







Bahasa
Halaman
Hukum


Complexin Has Opposite Eff
Current Biology 21, 97–105, January 25, 2011 ª2011 Elsevier Ltd All rights reserved DOI 10.1016/j.cub.2010.12.014
Articleects
on Two Modes of Synaptic Vesicle Fusion
Jesse A. Martin,1,3 Zhitao Hu,2,3 Katherine M. Fenz,1
Joel Fernandez,1 and Jeremy S. Dittman1,*1Department of Biochemistry, Weill Cornell Medical College,1300 York Avenue, New York, NY 10065, USA2Department of Molecular Biology, Simches 7, MassachusettsGeneral Hospital, 185 Cambridge Street, Boston, MA 02114,USA
Summary
Background: Synaptic transmission can occur in a binary orgraded fashion, depending on whether transmitter release istriggered by action potentials or by gradual changes inmembrane potential. Molecular differences of these two typesof fusion events and their differential regulation in a physiolog-ical context have yet to be addressed. Complexin is aconserved SNARE-binding protein that has been proposedto regulate both spontaneous and stimulus-evoked synapticvesicle (SV) fusion.Results: Here we examine complexin function at a gradedsynapse in C. elegans. Null complexin (cpx-1) mutants areviable, although nervous system function is significantlyimpaired. Loss of CPX-1 results in a 3-fold increase in therate of tonic synaptic transmission at the neuromuscular junc-tion, whereas stimulus-evoked SV fusion is decreased 10-fold.A truncated CPX-1 missing its C-terminal domain can rescuestimulus-evoked synaptic vesicle exocytosis but fails tosuppress tonic activity, demonstrating that these two modesof exocytosis can be distinguished at the molecular level. ACPX-1 variant with impaired SNARE binding also rescuesevoked, but not tonic, neurotransmitter release. Finally, tonic,but not evoked, release can be rescued in a syntaxin pointmutant by removing CPX-1. Rescue of either form of exocy-tosis partially restores locomotory behavior, indicating thatboth types of synaptic transmission are relevant.Conclusion: These observations suggest a dual role forCPX-1: suppressing SV exocytosis, driven by low levels ofendogenous neural activity, while promoting synchronousfusion of SVs driven by a depolarizing stimulus. Thus, patternsof synaptic activity regulate complexin’s inhibitory and permis-sive roles at a graded synapse.
Introduction
Neurotransmitter release is predominantly driven by actionpotentials in the vertebrate nervous system, whereas gradedneurotransmission is typically observed at sensory synapsesand at dendrodendritic connections [1–4]. Invertebratenervous systems display both impulse-driven and gradedsynaptic transmission, sometimes at the same synapse [5–7].It is widely thought that the underlying mechanism of synapticvesicle (SV) exocytosis is identical in these two forms ofsignaling, although the molecular differences have yet to be
*Correspondence: [email protected] authors contributed equally to this work
explored. Synaptic transmission is initiated by the fusion ofa synaptic vesicle with the presynaptic membrane, a highlyregulated process involving a host of specialized presynapticproteins [8–10]. SV fusion is catalyzed by the assembly ofa four-helix bundle containing three major SNARE proteins:synaptobrevin, SNAP-25, and syntaxin. The detailedmolecularchainof eventsprogressing fromSNAREassembly toSV fusionis not fully understood, but several SNARE-binding proteinshave been proposed to regulate this sequence [10, 11].In particular, the protein complexin has garneredmuch interestbecause of its ability to bind to the assembled ternary SNAREcomplex with high affinity. Complexin is a 15–18 kDa cyto-plasmic protein that contains a central a-helical region thatbinds within the groove between synaptobrevin and syntaxinin the assembled SNARE complex [12–16]. How the bindingof complexin to the SNARE complex regulates the probabilityof SV fusion is controversial.In vitro studies using SNARE-mediated fusion of proteolipo-
somes or cell-cell fusion have suggested that complexin eitherinhibits [17–19] or stimulates [20, 21] fusion. In several synapticpreparations, complexin function was explored in the contextof two types of SV exocytosis events: action potential-trig-gered fusion initiated by a depolarizing stimulus (evokedrelease) and stochastic fusion eventsmonitored in the absenceof activity (spontaneous release). Neuronal cultures and brainslices derived from complexin knockout mice exhibiteda reduction in evoked neurotransmitter release, as well asdecreased spontaneous fusion events [22–25]. In contrast,complexin knockdown by RNA interference led to elevatedspontaneous release in conjunction with decreased evokedrelease [26]. Genetic deletion of complexin inDrosophila mela-nogaster also increased tonic fusion at the neuromuscularjunction (NMJ) while simultaneously reducing evoked release[27, 28]. Cross-species rescue experiments and chimericvariants of complexin demonstrated that mouse and fly com-plexins share domains that can both enhance and inhibitexocytosis, indicating that complexin is a multifunctionalprotein and possibly has multiple duties at the synapse [25].In this study, we investigated the consequences of removing
the complexin ortholog CPX-1 on neurotransmission inC. elegans. We provide evidence that CPX-1 inhibits thecontinuous release of neurotransmitter driven by low levelsof endogenous neural activity (tonic release). In contrast,CPX-1 is required for SV fusion evoked by a depolarizing stim-ulus electrode. Both of these modes of synaptic transmissionare shown to be behaviorally relevant. Each mode of exocy-tosis could be independently rescued, suggesting that com-plexin operates through distinct mechanisms during synaptictransmission. Thus, CPX-1 can function to regulate synaptictransmission in an activity-dependent manner. A detailedunderstanding of complexin’s role may help delineate themolecular mechanisms underlying SV exocytosis.
Results
Mutants Lacking CPX-1 Have Motor System Defects
The C. elegans genome contains two complexin homologs:cpx-1 and cpx-2. Of these two genes, cpx-1 shares a higher

Figure 1. cpx-1 Mutants Display Motor System
Defects
(A) Protein sequence alignment for C. elegans
CPX-1 (worm), D. melanogaster dmCplx Gen-
Bank AAF69518.1 (fly), andmouse Cplx1 (mouse)
using a ClustalW algorithm. The N-terminal
domain (NTD), accessory domain (AD), central
helix (CH), and C-terminal domain (CTD) are indi-
cated in gray. Identically conserved residues
across all three species are shaded in red,
whereas similar residues are shaded in blue.
(B) Percent sequence identity for each domain is
indicated for each pairwise comparison: worm
(w), fly (f), and mouse (m).
(C) Representative trajectories for 20 animals
for wild-type, cpx-1(ok1552), cpx-1 expressing
a rescuing cDNA under a GABA promoter, and
cpx-1 expressing a rescuing cDNA in all neurons.
(D) Summary of average speed for the four
strains. The number of animals measured for
each average is indicated within the bar.
(E) Paralysis time course on 1.0 mM aldicarb for
wild-type (black circles), cpx-1 (red triangles),
rescue in GABA neurons (open green squares),
and rescue in all neurons (open blue circles).
Data are mean 6 standard error of the mean
(SEM). # denotes significantly different from
wild-type (p < 0.01) but not significantly different
from cpx-1. ** denotes significantly different from
cpx-1 (p < 0.01) but not significantly different from
wild-type. Significance determined by Tukey-
Kramer method.
Current Biology Vol 21 No 298
degree of homology with themouse (32% versus 19% identity)and fly (44% versus 28% identity) complexins (Figure 1A). Theprotein structure and function of mouse complexin suggeststhat it possesses four domains, with the central helix (CH)domain displaying the highest degree of conservation. Theworm complexin protein CPX-1 contains a CH domainwith 78% identity to the mouse Cplx1 and 91% identity tothe Drosophila complexin dmCplx (Figure 1B). In order tostudy the role of complexin in the worm nervous system, weutilized the mutant cpx-1(ok1552), which carries a 1.3 kb dele-tion of the cpx-1 gene, removing one of its two exons. cpx-1(ok1552) animals are viable but have significant defects inlocomotion, whereas heterozygous animals are indistinguish-able from wild-type. We also examined cpx-2 mutants, and,based on expression data and behavioral studies, we foundthat CPX-2 does not function redundantly with CPX-1 (see Fig-ure S1 available online). We thus focused on CPX-1 as themajor complexin in C. elegans.
We quantified the decrease in locomotion of cpx-1 mutantsby recording the center-of-mass speed of individual animalsmoving on an agar plate (Figure 1C). Compared to wild-typecontrols, cpx-1 mutants moved at significantly slower speeds(40% 6 2.2% of wild-type), consistent with a nervous system
defect (Figure 1D). We cloned the full-length cDNA encoding CPX-1 andexpressed a CPX-1::GFP fusion proteinunder a neuronal promoter (snb-1 syn-aptobrevin) in cpx-1 mutants. CPX-1::GFP restored locomotion (104% 65.7% of wild-type), confirming that thelocomotion defect was specific to lossof complexin in the nervous system(Figure 1D).
We also examined the sensitivity of cpx-1 mutants to aldi-carb, a cholinesterase inhibitor. Acetylcholine (ACh) levels inthe NMJ are rapidly reduced through the enzymatic actionof acetylcholinesterases, and inhibition of these enzymesresults in a buildup of ACh, followed by paralysis of the animal[29]. Animals with defects in ACh secretion are resistant toaldicarb, whereas animals secreting higher than normal levelsof ACh are hypersensitive and quickly paralyze upon expo-sure to aldicarb [30–32]. We examined the time course ofparalysis on 1.0 mM aldicarb for wild-type and cpx-1 mutantanimals (Figure 1E). cpx-1 mutants were paralyzed within20 min of aldicarb exposure, whereas wild-type animalsrequired 2 hr to paralyze. Neuronal expression of CPX-1::GFP completely restored normal aldicarb sensitivity, sug-gesting that CPX-1 normally inhibits the secretion of ACh.Alternatively, if CPX-1 specifically acts in GABA motorneurons to promote release, decreased GABA release in thecpx-1 mutant could also account for its hypersensitivity toaldicarb [33]. To test this hypothesis, we attempted to rescuecpx-1 mutants with CPX-1::GFP under a GABA-specificpromoter (unc-25 GAD). Neither locomotion nor aldicarbsensitivity were altered (Figures 1D and 1E), suggesting thatdefects in GABA neuron function in cpx-1 mutants cannot

Figure 2. CPX-1 Is Expressed throughout the Nervous
System and Localizes to Synapses
(A1) Fluorescent image of the head region expressing
GFP under the cpx-1 promoter.
(A2) Expression of GFP under the cpx-1 promoter in the
body region. Note the cell bodies in the ventral nerve
cord at the top with commissural fibers extending to
the dorsal nerve cord.
(B1) Expression ofmCherry under a cholinergic promoter
(unc-17 VAChT) and a nuclear-localized GFP under the
cpx-1 promoter in the ventral nerve cord.
(B2) Expanded view of the inset in (B1) showing indi-
vidual cholinergic (white arrows) and GABAergic (blue
arrowheads) motor neurons expressing cpx-1.
(C1 and C2) Fluorescent images of the dorsal nerve cord
expressing a CPX-1::GFP fusion protein (C1) andmCher-
ry::Rab3 (C2).
(C3) The two fluorescent images merged and color
coded.
Scale bars are 25 mm in (A1) and (A2), 30 mm in (B1), 10 mm
in (B2), and 5 mm in (C1)–(C3).
Complexin and Synaptic Vesicle Fusion99
account for the observed behavioral phenotypes. Consistentwith a role for CPX-1 in ACh release, we found that rescueof CPX-1::GFP under a cholinergic promoter (unc-17 VAChT)nearly completely rescued wild-type aldicarb sensitivity(Figure S2A).
CPX-1 Is a Synaptic Protein Broadly Expressed
in the Nervous SystemTo examine the expression of cpx-1 in C. elegans, we droveexpression of GFP under the control of the cpx-1 promoter(3 kb upstream region). Expression was observed in a largenumber of neurons in the head, ventral cord, and tail, includingall cholinergic and GABAergic motor neurons in the ventralcord (Figures 2A and 2B). In contrast, cpx-2 is expressed inonly a small number of neurons having little or no overlapwith cpx-1 expression (Figure S1B). To examine the subcel-lular localization of CPX-1, we imaged CPX-1::GFP in thedorsal nerve cord under the control of a modified unc-129promoter, which expresses in a subset of DA and DB cholin-ergicmotor neurons [34]. CPX-1::GFPwas diffusely expressedthroughout the axon with some enrichment at presynapticterminals based on colocalization with an SVmarker mCherry::Rab-3 (Figure 2C).
Tonic Activity Is Elevated, and Evoked ReleaseIs Decreased, in the Absence of Complexin
The behavioral assays and expression pattern indicate thatCPX-1 plays a major role in the nervous system. To examinethe effects of removing CPX-1 on synaptic transmission, werecorded synaptic activity at the NMJ in dissected animals.The worm NMJ is a graded synapse, and basal activity in thenervous system of the dissected animal continuously drivesneurotransmitter release, as measured by whole-cell patch-clamp recordings from body wall muscle [7]. This tonic excit-atory postsynaptic current (tEPSC) rate depended on externalcalcium, because over 90% of these fusion events were elim-inated in zero calcium external solution (Figure S3). cpx-1mutants display a large increase (290% of wild-type) in tEPSC
rate (Figures 3A and 3C), with no change in thetEPSC amplitude (Figure 3D). Even in lowcalcium, spontaneous fusion was significantlyincreased in cpx-1 mutants (Figure S3).
Surprisingly, EPSCs evoked by a depolarizing stimulus elec-trode (eEPSCs) were decreased by about 90% in cpx-1mutants (Figures 3B and 3E), suggesting a requirement forCPX-1 during SV fusion evoked by high levels of activity. Totalsynaptic charge was decreased to a similar degree, indicatingthat the eEPSC peak was not decreased simply because ofasynchrony of the evoked fusion events in the cpx-1 mutant(data not shown). Consistent with the behavioral assays,CPX-1 expression in GABA neurons failed to rescue eitherthe tEPSC rate or eEPSC, whereas expression in all neuronsrescued both modes of exocytosis. Thus, SV exocytosis issuppressed by CPX-1 during low levels of activity, whereashigh levels of activity require CPX-1 to promote synchronousexocytosis.
Synaptic Alterations of cpx-1 Mutants Are Not the Resultof Changes in Nervous System Development
Complexin has been proposed to play a role in synapsedevelopment in mouse and fly [27, 35], so it is important toexamine whether the nervous system has been altered incpx-1 mutants. In particular, developmental changes in themotor neurons and NMJsmay explain the behavioral and elec-trophysiological effects described above. The number ofcholinergic motor neurons in the ventral cord was identicalbetween wild-type and cpx-1 mutant animals (Figure 4A).Furthermore, we counted the number of active zones perunit length of axon by imaging the fluorescently tagged activezone (AZ) protein ELKS-1::CFP in wild-type and cpx-1 mutantanimals and found nearly identical AZ densities in the twostrains (Figure 4B). Thus, neither the number of motor neuronsnor the synapse number is altered appreciably in the absenceof cpx-1.
Overexpression of CPX-1 Does Not Affect the NMJThe experiments described above establish that loss of CPX-1at the synapse directly leads to dysregulation of tonic andstimulus-evoked neurotransmission. If complexin functionsas a fusion clamp by binding the trans-SNARE complex, then
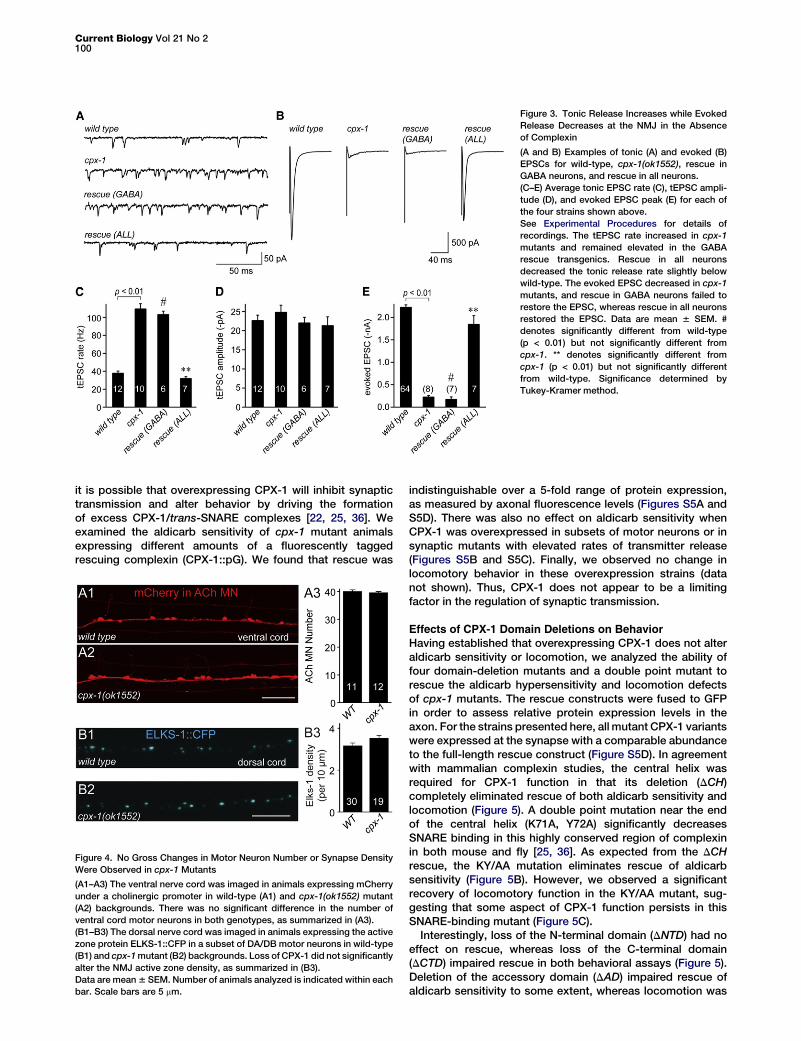
Figure 3. Tonic Release Increases while Evoked
Release Decreases at the NMJ in the Absence
of Complexin
(A and B) Examples of tonic (A) and evoked (B)
EPSCs for wild-type, cpx-1(ok1552), rescue in
GABA neurons, and rescue in all neurons.
(C–E) Average tonic EPSC rate (C), tEPSC ampli-
tude (D), and evoked EPSC peak (E) for each of
the four strains shown above.
See Experimental Procedures for details of
recordings. The tEPSC rate increased in cpx-1
mutants and remained elevated in the GABA
rescue transgenics. Rescue in all neurons
decreased the tonic release rate slightly below
wild-type. The evoked EPSC decreased in cpx-1
mutants, and rescue in GABA neurons failed to
restore the EPSC, whereas rescue in all neurons
restored the EPSC. Data are mean 6 SEM. #
denotes significantly different from wild-type
(p < 0.01) but not significantly different from
cpx-1. ** denotes significantly different from
cpx-1 (p < 0.01) but not significantly different
from wild-type. Significance determined by
Tukey-Kramer method.
Current Biology Vol 21 No 2100
it is possible that overexpressing CPX-1 will inhibit synaptictransmission and alter behavior by driving the formationof excess CPX-1/trans-SNARE complexes [22, 25, 36]. Weexamined the aldicarb sensitivity of cpx-1 mutant animalsexpressing different amounts of a fluorescently taggedrescuing complexin (CPX-1::pG). We found that rescue was
Figure 4. No Gross Changes in Motor Neuron Number or Synapse Density
Were Observed in cpx-1 Mutants
(A1–A3) The ventral nerve cord was imaged in animals expressing mCherry
under a cholinergic promoter in wild-type (A1) and cpx-1(ok1552) mutant
(A2) backgrounds. There was no significant difference in the number of
ventral cord motor neurons in both genotypes, as summarized in (A3).
(B1–B3) The dorsal nerve cord was imaged in animals expressing the active
zone protein ELKS-1::CFP in a subset of DA/DB motor neurons in wild-type
(B1) and cpx-1mutant (B2) backgrounds. Loss of CPX-1 did not significantly
alter the NMJ active zone density, as summarized in (B3).
Data are mean6 SEM. Number of animals analyzed is indicated within each
bar. Scale bars are 5 mm.
indistinguishable over a 5-fold range of protein expression,as measured by axonal fluorescence levels (Figures S5A andS5D). There was also no effect on aldicarb sensitivity whenCPX-1 was overexpressed in subsets of motor neurons or insynaptic mutants with elevated rates of transmitter release(Figures S5B and S5C). Finally, we observed no change inlocomotory behavior in these overexpression strains (datanot shown). Thus, CPX-1 does not appear to be a limitingfactor in the regulation of synaptic transmission.
Effects of CPX-1 Domain Deletions on BehaviorHaving established that overexpressing CPX-1 does not alteraldicarb sensitivity or locomotion, we analyzed the ability offour domain-deletion mutants and a double point mutant torescue the aldicarb hypersensitivity and locomotion defectsof cpx-1 mutants. The rescue constructs were fused to GFPin order to assess relative protein expression levels in theaxon. For the strains presented here, all mutant CPX-1 variantswere expressed at the synapse with a comparable abundanceto the full-length rescue construct (Figure S5D). In agreementwith mammalian complexin studies, the central helix wasrequired for CPX-1 function in that its deletion (DCH)completely eliminated rescue of both aldicarb sensitivity andlocomotion (Figure 5). A double point mutation near the endof the central helix (K71A, Y72A) significantly decreasesSNARE binding in this highly conserved region of complexinin both mouse and fly [25, 36]. As expected from the DCHrescue, the KY/AA mutation eliminates rescue of aldicarbsensitivity (Figure 5B). However, we observed a significantrecovery of locomotory function in the KY/AA mutant, sug-gesting that some aspect of CPX-1 function persists in thisSNARE-binding mutant (Figure 5C).Interestingly, loss of the N-terminal domain (DNTD) had no
effect on rescue, whereas loss of the C-terminal domain(DCTD) impaired rescue in both behavioral assays (Figure 5).Deletion of the accessory domain (DAD) impaired rescue ofaldicarb sensitivity to some extent, whereas locomotion was
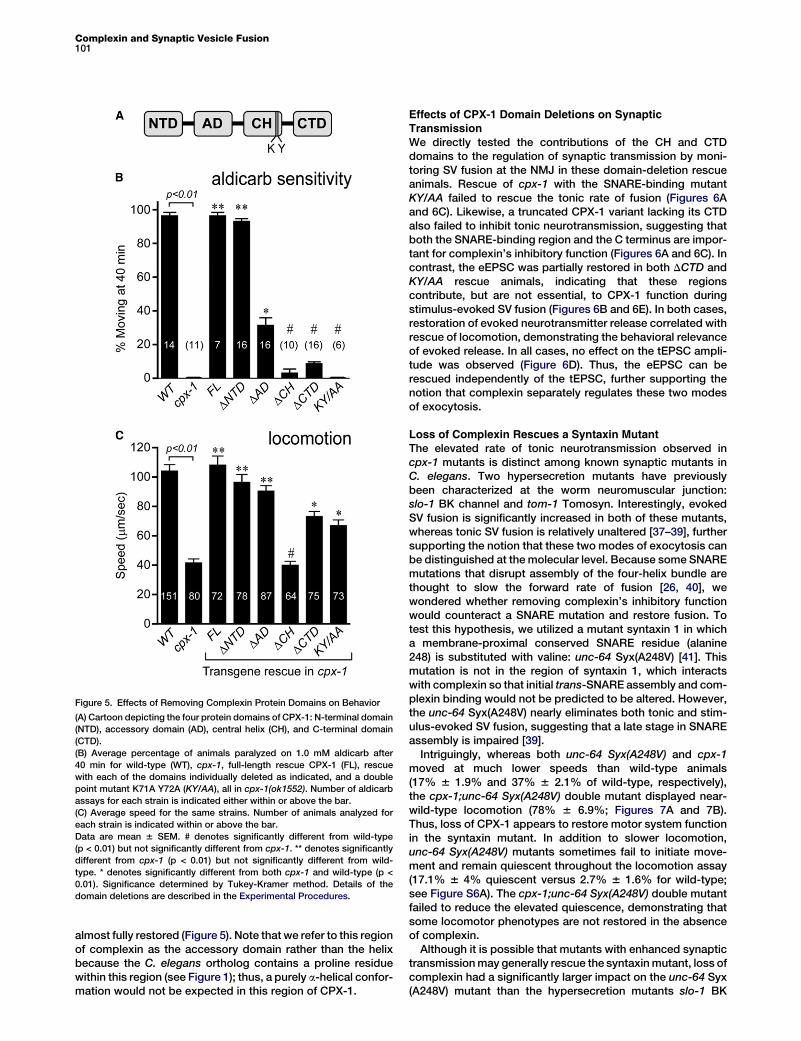
Figure 5. Effects of Removing Complexin Protein Domains on Behavior
(A) Cartoon depicting the four protein domains of CPX-1: N-terminal domain
(NTD), accessory domain (AD), central helix (CH), and C-terminal domain
(CTD).
(B) Average percentage of animals paralyzed on 1.0 mM aldicarb after
40 min for wild-type (WT), cpx-1, full-length rescue CPX-1 (FL), rescue
with each of the domains individually deleted as indicated, and a double
point mutant K71A Y72A (KY/AA), all in cpx-1(ok1552). Number of aldicarb
assays for each strain is indicated either within or above the bar.
(C) Average speed for the same strains. Number of animals analyzed for
each strain is indicated within or above the bar.
Data are mean 6 SEM. # denotes significantly different from wild-type
(p < 0.01) but not significantly different from cpx-1. ** denotes significantly
different from cpx-1 (p < 0.01) but not significantly different from wild-
type. * denotes significantly different from both cpx-1 and wild-type (p <
0.01). Significance determined by Tukey-Kramer method. Details of the
domain deletions are described in the Experimental Procedures.
Complexin and Synaptic Vesicle Fusion101
almost fully restored (Figure 5). Note that we refer to this regionof complexin as the accessory domain rather than the helixbecause the C. elegans ortholog contains a proline residuewithin this region (see Figure 1); thus, a purely a-helical confor-mation would not be expected in this region of CPX-1.
Effects of CPX-1 Domain Deletions on SynapticTransmission
We directly tested the contributions of the CH and CTDdomains to the regulation of synaptic transmission by moni-toring SV fusion at the NMJ in these domain-deletion rescueanimals. Rescue of cpx-1 with the SNARE-binding mutantKY/AA failed to rescue the tonic rate of fusion (Figures 6Aand 6C). Likewise, a truncated CPX-1 variant lacking its CTDalso failed to inhibit tonic neurotransmission, suggesting thatboth the SNARE-binding region and the C terminus are impor-tant for complexin’s inhibitory function (Figures 6A and 6C). Incontrast, the eEPSC was partially restored in both DCTD andKY/AA rescue animals, indicating that these regionscontribute, but are not essential, to CPX-1 function duringstimulus-evoked SV fusion (Figures 6B and 6E). In both cases,restoration of evoked neurotransmitter release correlated withrescue of locomotion, demonstrating the behavioral relevanceof evoked release. In all cases, no effect on the tEPSC ampli-tude was observed (Figure 6D). Thus, the eEPSC can berescued independently of the tEPSC, further supporting thenotion that complexin separately regulates these two modesof exocytosis.
Loss of Complexin Rescues a Syntaxin MutantThe elevated rate of tonic neurotransmission observed incpx-1 mutants is distinct among known synaptic mutants inC. elegans. Two hypersecretion mutants have previouslybeen characterized at the worm neuromuscular junction:slo-1 BK channel and tom-1 Tomosyn. Interestingly, evokedSV fusion is significantly increased in both of these mutants,whereas tonic SV fusion is relatively unaltered [37–39], furthersupporting the notion that these two modes of exocytosis canbe distinguished at the molecular level. Because some SNAREmutations that disrupt assembly of the four-helix bundle arethought to slow the forward rate of fusion [26, 40], wewondered whether removing complexin’s inhibitory functionwould counteract a SNARE mutation and restore fusion. Totest this hypothesis, we utilized a mutant syntaxin 1 in whicha membrane-proximal conserved SNARE residue (alanine248) is substituted with valine: unc-64 Syx(A248V) [41]. Thismutation is not in the region of syntaxin 1, which interactswith complexin so that initial trans-SNARE assembly and com-plexin binding would not be predicted to be altered. However,the unc-64 Syx(A248V) nearly eliminates both tonic and stim-ulus-evoked SV fusion, suggesting that a late stage in SNAREassembly is impaired [39].Intriguingly, whereas both unc-64 Syx(A248V) and cpx-1
moved at much lower speeds than wild-type animals(17% 6 1.9% and 37% 6 2.1% of wild-type, respectively),the cpx-1;unc-64 Syx(A248V) double mutant displayed near-wild-type locomotion (78% 6 6.9%; Figures 7A and 7B).Thus, loss of CPX-1 appears to restore motor system functionin the syntaxin mutant. In addition to slower locomotion,unc-64 Syx(A248V) mutants sometimes fail to initiate move-ment and remain quiescent throughout the locomotion assay(17.1% 6 4% quiescent versus 2.7% 6 1.6% for wild-type;see Figure S6A). The cpx-1;unc-64 Syx(A248V) double mutantfailed to reduce the elevated quiescence, demonstrating thatsome locomotor phenotypes are not restored in the absenceof complexin.Although it is possible that mutants with enhanced synaptic
transmissionmay generally rescue the syntaxinmutant, loss ofcomplexin had a significantly larger impact on the unc-64 Syx(A248V) mutant than the hypersecretion mutants slo-1 BK

Figure 6. Central Helix-SNARE Interactions and
the C-Terminal Domain of CPX-1 Are Required
for Its Inhibition of Tonic, but Not for Evoked SV
Fusion
(A and B) Representative traces for tonic (A) and
evoked (B) EPSCs are shown for wild-type,
cpx-1(ok1552), rescue with a central helix dele-
tion (DCH), rescuewith a C-terminal domain dele-
tion (DCTD), and rescue with a double point
mutant in the central helix predicted to decrease
SNARE binding (KY/AA), all in cpx-1(ok1552).
(C–E) Average values for the frequency (C) and
amplitude (D) of tEPSCs and peak eEPSC
amplitude (E) are shown for each strain. Data
aremean6 SEM. # denotes significantly different
from wild-type (p < 0.01) but not significantly
different from cpx-1. * denotes significantly
different from both cpx-1 and wild-type (p <
0.01). Significance determined by Tukey-Kramer
method.
Current Biology Vol 21 No 2102
channel and tom-1 Tomosyn. Although evoked SV exocytosisis increased in eachmutant [37–39], neither could restore loco-motion to the same extent as loss of CPX-1 (Figure 7C). Finally,rescue of locomotion in cpx-1 might arise simply from animpairment of SV fusion that protects a pool of vesicles fromdepletion. We examined three mutants that are defectivein distinct aspects of SV exocytosis (snb-1 synaptobrevin,unc-2Ca2+ voltage-dependent channel, and snt-1 synaptotag-min) and found that locomotion was not significantly rescued(Figure S6B).
We next examined the effects of removing CPX-1 in theA248V syntaxin mutant on SV fusion. Whereas unc-64 Syx(A248V) almost eliminated tEPSC fusion events, the cpx-1;unc-64 Syx(A248V) double mutant displayed wild-type rates(Figures 7D and 7F). In contrast, the eEPSC amplitude wasnot restored in the cpx-1;unc-64 Syx(A248V) double mutant,highlighting the distinct role of CPX-1 in supporting thismode of exocytosis (Figures 7E and 7H). The average tEPSCamplitude was decreased by about 43% in the unc-64 Syx(A248) mutant and restored in the cpx-1;unc-64 Syx(A248V)double mutant (Figure 7G). Although smaller tonic fusionevents might cause an apparent decrease in tEPSC frequencyif a significant number falls below detection threshold, a 43%decrease in tEPSC amplitude is expected to cause onlya 0.2% decrease in tEPSC frequency under our recordingconditions (Figures S6C and S6D). Thus, the decrease intEPSC rate (and its restoration in the double mutant) repre-sents a change in the frequency of fusion rather than a changein tEPSC amplitude. These observations suggest thatremoving CPX-1 in the unc-64 Syx(A248V) mutant can alle-viate the defect in tonic fusion, whereas evoked EPSCs arenot restored. Furthermore, restoration of tonic neurotransmis-sion was accompanied by an increase in locomotion, demon-strating that this mode of exocytosis is relevant to locomotorybehavior.
Discussion
The results of this study lead us toseveral conclusions about the roleof complexin in C. elegans. First,animals develop normally in theabsence of CPX-1. Second, CPX-1 hasboth negative and positive functions in
neurotransmission: it inhibits fusion of SVs driven by endog-enous activity while simultaneously promoting synchronousneurotransmitter release evoked by a strong depolarizingstimulus. Third, the positive and negative functions can beseparated within the structure of complexin. The N-terminaldomain does not appear to play a major role in CPX-1function, whereas the SNARE-interacting central helix isessential for both regulatory functions of CPX-1. The C-ter-minal domain and tight SNARE binding are required onlyfor complexin’s inhibitory role. Fourth, complexin’s functionin the final stages of fusion is underscored by its geneticinteractions with syntaxin. Specifically, a syntaxin ‘‘zipper-ing’’ mutant can be rescued by removing complexin. Thesedata suggest that complexin bound to the trans-SNAREcomplex raises the energy barrier for SV fusion. Theopposing effects of complexin removal on tonic and evokedrelease suggest that these are distinct exocytic events thatcan be separated at the molecular level (see model inFigure S7A).
Separating Functions of CPX-1 DomainsN-Terminal Domain
There is evidence from murine culture studies that the NTDfacilitates evoked SV fusion and that a few residues withinthe NTD are identical across several phyla (68% identitybetween worm and fly), suggesting a conserved function ofthis domain [24, 25]. The fly complexin NTD, however, doesnot appear to affect synaptic transmission when expressedin mouse neurons [25]. Similar to dmCplx, we observedcomplete rescue of function with a version of CPX-1 missingthe entire NTD. Thus, this domain does not play a major rolein complexin function in C. elegans.Accessory DomainPrior studies on the AD have suggested an inhibitory role infusion [22, 25, 42, 43]. The nematode CPX-1 AD contains
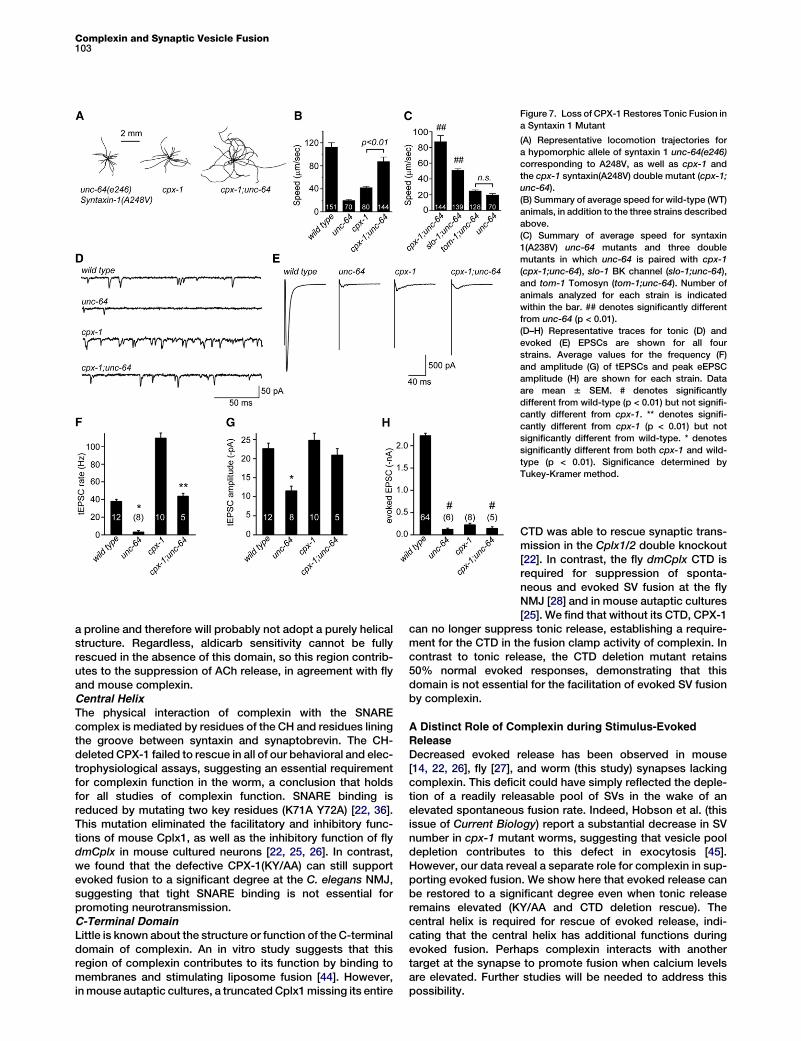
Figure 7. Loss of CPX-1 Restores Tonic Fusion in
a Syntaxin 1 Mutant
(A) Representative locomotion trajectories for
a hypomorphic allele of syntaxin 1 unc-64(e246)
corresponding to A248V, as well as cpx-1 and
the cpx-1 syntaxin(A248V) double mutant (cpx-1;
unc-64).
(B) Summary of average speed for wild-type (WT)
animals, in addition to the three strains described
above.
(C) Summary of average speed for syntaxin
1(A238V) unc-64 mutants and three double
mutants in which unc-64 is paired with cpx-1
(cpx-1;unc-64), slo-1 BK channel (slo-1;unc-64),
and tom-1 Tomosyn (tom-1;unc-64). Number of
animals analyzed for each strain is indicated
within the bar. ## denotes significantly different
from unc-64 (p < 0.01).
(D–H) Representative traces for tonic (D) and
evoked (E) EPSCs are shown for all four
strains. Average values for the frequency (F)
and amplitude (G) of tEPSCs and peak eEPSC
amplitude (H) are shown for each strain. Data
are mean 6 SEM. # denotes significantly
different from wild-type (p < 0.01) but not signifi-
cantly different from cpx-1. ** denotes signifi-
cantly different from cpx-1 (p < 0.01) but not
significantly different from wild-type. * denotes
significantly different from both cpx-1 and wild-
type (p < 0.01). Significance determined by
Tukey-Kramer method.
Complexin and Synaptic Vesicle Fusion103
a proline and therefore will probably not adopt a purely helicalstructure. Regardless, aldicarb sensitivity cannot be fullyrescued in the absence of this domain, so this region contrib-utes to the suppression of ACh release, in agreement with flyand mouse complexin.Central Helix
The physical interaction of complexin with the SNAREcomplex is mediated by residues of the CH and residues liningthe groove between syntaxin and synaptobrevin. The CH-deleted CPX-1 failed to rescue in all of our behavioral and elec-trophysiological assays, suggesting an essential requirementfor complexin function in the worm, a conclusion that holdsfor all studies of complexin function. SNARE binding isreduced by mutating two key residues (K71A Y72A) [22, 36].This mutation eliminated the facilitatory and inhibitory func-tions of mouse Cplx1, as well as the inhibitory function of flydmCplx in mouse cultured neurons [22, 25, 26]. In contrast,we found that the defective CPX-1(KY/AA) can still supportevoked fusion to a significant degree at the C. elegans NMJ,suggesting that tight SNARE binding is not essential forpromoting neurotransmission.C-Terminal DomainLittle is known about the structure or function of the C-terminaldomain of complexin. An in vitro study suggests that thisregion of complexin contributes to its function by binding tomembranes and stimulating liposome fusion [44]. However,inmouse autaptic cultures, a truncatedCplx1missing its entire
CTD was able to rescue synaptic trans-mission in the Cplx1/2 double knockout[22]. In contrast, the fly dmCplx CTD isrequired for suppression of sponta-neous and evoked SV fusion at the flyNMJ [28] and in mouse autaptic cultures[25]. We find that without its CTD, CPX-1
can no longer suppress tonic release, establishing a require-ment for the CTD in the fusion clamp activity of complexin. Incontrast to tonic release, the CTD deletion mutant retains50% normal evoked responses, demonstrating that thisdomain is not essential for the facilitation of evoked SV fusionby complexin.
A Distinct Role of Complexin during Stimulus-EvokedRelease
Decreased evoked release has been observed in mouse[14, 22, 26], fly [27], and worm (this study) synapses lackingcomplexin. This deficit could have simply reflected the deple-tion of a readily releasable pool of SVs in the wake of anelevated spontaneous fusion rate. Indeed, Hobson et al. (thisissue of Current Biology) report a substantial decrease in SVnumber in cpx-1 mutant worms, suggesting that vesicle pooldepletion contributes to this defect in exocytosis [45].However, our data reveal a separate role for complexin in sup-porting evoked fusion. We show here that evoked release canbe restored to a significant degree even when tonic releaseremains elevated (KY/AA and CTD deletion rescue). Thecentral helix is required for rescue of evoked release, indi-cating that the central helix has additional functions duringevoked fusion. Perhaps complexin interacts with anothertarget at the synapse to promote fusion when calcium levelsare elevated. Further studies will be needed to address thispossibility.

Current Biology Vol 21 No 2104
Possible Roles for Complexin in the Regulationof Synaptic Transmission
By demonstrating opposing roles for complexin in the controlof tonic release versus evoked responses, we have shown thatboth forms of synaptic transmission are distinct and differen-tially regulated. If the level of synaptic activity determineswhether complexin will inhibit or support SV fusion, complexincan serve as a temporal filter, suppressing low frequencyactivity while boosting higher frequencies (Figure S7B). Thetonic and phasic outputs seem to play different roles in theregulation of behavior. Tonic release in the absence of evokedrelease is sufficient for basal locomotory speed but not normallevels of foraging behavior. Thus, complexin can act asa master switch for temporal coding mechanisms at thesynapse, gating the balance of tonic and phasic output.
Experimental Procedures
Details of the behavioral assays, electrophysiology, and fluorescence
imaging, as well as a list of all strains used in this study, can be found in
the Supplemental Experimental Procedures.
Complexin Mutants and Rescue Constructs
C. elegans full-length complexin CPX-1 is 143 amino acids encoded by a
1.2 kb gene harboring a single 764 base intron. For the structural variants
of CPX-1 used in this study, we used the following residue positions to
define each domain. The first 30 amino acids are removed in the N-terminal
deletion CPX. Residues 31–50 are removed in the accessory domain
deletion. The central helix deletion corresponds to residues 51–79. The
C-terminal domain deletion removes the final 50 residues of CPX-1 and
includes a 12 residue linker between CPX-1 and paGFP.
Statistical Analysis
For data sets requiring a single pairwise comparison, we used Student’s t
test to compute significance. For all other comparisons, we used the Tukey-
Kramer method for multiple comparisons. This test assumes independent,
normal distributions with equal variance. In cases in which sample variance
was significantly different (calculated using Levene’s test), we employed the
Newman-Keulsmethod to test significance. Significancewas defined by the
criterion p < 0.01.
Supplemental Information
Supplemental Information includes seven figures and Supplemental Exper-
imental Procedures and can be found with this article online at doi:10.1016/
j.cub.2010.12.014.
Acknowledgments
We thank the C. elegans Stock Center and S. Mitani for strains. We thank
Miriam Goodman’s laboratory for use of their worm tracking software;
Cori Bargmann, Josh Kaplan, Jihong Bai, Peter Juo, Anant Menon, Erik
Jorgensen, Tim Ryan, and members of the Ryan laboratory for advice and
for critically reading the manuscript; and Kashyap Rajagopal for assisting
in developing the locomotion analysis. This work was supported by awards
from the Rita Allen Foundation (188423) and the March of Dimes (Basil
O’Connor Award 5-FY09-133) and by National Institutes of Health grant
GM095674 (J.S.D.).
Received: October 21, 2010
Revised: December 8, 2010
Accepted: December 8, 2010
Published online: January 6, 2011
References
1. Katz, B., and Miledi, R. (1967). Tetrodotoxin and neuromuscular trans-
mission. Proc. R. Soc. Lond. B Biol. Sci. 167, 8–22.
2. Schmitt, R.O., Dev, P., and Smith, B.H. (1976). Electrotonic processing
of information by brain cells. Science 193, 114–120.
3. Heidelberger, R. (2007). Mechanisms of tonic, graded release: Lessons
from the vertebrate photoreceptor. J. Physiol. 585, 663–667.
4. Shepherd, G.M., Chen, W.R., Willhite, D., Migliore, M., and Greer, C.A.
(2007). The olfactory granule cell: From classical enigma to central
role in olfactory processing. Brain Res. Rev. 55, 373–382.
5. Manor, Y., Nadim, F., Abbott, L.F., and Marder, E. (1997). Temporal
dynamics of graded synaptic transmission in the lobster stomatogastric
ganglion. J. Neurosci. 17, 5610–5621.
6. Burrows, M. (1979). Graded synaptic interactions between local premo-
tor interneurons of the locust. J. Neurophysiol. 42, 1108–1123.
7. Liu, Q., Hollopeter, G., and Jorgensen, E.M. (2009). Graded synaptic
transmission at the Caenorhabditis elegans neuromuscular junction.
Proc. Natl. Acad. Sci. USA 106, 10823–10828.
8. Jahn, R., Lang, T., and Sudhof, T.C. (2003). Membrane fusion. Cell 112,
519–533.
9. Sudhof, T.C. (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27,
509–547.
10. Sudhof, T.C., and Rothman, J.E. (2009). Membrane fusion: Grappling
with SNARE and SM proteins. Science 323, 474–477.
11. Melia, T.J., Jr. (2007). Putting the clamps on membrane fusion: How
complexin sets the stage for calcium-mediated exocytosis. FEBS Lett.
581, 2131–2139.
12. Chen, X., Tomchick, D.R., Kovrigin, E., Arac, D., Machius, M., Sudhof,
T.C., and Rizo, J. (2002). Three-dimensional structure of the com-
plexin/SNARE complex. Neuron 33, 397–409.
13. McMahon, H.T., Missler, M., Li, C., and Sudhof, T.C. (1995). Complexins:
Cytosolic proteins that regulate SNAP receptor function. Cell 83,
111–119.
14. Reim, K., Mansour, M., Varoqueaux, F., McMahon, H.T., Sudhof, T.C.,
Brose, N., and Rosenmund, C. (2001). Complexins regulate a late step
in Ca2+-dependent neurotransmitter release. Cell 104, 71–81.
15. Bracher, A., Kadlec, J., Betz, H., and Weissenhorn, W. (2002). X-ray
structure of a neuronal complexin-SNARE complex from squid.
J. Biol. Chem. 277, 26517–26523.
16. Pobbati, A.V., Razeto, A., Boddener, M., Becker, S., and Fasshauer, D.
(2004). Structural basis for the inhibitory role of tomosyn in exocytosis.
J. Biol. Chem. 279, 47192–47200.
17. Chicka, M.C., and Chapman, E.R. (2009). Concurrent binding of com-
plexin and synaptotagmin to liposome-embedded SNARE complexes.
Biochemistry 48, 657–659.
18. Schaub, J.R., Lu, X., Doneske, B., Shin, Y.K., and McNew, J.A. (2006).
Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I.
Nat. Struct. Mol. Biol. 13, 748–750.
19. Giraudo, C.G., Eng,W.S., Melia, T.J., andRothman, J.E. (2006). A clamp-
ing mechanism involved in SNARE-dependent exocytosis. Science 313,
676–680.
20. Malsam, J., Seiler, F., Schollmeier, Y., Rusu, P., Krause, J.M., and
Sollner, T.H. (2009). The carboxy-terminal domain of complexin I stimu-
lates liposome fusion. Proc. Natl. Acad. Sci. USA 106, 2001–2006.
21. Yoon, T.Y., Lu, X., Diao, J., Lee, S.M., Ha, T., and Shin, Y.K. (2008).
Complexin and Ca2+ stimulate SNARE-mediated membrane fusion.
Nat. Struct. Mol. Biol. 15, 707–713.
22. Xue, M., Reim, K., Chen, X., Chao, H.T., Deng, H., Rizo, J., Brose, N., and
Rosenmund, C. (2007). Distinct domains of complexin I differentially
regulate neurotransmitter release. Nat. Struct. Mol. Biol. 14, 949–958.
23. Xue, M., Stradomska, A., Chen, H., Brose, N., Zhang, W., Rosenmund,
C., and Reim, K. (2008). Complexins facilitate neurotransmitter release
at excitatory and inhibitory synapses in mammalian central nervous
system. Proc. Natl. Acad. Sci. USA 105, 7875–7880.
24. Xue, M., Craig, T.K., Xu, J., Chao, H.T., Rizo, J., and Rosenmund, C.
(2010). Binding of the complexin N terminus to the SNARE complex
potentiates synaptic-vesicle fusogenicity. Nat. Struct. Mol. Biol. 17,
568–575.
25. Xue, M., Lin, Y.Q., Pan, H., Reim, K., Deng, H., Bellen, H.J., and
Rosenmund, C. (2009). Tilting the balance between facilitatory and
inhibitory functions of mammalian and Drosophila complexins orches-
trates synaptic vesicle exocytosis. Neuron 64, 367–380.
26. Maximov, A., Tang, J., Yang, X., Pang, Z.P., and Sudhof, T.C. (2009).
Complexin controls the force transfer from SNARE complexes to
membranes in fusion. Science 323, 516–521.
27. Huntwork, S., and Littleton, J.T. (2007). A complexin fusion clamp regu-
lates spontaneous neurotransmitter release and synaptic growth. Nat.
Neurosci. 10, 1235–1237.

Complexin and Synaptic Vesicle Fusion105
28. Cho, R.W., Song, Y., and Littleton, J.T. (2010). Comparative analysis of
Drosophila and mammalian complexins as fusion clamps and facilita-
tors of neurotransmitter release. Mol. Cell. Neurosci. 45, 389–397.
29. Rand, J.B., and Russell, R.L. (1985). Molecular basis of drug-resistance
mutations in C. elegans. Psychopharmacol. Bull. 21, 623–630.
30. Miller, K.G., Alfonso, A., Nguyen, M., Crowell, J.A., Johnson, C.D., and
Rand, J.B. (1996). A genetic selection for Caenorhabditis elegans
synaptic transmission mutants. Proc. Natl. Acad. Sci. USA 93, 12593–
12598.
31. Dittman, J.S., and Kaplan, J.M. (2008). Behavioral impact of neurotrans-
mitter-activated G-protein-coupled receptors: Muscarinic and GABAB
receptors regulate Caenorhabditis elegans locomotion. J. Neurosci.
28, 7104–7112.
32. Nurrish, S., Segalat, L., and Kaplan, J.M. (1999). Serotonin inhibition of
synaptic transmission: Galpha(0) decreases the abundance of UNC-13
at release sites. Neuron 24, 231–242.
33. Vashlishan, A.B., Madison, J.M., Dybbs, M., Bai, J., Sieburth, D., Ch’ng,
Q., Tavazoie, M., and Kaplan, J.M. (2008). An RNAi screen identifies
genes that regulate GABA synapses. Neuron 58, 346–361.
34. Sieburth, D., Ch’ng, Q., Dybbs, M., Tavazoie, M., Kennedy, S., Wang, D.,
Dupuy, D., Rual, J.F., Hill, D.E., Vidal, M., et al. (2005). Systematic anal-
ysis of genes required for synapse structure and function. Nature 436,
510–517.
35. Yang, Y., Kim, A.H., Yamada, T.,Wu, B., Bilimoria, P.M., Ikeuchi, Y., de la
Iglesia, N., Shen, J., and Bonni, A. (2009). A Cdc20-APC ubiquitin
signaling pathway regulates presynaptic differentiation. Science 326,
575–578.
36. Tang, J., Maximov, A., Shin, O.H., Dai, H., Rizo, J., and Sudhof, T.C.
(2006). A complexin/synaptotagmin 1 switch controls fast synaptic
vesicle exocytosis. Cell 126, 1175–1187.
37. Gracheva, E.O., Burdina, A.O., Holgado, A.M., Berthelot-Grosjean, M.,
Ackley, B.D., Hadwiger, G., Nonet, M.L., Weimer, R.M., and
Richmond, J.E. (2006). Tomosyn inhibits synaptic vesicle priming in
Caenorhabditis elegans. PLoS Biol. 4, e261.
38. McEwen, J.M., Madison, J.M., Dybbs, M., and Kaplan, J.M. (2006).
Antagonistic regulation of synaptic vesicle priming by Tomosyn and
UNC-13. Neuron 51, 303–315.
39. Wang, Z.W., Saifee, O., Nonet, M.L., and Salkoff, L. (2001). SLO-1 potas-
sium channels control quantal content of neurotransmitter release at the
C. elegans neuromuscular junction. Neuron 32, 867–881.
40. Jahn, R., and Scheller, R.H. (2006). SNAREs—engines for membrane
fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643.
41. Saifee, O., Wei, L., and Nonet, M.L. (1998). The Caenorhabditis elegans
unc-64 locus encodes a syntaxin that interacts genetically with synap-
tobrevin. Mol. Biol. Cell 9, 1235–1252.
42. Giraudo, C.G., Garcia-Diaz, A., Eng, W.S., Chen, Y., Hendrickson, W.A.,
Melia, T.J., and Rothman, J.E. (2009). Alternative zippering as an on-off
switch for SNARE-mediated fusion. Science 323, 512–516.
43. Lu, B., Song, S., and Shin, Y.K. (2010). Accessory alpha-helix of com-
plexin I can displace VAMP2 locally in the complexin-SNARE quaternary
complex. J. Mol. Biol. 396, 602–609.
44. Seiler, F., Malsam, J., Krause, J.M., and Sollner, T.H. (2009). A role of
complexin-lipid interactions in membrane fusion. FEBS Lett. 583,
2343–2348.
45. Hobson, R.J., Liu, Q., Watanabe, S., and Jorgensen, E.M. (2011).
Complexin maintains vesicles in the primed state in C. elegans. Curr.
Biol. 20, this issue, 106–113.
Top Related
Copyright © 2022 FDOKUMEN

