







Bahasa
Halaman
Hukum


Binding of fatty acids facilitates oxidation of cysteine-34 andconverts copper–albumin complexes from antioxidants
to prooxidants
Y.A. Gryzunov,a,c A. Arroyo,a J.-L. Vigne,d Q. Zhao,a V.A. Tyurin,a C.A. Hubel,e,f
R.E. Gandley,a,e,f Y.A. Vladimirov,c R.N. Taylor,d and V.E. Kagana,b,*
a Department of Environmental and Occupational Health, University of Pittsburgh, 3343 Forbes Avenue, Pittsburgh, PA 15260, USAb Department of Pharmacology, University of Pittsburgh, 3343 Forbes Avenue, Pittsburgh, PA 15260, USA
c Department of Biophysics, Russian Medical University, Moscow, Russiad Department of Obstetrics/Gynecology and Reproductive Sciences, University of California, San Francisco, CA 94143, USA
e Department of Obstetrics and Gynecology, University of Pittsburghf Magee-Womens Research Institute, Pittsburgh, PA 15213, USA
Received 29 October 2002, and in revised form 3 February 2003
Abstract
As a transition metal capable of undergoing one-electron oxidation–reduction conversions, copper (Cu) is essential for life and
fulfills important catalytic functions. Paradoxically, the same redox properties of copper can make it extremely dangerous because it
can catalyze production of free radical intermediates from molecular oxygen. Factors involved in regulation of redox activity of
albumin-bound copper have not been well characterized. In the present study, effects of modification of the albumin cysteine-34
(Cys-34) and binding of nonesterified fatty acids on the redox-cycling activity of the complex of copper with human serum albumin
(Cu/HSA) were studied. Because ascorbate is the most abundant natural reductant/scavenger of free radicals in blood plasma, the
electron paramagnetic resonance assay of ascorbate radical formation was used as a method to monitor Cu/HSA redox-cycling
activity. At Cu/HSA ratios below 1:1, the bound Cu was virtually redox inactive, as long as Cys-34 was in reduced state (Cu/HSA–
SH). Alkylation, nitrosylation, or oxidation of Cu/HSA resulted in the appearance of redox-cycling activity. Experiments with
ultrafiltration of Cu/HSA alkylated with N-ethylmaleimide (Cu/HSA–NEM) showed that at Cu/HSA–NEM ratios below 1:1, the
ascorbate radicals were produced by Cu tightly bound to HSA rather than by Cu released in solution. The rate of ascorbate radical
production in HSA–NEM and S-nitrosylated HSA (HSA–NO) was, however, more than one order of magnitude lower than that in
a solution containing equivalent concentration of free copper ions. While Cu/HSA–SH was redox inactive, binding of oleic or li-
noleic acids induced Cu-dependent redox-cycling with maximal activity reached at a fatty acid to protein molar ratio of 3:1 for oleic
acid and 2:1 for linoleic acid. Binding of fatty acids caused profound conformational changes and facilitated oxidation of the Cys-34
SH-group at essentially the same ratios as those that caused redox-cycling activity of Cu/HSA. We conclude that fatty acids regulate
anti-/prooxidant properties of Cu–albumin via controlling redox status of Cys-34.
� 2003 Elsevier Science (USA). All rights reserved.
Keywords: Human serum albumin; Nitrosylation; Fatty acids; Copper; Redox-cycling; Ascorbate radical; Cysteine-34
As a transition metal capable of undergoing one-
electron oxidation–reduction conversions, copper is es-
sential for life and fulfills important catalytic functions
in a number of enzymes such as Cu,Zn–superoxide
dismutase, cytochrome oxidase, and ceruloplasmin. In
these enzymatic reactions, copper tightly binds to pro-
teins such that redox activity of the resulting chelate
formed is strictly regulated. Paradoxically, the same
redox properties of copper can make it extremely dan-
gerous for life: it can catalyze production of free radical
intermediates from molecular oxygen, particularly when
copper is released from or mishandled by proteins.
Disruption of the fine structure of copper-binding
Archives of Biochemistry and Biophysics 413 (2003) 53–66
www.elsevier.com/locate/yabbi
ABB
* Corresponding author. Fax: 1-412-383-2123.
E-mail address: [email protected] (V.E. Kagan).
0003-9861/03/$ - see front matter � 2003 Elsevier Science (USA). All rights reserved.
doi:10.1016/S0003-9861(03)00091-2

domains of mutated proteins enhances the redox activityof copper and results in cell damage and death. Not
surprisingly, delivery of copper to target enzymes is
rigorously controlled in cells and biological fluids. In
plasma, ceruloplasmin and albumin are the two major
proteins (along with transcuprein) responsible for
binding and safe-guarded transport of copper [1] and
prevention of its detrimental redox activity.
The major plasma pool of copper bound to cerulo-plasmin [2] remains apparently inactive with respect to
free radical formation—at least if the stoichiometry of
binding does not exceed 6:1 and/or unless copper is re-
duced by superoxide radicals [3]. The second largest
pool of plasma copper is associated with albumin. Hu-
man serum albumin (HSA)1 contains one high-affinity
site for copper, the N-terminal tripeptide (Cu2þ=Ni2þ
binding motif) Asp-Ala-His [4]. Although under usualcircumstances, less than 1% of total albumin (about
110mg=dl � 1:6lM) contains copper [2,5], HSA still
represents a large pool of bound copper as the human
body contains approximately 400 g of albumin (approx.
6mmol). In some pathologic conditions, e.g., Wilson�sdisease or arthritis, the level of albumin-bound copper
may be significantly increased (two- to fivefold) [6–8].
Human serum albumin contains one reduced cysteineresidue (Cys-34) that, due to the large amount of albu-
min in plasma, constitutes the largest pool of reactive
thiols in plasma [9,10]. The albumin Cys-34 SH-group is
believed to be important in protection against oxidative
stress [11,12], and its concentration has been reported
to decrease under disease conditions and with aging
[13–17].
While both Cu-binding and nucleophilic properties ofCys-34 are important for albumin�s antioxidant func-
tion, specific mechanisms involved in regulation of its
redox activity in human plasma are not fully under-
stood. Recent work has established that chemical
modification of albumin (such as its glycation in
diabetes) yields Cu–albumin molecular species with
elevated redox-cycling activity of Cu, a likely source
of oxidative stress in plasma [18–20]. In addition toposttranslational chemical modifications, albumin may
experience profound conformational changes upon
binding with its natural ligands and different hydro-phobic chemicals and drugs [9,21]. Albumin acts as the
major plasma carrier of nonesterified fatty acids (NE-
FAs), which are accommodated within its two to three
high-affinity binding sites for fatty acids [9]. Notably, as
a result of dyslipidemia typical of several cardiovascular
diseases such as diabetes and preeclampsia, the number
of fatty acid molecules bound to albumin increases [22].
Binding of NEFAs to albumin may be associated withdramatic conformational changes in the albumin mole-
cule that can affect its functional properties [9,23–25].
However, relationships between NEFA binding and
antioxidant properties of albumin (including redox sta-
tus of Cys-34 and redox-cycling activity of Cu-contain-
ing HSA molecular species) have not been quantitatively
characterized.
In the present work, we determined how binding ofNEFAs and/or modification of Cys-34 affect redox-cy-
cling activity of the Cu/HSA complex. Because ascor-
bate is the most abundant natural reductant and
scavenger of free radicals in plasma [26,27], we used
EPR spectroscopy of ascorbate radicals to monitor re-
dox-cycling activity of the Cu/HSA complex. We found
that binding of two to three NEFAs to albumin results
in profound conformational rearrangements that aredetectable by changes in its pI (a redistribution to the 4.8
isoform) and by changes in fluorescence of site-specific
probes. These conformational transitions are accompa-
nied by accelerated oxidation of Cys-34 and increased
redox-cycling activity of bound Cu; consequently,
NEFA-induced rearrangements convert HSA from an
effective antioxidant to a prooxidant, which, in the
presence of Cu, may act as a source of oxidative stress inplasma.
Methods
Reagents
Ascorbic acid (AA), 5,50-dithiobis(2-nitrobenzoicacid) (DTNB); CuSO4 � 5H2O, sodium nitrite (NaNO2),
human serum albumin (essentially fatty acid free), re-
duced glutathione (GSH), dihydrolipoic acid (DHLA),
dithiothreitol (DTT), 2,9-dimethyl-4,7-diphenyl-1,10-
phenanthrolinedisulfonate (bathocuproinedisulfonate,
BCS), ethylenediaminetetraacetic acid (EDTA), diethy-
lenetriaminepentaacetic acid (DTPA), N-ethylmaleimide
(NEM), oleic acid, linoleic acid, ethanol and methanol(HPLC grade), and trichloroacetic acid (TCA) were
obtained from Sigma (St. Louis, MO). Fluorescent
probes, dansyl amide (DNSA) and dansyl sarcosine
(DNSS), were purchased from Sigma. 4,5-Diaminoflu-
orescein (DAF-2) was purchased from Calbiochem (San
Diego, CA). A fluorogenic maleimide-based SH-reagent,
ThioGlo-1, was obtained from Calbiochem (LaJolla,
1 Abbreviations used: AA, ascorbic acid; BCS, 2,9-dimethyl-4,7-
diphenyl-1,10-phenanthrolinedisulfonate (bathocuproinedisulfonate);
Cu/HSA, complex of Cu2þ with HSA; Cu/HSA–NEM, complex of
Cu2þ with HSA–NEM; DAF-2, 4,5-diaminofluorescein; DHLA,
dihydrolipoic acid; DNSA, dansyl amide; DNSS, dansyl sarcosine;
DTNB, 5,50-dithiobis(2-nitrobenzoic acid); DTPA, diethylenetriamine-
pentaacetic acid; DTT, dithiothreitol; EDTA, ethylenediaminetetra-
acetic acid; GSH, reduced glutathione; HSA, human serum albumin;
HSA–NEM, human serum albumin with Cys-34 alkylated with N-
ethylmaleimide; HSA–NO, S-nitrosylated human serum albumin;
HSA–SH, human serum albumin with reduced Cys-34; NEFA,
nonesterified fatty acid; NEM, N-ethylmaleimide; TCA, trichloroacetic
acid; PBS, phosphate-buffered saline; GSNO, S-nitroso-glutathione;
ROS, reactive oxygen species.
54 Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66

CA). Phosphate-buffered saline (PBS; Ca2þ, Mg2þ free)was from Gibco BRL (Grand Island, NY). Chelex-100
resin, analytical grade 100–200 mesh, sodium salt was
from Bio-Rad (Hercules, CA). Cut-off filters were pur-
chased from Millipore Corp. (Bedford, MA). Double
distilled and deionized water was used in all experi-
ments.
Analytical methods
Concentration of copper. In the stock solution this
was quantified using the Cuþ chelator BCS. The BCS–
Cuþ complex exhibits a maximum absorbance at 480 nm
(molar absorbency is 13,500M�1 cm�1) [28]. Absorption
spectra were recorded immediately after addition of 5 llof CuSO4 solution to 1.0ml of PBS containing BCS
(400 lmol/L) and ascorbate (800 lmol/L). Ascorbateand BCS were added in 10- to 12-fold excess of Cu2þ.
Concentration of albumin. This was measured using
absorption at 279 nm. The absorbance at 279 nm for the
concentration of 1mg/ml in a 1-cm cuvette was taken as
E280 ¼ 0:609 [29].
Content of free SH-groups. In reduced HSA (HSA–
SH) and in HSA–NEM this was estimated with Ellman�sreagent, DTNB, using a molar extinction coefficient of13,600M�1 cm�1 at 412 nm [30].
Concentration of NO. This was determined fluoro-
metrically using DAF-2 as a specific probe [31]. To re-
lease NO from S-nitrosothiols, the samples were
irradiated with UV (Oriel UV light source, Model 66002)
using a cutoff filter (Balzers, Pittsburgh, PA) of >330 nm
for 10min (80 lW=cm2). Prior experiments showed that
under these conditions all NO was released from HSA[32]. Fluorescence intensity of DAF-2 was measured
under excitation at 495 nm and emission at 515 nm (slits
1.5 and 5.0 nm, respectively) using a Shimadzu RF-5301
spectrofluorimeter (Kyoto, Japan). A solution of S-nitr-
oso-glutathione (GSNO) was used for calibration of
DAF-2 assay as described in [33]. The concentration of
GSNO was determined photometrically using a molar
extinction coefficient of 900M�1 cm�1 at 336 nm [34].
Fluorescence assay of conformational changes in HSA
(binding sites I and II)
To assess fatty acid-induced conformational changes
of albumin, fluorescent probes DNSA and DNSS were
used as described [35]. DNSA and DNSS are specific
markers for principal albumin binding sites I and II,respectively [35]. Briefly, the fluorescent probes were
dissolved in 1:1 water/methanol mixture. Stock solutions
(10mM) were added to albumin samples (4.2 lM) at a
final concentration of 20 lM. The fluorescence emission
intensity of probes was determined at 483 nm, with ex-
citation at 370 nm (slit 5 nm) using a Shimadzu RF-5301
PC spectrofluorimeter. The data obtained were exported
and processed using Shimadzu RF-5301 PC personalsoftware.
Preparation of HSA derivatives
Several HSA derivatives were used in this study: (1)
HSA with fully reduced Cys-34 (HSA–SH), (2) alkyl-
ated HSA with Cys-34 blocked by N-ethylmaleimide
(HSA–NEM), and (3) HSA with S-nitrosylated Cys-34(HSA–NO). In a series of separate experiments, all
these HSA derivatives were modified with oleic or
linoleic acids.
HSA–SH. Commercial human albumin was 71–77%
oxidized (contained 0.23–0.29 mole of SH-groups/mole
protein). In all our experiments, HSA was, therefore,
reduced with dithiothreitol (1.5- to 2.0-fold excess) for
1.5 h at room temperature in PBS and the protein wasextensively separated (washed) from other solutes with
PBS (pH 7.4) using a 30,000-Da cutoff membrane filter.
After this treatment, approximately 92–98% HSA was
recovered in the reduced form. These results are in good
agreement with previously reported data [36]. This re-
duced HSA was used for further modifications (alkyl-
ation, nitrosylation, binding of Cu).
HSA–NEM. HSA–NEM was prepared by incubationof HSA–SH with N-ethylmaleimide (NEM=HSA–SH ¼5mole=mole), dissolved in PBS (pH 7.4) for 60min at
35 �C in the dark. The excess NEM was eliminated by
ultrafiltration as previously described.
HSA–NO. HSA–NO was prepared using a trans-
nitrosylation reaction with GSNO as a donor [37]. The
GSNO was prepared in a reaction of acidified NaNO2
with GSH. Briefly, 100mM GSH was mixed with100mM sodium nitrite in 20mM HCl at room temper-
ature [37]. The pH was adjusted to 7.4 with NaOH, and
the solution was used for S-trans-nitrosylation of HSA–
SH as rapidly as possible. (The solution was not stored
for more than 30min at 0 �C.) For S-nitrosylation of
HSA–SH, the protein (40mg/ml) was mixed with GSNO
and the solution was incubated for 12–15min at room
temperature. To prevent S-glutathiolation of HSA, thereaction was stopped with NEM, and the samples were
immediately and extensively washed to remove low-
molecular-weight components [37–40]. The resultant
HSA–NEM preparation was checked for the level of
residual HSA–SH and HSA–NO using DTNB and
DAF-2 assays, respectively. In typically used prepara-
tions of HSA–NO, the molar ratio of HSA–NO:HSA–
SH was 96:1.
Formation of complexes of Cu2+ with HSA–NEM (Cu/
HSA–NEM)
An aliquot of freshly prepared CuSO4 solution
in water was added to HSA–NEM and incubated for
45–60min at room temperature in the dark. Because
Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66 55

HSA–SH undergoes rapid oxidation in the presence ofCu, complexes of HSA–SH with Cu were prepared by
preincubation for 40min at 4 �C in the dark (an aliquot
of CuSO4 solution in water was added to freshly pre-
pared HSA–SH). The redox-cycling activity of the
complex was determined after subsequent addition of
ascorbate.
Incorporation of nonesterified fatty acids into HSA
Oleic or linoleic acids were dissolved in methanol–
H2O (1:1, v/v) or pure ethanol, respectively, and incu-
bated with HSA for 2–3min at 25 �C. Solutions of fatty
acids were prepared and used immediately before incu-
bation with HSA. In experiments to determine whether
binding of fatty acids to albumin accelerates copper-
mediated oxidation of HSA–SH, fatty acids (oleic orlinoleic) were dissolved in 1:1 methanol/ethanol mixture.
An appropriate amount of NEFA solution was placed
in an Eppendorf tube, and the solvent was evaporated at
room temperature. Then 100 lM HSA–SH in PBS (pH
7.4) was added and incubated at room temperature for
15min to allow fatty acid binding to albumin; 30 lMCu2þ (as CuSO4, 10mM in ddH2O) was slowly added
and the sample was gently stirred. After 15min of in-cubation with copper, an aliquot was diluted with PBS
(pH 7.4) to 3 lM HSA–SH; then 10 lM fluorescent
probe ThioGlo-1 and 3.3mM SDS were added, and the
kinetics of the fluorescence intensity of the sample were
immediately measured.
Removal of low-molecular-weight components
To remove low-molecular-weight components, sam-
ples were washed five or six times with a four- to fivefold
excess of PBS (pH 7.4) using 30,000-Da cutoff mem-
brane filters and centrifugation at 14,000g for 20–30min
at 4 �C.
Removal of trace metals from solutions
To remove adventitious divalent metals from solu-
tions, water and PBS were treated with Chelex-100 Re-
sin as described in the manufacturer�s instructions. After
decanting the buffer from the resin, the pH was adjusted
to 7.4.
EPR assay of ascorbate radicals (redox-cycling activity)
For these experiments, HSA (100 lM in PBS, pH 7.4)
was mixed with ascorbic acid (18 lM), and EPR signals
of ascorbate radical were scanned during a 30-min time
period. The measurements were performed in gas-per-
meable Teflon tubing (0.8mm internal diameter,
0.013mm thickness) obtained from Alpha Wire Corp.
(Elizabeth, NJ) on a JEOL-RE1X spectrometer at 25 �C.
The Teflon tube (approximately 8 cm in length) was fil-led with 65 ll of the reaction mixture, folded into halves,
and placed into an open EPR quartz tube (inner diam-
eter of 3.0mm) in such a way that the sample was en-
tirely within the microwave radiation area. In a typical
experiment, the spectra of ascorbate radicals were re-
corded under the following conditions: center field
3354G, power 20mW, field modulation 0.79G, sweep
times 10 and 20 s, sweep width 2.5G, receiver gain 4000,time constant 0.1 s. Spectra were collected using EPR-
ware software (Scientific Software Services, Blooming-
ton, IL).
Fresh human plasma
Samples of pooled human plasma were obtained
from Central Blood Bank (Pittsburgh, PA). Sodiumcitrate was used as an anticoagulant (3.15%, final con-
centration).
Isoelectric focusing
This was performed using a Multiphor II electro-
phoresis unit (LKB) connected to a MultiTemp II
thermostatic circulator set at 10 �C. Electrophoresis wasconducted according to the manufacturer�s protocol
using ampholine PAGplates cast on a plastic support
(pH range 4.0–6.5). The anode solution consists of 0.1M
glutamic acid in 0.5M phosphoric acid and the cathode
solution was 0.1M b-alanine. The running conditions
were as recommended: voltage ¼ 2000V, current ¼25mA, time ¼ 2:5 h. Immediately after isoelectric fo-
cusing, the gel was fixed in 10% TCA for 1 h, stained for10min in 0.2% Coomassie brilliant blue dissolved in
25% ethanol/8% acetic acid, and destained in 25% eth-
anol/8% acetic acid. Gels were scanned and the distri-
bution of pI ¼ 5:6 and pI ¼ 4:8 isoforms of albumin
was quantified, assigned arbitrary units, and plotted.
Gels were scanned using a Bio-Rad Fluor-S Multiim-
ager (Richmond, CA). Image capture and subsequent
densitometric analysis of the intensity of bands wasperformed using Bio-Rad Multi-Analyst Software ac-
cording to the manufacturer�s directions.
Results
Redox-cycling activity of Cu/HSA–SH complex is stim-
ulated by Cys-34 oxidation
To determine the extent to which catalytic activity of
Cu/HSA is dependent on the redox status (e.g., oxida-
tion or alkylation) of its Cys-34 residue, we comparedreactions of fatty acid-free HSA with completely re-
duced Cys-34 (HSA–SH) and HSA obtained by a
56 Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66
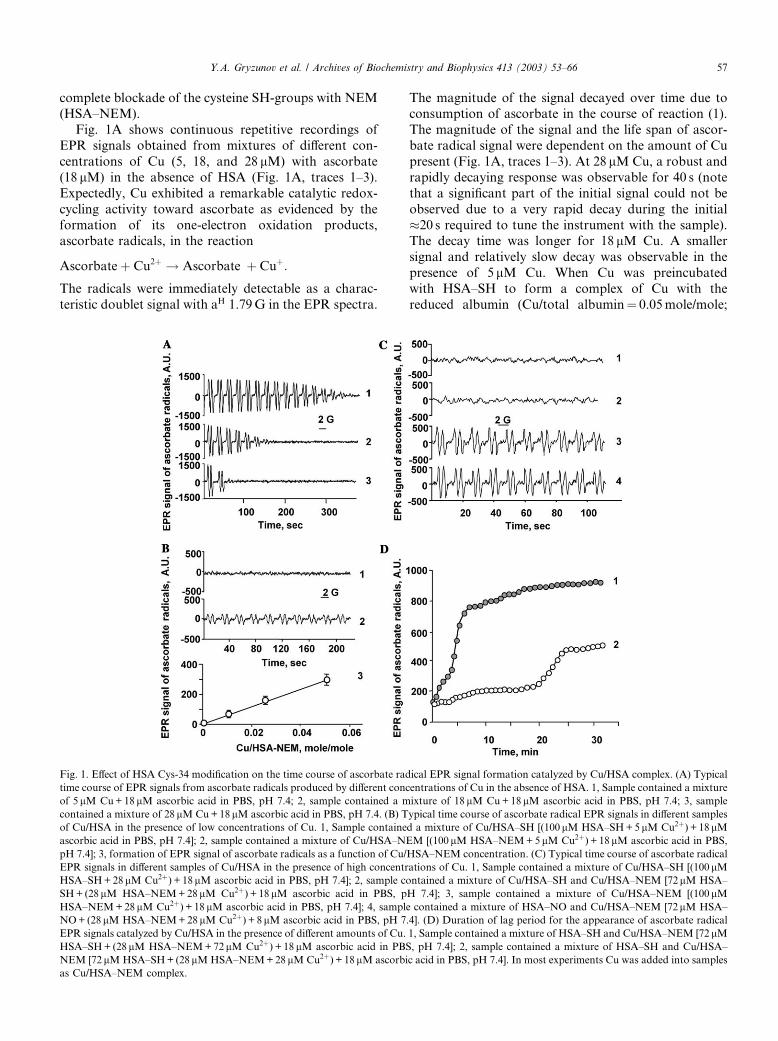
complete blockade of the cysteine SH-groups with NEM(HSA–NEM).
Fig. 1A shows continuous repetitive recordings of
EPR signals obtained from mixtures of different con-
centrations of Cu (5, 18, and 28 lM) with ascorbate
(18 lM) in the absence of HSA (Fig. 1A, traces 1–3).
Expectedly, Cu exhibited a remarkable catalytic redox-
cycling activity toward ascorbate as evidenced by the
formation of its one-electron oxidation products,ascorbate radicals, in the reaction
Ascorbateþ Cu2þ ! Ascorbate� þ Cuþ:
The radicals were immediately detectable as a charac-
teristic doublet signal with aH 1.79G in the EPR spectra.
The magnitude of the signal decayed over time due toconsumption of ascorbate in the course of reaction (1).
The magnitude of the signal and the life span of ascor-
bate radical signal were dependent on the amount of Cu
present (Fig. 1A, traces 1–3). At 28 lM Cu, a robust and
rapidly decaying response was observable for 40 s (note
that a significant part of the initial signal could not be
observed due to a very rapid decay during the initial
�20 s required to tune the instrument with the sample).The decay time was longer for 18 lM Cu. A smaller
signal and relatively slow decay was observable in the
presence of 5 lM Cu. When Cu was preincubated
with HSA–SH to form a complex of Cu with the
reduced albumin (Cu/total albumin¼ 0.05mole/mole;
Fig. 1. Effect of HSA Cys-34 modification on the time course of ascorbate radical EPR signal formation catalyzed by Cu/HSA complex. (A) Typical
time course of EPR signals from ascorbate radicals produced by different concentrations of Cu in the absence of HSA. 1, Sample contained a mixture
of 5 lM Cu+18 lM ascorbic acid in PBS, pH 7.4; 2, sample contained a mixture of 18 lM Cu+18lM ascorbic acid in PBS, pH 7.4; 3, sample
contained a mixture of 28 lM Cu+18lM ascorbic acid in PBS, pH 7.4. (B) Typical time course of ascorbate radical EPR signals in different samples
of Cu/HSA in the presence of low concentrations of Cu. 1, Sample contained a mixture of Cu/HSA–SH [(100 lM HSA–SH+5 lM Cu2þ) + 18 lMascorbic acid in PBS, pH 7.4]; 2, sample contained a mixture of Cu/HSA–NEM [(100 lM HSA–NEM+5 lM Cu2þ) + 18lM ascorbic acid in PBS,
pH 7.4]; 3, formation of EPR signal of ascorbate radicals as a function of Cu/HSA–NEM concentration. (C) Typical time course of ascorbate radical
EPR signals in different samples of Cu/HSA in the presence of high concentrations of Cu. 1, Sample contained a mixture of Cu/HSA–SH [(100lMHSA–SH+28lM Cu2þ) + 18lM ascorbic acid in PBS, pH 7.4]; 2, sample contained a mixture of Cu/HSA–SH and Cu/HSA–NEM [72lM HSA–
SH+ (28lM HSA–NEM+28lM Cu2þ) + 18lM ascorbic acid in PBS, pH 7.4]; 3, sample contained a mixture of Cu/HSA–NEM [(100lMHSA–NEM+28lM Cu2þ) + 18 lM ascorbic acid in PBS, pH 7.4]; 4, sample contained a mixture of HSA–NO and Cu/HSA–NEM [72lM HSA–
NO+ (28lM HSA–NEM+28 lM Cu2þ) + 8lM ascorbic acid in PBS, pH 7.4]. (D) Duration of lag period for the appearance of ascorbate radical
EPR signals catalyzed by Cu/HSA in the presence of different amounts of Cu. 1, Sample contained a mixture of HSA–SH and Cu/HSA–NEM [72 lMHSA–SH+ (28lM HSA–NEM+72 lM Cu2þ) + 18 lM ascorbic acid in PBS, pH 7.4]; 2, sample contained a mixture of HSA–SH and Cu/HSA–
NEM [72 lM HSA–SH+ (28lM HSA–NEM+28 lM Cu2þ) + 18lM ascorbic acid in PBS, pH 7.4]. In most experiments Cu was added into samples
as Cu/HSA–NEM complex.
Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66 57
HSA–SH=Cu ¼ 100lM=5lM), addition of ascorbatedid not reveal any signals of ascorbate radicals within at
least 20–25min of incubation. The same complex of Cu
prepared with alkylated albumin, HSA–NEM, gener-
ated the signal of ascorbate radicals immediately after
the addition of ascorbate (Fig. 1B, trace 2). The mag-
nitude of the EPR signal was significantly lower than
that obtained from Cu in the absence of albumin (Fig.
1A, trace 1); the magnitude of the EPR signal did notsignificantly change over the initial several minutes of
recording. Varying molar ratios of Cu in Cu/HSA–
NEM complex from 1 to 5 mole% caused proportional
changes in the magnitude of the EPR signal of ascorbate
radicals (Fig. 1B). Thus, binding of Cu by albumin de-
creases its redox-cycling activity. Fully reduced albumin,
HSA–SH, was remarkably more effective in quenching
redox-cycling activity of Cu than alkylated albumin(HSA–NEM).
Because Cys-34 of HSA–SH undergoes rapid oxida-
tion in the presence of Cu (see below), we further utilized
mixtures of Cu/HSA–NEM complex with HSA–SH to
study redox-cycling activity of albumin-bound Cu.
Again, HSA–SH coincubated with HSA–NEM complex
(Cu/total albumin¼ 0.28mole/mole; HSA–SH=HSA–
NEM=Cu ¼ 72lM=28lM=28lM) showed virtually noredox-cycling activity (no detectable EPR signals of
ascorbate radicals) during the initial incubation with Cu
and ascorbate and out to �30min (Fig. 1C, trace 2).
Note that the same amount of Cu bound by HSA–SH
(Cu/total albumin¼ 0.28mole/mole; HSA–SH=Cu ¼100lM=28lM) also did not exert any redox-cycling ac-
tivity within the initial several minutes of incubation with
ascorbate (Fig. 1C, trace 1). In contrast, HSA–NEMwas not able to prevent redox-cycling activity in the pres-
ence of Cu/HSA–NEM complex (Cu/total albumin¼0.28mole/mole; HSA–NEM=Cu ¼ 100lM=28lM) as
evidenced by an immediate appearance of typical
doublet signals of ascorbate radicals in the EPR spectra
(Fig. 1C, trace 3).
Overall, the data show that ascorbate radicals do not
appear if Cys-34 of fatty acid-free HSA is in the reducedstate. These results suggest that the SH-group of Cys-34
in albumin is critically involved in regulation of copper
redox-cycling activity.
We further tested different combinations of Cu/
HSA–SH and Cu/HSA–NEM to determine redox-cy-
cling activity of Cu at different ratios (combinations) of
fully reduced and fully alkylated Cu/HSA. We ob-
served a lag period in the appearance of ascorbateradical signal whose duration was dependent on the
amounts of HSA–SH and on Cu content in the com-
plex. Following a lag period, the signal of ascorbate
radical grew and reached a steady state level. A typical
time course of ascorbate radical for a relatively high
copper concentration (Cu/total albumin¼ 0.7mole/mole;
HSA–SH=HSA–NEM=Cu ¼ 72lM=28lM=72lM) is
shown in Fig. 1D (line 1). In this case, the steady statelevel of ascorbate radical was reached after a short lag
period of about 2–3min. At lower levels of Cu in the
Cu/total albumin complex (0.28mole/mole, HSA–SH=HSA–NEM=Cu ¼ 72lM=28lM=28lM), the steady
state levels of ascorbate radicals were achieved after
more prolonged incubations (�20min) (Fig. 1D, line
2). Decreases of the lag period at higher concentrations
of Cu and increases of the lag period at higher HSA–SH/HSA–NEM ratios suggest that Cu-catalyzed oxi-
dation of Cys-34 SH-groups in HSA, occurring during
the course of incubation, facilitate redox-cycling ac-
tivity of Cu/HSA, decreasing the lag period for ascor-
bate radical production.
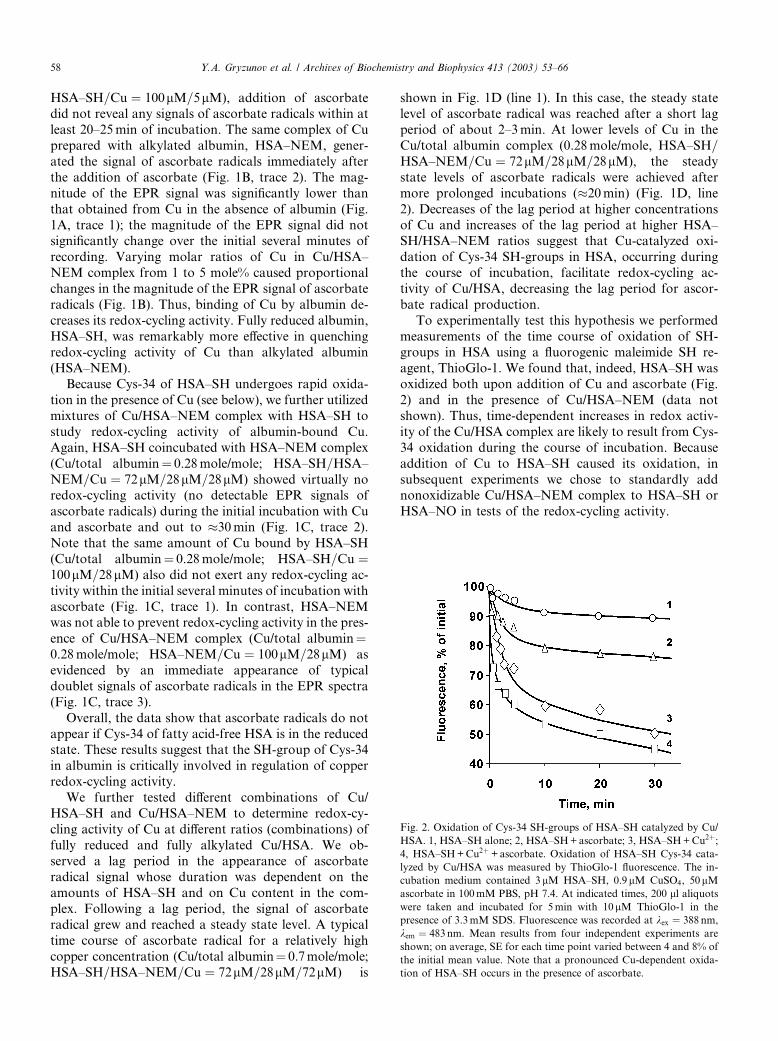
To experimentally test this hypothesis we performed
measurements of the time course of oxidation of SH-
groups in HSA using a fluorogenic maleimide SH re-agent, ThioGlo-1. We found that, indeed, HSA–SH was
oxidized both upon addition of Cu and ascorbate (Fig.
2) and in the presence of Cu/HSA–NEM (data not
shown). Thus, time-dependent increases in redox activ-
ity of the Cu/HSA complex are likely to result from Cys-
34 oxidation during the course of incubation. Because
addition of Cu to HSA–SH caused its oxidation, in
subsequent experiments we chose to standardly addnonoxidizable Cu/HSA–NEM complex to HSA–SH or
HSA–NO in tests of the redox-cycling activity.
Fig. 2. Oxidation of Cys-34 SH-groups of HSA–SH catalyzed by Cu/
HSA. 1, HSA–SH alone; 2, HSA–SH+ascorbate; 3, HSA–SH+Cu2þ;4, HSA–SH+Cu2þ +ascorbate. Oxidation of HSA–SH Cys-34 cata-
lyzed by Cu/HSA was measured by ThioGlo-1 fluorescence. The in-
cubation medium contained 3 lM HSA–SH, 0.9lM CuSO4, 50lMascorbate in 100mM PBS, pH 7.4. At indicated times, 200 ll aliquotswere taken and incubated for 5min with 10lM ThioGlo-1 in the
presence of 3.3mM SDS. Fluorescence was recorded at kex ¼ 388 nm,
kem ¼ 483nm. Mean results from four independent experiments are
shown; on average, SE for each time point varied between 4 and 8% of
the initial mean value. Note that a pronounced Cu-dependent oxida-
tion of HSA–SH occurs in the presence of ascorbate.
58 Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66
Nitrosylation of Cys-34–SH confers redox-cycling activ-
ity to HSA
Since HSA–NO represents the major reservoir of
bound nitric oxide in plasma [34], we further determined
whether S-nitrosylation of Cys-34 in HSA–SH confers
redox-cycling activity to its complex with copper. Upon
addition of ascorbate to HSA–NO and Cu/HSA–NEM
mixture, the ascorbate radical signal appeared immedi-ately without any lag period (Fig. 1C, trace 4); however,
in the mixture of HSA–SH and Cu/HSA–NEM, the
signal could be detected only following a lag period of
�20min (Fig. 1D). Thus, in the presence of HSA–NO,
albumin-bound Cu was catalytically as reactive in re-
dox-cycling of ascorbate as it was in Cu/HSA–NEM
complexes.
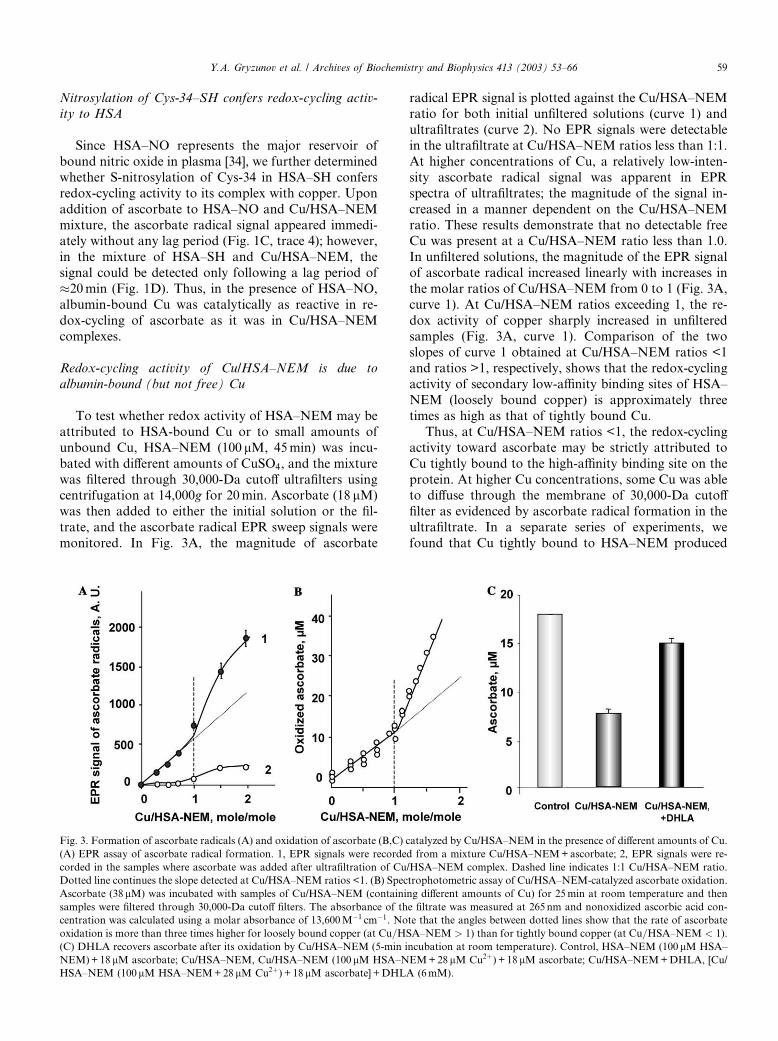
Redox-cycling activity of Cu/HSA–NEM is due to
albumin-bound (but not free) Cu
To test whether redox activity of HSA–NEM may be
attributed to HSA-bound Cu or to small amounts of
unbound Cu, HSA–NEM (100 lM, 45min) was incu-
bated with different amounts of CuSO4, and the mixture
was filtered through 30,000-Da cutoff ultrafilters usingcentrifugation at 14,000g for 20min. Ascorbate (18 lM)
was then added to either the initial solution or the fil-
trate, and the ascorbate radical EPR sweep signals were
monitored. In Fig. 3A, the magnitude of ascorbate
radical EPR signal is plotted against the Cu/HSA–NEMratio for both initial unfiltered solutions (curve 1) and
ultrafiltrates (curve 2). No EPR signals were detectable
in the ultrafiltrate at Cu/HSA–NEM ratios less than 1:1.
At higher concentrations of Cu, a relatively low-inten-
sity ascorbate radical signal was apparent in EPR
spectra of ultrafiltrates; the magnitude of the signal in-
creased in a manner dependent on the Cu/HSA–NEM
ratio. These results demonstrate that no detectable freeCu was present at a Cu/HSA–NEM ratio less than 1.0.
In unfiltered solutions, the magnitude of the EPR signal
of ascorbate radical increased linearly with increases in
the molar ratios of Cu/HSA–NEM from 0 to 1 (Fig. 3A,
curve 1). At Cu/HSA–NEM ratios exceeding 1, the re-
dox activity of copper sharply increased in unfiltered
samples (Fig. 3A, curve 1). Comparison of the two
slopes of curve 1 obtained at Cu/HSA–NEM ratios <1and ratios >1, respectively, shows that the redox-cycling
activity of secondary low-affinity binding sites of HSA–
NEM (loosely bound copper) is approximately three
times as high as that of tightly bound Cu.
Thus, at Cu/HSA–NEM ratios <1, the redox-cycling
activity toward ascorbate may be strictly attributed to
Cu tightly bound to the high-affinity binding site on the
protein. At higher Cu concentrations, some Cu was ableto diffuse through the membrane of 30,000-Da cutoff
filter as evidenced by ascorbate radical formation in the
ultrafiltrate. In a separate series of experiments, we
found that Cu tightly bound to HSA–NEM produced
Fig. 3. Formation of ascorbate radicals (A) and oxidation of ascorbate (B,C) catalyzed by Cu/HSA–NEM in the presence of different amounts of Cu.
(A) EPR assay of ascorbate radical formation. 1, EPR signals were recorded from a mixture Cu/HSA–NEM+ascorbate; 2, EPR signals were re-
corded in the samples where ascorbate was added after ultrafiltration of Cu/HSA–NEM complex. Dashed line indicates 1:1 Cu/HSA–NEM ratio.
Dotted line continues the slope detected at Cu/HSA–NEM ratios <1. (B) Spectrophotometric assay of Cu/HSA–NEM-catalyzed ascorbate oxidation.
Ascorbate (38lM) was incubated with samples of Cu/HSA–NEM (containing different amounts of Cu) for 25min at room temperature and then
samples were filtered through 30,000-Da cutoff filters. The absorbance of the filtrate was measured at 265 nm and nonoxidized ascorbic acid con-
centration was calculated using a molar absorbance of 13,600M�1 cm�1. Note that the angles between dotted lines show that the rate of ascorbate
oxidation is more than three times higher for loosely bound copper (at Cu=HSA–NEM > 1) than for tightly bound copper (at Cu=HSA–NEM < 1).
(C) DHLA recovers ascorbate after its oxidation by Cu/HSA–NEM (5-min incubation at room temperature). Control, HSA–NEM (100 lM HSA–
NEM)+18 lM ascorbate; Cu/HSA–NEM, Cu/HSA–NEM (100lM HSA–NEM+28 lM Cu2þ) + 18 lM ascorbate; Cu/HSA–NEM+DHLA, [Cu/
HSA–NEM (100lM HSA–NEM+28 lM Cu2þ) + 18 lM ascorbate] +DHLA (6mM).
Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66 59
ascorbate radical signals at least one order of magnitudelower than signals from equivalent amounts of free Cu
(Figs. 1A and B).
To further assess redox activities of tightly bound and
loosely bound copper (at Cu/HSA–NEM ratios >1),
spectrophotometric measurements of ascorbate oxida-
tion were performed. Fig. 3B shows that the amounts of
ascorbate oxidized during a 25-min incubation were
linearly dependent on the Cu/HSA–NEM ratio. Theslope of the curve was, however, approximately three
times greater for Cu/HSA–NEM ratios >1 than for Cu/
HSA–NEM ratios <1—in agreement with our EPR data
on enhanced ascorbate radical production by loosely vs
tightly bound Cu in HSA–NEM.
To determine the formation of dehydroascorbate
during Cu/HSA–NEM-catalyzed oxidation of ascor-
bate, we utilized dihydrolipoic acid known to reducedehydroascorbate back to ascorbate [41,42]. We found
that after 5-min incubation of ascorbate with Cu/HSA–
NEM (Cu/total albumin¼ 0.28mole/mole; HSA–NEM=Cu ¼ 100lM=28lM) 58.9% of ascorbate was oxidized
(Fig. 3C). After addition of dihydrolipoate, 72.0% of
oxidized ascorbate was reduced back to ascorbate, such
that 83.3% of reduced ascorbate was recovered. This
suggests that Cu/HSA–NEM-catalyzed oxidation ofascorbate via intermediate formation of ascorbate rad-
icals generates dehydroascorbate as the major reaction
product.
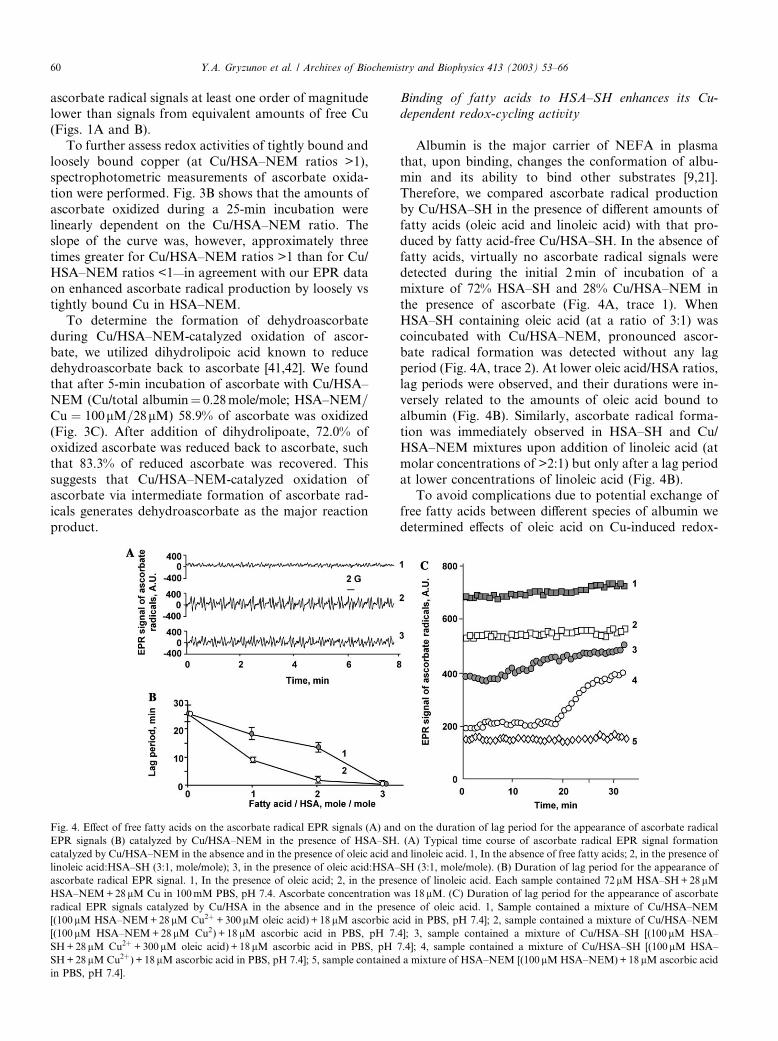
Binding of fatty acids to HSA–SH enhances its Cu-
dependent redox-cycling activity
Albumin is the major carrier of NEFA in plasma
that, upon binding, changes the conformation of albu-
min and its ability to bind other substrates [9,21].
Therefore, we compared ascorbate radical production
by Cu/HSA–SH in the presence of different amounts of
fatty acids (oleic acid and linoleic acid) with that pro-duced by fatty acid-free Cu/HSA–SH. In the absence of
fatty acids, virtually no ascorbate radical signals were
detected during the initial 2min of incubation of a
mixture of 72% HSA–SH and 28% Cu/HSA–NEM in
the presence of ascorbate (Fig. 4A, trace 1). When
HSA–SH containing oleic acid (at a ratio of 3:1) was
coincubated with Cu/HSA–NEM, pronounced ascor-
bate radical formation was detected without any lagperiod (Fig. 4A, trace 2). At lower oleic acid/HSA ratios,
lag periods were observed, and their durations were in-
versely related to the amounts of oleic acid bound to
albumin (Fig. 4B). Similarly, ascorbate radical forma-
tion was immediately observed in HSA–SH and Cu/
HSA–NEM mixtures upon addition of linoleic acid (at
molar concentrations of >2:1) but only after a lag period
at lower concentrations of linoleic acid (Fig. 4B).To avoid complications due to potential exchange of
free fatty acids between different species of albumin we
determined effects of oleic acid on Cu-induced redox-
Fig. 4. Effect of free fatty acids on the ascorbate radical EPR signals (A) and on the duration of lag period for the appearance of ascorbate radical
EPR signals (B) catalyzed by Cu/HSA–NEM in the presence of HSA–SH. (A) Typical time course of ascorbate radical EPR signal formation
catalyzed by Cu/HSA–NEM in the absence and in the presence of oleic acid and linoleic acid. 1, In the absence of free fatty acids; 2, in the presence of
linoleic acid:HSA–SH (3:1, mole/mole); 3, in the presence of oleic acid:HSA–SH (3:1, mole/mole). (B) Duration of lag period for the appearance of
ascorbate radical EPR signal. 1, In the presence of oleic acid; 2, in the presence of linoleic acid. Each sample contained 72lM HSA–SH+28lMHSA–NEM+28lM Cu in 100mM PBS, pH 7.4. Ascorbate concentration was 18lM. (C) Duration of lag period for the appearance of ascorbate
radical EPR signals catalyzed by Cu/HSA in the absence and in the presence of oleic acid. 1, Sample contained a mixture of Cu/HSA–NEM
[(100lM HSA–NEM+28lM Cu2þ +300lM oleic acid) + 18lM ascorbic acid in PBS, pH 7.4]; 2, sample contained a mixture of Cu/HSA–NEM
[(100lM HSA–NEM+28 lM Cu2) + 18 lM ascorbic acid in PBS, pH 7.4]; 3, sample contained a mixture of Cu/HSA–SH [(100lM HSA–
SH+28lM Cu2þ +300lM oleic acid) + 18lM ascorbic acid in PBS, pH 7.4]; 4, sample contained a mixture of Cu/HSA–SH [(100lM HSA–
SH+28lM Cu2þ) + 18lM ascorbic acid in PBS, pH 7.4]; 5, sample contained a mixture of HSA–NEM [(100lM HSA–NEM)+18 lM ascorbic acid
in PBS, pH 7.4].
60 Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66
cycling of ascorbate in simplified systems using HSA–SH alone or HSA–NEM alone. We found that Cu/
HSA–SH (28 lM Cu, 100 lM HSA–SH preincubated at
4 �C for 40min) did not catalyze one-electron oxidation
of ascorbate during the initial 20min as evidenced by the
absence of any detectable signals of ascorbate radicals in
the EPR spectra. After a lag period of approximately
20min, the EPR signals of ascorbate radicals were de-
tectable in the spectra. In contrast, preincubation of theCu/HSA–SH complex with oleic acid (Fig. 4C, trace 3)
resulted in an immediate appearance of typical doublet
signals of ascorbate radicals that slightly increased over
time. This suggests that oleic acid modifies redox-cycling
activity of Cu–albumin. To assess the role of Cys-34 in
this effect, we further determined to what extent this
could be observed in alkylated albumin. We found that
both in the absence and in the presence of oleic acid (at aratio of 3:1 NEFA/HSA), Cu/HSA–NEM complex was
able to generate ascorbate radicals immediately upon
addition of ascorbate without any lag period (Fig. 4C,
traces 1 and 2). Thus, in already modified (alkylated)
albumin (HSA–NEM), oleic acid did not significantly
change the appearance of redox-cycling response.
Comparison of the results indicates that the effect of
oleic acid is likely realized through its ability to modifyCys-34 in albumin.
Binding of fatty acids induces conformational changes of
HSA
Fatty acids are known to cause conformational
changes in serum albumin affecting the properties of
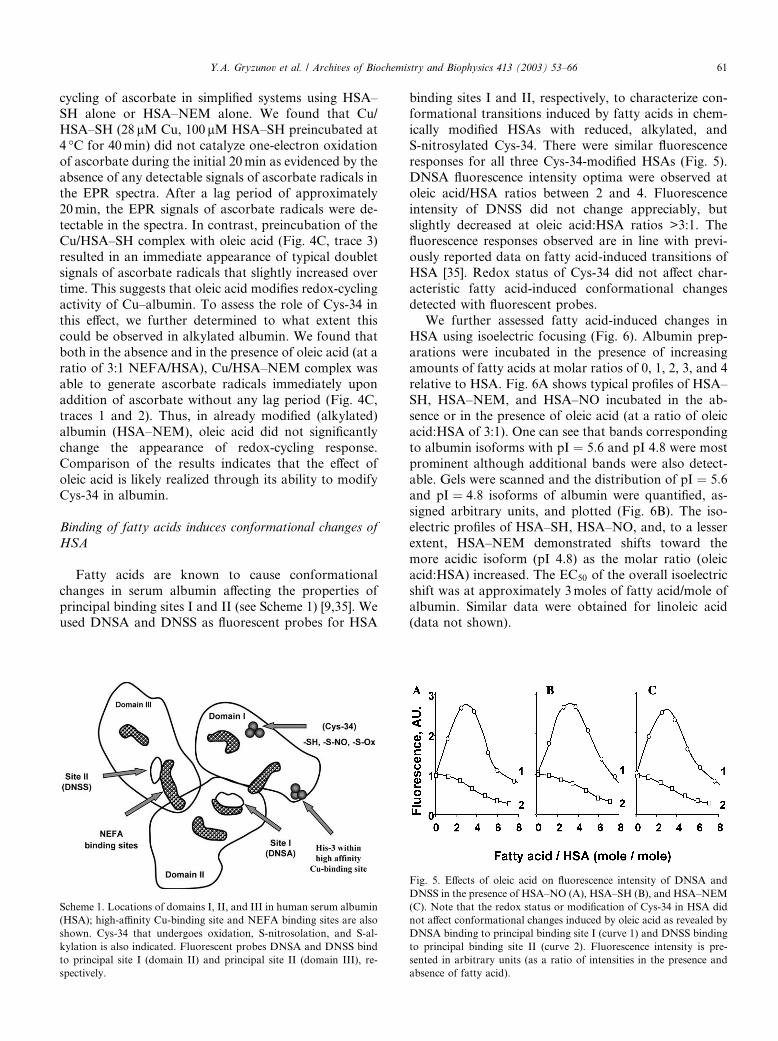
principal binding sites I and II (see Scheme 1) [9,35]. Weused DNSA and DNSS as fluorescent probes for HSA
binding sites I and II, respectively, to characterize con-formational transitions induced by fatty acids in chem-
ically modified HSAs with reduced, alkylated, and
S-nitrosylated Cys-34. There were similar fluorescence
responses for all three Cys-34-modified HSAs (Fig. 5).
DNSA fluorescence intensity optima were observed at
oleic acid/HSA ratios between 2 and 4. Fluorescence
intensity of DNSS did not change appreciably, but
slightly decreased at oleic acid:HSA ratios >3:1. Thefluorescence responses observed are in line with previ-
ously reported data on fatty acid-induced transitions of
HSA [35]. Redox status of Cys-34 did not affect char-
acteristic fatty acid-induced conformational changes
detected with fluorescent probes.
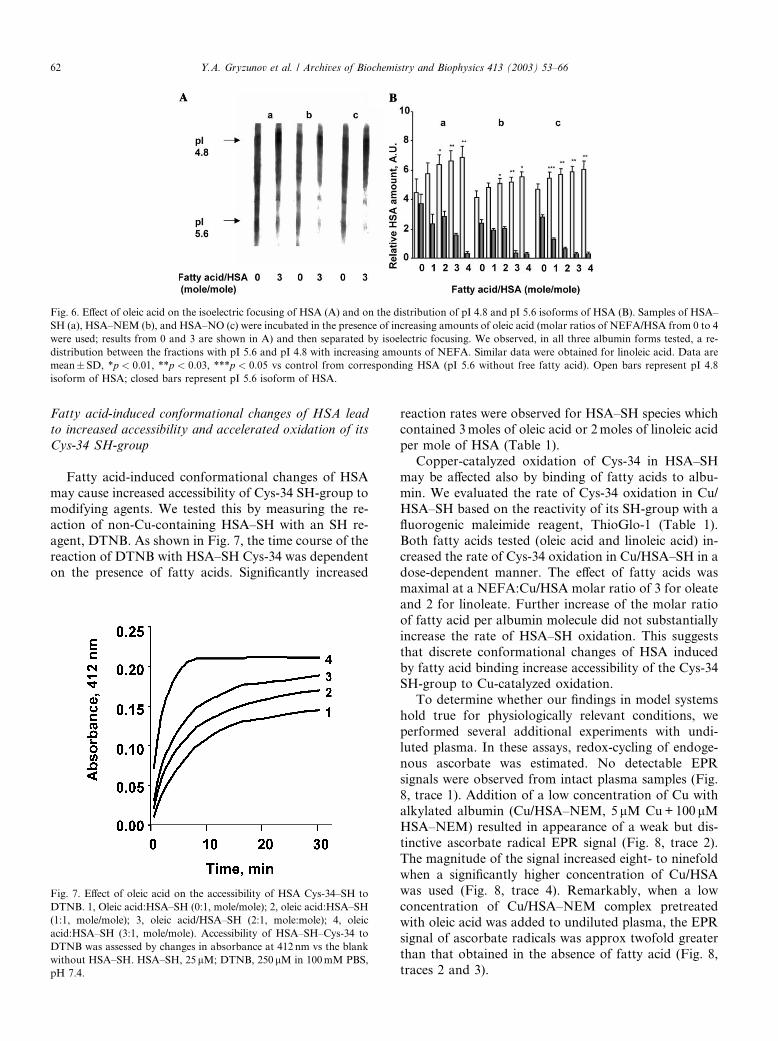
We further assessed fatty acid-induced changes in
HSA using isoelectric focusing (Fig. 6). Albumin prep-
arations were incubated in the presence of increasingamounts of fatty acids at molar ratios of 0, 1, 2, 3, and 4
relative to HSA. Fig. 6A shows typical profiles of HSA–
SH, HSA–NEM, and HSA–NO incubated in the ab-
sence or in the presence of oleic acid (at a ratio of oleic
acid:HSA of 3:1). One can see that bands corresponding
to albumin isoforms with pI ¼ 5:6 and pI 4.8 were most
prominent although additional bands were also detect-
able. Gels were scanned and the distribution of pI ¼ 5:6and pI ¼ 4:8 isoforms of albumin were quantified, as-
signed arbitrary units, and plotted (Fig. 6B). The iso-
electric profiles of HSA–SH, HSA–NO, and, to a lesser
extent, HSA–NEM demonstrated shifts toward the
more acidic isoform (pI 4.8) as the molar ratio (oleic
acid:HSA) increased. The EC50 of the overall isoelectric
shift was at approximately 3moles of fatty acid/mole of
albumin. Similar data were obtained for linoleic acid(data not shown).
Scheme 1. Locations of domains I, II, and III in human serum albumin
(HSA); high-affinity Cu-binding site and NEFA binding sites are also
shown. Cys-34 that undergoes oxidation, S-nitrosolation, and S-al-
kylation is also indicated. Fluorescent probes DNSA and DNSS bind
to principal site I (domain II) and principal site II (domain III), re-
spectively.
Fig. 5. Effects of oleic acid on fluorescence intensity of DNSA and
DNSS in the presence of HSA–NO (A), HSA–SH (B), and HSA–NEM
(C). Note that the redox status or modification of Cys-34 in HSA did
not affect conformational changes induced by oleic acid as revealed by
DNSA binding to principal binding site I (curve 1) and DNSS binding
to principal binding site II (curve 2). Fluorescence intensity is pre-
sented in arbitrary units (as a ratio of intensities in the presence and
absence of fatty acid).
Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66 61
Fatty acid-induced conformational changes of HSA lead
to increased accessibility and accelerated oxidation of its
Cys-34 SH-group
Fatty acid-induced conformational changes of HSA
may cause increased accessibility of Cys-34 SH-group to
modifying agents. We tested this by measuring the re-
action of non-Cu-containing HSA–SH with an SH re-
agent, DTNB. As shown in Fig. 7, the time course of thereaction of DTNB with HSA–SH Cys-34 was dependent
on the presence of fatty acids. Significantly increased
reaction rates were observed for HSA–SH species whichcontained 3moles of oleic acid or 2moles of linoleic acid
per mole of HSA (Table 1).
Copper-catalyzed oxidation of Cys-34 in HSA–SH
may be affected also by binding of fatty acids to albu-
min. We evaluated the rate of Cys-34 oxidation in Cu/
HSA–SH based on the reactivity of its SH-group with a
fluorogenic maleimide reagent, ThioGlo-1 (Table 1).
Both fatty acids tested (oleic acid and linoleic acid) in-creased the rate of Cys-34 oxidation in Cu/HSA–SH in a
dose-dependent manner. The effect of fatty acids was
maximal at a NEFA:Cu/HSA molar ratio of 3 for oleate
and 2 for linoleate. Further increase of the molar ratio
of fatty acid per albumin molecule did not substantially
increase the rate of HSA–SH oxidation. This suggests
that discrete conformational changes of HSA induced
by fatty acid binding increase accessibility of the Cys-34SH-group to Cu-catalyzed oxidation.
To determine whether our findings in model systems
hold true for physiologically relevant conditions, we
performed several additional experiments with undi-
luted plasma. In these assays, redox-cycling of endoge-
nous ascorbate was estimated. No detectable EPR
signals were observed from intact plasma samples (Fig.
8, trace 1). Addition of a low concentration of Cu withalkylated albumin (Cu/HSA–NEM, 5 lM Cu+100 lMHSA–NEM) resulted in appearance of a weak but dis-
tinctive ascorbate radical EPR signal (Fig. 8, trace 2).
The magnitude of the signal increased eight- to ninefold
when a significantly higher concentration of Cu/HSA
was used (Fig. 8, trace 4). Remarkably, when a low
concentration of Cu/HSA–NEM complex pretreated
with oleic acid was added to undiluted plasma, the EPRsignal of ascorbate radicals was approx twofold greater
than that obtained in the absence of fatty acid (Fig. 8,
traces 2 and 3).
Fig. 7. Effect of oleic acid on the accessibility of HSA Cys-34–SH to
DTNB. 1, Oleic acid:HSA–SH (0:1, mole/mole); 2, oleic acid:HSA–SH
(1:1, mole/mole); 3, oleic acid/HSA–SH (2:1, mole:mole); 4, oleic
acid:HSA–SH (3:1, mole/mole). Accessibility of HSA–SH–Cys-34 to
DTNB was assessed by changes in absorbance at 412 nm vs the blank
without HSA–SH. HSA–SH, 25 lM; DTNB, 250lM in 100mM PBS,
pH 7.4.
Fig. 6. Effect of oleic acid on the isoelectric focusing of HSA (A) and on the distribution of pI 4.8 and pI 5.6 isoforms of HSA (B). Samples of HSA–
SH (a), HSA–NEM (b), and HSA–NO (c) were incubated in the presence of increasing amounts of oleic acid (molar ratios of NEFA/HSA from 0 to 4
were used; results from 0 and 3 are shown in A) and then separated by isoelectric focusing. We observed, in all three albumin forms tested, a re-
distribution between the fractions with pI 5.6 and pI 4.8 with increasing amounts of NEFA. Similar data were obtained for linoleic acid. Data are
mean� SD, *p < 0:01, **p < 0:03, ***p < 0:05 vs control from corresponding HSA (pI 5.6 without free fatty acid). Open bars represent pI 4.8
isoform of HSA; closed bars represent pI 5.6 isoform of HSA.
62 Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66

Discussion
Dietary copper is required for normal functioning of
more than 30 mammalian enzyme systems [43] in which
its catalytic function is due to its ability to act as a
transition metal. Unfortunately, the same redox activity
of copper as both a donor and an acceptor of electrons
may cause oxidative stress and damage to mammalian
cells through catalysis of free-radical reactions. Free
copper readily catalyzes oxidation of the two mostabundant reductants in cells and biological fluids—
ascorbate and thiols—to generate their free radical in-
termediates, i.e., to catalyze their redox-cycling. For
ascorbate, the reaction scheme typically includes the
following reactions [44]:
Cu2þ þAscorbate ! Cuþ þAscorbate�; ð1Þ
O2 þ Cuþ ! Cu2þ þ �O�2 ; ð2Þ
2ð�O�2 Þ þ 2Hþ ! H2O2; and ð3Þ
H2O2 þ Cuþ ! Cu2þ þ �OHþOH�: ð4ÞOne-electron oxidation of ascorbate to its radical
triggers subsequent reactions in which ascorbate radicals
disproportionate to form ascorbate and dehydroascor-
bate, a two-electron oxidation product of ascorbate.Thus during Cu/HSA-catalyzed oxidation, there is a
balance between ascorbate, ascorbate radical, and de-
hydroascorbate. Within this balance, the concentration
of ascorbate radical that was produced from ascorbate
reflects the equilibrium between the fully reduced and
the fully oxidized forms of ascorbate. We further found
that addition of dihydrolipoic acid, a reducing agent
capable of two-electron reduction of dehydroascorbate[41,42], recovered most of the reduced ascorbate (83.3%)
after its oxidation by Cu/HSA–NEM. The unrecovered
part of ascorbate was, most likely, converted into sub-
sequent products of dehydroascorbate degradation such
as diketogulonic acid [45].
Redox-cycling of Cu by bioreductants is accompa-
nied by production of deleterious reactive oxygen spe-
cies (ROS). DNA and particularly its telomericsequences is susceptible to Cu-mediated ROS damage
attributed to hydroxyl radicals [46]. Therefore, it is not
surprising that in biological systems copper is normally
tightly bound to physiological chelators. The ‘‘redox-
cycling activity’’ of copper complexes, i.e., their ability
to undergo reduction of Cu2þ to Cuþ, depends, to a
large extent, on two factors: (i) properties/coordination
of copper in its microenvironment and (ii) accessibilityof bound copper to a reducing agent. Both intracellular
redox-catalysis by copper-containing enzymes and de-
livery of copper to target proteins by copper chaper-
ones are strictly regulated to prevent its uncontrolled
redox-cycling activity.
Table 1
Effects of oleic acid and linoleic acid on HSA–SH susceptibility to DTNB
FA/HSA SH susceptibility to DTNBa SH oxidation rate, k � 103b
Oleate Linoleate Oleate Linoleate
0 0:56� 0:04 0:56� 0:04 15:7� 2:0 13:7� 0:7
1 0:68� 0:07 0:72� 0:05 15:1� 0:6 15:1� 0:8
2 0:74� 0:02 0:81� 0:02 23:5� 1:8 23:3� 1:3
3 0:89� 0:03 0:84� 0:03 22:1� 0:4 27:5� 1:6
4 0:87� 0:05 0:86� 0:05 14:8� 1:8 20:2� 1:5
Data are mean�SD min�1 �103.a Susceptibility of HSA–SH was evaluated as A5=Amax, where A5 is the sample absorbance at 412 nm after 5min of incubation of HSA–SH with
DTNB and Amax is the sample absorbance corresponding to HSA–SH concentration as determined by Ellman�s method [28].b Fluorescent reagent ThioGlo-1 was used to measure SH-groups and the first order reaction rate constants k of HSA–SH oxidation were cal-
culated.
Fig. 8. Typical time course of EPR signals of ascorbate radicals gen-
erated from endogenous ascorbate in undiluted plasma samples in the
presence of Cu/HSA–NEM complex with different amounts of Cu in
the absence or in the presence of oleic acid. 1, Sample contained un-
diluted human plasma; 2, sample contained mixture of Cu/HSA–NEM
(100lM HSA–NEM+5lM Cu2þ) + undiluted human plasma; 3,
sample contained mixture of Cu/HSA–NEM/oleic acid (100 lM HSA–
NEM+5lM Cu2þ +300lM oleic acid)+ undiluted human plasma; 4,
sample contained a mixture of Cu/HSA–NEM (100 lM HSA–
NEM+100lM Cu2þ) + undiluted human plasma.
Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66 63

In plasma, specialized plasma proteins such as ceru-loplasmin, transcuprein, and albumin prevent redox-
cycling activity of copper and govern storage and
delivery of copper. In particular, albumin is believed to
play an important role in safeguarding copper in the
circulation. This is because of effective copper-chelating
properties of the peptide motif at the N terminus of
human albumin (see Scheme 1). In fact, Asp-Ala-His-
Lys, a specific copper-chelating tetrapeptide (d-analogof the N-terminus of human albumin), was shown to
attenuate DNA strand breaks in a dose-dependent
manner [46]. This important protective role of albumin
is determined not only by its ability to safely sequester
copper but also by its interactions with other copper-
binding proteins. Notably, albumin can limit redox-cy-
cling activity of a single copper atom at the surface of
ceruloplasmin, near His-426, which may cause oxidationof lipoproteins in the presence of superoxide [47]. Thus
albumin normally protects against redox-cycling activity
of copper and fulfills an important antioxidant function.
The antioxidant role of albumin in plasma is fortified by
its Cys-34 residue, which can directly participate in
radical scavenging [48,49]. Obviously, modifications of
Cys-34 resulting in loss of its electron-donating SH-
group preclude its direct radical-scavenging antioxidanteffects. In fact, it has been shown that the level of al-
bumin�s SH-groups may change during the course of
several diseases and in aging [13–17]. Several factors
may affect SH-groups in albumin, including free radi-
cals, glutathione, and nitric oxide [34,50]. For example,
enhanced S-nitrosylation of HSA–SH was demonstrated
in plasma from women with preeclampsia as compared
with plasma from women with normal pregnancies [32];elevated levels of albumin-bound fatty acids also are
noted in this condition [51].
Recent studies demonstrated that copper bound to
albumin can mediate the conversion of homocysteine to
its low-molecular-weight disulfide forms (homocystine
and homocysteine–cysteine mixed disulfide) by thiol/di-
sulfide exchange reactions [36]. Notably, albumin me-
diates the formation of disulfide forms of homocysteineby thiol/disulfide exchange, whereas ceruloplasmin
converts cysteine to cystine by copper-dependent au-
tooxidation. Moreover, Cys-34 of HSA–SH can be a
target of the disulfide exchange reactions itself resulting
in the formation of thiolated albumin. This modification
renders albumin–Cu complex highly redox-active, be-
cause removal of disulfide bonded to HSA–Cys-34 thiols
(cysteine and homocysteine) by treatment with dith-iothreitol to form albumin–Cys-34–SH (mercaptalbu-
min) completely blocks the conversion of homocysteine
to its disulfide forms. Since significant amounts of thi-
olated HSA may be present in plasma [61,62] this
modified albumin may function as a potent redox-cy-
cling source contributing to depletion of ascorbate and
induction of oxidative stress in plasma similarly to
S-nitrosylated or alkylated forms of albumin reportedhere.
Modifications of other sites in the HSA molecule may
be further associated with significant changes in its ability
to bind and handle copper. Glycation of albumin has
been shown to impair its ability to regulate redox-cycling
activity of bound Cu [19,20], resulting in cytotoxic effects
on cultured smooth muscle cells due to ROS production
in its presence [52]. This elevated redox-cycling activity ofCu/HSA may contribute to enhanced oxidative stress in
plasma typical of diabetes [18]. In preeclampsia, in-
creased binding of NEFA dramatically changes the
properties of albumin such that its pI is shifted from 5.6
to a more acidic 4.8 conformation [51]. The consequences
of these posttranslational modifications for regulation of
Cu handling by albumin has not been well understood.
Curiously, the two- to threefold molar excess of NEFA inthis clinical setting appears to correspond to the en-
hanced oxidation and redox-cycling of Cys-34.
In the present study, we demonstrated that chemical
modifications of Cys-34 by its alkylation, S-nitrosyla-
tion, or oxidation result in a dramatic enhancement of
the redox-cycling activity of the Cu/HSA complex as
evidenced by one-electron oxidation of ascorbate to
acsorbate radical. We found that HSA–SH and HSAwith chemically modified SH groups are distinct entities
with dramatically different copper redox-cycling activi-
ties. Cu complexes of modified albumins are potent
catalysts of oxidation of ascorbate to its radical. Cu
complexes of HSA–SH, however, do not support one-
electron oxidation of ascorbate until the SH-group is
oxidized during the incubation with ascorbate. We fur-
ther established that the elevated redox-cycling activityof HSA with modified Cys-34 is not due to release of Cu
from albumin but rather is associated with redox-cycling
activity of HSA-bound copper. In line with our findings
on dramatically enhanced redox-cycling activity of Cu in
albumin with modified Cys-34, the Cu binding affinity of
the N-terminal protein site has been recently shown to
be quantitatively higher in HSA–SH than in HSA–NEM
or in albumin with oxidized Cys-34 [53].Thus, the presence of an intact Cys-34 SH-group in
albumin is a prerequisite for its role as an efficient regu-
lator of redox-cycling activity of bound copper. Sup-
pression of the redox-cycling activity of albumin-bound
copper by Cys-34 indicates its potential involvement in
an electron transfer process likely to be accompanied by
the formation of immobilized thiyl radical. In fact, im-
mobilized adducts of thiyl radicals of albumin with a spintrap, 5,5-dimethyl-1-pyrroline-N-oxide, have been de-
tected in the course of Cys-34 oxidation induced by
peroxynitrite [54] or UV light [55]. In this regard, it is
important to elucidate potential mechanisms of reduc-
tion (recycling) of albumin Cys-34 in human plasma.
Fatty acids are the main structural and energy sour-
ces of the human body. Within the organism, they are
64 Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66

presented to cells as fatty acid:albumin complexes [56].In our experiments with oleic and linoleic acids, we
showed that fatty acids are effective modulators of al-
bumin�s redox-cycling activity. This effect is apparent at
fatty acid:albumin ratios exceeding 2:1 for linoleic acid
and 3:1 for oleic acid. In keeping with this, we found
that maximal conformational changes in HSA (revealed
by fluorescence spectroscopy with site-specific probes
and by isoelectric focusing) and maximal changes in therate of Cys-34 oxidation during incubation of Cu/HSA–
SH complex were achieved at approximately the same
molar ratios of fatty acid:albumin. Furthermore, fatty
acid-induced conformational changes in HSA are re-
versible [9]. This implies that decreases of plasma levels
of fatty acids due to their increased uptake and utiliza-
tion in tissues (skeletal muscle, heart) can, potentially,
restore regulation of Cu/HSA redox-cycling activity.Previous work has established that a high-affinity
binding site for NEFA is located in domain III of al-
bumin; the binding capacity of this site is one to two
fatty acid molecules. Subdomain IIB is believed to
harbor the second binding site [9,24,57,58]. The third
binding site for fatty acids with lower affinity than the
other two is likely located in the center of domain I
[24,25], near the active site for copper binding and inproximity to Cys-34 (see Scheme 1). It has been shown
that binding of fatty acids can affect the accessibility of
fluorescent labels covalently bound to Cys-34 [59].
However, there was no prior evidence that this confor-
mational change can affect redox-cycling activity of Cu/
HSA complex. Our results indicate that binding oleic or
linoleic acids to HSA–SH (the most abundant form of
HSA in blood and in tissues [10,17]) dramatically affectsthe susceptibility of its Cys-34 to oxidation.
We found that fatty acid-induced conformational
changes in the Cu/HSA–NEM complex did not affect its
redox-cycling activity. Our experiments with DTNB and
ThioGlo-1, however, showed that NEFA-induced con-
formational changes in HSA–SH increased accessibility
of the albumin SH-group to the SH reagent (DTNB).
This is in agreement with previous experiments whereinbinding of fatty acids by albumin was shown to affect
the thiol accessibility for acrylodan, which covalently
binds to Cys-34 to form a fluorescent complex [59].
Binding of oleic acid to HSA led to a red shift of
acrylodan fluorescence that was interpreted as an in-
creased accessibility of the label to water. Also, other
previous work [60] showed that binding of oleic acid to
HSA increased the accessibility of albumin SH-group toDTNB.
Importantly, similar relationships between the redox
status of Cys-34 in albumin and its redox-cycling ac-
tivity and the effects of fatty acids are inherent to plas-
ma. This was demonstrated in our experiments in which
addition of increasing concentrations of a complex of
alkylated albumin with Cu to undiluted plasma pro-
portionally induced redox-cycling activity detectable byEPR of ascorbate radicals. Moreover, the redox-cycling
activity of Cu/HSA–NEM complex in plasma was sig-
nificantly enhanced by oleic acid.
In summary, we have established that binding of fatty
acids causes conformational changes, facilitates oxida-
tion of Cys-34 SH-groups, and induces redox-cycling
activity of Cu/HSA–SH, i.e., converts HSA from a
radical scavenger and an antioxidant to a Cu-redox-cycling prooxidant. Obviously, only Cu-containing HSA
species that simultaneously harbor either a modified SH-
group and/or are loaded with two to three fatty acids
may be involved in Cu-dependent redox-cycling activity.
One may assume that the concentrations of such Cu/
HSA molecular species in plasma are relatively low.
Given that approximately 1% of HSA in plasma nor-
mally contains Cu [2,5] and only about 0.2–0.5% ofHSA may be S-nitrosylated [32], one can estimate that
only a relatively small fraction of HSA (�0.1%) may
participate in redox-cycling reactions. However, the
high concentration of HSA (�600 lM) and perpetual
catalytic nature of the redox-cycling process can make
these relatively ‘‘rare’’ molecular species of HSA very
powerful sources of oxidative stress. Moreover, our re-
sults suggest that not only S-nitrosylated albumin butalso other HSA molecular species with modified Cys-34
can reveal redox-cycling activity. The fraction of such
modified HSA (e.g., S-glutathionylated HSA) may be
markedly higher (up to 25%) and may further increase in
disease and during aging [17]. Consequently, the fraction
of copper–albumin complexes involved in redox-cycling
activity can exceed 1.0% of total HSA.
Finally, our data clearly indicate that reducing an-tioxidants such as ascorbate cannot be effectively used to
prevent or ameliorate oxidative stress induced by Cu/
HSA. Our results demonstrate that the continuous re-
dox-cycling process depletes any exogenously added
ascorbate, but does not remove the source of redox-cy-
cling. This observation may be of interest for thera-
peutic trials of antioxidant vitamins in clinical
syndromes associated with oxidative stress (e.g., pre-eclampsia) [63]. Clearly, use of Cu chelators may rep-
resent a promising approach to protect against Cu/
HSA-induced oxidative stress. The specific Cu-chelating
tetrapeptide d-analog of the N terminus of human al-
bumin Asp-Ala-His-Lys may be an excellent candidate
for this role as it binds Cu with high affinity, inhibits its
redox-cycling activity [46], and is not sensitive to fatty
acids and/or Cys-34 modifiers that confer redox-cyclingactivity to HSA–SH.
Acknowledgment
This study was supported by NIH 1RO1HL64145-
01A1.
Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66 65

References
[1] M.C. Linder, L. Wooten, P. Cerveza, S. Cotton, R. Shulze,
N. Lomeli, Am. J. Clin. Nutr. 67 (1998) 965S–971S.
[2] K. Inagaki, N. Mikuriya, S. Morita, H. Haraguchi, Y. Nakahara,
M. Hattori, T. Kinosita, H. Saito, Analyst 125 (2000) 197–203.
[3] C.K. Mukhopadhyay, P.L. Fox, Biochemistry 37 (1998)
14222–14229.
[4] J.P. Laussac, B. Sarkar, Biochemistry 23 (1984) 2832–2838.
[5] C. Harford, B. Sarkar, Acc. Chem. Res. 30 (1997) 123–130.
[6] G.W. Rafter, Med. Hypotheses 22 (1987) 245–249.
[7] M. Dastych, Vnitr. Lek. 45 (1999) 217–219.
[8] K.T. Suzuki, Y. Shiobara, A. Tachibana, Y. Ogra, K. Matsum-
oto, Res. Commun. Mol. Pathol. Pharmacol. 103 (1999) 189–194.
[9] D.C. Carter, J.X. Ho, Adv. Protein Chem. 45 (1994) 153–203.
[10] E. Schauenstein, F. Dachs, Z. Naturforsch. [C] 33 (1978) 803.
[11] B. Halliwell, J.M. Gutteridge, Arch. Biochem. Biophys. 280 (1990)
1–8.
[12] M.L. Hu, S. Louie, C.E. Cross, P. Motchnik, B. Halliwell, J. Lab.
Clin. Med. 121 (1993) 257–262.
[13] K. Kumano, S. Yokota, M. Go, K. Suyama, T. Sakai, S. Era,
M. Sogami, Adv. Perit. Dial. 8 (1992) 127–130.
[14] E. Suzuki, K. Yasuda, N. Takeda, S. Sakata, S. Era, K. Kuwata,
M. Sogami, K. Miura, Diabetes Res. Clin. Pract. 18 (1992)
153–158.
[15] M. Sogami, S. Era, S. Nagaoka, K. Kuwata, K. Kida, J. Shigemi,
K. Miura, E. Suzuki, Y. Muto, E. Tomita, et al., J. Chromatogr.
332 (1985) 19–27.
[16] A. Hayakawa, K. Kuwata, S. Era, M. Sogami, H. Shimonaka,
M. Yamamoto, S. Dohi, H. Hirose, J. Chromatogr. B Biomed.
Sci. Appl. 698 (1997) 27–33.
[17] S. Era, K. Kuwata, H. Imai, K. Nakamura, H. Tomoya,
M. Sogami, Biochim. Biophys. Acta 1247 (1995) 12–16.
[18] M. Qian, M. Liu, J.W. Eaton, Biochem. Biophys. Res. Commun.
250 (1998) 385–389.
[19] E. Bourdon, N. Loreau, D. Blache, FASEB J. 13 (2) (1999) 233–
244.
[20] J.W. Eaton, M. Qian, Mol. Cell. Biochem. 234–235 (1–2) (2002)
135–142.
[21] T.J. Peters, All About Albumin: Biochemistry, Genetics and
Medical Applications, Academic Press, San Diego, 1996.
[22] A.T. Hostmark, Med. Hypotheses 44 (1995) 539–541.
[23] I. Petitpas, T. Grune, A.A. Bhattacharya, S. Curry, J. Mol. Biol.
314 (2001) 955–960.
[24] A.A. Bhattacharya, T. Grune, S. Curry, J. Mol. Biol. 303 (2000)
721–732.
[25] S. Curry, P. Brick, N.P. Franks, Biochim. Biophys. Acta 1441
(1999) 131–140.
[26] B. Frei, L. England, B.N. Ames, Proc. Natl. Acad. Sci. USA 86
(1989) 6377–6381.
[27] B. Frei, in: B. Frei (Ed.), Free Radicals in Biology: Sources,
Reactivities, and Roles in the Etiology of Human Diseases,
Academic Press, New York, 1994.
[28] S.M. Lynch, B. Frei, J. Biol. Chem. 270 (1995) 5158–5163.
[29] G.D. Fasman, Practical Handbook of Biochemistry and Molec-
ular Biology, CRC Press Inc, Boston, 1990.
[30] G.L. Ellman, Arch. Biochem. Biophys. 82 (1959) 70.
[31] H. Kojima, N. Nakatsubo, K. Kikuchi, S. Kawahara, Y. Kirino,
H. Nagoshi, Y. Hirata, T. Nagano, Anal. Chem. 70 (1998) 2446–
2453.
[32] V.A. Tyurin, S.X. Liu, Y.Y. Tyurina, N.B. Sussman, C.A. Hubel,
J.M. Roberts, R.N. Taylor, V.E. Kagan, Circ. Res. 88 (2001)
1210–1215.
[33] V.A. Tyurin, Y.Y. Tyurina, S.X. Liu, H. Bayir, C.A. Hubel, V.E.
Kagan, Methods Enzymol. 352 (2002) 347–360.
[34] J.S. Stamler, O. Jaraki, J. Osborne, D.I. Simon, J. Keaney, J. Vita,
D. Singel, C.R. Valeri, J. Loscalzo, Proc. Natl. Acad. Sci. USA 89
(1992) 7674–7677.
[35] D.J. Birkett, S.P. Myers, G. Sudlow, Mol. Pharmacol. 13 (1977)
987–992.
[36] S. Sengupta, C. Wehbe, A.K. Majors, M.E. Ketterer, P.M.
DiBello, D.W. Jacobsen, J. Biol. Chem. 276 (50) (2001)
46896–46904.
[37] Y. Ji, T.P.M. Akeboom, H. Sies, J.A. Thomas, Arch. Biochem.
Biophys. 362 (1999) 67–78.
[38] J.W. Park, Biophys. Biochem. Res. Commun. 152 (1988) 916–920.
[39] E. Konorev, B. Kalyanaraman, H. Hogg, Free Radicals Biol.
Med. 28 (2000) 1671–1678.
[40] N. Hogg, Anal. Biochem. 272 (1999) 257–262.
[41] V.E.Kagan,A.A. Shvedova,E. Serbinova, S.Khan,C. Swansen,R.
Powell, L. Packer, Biochem. Pharmacol. 44 (8) (1991) 1637–1649.
[42] D.P. Xu, W.W. Wells, J. Bioenerg. Biomembr. 28 (1) (1996)
77–85.
[43] A.B. Lentsch, A. Kato, J.T. Saari, D.A. Schuschke, Am.
J. Physiol. Lung Cell. Mol. Physiol. 281 (2001) L387–L393.
[44] B. Halliwell, J.M. Gutteridge, Methods Enzymol. 186 (1990) 1–85.
[45] I. Koshiishi, Y. Mamura, T. Imaneri, Biochim. Biophys. Acta 379
(1998) 257–263.
[46] D. Bar-Or, G.W. Thomas, L.T. Rael, E.P. Lau, J.V. Winkler,
Biochem. Biophys. Res. Commun. 282 (2001) 356–360.
[47] P.L. Fox, B. Mazumder, E. Ehrenwald, C.K. Mukhopadhyay,
Free Radicals Biol. Med. 28 (2000) 1735–1744.
[48] H. Kondo, M. Takahashi, E. Niki, FEBS Lett. 413 (1997)
236–238.
[49] M. Soriani, D. Pietraforte, M. Minetti, Arch. Biochem. Biophys.
312 (1994) 180–188.
[50] D. Jourd�heuil, K. Hallen, M. Feelisch, M.B. Grisham, Free
Radicals Biol. Med. 28 (2000) 409–417.
[51] J.L. Vigne, J.T. Murai, B.W. Arbogast, W. Jia, S.J. Fisher, R.N.
Taylor, J. Clin. Endocrinol. Metab. 82 (1997) 3786–3792.
[52] N. Sakata, K. Miyamoto, J. Meng, Y. Tachikawa, Y. Imanaga,
S. Takebayashi, T. Furukawa, Atherosclerosis 136 (1998)
263–274.
[53] Y. Zhang, D.E. Wilcox, J. Biol. Inorg. Chem. 7 (2002) 327–337.
[54] J. Vasquez-Vivar, A.M. Santos, V.B.C. Junqurira, O. Augusto,
Biochem. J. 314 (1996) 869–876.
[55] J.A. Silvester, G.S. Timmins, M.J. Davies, Free Radicals Biol.
Med. 24 (5) (1998) 754–766.
[56] W. Stremmel, L. Pohl, A. Ring, T. Herrmann, Lipids 36 (2001)
981–989.
[57] H. Aki, M. Yamamoto, J. Pharm. Sci. 83 (1994) 1712–1716.
[58] C.B. Berde, B.S. Hudson, R.D. Simoni, L.A. Sklar, J. Biol. Chem.
254 (1979) 391–400.
[59] R. Narazaki, T. Maruyama, M. Otagiri, Biochim. Biophys. Acta
1338 (1997) 275–281.
[60] R. Narazaki, T. Maruyama, M. Otagiri, Biol. Pharm. Bull. 20
(1997) 452–454.
[61] A.K. Majors, S. Sengupta, B. Willard, M.T. Kinter, R.E. Pyeritz,
D.W. Jacobsen, Arterioscler. Thromb. Vasc. Biol. 22 (8) (2002)
1354–1359.
[62] S. Sengupta, H. Chen, T. Togawa, P.M. DiBello, A.K. Majors, B.
Budy, M.E. Ketterer, D.W. Jacobsen, J. Biol. Chem. 276 (32)
(2001) 30111–30117.
[63] L.C. Chappell, P.T. Seed, A.L. Briely, F.J. Kelly, R. Lee, B.J.
Hunt, K. Parmar, S.J. Bewley, A.H. Shennan, P.J. Steer, Lancet
354 (1999) 810–816.
66 Y.A. Gryzunov et al. / Archives of Biochemistry and Biophysics 413 (2003) 53–66
Top Related
Copyright © 2022 FDOKUMEN

