







Bahasa
Halaman
Hukum


0045-6535(95)00066-6
Chemosphere, Vol. 30, No. 10, pp. 1847-1860, 1995 Copyright 0 1995 Elsevier Science Ltd
Printed in Great Britain. All fights reserved 0045-6535/95 $9.50+0.00
LABORATORY-SCALE PHOTODEGRADATION OF PHENOL IN AQUEOUS SOLUTION BY
PHOTOCATALYTIC MEMBRANES IMMOBILIZING TITANIUM DIOXlDI~
Barbara Barni, Andrea Cavicchioli, Elisabetta Riva, Luca Zanoni, Feliee Bignoli and Ignazio Renato Bellobono*
Department of Physical Chemistry and Electrochemistry, University of Milan; via C. Goigi, 19, 1-20133 Milan, Italy
Franco Giantureo and Alessandra De Giorgi Chimia Prodotti e Processi, 1-20124 Milan, Italy
Herbert Muntau, Luca Montanarella and Sergio Facchetti Commission of the European Communities, Joint Research Centre, 1-21020 Ispra (VA), Italy
Leonardo Castellano Institute of General and Applied Physics, University of Milan, 1-20133 Milan, Italy
(Received in Germany 30 November 1994; accepted 14 February 1995)
ABSTRACT
The TiO2-mediated photodegradation of phenol was studied at 298 ± 2 or 308 ± 2 K (with the ratio between
the hydrogen peroxide added and the stoichiometric amount (N) in the range 0-10), using PHOTOPERM® CPPI313
membranes containing immobilized 30 ± 3 wt.% "130 2, with high or low pressure mercury arc lamps (radiant power in
the absorption range, 145 and 8 W respectively). The initial rate of photodegradation was studied as a function of the
initial concentration of substrate (1.0xl0 -5 - 0.12 IV 0 using the linearized form of the Langmuir-Hinshelwood equation,
from which the rate constants k and apparent equil~rium adsorption constants K were evaluated. The rationalization of
k and K values on the basis of their dependency on N, the apparent reaction order, the advantages of the membrane
process with respect to the use of colloidal suspensions of TiO 2, also tested in comparable conditions, and the kinetic
significance of K, on the basis of no measurable dark adsorption onto the photocatalytic membranes, are discussed.
§ Part 48 of the series "Photosynthetic Membranes".
1847

1848
INTRODUCTION
The utilization of semiconductor particulate systems to carry out photochemical transformations of organic and
inorganic compounds (heterogeneous photoeatalysis), particularly for the total oxidation of organic water contaminants
in the presence of titanium dioxide, has seen a remarkable growth during the past decade (1, 2). Even if expectations of
this advanced oxidation process are high, the performance of most of the mainly laboratory-scale experiments has been
rather deceiving, due to the need to filter a colloidal suspension of photocatalyst, as well as to problems of optical
absorption and design of photoreactors, in the presence of the particulate system given by semiconductors. As
supporting the photocatalytic system surely represents the first way towards a technological solution, some
immobilization experiments of the latter (2) have been carried out. Unfortunately, most of them have not produced
great effects, predominantly owing to the very modest amounts of semiconductor immobilized, and to consequent
diffusive and kinetic problems. Only lately, have photocatalytic systems been immobilized in massive amounts (up to
35 - 40 wt.%), in such a way as is essential to maintain full permeability through the supporting structure, while
keeping their surface area completely, or nearly completely active (3-7). To this purpose, photocatalytic membranes
have been devised (PHOTOPERM® membranes), in which the very fast photochemical method of preparation of these
composites could meet the requirements mentioned above. It also made possible to incorporate promoting
photocatalysts or photosensitizers together with the semiconductor, and exploiting at the same time all known
advantages of membrane processes (optimal photoreactor modelling, suitable permeability, continuous processing,
possible separations, and so on).
In previous papers (3, 5, 7), integral degradation, by these membranes, of some chloro-aliphaties and atrazine,
as model molecules, to carbon dioxide, water and chloride ions, has been systematically studied, in relation to kinetics,
at both a laboratory- and a pilot-plant-scale level. In the latter case with the aim of obtaining information on the most
relevant parameters, such as radiation kind and intensity, concentration of hydrogen peroxide added as oxygen supplier,
and influence of promoting photocatalysts (4, 6). In the present paper, systematic results for degradation of phenol in
aqueous solution during laboratory-scale experiments are presented, as a function of substrate concentration, as well as
of hydrogen peroxide, by operating alternatively with two different radiation sources (high and low pressure mercury
arc lam~), to compare the relative efficiency of the latter.
Contamination of water resources by a variety of organic substances is an increasingly common problem. The
nine general classes of environmental contaminants include phenols (8), which are widely used in a range of productive
processes, from fumigants, herbicides and insecticides to wood preservatives, to disinfectants, to polymers for finishing
in paper mills. The parent molecule, investigated in this work, is also a good model of the fate of the aromatic ring in
environmental photodegradations. The photooxidation of phenol in irradiated TiO 2 suspensions has been thoroughly
examined in earlier literature (9-21) under a variety of experimental conditions. Some immobilization experiments of
titanium dioxide have also been carried out (19, 22-26). Yet, owing to the very limited catalyst loadings obtained by the
methods which have been tested, diffusive and kinetic problems have not been substantially removed, as stated above,
and no significant increase on rates has been obtained.

1849
EXPERIMENTAL
Mater/a/s
The "nO 2 (mainly anatase) was Deguasa P-25 grade; its surface area, as determined by the B.E.T. method, was
69 ± 6 m2.g -1. Phenol was obtained from Aldrich (purity greater than 99.5%) and was used as received with no
further purification. Solutions were prepared with bidistilled water. No buffer system was added to solutions, either
initially or during photodegradation.
Photocata/yt/c membranes
Photocatalytic membranes (PHOTOPERM® membranes CPP/313 by Chimia Prodotti e Proceasi, Milan, I),
immobilizing 30 ± 3% of TiO2, were prepared industrially by a pilot plant operating continuously. Controlled amounts
of appropriate monomers and prepolymers, containing the semiconductor to be immobilized, and photoinitiated by a
proprietary photocatalytic system, were photografted onto both sides of a perforated polyester network possessing an
initial loading of 389 ± 1 g/m 2. After photografting, the overall loading was 930 ± 45 g/m 2. The void percentage of the
photografted membranes in the network was 45 _+ 2%. The final pore size of photosynthetized membranes was
regulated at 2.5 - 4.0 ttm, by controlling the theological and photochemical parameters during membrane manufacture.
Appara~ and procedures
The laboratory-scale reactor consisted of a pyrex glass, water jacketed, cylinder, in which both the radiation
source and the membrane were placed coaxially. The reacting solution was circulated continuously, at a constant flow
rate (51.1 ± 0.3 ml../h) from and to a reservoir and a sampling device, from which samples could be withdrawn for
analytical determinations. In the runs in which hydrogen peroxide was not employed as oxidant, solutions during
photodegradation were continuously saturated with oxygen. The distance between the axial lamp jacket, which was
made of quartz, and the membrane was 2 mm. The ratio between the overall reacting volume (in the photoreactor and
in the reservoir) and the apparent, geometrical surface area of the irradiated side of the membrane was 3.8 :e 0.I mL
crn -2. The optical path from the outer surface of the lamp cladding jacket and the inner surface of the reactor was 2.8
cm. Two radiation sources were employed alternatively: a 500 W high pressure mercury arc lamp and a 20 W low
pressure one (by Helios Italquartz, Milan, I; arc length 19.5 cm). For the former, in the UV range of the polychromatic
spectrum, from 400 nm downwards, emitted energy corresponded to 145 ± 5 W, as obtained from actinometry, by
measuring also the emission spectrum of the lamp as a function of wavelength. Except when indicated, in all these
runs, the lamp sheath made of quartz allowed no selective filtration of the radiation. For the low pressure lamp,
emitting 99% of UV energy at 254 nm, the output, in the range of absorption of photocatalytic membranes, was 8 ± 1
W, as measured actinometrically. Given the overall energy outputs of the lamps and the refrigerating water flow in the
rcactor jacket, the temperature of the photoreactor was regulated at 308 :e 2 K when using the high pressure lamp, and
at 298 _+ 2 K, when using the low pressure lamp.
The initial rate r 0 of photodegradation of phenol was evaluated from curves of their concentration vs. time in
the linear range, where zero-order kinetics was apparent. Experiments were repeated for each set of conditions,
essentially for each value of initial concentration C o of micropollutant, so that the mean initial rate and its standard
deviation could be estimated.
The disappearance of phenol was followed by its fluorimetric analysis (excitation at 268 nm and emission at
300 rim, a wavelength at which no interference by degradation products, such as catechol, hydroquinone,

1850
benzequinone, and pyrogallol, has been noticed). Calibration with standards was carried out in the range of
concentrations for which a satisfactory linearity between response and concentration was observed (from 1.00xl0 -6 to
1.00xl0 -4 M). To avoid any dissociation in the excited state, 1 mL of 0.1 N HCI was added to 2 mL of the sample (or
of the standard), before fluorimetric detections.
In order to obtain a direct measurement of membrane performance, with respect to the use of titanium dioxide
in aqueous suspension, a first systematic series of runs was effected by using varying amounts of the suspended
semiconductor, from 0.05 to 2.44 g, without employing the photocatalytic membrane, in the presence of oxygen alone,
and by using both the low and the high pressure mercury arc lamps. Results of this series were compared with those
resulting from photocatalytic membrane behaviour, by using, under the same experimental conditions and with the
same reactor volume, a membrane containing the maximum amount of titanium dioxide tested (2.44 g), immobilized in
the whole membrane structure. In a second series of runs, the aqueous suspensions of semiconductor were employed,
in the presence of stoichiometric hydrogen peroxide as oxidant, both with low and with high pressure mercury arc
lamps, and in the latter case by filtering wavelengths exceeding 310 nm (use of an optical pyrex lamp sheath) in
alternation with the use of the standard lamp sheath made of quartz, and these results were analogously compared with
those of photocatalytic membranes operating, as above, under the same conditions. The purely photolytic and photo-
oxidative degradations of phenol, in the absence of any dissolved oxygen or in the presence of either oxygen or
hydrogen peroxide respectively, but in the absence of any semiconductor, free or immobilized, were finally evaluated.
RESULTS AND DISCUSSION
As is known from the literature (1-7), and as has been done in our previous work (3-7), the Langmuir-
Hinshelwood kinetic rate law, combining apparent adsorption equilibrium and apparent zero-order surface reaction,
was used to interpret experimental results, in the linenrised form of eqn. (1):
1/r 0 = (1/k) + (1/kKCo) (1)
where r 0 is the initial rate, k the reaction rate constant, which has been checked to be directly proportional to the square
root of the absorbed radiation intensity (7), C o the initial concentration of the organic compound, which is being
photodegraded, and K the apparent equilibrium adsorption constant of this compound on to the photocatalytic system.
In the experimental conditions of the present work, three distinct photodegradation processes may occur
simultaneously:
a) direct photolysis of the micropollutant, independent of the presence of the photocatalytic membrane as well
as of the oxidizing agent (oxygen or hydrogen peroxide);
b) photodegradation deriving from photo-oxidative processes due to the action of UV radiation and oxidizing
agent, but without any participation of the photocatalytic system inunobilized in the membrane;
c) photocatalytic degradation of the organic molecule due to the action of UV radiation on the surface of the
semiconductor (titanium dioxide) immobilized in the membrane structure., in the presence of the oxidizing agent.
The contribution of these processes has been assessed, evaluated, and discussed in previous papers (4, 7, 27).
Experimental data for the whole degradation reaction, consisting of prooms~ a) - c) in our experimental conditions
have been found to fit eqn. (1) satisfactorily well, even if p r ~ a) and b) are not expected to follow the mechanism
of eqn. (1) itself. The latter processes, however, being substantially of zero-order in every given set of experimental
conditions, can modify the apparent k value, which anyway typifies a constant strongly dc~cndant on the choice of
experimental conditions (radiation intensity, membrane surface, membrane geometry, flow rate). The K adsorption
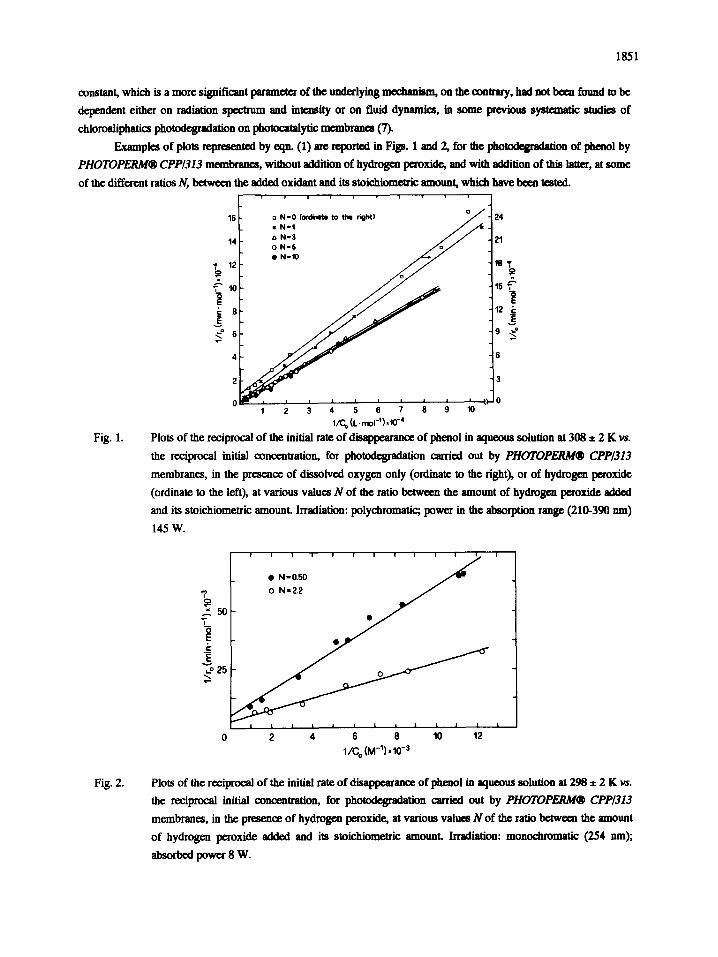
1851
constant, which is a more significant parameter of the underlying mechanism, on the contrary, had not bern found to be
dependent either on radiation spectrum and intensity or on fluid dynamics, in some previous systematic studies of
chloroaliphatics photodegradation on photocatalytic membranes (7).
Examples of plots represented by eqn. (1) are reported in Figs. 1 and 2, for the p h o ~ f i o n of phenol by
PHOTOPERM® CPP/313 membranes, without addition of hydroge~ peroxide, and with addition of this latter, at some
of the different ratios N, between the added oxidant and its stoichiometrif amount, which have been tested. i i i
16 o N-0 (ordirmte to the right) ~ 24 • N-I / . ~
X ' 3 / / 21 14 o N-s / . / / • N=10 = ~ ~
o10 ~ 15 T.~ 8 12 .~
~°
4
2
0 I I 1 2 3 4 5 6 7 8 9 10
1/C-o (L. mor ~) =10 -4
Fig. 1. Plots of the reciprocal of the initial rate of disappearance of phenol in aqueous solution at 308 ± 2 K vs.
the reciprocal initial concentlation, for photodegradation carried out by PHOTOPERM® CPP/313
membranes, in the presence of dissolved oxygen only (ordinate to the right), or of hydrogen peroxide
(ordinate to the left), at various values N of the ratio betweea the amount of hydrogen peroxide added
and its stoichiometric amounL Irradiation: polychromatic; power in the absorption range (210-390 am)
145 W.
'i' o ~50 7
o 25
. + o + . . . . . .
O N = 2.2 e S J
i I l t i i I i i I I I 2 4 6 8 10 12
1/C o {M -~) ,I0-3
Fig. 2. Plots of the reciprocal of the initial rate of disappearance of phenol in aqueouS solution at 298 ± 2 K vs.
the reciprocal initial concentration, for photodegradation carried out by PHOTOPERM® CPP/313
membranes, in the presence of hydrogen peroxide, at various values N of the ratio between the amount
of hydrogen peroxide ~ d ~ and its stoichiometric amounL Irradiation: monochromatic (254 tun);
absorbed power 8 W.

1852
The resulting values of k and K parameters, obtained by linear regression analysis of the l.angmuir-
Hinshelwood treatment of kinetic data (eqn. (1)), are collected in Table 1.
TABLE 1.
Kinetic constants k (mol.min -1) and apparent adsorption constants K (M-l), obtained by Langrnuir-Hinshelwood
treatment of kinetic data, over TiO 2 immobilized on to photografted membranes illuminated pelychromatically (from
210 nm up to the band gap wavelength) and monochromatically (254 rim), at temperatures of 308 ± 2 K and 298 ± 2 K
respectively, at various values (N) of the ratio between hydrogen peroxide added and the stoichiometric amount needed
for complete mineralization.
N k (mol.min -1) a x 104 K (M-l) a x 10 -3
0.0 b 0 . 8 0 ± 0 . ~ 5.7±0.3
0.50 c 2.2±0.3 0.8±0.1
1.0 b 1.8±0.3 4.3±0.5
2.2 c 3.8±0.4 1.0±0.3
3.0 b 4.3±0.5 1.9±0.3
6.0 b 5.0±0.4 1.9±0.2
l0 b 4.9±0.3 2.0±0.3
a Uncertainty, expressed as probable error, was calculated by evaluating the propagation of error in the regression
analysis of experimental data according to eqn. (1).
b Polyehromatic irradiation from 210 nm upwards.
c Monochromatic irradiation (254 nm).
The data in Table 1 clearly reflect the uncertainty connected with double--reciprocal-type plots such as that of
eqn. (1), which magnify the extrapolation error, and cause an error dependency of k, and consequently of K values, on
the range of concentrations selected for the experiments. Within the limits of this uncertainty, however, and by
considering that the plots of Fig. 2 are somewhat less extended in the concentration range than those of Fig. 1, a first
indication may be obtained. For the chloro-aliphatics examined in previous work (7), K values were without any doubt
independent of the radiation spectrum and intensity (as well as of membrane geometry and flow rate), but dependent on
N value (the ratio between the amount of hydrogen peroxide added and its stoichiometric quantity). On the contrary,
data of Table 1 show:/) a certain dependence of K on the irradiation wavelength;//) a dependence of K on N, for the
series of data relative to the polychromatic irradiation (this dependence is illustrated graphically, with mean values of
Table 1, in Fig. 3); iii) an independence of K on N, for the data relative to the monochromatic irradiation. Even if the
closeness of these values and the relative uncertainties do not entitle us to draw definitive conclusions, this behaviour
appears to be closely related to the true physical significance of K. Its narrow interpretation of a truly and/or solely
langmuirian adsorption has been questioned (28). The alternative explanation in terms of contributions of other factors,
such as weak surface complexation of labile surface oxygen species, or the rate of electron transfer to reducible species

1853
at the semiconductor surfaces subjected to radiation flux, in competition with eleetron-hole recombination at surface
traps, has been discussed and investigated experimentally (28). A purely kinetic interpretation of K in terms of kinetic
rate constants is also possible (29).
? o
N
f -
i [ i i , I I i I I I
I I I 1 I i I I I I I
2 4 6 8 10
N
Fig. 3. Apparent adsorption constants K (eqn. (1)) for phenol at 308 ± 2 K onto PHOTOPERM® CPP/313
membranes, containing 30 ± 3 % TiO2, as a function of the ratio N between the hydrogen peroxide
added and the stoichiometric amount required for complete degradation to carbon dioxide and water.
Polychromatie irradiation from 210 am upwards.
To examine the possible relevance of dark adsorption qualitatively, some specific experiments have been
carried out in the present work. It has been f'ast verified that adsorption on free, suspended TiO2, as measured by
fluorimetric analysis which is sensitive to variations of concentrations as low as 10 -6 M, is detectable, but very modest,
if compared to the more important adsorption of chloro-derivatives (28). On the photocatalytic membranes, on the
contrary, in the range of concentrations tested, no measurable adsorption could be observed. In relation to this effect,
the behaviour of photodegradation in the presence of hydrogen peroxide is instructive. For trichloroethene (7) K value
is decreased by about two orders of magnitude when N = 10. For phenol, on the contrary, (see Table 1 and Fig. 3) the
decrease is much less marked: the value of K is decreased to only about 35% of that observed in the absence of
hydrogen peroxide. Furthermore, this appears only when the higher intensity polychromatic radiation is used, with
prevailing energy peaks in the near UV spectrum. Thus, with all the care required by the limited number of cases which
have been examined, the influence of N on the K parameter may hypothetically reflect an adsorptive competition in the
ground state (dark adsorption), which strongly enhances rate when adsorption of an oxidizing species, such as
hydrogen peroxide, is effective. Alternatively, or in addition, when dark adsorption is less operative, the efficacy of
surface, photoproduced, radical species, such as *OH or 02-% will appear, and this may conceivably produce a certain
wavelength dependency, particularly in the presence of hydrogen peroxide, which is unable to absorb energy other than
at high frequencies in the UV spectrum. Experimental results, owing to the very complex nature of heterogeneous
photocatalysis even when the semiconductor is immobilized in highly permeating membrane structures allowing high
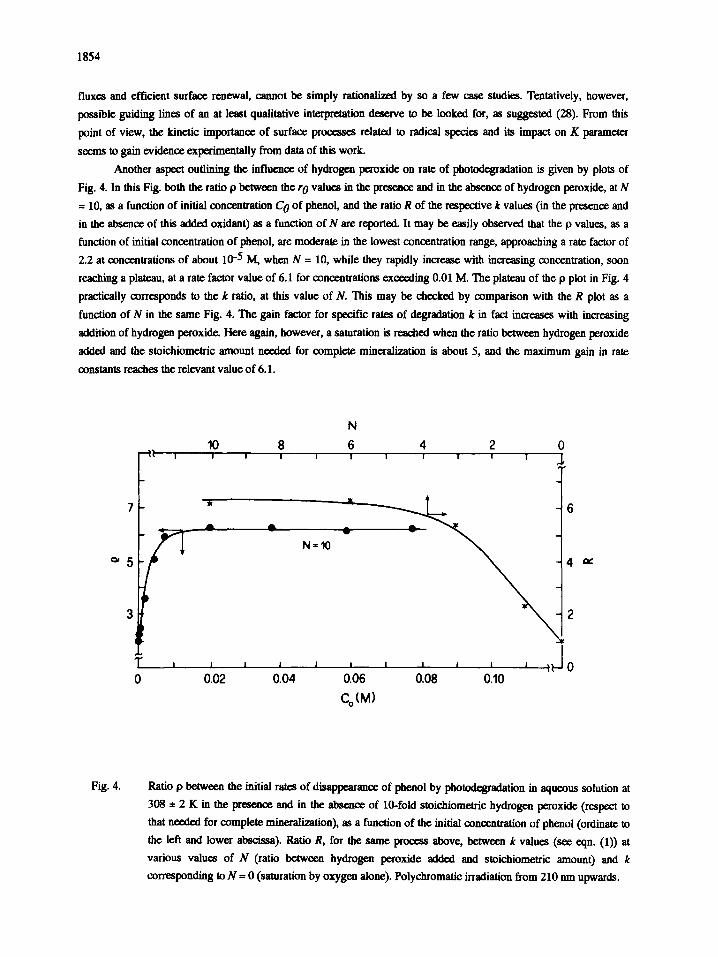
1854
fluxes and efficient surface renewal, cannot be simply rationalized by so a few case studies. Tentatively, however,
possible guiding lines of an at least qualitative interpretation deserve to be looked for, as suggested (28). From this
point of view, the kinetic importance of surface processes related to radical species and its impact on K parameter
seems to gain evidence experimentally from data of this work.
Another aspect outlining the influence of hydrogen peroxide on rate of photodegradation is given by plots of
Fig. 4. In this Fig. both the ratio p between the r 0 values in the presence and in the absence of hydrogen peroxide, at N
= 10, as a function of initial concentration C o of phenol, and the ratio R of the respective k values (in the presence and
in the absence of this added oxidant) as a function of N are reported. It may be easily observed that the p values, as a
function of initial concentration of phenol, are moderate in the lowest concentration range, approaching a rate factor of
2.2 at concentrations of about 10 -5 M, when N = 10, while they rapidly increase with increasing concentration, soon
reaching a plateau, at a rate factor value of 6.1 for concentrations exceeding 0.01 M. The plateau of the p plot in Fig. 4
practically corresponds to the k ratio, at this value of N. This may be checked by comparison with the R plot as a
function of N in the same Fig. 4. Tne gain factor for specific rates of degradation k in fact increases with increasing
addition of hydrogen peroxide. Here again, however, a saturation is reached when the ratio between hydrogen peroxide
added and the stoichiometric amount needed for complete mineralization is about 5, and the maximum gain in rate
constants reaches the relevant value of 6.1.
0, 5
10 8
I I
A A
T I J 1 I
0 0.02 0.04
N
6 4 2 0 I I I I I I I J~
I _ 6 I
v
N=10
l
0.06 0.08 0.10
Co(M)
Fig. 4. Ratio p between the initial rates of disappearance of phenol by photodegradation in aqueous solution at
308 e 2 K in the presence and in the absence of 10-fold stoichiomctric hydrogen peroxide (respect to
that needed for complete mineralization), as a function of the initial concentration of phenol (ordinate to
the left and lower abscissa). Ratio R, for the same process above, between k values (see eqn. (1)) at
various values of N (ratio between hydrogen peroxide added and stoichiometric amount) and k
corresponding to N = 0 (saturation by oxygen alone). Polychromatic irradiation from 210 nm upwards.

1855
As the double-reciprocal-type linearization of photodegradation data, particularly on photocatalytic membranes
tested in the present series of papers, holds effectively well in a very wide range of concentrations which has been
explored (3-7, 27), from 10 "1 to 10 .7 M, the importance and influence of K parameter on reaction order is also a
meaningful approach. In Fig. 5, plots of the relationship between decadic logarithms of both r 0 and C O are reported in
order to obtain information on the variation of reaction order with concentration.
E
e L.o
v
I
'T e-
, o
I
i i ! i ! ! i i
6 LOW PRIESSUIIE MERCURY AIIIC L A M P
• N - 0 . 5 0
o N-2.2
I I I I I I I I
H I G H PRESSURE MERCURY ARC LAMP
Y • N =0.00 • N=I.0 O N=3.0
i * 1 t i t [ i
2 3 4 5
- log (C O/M)
Fig. 5. Log-log plots of initial rates of phenol disappearance, by photodegradation in aqueous solution, vs.
initial concentration. Polychromatic and monochromatic irradiations, at 308 ± 2 and 298 ± 2 K
respectively.
As may be remarked in Fig. 5, the change of order with concentration appears when the concentration is about
(5-7)x10 -4 M, independent of kind and intensity of radiation, as well as of the presence or absence of hydrogen
peroxide. At concentrations lower than the above value, the reaction order approaches unity (1.0 and 0.86 with N equal
to 0.50 or 2.2 respectively when using the low pressure lamp for irradiation; substantially 0.9 for all N values, from 0.0
to 3.0, when using the high intensity, high pressure lamp). At concentrations higher than about 10 -3 M, the reaction
order approaches zero (it is about 0.1 at N = 0, and 0.2 - 0.3 in all other cases) down to the highest concentrations
reported in Fig. 5. This change of order has its origin in the linearity of the double-reciprocal plot of eqn. (1) and from
the value o f K C 0 respect to unity, as may be easily seen in the Langmuir-Hinshelwood mechanism of eqn. (1), as well
as on any other mechanism giving rise to this same kind of plot (28, 29).
In the light of these latter observations, let us now come back to the experimental facts of Fig. 4, illustrated and
partly commented on above. On the one hand, the behaviour of p and R plots of Fig. 4 is consistent with the
competition of adsorption of hydrogen peroxide in the ground state on to the semiconductor sites, which thus enhance
the probability of oxidation when radical species are photoproduced, if the mechanism is zero order,/.e, substantially in
the higher concentration range (r 0 - k). In the low concentration range, on the contrary, when a f'wst order mechanism

1856
is operative, (r 0 - kKCo), even at high N values, the effect on k is moderate, and furthermore a decrease of K with
increasing N is observed, so that the net benefit is lower or much lower than that of the preceding situation. Physically,
this means that if apparent adsorption of the substrate to be degraded is scarce or very scarce,, the gain in rate brought
about by adding chemically or photochamically active oxidants is moderately positive, but may even become negative
in some concentration range (as in the case of trichloroethene degradation (7)) if K decreases notably with increasing N.
On the other hand, the possible kinetic significance of the apparent adsorption constant (29) apart, this may not be the
sole interpretation, as stated above. Mainly two experimental facts, in the present work, require alternative or additional
explanation, in terms of surface photoproduction of reactive radicals, and/or their weak complexation: the K
independence of N for monochromatic irradiation, and the non strictly zero order in the presence of hydrogen peroxide,
even in highest range of concentrations (see Table 1 and Fig. 5). Much more work is needed to resolve the underlying
mechanism. The main objective of these papers is not directcxl towards fundamental aspects of immobilized
semiconductor catalysis, but rather to general understanding of its features in order to study and rationalize the
technological process and its applications.
E 0 E
,E 20 E
lO
l
• LOW pcessure klmp/2'54nm N=I.0
¢ Low pntnure lamp/254nm N = 0,0 ( o 2)
IllM}
• . . . . . . . --0 . . . . . . . . . . . ~(M)
I I e(M)
• High pressure klrnp/210-390nm N=I.0
• H~@~O.O~ ) ,imp/210-390nm
~ H ~ pteuum lamp/310-390nm N=I.0
~ e lamp/3'10-390 nm N = 0.0 (O 2)
.~ 'CI /
I I
1 2
mTioz(g )
Fig. 6. Initial rate of phenol disappearance by photodegradation onto aqueous suspension of TiO2, ro, as a
function of mass m of suspended semiconductor, in the presence of dissolved oxygen alone, or of
hydrogen peroxide, added in stoichiometric amount respect the full mineralization reaction (iV = 1.0).
Irradiation by low pressure and high pressure mercury arc lamps, and in the latter case also by filtrating
wavelengths shorter than 310 nm. Explanation of symbols inside the Fig.. Rate values measured on to
the photocatalytic membranes immobilizing the maximum amount of photocatalytst tested in these
runs (2.44 g) is also shown, for comparison, and the corresponding symbol marked with letter (M).
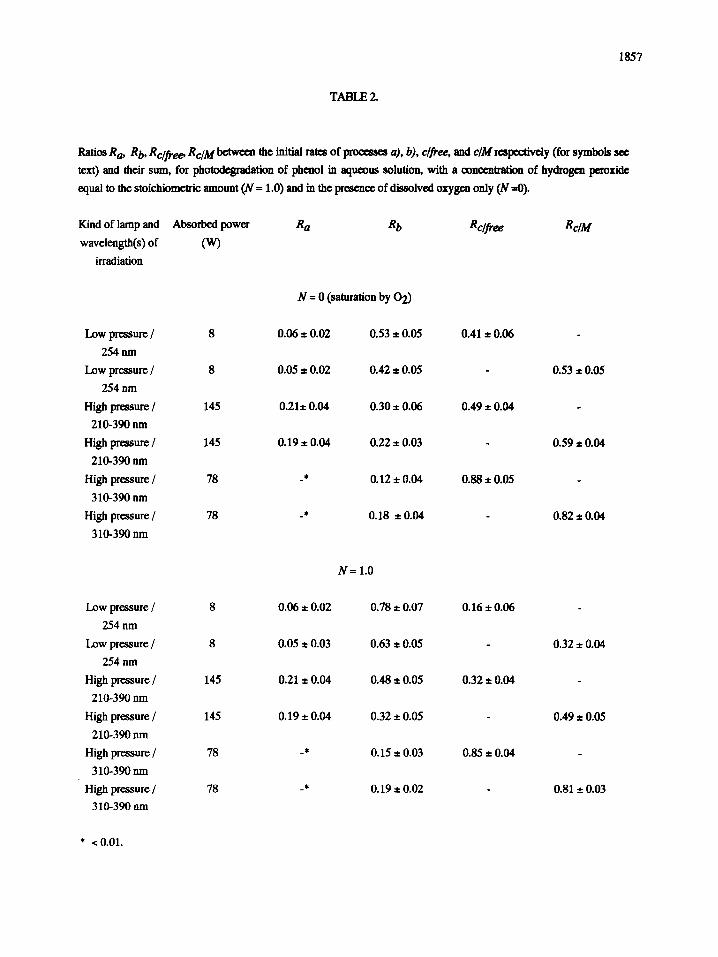
1857
TABLE 2.
Ratios Ra, Rb, Rc/free, Rc/M between the initial rates of processes a), b), c/free, and c/M respectively (for symbols see
text) and their sum, for photodegradation of phenol in aqueous solution, with a concentration of hydrogen peroxide
equal to the stoichiometric amount (N = 1.0) and in the presence of dissolved oxygen only (N =0).
Kind of lamp and Absorbed power
wavelength(s) of (3//)
irradiation
Ra Rb Rclfree Rc/M
N = 0 (saturation by 02)
Low pressure / 8
254 nm
Low pressure / 8
254 nm
High pressure / 145
210-390 nm
High pressure / 145
210-390 nm
High pressure / 78
310-390 nm
High pressure / 78
310-390 nm
0.06 ± 0.02 0.53 ± 0.05 0.41 ± 0.06
0.05 ± 0.02 0.42 ± 0.05
0.21± 0.04 0.30 ± 0.06 0.49 ± 0.04
0.19 ± 0.04 0.22 ± 0.03
-* 0.12 ± 0.04 0.88 ± 0.05
-* 0.18 ± 0.04
0.53 ± 0.05
0.59 ± 0.04
0.82 ± 0.04
N = 1.0
Low pressure / 8
254 nm
Low pressure / 8
254 nm
High pressure / 145
210-390 nm
High pressure / 145
210-390 nm
High pressure / 78
310-390 nm
High pressure / 78
310-390 nm
0.06 ± 0.02 0.78 ± 0.07 0.16 ± 0.06
0.05 ± 0.03 0.63 ± 0.05
0.21 ± 0.04 0.48 ± 0.05 0.32 ± 0.04
0.19 ± 0.04 0.32 ± 0.05
-* 0.15 :e 0.03 0.85 ± 0.04
-* 0.19 ± 0.02
0.32 ± 0.04
0.49 ± 0.05
0.81 ± 0.03
* < 0.01.

1858
From the technological point of view, the performance of the immobilized semiconductor may be evaluated by
comparing results obtained with the same experimental set-up, but operating with free, suspended titanium dioxide, in
order also to evaluate the relative performance of the three photodegradation processes a) - c) (see above) occurring
simultaneously. To this purpose, experiments (see the Experimental section) were performed first by evaluating the
purely photolytic process (a)). Afterwards, in the presence of dissolved oxygen alone, or of hydrogen peroxide at N
=1.0, various amounts m (g) of TiO 2 in suspension were used, up to the amount immobilized onto the membrane tested
in the same conditions. Both low pressure and high pressure lamps were employed, and in the latter case also by
filtering wavelengths shorter than 310 nm. Results are collected in Fig. 6. In some cases, the contribution of process a)
was evaluated as negligible, its rate being less than 1% of the other two processes. The rate of process b)
(photooxidation without any contribution by suspended semiconductor) has been evaluated at m = 0, by subtracting the
contribution of process a) when detectable. The plateaux of curves reported in Fig. 6, at the highest values of m tested,
clearly represent the rate of process c) (heterogeneous photocatalysis), because at high values of m, competition
towards energy absorption greatly favours the semiconductor possessing "infinite" absorption coefficients relatively to
those of any other species present, in the UV range of the spectrum. In Fig. 6 the rate values measured with the
photocatalytic membranes are also indicated in correspondence to the value of m immobilized in the overall surface of
the membranes. The rate maxima in curves of Fig. 6, when present, indicate a situation in which the three contributions
of processes a), b) and c) are integrally summed, and in fact correspond fairly well to values obtained by adding the
three rates evaluated separately. Rate maxima, on the contrary, are not obtained when rates of processes a) and b), on
one side, and c), o n the other, differ so strongly that great amounts of semiconductor are needed to be able to obtain a
measurable contribution by process c), a condition which minimizes or suppresses the contribution by process b) as
well as by process a) when operative.
For a direct comparison of these contributions, between themselves and with those of the photoeatalytic
membranes, in Table 2 the ratios R i between rate of/) process and the overall sum of rates for all processes occurring
have been calculated and reported, the/) process being one of the a), b), c/free, and c/M processes, where c/free and
c/M denote process c) in the presence of the free suspension of titanium dioxide or in the presence of immobilized
semiconductor in the membrane structure respectively. The operative conditions in which runs of Fig. 6 were carried
out are also specified and summarized in this Table. As the membranes tested in this work had no photocatalyst, in
their structure, able to promote photocatalytic activity of titanium dioxide, the comparison between Re~ M and Re~free
values corresponds strictly to relative performance of immobilized vs. suspended semiconductor, apart from any
marked advantage which may be further conferred to photoeatalytic membranes by immobilizing additional
photocatalysts, together with TiO2, as shown in previous work (4-7).
As may be seen in Table 2, in most cases, use of the photoeatalytie membrane gives a distinct advantage over
the use of semiconductor suspensions, this advantage reaching a factor of 2 for the 254 nm irradiation, and of 1.6 when
the high pressure, high intensity lamp is employed, in the presence of stoichiometrie hydrogen peroxide. Only when
UV radiation is limited to the near UV spectrum, performances of photocatalytic membranes somewhat decreases (but
nearly within the limits of experimental uncertainty), with respect to that of suspended semiconductor, in these cases
being 5% and 7% less than those of the free titanium dioxide, in the presence of stoiehiometrie hydrogen peroxide and
dissolved oxygen alone, respectively. This is not unexpected, since it has been shown that the polymeric structure of
the membrane dissipates a certain amount of energy (4), and this should occur mainly in the near UV region of the
spectrum. At high radiation frequencies, on the contrary, the appreciable gain in efficiency afforded by immobilization
is more than sufficient to compensate this, otherwise very limited, loss, being able to nearly double yields of the
photoeatalyzed degradation with respect to those of the suspended catalyst. Another possible or concomitant
explanation of this wavelength effect may arise from the differential contribution of photoproduction of reacting
radicals by oxidant molecules (oxygen or hydrogen peroxide), independent of the presence of semiconductor. These

1859
photooxidative processes, as a matter of fact, are driven by optical absorption, which depends on wavelength, and
which is more effectively hindered by TiO 2 suspensions than is possible for the composite membranes employed,
given their perforated parts which fully transmit radiation, and the membrane arrangement in the laboratory-scale
reactor used.
CONCLUSIONS
O Photocatalysis by titanium dioxide immobilized in massive amounts by membranes photografted on to the
support is a very useful and promising method for degrading organic contaminants in waste waters.
//) Even if fitting of kinetic data by the Langmuir-Hinshelwood equation is adequate, the variation of reaction
order seems to be correlated with a kinetic significance of the apparently langmuirian equilibrium constant K (see eqn.
(1)) in terms of surface photoproduction of reactive radicals and/or their surface reactivity. This is shown by the
correlation between the more or less marked effect on K, brought about by the addition of chemically or
photochemically active oxidants, such as hydrogen peroxide, (together with a wavelength dependence of this effect),
and the higher or lower concentration, of substrate being degraded, at which change of order occurs. The same
conclusions hold if the above kinetic processes pertain to diffusive layers adjacent to the surface, the outcome of which
would be kinetically undistinguishable.
///) No influence of flow rate either on k or on K values has been observed in runs of the present work, but the
operative flow rates used were relatively low and their range of variation too narrow to draw any definitive conclusion,
which may have a general validity, outside the confined extent of conditions tested for flow dynamics.
iv) Performance of immobilized vs. suspended semiconductor, particularly when using the low pressure
irradiation unit, is higher by a factor of about 2, any further factor (up to 20 or more), which may be conferred to
photocatalytic membranes by immobilizing additional photocatalysts (4-7), apart.
REFERENCES
(1) M. Schiavello (ed.), Photocatalysis andEnvironmenL Trends and Applications, NATO ASI Sex.C, vol. 237,
Kluwer, Dordrecht, 1988.
(2) D.F. Ollis and H. AI-Ekabi (eds.), Photocatalytic Purification and Treatment of Water andAir, Elsevier,
Amsterdam, 1993.
(3) I.R. Bellobono, M. Bonardi, L. Castellano, E. Selli and L. Righetto, J. Photoche~ PhotobioL A:Cher~, 67,
109 (1992).
(4) I.R. Bellobono and A. Carrara, in: Effective Membrane Processes. New Perspectfi~, (R. Paterson ed.)
BHR, Mech. Engineer. Publ., London (1993), pp. 257-274.
(5) I.R. Bellobono, B. Barni and F. Gianturco, in: Waste Waters and Sludges, (A. Frigerio and D. Rossi, eds.)
GSISR, Milan (1994) pp. 248-253.
(6) I.R. Bellobono and F. Gianturco, in: Potable Waters, (A. Frigerio and D. Rossi, eds.) GSISR, Milan ((1994)
pp. 206-214
(7) I.R. Bellobono, A. Carrara, B. Barni and A. Gazzotti, J. Photocher~ PhotobioL A:Chent, 84, 83 (1994).

1860
(8) M.A. Callahan, M. Slimak, N. Gabel, I. May, (2. Fowler, R. Freed, P. Jennings, R. Dupree, F. Whitemore, B.
Maestri, B. Holt and C. Gould, Water Related Environmental Fate of 129 Priority Pollutants, EPA,
Washington, DC (1979), Report 44014-79-029 a and b.
(9) K. Okamoto, Y. Yamamoto, H. Tanaka, and A. Itaya, Bull. Chem. Soc. Japan, 58, 2015, 2023 (1985).
(10) R.W. Matthcws, Austral. J. Chem., 40, 667 (1987).
(11) R.W. Matthcws, J. Solar Energy, 38, 405 (1987).
(12) R.W. Matthcws, J. Catal, 111,264(1988).
(13) R.W. Matthcws, Water Res., 2,4, 653 (1990).
(14) R.W. Matthcws and S.R. Mc Evoy, J. Photochent Photobiol. A:Chent, 64, 231 (1992).
(15) V. Augugliaro, L. Palmisano, A. Sclafani, C. Mincto and E. Pelizzctti, ToxicoL Environ. Chem., 16, 89
(1988).
(16) V. Augugliaro, E. Davl, L. Palmisano, M. Schiavello and A. Sclafani, Appl. C~m__!~,6$, 101 (1990).
(17) A. Sclafani, L Palmisano and E. Davt, New J. Chez~,14, 265 (1990).
(18) A. Sclafani, L. Palmisano and E. Day1, J. Photoche~ Photobiol. A:Chen~, 56, 113 (1991).
(19) H. AI-Ekabi and N. Scrpone, J. Phys. Chem., 92, 5726 (1988).
(20) J. Tscng and C.P. Huang, in: Emerging Technologies in Hazardous Waste Management (D.W. Tcdder
and F.G. Pohland, cds.) ACS Syrup. Scr., Amct. Chcra. Soc., Washington, D.C. 422 (1990) pp. 12-39.
(21) T.Y. Wei and C.C. Wan, Ind. Eng. Cher~ Res., 30, 1293 (1991).
(22) N. Scrpone, E. Borgarello, R. Harris, P. Cahill, M. Borgarello and E. Pelizzctti, Solar Energy Mater., 14, 121 (1986).
(23) R.W. Matthews, J. Phys. Chem.,91, 3328 (1987).
(24) S. Yamazaki-Nishida, K.J. Nagano, L.A. Phillips, S. Cervera-March and M.A. Anderson, J. Photochem.
Photobiol. A:Che~, 70, 95 (1993).
(25) M.A. Anderson, S. YamaT~ki-Nishida and S. Cervera-March, in: Photocatalytic Pur i~t ion and Treatment of
Water andA/r, (D.F. Ollis and H. AI-Ekabi, eds.), Elsevier, Amsterdam (1993) pp. 405-420.
(26) K. ~. Suzuki~ in: Ph~t~catalytic Puri~cation and TreamIent ~f Water and Air~(D.F. ~ i s and H. E~-Ekabi~ eds.)~
Elsevier, Amsterdam (1993) pp. 421-434.
(27) I.R. Bellobono, A. Carrara and H. Schwienbacher, in Waste Waters and Sludges (A. Frigerio, ed.), CSI,
Milan (1993), pp. 159-167.
(28) J. Cunningham and P. Sedlak, J. Photochem. Photobiol. A:Chem., 77, 255 (1994).
(29) C.S. Turchi and D..F. Ollis, J. Catal., 122, 178 (1990).
Top Related
Copyright © 2022 FDOKUMEN

