







Bahasa
Halaman
Hukum


Non-Steroidal Anti-Inflammatory Drugs
Nikita Shokur
25 April 2014
Medicinal Chemistry

Introduction:
Non-steroidal anti-inflammatory drugs (NSAIDs) are drugs
from diverse structural classes that show analgesic, anti-
inflammatory, and antipyretic activities. Ever since the
development of NSAIDs, the need for these drugs has been
exponentially growing due to the broad range of applications for
these medications and the recent development of selective COX-2
inhibitors. The use of NSAIDs is seen primarily in patients with
musculoskeletal illnesses. Their use relieves temporary
conditions such as sprains, strains, back pain, headaches,
menstrual cycle pains, colon polyps and long term conditions such
as rheumatoid arthritis, osteoarthritis, ankylosing spondylitis,
lupus, and clotting of blood.1 However, NSAIDs have also shown
extensive adverse effects including gastrointestinal bleeding,
peptic ulcer disease, hypertension, edema, and renal disease.2 In
2012, the use of NSAIDs has been associated with the risk of
acute myocardial infarction.3

NSAIDs act by blocking cyclooxygenase enzyme systems. In
1990, an important discovery was made differentiating between the
two forms of cyclooxygenase enzyme: the COX-1 that produces
prostaglandins and thromboxanes that regulate gastrointestinal,
renal, and vascular functions and COX-2 that regulates the
produces prostaglandins involved inflammation, pain, and fever.4
In the past two decades, an enormous commercial development of
selective COX-2 enzyme inhibitors, known as coxibs, has been
started. The idea was to reduce the stomach bleeding caused by
the inhibition of COX-1 enzyme, which protected the stomach
lining with mucous otherwise destroyed by acid.
Today, the use of NSAIDs continues to grow at an incredible
rate. Between 1984 and 1988, the number of patients receiving
prescriptions for NSAIDs jumped from 44 million to 70 million; by
1997 the worldwide
market for these medications was
over six billion dollars.2
Recent development of COX-2
selective inhibitor Celebrex has
brought in four billion
dollars since its debut in
1999 and became the sixth best-selling drug.5 Other classes of
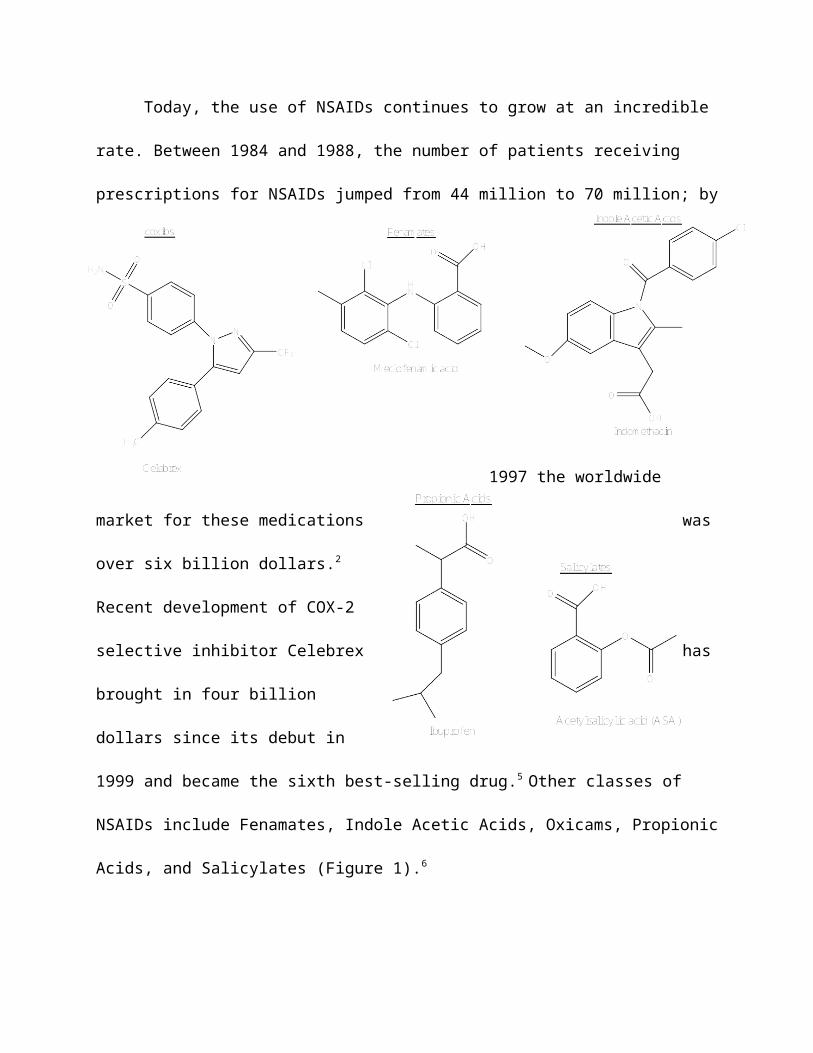
NSAIDs include Fenamates, Indole Acetic Acids, Oxicams, Propionic
Acids, and Salicylates (Figure 1).6

Figure 1. Major Classes of NSAIDs.
Discovery:
The first signs of NSAIDs use dates back about 3500 years
when Greek physician Hippocrates prescribed willow bark and
leaves to reduce fever and inflammation.7 In the 17th century,
salicin was identified as an active component of willow bark; in
the midst of the following century, Germany started producing
salicylic acid. It wasn’t until the 1899 that salicylic acid was
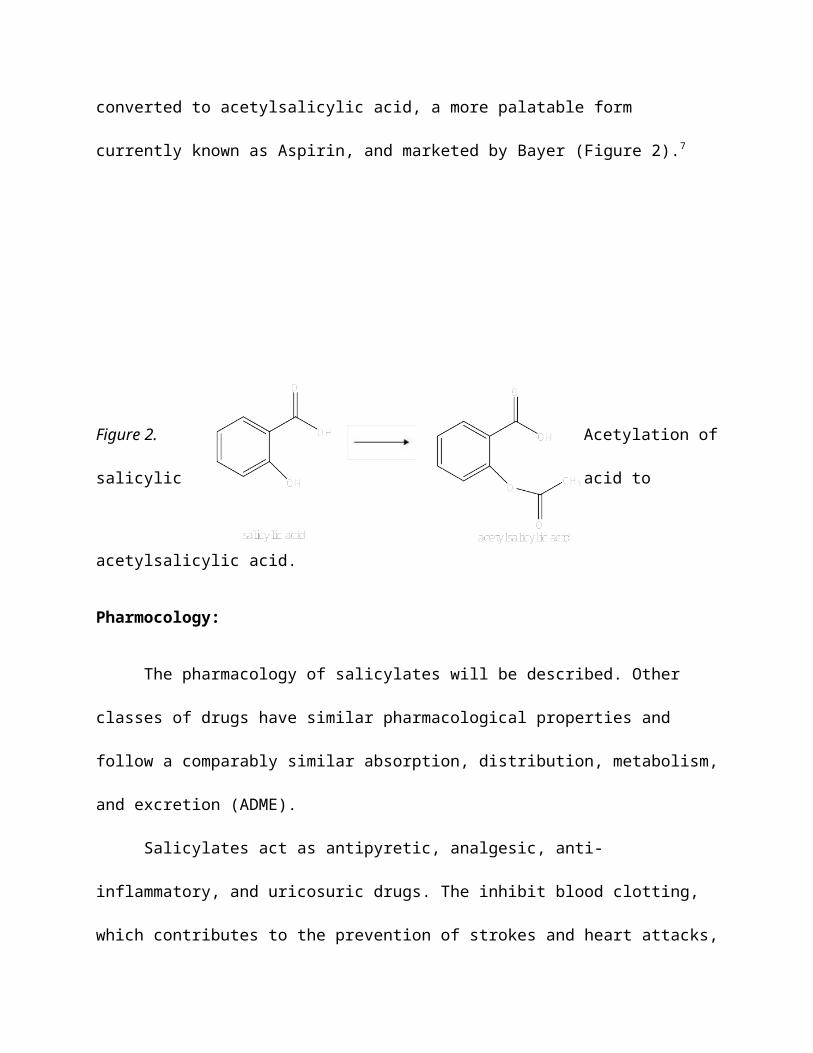
converted to acetylsalicylic acid, a more palatable form
currently known as Aspirin, and marketed by Bayer (Figure 2).7
Figure 2. Acetylation of
salicylic acid to
acetylsalicylic acid.
Pharmocology:
The pharmacology of salicylates will be described. Other
classes of drugs have similar pharmacological properties and
follow a comparably similar absorption, distribution, metabolism,
and excretion (ADME).
Salicylates act as antipyretic, analgesic, anti-
inflammatory, and uricosuric drugs. The inhibit blood clotting,
which contributes to the prevention of strokes and heart attacks,

due to inhibition of thromboxane A2. Salicylates are given orally
and have around a 55% bioavailability.2 Majority is absorbed in
the small intestine and some are absorbed in the stomach due to
passive diffusion. Absorption takes between 20-30 minutes after
oral intake as it is readily hydrolyzed in blood and liver .8
They are distributed throughout most of the body’s tissues.
Salicylates are initially converted to salicylic acid. Average
elimination time is about 24 hours; inactivation occurs in the
hepatic endoplasmic reticulum and mitochondria. Majority of it is
excreted after undergoing conjugation with glycine or with
glucuronic acid to form ether or ester, while around 10% of it is
excreted as a free acid, and trace amounts salicylates are
hydroxylated before excretion.2,8
SAR:
General Structure Activity Relationship (SAR) for all anti-
inflammatory non-steroidal drug has been identified.8 All agents
must possess a center of acidity represented by groups such as
carboxylic acids, enols, sulfanomide, or tetrazole groups. Amide
or ester derivatives of carboxylic acids are usually attributed
to the metabolic hydrolysis products. The center of acidity for

NSAIDs is generally located a distance of one carbon away from a
flat surface represented by an aromatic or hetero-aromatic ring.
This distance plays a critical role because as the distance
increases the activity of the drug diminishes. Aryl and hetero-
aryl derivatives are common since they correlate with the double
bond at the 5- and 8- position of the arachidonic acid (Figure
X).
Substitution of a methyl group on the alpha carbon has shown to
increase anti-inflammatory activity. The resulting derivatives,
alpha-methyl acetic, have been given a class name profen. Groups
larger than methyl show a decline in activity. The importance of
the methyl group comes from a creation of a new chiral center;
the anti-inflammatory activity is associated with the S (+)
enantiomer. The activity is further enhanced by an addition of a
second area of lipophilicity that is non-coplanar with the
original aromatic or hetero-aromatic ring. This second
lipophilic area corresponds to the double bond region at the
positon 11- of arachidonic acid.

Figure X. Arachidonic Acid.
Mechanism of Action:
The discovery of the mechanism of aspirin by John Vane in
the seventies has spiked our ability to develop NSAIDs. In the
early 90’s, Needleman, Simmons, and Herschman’s group reported a
presence of an isoform of cyclooxygenase, later named COX-2.7
While traditional NSAIDs inhibited both COX-1 and COX-2, the
adverse effects of gastrointestinal toxicity were attributed to
gastro protective prostaglandins and prostocyclins produced via
COX-1 pathway. Since then scientists and pharmaceutical companies
have shifted their focus on developing selective COX-2
inhibitors. Their efforts led to the first selective COX-2
inhibitor celecoxib and followed by rofexocib (Figure X).

Figure X. COX-2 with bound Celecoxib.
Arachidonic acid is produced from fatty acids by
Phosopholipadase A. Cyclooxygenase then catalyzes the oxidation
of arachidonic acid to prostoglandin G2, which is then reduced to
prostoglandin H2 (Figure X).7 COX-1 is compromised of 602 amino
acids whereas COX-2 has 604 amino acids. The two isoforms share
60-65% homology.7 The major difference is the substitution of
Isoleucine-523 in COX-1 for Valine-523 in COX-2.9 This opens a
new pocket for binding and allows for selectivity.

There are two types of inhibition of cyclooxygenase: the
irreversible ihibition and competitive inhibition of the
substrate. The irreversible inhibition is unique to Aspirin, the
drug permanently acetylates the key Serine-530 within the active
site.8 This inhibition is irreversible since plateles do not
carry out protein synthesis, thus inhibition lasts the cells
lifetime. The competitive inhibition of the substrate
(arachidonic acid) is reversible when the drug washes out or is
competing for the bidning site. The carboxylate part of the drug
bonds to Arginine-120 and Tyrosine-355.10 Methyl and the ring
structures fit into the hydrophobic pocket.
Cyclooxygenase 1 enzymes are thought to be constitutive,
being expressed in most of our bodily tissues. While
cyclooxygenase 2 enzymes is induced by cell-signaling proteins
called cytokines and is partially constitutive.Both are
responsible for the physiological production of prostaglandins.
Prostaglandins have a wide range of functions.8 PGI2 and PGE2 are
responsible for pain by sensitizing nerve endings to bradykinin,
histamine, and substance P. PGI2, PGD2, and PGE2 are vasodilators
responsible for inflammation. PGI2 protects the gastric mucosa.

PGE2 maintains renal blood flow and is responsible for fever. TXA2
stimulates platelet aggregation. PGD2 contracts uterus.
Figure X. Biosynthesis of prostaglandin from arachidonic acid via
COX isoform pathway.
NSAIDs can be classified into four groups.7 Group 1
includes NSAIDs that inhibit both COX-1 and COX-2 with very
little selectivity. Group 2 includes NSAIDs that inhibit COX-2
with 5-50 fold selectivity. Group 3 includes NSAIDs that inhibit
COX-2 with over 50 fold selectivity. And Group 4 shows weak
inhibition of both cyclooxygenase isoforms.

Figure X. Classification of NSAIDs according to their COX-1/2
activities.
Future Developments:
Despite the promise of therapeutic effectiveness of the
selective COX-2 inhibitors, there
have been cases reporting the arousal of severe cardiovascular
effects with the use of these inhibitors.8 Long-term clinical
trials of rofecoxib, a Group 3 coxib, that indicated an increased
risk of myocardial infarction. A later analysis of a vast
population has shown that the risk of heart problems grew many

folds while on rofecoxib compared to some older NSAIDs. These
effects may be related to the degree of selectivity of COX-1 and
COX-2 inhibitors. COX-1 mediates the production of prostaglandins
and thromboxanes, which are responsible for platelet aggregation,
and COX-2 produces prostacyclin, which regulates platelet
aggregation. The inhibition of only COX-2 significantly reduces
the production of prostacyclins, whereas the production of
thrombaxanes is unaffected. Thus, future developments of NSAIDs
may be aimed at preferentially inhibiting COX-2 while also having
an inhibition on COX-1 to a lesser extent.
Recent research has also shown a link between COX-2 and
cancer. The study revealed an over-expression of COX-2 mRNA in
colorectal cancer.12 Levels of COX-2 mRNA were found
overexpressed in almost 80% of the colorectal tumors compared to
normal colorectal mucosa. This suggests that COX-2 could be used
as a possible biomarker for the risk of cancer and that it could
be of value in chemoprevention of colon cancer.

References:
1. American College of Rheumatology Members. ‘‘NSAIDs:
Nonsteroidal Anti-inflammatory
Drugs’’ American College of Rheumatology, 2012, Web.
2. Saraf, S. Non-Steroidal Anti-Inflammatory Drugs, PharmaMed Press, 2008,
12-13,
168-169.
3. Krotz, F.; Schiele, T. M.; Klauss, V.; Sohn, H. Y.;
‘‘Selective COX-2 inhibitors and risk of

myocardial infarction’’ Journal of Vascular Research, 2005, 42(4),
312-24.
4. Rainsford, K. D. ‘‘Anti-Inflammatory Drugs In the 21st
century’’ Subcellular Biochemistry,
2007, 42, 3-27.
5. Halpern, G.M. ‘‘COX-2 Inhibitors: a story of greed, deception,
and death’’
Inflammopharmacology, 2005, 13(4), 419-25.
6. Monteleone, G. ‘‘Classes of NSAIDs’’ SportsMedicine. Web.
7. Pravee Rao, P.N.; Knaus, E.E. ‘‘Evolution of Nonsteroidal
Anti-Inflammatory Drugs
NSAIDs):Cyclooxygenase (COX) Inhibition and Beyond’’ Journal
of Pharmaceutical
Sciences, 2008, 11(2), 81-110.
8. Bkhaitan, M. ‘‘Non-Steroidal Anti-inflammatory Drugs’’
Department of Pharmaceutical
Chemistry, Web.

9. Gierse, J., McDonald, J., Hauser, S.; ‘‘A Single Amino Acid
Difference between
Cyclooxygenase-1 (COX-1) and −2 (COX-2) Reverses the Selectivity of COX-2 Specific
Inhibitors’’ Journal of Biological Chemistry, 1996, 271, 15810-15814.
10. Dunlap, N. ‘‘Enzyme Inhibitors’’ Medicinal Chemistry, Custom
Publishing, 2013, 88-93.
11. Billones, J.; Buenaobra, S. ‘‘Quantitative Structure-ActivityRelationship (QSAR)Study of
Cyclooxygenase-2 (COX-2) Inhibitors’’ Philippine Journal of Science,2008, 140(2),
125-132.
12. Roelofs, H. ‘‘Over-expression of COX-2 mRNA in colorectal
cancer’’ Biomedical Central
Gastroenterology, 2014, 14(1).
Top Related
Copyright © 2022 FDOKUMEN

