







Bahasa
Halaman
Hukum


A Classical Approach to Multichromophoric Resonance Energy Transfer
Sebastian Duque,1, 2 Paul Brumer,2 and Leonardo A. Pachon1
1Grupo de Fısica Atomica y Molecular, Instituto de Fısica, Facultad de Ciencias Exactas y Naturales,Universidad de Antioquia UdeA; Calle 70 No. 52-21, Medellın, Colombia.
2Chemical Physics Theory Group, Department of Chemistry and Center for Quantum Information and Quantum Control,University of Toronto, Toronto, Canada M5S 3H6
(Dated: January 26, 2015)
Enhanced rates in multichromophoric resonance energy transfer are shown to be well describedby a classical theory based on classical electrodynamics. In a coupling configuration between NA
acceptors and ND donors, the theory correctly predicts an enhancement of the energy transfer ratedependent on the total number of donor-acceptor pairs, NAND. As an example, the theory, appliedto the transfer rate in LH II, gives results in excellent agreement with experiment.
Introduction—Aspects of modern research on elec-tronic resonant energy transfer in photosynthetic light-harvesting systems have focussed on energy transfer as acoherent collective phenomenon. This feature has beenhighlighted as central to several transfer mechanisms,such as super transfer [1] and a network renormalizationscheme [2], and predicts dramatic enhancements of en-ergy transfer rates [3]. Qualitative arguments explainingsuch behavior often rely on interactions within donorsand acceptors that induce delocalization of the excitationand establish quantum correlations, such as entanglement,between chromophores. As a consequence, this observedunexpected rate enhancement has been widely attributedto quantum coherence of acceptors and donors.
This purportedly quantum behavior at ambient con-ditions in photosynthetic light-harvesting systems hascontributed to the view that quantum effects play animportant role in enhancing transport efficiency in photo-synthesis, and that these effects are somehow favored byevolutionary selection. For example, arguments to explaintransfer rate enhancements and irreversibility in light har-vesting complexes (such as LHII) as quantum processesinvolving superposition and process coherence have beenproposed [4–8], and the extent to which enhancement isquantum, and is therefore incapable of being accountedfor classically, is being extensively discussed [3, 7, 9].
In this letter we demonstrate that such enhanced ratesare readily explained by a classical theory that is reliantsolely upon classical electrodynamics. The resultant ex-pressions retain the simplicity of Forster energy transferformulae, while allowing a straightforward interpretationof the origin of the enhanced energy transfer rates. Weapply this approach to calculate the energy transfer ratein both a model system and in LHII and show that itaccurately describes enhanced multichromophoric energytransfer rates. Since multichromophoric electronic energytransfer is also prevalent in a large range of studies onmolecular systems such as DNA [10] and proteins [11],the theory is expected to be useful in a wide variety ofapplications.
Quantum Multichromopric Forster’s Resonance EnergyTransfer—Note first the current quantum perspective on
multichromophoric electronic energy transfer. Considerthe pairwise transfer of excitation from chromophore Dto A: D∗ + A → D + A∗, where D∗ (D) is the excited(ground) state donor and A (A∗) is the ground (excited)state acceptor. From the single chromophoric Forstertheory, the rate of energy transfer from D to A is givenby
kF =J2
2π~
∫ ∞−∞
dωED(ω)IA(ω) (1)
where J is the electronic coupling between D and A,ED(ω) is related to the normalized emission lineshape ofthe donor D, and IA(ω) to the linear absorption crosssection of the acceptor A [12].
As long as the D and A molecules are well separatedfrom one another, inter-D-A distances are larger thanintra-D and intra-A distances, and well-defined D and Asites exists. Therefore, use of the rate expression (1) isjustified. However, application of this single chromophorictheory to multichromophoric systems leads to errors be-cause transfer involves more than one pair of excitations,and because intra-D and intra-A coherences that allowexciton delocalization over multiple chromophores areneglected.
Motivated by these facts, a general quantum Forster-like rate expression for a set of Dj (j = 1, . . . , ND) donorsand Ak (k = 1, . . . , NA) acceptors with intra-D and intra-A coherences was formulated in Ref. [5]. The expressioncan be cast as
kMCF =
ND∑j′j′′
NA∑k′k′′
Jj′k′Jj′′k′′
2π~2
∞∫−∞
dωEj′′j′
D (ω)Ik′′k′
A (ω) ,
(2)
with the obvious generalization of the notation in Eq. (1).The intra-D and intra-A coherences are said to be quan-tum, arising from a superposition of energy eigenstates,and to be responsible for the enhanced transfer rate (e.g.,Ref. [6]). However, as shown below, the rate is well de-scribed by classical electrodynamics, and results in the
arX
iv:1
412.
8743
v1 [
phys
ics.
bio-
ph]
30
Dec
201
4

2
observed enhanced chromophoric energy transfer.Classical Multichromopric Forster’s Resonance Energy
Transfer—For the relevant configurations involved in light-harvesting systems, the distance between molecules is atmost a few nanometers, while the relevant wavelength oflight is on the order of several hundred nanometers. Henceenergy transfer will primarily be due to non-radiativecontributions, justifying a point dipole approximation.Moreover, the known inverse sixth-power distance depen-dence of the transfer mechanism owes its origin to thequadratic dependence of the rate on a coupling of transi-tion electric dipoles [13, 14]. In the quantum descriptionof the donor-acceptor energy transfer, deexcitation of thedonor is followed by emission of a virtual photon that isabsorbed by the acceptor [14].
Classically, the donor is envisioned as an oscillatingdipole of frequency ωD, and the acceptor as an absorberwith oscillation frequency ωA. The donor radiates anelectric field that permeates the acceptor and the acceptorabsorbs energy from this field [13, 15]. Adopting thisview, Kuhn [16] and Silbey et al. [15] derived, in the1970’s, Forster’s transfer rate using a completely classicalapproach. Specifically, they showed that the rate of energytransfer of a set of classically interacting dipoles can berecast in a form identical to that of Forster theory [15, 16].Here we significantly extend Refs. [15] and [16] to obtain aclassical description of multichromophoric energy transfer.
To do so, consider a set of Dj (j = 1, . . . , ND)donor molecules and NA (k = 1, . . . , NA) accep-tor molecules, located at rDj
and rAk, respectively.
In the frequency domain, the polarization of each
molecule is proportional to the applied field (linearresponse) pDj (ω) = ε0χDj (ω)E(rDj , ω) , pAk
(ω) =ε0χAk
(ω)E(rAk, ω) , where E(rDj , ω) is the ω frequency
component of the total electric field at the position ofthe donor (similarly for the acceptor) and χ(ω) is thepolarizability tensor of the molecule. The electric field atposition r can be decomposed into an externally incidentfield Eext and the sum of the fields produced by all othersmolecules in the aggregate. In the non-radiative approxi-mation, the electric field at point r due to the presenceof a dipole p at point r0 is
E(r, ω) =3nn− 1
4πε0|r− r0|3· p(ω) ≡ Φ(r− r0) · p(ω) , (3)
where n is the unit vector directed from r0 to r. Note thatp(ω) is a vector comprising all donors and acceptors. Ifthe external field is zero in the region of the acceptors, thepolarization of each of the donor and acceptor molecules ispDj(ω) = ε0χDj
(ω)Eext(rDj , ω) + ε0χDj(ω)
∑ND
j′ 6=j ΦDjj′ ·
pDj′(ω) + ε0χDj (ω)∑NA
k=1 Φkj · pAk(ω), and pAk(ω) =
ε0χAk(ω)
∑NA
k′ 6=k ΦAkk′ · pAk′(ω) + ε0χAk
(ω)∑ND
j=1 Φkj ·pDj(ω) where Φkj is the dipolar coupling between Dj
and Ak, ΦDjj′ is the Dj and D′j intra-D coupling and ΦA
kk′
the Ak and A′k intra-A coupling. Here Eext is the externalfield applied only to the donors.
To expose the interplay between donors and acceptors,it is convenient to subsitute the expression for pDj(ω)into pAk(ω), giving
pAk = ε0χAk
NA∑k′ 6=k
ΦAkk′ · pAk′ + ε20
ND∑j=1
χAkΦkj · χDj
Eext(rDj)
+ ε20
ND∑j=1
χAkΦkj · χDj
ND∑j′ 6=j
ΦDjj′ · pDj′ + ε20
ND∑j=1
χAkΦkj · χDj
NA∑k′=1
Φjk′ · pAk′ .
(4)
where the ω dependence of pDj , pAk, χDj and χAkis
implicit. Further iterations are, of course, possible butEq. (4) already displays a number of processes that en-hance the polarizability at Ak, and hence can affect theenergy transfer. (i) The first term in Eq. (4) will mediatethe transfer of energy between Ak′ and Ak via the inter-action term ΦA
kk′ . (ii) In the second term, the electricfield Eext(rDj) excites the donor Dj which can transferpart of the energy of the field to the acceptor Ak viathe interaction term Φkj . (iii) The third term describeshow energy in donor Dj′ can flow into donor Dj due tothe interaction term ΦD
jj′ , and how part of this energycan transfer to acceptor Ak via the interaction term Φkj .(iv) The last term describes transfer of energy stored in
acceptor Ak′ to donor Dj , assisted by the interaction Φjk′ ,and the subsequent transfer from Dj to Ak mediated byΦkj . Processes (i) and (ii) are first order in the inter-actions (via ΦA
kk′ and Φkj , respectively), while (iii) and(iv) are second order in the interactions (via ΦkjΦ
Djj′ and
ΦkjΦjk′ , respectively). If, in addition, the external fieldEext is allowed to interact with the acceptors (a situationnot relevant for the present discussion), energy can flowdirectly into the acceptors, and higher order processesin the transfer of energy from the field to acceptors willcontribute.
Although this description of energy transfer is formu-lated in the frequency domain, it is clear that in thetime domain these processes are oscillatory (see below)

3
and that the lifetime of the oscillations depends upon thestructure and values of χ. In the symmetric configuration,ΦAkk′ = ΦA, with identical acceptor response χAk
= χA,the first term in Eq. (4) is ε0χA(NA−1)ΦA ·pAk. Despitethe fact that this term already predicts an enhancementof the polarization of the k-acceptor, we show below thatthis interaction need not be the one responsible for thedramatic enhancement of the transfer rate. Rather, it isthe second term in Eq. (4) that allows every acceptor tointeract with every donor that is often significant.
Within classical electrodynamics, the Poynting vectorS = E×H describes the energy flux density of the elec-tromagnetic field. The rate of energy to or from a unitvolume free of current or charges is u(r, t) = −∇ · S and,using Maxwell’s equations and integrating over a volumeenclosing the acceptor region, the rate of energy flowabsorbed by the acceptors is [17–19]
Q(t) =
NA∑k=1
E(rAk, t) · pAk
(t) . (5)
By contrast to Eq. (5), pAk(t) denotes here the polarizabil-
ity in the time domain. The electric field E(rAk, t) labels
the total electric field at the position of the k-th acceptorin the time domain obtained by Fourier transforming thetotal electric field in Eq. (3).
Equation (5) provides the time dynamics of energytransfer. To see how, for example, Eq. (5) relatesto Forster rate theory [15], consider a set of ND
donors and NA acceptors. If each dipole is polariz-able along a single axis and the external field is ap-plied along this axis, then the acceptor polarization equa-tion, in the frequency domain, can be written in termof scalar quantities as pAk(ω) = ε0χAk
(ω)∑NA
k′ 6=k ΦAkk′ ·
pAk′(ω) + ε0χAk(ω)
∑ND
j=1 Φkj · pDj(ω). Defining the vec-
tors pD(ω), pA(ω) and Eext(ω) with components pDj ,pAk
and Eext(rDj , ω), respectively, and the matrices ΦA,Φ(ω), ΦA(ω) and Φ with components ΦA
kk′ , χAk(ω)Φkj ,χAk(ω)ΦA
kk and Φkj , respectively, the above equation canbe rewritten as pA(ω) = ε0ΦA(ω)pA(ω) + ε0Φ(ω)pD(ω).Hence, the linear relationship of the acceptor polarizationto the donor’s is pA(ω) = [1− ε0ΦA(ω)]−1ε0Φ(ω)pD(ω).
The rate of energy flow absorbed by the ac-ceptors within this configuration is Q(t) =[ΦApA(t) + ΦpD(t)] · pA(t). In order to com-pare with Forster’s rate, the time dependent rate[Eq. (5)] is transformed into the frequency domain˜Q(ω), the oscillations in the transfer rate inte-
grated out and the average value of the rate ˜Q(0)
obtained [18]. Here ˜Q(0) = 2ε0∫∞0
dω ωΦp∗D(ω) ·(Im{[1− ε0ΦA(ω)]−1Φ(ω)}pD(ω)
)+
2∫∞0
dω ω[ΦAp∗A(ω)
]· pA(ω), or, written explicitly
˜Q(0) =∑jj′
∑kk′
2ε0ΦkjΦk′j′
∞∫0
dω ω Ikk′
A (ω)Ejj′
D (ω)
+∑kk′
2ΦAkk′
∫ ∞0
dω ω p∗Ak(ω)pAk′(ω)
(6)
with Ikk′
A (ω) = Im{[1 − ε0ΦA(ω)]−1kk′χ′′Ak′(ω)} and
Ejj′
D (ω) = p∗Dj(ω)pDj′(ω) related to the emission andabsorption spectrum of the donors and acceptors. Thefirst term recovers the form of the multichromophoricForster expression (2). The second term is related to theenhancement due to intra-acceptor interaction and ap-pears here [and not in Eq. (2)] because there, the transferrate was derived for the particular case when no exci-tations are present at t = 0 in acceptor sector. As inEq. (2), the intra-donor interaction in Eq. (1) is encoded
in the definition of Ikk′
A (ω) and Ejj′
D (ω). Additionally, ifonly a single acceptor and a single acceptor are present,Eq. (1) coincides with a single donor transferring energyto a single acceptor (see Ref. [18]).
To show in detail how classical electrodynamics givesthe same transfer rate enhancement as predicted by quan-tum arguments, first consider a molecular aggregate modelcomprised of two donors and two acceptors in the configu-ration shown in the lower inset of Fig. 1. The main panel
0 0.2 0.4 0.6 0.8 1.0 ps
0.2
0.4
0.6
0.8
1.0
EA(t
)
All interactions ΦA =ΦD =0 Direct transfer
ΦA =ΦD =0 ΦA =0 ΦD =0
FIG. 1. Normalized total energy in the acceptors for donorsand acceptors in resonance at ω0 = 1.65 eV with dipole mo-ments of 2.6 D. All dipoles are separated 1 nm from each other.The radiative decay rate is γ = (0.8 ps)−1 for all dipoles. Notethat the blue dashed curve and dotted red curve lie atop oneanother.
of Fig. 1 shows the energy EA(t) absorbed by the accep-tors with a single excited state and Lorentzian lineshapesχ(ω) = 2(ε0~)−1ω0|µ|2/[(ω − ω0 + iγ/2)(ω + ω0 + iγ/2)],where µ is the transition dipole moment of the molecule,ω0 its transition frequency and γ is radiative decay rate.The donors are excited with a delta pulse in time. Eachmolecule is polarized along a single polarization axis and
4
all fields applied to the molecule are along this axis ofpolarization.
The rate of energy transfer when the excitation is sym-metrically delocalized over the interacting dipoles, i.e.,when the dipoles all interact (Φkj = ΦD
jj′ = ΦAkk′ = Φ)
is shown as a dashed blue line and is seen to be twiceas fast as the case where the dipoles only communicateindividually, i.e., no donor and no acceptor interaction ispresent (the so-called ”direct transfer” case, Φjk = Φδjk,ΦDjj′ = ΦA
kk′ = 0: continuous black line). Moreover, in theformer fully connected case, not only is the frequency ofthe energy oscillation (transfer rate) faster but the ampli-tude of the energy oscillations are larger as well. Thus,Fig. 1 shows that classical electrodynamics predicts thesame enhancement of a factor of two as found in quantumapproaches of excitonic energy transfer [1, 3].
To understand the origin of this enhancement, it isillustrative to compare to the case when there are nointra-interactions between donor or between acceptors,but where each acceptor can interact with each donor(Φjk = Φ, ΦD
jj′ = ΦAkk′ = 0: red dotted line). For this case,
the enhancement of the transfer rate is seen to be virtuallyidentical to the case where intra-interactions are allowed.Therefore, the enhancement here originates from the factthat all donors transfer to all acceptors and not from theintra-interactions between acceptors or between donors,an observation consistent with quantum results using the“diagonal (secular) Forster rate” model [5, 20, 21].
In the upper inset of Fig. 1, the case of vanishingintra-acceptor (or donor) interactions in the presence ofintra-donor (or acceptor) interactions is depicted by thedashed cyan curve (or dot-dashed green curve). Althoughthe effect here is small, it is clear that the transfer ratemay indeed benefit from the lack of intra-donor or intra-acceptor interactions helping the energy transfer pathway.
To test the predictions of this classical theory, the trans-fer rate of the Light Harvesting complex II is calculatedbelow.
Light Harvesting Complex II —LH II is formed by 27bacteriochlorophylls (BChls) arranged in two rings: eigh-teen of them form the B850 ring with nine forming αβ-heterodimer subunits (here referred as the acceptors), andthe other nine the B800 ring (as the donor). The LH IIcomplex is described here by a set of interacting dipoles.Specifically, the couplings between the BChls in the B800ring are much smaller than those in the B850 ring [4, 22]implying a monomeric structure for the B800 ring; hencethe donor is usually modelled as a single dipole [21]. Thealternating transition dipole moment orientations withinthe B850 ring, which gives rise to the nine-fold symmetry,is nicely shown in Fig. 1 of Ref. [23], as is the donor loca-tion. Interdimer, intradimer coupling and site energies inthe B850 ring are set as in [21]. The site energy of thetwo αβ-heterodimer subunits are E2n−1 = 12406 cm−1
and E2n = 12602 cm−1, the intradimer coupling isΦ2n−1,2n = Φ2n,2n−1 = 363 cm−1 and the interdimer cou-
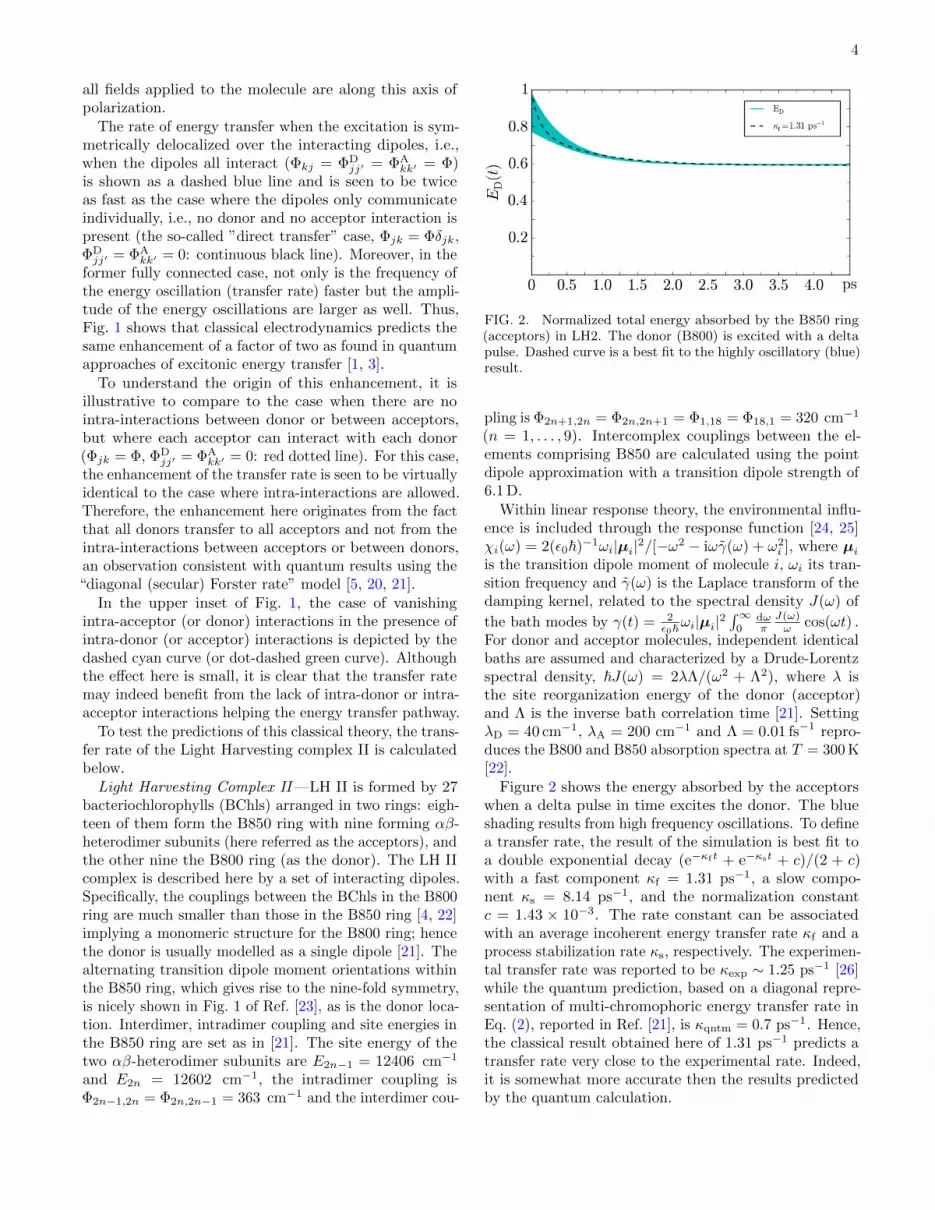
FIG. 2. Normalized total energy absorbed by the B850 ring(acceptors) in LH2. The donor (B800) is excited with a deltapulse. Dashed curve is a best fit to the highly oscillatory (blue)result.
pling is Φ2n+1,2n = Φ2n,2n+1 = Φ1,18 = Φ18,1 = 320 cm−1
(n = 1, . . . , 9). Intercomplex couplings between the el-ements comprising B850 are calculated using the pointdipole approximation with a transition dipole strength of6.1 D.
Within linear response theory, the environmental influ-ence is included through the response function [24, 25]χi(ω) = 2(ε0~)−1ωi|µi|2/[−ω2 − iωγ(ω) + ω2
i ], where µiis the transition dipole moment of molecule i, ωi its tran-sition frequency and γ(ω) is the Laplace transform of thedamping kernel, related to the spectral density J(ω) of
the bath modes by γ(t) = 2ε0~ωi|µi|
2∫∞0
dωπJ(ω)ω cos(ωt) .
For donor and acceptor molecules, independent identicalbaths are assumed and characterized by a Drude-Lorentzspectral density, ~J(ω) = 2λΛ/(ω2 + Λ2), where λ isthe site reorganization energy of the donor (acceptor)and Λ is the inverse bath correlation time [21]. SettingλD = 40 cm−1, λA = 200 cm−1 and Λ = 0.01 fs−1 repro-duces the B800 and B850 absorption spectra at T = 300 K[22].
Figure 2 shows the energy absorbed by the acceptorswhen a delta pulse in time excites the donor. The blueshading results from high frequency oscillations. To definea transfer rate, the result of the simulation is best fit toa double exponential decay (e−κf t + e−κst + c)/(2 + c)with a fast component κf = 1.31 ps−1, a slow compo-nent κs = 8.14 ps−1, and the normalization constantc = 1.43 × 10−3. The rate constant can be associatedwith an average incoherent energy transfer rate κf and aprocess stabilization rate κs, respectively. The experimen-tal transfer rate was reported to be κexp ∼ 1.25 ps−1 [26]while the quantum prediction, based on a diagonal repre-sentation of multi-chromophoric energy transfer rate inEq. (2), reported in Ref. [21], is κqntm = 0.7 ps−1. Hence,the classical result obtained here of 1.31 ps−1 predicts atransfer rate very close to the experimental rate. Indeed,it is somewhat more accurate then the results predictedby the quantum calculation.

5
The fact that classical theory provides better resultsthan does the quantum result arises from the dynamiccharacter of the classical description [see Eq. (6)] versusthe kinematic character of the quantum approach [seeEq. (2)]. Specifically, the classical simulation predictsthe transfer rate on the basis of the time dynamics whilethe multichromophoric rate equation adopts a number ofapproximations (see Ref. [22] for details) and is calculatedat t → ∞. Thus, the main dynamical features, such asthe correct transfer rate, are not incorporated into thequantum description. This suggests that a full dynamicquantum calculation for the LH II, at the same level ofthe classical one performed here, would be of interest.
Comments —It is of some interest to comment on therelationship of this approach to the quantum view ofmultichromophoric energy transfer.
(a) To reconcile the above result with the supertrans-fer mechanism [1], note the standard quantum argumentwhich proceeds as follows: if coherence is not presentwithin the donor region, the incoherent Fermi-golden-rule rate of a donor to transmit energy to the accep-tor is γD→A ∼ |µD · µA|2. Hence, for a pair of iden-tical donors and a pair of identical acceptors the to-tal rate reads Γinc
D→A = 2γD→A. However, if local co-herence is present and the donor is in the symmetricground state (µ1D + µ1D)/
√2 and communicates with
the corresponding state on the acceptor, the total rateΓcohD→A ∼ |(µ1D + µ2D) · (µ1A + µ2A)|2 /4, or explicitly,
ΓcohD→A ∼
1
4
∣∣∣∣µ1D ·µ1A+µ1D ·µ2A+µ2D ·µ1A+µ2D ·µ2A
∣∣∣∣2,so that Γcoh
D→A = 2ΓincD→A. Thus, the enhancement of
the coherent rate ΓcohD→A comes from the terms µ1D · µ2A
and µ2D · µ1A, which include the interactions betweenall donors and all acceptors. Therefore, the enhance-ment obtained above based on classical electrodynamicsis precisely the one predicted by supertransfer [1, 3] andcorresponds to the second term in Eq. (4). Specifically,this term contains ND donor contributions to each ac-ceptor polarization. A total number of NA such acceptorterms contribute to the rate [Eq. (5)], so that the overallenhancement effect is a function of NAND. Note that theclassical theory formulated here also predicts additionalprocesses that may enhance or diminish energy transfer[see discussion below Eq. (4)].
(b) Equations for the polarizations pDj(ω) and pAk(ω)can be solved via the matrix equation A−1p = Eext,where Eext
i is the external applied field on each polarizedmolecule pi, and the polarizability matrix A is defined as
Aij =[δijε0χi− Φij
]. Here i and j indices run over all the
aggregate molecules. The polarizability matrix dependssolely on the properties of the aggregate. The presenceof off-diagonal elements Aij implies that individual chro-mophores cannot be excited independently. Therefore,
the excitation at one site spreads over other sites and thistendency can be viewed as exciton delocalization. Indeed,although delocalization is often referred to as quantumcoherence, we see that delocalization is present in theclassical realm and enhances the energy transfer rate upto the number of interacting molecules (see Eq. 4).
Summary—A theory of multichromophoric electronicenergy transfer based on classical electrodynamics is de-veloped here and shown capable of producing the en-hancement predicted by quantum-based approaches. Theapproach constitutes the classical formulation of the quan-tum multichromophoric theory and allows for the iden-tification of a variety of processes of different order-in-the-interactions that contribute to the energy transferin molecular aggregates with intra-coupling in donorsand acceptor chromophores. When applied to model oflight harvesting systems, such as LH II, the results forthe transfer rate are similar to those that are based onquantum calculations. Results from the classical theoryderived here suggest that the mechanisms such as supertransfer may be robust against certain types of decoher-ence, since they have a classical origin. Further, theysuggest examination of a wide variety of cases, now ongo-ing, to reveal the role of various contributions in Eq. (4)to energy transfer.
Acknowledgements—The authors thank Professor Jian-shu Cao, MIT, for providing data on LH II, and P.B.thanks Professor Aephraim Steinberg for enlightening dis-cussions. This work was supported by NSERC Canada,by Comite para el Desarrollo de la Investigacion (CODI)of Universidad de Antioquia, Colombia under contractnumber E01651 and under the Estrategia de Sostenibili-dad 2014-2015 and by the Departamento Administrativode Ciencia, Tecnologıa e Innovacion (COLCIENCIAS)of Colombia under the contract number 111556934912.
[1] S. Lloyd and M. Mohseni, New J. Phys. 12, 075020 (2010).[2] A. K. Ringsmuth, G. J. Milburn, and T. M. Stace, Nature
Physics , 562 (2012).[3] I. Kassal, J. Yuen-Zhou, and S. Rahimi-Keshari, J. Phys.
Chem. Lett. 4, 362 (2013).[4] S. Jang, M. D. Newton, and R. J. Silbey, J. Phys. Chem.
B 111, 6807 (2007).[5] S. Jang, M. D. Newton, and R. J. Silbey, Phys. Rev. Lett.
92, 218301 (2004).[6] Y. C. Cheng and R. J. Silbey, Phys. Rev. Lett. 96, 028103
(2006).[7] F. Fassioli, R. Dinshaw, P. Arpin, and G. Scholes, J R
Soc Interface 11, 20130901 (2014).[8] A. Olaya-Castro, C. F. Lee, F. F. Olsen, and N. F.
Johnson, Phys. Rev. B 78, 085115 (2008).[9] K. Pelzer, T. Can, S. Gray, D. Morr, and G. Engel, J.
Phys. Chem. B 118, 2693 (2014).[10] T. Ha and J. Xu, Phys. Rev. Lett. 90, 223002 (2003).[11] E. A. Lipman, B. Schuler, O. Bakajin, and W. A. Eaton,

6
Science 301, 1233 (2003).[12] S. Jang, J. Chem. Phys. 127, 174710 (2007).[13] L. Novotny and B. Hecht, Principles of Nano-Optics
(Cambridge University Press, 2006) Chap. 8.[14] D. L. Andrews, in Tutorials in Complex Photonic Media,
edited by M. A. Noginov, G. Dewar, M. W. McCall, andN. I. Zheludev (SPIE press, Bellingham, WA, 2009).
[15] R. R. Chance, A. Prock, and R. Silbey, J. Chem. Phys.62, 2245 (1975).
[16] H. Kuhn, J. Chem. Phys. 53, 101 (1970).[17] L. D. Landau and Lifshitz, Electrodynamics of continous
media (Pergamon, Oxford, 1960) pp. 253–255.[18] E. N. Zimanyi and R. J. Silbey, J. Chem. Phys. 133,
144107 (2010).[19] S. Valleau, S. K. Saikin, D. Ansari-Oghol-Beig, M. Ros-
tami, H. Mossallaei, and A. Aspuru-Guzik, ACS Nano 8,
3884 (2014).[20] A. Mukai, S. Abe, and H. Sumi, J. Phys. Chem. B 103,
6096 (1999).[21] L. Cleary and J. Cao, New J. Phys. 15, 125030 (2013).[22] S. Jang and Y.-C. Cheng, Wiley Interdiscip. Rev. Comput.
Mol. Sci. 3, 84 (2013).[23] S. Jang and R. J. Silbey, J. Chem. Phys. 118, 9324 (2003).[24] G.-L. Ingold, in Coherent Evolution in Noisy Environ-
ments, Lecture Notes in Physics, Vol. 611, edited byA. Buchleitner and K. Hornberger (Springer Berlin Hei-delberg, 2002) pp. 1–53.
[25] H. Grabert et al., Phys. Rep. 168, 115 (1988).[26] Y.-Z. Ma, R. J. Cogdell, and T. Gillbro, J. Phys. Chem.
B 101, 1087 (1997).
Top Related
Copyright © 2022 FDOKUMEN

