(Set 1; sea, seb, sec, sed, see, femB: Set 2; seg, seh, sei,
rence strains
References
, selp, tst-1 Kuroda et al. [7]
, selo, tst-1 Kuroda et al. [7]
Baba et al. [19]
Novick [29]
Omoe et al. [9,17]
Omoe et al. [17]
, seln, selo, selr Omoe et al. [17]
Omoe et al. [17]
Su and Wong [11]
tst-1 Nakane et al. [30]
Omoe et al. [17]

Table 2
Nucleotide sequences and predicted size of PCR products for the staphylococcal superantigen-specific oligonucleotide primers
Gene Primer Oligonucleotides sequence (5 0–30) PCR product (bp) PCR set References
sea SEA-3 CCTTTGGAAACGGTTAAAACG 127 1 [12]
SEA-4 TCTGAACCTTCCCATCAAAAAC
seb SEB-1 TCGCATCAAACTGACAAACG 477 1 [12]
SEB-4 GCAGGTACTCTATAAGTGCCTGC
sec SEC-3 CTCAAGAACTAGACATAAAAGCTAGG 271 1 [12]
SEC-4 TCAAAATCGGATTAACATTATCC
sed SED-3 CTAGTTTGGTAATATCTCCTTTAAACG 319 1 [12]
SED-4 TTAATGCTATATCTTATAGGGTAAACATC
see SEE-3 CAGTACCTATAGATAAAGTTAAAACAAGC 178 1 [12]
SEE-2 TAACTTACCGTGGACCCTTC
seg SEG-1 AAGTAGACATTTTTGGCGTTCC 287 2 [17]
SEG-2 AGAACCATCAAACTCGTATAGC
seh SEH-1 GTCTATATGGAGGTACAACACT 213 2 [17]
SEH-2 GACCTTTACTTATTTCGCTGTC
sei SEI-1 GGTGATATTGGTGTAGGTAAC 454 2 [17]
SEI-2 ATCCATATTCTTTGCCTTTACCAG
selj SEJ-1 ATAGCATCAGAACTGTTGTTCCG 152 2 This study
SEJ-2 CTTTCTGAATTTTACCACCAAAGG
selk SEK-1 TAGGTGTCTCTAATAATGCCA 293 3 This study
SEK-2 TAGATATTCGTTAGTAGCTG
sell SEL-1 TAACGGCGATGTAGGTCCAGG 383 4 This study
SEL-2 CATCTATTTCTTGTGCGGTAAC
selm SEM-1 GGATAATTCGACAGTAACAG 379 3 This study
SEM-2 TCCTGCATTAAATCCAGAAC
seln SEN-1 TATGTTAATGCTGAAGTAGAC 282 4 This study
SEN-2 ATTTCCAAAATACAGTCCATA
selo SEO-1 TGTGTAAGAAGTCAAGTGTAG 214 3 This study
SEO-2 TCTTTAGAAATCGCTGATGA
selp SEP-3 TGATTTATTAGTAGACCTTGG 396 2 This study
SEP-4 ATAACCAACCGAATCACCAG
selq SEQ-1 AATCTCTGGGTCAATGGTAAGC 122 4 This study
SEQ-2 TTGTATTCGTTTTGTAGGTATTTTCG
selr SER-1 GGATAAAGCGGTAATAGCAG 166 4 This study
SER-4 GTATTCCAAACACATCTAAC
tst1 TST-3 AAGCCCTTTGTTGCTTGCG 447 3 [12]
TST-6 ATCGAACTTTGGCCCATACTTT
femA femA1 AAAAAAGCACATAACAAGCG 134 2, 3 [16]
FemA2 GATAAAGAAGAAACCAGCAG
femB femB1 TTACAGAGTTAACTGTTACC 651 1, 4 [24]
FemB2 ATACAAATCCAGCACGCTCT
K. Omoe et al. / FEMS Microbiology Letters 246 (2005) 191–198 193
selj, selp, femA: Set 3; selk, selm, selo, tst-1, femA: Set 4;
sell, seln, selq, selr, femB) of 10· primer master mixes
(containing 2 lM each primer) were prepared.
2.4. Uniplex PCR and sequencing analysis
To evaluate the specificity of the newly designed pri-
mer sets for detecting selj, selk, sell, selm, seln, selo, selp,selq and selr genes, uniplex PCR using each primer pair
was performed. The amplification was performed in an
automated thermalcycler with a hot bonnet (Takara
PCR Thermal Cycler MP). The reaction mixture
(50 ll) for uniplex PCR contained 0.4 lM of each pri-
mer, 2 mM MgCl2, 200 lM each of dGTP, dATP, dTTP
and dCTP (Takara Syuzo Co., Kyoto, Japan), 0.5U of
TaKaRa EX Taq DNA polymerase (Takara), and 5 llof 10· buffer (Takara). Thermal cycles of 94 �C for
30 s, 55 �C for 30 s, and 72 �C for 60 s were repeated
30 times. The DNA fragments obtained from uniplex
PCR were subcloned to pGEM-easy vector (Promega,
Madison, WI) and subjected to nucleotide sequencing
analysis using an ABI3100-avant automatic DNA se-
quencer (Applied Biosystems, Foster City, CA).
2.5. Multiplex PCR
Multiplex PCR of each primer set was performed
with QIAGEN Multiplex PCR Kit (QIAGEN) accord-
ing to manufacturer�s instructions. Each reaction mix
(50 ll) consisted of 25 ll of 2· QIAGEN Multiplex
PCR Master Mix (containing QIAGEN HotStartTaq
DNA polymerase, QIAGEN multiplex PCR buffer,
and dNTP mix), 0.2 lM of each primer, and 10–
100 ng of template DNA. DNA amplification was car-ried out with the following thermal cycling profile:
an initial denaturation of DNA and QIAGEN

194 K. Omoe et al. / FEMS Microbiology Letters 246 (2005) 191–198
HotStartTaq DNA polymerase activation at 95 �C for
15 min was followed by 35 cycles of amplification
(95 �C for 30 s, 57 �C for 90 s, and 72 �C for 90 s), end-
ing with a final extension at 72 �C for 10 min. PCR
products were resolved by electrophoresis in 3% NuSi-
eve 3:1 agarose gel (Cambrex Bio Science Rockland,Inc., Rockland, ME) in 0.5· TBE (Tris-boric acid-
EDTA) buffer, stained by 0.5 lg/ml of EtBr, and visual-
ized on a transilluminator.
3. Results and discussion
3.1. Development of multiplex PCR system for detection
of se and tst-1 genes
First of all, we tried amplifying target DNA of newly
designed PCR primers for selj, selk, sell, selm, seln, selo,
selp, selq and selr genes. Uniplex PCR using each primer
set with total DNA of reference S. aureus strains was
performed: S. aureus 196E total DNA for selj and selr;
S. aureus MW2 for selk and selq; and S. aureus N315for sell, selm, seln, selo and selp. The sizes of PCR prod-
ucts obtained by these uniplex PCRs were identical to
those predicted from the design of the primers (data
not shown). Then, these PCR products were subcloned
Fig. 1. Detection of staphylococcal superantigenic toxin genes by multiplex
and MW2 were amplified with 4 sets of multiplex PCR. SE gene negative refe
molecular size marker HaeIII digested /X174; 1, N315; 2, Mu50; 3, MW2; 4,
of S. aureus 196E, S6, FRI 326, FRI 569, N315. (b) Total DNAs from S. aure
M, molecular size marker HaeIII digested /X174; 1, 9, mixture of total DNA
361; 5, FRI-362, 6, FRI-569; 7, 834; 8, Saga1; 10, Milli-Q water (negative c
to pGEM-easy vector and subjected to nucleotide
sequencing analysis. The DNA sequences of these clones
of se genes were almost exactly identical to the published
DNA sequences of the respective se genes. These results
showed that the newly designed PCR primer sets could
amplify respective se genes with specificity.The combinations of primer sets and reaction condi-
tions for the multiplex PCR were optimized to ensure
that all PCR products of target genes were satisfactorily
amplified. We ultimately constructed four optimized
multiple primer sets, as described in Section 2. Fig. 1
shows the results of multiplex PCR when total DNAs
of reference S. aureus strains were used as a templates.
As a positive control, a mixture of total DNA of S. aur-eus 196E, S6, FRI-326, FRI-569 and N315 was used.
Reliable amplification of PCR products was observed
in all multiplex PCR reactions using the four primer
sets. The sizes of the PCR products obtained from the
positive control and the reference strains corresponded
to their predicted sizes (Table 2). Furthermore, the toxin
gene genotypes of all reference strains determined by
multiplex PCR were exactly identical to the toxin genegenotypes determined by full genome sequencing
(N315, Mu50 and MW2) or southern blot analysis
(196E, S6, FRI-361, FRI-326, FRI-569, 834 and Saga1)
(Table 1). When Milli-Q water was used as a negative
PCR. (a) Total DNA from full genome sequenced strains N315, Mu50
rence strain RN4220 was also included as a negative control. Lanes: M,
RN4220, 5, Milli-Q water (negative control); 6, mixture of total DNA
us reference strains were amplified with 4 sets of multiplex PCR. Lanes:
of S. aureus 196E, S6, FRI 326, FRI 569, N315; 2, 196E; 3, S6; 4, FRI-
ontrol).

K. Omoe et al. / FEMS Microbiology Letters 246 (2005) 191–198 195
control instead of template genomic DNA, no PCR
products were observed in any of the four sets of multi-
plex PCR.
3.2. Superantigenic toxin gene genotyping of S. aureus
isolates from food poisoning outbreaks and healthy human
nasal swabs using multiplex PCR
A total of 166 S. aureus isolates were subjected to
superantigenic toxin gene genotyping analysis. A total
of 32 superantigenic toxin genotypes were observed
among the 166 isolates (Table 3). All of the 166 isolates
tested harbored the femA and femB genes. Of the 69 iso-
lates that originated in food poisoning, all isolates werediagnosed as positive for se genes. Thirteen SE-geno-
types were observed in food poisoning-related isolates.
Table 3
S. aureus superantigenic toxin genotypes and relationship with mobile genet
S. aureus superantigenic toxin genotypes Prevalenc
SFPa
(n = 69)
S. aureus harboring classical superantigenic toxin genes 4 (5.8)
sea 4 (5.8)
seb
S. aureus harboring classical and new superantigenic toxin genes 44 (63.8)
sea, sec, sell
sea, seg, tst-1
sea, seb, selk, selq 2 (2.9)
sea, sed, selj, selr 2 (2.9)
sea, seh, selk, selq 5 (7.3)
sea, seb, seh, selk, selq 21 (30.4)
sea, seg, sei, seln, tst-1 2 (2.9)
sea, seg, sei, selm, seln, selo 4 (5.8)
seb, seh 4 (5.8)
seb, selp 4 (5.8)
seb, selk, selq
seb, selk, selq, selp
seb, seg, sei, selm, seln, selo
sec, seg, sei, sell, selm, seln, selo
sec, seg, sei, sell, selm, seln, selo, tst-1
sed, seg, sei, selj, selm, seln, selo, selp, selr
tst-1, seg
tst-1, seg, seh, seln
tst-1, seg, sei, seln
tst-1, seg, sei, selk, selm, seln, selo
S. aureus harboring new superantigenic toxin genes 21 (30.4)
seg, sei, selm, seln
seg, sei, selm, seln, selo 7 (10.2)
seg, sei, selm, seln, selo, selp 8 (11.6)
seg, sei, selj, selm, seln, selo, selr 3 (4.4)
seg, sei, selk, selm, seln, selo, selq
seh, selk, selq
selj, selr 3 (4.4)
selm, selo
seln
selp
S. aureus harboring no superantigenic toxin gene
a Isolates from Staphylococcal food poisoning outbreaks.
Forty (58%) isolates were associated with the sea gene,
and the majority of these isolates possessed other se
genes. Among the 97 healthy human isolates, only 77
isolates (79.4%) were diagnosed as se-positive. A total
of 25 genotypes were observed in healthy human iso-
lates. In contrast to the trend in food poisoning-relatedisolates, there were only 8 (8.3%) sea-associated isolates.
Twenty-one isolates from 6 SE-unidentified food poi-
soning outbreaks possessed newly identified se genes
(seg, sei, selj, selm, seln, selo, selp, or selr). The superan-
tigenic toxin genotypes of isolates within each outbreak
were the same (2 outbreaks: seg, sei, selm, seln, selo; 2
outbreaks: seg, sei, selm, seln, selo, selp; 1 outbreak:
seg, sei, selj, selm, seln, selo, selr; 1 outbreak: selj, selr).However, it is difficult to conclude that these newly iden-
tified SEs were responsible for these food poisoning
ic elements
e (%) Suspected genomic islands and plasmids
Human nasal
swab (n = 97)
Total
(n = 166)
3 (3.1) 7 (4.2)
1 (1.0) 5 (3.0) /Sa3mu
2 (2.1) 2 (1.2)
46 (47.4) 90 (54.2)
3 (3.1) 3 (1.8) /Sa3mu, Type II mSa31 (1.0) 1 (0.6) /Sa3mu + seg, tst-1
2 (1.2) /Sa3mu, mSa1(SaPI3) Or /Sa3mw + seb
2 (1.2) /Sa3mu, pIB485
2 (2.1) 7 (4.2) /Sa3mw + seh, or /Sa3mu +
seh, selk, selq
21 (12.7) /Sa3mu, mSa1(SaPI3) + seh or
/Sa3mw + seb, seh
2 (1.2) /Sa3mu + seg, sei, seln, tst-1
1 (1.0) 5 (3.0) /Sa3mu, Type I mSab4 (3.1)
7 (7.2) 11 (6.6) /Sa3n + seb
2 (3.4) 2 (1.6) mSa1 (SaPI3)
2 (2.1) 2 (1.2) / Sa3n, mSa1 (SaPI3)
6 (6.2) 6 (3.6) Type I mSab + seb
2 (2.1) 2 (1.2) Type II mSa3, Type I mSab2 (2.1) 2 (1.2) Type I mSa4 or mSa2, Type I mSab9 (9.3) 9 (5.4) /Sa3n, Type I mSab, pIB4851 (1.0) 1 (0.6)
1 (1.0) 1 (0.6)
6 (10.2) 6 (4.7)
1 (1.0) 1 (0.6) mSa1 (SaPI1), Type I mSab28 (28.9) 49 (29.5)
1 (1.0) 1 (0.6)
16 (16.5) 23 (13.9) Type I mSab3 (3.1) 11 (6.6) /Sa3n, Type I mSab
3 (1.8) Type I mSab, pF51 (1.7) 1 (0.8) Type I mSab + selk, selq
2 (3.4) 2 (1.2)
3 (1.8) pF5
1 (1.0) 1 (0.6)
1 (1.0) 1 (0.6)
3 (3.1) 3 (1.8) /Sa3n20 (20.6) 20 (12.1)

196 K. Omoe et al. / FEMS Microbiology Letters 246 (2005) 191–198
outbreaks. The emetic activity of the newly described
SEs has not been proved, except in the cases of SEG
and SEI [5]. To confirm the relationship between these
newly identified SEs and food poisoning, it is important
to demonstrate the emetic activity of these newly de-
scribed SEs using an experimental primate model. Re-cent studies have shown that specific non-primate
animal models, such as the ferret and the house musk
shrew, respond to SEs and exhibit emetic reactions
[25,26]. However, as recommended by INCSSN, the pri-
mate model is still the gold standard for estimating the
emetic activity of SEs. Moreover, detection of superan-
tigenic toxin genes in S. aureus isolates does not imply
expression of these genes by these isolates. Omoe et al.[17] have shown that seg- and sei-harboring S. aureus
isolates produce very low levels of SEG and SEI in vitro,
although transcription of mRNA of SEG and SEI in
these isolates was proven by reverse-transcriptase PCR
analysis. Demonstration of toxin production at levels
that are sufficient to cause diseases by strains harboring
these se genes is also needed. It has been reported that
the production of specific SEs may depend on the hostenvironment and may play a role in the adaptation of
S. aureus to the host [27]. SE production should be as-
sessed in vitro, in vivo and in food, using an immunolog-
ical detection method such as ELISA to confirm the
relationship between newly identified SEs and diseases.
3.3. The relationship between superantigenic toxin
genotypes and toxin gene-encoding mobile genetic
elements
In the present study, we have shown that there are
many superantigenic toxin genotypes in S. aureus iso-
lated from food poisoning outbreaks or healthy human
nasal swabs. It has been known that almost all superan-
tigenic toxin genes are associated with mobile genetic
elements such as genomic islands (pathogenicity islands,prophages and staphylococcal cassette chromosomes)
and plasmids. Thus, we analyzed the relationship be-
tween superantigenic toxin genotypes obtained in this
study and known superantigenic toxin gene-encoding
mobile genetic elements. Table 3 summarizes the rela-
tionship between superantigenic toxin genotypes and
known mobile genetic elements. One half (16/32) of
superantigenic toxin genotypes observed in this studycould be considered as combinations of known superan-
tigenic toxin gene-encoding profiles of genomic islands
or plasmids. For example, genotype sea, sec, sell could
be a combination of /Sa3mu (sea) and Type II mSa3(sec, sell), and genotype sed, seg, sei, selj, selm, seln, selp,
selr could be a combination of pIB485 (sed, selj, selr),
Type I mSab (seg, sei, selm, seln, selo) and /Sa3n (selp).
Of the remaining 16 genotypes, 7 could be considered ascombinations of known mobile genetic elements plus
particular se genes. For example, genotype sea, seh, selk,
selq could be a combination of /Sa3mw (sea, selk, selq)
plus seh or /Sa3mu (sea) plus seh, selk, selq. The
remaining 9 genotypes, such as ‘‘seb, seh’’, ‘‘seh, selk,
selq’’, ‘‘tst-1, seg, seh, seln’’, did not follow the rule of
known superantigenic toxin gene profiles of mobile ge-
netic elements. In these genotypes, we observed severalgene combinations that could be considered incomplete
Type I mSab, such as ‘‘seg, sei, selm, seln’’, ‘‘seg, sei,
seln’’, ‘‘selm, selo’’, and ‘‘seln’’. Becker et al. [13] re-
ported the prevalence of Type I mSab-related SE genes,
and showed that a substantial number of isolates were
found to harbor only one or two of the selm, seln, and
selo genes. These results suggest the possibility of the
existence of SE-encoding variants within the genomic is-land Type I mSab, or the existence of new types of mo-
bile genetic elements encoding seg, sei, selm, seln, or
selo genes. There is also the possibility of existence of
many new types of toxin-gene-encoding mobile genetic
elements. As shown above, it seems that the se genotype
of S. aureus may be determined by mobile genetic ele-
ments it harbors. To prove this hypothesis, an effort to
explore new types of mobile genetic element is needed,as well as detailed characterization of SE-encoding
genomic islands. Previously, Baba et al. [19] mentioned
that genomic island allotyping would be a useful ap-
proach to S. aureus genotyping and that this process
would enable the prediction of the pathogenic capability
of an S. aureus clinical strain. Our multiplex PCR sys-
tem for detecting superantigenic toxin genes will be use-
ful in determining genomic island allotypes. Recently,Sergeev et al. [28] reported a PCR-based microarray as-
say system for simultaneous detection of SE genes. This
microarray system would be a powerful method for
detecting several types of se genes simultaneously. How-
ever, equipment for microarrays is expensive, and
microarrays are not widely used in common laboratories
at present. By contrast, our PCR-based superantigenic
toxin gene detection system could be performed easilyin commonly equipped clinical laboratories.
In conclusion, the newly developedmultiplex PCR sys-
tem for comprehensive detection and identification of
staphylococcal superantigenic toxin genes described here
is a potentially powerful tool for diagnosis and epidemio-
logical study ofS. aureus.The data presented here suggest
the system�s potential role in determining superantigenic
toxin genotypes as combinations of toxin gene-encodingmobile genetic elements, such as genomic islands and
plasmids. Further exploration and characterization of
new types of mobile genetic elements are needed.
Acknowledgements
This work was partly supported by grants-in-aids forscientific research from the Japan Society for the Promo-
tion of Science (Grants 15580272 and 16380205).

K. Omoe et al. / FEMS Microbiology Letters 246 (2005) 191–198 197
We thank Dr. Keichi Hiramatsu (at Juntendo Uni-
versity) for kindly providing the S. aureus strains used
in this work.
References
[1] Bergdoll, M.S. (1989) Staphylococcus aureus In: Foodbone
Bacterial Pathogens (Doyle, M.P., Ed.), pp. 463–523. Marcel
Dekker, Inc., New York, NY.
[2] McCormick, J.K., Yarwood, J.M. and Schlievert, P.M. (2001)
Toxic shock syndrome and bacterial superantigens: an update.
Annu. Rev. Microbiol. 55, 77–104.
[3] Uchiyama, T., Kamagata, Y., Yan, X.-J., Kohno, M., Yoshik-
awa, M., Fujikawa, H., Igarashi, H., Okubo, M., Awano, F.,
Saito-Taki, T. and Nakano, M. (1987) Study of the biological
activities of toxic shock syndrome toxin-1. II. Induction of the
proliferative response and the interleukin 2 production by T cells
from human peripheral blood mononuclear cells stimulated with
the toxin. Clin. Exp. Immunol. 68, 638–647.
[4] Uchiyama, T., Yan, X.-J., Imanishi, K. and Yagi, J. (1994)
Bacterial superantigens – Mechanism of T cell activation by
superantigens and their role in the pathogenesis of infectious
diseases. Microbiol. Immunol. 38, 245–256.
[5] Munson, S.H., Tremaine, M.T., Beteley, M.J. and Welch, R.A.
(1998) Identification and characterization of staphylococcal
enterotoxin type G and I from Staphylococcus aureus. Infect.
Immun. 66, 3337–3348.
[6] Jarraud, S., Peyrat, M.A., Lim, A., Tristan, A., Bes, M., Mougel,
C., Etienne, J., Vandenesch, F., Bonneville, M. and Lina, G.
(2001) egc, a highly prevalent operon of enterotoxin gene, forms a
putative nursery of superantigens in Staphylococcus aureus. J.
Immunol. 166, 669–677.
[7] Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H.,
Kobayashi, I., Cui, L., Oguchi, A., Aoki, K., Nagai, Y., Lian, J.,
Ito, T., Kanamori, M., Matsumaru, H., Maruyama, A., Mura-
kami, H., Hosoyama, A., Mizutani-Ui, Y., Takahashi, N.K.,
Sawano, T., Inoue, R., Kaito, C., Sekimizu, K., Hirakawa, H.,
Kuhara, S., Goto, S., Yabuzaki, J., Kanehisa, M., Yamashita, A.,
Oshima, K., Furuya, K., Yoshino, C., Shiba, T., Hattori, M.,
Ogasawara, N., Hayashi, H. and Hiramatsu, K. (2001) Whole
genome sequencing of methicillin-resistant Staphylococcus aureus.
Lancet 357, 1225–1240.
[8] Letertre, C., Perelle, S., Dilasser, F. and Fach, P. (2003)
Identification of a new putative enterotoxin SEU encoded by
the egc cluster of S taphylococcus aureus. J. Appl. Microbiol. 95,
38–43.
[9] Omoe, K., Hu, D.-L., Takahashi-Omoe, H., Nakane, A.
and Shinagawa, K. (2003) Identification and characteriza-
tion of a new staphylococcal enterotoxin-related putative
toxin encoded by two kinds of plasmids. Infect. Immun.
71, 6088–6094.
[10] Omoe, K., Imanishi, K., Hu, D.-L., Kato, H., Takahashi-Omoe,
H., Nakane, A., Uchiyama, T. and Shinagawa, K. (2004)
Biological properties of staphylococcal enterotoxin-like toxin
type R. Infect. Immun. 72, 3664–3667.
[11] Su, Y.-C. and Wong, A.C.L. (1995) Identification and purification
of a new staphylococcal enterotoxin, H. Appl. Environ. Micro-
biol. 61, 1438–1443.
[12] Becker, K., Roth, R. and Peters, G. (1998) Rapid and specific
detection of toxigenic Staphylococcus aureus: Use of two multi-
plex PCR enzyme immunoassays for amplification and hybrid-
ization of staphylococcal enterotoxin genes, exfoliative toxin
genes, and toxic shock syndrome toxin1 gene. J. Clin. Microbiol.
36, 2548–2553.
[13] Becker, K., Friedrich, A.W., Peters, G. and von Eiff, C. (2004)
Systematic survey on the prevalence of genes coding for staph-
ylococcal enterotoxins SElM, SElO, and SElN. Mol. Nutr. Food
Res. 48, 488–495.
[14] Chen, T.-R., Chiou, C.-S. and Tsen, H.-Y. (2004) Use of novel
PCR primers specific to the genes of staphylococcal enterotoxin
G, H, I for the survey of Staphylococcus aureus strains isolated
from food-poisoning cases and food samples in Taiwan. Int. J.
Food Microbiol. 92, 189–197.
[15] Løvseth, A., Loncarevic, S. and Berdal, K.G. (2004) Modified
multiplex PCR method for detection of pyrogenic exotoxin
genes in staphylococcal isolates. J. Clin. Microbiol. 42, 3869–
3872.
[16] Mehrotra, M., Wang, G. and Johnson, W.M. (2000) Multiplex
PCR for detection for Staphylococcus aureus enterotoxins, exfo-
liative toxins, toxic shock syndrome toxin 1, and methicillin
resistance. J. Clin. Microbiol. 38, 1032–1035.
[17] Omoe, K., Ishikawa, M., Shimoda, Y., Hu, D.-L., Ueda, S. and
Shinagawa, K. (2002) Detection of seg, seh, and sei genes in
Staphylococcus aureus isolates and determination of the entero-
toxin productivities of S. aureus isolates harboring seg, seh, and
sei genes. J. Clin. Microbiol. 40, 857–862.
[18] Lina, G., Bohach, G.A., Nair, S.P., Hiramatsu, K., Jouvin-
Marche, E. and Mariuzza, R. (2004) Standard nomenclature for
the superantigens expressed by Staphylococcus. J. Infect. Dis. 189,
2334–2336.
[19] Baba, T., Takeuchi, F., Kuroda, M., Yuzawa, H., Aoki, K.,
Oguchi, A., Nagai, Y., Iwama, N., Asano, K., Naimi, T., Kuroda,
H., Cui, L., Yamamoto, K. and Hiramatsu, K. (2002) Genome
and virulence determinants of high virulence community-acquired
MRSA. Lancet 359, 1819–1827.
[20] Lindsay, J.A., Ruzin, A., Ross, H.F., Kurepina, N. and Novick,
R.P. (1998) The gene for toxic shock toxin is carried by a family of
mobile pathogenicity islands in Staphylococcus aureus. Mol.
Microbiol. 29, 527–543.
[21] Yarwood, J.M., McCormick, J.K., Paustian, M.L., Orwin, P.M.,
Kapur, V. and Schlievert, P.M. (2002) Characterization and
expression analysis of Staphylococcus aureus pathogenicity island
3. Implications for the evolution of staphylococcal pathogenicity
islands. J. Biol. Chem. 277, 13138–13147.
[22] Zhang, S., Iandolo, J.J. and Stewart, G.C. (1998) The enterotoxin
D plasmid of Staphylococcus aureus encodes a second enterotoxin
determinant (sej). FEMS Microbiol. Lett. 168, 227–233.
[23] Fitzgerald, J.R., Monday, S.R., Foster, T.J., Bohach, G.A.,
Hartigan, P.J., Meaney, W.J. and Smyth, C.J. (2001) Character-
ization of a putative pathogenicity island from bovine Staphylo-
coccus aureus encoding multiple superantigens. J. Bacteriol. 183,
63–70.
[24] Perez-Roth, E., Claverie-Martın, F., Villar, J. and Mendez-
Alvarez, S. (2001) Multiplex PCR for simultaneous identification
of Staphylococcus aureus and detection of methicillin and mup-
irocin resistance. J. Clin. Microbiol. 39, 4037–4041.
[25] Hu, D.-L., Omoe, K., Shimoda, Y., Nakane, A. and Shinagawa,
K. (2003) Induction of emetic response to staphylococcal entero-
toxins in the house musk shrew (Suncus murinus). Infect. Immun.
71, 567–570.
[26] Wright, A., Andrews, P.L.R. and Titball, R.W. (2000) Induc-
tion of emetic, pyrexic, and behavioral effects of Staphylococcus
aureus enterotoxin B in the ferret. Infect. Immun. 68, 2386–
2389.
[27] Banks, M.C., Kamel, N.S., Zabriskie, J.B., Larone, D.H., Ursea,
D. and Posnett, D.N. (2003) Staphylococcus aureus express unique
superantigens depending on the tissue source. J. Infect. Dis. 187,
77–86.
[28] Sergeev, N., Volokhov, D., Chizhikov, V. and Rasooly, A. (2004)
Simultaneous analysis of multiple staphylococcal enterotoxin

198 K. Omoe et al. / FEMS Microbiology Letters 246 (2005) 191–198
genes by an oligonucleotide microarray assay. J. Clin. Microbiol.
42, 2134–2143.
[29] Novick, R.P. (1991) Genetic systems in staphylococci. Methods
Enzymol. 204, 587–636.
[30] Nakane, A., Okamoto, M., Asano, M., Kohanawa, M. and
Minagawa, T. (1995) Endogenous gamma interferon, tumor
necrosis factor, and interleukin-6 in Staphylococcus aureus infec-
tion in mice. Infect. Immun. 63, 1165–1172.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 199–205
Passive immunisation of hamsters against Clostridiumdifficile infection using antibodies to surface layer proteins
Julie B. O�Brien a,*, Matthew S. McCabe a, Veronica Athie-Morales a,George S.A. McDonald b, Deirdre B. Nı Eidhin a, Dermot P. Kelleher a
a Department of Clinical Medicine, Trinity College Dublin and Dublin Molecular Medicine Centre, St. James�s Hospital, Dublin, Irelandb Department of Histopathology and Morbid Anatomy, Trinity College Dublin, Ireland
Received 17 February 2005; received in revised form 30 March 2005; accepted 6 April 2005
First published online 29 April 2005
Edited by J-I. Flock
Abstract
Clostridium difficile is a major cause of antibiotic-associated diarrhoea and the primary cause of psedomembraneous colitis in
hospitalised patients. We assessed the protective effect of anti-surface layer protein (SLP) antibodies on C. difficile infection in a
lethal hamster challenge model. Post-challenge survival was significantly prolonged in the anti-SLP treated group compared with
control groups (P = 0.0281 and P = 0.0283). The potential mechanism of action of the antiserum was shown to be through enhance-
ment of C. difficile phagocytosis. This report indicates that anti-SLP antibodies can modulate the course of C. difficile infection and
may therefore merit closer investigation for use as constituents of multi-component vaccines against C. difficile associated diarrhoea.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Clostridium difficile; Diarrhoea; Surface layer proteins; Hamster model
1. Introduction
Clostridium difficile is a Gram-positive, spore-form-ing, anaerobic bacterium that is recognised as the pri-
mary cause of pseudomembraneous colitis and a
frequent cause of antibiotic-associated diarrhoea [1].
Following disruption of the normal bowel flora by anti-
biotic therapy, C. difficile colonises the gut, resulting in a
spectrum of disease ranging from asymptomatic carriage
to fulminant colitis [1]. C. difficile-associated diarrhoea
(CDAD) is a worldwide problem with major incidencein the elderly and hospitalised populations. A recent
prospective study estimated the annual cost of managing
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.005
* Corresponding author. Fax: +353 1 4542043.
E-mail address: [email protected] (J.B. O�Brien).
CDAD in the United States of America at over US $1.1
billion [2]. The human and economic impacts highlight
the need for preventive approaches against CDAD.Two toxins secreted by the bacterium (toxins A and
B) mediate the pathogenesis of disease [1], and an IgG
response against toxin A is correlated with recovery in
humans [3]. Purified inactivated toxins and antibodies
against the toxins have been shown to protect against
CDAD in the hamster model [4]. However, these vac-
cines have not been shown to eradicate infection and
consequently permit persistence of the pool of asymp-tomatic carriers.
Adhesion to the intestinal epithelium is considered an
important primary step for gut colonisation by C. diffi-
cile. Direct binding of the bacterium to Caco-2 and
HT-29 colonic epithelial cell lines, as well as to primary
. Published by Elsevier B.V. All rights reserved.

200 J.B. O’Brien et al. / FEMS Microbiology Letters 246 (2005) 199–205
intestinal epithelial cells, has recently been demonstrated
[5,6]. Two surface layer proteins (SLPs) termed high-
and low-molecular weight (MW) SLPs, form a crystal-
line regular array that covers the surface of the bacterium
[7]. Based on their location, it has been proposed that
SLPs are involved in binding of C. difficile to the intes-tinal epithelium [8]. Native forms of SLPs and a recom-
binant high-MW SLP are able to bind directly to human
gastrointestinal tissue sections [9]. Moreover, antisera
against whole bacteria and the high-MW SLP can re-
duce binding of C. difficile to the gastric epithelial cell
lines HT-29 and Hep-2, respectively [6,9]. SLPs have
been reported to elicit a strong IgG response in patients
infected with C. difficile in some studies [10,11] and highIgM anti-SLP levels have been associated with a reduced
risk of recurrent CDAD in humans [11]. These data sup-
port the use of SLPs as potential candidates for a vac-
cine against CDAD.
We tested the protective capacity of anti-SLP serum
against CDAD in a lethal hamster challenge model,
and showed the ability of high-specificity, high-titre
anti-C. difficile SLP serum to delay the progression ofCDAD but not to prevent death following onset of
CDAD. The acute lethal outcome obtained in the ham-
ster does not directly parallel clinical presentation in hu-
mans, which results in severe disease (fulminant colitis
with ileus, toxic megacolon, perforation and death) in
only 3% of patients [1,12]. This model therefore provides
an extremely stringent test for any protective effect
against CDAD.
2. Materials and methods
2.1. Culture of C. difficile and preparation of SLPs
C. difficile (PCR Ribotype 1; toxin A and B posi-
tive; clindamycin resistant; PHLS UK referenceR13537, Anaerobe Reference Unit, Public Health
Laboratory, University Hospital of Wales) isolated
from a patient with CDAD was used for preparation
of SLPs and hamster challenges. SLPs were purified
from cultures grown anaerobically at 37 �C in BHI/
0.5% thioglycolate broth. Cultures were harvested
and crude SLP extracts made with 8 M urea comple-
mented with protease inhibitors (Complete�, Roche)[7]. The crude SLPs were further purified by anion ex-
change chromatography [13]. Briefly, the crude SLP
preparation was dialysed into 20 mM Tris–HCl pH
8.5 start buffer and applied to an anion exchange col-
umn attached to an AKTA FPLC system (MonoQ
HR 10/10 column, GE Healthcare). The pure SLPs
were eluted with a linear gradient of 0–0.3 M NaCl
at a flow rate of 4 ml/min. Peak fractions correspond-ing to pure SLPs were analysed on 12% SDS–PAGE
gels stained with Coomassie blue.
2.2. Antibody production and immunoblotting
Anti-SLP serum was raised in one New Zealand
White rabbit using 100 lg of purified SLPs at weeks 0,
2 and 4 in Freund�s complete and incomplete adjuvant,
respectively. For immunoblotting, 5 lg of a crude S-layer preparation was separated by SDS–PAGE, elec-
troblotted onto PVDF membrane, and incubated with
a range of dilutions (1:1,000, 1:5,000, 1:10,000,
1:15,000) of pre-immune or immune rabbit antiserum
followed by anti-rabbit HRP. Membranes were devel-
oped using ECL detection (Amersham Biosciences).
2.3. Agglutination assay
Twofold serial dilutions (1:2 to 1:4, 096) of anti-SLP
serum were prepared in duplicate in 96 well U-bottom
microtitre plates in PBS in 50 ll final volumes. C. diffi-
cile suspension was adjusted to an OD600 = 1, and 50
ll added to antiserum dilutions. PBS and rabbit pre-
immune serum served as negative controls. Plates were
incubated for 24 h at 4 �C and the degree of agglutina-tion scored. Endpoint titres were defined as the recipro-
cal of the highest dilution of serum causing strong
agglutination.
2.4. Hamster model of C. difficile infection
Female Golden Syrian hamsters (Charles River Lab-
oratories, UK), 6 to 7 weeks old with an average mass of134.6 g, were used for passive immunisation and chal-
lenge studies. Hamsters were assigned to treatment
groups on the basis of mass so that each group had sim-
ilar average mass. Animals were fed a standard labora-
tory diet ad libitum and caged individually in isolator
cages fitted with disposable air filters to prevent cross-
contamination among animals. Autoclaved food, bed-
ding, cages and filters were used. All animal procedureswere conducted under protocols approved by the Irish
Department of Health and Children and the Trinity
College BioResources Unit Committee.
Hamsters used for model optimisation were screened
for C. difficile carriage by culturing the bacterium from
their faeces. No C. difficile was found by this method
and all animal work thereafter was carried out under
identical conditions. Following infection, C. difficile iso-lated from perianal swabs was identified by culture in an
anaerobic environment. Presumptive C. difficile re-iso-
lated from hamsters in a preliminary experiment was
checked by immunoblotting with the rabbit anti-SLP
serum and invariably reacted identically to the infecting
type (data not shown).
Hamsters were given a 2 mg dose of clindamycin-HCl
(Sigma) orogastrically to predispose them to C. difficile
infection and challenged with 105 CFU 4 h later. For
experimental hamsters (n = 8), the challenge inoculum

J.B. O’Brien et al. / FEMS Microbiology Letters 246 (2005) 199–205 201
was pre-incubated with 100 ll of anti-SLP serum for 30
min at 37 �C and additional 100 ll antiserum doses were
given orogastrically at – 7, 6, 17 and 24 h of infection.
Antiserum was co-administered with 0.1 M sodium car-
bonate buffer, pH 9.6, to neutralise gastric acid. Control
hamsters (n = 8) were treated identically, but were givenan irrelevant rabbit antiserum raised against the malt-
ose-binding protein of Escherichia coli (anti-MBP ser-
um). A C. difficile only group, which received no
antiserum, was also included (n = 4).
From the day after infection, hamsters were observed
two-hourly in a blinded fashion by three individual
observers for 72 h, and four times a day at regular inter-
vals thereafter. Grading was as follows: 0, normal; 1,loose faeces or wet perianum, activity close to normal;
2, reduced activity, still responding to stimuli, tender
abdomen; 3, hunched, inactive, tender abdomen, loss
of balance, ruffled fur. Hamsters were sacrificed at grade
3. Time of sacrifice or last time seen alive (whichever was
earlier) was considered the endpoint.
To confirm C. difficile as the causative agent of dis-
ease, perianal swabs (no formed faeces due to diar-rhoea) were taken in a random order from a
representative number of symptomatic hamsters
(n = 5) and cultured anaerobically for four days on
blood agar plates containing 50 lg/ml clindamycin.
Caecum and colon samples were taken from a repre-
sentative number of animals (one hamster per group)
to confirm the typical epithelial damage seen in
CDAD. Tissues were fixed in 10% formalin andstained with haematoxylin and eosin.
2.5. C. difficile phagocytosis assay
C. difficile cultured on Columbia blood agar plates
was resuspended into 1 ml of RPMI-1640 medium (Gib-
coBRL) containing 5 lM 5-(and -6)-carboxyfluorescein
diacetate, succinimidyl ester (CFSE) (Molecular Probes)and stained for 10 min at 37 �C in the dark. The stained
bacteria were washed with RPMI/50% FCS and bacte-
rial concentration determined using OD600.
Human monocytic THP-1 cells were grown in RPMI
medium supplemented with 10% FCS, 2 mM L-gluta-
mine, 100 lg/ml penicillin and 100 U/ml streptomycin
at 37 �C in 5% CO2. THP-1 cells (1 · 106 cells per well
in 24-well plates) were induced to differentiate with100 nM PMA in complete medium for 72 h. Prior to
exposure to C. difficile, THP-1 cells were washed twice
with RPMI/10% FCS, mixed with the CFSE-C. difficile
(MOI of 400) and incubated at 37 �C in 5% CO2 for 2 h.
Cells were washed three times with PBS to remove de-
tached THP-1s and non-phagocytosed C. difficile.
THP-1s were detached with 250 ll per well of pre-cooleddetaching buffer (5 mM EDTA, 0.5% FCS in HBSS) for20 min at 200 rpm at 4 �C and washed three times with
pre-cooled PBS.
Green fluorescent emission of the phagocytosed C.
difficile was assessed by flow cytometry analysis. Single
THP-1 cells were gated using FSC and SSC. Events
(30,000) were acquired using a FACSCalibur flow
cytometer and data was analysed with Cell Quest soft-
ware (Becton Dickinson). The fluorescence of cell-surface attached C. difficile was quenched immediately
prior to acquisition by addition of 0.8 mg/ml crystal
violet.
The effects of anti-SLP and anti-MBP sera (12.5%) on
phagocytosis were assessed by co-incubation with the C.
difficile and THP-1 cells. To assess opsonising activity
due to complement, the anti-SLP serum was heat-trea-
ted at 56 �C for 30 min to inactivate complement. E. coliDH5a (pKFW408) expressing a modified GFP was used
as a positive control for phagocytosis (Fig. 3(d)).
2.6. Statistical analysis
Differences in mean survival time and mean time to
first symptoms between experimental and control
hamsters were tested for significance by a non-para-metric two-tailed Mann Whitney test (GraphPad
InStat). A P-value <0.05 was considered statistically
significant.
3. Results
3.1. SLP purification, antiserum production and
agglutination of C. difficile by anti-SLP
Based on their location on the outer bacterium sur-
face and their in vitro capacity to bind to human gastro-
intestinal tissues (9), SLPs represent a strong vaccine
candidate for targeting C. difficile colonisation and
CDAD. To develop an anti-SLP serum for passive
immunisation experiments, we initially purified SLPsto homogeneity from a crude S-layer preparation by an-
ion exchange chromatography (Fig. 1(a)). Pure SLPs
were then used to raise rabbit polyclonal antibodies,
which reacted strongly with both the high- and low-
MW SLPs by immunoblotting against a crude SLP
preparation (Fig. 1(b)). Pre-immune rabbit serum
showed no reactivity within a wide range of concentra-
tions (Fig. 1(b) and data not shown). Anti-SLP serumdid not recognise toxins as demonstrated by immuno-
blotting against a total C. difficile lysate (Fig. 1(b)).
The anti-MBP serum (diluted 1:100) showed no reactiv-
ity against a total C. difficile lysate by immunoblotting
(data not shown).
To corroborate that the anti-SLP serum was capable
of recognising SLPs on the surface of intact whole
C. difficile, we conducted in vitro agglutination experi-ments using twofold serial dilutions of anti-SLP or pre-
immune antiserum. The anti-SLP antibodies readily

Fig. 1. (a) Coomassie blue stained 12% SDS–PAGE gel showing purified C. difficile surface layer proteins (SLPs). MWmarker (lane 1), crude S-layer
preparation made with 8 M urea (lane 2), anion exchange purified SLPs (lane 3). (b) Immunoblot of the crude S-layer preparation probed with pre-
immune rabbit serum 1:15,000 (lane 1) and anti-SLP rabbit serum 1:15,000 (lane 2). C. difficile lysate probed with anti-SLP rabbit serum 1:15,000
(lane 3).
202 J.B. O’Brien et al. / FEMS Microbiology Letters 246 (2005) 199–205
agglutinated whole C. difficile cells, with a titre of 512 as
compared to a titre of 8 for the pre-immune serum.
3.2. Passive immunisation with anti-SLP prolonged
survival in C. difficile infected hamsters
We tested the ability of passively administeredanti-SLP serum to protect against CDAD in a lethal
hamster challenge model. All animals were predisposed
1 2 30
50
100
150
200
250*
anti-
SLP
seru
m
anti-
MB
Pse
rum
untr
eate
d
post
-hc
lale
ng
us erv
ilav,h
Fig. 2. Passive immunisation with anti-SLP serum prolongs survival in
C. difficile-infected hamsters. Following clindamycin treatment ham-
sters were given anti-SLP serum (group 1, n = 8) or anti-MBP serum
(group 2, n = 8) or no serum (group 3, n = 4). All groups received 105
C. difficile. T0 = time of administration of bacteria. Dots (�) representindividual hamsters, bars represent median values for each group.
Post-challenge survival was significantly prolonged in experimental
hamsters compared to hamsters treated with anti-MBP serum
(*P = 0.0281) and untreated hamsters (*P = 0.0283). Differences in
means were calculated using a non-parametric two-tailed Mann
Whitney test.
to C. difficile infection by clindamycin treatment, fol-
lowed by passive immunisation with anti-SLP serum,
anti-MBP serum or no serum. All three groups were
then challenged with C. difficile (105 CFU). Hamsters
from all three groups developed diarrhoea within two
to five days post-challenge, showing no significant differ-
ence in time to first symptoms (data not shown). How-ever, post-challenge survival (Fig. 2) was significantly
prolonged in animals treated with anti-SLP serum
(group 1) as compared to animals treated with anti-
MBP serum (group 2) (P = 0.0281) and untreated ani-
mals (group 3) (P = 0.0283) (Table 1). Seven out of eight
(group 2) and three out of four (group 3) control ham-
sters as compared to two out of eight (group 1) anti-
SLP treated hamsters had died at 72 h post-challenge.The pathology was confirmed as CDAD by the
typical epithelial damage observed on histological
examination of caecum and colon sections. C. difficile
was confirmed as the causative agent of the pathology
by isolation from perianal swabs. Since all hamsters,
independently of the treatment group, were sampled
Table 1
Post-challenge survival time of hamsters passively immunised with
anti-SLP serum and challenged with 105 C. difficile
Groupa Median post-challenge survival (h)b Range (h)
1 156.9 62.5, 221.5
2 76.6 55.2, 143.0
3 69.6 55.3, 126.4
a Group 1 received anti-SLP serum plus 105 C. difficile, group 2
received anti-MBP serum plus 105 C. difficile, group 3 received 105 C.
difficile only.b Post-challenge survival time was measured from time of adminis-
tration of C. difficile.

J.B. O’Brien et al. / FEMS Microbiology Letters 246 (2005) 199–205 203
at the same advanced stage of the infection and since
volume samples are physically impossible to standard-
ise, C. difficile counts in perianal swabs were not
determined. Since all hamsters eventually died from
CDAD, severe inflammation was observed to the same
Fig. 3. Anti-SLP serum enhances C. difficile phagocytosis by PMA differentia
(grey histogram), CFSE-C. difficile (open histogram). (b) Crystal violet q
histogram), plus 1.6 mg/ml crystal violet (open histogram, dashed line), plu
difficile (grey histogram). (c) C. difficile phagocytosis by THP-1s. THP-1s alon
E. coli DH5a (pKFW408) expressing a modified GFP was used as a pos
histogram). (e) In the presence of 12.5% anti-SLP serum (open histogram) o
(grey histogram). (f) In the presence of anti-SLP serum (open histogram) an
alone (grey histogram). Graphs are representative of at least three independ
extent in histological sections from all groups (data
not shown). The present data show that although pas-
sive immunisation with anti-SLP serum was unable to
delay the onset of CDAD and prevent animal death,
it did significantly prolong survival.
ted THP-1 cells. (a) CFSE staining of C. difficile. Unstained C. difficile
uenching of CFSE-C. difficile fluorescence. CFSE-C. difficile (open
s 0.8 mg/ml crystal violet (open histogram, dotted line), unstained C.
e (grey histogram), THP-1s and CFSE-C. difficile (open histogram). (d)
itive control for phagocytosis (open histogram), THP-1s alone (grey
r 12.5% anti-MBP serum (open histogram, dashed line), THP-1s alone
d heat-treated anti-SLP serum (open histogram, dotted line), THP-1s
ent experiments.

204 J.B. O’Brien et al. / FEMS Microbiology Letters 246 (2005) 199–205
3.3. Anti-SLP enhanced C. difficile phagocytosis
We considered that the effect of the anti-SLP serum
on post-challenge survival of the hamsters might be
mediated by increasing phagocytosis of C. difficile. To
investigate this possibility, phagocytosis of fluorescentstained C. difficile by differentiated THP-1 monocytes
was assessed in the presence and absence of anti-SLP
serum. Using this technique, 100% of C. difficile were
consistently stained with CFSE (Fig. 3(a)). CFSE-C. dif-
ficile fluorescence could be readily quenched with crystal
violet (Fig. 3(b)) to eliminate fluorescence from cell-
surface attached C. difficile. The levels of phagocytosis
of CFSE-C. difficile were low (17.97%) (Fig. 3(c)). E. coliDH5a (pKFW408) expressing a modified GFP was used
as a positive control for phagocytosis (Fig. 3(d)). The
addition of the anti-SLP serum resulted in markedly in-
creased phagocytosis of C. difficile (69.19%) as com-
pared to the anti-MBP serum (Fig. 3(e)). To eliminate
the possibility that this increase was due to complement,
the anti-SLP serum was heat-treated to inactivate com-
plement before the phagocytosis assay. No major contri-bution of complement to the phagocytosis was found
(Fig. 3(f)). These results suggest that the anti-SLP anti-
bodies may exert at least some of their protective effect
in vivo by enhancing phagocytosis of C. difficile.
4. Discussion
C. difficile toxins are known to mediate many of the
pathogenic features of CDAD. Previous studies have
shown protection of hamsters against CDAD using anti-
toxin antibodies through neutralisation of enterotoxicity
[4,14]. In one of these studies, administration of mouse
anti-toxin ascites protected 100% of hamsters from
death [4]. Although the role of anti-toxin immunity in
protection from CDAD in the hamster model is clear,vaccines based on toxins are unlikely to prevent coloni-
sation, and carriage and transmission of C. difficile will
therefore remain a persistent threat. Hence, a more com-
plete approach against CDAD should consider not only
the inhibition of toxicity but also the prevention of bac-
terial colonisation. The SLPs of C. difficile are potential
colonisation agents thought to be involved in bacteria–
host interactions [5,6,9]. It has been shown that anti-SLP antibodies decrease the direct binding of C. difficile
to human gastrointestinal tissue sections in vitro [9]. We
tested whether anti-SLP antibodies, assessed indepen-
dently of the toxins, could have a protective effect
against CDAD in vivo. Here we demonstrate that pas-
sive immunisation using anti-SLP antibodies signifi-
cantly delays progress of C. difficile infection in a
lethal hamster challenge model. To the authors� knowl-edge, this report identifies anti-SLP antibodies as poten-
tially playing a role in the modulation of disease
progression in an in vivo model of C. difficile infection
for the first time.
The recognition and interaction of the anti-SLP anti-
bodies with the SLPs on intact C. difficile could be hin-
dered by spatial and temporal constrains inherent to the
in vivo setting, allowing for the presence of free bacteria.To minimise this possibility in our system, the C. difficile
inoculum was pre-incubated with the respective antisera
prior to infection, an approach that has been success-
fully used elsewhere in passive immunisation for gastro-
intestinal pathogens [15].
The mechanism of action of the anti-SLP serum is
most likely multifactorial. Following C. difficile coloni-
sation of the gut epithelium and subsequent toxin pro-duction, the epithelial barrier is severely disrupted
resulting in an influx of phagocytic cells. Despite the
presence of phagocytic cells, C. difficile can persist in this
inflammatory exudate [16]. Using an in vitro assay, we
demonstrated a marked increase in phagocytosis of C.
difficile in the presence of the anti-SLP serum. These re-
sults are in agreement with Dailey et al. [16], who
showed that C. difficile requires opsonisation for signif-icant phagocytosis to occur. These data suggest that the
prolonged survival of anti-SLP treated hamsters might
be explained through an increase in phagocytosis of C.
difficile at damaged mucosal surfaces. However, as
shown in vitro, the anti-SLP serum might also inhibit
colonisation by agglutinating C. difficile in vivo and
therefore inhibiting binding of the bacterium to the epi-
thelium. A correlation between antibody-mediatedagglutination of C. difficile and protection against death
has previously been demonstrated in a hamster study
[17].
Hamsters are highly sensitive to the C. difficile toxins
and once their systemic action occurs the animals even-
tually die. In our study, all hamsters showed a similar
time to first symptoms and eventually died, indicating
that independently of treatment all were exposed tothe enterotoxins. Hence, the prolonged survival in the
anti-SLP treated group most likely reflects exposure to
lower toxin concentrations from a lower bacterial load
achieved by increased phagocytosis.
Based on our study it seems possible that the develop-
ment of a strong, specific mucosal immune response
against the SLPs has the potential to result in improved
long-term protection and elimination of bacterial car-riage. Although the antiserum used in this study was
IgG, it was delivered directly to the mucosal surface,
thereby mimicking a mucosal immune response. This
approach has been successfully used with IgG and IgY
anti-toxin antibodies in the hamster model of CDAD
[4,18].
In conclusion, our findings point to a role for SLPs in
the design of effective vaccines against CDAD. We pro-pose SLPs as candidate components of a multi-compo-
nent vaccine against C. difficile. In this study we used

J.B. O’Brien et al. / FEMS Microbiology Letters 246 (2005) 199–205 205
a C. difficile strain from the most commonly occurring
ribotype, Ribotype 1, which accounts for over half of
all the hospital isolates from England and Wales [19].
Sequencing of SLPs from three clinical isolates from
Ribotype 1 from different C. difficile episodes revealed
100% sequence identity (manuscript in preparation).However, given the well-known heterogeneity among
SLPs from different C. difficile strains [7,8], an effective
vaccine offering cross-protection between strains should
contain multiple SLPs. An active immunisation regimen
based on the SLPs combined with toxin neutralisation
could provide both a preventative and therapeutic vac-
cine strategy by reducing bacterial colonisation and car-
riage, as well as toxin-mediated pathology.
Acknowledgements
This work was supported by an Advanced Technolo-
gies Research Programme (Grant No. 01/165) fromEnterprise Ireland. Thanks to Dr. Kenneth F. Whelan,
Institute of Molecular Medicine, for providing E. coli
DH5a (pKFW408).
References
[1] Kelly, C.P. and LaMont, J.T. (1998) Clostridium difficile infection.
Annu. Rev. Med. 49, 375–390.
[2] Kyne, L., Hamel, M.B., Polavaram, R. and Kelly, C. (2002)
Health care costs and mortality associated with nosocomial
diarrhea due to Clostridium difficile. Clin. Infect. Dis. 34, 346–353.
[3] Kyne, L., Warny, M., Qamar, A. and Kelly, C.P. (2001)
Association between antibody response to toxin A and protection
against recurrent Clostridium difficile diarrhoea. Lancet 357, 189–
193.
[4] Giannasca, P.J., Zhang, Z., Lei, W., Boden, J.A., Giel, M.A.,
Monath, T.P. and J.R., W.D. (1999) Serum antitoxin antibodies
mediate systemic and mucosal protection from Clostridium
difficile disease in hamsters. Infect. Immun. 67, 527–538.
[5] Cerquetti, M., Serafino, A., Sebastianelli, A. and Mastrantonio,
P. (2002) Binding of Clostridium difficile to Caco-2 epithelial cell
line and to extracellular matrix proteins. FEMS Immunol. Med.
Microbiol. 32, 211–218.
[6] Drudy, D., O�Donoghue, D.P., Baird, A., Fenelon, L. and
O�Farrelly, C. (2001) Flow cytometric analysis of Clostridium
difficile adherence to human intestinal epithelial cells. J. Med.
Microbiol. 50, 526–534.
[7] Cerquetti, M., Molinari, A., Sebastianelli, A., Diociaiuti, M.,
Petruzzelli, R., Capo, C. and Mastrantonio, P. (2000) Character-
isation of surface layer proteins from different Clostridium difficile
clinical isolates. Microb. Pathog. 28, 363–372.
[8] McCoubrey, J. and Poxton, I.R. (2001) Variation in the surface
layer proteins of Clostridium difficile. FEMS Immunol. Med.
Microbiol. 31, 131–135.
[9] Calabi, E., Calabi, F., Phillips, A.D. and Fairweather, N.F. (2002)
Binding of Clostridium difficile surface layer proteins to gastro-
intestinal tissues. Infect. Immun. 70, 5770–5778.
[10] Pantosti, A., Cerquetti, M., Viti, F., Ortisi, G. and Mastrantonio,
P. (1989) Immunoblot analysis of serum immunoglobulin G
response to surface proteins of Clostridium difficile in patients
with antibiotic-associated diarrhea. J. Clin. Microbiol. 27, 2594–
2597.
[11] Drudy, D., Calabi, E., Kyne, L., Sougioultzis, S., Kelly, E.,
Fairweather, N. and Kelly, C.P. (2004) Human antibody response
to surface layer proteins in Clostridium difficile infection. FEMS
Immunol. Med. Microbiol. 41, 237–242.
[12] Borriello, S.P., Ketley, J.M., Mitchell, T.J., Barclay, F.E., Welch,
A.R., Price, A.B. and Stephen, J. (1987) Clostridium difficile – a
spectrum of virulence and analysis of putative virulence determi-
nants in the hamster model of antibiotic-associated colitis. J. Med.
Microbiol. 24, 53–64.
[13] Takeoka, A., Takumi, K., Koga, T. and Kawata, T. (1991)
Purification and characterisation of S layer proteins from Clos-
tridium difficile GAI 0714. J. Gen. Microbiol. 137, 261–267.
[14] Lyerly, D.M., Bostwick, E.F., Binion, S.B. and Wilkins, T.D.
(1991) Passive immunization of hamsters against disease caused
by Clostridium difficile by use of bovine immunoglobulin G
concentrate. Infect. Immun. 59, 2215–2218.
[15] Blanchard, T.G., Czinn, S.J., Maurer, R., Thomas, W.D., Soman,
G. and Nedrud, J.G. (1995) Urease-specific monoclonal antibod-
ies prevent Helicobacter felis infection in mice. Infect. Immun. 63,
1394–1399.
[16] Dailey, D.C., Kaiser, A. and Schloemer, R.H. (1987)
Factors influencing the phagocytosis of Clostridium difficile
by human polymorphonuclear leukocytes. Infect. Immun. 55,
1541–1546.
[17] Torres, J.F., Lyerly, D.M., Hill, J.E. and Monath, T.P. (1995)
Evaluation of formalin-inactivated Clostridium difficile vaccines
administered by parenteral and mucosal routes of immunization
in hamsters. Infect. Immun. 63, 4619–4627.
[18] Kink, J.A. and Williams, J.A. (1998) Antibodies to recombinant
Clostridium difficile toxins A and B are an effective treatment and
prevent relapse of C. difficile-associated disease in a hamster
model of infection. Infect. Immun. 66, 2018–2025.
[19] Brazier, J.S. (1998) The epidemiology and typing of Clostridium
difficile. J. Antimicrob. Chemother. 41 (Suppl. C), 47–57.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 207–212
Morphological and molecular taxonomy of Pythium longisporangiumsp. nov. isolated from the Burgundian region of France
Bernard Paul a,*, Kanak Bala a, Sabine Gognies b, Abdel Belarbi b
a Laboratoire des Sciences de la Vigne, Institut Jules Guyot, Universite de Bourgogne, B.P. 27877, 21078 Dijon, Franceb Laboratoire de Microbiologie Generale et Moleculaire, Universite de Reims, Europol Agro, B.P. 1039, 51687 Reims cedex 2, France
Received 16 March 2005; received in revised form 6 April 2005; accepted 7 April 2005
First published online 19 April 2005
Edited by R.C. Staples
Abstract
During the course of an investigation on the Pythiaceous oomycetes occurring in the Burgundian vineyards, some species of
Pythium possessing mainly hypogynous antheridia were found. These had been classified as oomycetes belonging to the ‘‘Pythium
rostratum’’ group for a long time. Three of these isolates, having similar structures and growth, are very closely related to a recently
described species, Pythium bifurcatum Paul. A close look at these, however, underlines some fundamental differences with the latter.
Not all of them produce zoospores but have very large sporangia. The type specimen is F-1200 (B 76a) which is a medium-slow
growing saprophyte. The sequence of the ITS region of the rDNA also shows a very close relationship with P. bifurcatum. On
the basis of morphological and molecular analysis, we now describe this species as Pythium longisporangium sp. nov. Morphological
features of this new species, the sequences of the ITS region of its nuclear ribosomal DNA, and its comparison with related species
are discussed.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Pythium longisporangium; Sporangia; Oogonia; Antheridia; Oospores; ITS region; rRNA
1. Introduction
The genus Pythium is a very common oomycete
found all over the world. In 1990, it was known to have
more then 130 species [1]. The first author of this man-
uscript has also added more then 20 species to this genussince that date. Many of the isolates belonging to this
genus are currently being studied for their structural
and molecular diversity. Some of these are also being
studied for their mycoparasitic activities on fungal
pathogens like Botrytis cinerea [2]. Most of the members
of the genus Pythium live in soil or aquatic environments
as saprophytes, however, some of them are known to be
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.004
* Corresponding author. Tel./fax: +33 3 80396326.
E-mail address: [email protected] (B. Paul).
very destructive plant pathogens, inflicting serious eco-
nomic losses of crops by destroying seed, storage or-
gans, roots, and other plant tissues [3]. Although, the
majority of these organisms produce biflagellate zoo-
spores, they are no longer considered as ‘‘aquatic fungi’’
and unlike most of the eumycetes, the members of thisgroup remain diploid throughout their life cycles with
meiosis occurring in the gametangia before fertilization.
The entire group of the oomycetes are now supposed to
be closer to algae (Phaeophyta and Chrysophyta) and
higher plants. These are now classified in the Kingdom
Stramenopila, one of the eukaryotic Kingdoms which
includes water molds and brown algae. The position of
the oomycetes as a unique lineage of stramenopileeukaryotes, unrelated to true fungi but closely related
to heterokont (brown) algae, has been well established
. Published by Elsevier B.V. All rights reserved.

208 B. Paul et al. / FEMS Microbiology Letters 246 (2005) 207–212
using molecular phylogenies that are based on ribo-
somal RNA (rRNA) sequences [4–6].
The taxonomy of the genus Pythium was mainly
based on the morphological descriptions like the size
and shape of oogonia, antheridia, and sporangia. Keys
provided by Middleton [7], Plaats-Niterink [8] andWaterhouse [9] are all based on these morphological
characters and are still indispensable. However these
are now being supplemented with molecular characteris-
tics. Comparative studies of the internal transcribed
spacer (ITS) regions of the ribosomal RNA genes
(rDNA) have become a useful tool in fungal taxonomy
as these regions evolve sufficiently rapidly to distinguish
different species within a genus [10,11]. The ITS se-quence data can provide valuable information on
‘‘new’’ or undescribed taxa, as sequence diversity may
support the erection of new species [12].
The morphology of Pythium longisporangium is typi-
cal for the genus Pythium. It is a slow growing oomy-
cete, having large spherical, globose, to cylindrical
sporangia; smooth-walled oogonia and mostly hypogy-
nous antheridia. Sexual structures are readily formedin water and on solid media within a week. The morpho-
logical details of the new species together with the se-
quences of its ITS region of the ribosomal nuclear
DNA, comparison of morphological and molecular
characteristics with related species are discussed in this
article.
2. Materials and methods
2.1. Fungal and oomyceteous material
P. longisporangium (Type strain, F-1201/B76a) was
isolated from soil samples taken in a vineyard in Mar-
sannay situated in the outskirts of the French city of Di-
jon in the Burgundian region by using the usual baitingtechniques [8,13]. It occurred thrice out of 65 such sam-
ples and was purified by repeated washing with sterile
distilled water and sub-culturing on solid media like po-
tato carrot agar (PCA) and corn meal agar (CMA). All
these isolates are maintained at ‘‘Institut Jules Guyot’’,
in Dijon, France. P. longisporangium was identified with
the help of keys provided by Middleton [7], Plaats-Nit-
erink [8] and Waterhouse [9] and also by its ITS se-quences using the BLAST search.
2.2. DNA isolation and PCR
All three isolates of P. longisporangium were grown in
PDB (potato dextrose broth) The culture conditions,
DNA isolation and the PCR of the ITS of the ribosomal
nuclear DNA was done using the procedures describedearlier [14,15]. Universal primers ITS1 (TCC GTA
GGT GAA CCT GCG G) and ITS4 (TCC TCC GCT
TAT TGA TAT GC) were synthesised and the DNA se-
quence was realised by Oligo Express (Paris). ITS1 is at
the 3 0 end of the 18S rDNA gene and ITS4 is at the 5 0
end of the 28S rDNA gene. The sequences obtained
were compared with the ITS1 sequences of related spe-
cies of Pythium: Pythium bifurcatum (Genbank accessionnumber AY083935), Pythium longandrum (AY039713),
P. sp. strain F-74 (AY455695), Pythium hypogynum
(AY455804),Pythium terrestris (AY039714), andPythium
segnitium (AY149173). Pythium sp. strain F-1216
(AY455697), Pythium canariense (AY06561) and Pythium
rostratum (AJ233456). The sequence of the ITS
region of the nuclear ribosomal DNA of P. longisporan-
gium (F-1200) has been deposited in Genbank.
3. Results
3.1. Morphological descriptions
P. longisporangium PAUL sp. nov. (Figs. 1–3).
Sporangia globosa, subglobosa, intercalaria, interdumterminalia, 25–50 lm diam., zoosporae non-observata.
Oogonia laevia, globosa, terminalia raro intercalaria,
17–36 lm diam, Antheridia hypogynata vel monoclinata.
cellulae antheridiales inflatae interdum bifurcatae, unam
vel duas in singulo oogonia. Oogonia continentia unam,
interdum duas oosporas pleroticas 16–35 lm diam, globo-
sas, 1.5–2 lm crassi tunicatas. Interdum oogonia et anthe-
ridia oriuntur ex appressorio. Incrementum radiale
quotiadianum 11 mm 25 �C in agaro Solani tuberosi et
Dauci carotae (PCA). Holotypus in herbario Universita-
tus Bourgogne conservatus (F-1200).
Etymology: The oomycete is being named as P. lon-
gisporangium because of the form of its sporangia which
often are pyriform to lemoniform and are formed in
abundance.
Mycelium hyaline, well branched. Main hyphae upto 6–8 lm wide. Colonies on PCA are submerged and
show a narrow chrysanthemal pattern. Average radial
growth of the oomycete at 25 �C on PCA is 11 mm/
day. It grows well in water on hemp-seed halves and
produces asexual and sexual structures at room temper-
atures (18–25 �C).Sporangia (Fig. 1, (a)–(d)) are globose to somewhat
cylindrical, oval, and at times peanut shaped mostlycatenulate and intercalary (Fig. 1(b)); measuring 15–55
lm in diameter (av. 32.6 lm) and up to 65 lm in length.
These structures are densely granulated and the larger
ones have a clear hyaline central zone which is sparsely
granulated (Fig. 1, (c)). Zoospores were not observed
despite repeated flooding of the cultures by distilled
water, tap water or pond water. The sporangia germi-
nate directly through germ tubes to produce a newmycelium (Fig. 1, (a, d)).

Fig. 1. (a)–(d): Sporangia of Pythium longisporangium. (a, c): terminal sporangia, (b): intercalary sporangia; (d): sporangia germinating through a
germ tube; (e)–(h): antheridia and oogonia of Pythium longisporangium. (e): Hypogynous antheridia with bulbous antheridial cells; (f): antheridia
showing intercalary antheridial cells; (g): intercalary oogonia with hypogynous antheridia and two bulbous antheridial cells; (h): fertilization. All
figures bar = 40 lm.
B. Paul et al. / FEMS Microbiology Letters 246 (2005) 207–212 209
The oomycete reproduces sexually by forming anthe-
ridia and oogonia plentifully in water cultures on hemp-seed halves and also on solid media like PCA. Oogonia
are smooth walled, spherical, terminal, sub-terminal and
intercalary (Fig. 1, (e)–(h)), measuring 17–36 lm in
diameter (av. 19.5 lm) and filled with dense, coarsely
granulated protoplasm.
All the oogonia are supplied with antheridia which
are usually hypogynous (Fig. 1, (e)–(h)) or monoclinous
sessile (Fig. 1, (c)). Each oogonium is supplied by 1–3antheridial cells which are at times borne in a catenulate
fashion on one antheridial branch (Fig. 1, (f), 2, (c, d)).
Very rarely diclinous antheridia are also present (Fig. 2,
(a, b)). After fertilization the oogonia is found attachedwith one or two balloon-shaped antheridial cells (Fig. 1,
(h), 2, (c, d, f)).
Oospores are plerotic or nearly so (Fig. 2, (e)–(h)),
spherical, usually one per oogonium (Fig. 2, (e)), occa-
sionally two per oogonium (Fig. 2, (f)) and rarely three
in a single oogonium (Fig. 2, (g)). In the intercalary
oogonia these can be aplerotic (Fig. 2, (h)); smooth-
walled, measuring 12–22 lm in diameter (av. 18.1 lm).The oospore wall is relatively thin, measuring 1–1.5
lm in thickness.

Fig. 2. Sexual reproduction of Pythium longisporangium. (a, b): Oogonia provided with diclinous antheridia; (c): oogonia provided with hypogynous
antheridia,antheridial branch bearing intercalary antheridial cells; (d): oogonia provided with hypogynous antheridia and bulbous antheridial cells;
(e): plerotic, single oospores; (f): two oospores per oogonium; (g): three oospores per oogonium; (h): intercalary oogonia having a single aplerotic
oospore. (a, c) bar = 50 lm, (b, d, e–h) bar = 25 lm.
210 B. Paul et al. / FEMS Microbiology Letters 246 (2005) 207–212
3.2. Internal transcribed spacer region
The ITS region of the nuclear ribosomal DNA of P.
longisporangium (Genbank accession number
AY455693) is comprised of 890 bases which are:
1 ccacacctaa aaactctcca cgtgaactgt ttgtatcaga ttagcgc-
caa gattttcgtg
61 cgtgtttgtg gtatcactat gtattcgtac gtggtgttag caag-
cattgt atggagcttg121 gctgatcgaa ggtcggtgcg caccttgtgt gtgtattggc tgat-
taacct tttaaaccct
181 ttcaataaat actgattata ctgtaaggac gaaagtcttt
gcttttatct agataacaac
241 tttcagcagt ggatgtctag gctcgcacat cgatgaagaa
cgctgcgaac tgcgatacgt301 aatgcgaatt gcagaattca gtgagtcatc gaaattttga acg-
catattg cactttcggg
361 ttatacctgg aagtatgtct gtatcagtgt ccgtacatca
aacttgcctc tttttgtcgg
421 tgtagtccga ttgagagtat ggcagacgtg aggtgtctcg
cgactcgtat atcattgtgt
481 gtgtaaatcg taagagatac atacataagg tagtatataa
cttgttgcga gtccctttaa541 aacgacacga tctttctatt tgctttctat ggagcgtcta
tttcgaacgc ggtggtcctc
601 ggatcgcttg cagtcggcag cgacttcagt gaagacatag
tgaagaaacc tctattcgcg

Fig. 3. CLUSTAL W, multiple sequence alignment of ITS1 regions of the rDNA of Pythium longisporangium, P. bifurcatum, P. longandrum, P.
hypogynum, P. terrestris, P. segnitium, P. rostratum, P. canariense, P. sp. (F-74), and P. sp. (F-1216).
B. Paul et al. / FEMS Microbiology Letters 246 (2005) 207–212 211
661 gtacgttagg cttcggctcg acaatgttgc gttctagtgt
gtggactccg ttttcgcttt
721 gaggtgtact gttcggttgt gggtttgagc cttggtattg ctttgt-
tagt agagatatgt
781 cgatatttct gtagtttgat tctgcataca cgcaagtgtgtgtgggtaga gagtatctat
841 ttgggaaatt ttgtactgcg tgcgctttcg agtgtgtgta
tgtatctcaa
1–236 = ITS1 complete sequence, 237–395 = 5.8 S
gene (in bold) complete sequence, 396–890 = ITS 2 com-plete sequence.

Table 1
Comparison of morphological features of Pythium longisporangium
and P. bifurcatum
Characters Pythium longisporangium Pythium bifurcatum
Sporangia Intercalary and catenulate,
15–55 lm (av. 32.6 lm)
Intercalary and
terminal, 25–50 lm(av. 35 lm)
Oogonia
diameter
13–23 lm (av. 19.5 lm) 17–36 lm(av. 26.7 lm)
Antheridia Hypogynous, monoclinous sessile,
diclinous, antheridial cells at times
catenulate
Hypogynous,
monoclinous sessile
Oospore Usually one, frequently 2 and
rarely 3 per oogonium,
12–22 lm (av. 18.1 lm).
Usually 1, rarely 2,
per oogonium,
16–35 lm(av. 24.9 lm)
212 B. Paul et al. / FEMS Microbiology Letters 246 (2005) 207–212
The comparison of the ITS1 sequences of P. longisp-
orangium and related species is given in Fig. 3 in the
form of CLUSTAL multiple alignments.
4. Discussion
P. longisporangium (F-1200) is a slow growing speciesthat falls within the category of species having spherical
non-proliferating, catenulate sporangia, smooth walled,
globose oogonia which are provided by hypogynous
antheridia. These characters bring this species very close
to a recently-described species, P. bifurcatum PAUL
from northern France. However the ITS sequences of
these species are very different, and a close look at the
morphological characters shows that they are distinctbut closely related (Table 1). The presence of triple
oospores and intercalary chained-antheridial cells are
unique for the taxon.
The ITS region of the nuclear ribosomal DNA of P.
longisporangium is comprised of 890 bases and a BLAST
search gives the closest resemblance of this oomycete
with P. bifurcatum having 99.6% similarity (Genbank
accession number AY083935); 96.2% with P. longan-
drum AY039713), 94.9% with an un-described species
P. sp strain F-74, AY455695) 84.6% with P. terrestris
(AY039714), P. hypogynum (AY455804), and another
un-described P. sp. Strain F-1216 (AY455697). Other
species with hypogynous type of antheridia that comes
close to P. longisporangium but having little resem-
blances as far as the ITS sequences concerned are:
P. segnitium (AY149173) 74.8% similarities, P. canar-iense (AY06561) having 60.7% similarity and P. rostra-
tum (AJ233456) having only 57.6% similarity with the
ITS 1 of P. longisporangium.
The morphological and molecular characteristics
indicate that the closest relative of P. longisporangium
is P. bifurcatum, a new species recently described from
northern France. However there are enough structural
as well as molecular differences between the two (Table
1) to justify the creation of a new taxon.
References
[1] Dick, M.W. (1990) Keys to Pythium. University of Reading Press,
Reading, UK.
[2] Paul, B. (1999) Pythium periplocum, an aggressive mycoparasite of
Botrytis cinerea causing the grey mould disease of grape-vine.
FEMS Microbiology Letters 227, 277–280.
[3] Hendrix, F.F. and Campbell, W. (1973) Pythiums as plant
pathogens. Annual Review of Phytopathology 11, 78–98.
[4] Kumar, C. and Rzhetsky, A. (1996) Evolutionary relationships of
eukaryotic kingdoms. Journal of Molecular Evolution 42, 183–
193.
[5] Van de Peer, Y. and De Wachter, R. (1997) Evolutionary
relationships among the eukaryotic crown taxa taking into
account site-to-site rate variation in 18S rRNA. Journal of
Molecular Evolution 45, 619–630.
[6] Paquin, B., Laforest, M.J., Forget, L., Roewer, I., Wang, Z.,
Longcore, J. and Lang, B.F. (1997) The fungal mitochondrial
genome project: evolution of fungal mitochondrial genomes and
their gene expression. Current Genetics 31, 380–395.
[7] Middleton, J.T. (1943) The taxonomy, host range and geographic
distribution of the genus Pythium. Mem. Torrey bot. Club. 20, 1–
171.
[8] Plaats-Niterink, A.J. Van der (1981) Monograph of the genus
Pythium. Studies in Mycology, Centraalbureau voor Schimmel-
cultures, Baarn 21, 1–242.
[9] Waterhouse, G.M. (1967) Key to Pythium Pringsheim. Mycolog-
ical Papers 109, 1–15.
[10] White, T.J., Bruns, T., Lee, S. and Taylor, J. (1990) Amplification
and direct sequencing of fungal ribosomal RNA genes for
phylogenetics. In: PCR Protocols: A Guide to Methods and
Applications (Innis, M.A., Gelfand, D.H., Sninsky, J.J. and
White, T.J., Eds.), pp. 315–322.
[11] Lee, S.B. and Taylor, J.W. (1992) Phylogeny of five fungus like
protoctistan Phytophthora species, inferred from the internal
transcribed spacer of ribosomal DNA. Molecular Biology and
Evolution 9, 636–653.
[12] Cooke Del, Jung, T., Williams, N.A., Schubert, R., Bahnweg, G.,
Oßwald, W. and Duncan, J.N. (1999) Molecular evidence
supports Phytophthora quercina as a new species. Mycological
Research 103, 799–804.
[13] Nechwatal, Jan. and Oßwald, W. (2003) Pythium montanum sp.
Nov., a new species from a spruce stand in the Bavarian Alps.
Mycological Progress 2 (1), 73–80.
[14] Paul, B., Galland, D. and Masih, I. (1999) Pythium prolatum
isolated from soil in the burgundy region: A new record for
Europe. FEMS Microbiology Letters 173, 69–75.
[15] Paul, B. (2001) ITS region of the rDNA of Pythium longandrum, a
new species; its taxonomy and its comparison with related species.
FEMS Microbiology Letters 202 (2), 239–242.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 213–219
Polymorphism and gene conversion of the 16S rRNA genesin the multiple rRNA operons of Vibrio parahaemolyticus
Narjol Gonzalez-Escalona, Jaime Romero, Romilio T. Espejo *
Laboratorio de Biotecnologıa, Instituto de Nutricion y Tecnologıa de los Alimentos, Universidad de Chile, El Lıbano 5524, Macul, Santiago, Chile
Received 3 September 2004; received in revised form 13 January 2005; accepted 8 April 2005
First published online 22 April 2005
Edited by J.M. Ketley
Abstract
The genome sequence of a strain of Vibrio parahaemolyticus holds 11 copies of rRNA operons (rrn) with identical 16S rRNA
genes (rrs). Conversely, the species type strain contains two rrs classes differing in 10 nucleotide sites within a short segment of
25 bp. Furthermore, we show here that the sequence of this particular segment largely differs between some strains of this species.
We also show that of the eleven rrn operons in the species type strain, seven contain one rrs class and four the other, indicating gene
conversion. Our results support the hypothesis that the rrs differences observed between strains of this species were caused by lateral
transfer of an rrs segment and subsequent conversion.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Gene conversion; Polymorphism; rrn operons; Vibrio parahaemolyticus; 16S rRNA
1. Introduction
Vibrio parahaemolyticus is a natural inhabitant of
coastal waters and one of the major seafood-borne gas-troenteritis-causing bacteria. Since 1996 an increasing
number of V. parahaemolyticus infections caused by
strains belonging to a clonal complex have been ob-
served throughout the world [1]. We have recently found
that a strain of this complex caused two large diarrhoea
outbreaks in Chile, in 1998 and 2004 [2]. The genome of
one of these strains, RIMD2210633 (VpKX), consists of
two circular chromosomes with 11 copies of rRNAoperons (rrn), 10 on chromosome 1 and 1 on chromo-
some 2 [3]. Analysis of the reported sequence [3] shows
that this strain contains almost identical 16S rRNA
genes (rrs) in their 11 rRNA operons. However, like
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.009
* Corresponding author. Tel.: +56 2 6781426; fax: +56 2 2214030.
E-mail address: [email protected] (R.T. Espejo).
many strains which have their genome sequenced [4],
most strains of the genus Vibrio show detectable se-
quence differences between their multiple rrs. The type
strain of the V. parahaemolyticus species, ATCC 17802(VpD), contains two rrs classes, differing in 10 nucleo-
tide sites of a 25 bp sequence that encodes a variable
stem loop of the 16S rRNA, including nucleotides
440–496 (Escherichia coli numbering) [5]. Furthermore,
the sequence of each of the segments in the two rrs clas-
ses of the type strain differ in 7 and 10 nucleotide sites
from the corresponding segment in the VpKX strain
(see sequences for VpD1, VpD2 and VpKX in Fig. 1).The presence of two different rrs classes in the genome
of the type strain and the difference between both rrs
classes of this strain with that in the pandemic strain
do not have a simple explanation.
The large number of mismatches together with the
compensating changes observed between the two rrs
segments found in VpD, implies that their divergence
. Published by Elsevier B.V. All rights reserved.
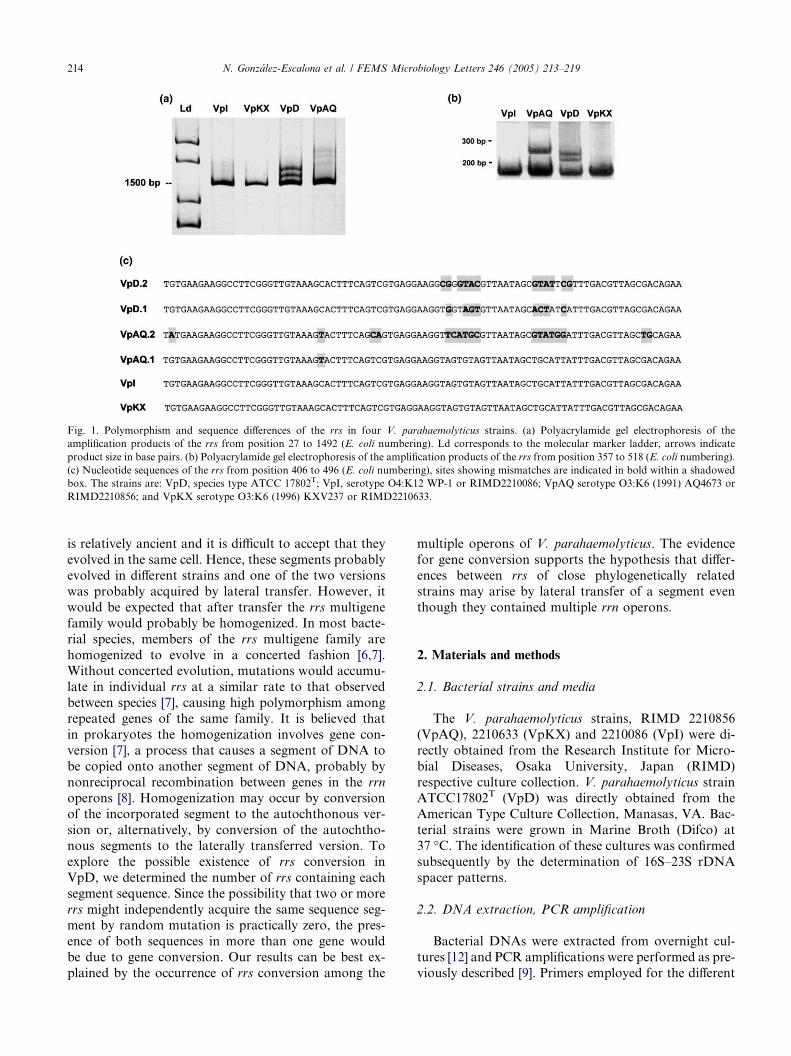
Fig. 1. Polymorphism and sequence differences of the rrs in four V. parahaemolyticus strains. (a) Polyacrylamide gel electrophoresis of the
amplification products of the rrs from position 27 to 1492 (E. coli numbering). Ld corresponds to the molecular marker ladder, arrows indicate
product size in base pairs. (b) Polyacrylamide gel electrophoresis of the amplification products of the rrs from position 357 to 518 (E. coli numbering).
(c) Nucleotide sequences of the rrs from position 406 to 496 (E. coli numbering), sites showing mismatches are indicated in bold within a shadowed
box. The strains are: VpD, species type ATCC 17802T; VpI, serotype O4:K12 WP-1 or RIMD2210086; VpAQ serotype O3:K6 (1991) AQ4673 or
RIMD2210856; and VpKX serotype O3:K6 (1996) KXV237 or RIMD2210633.
214 N. Gonzalez-Escalona et al. / FEMS Microbiology Letters 246 (2005) 213–219
is relatively ancient and it is difficult to accept that they
evolved in the same cell. Hence, these segments probably
evolved in different strains and one of the two versions
was probably acquired by lateral transfer. However, it
would be expected that after transfer the rrs multigene
family would probably be homogenized. In most bacte-
rial species, members of the rrs multigene family are
homogenized to evolve in a concerted fashion [6,7].Without concerted evolution, mutations would accumu-
late in individual rrs at a similar rate to that observed
between species [7], causing high polymorphism among
repeated genes of the same family. It is believed that
in prokaryotes the homogenization involves gene con-
version [7], a process that causes a segment of DNA to
be copied onto another segment of DNA, probably by
nonreciprocal recombination between genes in the rrn
operons [8]. Homogenization may occur by conversion
of the incorporated segment to the autochthonous ver-
sion or, alternatively, by conversion of the autochtho-
nous segments to the laterally transferred version. To
explore the possible existence of rrs conversion in
VpD, we determined the number of rrs containing each
segment sequence. Since the possibility that two or more
rrs might independently acquire the same sequence seg-ment by random mutation is practically zero, the pres-
ence of both sequences in more than one gene would
be due to gene conversion. Our results can be best ex-
plained by the occurrence of rrs conversion among the
multiple operons of V. parahaemolyticus. The evidence
for gene conversion supports the hypothesis that differ-
ences between rrs of close phylogenetically related
strains may arise by lateral transfer of a segment even
though they contained multiple rrn operons.
2. Materials and methods
2.1. Bacterial strains and media
The V. parahaemolyticus strains, RIMD 2210856
(VpAQ), 2210633 (VpKX) and 2210086 (VpI) were di-
rectly obtained from the Research Institute for Micro-
bial Diseases, Osaka University, Japan (RIMD)
respective culture collection. V. parahaemolyticus strainATCC17802T (VpD) was directly obtained from the
American Type Culture Collection, Manasas, VA. Bac-
terial strains were grown in Marine Broth (Difco) at
37 �C. The identification of these cultures was confirmed
subsequently by the determination of 16S–23S rDNA
spacer patterns.
2.2. DNA extraction, PCR amplification
Bacterial DNAs were extracted from overnight cul-
tures [12] and PCR amplifications were performed as pre-
viously described [9]. Primers employed for the different

N. Gonzalez-Escalona et al. / FEMS Microbiology Letters 246 (2005) 213–219 215
amplification protocols were: Eubac27F (5 0-AGAGTTT-
GATCCTGGCTCAG-3 0) and 1492R (5 0-GGTTACCT-
TGTTACGACTT-3 0) to amplify 16S rDNA, and
primers 357F (5 0 CTCCTACGGGAGGCAGCA-3 0)
and 518R (5 0-CGTATTACCGCGGCTGCTGG-3 0) to
amplify the shorter fragment containing the variable re-gion G1F (5 0-GAAGTCGTAACAAGG-3 0) and L1R
(5 0-AAGGCATCCACCGT-3 0) to amplify the 16S–23S
rDNA genes spacer [10]. PCR products were electropho-
resed and visualized as previously described [11].
2.3. Cloning and sequencing of rrs together with adjacent
spacer
Primers 357F and L1R were employed to amplify the
fragments containing both the 16S rDNA fragment and
the spacer. PCR products were purified using the Wiz-
ard system as indicated by the manufacturer (Promega)
and later cloned into pGEMT Easy Vector Systems
according to manufacturer�s instructions (Promega).
Plasmid DNA was obtained by a rapid alkaline extrac-
tion miniprep [12]. For analysis of the size of the spacersand the class of rrs by heteroduplex assay, the plasmid
DNA was diluted 1:100 (vol./vol.) in sterile distilled
water and 15 ll were used for PCR amplification of
either the 16S rDNA or the spacer, as described above.
The size of the spacer was subsequently determined by
polyacrylamide gel electrophoresis. The heteroduplex
assay was performed as described [13], except that the
electrophoresis was conducted at 150 V. For sequencing,plasmids were purified with E.Z.N.A. Plasmid Miniprep
Kit I (Omega Bio-tek) and the cloned segments were se-
quenced on an ABI 3100 Genetic Analyzer using Big
Dye Terminator Cycle Sequencing V2.0 Ready Reac-
tions Kit and recommended protocols with primers
M13F, 518R, G1F and L1R. DNA sequences were in-
spected individually and manually assembled. The align-
ments and sequence similarities were obtained usingBioEdit [14].
2.4. Pulsed-field gel electrophoresis and fragment analysis
Bacterial genomic DNA in agarose plugs was pre-
pared as described [15], and digested with the restriction
enzyme I-CeuI (New England Biolabs) for 16 h at 37 �C,using 50 U/plug. Electrophoresis was performed on aCHEF DRII System (BioRad,), using a 1% low melting
point agarose (Promega) gel in 0.5· TBE buffer
(0.45 mM Tris–borate, 1 mM EDTA-Na, pH 8.0). The
pulsed time employed was 6–60 s ramp time at 200 V
for 24 h, at a constant temperature of 14 �C. After elec-
trophoresis, the gel was stained with ethidium bromide
for 30 min and photographed. The observed bands were
excised from the gel with sterile razors and a slice ofeach band was then melted at 65 �C in 10 times its vol-
ume of 1· TE (10 mM Tris–Cl, 1 mM EDTA-Na, pH
8.0). Twelve liters of the solution containing DNA from
each band was then used for PCR, as described above
except that only 20 cycles were performed. Analysis of
both the size of the spacers and the class of rrs by het-
eroduplex assay was performed as described above.
2.5. Nucleotide sequences
Sequences have been deposited in GenBank under the
Accession Nos. AY298793, AY298798, AY298799–
AY298808, and AY527386–AY52735388.
3. Results
3.1. rrs polymorphism in the multi rrn operons of
V. parahaemolyticus strains
The polymorphism in the repeated rrs of the V.
parahaemolyticus species type strain was originally ob-
served by the formation of heteroduplexes after PCR
amplification of the rrs [5]. The presence of heterodu-plexes after PCR amplification of a single isolate with
multiple rrs occurs when these genes exhibit differences
in the nucleotide sequences. In polymorphic strains,
hybrids between synthesized copies with different se-
quences are formed, which show a retarded electropho-
retic migration in polyacrylamide gels. This assay was
employed for assessment of polymorphism in other
V. parahaemolyticus strains, including the pandemicstrain O3:K6 (VpKX), whose genome sequence shows
identical rrs genes [3]. Fig. 1(a) shows the results ob-
tained after PCR amplification of the 16S rRNA gene
from the species type strain VpD, the sequenced pan-
demic clone VpKX and two other non-pandemic
strains; VpAQ, an O3:K6 isolate obtained in 1991,
and VpI, an O4:K12 isolate found in 1968. After
amplification of the 16S rRNA gene, heteroduplexeswith retarded migration were observed for strains
VpD and VpAQ, but not for VpKX and VpI. The het-
eroduplex nature of the bands with retarded migration
was confirmed as previously described [13] (results not
shown).
Cloning and sequencing of the rrs in VpD has shown
the presence of two classes, called 1 and 2, which differ
in 10 nucleotide positions within a 25 bp segment in avariable stem loop of the 16S rRNA, including nucleo-
tides 440–496 (E. coli numbering) [5].To examine if the
polymophism observed in VpAQ occurred in this same
region, a shorter fragment of 161 bp encompassing the
variable segment was amplified by PCR and checked
for formation of heteroduplexes (Fig. 1(b)). The pres-
ence of extra bands observed above the main amplifica-
tion products in VpAQ and VpD indicated that thepolymorphism was in the same rrs region. The presence
of two bands above the main band in VpD is probably

216 N. Gonzalez-Escalona et al. / FEMS Microbiology Letters 246 (2005) 213–219
due to the differential migration of the reciprocal hy-
brids formed in this strain. Two different heterodu-
plexes, composed by plus and minus complementary
strands of each amplicon with different sequence, are
formed after annealing in the last PCR cycle. Although
the extent of dissimilarity in this hybrid pair is the same,non-paired regions may form distinct structural confor-
mations in each hybrid, decreasing the mobility to differ-
ent extents [5,13,16]. The sequences of the rrs variable
region in the different strains were determined after
amplification and cloning as described below. Fig. 1(c)
shows that the sequences found in both VpKX and
VpI are identical but they greatly differ from the two se-
quences found in the species type strain VpD and fromone of the two sequences found in VpAQ.
3.2. Operons with each rrs sequence in the species type
strain
To explore if a new sequence may be enforced in
every repeated gene, we looked for evidence of gene con-
version. Homogenization of the rrs requires that one ofthe versions convert the others. The occurrence of rrs
gene conversion was explored in VpD. The presence of
Fig. 2. Separation of the genomic fragments containing rrn operons and
parahaemolyticus strain ATCC 17802. (a) Pulsed-field gel electrophoresis of
indicated on the left side; the class of rrs and the approximate size of the space
the right side. (b) Polyacrylamide gel electrophoresis of the rrs–rrl spacer regio
on top of each lane indicates the analyzed band. VpD corresponds to the
ATCC17802T. Ld corresponds to a 100 bp molecular size marker. (c) Polyac
and annealing of the amplification products of the variable region of the rr
amplification products of clones containing rrs class 1, class 2 and with thems
and 2 in the last lanes to the right correspond to the self-annealing products fr
both sequence versions in more than one operon would
indicate gene conversion because the possibility that two
or more rrs might acquire the same segment sequence by
random mutation is practically zero. The sequence of
the variable segment of the rrs in the different operons
of VpD was determined by analysis of restriction frag-ments obtained by cleavage of the genomic DNA with
the restriction enzyme I-CeuI. This enzyme cleaves a
19-bp sequence in the 23S rRNA gene [17] and allows
for the separation of the rrn operons by gel electropho-
resis. Fig. 2(a) shows the result of the pulsed-field gel
electrophoresis with the resolved bands numbered from
1 to 8. The sequence of rrs (class 1 or 2) is shown to the
right of each band. The rrs class was defined by a hetero-duplex assay based on the retarded migration of the hy-
brids formed between rrs segments of different
nucleotide sequence. For this assay, a 161 bp segment
containing the rrs variable region was amplified from
each fragment and the product was hybridized with
those obtained from recombinant plasmids containing
rrs of either class 1 or 2 [5]. Analysis of the hybridization
products by gel electrophoresis shows exclusivelyhomoduplexes when amplicons are of the same class,
and homoduplex plus heteroduplexes with retarded
characterization of their rrs genes and rrs–rrl spacer regions in V.
the DNA digested with I-CeuI. The number assigned to each band is
rs (between parenthesis), observed in each DNA band, are indicated on
ns PCR amplified as described in Materials and Methods. The number
amplification product from the whole DNA of V. parahaemolyticus
rylamide gel electrophoresis of the products formed after denaturation
s contained in each band (indicated above every three lanes) with the
elves (indicated as 1, 2 and - above each lane, respectively). Numbers 1
om class 1 and 2 rrs clones, respectively. VpD and Ld as indicated in b.

Fig. 3. Schematic representation of the different putative operons identified both in the clones of the PCR amplification products and in the
restriction fragments. Nucleotides in polymorphic sites are in bold. Each operon is identified by the class of rrs (1 or 2) and the size of its neighbor
spacer in bp (first column on the left). The scheme shows the sequence of the variable region of the rrs followed by the tRNAs present in its adjacent
spacer. Pulsed field electrophoresis bands that may contain the operon are identified on the right side. The uncertainty is caused by the inability to
distinguish between spacers of almost equal size by gel electrophoresis, i.e., 706 and 669. ND, not detected among the pulsed field gel electrophoresis
bands.
N. Gonzalez-Escalona et al. / FEMS Microbiology Letters 246 (2005) 213–219 217
migration when the amplicons are of a different class.Fig. 2(c) shows the result of the heteroduplex assay for
each band. For some bands the presence of rrs of a sin-
gle sequence class is straightforward, as for the presence
of sequence class 1 in bands 1, 2 and 8 or class 2 in band
6 observed in this figure. Band 5 shows the presence of
heteroduplexes when hybridized with amplicons from
the clones of either class 1 or 2 and with itself. This kind
of result is expected when the band contains both rrs
classes. Heteroduplexes observed in minor proportion
after self-annealing of the amplicons of some bands,
e.g., band 7, are likely due to contamination in the gel
with other restriction fragments.
To define the number of different operons in each
band we determined the size of the 16S–23S rDNA
spacer regions by PCR amplification. The size of the
spacers allows for the placement of the operons of thisstrain into six groups [18]. Fig. 2(b) shows the spacers
observed after amplification of each band; the observa-
tion of more than one spacer in some bands (bands 1,
5 and 7) may be due to either the co-migration of two
restriction fragments or to the presence of two operons.
Fragments with two operons may be generated when
two neighbour operons are in opposite direction. The
sizes of the spacers found in each band are shown be-tween parenthesis in Fig. 2(a). Altogether, the determi-
nation of the size of the spacer and class of rrs
allowed for the identification of 11 putative rRNA oper-
ons, seven containing rrs 1 and four rrs 2.
The class of rrs was also determined on a complemen-
tary approach, independent of the location of the CeuI
restriction sites. This entailed the amplification and
cloning of about three-fourths of the rrs genes togetherwith their entire neighbouring spacers. The operons in
44 clones of the product were examined for both the size
of the spacer and the class of rrs. They were initially sep-
arated into four groups according to the estimated size
of the 16S–23S rDNA intergenic spacers by gel electro-
phoresis [18]. According to both the estimated size of the
spacer and the class of the rrs, six groups of clones were
distinguished. At least four clones from each group weresequenced. These sequences permitted the identification
of 8 groups of clones; six groups containing rrs with se-quence class 1 and two with sequence class 2. Fig. 3
shows a scheme of the rrs class and the size of the spacer
in the different groups. The sequence of the variable re-
gion of the rrs and the tRNAs deduced from the spacer
sequences are schematically shown on the right side. As
previously described by Maeda et al. [18], the sequences
we found allowed us to distinguish six groups of spacers
although only four bands are observed after polyacryl-amide electrophoresis.
3.3. Gene conversion in the 16S–23S rDNA spacer regions
To explore gene conversion in other regions of the rrn
operons, the sequence of the spacer in the different oper-
ons was analysed. The analysis of the spacer sequences
in VpD showed conserved sequence blocks in each rrn
operon corresponding to the 40 sites next to the 5 0 end
and the 208 sites next to the 3 0. Though these blocks
are highly conserved, a few polymorphic sites suggestive
of gene conversion were observed. Within the 40 sites
next to the 5 0 end, there is a polymorphic site with T
in two putative operons and A in the other six. Within
the 208 sites next to the 3 0 end there is a polymorphic
site with T in two putative operons and G in the othersix (results not shown).
4. Discussion
The high polymorphism observed between the multi-
ple rrs in VpD and VpAQ in a particular segment and
the differences between rrs of different strains in thissame segment is extraordinary. The segments with the
large changes found within the same strain are unlikely
to be due to accumulation of point mutations. It seems
more probable that each segment evolved independently
in different strains and that they came together by lateral
transfer. This transfer might have occurred via replace-
ment of a segment as proposed by Wang and Zhang
[19] in their simplified complexity hypothesis. Interest-ingly, despite their approximate 40% dissimilarity, the

218 N. Gonzalez-Escalona et al. / FEMS Microbiology Letters 246 (2005) 213–219
different versions of the 25 bp segment in the four V.
parahaemolyticus strains fully match with those in the
16S rRNA of strains of the genus Vibrio or with non-
classified marine Vibrionaceae, exclusively. Most isolates
containing any of the sequence versions found in V.
parahaemolyticus corresponded to the species V. vulnifi-cus, V. coralliilyticus, V. alginolyticus, V. fischeri, and V.
ponticus. Being an autochthonous marine bacterium, V.
parahaemolyticus is probably subjected to a high level of
recombination with the diverse, closely related bacterial
strains populating the seawater. Seawater is a particular
habitat where vibrios are exposed to high levels of gene
transfer by transduction [20]; two bacteriophage classes
that could potentially transfer rrs genes among Vibriorelated strains have been reported. These comprise
filamentous phages [21], including one that is able to
integrate into the host chromosome of V. parahaemolyt-
icus [22,23] and T4-related broad host range phages [24].
However, lateral transfer would probably change only
one of the multiple rrs. Changing more than one or
every rrs in the genome would require gene conversion.
Our analysis of the rrn operons showed the presence ofat least 7 operons containing rrs with sequence class 1
and class 4 with sequence class 2. Considering the num-
ber of different nucleotides together with compensatory
mutations required for the generation of the two rrs
classes, it is very unlikely that the redundant sequences
appeared independently in more than one operon.
Although the occurrence of reciprocal double exchanges
between sister chromosomes, as described by Segall andRoth [25] may generate redundancy of the sequences, it
seems more likely that the redundancy described above
was caused by gene conversion. The repeated mis-
matches found in the spacer regions are also suggestive
of the occurrence of gene conversion. If gene conversion
occurs at a higher rate than gene differentiation, the rrs
would become identical, as was observed in VpI and
VpKX. However, besides having identical segments intheir multiple rrs, these two strains contain a sequence
segment very different from that observed in the species
type strain. Therefore, it seems probable that the ob-
served differences between strains may have originated
by lateral transfer of the variable segment followed by
gene conversion of other rrs in the genome. A similar
mechanism for the polymorphism and redundancy ob-
served in the intervening sequences of the multiple rrl
copies present in Salmonella typhimurium and Salmo-
nella typhi has been postulated by Mattatall et al.
[26,27].
Acknowledgments
N. Gonzalez-Escalona acknowledges a scholarshipDr. Abraham Stekel from INTA-Nestle. This work
was supported in part by FONDECYT Grant 1040805.
References
[1] Chowdhury, N.R., Chakraborty, S., Ramamurthy, T., Nishibu-
chi, M., Yamasaki, S., Takeda, Y. and Nair, G.B. (2000)
Molecular evidence of clonal Vibrio parahaemolyticus pandemic
strains. Emerg. Infect. Dis. 6, 631–636.
[2] Gonzalez-Escalona, N., Cachicas, V., Acevedo, C., Rioseco,
M.L., Vergara, J.A., Cabello, F., Romero, J. and Espejo, R.T.
(2005) Vibrio parahaemolyticus diarrhea, Chile, 1998 and 2004.
Emerg. Infect. Dis. 11, 129–131.
[3] Makino, K., Oshima, K., Kurokawa, K., Yokoyama, K., Uda,
T., Tagomori, K., Iijima, Y., Najima, M., Nakano, M., Yamash-
ita, A., Kubota, Y., Kimura, S., Yasunaga, T., Honda, T.,
Shinagawa, H., Hattori, M. and Iida, T. (2003) Genome sequence
of Vibrio parahaemolyticus: a pathogenic mechanism distinct from
that of V cholerae. Lancet 361, 743–749.
[4] Acinas, S.G., Marcelino, L.A., Klepac-Ceraj, V. and Polz, M.F.
(2004) Divergence and redundancy of 16S rRNA sequences in
genomes with multiple rrn operons. J. Bacteriol. 186, 2629–2635.
[5] Moreno, C., Romero, J. and Espejo, R.T. (2002) Polymorphism
in repeated 16S rRNA genes is a common property of type strains
and environmental isolates of the genus Vibrio. Microbiology 148,
1233–1239.
[6] Elder Jr., J.F. and Turner, B.J. (1995) Concerted evolution of
repetitive DNA sequences in eukaryotes. Q. Rev. Biol. 70, 297–
320.
[7] Liao, D. (2000) Gene conversion drives within genic sequences:
concerted evolution of ribosomal RNA genes in bacteria and
archaea. J. Mol. Evol. 51, 305–317.
[8] Lan, R. and Reeves, P.R. (1998) Recombination between rRNA
operons created most of the ribotype variation observed in the
seventh pandemic clone of Vibrio cholerae. Microbiology 144,
1213–1221.
[9] Espejo, R.T. and Romero, J. (1997) Bacterial community in
copper sulfide ores inoculated and leached with solution from a
commercial-scale copper leaching plant. Appl. Environ. Micro-
biol. 63, 1344–1348.
[10] Jensen, M.A., Webster, J.A. and Straus, N. (1993) Rapid
identification of bacteria on the basis of polymerase chain
reaction-amplified ribosomal DNA spacer polymorphisms. Appl.
Environ. Microbiol. 59, 945–952.
[11] Pizarro, J., Jedlicki, E., Orellana, O., Romero, J. and Espejo, R.T.
(1996) Bacterial populations in samples of bioleached copper ore
as revealed by analysis of DNA obtained before and after
cultivation. Appl. Environ. Microbiol. 62, 1323–1328.
[12] Sambrook, J. and Russell, R.G. (2001) Molecular Cloning. A
Laboratory Manual, 3rd edn. Cold Spring Harbor Laboratory
Press, Cold Spring Harbor, NY.
[13] Espejo, R.T., Feijoo, C.G., Romero, J. and Vasquez, M. (1998)
PAGE analysis of the heteroduplexes formed between PCR-
amplified 16S rRNA genes: estimation of sequence similarity and
rDNA complexity. Microbiology 144, 1611–1617.
[14] Hall, T.A. (1999) BioEdit: a user-friendly biological sequence
alignment editor and analysis program for Windows 95/98/NT.
Nucleic Acids Symp. Ser. 41, 95–98.
[15] Iida, T., Suthienkul, O., Park, K.S., Tang, G.Q., Yamamoto,
R.K., Ishibashi, M., Yamamoto, K. and Honda, T. (1997)
Evidence for genetic linkage between the ure and trh genes in
Vibrio parahaemolyticus. J. Med. Microbiol. 46, 639–645.
[16] Jensen, M.A and Straus, N. (1993) Effect of PCR conditions on
the formation of heteroduplex and single-stranded DNA products
in the amplification of bacterial ribosomal DNA spacer regions.
PCR Methods Appl. 3, 186–194.
[17] Liu, S.L. and Sanderson, K.E. (1995) I-CeuI reveals conservation
of the genome of independent strains of Salmonella typhimurium.
J. Bacteriol. 177, 3355–3357.

N. Gonzalez-Escalona et al. / FEMS Microbiology Letters 246 (2005) 213–219 219
[18] Maeda, T., Takada, N., Furushita, M. and Shiba, T. (2000)
Structural variation in the 16S–23S rRNA intergenic spacers of
Vibrio parahaemolyticus. FEMS Microbiol. Lett. 192, 73–77.
[19] Wang, Y. and Zhang, Z. (2000) Comparative sequence analyses
reveal frequent occurrence of short segments containing an
abnormally high number of non-random base variations in
bacterial rRNA genes. Microbiology 146, 2845–2854.
[20] Jiang, S.C and Paul, J.H. (1998) Gene transfer by transduction in
themarine environment. Appl. Environ.Microbiol. 64, 2780–2787.
[21] Chang, B., Taniguchi, H., Miyamoto, H. and Yoshida, S. (1998)
Filamentous bacteriophages of Vibrio parahaemolyticus as a
possible clue to genetic transmission. J. Bacteriol. 180, 5094–5101.
[22] Iida, T., Makino, K., Nasu, H., Yokoyama, K., Tagomori, K.,
Hattori, A., Okuno, T., Shinagawa, H. and Honda, T. (2002)
Filamentous bacteriophages of vibrios are integrated into the dif-
like site of the host chromosome. J. Bacteriol. 184, 4933–4935.
[23] Nasu, H., Iida, T., Sugahara, T., Yamaichi, Y., Park, K.S.,
Yokoyama, K., Makino, K., Shinagawa, H. and Honda, T. (2000)
A filamentous phage associated with recent pandemic Vibrio
parahaemolyticus O3:K6 strains. J. Clin. Microbiol. 38, 2156–
2161.
[24] Miller, E.S., Heidelberg, J.F., Eisen, J.A., Nelson, W.C., Durkin,
A.S., Ciecko, A., Feldblyum, T.V., White, O., Paulsen, I.T.,
Nierman, W.C., Lee, J., Szczypinski, B. and Fraser, C.M. (2003)
Complete genome sequence of the broad-host-range vibriophage
KVP40: comparative genomics of a T4-related bacteriophage. J.
Bacteriol. 185, 5220–5233.
[25] Segall, A.M and Roth, J.R. (1994) Approaches to half-tetrad
analysis in bacteria: recombination between repeated, inverse-
order chromosomal sequences. Genetics 136, 27–39.
[26] Mattatall, N.R., Daines, D.A., Liu, S.L. and Sanderson, K.E.
(1996) Salmonella typhi contains identical intervening sequences in
all seven rrl genes. J. Bacteriol. 178, 5323–5326.
[27] Mattatall, N.R. and Sanderson, K.E. (1996) Salmonella typhimu-
rium LT2 possesses three distinct 23S rRNA intervening
sequences. J. Bacteriol. 178, 2272–2278.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 221–228
Induction of murine macrophage TNF-a synthesis by Mycobacteriumavium is modulated through complement-dependent interaction
via complement receptors 3 and 4 in relation to M. aviumglycopeptidolipid
Vida R. Irani a, Joel N. Maslow a,b,*
a Division of Infectious Diseases, School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, United Statesb Section of Infectious Diseases, VA Medical Center (151), Medical Research Building, 3900 Woodland Ave., Philadelphia, PA 19104, United States
Received 16 November 2004; received in revised form 17 February 2005; accepted 8 April 2005
First published online 20 April 2005
Edited by A.H.M. van Vliet
Abstract
We studied whether complement receptor (CR) mediated Mycobacterium avium interaction modulated macrophage TNF-aexpression. Compared to control conditions, infections performed with C3-depletion yielded significantly higher TNF-a levels.
Blockage of the CR4 iC3b site yielded increases in TNF-a for all morphotypic variants of a virulent serovar-8 strain (smooth trans-
parent (SmT), smooth opaque (SmO), serovar-specific glycopeptidolipid (ssGPL) deficient knockout mutant) whereas CR3 blockage
increased TNF-a only for SmT and ssGPL-deficient strains. Thus, complement-mediated binding of M. avium to CR3 and CR4 was
shown to modulate TNF-a expression. The differential activation of morphotypic and isogenic variants of a single strain provides an
excellent model system to delineate signaling pathways.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Mycobacterium avium; GPL; TNF-a; Macrophage; Complement receptor; Serum proteins
1. Introduction
Mycobacterium avium, a prevalent opportunistic
pathogen of immunocompromised patients, resides
and replicates inside the macrophages of the infected
host. Upon contact of M. avium with the host macro-
phage, reactions such as production of reactive oxygen
and nitrogen intermediates, release of pro-inflammatory(TNF-a, IL-6), and anti-inflammatory cytokines (IL-10)
are triggered [1–3]. It is widely accepted that the level of
macrophage TNF-a expression has important conse-
quences for host immunity [2,4]. In vivo, TNF-a induc-
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.008
* Corresponding author. Tel.: +1 215 823 6020; fax: +1 215 823 5171.
E-mail address: [email protected] (J.N. Maslow).
tion by M. avium correlates with increased granuloma
formation, resulting in rapid containment and clearance
of the infectious focus [5,6], whereas inhibition of TNF-
a expression by virulent strains of Mycobacterium tuber-
culosis or M. avium is associated with increased bacterial
survival [4,7,8].
Mycobacteria can bind to a number of macrophage
receptors such as the complement receptor types 1, 3and 4 (CR1, CR3, CR4), CD14, mannose receptor
(MR), transferrin receptor, and Fc receptors to gain en-
try into the macrophage [9,10]. CR3 (CD11b/CD18) in
particular, maintains intimate connections with the actin
cytoskeleton and is crucial to the uptake of intracellular
pathogens, including mycobacteria [11–13]. Opsonin-
mediated uptake is partially controlled by the bacterium
. Published by Elsevier B.V. All rights reserved.

222 V.R. Irani, J.N. Maslow / FEMS Microbiology Letters 246 (2005) 221–228
since M. avium has been shown to recruit complement
fragment C2a to form a C3 convertase and generate ac-
tive C3 breakdown products [14]. Receptor preference at
the time of bacterial entry is considered to determine
subsequent intracellular processing by the infected host
cell. In fact, utilization of CRs by the intracellularpathogens Leishmania donovani, Histoplasma capsula-
tum, Mycobacterium leprae, and Legionella pneumophila
to enter the macrophage, allows these organisms to
avoid phagocytic pathways that induce the respiratory
burst [15,16].
A link between receptor usage and early signal trans-
duction events has been recently demonstrated for M.
avium strain 2151 SmO. This strain utilizes CD14 tomanipulate TNF-a synthesis via the extracellular sig-
nal-regulated kinase (ERK) mitogen-activated protein
kinase (MAPK) pathway [17]. While it has been demon-
strated by Aderem and Underhill [18] and Forsberg
et al. [19] that TNF-a regulates CR3-mediated MAPK
activation, respiratory burst, and phagocytosis, the con-
verse i.e., a role for CR3 in regulating TNF-a has never
been studied although work done by Bohlson et al. [20]suggests that complement-mediated interactions may
not impact TNF-a activation.
Non-virulent strains of M. avium stimulate more
TNF-a compared to virulent strains [21], however, the
bacterial surface structures modulating TNF-a activa-
tion have not been identified. The glycopeptidolipids
(GPL) represent the most abundant cell wall component
of M. avium and are situated on the outermost surfacesof the cell [22], thus making them likely ligands to medi-
ate bacterial–host interactions. Studies have suggested a
role for serovar-specific GPL (ssGPL) in the pathogene-
sis of M. avium infection as highly antigenic molecules
affecting host immune function. These data have relied
on comparison of strains representing different serovars
or have used purified and/or chemically modified GPL
and GPL components [23].In a previous study, we disrupted the M. avium
rhamnosyltransferase (rtfA) gene via homologous
recombination to construct isogenic mutants devoid of
serovar-8 specific GPL using an allelic exchange vector
incorporating a temperature-sensitive plasmid origin of
replication and sacB [24]. In this study, we investigate
whether serum C3, and macrophage receptors such as
CR3, and CR4 are involved in M. avium–macrophageinteraction and whether they modulate TNF-a induction
among M. avium strains differing in ssGPL expression.
2. Materials and methods
2.1. Bacterial strains and reagents
M. avium 920A6 is a serovar-8 bloodstream isolate
cultured from a patient with AIDS that exists as smooth
opaque (SmO) and smooth transparent (SmT) morpho-
types [25,26]. 213R.4 is a ssGPL-null (serovar-null)
strain of 920A6 SmO generated by allelic exchange
mutagenesis for the rtfA gene [24]. Strain 233R.1 created
by transformation of 213R.4 with wild-type rtfA gene
incorporated into an integrative vector, contains onlya single-copy of rtfA, and demonstrates a pattern of
ssGPL and nsGPL similar to the parent wild-type strain
[24].
M. avium was grown in Middlebrook 7H9 sucrose
broth or 7H11 agar (Difco Laboratories, Detroit, MI)
supplemented with 10% oleic acid dextrose complex
(OADC) at 37 �C. Bacteria were grown to an optical
density at 600 nm of 0.4, diluted with an equal volumeof 10% glycerol, frozen on dry ice–ethanol, and stored
as 1 ml aliquots at �80 �C. One aliquot of frozen bacte-
ria of each type was serially diluted to determine base-
line colony forming units (cfu) and to confirm colony
morphotype. Prior to infections, bacteria were thawed
on ice and repeatedly passaged through a 27-gauge syr-
inge to disengage any clumps.
2.2. Macrophage infections and TNF-a measurements
The murine macrophage cell line, J774A.1 (ATCC,
TIB-67) was used in this study and propagated in RPMI
medium containing 10% fetal bovine serum (FBS, Sigma,
St. Louis, MO) without added antibiotics. To rule out
variability in results concerning the involvement of
serum proteins, only one source of FBS was usedthroughout the study to generate heat-inactivated and
complement-depleted serum. Heat-inactivated serum
was generated by heating FBS at 56 �C for 30 min. In
studies where C3-depleted serum was used, FBS was
incubated with fractionated C3 antiserum (Sigma, St.
Louis, MO) at a ratio of 8:1 (FBS:antiserum) for 30
min at 37 �C. To investigate the role of serum proteins
and serum opsonins in TNF-a synthesis, J774A.1 cellswere grown to confluence in RPMI with 10% heat-inac-
tivated FBS or 10% C3-depleted serum as indicated
above.
To derive murine bone-marrow derived macrophages
(BMDMs), bone marrow cells from femurs were cul-
tured (5 · 106 cells ml�1, 37 �C, 5% CO2), in complete
DMEM medium containing 10% fetal calf serum, 2
mM L-glutamine, penicillin (100 units ml�1), streptomy-cin (100 lg ml�1), and 30% (vol/vol) L-929 cell-condi-
tioned medium, to provide macrophage growth factor.
To eliminate contaminating fibroblasts, nonadherent
bone marrow cells were transferred after 24 h to nontis-
sue culture petri dishes and grown in L929 cell-condi-
tioned complete DMEM medium for 7 days. Adherent
BMDMs were lifted by incubation in PBS (20 min, 4
�C, 5% CO2) and used for this study.Endotoxin-free anti-CR3 Mac-1 and anti-CR4
monoclonal antibodies (moAb) M1/70 and HL3 against


0
50
100
150
200
250
300
350
Control SmO SmT 213R.4
M. avium 920A6 strains
TN
F-α
[pg
/ml]
0
50
100
150
200
250
300
350
400
control SmO 213R.4 233R.1M. avium 920A6 strains
TN
F-α
[pg
/ml]
0
100
200
300
400
500
600
Control SmO SmT 213R.4
M. avium 920A6 strains
TN
F-α
[pg
/ml]
(a)
(b)
(c)
Fig. 1. TNF-a induction in J774A.1 cells following infection with the
serovar-8 920A6 strains: (a) following the infection of J774A.1 murine
macrophage cell line in serum-containing culture media at a MOI of
5:1 with M. avium 920A6 SmO and SmT (serovar-8) and 213R.4
(serovar null, [24]), levels of TNF-a in supernatants were measured; (b)
TNF-a levels were measured following the infection of J774A.1 murine
macrophage cell line in serum-containing culture media at a MOI of
5:1 with M. avium 920A6 SmO, 213R.4 (serovar null, [24]), and strain
233R.1 (complemented serovar-null strain, [24]); (c) following the
infection of murine bone marrow derived macrophages (BMDMs) line
in serum-containing culture media at a MOI of 5:1 with M. avium
920A6 SmO and SmT (serovar-8) and 213R.4 (serovar null, [24]), levels
of TNF-a in supernatants were measured. Control = uninfected
J774A.1 (a), (b), or BMDMs (c). TNF-a measurements were done in
triplicates (three independent wells/test condition) and expressed as the
means ± SD. The results are representative of three separate
experiments.
0
200
400
600
800
1000
Control SmO SmT 213R.4
M. avium 920A6 strains
TN
F-α
[pg/
ml]
serum∆-inactivated serum
Fig. 2. TNF-a induction in J774A.1 cells following infection with the
serovar-8 strains in RPMI media supplemented with 10% heat-
inactivated serum. Following the infection of J774A.1 murine macro-
phage cell line in heat-inactivated serum-containing culture media at a
MOI of 5:1 with M. avium 920A6 SmO and SmT (serovar-8) and
213R.4 (serovar null), levels of TNF-a in supernatants were measured.
Control = uninfected J774A.1 macrophage cells. TNF-ameasurements
were done in triplicates (three independent wells/test condition) and
expressed as the means ± SD. The results are representative of three
separate experiments.
224 V.R. Irani, J.N. Maslow / FEMS Microbiology Letters 246 (2005) 221–228
overnight incubation of J774A.1 cells in medium sup-
plemented with heat-treated FBS did not yield maxi-
mal increases in TNF-a expression, suggesting that
residual bound serum proteins remained (data not
shown). Experiments performed with murine BMDMs
using shorter incubation periods in serum-free condi-
tions demonstrated results identical to the J774A.1
cells (data not shown), thus validating our use of this
cell line as a model of infection.
3.3. Increased TNF-a synthesis in heat-treated serum
conditions relates to loss of C3
It has been previously reviewed that C3 breakdown
components, C3b and iC3b bind to the mycobacterial
surface to initiate macrophage uptake via CR1, CR3,
and CR4 [12]. We next sought to determine whether
complement-mediated opsonization could be responsi-
ble for differences in TNF-a expression observed be-
tween serum and heat-inactivated serum conditions.As observed in Fig. 3, use of C3-depleted serum was
associated with increased levels of TNF-a for 920A6
SmO (�2-fold increase, p < 0.05), SmT (�8-fold in-
crease, p < 0.05), and 213R.4 (4-fold increase, p < 0.05)
relative to infections performed in the presence of nor-
mal serum. Between-strain comparisons in C3-depleted
serum were not significant. Similar to heat-treated ser-
um, we found that prolonged incubation of J774A.1cells with multiple changes of medium was necessary
to fully eliminate C3 (data not shown).
3.4. Opsonization of M. avium via CR3 and CR4 is
necessary to downregulate TNF-a synthesis
The observation that depletion of serum C3 resulted
in increased TNF-a expression for SmO, SmT, andserovar-null infected cells suggested that for all three
serovar-8 strains, the loss of opsonic interaction of

0
200
400
600
800
1000
Control SmO SmT 213R.4
M. avium 920A6 strains
TN
F-α
[pg/
ml]
serumM 1/70 anti-CR3 moAb
Fig. 4. TNF-a induction in J774A.1 cells following infection with the
serovar-8 strains. J774A.1 cells were blocked for 1 h with moAb M1/70
(15 lg ml�1) directed against the I domain of CD11b (CR3). Cells were
washed twice prior to infection of J774A.1 murine macrophage cell line
at a MOI of 5:1 with M. avium 920A6 SmO and SmT (serovar-8) and
213R.4 (serovar null), levels of TNF-a in supernatants were measured.
Control = uninfected J774A.1 macrophage cells. TNF-ameasurements
were done in triplicates (three independent wells/test condition) and
expressed as the means ± SD. The results are representative of three
separate experiments.
0
200
400
600
800
1000
Control SmO SmT 213R.4M. avium 920A6 strains
TN
F-α
[pg/
ml]
serumHL3 anti-CR4 moAb
Fig. 5. TNF-a induction in J774A.1 cells following infection with the
serovar-8 strains. J774A.1 cells were blocked for 1 h with moAb HL3
(15 lg ml�1) directed against the I domain of CD11c (CR4). Cells were
washed twice prior to infection of J774A.1 murine macrophage cell line
at a MOI of 5:1 with M. avium 920A6 SmO and SmT (serovar-8) and
213R.4 (serovar null), levels of TNF-a in supernatants were measured.
Control = uninfected J774A.1 macrophage cells. TNF-ameasurements
were done in triplicates (three independent wells/test condition) and
expressed as the means ± SD. The results are representative of three
separate experiments.
0
200
400
600
800
1000
1200
1400
SmO SmT 213R.4M. avium 920A6 strains
TN
F-α
[pg
/ml]
serumanti-C3 serum
Fig. 3. TNF-a induction in J774A.1 cells following infection with the
serovar-8 strains in RPMI media supplemented with C3-depleted
serum. Following the infection of J774A.1 murine macrophage cell line
in C3-depleted serum-containing culture media at a MOI of 5:1 with
M. avium 920A6 SmO and SmT (serovar-8) and 213R.4 (serovar null),
levels of TNF-a in supernatants were measured. TNF-a measurements
were done in triplicates (three independent wells/test condition) and
expressed as the means ± SD. The results are representative of three
separate experiments.
V.R. Irani, J.N. Maslow / FEMS Microbiology Letters 246 (2005) 221–228 225
M. avium via iC3b to macrophage receptors CR3 and/or
CR4 resulted in higher TNF-a levels. Interaction
through CR1 was not considered since murine macro-
phages do not express this receptor [29].Although CR3 has consistently been implicated as a
key macrophage receptor for M. avium uptake, its role
in TNF-a activation has not been studied. We, therefore
first assessed the role of CR3 in TNF-amodulation. The
I-domain of CD11b contains the iC3b opsonic binding
site [10]. To confirm that opsonic binding of M. avium
to CD11b suppressed TNF-a induction, bacterial infec-
tions were performed while blocking the iC3b bindingsite with anti-CD11b moAb M1/70. Blockage increased
TNF-a levels 4-fold for 920A-6 SmT and serovar-null
strains (Fig. 4, p < 0.05). In contrast, the SmO strain
demonstrated no difference in TNF-a induction when
CR3 is blocked. The increase in TNF-a was not due
to activation of CR3 by moAb M1/70 since control wells
containing moAb alone yielded TNF-a levels no differ-
ent than control wells containing medium alone (Fig. 4).Since all strains manifested increases in TNF-a
expression under opsonin-free conditions (Figs. 2 and
3) and CR3 blockade did not cause an increase in
TNF-a for SmO strains, this would suggest that for
the SmO strain, regulation of TNF-a expression was
mediated via CR4. We therefore investigated the role
of CR4 in TNF-a activation. Bacterial infections were
performed while blocking the iC3b binding site ofCR4 with anti-CD11c moAb HL3. Blockage of the
I-domain of CD11c with moAb HL3 significantly in-
creased TNF-a levels for all 920A6 strains (Fig. 5,
p > 0.05 for all intra-strain pair wise comparisons), com-
parable to levels obtained during heat-inactivated and
C3-depleted serum conditions. Between-strain compari-
sons were not statistically different (p > 0.05). To rule
out non-specific Fc-mediated activation in TNF-ainduction, control antibodies were used at 15 lg ml�1.The level of host TNF-a induction was not statistically
significant (date not shown, p > 0.05).
4. Discussion
Like M. tuberculosis, M. avium can survive within
macrophages and evade the host immune response.

226 V.R. Irani, J.N. Maslow / FEMS Microbiology Letters 246 (2005) 221–228
Because TNF-a appears to be involved in host immunity,
early bacterial down-regulation of TNF-a expression in
infected macrophages may be an important mechanism
for intracellular survival of virulent M. avium. One of
the goals of this research was to determine if serum pro-
teins, and receptors on the infected macrophage affectTNF-a expression and thus trigger pathways early on
during M. avium infection that could ultimately decide
the fate of the invading bacterium. Also, we wanted
to determine if strains differing in GPL expression differ-
entially induced TNF-a during the early phase of
M. avium–macrophage interaction and if possible, con-
struct consistent hypotheses and future experiments to
understand the variability in cytokine expression amongthe isogenic M. avium strains.
The role of serum proteins, in particular iC3b, to pre-
ventM. avium induced TNF-a expression is suggested by
our observation that M. avium infection of murine mac-
rophages in the presence of heat-inactivated serum re-
sulted in significantly higher levels of TNF-a than its
whole, active serum counterparts. Support for C3 opson-
ization in regulating TNF-a expression was demon-strated by our use of C3-depleted serum, where murine
macrophage cells infected with M. avium wt and
ssGPL-null strains produced higher levels of TNF-a.The complement-mediated opsonic interaction between
the mycobacterium and the infected macrophage
via the CR3 and/or CR4 could be advantageous to the
bacterium by avoidance of potentially harmful reactions,
such as TNF-a production, which could lead to increasedbacterial survival. Our results are in apparent contrast
with the study of Bohlson et al. [20] that demonstrated
no difference in TNF-a induction of BMDM macro-
phages derived from C3 +/+ and C3 �/� C57BL/6 mice
and J774A.1 macrophages. There are two important dif-
ferences between studies. First, the study by Bohlson
et al. studied two serovar-2 strains of M. avium,
TMC724 and 2-151, whereas our study investigated a vir-ulent serovar-8 strain and highlights the possibility of
GPL-related differences in the activation of macrophage
signaling pathways to affect the host immune response.
Moreover, unpublished results from our laboratory using
TMC724 have also demonstrated a lack of difference in
TNF-a induction in conditions varying in the presence
of complement. Second, Bohlson et al. studied TNF-ainduction of cells propagated in the presence of FBS.Prior to M. avium infection, cells were washed and then
incubated with C3-deficient medium for 2 h. We have
shown here that such short incubation times in C3-defi-
cient medium are insufficient to demonstrate differences
between conditions.
Intracellular pathogens, including mycobacteria,
bind CR1, CR3, CR4 as a first step in the invasion
of mammalian cells [10], linking the receptors cyto-plasmic domains to the actin cytoskeleton and proxi-
mal components of the cell signaling pathways [30].
While this process is regulated by TNF-a, we show
for the first time that C3 opsonization of M. avium
with subsequent binding to host macrophage receptors
CR3 and CR4 is necessary to decrease TNF-a secre-
tion by macrophages.
In addition to differences observed in TNF-a syn-thesis by M. avium infected J774A.1 cells during vari-
ous culture conditions, we also noted differences in
TNF-a induction among the three isogenic M. avium
serovar-8 strains. Effects were similar for the murine
macrophage cell line J774A.1 and the BMDMs demon-
strating the utility of this cell line to model in vivo
infections. We noted that the serovar-null mutant acti-
vated lower levels of TNF-a compared to the wt par-ent SmO strain under control conditions and that the
serovar-null strain complemented with wild-type
ssGPL induced macrophage TNF-a to levels identical
to the wild type M. avium parent strain. Absence of
M. avium ssGPL could be the reason for host TNF-
a suppression by the serovar-null strain since prior
studies have demonstrated the role of ssGPL as a po-
tent immunogen and its ability to trigger higher levelsof TNF-a [3]. Our results confirm the involvement of
M. avium ssGPL in macrophage TNF-a expression.
We also noted that the wt SmT strain activated lower
levels of TNF-a compared to wt SmO strain under
control (normal serum) conditions, this data is in
agreement with previous research [21]. One possible
explanation could be due to the differences in the
non-specific (ns) GPL/ssGPL ratios between the wtSmO and SmT strains. However, the full difference
in the expression profiles between SmO and SmT mor-
photypes is unknown, and thus we cannot rule out
other bacterial factors modulating host cell signaling
pathways. The latter may be likely since SmT morpho-
types typically express greater levels of ssGPL than
SmO strains [31].
Regulation of TNF-a induction mediated throughCR-interaction differed among strains. Wild-type
SmO parent strain and the derived serovar-null mu-
tant varied as to CR utilization responsible for regula-
tion of TNF-a levels. While the iC3b domain of CR4
was used by wt SmO for down-regulation of TNF-a,the serovar-null mutant was able to utilize the iC3b
binding domains of both CR3 and CR4 to downregu-
late TNF-a synthesis. Since these are isogenic strains,the differences in receptor binding and TNF-a modu-
lation between the parent SmO and the ssGPL-null
mutant are attributable to the absence of terminal
serovar specific sugars in the mutant strain. We have
demonstrated that the presence of M. avium ssGPL
triggers higher levels of host TNF-a. Whether altera-
tions in TNF-a expression correlate with survival is
unknown and is being investigated.Receptor preference has been shown to be crucial for
bacterial survival [15,16]. Little is known about the

V.R. Irani, J.N. Maslow / FEMS Microbiology Letters 246 (2005) 221–228 227
involvement of CR4 or whether it is preferred over CR3
during the M. avium infection process. In this study, we
have shown that the receptor preference depends on the
type of infecting M. avium strain. Preliminary results on
simultaneous blockade of the iC3b domains of CR3 and
CR4 suggests that on M. avium infection, modulation ofTNF-a synthesis via these macrophage receptors occurs
via independent MAPK p38 and p42/44 pathways (data
not shown), thus raising the possibility that these
M. avium strains could trigger different host MAPK sig-
naling pathways which could ultimately result in a dif-
ferent intracellular fate for each infecting strain.
In summary, serum protein C3, macrophage recep-
tors CR3, CR4, and M. avium ssGPL are involved inmodulation of TNF-a induction during the early stages
of M. avium–macrophage interaction. This is the first re-
ported study that demonstrates the involvement of CR3
and CR4 in suppression of TNF-a synthesis during M.
avium–macrophage interaction.
Acknowledgements
Y. Patterson and L. Buxbaum are gratefully
acknowledged for the gift of J774A.1 cells and BMPMs,
respectively. Paul M. Nealen is gratefully acknowledged
for assistance in statistical analyses. Thomas Glaze and
Andrea Rossi are acknowledged for technical assistance.
Support for this study was provided through Merit
Review and VISN 4 grant from the Veterans Affairs,and University Foundation Grant from the University
of Pennsylvania, 5-UO1-AI32783, P30-AI-045008-06 to
JNM.
References
[1] Appelberg, R., Sarmento, A. and Castro, A. (1995) Tumour
necrosis factor-alpha (TNF-alpha) in the host resistance to
mycobacteria of distinct virulence. Clin. Exp. Immunol. 101,
308–313.
[2] Eriks, I.S. and Emerson, C.L. (1997) Temporal effect of tumor
necrosis factor alpha on murine macrophages infection with
Mycobacterium avium. Infect. Immun. 65, 2100–2106.
[3] Barrow, W.W., Davis, T.L., Wright, E.L., Labrousse, V.,
Bachelet, M. and Rastogi, N. (1995) Immunomodulatory spec-
trum of lipids associated with Mycobacterium avium serovar 8.
Infect. Immun. 63, 126–133.
[4] Weiss, D., Evanson, O., Moritz, A., Deng, M. and Abrahamsen,
M. (2002) Differential responses of bovine macrophages to
Mycobacterium avium subsp. paratuberculosis and Mycobacterium
avium subsp. avium. Infect. Immun. 70, 5556–61.
[5] Ehlers, S., Kutsch, S., Ehlers, E., Benini, J. and Pfeffer, K. (2000)
Lethal granuloma disintegration in mycobacteria-infected TNFR
p55 �/� mice is dependent on T cells and IL-12. J. Immunol. 165,
483–92.
[6] Smith, D., Hansch, H., Bancroft, G. and Ehlers, S. (1997) T-cell-
independent granuloma formation, in response to Mycobacterium
avium: role of tumour necrosis factor-alpha and interferon-
gamma. Immunol. 92, 413–21.
[7] Greenwell-Wild, T., Vazquez, N., Sim, D., Schito, M., Chatterjee,
D., Orenstein, J. and Wahl, S. (2002) Mycobacterium avium
infection and modulation of human macrophage gene expression.
J. Immunol. 169, 6286–97.
[8] Blumenthal, A., Ehlers, S., Ernst, M., Flad, H-D. and Reiling, N.
(2002) Control of mycobacterial replication in human macro-
phages: role of extracellular signal-regulated kinases 1 and 2 and
p38 mitogen-activated protein kinase pathways. Infect. Immun.
70, 4961–4967.
[9] Roecklein, J.A., Swartz, R.P. and Yeager, H. (1992) Nonopsonic
uptake of Mycobacterium avium complex by human monocytes
and alveolar macrophages. J. Lab. Clin. Med. 119, 772–781.
[10] Ernst, J.D. (1998) Macrophage receptors for Mycobacterium
tuberculosis. Infect. Immun. 66, 1277–1281.
[11] Schlesinger, L.S., Bellinger-Kawahara, C.G., Payne, N.R. and
Horwitz, M.A. (1990) Phagocytosis of Mycobacterium tuberculo-
sis is mediated by human monocyte complement receptors and
complement component C3. J. Immunol. 144, 2771–80.
[12] Velasco-Velazquez, M., Barrera, D., Gonzalez-Arenas, A.,
Rosales, C. and Agramonte-Hevia, J. (2003) Macrophages–
Mycobacterium tuberculosis interactions – role of complement
receptor 3. Microb. Pathog. 35, 125–31.
[13] Agramonte-Hevia, J., Gonzalez-Arenas, A., Barrera, D. and
Velasco-Velazquez, M. (2002) Gram-negative bacteria and phag-
ocytic cell interaction mediated by complement receptor 3. FEMS
Immunol. Med. Microbiol. 34, 255–66.
[14] Schorey, J.S., Carroll, M.C. and Brown, E.J. (1997) A macro-
phage invasion mechanism of pathogenic mycobacteria. Science
277, 1091–1093.
[15] Wilson, M. and Pearson, R. (1988) Roles of CR3 and mannose
receptors in the attachment and ingestion of Leishmania donovani
by human mononuclear phagocytes. Infect. Immun. 56, 363–369.
[16] Wright, S. and Silverstein, S. (1983) Receptors for C3b and C3bi
promote phagocytosis but not the release of toxic oxygen from
human phagocytes. J. Exp. Med. 158, 2016–2023.
[17] Reiling, N., Blumenthal, A., Flad, H-D., Ernst, M. and Ehlers, S.
(2001) Mycobacteria-induced TNF-a and IL-10 formation by
human macrophages is differentially regulated at the level of
mitogen-activated protein kinase activity. J. Immunol. 167, 3339–
3345.
[18] Aderem, A. and Underhill, D.M. (1999) Mechanisms of phago-
cytosis in macrophages. Annu. Rev. Immunol. 17, 593–623.
[19] Forsberg, M., Lofgren, R., Zheng, L. and Stendahl, O. (2001)
Tumor necrosis factor-alpha potentiates CR3-induced respiratory
burst by activating p38 MAP kinase in human neutrophils.
Immunol. 103, 465–472.
[20] Bohlson, S.S., Strasser, J.A., Bower, J.J. and Schorey, J.S. (2001)
Role of complement in Mycobacterium avium pathogenesis: In
vivo and in vitro analyses of the host response to infection in the
absence of complement component C3. Infect. Immun. 69, 7729–
7735.
[21] Faldt, J., Dahlgren, C. and Ridell, M. (2002) Difference in
neutrophil cytokine production induced by pathogenic and non-
pathogenic mycobacteria. APMIS 100, 593–600.
[22] Chatterjee, D. and Khoo, K.-H. (2001) The surface glycopeptid-
olipids of mycobacteria:structures and biological properties. Cell
Mol. Life Sci. 58, 2018–2042.
[23] Newman, G.W., Gan, H.X., McCarthy Jr, P.L. and Remold,
H.G. (1991) Survival of human macrophages infected with
Mycobacterium avium-intracellulare correlates with increased
production of tumor necrosis factor-a and IL-6. J. Immunol.
147, 3942–3948.
[24] Irani, V., Lee, S., Eckstein, E., Inamine, J.M., Belisle, J.T. and
Maslow, J. (2004) Utilization of a ts-sacB selection system for the
generation of a Mycobacterium avium serovar-8 specific glyco-
peptidolipid allelic exchange mutant. Ann. Clin. Microbiol.
Antimicrob. 3, 18.

228 V.R. Irani, J.N. Maslow / FEMS Microbiology Letters 246 (2005) 221–228
[25] Arbeit, R.D., Slutsky, A., Barber, T.W., Maslow, J.N., Niemczyk,
S., Falkinham, J.O.I., O�Conner, G.T. and Von Reyn, C.F. (1993)
Genetic diversity among strains of Mycobacterium avium causing
monoclonal and polyclonal bacteremia in patients with AIDS. J.
Infect. Dis. 167, 1384–1390.
[26] Lee, B-Y. et al. (1991) Prevalence of serum antibody to the type-
specific glycopeptidolipid antigens of Mycobacterium avium in
human immunodeficiency virus-positive and -negative individu-
als. J. Clin. Microbiol. 29, 1026–1029.
[27] Bermudez, L.E., Young, L.S. and Enkel, H. (1991) Interaction of
Mycobacterium avium complex with human macrophages: role of
membrane receptors and serum proteins. Infect. Immun. 59,
1697–1702.
[28] Moehring, J.M. and Solotorovsky, M.R. (1965) Relationship of
colonial morphology to virulence for chickens of Mycobacterium
avium and the nonphotochromogens. Am. Rev. Respir. Dis. 92,
704–713.
[29] Martin, B.K. and Weis, J.H. (1993) Murine macrophages lack
expression of the Cr2-145 (CR2) and Cr2-190 (CR1) gene
products. Eur. J. Immunol. 23, 3037–3042.
[30] Gahmberg, C.G. (1997) CD11/CD18 integrins and intracel-
lular adhesion molecules. Curr. Opin. Cell. Biol. 9, 643–
650.
[31] Belisle, J. and Brennan, P. (1994) Molecular basis of colony
morphology in Mycobacterium avium. Res. Microbiol. 145, 237–
242.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 229–234
Overexpression of a hydrogenase gene in Clostridiumparaputrificum to enhance hydrogen gas production
Kenji Morimoto a, Tetsuya Kimura b, Kazuo Sakka b,*, Kunio Ohmiya c
a Rare Sugar Research Center, Kagawa University, 2393 Ikenobe, Miki-cho, Kita-gun, Kagawa 761-0795, Japanb Faculty of Bioresources, Mie University, Tsu 514-8507, Japan
c Agricultural High-Tech Research Center, Meijo University, Tenpaku, Nagoya 468-8502, Japan
Received 28 January 2005; received in revised form 11 April 2005; accepted 12 April 2005
First published online 29 April 2005
Edited by R.P. Gunsalus
Abstract
A [Fe]-hydrogenase gene (hydA) was cloned from Clostridium paraputrificum M-21 in Escherichia coli using a conserved DNA
sequence of clostridial hydrogenase genes amplified by PCR as the probe. The hydA gene consisted of an open reading frame of
1749 bp encoding 582 amino acids with an estimated molecular mass of 64,560 Da. It was ligated into a shuttle vector, pJIR751,
originally constructed for Clostridium perfringens and E. coli, and expressed in C. paraputrificum. Hydrogen gas productivity of
the recombinant increased up to 1.7-fold compared with the wild-type. In the recombinant, overexpression of hydA abolished lactic
acid production and increased acetic acid production by over-oxidation of NADH, which is required for reduction of pyruvic acid to
lactic acid in the wild-type.
� 2005 Published by Elsevier B.V. on behalf of the Federation of European Microbiological Societies.
Keywords: Clostridium paraputrificum; Hydrogenase; Hydrogen gas production
1. Introduction
Since hydrogen gas is an idealistic clean energy mate-
rial that does not generate carbon dioxide gas after com-
bustion, its sustainable production from biomass is in
demand worldwide [1]. To compensate for environmen-
tal problems by reducing carbon dioxide gas generationand overcome shortages of fossil energy in the future,
utilization of abundant biomasses such as chitin, a ma-
jor marine waste, is expected. Gaseous hydrogen is
widely produced by many microorganisms, but is virtu-
ally absent from higher organisms. Anaerobic microor-
ganisms are involved in hydrogen production,
especially photosynthetic microorganisms, and faculta-
0378-1097/$22.00 � 2005 Published by Elsevier B.V. on behalf of the Feder
doi:10.1016/j.femsle.2005.04.014
* Corresponding author. Tel.: +81 59 231 9621; fax: +81 59 231 9684.
E-mail address: [email protected] (K. Sakka).
tive and obligatory anaerobic bacteria have been re-
ported to produce hydrogen gas from soluble and
insoluble biomass such as agricultural by-products and
marine wastes [1–11]. Some chitin-degrading bacteria
such as Aeromonas [12], Serratia [13], and Bacillus [14]
have been reported, although hydrogen gas productivity
has not been characterized in detail in these microorgan-isms. Anaerobic bacteria generally have the ability to
produce hydrogen gas during catabolism of carbohy-
drates and [Fe]-hydrogenases (EC 1.12.7.2) are known
to release hydrogen gas from the reduced form of ferre-
doxin in Clostridium and Desulfovibrio species (Fig. 1)
[15–18].
It is well known that many clostridial species evolve
hydrogen gas as a fermentation product during growth.Information about hydrogenase genes and their
products has been reported from a few clostridia, for
ation of European Microbiological Societies.

glucose
NADH
Fd
pyruvate
H2
acetyl-CoA
butyrateacetate
lactate
1
2
Fd
ATP
CO2
H2
ATP
Fig. 1. Outline of biological hydrogen gas production from glucose by
fermentative microorganisms: 1, ferredoxin–NAD+ reductase (EC
1.18.1.3); 2, ferredoxin hydrogenase (EC 1.12.7.2).
230 K. Morimoto et al. / FEMS Microbiology Letters 246 (2005) 229–234
example, [Fe]-hydrogenase I of Clostridium pasteuria-
num [15,16], hydrogenase A of Clostridium perfringens
[17], and hydrogenase A of Clostridium acetobutylicum
P262 [18]. A number of clostridial species are also
known to degrade and ferment various biomass poly-
mers such as polysaccharides and proteins to obtain en-ergy and reducing powers such as the proton/electron
and reduced compounds in cells. Since anaerobic bacte-
ria possess a mechanism to remove excessive reducing
powers as hydrogen gas using hydrogenase, it is possible
that they produce huge amounts of hydrogen gas during
growth on biomass materials.
Clostridium paraputrificum M-21 was isolated and
characterized as a chitin-degrading hydrogen-producinganaerobe in our laboratory [8,9]. Two chitinase and two
b-N-acetylglucosaminidase genes of this bacterium were
characterized along with their translated products [19–
22]. In our preceding paper [23], we reported the con-
struction of the host–vector system of C. paraputrificum
M-21, allowing us to improve its biomass-degrading and
hydrogen gas-producing abilities.
In the present study, we isolated the hydA geneencoding a [Fe]-hydrogenase from C. paraputrificum
M-21, which was isolated from soil as a chitin-degrading
hydrogen-producing anaerobe, and studied the effect of
hydA overexpression on the production of hydrogen gas.
As a result, we found that the recombinant clone over-
expressing the hydA gene produced 1.7 times as much
hydrogen gas as the parental clone, along with a drastic
reduction in lactic acid production.
2. Materials and methods
2.1. Bacterial strains, plasmids, and growth conditions
C. paraputrificum M-21 was isolated and character-
ized as a chitin-degrading hydrogen-producing bacte-rium, was described previously [8]. Plasmid pJIR751
was obtained from Dr. J.I. Rood (Monash University,
Australia) [24] and used to overexpress the hydA gene.
C. paraputrificum was grown anaerobically at 45 �C in
modified GS medium (pH 6.5) supplemented with 1%
N-acetylglucosamine (GlcNAc). Cultivations were con-
ducted in test tubes under static condition or in a 1-ljar fermenter (B.E. Marubishi Lab., Tokyo) containing
500 ml of the medium with agitation at 250 rpm. Re-
combinant C. paraputrificum was cultivated under the
same conditions but with erythromycin (10 lg/ml).
The following plasmids were used as the cloning and
sequencing vectors for Escherichia coli: pT7Blue (Nova-
gen, Madison, WI), pBluescript II KS (�) and pBlue-
script II KS (+) (Stratagene, La Jolla, CA), andCharomid 9-28 (Nippon Gene, Tokyo). E. coli XL1-
Blue and DH5a were grown aerobically at 37 �C in Lur-
ia–Berrani (LB) medium supplemented with ampicillin
(100 lg/ml) and IPTG (50 lg/ml) when necessary.
2.2. Cloning of the C. paraputrificum hydA gene
Chromosomal DNA of C. paraputrificum M-21 wasisolated according to the procedure of Silhavy et al.
[25], partially digested with EcoRI, and separated on
0.4% Agarose H gel (Nippon Gene). DNA fragments
with appropriate sizes were recovered from the agarose
gel using the GeneClean (Bio101, La Jolla, CA) proce-
dure, and ligated into the EcoRI site of Charomid 9-
28. Ligation and transformation of E. coli DH5a were
carried out according to the protocol of the Charomidcloning kit. A pair of PCR primers were designed
according to the sequences highly conserved in the
[Fe]-hydrogenase genes from Clostridium and Desulf-
ovibrio species to obtain a partial region of a hydroge-
nase gene from C. paraputrificum M-21. The following
forward and reverse primers were used: 5 0-
TTYGGNGCNGAYATGACNATHATGGARGA-3 0
and 5 0-CANCCNCCNKGRCANGCCATNACYTC-3 0, respectively. A 700-bp PCR fragment amplified
from C. paraputrificum chromosomal DNA was li-
gated into pT7Blue and introduced into E. coli XL1-
Blue. The cloned DNA fragment was then amplified
and labeled with digoxigenin-11 dUTP (Roche Diag-
nostics GmbH, Penzberg) by PCR with the primers de-
scribed above. The labeled DNA fragment was used as
a probe for colony hybridization to clone the full-length [Fe]-hydrogenase gene (hydA) from the C.
paraputrificum genome library.
2.3. DNA sequencing
Nucleotide sequencing was carried out on a LICOR
model 4000L automated DNA sequencer (Lincoln,
Neb.), with appropriate dye primers and a series of sub-clones. Nucleotide sequence data was analyzed with
GENETYX computer software (Software Development

K. Morimoto et al. / FEMS Microbiology Letters 246 (2005) 229–234 231
Co. Ltd., Tokyo, Japan). Homology searches in DDBJ
were carried out with the BLAST program.
2.4. Construction of pJIR751–hyd
For overexpression of hydA in C. paraputrificum M-21, the hydA gene was subcloned into an E. coli–C. per-
fringens shuttle vector, pJIR751, as follows: plasmid
pHYD101 containing the full-length hydA gene was di-
gested with XbaI and SpeI then a 2.3-kbp DNA frag-
ment containing hydA was ligated into pJIR751, which
had been digested with XbaI in advance, yielding
pJIR751–hyd.
2.5. Electroporation of C. paraputrofocum M-21
C. paraputrificum M-21 was transformed with
pJIR751–hyd according to the electroporation proce-
dure described previously [23].
2.6. Analysis of hydrogen gas production
The total amount of gas produced by the wild-type
and recombinant strains from a 500-ml culture in a 1-l
jar fermenter was measured with a wet gas meter (W-
NK Da-0.5A, Shinagawa, Co., Tokyo) connected to
the jar fermenter by rubber tubing during cultivation.
The absorbance was measured at 600 nm for evaluating
bacterial cell growth with a double-beam spectropho-
tometer (UV-150-02, Shimadzu Co., Kyoto). The com-position of the fermentation gas was analyzed using a
gas chromatography system (model GC-323 equipped
with Molecular Sieve 5A and Porapak Q columns and
a TCD detector; GL Sciences Inc., Tokyo). Separation
was carried out at 50 �C using argon gas as the carrier
gas. Organic acids produced were analyzed using an
HPLC system, GL Sciences EZ Chrom Elite analyzer
equipped with a Shodex Rspak KC-811 column and aGL Sciences UV 620 detector. Separation was con-
ducted at 40 �C using 1 mM HClO4 as an eluent and
ST3-R as a regent at a flow rate of 1.0 ml/min.
2.7. Nucleotide sequence accession number
The nucleotide sequence reported in this paper is
available in the DDBJ, EMBL, and GenBank nucleotidesequence databases under accession number AB159510.
3. Results
3.1. Cloning and sequencing of the hydA gene from
C. paraputrificum M-21
Employing PCR and colony hybridization methods,
a 9.0-kbp DNA fragment expected to contain the full-
length hydA gene and its flanking region was cloned
from C. paraputrificum M-21 using Charomid 9-28.
Sequencing of the inserted DNA fragment identified
an open reading frame of 1749 bp, which encoded a no-
vel hydrogenase (HydA) of 582 amino acids with a pre-
dicted molecular mass of 64,560 Da. The predictedmolecular mass was in good agreement with that of
many clostridial [Fe]-hydrogenases [15,17,18]. A puta-
tive ribosomal-binding site (GGAGG) and �35 and
�10 regions (TTGAAC and AAAAAT with a 18-bp
spacing) were located upstream of the hydA ATG start
codon. Transcription of the hydA gene was expected to
end in rho-factor dependence, because there was no
clear stem-loop structure downstream of the stop codon.Comparisons of the amino acid sequence of HydA
with entries in the DDBJ database indicated that this en-
zyme is highly homologous with some clostridial [Fe]-
hydrogenases (EC 1.12.7.2) as expected; for example,
HydA of C. acetobutylicum P262 (sequence identity
75.1%) [18], HydI of C. pasteurianum W5 (69.4%) [15],
HydA of C. perfringens NTCT8237 (71.3%) [17], and
HydA of Clostridium thermocellum (47.6%, DDBJ acces-sion no. AAD33071). Fig. 2 shows alignment of amino
acid sequences of clostridial hydrogenases. In addition,
C. paraputrificum HydA showed a certain similarity to
some large subunits of [Fe]-hydrogenases in Desulfovib-
rio species such as Desulfovibrio vulgaris subsp. oxami-
cus Monticell (sequence identity 45.4%) [26] and D.
vulgaris subsp. vulgaris str. Hildenborough (45.1%) [27].
3.2. Hydrogen gas production by C. paraputrificum M-21
carrying multiple copies of hydA
When C. paraputrificum M-21 was cultivated in the
fermenter containing 500 ml of GS medium (pH 6.5)
with 1% GlcNAc as the carbon source at 45 �C, the totalvolume of fermentation gas evolved was about 2 l per li-
tre of medium. The ratio of H2 to CO2 was 2:1 and thehydrogen gas yield was 1.4 mol/mol GlcNAc (Fig. 3(a)).
Plasmid pJIR751–hyd contained the hydA structural
gene along with its flanking region containing the possi-
ble promoter region. This plasmid was introduced and
expressed in C. paraputrificum M-21. Although the ratio
of H2 to CO2 in the fermentation gas of the wild-type
and recombinant strains was constantly 2:1, the total
volume of fermentation gas produced by the recombi-nant was about 3.5 l per litre of the medium; hydrogen
gas yield was 2.4 mol/mol GlcNAc, 1.7-fold higher than
that of the wild type (Fig. 3(b)). These results suggested
that enforced hydrogenase activity accelerated oxidation
of ferredoxin to release hydrogen gas. The composition
of organic acids produced by the recombinant and host
was determined and compared (Table 1). The amount of
acetic acid remarkably increased in parallel with an in-crease in hydrogen gas evolution, while on the contrary,
the amount of lactic acid drastically decreased (Table 1).

Fig. 2. Alignment of [Fe]-hydrogenases of C. paraputrificum (C. par), C. acetobutylicum P262 (C. ace), C. pasteurianum W5 (C. pas), C. perfringens
NTCT8237 (C. per), and C. thermocellum (C. the). Amino acids which are conserved in all sequences are highlighted. A His residue and 19 Cys
residues responsible for holding Fe–S clusters are shown with sharp signs (#). –, gap left to improve alignment. Numbers refer to amino acid residues
at the start of the respective lines; all sequences are numbered from Met-1 of the peptide.
232 K. Morimoto et al. / FEMS Microbiology Letters 246 (2005) 229–234
An increase in acetic acid with an increase in hydrogen
gas evolution seems to be reasonable since their produc-
tions result from the same metabolic pathway (Fig. 1).
The production of lactic acid was negligible in the
hydrogenase-fortified recombinant (Table 1), indicatingthat the conversion of pyruvic acid to lactic acid was al-
most shut down. The growth rate of the recombinant
clone was identical to that of the host and the trans-
formant containing pJIR751–hyd as judged by measure-
ment of absorbance at 600 nm (Fig. 3).
4. Discussion
In our previous papers, we reported the hydrogen gas
productivity of C. paraputricum M-21 from chitinous
materials [8,9] and suggested that the enhancement of
hydrogenase activity using genetic engineering would
improve hydrogen gas production. Kaji et al. [17] re-
ported that disruption of the hydA gene encoding a
[Fe]-hydrogenase by homologous recombination in C.
perfringens strain 13 abolished its hydrogen gas produc-
tion from glucose, suggesting that this gene was respon-
sible for hydrogen gas production. Therefore, we cloned
a C. paraputrificum M-21 [Fe]-hydrogenase gene that
was a homologue of the C. perfringens hydA gene.
[Fe]-hydrogenases belong to a category of metal-con-
taining hydrogenases including [Ni–Fe] and [Ni–Fe–Se]
hydrogenases [28]. The amino acid sequence ofC. paraputrificum M-21 HydA showed strong similarity
to that of clostridial [Fe] hydrogenases, particularly that
from C. acetobutylicum P262, and moderate similarity to

Table 1
Composition of organic acids produced from GlcNAc by C. parapu-
trificum M-21
Plasmid Organic acid (mM)
Lactic
acid
Acetic
acid
Butyric
acid
Formic
acid
Propionic
acid
None 29.3 38.1 12.7 4.87 0.03
pJIR751 28.6 33.9 14.4 5.13 0.04
pJIR751–hyd 0.42 51.8 13.1 5.90 0.02
Fig. 3. Enhanced hydrogen gas production by the overexpression of
the hydA gene in C. paraputrificum. Non-transformant cells (a) and
recombinant cells harboring pJIR751–hyd (b) were cultivated in 500
ml of GS medium containing 1% GlcNAc.
K. Morimoto et al. / FEMS Microbiology Letters 246 (2005) 229–234 233
those of a large subunit of hydrogenases from Desulf-
ovibrio species; no sequence similarity with those of
[Ni–Fe] or [Ni–Fe–Se] hydrogenases was seen. The
three-dimensional structure of a [Fe]-hydrogenase
(HydI) of C. pasteurianum was previously reported
[16]. In HydI and some other clostridial hydrogenases,
19 cysteine residues and a histidine residue are conserved
as shown in Fig. 2, and these residues are known to fas-ten one [2Fe–2S] cluster, three [4Fe–4S] clusters, and one
H cluster that functions as an active center [16].
Hydrogen gas production has been discussed in a
number of studies using various clostridia, especially in
C. acetobutylicum. Sugar metabolism of this bacterium
is composed of two phases: an acidogenesis phase and
solventogenesis phase [18]. During acidogenesis in clos-
tridia, a large amount of electron flow is directed tohydrogen gas production while sugars are converted to
organic acids such as acetic acid (Fig. 1). Kim et al.
[29] reported that a significant decrease in the rate of
hydrogen gas production correlated with the shift of
metabolic phase from acidogenesis to solventogenesis.
The metabolic pathway in acidogenic Clostridium has
several possible end products, including butyrate, ace-
tate, lactate, CO2, and H2 (Fig. 1). Acetate and butyratefermentation on glucose by typical acidogenic Clostrid-
ium species ideally proceeds according to the following
equation [30]:
Glucose ! 0.8 butyrateþ 0.4 acetate þ 2 CO2 þ 2.4 H2
Although C. paraputrificum M-21 also produced anenormous volume of hydrogen gas during the exponen-
tial growth phase [8,9], the yield of hydrogen gas from 1
mol of glucose did not reach 2.4 mol (1.4 mol). When C.
paraputrificum M-21 was cultured with 1% GlcNAc, not
only acetic and butyric acids but also lactic acid was
produced from GlcNAc (Table 1). Since hydrogen gas
is not coproduced with lactic acid (Glucose ! 2 lactic
acid), the production of lactic acid would reduce theyield of hydrogen gas production. On the other hand,
when the C. paraputrificum M-21 recombinant over-
expressing hydA was cultured with 1% GlcNAc, the re-
combinant produced higher amounts of acetic acid
and negligible amounts of lactic acid in the culture fluid
compared with the host organism: the yield of acetic
acid, lactic acid and butyric acid from 1 mol of glucose
was calculated as 0.93, 0.01, and 0.24 mol, respectively.Improvement of hydrogen gas production in the recom-
binant was caused by reduction in the amount of lactic
acid and enhancement in the amount of acetic acid. It
seems apparent that the enhanced hydrogenase activity
caused over-oxidation of NADH to NAD+, and conse-
quently the depletion of NADH to reduce pyruvic acid
to lactic acid (Fig. 1). However, the production of buty-
ric acid was not affected by the overexpression of hydA.Further improvement of hydrogen gas production might
be achieved by the inhibition of electron flow to butyric
acid by the disruption of the gene responsible for butyric
acid production.
In conclusion, the hydA-expressing recombinant of C.
paraputrificum M-21 produced an increased amount of
hydrogen gas from GlcNAc, along with increased acetic
acid production and reduced production of lactic acid.Further studies are necessary to further improve the
hydrogen gas productivity of C. paraputrificum M-21
by gene disruption leading to the inhibition of butyric
acid production.
Acknowledgements
This work was supported in part by a Grant-in-Aid
for University and Society Collaboration (Grant No.
12794004), the Ministry of Education, Culture, Sports,

234 K. Morimoto et al. / FEMS Microbiology Letters 246 (2005) 229–234
Science, and Technology of Japan, and by the NEDO
project ‘‘High efficiency bioenergy conversion prohect
development of high efficiency hydrogen–methane fer-
mentation process using organic wastes’’.
References
[1] Hawkes, F.R., Dinsdale, R., Hawkes, D.L. and Hussy, I. (2002)
Susustainable fermentative hydrogen production: challenges for
process optimization. Int. J. Hydrogen Energy 27, 1339–1347.
[2] Miyake, J., Mao, X. and Kawamura, S. (1984) Photoproduction
of hydrogen from glucose by a co-culture of a photosynthetic
bacterium and Clostridium butyricum. J. Ferment. Technol. 62,
531–535.
[3] Yokoi, H., Ohkawara, T., Hirose, J., Hayashi, S. and Takasaki,
Y. (1995) Characteristics of hydrogen production by aciduric
Enterobacter aerogenes strain HO-39. J. Ferment. Bioeng. 80,
571–574.
[4] Ueno, Y., Haruta, S., Ishii, M. and Igarashi, Y. (2001) Microbial
community in anaerobic hydrogen-producing microflora enriched
from sludge compost. Appl. Microbiol. Biotechnol. 57, 555–562.
[5] Pel, R., Wessels, G., Aalfs, H. and Gottschal, J.C. (1989) Chitin
degradation by Clostridium sp. strain 9.1 in mixed cultures with
saccharolytic and sulphate-reducing bacteria. FEMS Microbiol.
Ecol. 62, 191–200.
[6] Taguchi, F., Mizukami, N., Yamada, K., Hasegawa, K. and
Saito-Taki, T. (1995) Direct conversion of cellulosic materials to
hydrogen by Clostridium sp. strain no. 2. Enzyme Microb.
Technol. 17, 147–150.
[7] Yokoi, H., Tokushige, T., Hirose, J., Hayashi, S. and Takasaki,
Y. (1997) Hydrogen production by immobilized cells of aciduric
Enterobacter aerogenes strain HO-39. J. Ferment. Bioeng. 83,
481–484.
[8] Evvyernie, D., Yamazaki, S., Morimoto, K., Karita, S., Kimura,
T., Sakka, S. and Ohmiya, K. (2000) Identification and charac-
terization of Clostridium paraputrificum M-21, a chitinolytic,
mesophilic and hydrogen-producing bacterium. J. Biosci. Bioeng.
89, 596–601.
[9] Evvyernie, D., Morimoto, K., Karita, S., Kimura, T., Sakka, K.
and Ohmiya, K. (2001) Conversion of chitinous wastes to
hydrogen gas by Clostridium paraputrificum M-21. J. Biosci.
Bioeng. 91, 339–343.
[10] Hussy, I., Hawkes, F.R., Dinsdale, R. and Hawkes, D.L. (2003)
Continuous fermentative hydrogen production from a wheat
starch co-product by mixed microflora. Biotechnol. Bioeng. 84,
619–626.
[11] Lin, C.Y. and Lay, C.H. (2004) Carbon/nitrogen-ratio effect on
fermentative hydrpgen production by mixed microflora. Int. J.
Hydrogen Energy 29, 41–45.
[12] Ueda, M., Shiro, M., Kawaguchi, T. and Arai, M. (1996)
Expression of the chitinase III gene of Aeromonas sp. No. 10S-
24 in Escherichia coli. Biosci. Biotechnol. Biochem. 60, 1195–1197.
[13] Suzuki, K., Sugawara, N., Suzuki, M., Uchiyama, T., Katouno,
F., Nikaidou, N. and Watanabe, T. (2002) Chitinases A, B, and
C1 of Serratia marcescens 2170 produced by recombinant
Escherichia coli: enzymatic properties and synergism on chitin
degradation. Biosci. Biotechnol. Biochem. 66, 1075–1083.
[14] Alam, M.M., Nikaido, N., Tanaka, H. and Watanabe, T. (1995)
Cloning and sequencing of chiC gene of Bacillus circulans WL-12
and relationship of its product to some other chitinases and
chitinase-like proteins. J. Ferment. Bioeng. 80, 454–461.
[15] Meyer, J. and Gagnon, J. (1991) Primary structure of hydroge-
nase I from Clostridium pasteurianum. Biochemistry 30, 9697–
9704.
[16] Peters, J.W., Lanzilotta, W.N., Lemon, B.J. and Seefeldt, L.C.
(1998) X-ray crystal structure of the Fe only hydrogenase (CpI)
from Clostridium pasteurianum to 1.8 angstrom resolution.
Science 282, 1853–1858.
[17] Kaji, M., Taniguchi, Y., Matsushita, O., Katayama, S., Miyata,
S., Morita, S. and Okabe, A. (1999) The hydA gene encoding the
H2-evolving hydrogenase of Clostridium perfringens: molecular
characterization and expression of the gene. FEMS Microbiol.
Lett. 181, 329–336.
[18] Santangelo, J.D., Durre, P. and Woods, R.D. (1995) Charac-
terization and expression of the hydrogenase-encoding gene
from Clostridium acetobutylicum P262. Microbiology 141, 171–
180.
[19] Morimoto, K., Karita, S., Kimura, T., Sakka, K. and Ohmiya, K.
(1997) Cloning, sequencing, and expression of the gene encoding
Clostridium paraputrificum chitinase ChiB and analysis of the
functions of novel cadherin-like domains and a chitin-binding
domain. J. Bacteriol. 179, 7306–7314.
[20] Morimoto, K., Karita, S., Kimura, T., Sakka, K. and Ohmiya, K.
(1999) Sequencing, expression, and transcription analysis of the
Clostridium paraputrificum chiA gene encoding chitinase ChiA.
Appl. Microbiol. Biotechnol. 51, 340–347.
[21] Li, H., Morimoto, K., Katagiri, N., Kimura, T., Sakka, K., Lun,
S. and Ohmiya, K. (2002) A novel b-N-acetylglucosaminidase of
Clostridium paraputrificum M-21 with high activity on chitobiose.
Appl. Microbiol. Biotechnol. 60, 420–427.
[22] Li, H., Morimoto, K., Kimura, T., Sakka, K. and Ohmiya, K.
(2003) A new type of b-N-acetylglucosaminidase from hydrogen-
producing Clostridium paraputrificum M-21. J. Biosci. Bioeng. 96,
268–274.
[23] Sakka, K., Kawase, M., Baba, D., Morimoto, K., Karita, S.,
Kimura, T. and Ohmiya, K. (2003) Electrotransformation of
Clostridium paraputrificum M-21 with some plasmids. J. Biosci.
Bioeng. 96, 304–306.
[24] Sloan, J., Warner, T.A., Scott, P., Bannam, T.I., Berryman, D.I.
and Rood, J.I. (1992) Construction of a sequenced Clostridium
perfringens–Escherichia coli shuttle plasmid. Plasmid 27, 207–219.
[25] Silhavy, T.J., Berman, M.L. and Enquist, L.W. (1984) Experi-
ments with Gene Fusions. Cold Spring Harbor Laboratory, Cold
Spring Harbor, New York.
[26] Voordouw, G., Strang, J.D. and Wilson, F.R. (1989) Organiza-
tion of the genes encoding [Fe] hydrogenase in Desulfovibrio
vulgaris subsp. oxamicus Monticello. J. Bacteriol. 171, 3881–3889.
[27] Voordouw, G. and Brenner, S. (1985) Nucleotide sequence of the
gene encoding the hydrogenase from Desulfovibrio vulgaris
(Hildenborough). Eur. J. Biochem. 148, 515–520.
[28] Vignais, P.M. and Colbeau, A. (2004) Molecular biology of
microbial hydrogenases. Curr. Issues Mol. Biol. 6, 159–188.
[29] Kim, B.H., Bellows, P., Datta, R. and Zeikus, J.G. (1984) Control
of carbon and electron flow in Clostridium acetobutylicum
fermentations: utilization of carbon monoxide to inhibit hydrogen
production and enhance butanol yield. Appl. Environ. Microbiol.
48, 764–770.
[30] Rogers, P. and Gottschalk, G. (1993) Biochemistry and regulation
of acid and solvent production in Clostridia In: The Clostridia and
Biotechnology (Woods, D.R., Ed.), pp. 25–50. Butterworth-
Heinemann, Stoneham, MA.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 235–242
RirA is the iron response regulator of the rhizobactin1021 biosynthesis and transport genes in Sinorhizobium meliloti 2011
Caroline Viguier, Paraic O Cuıv, Paul Clarke, Michael O�Connell *
School of Biotechnology, National Centre for Sensor Research, Dublin City University, Dublin 9, Ireland
Received 3 March 2005; received in revised form 24 March 2005; accepted 12 April 2005
First published online 22 April 2005
Edited by K. Hantke
Abstract
The genes encoding the biosynthesis and transport of rhizobactin 1021, a siderophore produced by Sinorhizobium meliloti, are
negatively regulated by iron. Mutagenesis of rirA, the rhizobial iron regulator, resulted in abolition of the iron responsive regulation
of the biosynthesis and transport genes. Bioassay analysis revealed that the siderophore is produced in the presence of iron in a rirA
mutant. RNA analysis and GFP fusions supported the conclusion that RirA is the mediator of iron-responsive transcriptional
repression of the two transcripts encoding the biosynthesis and transport genes. RirA in S. meliloti appears to fulfil the role often
observed for Fur in other bacterial species. The regulator was found to mediate the iron-responsive expression of two additional
genes, smc02726 and dppA1, repressing the former while activating the latter. The rirA mutant nodulated the host plant Medicago
sativa (alfalfa) and fixed nitrogen as effectively as the wild type.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Siderophore; Iron response; RirA; Fur
1. Introduction
Most bacteria possess a variety of mechanisms that
enable them to obtain iron from the environment. To
ensure that the various mechanisms are employed effec-tively and in a manner that does not result in oversupply
of iron, there is a need for coordinated regulation [1].
This is frequently achieved by regulating gene expression
at the transcriptional level. In the presence of adequate
amounts of iron, the transcriptional repressor Fur binds
ferrous iron and binds to DNA at a Fur box, preventing
transcription [2]. However, the binding of ferrous iron to
Fur is relatively weak and under iron-deplete conditionsthe metal does not bind the regulator preferentially, in
which case Fur does not bind DNA and transcription
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.012
* Corresponding author. Tel.: +353 1 7005318; fax: +353 1 7005412.
E-mail address: [email protected] (M. O�Connell).
can occur, often under the control of transcriptional
activators [3,4]. Fur has been found widely in bacteria
[5–7]. However, in some rhizobia, while there is a fur
homologue present, the encoded protein appears to
function in the regulation of manganese acquisitionand not in iron acquisition [8–10].
Rhizobia are found free-living in soil and also as
endosymbionts of legumes, where they induce the for-
mation of nitrogen fixing nodules. Rhizobia infect their
host plants in a species-specific manner; for example
Sinorhizobium meliloti is the endosymbiont of Medicago
sativa (alfalfa). In all cases, the functioning nodule con-
tains an abundance of iron-containing proteins, includ-ing nitrogenase, the central enzyme in nitrogen
fixation. Consequently, the acquisition of iron must be
essential for an effective nitrogen fixing symbiosis. Fur-
thermore, in many soils, free-living rhizobia would be
likely to encounter competition from other microbes
. Published by Elsevier B.V. All rights reserved.

236 C. Viguier et al. / FEMS Microbiology Letters 246 (2005) 235–242
for iron. The bacteria must employ efficient mechanisms
to satisfy the iron requirement in these diverse environ-
ments. Some strains of S. meliloti produce the sidero-
phore rhizobactin 1021, which probably contributes to
their competitive ability while growing under free-living
conditions in soil. However, the siderophore is not pro-duced in mature nodules [11] and the mechanism by
which the bacteria satisfy their iron requirement in sym-
biosis is not as yet clearly understood.
Rhizobactin 1021 is a citrate hydroxamate sidero-
phore, biosynthesis of the core structure being encoded
by six genes, rhbABCDEF, which are contiguous in a
single operon with the gene encoding the novel permease
RhtX [12]. The gene encoding the outer membranereceptor RhtA is expressed on a separate transcript.
rhtA and the operon encoding the biosynthesis genes
have been shown previously to be iron-regulated [11].
Chao et al. [10] isolated a fur mutant of S. meliloti and
showed by microarray analysis that in the fur mutant
the rhizobactin 1021 biosynthesis genes and rhtA were
not upregulated, in comparison with the wild type, un-
der iron-replete conditions. Contrary to expectation,they observed that the genes were downregulated,
although it was suggested that this unexpected result
may be due to the effect of Fur regulation on the SitA-
BCD transport system, which transports manganese pri-
marily and would influence metal homeostasis.
However, the observation that the fur mutation does
not result in upregulation of the genes involved in rhi-
zobactin 1021 production and utilisation implies thatan alternative iron response regulator to Fur is present
in S. meliloti, a conclusion that concurs with that ob-
served for Rhizobium leguminosarum [13]. In contrast,
a role for Fur in iron-responsive regulation has been
established in another member of the rhizobia, Brady-
rhizobium japonicum [8].
A novel iron response regulator (RirA) has been re-
ported in R. leguminosarum and has been shown to reg-ulate at least eight operons including operons involved
in the production and utilisation of vicibactin [13]. In
view of the evidence that Fur is not the iron response
regulator for siderophore production and utilisation in
S. meliloti, it was of interest to determine if RirA fulfils
the role in this species. Here, we report that RirA is the
regulator controlling the iron response of genes involved
in the biosynthesis and transport of rhizobactin 1021and, in addition, other iron-related genes in S. meliloti.
2. Materials and methods
2.1. Bacterial strains and growth conditions
The bacterial strains and plasmids used are describedin Table 1. S. meliloti was cultured on TY medium [19]
at 30 �C and Escherichia coli on LB [14] at 37 �C. Anti-
biotics were used at the following concentrations: for S.
meliloti, kanamycin at 100 lg · ml�1, gentamicin at
30 lg · ml�1, tetracycline at 10 lg · ml�1 and strepto-
mycin at 1 mg · ml�1; for E. coli, kanamycin at
30 lg · ml�1, gentamicin at 20 lg · ml�1, tetracycline
at 20 lg · ml�1, ampicillin at 100 lg · ml�1 and strepto-mycin at 100 lg · ml�1.
2.2. Construction of mutants and plasmid fusions
To construct the mutant S. meliloti 2011rirA2 carry-
ing a cassette encoding resistance to kanamycin inserted
in the rirA gene (smc00785 in the S. meliloti genome
available at http://bioinfo.genopole-toulouse.prd.fr/annotation/iANT/bacteria/rhime/), a region approxi-
mately 2 kb in length containing the gene and its flank-
ing sequences was amplified by PCR from genomic
DNA using the forward and reverse primers onto which
sites for XhoI and SpeI were added (Table 1). The frag-
ment was ligated to the pCR2.1 vector and then sub-
cloned into the vector pJQ200ks, which had been
restricted with XhoI and SpeI. This construct was thenrestricted with NcoI, which made a single cut within
the rirA gene into which a cassette encoding kanamycin
resistance, could be inserted by ligation. The cassette
was obtained as an NcoI fragment by PCR amplifica-
tion using plasmid pUC4K as a source (Table 1). The
pJQ200ks derivative carrying the rirA gene with the
kanamycin resistance gene insertion was mobilised into
S. meliloti 2011 by triparental mating with pRK600.Selection was made for integration of the narrow host
range plasmid into the S. meliloti genome by plating
on medium containing gentamicin. After purification
of the transconjugant, a second recombination and alle-
lic replacement were selected on medium containing 5%
sucrose and kanamycin. Gentamicin sensitivity was
checked and Southern hybridisation was used to con-
firm the loss of the vector and correct insertion of thecassette. The mutation was named 2011rirA2.
Construction of the pOTCV1 plasmid in which the
promoter for the rhtXrhbABCDEF operon is fused to
GFP was undertaken as follows: the promoter region
upstream of rhtX was amplified as a HindIII/PstI frag-
ment from S. meliloti genomic DNA by PCR, using
the forward and reverse primers described in Table 1.
The amplified product was cleaned using the PCR prod-uct purification kit (Eppendorf), restricted with HindIII
and PstI and ligated to the vector pOT1, which had been
restricted with the same enzymes.
2.3. Molecular biology techniques
Genomic DNA was prepared by the method of
Meade et al. [15]. RNA was prepared using RNA Whiz(Ambion) as directed by the manufacturers. Plasmid
DNA was isolated by the alkaline lysis method [20].

Table 1
Bacterial strains, plasmid and primers
Strain/plasmids/genes Relevant characteristic(s)/primers Source/reference
Strains
Escherichia coli
DH5a endA1 gyrA96 thi-1 hsdR17 supE44 relA1 recA1 DlacU169 (/80DlacZ DM15) [14]
Sinorhizobium meliloti
2011 Wild type, Strr, Nod+, Fix+ [15]
2011rhbA62 Tn5lac insertion in rhbA [10]
2011rirA2 Kanamycin resistance insertion in rirA This study
Plasmids
pJQ200ks Gmr mob sacB [16]
pRK600 Tra Cmr [17]
pCR2.1 Apr Invitrogen
pOT1 Gmr [18]
pOTCV1 Gmr with rhtXrhbABCDEF promoter region cloned upstream gfp This study
Primers 50 ! 3 0
rirA (mutation) rirAM-F: CTCGAG TCG CCG AGG CCC ATT CCT TCT This study
rirAM-R A: CTAGT GAA GTC GGC TGT AAA CGG TAT GCG
Kanamycin resistance cassette (rirA mutation) kanNcoI-F: CCATGG GAC GTT GTA AAA CGA CGG CCA GTG This study
kanNcoI-R: CCATGG GGA AAC AGC TAT GAC CAT GAT TAC G
pOTCV1 construction pOTCV1-F: CCCAAGCTTCCCTGGAGGCGTCCTATCGCC This study
pOTCV1-R: AAACTGCAGGGCAACATTGTCTGACGATAAACATG
16S rRNA 16S rRNA-F: TCT TTC CCC CGA AGG GCT C16S This study
rRNA-R: ACT TGA GAG TTT GAT CCT GGC
rhbA rhbA-F: ATG CCG GCC GAT TTA GCC This study
rhbA-R: TCG CGT CTT TCC TGT CGG
rhtA rhtA-F: CTATGGAATTGGCAACTACTC This study
rhtA:R: CGATGATCTCAACGGCAAGC
rirA rirA-F: GCG TCT GAC GAA GCA AAC C This study
rirA-R: TAC CGT CTC GAC CAG GCC
dppA1 dppA1-F: CAC TAC TCT CTT GGC AGC G This study
dppA1-R: ACG GCT GTA AAC GGT ATG CG
smc02726 smc02726-F: ATGCTCAACCGGCATCATCGCCTGGC This study
smc02726-R: CGCGACGATCTTCTTCAGCACGGTCG
C. Viguier et al. / FEMS Microbiology Letters 246 (2005) 235–242 237
Restriction, ligation and Southern hybridisation were
carried out by standard procedures [14]. Transformation
was by the method of Inoue et al. [21] and conjugation
by the method of O�Connell et al. [22]. Primers were ob-
tained from MWG Biotech (Milton Keynes) and PCRs
were undertaken using a Thermo Hybaid PCR Express
thermal cycler.
2.4. Fluorescence detection and microscopy
Green fluorescent protein activity was detected qual-
itatively using a UV microscope to view cells from cul-
tures grown in TY broth (iron-replete conditions) or
made iron-deplete with 200 lM 2,2 0-dipyridyl. For
quantitative measurements, 100 ll of culture was trans-
ferred to a microtitre plate and fluorescence was evalu-ated with a luminescence spectrometer LB 50 at
490 nm excitation and 520 nm emission. Fluorescence
was calculated according to Tang et al. [23].
2.5. Real time (quantitative) PCR
RNA was treated with DNAse prior to the RT-reac-
tion. For the RT-reaction, 2 lg of RNA was incubated
with a mix of random primers for 10 min at 95 �C before
addition of reverse transcriptase and incubation for 1 h
at 37 �C. The reaction was completed by incubation at
72 �C for 10 min.A 2 ll aliquot from the RT reaction was used for
PCR. Each reaction contained 12.5 ll of a SYBR Green
PCR Master mix in a final volume of 25 ll. Primers, as
described in Table 1, were added at a concentration of
0.4 lmol. PCR reactions were heated to 95 �C for
10 min and then for 50 cycles with steps of 95 �C for
20 s, 56 �C for 30 s and 72 �C for 30 s. The evolution
of the fluorescent intensity of each dye was recordedcontinuously by the Rotor Gene 3000 multiplex system
(Corbett Research). Data were normalised using the del-
ta–delta CT method with respect to 16S rRNA as the

238 C. Viguier et al. / FEMS Microbiology Letters 246 (2005) 235–242
housekeeping gene. Samples for which the RT reaction
was omitted were used as negative controls. The gener-
ation of specific PCR products was confirmed by melt-
ing curve analysis and gel electrophoresis.
2.6. Siderophore detection by bioassay
Production of siderophore under iron-replete condi-
tions was determined by concentration of the culture
supernatant and detection of the presence of the sidero-
phore by bioassay, as previously described [11].
2.7. Plant nodulation and nitrogen fixation assays
Symbiotic properties of the wild type and mutant
strains were assayed as previously described [11].
Fig. 1. Siderophore plate bioassay (a) iron-replete conditions, S.
meliloti 2011 siderophore preparation. (b) Iron-deplete conditions, S.
meliloti 2011 siderophore preparation. (c) Iron-replete conditions,
S. meliloti 2011rirA2 siderophore preparation. Haloes of growth are
arrowed.
3. Results
3.1. Identification and mutagenesis of rirASm
In view of the observation that Fur is not the iron re-
sponse regulator in S. meliloti [10], it was of interest to
identify the regulator that mediates the response to iron
stress that has clearly been observed for a number of
genes [11]. The protein encoded by smc00785 on the
chromosome of S. meliloti (as annotated on the genome
referenced above) shows 84% identity to the sequence of
RirA, the novel iron response regulator identified in R.
leguminosarum. This gene was mutated, constructing
the mutant S. meliloti 2011rirA2 as described in Section
2 and the mutant was phenotypically characterised with
respect to the production of rhizobactin 1021. Superna-
tants were prepared by growing the wild type and mu-
tant strains in TY medium in the presence and absence
of iron.
The supernatants were then analysed for the presenceof the siderophore using the plate bioassay with the non-
producing mutant S. meliloti 2011rhbA62 as an indica-
tor. In contrast to the wild type, the mutant produced
siderophore when grown in the presence of iron indicat-
ing that the smc00785 mutation resulted in the abolition
of the iron regulated repression of rhizobactin 1021 bio-
synthesis (Fig. 1).
Fig. 2. The regulon encoding rhizobactin 1021 biosynthesis, transport an
promoter of the rhtXrhbABCDEF operon that was cloned in pOTCV1.
To determine whether the mutation was affecting
the expression of genes involved in symbiosis, for
example genes that may function in iron acquisition
within the plant root nodule, alfalfa plants were in-
fected with the mutant and wild type strains. Theplants nodulated and fixed nitrogen, as measured by
the acetylene reduction technique, with no difference
being observed between the performance of the wild
type and the mutant.
3.2. RirA negatively regulates expression from the
promoter upstream from the rhtXrhbABCDEF operon
To determine if RirA was acting on the promoter of
the rhizobactin 1021 biosynthesis gene cluster (Fig. 2), a
sequence extending 138 base pairs upstream from the
ribosome-binding site of rhtX, and known to contain
the promoter for the operon, was cloned in the promoter
probe vector pOT1, to form pOTCV1, allowing expres-
sion from the promoter to be monitored by the level of
GFP activity. The plasmid was introduced into the S.
d activation. The striped region indicates the region containing the

C. Viguier et al. / FEMS Microbiology Letters 246 (2005) 235–242 239
meliloti 2011rirA2 mutant and the S. meliloti 2011 wild
type and cultures were analysed for levels of GFP activ-
ity when grown under iron-replete and iron-deplete con-
ditions. Microscopic analysis indicated that the
promoter was repressed under iron-replete conditions
in the wild type but not in the mutant. Levels of fluores-cence normalised with the strain containing the empty
vector were calculated to have a 9-fold increase in
S. meliloti 2011rirA2 pOTCV1 (Fig. 3) compared to
S. meliloti 2011 pOTCV1.
3.3. RirA regulates other iron responsive genes, in
addition to those involved in rhizobactin 1021 biosynthesis
and can activate, as well as repress, gene expression
Using ribonuclease protection assays we had previ-
ously analysed the abundance of RNA transcripts in
response to iron for genes involved in rhizobactin
1021 production and utilisation [11]. Here we report
the use of real-time RT PCR as the technique of
choice to detect transcripts, using the 16S ribosomal
RNA gene as a housekeeping gene, to assess the rela-tive abundance of the transcripts of interest. Initially
we confirmed that the biosynthesis gene rhbA and
the gene encoding the rhizobactin 1021 outer mem-
brane receptor, rhtA, are iron-regulated, demonstrating
the correlation of results obtained by the real-time RT
PCR and ribonuclease protection assays. Subse-
quently, we assessed the level of expression of these
Fig. 3. Cultures of S. meliloti 2011 [pOTCV1] (A and B) and S. meliloti 2011r
the bacteria (A and C) and UV light to detect green fluorescent protein (B a
genes in the S. meliloti 2011rirA2 mutant by compar-
ison with the wild type. In agreement with the results
obtained using the promoter probe plasmid, we de-
tected the transcripts under iron-replete conditions in
the mutant but not in the wild type (Fig. 4).
We investigated the expression of the gene encoding aputative iron transporter, smc02726. It was determined
that the gene is iron responsive and that it is expressed
in the rirA2 mutant but not in the wild type under iron
replete conditions, implying that RirA is a repressor of
the gene (Fig. 4). Furthermore, the iron-responsive
expression of the smc02726 gene was observed in
S. meliloti Rm818, a strain that is cured of the pSymA
megaplasmid and that therefore lacks rhrA, the geneencoding the AraC-like activator of rhizobactin 1021
biosynthesis and transport genes. This result is signifi-
cant in that it decouples the iron-responsive activity of
RirA from any effect of RhrA.
Interestingly, we observed that RirA appears to act as
an activator in the case of dppA1, the homologue of a
gene encoding an ABC transporter of d-aminolevulinic
acid, a precursor of heme, which was previously charac-terised in R. leguminosarum [24], and which is located
adjacent to rirA on the S. meliloti chromosome. Under
iron-replete conditions, dppA1 is expressed in the wild
type but the level of expression is significantly reduced
in the rirA2 mutant (Fig. 4).
Finally, we determined that rirA is itself downregu-
lated under iron deplete conditions.
irA2 [pOTCV1] (C and D) under bright light to confirm the presence of
nd D). Magnification 1000·.

Fig. 4. In vivo analysis of iron and RirA responsive genes by Real-Time PCR. (a) Iron regulation of rhbA, rhtA, smc02726 and dppA1 in the wild
type. (b) Iron regulation of rirA in the wild type. (c) Comparisons of rhbA, rhtA and Smc02726 expression in the wild type and the rirA2 mutant
under iron-replete conditions. (d) Comparison of dppA1 expression in the wild type and the rirA2 mutant under iron-replete conditions.
240 C. Viguier et al. / FEMS Microbiology Letters 246 (2005) 235–242
4. Discussion
Rhizobactin 1021 is the only siderophore producedby S. meliloti 2011, although an analysis of the genome
sequence (strain 1021 for which the genome sequence
was determined is a derivative of strain 2011) suggests
that the organism possesses a number of additional
mechanisms by which it can obtain iron. The genes
smc02889 and smc02726, for example, are two candi-
dates that would be predicted to function in iron trans-
port. The Fur homologue in S. meliloti has beeninvestigated regarding its role in iron-responsive gene
expression and it was concluded that it is the regulator
of the sitABCD operon, that functions primarily in man-
ganese acquisition, but it is not the general regulator of
the iron response [9,10]. Here we report that RirA is the
iron response regulator of rhizobactin 1021 biosynthesis
and transport genes, as well as other iron responsive
genes. RirA is the regulator of iron responsive genes inR. leguminosarum, including the genes encoding vicibac-
tin production and transport [13]. In contrast, in Brady-
rhizobium japonicum it has been discovered that an
additional protein, Irr, functions along with Fur in the
iron response [25]. There is no obvious reason why some
rhizobia have recruited RirA as an alternative to Fur as
the general iron response regulator.
Rhizobactin 1021 biosynthesis and transport are neg-atively regulated by iron and positively regulated by the
AraC-like activator RhrA. Under iron-replete condi-
tions but in the absence of RirA, expression of both
the rhtXrhbABCDEF operon and rhtA are de-repressed.
It is not clear however whether RirA mediates this effect
by directly affecting the individual promoters, or indi-
rectly by modulating the activity of RhrA. It is likelythat RhrA acts as a �local� regulator of the rhizobactin
1021 regulon and no other iron acquisition systems have
been detected that are affected either positively or nega-
tively by RhrA (O Cuıv and O� Connell, unpublishedobservation). In this study the expression of smc02726,
which encodes a protein (designated ShmR) that is a
hemin-binding iron regulated outer membrane protein
from S. meliloti 242 [26], was shown to be RirA-regulated. The fact that the iron-responsive regulation
of smc02726 is maintained in a pSymA cured strain indi-
cates that RirA can function independently of RhrA and
implies that RirA likely acts as a global regulator of iron
responsive genes.
In addition to our observation that RirA acts as a
negative regulator of gene expression in response to
the presence of iron, we also observed that it could actas a positive regulator in the case of the dppA1 gene. Re-
cently, Delany et al. [27] reported that Fur could act as
an activator of putative virulence genes in Neisseria
meningitidis. In E. coli, Fur is known to indirectly regu-
late gene expression in a positive manner through its ef-
fect on the expression of small RNAs [28]. RirA may be
acting through DNA binding or it may be acting indi-
rectly to positively regulate activity. Under iron-repleteconditions, the cell down-regulates the expression of
high affinity iron acquisition systems, and depends on
lower affinity iron acquisition systems to satisfy its iron
demands. By extension, under iron-deplete conditions,
the cell down-regulates the synthesis of many iron-
containing proteins that place an increased iron burden

C. Viguier et al. / FEMS Microbiology Letters 246 (2005) 235–242 241
on the cell. DppA1 was characterised as a member of a
heme precursor transporter system in R. leguminosarum
[24] and may function in a similar manner in S. meliloti.
The properties of heme enable it to function as a pros-
thetic group for many haemoproteins, however under
iron-deplete conditions it is likely that many of theseproteins are down-regulated as the cell switches to a
�low iron mode�. The dppA1 gene is located adjacent to
rirA and is transcribed in the same direction. As both
genes are positively regulated in the presence of iron it
is possible that they are regulated in a coordinated
manner.
Plants inoculated with the rirA2 mutant formed effec-
tive nitrogen fixing nodules indicating that the activa-tion properties of the regulator are not involved in
regulating the availability of iron during the nitrogen
fixing stage of symbiosis. Indeed, the mechanism by
which the bacterium acquires iron in symbiosis remains
to be elucidated, as does the regulator or signal that may
be controlling such a mechanism.
Acknowledgement
This work was supported by Science Foundation Ire-
land and Enterprise Ireland.
References
[1] Crosa, J.H. (1997) Signal transduction and transcriptional and
posttranscriptional control of iron-regulated genes in bacteria.
Microbiol. Mol. Biol. Rev. 61, 319–336.
[2] Escolar, L., Perez-Martin, J. and De Lorenzo, V. (1999) Opening
the iron box, transcriptional metalloregulation by the Fur protein.
J. Bacteriol. 181, 6223–6229.
[3] Beaumont, F.C., Kang, H.Y., Brickman, T.J. and Armstrong,
S.K. (1998) Identification and characterization of alcR, a gene
encoding an AraC-like regulator of alcaligin siderophore biosyn-
thesis and transport in Bordetella pertussis and Bordetella
bronchiseptica. J. Bacteriol. 180, 862–870.
[4] Heinrichs, D.E. and Poole, K. (1996) PchR, a regulator of
ferripyochelin receptor gene (fptA) expression in Pseudomonas
aeruginosa, functions both as an activator and as a repressor. J.
Bacteriol. 178, 2586–2592.
[5] De Lorenzo, V., Wee, S., Herrero, M. and Neilands, J.B. (1987)
Operator sequences of the aerobactin operon of plasmid ColV-
K30 binding the ferric uptake regulation (fur) repressor. J.
Bacteriol. 169, 2624–2630.
[6] Prince, R.W., Cox, C.D. and Vasil, M.L. (1993) Coordinate
regulation of siderophore and exotoxin A production, molecular
cloning and sequencing of the Pseudomonas aeruginosa fur gene. J.
Bacteriol. 175, 2589–2598.
[7] Baichoo, N., Wang, T., Ye, R. and Helmann, J.D. (2002) Global
analysis of the Bacillus subtilis Fur regulon and the iron starvation
stimulon. Mol. Microbiol. 45, 1613–1629.
[8] Diaz-Mireles, E., Wexler, M., Sawers, G., Bellini, D., Todd, J.D.
and Johnston, A.W.B. (2004) The Fur-like protein Mur of
Rhizobium leguminosarum is a Mn(II)-responsive transcriptional
regulator. Microbiology 150, 1447–1456.
[9] Platero, R., Peixoto, L., O�Brian, M.R. and Fabiano, E. (2004)
Fur is involved in manganese-dependent regulation of mntA (sitA)
expression in Sinorhizobium meliloti. Appl. Environ. Microbiol.
70, 4349–4355.
[10] Chao, T.C., Becker, A., Buhrmester, J., Puhler, A. and Weidner,
S. (2004) The Sinorhizobium meliloti fur gene regulates, with
dependence on Mn(II), transcription of the sitABCD operon,
encoding a metal-type transporter. J. Bacteriol. 186, 3609–3620.
[11] Lynch, D., O�Brien, J., Welch, T., Clarke, P., O Cuıv, P., Crosa,
J.H. and O�Connell, M. (2001) Genetic organisation of the region
encoding regulation, biosynthesis and transport of rhizobactin
1021, a siderophore produced by Sinorhizobium meliloti. J.
Bacteriol. 183, 2576–2585.
[12] O Cuıv, P., Clarke, P., Lynch, D. and O�Connell, M. (2004)
Identification of rhtX and fptX, novel genes encoding proteins
that show homology and function in the utilization of the
siderophores rhizobactin 1021 by Sinorhizobium meliloti and
pyochelin by Pseudomonas aeruginosa. J. Bacteriol. 186, 2996–
3005.
[13] Todd, J.D., Wexler, M., Sawers, G., Yeoman, K.H., Poole, P.S.
and Johnston, A.W.B. (2002) RirA, an iron-responsive regulator
in the symbiotic bacterium Rhizobium leguminosarum. Microbiol-
ogy 148, 4059–4071.
[14] Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular
Cloning: a Laboratory Manual, 2nd ed. Cold Spring Harbor
Laboratory Press, Cold Spring Harbor, NY.
[15] Meade, H.M., Long, S.R., Ruvkun, G.B., Brown, S.E. and
Ausubel, F.M. (1982) Physical and genetic characterisation of
symbiotic and auxotrophic mutants of Rhizobium meliloti induced
by transposon Tn5 mutagenesis. J. Bacteriol. 149, 114–122.
[16] Quandt, J. and Hynes, M.F. (1993) Versatile suicide vectors which
allow direct selection for gene replacement in Gram-negative
bacteria. Gene 127, 15–21.
[17] Finan, T.M., Kunkel, B., De Vos, G.F. and Signer, E.R. (1986)
Second symbiotic megaplasmid in Rhizobium meliloti carrying
exopolysaccharide and thiamine synthesis genes. J. Bacteriol. 167,
66–72.
[18] Allaway, D., Schofield, N.A., Leonard, M.E., Gilardoni, L.,
Finan, T.M. and Poole, P.S. (2001) Use of differential fluorescence
induction and optical trapping to isolate environmentally induced
genes. Environ. Microbiol. 3, 397–406.
[19] Beringer, J.E. (1974) R factor transfer in Rhizobium leguminosa-
rum. J. Gen. Microbiol. 84, 188–198.
[20] Birnboim, H. and Doly, J. (1979) A rapid alkaline extraction
procedure for screening recombinant plasmid DNA. Nucl. Acids
Res. 7, 1513–1523.
[21] Inoue, H., Nojima, H. and Okayama, H. (1990) High efficiency
transformation of Escherichia coli with plasmids. Gene 96, 23–28.
[22] O�Connell, M., Hynes, M.F. and Puehler, A. (1987) Incompat-
ibility between a Rhizobium Sym plasmid and a Ri plasmid of
Agrobacterium. Plasmid 18, 156–163.
[23] Tang, X., Lu, B.F. and Pan, S.Q. (1999) A bifunctional
transposon mini-Tn5gfp-km which can be used to select for
promoter fusions and report gene expression levels in Agrobac-
terium tumefaciens. FEMS Microbiol. Lett. 179, 37–42.
[24] Carter, R.A., Yeoman, K.H., Klein, A., Hosie, A.H., Sawers, G.,
Poole, P.S. and Johnston, A.W.B. (2002) dpp genes of Rhizobium
leguminosarum specify uptake of d-aminolevulinic acid. Mol.
Plant Microbe Interact. 15, 69–74.
[25] Hamza, I., Qi, Z., King, N.D. and O�Brian, M.R. (2000) Fur-
independent regulation of iron metabolism by Irr in Bradyrhiz-
obium japonicum. Microbiology 146, 669–676.
[26] Battistoni, F., Platero, R., Duran, R., Cervenansky, C., Batti-
stoni, J., Arias, A. and Fabiano, E. (2002) Identification of an
iron-regulated, hemin-binding outer membrane protein in Sino-
rhizobium meliloti. Appl. Environ. Microbiol. 68, 5877–5881.

242 C. Viguier et al. / FEMS Microbiology Letters 246 (2005) 235–242
[27] Delany, I., Rappuoli, R. and Scarlato, V. (2004) Fur functions as
an activator and as a repressor of putative virulence genes in
Neisseria meningitidis. Mol. Microbiol. 52, 1081–1090.
[28] Masse, E. and Gottesman, S. (2002) A small RNA regulates
expression of genes involved in iron metabolism in Escherichia
coli. Proc. Natl. Acad. Sci. USA 99, 4620–4625.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 243–249
Polymerase chain reaction for identification of aldoximedehydratase in aldoxime- or nitrile-degrading microorganisms
Yasuo Kato, Satoshi Yoshida, Yasuhisa Asano *
Biotechnology Research Center, Faculty of Engineering, Toyama Prefectural University, 5180 Kurokawa, Kosugi, Toyama 939-0398, Japan
Received 18 February 2005; received in revised form 7 April 2005; accepted 12 April 2005
First published online 29 April 2005
Edited by H-P.E. Kohler
Abstract
We developed a molecular screening procedure using Southern hybridization and polymerase chain reaction (PCR) to identify
aldoxime dehydratase (Oxd) encoding genes (oxds) among 14 aldoxime- or nitrile-degrading microorganisms. When an oxd gene of
Rhodococcus erythropolis N-771 was used as a probe, positive hybridization signals were seen with the chromosomal DNA of eight
strains, suggesting that these strains have similar oxd genes to R. erythoropolis N-771. By analyzing the PCR-amplified fragments
with degenerate consensus primers, the occurrence of homologous Oxd coexisting with Fe-containing NHase in the active eight
strains was demonstrated coinciding with the results of Southern hybridization. Whole length of oxd gene was cloned as an example
from one of the positive strains, Pseudomonas sp. K-9, sequenced, and expressed in E. coli. Analysis of the primary structure of the
protein (OxdK) encoded by the oxd gene of Pseudomonas sp. K-9 led to identify an Oxd having a new primary structure. Thus, the
PCR-based analysis of oxd gene is a useful tool to detect and analyze the ‘‘aldoxime-nitrile pathway’’ in nature, since Oxd is the key
enzyme for the pathway.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Aldoxime dehydratase; Nitrile hydratase; PCR; Aldoxime-nitrile pathway; Screening
1. Introduction
Nitrile compounds, which are extensively used in the
chemical industry, are degraded by microorganisms to
carboxylic acids by nitrilase (Nit; EC 3.5.5.1) or by com-
bination of nitrile hydratase (NHase; EC 4.2.1.84) and
amidase (Ami; EC 3.5.1.4) [1–3]. Starting from the pio-
neering research for the first isolation of NHase from R.
rhodochrous J-1 [4,5], the enzyme has been extensively
studied and used for manufacturing acrylamide, nicotin-amide, and 5-cyanovaleroamide [1,6]. NHases are classi-
fied into two groups based on the prosthetic metal
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.011
* Corresponding author. Tel.: +81 766 56 7500; fax: +81 766 56 2498.
E-mail address: [email protected] (Y. Asano).
group, non-heme Fe [NHase(Fe)] or non-corrinoid Co
[NHase(Co)] [1–3]. Despite its important uses, however,the physiological function of NHase in nature remains
unclear. We have isolated several microbial aldoxime
degraders [1], which can convert aldoximes, such as pyr-
idine-3-aldoxime and phenylacetaldoxime as a model for
aryl- and arylalkyl-aldoximes, respectively, to the corre-
sponding carboxylic acids, from soil samples. The
metabolism occurs via intermediate nitrile and involves
a combination of enzymes including a novel heme-con-taining enzyme, aldoxime dehydratase (Oxd; EC
4.99.1.-), and nitrile-hydrolyzing enzymes, such as
NHase and Ami, and/or Nit (1, 5, 7): the pathway could
be named as ‘‘aldoxime-nitrile pathway’’ (Fig. 1). From
some aldoxime- or nitrile-degraders, Oxds were purified
. Published by Elsevier B.V. All rights reserved.

R N
H
OH
R C N R CO
OHNitrile Carboxylic acid
Nitrilase
Nitrilehydratase R C
ONH2
Aldoximedehydratase
AldoximeAmidase
Amide
H2O
Fig. 1. The aldoxime-nitrile pathway in microorganisms.
244 Y. Kato et al. / FEMS Microbiology Letters 246 (2005) 243–249
and characterized and the genes (oxd) were cloned, se-
quenced and overexpressed in E. coli [8–11]. The oxd
genes were linked with genes for Nit and NHase/Ami
in the genome of the strains [8–11], confirming the genetic
relationship of the pathway. Oxds were used for the
enzymatic synthesis of nitriles from the corresponding
aldoximes [1,12]. Since Oxd is located upstream of the
‘‘aldoxime-nitrile pathway’’ (Fig. 1), the enzyme can be-
come the key enzyme for the pathway. It is of our inter-
est to accumulate the genetic information of thepathway from different sources in order to know diver-
sities and evolution of the pathway in nature. The aim
of this study is to develop a molecular screening proto-
col based on Southern hybridization and PCR to rapidly
identify genes coding Oxds in several aldoxime- and ni-
trile-degrading microorganisms, and to study the use of
the method as a tool to detect and analyze the pathway.
2. Materials and methods
2.1. Materials
Restriction enzymes and DNA-modifying enzymes
were purchased from Takara (Tokyo, Japan), Toyobo
(Osaka, Japan), New England Biolabs. (Beverly, MA,USA), Roche (Mannheim, Germany), and MBI Fer-
mentas (Vilnius, Lithuania) and used according to the
manufacturers� protocols. All other chemicals were from
commercial sources and used without further
purification.
2.2. Bacterial strains, plasmids and culture conditions
Strains used for screening oxd gene were from culture
collections (TPU) of our own laboratory [7] and were
grown at 30 �C in TGY medium consisted of 0.5% yeast
extract (Nippon Seiyaku, Tokyo, Japan), 0.5% Bacto
Tryptone (Difco, Detroit, WI, USA), 0.1% of K2H-
PO4, and 0.1% D-glucose, pH 7.0. The E. coli strains,
JM109 {recA1 endA1 gyrA96 thi hsdR17 supE44 relA1
D(lac-proAB)/F 0[traD36 proAB+ lacIq lacZ M15]} andBL21 Stare (DE3) {F� ompT hsdSB ðr�B ; m�
B Þ gal dcm
rne131 (DE3)}, were used as hosts. Plasmids pT7-Blue
(Novagen, Madison, WI, USA) and pRSETB (Invitro-
gen, Carlsbad, CA, USA) were used as cloning and
expression vectors, respectively. Recombinant E. coli
cells were cultured in a Luria–Bertani (LB) medium
(1% Bacto Tryptone, 0.5% Bacto yeast extract (Difco),
and 1% NaCl, pH 7.5) containing 100 lg ml�1 of ampi-
cillin. Pseudomonas sp. K-9 [13] was used as the source
of the DNA to clone whole oxd gene. The partial
sequencing of the 16S rDNA fragment (1.6 kbp) of the
strain showed 100% identity with that of Pseudomonas
synxantha DSM 13080 (GenBank accession no.AF267911).
2.3. General recombinant DNA techniques
The plasmid DNA was isolated by a PI-100 Auto-
matic Plasmid Isolation System (Kurabo, Osaka, Ja-
pan). The other general procedures were performed as
described by Sambrook et al. [14]. The nucleotide se-quence was determined with an ABI PRISM 310 auto-
mated sequencer (Applied Biosystems, Foster City,
CA, USA) using the dideoxy chain termination method.
A homology search was performed with the programs
FASTA [15] and BLAST [16], and the ClustalW method
[17] was used to align the sequence.
2.4. Southern hybridization
Chromosomal DNAs of the strains were extracted
by the method as described by Saito and Miura [18]
and digested with EcoRI followed by fractionation
with a 0.7% agarose gel. The digested DNAs were
blotted onto a nylon membrane, GeneScreen Pluse
(Dupont, Boston, MA, USA), and the membrane
was hybridized with the oxd gene of R. erythropolis
N-771 [11] and Bacillus sp. OxB-1 [9], labelled with
the digoxigenin (DIG) system (Roche), at 37–42 �Caccording to the procedure recommended by the man-
ufacturer. Optimum conditions for stringency washing
of the blotted DNA were sought by increasing the
washing temperature (60–68 �C) or by raising the con-
centrations (0.1–2·) of SSC [14] in a washing solution.
The membrane was visualized with alkaline phospha-tase-conjugated anti-DIG and nitro blue tetrazolium
(NBT)/5-bromo-4-chloro-3-indolyl phosphate (BCIP)
reagents (Roche).

Y. Kato et al. / FEMS Microbiology Letters 246 (2005) 243–249 245
2.5. PCR amplification method
One colony of the strains grown on a TGY agar plate
was picked-up with a sterile pipette tip and transferred it
directly to a PCR reaction mixture (50 ll) comprising
50 pmol each of primers, 500 lM dNTPs, 5 ll of 10·PCR buffer and 1–2.5 units of various DNA polymer-
ases, such as Taq (Takara), ExTaq (Takara), Blend
Taq (Toyobo), Pwo (Roche), KOD-Plus (Toyobo),
and Vent (New England Biolabs.) polymerases. The
PCR reaction was performed with a PCT 200 thermocy-
cler (MJ Research, Watertown, MA, USA) with the fol-
lowing program: 35 cycles of denaturation at 96 �C for
0.5 min, primer annealing at 50–55 �C for 0.5 min andextension at 72 �C for 1 min. The fragment was purified
with Qiaquick gel extraction kit (Qiagen, Valencia, CA,
USA) and ligated with pT7Bule vector for sequencing.
To amplify genes coding consensus regions for a-sub-unit (nha1) of NHase(Co) and NHase(Fe), the primer
pairs [19] NHCo1 [5 0-GTCGTGGCGAAGGCCTGG-
3 0]/NHCo2 [5 0-GTCGCCGATCATCGAGTC-3 0] and
NHFe1 [5 0- CCCGACGGTTACGTCGAG-3 0]/NHFe2[5 0-CCATGTAGCGAGTTTCGGCG-3 0], were used,
respectively.
2.6. Cloning of whole oxd gene from Pseudomonas sp.K-9
by an inverse PCR and expression of the gene in E. coli
Five micrograms of genomic DNA of Pseudomonas
sp. K-9 was digested with BamHI and the digestedDNA was electrophoresed through a 0.7% agarose
gel. Appropriate fragments (2.5–3.0 kb), which ex-
pected to contain oxd gene based on the results of
Southern hybridization with the PCR-amplified oxd
gene as a probe, were extracted from gel and purified.
The fragment was self-circularized as described by
Sambrook et al. [14]. The PCR was performed in
reactions (25 ll) containing 0.001–0.1 lg of circular-ized DNA, 50 pmol each of the primer InvR2
[5 0-GCGGTCGCGCATCGAGCCCCAATAACC-3 0]
and InvF2 [5 0-TCGGTGGAGAAACTCGAACGC-
TGGACCGAA-3 0], 500 lM dNTPs, 1· PCR buffer,
and 2.5 units of Vent polymerase. The PCR reaction
was 35 cycles of denaturation at 95 �C for 0.5 min,
primer annealing at 55 �C for 0.5 min, and exten-
sion at 72 �C for 6 min. The amplified band, ex-tracted from an agarose gel, was ligated with
pT7Blue vector and used for further sequencing.
A 1.0-kb NdeI-HindIII fragment containing the
whole oxd gene was PCR-amplified by Vent polymer-
ase using the primers K9OxdKNde (5 0-GCTCACA-
TATGAATCTGCAATC-3 0; the restriction site is
underlined) and K9OxdKHindR (5 0-AGGGAAGCT-
TTCAGGCGGGGCATACT-3 0) and the genomicDNA of Pseudomonas sp. K-9 as a template, then
subjected to enzyme digestion with NdeI and HindIII.
The fragment was cloned into the same site of
pRSETB to give pOxdKInt and the plasmid was used
to transform E. coli BL21 Stare (DE3). A 1% aliquot
of the overnight culture of E. coli BL21 Stare (DE3)/
pOxdKInt was added into 8 ml of LB medium con-
taining 100 lg ml�1 of ampicillin in a test tube(18 · 170 mm) and incubated with shaking (200 rpm)
at 37 �C for 3–4 h. When the optical density at
610 nm of the medium reached 1.0, isopropyl b-D-thio-galactopyranoside (IPTG) was added as an inducer to
a final concentration of 1 mM, and the culture was
further incubated at 20 �C for 60 h. The cells were
harvested by centrifugation (3500g, 10 min) at appro-
priate intervals, suspended in 0.1 M potassium phos-phate buffer (KPB, pH 7.0), and disrupted by
ultrasonication as described [8]. The Oxd activity in
a cell-free extract obtained by centrifugation
(15,000g, 10 min) was measured according to our pre-
vious report [1,7,9,11]. The electronic absorbance spec-
tra of the cell-free extract were recorded on a JASCO
V-530 spectrophotometer (JASCO, Tokyo, Japan).
3. Results
3.1. Screening for microorganisms carrying oxd genes by
Southern hybridization
The existence of oxd genes among 14 aldoxime- or ni-
trile-degraders shown in Table 1 was examined bySouthern hybridization with the oxd gene from R. ery-
thropolis N-771 and Bacillus sp. OxB-1 as probes. It
had been suggested that the strains had ‘‘aldoxime-
nitrile pathway’’ by activity measurement [7]: i.e., the
activities of Oxd and nitrile-degrading enzymes, NHase
and Ami and/or Nit, were detected in the strains. By
using R. erythropolis N-771oxd gene as a probe, the po-
sitive hybridization signals were found with the chromo-somal DNA of six strains, i.e., R. erythropolis JCM
3201, Rhodococcus sp. NCIBM 11215, B. butanicum
ATCC 21196, R. erythropolis BG 13, R. erythropolis
BG 16, and Pseudomonas sp. K-9, in addition to control
strains R. globerulus A-4 [10] and Rhodococcus sp. N-771
[11], even after stringency washing of the hybridized
membrane with 0.5· SSC containing 0.1% SDS at
68 �C for 30 min. The result suggests that these strainshave similar oxd gene to that of R. erythropolis N-771.
The positive signals were seen mainly with the genome
of the strains having NHase. Although various condi-
tions for hybridization and stringency washing were
examined, the other NHase-containing strains, R. rho-
dochrous NCIMB 11216, R. rhodochrous J-1 and Rhodo-
coccus sp. YH3-3 did not show any hybridization signals
with the Rhodococcus oxd gene despite the detection ofOxd activity in the strains [7]. The oxd gene of Bacillus
sp. OxB-1 did not hybridize with the genome of any

Table 1
Summary of activity measurement of aldoxime dehydratase, nitrile hydratase, and nitrilase, and Southern hybridization and PCR-amplification
analysis of their genes, in several aldoxime- or nitrile-degrading microorganisms
TPU no. Strain Oxd NHase(Fe) NHase(Co) Nit
Aa Hb Pc Aa Pd Aa Pe Aa Pf
3207 Rhodococcus erythropolis JCM 3201 + + + + + – – – NE
3311 Rhodococcus rhodochrous J-1 + – – – – + + + NE
3451 Rhodococcus sp. NCIBM 11215 + + + – + – – + NE
3452 Rhodococcus sp. NCIMB 11216 + + + – – – – + NE
3453 Rhodococcus sp. YH3-3 + – – – – + – – NE
3466 Rhodococcus sp. AK32 + – – – – – – + NE
5710 Brevibacterium butanicum ATCC 21196 + + + + + – – – NE
6007 Rhodococcus erythropolis BG 13 + + + * + * – – NE
6008 Rhodococcus erythropolis BG 16 + + + * + * – – NE
6015 Corynebacterium sp. C5 + – – – – – – + NE
7177 Pseudomonas sp. K-9 + + + * + * – – NE
3383 Rhodococcus globerulus A-4g + + + + + – – – NE
3467 Rhodococcus erythropolis N-771g + + + + + – – – NE
5563 Bacillus sp. OxB-1g + – + – – – – + NE
a ‘‘A’’ denotes activity measurement. Aldoxime dehydratase (Oxd), nitrile hydratase (NHase), and nitrilase (Nit) activities were measured in each
strain by our previous study [7]. Prosthetic metal group of NHase was suggested by an enzyme purification or a gene analysis. An asterisk (*)
indicates that the strain showed NHase activity but its metal group was not identified.b ‘‘H’’ denotes Southern hybridization. Positive (+) and no (�) hybridization signals were seen by Southern hybridization with the genomic DNA
of each strain by using oxd from R. erythropolis N-771 as a probe.c ‘‘P’’ denotes PCR amplification. A plus (+) indicates that the estimated length of PCR product was amplified with primers OxB4-S3/OxB4-AS2.d ‘‘P’’ denotes PCR amplification. A plus (+) indicates that the estimated length of PCR product was amplified with primers NHFe1/NHFe2.e ‘‘P’’ denotes PCR amplification. A plus (+) indicates that the estimated length of PCR product was amplified with primers NHCo1/NHCo2.f NE – not examined.g The strains were used as control: Oxd, NHase and Nit of the strains had been isolated, characterized, and their genes were cloned and sequenced
in our previous research [8–11].
246 Y. Kato et al. / FEMS Microbiology Letters 246 (2005) 243–249
strains including ones having Nit used in this study un-
der the examined conditions, suggesting that Bacillus
oxd gene had low similarities with oxds of the strains
tested.
3.2. PCR-based analysis of oxd genes
In order to know the genetic information of oxd iden-tified in the strains, we amplified oxd gene from the
strains by using PCR with degenerated consensus prim-
ers under various PCR conditions. The primers OxB4-S3
(5 0-CAYGRHTAYTGGGGHKCRATGCGCGA-3 0)
and OxB4-AS2 (5 0-ACCGADACYTCRTGSYA) used
were designed based on the conserved sequences among
the known Oxds, H(G/E)YMG(S/A)MRE/D and
HEVSV(F/S/L), respectively. A strong band of the ex-pected size (450 bp) was seen on an agarose gel when
Blend-Taq polymerase (Toyobo, Osaka) was used at
an annealing temperature of 55 �C. The PCR product
was amplified from the eight strains including the con-
trol strains (Table 1), all of which showed positive
Southern hybridization signals with R. erythropolis
N-771oxd gene. In addition, PCR product was also ob-
tained from Bacillus sp. OxB-1 that has Oxd and Nit[1,7]. As shown in Fig. 2, the comparison of amino acid
sequences deduced from the amplified genes from the 6
positive strains suggest the existence of similar (80.6%
identities) Oxds in the strains to those linked with NHa-
se(Fe) found in the control strains, R. globerulus A-4 [10]
and R. erythropolis N-771 [11]. The primers are shown
to be suitable for amplifying the oxd gene from the
microorganisms having NHase(Fe). We have previously
clarified that these strains had Oxd activities [7] but this
study gave us genetic information of the enzymes for
the first time. Under the conditions tested, no PCRproduct was obtained from the other strains except
Bacillus sp. OxB-1 that has Oxd and Nit and/or NHase
[1,7].
3.3. PCR amplification of genes coding for NHase
In order to confirm genetically the co-existence of
NHase in the positive strains, genes coding for NHase-(Co) and NHase(Fe) were amplified by PCR with the
primer pairs NHCo1/NHCo2 and NHFe1/NHFe2,
respectively, which were designed based on the con-
served sequences of a-subunit (nha1) of NHase(Co)
(PDGYVE/AETRYM) and NHase(Fe) (VVAKAW/
DSMIGD), respectively, as reported by Duran et al.
[19]. As shown in Table 1 and Fig. 3, NHFe1/NHFe2
primers allowed the amplification of a 400-bp DNAfragment from the positive six strains and proteins en-
coded by the fragments showed similarities (63.7% iden-
tities) to the known NHases (Fe) from P. chlororaphis

1 10 20 30 40 50 60 70 80
BG13,BG16 HGYWGAMRERFPISQTDWMQASGELRVVAGDPAVGGRVVVRGHDNIALIRSGQDWADAEADERSLYLDEILPTLQSGMDF
OxdRG, Bb21196 HGYWGSMRERFPISQTDWMQASGELRVVAGDPAVGGRVVVRGHDNIALIRSGQDWADAEADERSLYLDEILPTLQSGMDF
OxdRE, Re3201 HGYWGSMRERFPISQTDWMQASGELRVIAGDPAVGGRVVVRGHDNIALIRSGQDWADAEADERSLYLDEILPTLQSGMDF
Rs11215 HGYWGSMRERFPISRTDWTHASGELRVVAGDPAAGGRVVVRGHDNIALIRSGQDWADAEADERSLYLDEILPTLQSGMGF
K-9 HGYWGSMRDRFPISQTDWMKPTSELQVIAGDPAKGGRVVVLGHGNLTLIRSGQDWADAEAEERSLYLDEILPTLQDGMDF
***** ** ***** * * ** * ***** ****** ** * ************* ************** ** *
90 100 110 120 130 140 150
BG13, BG16 LRDNGPAVGCYSNRFVRNIDIDGNFLDLSYNIGHWASLDQLERWSESHPTHLRIFTTFFRVAEGLSKLRLYHEVSV
OxdRG, Bb21196 LRDNGPAVGCYSNRFVRNIDIDGNFLDLSYNIGHWASLDQLERWSESHPTHLRIFTTFFRVAEGLSKLRLYHEVSV
OxdRE, Re3201 LRDNGPAVGCYSNRFVRNIDIDGNFLDLSYNIGHWASLDQLERWSESHPTHLRIFTTFFRVAAGLSKLRLYHEVSV
Rs11215 LRDNGPAVGCYSNRFVRNIDIDGNFLDLSYNIGHWASLDQLERWSESHPTHLRIFTTFFRVAEGLSKLRLYHEVSV
K-9 LRDNGQPLGCYSNRFVRNIDLDGNFLDVSYNIGHWRSVEKLERWTESHPTHLRIFVTFFRVAAGLKKLRLYHEVSV
***** ************ ****** ******* * *************** ****** ** **********
Fig. 2. Amino acid sequence comparison of Oxds deduced from the PCR-amplified fragments from R. erythropolis BG 13 (BG13), R. erythropolis
BG 16 (BG16), R. globerulus A-4 (OxdRG), B. butanicum ATCC 21196 (Bb21196), Rhodococcus sp. N-771 (OxdRE), R. erythropolis JCM 3201
(Re3201), Rhodococcus sp. NCIBM 11215 (Rs11215), and Pseudomonas sp. K-9 (K-9). Identical amino acids are indicated by asterisks. The residues
used for designing primers for inverse PCR are underlined.
Y. Kato et al. / FEMS Microbiology Letters 246 (2005) 243–249 247
B23 (accession no. D90216), Brevibacterium sp. R312
(B37806), and Acinetobacter sp. ADP1 (CR543861)including control strains, R. globerulus A-4 [10] and R.
erythropolis N-771 (S04472). There is no report on a
detection of NHase activity in Rhodococcus sp. NCIMB
11215, which shows Oxd and Nit activities [7], but we
could suggest here the occurrence of NHase(Fe) in the
strain for the first time. No fragment was amplified with
NHCo1/NHCo2 primers except NHase(Co)-containing
R. rhodochrous J-1. We did not amplify Nit gene byPCR in this study since we could not identify consensus
sequences among the reported Nits.
1 10 20
Bb21196, Re3201 PDGYVEGWKKTFEEDFSPRRGAELVAR
Rs11215, BG16, A-4
BG13, N-771, 312 PDGYVEGWKKTFEEDFSPRRGAELVAR
K-9 PQGYVEQLTQLMEHGWSPENGARVVAK
B23 PEGYVEQLTQLMAHDWSPENGARVVAK
ADP PDGYVEGWKKTFEEDFSPRRGAELVAR
* **** ** ** ** *
70 80 90
Bb21196, Re3201 LKNVIVCSLCSCTAWPILGLPPTWYKS
Rs11215, BG16, A-4
BG13, N-771, 312 LKNVIVCSLCSCTAWPILGLPPTWYKS
K-9 LKNVIVCSLCSCTNWPVLGLPPEWYKG
B23 VKNVIVCSLCSCTNWPVLGLPPEWYKG
ADP LKNVIVCSLCSCTAWPILGLPPTWYKS
************* ** ***** ***
Fig. 3. Amino acid sequence comparison of a-subunit of NHases deduced f
Rhodococcus sp. N-771 (N-771), Pseudomonas sp. K-9 (K-9), B. butanicum
NCIBM 11215 (Rs11215), R. erythropolis BG 16 (BG16), and R. erythro
chlororaphis B23 (B23), Brevibacterium sp. R312 (312), and Acinetobacter sp
3.4. Cloning of whole oxd gene from Pseudomonas sp.
K-9 by an inverse PCR and expression of the enzyme in
E. coli
To show the effectiveness to identify oxd gene as a key
enzyme of the ‘‘aldoxime-nitrile pathway’’, we cloned
the whole oxd gene as a typical example from one of
the positive strain, Pseudomonas sp. K-9, which had
been isolated as a glutaronitrile-degrader [13], by an in-
verse PCR approach [14]. The primers, InvR2 andInvF2, used were designed based on the identified oxd
sequence of the strain by PCR, GYWGSMRDR and
30 40 50 60
AWTDPDFRQLLLTDGTAAVAQYGYLGPQGEYIVAVEDTPT
AWTDPEFRQLLLTDGTAAVAQYGYLGPQGEYIVAVEDTPT
AWVDPQFRALLLKDGTAACAQFGYTGPQGEYIVALEDTPQ
AWVDPQFRALLLKDGTAACAQFGYTGPQGEYIVALEDTPG
AWTDPDFRQLLLTDGTAAVAQYGYLGPQGEYIVAVEDTPT
* ** ** *** ***** ** ** ********* ****
100 110 120 130
FEYRARVVREPRKVLSEMGTEIASDVEIRVYDTTAETRYM
FEYRARVVREPRKVLSEMGTEIASDVEIRVYDTTAETRYM
FEFRARLVREGRTVLRELGTELPNDMVVKVWDTSAESRYL
FEFRARLVREGRTVLRELGTELPSDTVIKVWDTSAESRYL
FEYRARVVREPRKVLSEMGTEIASDVEIRVYDTTAETRYM
** *** *** * ** * *** * ** ** **
rom the amplified genes by PCR from R. erythropolis BG 13 (BG13),
ATCC 21196 (Bb21196), R. globerulus A-4 (A-4), Rhodococcus sp.
polis JCM 3201 (Re3201), and with the known NHase(Fe) from P.
. ADP1 (ADP1). Identical amino acids are indicated by asterisks.

1 10 20 30 40 50 60 70 80 90 100 110 120OxdRE MESAIGEHLQCPRTLTRRVPDTYTPPFPMWVGRADDALQQVVMGYLGVQFRDEDQRPAALQAMRDIVAGFDLPDGPAHHDLTHHIDNQGYENLIVVGYWKDVSSQHRWSTSTPIASWWESEDR-LOxdRG MESAIGEHLQCPRTLTRRVPDTYTPPFPMWVGRADDTLHQVVMGYLGVQFRGEDQRPAALRAMRDIVAGFDLPDGPAHHDLTHHIDNQGYENLIVVGYWKDVSSQHRWSTSPPVSSWWESEDR-LOxdK MESAIDTHLKCPRTLSRRVPDEYQPPFAMWMARADEHLEQVVMAYFGVQYRGEAQRAAALQAMRHIVESFSLADGPQTHDLTHHTDNSGFDNLIVVGYWKDPAAHCRWLRSAPVNAWWASEDR-LOxdA MESAIDTHLKCPRTLSRRVPEEYQPPFPMWVARADEQLQQVVMGYLGVQYRGEAQREAALQAMRHIVSSFSLPDGPQTHDLTHHTDSSGFDNLMVVGYWKDPAAHCRWLS-AEVNDWWTSQDR-LOxdB ----------------KNMPENHNPQANAWTAEFPPEMSYVVFAQIGIQSK---SLDHAAEHLGMMKKSFDLRTGPKHVDRALHQGADGYQDSIFLAYWDEPETFKSWVADPEVQKWWSGKKIDE
130 140 150 160 170 180 190 200 210 220 230 240 250OxdRE SDGLGFFREIVAPRAEQFETLYAFQED-LPGVGAVMDGISGEINEHGYWGSMRERFPISQTDWMQAS--GELRVIAGDPAVGGRVVVR-GHDNIALIRSGQDWADAEADERSLYLDEILPTLQSGOxdRG SDGLGFFREIVAPRAEQFETLYAFQDD-LPGVGAVMDGVSGEINEHGYWGSMRERFPISQTDWMQAS--GELRVVAGDPAVGGRVVVR-GHDNIALIRSGQDWADAEADERSLYLDEILPTLQSGOxdK NDGLGYFREISAPRAEQFETLYAFQDN-LPGVGAVMDRISGEIEEHGYWGSMRDRFPISQTDWMKPT--SELQVIAGDPAKGGRVVVL-GHGNLTLIRSGQDWADAEAEERSLYLDEILPTLQDGOxdA GEGLGYFREISAPRAEQFETLYAFQRDNLPGVGAVMDSTSGEIEEHGYWGSMRDRFPISQT-WMKPT--NELQVVAGDPAKGGRVVIM-GHDNIALIRSGQDWADAEAEERSLYLDEILPTLQDGOxdB NSPIGYWSEVTTIPIDHFETLHSGENY-DNGVSHFVP--IKHTEVHEYWGAMRDRMPVSASSDLESPLGLQLPEPIVRESFGKRLKVT-APDNICLIRTAQNWSKCGSGERETYIGLVEPTLIKA
260 270 280 290 300 310 320 330 340 350 360 370 OxdRE MDFLRDNGPAVGCYSNRFVRNIDIDGNFLDLSYNIGHWASLDQLERWSESHPTHLRIFTTFFRVAAG---LSKLRLYHEVSVFDAADQLYEYINCHPGTGMLRDAVTIAEHOxdRG MDFLRDNGPAVGCYSNRFVRNIDIDGNFLDLSYNIGHWASLDQLERWSESHPTHLRIFTTFFRVAEG---LSKLRLYHEVSVFDAADQLYEYINCHPGTGMLRDAVITAEHOxdK MDFLRDNGQPLGCYSNRFVRNIDLDGNFLDVSYNIGHWRSVEKLERWTESHPTHLRIFVTFFRVAAG---LKKLRLYHEVSVSDAKSQIFGYINCHPQTGMLRDAQVSPAOxdA MDFLRDNGQPLGCYSNRFVRNIDLDGNFLDVSYNIGHWRSLEKLERWAESHPTHLRIFVTFFRVAAG---LKKLRLYHEVSVSDAKSQVFEYINCHPHTGMLRDAVVAPTOxdB NTFLRENASETGCISSKLVYEQTHDGEIVDKSCVIGYYLSMGHLERWTHDHPTHKAIYGTFYEMLKRHDFKTELALWHEVSVLQSKDIELIYVNCHPSTGFLPFFEVTEIQEPLLKSPSVRI
Fig. 4. Amino acid sequence comparison of Oxds from Rhodococcus sp. N-771 (OxdRE), R. globerulus A-4 (OxdRG), Pseudomonas sp. K-9 (OxdK),
P. chlororaphis B23 (OxdA), and Bacillus sp. OxB-1 (OxdB). Residues in boxes indicate identical sequences among the Oxds.
248 Y. Kato et al. / FEMS Microbiology Letters 246 (2005) 243–249
SVEKLERWTE, respectively (Fig. 2). By sequencing
the amplified fragment, whole oxd gene sequence ofPseudomonas sp. K-9 was identified. Fig. 4 shows the
amino acid sequence similarities of a polypeptide en-
coded by the oxd gene with the known Oxds. It showed
identity with the Oxds of P. chlororaphis B23 (OxdA)
[20], R. erythropolis N-771 (OxdRE) [11], R. globerulus
A-4 (OxdRG) [10], and Bacillus sp. OxB-1 (OxdB) [8]
at 90.3%, 76.9%, 76.0%, and 32.7%, respectively. The se-
quence data have been submitted to DDBJ/EMBL/Gen-Bank databases under accession no. AB193508. A
plasmid, pOxdKInt, was constructed to express the
oxd gene in E. coli under the control of T7 promoter.
The protein was expressed in E. coli BL21 Star (DE3)/
pOxdKInt as we did for OxdRE [11]. The cell-free ex-
tract of the recombinant strain had an absorbance max-
imum at 410 nm, a characteristic Soret band for Oxds
[8–11], and exhibited a stoichiometric dehydration activ-ity of Z-phenylacetaldoxime into phenylacetonitrile
(320 U (lmol min�1) l�1 culture). Although further
investigations on the detailed properties of OxdK are
in progress, it is evidently clear that the enzyme is an
Oxd having a new primary structure and we tentatively
named it as OxdK.
4. Discussion
Here, we newly identified similar Oxd and NHase in
the 6 microbial nitrile- or aldoxime-degraders. The
PCR-based method shown in this study is quite useful
for the rapid identification of Oxds from various micro-
organisms without isolating Oxd protein. Indeed, the
newly identified OxdK has a new primary structureand the results encourage us to use this method to iden-
tify new types of Oxds from various microorganisms.
We could not amplify Oxd genes from the strains having
Nit and NHase(Co) probably because they might con-
tain Oxd having low similarities with the known ones.
Also, we [21] and the other group [22] recently reported
primitive studies on elucidating reaction mechanisms of
OxdB and OxdA, respectively, but the details of themechanism have not yet been understood. Further stud-
ies on purification and gene cloning of Oxds from a vari-
ety of microorganisms would accumulate genetic and
enzymatic information of the new types of Oxds and
comparison of the characters of the Oxds may help to
explain the mechanisms.
As reported by Duran et al. [19] and very recently by
Novo et al. [23], parts of NHases were amplified by PCRwith primers designed based on homologies of NHase to
show the existence of NHase(Fe) and NHase(Co) in
some bacterial strains. By comparing the results shown
in Figs. 2 and 3, it is possible to say that the primary
structure of Oxds is much similar each other (80.6%
identity) than those of NHase(Fe) (63.7% identity)
found among NHase(Fe)-containing strains. Thus, we
claim that the genetic analysis of oxd gene becomes amuch better tool to identify not only Oxd, but also
NHase(Fe).
The direct cloning of genes from environmental DNA
– the metagenome [24] has recently been paid much
attentions to obtain enzymes having novel primary
structures. Since enzymes comprising ‘‘aldoxime-nitrile
pathway’’, i.e., Oxd, NHase, Nit, and Ami, have become
potential catalysts in chemical industries [1,2], it isimportant to rapidly clone and identify genes of the
pathway in nature including metagenomes. It would be
advantageous to use Oxd as a key enzyme in the path-
way, because Oxd locates at an upstream of the
‘‘branched’’ nitrile-degradation which is catalyzed by di-
verse enzymes, Nit, NHase(Fe), and NHase(Co). In
practice, we have been focusing on Oxd and have shown
that all the microorganisms having Oxd also had nitrile-degrading enzymes and Oxd and the nitrile-degrading
enzymes are linked genetically as well as enzymatically
[1,7–11]. In a separate experiment from this study, we se-
quenced flanking regions of oxd gene in the genome of
Pseudomonas sp. K-9 and found that the oxd gene was
clustered with genes coding NHase(Fe), Ami, and their

Y. Kato et al. / FEMS Microbiology Letters 246 (2005) 243–249 249
activators and regulatory proteins, as seen in NHa-
se(Fe)-containing microorganisms, such as P. chlorora-
phis B23, R. erythropolis N-771, and R. globerulus A-4
(data not shown). Based on the results, we can conclude
here that the PCR-based analysis of oxd gene is a useful
tool to detect and analyze the ‘‘aldoxime-nitrile path-way’’ in nature because Oxd is important as a key en-
zyme. The structures of the ‘‘aldoxime-nitrile
pathway‘‘ gene cluster in Pseudomonas sp. K-9 and the
enzymatic properties of OxdK will be reported
elsewhere.
References
[1] Asano, Y. (2002) Overview of screening for new microbial
catalysts and their uses in organic synthesis – selection and
optimization of biocatalysts. J. Biotechnol. 94, 65–72.
[2] Banerjee, A., Sharma, R. and Banerjee, U.C. (2002) The nitrile-
degrading enzymes: current status and future prospects. Appl.
Microbiol. Biotechnol. 60, 33–44.
[3] Cowan, D.A., Cameron, R.A. and Tsekoa, T.L. (2003) Compar-
ative biology of mesophilic and thermophilic nitrile hydratases.
Adv. Appl. Microbiol. 52, 123–158.
[4] Asano, Y., Tani, Y. and Yamada, H. (1980) A new enzyme nitrile
hydratase which degrades acetonitrile in combination with
amidase. Agric. Biol. Chem. 44, 2251–2252.
[5] Asano, Y., Fujishiro, K., Tani, Y. and Yamada, H. (1982)
Aliphatic nitrile hydratase from Arthrobacter sp. J-1. Purification
and characterization. Agric. Biol. Chem. 46, 1165–1174.
[6] Asano, Y., Yasuda, T., Tani, Y. and Yamada, H. (1982) A new
enzymatic method of acrylamide production. Agric. Biol. Chem.
46, 1183–1189.
[7] Kato, Y., Ooi, R. and Asano, Y. (2000) Distribution of aldoxime
dehydratase in microorganisms. Appl. Environ. Microbiol. 66,
2290–2296.
[8] Kato, Y., Nakamura, K., Sakiyama, H., Mayhew, S.G. and
Asano, Y. (2000) A novel heme-containing lyase, phen-
ylacetaldoxime dehydratase from Bacillus sp. strain OxB-1:
purification, characterization, and molecular cloning of the gene.
Biochemistry 39, 800–809.
[9] Kato, Y. and Asano, Y. (2003) High-level expression of a novel
FMN-dependent heme-containing lyase, phenylacetaldoxime
dehydratase of Bacillus sp. strain OxB-1, in heterologous hosts.
Protein Exp. Purif. 28, 131–139.
[10] Xie, S.-X., Kato, Y., Komeda, H., Yoshida, S. and Asano, Y.
(2003) A novel gene cluster responsible for alkylaldoxime metab-
olism coexisting with nitrile hydratase and amidase in Rhodococ-
cus globerulus A-4. Biochemistry 42, 12056–12066.
[11] Kato, Y., Yoshida, S., Xie, S.-X. and Asano, Y. (2004) Aldoxime
dehydratase co-existing with nitrile hydratase and amidase in
iron-type nitrile hydratase producer Rhodococcus sp. N-771. J.
Biosci. Bioeng. 97, 250–259.
[12] Xie, S.X., Kato, Y. and Asano, Y. (2001) High yield synthesis of
nitriles by a new enzyme, phenylacetaldoxime dehydratase, from
Bacillus sp. strain OxB-1. Biosci. Biotechnol. Biochem. 65, 2666–
2672.
[13] Yamada, H., Asano, Y. and Tani, Y. (1980) Microbial utilization
of glutaronitrile. J. Ferment. Technol. 58, 495–500.
[14] Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular
Cloning: a Laboratory Manual, 3rd edn. Cold Spring Harbor
Laboratory Press, Cold Spring Harbor, NY.
[15] Pearson, W.R. and Lipman, D.J. (1988) Improved tools for
biological sequence comparison. Proc. Natl. Acad. Sci. USA 85,
2444–2448.
[16] Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman,
D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215,
403–410.
[17] Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUS-
TAL W: improving the sensitivity of progressive multiple
sequence alignment through sequence weighting, position-specific
gap penalties and weight matrix choice. Nucleic Acid. Res. 22,
4673–4680.
[18] Saito, H. and Miura, K. (1963) Preparation of transforming
deoxyribonucleic acid by phenol treatment. Biochim. Biophys.
Acta 72, 619–629.
[19] Precigou, S., Goulas, P. and Duran, R. (2001) Rapid and specific
identification of nitrile hydratase (NHase)-encoding genes in soil
samples by polymerase chain reaction. FEMS Microbiol Lett.
204, 155–161.
[20] Oinuma, K., Hashimoto, Y., Konishi, K., Goda, M., Noguchi, T.,
Higashibata, H. and Kobayashi, M. (2003) Novel aldoxime
dehydratase involved in carbon-nitrogen triple bond synthesis of
Pseudomonas chlororaphis B23. Sequencing, gene expression,
purification, and characterization. J. Biol. Chem. 278, 29600–
29608.
[21] Lourenco, P.M., Almeida, T., Mendonca, D., Simoes, F. and
Novo, C. (2004) Searching for nitrile hydratase using the
Consensus-Degenerate Hybrid Oligonucleotide Primers strategy.
J. Basic Microbiol. 44, 203–214.
[22] Kobayashi, K., Yoshioka, S., Kato, Y., Asano, Y. and Aono, S.
(2005) Regulation of aldoxime dehydratase activity by redox-
dependent change in the coordination structure of the aldoxime-
heme complex. J. Biol. Chem. 280, 5486–5490.
[23] Konishi, K., Ishida, K., Oinuma, K., Ohta, T., Hashimoto, Y.,
Higashibata, H., Kitagawa, T. and Kobayashi, M. (2004)
Identification of crucial histidines involved in carbon-nitrogen
triple bond synthesis by aldoxime dehydratase. J. Biol. Chem.
279, 47619–47625.
[24] Streit, W.R. and Schmitz, R.A. (2004) Metagenomics – the key to
the uncultured microbes. Curr. Opin. Microbiol. 7, 492–498.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 251–257
The gene encoding xylulose-5-phosphate/fructose-6-phosphatephosphoketolase (xfp) is conserved among Bifidobacterium
species within a more variable region of the genome and bothare useful for strain identification
Xianhua Yin, James R. Chambers, Kathleen Barlow, Aaron S. Park, Roger Wheatcroft *
Agriculture and Agri-Food Canada, Food Research Program, 93 Stone Road West, Guelph, Ont., Canada N1G 5C9
Received 31 December 2004; received in revised form 23 March 2005; accepted 12 April 2005
First published online 29 April 2005
Edited by R.Y.C. Lo
Abstract
The nucleotide sequence of the xfp-gene region in six known and two unknown species of Bifidobacterium was determined and
compared with the published sequences of B. animalis subsp. lactis DSM10140 and B. longum biovar longum NCC2705. The xfp
coding sequences were 73% identical and coded for 825 amino acids in all 10 sequences. Partial sequences of an adjacent gene, guaA,
were 61% identical in six sequences for which data were available. The region between xfp and guaA was variable in both length and
sequence. Oligonucleotide sequences from the conserved and variable xfp regions were used as PCR primers, in combinations of
appropriate specificity, for the detection and identification of Bifidobacterium isolates.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Bifidobacterium; Phosphoketolase; xfp; Strain identification; Detection
1. Introduction
Bacteria of the genus Bifidobacterium are anaerobic,
Gram-positive, non-spore-forming, non-motile bacilli
[1]. They are found in sewage and in the internal tracts
of animals, including insects and humans [2,3]; about
30 species are currently recognized [4]. Some species
are used in industry for the preparation of fermentedfoods and dietary supplements [2,5]. In the human gut,
bifidobacteria are generally regarded as safe and consis-
tent with good health [6–8]. They possess a wide range of
catabolic pathways which break down undigested food
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.013
* Corresponding author. Tel: +1 519 780 8025; fax: +1 519 829 2600.
E-mail address: [email protected] (R. Wheatcroft).
and secretions of the host [9,10]. They are at an advan-
tage as scavengers in the large intestine where readily
fermentable carbohydrates are in short supply [11,12].
A characteristic pathway is the �bifid shunt�, by which
bifidobacteria convert hexoses to acetic acid and lactic
acid, as chief products, in a theoretical molar ratio of
3:2 [13]. Secretion of acid into the gut is likely to affect
the growth and composition of the local microflora[14,15]. When acetic acid is undissociated, for example,
it acts synergistically with lactic acid to inhibit growth
of many enteric bacteria [16]. There is evidence to sug-
gest that bifidobacteria provide protection against some
pathogens, by this mechanism, in both humans and live-
stock [17–19]: a possibility that is of considerable inter-
est for public health and food safety. Clearly, there is a
need to establish whether bifidobacteria can be reliably
. Published by Elsevier B.V. All rights reserved.

Fig. 1. Southern hybridization of genomic DNA digested with EcoRI
probed with chemiluminescent xfp probe, P1. m: Size marker; lane 1:
B. animalis subsp. animalis ATCC27674; lane 2: B. gallinarum
ATCC33777, lane 3: B. longum biovar infantis ATCC15697, lane 4:
B. longum biovar longum ATCC15707, lane 5: B. pseudolongum subsp.
pseudolongum ATCC25526, lane 6: B. pullorum ATCC49618, lane 7: B.
thermophilum ATCC25525, lane 8: B. sp. BcRW10.
Table 1
Strains of bifidobacteria
Species Strain Source
B. adolescentis CFAR335 Human infant
B. animalis subsp. animalis ATCC27536 Chicken faeces
B. animalis subsp. animalis ATCC27674 Rabbit faeces
B. bifidum CFAR115 Wheat germ
B. boum ATCC27917 Bovine rumen
B. breve CFAR118 Morinaga Institute
252 X. Yin et al. / FEMS Microbiology Letters 246 (2005) 251–257
used as an adjunct or effective alternative to antibiotics
[20]; and, if so, whether natural populations can be stim-
ulated [21], supplemented [22] or genetically modified
for this purpose [23].
An early step in the �bifid shunt� is the phosphoketo-
lase reaction [EC: 4.1.2.22] by which D-fructose-6-phos-phate (F6P) is converted to erythrose-4-phosphate and
acetyl-1-phosphate [13]. This reaction is used to test
for Bifidobacterium species [24] though it is not exclusive
to them [25]. There is evidence for the existence of two
distinct F6P-phosphoketolase enzymes in bifidobacteria
[26,27]. One is specific solely for F6P; the other is less
stringent and is able to utilize D-xylulose-5-phosphate
(X5P) as an alternative substrate [EC: 4.1.2.9]. The latterreaction, which yields glyceraldehyde-3-phosphate and
acetyl-1-phosphate, is a later step in the �bifid shunt�[13]. Thus, it would seem that the same enzyme is able
to play an important double role in this pathway.
The dual-specificity X5P/F6P-phosphoketolase is en-
coded by the gene xfp, first described in B. animalis
subsp. lactis [25]. In the human isolate B. longum biovar
longum NCC2705, the genome has been completelysequenced [10] and a single copy of xfp is identified at lo-
cus BLO959. In another important contribution, a 503-
bp amplicon of xfp has been sequenced in most, if not
all, Bifidobacterium species; see GenBank Accessions:
AY574091 and AY377393 to AY377424, inclusive [28].
In the present study, we have sequenced xfp and its
neighbouring region in a selection of Bifidobacterium
species to explain the restriction-fragment-length poly-morphism (RFLP) observed (Fig. 1). We have selected
and tested primer and target sequences for the detection
of these species, and for the identification of isolates, by
PCR. This approach is but one of many molecular
methods now available for the detection and identifica-
tion of bifidobacteria [29–33].
(Japan)B. choerinum ATCC27686 Pig faeces
B. cuniculi ATCC27916 Rabbit faeces
B. dentium ATCC27534 Human dental caries
B. gallinarum ATCC33777 Chicken caecum
B. gallinarum ATCC33778 Chicken caecum
B. longum biovar infantis ATCC15697 Human infant intestine
B. longum biovar longum ATCC15707 Human adult intestine
B. longum biovar suis ATCC27533 Pig faeces
B. magnum ATCC27540 Rabbit faeces
B. merycicum CFAR339 Bovine rumen
B. minimum ATCC27538 Sewage
B. pseudolongum
subsp. globosum
ATCC25865 Bovine rumen
B. pseudolongum
subsp. pseudolongum
ATCC25526 Pig faeces
B. pullorum ATCC27685 Chicken faeces
B. pullorum ATCC49618 Chicken faeces
B. thermophilum ATCC25525 Pig faeces
B. sp. CFAR172 Calf-Guard (Pfizer)
B. sp. BcRW10 Pig faeces
ATCC, American Type Culture Collection, Rockville, MD, USA.
CFAR, Centre for Food and Animal Research (Agriculture and Agri-
Food Canada).
2. Materials and methods
2.1. Bacterial strains, plasmids and primers
Strains of bifidobacteria used in this study are listed
in Table 1. Cultures were grown anaerobically at 37
�C, in MRS medium (Difco) supplemented with 0.05%
cysteine hydrochloride, 0.02% Na2CO3 and 0.01%CaCl2. Tests were also made on the bacteria listed in
Table 2, which were grown according to ATCC recom-
mendations. Plasmids, listed in Table 3, were propa-
gated in Escherichia coli TOP10 (Invitrogen) or
GM2163 dam cells (NEB) at 37 �C, in LB broth [34] sup-
plemented with ampicillin (50 lg ml�1). T3 and T7 prim-
ers were used to sequence pRWBl10 and pRWBp10;
16S-rRNA primers (P0 and 338F), which typically pro-duce a PCR amplicon of 332 bp from genomic DNA,
were used as positive controls [35]; other oligonucleo-

Table 2
Other bacterial strains tested
Species Strain or source PCR product
obtained with
primers U1R/U2L
Actinomyces bovis ATCC13683 +
Actinomyces israelii ATCC12102 +
Arthrobacter ureafaciens ATCC7562 +
Bacillus cereus ATCC14579 �Bacillus subtilis ATCC6051 �Citrobacter freundii ATCC8090 �Clostridium lituseburense DSM797 �Clostridium perfringens D. Barnuma �Enterobacter aerogenes ATCC13048 �Enterococcus faecalis ATCC19433 �Escherichia coli ATCC11775 �Gardnerella vaginalis ATCC14018 +
Gluconacetobacter hansenii ATCC23769 �Gluconacetobacter xylinus ATCC23767 +
Klebsiella pneumoniae ATCC13883 �Lactobacillus amylovorus DSM20531 �Lactobacillus salivarius DSM20555 �Leuconostoc mesenteroides ATCC8293 +
Listeria innocua B. Blaisb �Listeria monocytogenes B. Blaisb �Propionibacterium acnes ATCC6919 �Propionibacterium
freudenreichii
ATCC8262 �
Proteus hauseri ATCC13315 �Pseudomonas aeruginosa ATCC10145 �Rhodococcus fascians ATCC12974 +
Ruminococcus torques ATCC27756 �Salmonella choleraesuis
subsp. choleraesuis
Montevideo
ATCC8387 �
Salmonella choleraesuis
subsp. choleraesuis
Typhimurium
ATCC14028 �
Serratia marcescens ATCC13880 �Shigella sonnei ATCC29930 �Staphylococcus aureus ATCC12600 �Staphylococcus epidermidis ATCC12228 �Yersinia enterocolitica ATCC9610 �ATCC, American Type Culture Collection, Rockville, MD, USA.
CFAR, Centre for Food and Animal Research (Agriculture and Agri-
Food Canada). DSM, German Collection of Microorganisms and Cell
Cultures, Braunschweig, Germany.a University of Guelph, Ont., Canada.b Canadian Food Inspection Agency, Ottawa, Canada.
X. Yin et al. / FEMS Microbiology Letters 246 (2005) 251–257 253
tides used in this study are listed in Table 3 and mapped
in Fig. 2.
2.2. DNA manipulation and analysis
Genomic DNA was isolated from 5-ml bacterial cul-
tures as previously described [36]. A commercial kit
(Qiagen) was used to extract plasmids from cells andto purify DNA from agarose gels. DNA restriction,
cloning, and PCR were carried out by routine methods
[34]. The PCR programme consisted of an initial step
at 94 �C for 4 min, followed by 35 cycles at 94 �C for
30 s, 60 �C for 30 s, and 72 �C for 1 min, followed by
a final step at 72 �C for 10 min. DNA probes, P1 and
P2, were digoxigenin labelled by PCR, as previously
described [37], using B. animalis ATCC27536 DNA as
substrate with primers A1R/A1L and A2R/A2L, respec-
tively. Southern blot and colony hybridizations werecarried out on HyBond filters (Amersham–Pharmacia
Biotech). Hybridization was detected using a commer-
cial chemiluminescence kit (Roche Diagnostic) and
XAR-2 film (Kodak). Nucleotide sequences of DNA
were determined using a Prism 377 automated sequencer
(ABI); alignments and analysis were carried out using
DNAman software (Lynnon BioSoft). GenBank acces-
sion numbers are included in Fig. 2.
3. Results
3.1. RFLP of the xfp-gene region
Southern hybridization profiles of Bifidobacterium
genomic DNA digested with EcoRI and probed withP1 are shown in Fig. 1. A single band indicates the pres-
ence of one copy of xfp in the genome and the absence
of any EcoRI-cleavage sites in the region covered by
the probe. The relative position of bands shows that
the location of EcoRI sites in the xfp region and the size
of hybridizing fragments are variable among genomes
(RFLP). A set of single- or multi-banded xfp-hybridiza-
tion profiles were obtained for several endonucleases(data not shown), whose cleavage sites were subse-
quently confirmed by DNA sequencing (Fig. 2).
3.2. DNA-sequence analysis
Blots were probed successively, with P1 and P2, to
identify the shortest restriction fragments that contained
both ends of xfp and, therefore, might be expected tocontain its full length. Thus, the 4.96-kb-HindIII/EcoRI
fragment of B. longum biovar longum ATCC15707
(pRWBl10) and the 4.43-kb-HindIII/XbaI fragment of
B. pullorum ATCC49618 (pRWBp10) were identified,
cloned and sequenced. The sequences were compared
with the published sequences of B. animalis subsp. lactis
DSM10140 and B. longum biovar longum NCC2705
(Fig. 2). Suitable oligonucleotides were synthesized (Ta-ble 3) to sequence the whole of xfp and its 5 0-neighbour-
ing region in B. pullorum ATCC49618 and in six other
Bifidobacterium isolates, including two unknown species
(Fig. 2). The 10 xfp coding sequences were found to be
73% identical at the nucleotide level. Their relatedness
is shown in Fig. 3. Each was found to encode 825 amino
acids of which 77% are conserved. By comparison, a
395-nucleotide partial sequence of guaA, a neighbouringgene encoding GMP synthase, was shown to be 61%
conserved at the nucleotide level, in six sequences for

Table 3
Plasmids and oligonucleotides
Source Designation Description or sequence (5 0–30)
Stratagene BlueScriptII SK+ Cloning vehicle for restriction fragments; Apr
Invitrogen pCR4-TOPO Cloning vehicle for PCR amplicons; Apr
B. longum biovar longum ATCC15707 pRWBl10 4.96-kb HindIII–EcoRI fragment containing xfp gene in BlueScriptII SK+; Apr
B. pullorum ATCC49618 pRWBp10 4.43-kb HindIII–XbaI fragment containing truncated xfp gene in
BlueScriptII SK+; Apr
B. animalis subsp. lactis DSM10140 [25] A1R catggcagaagctggatcgt
A2R gtcaccaagaagcagtgggac
A3R gctcaagcactgcaatcacaag
A1L ctccggcttgtaggattcca
A2L ggcctttcatcggctaagc
B. longum biovar longum NCC2705 [10] L1R gccactgcacaccatagagcttg
L2R tggctcatccacgtggtctgctc
L3R gatcacgtgcaggagtacagg
L4R atcctgcacctcaacggctac
L5R aagggctggacctgcccgaag
L1L agccctcggtcttcttgccgtc
L2L taggactcgagccagttcttgag
L3L tcactcgttgtcgccagcgg
B. pullorum ATCC49618 [this work] P1R gtctattgtggcggttcaagg
U1R acctgcccgaagtacatcgac
U1L tgtactcctgtactcctgcac
U2L gagctccagatgccgtgacg
B. sp. CFAR172 [this work] B1R cgactcagtactgattgatacc
B. sp. BcRW10 [this work] B1L tgcagcttcaggaggtcaacg
Apr, ampicillin resistant.
Fig. 2. Restriction maps to show variation (RFLP) in the xfp-gene region of Bifidobacterium species (including two cloned segments, pRWBp10 and
pRWBl10). Thick lines indicate coding sequences of xfp and part of guaA. Small arrowheads indicate the 5 0-ends of oligonucleotides (Table 3) used as
primers for PCR and sequencing (underlined). Primers, A1R/A1L and A2R/A2L, were used to make probes, P1 and P2, respectively. U1R/U2L
produced a 593-bp amplicon with all Bifidobacterium and some other species tested (Table 2). A3R, B1R and P1R with U1L produced amplification
products only with those species indicated to contain them. Dotted lines indicate the 5 0-ends of xfp and guaA delimiting the variable region between
them. Restriction sites: B, BamHI; C, BclI; G, BglII; H, HindIII; S, SphI; X, XbaI; Y, XmnI. GenBank accession numbers for the nucleotide
sequences determined in this work are given at right.
254 X. Yin et al. / FEMS Microbiology Letters 246 (2005) 251–257

Fig. 3. Homology tree of xfp-gene coding sequences of Bifidobacterium species. DNA homology is expressed as the number of identical nucleotide
residues between sequences as a percentage of the length of the aligned sequences (2475 nucleotides). The 10 sequences studied were 73% identical
overall.
X. Yin et al. / FEMS Microbiology Letters 246 (2005) 251–257 255
which data were available. In these species, the region
between xfp and guaA was found to be variable both
in sequence and in length (Fig. 2).
3.3. Specific detection and identification of bifidobacteria
by PCR
Primer sequences were selected from the xfp region
to use in PCR for diagnostic purposes (Table 3; Fig.
2). The combination, U1R/U2L, from the coding se-
quence of B. pullorum, generated a 593-bp amplicon
from genomic DNA of all Bifidobacterium strainstested (Table 1). The DNA of 33 strains of non-Bifido-
bacterium species was also tested. Seven species, which
were reported in the literature to possess a F6P-phos-
phoketolase, tested positive with U1R/U2L (Table 2).
The remaining strains were negative with U1R/U2L
but all tested positive with 16S-rRNA primers P0/
338F, used in controls of the PCR and template
DNA (data not shown).The Bifidobacterium strains were further resolved by
using combinations of primers chosen from both con-
served and variable sequences. For example, the combi-
nation P1R/U1L generated a 410-bp amplicon with both
B. gallinarum and B. pullorum, which are closely related
species (Fig. 3). Primers B1R/U1L generated a 565-bp
amplicon only with isolate B. sp. CFAR172, whereas
A3R/U1L gave a 362-bp amplicon only with B. animalis
strains. None of these primer combinations gave posi-
tive PCR results with any other bacterial species tested(Tables 1 and 2). These primers are now used routinely
to test directly for their respective species in samples
(Fig. 4).
4. Discussion
The gene xfp encodes one of two F6P-phosphoketo-lases reported to occur in bifidobacteria [26,27]. There
is evidence to suggest that one, if not both of these en-
zymes, is polymorphic, differing in human and animal
Bifidobacterium species [27,38]. In this work, we have
investigated the xfp region in several examples but have
been unable to detect any sequence differences that
would simply explain the enzyme polymorphism
reported. We conclude, therefore, that a species-dependent modification of the xfp-gene product takes
place or, alternatively, that the polymorphism reported
does not apply to the phosphoketolase encoded by
xfp.
We showed that the length (2475 nucleotides) and
73% of the xfp coding sequence are absolutely conserved

Fig. 4. Detection of diagnostic PCR products in Bifidobacterium species. m: size marker; and for each set, lane 1: B. pullorum ATCC49618; lane 2: B.
gallinarum ATCC33778; lane 3: B. longum biovar longum ATCC15707; lane 4: B. animalis subsp. animalis ATCC27536. Set A: 593-bp amplicon
obtained with all Bifidobacterium and some other species (Table 2), using primers U1R/U2L; set B: 410-bp amplicon only obtained with B. pullorum
and B. gallinarum, using primers P1R/U1L; set C: 362-bp amplicon obtained with all B. animalis strains tested, using primers A3R/U1L.
256 X. Yin et al. / FEMS Microbiology Letters 246 (2005) 251–257
in the 10 examples studied; whereas an adjacent house-
keeping gene, guaA, appeared to be less conserved. This
suggests that the function of xfp, if not the whole �bifidshunt� pathway, confers a significant advantage inBifidobacterium.
The xfp gene is characteristic of Bifidobacterium, yet
some non-Bifidobacterium species also gave positive
PCR results with primers designed to amplify its core,
for example U1R and U2L. The usefulness of these
primers for the detection of bifidobacteria must there-
fore be circumscribed; they are likely to be most useful
in the study of exclusive habitats like the gut, for exam-ple. By comparing amplicon sequences generated by
U1R/U2L from purified isolates, it is possible to differ-
entiate between species and assign them to homology
groups consistent with current taxonomy (Fig. 3). It is
also possible, and most convenient, to make a positive
species identification by matching the nucleotide se-
quence of an xfp amplicon with that of an authenticated
species [28]. We have, for example, used the currentGenBank database to identify isolates B. sp. BcRW10
and B. sp. CFAR172 precisely, as B. choerinum and B.
thermophilum, respectively.
Since 5 0-sequences adjacent to xfp are variable in
Bifidobacterium, they present species-specific targets
for diagnostic PCR. The primer combination A3R/
U1L, for example, is useful for the detection of B. ani-
malis strains. In our current work in progress, we areusing the combination P1R/U1L to detect strains of
B. pullorum [39], including mutant derivatives, in a ge-
netic study of their purported probiotic effects in
chickens.
Acknowledgements
We are grateful to our AAFC colleagues for help, ad-
vice and comments on the manuscript.
References
[1] Scardovi, V. (1986) Genus Bifidobacterium Orla-Jensen, 1924 In:
Bergey�s Manual of Systematic Bacteriology (Sneath, P.H.A.,
Mair, N.S., Sharpe, M.E. and Holt, J.G., Eds.), Vol. 2, pp. 1418–
1434. Williams and Wilkins, Baltimore, MD.
[2] Rasic, J. and Kurmann, J. (1983). Bifidobacteria and Their Role.
Experientia, Vol. 39. Birkhauser Verlag, Basel, Supplementum
278 pp..
[3] Biavati, B., Vescovo, M., Torriani, S. and Bottazzi, V. (2000)
Bifidobacteria: history, ecology, physiology and applications.
Ann. Microbiol. 50, 117–131.
[4] Ventura, M., van Sinderen, D., Fitzgerald, G.F. and Zink, R.
(2004) Insights into the taxonomy, genetics and physiology of
bifidobacteria. Anton. Leeuw. 86, 205–223.
[5] Kurmann, J.A. and Rasic, J.L. (1991) The health potential of
products containing bifidobacteria In: Therapeutic Properties of
Fermented Milks (Robinson, R.K., Ed.). Elsevier, London.
[6] Tannock, G.W. (1995) Normal Microflora: An Introduction to
Microbes Inhabiting the Human Body. Chapman & Hall,
London, 115 pp..
[7] Mitsuoka, T. (1990) Bifidobacteria and their role in human
health. J. Ind. Microbiol. 6, 263–268.
[8] Ishibashi, N., Yaeshima, T. and Hayasawa, H. (1997) Bifidobac-
teria: their significance in human intestinal health. Mal. J. Nutr. 3,
149–159.
[9] Crociani, F., Alessandrini, A., Mucci, M.M. and Biavati, B.
(1994) Degradation of complex carbohydrates by Bifidobacterium
spp.. Int. J. Food Microbiol. 24, 199–210.
[10] Schell, M.A., Karmirantzou, M., Snel, B., Vilanova, D., Berger,
B., Pessi, G., Zwahlen, M.C., Desiere, F., Bork, P., Delley, M.,
Pridmore, R.D. and Arigoni, F. (2002) The genome sequence of
Bifidobacterium longum reflects its adaptation to the human
gastrointestinal tract. Proc. Natl. Acad. Sci. USA 99, 14422–
14427.
[11] Macfarlane, G.T. and Macfarlane, S. (1997) Human colonic
microbiota: ecology physiology and metabolic potential of
intestinal bacteria. Scand. J. Gastroenterol. 32 (Suppl. 222), 3–9.
[12] Hopkins, M.J., Cummings, J.H. and Macfarlane, G.T. (1998)
Inter-species differences in maximum specific growth rates and cell
yields of bifidobacteria cultured on oligosaccharides and other
simple carbohydrate sources. J. Appl. Microbiol. 85, 381–386.
[13] Scardovi, V. and Trovatelli, L.D. (1965) The fructose-6-phos-
phate shunt as peculiar pattern of hexose degradation in the genus
Bifidobacterium. Ann. Microbiol. 15, 19–29.

X. Yin et al. / FEMS Microbiology Letters 246 (2005) 251–257 257
[14] Benno, Y. and Mitsuoka, T. (1992) Impact of Bifidobacterium
longum on human fecal microflora. Microbiol. Immunol. 36, 683–
694.
[15] Gibson, G.R. and Wang, X. (1994) Regulatory effects of
bifidobacteria on the growth of other colonic bacteria. J. Appl.
Bacteriol. 77, 412–420.
[16] Adams, M.R. and Hall, C.J. (1988) Growth inhibition of food-
borne pathogens by lactic and acetic acids and their mixtures. Int.
J. Food Sci. Technol. 23, 287–292.
[17] Savage, D.C. (1987) Factors influencing biocontrol of bacterial
pathogens in the intestine. Food Technol. 41, 82–87.
[18] Ibrahim, S.A. and Bezkorovainy, A. (1993) Inhibition of Esch-
erichia coli by bifidobacteria. J. Food Prot. 56, 713–715.
[19] Araya-Kojima, T., Yaeshima, T., Ishibashi, N., Shimamura, S.
and Hayasawa, H. (1995) Inhibitory effects of Bifidobacterium
longum BB536 on harmful intestinal bacteria. Bifidobacteria
Microflora 14, 59–66.
[20] Gomes, A.M.P. and Malcata, F.X. (1999) Bifidobacterium spp.
and Lactobacillus acidophilus: biological, biochemical, technolog-
ical and therapeutical properties relevant for use as probiotics.
Trends Food Sci. Technol. 10, 139–157.
[21] Gibson, G.R. and Wang, X. (1994) Enrichment of bifidobacteria
from human gut contents by oligofructose using continuous
culture. FEMS Microbiol. Lett. 118, 121–128.
[22] Ballongue, J., Grill, J.P. and Baratte-Euloge, P. (1993) Action sur
la flore intestinale de laits fermentes au Bifidobacterium. Lait 73,
249–256.
[23] Kullen, M.J. and Klaenhammer, T.R. (2000) Genetic modifica-
tion of intestinal lactobacilli and bifidobacteria. Curr. Issues Mol.
Biol. 2, 41–50.
[24] Orban, J.I. and Patterson, J.A. (2000) Modification of the
phosphoketolase assay for rapid identification of bifidobacteria.
J. Microbiol. Methods 40, 221–224.
[25] Meile, L., Rohr, L.M., Geissmann, T.A., Herensperger, M. and
Teuber, M. (2001) Characterization of the D-xylulose 5-phos-
phate/D-fructose 6-phosphate phosphoketolase gene (xfp) from
Bifidobacterium lactis. J. Bacteriol. 183, 2929–2936.
[26] Sgorbati, B., Lenaz, G. and Casalicchio, F. (1976) Purification
and properties of two fructose-6-phosphate phosphoketolases in
Bifidobacterium. Anton. Leeuw. 42, 49–57.
[27] Grill, J.P., Crociani, J. and Ballongue, J. (1995) Characterization
of fructose 6 phosphate phosphoketolases purified from Bifido-
bacterium species. Curr. Microbiol. 31, 49–54.
[28] Berthoud, H., Chavagnat, F., Haueter, M. and Casey, M.G.
(2005) Comparison of partial gene sequences encoding a phos-
phoketolase for the identification of bifidobacteria. Lebensm.
Wiss. Technol. 38, 101–105.
[29] Zavaglia, A.G., de Urraza, P. and De Antoni, G. (2000)
Characterization of Bifidobacterium strains using box primers.
Anaerobe 6, 169–177.
[30] Satokari, R.M., Vaughan, E.E., Smidt, H., Saarela, M., Matto, J.
and de Vos, W.M. (2003) Molecular approaches for the detection
and identification of bifidobacteria and lactobacilli in the human
gastrointestinal tract. Syst. Appl. Microbiol. 26, 572–584.
[31] Mullie, C., Odou, M.F., Singer, E., Romond, N.B. and Izard, D.
(2003) Multiplex PCR using 16S rRNA gene-targeted primers for
the identification of bifidobacteria from human origin. FEMS
Microbiol. Lett. 222, 129–136.
[32] Zhu, L., Li, W. and Dong, X. (2003) Species identification of
genus Bifidobacterium based upon partial HSP60 gene
sequences and proposal of Bifidobacterium thermacidophilum
subsp. porcinum subsp. nov. Int. J. Syst. Evol. Microbiol. 53,
1619–1623.
[33] Delcenserie, V., Bechoux, N., Leonard, T., China, B. and Daube,
G. (2004) Discrimination between Bifidobacterium species from
human and animal origin by PCR-restriction fragment length
polymorphism. J. Food Protection 67, 1284–1288.
[34] Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular
Cloning: A Laboratory Manual, second ed. Cold Spring Harbor
Laboratory Press, Cold Spring Harbor, NY.
[35] Ventura, M., Reniero, R. and Zink, R. (2001) Specific identifi-
cation and targeted characterization of Bifidobacterium lactis
from different environmental isolates by a combined multiplex-
PCR approach. Appl. Environ. Microbiol. 67, 2760–2765.
[36] Wheatcroft, R. and Watson, R.J. (1988) A positive strain
identification method for Rhizobium meliloti. Appl. Environ.
Microbiol. 54, 574–576.
[37] Anon (1995) The DIG System User�s Guide for Filter Hybrid-
ization. Boehringer Mannheim GmbH, Mannheim, 100 pp..
[38] Scardovi, V., Sgorbati, B. and Zani, G. (1971) Starch gel
electrophoresis of fructose-6-phosphate phosphoketolase in the
genus Bifidobacterium. J. Bacteriol. 106, 1036–1039.
[39] Trovatelli, L.D., Crociani, F., Pedinotti, M. and Scardovi, V.
(1974) Bifidobacterium pullorum sp. nov.: a new species isolated
from chicken feces and a related group of bifidobacteria isolated
from rabbit feces. Arch. Microbiol. 98, 187–198.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 259–264
The stabilization of housekeeping transcripts in Trypanosoma cruziepimastigotes evidences a global regulation of RNA decay
during stationary phase
Ana Marıa Cevallos, Mariana Perez-Escobar, Norma Espinosa, Juliana Herrera,Imelda Lopez-Villasenor, Roberto Hernandez *
Departamento de Biologıa Molecular y Biotecnologıa, Instituto de Investigaciones Biomedicas, Universidad Nacional Autonoma de Mexico,
Apartado Postal 70-228, 04510 Mexico D.F., Mexico
Received 24 February 2005; received in revised form 17 March 2005; accepted 13 April 2005
First published online 27 April 2005
Edited by D.P. Wakelin
Abstract
The relative steady state concentration of mRNAs of four housekeeping single-copy type Trypanosoma cruzi genes (actin, triose-
phosphate isomerase, trypanothion reductase and the ribosomal protein S4) was analyzed throughout the growth curve. A distin-
guishable pattern was observed with maximal levels occurring at the logarithmic phase of growth and minimum levels occurring at
the stationary phase. The half-lives of all analyzed messenger RNAs, and also of three molecular species of immature ribosomal
RNAs were increased in cells isolated from stationary phase. These results suggest the occurrence of a novel global regulation mech-
anism that might protect transcripts from degradation in stationary epimastigotes, probably as a strategy to perpetuate through this
quiescent stage.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Protozoa; Kinetoplastid; RNA stability; Gene expression
1. Introduction
Trypanosoma cruzi is a parasitic protozoa causative
agent of Chagas disease or American trypanosomiasis.
During its life cycle this parasite can alternate throughvertebrate and invertebrate hosts. In both hosts, T. cruzi
goes through a cycle that includes infective and non-
infective forms and these forms are morphologically
identifiable (see [1] for review). Differential gene expres-
sion occurs during development of this organism, espe-
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.017
* Corresponding author. Tel.: +52 55 5622 3872; fax: +52 55 5550
0048.
E-mail address: [email protected] (R. Hernandez).
cially through post-transcriptional regulation of
mRNAs [2]. The extracellular epimastigote forms (pres-
ent in the digestive tract of the reduvid vector) can be
readily cultured in axenic media, and are hence amena-
ble to experimentation. The growth curve of epimastig-otes represents a useful tool to analyze differential gene
expression throughout a cellular population that
changes its morphology, from being rounded or oval
cells with a short flagellum during the logarithmic phase
of growth to elongated cells with an extended flagellum
(over 30 lm) during the stationary phase [1]. Cells in the
latter phase can perpetuate for long periods under cul-
ture conditions. The stationary phase has been consid-ered an environmental condition where differentiation
. Published by Elsevier B.V. All rights reserved.

260 A.M. Cevallos et al. / FEMS Microbiology Letters 246 (2005) 259–264
towards non-dividing metacyclic trypomastigotes is trig-
gered [3]. The mechanisms that allow epimastigotes to
survive in the stationary phase are unknown. The search
for different steady state concentrations of specific
mRNAs during the growth curve showed that a and btubulin [4] and actin mRNAs [5] decrease prior to andduring the stationary phase. In order to gain insight in
the biology of epimastigotes at the stationary phase,
and to further analyze a potential modulation of addi-
tional RNAs during non proliferative conditions, we
investigated: (1) the steady state concentrations of four
mRNAs from single-copy type housekeeping genes
along the growth curve, and (2) the relative stability of
these mRNAs and of three ribosomal RNA precursormolecules from both logarithmic and stationary cells.
2. Materials and methods
2.1. Parasites and culture conditions
T. cruzi epimastigotes from the CL Brener strain weregrown at 28 �C in liver infusion triptone medium supple-
mented with 10% heat inactivated fetal bovine serum [3].
In order to obtain reproducible results, the cellular pop-
ulation was homogenized as follows: epimastigotes were
maintained in logarithmic growth during at least 3 cy-
cles from 1 · 106 to 30 · 106 cells per ml, and then di-
luted to 1 · 106 cells per ml to start the actual time
course analysis. The number of cells was registered at24–48 h intervals; the cells were harvested at selected
days of the culture during the logarithmic and stationary
phases. Mid-logarithmic phase was defined as the time
when epimastigotes reached a concentration of approx-
imately 8–12 · 106 cells per ml (days 3–4 post-inocula-
tion). Stationary phase was defined as the time when
parasites stopped their growth for 72 h, (90–100 · 106
cells per ml; days 12–14 post-inoculation). Mid-logarith-mic cellular populations were devoid of metacyclic
forms while stationary phase cultures had about 5% of
metacylic trypomastigotes as estimated from fixed
stained preparations.
2.2. Gene probes
To prepare gene probes, DNA fragments were gelpurified from the following T. cruzi plasmid clones:
pD-4, actin genomic clone [5]; pBTR, plasmid bearing
a PCR derived coding sequences from the tripanothion
reductase (TR) gene [6]; recombinant plasmid composed
the pCR II vector carrying a genomic derived PCR
amplification product from the triosephosphate isomer-
ase (TIM) gene [7]; pS4-2, cDNA clone from the ribo-
somal protein S4 (S4) locus [8]. In northern blotanalysis of total RNA obtained from epimastigotes in
culture, each of these probes recognizes a single mRNA
band at every stage during growth (data not shown).
Three ribosomal genomic clones pRTC20, pRTC42
and pRTC32 were also used [9]. All DNA probes were
labeled with [a-32P] dCTP using a random prime label-
ing system (Redi prime II, Amersham Pharmacia
Biotechnology).
2.3. Northern blot analyses
Total RNA preparations and northern blot hybrid-
izations were carried out as earlier described [5]. The
amount of bound radioactivity on the membranes was
detected with the Molecular Imager FX system (Bio-
Rad), and quantitation of the samples was done usingthe Quantity One software (BioRad). The levels of spe-
cific mRNAs in each lane were normalized as a ratio to
an independent rRNA probe to correct for potential dif-
ferences in loading (mRNA/rRNA ratio). Values are
thereby expressed as a percentage of the maximal ratio
obtained for each probe. All RNA size determinations
were estimated with the 0.24–9.5 kb RNA ladder (Gibco
BRL).
2.4. Analysis of RNA precursors’ half-life
The half-life of mRNAs was quantitated in northern
blots as above with RNA isolated at several time points
from cells incubated in the presence of the transcription
inhibitor actinomycin D (10 lg/ml) [10]. Due to their cel-
lular abundance the amounts of rRNA precursors weredetected directly from ethidium bromide stained gels
with the Molecular Imager FX system. Their quantifica-
tion was done using the Quantity One software
(BioRad).
3. Results and discussion
3.1. Steady state concentrations of mRNAs along
epimastigotes growth curve
In the early stages of this work, we were interested to
find out whether in culture derived epimastigotes
mRNAs other than actin transcripts showed the increase
and decrease pattern observed in our previous work [5].
The mRNAs for the TIM, TR and S4 genes were thenanalyzed in this context. Reproducible results were ob-
tained only when cultures were passaged while in loga-
rithmic growth prior to the start of the experiment.
Actin, TIM, TR and S4 mRNAs were analyzed by
northern hybridizations of total RNA isolated from
the cultures at different time points. Fig. 1 is representa-
tive of three independent experiments and shows an
increment in the concentration of mRNAs during earlystages of the curve followed by a decrease in the station-
ary phase. In order to analyze the viability of cells in

Fig. 1. mRNAs levels during growth of Trypanosoma cruzi epimastig-
otes. Total RNA was isolated from cultured epimastigotes at the
indicated days in culture and analyzed with gene specific probes in
northern hybridizations (�10 lg/lane, panel A). The observed levels of
mRNAs (actin, closed triangles, straight line; S4, closed squares,
straight line; TIM, crosses, dotted line; and TR, empty diamonds,
dotted line) are depicted in panel B as the ratio of radioactivity in the
specific mRNA band/radioactivity of rRNA in the corresponding lane
(right, Y-axis). Values were normalized to 100% with the higher ratio
for each mRNA series. Solid circles represent the density of
epimastigotes per ml (left, Y-axis).
Fig. 2. Differential stability of mRNAs in Trypanosoma cruzi epim-
astigotes from the mid logarithmic and stationary phase. Panel A:
northern blots of total RNA were carried out at different time points
after the addition of actinomycin D. Gene specific probes are the same
as those depicted in Fig. 1. Panel B, graphs corresponding to half-life
of actin (closed triangles, straight line), S4 (closed squares, straight
line) TIM (crosses, dotted line) and TR open diamonds, dotted line)
mRNAs at both mid-logarithmic and stationary phases of growth.
A.M. Cevallos et al. / FEMS Microbiology Letters 246 (2005) 259–264 261
well established stationary phase, the inoculum used to
start one of the experimental growth curves were cells
from aged cultures, that is, cells that remained in sta-
tionary phase for 14 days after growth had stopped.
This experiment reproduced a similar pattern albeitthere was a delay in the accumulation of steady state
concentration of mRNAs in correlation with the ob-
served extended lag phase of the growth curve (data
not shown).
3.2. Stability of RNAs during growth and stationary
phases
Non-dividing epimastigotes from stationary phase
can survive for few weeks when transcription of Pol I
and potentially all Pol II transcripts are down regulated
[11]. To investigate mechanisms that would support a
minimal level of transcripts needed for cellular survival
during stationary phase, the stability of the four
mRNAs here studied was analyzed from cells at both
the mid-logarithmic phase of growth and the stationaryphase of growth. A kinetics profile of RNA decay was
carried out in cells treated with actinomycin D. The pat-
terns depicted in Fig. 2B are consistent with a heteroge-
neous RNA population. In any case, it is interesting that
all four mRNAs analyzed showed an expanded half-life
(at least threefold) in cells from the stationary phase as
compared with cells from logarithmic phases of growth
(Fig. 2 and Table 1). This result agrees with the reportthat the half-lives of histone H2A mRNAs are about
twice as long in T. cruzi epimastigotes from the station-
ary phase when compared to cells at the logarithmic
phase of growth [12]. As a not understood phenomenon,
the presence of actinomycin D correlated with an initial
short and temporal increase in the concentration of
some RNA species. An analogous unexplained effect in
mRNAs from Trypanosoma brucei has been observed[13].

Table 1
Half lives of mRNAs and immature rRNAs at both mid-logarithmic
(ML) and stationary phases (S) of growtha
ML S S/ML
mRNAs
Actin 5.7 40.6 7.1
S4 4.2 18.3 4.3
TIM 6.9 22.1 3.2
TR 3.8 16.6 4.3
Immature rRNAs
7.6 kb 1.7 10.8 6.2
6.7 kb 6.8 17.9 2.6
5.3 kb 2.0 11.6 5.7
a Results were derived from the lineal regression calculations
obtained from the lineal segment of the decay curve and are expressed
in hours. The relative stabilization of transcripts was determined by the
stationary to mid-logarithmic ratio.
262 A.M. Cevallos et al. / FEMS Microbiology Letters 246 (2005) 259–264
Immature and mature rRNAs species are so abun-
dant that can be visualized from total RNA electropher-
ograms stained with ethidium bromide. This is
demonstrated in Fig. 3 where major bands are shown
to hybridize with rRNA gene probes. The relative stabil-
Fig. 3. (Top) Diagram of the rRNA cistron. The coding regions are
depicted as solid boxes interconnected by a thin line that represents
transcribed spacer regions. The transcription start point is indicated by
an arrow [19]. The bars marked as pRTC show three genomic
fragments of this region previously cloned [9]. (Bottom) Ethidium
bromide profile of total RNA of growing epimastigotes. Total RNA
from parasites in the mid-logarithmic phase of growth was extracted
and loaded into a non-denaturing TBE, 1%, agarose gel in three
identical lanes. The whole gel was stained with ethidium bromide and
the negative version of the image was registered using a phosphor
imager (Molecular Imager FX, BioRad), only one lane is shown. For
the hybridization analysis, the stained gel was equilibrated with the
standard MOPS/formaldehyde solution and transferred to a nylon
membrane. Each lane was cut separately and hybridized to probes
pRTC20, pRTC42, and pRTC32 as indicated (lanes 20, 42 and 32).
ity analysis of the pre RNAs was therefore carried out
from ethidium bromide stained gels (Fig. 4). Similarly
to data from mRNAs, the immature rRNAs were found
to be more stable in cells from stationary phase (Fig. 4,
Table 1). Whether the observed stabilization of pre
rRNAs is due to a slower decay and/or a decrease inthe rate of processing cannot be distinguished with this
experimental approach.
All together, the stabilization of specific mRNAs and
immature rRNAs during the stationary phase suggests
the occurrence of a post-transcriptional control point
associated to the growth phase of T. cruzi epimastigotes
(at least under culture conditions). Differential stability
of mRNAs has been well documented to occur as animportant mechanism to regulate differential gene
expression during development in kinetoplastids, mainly
involving sequences present in their 3 0 untranslated re-
gions (3 0 UTR) [14]. In the case of T. cruzi, at least
two RNA sequence elements present in the 3 0 UTR of
the small type mucin mRNAs either destabilize (AU rich
sequences, ARE), or stabilize (G rich elements, GREs)
with a cis effect on their coding flanking sequences.
Fig. 4. Half-life of rRNA precursors. (A) Negative image of ethidium
bromide stained gels of total RNA (�1 lg/lane) from epimastigotes in
mid-logarithmic and stationary phases of growth, at different time
intervals after the addition of actinomycin D (10 lg/ml). (B) Kinetics
of the rRNA precursors decay after the addition of actinomycin D in
mid-logarithmic and stationary phase epimastigotes. Values represent
the mean of four independent assays; standard errors are depicted as
vertical lines. Open circles correspond to data from the 7.6 kb
precursor, closed squares to the 6.7 kb precursor and closed triangles
to the 5.3 kb precursor.

A.M. Cevallos et al. / FEMS Microbiology Letters 246 (2005) 259–264 263
Interestingly these two elements seem to function at dif-
ferent developmental stages, ARE functions in trypom-
astigotes while GREs function in epimastigotes [15,16].
We therefore explored the 3 0 UTR sequences for the
presence of common motifs either in their primary se-
quence or in the secondary structure using the RNAanalyzer software described by Bengert and Dandekar
[17]. The analyzed 3 0 UTRs varied in length and in the
percentage of A + U content. No regions with signifi-
cant homology were identified and in no case were the
ARE or GRE motifs found. We also studied the 5 0
UTRs as they could also be involved in gene expression.
The 5 0 UTR also varied in length an in percentage of
A + U content and no regions of homology were identi-fied. Therefore, no correlation between UTR�s and dif-
ferential mRNA stability could be found. It is to point
out that this phenomenon includes at least five types
of mRNAs (including histone H2A [12]) and three
immature rRNA molecular species, therefore it is
improbable that a single RNA motif could be involved
as the main recognition element. A more likely mecha-
nism may be a general down regulation of the RNA de-cay (or processing) at stationary phase. It has been
demonstrated that transcription by both Pol I and Pol
II polymerases is down regulated in non-dividing
T. cruzi stages and in non-infective cells from stationary
phase cultures [11]. Data from our laboratory is in
accordance with this observation: the incorporation rate
of labeled uridine is about sixfold higher in growing
epimastigotes than in cells from the stationary phase,whose activity is registered well above background (data
not shown). In T. brucei species the RNA synthesis is
down regulated in the stationary phase that occurs dur-
ing differentiation of bloodstream to procyclic forms
[18]. In this situation, the stabilization of housekeeping
transcripts observed in the present work may be part
of a global regulation strategy to compensate for a
reduction in transcription.It is widely accepted that during the stationary
phase of culture a small proportion of epimastigotes
spontaneously transform into metacyclic trypomastig-
otes [3]. Therefore the entrance of epimastigotes into
the stationary phase can be considered as an onset
for differentiation. Metacyclic trypomastigotes are
non-dividing infective forms of the T. cruzi parasite,
that in natural conditions reside in the cloacal regionof the alimentary tract of the Triatome vector. Metacy-
clic trypomastigotes remain there until the insect finds
an appropriate host to feed. At the time of feeding
the parasites are excreted and upon entrance into a sus-
ceptible host they differentiate into dividing amastig-
otes. A general stabilization of transcripts may
therefore be a selected mechanism in these non-dividing
forms of T. cruzi to maintain a minimal level of expres-sion of housekeeping genes that would allow the para-
site to resume growth if environmental conditions turn
favorable for proliferation. The mechanism for this sta-
bilization of RNAs during the stationary phase re-
mains to be determined.
Acknowledgements
The authors thank Dr. Jorge Tovar and Dr. Ruy
Perez-Montfort for their kind donations of recombinant
plasmids used in this study, and Dr. Joaquın Sanchez
for the critical reading of the manuscript. We acknowl-
edge Lorena Lopez-Griego for technical help. This work
was supported by grant IN209302 from PAPIIT and
Grants 28036M, 37620M and 45037Q from CONACyT,Mexico. Mariana Perez-Escobar was supported by a
scholarship from CONACyT during her Ph.D. Thesis
program.
References
[1] Tyler, K.M. and Engman, D.M (2001) The life cycle of Trypan-
osoma cruzi revisited. Int. J. Parasitol. 31, 472–481.
[2] Teixeira, S.M. (1998) Control of gene expression in Trypanoso-
matidae. Braz. J. Med. Biol. Res. 31, 1503–1516.
[3] Camargo, E.P. (1964) Growth and differentiation of Trypanosoma
cruzi, origin of metacyclic trypanosomes in liquid medium. Rev.
Inst. Med. Trop. Sao Paulo 12, 93–100.
[4] Gonzalez-Pino, M.J., Rangel-Aldao, R. and Slezynger, T.C.
(1999) Expression of alpha- and beta-tubulin genes during growth
of Trypanosoma cruzi epimastigotes. DNA Cell Biol. 18, 449–455.
[5] Cevallos, A.M., Lopez-Villasenor, I., Espinosa, N., Herrera, J.
and Hernandez, R. (2003) Trypanosoma cruzi: allelic comparisons
of the actin genes and analysis of their transcripts. Exp. Parasitol.
103, 27–34.
[6] Borges, A., Cunningham, M.L., Tovar, J. and Fairlamb, A.H.
(1995) Site-directed mutagenesis of the redox-active cysteines of
Trypanosoma cruzi trypanothione reductase. Eur. J. Biochem.
228, 745–752.
[7] Ostoa-Saloma, P., Garza-Ramos, G., Ramirez, J., Becker, I.,
Berzunza, M., Landa, A., Gomez-Puyou, A., Tuena de Gomez-
Puyou, M. and Perez-Montfort, R. (1997) Cloning, expression,
purification and characterization of triosephosphate isomerase
from Trypanosoma cruzi. Eur. J. Biochem. 244, 700–705.
[8] Hernandez, R., Palacios, S., Herrera, J., Martinez-Calvillo, S. and
Lopez, I. (1998) The deduced primary structure of a ribosomal
protein S4 from Trypanosoma cruzi. Biochim. Biophys. Acta 1395,
321–325.
[9] Hernandez, R., Diaz-de Leon, F. and Castaneda, M. (1988)
Molecular cloning and partial characterization of ribosomal RNA
genes from Trypanosoma cruzi. Mol. Biochem. Parasitol. 27, 275–
279.
[10] Charest, H., Zhang, W.W. and Matlashewski, G. (1996) The
developmental expression of Leishmania donovani A2 amastigote-
specific genes is post-transcriptionally mediated and involves
elements located in the 30-untranslated region. J. Biol. Chem. 271,
17081–17090.
[11] Elias, M.C., Marques-Porto, R., Freymuller, E. and Schenkman,
S. (2001) Transcription rate modulation through the Trypano-
soma cruzi life cycle occurs in parallel with changes in nuclear
organisation. Mol. Biochem. Parasitol. 112, 79–90.
[12] Maranon, C., Thomas, M.C., Puerta, C., Alonso, C. and Lopez,
M.C. (2000) The stability and maturation of the H2A histone

264 A.M. Cevallos et al. / FEMS Microbiology Letters 246 (2005) 259–264
mRNAs from Trypanosoma cruzi are implicated in their post-
transcriptional regulation. Biochim. Biophys. Acta 1490, 1–10.
[13] Irmer, H. and Clayton, C. (2001) Degradation of the unstable
EP1 mRNA in Trypanosoma brucei involves initial destruction of
the 3 0-untranslated region. Nucleic Acids Res. 29, 4707–4715.
[14] Clayton, C.E. (2002) Life without transcriptional control? From
fly to man and back again. EMBO J. 21, 1881–1888.
[15] Di Noia, J.M., D�Orso, I., Sanchez, D.O. and Frasch, A.C. (2000)
AU-rich elements in the 30-untranslated region of a new mucin-
type gene family of Trypanosoma cruzi confers mRNA instability
and modulates translation efficiency. J. Biol. Chem. 275, 10218–
10227.
[16] D�Orso, I. and Frasch, A.C. (2001) Functionally different AU-
and G-rich cis-elements confer developmentally regulated mRNA
stability in Trypanosoma cruzi by interaction with specific RNA-
binding proteins. J. Biol. Chem. 276, 15783–15793.
[17] Bengert, P. and Dandekar, T. (2003) A software tool-box for
analysis of regulatory RNA elements. Nucleic Acids Res. 31,
3441–3445.
[18] Pays, E., Hanocq-Quertier, J., Hanocq, F., Van Assel, S., Nolan,
D. and Rolin, S. (1993) Abrupt RNA changes precede the first cell
division during the differentiation of Trypanosoma brucei blood-
stream forms into procyclic forms in vitro. Mol. Biochem.
Parasitol. 61, 107–114.
[19] Figueroa-Angulo, E., Martınez Calvillo, S., Lopez-Villasenor, I.
and Hernandez, R. (2003) Evidence supporting a major promoter
in the Trypanosoma cruzi rRNA gene. FEMS Microbiol. Lett.
225, 221–225.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 265–272
Genotypic and phenotypic characterization of a biofilm-formingSerratia plymuthica isolate from a raw vegetable processing line
Rob Van Houdt *, Pieter Moons, An Jansen, Kristof Vanoirbeek, Chris W. Michiels
Laboratory of Food Microbiology, Katholieke Universiteit Leuven, Kasteelpark Arenberg 22, B-3001 Leuven, Belgium
Received 11 March 2005; received in revised form 8 April 2005; accepted 13 April 2005
First published online 27 April 2005
Edited by J.A. Cole
Abstract
Recently, we isolated from a raw vegetable processing line a Serratia strain with strong biofilm-forming capacity and which pro-
duced N-acyl-L-homoserine lactones (AHLs). Within the Enterobacteriaceae, strains of the genus Serratia are a frequent cause of
human nosocomial infections; in addition, biofilm formation is often associated with persistent infections. In the current report,
we describe the detailed characterization of the isolate using a variety of genotypic and phenotypic criteria. Although the strain
was identified as Serratia plymuthica on the basis of its small subunit ribosomal RNA (16S rRNA) gene sequence, it differed from
the S. plymuthica type strain in production of pigment and antibacterial compounds, and in AHL production profile. Nevertheless,
the identification as S. plymuthica could be confirmed by gyrB phylogeny and DNA:DNA hybridization.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Serratia; Identification natural isolate; gyrB; Phylogeny; Quorum sensing; N-Acyl-L-homoserine lactone
1. Introduction
The genus Serratia, named after the Italian physicistSerafino Serrati, belongs to the family Enterobacteria-
ceae and consists of the recognized species: Serratia mar-
cescens, S. liquefaciens, S. ficaria, S. rubidaea, S.
fonticola, S. odorifera, S. plymuthica, S. grimesii, S. pro-
teamaculans, S. quinivorans, and S. entomophila [1]. All
species except S. entomophila have been frequently iso-
lated from clinical samples, and S. marcescens in partic-
ular is recognized as an important nosocomial pathogencapable of causing pneumonia, intravenous catheter-
associated infections, urinary tract infections, osteomy-
elitis and endocarditis [2]. However, the recently
described virulence-associated properties in Serratia
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.016
* Correspondent author. Tel.: +32 16 321752; fax: +32 16 321960.
E-mail address: [email protected] (R. Van Houdt).
strains other than S. marcescens, and the increasing
number of documented infections caused by such
strains, together with the difficult identification of thesebacteria by commercial systems urges for a more de-
tailed investigation of the physiology, virulence and tax-
onomy of this genus [3]. As ubiquitous inhabitants of
soil, air and water, Serratia species are commonly asso-
ciated with food raw materials and are implicated in the
spoilage of various foods of plant and animal origin. In
addition, as opportunistic pathogens, they may pose a
foodborne health hazard. We have recently conductedan investigation on the biofilm-forming capacity and
the production of quorum-sensing signalling molecules
in Gram-negative bacteria isolated from a raw vegetable
processing line [4]. Five out of 68 isolates produced N-
acyl-L-homoserine lactones (AHLs), and two of these,
one with strong and one with weak biofilm-forming
capacity, were tentatively identified as S. plymuthica
. Published by Elsevier B.V. All rights reserved.

266 R. Van Houdt et al. / FEMS Microbiology Letters 246 (2005) 265–272
using Biolog carbon utilization patterns. S. plymuthica
has been described as a non-motile, prodigiosin pig-
ment-producing Serratia and is regarded as a significant
pathogen [5] to which a variety of infections including
peritonitis, pneumonia, sepsis and wound infections
have been attributed [6–10]. The capacity to formbiofilms often contributes to pathogen virulence because
it provides protection against host defense and antibiotic
therapy, it allows cells to survive in hostile environments
and from there to disperse and colonize new niches, and
may facilitate the spread of antibiotic resistance by hor-
izontal gene transfer [reviewed in 11,12]. In the current
report, we describe the detailed identification and char-
acterization of the tentative S. plymuthica isolate withstrong biofilm-forming capacity, using a variety of geno-
typic and phenotypic criteria.
2. Materials and methods
2.1. Bacterial strains, plasmids, and media
Strains used in this study are listed in Table 1. All
Serratia species type strains were obtained from the
Deutsche Sammlung von Mikroorganismen und Zellk-
ulturen (DSM, Braunschweig, Germany), except S. liq-
uefaciens DSM 4487 (=LMG 7884), which was
obtained from the Belgian Co-ordinated Collections of
Micro-organisms (BCCMe/LMG). Escherichia coli
ESS [13] and Chromobacterium violaceum CV026 [14]were obtained from Dr. Susan E. Jensen (University of
Alberta) and Dr. Rene De Mot (Katholieke Universiteit
Leuven), respectively. All strains were routinely grown
in Luria–Bertani (LB) medium at 30 �C.
Table 1
Strains used in this study
Species Straina
Chromobacterium violaceum CV026 (cviI::mini-Tn5 derivative of A
Escherichia coli ESS
Plesiomonas shigelloides DSM 8224T = ATCC 14029T
Serratia entomophila DSM 12358T = ATCC 43705T
Serratia ficaria DSM 4569T = ATCC 33105T
Serratia fonticola DSM 4576T = ATCC 29844T
Serratia grimesii DSM 30063T = ATCC 14460T
Serratia liquefaciens DSM 4487T = ATCC 27592T
Serratia marcescens DSM 30121T = ATCC 13880T
Serratia odorifera DSM 4582T = ATCC 33077T
Serratia plymuthica DSM 4540T = ATCC 183T
Serratia proteamaculans DSM 4543T = ATCC 19323T
Serratia quinivoransb DSM 4597T = ATCC 33765T
Serratia rubidaea DSM 4480T = ATCC 27593T
Serratia sp. RVH1
TType strain.a DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen,
Manassas, VA, USA.b Originally classified as S. proteamaculans subsp. quinovora the transfer
proteamaculans to Serratia proteamaculans.
2.2. Phenotypic analysis
2.2.1. Swimming and swarming motility
Motility was tested by stab inoculating the strain to
be tested in both LB and minimal AB [15] medium solid-
ified with either 0.3% agar to examine swimmingthrough the water-filled channels in the agar, or 0.7%
agar to examine swarming over the agar surface [16].
2.2.2. Proteolytic activity
Production of extracellular proteolytic enzymes was
evaluated by observation of clearing zones around stab
inoculated bacteria on LB agar supplemented with 10%
skimmed milk after 24 h of incubation at 30 �C.
2.2.3. Production of antibacterial factors
Bacteria were checked for the production of antibac-
terial compounds active against various target strains by
scoring inhibition or lysis zones. Briefly, 100 ll of an
overnight LB broth culture of the target strain was
mixed with liquid 0.7% LB agar at 50 �C and poured
into a petri dish. After solidification, potential antibacte-rial producer strains were stab inoculated onto this
lawn, and plates were scored after overnight incubation
at 30 �C for the presence of inhibition or lysis zones.
2.2.4. Analysis of the N-acyl-L-homoserine lactone
production pattern
Analysis of the AHL production pattern was per-
formed by thin-layer chromatography (TLC) on C18 re-versed-phase plates (VWR International, Leuven,
Belgium) using a methanol/water (60:40 v/v) solvent sys-
tem essentially as described by Shaw et al. [17]. Briefly,
cell-free culture supernatants from 21 h LB broth
GenBank Accession No.
16S rDNA gyrB
TCC 31532, Kmr, AHL�)
M59159 AJ300545
AJ233427 AJ300543
AJ233428 AJ300541
AJ233429 AJ300539
AJ233430 AJ300538
AJ306725 AJ300537
AJ233431 AJ300536
AJ233432 AJ300533
AJ233433 AJ300532
AJ233434 AJ300531
AJ233435
AJ233436 AJ300530
AY394724 AY787168
Braunschweig, Germany; ATCC, American Type Culture Collection,
to Serratia quinivorans [30] reduces Serratia proteamaculans subsp.

R. Van Houdt et al. / FEMS Microbiology Letters 246 (2005) 265–272 267
stationary-phase cultures (500 ml) of the Serratia spp.
were extracted twice with the same volume of ethyl ace-
tate, dried over anhydrous MgSO4, evaporated to dry-
ness, and the residue was dissolved in a small volume
of ethyl acetate and loaded onto the TLC-plates. After
chromatographic separation, the presence of AHLswas detected by overlaying the dried TLC-plates with
a thin film of AHL sensor strain C. violaceum CV026
in 1.4% LB agar, and looking for the appearance of pur-
ple spots indicative of induction of violacein production
after incubation at 30 �C for 24 h.
2.3. 16S rDNA analysis
Analysis of 16S rDNA of S. plymuthica RVH1 was
performed by BCCMe/LMG (Gent, Belgium). Briefly,
genomic DNA was extracted following the protocol of
Pitcher et al. [18] and the part of the 16S rRNA gene
corresponding to positions 28–1521 of the E. coli 16S
rRNA gene was PCR amplified with the primers
16F27 (5 0-AGAGTTTGATCCTGGCTCAG-3 0) and
16R1522 (5 0-AAGGAGGTGATCCAGCCGCA-3 0).The PCR product was purified using the QIAquick
PCR Purification Kit (Qiagen GmbH, Hilden, Ger-
many) and sequenced using five forward primers and
three reverse primers annealing to universally conserved
regions, with the ABI PRISM TM BigDye TM Termi-
nator Cycle Sequencing Ready Reaction Kit (Perkin–El-
mer, Applied Biosystems Div., Foster City, CA, USA)
and an Applied Biosystems 377 DNA Sequencer (Ap-plied Biosystems, Foster City, CA, USA). The sequence
assembly was performed using the program AutoAssem-
bler (Perkin–Elmer).
2.4. DNA:DNA hybridizations
DNA:DNA hybridizations were performed by
BCCMe/LMG (Gent, Belgium). Briefly, DNA was pre-pared according to a slightly modified procedure of Wil-
son [19] and hybridizations were performed at 46 �Cusing the method described by Ezaki et al. [20] with
some modifications.
2.5. gyrB gene amplification and sequencing
The gyrB gene of S. plymuthica RVH1 was PCRamplified as described by Dauga [21]. Briefly, 50 pmol
of each primer gyr-320 (5 0-TAARTTYGAYGAYAA-
CTCYTAYAAAGT-3 0) and rgyr-1260 (5 0-CMCCYTC-
CACCARGTAMAGTTC-3 0) were used in a reaction
mixture (100 ll) containing 10 mM Tris–HCl (pH 8.3),
50 mM KCl, 2.5 mM MgCl2. PCR amplification was
carried out as follows: 94 �C for 4 min, followed by 35
cycles of 94 �C for 1 min, 55 �C for 1 min and 72 �Cfor 2 min, with a final incubation at 72 �C for 10 min.
The amplification product was purified using the High
Pure PCR Purification Kit (Roche Diagnostics, Vilvo-
orde, Belgium) and sequenced in both directions using
the same primers as used for amplification at a commer-
cial sequencing facility (MWG-Biotech AG, Ebersberg,
Germany).
2.6. Phylogenetic data analysis
Multiple-sequence alignments were performed using
the CLUSTAL W algorithm from the European Bio-
informatics Institute (EBI) toolbox (http://www.ebi.
ac.uk/clustalw/) and were further refined by eye, intro-
ducing gaps to improve overall alignment. Sequence
distance matrices were established in pairwise compar-isons by use of the Kimura algorithm [22]. Phyloge-
netic trees were constructed by the neighbour-joining
method [23] using the PHYLIP version 3.5 software
package [24]. Statistical significance was evaluated by
bootstrap analysis [25] with 100 repeats of bootstrap
samplings.
3. Results and discussion
3.1. Phenotyping of strain RVH1
In a screening of 68 biofilm-forming Gram-negative
bacteria from a raw vegetable processing line, one of
the strongest biofilm-forming isolates that also produced
different AHLs and AI-2 as quorum signalling com-pounds, designated RVH1, was a catalase negative and
oxidase positive rod-shaped organism (1.0 lm width;
1.2–1.5 lm length), and was tentatively identified as S.
plymuthica based on phenotypic analysis with the Biolog
GN2 Microplate System [4]. The results of additional
phenotypic analysis of this strain, in comparison to type
strains of the nine Serratia species most closely related
to S. plymuthica, are described below and summarizedin Table 2 and Fig. 1.
3.1.1. Proteolytic activity and swimming and swarming
motility
All bacteria, including RVH1, showed proteolytic
activity on skimmed milk plates except S. proteamacu-
lans and S. grimesii. Since all strains except S. fonticola
score positive in gelatin hydrolysis assays [26], it is pos-sible that the proteases produced by S. proteamaculans
and S. grimesii are not able to hydrolyse caseins.
All strains showed swimming motility, as expected
for the genus Serratia, but only S. ficaria showed
swarming motility on both LB and AB medium with
0.7% agar. The S. liquefaciens type strain showed no
swarming motility; contrary to S. liquefaciens strain
MG1, which we used as an internal control for ourswarming motility assay because it is a model organism
in many studies of swarming motility [16].

Table 2
Summary of phenotypic tests
Strain Pigment production Swimming motility Swarming motility Proteolytic activity ESSa RVH1b
S. entomophila � + � + � +
S. ficaria � + + + � +
S. fonticola � + � + � �S. grimesii � + � � � +
S. liquefaciens � + � + � �S. odorifera � + � + � +
S. plymuthica Red + � + � +
S. proteamaculans � + � � � +
S. quinivorans � + � + � �RVH1 � + � + + �a ESS: Production of antibacterial factor scored on E. coli ESS overlay plates.b RVH1: Production of antibacterial factor by RVH1 scored on overlay plates of listed strains.
Fig. 1. N-Acyl-L-homoserine production profile as indicated by biosensor strain Chromobacterium violaceum CV026. (a) S. ficaria DSM 4569; (b) S.
liquefaciens DSM 4487; (c) S. quinivorans DSM 4597; (d) strain RHV1; (e) S. plymuthica DSM 4540; (f) S. entomophila DSM 12358; (g) S. odorifera
DSM 4582; (h) S. proteamaculans DSM 4543; (i) S. grimesii DSM 30063; and (j) S. fonticola DSM 4576.
268 R. Van Houdt et al. / FEMS Microbiology Letters 246 (2005) 265–272
3.1.2. Production of antibacterial factor
Serratia strains have been reported to produce certaincompounds with antibacterial activity, such as the sim-
ple carbapenem, 1-carbapen-2-em-3-carboxylic acid,
identified in Serratia sp. strain ATCC 39006 [27]; Serra-
cin P, a phage-tail-like bacteriocin, produced by S. ply-
muthica J7 [28]; and bacteriocin 28b produced by most
S. marcescens biotypes [29]. Therefore, the production
and activity spectrum of possible antibacterial factors
produced by RVH1 and the type strains was analyzedby stab inoculating each strain onto a series of plates
each containing a lawn of one of the other Serratia
strains or of E. coli ESS, a b-lactam supersensitive strain
used in carbapenem production analysis. None of the
Serratia spp. type strains produced a halo in this test ex-
cept for S. proteamaculans, which caused a weak inhibi-
tion of S. grimesii. However, strain RVH1 caused
complete inhibition (clear halo) of S. entomophila, S. fi-caria, S. grimesii, S. odorifera, S. plymuthica, S. protea-
maculans, and E. coli ESS, but not of S. fonticola, S.
liquefaciens, and S. quinivorans. The activity spectrum
of the antibacterial factor produced by RVH1 differs
from Serracin P, which shows no activity towards the
E. coli strains tested [28]. Preliminary tests suggest that
the antibacterial activity can be ascribed to a protein,
but the presence of other compounds cannot be ex-cluded at this stage (data not shown). The antibacterial
spectra of the type strains in this study differ from thosereported by Ashelford et al. [30], possibly due to differ-
ences in growth temperature and other experimental
parameters.
3.1.3. N-Acyl-L-homoserine lactones
N-Acyl-L-homoserine lactone mediated quorum-sens-
ing is a widespread communication system in Gram-neg-
ative bacteria, in which small diffusible AHL signallingmolecules, synthesized by a LuxI homologue, interact
with a LuxR homologue and activate or repress the tar-
get genes when their concentration reaches a certain
threshold, related to population density [31]. Since quo-
rum sensing regulates a range of important biological
functions, such as antibiotic production, plasmid trans-
fer, motility, virulence and biofilm formation [reviewed
in 31], it is considered as a possible target for antibacte-rial treatment, and several studies have demonstrated
the feasibility of interfering with quorum sensing by
the use of specific antagonists of the signalling mole-
cules, an approach known as �quorum quenching�[32,33].
Different AHL production profiles and target genes
have been described in a number of Serratia spp.,
showing the specificity and diversity of quorum sensingsignal molecules and regulation in this genus. For
example, Serratia proteamaculans strain B5a produces

R. Van Houdt et al. / FEMS Microbiology Letters 246 (2005) 265–272 269
3-oxo-N-hexanoyl-L-homoserine lactone (3-oxo-C6-
HSL) and N-hexanoyl-L-homoserine lactone (C6-HSL)
[34], S. liquefaciens strain MG1 produces primarily N-
butanoyl-L-homoserine lactone (C4-HSL) and also C6-
HSL [35], while S. marcescens strain SS-1 produces at
least four AHLs, namely 3-oxo-C6-HSL, C6-HSL, N-heptanoyl-L-homoserine lactone (C7-HSL) and N-octa-
noyl-L-homoserine lactone (C8-HSL) [36]. Recently,
three AHLs produced by S. plymuthica IC1270 were
tentatively identified as 3-hydroxy-N-hexanoyl-L-homo-
serine lactone (3-hydroxy-C6-HSL), 3-hydroxy-N-octa-
noyl-L-homoserine lactone (3-hydroxy-C8-HSL) and
an unidentified compound by comigration with syn-
thetic compounds in thin layer chromatography [37].These AHL molecules are produced by the AHL syn-
thase from the substrates S-adenosyl-L-methionine
(SAM) and acylated acyl carrier protein (acyl-ACP)
[38] and can vary in acyl chain length (from C4 to
C14), oxidation at the C3 position and saturation
[39,40] due to the enzyme acyl chain specificity and the
available cellular pool of acyl-ACPs [39,41].
As additional phenotype, we examined the AHL pro-duction profile of strain RVH1 and the Serratia sp. type
strains by TLC analysis in combination with C. viola-
ceum CV026 biosensor overlay, and compared the
AHL profiles to those already described in Serratia
spp. In C. violaceum CV026, the proper production of
AHL molecules has been blocked by mutation of the
AHL synthase but the gene encoding the production
of the purple pigment violacein remains AHL-respon-sive. In the presence of specific AHLs with acyl chain
lengths shorter than C10, this strain will therefore pro-
duce purple pigment due to violacein production [14].
The results of the AHL profile analysis are shown in
Fig. 1, and reveal at least four different AHLs with a dif-
ferent TLC migration. The two farthest migrating spots
(spots 3 and 4 in Fig. 1) are the most common AHLs,
being present in S. ficaria, S. quinovorans, S. entomo-
phila, S. odorifera, S. proteamaculans, and RVH1. Since
another strain of S. proteamaculans (B5a) was previ-
ously reported to produce 3-oxo-C6-HSL and C6-HSL
[34], these two spots most likely correspond to these
two AHL molecules, although the existence of other
AHLs with the same migration cannot be excluded at
this stage. Strain RVH1 shows a third spot (spot 1 in
Fig. 1) which did not migrate from the point of applica-tion and which was not seen in any of the other Serratia
species. The slow migration could indicate the presence
of an AHL with a long-chain hydrophobic fatty acid res-
idue that binds strongly to the C-18 solid phase, but
such an AHL should not be able to elicit violacein pro-
duction. Even so, when a lot of material is loaded onto a
TLC-plate, molecules with shorter acyl chains some-
times get blocked, but on the other hand preliminarymass spectrometry analysis suggests indeed the presence
of 3-oxo-C12-HSL in the ethyl acetate extracts of RVH1
(data not shown). Finally, one TLC spot (spot 2 in Fig.
1) was observed only in S. ficaria and S. odorifera, but
its nature remains unknown. No AHLs capable to in-
duce violacein production were found for the S. grimesii,
S. plymuthica, and S. liquefaciens type strains, although
AHL production has been described for S. plymuthica
IC1270 [37] and S. liquefaciensMG1 [35], indicating that
the AHL synthases in S. plymuthica and S. liquefaciens
type strains are absent or mutated, resulting in the loss
of AHL production.
In spite of the tentative biochemical identification of
strain RVH1 as S. plymuthica, the difficulties in precise
phylogenetic positioning of Serratia strains combined
with the phenotypic differences between strain RVH1and the S. plymuthica type strain (see Table 2) motivated
us to perform a detailed phylogenetic study based on
16S rDNA and gyrB sequence comparison, and on
DNA:DNA hybridization.
3.2. 16S rRNA-based phylogeny
Part of the 16S rDNA gene sequence of strain RVH1was amplified and analysed (GenBank Accession No.
AY394724). Fig. 2(a) shows a neighbour-joining phylo-
genetic tree based on the alignment of the nearly com-
plete 16S rDNA gene sequence of strain RVH1 with
16S rDNA sequences of the 11 described Serratia type
strains available in GenBank and EMBL databases (see
Table 1 for corresponding accession numbers), and
rooted by using Plesiomonas shigelloides, which is themost closely related species to the Enterobacteriaceae
family [42]. The 16S rDNA sequence similarity between
strain RVH1 and the 11 described Serratia species ranged
between 99.3% and 96.3%, with the highest similarity to
S. plymuthica (99.3%) and S. ficaria (99.2%) and the low-
est to S. rubidaea (96.4%) and S. marcescens (96.3%).
Two separate clusters were obtained as described by
Sproer et al. [43]. One cluster comprised S. rubidaea, S.marcescens, S. odorifera, S. entemophila, and S. ficaria
and the second cluster comprised S. plymuthica, S. fonti-
cola, S. liquefaciens, S. quinivorans, S. grimesii, and S.
proteamaculans. The phylogenetic information obtained
from these sequences is poor due to the low rate of vari-
ation of 16S rDNA sequences. Therefore, we also deter-
mined a phylogeny based on the gyrB sequence.
3.3. gyrB-based phylogeny
Dauga [21] described the use of the gyrB sequence for
determining relationships among Serratia species. In
general, phylogenetic trees based on gyrB sequences ap-
pear to be more reliable for closely related bacterial spe-
cies than trees based on 16S rDNA. The gyrB nucleotide
sequence from strain RVH1 was determined from thePCR-amplified gyrB gene, revealing a 910 bp open read-
ing frame (GenBank Accession No. AY787168). The

Fig. 2. Neighbour-joining phylogenetic tree obtained from (a) 16S rRNA gene sequences, with the scale bar representing an estimated five base
substitutions per 1000 nt positions and (b) gyrB sequences, with the scale bar representing an estimated 25 substitutions per 1000 nt positions.
Numbers refer to significant bootstrap values of 100 calculated trees.
270 R. Van Houdt et al. / FEMS Microbiology Letters 246 (2005) 265–272
translated amino acid sequence had a lysine (K) at co-
don 206 (E. coli amino acid numbering system, Acces-
sion No. X04341) in a b-sheet-shaped region of the
ATP binding site, which is a Serratia signature sequence
[21]. The sequence similarity of strain RVH1 to the 10
Serratia species examined (the gyrB sequence of S. qui-
novorans was not available in a database) ranged be-
tween 98.9% and 86.5%, with the highest similarity toS. plymuthica (98.9%) and S. liquefaciens (94.2%) and
the lowest to S. rubidaea (87.8%) and S. fonticola
(86.5%). Fig. 2(b) represents a phylogenetic tree based
on the alignment of the gyrB gene sequence of strain
RVH1 and the Serratia sp. type strain gyrB sequences
available in GenBank and EMBL databases (see Table
1 for corresponding accession numbers) and rooted by
using P. shigelloides. Two phylogenetic clusters with sig-nificant bootstrap values were again found. The first
cluster (bootstrap value 93%) contained S. rubidaea, S.
marcescens, S. entomophila, and S. ficaria. The second
cluster (bootstrap value 99%) contained strain RVH1,
S. grimesii, S. proteamaculans, S. liquefaciens, and S.
plymuthica. Within this cluster strain RVH1 and S. ply-
muthica formed a coherent group validated by a signifi-
cant bootstrap value of 100%.
3.4. DNA:DNA hybridization
Bacterial strains are generally considered to belong tothe same species if they share a 16S rDNA sequence
identity of >97% and/or 70% or greater DNA–DNA
relatedness with 5 �C or less difference of melting tem-
perature (DTm), with the latter criterion being decisive
[44]. Therefore, to conclusively confirm the identity of
RVH1, we performed DNA:DNA hybridization be-
tween RVH1 and the two most closely related type
strains based on 16S rDNA sequence identity, i.e., S.plymuthica and S. ficaria. We found 100% DNA–DNA
hybridization with the S. plymuthica type strain and
46% with the S. ficaria type strain, confirming the iden-
tity of RVH1 as S. plymuthica.

R. Van Houdt et al. / FEMS Microbiology Letters 246 (2005) 265–272 271
4. Conclusions
We have performed a comparative characterization
of Serratia sp. RVH1, which was previously isolated
as a biofilm-forming strain from a raw vegetable pro-
cessing line [4], with the type strains of the 9 or 10 mostclosely related Serratia species. Phenotypically, the iso-
late could not be clearly assigned to any of the described
Serratia species, but 16S rRNA and gyrB sequence com-
parison and DNA:DNA hybridization unequivocally
identified the strain as S. plymuthica. Furthermore, these
observations add new phenotypic and genotypic infor-
mation to the Serratia genus, thus contributing to a
more precise phylogenetic positioning of Serratia
strains.
Acknowledgement
Rob Van Houdt is a research assistant of the Fund
for Scientific Research-Flanders (F.W.O.-Vlaanderen).
References
[1] Euzeby, J.P. (2004) List of bacterial names with standing in
nomenclature Available from: <http://www.bacterio.cict.fr> .
[2] Eisenstein, B.I. (1990) Enterobacteriaceae In: Principles and
Practice of Infectious Disease (Mandell, G.L., Douglas, R.G.
and Bennett, J.E., Eds.), 3rd edn, pp. 1658–1673. Churchill
Livingstone, New York.
[3] Stock, I., Burak, S., Sherwood, K.J., Gruger, T. and Wiedemann,
B. (2003) Natural antimicrobial susceptibilities of strains of
�unusual� Serratia species: S. ficaria, S. fonticola, S. odorifera, S.
plymuthica and S. rubidaea. J. Antimicrob. Chemother. 51, 865–
885.
[4] Van Houdt, R., Aertsen, A., Jansen, A., Quintana, A.L. and
Michiels, C.W. (2004) Biofilm formation and cell-to-cell signalling
in Gram-negative bacteria isolated from a food processing
environment. J. Appl. Microbiol. 96, 177–184.
[5] Berg, G. (2000) Diversity of antifungal and plant-associated
Serratia plymuthica strains. J. Appl. Microbiol. 88, 952–960.
[6] Carrero, P., Garrote, J.A., Pacheco, S., Garcia, A.I., Gil, R. and
Carbajosa, S.G. (1995) Report of six cases of human infection by
Serratia plymuthica. J. Clin. Microbiol. 35, 2531–2536.
[7] Clark, R.B. and Janda, J.M. (1985) Isolation of Serratia
plymuthica from human burn site. J. Clin. Microbiol. 21, 656–657.
[8] Domingo, D., Limia, A., Alarcon, T., Sanz, J.C., Del Rey, M.C.
and Lopez-Brea, M. (1994) Nosocomial septicemia caused by
Serratia plymuthica. J. Clin. Microbiol. 32, 575–577.
[9] Nough, F. and Bhandari, S. (2000) CAPD peritonitis due to
Serratia plymuthica. Periton. Dialysis Int. 20, 349.
[10] Reina, J., Borrell, N. and Llompart, I. (1992) Community-
acquired bacteremia caused by Serratia plymuthica: case report
and review of the literature. Diagn. Microbiol. Infect. Dis. 15,
449–452.
[11] Fux, C.A., Costerton, J.W., Stewart, P.S. and Stoodley, P. (2005)
Survival strategies of infectious biofilms. Trends Microbiol. 13,
34–40.
[12] Hall-Stoodley, L., Costerton, J.W. and Stoodley, P. (2004)
Bacterial biofilms: from the natural environment to infectious
diseases. Nat. Rev. Microbiol. 2, 95–108.
[13] Jensen, S.E., Elder, K.J., Aidoo, K.A. and Paradkar, A.S. (2000)
Enzymes catalyzing the early steps of clavulanic acid biosynthesis
are encoded by two sets of paralogous genes in Streptomyces
clavuligerus. Antimicrob. Agents Chemother. 44, 720–726.
[14] McClean, K.H., Winson, M.K., Fish, L., Taylor, A., Chhabra,
S.R., Camara, M., Daykin, M., Lamb, J.H., Swift, S., Bycroft,
B.W., Stewart, G.S.A.B. and Williams, P. (1997) Quorum sensing
and Chromobacterium violaceum: exploitation of violacein pro-
duction and inhibition for the detection of N-acylhomoserine
lactones. Microbiology 143, 3703–3711.
[15] Clark, J.D. and Maaloe, O. (1967) DNA replication and the
division cycle in Escherichia coli. J. Mol. Biol. 23, 99–112.
[16] Eberl, L., Molin, S. and Givskov, M. (1999) Surface motility of
Serratia liquefaciens MG1. J. Bacteriol. 181, 1703–1712.
[17] Shaw, P.D., Ping, G., Laly, S.L., Cha, C., Cronan, J.E., Rinehart,
K.L. and Farrand, S.K. (1997) Detecting and characterizing N-
acyl-homoserine lactone signal molecules by thin-layer chroma-
tography. Proc. Natl. Acad. Sci. USA 94, 6036–6041.
[18] Pitcher, D.G., Saunders, N.A. and Owen, R.J. (1989) Rapid
extraction of bacterial genomic DNA with guanidium thiocya-
nate. Lett. Appl. Microbiol. 8, 151–156.
[19] Wilson, K. (1987) Preparation of genomic DNA from bacteria In:
Current Protocols in Molecular Biology (Ausubel, F.M., Brent,
R.E., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A.
and Struhl, K., Eds.), pp. 2.4.1–2.4.5. Greene Publishing and
Wiley–Interscience, New York.
[20] Ezaki, T., Hashimoto, Y. and Yabuuchi, E. (1989) Fluorometric
deoxyribonucleic acid-deoxyribonucleic acid hybridization in
microdilution wells as an alternative to membrane filter hybrid-
ization in which radioisotopes are used to determine genetic
relatedness among bacterial strains. Int. J. Syst. Bacteriol. 39,
224–229.
[21] Dauga, C. (2002) Evolution of the gyrB gene and the molecular
phylogeny of Enterobacteriaceae: a model molecule for molecular
systematic studies. Int. J. Syst. Evol. Microbiol. 52, 531–547.
[22] Kimura, M. (1980) A simple method for estimating evolutionary
rates of base substitutions through comparative studies of
nucleotide sequences. J. Mol. Evol. 16, 111–120.
[23] Saitou, N. and Nei, M. (1987) The neighbor-joining method: a
new method for reconstructing phylogenetic trees. Mol. Biol.
Evol. 4, 406–425.
[24] Felsenstein, J. (1993). Phylogenies Inference Package (PHYLIP),
Version 3.53c. Department of Genetics, University of Washing-
ton, Seattle.
[25] Felsenstein, J. (1985) Confidence limits on phylogenies: an
approach using the bootstrap. Evolution 39, 783–791.
[26] Holt, J.G., Krieg, N.R., Sneath, P.H.A., Staley, J.T. and
Williams, S.T. (1994) Genus Serratia In: Bergey�s Manual of
Determinative Bacteriology (Holt, J.G., Krieg, N.R., Sneath,
J.T., Staley, J.T. and Williams, S.T., Eds.), 9th edn, p. 187.
Williams & Wilkins, Baltimore.
[27] Bycroft, B.W., Maslen, C., Box, S.J., Brown, A. and Tyler, J.W.
(1987) The isolation and characterisation of (3R,5R)- and
(3S,5R)-carbapenem-3-carboxylic acid from Serratia and Erwinia
species and their putative biosynthetic role. J. Chem. Soc., Chem.
Commun. 21, 1623–1625.
[28] Jabrane, A., Sabri, A., Compere, P., Jacques, P., Vandenberghe,
I., Van Beeumen, J. and Thonart, P. (2002) Characterization of
serracin P, a phage-tail-like bacteriocin, and its activity against
Erwinia amylovora, the fire blight pathogen. Appl. Environ.
Microbiol. 68, 5704–5710.
[29] Guasch, J.F., Enfedaque, J., Ferrer, S., Gargallo, D. and Regue,
M. (1995) Bacteriocin 28b, a chromosomally encoded bacteriocin
produced by most Serratia marcescens biotypes. Res. Microbiol.
146, 477–483.
[30] Ashelford, K.E., Fry, J.C., Bailey, M.J. and Day, M.J. (2002)
Characterization of Serratia isolates from soil, ecological impli-

272 R. Van Houdt et al. / FEMS Microbiology Letters 246 (2005) 265–272
cations and transfer of Serratia proteamaculans subsp. quinovora
Grimont et al. 1983 to Serratia quinivorans corrig., sp. nov. Int. J.
Syst. Evol. Microbiol. 52, 2281–2289.
[31] Whitehead, N.A., Barnard, A.M., Slater, H., Simpson, N.J. and
Salmond, G.P. (2001) Quorum-sensing in Gram-negative bacte-
ria. FEMS Microbiol. Rev. 25, 365–404.
[32] Williams, P. (2002) Quorum sensing: an emerging target for
antibacterial chemotherapy. Expert Opin. Ther. Targets 6, 1–18.
[33] Zhang, L.H. and Dong, Y.H. (2004) Quorum sensing and signal
interference: diverse implications. Mol. Microbiol. 53, 1563–1571.
[34] Christensen, A.B., Riedel, K., Eberl, L., Flodgaard, L.R., Molin,
S., Gram, L. and Givskov, M. (2003) Quorum-sensing-directed
protein expression in Serratia proteamaculans B5a. Microbiology
149, 471–483.
[35] Eberl, L., Winson, M.K., Sternberg, C., Stewart, G.S.A.B.,
Christiansen, G., Chhabra, S.R., Bycroft, B., Williams, P., Molin,
S. and Givskov, M. (1996) Involvement of N-acyl-L-homoserine
lactone autoinducers in controlling the multicellular behaviour of
Serratia liquefaciens. Mol. Microbiol. 20, 127–136.
[36] Horng, Y.T., Deng, S.C., Daykin, M., Soo, P.C., Wei, J.R., Luh,
K.T., Ho, S.W., Swift, S., Lai, H.C. and Williams, P. (2002) The
LuxR family protein SpnR functions as a negative regulator of N-
acylhomoserine lactone-dependent quorum sensing in Serratia
marcescens. Mol. Microbiol. 45, 1655–1671.
[37] Ovadis, M., Liu, X., Gavriel, S., Ismailov, Z., Chet, I. and
Chernin, L. (2004) The global regulator genes from biocontrol
strain Serratia plymuthica IC1270: cloning, sequencing, and
functional studies. J. Bacteriol. 186, 4986–4993.
[38] Parsek, M.R., Val, D.L., Hanzelka, B.L., Cronan, J.E.J. and
Greenberg, E.P. (1999) Acyl homoserine-lactone quorum-sensing
signal generation. Proc. Natl. Acad. Sci. USA 96, 4360–4365.
[39] Fuqua, C. and Eberhard, A. (1999) Signal generation in autoin-
duction systems: synthesis of acylated homoserine lactones by
LuxI-type proteins In: Cell–cell Communication in Bacteria
(Dunny, G. and Winans, S.C., Eds.), pp. 211–230. ASM Press,
Washington.
[40] Kuo, A., Blough, N.V. and Dunlap, P.V. (1994) Multiple N-acyl-
L-homoserine lactone autoinducers of luminescence in the marine
symbiotic bacterium Vibrio fischeri. J. Bacteriol. 176, 7558–7565.
[41] Fray, R.G., Throup, J.P., Daykin, M., Wallace, A., Williams, P.,
Stewart, G.S. and Grierson, D. (1999) Plants genetically modified
to produce N-acylhomoserine lactones communicate with bacte-
ria. Nat. Biotechnol. 171, 1017–1020.
[42] Brenner, D.J. (1984) Enterobacteriaceae, 2nd edn In: Bergey�sManual of Systematic Bacteriology (Krieg, N.R. and Holt, J.G.,
Eds.), Vol. 1, pp. 408–420. Williams & Wilkins, Baltimore.
[43] Sproer, C., Mendrock, U., Swiderski, J., Lang, E. and Stacke-
brandt, E. (1999) The phylogenetic position of Serratia, Butti-
auxella and other genera of the family Enterobacteriaceae. Int. J.
Syst. Bacteriol. 49, 1433–1438.
[44] Wayne, L.G., Brenner, D.J., Colwell, R.R., Grimont, P.A.D.,
Kandler, O., Krichevsky, M.I., Moore, L.H., Moore, W.E.C.,
Murray, R.G.E., Stackebrandt, E., Strarr, M. and Truper, H.G.
(1987) Report of the ad hoc committee on reconciliation of
approaches to bacterial systematics. Int. J. Syst. Bacteriol. 37,
463–464.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 273–278
Chemotypes significance of lichenized fungi bystructural characterization of heteropolysaccharides from the genera
Parmotrema and Rimelia
Elaine Rosechrer Carbonero a, Caroline Grassi Mellinger a, Sionara Eliasaro b,Philip Albert James Gorin a, Marcello Iacomini a,*
a Departamento de Bioquımica e Biologia Molecular, Universidade Federal do Parana, C.P. 19046, CEP 81531-990 Curitiba, PR, Brazilb Departamento de Botanica, Universidade Federal do Parana, C.P. 19031, CEP 81531-990 Curitiba, PR, Brazil
Received 2 September 2004; accepted 14 April 2005
First published online 27 April 2005
Edited by G.M. Gadd
Abstract
Galactoglucomannans were isolated from the lichenized fungi of the genus Parmotrema (Parmotrema austrosinense, Parmotrema
delicatulum, Parmotrema mantiqueirense, Parmotrema schindlerii, and Parmotrema tinctorum and that of Rimelia (Rimelia cetrata
and Rimelia reticulata) via successive hot alkaline extraction and precipitation with Fehling solution. The structure of each polysac-
charide was investigated using 13C NMR and HSQC-DEPT spectroscopy, methylation analysis, and HPSEC-MALLS. The galac-
toglucomannans had a (1 ! 6)-linked main chain of a-Manp units, substituted preferentially at O-2 and O-4 by a-Galp and b-Galp
nonreducing end-units, respectively. The C-1 region of the 13C NMR spectra of these heteropolysaccharides is typical of the lichen
species, and is an additional tool in lichenized fungi classification.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Lichenized fungi; Parmeliaceae; Galactoglucomannan; Chemical structure; 13C NMR
1. Introduction
The identification and classification of lichenized fun-
gi was originally carried out on the basis of morphology.
Since the 1860s, species differentiation was aided by the
specific color reactions of their components [1,2], pres-
ent at concentrations of 0.15% to 10%, or carotenoids
[3,4]. The chemical analysis of compounds for taxo-
nomic purposes was carried out by microcrystallization,chromatography, fluorescence and mass spectroscopy
analysis [5]. Recently, advances in DNA technology
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.019
* Corresponding author. Tel.: +55 41 361 1655; fax: +55 41 266 2042.
E-mail address: [email protected] (M. Iacomini).
and fine chemical characterization of macromoleculesserved as a useful tools in the classification of lichens.
The use of structurally different mannose-containing
polysaccharides for the classification and identification
of yeasts [6] led to the investigation of related polysac-
charides isolated from ascomycetous lichens via Fehling
precipitation. Their structure, as evidenced by chemical
and 13C NMR studies, proved to be typical of the parent
lichen and could thus be utilized in chemotyping studies[7–11].
In terms of macromolecules, the study of mannose-
containing polysaccharides as a taxonomic tool involves
the structural diversity of the galactomannans from
several lichenized fungi, and depends on their side-chain
. Published by Elsevier B.V. All rights reserved.

274 E.R. Carbonero et al. / FEMS Microbiology Letters 246 (2005) 273–278
substituents on (1 ! 6)-linked a-D-mannopyranosyl
main-chains [12]. These generally include monosubstitu-
ents at O-2 of a-D-Manp or a-D-Galp, at O-4 by b-Galp
and sometimes with disubstitution occurring at O-2 and
O-4 by a-D-Galp and b-D-Galp, or a-D-Manp and b-D-Galp, respectively, although some of the main-chainunits are frequently not substituted.
Studies involving the taxonomy of lichenized fungi
from Cladonia and Cladina species were carried out
since classical taxonomy considered Cladina to be a sub-
genus of Cladonia, but thereafter lichenologists decided
to be a distinct genus. Woranovicz-Barreira et al. [11]
showed that galactoglucomannans are chemotypes
which could be significant in aiding the taxonomy ofCladonia spp. and those of related genera. Ahti and
Depriest [13] proposed, based on molecular phyloge-
netic results, that Cladina becomes a synonym of Clado-
nia. Subsequently, Carbonero et al. [14] studied the
structures of polysaccharides of Cladina spp. and, based
on their chemical characterization and when compared
to those of Cladonia species, agreed with results ob-
tained with DNA studies.Another suitable example of conflicting taxonomic
data concerns the segregation of the genus Rimelia from
the earlier Parmotrema, which was proposed by Hale
and Fletcher [15]. Studies on the chemical elucidation
of polysaccharides of species from these two genera
are now reported as a taxonomic aid.
Aq. 2% KOH at 100ºC for 3 h (x3)
LICHENIZED FUNGUS Cleaned, dried and powdered
CHCl3: MeOH (2:1; v/v) at 60ºC, 3 h (x3)
MeOH: H2O (4:1; v/v) at 80ºC for 3 h (x3)
Lipid extractLichen residue I
Extract of low molecular mass
Alcaline extractLichen residue III
Lichen residue II
Treatment with Fehling solutionCentrifugation
Felhing supernatant Fehling precipitate
Freeze-Thawing Centrifugation
PrecipitateSupernatant
GALACTOGLUCOMANNAN
Fig. 1. Scheme of extraction and purification of the galactogluco-
mannans obtained from Parmotrema spp. and Rimelia spp.
2. Materials and methods
2.1. Lichenized fungi (family, Parmeliaceae)
Parmotrema austrosinense (Zahlbr.) Hale, Parmo-
trema delicatulum (Vain.) Hale, Parmotrema schindlerii
Hale, Parmotrema mantiqueirense Hale, Parmotrema
tinctorum (Nyl.) Hale, Rimelia cetrata (Ach.) Hale andFletcher and Rimelia reticulata (Taylor) Hale and
Fletcher were examined. Parmotrema spp. were col-
lected in 1996, in Lapa, State of Parana, Brazil, while
Rimelia spp. are from Curitiba, State of Parana, and
have their vouchers (no. 33886, 33354, 33890, 33355,
28838, 38057, 38118, respectively) deposited in the
UPCB (Herbarium name follows Holmgren et al. [16]).
2.2. Isolation and purification of polysaccharides
Lichenized fungus samples (P. austrosinense, 41 g;
P. delicatulum, 32 g; P. schindlerii, 35 g; P. mantiquei-
rense, 43 g; P. tinctorum, 60 g; R. cetrata, 31 g; and
R. reticulata, 26 g) were successively refluxed in
CHCl3-MeOH (2:1 v/v; 300 ml) and 80% aqueous
MeOH (300 ml), in order to extract low molecularcomponents. The residual material was then extracted
three times with 2% aq. KOH containing traces of
NaBH4 at 100 �C for 3 h. The alkaline extract was
neutralized with HOAc, dialyzed against tap water,
and after 48 h was freeze dried. The crude fraction
obtained from alkaline extraction was submitted to a
freeze-thawing process, which furnished insoluble and
soluble material, which were separated by centrifuga-tion (15 min, 9000 rpm, 25 �C). The soluble fraction
was submitted to a second purification process using
Fehling solution [17], resulting in a precipitate
(Cu2+-ppt) and a soluble fraction (Cu2+-sup) which
were separated by centrifugation under the above con-
ditions. Each fraction was neutralized with HOAc,
dialyzed against tap water and deionized with mixed
ion exchange resins.
2.3. Monosaccharide composition
Hydrolysis of the fractions were carried out with 1
M TFA at 100 �C for 8 h and the hydrolyzates then
evaporated to dryness, followed by successive reduc-
tion with NaBH4 and acetylation with Ac2O–pyridine
(1:1 v/v; 2 ml) at room temperature for 12 h [18,19].The resulting alditol acetates were analyzed by GC-
MS using a Varian model 3300 gas chromatograph
linked to a Finnigan Ion-Trap, model 810 R-12 mass
spectrometer, using a DB-225 capillary column (30
m · 0.25 mm i.d.), with helium as carrier gas. The
analysis was carried out from 50–220 �C at 40 �C/min maintaining the temperature constant to the end
of analysis (18 min). The products were identified bytheir typical retention times and electron impact
profiles.

Table 1
Yield of Fehling precipitates obtained from Parmotrema spp. and
Rimelia spp. and their monosaccharide composition
Lichenized fungus Yield (%)a Monosaccharide
composition (%)b
Man Gal Glc
Parmotrema austrosinense 6.7 50 44 5
P. delicatulum 3.8 49 44 6
P. mantiqueirense 5.6 50 43 6
P. schindlerii 3.2 50 43 7
P. tinctorum 5.4 51 42 6
Rimelia cetrata 3.4 53 40 7
R. reticulata 5.2 52 40 8
a Yields based on dry material.b Alditol acetates obtained on successive hydrolysis, NaBH4 reduc-
tion, and acetylation, analyzed by GC-MS (DB-225 column).
E.R. Carbonero et al. / FEMS Microbiology Letters 246 (2005) 273–278 275
2.4. Methylation analysis
Each sample (5 mg) was per-O-methylated according
to the method of Ciucanu and Kerek [20], using pow-
dered NaOH in Me2SO–MeI. The per-O-methylated
derivatives were hydrolyzed with 50% v/v sulfuric acid
(1 h, 0 �C), followed by dilution to 5.5% v/v (5 h, 100
�C), neutralization (BaCO3) and filtration [21]. Theresulting mixture of O-methylaldoses was reduced with
NaBH4 or NaBD4 and acetylated as cited above to give
Table 2
Partially O-methylated alditol acetates obtained from methylated galactoglu
O-Me-alditol acetatesa Molar % b,c
Pa Pd Pm
2,3,4,6-Me4Man 1.1 0.9 0.9
2,3,4,6-Me4Glc 1.6 1.4 3.1
2,3,5,6-Me4Gal – 0.6 0.5
2,3,4,6-Me4Gal 40.0 40.3 39.6
2,4,6-Me3Glc 3.3 3.6 2.5
2,4,6-Me3Man 0.4 – 0.4
2,4,6-Me3Gal 0.4 0.3 –
2,3,6-Me3Man 0.2 0.1 –
3,4,6-Me3Gal – – 1.7
2,3,4-Me3Man 22.0 21.4 15.7
2,3,4-Me3Gal 1.1 0.5 0.7
2,6-Me2Man 0.2 0.2 0.3
4,6-Me2Gal 0.6 0.2 0.3
3,6-Me2Gal 0.5 0.5 0.9
2,3-Me2Man 1.9 4.4 3.8
3,4-Me2Man 7.2 7.8 6.1
2,4-Me2Man 0.2 0.2 0.1
2,3-Me2Gal 0.4 0.1 0.2
2-MeMan 0.4 0.1 0.8
3-MeMan 18.1 16.4 19.3
Man 0.4 0.2 0.7
a O-Me-alditol acetates obtained by methylation analysis, followed by su
(column DB-225).b % of peak area relative to total peak area.c The symbols are: Pa, P. austrosinense; Pd, P. delicatulum; Pm, P. mant
reticulata).
a mixture of partially O-methylated alditol acetates,
which was analyzed by GC-MS. The analysis was car-
ried out from 50–215 �C at 40 �C/min maintaining the
temperature constant to the end analysis (31 min), and
the resulting partially O-methylated alditol acetates
identified by their typical electron impact breakdownprofiles and retention times [22,23].
2.5. Determination of homogeneity and molar mass
The elution profiles of fractions were determined by
high performance size-exclusion chromatography
(HPSEC), using a WATERS 510 HPLC pump at 0.6
ml/min with four gel permeation columns in series withexclusion sizes of 7 · 106, 4 · 105, 8 · 104, and 5 · 103
Da, using a refraction index (RI) detector. The eluent
was 0.1 mol/l aq. NaNO3 containing 200 ppm aq.
NaN3. Samples, previously filtered through a membrane
(0.22 lm; Millipore), were injected (250 ll loop) at 2 mg/
ml.
The specific refractive index increment (dn/dc) was
determined, with the samples being dissolved in 50mM NaNO3 and five increasing concentrations, rang-
ing from 0.2 to 1.0 mg/ml, were used to determine the
slope of the increment. Results were processed in
software provided by the manufacturer (Wyatt
Technologies).
comannans
Ps Pt Rc Rr
1.3 1.3 1.0 0.9
2.4 1.7 3.8 4.1
0.7 0.2 0.3 0.7
39.7 39.2 38.4 38.1
3.8 3.2 4.1 3.9
0.3 – – –
0.3 0.5 0.2 0.5
0.4 0.9 – –
– – 1.3 1.7
19.7 20.9 17.9 14.1
0.8 1.2 0.6 1.1
0.5 0.3 0.3 0.2
0.4 0.4 0.1 0.3
0.4 0.7 0.3 1.0
3.6 2.4 6.2 6.5
6.6 7.4 7.6 8.7
– 0.4 – –
– 0.4 0.3 0.4
0.3 0.3 0.2 0.3
18.4 18.3 17.1 16.8
0.4 0.3 0.3 0.7
ccessive hydrolysis, reduction and acetylation, analyzed by GC-MS
iqueirense; Ps, P. schindlerii; Pt, P. tinctorum; Rc, R. cetrata; Rr, R.

276 E.R. Carbonero et al. / FEMS Microbiology Letters 246 (2005) 273–278
2.6. Nuclear magnetic resonance spectroscopy
NMR spectra were obtained using a 400 MHz Bru-
ker model DRX Avance spectrometer with a 5 mm
inverse probe. 13C NMR (100.6 MHz) and HSQC-
1DEPT analyses were performed at 50 or 30 �C, withsamples being dissolved in D2O, the OH groups being
exchanged with D2O followed by freeze-drying. Chem-
ical shifts of samples are expressed in ppm (d) relativeto acetone at d 30.20 and 2.22 for 13C and 1H signals,
respectively.
Fig. 2. 13C NMR spectra of heteropolysaccharide from Parmotrema austrosi
tinctorum (e), Rimelia cetratum (f), and R. reticulata (g).
3. Results and discussion
Samples of seven species of lichenized fungi from
Parmotrema and Rimelia genus were submitted to puri-
fication procedures, according to Fig. 1, and after the
treatment with Fehling solution supernatant and precip-itated fractions were obtained. Table 1 shows all Fehling
precipitated fractions to contain mannose, galactose and
glucose as monosaccharide components. The average
obtained from all the fractions was 51% Man, 42%
Gal and 7% of Glc, and it is important to observe that
nense (a), P. delicatulum (b), P. mantiqueirense (c), P. schindlerii (d), P.

Fig. 3. HSQC-DEPT of heteropolysaccharide from Parmotrema
austrosinense in D2O at 30 �C (chemical shifts are expressed as d, ppm).
E.R. Carbonero et al. / FEMS Microbiology Letters 246 (2005) 273–278 277
no species showed a significant variation of monosac-
charide composition when compared to the average
data.
All Fehling precipitate fractions showed homoge-
neous elution profiles when analyzed by HPSEC-
MALLS, and as all the elution profiles were similar,the averaged specific refractive index increment was
dn/dc = 0.148. The samples had Mw of 53.7 kDa for P.
austrosinense, 68.2 kDa for P. delicatulum, 62.7 kDa
for P. schindlerii, 64.5 kDa for P. mantiqueirense, 39.4
kDa for P. tinctorum, 38.5 kDa for R. cetrata and 59.7
kDa for R. reticulata.
Methylation analysis of Fehling precipitate fractions
(Table 2) showed highly branched structures based onresulting partially O-methylated alditol acetates (GC-
MS) with high proportion of non-reducing ends of Galp,
besides small percentages of Manp, Galf and Glcp. They
also showed 2,3,4-Me3Man, 2,3-Me2Man, 3,4-Me2Man,
and 3-MeMan, corresponding to the main chains
formed by a-Manp-(1 ! 6) units, which were non-
substituted, substituted at O-2, O-4, and disubstituted
at O-2,4. Small amounts of Manp fully substituted unitswere also observed. Substitutions at O-2, O-3, and O-6;
disubstitutions at O-2,3; O-2,4; and O-4,6 were observed
for Galp units. Glcp was 3-O-substituted, besides its
nonreducing end-units.
The 13C NMR spectra of the galactoglucomannan
(Fig. 2) contained major signals in common, but there
were minor differences typical of the species. In gen-
eral, we have found that such 13C NMR spectra cor-respond to the lichen species [10,11,14,24], to the
extent that have been used for classification and iden-
tification [9–11].
The 13C NMR spectra of all species (Fig. 2), con-
tained C-1 signals that indicated predominant branched
structures with nonreducing end-units of b-D-Galp-
(1 ! 4)-a-D-Manp (d 104.6), a-D-Galp-(1 ! 2)-a-D-Manp (d 102.8) [12,25], along with 6-O-(d 101.6) and2,6-di-O-and 2,4,6-tri-O-substituted (d 99.8) units of a-D-Manp from the polysaccharide core [12,26]. The signal
at d 80.8 arose from 2-O-substituted a-D-Manp units
[27].
The HSQC spectrum of the P. austrosinense galacto-
glucomannan (Fig. 3) defined its a-and b-glycosidic con-figurations: the nonreducing end-units of Galp that had
a b-configuration by virtue of a high-field H-1 signal at4.42 (C-1 d 104.6), and an a-configuration due to a low
field H-1 signal at d 5.17 (102.8). The low-field H-1 sig-
nals in d 4.98 (101.6) and 5.23 (99.8) indicated that the
units of Manp had the a-configuration.Its HSQC-DEPT spectrum showed inverted signals in
d 67.9 and d 67.3 suggesting a substituted CH2 group,
probably from C-6 of a-Manp units, data in agreement
with the methylation analysis that gave mainly 3-Meand 2,3,4-Me3Man derivatives. The non substituted C-
6�s appeared at d 62.4 (3.92), d 62.7 (3.78) and d 62.9
(3.74) from the non reducing ends of Galp, Glcp, andManp units.
According to the present data, we can conclude that
the galactoglucomannans showed much similarity be-
tween the two distinct genera Parmotrema and Rimelia,
but with minor differences, typical of the species. The
galactoglucomannans have main chains of (1! 6)-
linked a-D-mannopyranosyl residues, that are mainly
unsubstituted and disubstituted at O-2 and O-4 witha-Galp and b-Galp side-chains, respectively. These re-
sults agree with previous data on other species of these
genera [9,27], and show the chemical method based on
polysaccharides to be useful as an additional tool in lich-
enized fungi classification.
Acknowledgements
The authors thank the Brazilian agencies, Coor-
denacao de Aperfeicoamento de Pessoal de Nıvel Supe-
rior (CAPES), Conselho Nacional de Desenvolvimento
Cientıfico e Tecnologico (CNPq), and Fundacao
Araucaria for financial assistance, and Dr. G. Torri,
from the Istituto di Ricerche Chimiche e Biochimiche
‘‘G. Ronzoni’’, Milan, Italy, for preparation of theHSQC-DEPT spectrum.
References
[1] Purvis, W. (2000) Lichens 122 p. Craft Print, Singapore.
[2] Aghoramurth, K., Sarma, K.G. and Seshadri, T.R. (1961)
Chemical investigation of Indian lichens. J. Sci. Ind. Res. 20,
166–168.

278 E.R. Carbonero et al. / FEMS Microbiology Letters 246 (2005) 273–278
[3] Czezuga, B. and Skult, H. (1988) Carotenoids in lichens of
Southern Finland. Ann. Bot. Fennici 25, 229–232.
[4] Czezuga, B. and Xavier Filho, L. (1987) Investigations on
carotenoids in lichens. VII. Some lichens from Brazil. Rev. Bras.
Biol. 47, 243–246.
[5] Honda, N.K. and Vilegas, W. (1988) A quımica dos liquens.
Quım. Nova 21 (6), 110–125.
[6] Gorin, P.A.J. and Spencer, J.F.T. (1970) Proton magnetic
resonance spectroscopy: an aid in identification and chemotax-
onomy of yeasts. Adv. Appl. Microbiol. 13, 25–89.
[7] Gorin, P.A.J., Baron, M. and Iacomini, M. (1988) Storage
products of lichens In: CRC Handbook of Lichenology
(Galun, M., Ed.), vol. III, pp. 9–23. CRC Press, Boca Raton,
FL.
[8] Gorin, P.A.J., Baron, M., da Silva, M.L.C., Teixeira, A.Z.A. and
Iacomini, M. (1993) Lichen carbohydrates. Ciencia e Cultura
(Braz.) 45, 27–36.
[9] Teixeira, A.Z.A., Iacomini, M. and Gorin, P.A.J. (1995) Chem-
otypes of mannose-containing polysaccharides of lichens myco-
bionts: a possible aid in classification and identification.
Carbohydr. Res. 266, 309–314.
[10] Woranovicz, S.M., Pinto, B.M., Gorin, P.A.J. and Iacomini, M.
(1999) Novel structures in galactoglucomannans of the lichens
Cladonia substellata and Cladonia ibitipocae: significance as
chemotypes. Phytochemistry 51, 395–402.
[11] Woranovicz-Barreira, S.M., Gorin, P.A.J., Sassaki, P.L., Mar-
celli, M.P. and Iacomini, M. (1999) Galactomannoglucans of
lichenized fungi of Cladonia spp.: significance as chemotypes.
FEMS Microbiol. Lett. 52, 313–317.
[12] Gorin, P.A.J. and Iacomini, M. (1985) Structural diversity
of D-galacto–D-mannan components isolated from lichens
having ascomycetous mycosymbionts. Carbohydr. Res. 142,
253–267.
[13] Ahti, T. and Depriest, P.T. (2001) New combination of Cladina
epithets in Cladonia (Ascomycotina, Cladoniaceae). Mycotaxon
78, 499–502.
[14] Carbonero, E.R., Montai, A.V., Woranovicz-Barreira, S.M.,
Gorin, P.A.J. and Iacomini, M. (2002). Phytochemistry 61, 681–
686.
[15] Hale, M.E. and Fletcher, A. (1990) Rimelia Hale & Fletcher, a
new lichen genus (Ascomycotina: Parmeliaceae). Bryologist 93,
23–29.
[16] Holmgren, P.K., Holmgren, N.H. and Barnett, L.C. (1990) Index
Herbariorum. 8 th ed. Part I: The herbaria of the world. Regnum
Veg. 120, 1–693.
[17] Jones, J.K.N. and Stoodley, R.J. (1965) Fractionation using
copper complexes. Meth. Carbohydr. Chem. 5, 36–38.
[18] Wolfrom, M.L. and Thompson, A. (1963) Reduction with sodium
borohydride. Meth. Carbohydr. Chem. 2, 65–67.
[19] Wolfrom, M.L. and Thompson, A. (1963) Acetylation. Meth.
Carbohydr. Chem. 2, 211–215.
[20] Ciucanu, I. and Kerek, F. (1984) A simple and rapid method for
the permethylation of carbohydrates. Carbohydr. Res. 131, 209–
217.
[21] Saeman, J.F., Moore, W.E., Mitchell, R.L. and Millet, M.A.
(1954) Techniques for the determination of pulp constituents by
quantitative paper chromatography. Tech. Assoc. Pulp Pap. Ind.
37, 336–343.
[22] Jansson, P., Kennec, L., Liedgren, H., Lindberg, B. and Lonn-
gren, J. (1976) A pratical guide to the methylation analysis of
carbohydrates. Chem. Commun. (Univ. of Stockholm) 48, 1–70.
[23] Carpita, N.C. and Shea, E. (1989) Linkage structure of carbohy-
drates by gas chromatography-mass spectrometry (GC-MS) of
partially methylated alditol acetates In: Analysis of Carbohy-
drates by GLC and MS (Biermann, C.J. and McGinnis, G.D.,
Eds.), pp. 157–216. CRC Press, Boca Raton, FL.
[24] Woranovicz, S.M., Gorin, P.A.J., Marcelli, M., Torri, G. and
Iacomini, M. (1997) Structural studies on the galactomannans of
lichens of the genus Cladonia. Lichenologist 29, 471–481.
[25] Gorin, P.A.J. and Iacomini, M. (1984) Polysaccharides of the
lichens Cetraria islandica and Ramalina usnea. Carbohydr. Res.
128, 129–132.
[26] Gorin, P.A.J. (1973) Rationalization of carbon-13 magnetic
resonance spectra of yeast mannans and structurally related
oligosaccharides. Can. J. Chem. 51, 2375–2383.
[27] Corradi da Silva, M.L., Gorin, P.A.J. and Iacomini, M. (1993)
Unusual carbohydrates from the lichen, Parmotrema cetratum.
Phytochemistry 34 (3), 715–717.

www.fems-microbiology.org
FEMS Microbiology Letters 246 (2005) 279–284
Isolation of genes differentially expressed during the fruitbody development of Pleurotus ostreatus by differential display
of RAPD
Masahide Sunagawa *, Yumi Magae
Department of Applied Microbiology, Forestry and Forest Products Research Institute, Tsukuba, Ibaraki 305-8687, Japan
Received 8 December 2004; received in revised form 12 April 2005; accepted 14 April 2005
First published online 27 April 2005
Edited by G.M. Gadd
Abstract
To analyze genes involved in fruit body development of Pleurotus ostreatus, mRNAs from three different developmental stages:
i.e., vegetative mycelium, primordium, and mature fruit body, were isolated and reverse-transcribed to cDNAs. One hundred and
twenty random PCR amplifications were performed with the cDNAs, which generated 382, 394, 393 cDNA fragments from each
developmental stage. From these fragments, four cDNA clones specifically expressed in primordium or mature fruit body were
detected. Sequence analysis and database searches revealed significant similarity with triacylglycerol lipase, cytochrome P450 sterol
14 a-demethylase and developmentally regulated genes of other fungi. Northern blot analyses confirmed that all of the four cDNAs
were unexpressed in mycelium, thus stage-specific genes for fruit body formation of P. ostreatus were successfully isolated.
� 2005 Federation of European Microbiological Societies. Published by Elsevier B.V. All rights reserved.
Keywords: Differential display; Pleurotus ostreatus; RAPD
1. Introduction
Fruit body morphogenesis is an important subject in
both basic and applied fields of mycological research.
The shift from vegetative growth to fruit body develop-
ment is a very interesting biological phenomenon for ba-
sic research; moreover, understanding the mechanism offruit body development will contribute to the advance-
ment of commercial mushroom production. Several
genes related to fruit body development have been iden-
tified in Coprinus cinereus [1,2], Schizophyllum commune
[3,4], Tuber borchii [5,6], Agaricus bisporus [7], Agrocybe
0378-1097/$22.00 � 2005 Federation of European Microbiological Societies
doi:10.1016/j.femsle.2005.04.018
* Corresponding author. Tel.: +81 298 733211; fax: +81 298 743720.
E-mail address: [email protected] (M. Sunagawa).
aegerita [8], Lentinula edodes [9–13], and Flammulina
velutipes [14]. In Pleurotus ostreatus, Lee et al. [15] ana-
lyzed expressed sequence tags (ESTs) of cDNAs library
derived from liquid-culture mycelia and fruit bodies of
P. ostreatus.
The method of differential mRNA display (DD) [16]
has mostly been used to study differential gene expres-sions in plants [17–19]. In fungi, Leung et al. [11], have
used this method to identify differentially expressed
genes in RNA populations of four developmental stages
of L. edodes: vegetative mycelium, primordium, young
fruit body and mature fruit body. Also, DD has been
used to identify putative genes involved in the develop-
ment of fruit bodies of T. borchii [6]. In the present
study, genes expressed during fruit-body developmentof P. ostreatus were examined by DD.
. Published by Elsevier B.V. All rights reserved.

280 M. Sunagawa, Y. Magae / FEMS Microbiology Letters 246 (2005) 279–284
2. Materials and methods
2.1. Strain and culture conditions
A dikaryotic strain, P. ostreatus ASI2029, was pro-
vided by Dr. Beom-Gi Kim (National Institute of Agri-culture Science and Technology, Korea). ASI2029 was
cultivated in a sawdust-medium containing beech saw-
dust and rice bran 3:1 (v/v). The sawdust-medium was
adjusted to a hydrous rate of 65% with tap water and
packed into a culture bottle (850 ml). After autoclaving,
5 ml of liquid inoculum was inoculated. The cultures
were grown at 20 �C for 30 days, and then the tempera-
ture was lowered to 15 �C to induce fruit body develop-ment. During the cultivation of ASI2029, samples from
three stages of development, i.e., mycelium, primordium
(3–7 mm in diameter), and mature fruit body (Fig. 1)
were collected. These samples were immediately frozen
in liquid nitrogen and stored at �80 �C until use.
2.2. RNA preparation
Total RNAs were isolated using a Qiagen RNA Prep-
aration Kit (Qiagen K.K., Tokyo, Japan). Poly(A)+–
RNA was prepared by Oligotex-dT30 super (Takara
Bio Co., Shiga, Japan). Both procedures were carried
out according to the manufacturer�s instructions.
2.3. Differential display of mRNA
Poly(A)+–RNA (0.5 lg) was heated at 65 �C for 10
min and immediately chilled on ice. First-strand cDNA
synthesis was performed in a reaction mixture contain-
ing 50 mM Tris–HCl (pH 8.5), 40 mM KCl, 5 mM
MgCl2, 2 mM DTT, 850 lM each dNTP, 95 units of
RNAase Inhibitor (Takara Bio Co.), 0.2 mM random
primer (Takara Bio Co.), and 40 units of Superscript
II Reverse Transcriptase (Invitrogen, Tokyo, Japan).The reaction was carried out for 1 h at 42 �C. After
heat-denatureation of the enzyme at 95 �C for 5 min,
the random primers were removed by ultrafiltration with
Super-02 (Takara Bio Co.). 10-mer RAPD primers (Op-
eron Technologies, Inc., Alameda, CA) were used to
Fig. 1. Samples used for the RNA preparation. A: myc
PCR amplify the second-strand cDNA. PCR was car-
ried out as 45 cycles of the following thermal cycle: 30
s at 95 �C, 1 min at 50 �C, and 2 min at 72 �C [20].
2.4. Cloning and sequence of specific cDNA fragments
Specific cDNA fragments found in RAPD were cut
out from the agarose gel and purified with Gel purifica-
tion column (Nippon Bio-Rad Lab., Tokyo, Japan).
The purified DNA was cloned into vector pCR 2.1 with
the TA cloning System Kit (Invitrogen). DNA sequenc-
ing of the clones were performed by Dynamic ET Ter-
minater Sequencing Kit (Amersham Biosciences K.K.,
Tokyo, Japan) and Mega Base 1000 Sequencer (Amer-sham Biosciences K.K.). Homology search was done
using BlastX program [21] for the translated protein
and EST sequences in the NCBI data bank.
2.5. Northern blotting analyses
Total RNA (20 lg) of each developmental stage used
for the isolation of mRNA was fractionated in a 1.0%agarose gel containing formaldehyde, and transferred
to Hybond-N (Amersham Bioscience K.K.). Northern
blots were hybridized with DIG (Roche Diagnostics
K.K., Tokyo, Japan) labeled cDNA at 42 �C according
to the manufacture�s instructions. As a control, North-
ern blots were probed with 18S rDNA fragment of the
P. ostreatus (ASI2029). The 18S rDNA was isolated as
described by White et al. [22].
2.6. Nucleotide sequence accession numbers
All the clones have been deposited with the DDBJ
data banks under the Accession No. AB19629,
AB196292, AB196293 and AB196294.
3. Results and discussion
In the present study, four genes specifically expressed
during the fruit body development of P. ostreatus were
isolated by means of differential displays of randomly
elium (My), B: primordium (P), C: fruit body (F).

M. Sunagawa, Y. Magae / FEMS Microbiology Letters 246 (2005) 279–284 281
amplified cDNA. To detect changes in transcripts dur-
ing fruit body development of P. ostreatus, mRNAs
were isolated from three stages of development: myce-
lium, primordium, and mature fruit body (Fig. 1). Then,
reverse-transcribed cDNAs were used as templates for
the following PCR. A total of 120 PCR amplificationswere performed with 10-mer RAPD primers. Each
PCR product separated by the agarose gel was resolved
into 1–9 distinct DNA bands in agarose gel. A total of
382, 394, and 393 cDNA fragments were identified in
the mycelium, primordium, and mature fruit body,
respectively. The electrophoresis patterns of the PCR-
amplified cDNA were confirmed as reproducible in three
independent experiments.Of the 120 random primers tested, three primers gen-
erated cDNA fragments analyzed in the present study
(Fig. 2). Ninety-five PCR amplifications (79%) gave
identical cDNA patterns between the three developmen-
tal stages. In 16 PCR, the same cDNA patterns were ob-
tained with primordium and fruit body. In one case,
specific cDNA was detected only in the mycelium stage.
All of the differentially expressed cDNA fragments wererecovered from the agarose gel and cloned into the vec-
tor pCR2.1. As a result, two fruit body-specific and the
Fig. 2. RAPD patterns of cDNA generated from mRNA of each
developmental stage. cDNA indicated by arrowheads were isolated,
cloned and analyzed in this study. M: molecular standard k/HindIII,
My: mycelium, P: primordium, F: fruit body.
Table 1
Characterization of differentially expressed cDNA clones of P. ostreatus
Levels Accession No. Homology
Clone Size
A8-U 777 AB196291 Cytochrome P450 sterol 14 a-demethyla
A8-D 574 AB196294 cDNA clone expressed during carbon st
T19-4 506 AB196294 Triacylglycerol lipase (E.C.3.1.1.3) [Cand
W3-7 415 AB196293 Early developmental cDNA clone GM57
two primordium-specific cDNA fragments were success-
fully cloned. Two specifically expressed cDNA were
detected in mature fruit body using primer A8
(5 0-GTGACGTAGG-3 0) (Fig. 2). The sizes of the frag-
ments were 777 and 574 bps, and were designated as
A8-U and A8-D, respectively. In the cases of primerT19 (5 0-GTCCGTATGG-3 0) and W3 (5 0-GTCC-
GGAGTG-3 0), two differentially amplified cDNA
fragments, 506 and 415 bps, were identified in the pri-
mordium (Fig. 2). They were designated as T19-4 and
W3-7, respectively. The cDNA clones were subjected
to sequence analysis.
Homology of the deduced amino acid sequences of
A8-U, A8-D, T19-4, and W3-7 with the database wassearched using the BlastX program. When no significant
homology was found with the protein databases, the
homology search was performed with EST databases
with the tBlastX program. The results are summarized
in Table 1. Predicted protein of T19-4 (+2 frame)
showed significant homology with triacylglycerol lipase
and contained a conserved domain of esterase-lipase.
The highest homology was found with triacylglycerol
E-value mRNA
My P F
se [Aspergillus fumigatus, AAF32372] 0.001 � � +
arvation [Trichoderma reesei, CF869295] 2e � 05 � � +
ida rugosa, 1LPP] 9e � 26 � + +
8 [Glomus mosseae, AJ315727] 0.008 � + +
Fig. 3. Northern analysis of four cDNA that are differentially
expressed during fruit body development of P. ostreatus. My:
mycelium, P: primordium, F: fruit body. Total RNA isolated from
each sample was hybridized with DIG-labeled cDNA clone and as a
control by DIG-labeled 18S rDNA. EtBr-stained ribosomal RNA
bands are loaded as quantitative control.

Fig. 4. Alignment of lipase sequences. The deduced amino acid sequence of T19-4 and three fungal lipase sequences were compared using Clustal W.
Residues, which are identical to P. ostreatus T19-4 are marked by asterisks. Underline shows the conserved domain of esterase and lipase. ILPP:
Triacyl glycerol Lipase (E.C.3.1.1.3) of Candida rugosa (GI:1064964), 1THG: Lipase of Galactomyces geotrichum (GI:443280), NCHP: Hypothetical
protein of Neurospora crassa (XM_322879).
282 M. Sunagawa, Y. Magae / FEMS Microbiology Letters 246 (2005) 279–284
lipase of Candida rugosa (9e � 26). The multiple align-
ment of the deduced T19-4 polypeptide with putative
amino acid sequences of lipase from other fungi is
shown in Fig. 4. Triacyl glycerol (TAG) is the predom-
inant acyl lipid in cultures undergoing sexual develop-
ment of Neurospora crassa [23]. In addition, as
described withMagnaporthe grisea, triacylglycerol lipase
activity increased during appressorium maturation [24].Mass transfer of storage lipid reserve to the aspersorium
occurred under the control of the MAP kinase and tur-
gor generation proceeded under the control of protein
kinase A [24]. Since TAG is known as an energy dense
substance [25], it is plausible that TAG is used as an en-
ergy source for the rapid development of P. ostreatus
fruit body while triacylglycerol lipase plays a role in lipid
degradation.As with A8-U, similarity (51%) was found with the
cytochrome P450 CYP51 (sterol 14 a-demethylase) of
Aspergillus fumigatus. Gene of cytochrome P450 has
been identified as involved in fruit body development
of A. bisporus [7], C. cinereus [26] and L. edodes [9],
but this is the first case of P450 CYP51, which is an
essential enzyme required in sterol biosynthesis and pri-
mary target of azole antimycotic drugs, isolated as agene related to fruit body development of mushroom.
The multiple alignment of the deduced A8-U polypep-
tide with putative amino acid sequences of CYP51 from
A. fumigatus and Penicillium italicum is shown in Fig. 5.
Fig. 5. The deduced amino acid sequence of A8-U was aligned with cytochro
and Penicillium italicum (GI:836642). Residues, which are identical to P. ost
No highly significant homology was found between
the function-known genes and the deduced polypep-
tide of A8-D and W3-7. But when they were com-
pared with genes deposited in the EST database,
high similarity was found with developmentally regu-
lated cDNA of other fungi. W3-7 may encode a pro-
tein with 84% similarity to the early developmental
gene of arbuscular mycorrhizal fungus Glomus mos-
seae (Table 1). On the other hand, the deduced poly-
peptide of A8-D showed significant homology with the
predicted protein of Trichoderma reesei cDNA clone
(2e � 05), expressed during carbon starvation. Interest-
ingly, A8-D codes a common gene involved in the
development of T. reesei and P. ostreatus although
its function is yet unknown.
Northern analysis showed that T19-4 and W3-7that were detected in primordium by the RAPD anal-
ysis, hybridized to total RNA in both primordium and
mature fruit body. The fact that T19-4 and W3-7
hybridized to the RNA of both primordium and ma-
ture fruit body shows that these mRNAs encode pro-
teins that play specific roles during both stages of fruit
body development. One possible reason for why T19-4
and W3-7 were undetected in the RAPD of fruit bodystage is that most of the RAPD primers were con-
sumed for amplification of more highly expressed
cDNA. In contrast, A8-U and A8-D hybridized only
to the total RNA of mature fruit body indicating that
me P450 sterol 14 a-demethylase of Aspergillus fumigatus (GI:6942241)
reatus are marked by asterisks.

M. Sunagawa, Y. Magae / FEMS Microbiology Letters 246 (2005) 279–284 283
they are specific genes in the latter stage of the devel-
opment. None of the four cDNA isolated in the pres-
ent study, hybridized to the RNA derived from
mycelium. The control probe, 18S rRNA gene of the
P. ostreatus, hybridized to RNA of all the stages
(Fig. 3).Liang and Pardee [16] reported about differential dis-
plays of mRNA using 3 0-anchored oligo-dT for the first-
strand cDNA synthesis and 10-mer arbitrary primers for
the second-strand synthesis by PCR. In their study,
numerous amplified fragments were visible after autora-
diography of labeled PCR products and the subsequent
isolation of the specific DNA band was rather difficult.
Leung et al. [11] and Zeppa et al. [6] also used 3 0-an-chored oligo-dT and various random primers for the
isolation of differentially expressed gene fragments.
The difference between the previous DD studies and
ours is that we did not use 3 0-anchored oligo-dT. The
number of cDNA fragments obtained in one PCR reac-
tion was not too large and each cDNA could be resolved
as a separate band in agarose gel.
With the basidiomycetes, not many genes related tofruit body development have been isolated but they of-
ten share common genes. For instance, Hydrophobin
is one of the most abundant genes in fruit bodies of
basidiomycetes, as reported by Penas et al. [27] and
Asgeirsdottir et al. [28] and has been isolated as fruiting
related gene with A. bisporus [7], F. velutipes [14] and L.
edodes [13]. But we did not detect hydrophobin as a spe-
cific gene for fruit body development of P. ostreatus inthe present study. ATPase has been detected in L. edodes
[11] and T. borchii [6]. PriA was detected in L. edodes
[10], Agrocybe aegerita [8] and EST of P. ostreatus
[15]. In addition, Septin has been isolated as a gene re-
lated to fruit body development [15,29,6]. However,
none of these genes were detected in this study. Presum-
ably because the method used in the previous studies
screen the elevated expression of genes, abundant genesinstead of unique genes tended to be isolated. But by
comparing RAPD patterns of ca. 1200 cDNA frag-
ments, abundant but not stage specific gene was easily
eliminated. All four cDNAs isolated in this study were
novel fruiting genes for basidiomycetes. The Northern
analysis confirmed that they were not expressed during
the mycelium stage, but expressed after the primordium
development. In conclusion, the DD technique was asimple and efficient method to detect fruit body stage-
specific cDNA of P. ostreatus.
Acknowledgements
We are grateful to Dr. Beom-Gi Kim of the National
Institute of Agriculture Science and Technology for pro-
viding the Pleurotus ostreatus strain (ASI2029).
References
[1] Boulianne, R.P., Liu, Y., Aebi, M., Lu, B.C. and Kues, U. (2000)
Fruiting body development in Coprinus cinereus: regulated
expression of two galectins secreted by a non-classical pathway.
Microbiology 146, 1841–1853.
[2] Kamada, T. (2002) Molecular genetics of sexual development in
the mushroom Coprinus cinereus. Bioessays 24, 449–459.
[3] Horton, J.S., Palmer, G.E. and Smith, W.J. (1999) Regulation of
dikaryon-expressed genes by FRT1 in the basidiomycete Schizo-
phyllum commune. Fungal Genet. Biol. 26, 33–47.
[4] Schuren, F.H., Asgeirsdottir, S.A., Kothe, E.M., Scheer, J.M.
and Wessels, J.G. (1993) The Sc7/Sc14 gene family of Schizo-
phyllum commune codes for extracellular proteins specifically
expressed during fruit-body formation. J. Gen. Microbiol. 139,
2083–2090.
[5] Lacourt, I., Duplessis, S., Abba, S., Bonfante, P. and Martin, F.
(2002) Isolation and characterization of differentially expressed
genes in the mycelium and fruit body of Tuber borchii. Appl.
Environ. Microbiol. 68, 4574–4582.
[6] Zeppa, S., Guidi, C., Zambonelli, A., Potenza, L., Vallorani, L.,
Pierleoni, R., Sacconi, C. and Stocchi, V. (2002) Identification of
putative genes involved in the development of Tuber borchii fruit
body by mRNA differential display in agarose gel. Curr. Genet.
42, 161–168.
[7] De Groot, P.W., Schaap, P.J., Van Griensven, L.J. and Visser, J.
(1997) Isolation of developmentally regulated genes from the
edible mushroom Agaricus bisporus. Microbiology 143, 1993–
2001.
[8] Sirand-Pugnet, P. and Labarere, J. (2002) Molecular character-
ization of the Pri3 gene encoding a cysteine-rich protein,
specifically expressed during fruiting initiation within the Agro-
cybe aegerita complex. Curr. Genet. 41, 31–42.
[9] Hirano, T., Sato, T. and Enei, H. (2004) Isolation of genes
specifically expressed in the fruit body of the edible basidio-
mycete Lentinula edodes. Biosci. Biotechnol. Biochem. 68, 468–
472.
[10] Kajiwara, S., Yamaoka, K., Hori, K., Miyazawa, H., Saito, T.,
Kanno, T. and Shishido, K. (1992) Isolation and sequence of a
developmentally regulated putative novel gene, priA, from the
basidiomycete Lentinus edodes. Gene 114, 173–178.
[11] Leung, G.S., Zhang, M., Xie, W.J. and Kwan, H.S. (2000)
Identification by RNA fingerprinting of genes differentially
expressed during the development of the basidiomycete Lentinula
edodes. Mol. Gen. Genet. 262, 977–990.
[12] Nishizawa, H., Miyazaki, Y., Kaneko, S. and Shishido, K. (2002)
Distribution of hydrophobin 1 gene transcript in developing
fruiting bodies of Lentinula edodes. Biosci. Biotechnol. Biochem.
66, 1951–1954.
[13] Ng, W.L., Ng, T.P. and Kwan, H.S. (2000) Cloning and
characterization of two hydrophobin genes differentially
expressed during fruit body development in Lentinula edodes.
FEMS Microbiol. Lett. 185, 139–145.
[14] Ando, A., Harada, A., Miura, K. and Tamai, Y. (2001) A gene
encoding a hydrophobin, fvh1, is specifically expressed after the
induction of fruit in the edible mushroom Flammulina velutipes.
Curr. Genet. 39, 190–197.
[15] Lee, S.H., Kim, B.G., Kim, K.J., Lee, J.S., Yun, D.W.,
Hahn, J.H., Kim, G.H., Lee, K.H., Suh, D.S., Kwon, S.T.,
Lee, C.S. and Yoo, Y.B. (2002) Comparative analysis of
sequences expressed during the liquid-cultured mycelia and
fruit body stages of Pleurotus ostreatus. Fungal Genet. Biol.
35, 115–134.
[16] Liang, P. and Pardee, A.B. (1992) Differential display of eukary-
otic messenger RNA by means of the polymerase chain reaction.
Science 257, 967–971.

284 M. Sunagawa, Y. Magae / FEMS Microbiology Letters 246 (2005) 279–284
[17] Stein, S., Cho, Y.J., Jackson, R.S. and Liang, P. (2003) Identi-
fication of p53 target genes by fluorescent differential display.
Methods Mol. Biol. 234, 51–63.
[18] Ueda, A., Shi, W., Nakamura, T. and Takabe, T. (2002) Analysis
of salt-inducible genes in barley roots by differential display. J.
Plant Res. 115, 119–130.
[19] Wu, L.M., Ni, Z.F., Meng, F.R., Lin, Z. and Sun, Q.X. (2003)
Cloning and characterization of leaf cDNAs that are differentially
expressed between wheat hybrids and their parents. Mol. Genet.
Genomics. 270, 281–286.
[20] Sunagawa, M., Tamai, Y., Neda, H., Miyazaki, K. and Miura, K.
(1995) Application of random amplified polymorphic DNA
(RAPD) markers (I): analyses of fussants in edible mushrooms.
Nihon Mokuzai Gakkaishi 41, 945–948.
[21] Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang,
Z., Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI-
BLAST: a new generation of protein database search programs.
Nucleic Acids Res. 25, 3389–3402.
[22] White, T.J., Bruns, T., Lee, S. and Taylor, J. (1990) Amplification
and direct sequencing of fungal ribosomal RNA genes for
phylogenetics In: PCR Protocols (Innis, M.A., Gelfand, D.H.,
Sninsky, J.J. and White, T.J., Eds.), pp. 315–322. Academic Press,
San Diego, CA.
[23] Goodrich-Tanrikulu, M., Howe, K., Stafford, A. and Nelson,
M.A. (1998) Changes in fatty acid composition of Neurospora
crassa accompany sexual development and ascospore germina-
tion. Microbiology 144, 1713–1720.
[24] Thines, E., Weber, R.W. and Talbot, N.J. (2000) MAP kinase and
protein kinase A-dependent mobilization of triacylglycerol and
glycogen during appressorium turgor generation by Magnaporthe
grisea. Plant Cell 12, 1703–1718.
[25] Gibbons, G.F., Islam, K. and Pease, R.J. (2000) Mobilisation of
triacylglycerol stores. Biochim. Biophys. Acta 1483, 37–57.
[26] Muraguchi, H. and Kamada, T. (2000) A mutation in the
elen2 gene encoding a chtochrome P450 of Coprinus cinereus
affects mushroom morphogenesis. Fungal Genet. Biol. 29, 49–
59.
[27] Penas, M.M., Asgeirsdottir, S.A., Lasa, I., Culianez-Macia, F.A.,
Pisabarro, A.G., Wessels, J.G. and Ramirez, L. (1998) Identifi-
cation, characterization, and In situ detection of a fruit-body-
specific hydrophobin of Pleurotus ostreatus. Appl. Environ.
Microbiol. 64, 4028–4034.
[28] Asgeirsdottir, S.A., de Vries, O.M. and Wessels, J.G. (1998)
Identification of three differentially expressed hydrophobins in
Pleurotus ostreatus (oyster mushroom). Microbiology 144, 2961–
2969.
[29] Stoop, J.M.H. and Mooibroek, H. (1999) Advances in
genetic analysis and biotechnology of the cultivated button
mushroom, Agaricus bisporus. Appl. Microbiol. Biotechnol.
52, 474–483.

FEMS Microbiology Letters
Author Index Volume 246
Alves, A., see Tacao, M. (246) 11
Ando, S., see Goto, M. (246) 33
Anne, J., see Barbe, S. (246) 67
Arciola, C.R., Campoccia, D., Gamberini, S., Baldassarri, L. and
Montanaro, L. Prevalence of cna, fnbA and fnbB adhesin genes
among Staphylococcus aureus isolates from orthopedic infections
associated to different types of implant (246) 81
Asano, Y., see Kato, Y. (246) 243
Athie-Morales, V., see O�Brien, J.B. (246) 199Azcarate-Peril, M.A., see Bruno-Barcena, J.M. (246) 91
Bala, K., see Paul, B. (246) 207
Baldassarri, L., see Arciola, C.R. (246) 81
Barbe, S., Van Mellaert, L., Theys, J., Geukens, N., Lammertyn, E.,
Lambin, P. and Anne, J. Secretory production of biologically active
rat interleukin-2 by Clostridium acetobutylicum DSM792 as a tool
for anti-tumor treatment (246) 67
Barlow, K., see Yin, X. (246) 251
Behr, T., see Lehner, A. (246) 133
Belarbi, A., see Paul, B. (246) 207
Bera, R., Nayak, A., Sen, A.K., Chowdhury, B.P. and Bhadra, R.
Isolation and characterisation of the lipopolysaccharide from
Acidiphilium strain GS18h/ATCC55963, a soil isolate of Indian
copper mine (246) 183
Bevivino, A., see Dalmastri, C. (246) 39
Bhadra, R., see Bera, R. (246) 183
Blanch, A.R., see Garcıa-Aljaro, C. (246) 55
Blanco, J., see Garcıa-Aljaro, C. (246) 55
Blanco, J.E., see Garcıa-Aljaro, C. (246) 55
Blanco, M., see Garcıa-Aljaro, C. (246) 55
Bordons, A., see Reguant, C. (246) 111
Bruno-Barcena, J.M., Azcarate-Peril, M.A., Klaenhammer, T.R. and
Hassan, H.M. Marker-free chromosomal integration of the
manganese superoxide dismutase gene (sodA) from Streptococcus
thermophilus into Lactobacillus gasseri (246) 91
Campoccia, D., see Arciola, C.R. (246) 81
Carbonero, E.R., Mellinger, C.G., Eliasaro, S., Gorin, P.A.J. and
Iacomini, M. Chemotypes significance of lichenized fungi by
structural characterization of heteropolysaccharides from the
genera Parmotrema and Rimelia (246) 273
Carrascosa, A.V., see Cebollero, E. (246) 1
Carrete, R., see Reguant, C. (246) 111
Cebollero, E., Martinez-Rodriguez, A., Carrascosa, A.V. and
Gonzalez, R. Overexpression of csc1-1. A plausible strategy to
obtain wine yeast strains undergoing accelerated autolysis (246) 1
Cevallos, A.M., Perez-Escobar, M., Espinosa, N., Herrera, J.,
Lopez-Villasenor, I. and Hernandez, R. The stabilization of
housekeeping transcripts in Trypanosoma cruzi epimastigotes
evidences a global regulation of RNA decay during stationary
phase (246) 259
Chambers, J.R., see Yin, X. (246) 251
Chiarini, L., see Dalmastri, C. (246) 39
Chowdhury, B.P., see Bera, R. (246) 183
Clarke, P., see Viguier, C. (246) 235
Constantı, M., see Reguant, C. (246) 111
Cornelis, P., see Ghysels, B. (246) 167
Correia, A., see Tacao, M. (246) 11
Dalmastri, C., Pirone, L., Tabacchioni, S., Bevivino, A. and
Chiarini, L. Efficacy of species-specific recA PCR tests in the
identification of Burkholderia cepacia complex environmental
isolates (246) 39
Dunn, M.F., see Guillen-Navarro, K. (246) 159
Eliasaro, S., see Carbonero, E.R. (246) 273
Encarnacion, S., see Guillen-Navarro, K. (246) 159
Espejo, R.T., see Gonzalez-Escalona, N. (246) 213
Espinosa, N., see Cevallos, A.M. (246) 259
Fan, K.-Q., see Wu, X.-B. (246) 103
Forster-Fromme, K. and Jendrossek, D. Malate:quinone
oxidoreductase (MqoB) is required for growth on acetate and
linear terpenes in Pseudomonas citronellolis (246) 25
Fu, X., see Qu, Y. (246) 143
Gaenge, H., see Lehner, A. (246) 133
Gamberini, S., see Arciola, C.R. (246) 81
Garcıa-Aljaro, C., Muniesa, M., Blanco, J.E., Blanco, M., Blanco, J.,
Jofre, J. and Blanch, A.R. Characterization of Shiga toxin-
producing Escherichia coli isolated from aquatic environments
(246) 55
Geukens, N., see Barbe, S. (246) 67
Ghysels, B., Ochsner, U., Mollman, U., Heinisch, L., Vasil, M.,
Cornelis, P. and Matthijs, S. The Pseudomonas aeruginosa pirA
gene encodes a second receptor for ferrienterobactin and synthetic
catecholate analogues (246) 167
Gognies, S., see Paul, B. (246) 207
Gonzalez, R., see Cebollero, E. (246) 1
Gonzalez-Escalona, N., Romero, J. and Espejo, R.T. Polymorphism
and gene conversion of the 16S rRNA genes in the multiple rRNA
operons of Vibrio parahaemolyticus (246) 213
Gorin, P.A.J., see Carbonero, E.R. (246) 273
Goto, M., Ando, S., Hachisuka, Y. and Yoneyama, T. Contamination
of diverse nifH and nifH-like DNA into commercial PCR primers
(246) 33
doi:10.1016/S0378-1097(05)00268-5
FEMS Microbiology Letters 246 (2005) 285–287
www.fems-microbiology.org

Guillen-Navarro, K., Encarnacion, S. and Dunn, M.F. Biotin
biosynthesis, transport and utilization in rhizobia (246) 159
Guo, J., see Lai, X. (246) 87
Hachisuka, Y., see Goto, M. (246) 33
Hassan, H.M., see Bruno-Barcena, J.M. (246) 91
Heinisch, L., see Ghysels, B. (246) 167
Henriques, I., see Tacao, M. (246) 11
Hernandez, R., see Cevallos, A.M. (246) 259
Herrera, J., see Cevallos, A.M. (246) 259
Hirai, H., Sugiura, M., Kawai, S. and Nishida, T. Characteristics of
novel lignin peroxidases produced by white-rot fungus
Phanerochaete sordida YK-624 (246) 19
Hu, D.-L., see Omoe, K. (246) 191
Hubalek, M., see Lenco, J. (246) 47
Iacomini, M., see Carbonero, E.R. (246) 273
Irani, V.R. and Maslow, J.N. Induction of murine macrophage TNF-asynthesis by Mycobacterium avium is modulated through
complement-dependent interaction via complement receptors 3
and 4 in relation to M. avium glycopeptidolipid (246) 221
Jansen, A., see Van Houdt, R. (246) 265
Jendrossek, D., see Forster-Fromme, K. (246) 25
Jofre, J., see Garcıa-Aljaro, C. (246) 55
Jovcic, B., see Kojic, M. (246) 175
Kato, Y., Yoshida, S. and Asano, Y. Polymerase chain reaction for
identification of aldoxime dehydratase in aldoxime- or nitrile-
degrading microorganisms (246) 243
Kawai, S., see Hirai, H. (246) 19
Kelleher, D.P., see O�Brien, J.B. (246) 199Kimura, T., see Morimoto, K. (246) 229
Klaenhammer, T.R., see Bruno-Barcena, J.M. (246) 91
Kojic, M., Jovcic, B., Vindigni, A., Odremanp, F. and Venturi, V.
Novel target genes of PsrA transcriptional regulator of
Pseudomonas aeruginosa (246) 175
Kuhl, M., see Thar, R. (246) 75
Lai, X., Guo, J., Zhang, X. and Wang, H. Identification of a novel
domain – DIM, which defines a new family composed mainly of
bacterial membrane proteins (246) 87
Lambin, P., see Barbe, S. (246) 67
Lammertyn, E., see Barbe, S. (246) 67
Lehner, A., Loy, A., Behr, T., Gaenge, H., Ludwig, W., Wagner, M.
and Schleifer, K.-H. Oligonucleotide microarray for identification
of Enterococcus species (246) 133
Lenco, J., Pavkova, I., Hubalek, M. and Stulik, J. Insights into the
oxidative stress response in Francisella tularensis LVS and its
mutant DiglC1+2 by proteomics analysis (246) 47
Lopez-Villasenor, I., see Cevallos, A.M. (246) 259
Loy, A., see Lehner, A. (246) 133
Ludwig, W., see Lehner, A. (246) 133
Magae, Y., see Sunagawa, M. (246) 279
Martinez-Rodriguez, A., see Cebollero, E. (246) 1
Maslow, J.N., see Irani, V.R. (246) 221
Matthijs, S., see Ghysels, B. (246) 167
McCabe, M.S., see O�Brien, J.B. (246) 199McDonald, G.S.A., see O�Brien, J.B. (246) 199Melin, P., see Strom, K. (246) 119
Mellinger, C.G., see Carbonero, E.R. (246) 273
Michiels, C.W., see Van Houdt, R. (246) 265
Mollman, U., see Ghysels, B. (246) 167
Montanaro, L., see Arciola, C.R. (246) 81
Moons, P., see Van Houdt, R. (246) 265
Morimoto, K., Kimura, T., Sakka, K. and Ohmiya, K. Overexpression
of a hydrogenase gene in Clostridium paraputrificum to enhance
hydrogen gas production (246) 229
Moura, A., see Tacao, M. (246) 11
Muniesa, M., see Garcıa-Aljaro, C. (246) 55
Nakane, A., see Omoe, K. (246) 191
Nayak, A., see Bera, R. (246) 183
Nı Eidhin, D.B., see O�Brien, J.B. (246) 199Nishida, T., see Hirai, H. (246) 19
O�Brien, J.B., McCabe, M.S., Athie-Morales, V., McDonald, G.S.A.,
Nı Eidhin, D.B. and Kelleher, D.P. Passive immunisation of
hamsters against Clostridium difficile infection using antibodies to
surface layer proteins (246) 199
O Cuıv, P., see Viguier, C. (246) 235
Ochsner, U., see Ghysels, B. (246) 167
O�Connell, M., see Viguier, C. (246) 235
Odremanp, F., see Kojic, M. (246) 175
Ohmiya, K., see Morimoto, K. (246) 229
Omoe, K., Hu, D.-L., Takahashi-Omoe, H., Nakane, A. and
Shinagawa, K. Comprehensive analysis of classical and newly
described staphylococcal superantigenic toxin genes in
Staphylococcus aureus isolates (246) 191
Ona, O., Van Impe, J., Prinsen, E. and Vanderleyden, J. Growth and
indole-3-acetic acid biosynthesis of Azospirillum brasilense Sp245 is
environmentally controlled (246) 125
Park, A.S., see Yin, X. (246) 251
Paul, B., Bala, K., Gognies, S. and Belarbi, A. Morphological and
molecular taxonomy of Pythium longisporangium sp. nov. isolated
from the Burgundian region of France (246) 207
Pavkova, I., see Lenco, J. (246) 47
Perez-Escobar, M., see Cevallos, A.M. (246) 259
Pirone, L., see Dalmastri, C. (246) 39
Prinsen, E., see Ona, O. (246) 125
Qu, Y., Zhou, J., Wang, J., Fu, X. and Xing, L. Microbial community
dynamics in bioaugmented sequencing batch reactors for
bromoamine acid removal (246) 143
Reguant, C., Carrete, R., Constantı, M. and Bordons, A. Population
dynamics of Oenococcus oeni strains in a new winery and the effect
of SO2 and yeast strain (246) 111
Romero, J., see Gonzalez-Escalona, N. (246) 213
Saavedra, M.J., see Tacao, M. (246) 11
Sakka, K., see Morimoto, K. (246) 229
Schleifer, K.-H., see Lehner, A. (246) 133
Schnurer, J., see Strom, K. (246) 119
Sen, A.K., see Bera, R. (246) 183
Shinagawa, K., see Omoe, K. (246) 191
Strom, K., Schnurer, J. and Melin, P. Co-cultivation of antifungal
Lactobacillus plantarum MiLAB 393 and Aspergillus nidulans,
evaluation of effects on fungal growth and protein expression
(246) 119
Stulik, J., see Lenco, J. (246) 47
Sugiura, M., see Hirai, H. (246) 19
Sunagawa, M. and Magae, Y. Isolation of genes differentially
expressed during the fruit body development of Pleurotus
ostreatus by differential display of RAPD (246) 279
Tabacchioni, S., see Dalmastri, C. (246) 39
Tacao, M., Moura, A., Alves, A., Henriques, I., Saavedra, M.J. and
Correia, A. Evaluation of 16S rDNA- and gyrB-DGGE for typing
members of the genus Aeromonas (246) 11
Takahashi-Omoe, H., see Omoe, K. (246) 191
286 Author Index Volume 246

Thar, R. and Kuhl, M. Complex pattern formation of marine gradient
bacteria explained by a simple computer model (246) 75
Theys, J., see Barbe, S. (246) 67
Van Houdt, R., Moons, P., Jansen, A., Vanoirbeek, K. and Michiels,
C.W. Genotypic and phenotypic characterization of a biofilm-
forming Serratia plymuthica isolate from a raw vegetable
processing line (246) 265
Van Impe, J., see Ona, O. (246) 125
Van Mellaert, L., see Barbe, S. (246) 67
Vanderleyden, J., see Ona, O. (246) 125
Vanoirbeek, K., see Van Houdt, R. (246) 265
Vasil, M., see Ghysels, B. (246) 167
Venturi, V., see Kojic, M. (246) 175
Viguier, C., O Cuıv, P., Clarke, P. and O�Connell, M. RirA is the iron
response regulator of the rhizobactin 1021 biosynthesis and
transport genes in Sinorhizobium meliloti 2011 (246) 235
Vindigni, A., see Kojic, M. (246) 175
Wagner, M., see Lehner, A. (246) 133
Wang, H., see Lai, X. (246) 87
Wang, J., see Qu, Y. (246) 143
Wang, Q.-H., see Wu, X.-B. (246) 103
Wheatcroft, R., see Yin, X. (246) 251
Wu, X.-B., Fan, K.-Q., Wang, Q.-H. and Yang, K.-Q. C-terminus
mutations of Acremonium chrysogenum deacetoxy/deacetyl-
cephalosporin C synthase with improved activity toward
penicillin analogs (246) 103
Xing, L., see Qu, Y. (246) 143
Yang, K.-Q., see Wu, X.-B. (246) 103
Yin, X., Chambers, J.R., Barlow, K., Park, A.S. and Wheatcroft, R.
The gene encoding xylulose-5-phosphate/fructose-6-phosphate
phosphoketolase (xfp) is conserved among Bifidobacterium species
within a more variable region of the genome and both are useful for
strain identification (246) 251
Yoneyama, T., see Goto, M. (246) 33
Yoshida, S., see Kato, Y. (246) 243
Zhang, X., see Lai, X. (246) 87
Zhou, J., see Qu, Y. (246) 143
Zwirglmaier, K. Fluorescence in situ hybridisation (FISH) – the next
generation (246) 151
Author Index Volume 246 287

FEMS Microbiology Letters
Subject Index Volume 246
Acidiphilium ATCC55963
Lipopolysaccharide; Lipid A; Lethal toxicity (Bera, R. (246) 183)
Aeromonas
DGGE; gyrB; 16S rRNA (Tacao, M. (246) 11)
Aldoxime dehydratase
Nitrile hydratase; PCR; Aldoxime-nitrile pathway; Screening (Kato,
Y. (246) 243)
Aldoxime-nitrile pathway
Aldoxime dehydratase; Nitrile hydratase; PCR; Screening (Kato, Y.
(246) 243)
Antheridia
Pythium longisporangium; Sporangia; Oogonia; Oospores; ITS region;
rRNA (Paul, B. (246) 207)
Anti-bacterial drugs
DIM; Protein domain; Bacteria; Transmembrane region (Lai, X. (246)
87)
Anti-cancer treatment
Clostridium acetobutylicum; Protein secretion; Interleukin-2 (Barbe, S.
(246) 67)
Antifungal activity
Two dimensional gel electrophoresis; 3-Phenyl lactic acid; Cyclic
dipeptide (Strom, K. (246) 119)
Autolysis
Sparkling wine; Genetic engineering; Autophagy; Saccharomyces
cerevisiae (Cebollero, E. (246) 1)
Autophagy
Sparkling wine; Autolysis; Genetic engineering; Saccharomyces
cerevisiae (Cebollero, E. (246) 1)
Bacteria
DIM; Protein domain; Transmembrane region; Anti-bacterial drugs
(Lai, X. (246) 87)
Bacterial adhesion
Collagen adhesion gene (cna); Fibronectin-binding protein;
Biomaterial-associated infections; Staphylococcus aureus (Arciola,
C.R. (246) 81)
Bifidobacterium
Phosphoketolase; xfp; Strain identification; Detection (Yin, X. (246)
251)
Bioaugmentation
Bromoamine acid; Community dynamics; Ribosomal intergenic
spacer; Sphingomonas xenophaga (Qu, Y. (246) 143)
Biomaterial-associated infections
Collagen adhesion gene (cna); Fibronectin-binding protein; Bacterial
adhesion; Staphylococcus aureus (Arciola, C.R. (246) 81)
Biotin biosynthesis
Rhizobia; Rhizobia–legume symbiosis (Guillen-Navarro, K. (246) 159)
Branched-chain carbon metabolism
Citronellol pathway; Malate:quinone oxidoreductase; Geranyl-
coenzymeA carboxylase; Pseudomonas citronellolis (Forster-Fromme,
K. (246) 25)
Bromoamine acid
Bioaugmentation; Community dynamics; Ribosomal intergenic spacer;
Sphingomonas xenophaga (Qu, Y. (246) 143)
Burkholderia cepacia complex identification
recA specific PCR; Cystic fibrosis; Rhizosphere environment
(Dalmastri, C. (246) 39)
Catecholate siderophores
Pseudomonas aeruginosa; Enterobactin; pfeA; pirA; TonB-dependent
receptors (Ghysels, B. (246) 167)
Chemical structure
Lichenized fungi; Parmeliaceae; Galactoglucomannan; 13C NMR
(Carbonero, E.R. (246) 273)
Chemotaxis
Self-organisation; Thiovulum; Microbial pattern formation; Quorum
sensing; Computer model (Thar, R. (246) 75)
Citronellol pathway
Branched-chain carbon metabolism; Malate:quinone oxidoreductase;
GeranylcoenzymeA carboxylase; Pseudomonas citronellolis (Forster-
Fromme K. (246) 25)
Clostridium acetobutylicum
Protein secretion; Interleukin-2; Anti-cancer treatment (Barbe, S. (246)
67)
doi:10.1016/S0378-1097(05)00269-7
FEMS Microbiology Letters 246 (2005) 289–294
www.fems-microbiology.org

Clostridium difficile
Diarrhoea; Surface layer proteins; Hamster model (O�Brien, J.B. (246)199)
Clostridium paraputrificum
Hydrogenase; Hydrogen gas production (Morimoto, K. (246) 229)
13C NMR
Lichenized fungi; Parmeliaceae; Galactoglucomannan; Chemical
structure (Carbonero, E.R. (246) 273)
Collagen adhesion gene (cna)
Fibronectin-binding protein; Bacterial adhesion; Biomaterial-
associated infections; Staphylococcus aureus (Arciola, C.R. (246) 81)
Community dynamics
Bioaugmentation; Bromoamine acid; Ribosomal intergenic spacer;
Sphingomonas xenophaga (Qu, Y. (246) 143)
Complement receptor
Mycobacterium avium; GPL; TNF-a; Macrophage; Serum proteins
(Irani and J.N. Maslow V.R. (246) 221)
Computer model
Self-organisation; Chemotaxis; Thiovulum; Microbial pattern
formation; Quorum sensing (Thar, R. (246) 75)
Contamination
nifH; PCR; Primer (Goto, M. (246) 33)
C-terminus
DAOC; DAC; Deacetoxy/deacetylcephalosporin C synthase;
Mutagenesis; Acremonium chrysogenum; Kinetics (Wu, X.-B. (246)
103)
Cyclic dipeptide
Antifungal activity; Two dimensional gel electrophoresis; 3-Phenyl
lactic acid (Strom, K. (246) 119)
Cystic fibrosis
Burkholderia cepacia complex identification; recA specific PCR;
Rhizosphere environment (Dalmastri, C. (246) 39)
DAC
DAOC; Deacetoxy/deacetylcephalosporin C synthase; C-terminus;
Mutagenesis; Acremonium chrysogenum; Kinetics (Wu, X.-B. (246)
103)
DAOC
DAC; Deacetoxy/deacetylcephalosporin C synthase; C-terminus;
Mutagenesis; Acremonium chrysogenum; Kinetics (Wu, X.-B. (246)
103)
Deacetoxy/deacetylcephalosporin C synthase
DAOC; DAC; C-terminus; Mutagenesis; Acremonium chrysogenum;
Kinetics (Wu, X.-B. (246) 103)
Detection
Bifidobacterium; Phosphoketolase; xfp; Strain identification (Yin, X.
(246) 251)
DGGE
Aeromonas; 16S rRNA; gyrB (Tacao, M. (246) 11)
Diarrhoea
Clostridium difficile; Surface layer proteins; Hamster model (O�Brien,J.B. (246) 199)
Differential display
Pleurotus ostreatus; RAPD (Sunagawa, M. (246) 279)
DIM
Protein domain; Bacteria; Transmembrane region; Anti-bacterial
drugs (Lai, X. (246) 87)
Enterobactin
Pseudomonas aeruginosa; Catecholate siderophores; pfeA; pirA; TonB-
dependent receptors (Ghysels, B. (246) 167)
Enterococcus
rRNA gene; Oligonucleotide; Microarray; Probe; Microbial
diagnostics (Lehner, A. (246) 133)
Enterotoxin
Staphylococcus aureus; Multiplex PCR; Genotyping; Mobile genetic
elements (Omoe, K. (246) 191)
Escherichia coli
STEC; stx2; VT2 verotoxin; VTECWater (Garcıa-Aljaro, C. (246) 55)
Fermentor
Azospirillum brasilense; Indole-3-acetic acid (Ona, O. (246) 125)
Fibronectin-binding protein
Collagen adhesion gene (cna); Bacterial adhesion; Biomaterial-
associated infections; Staphylococcus aureus (Arciola, C.R. (246) 81)
Fluorescence in situ hybridisation
Tyramide signal amplification; Polynucleotide probes (Zwirglmaier K.
(246) 151)
Francisella tularensis
Oxidative stress; Quantitative proteomics; Two-dimensional
electrophoresis (Lenco, J. (246) 47)
Functional gene replacement
Manganese superoxide dismutase; Oxidative stress; Lactobacillus
gasseri; Lactic acid bacteria; Probiotics (Bruno-Barcena, J.M. (246) 91)
Fur
Siderophore; Iron response; RirA (Viguier, C. (246) 235)
Galactoglucomannan
Lichenized fungi; Parmeliaceae; Chemical structure; 13C NMR
(Carbonero, E.R. (246) 273)
Gene conversion
Polymorphism; rrn operons; Vibrio parahaemolyticus; 16S rRNA
(Gonzalez-Escalona, N. (246) 213)
Gene expression
Protozoa; Kinetoplastid; RNA stability (Cevallos, A.M. (246) 259)
Genetic engineering
Sparkling wine; Autolysis; Autophagy; Saccharomyces cerevisiae
(Cebollero, E. (246) 1)
Genotyping
Staphylococcus aureus; Enterotoxin; Multiplex PCR; Mobile genetic
elements (Omoe, K. (246) 191)
GeranylcoenzymeA carboxylase
Citronellol pathway; Branched-chain carbon metabolism;
Malate:quinone oxidoreductase; Pseudomonas citronellolis (Forster-
Fromme, K. (246) 25)
290 Subject Index Volume 246

GPL
Mycobacterium avium; TNF-a; Macrophage; Complement receptor;
Serum proteins (Irani, V.R. (246) 221)
gyrB
Aeromonas; DGGE; 16S rRNA (Tacao, M. (246) 11)
Serratia; Identification natural isolate; Phylogeny; Quorum sensing; N-
Acyl-L-homoserine lactone (Van Houdt, R. (246) 265)
Hamster model
Clostridium difficile; Diarrhoea; Surface layer proteins (O�Brien, J.B.(246) 199)
Hydrogen gas production
Clostridium paraputrificum; Hydrogenase (Morimoto, K. (246) 229)
Hydrogen peroxide
Phanerochaete sordida YK-624; Lignin peroxidase; Ordered bi-bi ping-
pong mechanism; Lignin substructure model compounds (Hirai, H.
(246) 19)
Hydrogenase
Clostridium paraputrificum; Hydrogen gas production (Morimoto, K.
(246) 229)
Identification natural isolate
Serratia; gyrB; Phylogeny; Quorum sensing; N-Acyl-L-homoserine
lactone (Van Houdt, R. (246) 265)
Indole-3-acetic acid
Azospirillum brasilense; Fermentor (Ona, O. (246) 125)
Interleukin-2
Clostridium acetobutylicum; Protein secretion; Anti-cancer treatment
(Barbe, S. (246) 67)
Iron response
Siderophore; RirA; Fur (Viguier, C. (246) 235)
ITS region
Pythium longisporangium; Sporangia; Oogonia; Antheridia; Oospores;
rRNA (Paul, B. (246) 207)
Kinetics
DAOC; DAC; Deacetoxy/deacetylcephalosporin C synthase; C-
terminus; Mutagenesis; Acremonium chrysogenum (Wu, X.-B. (246)
103)
Kinetoplastid
Protozoa; RNA stability; Gene expression (Cevallos, A.M. (246) 259)
Lactic acid bacteria
Functional gene replacement; Manganese superoxide dismutase;
Oxidative stress; Lactobacillus gasseri; Probiotics (Bruno-Barcena,
J.M. (246) 91)
Lactobacillus gasseri
Functional gene replacement; Manganese superoxide dismutase;
Oxidative stress; Lactic acid bacteria; Probiotics (Bruno-Barcena,
J.M. (246) 91)
Lethal toxicity
Acidiphilium ATCC55963; Lipopolysaccharide; Lipid A (Bera, R.
(246) 183)
Lichenized fungi
Parmeliaceae; Galactoglucomannan; Chemical structure; 13C NMR
(Carbonero, E.R. (246) 273)
Lignin peroxidase
Phanerochaete sordida YK-624; Ordered bi-bi ping-pong mechanism;
Lignin substructure model compounds; Hydrogen peroxide (Hirai, H.
(246) 19)
Lignin substructure model compounds
Phanerochaete sordida YK-624; Lignin peroxidase; Ordered bi-bi ping-
pong mechanism; Hydrogen peroxide (Hirai, H. (246) 19)
Lipid A
Acidiphilium ATCC55963; Lipopolysaccharide; Lethal toxicity (Bera,
R. (246) 183)
Lipopolysaccharide
Acidiphilium ATCC55963; Lipid A; Lethal toxicity (Bera, R. (246) 183)
Macrophage
Mycobacterium avium; GPL; TNF-a; Complement receptor; Serum
proteins (Irani, V.R. (246) 221)
Malate:quinone oxidoreductase
Citronellol pathway; Branched-chain carbon metabolism;
GeranylcoenzymeA carboxylase; Pseudomonas citronellolis (Forster-
Fromme, K. (246) 25)
Malolactic fermentation
Oenococcus oeni; RAPD multiplex; Strain typification; Sulphur
dioxide; Wine (Reguant, C. (246) 111)
Manganese superoxide dismutase
Functional gene replacement; Oxidative stress; Lactobacillus gasseri;
Lactic acid bacteria; Probiotics (Bruno-Barcena, J.M. (246) 91)
Microarray
Enterococcus; rRNA gene; Oligonucleotide; Probe; Microbial
diagnostics (Lehner, A. (246) 133)
Microbial diagnostics
Enterococcus; rRNA gene; Oligonucleotide; Microarray; Probe
(Lehner, A. (246) 133)
Microbial pattern formation
Self-organisation; Chemotaxis; Thiovulum; Quorum sensing;
Computer model (Thar, R. (246) 75)
Mobile genetic elements
Staphylococcus aureus; Enterotoxin; Multiplex PCR; Genotyping
(Omoe, K. (246) 191)
Multiplex PCR
Staphylococcus aureus; Enterotoxin; Genotyping; Mobile genetic
elements (Omoe, K. (246) 191)
Mutagenesis
DAOC; DAC; Deacetoxy/deacetylcephalosporin C synthase; C-
terminus; Acremonium chrysogenum; Kinetics (Wu, X.-B. (246) 103)
Mycobacterium avium
GPL; TNF-a; Macrophage; Complement receptor; Serum proteins
(Irani and J.N. Maslow V.R. (246) 221)
Subject Index Volume 246 291

N-Acyl-L-homoserine lactone
Serratia; Identification natural isolate; gyrB; Phylogeny; Quorum
sensing (Van Houdt, R. (246) 265)
nifH
Contamination; PCR; Primer (Goto, M. (246) 33)
Nitrile hydratase
Aldoxime dehydratase; PCR; Aldoxime-nitrile pathway; Screening
(Kato, Y. (246) 243)
Oenococcus oeni
Malolactic fermentation; RAPD multiplex; Strain typification;
Sulphur dioxide; Wine (Reguant, C. (246) 111)
Oligonucleotide
Enterococcus; rRNA gene; Microarray; Probe; Microbial diagnostics
(Lehner, A. (246) 133)
Oogonia
Pythium longisporangium; Sporangia; Antheridia; Oospores; ITS
region; rRNA (Paul, B. (246) 207)
Oospores
Pythium longisporangium; Sporangia; Oogonia; Antheridia; ITS
region; rRNA (Paul, B. (246) 207)
Ordered bi-bi ping-pong mechanism
Phanerochaete sordida YK-624; Lignin peroxidase; Lignin substructure
model compounds; Hydrogen peroxide (Hirai, H. (246) 19)
Oxidative stress
Francisella tularensis; Quantitative proteomics; Two-dimensional
electrophoresis (Lenco, J. (246) 47)
Functional gene replacement; Manganese superoxide dismutase;
Lactobacillus gasseri; Lactic acid bacteria; Probiotics (Bruno-
Barcena, J.M. (246) 91)
Parmeliaceae
Lichenized fungi; Galactoglucomannan; Chemical structure; 13C
NMR (Carbonero, E.R. (246) 273)
PCR
Contamination; nifH; Primer (Goto, M. (246) 33)
Aldoxime dehydratase; Nitrile hydratase; Aldoxime-nitrile pathway;
Screening (Kato, Y. (246) 243)
pfeA
Pseudomonas aeruginosa; Enterobactin; Catecholate siderophores;
pirA; TonB-dependent receptors (Ghysels, B. (246) 167)
Phanerochaete sordida YK-624
Lignin peroxidase; Ordered bi-bi ping-pong mechanism; Lignin
substructure model compounds; Hydrogen peroxide (Hirai, H. (246)
19)
3-Phenyl lactic acid
Antifungal activity; Two dimensional gel electrophoresis; Cyclic
dipeptide (Strom, K. (246) 119)
Phosphoketolase
Bifidobacterium; xfp; Strain identification; Detection (Yin, X. (246)
251)
Phylogeny
Serratia; Identification natural isolate; gyrB; Quorum sensing; N-Acyl-
L-homoserine lactone (Van Houdt, R. (246) 265)
pirA
Pseudomonas aeruginosa; Enterobactin; Catecholate siderophores;
pfeA; TonB-dependent receptors (Ghysels, B. (246) 167)
Pleurotus ostreatus
Differential display; RAPD (Sunagawa, M. (246) 279)
Polymorphism
Gene conversion; rrn operons; Vibrio parahaemolyticus; 16S rRNA
(Gonzalez-Escalona, N. (246) 213)
Polynucleotide probes
Fluorescence in situ hybridisation; Tyramide signal amplification
(Zwirglmaier K. (246) 151)
Primer
Contamination; nifH; PCR (Goto, M. (246) 33)
Probe
Enterococcus; rRNA gene; Oligonucleotide; Microarray; Microbial
diagnostics (Lehner, A. (246) 133)
Probiotics
Functional gene replacement; Manganese superoxide dismutase;
Oxidative stress; Lactobacillus gasseri; Lactic acid bacteria (Bruno-
Barcena, J.M. (246) 91)
Protein domain
DIM; Bacteria; Transmembrane region; Anti-bacterial drugs (Lai, X.
(246) 87)
Protein secretion
Clostridium acetobutylicum; Interleukin-2; Anti-cancer treatment
(Barbe, S. (246) 67)
Protozoa
Kinetoplastid; RNA stability; Gene expression (Cevallos, A.M. (246)
259)
Pseudomonas aeruginosa
Enterobactin; Catecholate siderophores; pfeA; pirA; TonB-dependent
receptors (Ghysels, B. (246) 167)
Pseudomonas citronellolis
Citronellol pathway; Branched-chain carbon metabolism;
Malate:quinone oxidoreductase; GeranylcoenzymeA carboxylase
(Forster-Fromme, K. (246) 25)
PsrA regulon
Stationary phase (Kojic, M. (246) 175)
Pythium longisporangium
Sporangia; Oogonia; Antheridia; Oospores; ITS region; rRNA (Paul,
B. (246) 207)
Quantitative proteomics
Francisella tularensis; Oxidative stress; Two-dimensionalelectro-
phoresis (Lenco, J. (246) 47)
292 Subject Index Volume 246

Quorum sensing
Self-organisation; Chemotaxis; Thiovulum; Microbial pattern
formation; Computer model (Thar, R. (246) 75)
Serratia; Identification natural isolate; gyrB; Phylogeny; N-Acyl-L-
homoserine lactone (Van Houdt, R. (246) 265)
RAPD
Differential display; Pleurotus ostreatus (Sunagawa, M. (246) 279)
RAPD multiplex
Malolactic fermentation; Oenococcus oeni; Strain typification; Sulphur
dioxide; Wine (Reguant, C. (246) 111)
recA specific PCR
Burkholderia cepacia complex identification; Cystic fibrosis;
Rhizosphere environment (Dalmastri, C. (246) 39)
Rhizobia
Biotin biosynthesis; Rhizobia–legume symbiosis (Guillen-Navarro, K.
(246) 159)
Rhizobia–legume symbiosis
Biotin biosynthesis; Rhizobia (Guillen-Navarro, K. (246) 159)
Rhizosphere environment
Burkholderia cepacia complex identification; recA specific PCR; Cystic
fibrosis (Dalmastri, C. (246) 39)
Ribosomal intergenic spacer
Bioaugmentation; Bromoamine acid; Community dynamics;
Sphingomonas xenophaga (Qu, Y. (246) 143)
RirA
Siderophore; Iron response; Fur (Viguier, C. (246) 235)
RNA stability
Protozoa; Kinetoplastid; Gene expression (Cevallos, A.M. (246) 259)
rrn operons
Gene conversion; Polymorphism; Vibrio parahaemolyticus; 16S rRNA
(Gonzalez-Escalona, N. (246) 213)
rRNA
Pythium longisporangium; Sporangia; Oogonia; Antheridia; Oospores;
ITS region (Paul, B. (246) 207)
rRNA gene
Enterococcus; Oligonucleotide; Microarray; Probe; Microbial
diagnostics (Lehner, A. (246) 133)
Saccharomyces cerevisiae
Sparkling wine; Autolysis; Genetic engineering; Autophagy
(Cebollero, E. (246) 1)
Screening
Aldoxime dehydratase; Nitrile hydratase; PCR; Aldoxime-nitrile
pathway (Kato, Y. (246) 243)
Self-organisation
Chemotaxis; Thiovulum; Microbial pattern formation; Quorum
sensing; Computer model (Thar, R. (246) 75)
Serratia
Identification natural isolate; gyrB; Phylogeny; Quorum sensing; N-
Acyl-L-homoserine lactone (Van Houdt, R. (246) 265)
Serum proteins
Mycobacterium avium; GPL; TNF-a; Macrophage; Complement
receptor (Irani, V.R. (246) 221)
Siderophore
Iron response; RirA; Fur (Viguier, C. (246) 235)
Sparkling wine
Autolysis; Genetic engineering; Autophagy; Saccharomyces cerevisiae
(Cebollero, E. (246) 1)
Sphingomonas xenophaga
Bioaugmentation; Bromoamine acid; Community dynamics;
Ribosomal intergenic spacer (Qu, Y. (246) 143)
Sporangia
Pythium longisporangium; Oogonia; Antheridia; Oospores; ITS region;
rRNA (Paul, B. (246) 207)
16S rRNA
Aeromonas; DGGE; gyrB (Tacao, M. (246) 11)
Gene conversion; Polymorphism; rrn operons; Vibrio parahaemolyticus
(Gonzalez-Escalona, N. (246) 213)
Staphylococcus aureus
Collagen adhesion gene (cna); Fibronectin-binding protein;
Bacterial adhesion; Biomaterial-associated infections (Arciola,
C.R. (246) 81)
Enterotoxin; Multiplex PCR; Genotyping; Mobile genetic elements
(Omoe, K. (246) 191)
Stationary phase
PsrA regulon (Kojic, M. (246) 175)
STEC
Escherichia coli; stx2; VT2 verotoxin; VTECWater (Garcıa-Aljaro, C.
(246) 55)
Strain identification
Bifidobacterium; Phosphoketolase; xfp; Detection (Yin, X. (246) 251)
Strain typification
Malolactic fermentation; Oenococcus oeni; RAPD multiplex; Sulphur
dioxide; Wine (Reguant, C. (246) 111)
stx2Escherichia coli; STEC; VT2 verotoxin; VTECWater (Garcıa-Aljaro,
C. (246) 55)
Sulphur dioxide
Malolactic fermentation; Oenococcus oeni; RAPD multiplex; Strain
typification; Wine (Reguant, C. (246) 111)
Surface layer proteins
Clostridium difficile; Diarrhoea; Hamster model (O�Brien, J.B. (246)199)
Subject Index Volume 246 293

Thiovulum
Self-organisation; Chemotaxis; Microbial pattern formation; Quorum
sensing; Computer model (Thar, R. (246) 75)
TNF-aMycobacterium avium; GPL; Macrophage; Complement receptor;
Serum proteins (Irani, V.R. (246) 221)
TonB-dependent receptors
Pseudomonas aeruginosa; Enterobactin; Catecholate siderophores;
pfeA; pirA (Ghysels, B. (246) 167)
Transmembrane region
DIM; Protein domain; Bacteria; Anti-bacterial drugs (Lai, X. (246) 87)
Two dimensional gel electrophoresis
Antifungal activity; 3-Phenyl lactic acid; Cyclic dipeptide (Strom, K.
(246) 119)
Two-dimensional electrophoresis
Francisella tularensis; Oxidative stress; Quantitative proteomics
(Lenco, J. (246) 47)
Tyramide signal amplification
Fluorescence in situ hybridisation; Polynucleotide probes (Zwirglmaier
K. (246) 151)
Vibrio parahaemolyticus
Gene conversion; Polymorphism; rrn operons; 16S rRNA (Gonzalez-
Escalona, N. (246) 213)
VTECWater
Escherichia coli; STEC; stx2; VT2 verotoxin (Garcıa-Aljaro, C. (246)
55)
VT2 verotoxin
Escherichia coli; STEC; stx2; VTECWater (Garcıa-Aljaro, C. (246) 55)
Wine
Malolactic fermentation; Oenococcus oeni; RAPD multiplex; Strain
typification; Sulphur dioxide (Reguant, C. (246) 111)
xfp
Bifidobacterium; Phosphoketolase; Strain identification; Detection
(Yin, X. (246) 251)
294 Subject Index Volume 246
Top Related
Copyright © 2022 FDOKUMEN

