







Bahasa
Halaman
Hukum


ORIGINAL ARTICLE
© Copyright 2006 by Humana Press Inc.All rights of any nature whatsoever reserved.1085-9195/(Online)1559-0283/06/44:395–404/$30.00
Cell Biochemistry and Biophysics 395 Volume 44, 2006
INTRODUCTION
In this work, we present Molecular Modeling (MM)and Dynamics (MD) techniques applied to the study ofHIV-1 protease complexed with the inhibitor ritonavir.We intend to evaluate similarities and differencesbetween B and non-B subtypes of HIV-1 protease.Derived from our MD simulations, the most importantresult that we can highlight is the decline in bindingaffinity for inhibitor relative to non-B subtypes com-pared with subtype B, in accordance with some previ-ous experimental results, and that can, in due course,favor the emergence of drug resistance.
One of the major characteristics of HIV-1 is its exten-sive genetic diversity as a result of the high error rate,the recombinogenic properties of the reverse transcrip-tase enzyme (1,2), and the extremely high turnover ofvirions (range of 109/d) in HIV-infected individuals (3).Phylogenetic analyses have classified three classes ofHIV-1 in the world: M (major), O (outline), and N (new)(4). Within HIV-1 group M (~90% of reported HIV AIDScases), at least nine distinct subtypes (A, B, C, D, F, G, H,J, and K) and 14 circulating recombinant forms (CRF)have been identified (reviewed in refs. 5,6). Of the esti-mated 40 million people infected with HIV-1 world-wide, more than 26 million are in Africa and a veryfrightful statistic has been seen last year: Asia housedone quarter of the world’s newly infected HIV positiveindividuals (7).
Molecular Dynamics Simulations Applied to the Study of Subtypes of HIV-1 Protease Common to Brazil, Africa, and Asia
Paulo R. Batista,1,* Alan Wilter,2 Elza H. A. B. Durham,3 and Pedro G. Pascutti1
1Laboratório de Modelagem e Dinâmica Molecular, Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil; 2Laboratório Nacional de Computação Científica,
Rio de Janeiro, Brazil; 3Instituto Ludwig de Pesquisa sobre o Câncer, São Paulo, Brazil; and 3Instituto de Matemática eEstatística, Universidade São Paulo, São Paulo, Brazil
Abstract
Africa accounts for the majority of HIV-1 infections worldwide caused mainly by the A and C viral subtypesrather than B subtype, which prevails in the United States and Western Europe. In Brazil, B subtype is the majorsubtype, but F, C, and A also circulate. These non-B subtypes present polymorphisms, and some of them occurat sites that have been associated with drug resistance, including the HIV-1 protease (PR), one important drugtarget. Here, we report a Molecular Dynamics study of the B and non-B PR complexed with the inhibitor riton-avir to delineate the behavior of each subtype. We compare root mean squared deviation, binding free energyby linear interaction energy approach, hydrogen bonds, and intermolecular contact surface area betweeninhibitor and PR. From our results, we can provide a basis to understand the molecular mechanism of drug resis-tance in non-B subtypes. In this sense, we found a decrease of approx 4 kcal/mol in ∆G of binding between Band non-B subtypes. This corresponds to the loss of one hydrogen bond, which is in agreement with our H-bondanalysis. Previous experimental affinity studies reported analogous results with inhibition constant values fornon-B PR.
Index Entries: Molecular Dynamics; Gromacs; HIV-1 protease; ritonavir; subtypes; non-B; modelling; freeenergy; LIE.
*Author to whom all correspondence and reprint requestsshould be addressed. E-mail: [email protected]

The HIV-1 subtypes prevalent in Africa and Asia aredistinct from the one prevailing in North America andWestern Europe. In these developed regions, subtype Bis responsible for the majority of HIV infections,whereas in sub-Saharan Africa and Asia, subtypes Aand C account for most of the infections. Non-B sub-types accounted for 88% of new worldwide infections in2000 (A, 30% and C, 47%) (8). In Brazil, subtype B is themajor subtype, but F (18% of infections), C (30% ofinfections in southern region), and D can also be found(9). These subtypes exhibit differences with respect tosubtype B, which involves current drug targets, such asthe HIV-1 protease (PR).
Extensive research in the past decade has been dedi-cated to designing resistance-evading drugs for PR,which is critical for the maturation of viral structural(gag) and enzymatic (pol) proteins. The PR is an aspartylprotease and is composed of two symmetric subunits,each with 99 residues that can recognize either symmet-ric or asymmetric substrates (10).
One important region in PR for interaction with thesubstrate is the flap (residues 45–58) that must open forthe substrate to access the active site and close for cleav-age. The substrate stabilization in the active siteinvolves hydrogen bonds with the PR flap, mainlythrough a structured water molecule close to ILE50. Thebinding of inhibitors to the flaps, and likewise substrate,helps to keep the flaps stable (11).
Many crystal structures of the PR and its complexeswith inhibitors are currently available, but there is nostructural information for non-B subtypes. The firstcrystallization of a non-B PR was reported recently, butits structure has not been solved yet (12).
The major success of structure-assisted drug designwas the development of PR inhibitors (13). Ritonavirwas the first Food and Drug Administration-approvedprotease inhibitor reached by this methodology, and, inthis work, it was the chosen inhibitor to study interac-tions, at the atomic level, with the main PR subtypespresent in Brazil, Africa, and Asia (B, F, C, and A sub-types). To understand similarities and differencesbetween B and non-B subtypes of PR complexed withritonavir, we made use of MM because there is no struc-ture of non-B PR from nuclear magnetic resonance andX-ray crystallography methods. Furthermore, althoughthese experimental methods are the ideal modelingapproach, they are also more time-consuming andexpensive, and they involve hazardous biological mate-rials compared with MM approaches.
Computational simulations are powerful tools usedto investigate ligand–protein interactions, besides show-ing a nonstatic behavior of the system (as opposed tocrystallographic diffraction). MD allows the estimationof several thermodynamics properties (e.g., binding free
energy calculations), and is a low cost and practicalmethod as well. With comparative MM technique, byusing available template three-dimensional (3D) struc-tures found in Protein Data Bank (PDB) (14), we wereable to construct models for each PR subtype in com-plex with PR inhibitor ritonavir.
Because protease inhibitors have been developed andtested against the HIV-1 B subtype, and PR from othersubtypes carry up to 10 amino acid polymorphisms, it isimportant to assess the influence of these naturallyoccurring polymorphisms on the potency of existinginhibitors, as well as their synergistic interactions withmutations known to cause drug resistance (15). At thebiochemical level, non-B-subtype polymorphisms lowerthe binding affinities of existing clinical inhibitors, butnot to the point of causing drug resistance (15,16).However, these polymorphisms amplify the effects ofmutations causing drug resistance and may play a rolein the long-term viability of these inhibitors. MD stud-ies of PR can help to provide the grounds for under-standing the molecular basis of drug resistance.
MATERIALS AND METHODS
Construction of Models: Comparative Molecular Modeling
To ascertain the 3D coordinates of each system, weconstruct the target models of the PR consensus sub-types A (17), C (18), and F (19) by using comparative MMtechniques with SwissPDBViewer program (20). As tem-plate, we used the X-ray crystallography structure of thePR consensus B complexed with ritonavir (PDB code1HXW) (21), including the crystallographic water mole-cules and inhibitor coordinates. To achieve our models,we had only to cope with residues substitution, becausePRs display high sequence similarity, with the samenumber of residues, not varying more than nine aminoacids among consensus. The consensus sequences of PRsubtypes present these differences from the PR consen-sus B sequence (9): A (L10V, I13V, K14R, I15V, K20I,M36I, R41K, H69K, and L89M), C (T12S, I15V, L19I,M36I, R41K, H69K, L89M, and I93L), and F (I15V, E35D,M36I, R41K, R57K, Q61N, and L89M). Additionally, Fig. 1features non-B polymorphism with localization of theirmutations referent to 1HXW on the 3D structure of PR.All models were validated by stereochemistry withProcheck program (22).
Inhibitor TopologyFor the inhibitor’s (ritonavir) topology, which is not
publicly available for GROMOS96 force field (23), wehad to build its required parameters to run MD simula-tions. We started with the server PRODRG (24), which
396 Batista et al.
Cell Biochemistry and Biophysics Volume 44, 2006

was used to generate a first set of parameters for bonds,angles, and charges based on GROMOS87 force field(25). Thus, eventually we had to fit such parameters toGROMOS96, appealing to a set of parameters previ-ously determined for some groups similar to the ones ofritonavir, or, when the reference was not found, to the abinitio calculation by means of GAUSSIAN94 (26), usingthe base B3LYP/6-31G** with option CHELPG, assum-ing null total charge for such drug.
Molecular DynamicsThe molecular mechanics potential energy minimiza-
tions and MD simulations were carried out with the pro-gram package GROMACS, version 3.2.1 (27,28) by usingGROMOS96 force field. For all the systems, we usedvisual MD (VMD) (29) and SwissPDBViewer, programsfor virtual molecular visualization and manipulation, toset up spatial orientation of complexes and to have theirprincipal axes aligned to the Cartesian axes. The solva-tion procedure was performed with a layer of at least15 Å around protease–ligand complex, in an orthorhom-
bic geometry box (for periodic boundary conditions).The model of solvent was single point charge (SPC)water (30) (about 12,000 molecules). To neutralize the sys-tem charge, chloride (Cl–) counterions were inserted. Weended up with four systems for MD simulations withtheir final volumes ranged from 400 to 420 nm3, and eachwith the number of atoms between 35,000 and 40,000.
For energy minimization, we used the algorithmssteepest descent (preceded by a position restrained stagefor protein atoms), conjugate gradient and a quasi-Newton low-memory minimizer in sequence until reach-ing an energy gradient lower than 2.39 kcal/mol/Å. TheMD simulations were performed according to the fol-lowing procedure: 500 ps with positions of protein’satoms restrained, to allow the solvent equilibration, andthen a full MD for 3.5 ns, with no restrictions. The MDintegration time was 2 fs. LINCS (31) and SETTLE (32)constraints were used for protein/ritonavir (all bonds)and solvent, respectively. Temperature was maintained at300 K, and pressure maintained at 1 atm by the Berendsenweak-coupling approach (33). For long-range interac-tions, reaction field method (34) was used, with dielectricconstant set to 54 (35). Non-bonded cutoffs were 1.2 Å forvan der Waals and 1.4 Å for Coulomb interactions.According to kinetic and MD studies of PR (36–38),only the catalytic ASP124 was protonated (not ASP25).Simulations demanded 7 to 9 d of computation at ClusterXML (BioPAUÁ Project, LNCC, IBCCF/ UFRJ, and HPBrazil R&D).
Binding Free Energy CalculationTo estimate the binding free energies of ritonavir to
the PR receptors cited in this work, we used a semi-empirical MD method, based on linear responseassumptions, entitled linear interaction energy (LIE),conceived by Åqvist and collaborators (39). It is a fastermethod than free energy perturbation or thermody-namic integration because it does not need any uninter-esting intermediate state between the initial and finalstates, and the results of its application are in goodagreement with experimental data (40–42). LIE, asdepicted in Eq. 1, divides the interaction between theligand and its environment into electrostatic and vander Waals terms:
[1]
where � � denotes MD averages of nonbonded potentialenergy differences between two states of the inhibitor:bonded to the PR active site and free in solution. Thenonbonded potential energy corresponds to the van derWaals (vdW) and electrostatic (el) interaction of the lig-and and its surrounding environment, the enzyme,
∆ ∆ ∆G G G V V V Vbind bindel
bindvdW
boundel
freeel
boundvdW
freevdW= + ≈ − + − +α β γ
Computational Studies of HIV-1 Protease of B and Non-B Subtypes 397
Cell Biochemistry and Biophysics Volume 44, 2006
Fig. 1. Non-B polymorphisms along the PR 3D structure.Left, common substitution among the non-B sequences.Right, particular substitutions (little spheres) of each sub-type are labeled.
ions, and solvent in one situation (bonded), and the sol-vent only in the other situation (free). The additionalconstant term γ can be used to adjust LIE to reproduceexperimental binding free energy data (39).
Intermolecular SurfaceWe developed a special program called “Surfmds” to
calculate intermolecular contact surface area from MDtrajectories, based on Connolly’s algorithm (43). From sol-vent accessible surface (SAS) of protein and ligand, it ispossible to determine the intermolecular surface as beingthe intersection between the SAS of ligand and the SAS ofprotein, i.e., the sum of the areas of protein and ligand,close enough to avoid the allocation of a water molecule.
RESULTS
Properties of the Global StructuresThe four systems—1HXW (consB), consA, consC,
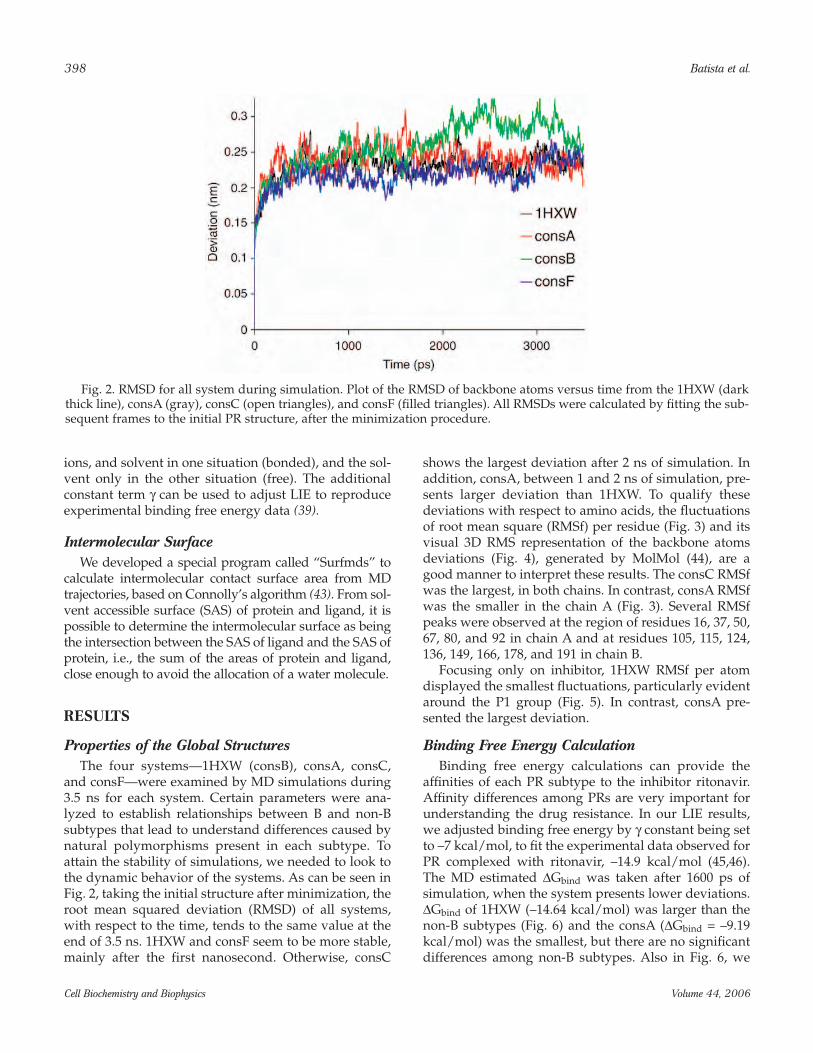
and consF—were examined by MD simulations during3.5 ns for each system. Certain parameters were ana-lyzed to establish relationships between B and non-Bsubtypes that lead to understand differences caused bynatural polymorphisms present in each subtype. Toattain the stability of simulations, we needed to look tothe dynamic behavior of the systems. As can be seen inFig. 2, taking the initial structure after minimization, theroot mean squared deviation (RMSD) of all systems,with respect to the time, tends to the same value at theend of 3.5 ns. 1HXW and consF seem to be more stable,mainly after the first nanosecond. Otherwise, consC
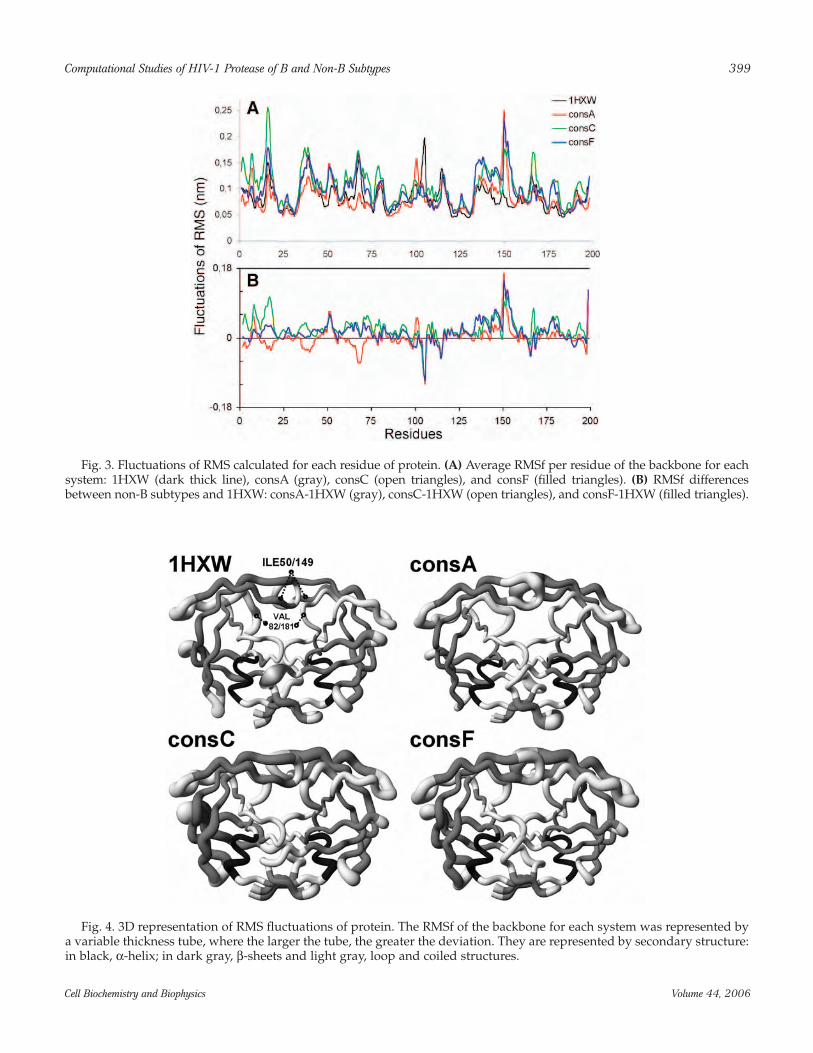
shows the largest deviation after 2 ns of simulation. Inaddition, consA, between 1 and 2 ns of simulation, pre-sents larger deviation than 1HXW. To qualify thesedeviations with respect to amino acids, the fluctuationsof root mean square (RMSf) per residue (Fig. 3) and itsvisual 3D RMS representation of the backbone atomsdeviations (Fig. 4), generated by MolMol (44), are agood manner to interpret these results. The consC RMSfwas the largest, in both chains. In contrast, consA RMSfwas the smaller in the chain A (Fig. 3). Several RMSfpeaks were observed at the region of residues 16, 37, 50,67, 80, and 92 in chain A and at residues 105, 115, 124,136, 149, 166, 178, and 191 in chain B.
Focusing only on inhibitor, 1HXW RMSf per atomdisplayed the smallest fluctuations, particularly evidentaround the P1 group (Fig. 5). In contrast, consA pre-sented the largest deviation.
Binding Free Energy CalculationBinding free energy calculations can provide the
affinities of each PR subtype to the inhibitor ritonavir.Affinity differences among PRs are very important forunderstanding the drug resistance. In our LIE results,we adjusted binding free energy by γ constant being setto –7 kcal/mol, to fit the experimental data observed forPR complexed with ritonavir, –14.9 kcal/mol (45,46).The MD estimated ∆Gbind was taken after 1600 ps ofsimulation, when the system presents lower deviations.∆Gbind of 1HXW (–14.64 kcal/mol) was larger than thenon-B subtypes (Fig. 6) and the consA (∆Gbind = –9.19kcal/mol) was the smallest, but there are no significantdifferences among non-B subtypes. Also in Fig. 6, we
398 Batista et al.
Cell Biochemistry and Biophysics Volume 44, 2006
Fig. 2. RMSD for all system during simulation. Plot of the RMSD of backbone atoms versus time from the 1HXW (darkthick line), consA (gray), consC (open triangles), and consF (filled triangles). All RMSDs were calculated by fitting the sub-sequent frames to the initial PR structure, after the minimization procedure.
Computational Studies of HIV-1 Protease of B and Non-B Subtypes 399
Cell Biochemistry and Biophysics Volume 44, 2006
Fig. 3. Fluctuations of RMS calculated for each residue of protein. (A) Average RMSf per residue of the backbone for eachsystem: 1HXW (dark thick line), consA (gray), consC (open triangles), and consF (filled triangles). (B) RMSf differencesbetween non-B subtypes and 1HXW: consA-1HXW (gray), consC-1HXW (open triangles), and consF-1HXW (filled triangles).
Fig. 4. 3D representation of RMS fluctuations of protein. The RMSf of the backbone for each system was represented bya variable thickness tube, where the larger the tube, the greater the deviation. They are represented by secondary structure:in black, α-helix; in dark gray, β-sheets and light gray, loop and coiled structures.
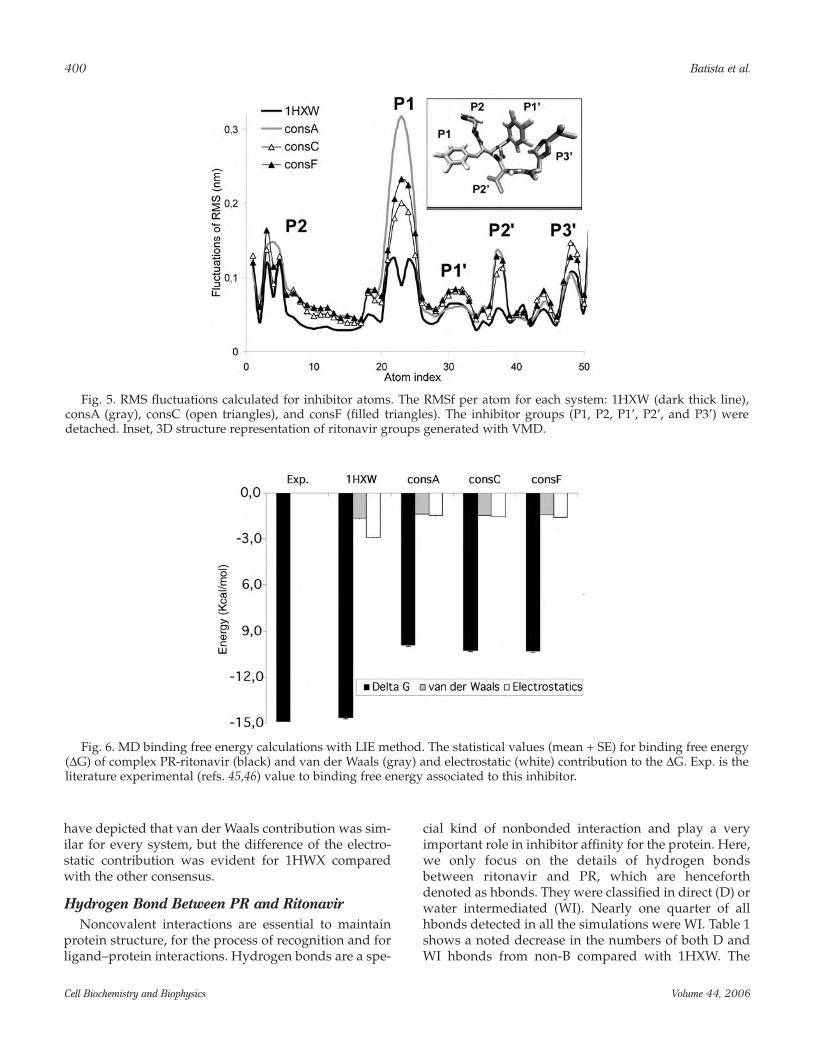
have depicted that van der Waals contribution was sim-ilar for every system, but the difference of the electro-static contribution was evident for 1HWX comparedwith the other consensus.
Hydrogen Bond Between PR and RitonavirNoncovalent interactions are essential to maintain
protein structure, for the process of recognition and forligand–protein interactions. Hydrogen bonds are a spe-
cial kind of nonbonded interaction and play a veryimportant role in inhibitor affinity for the protein. Here,we only focus on the details of hydrogen bondsbetween ritonavir and PR, which are henceforthdenoted as hbonds. They were classified in direct (D) orwater intermediated (WI). Nearly one quarter of allhbonds detected in all the simulations were WI. Table 1shows a noted decrease in the numbers of both D andWI hbonds from non-B compared with 1HXW. The
400 Batista et al.
Cell Biochemistry and Biophysics Volume 44, 2006
Fig. 5. RMS fluctuations calculated for inhibitor atoms. The RMSf per atom for each system: 1HXW (dark thick line),consA (gray), consC (open triangles), and consF (filled triangles). The inhibitor groups (P1, P2, P1’, P2’, and P3’) weredetached. Inset, 3D structure representation of ritonavir groups generated with VMD.
Fig. 6. MD binding free energy calculations with LIE method. The statistical values (mean + SE) for binding free energy(∆G) of complex PR-ritonavir (black) and van der Waals (gray) and electrostatic (white) contribution to the ∆G. Exp. is theliterature experimental (refs. 45,46) value to binding free energy associated to this inhibitor.
average of total number (D plus WI) for consB wasapprox 1 hbond higher than non-B PRs. To evaluatequalitatively the nature of these hbonds, we calculatedthe prevalence of each one along dynamics (Table 2),and we can point out that hbond RIT199N34-ASP25OD2 is present only in 1HXW simulation(82.5%). Although we have almost 90% of ILE149 WIhbond in 1HXW, this value goes to about half or less forthe others groups.
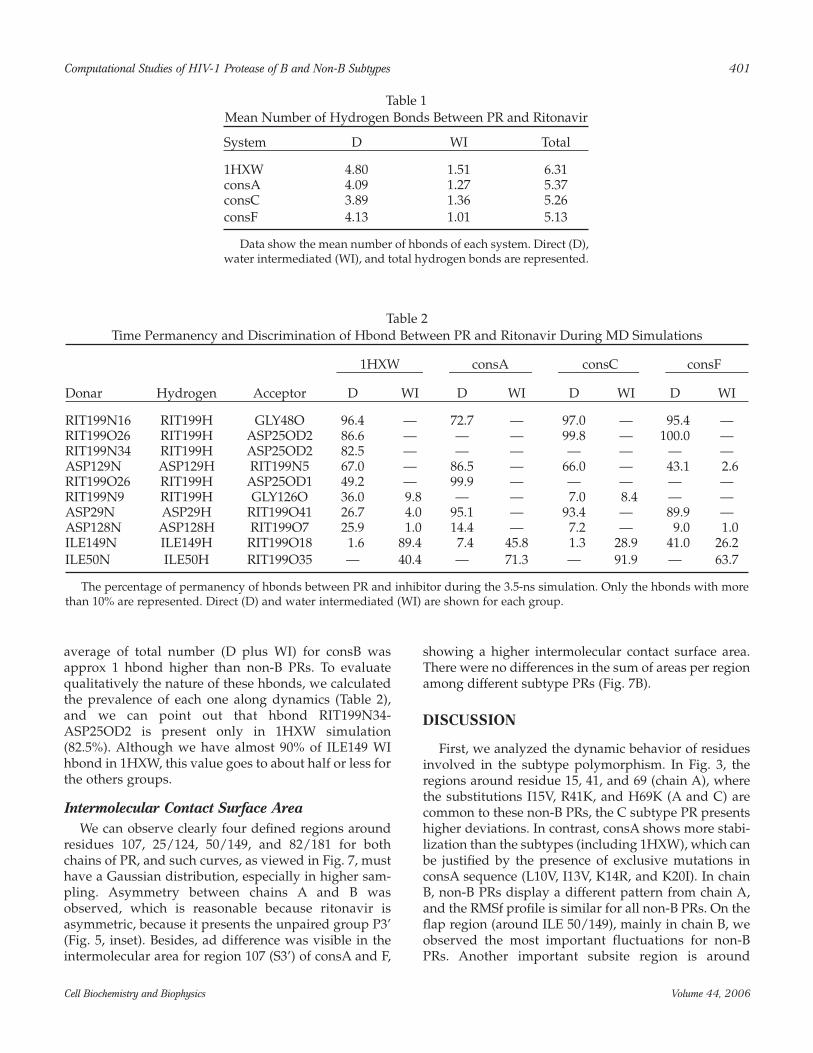
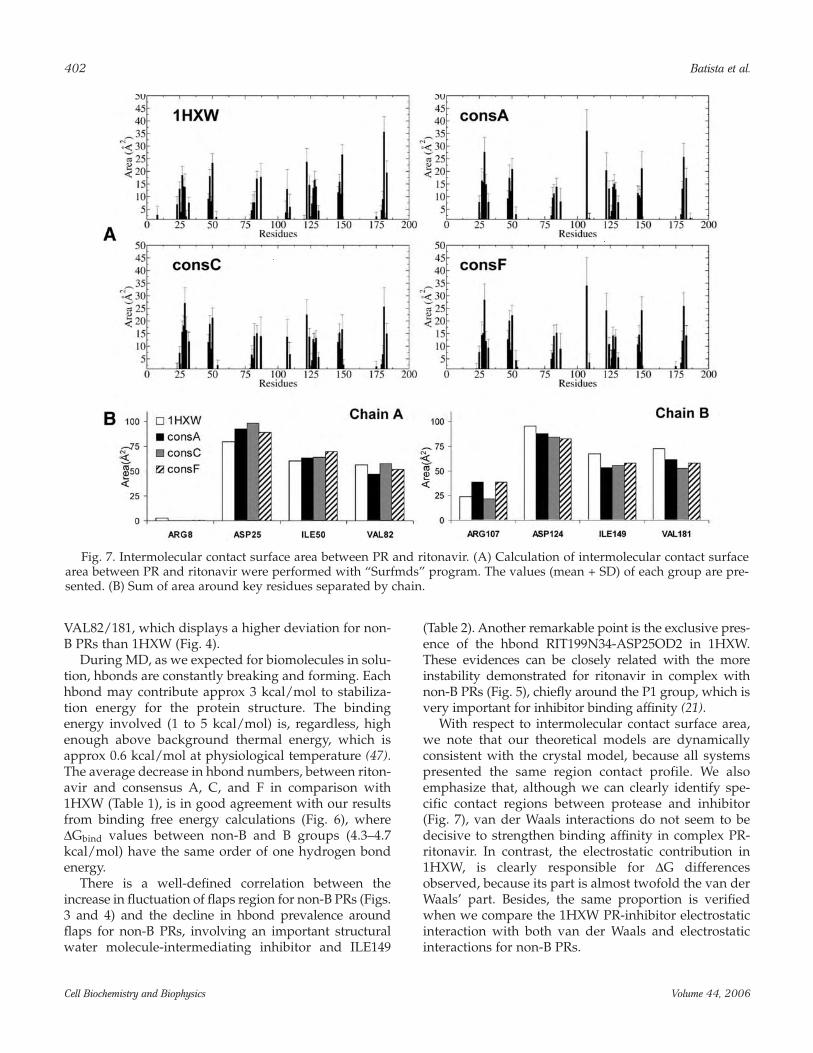
Intermolecular Contact Surface AreaWe can observe clearly four defined regions around
residues 107, 25/124, 50/149, and 82/181 for bothchains of PR, and such curves, as viewed in Fig. 7, musthave a Gaussian distribution, especially in higher sam-pling. Asymmetry between chains A and B wasobserved, which is reasonable because ritonavir isasymmetric, because it presents the unpaired group P3’(Fig. 5, inset). Besides, ad difference was visible in theintermolecular area for region 107 (S3’) of consA and F,
showing a higher intermolecular contact surface area.There were no differences in the sum of areas per regionamong different subtype PRs (Fig. 7B).
DISCUSSION
First, we analyzed the dynamic behavior of residuesinvolved in the subtype polymorphism. In Fig. 3, theregions around residue 15, 41, and 69 (chain A), wherethe substitutions I15V, R41K, and H69K (A and C) arecommon to these non-B PRs, the C subtype PR presentshigher deviations. In contrast, consA shows more stabi-lization than the subtypes (including 1HXW), which canbe justified by the presence of exclusive mutations inconsA sequence (L10V, I13V, K14R, and K20I). In chainB, non-B PRs display a different pattern from chain A,and the RMSf profile is similar for all non-B PRs. On theflap region (around ILE 50/149), mainly in chain B, weobserved the most important fluctuations for non-BPRs. Another important subsite region is around
Computational Studies of HIV-1 Protease of B and Non-B Subtypes 401
Cell Biochemistry and Biophysics Volume 44, 2006
Table 1Mean Number of Hydrogen Bonds Between PR and Ritonavir
System D WI Total
1HXW 4.80 1.51 6.31consA 4.09 1.27 5.37consC 3.89 1.36 5.26consF 4.13 1.01 5.13
Data show the mean number of hbonds of each system. Direct (D),water intermediated (WI), and total hydrogen bonds are represented.
Table 2Time Permanency and Discrimination of Hbond Between PR and Ritonavir During MD Simulations
1HXW consA consC consF
Donar Hydrogen Acceptor D WI D WI D WI D WI
RIT199N16 RIT199H GLY48O 96.4 — 72.7 — 97.0 — 95.4 —RIT199O26 RIT199H ASP25OD2 86.6 — — — 99.8 — 100.0 —RIT199N34 RIT199H ASP25OD2 82.5 — — — — — — —ASP129N ASP129H RIT199N5 67.0 — 86.5 — 66.0 — 43.1 2.6RIT199O26 RIT199H ASP25OD1 49.2 — 99.9 — — — — —RIT199N9 RIT199H GLY126O 36.0 9.8 — — 7.0 8.4 — —ASP29N ASP29H RIT199O41 26.7 4.0 95.1 — 93.4 — 89.9 —ASP128N ASP128H RIT199O7 25.9 1.0 14.4 — 7.2 — 9.0 1.0ILE149N ILE149H RIT199O18 1.6 89.4 7.4 45.8 1.3 28.9 41.0 26.2ILE50N ILE50H RIT199O35 — 40.4 — 71.3 — 91.9 — 63.7
The percentage of permanency of hbonds between PR and inhibitor during the 3.5-ns simulation. Only the hbonds with morethan 10% are represented. Direct (D) and water intermediated (WI) are shown for each group.
VAL82/181, which displays a higher deviation for non-B PRs than 1HXW (Fig. 4).
During MD, as we expected for biomolecules in solu-tion, hbonds are constantly breaking and forming. Eachhbond may contribute approx 3 kcal/mol to stabiliza-tion energy for the protein structure. The bindingenergy involved (1 to 5 kcal/mol) is, regardless, highenough above background thermal energy, which isapprox 0.6 kcal/mol at physiological temperature (47).The average decrease in hbond numbers, between riton-avir and consensus A, C, and F in comparison with1HXW (Table 1), is in good agreement with our resultsfrom binding free energy calculations (Fig. 6), where∆Gbind values between non-B and B groups (4.3–4.7kcal/mol) have the same order of one hydrogen bondenergy.
There is a well-defined correlation between theincrease in fluctuation of flaps region for non-B PRs (Figs.3 and 4) and the decline in hbond prevalence aroundflaps for non-B PRs, involving an important structuralwater molecule-intermediating inhibitor and ILE149
(Table 2). Another remarkable point is the exclusive pres-ence of the hbond RIT199N34-ASP25OD2 in 1HXW.These evidences can be closely related with the moreinstability demonstrated for ritonavir in complex withnon-B PRs (Fig. 5), chiefly around the P1 group, which isvery important for inhibitor binding affinity (21).
With respect to intermolecular contact surface area,we note that our theoretical models are dynamicallyconsistent with the crystal model, because all systemspresented the same region contact profile. We alsoemphasize that, although we can clearly identify spe-cific contact regions between protease and inhibitor(Fig. 7), van der Waals interactions do not seem to bedecisive to strengthen binding affinity in complex PR-ritonavir. In contrast, the electrostatic contribution in1HXW, is clearly responsible for ∆G differencesobserved, because its part is almost twofold the van derWaals’ part. Besides, the same proportion is verifiedwhen we compare the 1HXW PR-inhibitor electrostaticinteraction with both van der Waals and electrostaticinteractions for non-B PRs.
402 Batista et al.
Cell Biochemistry and Biophysics Volume 44, 2006
Fig. 7. Intermolecular contact surface area between PR and ritonavir. (A) Calculation of intermolecular contact surfacearea between PR and ritonavir were performed with “Surfmds” program. The values (mean + SD) of each group are pre-sented. (B) Sum of area around key residues separated by chain.

There are four common polymorphisms among PRconsensus subtypes compared with B consensus.Among them, there is M36I, a secondary mutation asso-ciated with drug resistance against ritonavir. Besidesthat, there are two more secondary mutations in consAalso associated with drug resistance: L10V and K20I.Such mutations alone cannot cause drug resistance;they must come in connection with a primary or othersecondary mutations (19,48,49). Only consA revealsthree secondary mutations, but for consC and consF,they show such a polymorphism that could also lead toresistance. In fact, our results of binding free energy(Fig. 5) corroborate the Velazquez-Campoy data (16),where Ki of non-B PR indicates reduction in their affini-ties to ritonavir. However, these affinity differencesalone are not sufficient to cause drug resistance.Nevertheless, they can intensify the effects of mutationsand eventually lead to drug resistance.
ACKNOWLEDGMENTS
We thank the individuals who developed and main-tained the GPL software. This work was supported byHP Brazil R&D, CAPES, FAPERJ, and CNPq.
REFERENCES
1. Hu, W. S. and Temin, H. M. (1990) Retroviral recombina-tion and reverse transcription. Science 250, 1227–1233.
2. Preston, B. D., Poiesz, B. J., and Loeb, L. A. (1988) Fidelityof HIV-1 reverse transcriptase. Science 242, 1168–1171.
3. Ho, D. D., Neumann, A. U., Perelson, A. S., Chen, W.,Leonard, J. M., and Markowitz, M. (1995) Rapid turnoverof plasma virions and CD4 lymphocytes in HIV-1 infec-tion. Nature 373, 123–126.
4. Simon, F., Mauclere, P., Roques, P., et al. (1998) Identificationof a new human immunodeficiency virus type 1 distinctfrom group M and group O. Nat. Med. 4, 1032–1037.
5. Kantor, R. and Katzenstein, D. (2003) Polymorphism inHIV-1 non-subtype B protease and reverse transcriptaseand its potential impact on drug susceptibility and drugresistance evolution. AIDS Rev. 5, 25–35.
6. Wainberg, M. A. (2004) HIV-1 subtype distribution and theproblem of drug resistance. AIDS 18 (Suppl.) 3, S63-S68.
7. UNAIDS (2004) AIDS epidemic update: 2004. UNAIDS/WHO, Geneva, Switzerland.
8. Osmanov, S., Pattou, C., Walker, N., Schwardlander, B.,Esparza, J., and Charact, W.-U.N.H.I. (2002) Estimatedglobal distribution and regional spread of HIV-1 geneticsubtypes in the year 2000. J. Acquir. Immun. Defic. Syndr.29, 184–190.
9. Soares, M. A., Brindeiro, R. M., and Tanuri, A. (2004)Primary HIV-1 drug resistance in Brazil. AIDS 18 (Suppl.)3, S9–S13.
10. Prabu-Jeyabalan, M., Nalivaika, E., and Schiffer, C. A.(2000) How does a symmetric dimer recognize an asym-
metric substrate? A substrate complex of HIV-1 protease.J. Mol. Biol. 301, 1207–1220.
11. Freedberg, D. I., Ishima, R., Jacob, J., et al. (2002) Rapidstructural fluctuations of the free HIV protease flaps insolution: relationship to crystal structures and compar-ison with predictions of dynamics calculations. ProteinSci. 11, 221–232.
12. Sanches, M., Martins, N. H., Calazans, A., et al. (2004)Crystallization of a non-B and a B mutant HIV protease.Acta Crystallogr. D. Biol. Crystallogr. 60, 1625–1627.
13. Wlodawer, A. and Vondrasek, J. (1998) Inhibitors of HIV-1protease: a major success of structure-assisted drugdesign. Annu. Rev. Biophys. Biomol. Struct. 27, 249–284.
14. Berman, H. M., Westbrook, J., Feng, Z., et al. (2000) TheProtein Data Bank. Nucleic Acids Res. 28, 235–242.
15. Velazquez-Campoy, A., Vega, S., Fleming, et al. (2003)Protease inhibition in African subtypes of HIV-1. AIDSRev. 5, 165–171.
16. Velazquez-Campoy, A., Todd, M. J., Vega, S., and Freire, E.(2001) Catalytic efficiency and vitality of HIV-1 proteasesfrom African viral subtypes. Proc. Natl. Acad. Sci. USA 98,6062–6067.
17. Vicente, A. C., Agwale, S. M., Otsuki, K., et al. (2001)Genetic variability of HIV-1 protease from Nigeria andcorrelation with protease inhibitors drug resistance. VirusGenes 22, 181–186.
18. Soares, M. A., De Oliveira, T., Brindeiro, R. M., et al. (2003)A specific subtype C of human immunodeficiency virustype 1 circulates in Brazil. AIDS 17, 11–21.
19. Caride, E., Hertogs, K., Larder, B., et al. (2001)Genotypic and phenotypic evidence of different drug-resistance mutation patterns between B and non-B sub-type isolates of human immunodeficiency virus type 1found in Brazilian patients failing HAART. Virus Genes23, 193–202.
20. Guex, N. and Peitsch, M. C. (1997) SWISS-MODEL and theSwiss-PdbViewer: an environment for comparative pro-tein modeling. Electrophoresis 18, 2714–2723.
21. Kempf, D. J., Marsh, K. C., Denissen, J. F., et al. (1995) ABT-538 is a potent inhibitor of human immunodeficiencyvirus protease and has high oral bioavailability inhumans. Proc. Natl. Acad. Sci. USA 92, 2484–2488.
22. Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W.,Kaptein, R., and Thornton, J. M. (1996) AQUA andPROCHECK-NMR: programs for checking the quality ofprotein structures solved by NMR. J. Biomol. NMR 8,477–486.
23. van Gunsteren, W. F., Billeter, S. R., Eising, A. A., et al.(1996) Biomolecular Simulation: The GROMOS96 Manualand User Guide. vdf Hochschulverlag AG an der ETHZürich and BIOMOS b.v., Zürich, Groningen.
24. van Aalten, D. M., Bywater, R., Findlay, J. B., Hendlich, M.,Hooft, R. W., and Vriend, G. (1996) PRODRG, a programfor generating molecular topologies and unique moleculardescriptors from coordinates of small molecules. J. Comput.Aided Mol. Des, 10, 255–262.
25. van Gunsteren, W. F. and Berendsen, H.J.C. (1987)Groningen Molecular Simulation (GROMOS) Library Manual.BIOMOS b.v., Groningen.
Computational Studies of HIV-1 Protease of B and Non-B Subtypes 403
Cell Biochemistry and Biophysics Volume 44, 2006

26. Frisch, M. J., Trucks, G. W., Schlegel, H. B., et al. (1995)GAUSSIAN94, Revision B.1. Gaussian, Inc., Pittsburgh, PA.
27. van der Spoel, D., van Buuren, A. R., Apol, E., et al. (2001)Gromacs User’s Manual version 3.0, Groningen.
28. Berendsen, H.J.C., van der Spoel, D., and van Drunen, R.(1995) GROMACS: A message-passing parallel moleculardynamics implementation. Comp. Phys. Commun. 91, 43–56.
29. Humphrey, W., Dalke, A., and Schulten, K. (1996) VMD:visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–38.
30. Berendsen, H. J. C., Postma, J. P. M., Gunsteren, W. F. V.,and Hermans, J. (1981) Interaction models for water in rela-tion to protein hydration, in Intermolecular Forces (Pullman,B., ed.), Reidel, Dordrecht, The Netherlands, pp. 331–342.
31. Hess, B., Bekker, H., Berendsen, H. J. C., and Fraaije, J. G.E. M. (1997) LINCS: a linear constraint solver for molecu-lar simulations. J. Comput. Chem. 18, 1463–1472.
32. Miyamoto, S. and Kollman, P. A. (1992) Settle - an analyti-cal version of the shake and rattle algorithm for rigidwater models. J. Comput. Chem. 13, 952–962.
33. Berendsen, H. J. C., Postma, J. P. M., Vangunsteren, W. F.,Dinola, A., and Haak, J. R. (1984) Molecular-dynamics withcoupling to an external bath. J. Chem. Phys. 81, 3684–3690.
34. Schreiber, H. and Steinhauser, O. (1992) Taming cut-offinduced artifacts in molecular dynamics studies of sol-vated polypeptides. The reaction field method. J. Mol. Biol.228, 909–923.
35. Smith, P. E. and Vangunsteren, W. F. (1994) Consistentdielectric-properties of the simple point-charge andextended simple point-charge water models at 277 and300 K. J. Chem. Phys. 100, 3169–3174.
36. Hyland, L. J., Tomaszek, T. A., Jr., Roberts, G. D., et al.(1991) Human immunodeficiency virus-1 protease. 1.Initial velocity studies and kinetic characterization of reac-tion intermediates by 18O isotope exchange. Biochemistry30, 8441–8453.
37. Hyland, L. J., Tomaszek, T. A., Jr. and Meek, T. D. (1991)Human immunodeficiency virus-1 protease. 2. Use of pHrate studies and solvent kinetic isotope effects to eluci-date details of chemical mechanism. Biochemistry 30,8454–8463.
38. Okimoto, N., Tsukui, T., Hata, M., Hoshino, T., and Tsuda,M. (2000) Molecular dynamics study of HIV-1 protease-substrate complex: roles of the water molecules at the loopstructures of the active site. J. Am. Chem. Soc. 122,5613–5622.
39. Aqvist, J., Medina, C., and Samuelsson, J. E. (1994) A newmethod for predicting binding affinity in computer-aideddrug design. Protein Eng. 7, 385–391.
40. Hulten, J., Bonham, N. M., Nillroth, U., et al. (1997) CyclicHIV-1 protease inhibitors derived from mannitol: synthe-sis, inhibitory potencies, and computational predictions ofbinding affinities. J. Med. Chem. 40, 885–897.
41. Wang, W., Wang, J., and Kollman, P. A. (1999) What deter-mines the van der Waals coefficient beta in the LIE (linearinteraction energy) method to estimate binding free energiesusing molecular dynamics simulations? Proteins 34, 395–402.
42. Aqvist, J., Luzhkov, V. B., and Brandsdal, B. O. (2002)Ligand binding affinities from MD simulations. AccountsChem. Res. 35, 358–365.
43. Connolly, M. L. (1983) Solvent-accessible surfaces of pro-teins and nucleic-acids. Science 221, 709–713.
44. Koradi, R., Billeter, M., and Wuthrich, K. (1996) MOL-MOL: a program for display and analysis of macromolec-ular structures. J. Mol. Graph. 14, 51–55, 29–32.
45. Wang, W. and Kollman, P. A. (2001) Computational studyof protein specificity: the molecular basis of HIV-1 pro-tease drug resistance. Proc. Natl. Acad. Sci. USA 98,14,937–14,942.
46. Brandsdal, B. O., Osterberg, F., Almlof, M., Feierberg, I.,Luzhkov, V. B., and Aqvist, J. (2003) Free energy calcula-tions and ligand binding. Adv. Protein Chem. 66, 123–158.
47. Garrett, R. and Grisham, C. M. (1995) Biochemistry,Saunders College Pub., Fort Worth, TX.
48. Ala, P. J., Huston, E. E., Klabe, R. M., et al. (1997)Molecular basis of HIV-1 protease drug resistance: struc-tural analysis of mutant proteases complexed with cyclicurea inhibitors. Biochemistry 36, 1573–1580.
49. Deeks, S. G., Smith, M., Holodniy, M., and Kahn, J. O.(1997) HIV-1 protease inhibitors - a review for clinicians. J.Am. Med. Assoc. 277, 145–153.
404 Batista et al.
Cell Biochemistry and Biophysics Volume 44, 2006
Top Related
Copyright © 2022 FDOKUMEN

