
Multilocus sequence typing and phylogenetic analyses of Pseudomonas aeruginosa isolates from the...
13
Published Ahead of Print 29 August 2008. 10.1128/AEM.02322-07. 2008, 74(20):6194. DOI: Appl. Environ. Microbiol. Shoichi Hosoya, Akira Yokota and Kazuhiro Kogure Nurul Huda Khan, Mahbuba Ahsan, Susumu Yoshizawa, Isolates from the Ocean aeruginosa Pseudomonas Phylogenetic Analyses of Multilocus Sequence Typing and http://aem.asm.org/content/74/20/6194 Updated information and services can be found at: These include: REFERENCES http://aem.asm.org/content/74/20/6194#ref-list-1 at: This article cites 52 articles, 26 of which can be accessed free CONTENT ALERTS more» articles cite this article), Receive: RSS Feeds, eTOCs, free email alerts (when new http://journals.asm.org/site/misc/reprints.xhtml Information about commercial reprint orders: http://journals.asm.org/site/subscriptions/ To subscribe to to another ASM Journal go to: on September 17, 2014 by guest http://aem.asm.org/ Downloaded from on September 17, 2014 by guest http://aem.asm.org/ Downloaded from
Transcript of Multilocus sequence typing and phylogenetic analyses of Pseudomonas aeruginosa isolates from the...

Published Ahead of Print 29 August 2008. 10.1128/AEM.02322-07.
2008, 74(20):6194. DOI:Appl. Environ. Microbiol. Shoichi Hosoya, Akira Yokota and Kazuhiro KogureNurul Huda Khan, Mahbuba Ahsan, Susumu Yoshizawa,
Isolates from the Ocean aeruginosaPseudomonasPhylogenetic Analyses of
Multilocus Sequence Typing and
http://aem.asm.org/content/74/20/6194Updated information and services can be found at:
These include:
REFERENCEShttp://aem.asm.org/content/74/20/6194#ref-list-1at:
This article cites 52 articles, 26 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Oct. 2008, p. 6194–6205 Vol. 74, No. 200099-2240/08/$08.00�0 doi:10.1128/AEM.02322-07Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Multilocus Sequence Typing and Phylogenetic Analyses ofPseudomonas aeruginosa Isolates from the Ocean�
Nurul Huda Khan,1* Mahbuba Ahsan,1 Susumu Yoshizawa,1 Shoichi Hosoya,2Akira Yokota,2 and Kazuhiro Kogure1
Marine Microbiology Laboratory, Ocean Research Institute, University of Tokyo, Tokyo, Japan,1 and Institute ofMolecular and Cellular Biosciences, University of Tokyo, Tokyo, Japan2
Received 1 October 2007/Accepted 15 August 2008
Recent isolation of Pseudomonas aeruginosa strains from the open ocean and subsequent pulsed-field gel elec-trophoresis analyses indicate that these strains have a unique genotype (N. H. Khan, Y. Ishii, N. Kimata-Kino, H.Esaki, T. Nishino, M. Nishimura, and K. Kogure, Microb. Ecol. 53:173–186, 2007). We hypothesized that ocean P.aeruginosa strains have a unique phylogenetic position relative to other strains. The objective of this study was toclarify the intraspecies phylogenetic relationship between marine strains and other strains from various geograph-ical locations. Considering the advantages of using databases, multilocus sequence typing (MLST) was chosen forthe typing and discrimination of ocean P. aeruginosa strains. Seven housekeeping genes (acsA, aroE, guaA, mutL,nuoD, ppsA, and trpE) were analyzed, and the results were compared with data on the MLST website. These geneswere also used for phylogenetic analysis of P. aeruginosa. Rooted and unrooted phylogenetic trees were generated foreach gene locus and the concatenated gene fragments. MLST data showed that all the ocean strains were new. Treesconstructed for individual and concatenated genes revealed that ocean P. aeruginosa strains have clusters distinctfrom those of other P. aeruginosa strains. These clusters roughly reflected the geographical locations of the isolates.These data support our previous findings that P. aeruginosa strains are present in the ocean. It can be concludedthat the ocean P. aeruginosa strains have diverged from other isolates and form a distinct cluster based on MLSTand phylogenetic analyses of seven housekeeping genes.
The ubiquitously distributed gammaproteobacterium Pseudo-monas aeruginosa is capable of thriving in a large number ofdissimilar ecological niches (12, 15, 16, 21, 22, 24, 33, 37, 38, 39,41, 42, 44, 50, 52, 54, 55, 56). Our recent study documented itssurvival and existence in the open ocean (22, 24). Molecularfingerprinting techniques, such as pulsed-field gel electrophoresis,have demonstrated that oceanic P. aeruginosa strains harbor aunique genotype. Their growth and survival under nutrient-free,high-NaCl, and high-pH stress conditions in laboratory-based mi-crocosms have been evaluated, and it was found that they couldsurvive and thrive longer than clinical and freshwater strains (22).In light of these results, we hypothesized that marine P. aerugi-nosa possesses new sequence types and assumes unique positionsin intraspecies phylogenetic trees.
The genome sizes of P. aeruginosa range from 5.2 to 7.1 Mbp(46). Recent work suggests that more than 80% of the genome ofthe sequenced strain (strain PAO1) is shared (with only 0.5%nucleotide divergence) with the isolate from cystic fibrosis pa-tients and with environmental strains (47), indicating that thegenome of this bacterium is characterized by a combination ofrelatively conserved core genes and some variable accessorygenes. Therefore, depending on the genes analyzed and the meth-ods used, apparently various genomic divergences are obtained.
Denamur et al. (6) and Picard et al. (40) argued that thespecies had a panmictic population structure, but Kiewitz andTummler (23) proposed a net-like structure characterized by
high frequencies of recombination. Lomholt et al. (29) favoredan epidemic structure, and Pirnay et al. (41) combined se-quencing-based techniques, such as sequencing of the outermembrane lipoprotein, with serotyping and pyoverdine typedetermination in a polyphasic approach to reveal extensivegenetic mosaicism, particularly in the oprD gene.
Multilocus sequence typing (MLST) is a recently devel-oped strain-typing system that focuses strictly on conservedhousekeeping genes (5, 10, 31, 53). The choice of seven lociensures adequate variability such that one can distinguishbetween the most closely related strains and still be able totrack the global clonal history of the species with the highestpossible accuracy. Because of these advantages, we used anMLST scheme to characterize strains of P. aeruginosa iso-lated from the open ocean, which enabled us to compare ourdata with those in the database generated from variousclinical and environmental P. aeruginosa strains. In addition,we attempted to clarify the phylogenetic relationships ofthese isolates by concatenating seven genes used for MLST.These genes are generally conserved and less likely to un-dergo horizontal gene transfer and are therefore suitable forphylogenetic analysis.
In order to test the hypothesis that marine P. aeruginosacomprises a population uniquely adapted to the ocean envi-ronment, seven housekeeping genes (acsA, aroE, guaA, mutL,nuoD, ppsA, and trpE) from 34 P. aeruginosa strains weresequenced, and the data were compared with those for MLSTalleles in the existing database. The concatenated sequenceswere used for phylogenetic analysis to determine the positionof ocean P. aeruginosa strains relative to other strains. To ourknowledge, this is the first phylogenetic study of marine P.
* Corresponding author. Mailing address: Marine MicrobiologyLaboratory, Ocean Research Institute, University of Tokyo, 1-15-1Minamidai, Nakano-Ku, Tokyo 164-8639, Japan. Phone: 81-3-5351-6834. Fax: 81-3-5351-6482. E-mail: [email protected].
� Published ahead of print on 29 August 2008.
6194
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from
aeruginosa and will contribute significantly to a new ecologicalperspective on these well-known bacterial strains.
MATERIALS AND METHODS
Bacterial strains. Thirty-four environmental and clinical strains were used.The environmental strains were isolated from the open ocean, coastal areas, andfreshwater environments in Japan (Table 1 and Fig. 1). The coastal strains (12strains) included those from Tokyo Bay, Sagami Bay (station S1), Suruga Bay(station N2), and station S4. Although stations N2 and S4 were at least 50 kmaway from the shore, both were north of the Kuroshio Current, and the possibleinfluence of terrestrial environments was not completely rejected; thus, strainsfrom these areas were considered coastal. Two strains each from Kumamoto Bayand Aburatsubo Bay were also included in the coastal category. Kumamoto Bayis far from Tokyo Bay, whereas Aburatsubo Bay is close to Tokyo Bay andlocated between Tokyo Bay and Sagami Bay. Ocean strains (9 strains) wereisolated from the south of the Kuroshio Current (22), where the influence of
human activities is negligible. Freshwater strains were isolated from ponds, lakes,and rivers near the greater Tokyo metropolitan area (22). The sampling locationsfor freshwater strains are shown in Fig. 1. Five strains were isolated from humanblood, sputum, and urine samples and collected from different hospitals in Japan.One strain was isolated from a dead dolphin from Otsuchi Bay, Japan. Thepossible source of the P. aeruginosa strain isolated from the dolphin could not beclarified. As the strain was isolated from a dead dolphin, a possible secondaryinfection could not be rejected, as there are reports of fatal bronchopneumoniaand dermatitis of bottlenose Atlantic dolphins induced by P. aeruginosa (7). Thesources, origins, dates of isolation, and serotypes of the strains are given in Table1. The origins and geographical locations of the sampling points are indicated inFig. 1A and B. Except for the strains numbered Coastal-54 to Coastal-252 (Table1), all strains were analyzed in our previous study (22).
Isolation and identification of P. aeruginosa strains. The isolation methods forP. aeruginosa were described previously (22). Several methods were applied forthe identification of P. aeruginosa. Selective agar culture media were used for theidentification of P. aeruginosa at the initial stage. Suspected colonies that showed
TABLE 1. Strain numbers and corresponding groups, origins, times of isolation, and serotypes for P. aeruginosa and other pseudomonadsused for phylogenetic and MLST analyses
Strain Groupa Origin (station, depth �m�) Time of isolation Serotype Source orreference
P. aeruginosaCoastal-54 Coastal Suruga Bay (N2, 200) March 2004 I This studyCoastal-60 Coastal Suruga Bay (N2, 200) March 2004 I This studyCoastal-86 Coastal Suruga Bay (N2, 0) March 2004 I This studyOcean-100 Ocean Open ocean (N7, 0) March 2004 B This studyArakawa River-206 Freshwater Arakawa River, Saitama (AK1) May 2004 F This studyOcean-222 Ocean Open ocean (S2, 0) May 2004 I This studyOcean-224 Ocean Open ocean (S2, 0) May 2004 E This studyCoastal-235 Coastal Sagami Bay (S1, 0) May 2004 G This studyOcean-238 Ocean Open ocean (S2, 0) May 2004 I This studyCoastal-241 Coastal Sagami Bay (S1, 0) May 2004 G This studyCoastal-242 Coastal Sagami Bay (S1, 0) May 2004 G This studyCoastal-243 Coastal Sagami Bay (S1, 0) May 2004 G This studyDolphin-245 Animal Dolphin May 2004 E This studyCoastal-252 Coastal Aburatsubo Bay, close to Tokyo Bay (0) May 2004 NDb This studyCoastal-601 Coastal Kumamoto Bay (0) December 2003 ND 22Coastal-963 Coastal Tokyo Bay (T1, 0) May 2003 F 22Ocean-1155 Ocean Open ocean (S2, 0) June 2003 E 22Ocean-1170 Ocean Open ocean (S2, 0) June 2003 E 22Ocean-1175 Ocean Open ocean (S2, 0) June 2003 E 22Ocean-1187 Ocean Open ocean (S2, 0) June 2003 E 22Ocean-1206 Ocean Open ocean (S2, 0) June 2003 E 22Coastal-1276 Coastal Tokyo Bay (0) June 2003 B 22Coastal-1303 Coastal North of Kuroshio (S4, 0) June 2003 K 22Arakawa River-1504 Freshwater Arakawa River, Tokyo (AK2) November 2003 K 22Arakawa River-1508 Freshwater Arakawa River, Tokyo (AK4) November 2003 B 22Clinical-1529 Clinical Urine, Tokyo 2002 M 22Pond-1540 Freshwater Zenpukujii Pond, Tokyo September 2003 M 22Pond-1549 Freshwater Zenpukujii Pond, Tokyo September 2003 G 22Tama Lake-1559 Freshwater Tama Lake, Tokyo September 2003 G 22Tama Lake-1563 Freshwater Tama Lake, Tokyo September 2003 G 22Clinical-1683 Clinical Sputum, Okayama 2002 E 22Clinical-1732 Clinical Urine, Myagi 2002 E 22Clinical-1920 Clinical Blood, Saitama 2002 E 22Clinical-1921 Clinical Blood, Saitama 2002 E 22JCM 5962Tc,d Unknown Unknown Unknown ND 8PAO1e Clinical Wound, Australia 1955 ND 49
Other speciesP. fluorescens JCM 5963Tc Environmental Prefilter tanks 1961 ND 9P. putida KT2440e Plant Osaka, Japan 1961 ND 34P. syringae B728ae Plant United States 1987 ND 30P. fluorescens Pf-5e Rhizosphere United States Unknown ND 17
a The strains were assigned to different groups depending on their sources.b ND, not done.c These strains were used for DNA-DNA hybridization.d This strain was used for sequencing and comparative analyses.e These strains were used for comparative analyses, and the data were taken from the GenBank database.
VOL. 74, 2008 PHYLOGENETIC ANALYSES OF OCEAN PSEUDOMONAS AERUGINOSA 6195
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from
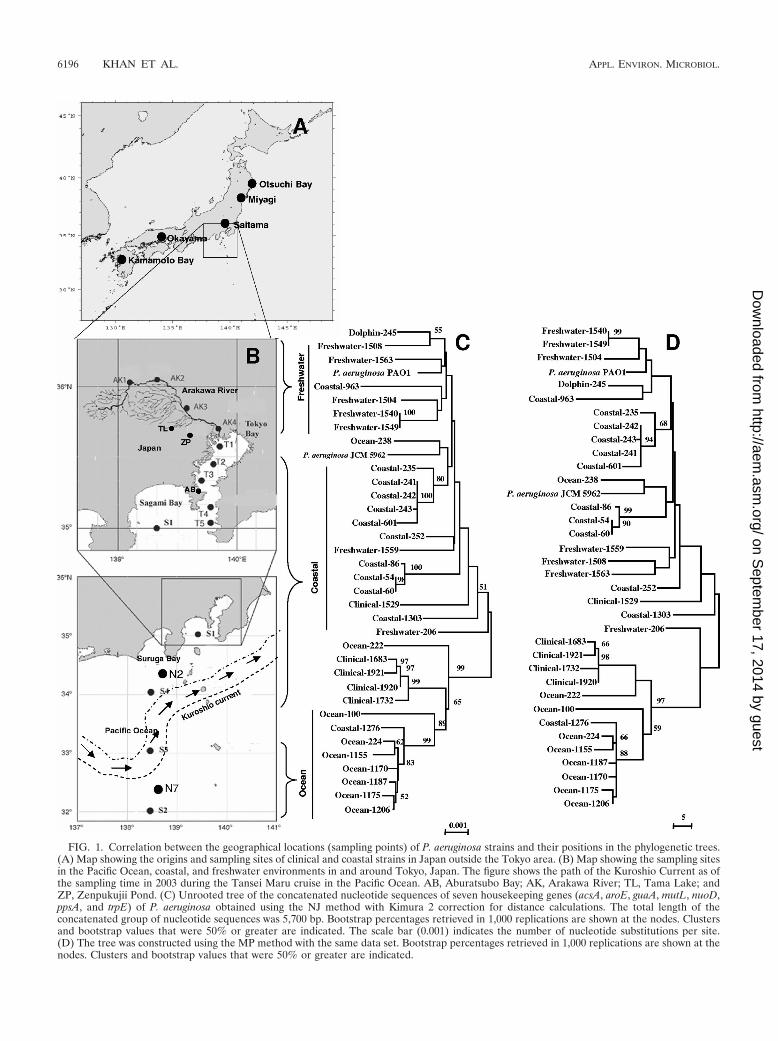
FIG. 1. Correlation between the geographical locations (sampling points) of P. aeruginosa strains and their positions in the phylogenetic trees.(A) Map showing the origins and sampling sites of clinical and coastal strains in Japan outside the Tokyo area. (B) Map showing the sampling sitesin the Pacific Ocean, coastal, and freshwater environments in and around Tokyo, Japan. The figure shows the path of the Kuroshio Current as ofthe sampling time in 2003 during the Tansei Maru cruise in the Pacific Ocean. AB, Aburatsubo Bay; AK, Arakawa River; TL, Tama Lake; andZP, Zenpukujii Pond. (C) Unrooted tree of the concatenated nucleotide sequences of seven housekeeping genes (acsA, aroE, guaA, mutL, nuoD,ppsA, and trpE) of P. aeruginosa obtained using the NJ method with Kimura 2 correction for distance calculations. The total length of theconcatenated group of nucleotide sequences was 5,700 bp. Bootstrap percentages retrieved in 1,000 replications are shown at the nodes. Clustersand bootstrap values that were 50% or greater are indicated. The scale bar (0.001) indicates the number of nucleotide substitutions per site.(D) The tree was constructed using the MP method with the same data set. Bootstrap percentages retrieved in 1,000 replications are shown at thenodes. Clusters and bootstrap values that were 50% or greater are indicated.
6196 KHAN ET AL. APPL. ENVIRON. MICROBIOL.
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from

the characteristic appearance and color on nalidixic acid cetrimide agar plateswere inoculated onto cetrimide kanamycin nalidixic acid agar (26) and incubatedat 42°C overnight. Isolates that showed growth at 42°C were primarily identifiedas P. aeruginosa. Colonies were transferred to other selective agar plates andincubated at 4°C for 3 to 4 days. No colonies appeared at 4°C. Sequences of the16S rRNA gene were determined by direct sequencing using the primers 27f and1492r (28). The PCR products were purified with an EXOSAP-IT kit (USBCorp., Cleveland, OH), sequenced using an ABI Prism BigDye Terminator cyclesequencing ready reaction kit (Applied Biosystems, Foster city, CA), and run onan ABI 3100 genetic analyzer (Applied Biosystems). BLAST programs were usedto find the most closely related sequences (2). The isolates were also identifiedby the BD (Becton, Dickinson and Company, MD) Phoenix automated micro-biology system according to the method previously described by Khan et al. (22).It is necessary to perform DNA-DNA hybridization to judge whether a novelisolate belongs to a species when the isolate and species representatives sharemore than 97% 16S rRNA gene sequence similarity (14, 48). Thus, two marinestrains were randomly selected for DNA-DNA hybridization. P. aeruginosa JCM5962T and P. fluorescence JCM 5963T were selected as the same species and aclosely related species, respectively. DNA-DNA hybridization was determined byusing photobiotin-labeled probes in a polystyrene 96-well plate (Nunc Maxisorp)as described by Ezaki et al. (11), utilizing Bacteroides sp. strain IAM 15343 as anegative control.
Culture of isolates and preparation of chromosomal DNA. Bacterial strainswere maintained at �80°C in 40% (vol/vol) glycerol in nutrient broth, streakedto single colonies, and cultured on Mueller-Hinton agar at 37°C under aerobicconditions. Chromosomal DNA was extracted from 1-ml cultures of strainsgrown overnight in nutrient broth by using a Qiagen DNeasy tissue extraction kit(Valencia, CA).
Locus selection. The seven genes acsA, aroE, guaA, mutL, nuoD, ppsA, andtrpE (see Table 3) were selected according to the MLST scheme for P. aeruginosa(http://pubmlst.org/paeruginosa/). The criteria governing locus selection in-cluded biological role (comprising genes with diverse central housekeeping roles,such as mismatch repair, DNA replication, and amino acid biosynthesis), size(�600 bp), location (i.e., a minimum of 6 kbp upstream or downstream fromknown virulence factors, lysogenic phages, or insertion sequence elements), andsuitability for nested primer design and sequence diversity (5). All phylogeneticanalyses were performed with the same criteria.
Amplification of the loci. The seven housekeeping genes were amplified withZ-Taq polymerase (Takara Bio, Inc., Shiga, Japan), using the primers describedby Curran et al. (5). The primers had variable melting temperatures. The 25-�lamplification reaction mixture comprised 0.5 to 1.0 �l of chromosomal DNA(final concentration, 4 to 8 ng/�l), 0.5 �l each of the 10-pmol primers, 2.5 �l 10�Z-Taq buffer, 0.25 �l of Takara Z-Taq (2.5 units/�l), and 2 �l of deoxynucleosidetriphosphate mixture (2.5 mM) and was brought up to the correct reactionvolume with sterilized distilled water. The following PCR protocol was used: 1 sof denaturing at 98°C and 30 to 33 cycles (depending on the gene) of 1 s at 98°C(denaturing), 10 s at 57°C to 60°C (primer annealing), and 20 s at 72°C (poly-merase extension). Reactions were performed with a PTC-100 thermal cycler(Bio-Rad Laboratories, Inc., Hercules, CA).
BigDye reaction and sequencing. Two microliters of ExoSAP-IT was addeddirectly to the 7-�l reaction products following PCR. ExoSAP-IT was inactivatedat 80°C for 15 min, and the treated PCR products were ready for subsequentanalysis in BigDye reactions, which required the DNA to be free of excessprimers and nucleotides. BigDye reactions were carried out using an ABI PrismBigDye Terminator cycle sequencing ready reaction kit (Applied Biosystems,Foster City, CA). DNA sequencing was performed with an ABI 3100 geneticanalyzer (Applied Biosystems) with a 50-cm array. Both forward and reversesequences were obtained by using the PCR primer or internal sequence primers(specifically for MLST sequences) for each locus.
Sequence data analysis. Both the forward and the reverse primer sequences ofacsA, aroE, guaA, mutL, nuoD, ppsA, and trpE were verified with the Chromassoftware program, version 2.32 (Technelysium Pty., Ltd.). Several representativeDNA sequences were randomly selected from each group of isolates and con-verted into amino acid sequences, and multiple alignments of these sequenceswere calculated. These results and the published alignment data were used astemplates to conduct multiple alignments of all DNA sequences determined(45). Positions in which gaps were present in any of the aligned sequences wereexcluded from the analysis. All sequences were confirmed by individual BLASTsearches to determine whether the corresponding data matched well with theappropriate gene of P. aeruginosa. Both forward and reverse primer sequenceswere assembled by using ATGC, version 4.0 (Genetyx Corporation, Tokyo,Japan). The assembled sequences were saved with Genetyx-Win, version 5.0(Genetyx Corporation, Tokyo, Japan) in Fasta format. The alignment was per-
formed using a hierarchical method for multiple alignments implemented in theCLUSTAL-W tool in MEGA 3.1 (27, 51). Automatically aligned sequences werechecked manually.
Allele and strain type assignment. A number was assigned to each distinctallele within a locus according to the number available in the P. aeruginosa MLSTdatabase. Any allele that did not match with existing data was designated “new.”Each isolate was therefore given seven numbers that represented its strain type.Any strain type that did not match with the existing database was numbered asa “new” type. MLST analysis yielded alleles for individual loci of seven house-keeping genes. The alleles were numbered according to their entries in theMLST database. Alleles that did not exist in the database were termed “new.” Atable was prepared (see Table 4), and all the alleles were provided separatenumbers. Strains having the same allele were given the same number.
Phylogenetic analysis. Similarity and evolutionary distance were calculatedwith different software programs. Gene distance matrices were calculated fromnucleotide sequences by the Jukes-Cantor method (20). Mann-Whitney’s U testwas employed for within-group and between-group mean distances, and signif-icance levels were calculated (32). Dendrograms were generated by neighborjoining (NJ), maximum parsimony (MP), minimum evolution, the unweighted-pair group method with arithmetic means, and split decomposition (SD) withbootstrap analysis (1,000 replications) using MEGA (27) and PAUP* (version 4;D. L. Swofford, Sinauer Associates, Sunderland, MA). Analyses were performedon individual gene sequences as well as on the concatenated data set. Rooted andunrooted NJ trees of the concatenated seven-gene sequence data were generatedwith MEGA, version 3.1 (27), using the Kimura 2 parameter model (25) withgamma correction and 1,000 bootstrap replicates for all sequences. In NJ anal-yses, closely related pseudomonads were used as an outgroup. The data foroutgroups were collected from the NCBI GenBank database. An MP tree wasgenerated from the concatenated gene sequence data from which bootstrappercentages were retrieved for 1,000 replicates. The number of polymorphicnucleotide sites and the ratio of the number of nonsynonymous substitutions tothe number of synonymous substitutions (the dN/dS ratio) were calculated withthe MEGA and START programs (18, 19, 27, 35).
Nucleotide sequence accession numbers. The nucleotide sequences deter-mined in this study were submitted to GenBank. The accession numbers for the16S rRNA gene, acsA, aroE, guaA, mutL, nuoD, ppsA, and trpE are EU710848 toEU710881, EU561186 to EU561219, EU56084 to EU561117, EU561118to EU561151, EU561152 to EU561185, EU590764 to EU590797, EU590730 toEU590763, and EU590696 to EU590729, respectively.
RESULTS
Identification of P. aeruginosa isolates. The marine strainswere confirmed as P. aeruginosa following observation of theirphysical properties (22) by BD Phoenix automated microbiol-ogy systems (22) and 16S rRNA sequence data. The 16S rRNAdata revealed 99 to 100% similarity to the existing P. aerugi-nosa database in GenBank. When a dendrogram was preparedfrom the 16S rRNA sequence data and other P. aeruginosastrains were used as the outgroup, all P. aeruginosa strainsformed a single cluster. The distance between P. aeruginosaand other Pseudomonas spp. was supported by a 99% boot-strap value (Fig. 2). All P. aeruginosa strains belonged to asingle cluster, in which P. aeruginosa PAO1 and type strain P.aeruginosa JCM 5962T were included. Individual gene se-quence data for each of the seven housekeeping genes alsoshowed 98 to 100% similarity to the sequences in the P. aerugi-nosa database in GenBank when BLAST searches were carriedout. For further confirmation of the P. aeruginosa strains, astandard DNA-DNA hybridization method was employed. Thelevels of DNA-DNA relatedness at the optimal temperature(51.6°C for P. aeruginosa, which was calculated based on G�Cmolecule contents) within each species were 89.8 to 93.2%(Table 2). All DNA-DNA relatedness values were above thethreshold value that has been suggested as delineating thesame bacterial species (approximately 70%) under optimalhybridization conditions (12). On the other hand, the values
VOL. 74, 2008 PHYLOGENETIC ANALYSES OF OCEAN PSEUDOMONAS AERUGINOSA 6197
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from

for DNA-DNA relatedness among the selected strains andPseudomonas fluorescens were less than 70% (Table 2). Thisconfirmed the view that strains Ocean-1187 and Coastal-54represent the species P. aeruginosa.
Nucleotide fragments and allelic profiles. The fragmentlengths for MLST data (obtained from MLST primers) andwhole-gene sequence data (obtained from PCR amplificationprimers) for the seven housekeeping genes and their variablesites are given in Table 3. The allelic sequences obtained weresearched for in the P. aeruginosa MLST database. For aroE, allprofiles were found in the existing database, whereas one
FIG. 2. Rooted tree of 16 rRNA gene sequence data of P. aeruginosa along with data on other Pseudomonas spp. obtained using the NJ methodwith Kimura 2 correction for distance calculations. Bootstrap percentages retrieved in 1,000 replications are shown at the nodes. Clusters andbootstrap values that were 50% or greater are indicated. The scale bar (0.005) indicates the number of nucleotide substitutions per site. Data forP. aeruginosa JCM 5962T were collected from the DDBJ database, and data for all outgroup strains were collected from the NCBI GenBankdatabase.
TABLE 2. DNA-DNA relatedness between strains Ocean-1187,Coastal-54, P. aeruginosa JCM 5962T, and P. fluorescens 5963T
Strain
DNA-DNA relatedness (%) for indicated strain
Ocean-1187
Coastal-54
P. aeruginosaJCM 5962T
P. fluorescensJCM 5963T
Ocean-1187 100 89.8 89.8 10.9Coastal-54 100 93.2 10.1P. aeruginosa JCM
5962T100 12.7
P. fluorescens JCM5963T
100
6198 KHAN ET AL. APPL. ENVIRON. MICROBIOL.
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from
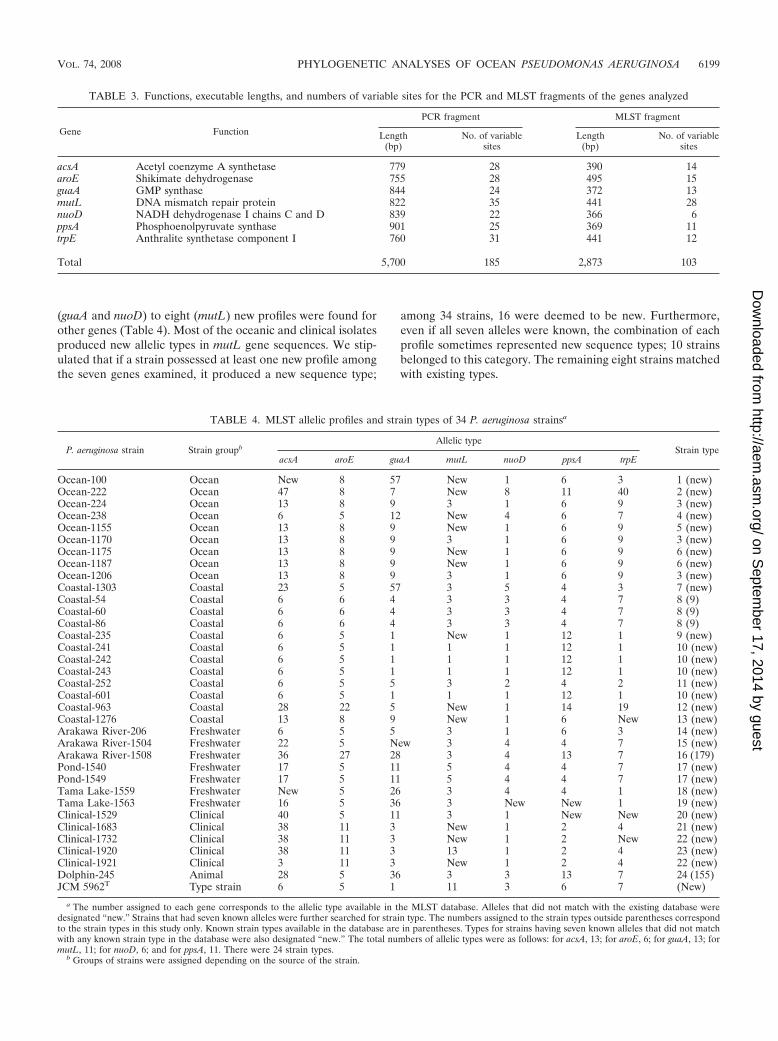
(guaA and nuoD) to eight (mutL) new profiles were found forother genes (Table 4). Most of the oceanic and clinical isolatesproduced new allelic types in mutL gene sequences. We stip-ulated that if a strain possessed at least one new profile amongthe seven genes examined, it produced a new sequence type;
among 34 strains, 16 were deemed to be new. Furthermore,even if all seven alleles were known, the combination of eachprofile sometimes represented new sequence types; 10 strainsbelonged to this category. The remaining eight strains matchedwith existing types.
TABLE 3. Functions, executable lengths, and numbers of variable sites for the PCR and MLST fragments of the genes analyzed
Gene Function
PCR fragment MLST fragment
Length(bp)
No. of variablesites
Length(bp)
No. of variablesites
acsA Acetyl coenzyme A synthetase 779 28 390 14aroE Shikimate dehydrogenase 755 28 495 15guaA GMP synthase 844 24 372 13mutL DNA mismatch repair protein 822 35 441 28nuoD NADH dehydrogenase I chains C and D 839 22 366 6ppsA Phosphoenolpyruvate synthase 901 25 369 11trpE Anthralite synthetase component I 760 31 441 12
Total 5,700 185 2,873 103
TABLE 4. MLST allelic profiles and strain types of 34 P. aeruginosa strainsa
P. aeruginosa strain Strain groupbAllelic type
Strain typeacsA aroE guaA mutL nuoD ppsA trpE
Ocean-100 Ocean New 8 57 New 1 6 3 1 (new)Ocean-222 Ocean 47 8 7 New 8 11 40 2 (new)Ocean-224 Ocean 13 8 9 3 1 6 9 3 (new)Ocean-238 Ocean 6 5 12 New 4 6 7 4 (new)Ocean-1155 Ocean 13 8 9 New 1 6 9 5 (new)Ocean-1170 Ocean 13 8 9 3 1 6 9 3 (new)Ocean-1175 Ocean 13 8 9 New 1 6 9 6 (new)Ocean-1187 Ocean 13 8 9 New 1 6 9 6 (new)Ocean-1206 Ocean 13 8 9 3 1 6 9 3 (new)Coastal-1303 Coastal 23 5 57 3 5 4 3 7 (new)Coastal-54 Coastal 6 6 4 3 3 4 7 8 (9)Coastal-60 Coastal 6 6 4 3 3 4 7 8 (9)Coastal-86 Coastal 6 6 4 3 3 4 7 8 (9)Coastal-235 Coastal 6 5 1 New 1 12 1 9 (new)Coastal-241 Coastal 6 5 1 1 1 12 1 10 (new)Coastal-242 Coastal 6 5 1 1 1 12 1 10 (new)Coastal-243 Coastal 6 5 1 1 1 12 1 10 (new)Coastal-252 Coastal 6 5 5 3 2 4 2 11 (new)Coastal-601 Coastal 6 5 1 1 1 12 1 10 (new)Coastal-963 Coastal 28 22 5 New 1 14 19 12 (new)Coastal-1276 Coastal 13 8 9 New 1 6 New 13 (new)Arakawa River-206 Freshwater 6 5 5 3 1 6 3 14 (new)Arakawa River-1504 Freshwater 22 5 New 3 4 4 7 15 (new)Arakawa River-1508 Freshwater 36 27 28 3 4 13 7 16 (179)Pond-1540 Freshwater 17 5 11 5 4 4 7 17 (new)Pond-1549 Freshwater 17 5 11 5 4 4 7 17 (new)Tama Lake-1559 Freshwater New 5 26 3 4 4 1 18 (new)Tama Lake-1563 Freshwater 16 5 36 3 New New 1 19 (new)Clinical-1529 Clinical 40 5 11 3 1 New New 20 (new)Clinical-1683 Clinical 38 11 3 New 1 2 4 21 (new)Clinical-1732 Clinical 38 11 3 New 1 2 New 22 (new)Clinical-1920 Clinical 38 11 3 13 1 2 4 23 (new)Clinical-1921 Clinical 3 11 3 New 1 2 4 22 (new)Dolphin-245 Animal 28 5 36 3 3 13 7 24 (155)JCM 5962T Type strain 6 5 1 11 3 6 7 (New)
a The number assigned to each gene corresponds to the allelic type available in the MLST database. Alleles that did not match with the existing database weredesignated “new.” Strains that had seven known alleles were further searched for strain type. The numbers assigned to the strain types outside parentheses correspondto the strain types in this study only. Known strain types available in the database are in parentheses. Types for strains having seven known alleles that did not matchwith any known strain type in the database were also designated “new.” The total numbers of allelic types were as follows: for acsA, 13; for aroE, 6; for guaA, 13; formutL, 11; for nuoD, 6; and for ppsA, 11. There were 24 strain types.
b Groups of strains were assigned depending on the source of the strain.
VOL. 74, 2008 PHYLOGENETIC ANALYSES OF OCEAN PSEUDOMONAS AERUGINOSA 6199
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from

Of the 34 P. aeruginosa strains isolated from differentsources, we found 24 distinct strain types. Among them, three(types 9, 155, and 179) existed in the MLST database (Table 4),while the other 21 were new. Three coastal strains (Coastal-54,Coastal-60, and Coastal-86) belonged to type 9. Three strainswith this type had been deposited, one from an unknownsource from France (strain 5757) and two from sputum sam-ples from Canada (strains R1 and T1). The strain Dolphin-245,isolated from a dolphin in Otsuchi Bay, Iwate Prefecture, Ja-pan, matched with type 155, which comprises two clinicalstrains from Canada (strains B5 and S3). The Arakawa River-1508 strain, from Arakawa River, matched with type 179,which includes five clinical strains from Canada (strains B4,E4SIB1, C1, W4, and C2). Other than for these three types, thestrains did not match with any types available in the 409-straindatabase. Among ocean (9 strains) and coastal (12 strains)isolates, six and six new strain types, respectively, were found.
Genetic distances between the P. aeruginosa groups. Theaverage mean genetic distances between and within the de-signed P. aeruginosa groups were calculated by MEGA. Mann-Whitney’s U test was carried out to see whether the differenceswithin and between the group average means were statisticallysignificant (32). In concatenated gene sequences (5,700 bp),the distances within groups were lower than the distances be-tween groups (P � 0.05). The average mean distance betweenocean-derived and other P. aeruginosa groups varied from0.0075 to 0.0092, which was higher than the within-group av-erage mean distance (P � 0.05) (Table 5).
Phylogenetic analysis and comparison with the geographicallocations of the origins of the strains. In order to assess thephylogenetic relationships between all the strains tested, PCRfragments for each gene, as well as the concatenated sequencesof all PCR fragments, were analyzed to construct trees basedon NJ, MP, minimum evolution, the unweighted-pair groupmethod with arithmetic means, or SD. Thirty-nine strains wereused, including P. aeruginosa PAO1, P. aeruginosa JCM 5962T,and three outgroup species strains (Pseudomonas putidaKT2440, P. fluorescens Pf-5, and Pseudomonas syringae B728a)obtained from the NCBI database. Because the most conser-vative segments of the housekeeping genes were used in thisstudy, the distance to the outgroups was too great to display inthe tree. Only the unrooted trees are shown (Fig. 1 and 4).Figure 3 shows the rooted NJ tree of the concatenated data setof seven housekeeping genes with the outgroup.
In this study, we present only the data derived by the NJ and
MP methods, as other methods showed similar patterns for thetrees, so they were eliminated. Figure 1 shows two unrootedtrees constructed by NJ and MP methods from concatenatedgene sequence data together with a map identifying the geo-graphical locations of the isolates. All open-ocean strainsshowed a trend of forming separate clusters in the trees excepttwo ocean P. aeruginosa strains. Except for strains Ocean-222and Ocean-238, all ocean strains formed a single cluster, withone coastal strain (Coastal-1276) included. Although the fresh-water strains did not show a distinct cluster, they were mostlyseparated from the other strains, with the exception of strainsFreshwater-206 and -1559. Strains P. aeruginosa PAO1, Dol-phin-245, and Coastal-963 were included in the freshwatergroup. Mixing of coastal strains with freshwater strains mightindicate the influence of freshwater outfalls on coastal systems.Although the coastal strains are rather scattered in the tree,they were mostly close to each other and formed two separateclusters (one from the strains of Sagami Bay and one fromthose of Suruga Bay) that were always prominent. Four of thefive clinical isolates also clustered together.
Phylogenetic analysis of individual loci. The relationshipsbetween the 34 strains were further analyzed by constructingunrooted trees using the NJ method based on the nucleotidesequences of PCR fragments for each gene (data not shown).Ocean alleles were clustered with respect to acsA, aroE, guaA,ppsA, and trpE. This clustering was supported by bootstrapvalues higher than 50%, indicating their distinct positions inthe trees.
Phylogenetic analysis of the MLST sequences. To clarify thephylogenetic positions of marine isolates among those fromvarious sources, concatenated allelic data on 28 strains ran-domly selected from the MLST database were obtained. Inaddition, the corresponding sequence of P. aeruginosa PAO1was taken from the NCBI database, and the sequence of typestrain P. aeruginosa JCM 5962T was added. Unrooted treeswere constructed for all strains through different methods.Regardless of the methodology used for designing phyloge-netic trees, all open-ocean strains except two (Ocean-238 andOcean-222) invariably formed clusters distinct from those ofother strains. Here, we present a radiation tree prepared by theNJ method, using MEGA as the representative method (Fig.4). The clinical and soil strains taken from the database tendedto scatter throughout the entire tree. The freshwater strainsshowed loose association with some clinical strains. Three sep-arate clusters were observed for marine strains, two for coastal
TABLE 5. Average genetic distances within and between P. aeruginosa groupsa
Group Distance withingroup
Distance between groups
Ocean Coastal Freshwater Clinical Animal PAO1 Outgroup
Ocean 0.0046 0 0.0077 0.0086 0.0075 0.0092 0.0078 0.2439Coastal 0.0047 0 0.0052 0.0075 0.0053 0.0047 0.2434Freshwater 0.0047 0 0.0080 0.0046 0.0035 0.2438Clinical 0.0042 0 0.0087 0.0076 0.2426Animal NCb 0 0.0031 0.2442PAO1 NCb 0 0.2431Outgroup 0.1800 0
a Distances were calculated for the concatenated PCR sequences (5,700 bp) by using MEGA. Rates among sites were uniform. The pattern among lineages washomogeneous. The total numbers of strains are as follows: open ocean, 9; coastal, 12; freshwater, 7; clinical, 5; animal, 1; PAO1, 1; and outgroup, 3.
b NC, could not be calculated.
6200 KHAN ET AL. APPL. ENVIRON. MICROBIOL.
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from

isolates and one for ocean strains. Each coastal cluster wascomposed of isolates from different geographical areas. Thebiggest and most distinct cluster was represented by the P.aeruginosa strains from the ocean.
DISCUSSION
Our previous studies revealed that the oceanic P. aeruginosastrains have advantages over other strains with regard to theirphysiological adaptability to high-NaCl and pH stresses in lab-oratory-based microcosms. Molecular typing studies (pulsed-
field gel electrophoresis) also indicated a clonal structure foroceanic P. aeruginosa strains that was distinct from those ofother strains (22). Based on these results, we hypothesized thatthere are new sequence types of ocean P. aeruginosa strains notfound in other strains, which confirms that the ocean P. aerugi-nosa strains have diverged from other isolates and form adistinct cluster, as determined by MLST and phylogeneticanalyses of seven housekeeping genes.
In the present study, we applied MLST analysis to 34 strainsand found that most marine strains represent new types. Inaddition, analysis using concatenated gene sequences indicated
FIG. 3. Rooted tree of the concatenated nucleotide sequences of seven housekeeping genes (acsA, aroE, guaA, mutL, nuoD, ppsA, and trpE)of P. aeruginosa obtained using the NJ method with Kimura 2 correction for distance calculations. The total length of the concatenated group ofnucleotide sequences was 5,700 bp. Bootstrap percentages retrieved in 1,000 replications are shown at the nodes. Clusters and bootstrap valuesthat were 50% or greater are indicated. The scale bar (0.02) indicates the number of nucleotide substitutions per site.
VOL. 74, 2008 PHYLOGENETIC ANALYSES OF OCEAN PSEUDOMONAS AERUGINOSA 6201
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from

that ocean and coastal strains form their own phylogeneticclusters (Fig. 1, 3, and 4). Although freshwater strains tend tomix with soil or clinical strains (Fig. 4), the phylogenetic posi-tions of ocean, coastal, and freshwater strains roughly reflecttheir geographic distributions, with some exceptions (Fig. 1).To our knowledge, this is the first report on phylogeneticanalysis of marine P. aeruginosa and the first finding of apossible connection of the phylogeny of this bacterium to geo-graphical distribution in aquatic environments.
We used MLST in this study primarily because it enabled usto compare our data with those on other strains, especiallyclinical isolates and environmental isolates from various areas.In addition, phylogenetic analysis of each gene and concate-
nated gene fragments clarifies the intraspecies phylogeneticpositions of marine strains. The MLST database so far includes409 types of P. aeruginosa (4) (http://www.mlst.org/) but doesnot yet include any marine or coastal P. aeruginosa strain. Asthis study is the first application to marine isolates, we used theanalytical methods with care. Genes were sequenced usingestablished primers (5), and only assembled sequences fromforward and reverse primers were used.
In addition to the type determination, the genetic related-ness among strains was calculated from the MLST allelic pro-files and the whole-gene sequence data by the phylogeneticanalyses shown in Table 4 and Fig. 1, 3, and 4, which indicatesthat the relatedness is primarily dependent on the origins. The
FIG. 4. Unrooted and NJ radiation tree based on concatenated MLST sequences of seven gene loci. acsA, aroE, guaA, mutL, nuoD, ppsA, andtrpE gene sequences (2,873 bp) from 63 taxa (34 from the present study, 28 from the MLST database, and 1 P. aeruginosa PAO1 strain from thedatabase) were concatenated and constructed by the MEGA 3.1 program. Sequence similarity was corrected for by using the Kimura 2 parameter.All nodes were supported by 1,000 bootstrap replications. The largest cluster was marked as open ocean, as all strains were from the ocean, andtwo other clusters were marked as coastal. The scale bar (0.001) indicates the number of nucleotide substitutions per site.
6202 KHAN ET AL. APPL. ENVIRON. MICROBIOL.
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from

ocean isolates formed one cluster (except Ocean-222 andOcean-238), which includes only one coastal strain (Coastal-1276) and none from other environments. The presence ofsuch clusters is also implied by the trees constructed for eachgene (data not shown), a result further supported by MEGAanalysis. The unrooted phylogenetic trees shown in Fig. 1 dem-onstrate a rough relationship between the geographic distribu-tions and phylogenetic positions of the strains. The unrootedtree constructed from concatenated MLST data further clari-fied the presence of three clusters unique to ocean and coastalenvironments (Fig. 4). These results confirmed our hypothesisregarding the presence of distinct marine types. As the possi-bility of mixing of freshwater with the coastal environmentscould not be rejected, it is assumed that the influence of fresh-water and terrestrial environments reflected on the distribu-tions of the coastal strains between clinical and freshwaterenvironments and the separation of freshwater strains towardthe other parts of the trees. On the other hand, the uniqueocean environment retained a distinct group of P. aeruginosastrains, which always formed a single cluster, separated fromthe other isolates, with the exception of one coastal strain(Coastal-1276). A possible explanation for this kind of distri-bution is difficult to infer. We sampled seawater across theKuroshio Current, primarily because the influence of freshwa-ter inflow is hardly expected in the south part of the current. Inother words, those strains are not recent contaminants of ter-restrial origin. However, this does not necessarily mean thatcoastal strains should never be found in the open ocean or thatthose strains should never survive for a considerable time inthe open ocean. The ocean is composed of one huge watermass without physical barriers. Although the physical processis unknown, coastal strains may be transferred to the openocean and survive for years. As P. aeruginosa was regarded asa potential pathogen of marine mammals causing broncho-pneumonia and dermatitis (7), it may be possible for the patho-gen to travel with an ocean mammal from the open ocean tothe coastal region or vice versa. If there are certain carriers,such as dolphins, they may release strains that are apparentlyof coastal origins. The distributions of ocean strains 222 and238 may also be clarified in a similar way.
The Kuroshio Current is also a variable phenomenon in theSouth Pacific Ocean regarding its flow path. It always changesits path. The samples were collected twice from inside andoutside the Kuroshio Current, which indicated a clear distinc-tion among the strains inside and outside the Kuroshio Cur-rent. At this stage, we cannot conclude that Kuroshio is theonly factor that differentiated coastal and open-ocean P.aeruginosa strains. Further studies are needed to make such acomment.
Our idea on the biogeography of P. aeruginosa is that themajority of strains from a certain environment are composedof one or a few particular groups that are unique to the envi-ronment. The opposite idea is “random” distribution regard-less of environment. We believe that the data shown in Fig. 1support the former idea but never support the latter.
Recently, the biogeography of free-living bacteria has drawnconsiderable attention because of the accumulation of datafrom various environments. Theoretically, biogeographical dis-tributions may be explained either by environmental variationsor by historical events. We believe that our present results
indicate that environmental variations may be more importantthan the historical aspects. We also believe that our work is ofconsiderable impact on such scientific issues because this studyis based on multiple gene sequences of many isolates, whereasmost works on biogeography are solely dependent on geneticinformation without any culture.
MLST has been also applied to other bacterial species forsimilar purposes. Sarkar and Guttman (45) carried out a studyof the evolution of core genes of P. syringae, demonstrating thepresence of a distinct group of strains from the SD of theircombined phylogenetic tree. Cladera et al. (4) compared sevenmarine Pseudomonas stutzeri strains isolated from Barcelona,Spain, with strains from other sources by MLST. This analysisindicated that among the 25 total strains investigated, therewere 24 alleles, which included seven marine types. The NJtree drawn by the data indicated that marine P. stutzeri strainswere distributed throughout the tree, suggesting that the di-vergence patterns of P. aeruginosa and P. stutzeri may be dif-ferent. However, it should also be noted that the presentMLST method was originally developed primarily to type clin-ical strains. Selecting a greater number of variable sites orgenes, which increases the sensitivity toward aquatic strains,may offer better resolution and reliability as a typing method.For phylogenetic work, the analysis of more gene sequenceswill definitely be required.
An understanding of the community structure and geneticrelatedness among strains of P. aeruginosa present in nature(sea, river, soil, plants, and animals) is essential for gaining agreater insight into the ecology and distribution of this bacte-rium. There have been debates on the extent to which theyshare common niches in the environment and the extent towhich environmental and clinical strains are genetically similar.Recently, Pirnay et al. (42) aimed to determine the biodiversityamong the P. aeruginosa population of a limited natural envi-ronment, the Woluwe River in Belgium, and compared it withthe global biodiversity. They found that the population struc-ture in that particular area, a small river, seemed to be com-parable with that on the global scale. P. aeruginosa residesprimarily in an aquatic environment, yet the organism is indeeda typical opportunistic bacterium and thrives well, eventuallycausing disease, independent of geographical location or host-specific niche. They concluded that P. aeruginosa is an autoch-thonous member of the bacterial community of the WoluweRiver, which strongly suggests a worldwide spread of a homo-geneously diverse population of this bacterium. In the presentstudy, the strong support of the NJ tree by the MP tree re-garding formation of a single cluster of the ocean strains andthe correlation between phylogenetic position and geographi-cal location give insight into the adaptation of these strains toa particular environment.
Aoi et al. (3) proposed that P. aeruginosa strains isolatedfrom the Ushubetsu River in Japan originated primarily fromenvironments of human activity. Many other scientists haveobserved that environmental P. aeruginosa isolates are geno-typically, chemotaxonomically, and functionally indistinguish-able from clinical isolates (1, 13, 36, 43, 44). Together, theseworks indicate that the populations present in different envi-ronments are highly intermingled or that high rates of geneticrecombination lead to relatively homogeneous genetic struc-tures among the strains.
VOL. 74, 2008 PHYLOGENETIC ANALYSES OF OCEAN PSEUDOMONAS AERUGINOSA 6203
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from

Our results, however, indicate a divergence of the marine P.aeruginosa strains from the other strains. The open ocean isgenerally characterized by low nutrient levels, high salinity, andalkaline pH (�8). Therefore, it seems reasonable to assumethat these conditions select certain types of strains that differfrom those in terrestrial or freshwater environments. In addi-tion, it is difficult to imagine how terrestrial strains can betransported to the open ocean several hundred kilometersaway from the shore, cross the Kuroshio Current (22), and mixwith autochthonous populations. However, our results do notreject the possibility that further analysis of more genes ofmore strains or an entire genome may show high genetic sim-ilarities between marine strains and other strains. It is sug-gested that at least the core genes of P. aeruginosa are ex-tremely conserved. Further work with a greater number ofenvironmental strains from diversified geographical locationsand with various genes is required to answer this question.Currently, we are sequencing the entire genome of one of themarine strains. The data will be published elsewhere soon.
In conclusion, the presence of marine P. aeruginosa strainsthat have unique genotypes and distinct clusters was confirmedby MLST and phylogenetic analysis for the first time. In addi-tion, the geographic distributions of these marine strains havea roughly close relation to intraspecies phylogenetic position,which needs to be clarified by further analyses with morestrains from various geographical locations and origins. Thesefindings will have a considerable impact on the study of theorigin, ecology, physiology, and genetic characteristics of thiswell-known bacterium.
ACKNOWLEDGMENTS
We thank the Japanese Society for the Promotion of Sciences forsponsoring Nurul Huda Khan with a postdoctoral fellowship. Thestudy was supported by Grants-in-Aid for Creative Basic Research, no.12NP0201 (DOBIS) and no. 14208063, from the Ministry of Educa-tion, Culture, Sports, Science and Technology (MEXT), Japan.
We are grateful to Yoshikazu Ishii, Department of Microbiologyand Infectious Diseases, Toho University School of Medicine, forproviding clinical P. aeruginosa strains. We remain indebted to Rita R.Colwell, University of Maryland, for her valuable comments and in-spiration regarding this study. This publication made use of the P.aeruginosa MLST website (http://pubmlst.org/paeruginosa/), devel-oped by Keith Jolley and located at the University of Oxford (18). Thedevelopment of this site has been funded by the Wellcome Trust.
REFERENCES
1. Alonso, A., F. Rojo, and J. L. Martínez. 1999. Environmental and clinicalisolates of Pseudomonas aeruginosa show pathogenic and biodegradativeproperties irrespective of their origin. Environ. Microbiol. 1:421–430.
2. Altschul, S. F., L. M. Thomas, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller,and D. J. Lipman. 1997. Gapped BLAST & PSI-BLAST: a new generationof protein database search programs. Nucleic Acids Res. 25:3389–3402.
3. Aoi, Y., H. Nakata, and H. Kida. 2000. Isolation of Pseudomonas aeruginosafrom Ushubetsu River water in Hokkaido, Japan. Jpn. J. Vet. Res. 48:29–34.
4. Cladera, A. M., A. Bennasar, M. Barcelo, L. Lalucat, and E. Garcia-Valdes.2004. Comparative genetic diversity of Pseudomonas stutzeri genomovars,clonal structure, and phylogeny of the species. J. Bacteriol. 186:5239–5248.
5. Curran, B., D. Jonas, H. Grundmann, T. Pitt, and C. G. Dowson. 2004.Development of a multilocus sequence typing scheme for the opportunisticpathogen Pseudomonas aeruginosa. J. Clin. Microbiol. 42:5644–5649.
6. Denamur, E., B. Picard, G. Decoux, J. B. Denis, and J. Elion. 1993. Theabsence of correlation between allozyme and rrn RFLP analysis indicates ahigh gene flow rate within human clinical Pseudomonas aeruginosa isolates.FEMS Microbiol. Lett. 110:275–280.
7. Diamond, S. S., D. E. Ewing, and G. A. Cadwell. 1979. Fatal bronchopneu-monia and dermatitis caused by Pseudomonas aeruginosa in an Atlanticbottle nosed dolphin. J. Am. Vet. Med. Assoc. 175:984–987.
8. Editorial Secretary for the Judicial Commission of the International Com-
mittee on Nomenclature of Bacteria. 1970. Opinion 36: designation of strainATCC 10145 as the neotype strain of Pseudomonas aeruginosa (Schroeter)Migula. Int. J. Syst. Bacteriol. 20:15–16.
9. Editorial Secretary for the Judicial Commission of the International Com-mittee on Nomenclature of Bacteria. 1970. Opinion 37: designation of strainATCC 13525 as the neotype strain of Pseudomonas fluorescens Migula. Int.J. Syst. Bacteriol. 30:17–18.
10. Enright, M. C., and B. G. Spratt. 1999. Multilocus sequence typing. TrendsMicrobiol. 7:482–487.
11. Ezaki, T., Y. Hashimoto, and E. Yabuuchi. 1989. Fluorometric deoxy-rinucleic acid-deoxyribonucleic acid hybridization in micro-dilution wells asan alternative to membrane filter hybridization in which radioisotopes areused to determine genetic relatedness among bacterial strains. Int. J. Syst.Bacteriol. 39:224–229.
12. Finnan, S., J. P. Morrissey, F. O’Gara, and E. F. Boyd. 2004. Genomediversity of Pseudomonas aeruginosa isolates from cystic fibrosis patients andthe hospital environment. J. Clin. Microbiol. 42:5783–5792.
13. Foght, J. M., D. W. S. Westlake, W. M. Johnson, and H. F. Ridgway. 1996.Environmental gasoline-utilizing isolates of Pseudomonas aeruginosa aretaxonomically indistinguishable by chemotaxonomic and molecular tech-niques. Microbiology 142:2333–2340.
14. Grimont, P. A. D. 1999. Taxonomy and classification of bacteria, p. 249–259In P. R. Murray, E. J. Baron, M. A. Pfaller, F. C. Tenover, and R. H. Yolken(ed.), Manual of clinical microbiology, 7th ed. American Society for Micro-biology, Washington, DC.
15. Hoadley, A. W. 1977. Potential health hazards associated with Pseudomonasaeruginosa in water, p. 80–114. In A. W. Hoadley and B. J. Dutka (ed.),Bacterial indicators/health hazards associated with water. American Societyfor Testing and Materials, Philadelphia, PA.
16. Hoadley, A. W. 1968. On the significance of Pseudomonas aeruginosa insurface waters. J. N. Engl. Water Works Assoc. 82:99–111.
17. Howell, C. R., and R. D. Stipanovic. 1979. Control of Rhizoctonia solani oncotton seedlings with Pseudomonas fluorescens and with an antibiotic pro-duced by the bacterium. Phytopathology 69:480–482.
18. Jolley, K. A., M. S. Chan, and M. C. Maiden. 2004. mlstdbNet—distributedmulti-locus sequence typing (MLST) databases. BMC Bioinform. 5:86.
19. Jolley, K. A., F. J. Feil, M. S. Chan, and M. C. Maiden. 2001. Sequence typeanalysis and recombinational tests (START). Bioinformatics 17:1230–1231.
20. Jukes, T. H., and C. R. Cantor. 1969. Evolution of protein molecules, p.21–132. In H. N. Munro (ed.), Mammalian protein metabolism. AcademicPress, Inc., New York, NY.
21. Khan, A. A., and C. E. Cerniglia. 1994. Detection of Pseudomonas aeruginosafrom clinical and environmental samples by amplification of the exotoxin Agene using PCR. Appl. Environ. Microbiol. 60:3739–3745.
22. Khan, N. H., Y. Ishii, N. Kimata-Kino, H. Esaki, T. Nishino, M. Nishimura,and K. Kogure. 2007. Isolation of Pseudomonas aeruginosa from open oceanand comparison with freshwater, clinical, and animal isolates. Microb. Ecol.53:173–186.
23. Kiewitz, C., and B. Tummler. 2000. Sequence diversity of Pseudomonasaeruginosa: impact on population structure and genome evolution. J. Bacte-riol. 182:3125–3135.
24. Kimata, N., T. Nishino, S. Suzuki, and K. Kogure. 2004. Pseudomonasaeruginosa isolated from marine environments in Tokyo Bay. Microb. Ecol.47:41–47.
25. Kimura, M. 1980. A simple method for estimating evolutionary rates of basesubstitutions through comparative studies of nucleotide sequences. J. Mol.Evol. 16:111–120.
26. Kodaka, H., M. Iwata, S. Yumoto, and F. Kashitani. 2003. Evaluation of anew agar medium containing cetrimide, kanamycin and nalidixic acid forisolation and enhancement of pigment production of Pseudomonas aerugi-nosa in clinical samples. J. Basic Microbiol. 43:407–413.
27. Kumar, S., K. Tamura, and M. Nei. 1994. MEGA: molecular evolutionarygenetics analysis software for microcomputers. Comput. Appl. Biosci. 10:189–191.
28. Lane, D. J. 1991. 16S/23S rRNA sequencing in nucleic acid techniques, p.115–148. In E. Stackebrandt and M. Goodfellow (ed.), Bacterial systematics.John Wiley and Sons, New York, NY.
29. Lomholt, J. A., K. Poulsen, and M. Kilian. 2001. Epidemic populationstructure of Pseudomonas aeruginosa: evidence for a clone that is pathogenicto the eye and that has a distinct combination of virulence factors. Infect.Immun. 69:6284–6295.
30. Loper, J. E., and S. E. Lindow. 1987. Lack of evidence for in situ fluorescentpigment production by Pseudomonas syringae pv. syringae on bean leaf sur-faces. Phytopathology 77:1449–1454.
31. Maiden, M. C. J., J. A. Bygraves, E. Feil, G. Morelli, J. E. Russell, R. Urwin,Q. Zhang, J. J. Zhou, K. Zurth, D. A. Caugant, I. M. Feavers, M. Achtman,and B. G. Spratt. 1998. Multilocus sequence typing: a portable approach tothe identification of clones within populations of pathogenic microorgan-isms. Proc. Natl. Acad. Sci. USA 95:3140–3145.
32. Mann, H. B., and D. R. Whitney. 1947. On a test of whether one of tworandom variables is stochastically larger than the other. Ann. Math. Stat.18:50–60.
6204 KHAN ET AL. APPL. ENVIRON. MICROBIOL.
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from

33. Mates, A. 1992. The significance of testing for Pseudomonas aeruginosa inrecreational seawater beaches. Microbios 71:89–93.
34. Nakazawa, T. 2002. Travels of a Pseudomonas, from Japan around the world.Environ. Microbiol. 4:782–786.
35. Nei, M., and T. Gojobori. 1986. Simple methods for estimating the numbersof synonymous and nonsynonymous nucleotide substitutions. Mol. Biol.Evol. 3:418–426.
36. Nicas, T. I., and B. H. Iglewski. 1986. Production of elastase and otherexoproducts by environmental isolates of Pseudomonas aeruginosa. J. Clin.Microbiol. 23:967–969.
37. Panagea, S., C. Winstanley, M. J. Walshaw, M. J. Ledson, and C. A. Hart.2005. Environmental contamination with an epidemic strain of Pseudomonasaeruginosa in a Liverpool cystic fibrosis centre, and study of its survival on drysurfaces. J. Hosp. Infect. 59:102–107.
38. Papapetropourou, M., and G. Rodopoulou. 1994. Occurrence of enteric andnon-enteric indicators in coastal waters of southern Greece. Bull. Mar. Sci.54:63–70.
39. Pellett, S., D. V. Bigley, and D. J. Grimes. 1983. Distribution of Pseudomonasaeruginosa in a riverine ecosystem. Appl. Environ. Microbiol. 45:328–332.
40. Picard, B., E. Denamur, A. Barakat, J. Elion, and P. Goullet. 1994. Geneticheterogeneity of Pseudomonas aeruginosa clinical isolates revealed by ester-ase electrophoretic polymorphism and restriction fragment length polymor-phism of the ribosomal RNA gene region. J. Med. Microbiol. 40:313–322.
41. Pirnay, J. P., D. De Vos, C. Cochez, F. Bilocq, A. Vanderkelen, M. Zizi, B.Ghysels, and P. Cornelis. 2002. Pseudomonas aeruginosa displays an epi-demic population structure. Environ. Microbiol. 4:898–911.
42. Pirnay, J. P., S. Matthijs, H. Colak, P. Chablain, F. Bilocq, V. Eldere, D. D.Vos, M. Zizi, L Triest, and P. Cornelis. 2005. Global Pseudomonas aerugi-nosa biodiversity as reflected in a Belgian river. Environ. Microbiol. 7:969–980.
43. Rahme, L. G., E. J. Stevens, S. F. Wolfort, J. Shao, R. G. Tompkins, andF. M. Ausubel. 1995. Common virulence factors for bacterial pathogenicity inplants and animals. Science 268:1899–1902.
44. Romling, U., J. Wingender, H. Muller, and B. Tummler. 1994. A majorPseudomonas aeruginosa clone common to patients and aquatic habitats.Appl. Environ. Microbiol. 60:1734–1738.
45. Sarkar, S. F., and D. S. Guttman. 2004. Evolution of the core genome ofPseudomonas syringae, a highly clonal, endemic plant pathogen. Appl. Envi-ron. Microbiol. 70:1999–2012.
46. Schmidt, K. D., B. Tummler, and U. Romling. 1996. Comparative genomemapping of Pseudomonas aeruginosa PAO with P. aeruginosa C, which be-
longs to a major clone in cystic fibrosis patients and aquatic habitats. J.Bacteriol. 178:85–93.
47. Spencer, D. H., A. Kas, E. E. Smith, C. K. Raymond, E. H. Sims, M.Hastings, J. L. Burns, R. Kaul, and M. V. Olson. 2003. Whole-genomesequence variation among multiple isolates of Pseudomonas aeruginosa. J.Bacteriol. 185:1316–1325.
48. Stackebrandt, E., and B. M. Goebel. 1994. Taxonomic note: a place forDNA-DNA reassociation and 16S rRNA sequence analysis in the presentspecies definition in bacteriology. Int. J. Syst. Bacteriol. 44:846–849.
49. Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J.Hickey, F. S. L. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L.Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L.Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D.Spencer, G. K.-S. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. F. W.Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence ofPseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964.
50. Szoboszlay, S., B. Atzel, and B. Kriszt. 2003. Comparative biodegradationexamination of Pseudomonas aeruginosa (ATCC 27853) and other oil de-graders on hydrocarbon contaminated soil. Commun. Agric. Appl. Biol. Sci.68:207–210.
51. Thompson, J., D. G. Huggins, and T. J. Gibson. 1994. Clustal W: improvingthe sensitivity of progressive multiple sequence alignment through sequenceweighting, position-specific gap penalties and weight matrix choice. NucleicAcids Res. 22:4673–4680.
52. Velammal, A., B. Aiyamperumal, V. K. Venugopalan, and S. Ajmalkhan.1994. Distribution of Pseudomonas aeruginosa in Pondicherry coastal envi-ronments. Ind. J. Mar. Sci. 23:239–241.
53. Vernez, I., P. Hauser, M. V. Bernasconi, and D. S. Blanc. 2005. Populationgenetic analysis of Pseudomonas aeruginosa using multilocus sequence typ-ing. FEMS Immunol. Med. Microbiol. 43:29–35.
54. Walker, T. S., H. P. Bais, E. Deziel, H. P. Schweizer, L. G. Rahme, R. Fall,and J. M. Vivanco. 2004. Pseudomonas aeruginosa-plant root interactions.Pathogenicity, biofilm formation, and root exudation. Plant Physiol. 34:320–331.
55. Yoshpe-Purer, Y., and S. Golderman. 1987. Occurrence of Staphylococcusaureus and Pseudomonas aeruginosa in Israeli coastal water. Appl. Environ.Microbiol. 53:1138–1141.
56. Young, A. L., A. T. Leung, L. L. Cheng, R. W. Law, A. K. Wong, and D. S.Lam. 2004. Orthokeratology lens-related corneal ulcers in children: a caseseries. Ophthalmology 111:590–595.
VOL. 74, 2008 PHYLOGENETIC ANALYSES OF OCEAN PSEUDOMONAS AERUGINOSA 6205
on Septem
ber 17, 2014 by guesthttp://aem
.asm.org/
Dow
nloaded from




















