







Bahasa
Halaman
Hukum


Myocardial tissue troponins T and I
An immunohistochemical study in experimental
models of myocardial ischemia
Michael C. Fishbeina,*, Tiffany Wanga, Maria Matijasevica,Longsheng Honga, Fred S. Appleb
aDivision of Anatomic Pathology, Department of Pathology and Laboratory Medicine, UCLA Center for the Health Sciences,
A7-149 CHS, 10833 Le Conte Avenue, Los Angeles, CA 90095-1732, USAbDepartment of Laboratory Medicine and Pathology, Hennepin County Medical Center, University of Minnesota School of Medicine,
914 S. 8th Street, Minneapolis, MN 55404, USA
Received 4 August 2002; received in revised form 30 September 2002; accepted 12 November 2002
Abstract
Background: Cardiac troponins T (cTnT) and I (cTnI) are proven diagnostic and risk stratification biomarkers in patients with acute
coronary syndromes. To date, no immunohistochemical studies have been performed which allow visualization of the time course and pattern
of myocardial troponin egress from the myocardium during the early evolution of ischemic injury in experimental systems. Methods: We
studied archival formalin-fixed, paraffin-embedded myocardium from 50 experimental animals (dogs, pigs and rats) that had undergone
permanent coronary occlusion (n = 34) for 0.5–6 h or occlusion of 0.75–6 h followed by reperfusion (n = 16). Histologic sections that
included ischemic and nonischemic myocardium were studied by immunohistochemistry with three different antibodies to human cTnI and
one to cTnT, using a standard avidin–biotin–peroxidase system. Results: All antibodies detected cTnT or cTnI in normal myocardium and its
loss from necrotic myocardium, in some cases as early as 30 min after coronary occlusion, before histologic evidence of necrosis was present.
Loss was nonuniform, being greater at the periphery of the infarcts then at their central regions. Usually, loss of cTnT appeared greater
than loss of cTnI. With reperfusion, findings were similar to those after permanent occlusion, except that there was a greater contrast between
loss at the periphery compared to the loss in the central region. Considerable residual staining persisted for hours after occlusion, indicating
delayed release over time, concordant with sustained serum elevations in patients with acute myocardial infarction. No loss of staining was
observed in nonnecrotic myocardium. Conclusions: Immunohistochemical staining using antibodies to human cTnT and cTnI can be used to
visualize cardiac troponins and document their loss in histologic sections of myocardium in different animal species. Loss of cTnT and cTnI
occurs very early following ischemic injury and may precede histologic evidence of necrosis, but does not occur in myocardium that is not
necrotic. Immunohistochemical staining of hearts for cTnT and cTnI can assist in the often difficult recognition of myocardial necrosis at
autopsy, in patients suspected of dying from acute myocardial ischemia. D 2003 Elsevier Inc. All rights reserved.
Keywords: Immunohistochemistry; Troponins; Myocardial Necrosis
1. Introduction
Until recently, measurement of creatine kinase (CK)
and its myocardial isoenzyme (CK-MB) in the blood have
been the gold standard for the enzymatic diagnosis of
acute myocardial infarction [1]. Recent data indicate
measurement of cardiac troponins T (cTnT) and I (cTnI)
may be even more useful in diagnosing acute myocardial
infarction and for risk stratification in acute myocardial
ischemia [2–5]. Indeed, a joint committee of the Amer-
ican College of Cardiology and the European Society of
Cardiology has recommended that cardiac troponin
replace CK-MB in the evaluation of patients for myocar-
dial ischemia/infarction, as cardiac troponin measurements
appear to be more sensitive, myocardial-tissue specific,
1054-8807/03/$ – see front matter D 2003 Elsevier Inc. All rights reserved.
doi:10.1016/S1054-8807(02)00188-6
Abbreviations: CK, creatine kinase; cTnI, cardiac troponin I; cTnT,
cardiac troponin T; EM, electron microscopy; LDH, lactic dehydrogenase;
NHS, normal horse serum; PAS, periodic acid Schiff; PBS, phosphate-
buffered saline; TTC, triphenyl tetrazolium chloride
* Corresponding author. Tel.: +1-310-825-9731; fax: +1-310-794-4161.
E-mail address: [email protected] (M.C. Fishbein).
Cardiovascular Pathology 12 (2003) 65–71

and remain elevated longer after the onset of myocardial
infarction [6].
We have previously shown that the presence and
absence of the myocardial proteins CK-M, CK-B, myo-
globin, LDH-1, and cytosolic and mitochondrial aspartate
aminotransferase can be demonstrated in formalin-fixed,
paraffin-embedded tissues by immunohistochemical stain-
ing techniques [7–11]. Furthermore, we demonstrated loss
of these enzymes in necrotic human myocardium from
autopsied patients dying with myocardial infarction [8–
12]. In experimental models of myocardial infarction,
using electron microscopy (EM), gross triphenyl tetrazo-
lium chloride (TTC) staining, and periodic acid-Schiff
(PAS) staining for glycogen, we showed that loss of these
proteins did not occur with ischemia alone, but could be
demonstrated in necrotic myocardium as early as 3 h after
coronary occlusion [13].
Since we have retained paraffin blocks from our previous
immunohistochemical studies of well-characterized experi-
mental myocardial ischemic injury, we were able to restudy
these cases using antibodies to cTnI and cTnT to character-
ize the patterns of staining in experimental models in three
different animal species. The goals were to: (1) examine the
time course and pattern of loss of troponins from ischemic
myocardium; (2) to determine if troponin loss precedes
histologic evidence of necrosis; (3) to determine the spe-
cificity of loss of troponins as a marker of myocardial
necrosis; and (4) to determine if immunohistochemical
visualization of cardiac troponins would, therefore, be a
useful tool for the evaluation of myocardial ischemic injury
in experimental studies and in human autopsy material.
Preliminary immunohistochemical studies in autopsied
patients with well-developed infarcts have shown dimin-
ished staining in necrotic regions [14,15].
2. Materials and methods
2.1. Tissues
We reviewed our log books of experiments performed
from 1979 to 1997 to identify animal experiments in which
well-characterized myocardial injury was produced in open
and closed chest models of myocardial ischemia. These
consisted of archival formalin-fixed, paraffin-embedded
myocardium from 50 experimental animals (dogs, pigs
and rats) that had undergone permanent coronary occlusion
(n = 34) for 0.5–6 h or occlusion of 0.75–6 h followed by
reperfusion (n = 16). Ischemic injury was characterized by a
number of methods including EM, PAS staining for gly-
cogen loss, and TTC staining and evaluation of H&E-
stained sections to demonstrate myocardial necrosis. In
studies in larger animals, alternate gross slices of myocar-
dium (slices 1, 3 and 5) were stained with TTC and
sampled for histologic evaluation (H&E stain) from TTC
positive and negative regions. Adjacent surfaces from slices
2 and 4 were sampled and processed for EM and PAS
staining. This protocol allowed comparison of staining for
troponins with ultrastructural and histochemical findings in
the same regions of myocardium. The experimental meth-
ods of producing myocardial ischemia and evaluating the
myocardial ischemic injury have been described in detail
[8–11,16–18]. All experiments were done in compliance
with institutional and national research council guidelines
for the care and use of laboratory animals. All animals were
sacrificed with an overdose of intravenous KCl.
We identified 50 animal experiments to restudy. These
50 cases were selected from 145 studies reviewed, because
they provided a range of coronary occlusion from 0.5 to
6.0 h, and there was adequate paraffin-embedded tissue
remaining to perform immunohistochemical studies. The
archival paraffin blocks were retrieved and recut. One H&E-
stained section of each block was prepared and examined
for histologic evidence of myocardial ischemia/necrosis.
Necrosis was defined as hypereosinophilia of myocytes
with or without contraction bands, with surrounding edema.
In many of the cases, there were varying degrees of
neutrophilic infiltrate in the necrotic regions. Some sections
showed only edema and wavy fibers and only subtle hyper-
eosinophilia of fibers [19]. These regions were judged not to
show definite histologic evidence of necrosis; however, our
previous EM studies had demonstrated necrosis in these
regions. Numerous unstained slides were prepared from
each paraffin block for immunohistochemical staining.
2.2. Immunohistochemical staining
All tissues used for immunohistochemical studies came
from transverse slices of myocardium fixed in 10%
neutral-buffered formalin for 24–36 h, processed routinely
and embedded in paraffin. Four-micron-thick tissue sec-
tions were deparaffinized in xylene for 5 min. Slides were
rehydrated in graded alcohols. Endogenous peroxidase
Table 1
Troponin loss with permanent occlusion
Duration of
occlusion
Degree of
troponin loss
(0 to � 3)
Histologic
evidence
of necrosis
Species Hours n Scorea Mean n
Dogs 0.5 6 � 1 to� 3 � 2.0 3 of 6
0.75 5 � 1 to� 3 � 1.4 1 of 5
3.0 3 � 1 to� 3 � 1.7 3 of 3
5.0 1 � 3 – 1 of 1
6.0 10 � 1 to� 3 � 2.3 10 of 10
Pigs 4.0 5 � 1 to� 3 � 2.6 5 of 5
Rats 1.0 2 � 3 � 3.0 0 of 2
3.0 1 � 3 – 0 of 1
0.0b 1 0 – 0 of 1
a Maximum score for any antibody tested from center or periphery
of infarct.b Sham-operated positive control.
M.C. Fishbein et al. / Cardiovascular Pathology 12 (2003) 65–7166
activity was inhibited by incubation in 3% hydrogen
peroxidase in methanol for 15 min followed by two 5-min
washes in phosphate-buffered saline (PBS). Sections were
then incubated for 30 min with 3% normal horse serum
(NHS, diluted with PBS) and then incubated for 1.5–2 h
with primary antibodies: cTnI 3302 (1:200), cTnI 3350-1D
(1:50), cTnI 3350-2F (1:100) and cTnT (1:100). After
removal of the primary antibodies with three 5-min
washes in PBS, sections were incubated for 40 min with
biotinylated horse antimouse IgG (Vector) diluted 1:200 in
1% NHS. After three 5-min washes in PBS, sections were
incubated for 30 min with horseradish peroxidase avidin D
(HRP, Vector) diluted 1:1000 with PBS. After three 5-min
washes of PBS, the sections were developed with DAB kit
(Vector), stopped with rinses of double-distilled water.
Slides were counterstained with dilute hematoxylin, rinsed
with ammonia and then with tap water. Sections were
dehydrated with graded ethanol, cleared in xylene and
then coverslipped.
The 33-2 (IgG1), 3350-ID (IgG1) and 3350-2F (IgG2A)
mouse antihuman cTnI antibodies were produced for West-
ern blot assays by Main Biotechnology Services and kindly
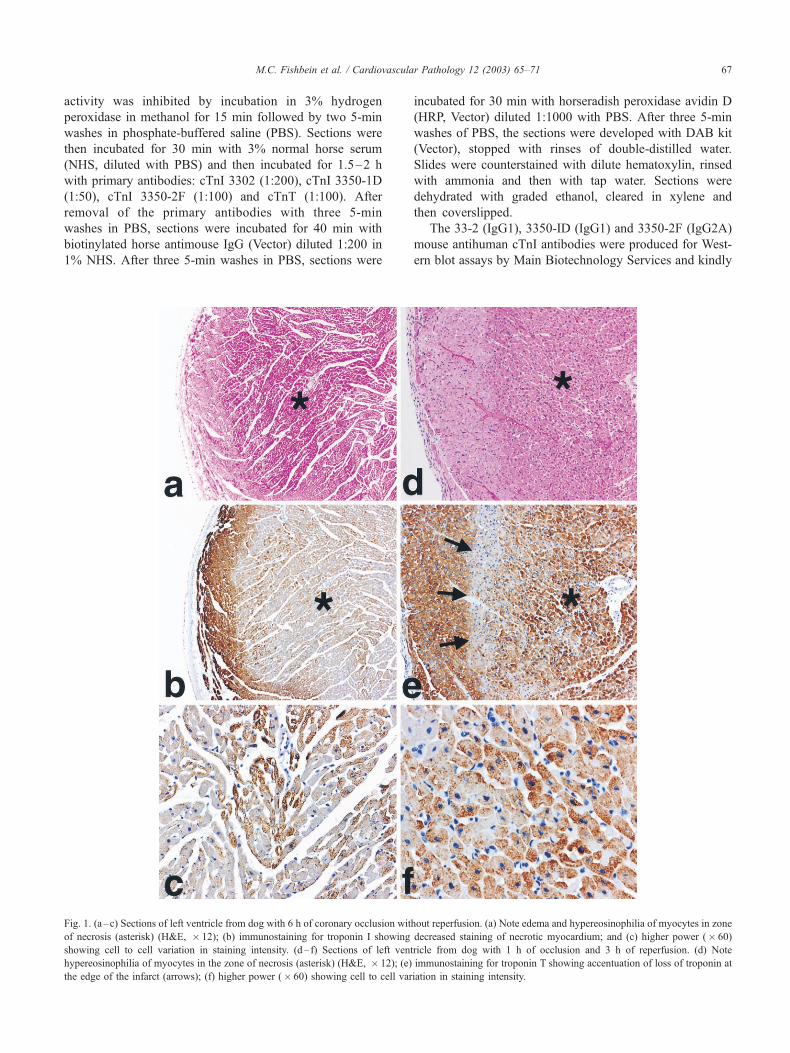
Fig. 1. (a–c) Sections of left ventricle from dog with 6 h of coronary occlusion without reperfusion. (a) Note edema and hypereosinophilia of myocytes in zone
of necrosis (asterisk) (H&E, � 12); (b) immunostaining for troponin I showing decreased staining of necrotic myocardium; and (c) higher power (� 60)
showing cell to cell variation in staining intensity. (d– f) Sections of left ventricle from dog with 1 h of occlusion and 3 h of reperfusion. (d) Note
hypereosinophilia of myocytes in the zone of necrosis (asterisk) (H&E, � 12); (e) immunostaining for troponin T showing accentuation of loss of troponin at
the edge of the infarct (arrows); (f) higher power (� 60) showing cell to cell variation in staining intensity.
M.C. Fishbein et al. / Cardiovascular Pathology 12 (2003) 65–71 67
donated to us by Jack Ladenson, PhD at Washington
University School of Medicine in St. Louis. All three
antibodies are directed against the N terminus cTnI region.
We used these three antibodies available to us to evaluate if
they were equal in sensitivity and specificity for detecting
cTnI loss from necrotic myocardium. We also wished to
determine if they differed in cross-reactivity to cardiac cTnI
from myocardium of species other than human beings. The
cTnT antiserum was purchased from Neomarkers (Fremont,
CA). Antibody dilutions and incubation times were chosen
to optimize staining of tissues.
Positive control slides came from a sham-operated rat
heart. Negative control slides consisted of animal tissues
with smooth muscle and skeletal muscle but no cardiac
muscle. A positive and negative control slide were stained
every time immunohistochemical staining was done. In
addition, each slide had its own built in negative and
positive controls: arteries within the myocardium were
expected to be negative for cTnT and cTnI, and nonischemic
regions of myocardium were expected to be positive. Strong
positive staining in the nonischemic regions also showed
that the integrity of the tissue antigenicity was preserved,
even in tissues stored up to 20 years in paraffin blocks. An
additional negative control for our staining procedure con-
sisted of replacing the primary antisera with saline.
Each immunostained slide was reviewed by two observ-
ers. Foci of ischemic injury were divided into peripheral and
central regions. The peripheral region consisted of several
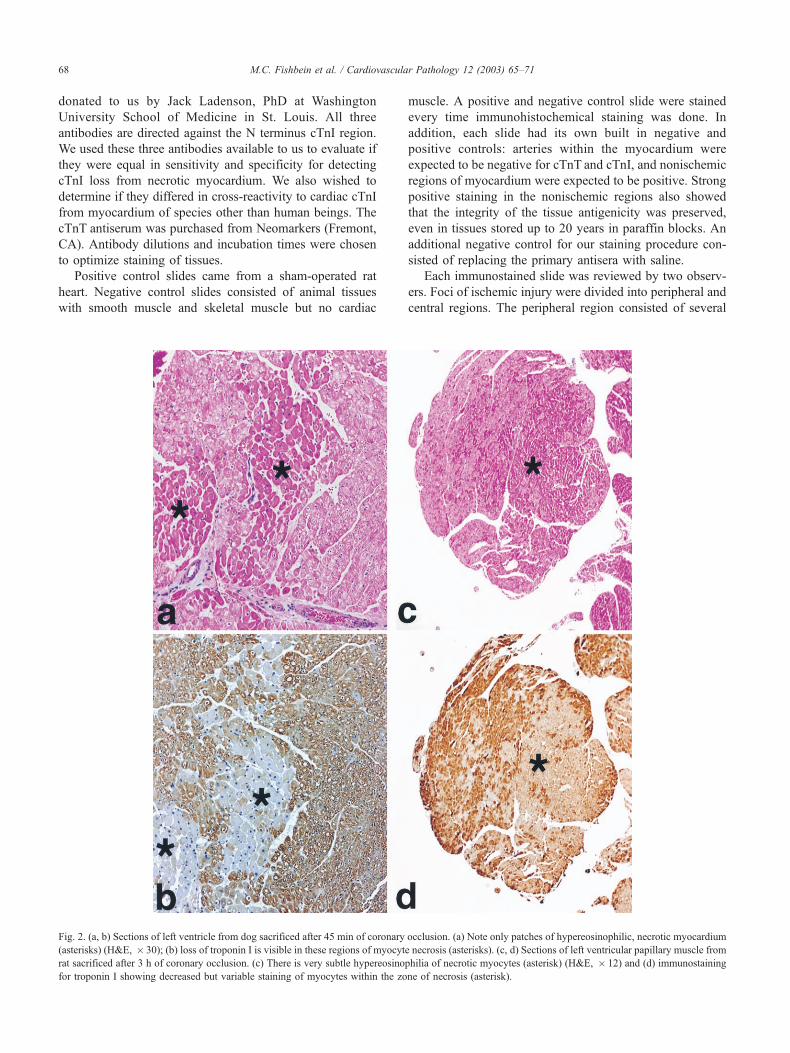
Fig. 2. (a, b) Sections of left ventricle from dog sacrificed after 45 min of coronary occlusion. (a) Note only patches of hypereosinophilic, necrotic myocardium
(asterisks) (H&E, � 30); (b) loss of troponin I is visible in these regions of myocyte necrosis (asterisks). (c, d) Sections of left ventricular papillary muscle from
rat sacrificed after 3 h of coronary occlusion. (c) There is very subtle hypereosinophilia of necrotic myocytes (asterisk) (H&E, � 12) and (d) immunostaining
for troponin I showing decreased but variable staining of myocytes within the zone of necrosis (asterisk).
M.C. Fishbein et al. / Cardiovascular Pathology 12 (2003) 65–7168

layers of myocytes immediately adjacent to viable myocar-
dium. The central region consisted of the remaining myo-
cardium within the ischemic zone. Loss of cardiac troponin
staining was scored for each region from 0 to � 3: 0 = no
loss of staining; � 1 =minimal decrease in staining, com-
pared to normally stained tissue; � 2 = clear decrease in
staining with some positivity (brown color) remaining; and
� 3 = no positive (brown) staining. The grade was based on
the maximum loss of cardiac troponin noted in the region.
3. Results
Permanent occlusion (no reperfusion) (Table 1): There
were 34 animals that had undergone permanent occlusions:
25 dogs with occlusions from 0.5 to 6 h; 5 pigs with 4-h
occlusions; and 3 rats, 2 with 1-h occlusion and 1 with a
3-h occlusion.
In all dogs with occlusions ranging from 0.5 to 6 h, if
there was histologic evidence of necrosis, there was some
loss of cTnT and cTnI from the tissue (Figs. 1 and 2). In
general, cTnT loss was more prominent than loss of cTnI
and, for all antisera, loss of troponin tended to be greater at
the periphery of the regions than at their centers. Two dogs
with 30-min occlusions and no histologic evidence of
necrosis showed no loss (score = 0) of cTnI (with all three
antibodies), but loss of cTnT (score =� 2). In three dogs
with 45-min occlusions and no histologic evidence of
necrosis (normal or edema only), all four antisera studied
demonstrated mild loss of cTnT and cTnI (score =� 1). In
all of these, dogs with brief periods of occlusion tissues
from the ischemic zone sampled for EM showed ultrastruc-
tural evidence of necrosis. Myocardium outside of the area
at risk showed no troponin loss and no morphologic
evidence of necrosis.
Four pigs each had 4 h of coronary occlusion accom-
panied by histologic evidence of myocardial necrosis. Three
had at least focal complete loss of cTnI and cTnT
(score =� 3); one had only mild loss (score =� 1).
Of the three rat hearts studied with 1 or 3 h of occlusion,
all had histologic evidence of myocardial edema. In addi-
tion, two had very subtle hypereosinophilia of myocytes.
These two showed focal marked loss of cTnI and cTnT
(score =� 3). One rat heart with a 1-h occlusion that had no
histologic evidence of necrosis showed no loss of either
cTnT or cTnI. Ultrastructural studies were not performed in
the rat hearts.
To summarize the findings with permanent occlusion,
all antibodies could detect both cTnT and cTnI in normal
myocardium and their loss in necrotic myocardium. Most
of the time, there was greater loss at the periphery than at
the center of the lesions; however, in all cases, the loss
was nonuniform in that some fibers showed complete loss
(score =� 3) with other fibers having varying degrees of
incomplete loss. Some fibers clearly within the necrotic
zone stained normally (score = 0). Usually, loss of cTnT
appeared to be greater than and appeared earlier than, loss
of cTnI. All three antibodies to cTnI gave similar results.
In some animals, primarily those with relatively brief
coronary occlusion (30–45 min) and ultrastructural evid-
ence of irreversible injury, loss of cardiac troponin stain-
ing could be identified even in the absence of histologic
proof of necrosis. The H&E-stained sections of these
slides showed only edema and, at most, subtle hyper-
eosinophilia. In our prior studies, EM evidence of nec-
rosis was present in the regions of troponin loss. No loss
of troponins was seen in myocardium that was viable by
H&E and TTC staining, and EM.
Occlusion followed by reperfusion (Table 2): In this
group, there were 10 dogs with occlusions of 0.75–6 h
followed by 15–30 min of reperfusion, and 6 pigs with 1 or
1.5 h of occlusion followed by 15 min of reperfusion. All of
these dogs and pigs had histologic evidence of myocardial
necrosis. All demonstrated at least focal loss of cardiac
troponins: 13 had a score of � 3, 1 had a score of � 2, and
2 had a score of � 1. As with animals with permanent
occlusion, there appeared to be more loss of cTnT than
cTnI. However, the marked difference observed between the
center and periphery of infarction in the group was much
more striking. The loss of staining at the periphery was
much greater than the loss observed in the center of the
infarct after reperfusion compared to permanent occlusion
(see Figs. 1 and 2). No loss of troponins was seen in
myocardium that was viable by H&E and TTC staining,
and EM.
4. Discussion
There have been a number of studies in a variety of
experimental models indicating that measurement of cardiac
troponins in serum might be useful for the diagnosis of
myocardial necrosis. O’Brien et al. showed that in both dog
and rat models of myocardial infarction, serum elevations of
cTnT were highly correlated with infarct size, 3 h after
occlusion. Further, using several animal species (dog, rat,
mouse and ferret), these authors showed that serum cTnT
Table 2
Troponin loss with occlusion followed by reperfusion
Duration
of occlusion
before reperfusiona
Degree of
troponin loss
(0 to � 3)
Histologic
evidence
of necrosis
Species Hours n Scorea Mean n
Dogs 0.75 1 � 3 – 1 of 1
1.0 3 � 3 � 3.0 3 of 3
2.0 4 � 2 to� 3 � 2.8 4 of 4
3.0 1 � 3 – 1 of 1
6.0 1 � 1 – 1 of 1
Pigs 1.0 5 � 1 to� 3 � 2.6 5 of 5
1.5 1 � 3 � 1 of 1
a Maximum score from center or periphery of infarct.
M.C. Fishbein et al. / Cardiovascular Pathology 12 (2003) 65–71 69

was a useful biomarker to detect myocardial injury associ-
ated with doxorubicin and trauma, as well as ischemic injury
[20]. Furthermore, Ricchiuti et al. [21] demonstrated that in
dogs with experimental myocardial infarction, increases in
serum troponins occurred in parallel with loss of troponins
from the ischemic myocardium. In rats, alterations in cTnT
and cTnI occurred with severe left ventricular remodeling
[22] and after prolonged intense exercise [23]. Direct
measurement of cTnT and cTnI in dogs, goats, cows, sheep,
horses, rabbits, turkeys, chicken and fish have confirmed
tissue reactivity of varying degrees indicating that elevations
in the blood would serve as useful biomarkers of myocardial
injury in many animal species [24,25]. Directly relevant to
man, Voss et al. [26] showed a 35–50% loss of cytosolic
myofibrillar cTnT in human myocardium from three patients
who died after myocardial infarction. All of the above
studies were biochemical rather than morphologic. None
examined the tissue microscopically to visualize the pattern
of cardiac troponin loss from the injured myocardium.
Initial observations suggest that cardiac troponin immu-
nostaining might be useful for identifying myocardial
necrosis in human autopsy hearts [14,15]. Our study, in
well-characterized animal models of myocardial infarction,
demonstrates that loss of tissue cTnT and cTnI can be seen
in infarcts only 30 min old, even before histologic evidence
of necrosis is apparent. It is well known that while EM can
detect necrotic myocytes 15–20 min after occlusion, histo-
logic changes take longer to evolve, often several hours
[27]. Thus, our studies indicate that immunohistochemical
staining for cTnI and cTnT may be more sensitive than
routine H&E staining for the recognition of myocardial
necrosis in experimental animals and human hearts at
autopsy. In addition, our study demonstrates that all of the
antibodies to human cardiac troponins we used cross-react
with canine, porcine and rodent cTnT and cTnI and can
therefore be applied to experimental animal studies of
myocardial ischemia.
In this study, we were not able to precisely map entire
regions that were proven to be ischemic, but not necrotic.
However, whenever we did observe loss of staining, there
was histologic or ultrastructural evidence of irreversibly
injured myocytes in the tissue. In the dog studies, we had
performed ultrastructural studies on samples of tissue within
the zone at risk of ischemic injury. In regions of myocar-
dium that did not demonstrate irreversible injury by EM, we
did not observe decreased staining for troponins.
We observed greater loss of cardiac troponin at the
periphery of an infarct than at its center. This likely reflects
greater antegrade and retrograde flow at the periphery with
‘‘washout’’ of proteins from the necrotic myocardium [28].
There was accentuation of this phenomenon with reperfu-
sion. Since reflow is known to be associated with the
‘‘washout phenomenon,’’ and greater, earlier blood pro-
tein/enzyme elevations in man, it is likely that the greater
loss of cardiac troponin seen at the periphery, especially
with reperfusion, represents a manifestation of this washout.
We observed heterogeneous loss of cTnT and cTnI in
necrotic myocardium. Even with relatively long periods of
occlusion (6 h), with or without reperfusion, there was a
great deal of variability of intensity of residual staining for
cardiac troponins within the necrotic region. There are
several explanations for this. First, since the distribution
of flow within the ischemic tissue is heterogeneous, necrosis
may evolve faster in some parts of the infarct than others.
Second, flow away from the necrotic fibers may also be
variable, resulting in different rates of ‘‘washout’’ from
different fibers. Since cell to cell variation in staining
occurred within small regions, as well as the entire infarcts,
these two mechanisms do not seem to explain the observed
findings. It is known that cardiac troponins are cleared from
the blood faster than CK-MB, but levels stay elevated
longer [29]. These observations suggest continued, delayed
release from the necrotic myocytes. We found substantial
positive staining of necrotic myocytes hours after coronary
occlusion in our experimental infarcts. This finding may
explain the observation in patients with myocardial infarcts
that cardiac troponin levels in the blood remain elevated for
several days, as opposed to CK-MB levels which return to
baseline more quickly. Our findings support the notion that
the sustained elevation of cardiac troponin in the blood,
days after coronary occlusion, is not the result of slow
elimination from the circulation, but rather, ongoing release
during infarct evolution. This persistent elevation in the
serum does not appear to be eliminated by reperfusion.
Concordant with the clinical observation, considerable posi-
tivity in necrotic myocardium persisted, even in animals
with coronary occlusion followed by reperfusion.
This study, as well as our previous studies of other
myocardial proteins in these models [7–11], indicate that
troponins are not lost from ischemic but nonnecrotic myo-
cardium. These findings are in concordance with the major-
ity of experimental work and clinical studies. Carlson et al.
[30] have reported that in patients undergoing dobutamine
stress echocardiography, stress-related reversible ischemia
was not accompanied by increased troponin levels in the
blood, even when using very sensitive methods and strin-
gent criteria. On the other hand, Venge et al. [31] have
shown that patients with unstable angina who do manifest
elevated c troponin in blood are at higher risk of adverse
outcome than those patients who do not. These patients who
presumably are experiencing episodes of myocardial nec-
rosis may benefit from more aggressive antithrombotic and
revascularization strategies. In our studies, we did not
sample blood for markers of necrosis; however, our mor-
phologic studies in well-characterized models of ischemic
injury support the concept that troponin release is specific
and sensitive for myocardial necrosis.
Lastly, in these studies, antibodies generated against
human cardiac troponins, stained normal rat, dog and pig
myocardium and were diminished in necrotic myocardium
in these animals. Thus, these antibodies should be useful in
experimental studies of myocardial ischemic injury in a
M.C. Fishbein et al. / Cardiovascular Pathology 12 (2003) 65–7170

variety of animal models. The ability to detect loss of
cardiac troponins so early in the evolution of myocardial
necrosis also suggests a role for immunohistochemical
studies for cardiac troponins in human autopsies. Such
studies might assist pathologists in the recognition and
verification of myocardial infarction at autopsy, which has
been a challenge for pathologists to date. In this regard, a
preliminary report appears encouraging [14].
Acknowledgments
The authors are grateful to Dr. Jack Ladenson for
donating antisera to troponin I. The authors also wish to
recognize the expert secretarial assistance and word
processing skills of Ms. Judy Wiltz and the photographic
expertise of Ms. Carol Appleton. This study was support-
ed by a generous endowment from the Piansky Family
Trust (MCF).
References
[1] Jaffe AS, Ravkilde J, Roberts R, Naslund U, Apple FS, Galvani M,
Katus H. Its time for a change to a troponin standard. Circulation
2000;102:1216–20.
[2] Antman EM, Tanasijevic MJ, Thompson B, Schactman M,
McCabe CH, Cannon CP, Fischer GA, Fung AY, Thompson C,
Wybenga D, Braunwald E. Cardiac-specific troponin I levels to
predict the risk of mortality in patients with acute coronary syn-
dromes. N Engl J Med 1996;335:1342–9.
[3] Ohman ME, Armstrong PW, Christenson RH, Granger CB, Katus HA,
Hamm W, O’Hanesian MA, Wagner GS, Kleiman NS, Harrell FE,
Califf RM, Topol EJ. Cardiac troponin T levels for risk stratification in
acute myocardial ischemia. N Engl J Med 1996;335:1333–41.
[4] Apple FS, Falahati A, Paulson PR, Miller E, Sharkey SW. Improved
detection of minor ischemic myocardial injury with measurement of
serum cardiac troponin I. Clin Chem 1997;43:2047–51.
[5] Giannitsis E, Muller-Bardorff M, Lehrke S, Wiegand U, Tolg R,
Weidtmann B, Hartmann F, Richardt G, Katus HA. Admission tro-
ponin T level predicts clinical outcome, TIMI flow, and myocardial
tissue perfusion after primary percutaneous intervention for acute
ST-segment elevation myocardial infarction. Circulation 2001;104:
630–5.
[6] Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction
redefined—a consensus of the Joint European Society of Cardiology/
American College of Cardiology Committee for the Redefinition of
Myocardial Infarction. J Am Coll Cardiol 2000;36:959–69.
[7] Moran MM, Siegel RJ, Said JW, Fishbein MC. Demonstration of
myoglobin CK-M in myocardium. J Histochem Cytochem 1985;33:
1110–5.
[8] Siegel RJ, Said JW, Shell WE, Corson G, Fishbein MC. Identification
and localization of creatine kinase B and M in normal, ischemic and
necrotic myocardium: an immunohistochemical study. J Mol Coll
Cardiol 1984;16:95–103.
[9] Block MI, Said JW, Siegel RJ, Fishbein MC. Myocardial myoglobin
following coronary artery occlusion: an immunohistochemical study.
Am J Pathol 1983;111:374–9.
[10] Herscher LL, Siegel RJ, Said JW, Edwalds LL, Moran MM, Fishbein
MC. Distribution of LDH-1 in normal, ischemic, and necrotic myo-
cardium. Am J Clin Pathol 1984;81:198–203.
[11] Siegel RJ, Edwalds G, Rej R, Fishbein MC. Distribution of cytosolic
and mitochondrial aspartate aminotransferase in normal, ischemic, and
necrotic myocardium: an immunohistochemical study. Lab Invest
1984;51:648.
[12] Edwalds GM, Said JW, Block MI, Herscher LL, Siegel RJ, Fishbein
MC. Myocytolysis (vacuolar degeneration) of myocardium: immuno-
histochemical evidence of viability. Hum Pathol 1984;15:753–6.
[13] FishbeinMC,Meerbaum S, Rit J, LandoU, Kanmatsuse K,Mercier JC,
Corday E, Ganz W. Early phase acute myocardial infarct size quantifi-
cation: validation of the triphenyl tetrazolium chloride tissue enzyme
staining technique. Am Heart J 1981;101:593–600.
[14] Hansen SH, Rossen K. Evaluation of cardiac troponin I immunoreac-
tion in autopsy hearts: a possible marker of early myocardial infarc-
tion. Forensic Sci Int 1999;99:189–96.
[15] Lambie WK, Afify AM. Immunohistochemical evaluation of recent
myocardial infarction using troponin I, troponin T, and complement 9.
Lab Invest 2002;82:6A.
[16] Hatori N, Tadokoro H, Satomura K, Miyazaki A, Fishbein MC,
Ryden L, Corday E, Drury JK. Beneficial effects of coronary venous
retroinfusion but not left atrial administration of superoxide dismu-
tase on myocardial necrosis in pigs. Eur Heart J 1991;12:442–50.
[17] HatoriN,RobertsRL, TadokoroH,RydenL, SatomuraK, FishbeinMC,
Stiehm ER, Corday E, Drury JK. Differences of infarct size with
lidocaine as compared with bretylium tosylate in acute myocardial
ischemia and reperfusion in pig. J Cardiol Pharm 1991;18:581–8.
[18] McElroy CL, Gissen SA, Fishbein MC. Exercise-induced reduction in
myocardial infarct size after coronary artery occlusion in the rat. Cir-
culation 1978;57:5.
[19] Bouchardy B, Majno G. Histopathology of early myocardial infarcts.
Am J Pathol 1974;74:301–30.
[20] O’Brien PJ, Dameron GW, Beck ML, Kang YJ, Erickson BK, Di
Battista TH, Miller KE, Jackson KN, Mittelstadt S. Cardiac troponin
T is a sensitive specific biomarker of cardiac injury in laboratory
animals. Lab Anim Sci 1997;47:5.
[21] Ricchiuti V, Sharkey SW, Murakami MM, Voss EM, Apple FS. Car-
diac troponin I and T alterations in dog hearts with myocardial in-
farction. Am J Clin Pathol 1998;110:241–7.
[22] Ricchiuti V, Zhang J, Apple FS. Cardiac troponin I and T alterations in
hearts with severe left ventricular remodeling. Clin Chem 1997;43–
46:990–5.
[23] Chen Y, Serfass RC, Mackey-Bojack S, Kelly KL, Titus JL, Apple
FS. Cardiac troponin T alterations in myocardium and serum of rats
after stressful, prolonged intense exercise. J Appl Physiol 2000;88:
1949–55.
[24] O’Brien PJ, Dameron GW, Beck ML, Brandt M. Differential reactivity
of cardiac and skeletal muscle from various species in two generations
of cardiac troponin T immunoassays. Res Vet Sci 1998;65:135–7.
[25] O’Brien PJ, Landt Y, Ladenson JH. Differential reactivity of cardiac
and skeletal muscle from various species in a cardiac troponin I im-
munoassay. Clin Chem 1997;43:2333–8.
[26] Voss EM, Sharkey SW, Gernert AE, Murakami MM, Johnston RB,
Hsieh CC, Apple FS. Human and canine cardiac troponin T and
creatine kinase-MB distribution in normal and diseased myocardium:
infarct sizing using serum profiles. Arch Pathol Lab Med 1995;119.
[27] Jennings RB, Ganote CE. Structural changes in myocardium during
acute ischemia. Circ Res 1974;34–35(suppl III):156–72.
[28] Fishbein MC. Reperfusion injury. Clin Cardiol 1990;13:213–7.
[29] Katus HA, Remppis A, Scheffold T, Diederich KW, Kuebler W. Intra-
cellular compartmentation of cardiac troponin T and its release ki-
netics in patients with reperfused and nonreperfused myocardial
infarction. Am J Cardiol 1991;67:1360–7.
[30] Carlson RJ, Navone A, McConnell JP, Burritt M, Castle MC, Grill D,
Jaffe AS. Effect of myocardial ischemia on cardiac troponin I and T.
Am J Cardiol 2002;89:224–6.
[31] Venge P, Lagerqvist B, Diderholm E, Lindahl B, Wallentin L. Clinical
performance of three cardiac troponin assays in patients with unstable
coronary artery disease (a FRISC II substudy). Am J Cardiol 2002;
89:1035–41.
M.C. Fishbein et al. / Cardiovascular Pathology 12 (2003) 65–71 71
Top Related
Copyright © 2022 FDOKUMEN

