







Bahasa
Halaman
Hukum


SYLVIE DESJARDINS
ANALYSE DE GENES CANDIDATS AU CANCER DU SEIN IMPLIQUÉS DANS LES INTERACTIONS AVEC
BRCA1 ET BRCA2.
Thèse présentée à la Faculté des études supérieures de l'Université Laval
dans le cadre du programme de doctorat en Physiologie-Endocrinologie pour l'obtention du grade de Philosophiae Doctor (Ph.D.)
DEPARTEMENT DE MEDECINE MOLECULAIRE FACULTÉ DE MÉDECINE
UNIVERSITÉ LAVAL QUÉBEC
2010
© Sylvie Desjardins, 2010

RESUME La susceptibilité d'un individu à développer un cancer du sein est le résultat d'une
interaction complexe de facteurs reliés au style de vie, à l'histoire reproductive et aux
déterminants génétiques propres à chaque individu. Jusqu'à présent, un nombre limité de
gènes ont été impliqués dans une telle susceptibilité. Il est présentement estimé que des
mutations et variations de l'ensemble des gènes de susceptibilité connus (par exemple
BRCA1, BRCA2, TP53, STK11, PTEN, ATM, PALB2, CHEK2, BR1P1 et les alleles de
faible penetrance identifiés récemment) ne seraient responsables que d'un maximum de
30% des cas de cancers clairement familiaux. Dans cette thèse, la contribution de certains
gènes a été investiguée dans une cohorte de femmes provenant de la population canadienne
française et présentant des évidences claires de l'implication forte de facteurs génétiques de
susceptibilité non reliés à BRCA1 ou BRCA2. Notre étude concerne les gènes candidats
ZBRK1 (Zinc finger and BRCA1-interacting protein with KRAB domain 1), GADD45A
(Growth arrest and DNA-damage-inducible alpha) et NBS1 (Nijmegen breakage syndrome
1). Notre analyse de ZNF350/ZBRK1 a permis de mettre en évidence trois haplotypes
modulant de façon significative le risque de cancer du sein dans notre population. Parmi
ceux-ci, deux pourraient être associés à un effet protecteur (p=0.01135 et p=0.00268) alors
qu'un autre haplotype est lié à une augmentation du risque (p=0.00143). Dans le cas de
GADD45A, nous avons identifié un haplotype commun démontrant une fréquence plus
élevée dans le groupe contrôle, bien que cette association soit faible. En ce qui concerne
NBN, le variant du promoteur c.-242-l lOdelAGTA est significativement surreprésenté dans
notre groupe de femmes atteintes. Des essais de type gène luciférase rapporteur n'ont pas
démontré de variation d'expression dans la lignée cellulaire de cancer du sein MCF-7, mais
ont indiqué une réduction de l'expression dans les lignées cellulaires HEK293 et LNCaP.
Ces résultats indiquent qu'il est possible que des variations de ces gènes, bien que n'étant
pas très fréquentes, soient impliquées dans la susceptibilité au cancer du sein dans notre
population et des études plus approfondies sur de grandes cohortes seront nécessaires afin
de confirmer ou infirmer nos résultats.

11
ABSTRACT An individual's susceptibility to develop breast cancer is the result of a complex interaction
between lifestyle factors, reproductive history and genetic determinants unique to each
individual. To date, a limited number of genes have been implicated in such susceptibility.
It is estimated that mutations and variations in all known susceptibility genes (for example
BRCA1, BRCA2, TP53, STKll, PTEN, ATM, PALB2, CHEK2, BR1P1 and low penetrance
alleles recently discovered) account for less than 30% of clearly familial cancer cases. In
this thesis, the contribution of certain genes has been investigated in a cohort of women
from the French Canadian population and presenting clear evidence of strong non-BRCA 1
or -BRCA2 genetic factors. The focus of my thesis is on candidate genes such as ZBRK1
(Zinc finger and BRCA1-interacting protein with KRAB domain 1), GADD45A (Growth
arrest and DNA-damage-inducible alpha) and NBS1 (Nijmegen breakage syndrome 1). Our
analysis of ZNF350/ZBRK1 allowed the identification of three haplotypes modulating
breast cancer risk in a significant manner in our population. Among those, two could be
associated with a protective effect (p=0.01135 et p=0.00268) while another haplotype is
linked to an increased risk (p=0.00143). In the case of GADD45A, we identified a common
haplotype displaying an increased frequency in the control group, although this association
is weak. Regarding NBN, the promoter variant c.-242-110delAGTA is significantly over-
represented in our group of affected women. Luciférase reporter gene assays failed to
indicate a variation in promoter activity in the breast cancer cell line MCF-7, although a
reduced activity was observed in both the HEK293 and LNCaP cell lines. These results
indicate that variations in those genes may be implicated in breast cancer susceptibility in
our population, albeit at low frequency. Further studies in larger cohorts are warranted to
confirm or infirm our results.

AVANT-PROPOS Le cancer, en particulier le cancer du sein chez la femme, est l'une des causes principales
de maladie dans les pays occidentaux, occasionnant un fardeau émotionnel et financier
important pour les individus, familles et sociétés. La compréhension des facteurs pouvant
affecter l'initiation et la progression du cancer, par exemple les facteurs génétiques pouvant
être associés à une prédisposition à développer un cancer, est primordiale. Ultimement, ces
connaissances devraient permettre de cibler la population à risque et de fournir des soins de
santé adaptés. Cependant, bien du chemin reste à faire puisque les alleles de risque connus
actuellement ne peuvent expliquer qu'une minorité des cas de cancer du sein d'origine
clairement héréditaire.
L'objet de la présente thèse est donc d'évaluer la contribution à la susceptibilité au cancer
du sein de gènes candidats sélectionnés sur la base de leur interaction avec les deux gènes
majeurs de susceptibilité connus, soit BRCA1 et BRCA2. Il s'agit d'une thèse avec insertion
d'articles, qui sont rédigés en langue anglaise afin de se conformer aux exigences des
revues dans lesquelles ils ont été publiés. Ces articles sont présentés dans leur version
intégrale. Tous les articles présentés dans cette thèse sont des manuscrits pour lesquels je
suis l'auteure principale.
La première partie de la thèse sera constituée d'une introduction générale, qui sera suivie
aux chapitres I à III par les manuscrits publiés. Finalement, les conclusions de mes
recherches entreprises pour l'obtention de mon grade de Philosophiae Doctor constitueront
la 3 e et dernière partie de la thèse. Le corps de la thèse sera constitué de 3 chapitres. Les
résultats de mes travaux effectués sur les gènes ZNF350/ZBRK1 et GADD45A constitueront
les chapitres I et II, respectivement. Le chapitre III présente les résultats de l'analyse du
gène NBN dans notre cohorte de familles à risque élevé de cancer du sein. Les résultats de
l'analyse des gènes ZNF350/ZBRK1, GADD45A et NBN font chacun l'objet d'une
publication. Les détails de l'état de chacun des manuscrits est précisé dans la liste suivante:
Chapitre I : Desjardins S, Belleau P, Labrie Y, Ouellette G, Bessette P, Chiquette J, Laframboise R, Lépine J, Lesperance B, Pichette R, Plante M; INHERIT BRCAs, Durocher

IV
F. Genetic Variants and Haplotype Analyses of the ZBRK1/ZNF350 Gene in High-Risk non BRCA1/2 French Canadian Breast and Ovarian Cancer Families. Int J Cancer. 2008; 122(1): 108-16.
Chapitre II : Desjardins S, Ouellette G, Labrie Y, Simard J; INHERIT BRCAs, Durocher F. Analysis of GADD45A sequence variations in French Canadian families with high risk of breast cancer. J Hum Genet. 2008; 53(6):490-8.
Chapitre III : Desjardins S, Joly Beauparlant C, Labrie Y, Ouellette G, INHERIT BRCAs, Durocher F. Variations in the NBN/NBS1 gene and the risk of breast cancer in non-BRCA1/2 French Canadian families with high risk of breast cancer. BMC Cancer. 2009:9:181.
Les résultats présentés dans ces chapitres sont l'aboutissement d'un travail d'équipe. Les
recherches décrites dans ces chapitres ont toutes été effectuées dans le cadre du programme
interdisciplinaire INHERIT BRCAs, dont le Dr Jacques Simard est l'instigateur, et ces
travaux ont été effectués sous la direction du Dre Francine Durocher au Laboratoire de
Génomique des cancers du centre de recherche du CHUL, faisant partie intégrante du
Centre hospitalier universitaire de Québec (CHUQ). Par conséquent, Dre Francine
Durocher a été responsable, au sein des divers projets élaborés dans cette thèse, de la
conception et de la coordination des études, en plus de son aide précieuse lors de la
préparation et de la révision critique des manuscrits. Le Dr Yvan Labrie a, quant à lui,
fourni une expertise technique de premier plan et a participé activement à la préparation des
manuscrits et aux analyses bio-informatiques présentées au Chapitre II. Mme Geneviève
Ouellette a été responsable des extractions d'ADN, d'ARN, ainsi que du maintien des
cellules en culture, matériel essentiel à la réalisation de chacun des projets traités dans cette
thèse. De plus, soulignons l'aide précieuse de Mme Ouellette dans les études moléculaires
par séquençage (Chapitre II), la réalisation des transfections nécessaires aux essais gène
rapporteur luciférase (Chapitre III). Notons aussi l'apport de M. Pascal Belleau, qui a
fourni une aide précieuse pour l'ensemble des analyses bioinformatiques présentées au
Chapitre I et l'aide de M. Charles Joly Beauparlant qui a été en charge du séquençage chez
les contrôles au Chapitre III. Au Chapitre I, les autres auteurs sont des chercheurs cliniciens
dont l'apport au projet est essentiel dans le cadre d'une étude effectuée chez des sujets
humains.

À titre de premier auteur de tous les manuscrits présentés aux chapitres I à III, j 'ai été en
charge de la majeure partie des analyses moléculaires au laboratoire. En plus du séquençage
de ces gènes, j 'ai aussi effectué les expériences de gène rapporteur luciférase incluses au
Chapitre III. Dans le cadre de ces 3 chapitres, j'ai aussi été responsable des analyses bio
informatiques et statistiques des résultats, et de leur interprétation. J'ai rédigé les
manuscrits présentés dans le corps de la thèse, avec les commentaires et suggestions des
Drs Francine Durocher et Yvan Labrie. J'ai été en charge de la soumission et de la révision
de chacun de ces manuscrits, avec le support de ma directrice de recherche. Il est de plus
important de mentionner que tous les contributeurs ont lu et approuvé la version finale des
manuscrits présentés dans le cadre de cette thèse.

REMERCIEMENTS C'est avec une grande fierté, beaucoup de soulagement, une touche de mélancolie et une
énorme gratitude que s'achève mon aventure doctorale. Par conséquent, je tiens à utiliser
ces quelques lignes afin d'articuler mes remerciements aux personnes qui m'ont offert le
soutien moral et technique ayant rendu ce projet possible. Il s'agit d'une tâche dont la
complexité réside à la fois à trouver les mots adéquats mais surtout à n'oublier personne. Je
vais donc accuser tout oubli de ma part sur la faute d'un épuisement cérébral, et je remercie
sincèrement à l'avance toute personne m'ayant apporté un soutien et dont le nom a été
accidentellement omis. Toutes mes excuses et merci de votre compréhension.
Je tiens en premier lieu à remercier ma directrice de thèse, Dre Francine Durocher, pour
m'avoir accueillie au sein de son laboratoire à la maîtrise, puis au doctorat. Elle a su
m'offrir l'encadrement nécessaire à la réalisation des travaux présentés dans cet ouvrage.
Ses encouragements, ses directives et ses conseils m'ont été d'une grande aide tout au long
de ces années, au cours desquelles elle m'a donné l'opportunité de voyager et m'a confié
des responsabilités qui se sont avérées des atouts précieux dans ma croissance personnelle
et professionnelle. Par conséquent, je tiens à lui réitérer mes remerciements pour la
confiance et l'attention qu'elle m'a accordée au cours de ces années ainsi que pour son
énergie et sa détermination.
Je tiens également à reconnaître l'apport du Dr Jacques Simard à mes études doctorales, et
à le remercier de m'avoir dirigée dans un premier temps vers Dre Durocher lors de notre
première rencontre à la fin de mes études de baccalauréat, ce qui aura lancé ma trajectoire
aux études graduées. Je le remercie pour ses conseils, et la diligence avec laquelle il a su
répondre à mes demandes au cours de ces années.
Mes plus sincères remerciements aux membres de l'équipe du Dr Durocher : Dr Yvan
Labrie, Geneviève Ouellette, Charles Joly Beauparlant et Frédéric Guénard, à qui j'adresse
une pensée particulière pour toute leur patience à mon égard. J'aimerais aussi exprimer
mon respect pour tous les membres présents et passés de l'équipe du Dr Simard, avec
laquelle j 'ai collaboré durant ces années. Je tiens à mentionner le plaisir particulier que j'ai

Vil
eu à côtoyer Dr Anne-Marie Moisan, en qui j'ai trouvé une amie précieuse qui apporte une
couleur particulière à notre amitié. Merci pour tes encouragements répétés !
J'aimerais aussi souligner l'appui répété que j'ai reçu de la part de toute l'équipe de la
Plateforme de séquençage et de génotypage du CHUL, Marc-André Rodrigue, Annie-
Claude Collin-Deschesnes et Guy Reimnitz, ainsi que toute l'équipe du Dr Vincent
Raymond, qui ont su m'encourager à terminer ce projet.
Je tiens de plus à remercier ma famille et mes amis. Paul et Monique : ils m'ont offert un
soutien et des encouragements constants au cours de ces années et ils ont toujours été fiers
de moi. Mon grand frère Vincent, sa conjointe Véro et les deux petits amours Victor et
César : je vous aime. Mes frères Benoît et André, pour leur support. A mes amies Caroline,
et Manon : dix ans d'amitié et bien d'autres à venir !
Finalement, je ne veux pas passer sous silence la contribution financière de la Fondation
Desjardins, qui m'a offert une bourse mais surtout de la Fondation René Bussières qui m'a
accompagnée sans faille au cours de toutes ces années de doctorat. Votre intérêt à mon
égard a été une grande source d'inspiration.
La réalisation d'une thèse nous paraît parfois bien solitaire. À la lumière de ces
remerciements, je lève mon chapeau à tous mes compagnons de fortune (ou d'infortune).
Votre présence a été précieuse et cet ouvrage vous appartient aussi...

A ma famille : Monique et Paul; Véronique, Vincent, Victor et César; Benoît; André. Pour m'avoir accompagnée et soutenue au cours de toutes ces années, chacun à sa façon...
A Caroline et Manon. Lorsque j ' a i besoin d'un rire ou d'un simple sourire; d'un encouragement ou d'une épaule sympathique; de mettre de l'ordre dans mes pensées ou de me changer les idées. Votre amitié vaut de l'or...

TABLE DES MATIERES RÉSUMÉ i ABSTRACT ii AVANT-PROPOS iii REMERCIEMENTS vi TABLE DES MATIÈRES ix LISTE DES TABLEAUX xii LISTE DES FIGURES xiv LISTE DES ABRÉVIATIONS xvi INTRODUCTION 1 1. Le cancer 1
1.1 Epidemiologic : les conséquences du cancer à travers le monde et au Canada 2 1.1.1 L'épidémiologie du cancer du sein 2
1.2 Le cancer du sein 4 1.2.1 Le sein normal 4 1.2.2 Lésions et maladies du sein 5
1.3 Les facteurs de risque du cancer du sein 6 2. Déterminants génétiques du cancer du sein 12
2.1 Anecdotes historiques : cancer du sein, histoire familiale et prédisposition 12 2.2 Gènes de susceptibilité au cancer du sein connus 14
2.2.1 Alleles de forte penetrance 17 2.2.1.1 Le syndrome de cancer du sein et de l'ovaire héréditaire, BRCA1 et BRCA2
17 2.2.1.2 Le syndrome de Li-Fraumeni et TP53 22
2.2.2 Alleles de penetrance incertaine 24 2.2.2.1 Le syndrome de Cowden et PTEN. 24 2.2.2.2 Le syndrome de Peutz-Jegher et STK11 25 2.2.2.3 Le syndrome du cancer gastrique héréditaire diffus et CDH1 26
2.2.3 Alleles de penetrance modérée 27 2.2.3.1 L'ataxie-télangiectasieetj4rM 27 2.2.3.2 CHEK2 28 2.2.3.3 BRIP1/BACH 1/FANCJ 30 2.2.3.4 PALB2/FANCN 31
2.2.4 Alleles de faible penetrance 32 2.3 Modèles de la susceptibilité génétique au cancer du sein 37
3. Fonctions et partenaires cellulaires deBRCAl etBRCA2 41 3.1 Structure des protéines BRCA1 et BRCA2 41 3.2 Fonctions de BRCA1 et BRCA2 44
3.2.1 Régulation de la transcription 45 3.2.1.1 Régulation transcriptionnelle de GADD45A par BRCA1 et ZNF350 46
3.2.2 Détection, signalisation et réparation des dommages de l'ADN 49 3.2.2.1 Interaction entre la nibrine et BRCA1 52 3.2.2.2 Implication de BRCA1 et BRCA2 dans l'Anémie de Fanconi 54
3.2.3 Autres fonctions de BRCA1 et BRCA2 61 4. Types de variations de séquence 63

5. Problématique 66 5.1 Collection familiale étudiée : les familles à risque élevé de la population canadienne française 66 5.2 Objectifs de l'étude 67
CHAPITRE I : Genetic Variants and Haplotype Analyses of the ZBRK1/ZNF350 Gene in High-Risk non BRCA1/2 French Canadian Breast and Ovarian Cancer Families 69
Résumé 72 Abstract 73 Introduction 74 Materials and Methods 76 Results 80 Discussion 85 Acknowledgements 90 Appendix 1 91 References 92 Figure legends 103
CHAPITRE II : Analysis of GADD45A Sequence Variations in French Canadian Families with High Risk of Breast Cancer 106
Résumé 108 Abstract 109 Introduction 110 Materials and methods 112 Results 114 Discussion 118 Acknowledgements 122 Appendix 123 References 124 Legends to Figures 130
CHAPITRE III : Variations in the NBN/NBS1 gene and the risk of breast cancer in non-BRCA1/2 French Canadian families with high risk of breast cancer. 133
Résumé 135 Abstract 136 Background 138 Methods 141 Results 143 Discussion 148 Conclusion 153 Abbreviations 154 Competing interests 154 Authors' contributions 154 Acknowledgements 155 Appendix 156 References 157 Figure legends 162
DISCUSSION ET CONCLUSION 174 1. CHAPITRE I : Analyse du gène ZNF350/ZBRK1 174 2. CHAPITRE II : Investigation du gène GADD45A 177

XI
3. CHAPITRE III : Étude du gène NBN 179 4. L'approche gène candidat dans la population canadienne française : considérations méthodologiques 181 5. Conclusions et perspectives 183 BIBLIOGRAPHIE 185

LISTE DES TABLEAUX INTRODUCTION Tableau 1. Classification des lésions bénignes du sein suite à l'examen histologique,
selon le risque relatif de cancer du sein 6 Tableau 2. Facteurs de risque modifiant le risque de développer un cancer du sein 8 Tableau 3. Principaux alleles de susceptibilité au cancer du sein connus 15 Tableau 4. Caractéristiques des familles présentant des cas de cancer du sein
héréditaires, familiaux et sporadiques 16 Tableau 5. Pourcentage des familles présentant des mutations de BRCA1 ou BRCA2,
selon le type de cas sélectionné 18 Tableau 6. Risque d'autres types de cancers associé à une mutation délétère de
BRCA1 ou BRCA2 22 Tableau 7. Critères de diagnostic des familles Li-Fraumeni 24 Tableau 8. Caractéristiques des gènes associés à l'Anémie de Fanconi 55
CHAPITRE I Table I. Coding and intronic sequence variations and genotype frequencies in familial
breast cancer cases and controls 97 Table IL Estimated haplotype frequencies in cases and controls using PHASE. 99 Table III. Estimated haplotype numbers and associated breast cancer risks for cases and
controls using PHASE and Haplostats 100 Supplemental Table I. Oligonucleotide primers used for amplification and sequence
analysis of the ZBRK1 gene 101 Supplemental Table II. Global p-value for case and control comparison using PHASE,
Cocaphase and Haplostats programs 102
CHAPITRE II Table 1. GADD45 sequence variations and genotype distribution in familial breast cancer
cases and controls 127 Table 2. Haplotypes as estimated by PHASE and WHAP, and their estimated frequencies in
cases and controls using all SNPs genotyped in both case and control series. ...128 Supplemental Table 1. PCR and sequencing primers 729
CHAPITaRE III Table 1. Oligonucleotides used for NBN PCR amplification and sequencing. 164 Table 2. Exonic sequence variations of the NBN gene in French Canadian breast cancer
cases and healthy controls 166 Table 3. Promoter and intronic variations of the NBN gene in French Canadian breast
cancer cases and healthy controls 167

Xlll
Table 4. Estimated haplotypes using the Whap program and all variants identified, for cases and controls combinend. 168
Table 5. Estimated haplotypes using the WHAP program and SNPs with a frequency greater than 5% for cases and controls combined. 169

LISTE DES FIGURES INTRODUCTION Figure 1. Le cancer est un processus à étapes multiples 1 Figure 2. Répartition relative des 5 types de cancers les plus fréquents au monde et au
Canada 3 Figure 3. Anatomie du sein humain normal 4 Figure 4. Représentation du taux de cancer du sein par pays, par 100 000 habitants 7 Figure 5. Événements sélectionnés de l'histoire de la génétique et du cancer 13 Figure 6. Estimation de la proportion des familles présentant une forte histoire familiale
pouvant être associées à des mutations de BRCA1, BRCA2 ou aucun de ces deux gènes. D'après les données de Ford et collaborateurs.66 19
Figure 7. Modèle polygénique de la répartition du risque de cancer du sein chez les cas et dans la population 38
Figure 8. Proportion des cas de cancer du sein expliqués par la proportion de la population à risque élevé 38
Figure 9. Variation de la taille de l'échantillon 40 Figure 10. Représentation des domaines des protéines BRCA1 et BRCA2 humaines 42 Figure 11. Structures chromosomiques aberrantes de cellules déficientes en Brca2 44 Figure 12. Mécanismes de la régulation de GADD45A par p53, BRCA1 et ZBRK1 47 Figure 13. Recrutement des premières protéines de détection/réparation au site de bris 50 Figure 14. Mécanisme de la recombinaison homologue 51 Figure 15. Implication de BRCA1 et BRCA2 dans la voie Fanconi/BRCA 57 Figure 16. La réparation des ICLs rencontrés lors de la replication par la voie
Fanconi/BRCA 57 Figure 17. Implication de BRCA1 dans la régulation du cycle cellulaire 62 Figure 18. Position des altérations pouvant affecter l'expression d'un gène 64
CHAPITRE I Figure 1. Proportional genomic structure of the ZBRK1 gene and protein structure of
ZBRK1 illustrating domains involved in function/regulation of the protein 104 Supplemental Figure 1. Pairwise linkage disequilibrium (LD) measures for SNPs identified
in ZBRK1 and LD measures according to HapMap for ZBRK1 chromosome region 105
CHAPITRE II Figure 1. Pairwise LD measures of\D'\ and r2 for the 12 SNPs identified in the breast
cancer and control series 131 Figure 2. Haploview predictions of haplotype blocks using SNPs displaying a MAF higher
than 5% 132

XV
CHAPITRE III Figure 1. Schematic representation of the NBN gene on chromosome 8q21 770 Figure 2. In silico analysis of the effect of coding variants on putative exonic splicing
enhancer (ESE) motifs by the ESEfinder program 777 Figure 3. Effect of the c.-242-l WdelAGTA deletion on NBN promoter activity 772 Figure 4. Linkage disequilirium across the NBN gene and neighbouring chromosome
region 773

LISTE DES ABREVIATIONS
3 75'UTR ADN/DNA ADNc/cDNA ADNâb/dsDNA ADNsb/ssDNA AF/FA aa AIR AKAP9 ALL ANGPT1 ARNm/mRNA ATBF1 ATM BACH1 BARD1 BDB/DSB BOC bp BRCA1/2 BRCT BR1P1 C5orf35 CASP8 CDX2 CDK CEPH CEU CDH1 CDX2 CHEK2 cSNP CtlP dbSNP ECHDC1 EM ER+/-ESE/ESS ESRI FAAP FGFR2 FHA GADD45A
3 75 ' untranslated region Acide Désoxyribonucléique/DeoxvnTjortwc/e/c acid ADN complémentaire/complementary DNA ADN double brins/double stranded DNA ADN simple brin/single stranded DNA Anémie de Fanconi/Fanconi Anemia Acides aminés/a/m'Ho acids ATM interacting region A kinase anchor protein 9 Acute lymphoblastic leukemia Angiopoietin 1 Acide ribonucléique messager/messenger Ribonucleic acid AT motif-binding factor 1 Ataxia Telangiectasia Mutated BRCA1-associated C-terminal helicase 1 BRCA1 -associated RING domain 1 Bris double brinslDouble strand breaks Breast and ovarian cancer Base pair Breast cancer 1/2, early onset BRCA1 c-terminal BRCA 1-interacting protein 1 Chromosome 5 orf35 Caspase 8 Caudal-type homeobox 2 Cyclin-dependent kinase Centre d'Étude du Polymorphisme Humain CEPH-Utah residents of Northern and Western Europe ancestry Cadherin 1 Caudal-type homeobox 2 Checkpoint Kinase 2 Coding SNP CTBP interacting protein SNP database Enoyl coenzyme A hydratase domain containing 1 Expectation maximized Estrogen receptor positive/negative Exonic splicing enhancer/silencer Estrogen receptor 1 Fanconi anemia associated protein Fibroblast growth factor receptor 2 Forkhead-associated Growth arrest and DNA damage-inducible alpha

XV11
Y-H2AX GNG12 GWAS HaR HBOC HMGA2 HR HWE IC/CI ICL ID IGF2 1RES ISE/ISS kb kDa LFL LFS LKB1 LD LDA LSP1 MAF MAP3K1 Mf MER3 M R MLPA MMC MMP9 MRE11A M/RJN MRPL23 MRPS30 MTBFl MYB N-/C-terminus NBN/NBS1 NBS NCBI NER NHEJ NKX3-1 OB OCCR OR PALB2
Gamma-H2AX G-Protein gamma 12 subunit Genome-wide association study Hazard ratio/Rapport de risque Hereditary breast and ovarian cancer High mobility group A T-hook 2 Homologous recombinationAtecombinaison homologue Hardy- Weinberg equilibrium Intervalle de confiance/confidence interval Interstrand crosslink Identification number Insulin-like growth factor 2 Internal ribosome entry site Intronic splicing enhancer/silencer Kilobase Kilo Daltons Li-Fraumeni-like syndrome classic Li-Fraumeni syndrome Liver kinase BI Linkage disequilibrium LD analyzsis program Lymphocyte-specific protein 1 Minor allele frequency Mitogen-activated protein kinase kinase kinase 1 Macaca fascicularis Mesoderm induction early response family member 3 MRE11A interacting region Multiplex ligation-dependent probe amplification Mitomycin C Matrix metallopeptidase 9 MRE11 meiotic recombination 77 homolog A MRE11A/RAD50/Nibrin Mitochondrial ribosomal protein L23 Mitochondrial ribosomal protein S30 Muscle-specific MT binding factor v-myb Myeloblastosis viral oncogene homolog Amino-ZCarboxy-terminus Nibrin/ Nijmegen breakage syndrome 1 Nijmegen breakage syndrome National center for biotechnology information Nucleotide excision repair Non-homologous end joining NK3 homeobox 1 Oligonucleotide/oligosaccharide binding Ovarian cancer cluster region Odd ratio/i\appori de cotes Partner and localizer ofBRCA2

xviu
PCR PHTS PI3K PPP2R1A PTEN QRT-PCR RAD50/51 RC RING RNF146 RR RT-PCR SEN SERBP1 SIR SLN SNP SR SSPNN STK 11 SUMO TGFB1 TNNT3 TLS TNS T0X3 TP53 tSNP UCSC USP1 ZBRK1 ZFHX3 ZNF350 ZNF432 ZNF613 ZNF614 ZNF615
Polymerase chain reaction PTEN hamartoma tumor syndrome Phosphatidylinositol-3-kinase Protein phosphatase 2 regulatory sub-unit 1, alpha Phosphatase and tensin homolog Quantitative real-time PCR Rad50/5l homolog Risque cumulatif Really interesting new gene Ring finger protein 146 Risque relatif/Relative risk Real-Time PCR Séquence d'exportation nucléaire SERPINE1 mRNA binding protein 1 Standardized incidence ratio Séquence de localisation nucléaire Single nucleotide polymorphism Serine/arginine-rich Splice site prediction program using neural network Serine/threonine kinase 11 Small ubiquitin-like modifier Transforming growth factor beta 1 Troponin T type 3 Translesion synthesis Type non spécifique TOXhigh mobility group box family member 3 Tumor Protein 53 Tagging SNP University of California Santa Cruz Ubiquitin specific protein 1 Zinc finger and BRCA1-interacting protein with a KRAB domain 1 Zinc finger homeobox 3 Zinc finger protein 350 Zinc finger protein 432 Zinc finger protein 613 Zinc finger protein 614 Zinc finger protein 615

INTRODUCTION
1. Le cancer Le cancer est un groupe hétérogène de maladies : plus de 100 sous-types de cancers sont connus chez l'humain, dont plusieurs peuvent affecter un même organe.1 Le cancer est associé à une croissance anormale des cellules, qui présentent une dérégulation des processus de prolifération et de mort cellulaire. Ces cellules peuvent acquérir de nouvelles capacités, telle la propriété d'envahir les tissus adjacents et de former des métastases à d'autres tissus ou organes, pouvant ainsi entraîner la morbidité ou la mort de l'hôte. Les changements observés au niveau de ces cellules sont ultimement le résultat d'une expression anormale de gènes. Ce dérèglement peut avoir plusieurs sources : l'hérédité, l'action d'un composé mutagène, la translocation d'une partie d'un chromosome, l'amplification ou la perte d'hétérozygosité d'une région du génome, ou tout autre mécanisme pouvant mener à une transcription anormale ou à une translocation d'une partie d'acide désoxyribonucléique (ADN ou desoxyribonucleic acid; DNA). ' Dans plusieurs cas, les causes exactes du cancer sont inconnues, mais il reste clair qu'il s'agit de l'action conjuguée de facteurs environnementaux et génétiques agissant ensemble pour l'initiation (modification initiale de l'ADN) ou la promotion (prolifération des cellules initiées) du cancer (Figure 1). Les changements de l'ADN peuvent suivre soit un modèle génétique (changement dans la séquence même de l'ADN) ou un modèle épigénétique (modification de l'expression d'un gène sans que la séquence de l'ADN soit affectée), bien que ces deux modèles ne soient pas mutuellement exclusifs.3'4
4e -.Hautrw S j \^m\ normale mutation mutation
Figure 1. Le cancer est un processus à étapes multiples. La cellule accumule des mutations lui permettant de passer les étapes d'initiation, de promotion, de multiplication clonale et de progression. Dans le cas du cancer du sein, les métastases sont retrouvées le plus fréquemment au niveau des poumons, du foie et des os.

1.1 Epidémiologie : les conséquences du cancer à travers le monde et au Canada. Le cancer est l'une des causes principales de mortalité au monde, arrivant au second rang
derrière les maladies coronariennes. Pour l'année 2007, on estime qu'il y a eu 12 millions
de nouveaux cas et 7,6 millions de morts dus au cancer à travers le monde.5 D'ici 2050, on
estime que l'incidence du cancer grimpera à 27 millions de nouveaux cas par an et sera
associée à 17,5 millions de décès annuels, et ce dû seulement à des facteurs
démographiques de croissance et de vieillissement de la population. Dans les pays en voie
de développement, on s'attend à observer un changement de l'incidence et des types de
cancers retrouvés. Ce phénomène serait associé à un prolongement de l'espérance de vie
(baisse de la mortalité infantile et de la mortalité associée aux maladies infectieuses) et à
l'adoption d'un mode de vie «occidental» (hausse du taux de fumeurs, adoption d'une diète
riche en gras saturés et en calories, baisse de l'activité physique).5
Les types de cancers et leur incidence varient selon la région géographique et le statut
socio-économique des pays (Figure 2). Chez les hommes, le cancer du poumon est le plus
fréquemment diagnostiqué, bien qu'il soit devancé par le cancer de la prostate dans les pays
économiquement développés. Le cancer du poumon est aussi le type de cancer responsable
de la plus grande proportion de décès, et ce quel que soit le statut socio-économique. Chez
la femme, le cancer du sein est à la fois le plus diagnostiqué et le responsable de la plus
grande proportion des décès mondialement, bien qu'il arrive au second rang derrière le
cancer du poumon en termes de causes de décès par cancer au Canada.
1.1.1 L'épidémiologie du cancer du sein En raison de son incidence élevée et de son pronostic relativement bon (les taux de survie
sont en moyenne de 73% dans les pays à économie développée et de 57% dans les pays en
voie de développement), le cancer du sein est le cancer le plus prévalent au monde. La
prévalence est habituellement calculée sur 5 ans puisque le risque encouru par les
survivants après cette période est généralement près de celui de la population en général.
Les individus toujours en vie après cette période sont donc considérés «guéris», bien que
dans le cas du cancer du sein, les femmes ayant été atteintes présentent un risque plus élevé
que la population pour une longue période (voir la section 1.3 Les facteurs de risque du

cancer du sein). On estime que 4,4 millions de femmes vivent actuellement avec un cancer
du sein diagnostiqué au cours des 5 dernières années mondialement, ce qui est trois fois
plus que la prévalence du cancer du poumon pour la même période, hommes et femmes
confondus.7 La prévalence élevée du cancer du sein a eu pour effet d'attirer l'attention sur
ce type de cancer. Cette attention s'est à son tour traduite par des améliorations des
techniques de prévention, de diagnostic et de traitement, permettant d'obtenir une meilleure
définition de la biologie du cancer du sein et des facteurs pouvant modifier le risque d'une
femme de développer cette maladie.
NUII*.eaux cas
Pays à économie dt-aeloppée
Pays ra voie de développement
Canada
• 4. •
c-Çdç ■ Poumon ■ Prostate ■ Estomac "Colorectal ■Foie ■Sein
Col de l'utérus ■ Oesophage -"Thyroïde ■ Pancréas ■ Corps de
l'utérus m Lymphome non
Hodgkinien ■ Vessie ■Ovaire J Autres
Figure 2. Répartition relative des 5 types de cancers les plus fréquents au monde et au Canada. La partie centrale de la figure montre la sous-division de ces cancers en fonction du statut socio-économique des pays. Les pays à économie développée comprennent l'Amérique du Nord, le Japon, l'Europe, l'Australie et la Nouvelle-Zélande. Les pays en voie de développement comprennent le reste du monde. D'après les données de Global Cancer Facts and Figures 20075 et Canadian Cancer Statistics 20086.
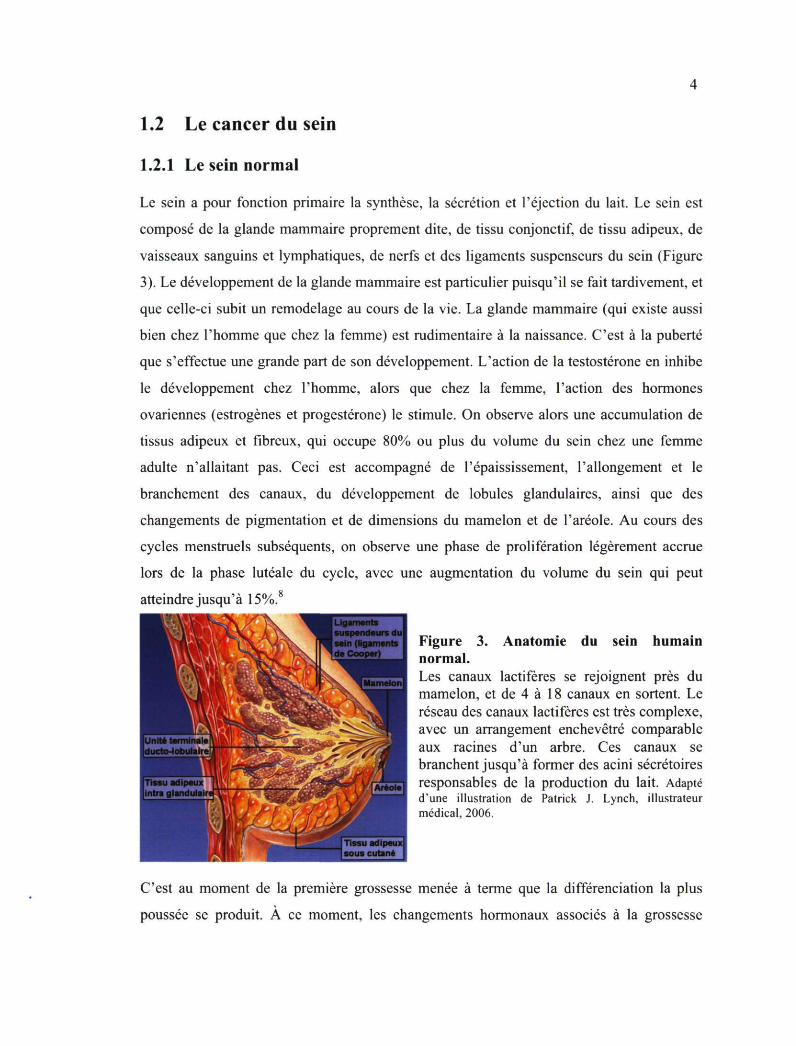
1.2 Le cancer du sein
1.2.1 Le sein normal
Le sein a pour fonction primaire la synthèse, la sécrétion et l'éjection du lait. Le sein est composé de la glande mammaire proprement dite, de tissu conjonctif, de tissu adipeux, de vaisseaux sanguins et lymphatiques, de nerfs et des ligaments suspenseurs du sein (Figure 3). Le développement de la glande mammaire est particulier puisqu'il se fait tardivement, et que celle-ci subit un remodelage au cours de la vie. La glande mammaire (qui existe aussi bien chez l'homme que chez la femme) est rudimentaire à la naissance. C'est à la puberté que s'effectue une grande part de son développement. L'action de la testosterone en inhibe le développement chez l'homme, alors que chez la femme, l'action des hormones ovariennes (estrogènes et progestérone) le stimule. On observe alors une accumulation de tissus adipeux et fibreux, qui occupe 80% ou plus du volume du sein chez une femme adulte n'allaitant pas. Ceci est accompagné de l'épaississement, l'allongement et le branchement des canaux, du développement de lobules glandulaires, ainsi que des changements de pigmentation et de dimensions du mamelon et de l'aréole. Au cours des cycles menstruels subséquents, on observe une phase de prolifération légèrement accrue lors de la phase lutéale du cycle, avec une augmentation du volume du sein qui peut atteindre jusqu'à 15%.8
Figure 3. Anatomie du sein humain normal. Les canaux lactifères se rejoignent près du mamelon, et de 4 à 18 canaux en sortent. Le réseau des canaux lactifères est très complexe, avec un arrangement enchevêtré comparable aux racines d'un arbre. Ces canaux se branchent jusqu'à former des acini sécrétoires responsables de la production du lait. Adapté d'une illustration de Patrick J. Lynch, illustrateur médical, 2006.
C'est au moment de la première grossesse menée à terme que la différenciation la plus
poussée se produit. À ce moment, les changements hormonaux associés à la grossesse

produisent une croissance et une prolifération intenses, et induisent la formation des acini
sécrétoires (produits par un branchement plus poussé des canaux et leur différenciation),
une augmentation de la quantité de tissu adipeux et une augmentation du flux sanguin. Au
moment du sevrage, la glande mammaire involue (arrêt de l'activité sécrétoire et retour à
un état moins différencié qui implique majoritairement les acini retrouvés au bout des
canaux) jusqu'à la grossesse suivante. Le dernier changement important de la glande
mammaire survient après la ménopause. À ce moment, il y a arrêt de la production des
hormones ovariennes, ce qui provoque une involution de la glande mammaire.
Contrairement à l'involution observée après une période d'allaitement, cette involution
touche à la fois les lobules et les canaux. Ces structures régressent et sont progressivement
remplacées par du collagène et du tissu adipeux. Chez la femme âgée, il ne reste plus que
quelques acini et canaux dispersés à travers le tissu adipeux.9
1.2.2 Lésions et maladies du sein
Il existe plusieurs lésions et maladies du sein, bénignes ou malignes. La majorité de ces
lésions sont retrouvées au niveau de l'unité fonctionnelle de base du sein, l'unité terminale
ducto-lobulaire (Figure 3). Un nombre relativement grand de lésions bénignes du sein
existent, et il semble que certaines d'entre elles peuvent êtres asymptomatiques et
relativement courantes dans la population.10 La compréhension des lésions bénignes est
essentielle, car il a été démontré que le développement de certaines d'entre elles constitue
un facteur de risque de développer un cancer du sein (voir la section 1.3 Les facteurs de
risque du cancer du sein). De façon pratique, ces lésions sont classées en trois groupes,
selon le risque relatif de cancer du sein qu'elles confèrent: les lésions non prolifératives, les
lésions prolifératives sans atypie et les lésions prolifératives avec atypie (Tableau 1).
Selon leur histologie, on reconnaît plusieurs types de lésions mammaires malignes. Dans un
premier temps, on distingue les lésions sur la base de leur degré d'invasion. La principale
lésion pré-invasive est le carcinome mammaire in situ, dans lequel les cellules anormales
demeurent confinées au système ducto-lobulaire et ne présentent pas d'invasion au stroma.
Le recours aux programmes de mammographies de dépistage a grandement augmenté la
fréquence du diagnostic de ce type de lésion.11'12 Les carcinomes infiltrants sont divisés en
carcinomes canalaires de type non spécifique (TNS) et en tous ceux qui possèdent des

caractéristiques morphologiques distinctes qui permettent une caractérisation reproductible
en un type spécifique. Par conséquent, la classification des carcinomes canalaires en est une
d'exclusion. Les carcinomes canalaires TNS représentent entre 70% et 80% des tumeurs
malignes de la glande mammaire, les autres types de carcinomes canalaires (tubuleux,
crubriforme infiltrant, mucineux, papillaire infiltrant, sécrétant, apocrine infiltrant,
adénoïde kystique, médullaire) étant beaucoup moins fréquents. Le dernier type de
carcinome infiltrant est de type lobulaire, et représente de 5% à 14% de tous les carcinomes
infiltrants.11'12
Tableau 1. Classification des lésions bénignes du sein suite à l'examen histologique, selon le risque relatif de cancer du sein. Risque Prolifération Résultats de l'examen histologique Pas d'augmentation Minimale
Faible augmentation Prolifératif (risque relatif : 1,5 à 2,0) sans atypie
Augmentation modérée Prolifératif (risque relatif : >2,0) avec atypie
Changements fibrokystiques : kyste et ectasie canalaire (72%), hyperplasie épithéliale simple (40%), adénose non-sclérosante (22%), et fibrose périnanalaire (16%)*; adénofibrome simple (15-23%)+; et lésions diverses (hyperplasie lobulaire, hypertrophie juvénile et hyperplasie du stroma) Tumeurs bénignes : hamartome, lipome, tumeur phyllodeî, papillome solitaire, neurofibrome, adénome géant et adénome myoepithelial Lésions traumatiques : hématome, nécrose adipeuse et lésions résultant de la pénétration d'un corps étranger Infections : granulome et mastites Sarcoïdose Métaplasies : squameuse ou apocrine Mastopathie diabétique Hyperplasie canalaire sans atypie, adénofibrome complexe (kystes de plus de 3mm de diamètre, adénose sclérosante, calcifications épithéliales ou changements papillaires apocrines), papillome ou papillomatose, cicatrice radiale et métaplasie cylindrique) Hyperplasie atypique canalaire et hyperplasie atypique lobulaire
*Les pourcentages indiquent la proportion de seins examinés lors d'autopsies présentant ce type de lésion. Données de Sandison13 fDonnées de Goehring et Morabia.14 JLa plupart des tumeurs phyllodes sont considérées comme des tumeurs fibroépithéliales, mais certaines ont des caractéristiques cliniques et histologiques malignes. Adapté de9
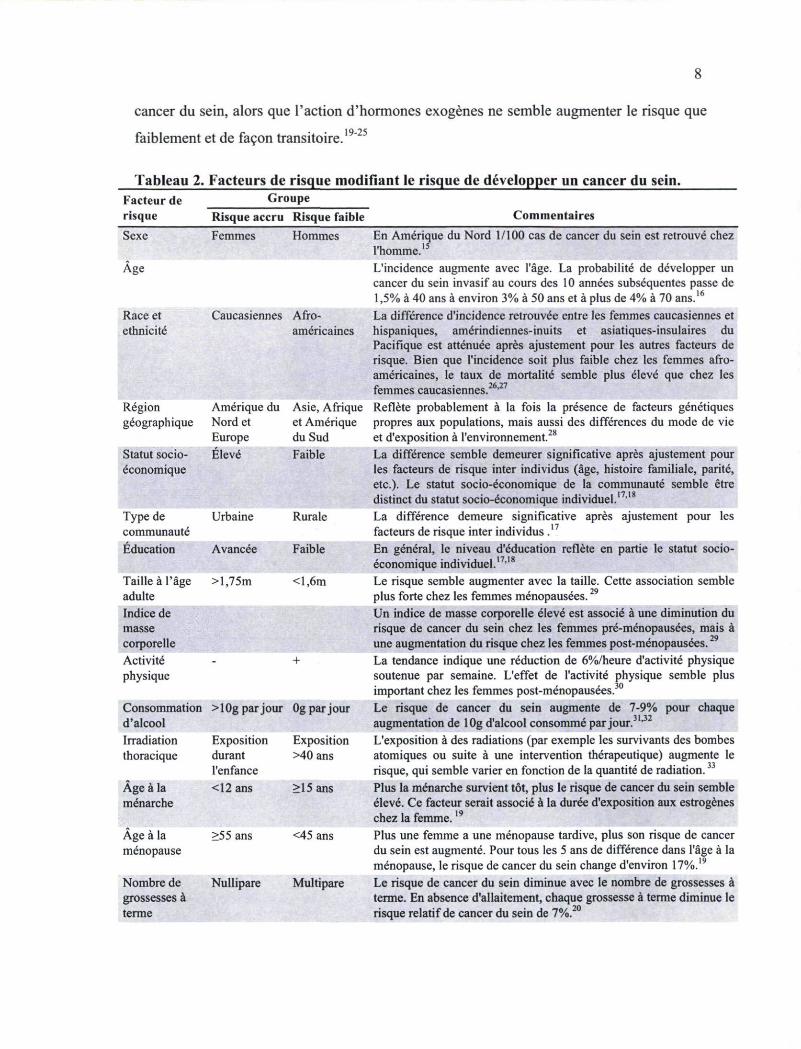
1.3 Les facteurs de risque du cancer du sein
Le cancer du sein est une maladie complexe influencée par l'effet de plusieurs facteurs
génétiques et environnementaux (Tableau 2). Au sens strict, les deux facteurs de risque les
plus importants sont le sexe (bien qu'il soit rare, le cancer du sein existe chez les hommes)

et l'âge.15'16 Outre ces deux éléments, plusieurs facteurs de risque ont été étudiés en ce qui
concerne le cancer du sein, plusieurs d'entre eux étant interreliés.
Région géographique :
De façon similaire à ce qui est observé pour l'incidence et la mortalité associées au cancer
en général, on observe une grande variabilité dans l'incidence du cancer du sein par pays
(Figure 4). Bien que l'on parle de régions géopolitiques en tant que facteurs de risque,
celles-ci demeurent une mesure indirecte reflétant à la fois des particularités génétiques
propres à certaines populations et ethnies, des différences de modes de vie et des variations
d'exposition à l'environnement. De plus, bien que des corrélations aient été faites entre
l'incidence observée et le statut socio-économique des pays, il est aussi possible de voir
cette même variation au niveau d'individus vivant dans une même communauté17'18, ainsi
qu'entre des individus vivant dans des communautés urbaines par opposition à des 17
communautés rurales .
Figure 4. Représentation du taux de cancer du sein par pays, par 100 000 habitants. A. Incidence B. Mortalité. Adapté de la base de données GLOBOCAN 2002 (http://www-dep. iarc. fr/globocan/database .htm).
Facteurs hormonaux :
Il a été démontré que les hormones ovariennes sont impliquées dans le développement du
cancer du sein. Plusieurs facteurs de risque connus influencent la durée de l'exposition de
la femme à ces hormones au cours de sa vie, en particulier durant sa période reproductive.
En effet, une ménarche (premières menstruations) tardive, une ménopause précoce, la
multiparité (plusieurs grossesses à terme), un jeune âge à la première grossesse,
l'allaitement et des taux bas de prolactine dans le sang sont associés à un risque moindre de
cancer du sein, alors que l'action d'hormones exogènes ne semble augmenter le risque que
faiblement et de façon transitoire 19-25
Tableau 2. Facteurs de risque modifiant le risque de développer un cancer du sein. Facteur de Groupe risque Risque accru Risque faible Commentaires Sexe
Age
Race et ethnicité
Femmes Hommes
Caucasiennes Afro-américaines
Région géographique
Amérique du Nord et Europe
Asie, Afrique et Amérique du Sud
Statut socio-économique
Elevé Faible
Type de communauté
Urbaine Rurale
Education Avancée Faible
Taille à l'âge adulte
>l,75m <l,6m
Indice de masse corporelle Activité physique
+
Consommation d'alcool
> 10g par jour Og par jour
Irradiation thoracique
Exposition durant l'enfance
Exposition >40 ans
Age à la ménarche
<12 ans >15 ans
Age à la ménopause
>55 ans <45 ans
Nombre de grossesses à terme
Nullipare Multipare
En Amérique du Nord 1/100 cas de cancer du sein est retrouvé chez l'homme.15
L'incidence augmente avec l'âge. La probabilité de développer un cancer du sein invasif au cours des 10 années subséquentes passe de 1,5% à 40 ans à environ 3% à 50 ans et à plus de 4% à 70 ans.16
La différence d'incidence retrouvée entre les femmes caucasiennes et hispaniques, amérindiennes-inuits et asiatiques-insulaires du Pacifique est atténuée après ajustement pour les autres facteurs de risque. Bien que l'incidence soit plus faible chez les femmes afro-américaines, le taux de mortalité semble plus élevé que chez les femmes caucasiennes.26,27
Reflète probablement à la fois la présence de facteurs génétiques propres aux populations, mais aussi des différences du mode de vie et d'exposition à l'environnement.28
La différence semble demeurer significative après ajustement pour les facteurs de risque inter individus (âge, histoire familiale, parité, etc.). Le statut socio-économique de la communauté semble être distinct du statut socio-économique individuel.1
La différence demeure significative après ajustement pour les facteurs de risque inter individus ,17
En général, le niveau d'éducation reflète en partie le statut socio-économique individuel.17,18
Le risque semble augmenter avec la taille. Cette association semble plus forte chez les femmes ménopausées.29
Un indice de masse corporelle élevé est associé à une diminution du risque de cancer du sein chez les femmes pré-ménopausées, mais à une augmentation du risque chez les femmes post-ménopausées.29
La tendance indique une réduction de 6%/heure d'activité physique soutenue par semaine. L'effet de l'activité physique semble plus important chez les femmes post-ménopausées.30
Le risque de cancer du sein augmente de 7-9% pour chaque augmentation de 10g d'alcool consommé par jour.31,32
L'exposition à des radiations (par exemple les survivants des bombes atomiques ou suite à une intervention thérapeutique) augmente le risque, qui semble varier en fonction de la quantité de radiation.33
Plus la ménarche survient tôt, plus le risque de cancer du sein semble élevé. Ce facteur serait associé à la durée d'exposition aux estrogènes chez la femme.:
Plus une femme a une ménopause tardive, plus son risque de cancer du sein est augmenté. Pour tous les 5 ans de différence dans l'âge à la ménopause, le risque de cancer du sein change d'environ 17%.19
Le risque de cancer du sein diminue avec le nombre de grossesses à terme. En absence d'allaitement, chaque grossesse à terme diminue le risque relatif de cancer du sein de 7%.20
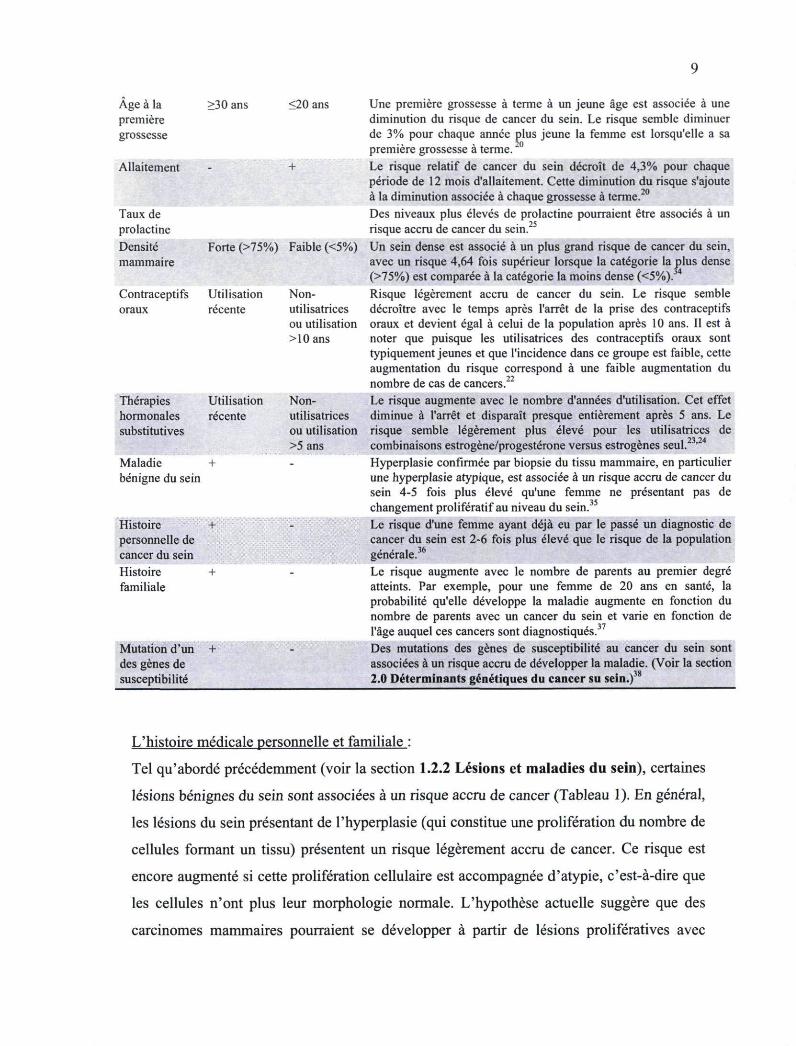
Age à la >30 ans <20 ans premiere grossesse
Allaitement +
Taux de prolactine Densité Forte (>75%) Faible (<5%) mammaire
Contraceptifs Utilisation Non-oraux récente utilisatrices
ou utilisation >10ans
Thérapies Utilisation Non-hormonales récente utilisatrices substitutives ou utilisation
>5 ans Maladie + -bénigne du sein
Histoire ' - .':,«VSS| personnelle de cancer du sein Histoire + -familiale
Mutation d'un + des gènes de susceptibilité
Une première grossesse à terme à un jeune âge est associée à une diminution du risque de cancer du sein. Le risque semble diminuer de 3% pour chaque année plus jeune la femme est lorsqu'elle a sa première grossesse à terme.20
Le risque relatif de cancer du sein décroît de 4,3% pour chaque période de 12 mois d'allaitement. Cette diminution du risque s'ajoute à la diminution associée à chaque grossesse à terme.2
Des niveaux plus élevés de prolactine pourraient être associés à un risque accru de cancer du sein. Un sein dense est associé à un plus grand risque de cancer du sein, avec un risque 4,64 fois supérieur lorsque la catégorie la plus dense (>75%) est comparée à la catégorie la moins dense (<5%). 4
Risque légèrement accru de cancer du sein. Le risque semble décroître avec le temps après l'arrêt de la prise des contraceptifs oraux et devient égal à celui de la population après 10 ans. Il est à noter que puisque les utilisatrices des contraceptifs oraux sont typiquement jeunes et que l'incidence dans ce groupe est faible, cette augmentation du risque correspond à une faible augmentation du nombre de cas de cancers.22
Le risque augmente avec le nombre d'années d'utilisation. Cet effet diminue à l'arrêt et disparaît presque entièrement après 5 ans. Le risque semble légèrement plus élevé pour les utilisatrices de combinaisons estrogène/progestérone versus estrogènes seul.' Hyperplasie confirmée par biopsie du tissu mammaire, en particulier une hyperplasie atypique, est associée à un risque accru de cancer du sein 4-5 fois plus élevé qu'une femme ne présentant pas de changement prolifératif au niveau du sein. Le risque d'une femme ayant déjà eu par le passé un diagnostic de cancer du sein est 2-6 fois plus élevé que le risque de la population générale.36
Le risque augmente avec le nombre de parents au premier degré atteints. Par exemple, pour une femme de 20 ans en santé, la probabilité qu'elle développe la maladie augmente en fonction du nombre de parents avec un cancer du sein et varie en fonction de l'âge auquel ces cancers sont diagnostiqués.37
Des mutations des gènes de susceptibilité au cancer du sein sont associées à un risque accru de développer la maladie. (Voir la section 2.0 Déterminants génétiques du cancer su sein.)38
L'histoire médicale personnelle et familiale :
Tel qu'abordé précédemment (voir la section 1.2.2 Lésions et maladies du sein), certaines
lésions bénignes du sein sont associées à un risque accru de cancer (Tableau 1). En général,
les lésions du sein présentant de l'hyperplasie (qui constitue une prolifération du nombre de
cellules formant un tissu) présentent un risque légèrement accru de cancer. Ce risque est
encore augmenté si cette prolifération cellulaire est accompagnée d'atypie, c'est-à-dire que
les cellules n'ont plus leur morphologie normale. L'hypothèse actuelle suggère que des
carcinomes mammaires pourraient se développer à partir de lésions prolifératives avec

10
atypie, et que ces dernières pourraient avoir pour origine les lésions prolifératives sans
atypie.11 C'est pourquoi les lésions du sein présentant des caractéristiques de prolifération,
en particulier celles avec de l'atypie, sont généralement considérées comme des lésions pré
cancéreuses.
En plus de ces maladies bénignes du sein, une histoire personnelle de cancer du sein
confère 2 à 6 fois le risque de la population en général de développer une seconde tumeur
primaire du sein. Une tumeur est dite «primaire» si elle se développe à partir des cellules
du tissu en question, dans le cas présent le sein, qui sont devenues cancéreuses. Une tumeur
sera dite «secondaire» si elle a pour origine des cellules détachées d'une tumeur dans un
autre organe, qui se sont propagées dans le corps (métastases). Parmi les facteurs affectant
l'incidence de cancer contralateral, on reconnaît, outre l'histoire familiale de cancer, l'âge
au diagnostic du premier cancer primaire. Par exemple, l'incidence «âge spécifique» du
cancer du sein contralateral est 50 fois plus élevée dans le groupe 30-34 ans que l'incidence
de cancer unilatéral au même âge.39 Ensuite, l'incidence de cancer contralateral diminue
avec l'accroissement de l'âge au diagnostic du premier cancer, peut-être dû en partie à
l'épuisement d'une sous-population à risque.39 Il est à remarquer que l'épidémiologie du
second cancer est tributaire de facteurs tels l'hérédité, le mode de vie, le traitement
administré suite au premier cancer, mais aussi à la surveillance accrue faisant partie
intégrante du suivi de la patiente suite au premier cancer.
De la même façon qu'une histoire personnelle de maladie du sein (bénigne ou maligne) est
associée à un risque accru de cancer du sein, l'histoire familiale joue aussi un rôle dans la
susceptibilité à ce cancer, et représente un facteur de risque majeur. Une femme ayant une
parente au premier degré (mère, sœur, fille) ayant été diagnostiquée d'un cancer du sein
présente un risque relatif de cancer du sein deux fois plus élevé.37 Le risque conféré
augmente s'il s'agit d'un cancer bilatéral et/ou si ce cancer est précoce (avant la
ménopause). Le risque est aussi augmenté avec le nombre de parentes au premier degré
atteintes (et de façon moins importante le nombre de parentes au deuxième degré), ainsi
qu'avec une histoire familiale de maladie bénigne du sein.36'37,40 Le regroupement de cas de
cancer du sein à l'intérieur d'une même famille met en évidence le partage de facteurs de
risque liés au mode de vie, mais surtout l'importance de l'hérédité dans la susceptibilité au

11
cancer du sein. Que ce soit de façon directe (par la transmission d'allèles de susceptibilité
au cancer du sein; voir la section 2. Déterminants génétiques du cancer du sein) ou
indirecte (par exemple des variants de gènes régulant un facteur de risque du cancer du sein
tel que la densité mammaire41), l'hérédité représente fort probablement une composante
pivot dans la détermination de quelle femme développera un cancer du sein au cours de sa
vie.

12
2. Déterminants génétiques du cancer du sein
2.1 Anecdotes historiques : cancer du sein, histoire familiale et prédisposition Le cancer, en particulier le cancer du sein, est connu depuis longtemps (Figure 5). Déjà
parmis les plus vieux traités médicaux connus, le papyrus Edwin Smith et le papyrus Georg
Ebers de l'époque pharaonique font mention de cancers, en particulier le cancer du sein.
Toutefois, sa prévalence devait être moindre que celle d'aujourd'hui, dû entre autres à des
changements importants du mode de vie, de l'exposition aux facteurs de risque
environnementaux, de l'espérance de vie, ainsi que des options de traitement disponibles.
L'idée d'un regroupement familial des cas de cancer du sein, bien que beaucoup plus
récente, n'est pas nouvelle. Rapporté comme une curiosité dans la littérature médicale de
l'époque gréco-romaine,42 le diagnostic de plusieurs cas au sein d'une même famille était
plutôt associé à une exposition à des facteurs environnementaux communs. Hippocrate
suggérait que la maladie est un état lors duquel le corps expérimente une difficulté à
maîtriser son environnement. Galien, un médecin réputé du Ile siècle, notait au sujet des
maladies «qu'aucune cause externe n'est efficace sans une prédisposition du corps lui-
même. Autrement, les causes externes qui affectent l'un nous affecteraient tous».
Il fallut attendre les 18e et 19e siècles avant de retrouver dans la littérature médicale des
anecdotes reliant l'histoire familiale et le cancer du sein. En 1757, le chirurgien français Le
Dran est appelé à consulter au sujet d'une religieuse de 19 ans atteinte d'un cancer du sein.
Convaincue de la nature héréditaire et systémique de sa maladie puisque sa grand-mère et
un grand-oncle en étaient décédés, elle affirmait que son sang «était porteur d'un ferment
cancéreux naturel à sa famille».43 La première étude détaillée d'une famille présentant de
nombreux cas de cancer du sein ne devait paraître que plus de 100 ans plus tard. Un autre
chirurgien français, Paul Broca, étudie l'histoire médicale d'une famille présentant 16 cas
de mort par cancer sur 4 générations, en particulier 10 cas de cancer du sein. Ses
observations, publiées en 1866, indiquent que l'influence de l'hérédité sur le
développement de tumeurs demeurait un sujet de controverse, bien que lui-même soit
convaincu qu'un tel lien puisse être établi, du moins dans certains cas.44 Selon les

13
estimations de son contemporain Lebert, un cancéreux sur sept rapporte une histoire
familiale de cancer 45
de fossiles indique la présence de tumeurs (os;
25sv .JC-5*Ccisas Première classification de tumeurs du sein 1713 Observe une fréquence accrue de
cancer du sein chez les nonnes 1*51 Heraaaaa Lebert
Traité pratique des cancéreuses et affections curables 1866 Paul Broca Traité des tumeurs 1*90 David voa Haasemann Le cancer est un désordre génétique 1900 DeVries/Correns/Tsctiennak Redécouverte des travaux de Mendel
1913 Alfred Scott Warthin Etude de famille» présentant une féquence élevée de cancen
1953 Ni L'accumulatiôô de mutations peut expliquer
la distribution de l'incidence de cancers 1M0 HwreU et Haaterford Chromosome de Phil
1976 Peter \owell Accumulation et selection de changements
génétique» lors de la tirrnorigenése
19*3 Cavanee et collaborateurs Découverte du premier gène supresseur de tumeurs
l9»Uviag»oaetHariow Point» de restriction du cycle cellulaire
1994 M U ef ca-aaaboratean i du gène BRCA1
1995 Waaster et eoUaboraSmrtr Identification du gène BRCA2
—30*4-15*0 av. JC Papyrus Smith et Ebers
4*»-375ar.JC Le cancer ressemble i un crabe (catemos)
Caiea 131-200
^ W T ^ ë S T Û D Ï - ï r
Gregor Mendd 1865 Lois de l'hérédité
Gearge Beatsoa 18 Réduction d'une tumeur chez une patiente c
1901-1914 Theodor I Les chromosomes sont des unités de 1 '1 et des abandons de ceux-ci causent le cancer
LathrapetL«ebl91S| htfhimcn hormonales sur le développement du c
Watson et Crick 1953 Structure de l'ADN
Alfred Kaadsoa 1971 Hypothèse des deux «coups»
1977 Méthode de séquençage dé l'ADN
Mallk,SaildetcaUaborateanl9M Réaction de polymérisation en chaîne (PCR)
Hal et collaborate»!-! 199* Premier locus de suceptibilité au cancer du sein
Wooster et collaborateurs 1994 Deuxième locus de suceptibilité au cancer du sein
Eastaa et colaboratean 2 « 7 Association de variants communs avec le cancer du sein
(FGFR2, TNRC9/LOC643714, MAP3K, LSP1.8q)
Figure 5. Événements sélectionnés de l'histoire de la génétique et du cancer.
Près de 30 ans après Broca, Alfred Scott Warthin s'intéresse à la question de l'hérédité en
relation avec le cancer lorsqu'il remarque en 1895 l'état dépressif de sa couturière,
convaincue de sa mort éventuelle du cancer dû à la fréquence très élevée de cette affliction
dans sa famille. Cela le conduit à faire l'étude détaillée de l'histoire médicale de cette
famille, ainsi que d'autres familles présentant plusieurs cas de cancer. Il publiera, en 1913,
la première étude comprehensive d'histoires familiales de cancer et mettant en relief leur
caractère apparemment héréditaire, ce qui devait paver la voie à l'étude moderne des

14
familles à risque élevé de cancer.4 Entre autres il concluait que la tendance héréditaire
semblait plus marquée lorsque la famille présentait une histoire familiale de cancer à la fois
du côté maternel et paternel. De plus, il observait que dans une famille présentant des
carcinomes sur plusieurs générations, il existe une tendance à développer les néoplasmes à
un âge précoce chez les membres des générations les plus récentes.
Les décennies subséquentes voient se multiplier les descriptions de familles présentant de
nombreux cas de cancer. À travers la quantité brute de cas de cancer du sein diagnostiqué,
on pouvait maintenant identifier, comme les familles décrites par Broca et Warthin, des
familles présentant des syndromes de cancer du sein héréditaires, chacun caractérisé par
une combinaison de tumeurs (Tableau 3). Le plus commun de ceux-ci est le syndrome de
cancers du sein et/ou de l'ovaire héréditaire (hereditary breast and ovarian cancer
syndrome), décrit dans la littérature au début des années 70.47'48
En parallèle de ces études, le 20e siècle voit se développer une nouvelle science dérivée de
la biologie, la génétique. Peu à peu, tel que prédit par les travaux de Boveri au début du 20e
siècle, les bases génétiques de l'hérédité et de plusieurs maladies, dont le cancer, sont
découvertes. Entre autres, on assiste à l'identification de gènes suppresseurs de tumeurs,
dont le dérèglement est associé au développement de tumeurs. Ainsi, un modèle de
développement du cancer dans lequel la cellule accumule des mutations devant
éventuellement mener au développement tumoral est articulé par Knudson au début des
années 70.49 Selon ce modèle, le premier «hit» peut être acquis ou hérité, d'où la
transmission d'une susceptibilité telle que celle qui est observée dans les syndromes de
cancers héréditaires. C'est à travers l'étude de ces familles, en particulier les familles du
syndrome de cancer du sein et de l'ovaire héréditaire, que l'identification de gènes de
susceptibilité de forte penetrance a été effectuée.
2.2 Gènes de susceptibilité au cancer du sein connus
Tel qu'illustré précédemment, l'accumulation de certains types de cancers à l'intérieur de
certaines familles a apporté un intérêt particulier à comprendre la place de la susceptibilité
héréditaire de l'individu dans la carcinogenèse. En effet, le cancer du sein tend à se
présenter en regroupement à l'intérieur de certaines familles et plus de 12% des femmes
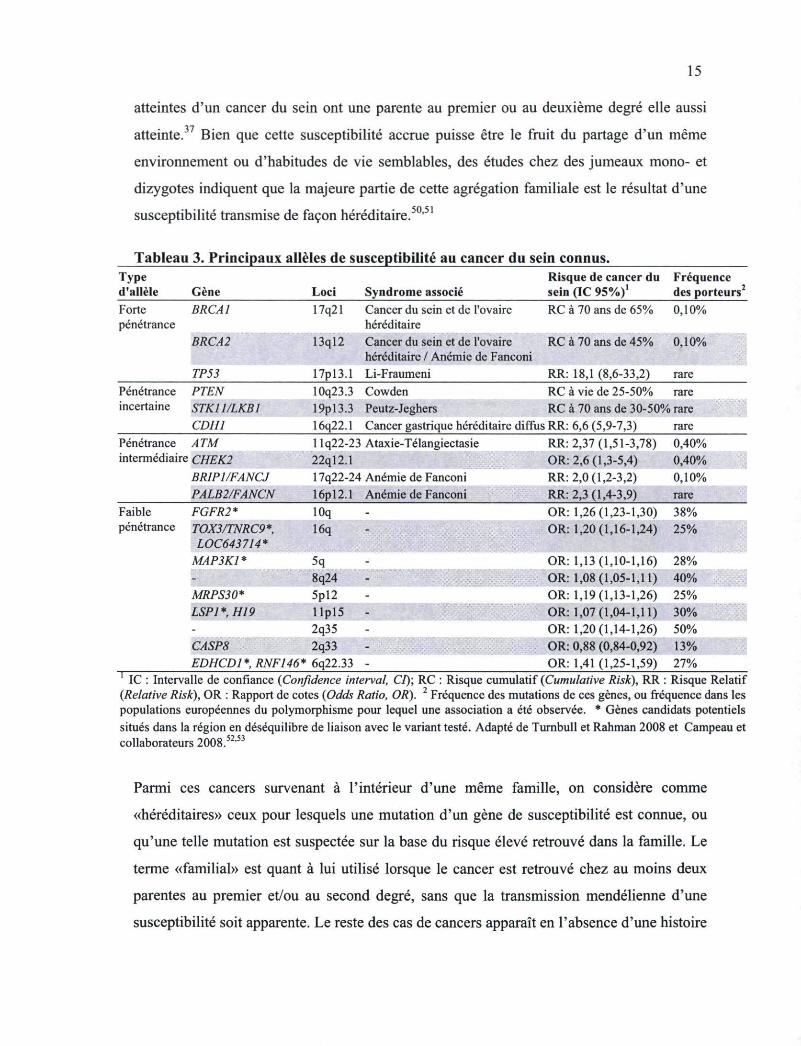
15
atteintes d'un cancer du sein ont une parente au premier ou au deuxième degré elle aussi
atteinte.37 Bien que cette susceptibilité accrue puisse être le fruit du partage d'un même
environnement ou d'habitudes de vie semblables, des études chez des jumeaux mono- et
dizygotes indiquent que la majeure partie de cette agrégation familiale est le résultat d'une
susceptibilité transmise de façon héréditaire.50,51
Tableau 3. Principaux alleles de susceptibilité au cancer du sein connus. Type (l'allèle Gène Loci Syndrome associé
Risque de cancer du Fréquence sein (IC 95%)a des porteurs
Forte BRCA1 penetrance
BRCA2
17q21 Cancer du sein et de l'ovaire RC à 70 ans de 65% 0,10% héréditaire
13ql2 Cancer du sein et de l'ovaire RC à 70 ans de 45% 0,10% héréditaire / Anémie de Fanconi
TP53 17pl3.1 Li-Fraumeni RR: 18,1 (8,6-33,2) rare Penetrance PTEN 10q23.3 Cowden RC à vie de 25-50% rare incertaine STK11/LKB1 19pl3.3 Peutz-Jeghers RC à 70 ans de 30-50% rare :;; v
CDH1 16q22.1 Cancer gastrique héréditaire diffus RR: 6,6 (5,9-7,3) rare Penetrance A TM llq22-23 Ataxie-Télangiectasie intermédiaire CHEK2 22ql2.1
BRIP1/FANCJ 17q22-24 Anémie de Fanconi PALB2/FANCN 16pl2.1 Anémie de Fanconi
Faible FGFR2* lOq penetrance TOX3/TNRC9*.
LOC643714* 16q
MAP3K1 * 5q
RR: 2,37 (1,51-3,78) 0,40% OR: 2,6 (1,3-5,4) 0,40% RR: 2,0 (1,2-3,2) 0,10% RR: 2,3 (1,4-3,9) rare OR: 1,26(1,23-1,30) 38% OR: 1,20(1,16-1,24) 25%
MAP3K1 * 5q 8q24 - ^ i m ^ ^ Ê m S -
MRPS30* 5pl2 LSP1*,H19 l lp l5 -
2q35 2q33
OR: 1,13(1,10-1,16) 28% OR: 1,08 (1,05-1,11) 40% OR: 1,19(1,13-1,26) 25% OR: 1,07 (1,04-1,11) 30% OR: 1,20(1,14-1,26) 50% OR: 0,88 (0,84-0,92) 13%
EDHCD1*,RNF146* 6q22.33 OR: 1,41 (1,25-1,59) IC : Intervalle de confiance (Confidence interval, CI); RC : Risque cumulatif (Cumulative Risk), RR : Risque Relatif
(Relative Risk), OR : Rapport de cotes (Odds Ratio, OR). 2 Fréquence des mutations de ces gènes, ou fréquence dans les populations européennes du polymorphisme pour lequel une association a été observée. * Gènes candidats potentiels situés dans la région en déséquilibre de liaison avec le variant testé. Adapté de Turnbull et Rahman 2008 et Campeau et collaborateurs 2008.52'53
Parmi ces cancers survenant à l'intérieur d'une même famille, on considère comme
«héréditaires» ceux pour lesquels une mutation d'un gène de susceptibilité est connue, ou
qu'une telle mutation est suspectée sur la base du risque élevé retrouvé dans la famille. Le
terme «familial» est quant à lui utilisé lorsque le cancer est retrouvé chez au moins deux
parentes au premier et/ou au second degré, sans que la transmission mendélienne d'une
susceptibilité soit apparente. Le reste des cas de cancers apparaît en l'absence d'une histoire

16
familiale de cancer du sein et sont habituellement appelés des cas «sporadiques» (Tableau
4).54 Cependant, la découverte de nouveaux alleles de susceptibilité et l'étude exhaustive
des antécédents familiaux liés à certains cas pourraient permettre de reclassifier une partie
des cancers familiaux (et même certains cancers sporadiques) en tant que cancers
héréditaires.
Tableau 4. Caractéristiques des familles présentant des cas de cancer du sein héréditaires, familiaux et sporadiques.
Classification Caractéristiques des familles Cancer Transmission autosomique dominante apparente de type spécifique de cancer héréditaire Âge p*us j e u n e a u diagnostic du cancer que ce qui est attendu
Multiples cancers primaires chez un même individu Regroupement de cancers rares Cancer bilatéral ou multifocal Parents au premier degré des individus atteints ont un risque de 50% d'être porteurs de la même mutation Penetrance incomplète et expression variable, de telle façon que les porteurs obligatoires de la mutation familiale peuvent ne pas être affectés par le cancer et que l'âge au diagnostic du cancer parmi les parents sera variable Les individus qui n'ont pas la mutation familiale ont le même risque que la population générale de développer un cancer
Cancer Plus de cas d'un ou plusieurs type(s) de cancer(s) à l'intérieur d'une même famille que ce qui est familial statistiquement attendu, mais pas de patron d'héritabilité clair
Âge variable au diagnostic Peut résulter du regroupement par chance de cas sporadiques Peut résulter de facteurs génétiques communs, d'un environnement et/ou d'habitudes de vie similaires Ne présente pas habituellement les caractéristiques classiques des syndromes de cancers héréditaires
Cancer Les cancers dans la famille sont probablement dus à des causes non héréditaires sporadique Âge du diagnostic typique
Même s'il y a plus d'un cas dans la famille, il n'y a pas de patron de transmission héréditaire clair La probabilité est très basse que la recherche de mutations de gènes de susceptibilité sera positive, le test génétique n'offrira pas d'information supplémentaire sur le risque de cancer
Adapté de Berliner et collaborateurs 2007.
L'étude de l'héritabilité d'une prédisposition au cancer du sein a permis de mettre en
évidence trois catégories d'allèles de susceptibilité, chacune de ces catégories étant associée
à un risque relatif et à une prévalence dans la population.38 La première classe est
constituée d'allèles rares de forte penetrance (BRCA1, BRCA2, TP53) et de penetrance
incertaine (PTEN, STK11, CDH1) associés à un risque élevé de cancer du sein, et qui ont
été identifiés par l'intermédiaire de syndromes familiaux. La seconde catégorie d'allèles de
susceptibilité regroupe des alleles relativement rares et de penetrances modérées (ATM,

17
CHEK2, BR1P1IFANCJ, PALB2IFANCN), associés à un risque moins grand de développer
un cancer du sein. Ces gènes ont été identifiés principalement par l'intermédiaire d'une
approche de type «gène candidat» dans laquelle la recherche de mutations est effectuée
directement dans un gène sélectionné sur la base d'une probabilité antérieure liée à sa
fonction. La toute dernière catégorie d'allèles récemment identifiés est constituée d'allèles
communs de faible penetrance, associés à un risque faible de cancer du sein. Puisque ces
alleles ne confèrent qu'une augmentation très faible de la susceptibilité au cancer du sein,
leur identification nécessite l'utilisation d'études de grande envergure regroupant plusieurs
milliers d'individus provenant de plusieurs pays et représentant habituellement plusieurs
ethnies. Les caractéristiques de ces gènes et de ces syndromes sont résumées au Tableau 3.
2.2.1 Alleles de forte penetrance
2.2.1.1 Le syndrome de cancer du sein et de l'ovaire héréditaire, BRCA1 et BRCA2 Ce syndrome de cancer du sein héréditaire le plus couramment retrouvé chez les familles
présentant une forte histoire familiale. On doit à Henry Lynch, au début des années 1970, la
réalisation qu'une agrégation de cas de cancers du sein et de l'ovaire47'48 semble souvent
apparaître à un jeune âge dans certaines familles.55 Le syndrome HBOC est caractérisé par
la présence de cancers du sein avant la ménopause, de cancers de l'ovaire (peu importe
l'âge), de cancers du sein bilatéraux, de cancers du sein chez l'homme, et par la présence
chez un même individu de cancer du sein et de l'ovaire. L'analyse de ces familles semblait
suggérer l'existence d'un (de) gène(s) de susceptibilité au cancer du sein et/ou de l'ovaire
se transmettant dans ces familles selon un mode autosomique dominant et présentant une
penetrance élevée.56 La première localisation convaincante d'un locus de susceptibilité au
cancer du sein a été présentée par Hall et collaborateurs en 1990.57 L'identification de ce
locus au chromosome 17q21, appelé Breast Cancer locus 1, a précédé de quelques années
la localisation d'un second locus au chromosome 13ql2-1358 et l'identification des deux
gènes impliqués, en 1994-1995 : BRCA1 e\BRCA2 (Breast cancer 1/2, early onset).59'60
Prévalence des mutations de BRCA1 et BRCA2
La détermination de la prévalence exacte des mutations de BRCA1 et BRCA2 est un
problème complexe, puisque cette dernière est influencée par la population (ou le sous-

18
groupe de patients) et la méthode de détection employée. En effet, concernant les cas de
cancers du sein et de l'ovaire provenant de la population générale, la fréquence des
mutations de BRCA1 est estimée à 0-7% alors que celle de BRCA2 est estimée à l-3%.61
Lorsque la recherche de mutations est restreinte à des patients présentant des
caractéristiques associées à des syndromes héréditaires (cancer de l'ovaire, cancer du sein
précoce, cancer du sein chez l'homme) mais en l'absence d'histoire familiale établie ou
rapportée, la prévalence des mutations BRCA1 et BRCA2 augmente (Tableau 5). En
général, on estime que des mutations de BRCA1 et BRCA2 sont retrouvées dans 16-24%
des familles présentant une histoire familiale,62"65 et on observe une proportion variable des
cas reliés à BRCA1 et BRCA2 en fonction du type d'histoire familiale (Figure 6).66'67 Ainsi,
des mutations de BRCA1 sont retrouvées dans une grande proportion des familles chez
lesquelles on observe à la fois des cancers du sein et de l'ovaire, alors qu'il est plus
probable de retrouver des mutations de BRCA2 au sein de familles présentant des cas de
cancer du sein chez l'homme.
Tableau 5. Pourcentage des familles présentant des mutations de BRCA1 ou BRCA2, selon le type de cas sélectionné.
Type de cas BRCA1 (%) BRCA2 (%) Cancers du sein et de l'ovaire, non sélectionnés sur la base de l'histoire familiale Cancers de l'ovaire, sans indication d'histoire familiale Cancers du sein précoces, sans indication d'histoire familiale Cancers du sein chez l'homme, sans indication d'histoire familiale Cancers du sein avec la présence d'une histoire familiale
Adapté des données de Fackenthal et Olopade 2007.
Penetrance des mutations de BRCA1 et BRCA2
Suite à l'identification de BRCA1 et BRCA2, il devint rapidement évident que des
mutations de ces deux gènes conféreraient un risque élevé de cancer du sein et de l'ovaire.
Les premières estimations du risque encouru par les porteurs de mutations de BRCA1 et
BRCA2 ont été effectuées chez des familles sélectionnées sur la base de leur histoire
familiale très sévère de cancer du sein et/ou de l'ovaire. Ces études ont permis de
déterminer un risque associé à des mutations de BRCA1 de 87% pour le cancer du sein et
entre 44% pour le cancer de l'ovaire.68 Pour ce qui est des mutations de BRCA2, le risque a
été estimé au sein de ces familles à 84% (IC 95% 43-95%) et le risque de cancer de l'ovaire
à 27% (IC 95% 0-47%).69 Les hommes porteurs de mutations de BRCA2 sont à risque plus
1-7 1-3 4-29 0,6-16 0,7-10 1-6 - 7-14 0-29 1,5-25

19
élevé de développer un cancer du sein (6,9% à 80 ans ; IC 95% l,2-38,6%).70 Bien que les
hommes porteurs de mutations de BRCA1 soient à risque moins élevé, le risque de cancer
du sein atteint tout de même 5,8% (IC 95% 1,3-10,4%), ce qui est significativement plus
élevé que le risque encouru par la population masculine générale.71
«Caa»
Toutes les famille*; >6 1 4-S
Toutes les famille*; >6 1 4-S
Toutes les famille*;
Familles avec uniquement des cas de cancer du sein chez la femme
?6
4-5 aaaaaaaH Familles avec uniquement des cas de
cancer du sein chez la femme ?6
4-5
Toutes les familles sauf celles avec des cas dc cancer du 1 sein chez l'homme (incluant des cancers ovariens)
Familles avec des cas de cancer du sein et de l'ovaire. mais sans cancer du sein chez l'homme
Familles a\cc des cas de cancer du sein chez l'homme
?6 Toutes les familles sauf celles avec des cas dc cancer du 1 sein chez l'homme (incluant des cancers ovariens)
Familles avec des cas de cancer du sein et de l'ovaire. mais sans cancer du sein chez l'homme
Familles a\cc des cas de cancer du sein chez l'homme
?6 Toutes les familles sauf celles avec des cas dc cancer du 1 sein chez l'homme (incluant des cancers ovariens)
Familles avec des cas de cancer du sein et de l'ovaire. mais sans cancer du sein chez l'homme
Familles a\cc des cas de cancer du sein chez l'homme
4-5 >-» 1
Toutes les familles sauf celles avec des cas dc cancer du 1 sein chez l'homme (incluant des cancers ovariens)
Familles avec des cas de cancer du sein et de l'ovaire. mais sans cancer du sein chez l'homme
Familles a\cc des cas de cancer du sein chez l'homme
4-5 >-» 1
Toutes les familles sauf celles avec des cas dc cancer du 1 sein chez l'homme (incluant des cancers ovariens)
Familles avec des cas de cancer du sein et de l'ovaire. mais sans cancer du sein chez l'homme
Familles a\cc des cas de cancer du sein chez l'homme
4-5 >-» 1
Toutes les familles sauf celles avec des cas dc cancer du 1 sein chez l'homme (incluant des cancers ovariens)
Familles avec des cas de cancer du sein et de l'ovaire. mais sans cancer du sein chez l'homme
Familles a\cc des cas de cancer du sein chez l'homme
4-5 >-» 1
>- 1 L- «
' ? - • ::•-• ■SC D ÔQ-.'c 50° c 100%
■ BRCA1 ■ BRCA2 Autre
Figure 6. Estimation de la proportion des familles présentant une forte histoire familiale pouvant être associées à des mutations de BRCA1, BRCA2 ou aucun de ces deux gènes. D'après les données de Ford et collaborateurs.**
Cependant, les études subséquentes, basées en majeure partie sur des méthodologies
différentes, ont résulté en des estimations variables du risque et souvent inférieures. Par
exemple, une méta-analyse regroupant 22 études a estimé un risque cumulatif de cancer du
sein à 70 ans de 65% (IC 95% 51-75%) pour les porteurs de mutations de BRCA1 et de
45% (IC 95% 33-54%) pour les porteurs de mutations de BRCA2. Le risque de cancer de
l'ovaire au même âge est de 39% (IC 95% 22-51%) pour BRCA1 et de 11% (IC 95% 4,1-
18%) pour BRCA2.72 Souvent biaisées par l'utilisation de l'histoire familiale, et non le test
moléculaire direct des porteurs présumés et la validation de leur statut clinique, les
estimations basées sur des cohortes de patients (au lieu de familles) tendent à estimer un
risque inférieur à celui obtenu avec des familles présentant une histoire médicale chargée.
Cependant, les ICs associés à ces estimations se chevauchent et, dans plusieurs cas, ne
présentent pas de différences significatives. Bien qu'une part de la variabilité associée à ces
estimations est clairement due à des différences méthodologiques, il semble que la
penetrance d'une mutation en particulier puisse être influencée par des facteurs génétiques
et environnementaux propres aux patients, comme c'est le cas par exemple pour certaines
mutations fondatrices.

20
Il est maintenant clair que la penetrance peut être influencée par une corrélation génotype-
phénotype puisque les estimations présentées sont la plupart du temps des moyennes d'un
ensemble de mutations. Cependant, certaines études semblent démontrer un effet
hétérogène dans lequel la position de certaines mutations pourraient influencer le risque
conféré. L'analyse des mutations de BRCA2 a permis de démontrer que la présence de
mutations dans la région centrale du gène (appelée le ovarian cancer cluster region ;
OCCR) est associée à un ratio plus élevé de cancers de l'ovaire que de cancers du sein,
contrairement aux mutations retrouvées dans les régions 5' et 3' du gène.70'73 Une
association similaire a aussi été démontrée pour BRCA1.74 Il semble aussi probable que
certaines mutations spécifiques puissent avoir un profil de risque particulier, bien que cette
hypothèse demeure à être confirmée. Les études visant à déterminer le risque de cancer du
sein et de l'ovaire chez les porteurs de mutations de BRCA1 et BRCA2 ont mis en évidence
la présence d'une variabilité du risque entre les familles de même qu'à l'intérieur d'une
même famille.64'69 La raison exacte de cette variabilité du risque entre les porteurs n'est pas
claire, mais elle peut conceptuellement être le résultat du partage d'allèles de gènes de
modification ou de facteurs de risque environnementaux.
Bien que plusieurs facteurs de risque environnementaux aient été associés au risque général
de développer un cancer du sein (voir la section 1.3 Facteurs de risque de développer un
cancer du sein), leur influence sur le risque encouru par les porteurs de mutations de
BRCA1 et BRCA2 est moins claire. Certains facteurs de risque (e.g. la densité mammaire ou
l'effet de la parité) semblent avoir le même effet sur le risque des porteurs que dans la
population générale,75'76 alors que d'autres facteurs (e.g. l'âge de la ménarche, l'utilisation
de contraceptifs oraux ou l'allaitement) semblent agir différemment.77"79 Il est à noter que la
densité mammaire est déterminée en forte partie par des composantes génétiques,80 ce qui
offre des évidences de l'impact potentiel d'autres gènes sur le risque auquel un porteur peut
être sujet.
La variabilité du risque observé peut aussi être attribuée à des polymorphismes ou des
mutations d'autres gènes. Une évidence de ce phénomène est la présence d'un
regroupement familial du site du cancer (sein ou ovaire) en fonction du type de cancer
retrouvé chez le cas de référence. On remarque que dans une famille où le cas de référence

21
est un cas de cancer de l'ovaire, les porteurs ont un risque plus élevé de développer un
cancer de l'ovaire qu'un cancer du sein. De la même façon, dans une famille où le cas de
référence est un cancer du sein, le risque de cancer du sein est plus grand chez les porteurs
que le risque de cancer de l'ovaire. ' Cette corrélation est observée sur de multiples
générations, avec des facteurs environnementaux variables, ce qui suggère fortement
l'influence de gènes pouvant modifier le risque conféré par les mutations de BRCA1 et
BRCA2. De plus, dans les familles où l'on retrouve une mutation de BRCA1 ou de BRCA2,
il est souvent assumé que les individus qui obtiennent un test négatif pour la présence de la
mutation sont au même risque que la population générale. Cependant, certaines études
semblent démontrer que le risque chez ces individus serait de 2 à 5 fois supérieur à celui de
la population.82'83 Cette augmentation pourrait être le résultat de facteurs génétiques et/ou
environnementaux se regroupant dans ces familles. Jusqu'à présent, l'association la plus
claire d'un allele de modification semble être retrouvée pour le variant de la région 5' non-
codante de RAD51 (Rad51 homolog, S. cerevisiae) 135G>C, qui semble en altérer
l'expression. Ce polymorphisme augmente le risque chez les porteurs de mutations de
BRCA2 (HaR (Rapport de risque, Hazard ratio) 1.17 IC 95% 0,91-1,51 chez les
hétérozygotes et de 3,18 IC 95% 1,39-7,27 chez les homozygotes).84 Certains variants dans
d'autres gènes pourraient aussi modifier le risque de cancer chez les porteurs BRCA1/2, du
moins dans la population ashkénaze.85'86 Récemment, des études ont permis de mettre en
évidence certains variants communs de susceptibilité au cancer du sein, identifiés suite a
des études de grande envergure de type tour du génome (genome-wide association study,
GWAS).*1'90 Certains de ces variants ont par la suite été identifiés comme modificateurs du
risque chez les porteurs de mutations de BRCA1 et/ou de BRCA2 (voir la section 2.2.4
Alleles de faible penetrance).91
En plus du risque de cancer du sein et de l'ovaire, des mutations de BRCA1 et BRCA2 sont
aussi associées à un risque accru de cancers à d'autres sites anatomiques. Un résumé des
principales études évaluant le risque d'autres cancers associé à ces mutations est présenté
au Tableau 6. Le type de cancer pour lequel les évidences d'une susceptibilité accrue sont
les plus fortes est le cancer du pancréas, chez les porteurs de mutations de BRCA2.92'95 Le
risque conféré est cependant beaucoup plus faible que celui associé au cancer du sein et de
l'ovaire et varie entre les études, probablement dû à des différences méthodologiques. En

22
plus d'être associé à d'autres types de cancers, certaines évidences semblent suggérer que
des mutations de BRCA2 pourraient être impliquées dans certains cas du syndrome de Li-
Fraumeni96, bien que l'occurrence de familles où l'on retrouve une mutation de BRCA2
présentant les caractéristiques de ce syndrome (voir la section 2.2.1.2 Le syndrome de Li-
Fraumeni et 77*5.?) soit relativement rare. Lorsque les deux alleles de BRCA2 sont
inactivés, les patients souffrent d'Anémie de Fanconi (AF ; Fanconi Anemia, FA) de sous-
groupe Dl, qui se manifeste par défaillance progressive de la moelle osseuse et une
instabilité chromosomique (voir la section 3.2.2.2 Implication de BRCA1 et BRCA2 dans
l'Anémie de Fanconi).97 De plus, les patients du sous-groupe Dl présentent une
susceptibilité marquée à certains types de cancers infantiles.98"101
Tableau 6. Risque d'autres types de cancers associé à une mutation délétère de BRCA1 ou BRCA2. Organe RR (IC 95%)
BRCA1 Thompson et coll. 200274 Brose et coll., 200271 Ford et coll. 199466
Pancréas 2,26(1,26-4,06) 2,8(1,46-4,07) Col de l'utérus 3,72 (2,26-6,10) Corps de l'utérus 2,65 (1,69-4,16) Prostate 1,82(1,01-3,29) 3,33 (1,78-6,20) Colon 2,03(1,45-2,85) 2,0(1,5-2,5) 4,11(2,36-7,15) Foie 4,06(1,77-9,34) Gastrique 6,9 (4,25-9,38)
BRCA2 BCLC 1999102 van Asperen et coll. 2005103
Pharynx et cavité buccale 2,26(1,09-4,58) 7,3(2,0-18,6) Pancréas 3,51 (1,87-6,58) 5,9(3,2-10,0) Prostate 4,65 (3,48-6,22) 2,5(1,6-3,8) Os 14,4(2,9-41,1) Mélanomes malins 2,58(1,28-5,17) Vésicule biliaire 4,97(1,50-16,52) Estomac 2,59(1,46-4,61) Foie 4,18(1,56-11,23)
2.2.1.2 Le syndrome de Li-Fraumeni et TP53 En 1969, Frederick Li et Joseph Fraumeni décrivent pour la première fois un syndrome de
sarcomes se développant à l'enfance, chez 4 familles présentant aussi une histoire familiale
médicale importante de cancers, dont des cancers du sein.104 Le syndrome de Li-Fraumeni
est caractérisé par une hérédité de type autosomique dominante, l'apparition de tumeurs à
un âge précoce, ainsi que le développement de tumeurs primaires multiples chez un même
individu. Plusieurs types de tumeurs sont retrouvés chez ces individus et les types les plus

23
courants sont des sarcomes, des cancers du sein, des tumeurs au cerveau, des leucémies et
des carcinomes des glandes surrénales. Ce n'est qu'en 1990 que Malkin et collaborateurs,
en se basant sur l'implication de TP53 (Tumor protein P53) dans la version somatique de
plusieurs des tumeurs associées au syndrome, identifient la présence de mutations du gène
TP53 au sein de certaines de ces familles.105'106 Les déterminants génétiques au sein des
autres familles restent à déterminer, bien que l'implication de mutations des gènes CHEK2
et BRCA2 aient été suggérées dans certaines de ces familles (voir les sections 2.2.3.2
CHEK2 et 2.2.1.1 HBOC, BRCA1 et BRCA2).
Le gène TP53 encode une protéine hautement conservée entre les espèces et qui agit dans le
choix de la cellule entre l'arrêt du cycle cellulaire pour la réparation de l'ADN ou
l'enclenchement du programme de mort cellulaire (apoptose).107 Cliniquement, on
reconnaît deux formes du syndrome de Li-Fraumeni : la forme classique (classic Li-
Fraumeni syndrome) et une forme évocatrice du syndrome, sans pour autant en remplir
exactement les caractères cliniques (Li-Fraumeni-like syndrome). Le diagnostic différentiel
entre ces deux formes est détaillé au Tableau 7. Des mutations de TP53 sont retrouvées
dans environ 70% des familles de la forme classique, alors qu'elles ne sont retrouvées que
dans environ 40% des familles du syndrome évocateur. Parmi les familles chez lesquelles
une mutation de TP53 a été identifiée, le risque de développer un cancer est de plus de 50%
à 30 ans et plus de 90% des porteurs de ces mutations développeront un cancer avant 70
ans.106 Ce risque très élevé à l'âge adulte est en majeure partie dû au risque important de
cancer du sein. On estime que le RR de cancer du sein associé à une mutation de TP53 est
de 18,1 (IC 95% 8,6-33,2).108 La majorité de ces cancers du sein surviennent entre 20 et 44
ans.109'110
Chez les familles du syndrome de Li-Fraumeni et porteuses de mutations du gène TP53, on
remarque que l'âge moyen au diagnostic de cancer du sein est inférieur comparé aux
familles de la forme évocatrice du syndrome sans mutation de TP53.n] On note aussi que
parmi ces familles porteuses de mutations TP53, l'âge moyen d'apparition du cancer du
sein est plus élevé lorsque la mutation est un changement faux-sens situé dans la boucle de
la protéine ne liant pas l'ADN ou dans le domaine d'oligomérisation, comparé aux
mutations faux sens des boucles liant l'ADN et aux mutations troncantes.111 Bien que les

24
mutations de TP53 soient hautement pénétrantes, le syndrome de Li-Fraumeni est un
syndrome rare, avec moins de 400 familles rapportées mondialement. Des mutations
germinales de TP53 sont rarement retrouvées chez des familles ne présentant que des cas de
cancer du sein et/ou de l'ovaire.110 Par conséquent, on estime que les mutations de TP53 ne
sont responsables que d'environ 1% des cas de cancer du sein.113
Tableau 7. Critères de diagnostic des familles Li-Fraumeni. Critères du syndrome de Li-Fraumeni
1 Cas de référence avec un sarcome osseux ou du tissu mou, diagnostiqué avant 45 ans ; 2 un parent au premier degré avec un cancer avant 45 ans ; 3 et un parent du premier ou du second degré de la même branche parentale avec un cancer avant 45 ans
ou un sarcome, peu importe l'âge. Critères du syndrome similaire au syndrome de Li-Fraumeni
1 Cas de référence avec un cancer de l'enfance ou un sarcome, une tumeur au cerveau ou un carcinome des glandes surrénales diagnostiqué avant 45 ans ;
2 avec un parent au premier ou au second degré avec un cancer typique de LFS (sarcomes, tumeurs au cerveau, un cancer du sein, une leucémie ou un carcinome surrénal), peu importe l'âge ;
3 et un parent au premier ou au second degré de la même branche parentale avec un cancer diagnostiqué avant 60 ans.
2.2.2 Alleles de penetrance incertaine 2.2.2.1 Le syndrome de Cowden et PTEN Le syndrome de Cowden est une maladie héritée de façon autosomique dominante dans
laquelle les patients présentent de multiples lésions hamartomateuses (peau, seins, intestins,
thyroïde) ainsi qu'un risque accru de cancers du sein, de la thyroïde et de l'endomètre. Les
individus atteints présentent habituellement une macrocéphalie et une atteinte de la peau et
des muqueuses. Bien que rapporté pour la première fois en 1963114, l'importance du cancer
du sein dans ce syndrome n'a été comprise que plus tard.115 Jusqu'à 75% des femmes
atteintes du syndrome de Cowden développeront une maladie bénigne du sein et le risque
de cancer du sein invasif est estimé à 25-50% au cours de la vie.116'117 L'âge moyen
d'apparition du cancer du sein est de 40-45 ans,116'117 avec quelques cas de cancer du sein
chez l'homme.118 Environ 5-10% des femmes atteintes du syndrome développeront un
cancer de l'endomètre, et le risque de cancer de la thyroïde est d'environ 10%.116
Des mutations de PTEN (Phosphatase tensin homolog on chromosome ten) ont été
identifiées dans environ 80-85% des cas de syndrome de Cowden, suggérant la présence de

25
mutations dans des régions non étudiées (le promoteur par exemple) ou alternativement,
l'implication d'autres gènes.119"121 PTEN encode une protéine suppresseur de tumeurs et
phosphatase à double spécificité (tyrosine et sérine/thréonine) impliquée dans la régulation
de la croissance en exerçant une régulation négative de la voie de signalisation de PI3K
(Phosphatidylinositol-3-kinase)/'AKT. Cette voie de signalisation est une composante
principale de plusieurs processus dérégulés lors de la progression tumorale tels
l'angiogenèse, la survie cellulaire et la transition épithelio-mésenchymateuse. PTEN
jouerait aussi un rôle important dans l'inhibition de la migration cellulaire et dans
l'inhibition de cyclines.120
Il semble que les mutations fronçantes de PTEN soient particulièrement associées à un
risque de cancer,123 bien qu'il ait été suggéré que les mutations de PTEN ne soient pas
associées à un risque accru de cancer du sein dans les familles ne présentant pas de
symptômes classiques du syndrome de Cowden.124"126 Depuis l'identification de mutations
de PTEN dans le syndrome de Cowden, des mutations de ce gène ont aussi été associées à
d'autres maladies ne présentant pas de lien évident avec ce syndrome. Des mutations de
PTEN ont jusqu'à présent été retrouvées dans environ 60% des familles avec le syndrome
de Bannayan-Riley-Rubalcava, 20% des familles avec le syndrome de Proteus et 50% des
familles avec un syndrome similaire au syndrome de Proteus.120 Les différences
phénotypiques observées entre ces syndromes sont jusqu'à présent difficilement
réconciliables avec une relation génotype/phénotype. Il est possible que le phénotype soit
influencé par l'action de gènes de modification, ou par une interaction avec
l'environnement.120 Il a récemment été suggéré que le syndrome de Bannayan-Riley-
Rubalcava constitue une manifestation juvénile du syndrome de Cowden.127 Puisque ces
syndromes (Cowden, Bannayan-Riley-Rubalcava, Proteus et Proteus-like) présentent des
symptômes cliniques se chevauchant et qu'ils sont en partie causés par des mutations de
PTEN, il a été suggéré de les regrouper sous la désignation de PTEN hamartoma tumor
syndrome.
2.2.2.2 Le syndrome de Peutz-Jegher et STK11 La présence de multiples polypes hamartomateux du système gastro-intestinal et des taches
pigmentaires bleues ou brunes des lèvres, de la muqueuse buccale et de la peau constituent

26
des symptômes clés du syndrome de Peutz-Jeghers, une maladie autosomique dominante
rare.128'129 Les patients atteints de ce syndrome présentent de plus un risque accru de
cancers du sein, du pancréas, du testicule, de l'endomètre, de l'ovaire, de l'utérus, du col,
des poumons, de l'estomac et du côlon.130"131 Il est estimé qu'entre 37% et 93% des patients
atteints du syndrome de Peutz-Jeghers développeront un cancer avant 65 ans.132 On estime
qu'entre 29 et 54% des patientes atteintes du syndrome de Peutz-Jeghers développeront un
cancer du sein avant 65 ans.130'132
Le syndrome de Peutz-Jeghers est causé par des mutations du gène STKll/LKBl
(Serine/threonine kinase 11/Liver kinase 57)133'134, encodant une sérine/thréonine kinase
impliquée dans l'inhibition de la prolifération cellulaire, la réponse cellulaire au stress
énergétique et le contrôle de la polarité cellulaire.135'136 Il est à noter que des mutations de
STK11 ne sont pas identifiées chez tous les patients, bien que les estimations aient pu être
faussées par la présence de grands réarrangements non-détectés.137"139 Puisque les patients
présentent une variabilité phénotypique intra- et inter-familiale, spécialement en ce qui
concerne le risque de cancer, il est possible que le phénotype soit influencé par des facteurs
environnementaux ou par l'action d'autres gènes. Des mutations somatiques de STK11 ont
été rapportées dans certains types de cancers, bien que ce ne semble pas être un phénomène
fréquent.140 L'inactivation épigénétique (hyperméthylation du promoteur) de STK 11 a aussi
été notée dans 50% des tumeurs papillaires sporadiques du sein, bien que le phénomène
semble peu commun ou absent dans les autres types de cancers du sein.141
2.2.2.3 Le syndrome du cancer gastrique héréditaire diffus et CDH1 Le cancer gastrique est une forme prévalente de cancer, en particulier dans les pays
asiatiques. On estime qu'entre 5% et 10% de tous les cancers gastriques seraient
familiaux.142 Le cancer héréditaire gastrique diffus constitue la forme autosomique
dominante de la susceptibilité à ce type de cancers. Il s'agit d'un adénocarcinome peu
différencié infiltrant la paroi de l'estomac, en causant l'épaississement sans pour autant
former une masse distincte. Ce syndrome s'accompagne aussi d'un risque accru de cancer
du sein de type lobulaire.143 Chez les patients atteints du syndrome, le risque cumulatif de
développer un cancer gastrique avant 80 ans est de 67% chez les hommes et de 83% chez
les femmes, avec un âge moyen d'apparition de la maladie de 38 ans.144'145

27
Des mutations de CDH1 (Cadherin 1), qui encode la cadhérine E, ont été associées à des
cas de cancers gastriques héréditaires diffus. Les cadherines sont des protéines impliquées
dans l'adhésion cellule à cellule dépendante du calcium.146 La fonction de la cadhérine E
est essentielle dans le maintien d'un epithelium polarisé et différencié au cours du
développement. Cette protéine joue un rôle important dans la transmission des signaux, la
différenciation, l'expression génique, la mobilité cellulaire et l'inflammation.146 Des
mutations de CDH1 ne sont identifiées que dans 30% à 50% des cas de cancer gastrique
héréditaire diffus.1 ' 7 Bien que l'incidence du cancer gastrique soit plus élevée dans les
pays asiatiques (Japon et Chine), la plupart des mutations de CDH1 ont été retrouvées dans
les populations européennes.147 Entre 39% et 52% des femmes porteuses d'une mutation de
CDHl ou atteintes du syndrome de cancer gastrique héréditaire diffus développeront un
cancer du sein de type lobulaire.148'149 On note aussi que dans de rares cas, des familles
porteuses d'une mutation de CDHl présentent uniquement, ou presque exclusivement, des
cas de cancers du sein de type lobulaire,150'151 ce qui peut suggérer que certaines mutations
de CDHl ne seraient pas identifiées en utilisant uniquement une sélection des cas basée sur
la présence de cancers gastriques diffus. L'expression de la cadhérine E est perdue dans la
plupart des cancers du sein de types lobulaires sporadiques, bien que son expression soit
maintenue dans les cancers du sein de type canalaire.152
2.2.3 Alleles de penetrance modérée
2.2.3.1 L'ataxie-télangiectasie et A TM L'ataxie-télangiectasie est une maladie autosomique récessive rare aussi connue sous le
nom de syndrome de Louis-Bar et, plus rarement, de syndrome de Boder-Segwick. Ce
syndrome a été décrit pour la première fois en 1926 par Syllaba et Henner,153 mais ce n'est
qu'en 1958 que Boder et Segwick étudient 8 cas provenant de 5 familles et introduisent la
dénomination d'ataxie-télangiectasie.154 Cette maladie se manifeste par une ataxie
cérébelleuse, des télangiectasies oculaires et cutanées, un déficit immunitaire et des
infections sino-pulmonaires à répétition. En plus de ces symptômes, les individus atteints
d'ataxie-télangiectasie ont un risque 100 fois plus élevé de développer un cancer que le
reste de la population.155 Les leucémies prédominent à l'enfance, alors que des cancers
épithéliaux, dont le cancer du sein, sont observés chez l'adulte.156

28
L'ataxie-télangiectasie est causée par des mutations du gène ATM (Ataxia-telangiectasia
mutated), clone en 1995.157 ATM joue un rôle central dans la réponse de la cellule aux
agents induisant des bris double-brins (BDBs) de l'ADN. ATM initie une cascade de
signalisation en induisant la phosphorylation de plusieurs protéines telles p53, BRCA1 et
CHEK2. En 1976 une étude épidémiologique conduite au sein de familles de patients
atteints d'ataxie-télangiectasie a démontré que les femmes apparentées à ces patients
présentent un risque accru de cancer,158 dont le cancer du sein.159 Une étude récente
regroupant 1160 individus de 169 familles de patients atteints d'ataxie-télangiectasie a
permis d'estimer que les porteurs d'une mutation du gène ATM ont un risque relatif de
cancer du sein de 2,23 (IC 95% 1,16-4,28) comparé au reste de la population. De plus, ce
risque semble plus élevé chez les femmes de moins de 50 ans (RR 4,94; IC 95% 1,90-
12,9).160 Ces résultats sont aussi consistants avec ceux de Olsen et collaborateurs
regroupant 1445 individus de 66 familles de patients atteints d'ataxie-télangiectasie.161
Une hypothèse découlant de ces études épidémiologiques veut que des mutations
hétérozygotes du gène ATM devraient être plus fréquemment observées chez les individus
atteints de cancer du sein que chez la population. En conséquence, l'étude de mutations du
gène ATM chez des individus atteints de cancer du sein a été effectuée par plusieurs
investigateurs avec des résultats mitigés.156'162 Dans une étude de cas de cancers du sein
familiaux comparés à des contrôles sains, Renwick et collaborateurs163 ont démontré une
association entre des mutations d'ATM et le cancer du sein, avec un risque relatif estimé à
2,37 (IC 95% 1,58-3,78), ce qui est similaire au risque estimé suite aux études
d'association. En conséquence, des mutations d'ATM semblent conférer un risque
d'environ deux fois, ce qui est généralement considéré comme un allele de susceptibilité de
penetrance intermédiaire.
2.2.3.2 CHEK2 En 1999, Bell et collaborateurs identifient des mutations du gène CHEK2 (Checkpoint
kinase 2) chez des familles atteintes de LFS pour lesquelles la recherche de mutations de
TP53 s'était avérée négative.164 Le rôle de mutations de CHEK2 en tant qu'allèles pouvant
causer un syndrome de cancer de forte penetrance tel le LFS a cependant été remis en
question par la suite. En effet, ces mêmes mutations ont aussi été retrouvées chez des

29
patientes atteintes de cancer du sein et des individus sains.1 Bien que le rôle de mutations
de CHEK2 comme agent causant le LFS soit disputé, il demeure possible que ces mutations
soient responsables de certains cas de cancer de sein dans les familles du syndrome de Li-
Fraumeni ou qu'il agisse comme gène de modification en contribuant au phénotype
observé.165
Le gène CHEK2 encode une protéine sérine/thréonine kinase qui joue un rôle clé dans la
transmission de signaux intracellulaires suite à son activation par phosphorylation par
ATM.166 Une fois activée, CHEK2 phosphoryle à son tour p53 et BRCA1, deux protéines
impliquées dans la tumorigénèse mammaire, ce qui permet la régulation de la réponse
cellulaire aux bris de l'ADN.167'168 CHEK2 est aussi impliqué dans le contrôle du cycle
cellulaire en empêchant l'entrée de la cellule en mitose.166
La mutation 1 lOOdelC de CHEK2, décrite à l'origine par Bell et collaborateurs164, constitue
la mutation de CHEK2 la plus couramment étudiée en tant qu'allèle de susceptibilité au
cancer du sein. Une méta-analyse récente regroupant plus de 26000 cas de cancers du sein
et plus de 27000 contrôles provenant de 16 études a mis en évidence un risque accru de
cancer à la fois chez les cas non sélectionnés (OR 2,7; IC 95% 2,1-3,4), chez les cas de
cancers du sein précoces (OR 2,6; IC 95% 1,3-5,5), ainsi que chez les cas familiaux (OR
4,8; IC 95% 3,3-7,2).169 Ces estimations sont comparables à celles obtenues par le
consortium CHEK2170 chez les populations d'ascendance nord européenne. De façon
intéressante, cette mutation de CHEK2 semble absente dans les populations non-
caucasiennes. 71 Le risque de cancer du sein chez l'homme associé à IlOOdelC demeure
controversé, avec certaines études rapportant un risque accru (OR 4,1-10)172'173, alors que
d'autres études demeurent non concluantes.174"176 Récemment, Cybulski et collaborateurs177
ont étudié la contribution d'autres variants de CHEK2 dans la susceptibilité au cancer du
sein dans une large étude. Leur étude rapporte un risque accru de cancer du sein chez les
porteurs de certaines mutations fondatrices polonaises del5395 (OR 2,4; IC 95% 1,5-3,8),
IVS2+1G>A (OR 3,6; IC 95% 2,3-5,5) et I157T (OR 1,6; IC 95% 1,4-1,9). Un risque accru
de cancers de la prostate et de cancer colorectal a aussi été suggéré,178"181 bien que cette
association demeure controversée.

30
2.2.3.3 BRIP1/BACH1/FANCJ A la recherche de partenaires cellulaires interagissant spécifiquement avec le domaine C-
terminal de BRCA1, Cantor et collaborateurs182 identifient une nouvelle protéine, BACH1
(BRCA1-associated c-termial helicase 1), membre de la famille des hélicases DEAH. Aussi
connu sous le nom de BRIP1 (BRCA1-interacting protein 1), l'association de la forme
phosphorylée de cette protéine à BRCA1 serait nécessaire au point de restriction cellulaire
G2/M suite aux dommages à l'ADN.183'184 Jusqu'à présent, les évidences suggèrent que
l'activité helicase de BRIP1/FANCJ (déroulement des duplexes d'ADN en direction 5' vers
3') serait nécessaire à son activité en aval de la monoubiquitination de FANCD2 dans la
voie Fanconi/BRCA, au niveau des foci de réparation. À cet endroit, BRIP1/FANCJ serait
essentielle au passage de la polymerase aux sites de blocage de la fourche de replication.
Ces notions seront abordées plus à fond dans la section 3.2.2.2 Implication de BRCA1 et
BRCA2 dans l'Anémie de Fanconi.
La première analyse du gène BRIP1 chez des cas de cancers du sein et de l'ovaire a permis
d'identifier deux variants faux-sens affectant le domaine helicase (1/35 cas de cancers du
sein familial et 1/30 cas de cancers du sein précoces, ces mutations n'étant pas observées
chez 200 contrôles sains), ce qui semblait indiquer que BRIP1 pouvait être la cible de
mutations augmentant le risque de cancer du sein.182 L'association convaincante d'une
susceptibilité au cancer du sein avec des mutations de BRIP1 a été démontrée en 2006
lorsque Seal et collaborateurs185 ont identifié des mutations fronçantes hétérozygotes chez
9/1212 individus atteints de cancer du sein ne présentant pas de mutations de BRCA1 ou
BRCA2 et chez seulement 2/2081 contrôles sains (RR 2,0; IC 95% 1,2-3,2). Le risque
conféré semble plus élevé chez les porteurs de moins de 50 ans (RR 3,5; IC 95% 1,9-5,7).
Bien que le risque de cancer du sein associé à des mutations faux-sens de BRIP1 n'a pu être
confirmé par cette étude, il demeure possible que celles-ci confèrent un risque moindre
nécessitant une étude de plus grande envergure. À cet égard, il est intéressant de noter que
l'étude de variants de séquence de BRIP1 chez des familles non-BRCAl/2 de la population
canadienne française n'a pas permis de mettre en évidence des mutations délétères de ce
gène.186 En plus du cancer du sein, des évidences récentes suggèrent que certaines
mutations fronçantes et variants de séquence de BRIP1 pourraient être associés à un risque
accru de cancer de la prostate.187

31
En 2005, les équipes de Cantor, Auerbach et Joenje188"190 identifient indépendamment des
mutations troncantes bi-alléliques de BRIP1 chez des patients atteints d'AF de groupe de
complémentation J (d'où l'appellation « FANCJ »). Ainsi, comme BRCA2, des mutations
mono-alléliques sont associées à un risque accru de cancer du sein alors que des mutations
bi-alléliques sont associées à un syndrome de pancytopénie (diminution des globules
blancs, des globules rouges et des plaquettes associée à une défaillance progressive de la
moelle osseuse) infantile (voir la section 3.2.2.2 Implication de BRCA1 et BRCA2 dans
l'Anémie de Fanconi).
2.2.3.4 PALB2/FANCN A l'aide d'expériences de co-immunoprécipitation, PALB2 (Partner and localizer of
BRCA2) a été identifié comme nouveau partenaire cellulaire de BRCA2 impliqué dans la
localisation/accumulation nucléaire de BRCA2 et promouvant sa stabilité.191'192 PALB2
serait probablement impliqué dans l'association de BRCA2 avec les structures nucléaires,
ce qui empêcherait sa dégradation par le protéasome et faciliterait ses fonctions dans la
réparation de l'ADN et la suppression tumorale. Puisque les cellules déficientes en PALB2
présentent des caractéristiques cellulaires similaires à celles des cellules déficientes en
BRCA1 ou BRCA2}91 une fonction de protéine supresseure de tumeurs a également été
suggérée pour PALB2. Dans le contexte de la voie Fanconi/BRCA, PALB2/FANCN agit
tout comme BRIP1/FANCJ en aval de la monoubiquitination de FANCD2 aux sites de
réparation des bris double brins. À ce niveau, l'interaction FANCN-BRCA2 serait
essentielle à la réparation par recombinaison homologue. Les détails de la voie
Fanconi/BRCA 1 seront abordés plus en détails dans la section 3.2.2.2 Implication de
BRCA1 et BRCA2 dans l'Anémie de Fanconi.
L'analyse de PALB2 chez des individus atteints de cancer du sein et présentant une histoire
familiale a permis de mettre en évidence la présence de mutations troncantes à l'état
hétérozygote, qui semblent conférer un risque relatif de 2,3 (95% IC 1,4-3,9).192 Ces
résultats ont été confirmés par une seconde étude chez des familles finlandaises avec une
histoire familiale de cancer du sein, qui a permis d'identifier chez ces familles une mutation
fondatrice récurrente introduisant un changement du cadre de lecture et un codon d'arrêt
prématuré.193 Cette mutation semble enrichie chez les cas de cancers du sein familiaux

32
finlandais et semble associée à un phénotype tumoral aggressif et un risque d'environ 40%
de développer un cancer du sein avant 70 ans.194,195 En plus d'avoir identifié cette mutation
au sein d'une famille de cancer de la prostate, des mutations troncantes de PALB2 ont été
rapportées chez des individus atteints de cancer du pancréas de type familial et du cancer de
l'ovaire, ce qui suggère l'implication potentielle de mutations de PALB2 dans d'autres
types de cancers.193'196'197 D'autres variants fréquents (fréquence de l'allèle mineur de plus
de 5%) ont aussi été associés à un risque accru de cancer du sein (OR 1,21-1,36) dans la
population chinoise.198
En parallèle de la découverte de l'implication de PALB2 dans la susceptibilité au cancer du
sein, les groupes de Rahman et de de Winter annonçaient indépendamment, dans le même
numéro du magazine Nature Genetics, la découverte de mutations bi-alléliques de PALB2
chez des patients atteints d'AF de groupe N (voir la section 3.2.2.2 Implication de BRCA1
et BRCA2 dans l'Anémie de Fanconi).199'200 PALB2 devenait ainsi le troisième gène
associé par des mutations mono-alléliques à un risque accru de cancer du sein, alors que des
mutations bi-alléliques causent l'AF.
2.2.4 Alleles de faible penetrance
Au cours des deux dernières années, la conduite d'études d'association de grande envergure
a permis de mettre en évidence certains variants conférant un risque modeste, mais très
fréquents dans la population. Ces études, qui adressent l'implication de polymorphismes
d'un gène en particulier ou qui investiguent une multitude de variants par un tour du
génome, ont mené à l'identification d'allèles dont l'association avec le cancer du sein a pu
être confirmée par des études subséquentes. De façon intéressante, les polymorphismes
identifiés sont situés dans des régions régulatrices, introniques, ou intergéniques et leur
impact biologique n'est pas clairement apparent. Ces alleles sont retrouvés sur des blocs
comprenant plusieurs polymorphismes adjacents qui sont souvent corrélés (en déséquilibre
de liaison; linkage disequilibrium, LD) les uns avec les autres. Il est par conséquent
possible que le variant identifié initialement par l'étude d'association ne soit pas à l'origine
de l'effet observé, mais que celui-ci soit dû à d'autres gènes ou éléments cryptiques
adjacents.

33
Locus lQq25.3-q26 (intron 2 du gène FGFR2)
L'un des alleles de susceptibilité de faible penetrance pour lequel les évidences actuelles
d'association avec le cancer du sein sont les plus convaincantes est un polymorphisme d'un
nucleotide (single nucleotide polymorphism; SNP), rs2981582, sur un bloc de LD de 25
kilobases (kb) situé dans l'intron 2 du gène FGFR2 encodant un récepteur du facteur de
croissance des fibroblastes (Fibroblast growth factor receptor 2).87'88'90 Ce variant a une
fréquence de l'allèle mineur (minor allele frequency, MAF) de 0,38 dans la population du
Royaume-Uni, avec une fréquence et un risque semblables estimés dans d'autres
populations caucasiennes.87,88'90 Des données récentes indiquent que cette association
pourrait être prédominante dans le cas de tumeurs exprimant le récepteur des estrogènes
(Estrogen receptor positive, ER+).90'20* De plus, le variant rs2981582 pourrait agir comme
gène de modification chez les individus porteurs d'une mutation de BRCA2 (HaR par allele
1,32; IC 95% 1,20-1,45), mais pas chez les individus porteurs d'une mutation du gène
BRCAl.9i II est de plus intéressant de souligner que l'association de FGFR2 avec un risque
accru de cancer du sein a aussi été retrouvée dans une étude regroupant des femmes afro-
américaines.2013
Des altérations somatiques de FGFR2 ont été rapportées dans plusieurs cancers (par
exemple dans certains cas de cancers de la prostate, de cancers urothéliaux et de cancers du
sein202"204) et résultent en une suractivité de la protéine produite. L'intron 2 de FGFR2 est
conservé au cours de l'évolution des mammifères et contient plusieurs sites potentiels de
liaison à des facteurs de transcription. Émettant l'hypothèse que l'action de ces variants
pouvait se produire par une régulation de la transcription de FGFR2, Meyer et
collaborateurs ont examiné les interactions protéines-ADN dans cette région. Leurs
analyses ont mis en évidence deux SNPs (rs7895676 et rs2981578) en LD avec ceux
identifiés précédemment, qui modifient l'affinité de facteurs de transcription (Oct-l/Runx2
et C/EBPB) pour leur site de reconnaissance, ce qui amènerait une augmentation de
l'expression de FGFR2 et une plus grande propension à former des tumeurs.
Locus 16ql2.1 (TOX3/TNRC9 ou LOC643714)
La plus forte association retrouvée par Easton et collaborateurs au locus 16q implique le
SNP rs3803662 situé sur un bloc de LD de 160 kb87, et cette association a été confirmée

34
dans une seconde étude dans la population islandaise.89 Ce variant encode un changement
synonyme du gène putatif LOC643714 situé 8 kb en amont du gène TOX3 (TOX high
motility group box family member 3) aussi connu sous le nom de TRNC9. Basé sur son
homologie de séquence, on assume que TOX3 serait impliqué dans la modification de la
structure de l'ADN et agirait comme facteur de transcription.206 L'augmentation de
l'expression de TOX3 a récemment été suggéré comme facteur prédictif de la production de
métastases du sein vers les os207 et rs3803662 semble être particulièrement associé à des
tumeurs ER+.89'201 De façon intéressante, la classification ER+ constitue le plus important
facteur histo-pathologique prédictif associé à la formation de métastases aux os.208 TOX3
pourrait de plus agir comme gène de modification puisque le SNP rs3803662 est associé à
une augmentation du risque chez les porteurs de mutations de BRCA1 et BRCA2 (HaR par
allele 1,13; IC 95% 1,06-1,20).91
Locus 5qll (MAP3K1/MEKK ou MGC33648 ou MIER3)
Une association a été retrouvée dans l'étude de Easton et collaborateurs sur le bras long du
chromosome 5 pour le SNP rs889312, qui se situe sur un bloc de LD de 280 kb couvrant les
gènes MAP3K1 (Mitogen-activated protein kinase kinase kinase 1), le gène hypothétique
C5orf35 (Chromosome 5 orf 35) et M1ER3 (Mesoderm induction early response 1, family
member 3).87 MAP3K1 est présentement considéré comme le candidat le plus probable, et il
encode une protéine sérine/thréonine kinase qui est impliquée dans une cascade d'enzymes
de phosphorylation intégrant les réponses cellulaires suite à des signaux de croissance et du
métabolisme.209 Tout comme dans le cas de FGFR2, le variant rs889312 est associé à une
augmentation du risque chez les porteurs de mutations de BRCA2 (HaR par allele 1,12; IC
95% 1,02-1,24), mais pas chez les porteurs de mutations de BRCA1.91
Locus 11 pi5 (LSP1 ou H19 ou TNNT3 ou MRPL23 ou LOC728008)
Le SNP rs3817198 situé sur un bloc de 180 kb sur le chromosome 11 a été associé à un
risque accru de cancer du sein.87 Ce polymorphisme est situé dans l'intron 10 du gène LSP1
(Lymphocyte-specific protein 1), qui est exprimé dans la lignée hématopoïétique et dans les
cellules endothéliales. Ce gène serait responsable de la régulation de la mobilité des
neutrophiles, leur adhésion aux protéines de la matrice de fibrinogène et leur migration à
travers l'épithélium endothelial.210 L'allèle mineur du variant rs3817198 a été associé à un

35
risque accru de cancer chez les porteurs de mutations dans BRCA2 (HaR par allele 1,16; IC 95% 1,07-1,25) uniquement 211
Un autre variant (rs2107425) est retrouvé 110 kb plus loin sur le même bloc dans la région
couverte par H19. L'association de ce variant avec le cancer du sein est moins prononcée
que celle observée pour LSP1. H19 est un gène non-traduit soumis à l'empreinte
parentale, dont l'allèle maternel est exprimé. L'ARNm produit est impliqué dans la
régulation d,IGF2 (Insulin-like growth factor 2). Bien que H19 soit un petit gène, des
études chez son orthologue murin ont démontré qu'il subit une régulation complexe avec
un ensemble de séquences promotrices tissus-spécifiques situées en aval du gène.212 Les
autres gènes potentiels retrouvés sur ce même bloc de déséquilibre de liaison sont TNNT3
(Troponin T type 3), MRPL23 (Mitochondrial ribosomal protein L23) et le gène
hypothétique LOC728008*7
Locus 2q35
Dans une étude de grande envergure de la population islandaise, Stacey et collaborateurs
ont retrouvé une association avec le variant rsl3387042 situé dans une région de LD de 117
kb du chromosome 2.89 Aucun gène n'a jusqu'à présent été identifié sur ce bloc de LD. Le
risque conféré par le variant rsl3387042 semble lié aux tumeurs ER+.89 Bien que ce locus
n'ait pas été identifié dans d'autres études, cette association a été confirmée dans une étude
indépendante de la population du Royaume-Uni, avec un risque estimé similaire (OR par
allele 1,17; 95% IC 1,07-1,27).50 Ce variant serait associé avec une augmentation du risque
à la fois chez les porteurs de mutations de BRCA1 et de BRCA2.2U
Locus 5pl2 (MRPS30)
Basé sur les résultats de leur première étude de grande envergure89, ainsi que ceux de deux
autres études préalables,87'88 Stacey et collaborateurs ont identifié une région de 310 kb du
chromosome 5 présentant certaines évidences d'une association avec le cancer du sein.90
Une seconde étude de cette région dans des populations d'ascendance européenne ont
permis d'identifier les variants rs4415084 (OR par allele 1,16; 95% IC 1,10-1,21) et
rsl0941679 (OR par allele 1,19; 95% IC 1,13-1,26) qui semblent associés à des tumeurs
ER+.90 Le seul gène dans cet intervalle est MRPS30 (Mitochondrial ribosome protein S30),
un gène encodant l'une des composantes de la petite sous unité du ribosome mitochondrial,

36
qui est impliqué dans certains événements pro-apoptotiques. De façon intéressante,
MRPS30 fait partie du profil d'expression génique permettant la distinction entre les
tumeurs ER+ et ER-.90'213
Locus 8q24
Parmi les variants identifiés dans l'étude de Easton et collaborateurs, on retrouve le SNP
rsl3281615 situé dans un «désert génique» (grande région chromosomique où aucun gène
n'a été identifié jusqu'à présent) sur la bande q24.21 du chromosome 8.87 Ce variant est
retrouvé sur un bloc de LD de 110 kb qui ne comprend aucun gène connu. De façon
importante, l'association de ce bloc de LD avec le cancer du sein a été reproduite et semble
spécifique au cancer du sein,214"217 bien que des régions adjacentes de 8q24.21 présentent
de multiples variants indépendamment associés à une susceptibilité aux cancers colorectal,
de la prostate, de l'ovaire et de la vessie. 214-218"226 Le locus 8q24 serait particulièrement
impliqué dans la formation de tumeurs ER+.201 Plusieurs déserts géniques ont été associés à
des maladies (par exemple la maladie de Parkinson,227 la maladie de Crohn,228 la fibrillation
auriculaire, et le cancer ), ce qui laisse présager que ces régions pourraient avoir
certaines fonctions. Des évidences suggèrent que cette région puisse arborer des régulateurs
tissus-spécifiques de la transcription, qui pourraient agir sur de grandes distances.230
Locus 6q22.33 ŒDHCD1 ou RNF146)
En utilisant la population fondatrice juive ashkénaze, chez laquelle le LD entourant les
alleles de susceptibilité potentiel est théoriquement accru, Gold et collaborateurs ont
identifié quatre variants (dont la plus forte association avec rs2180341) situés sur un bloc
de LD d'environ 200 kb sur le chromosome 6. : Ces variants sont fortement associés à
deux gènes de cette région : RNF146 (Ring finger protein 146), qui encode probablement
une E3 ubiquitine ligase, et ECHDC1 (Enoyl Coenzyme A hydratase domain containing 1),
qui est impliqué dans l'oxydation mitochondriale des acides gras.
Locus 2q33 (CASP8)
Dans une étude de type gènes candidats, Cox et collaborateurs ont génotype neuf variants
de séquence pour lesquels il existait des évidences préalables d'association avec le cancer
du sein.232"234 Parmi ceux-ci, le SNP rsl045485, qui est associé à un changement faux-sens
p.Asp302His du gène CASP8 (Caspase 8), présente un effet protecteur qui semble associé

37
aux tumeurs ER+.234 La caspase 8 est un initiateur important de la mort cellulaire
programmée (apoptose) en réponse à des dommages importants de l'ADN ou à des signaux
apoptotiques extracellulaires. De façon intéressante, une étude de CASP8 dans la
population chinoise, chez laquelle le variant p.Asp302His est très rare, a démontré une
association d'une deletion de 6 nucleotides de la région promotrice avec un risque réduit de
plusieurs cancers dont le cancer du sein (OR hétérozygotes 0,65; IC 5% 0,54-0,78)235 et
l'effet de ces deux variants de CASP8 a été confirmé dans une méta-analyse récente.236
En plus des variants identifiés dans les sections précédentes, il existe des évidences que
certains autres gènes pourraient être impliqués dans la susceptibilité au cancer du sein, tels "yxn TIR
que certains alleles de TGFB1 (Transforming growth factor beta 1), ' ESRI (Estrogen
receptor l),2i9 AKAP9 (A kinase anchor protein 9).240 Cependant, les résultats de ces études
d'association demeurent à être reproduits dans d'autres cohortes.
2.3 Modèles de la susceptibilité génétique au cancer du sein
Depuis plusieurs années, des outils de modélisation génétique ont été utilisés afin de tenter
de mieux comprendre les effets des déterminants génétiques de la susceptibilité au cancer
du sein. Ces analyses consistent à simuler de façon mathématique l'occurrence de la
maladie en combinant l'action de gènes connus et/ou hypothétiques ayant chacun sa
prévalence, sa penetrance et son mode de transmission. Ces simulations sont ensuite
comparées à la fréquence observée du cancer dans la population. Bien que les premiers
modèles (établis avant l'identification de BRCA1 et BRCA2) aient favorisé un type de
transmission autosomique dominant56, les simulations ultérieures (où l'effet de ces deux
gènes est ajouté) favorisent un modèle polygénique.64'72'241"243 Ce modèle suggère que chez
les cas tirés de la population, le risque de cancer du sein pourrait être expliqué par plusieurs
variants génétiques agissant de façon indépendante sur le risque. Il existerait ainsi dans la
population une répartition étendue du risque, présentant une distribution normale à l'échelle
logarithmique et un écart type estimé à 1,2.244 Sous ce modèle, dans l'éventualité où tous
les alleles de susceptibilité seraient identifiés, plus de 50% des cas de cancer du sein
pourraient en théorie survenir dans un 12% de la population démontrant une prédisposition
plus forte pour la maladie (Figure 7).244 De façon intéressante, les alleles communs de
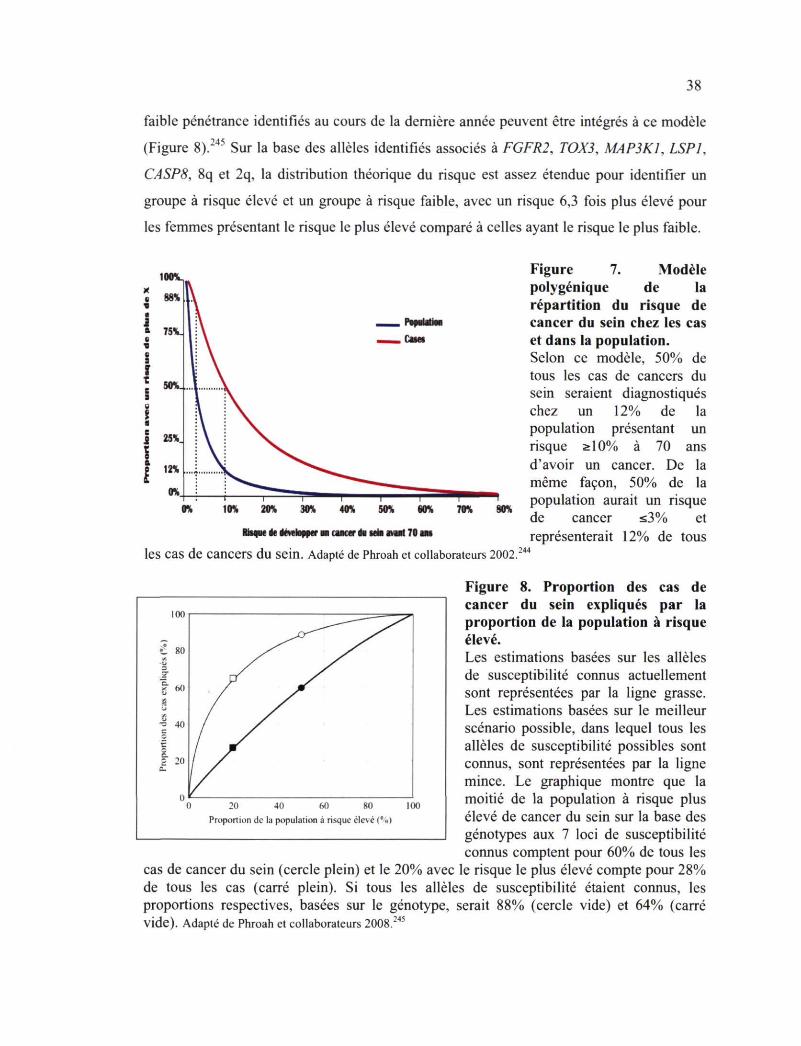
38
faible penetrance identifiés au cours de la dernière année peuvent être intégrés à ce modèle
(Figure 8).245 Sur la base des alleles identifiés associés à FGFR2, TOX3, MAP3K1, LSP1,
CASP8, 8q et 2q, la distribution théorique du risque est assez étendue pour identifier un
groupe à risque élevé et un groupe à risque faible, avec un risque 6,3 fois plus élevé pour
les femmes présentant le risque le plus élevé comparé à celles ayant le risque le plus faible.
o% 70% 80% 20% 30% 40% 50% 60%
Roque fc it\ttmtt M CMCtr fc sein avant 70 u*
les cas de cancers du sein. Adapté de Phroah et collaborateurs 2002.
Figure 7. Modèle polygénique de la répartition du risque de cancer du sein chez les cas et dans la population. Selon ce modèle, 50% de tous les cas de cancers du sein seraient diagnostiqués chez un 12% de la population présentant un risque a 10% à 70 ans d'avoir un cancer. De la même façon, 50% de la population aurait un risque de cancer s3% et représenterait 12% de tous
Figure 8. Proportion des cas de cancer du sein expliqués par la proportion de la population à risque élevé. Les estimations basées sur les alleles de susceptibilité connus actuellement sont représentées par la ligne grasse. Les estimations basées sur le meilleur scénario possible, dans lequel tous les alleles de susceptibilité possibles sont connus, sont représentées par la ligne mince. Le graphique montre que la moitié de la population à risque plus élevé de cancer du sein sur la base des génotypes aux 7 loci de susceptibilité connus comptent pour 60% de tous les
cas de cancer du sein (cercle plein) et le 20% avec le risque le plus élevé compte pour 28% de tous les cas (carré plein). Si tous les alleles de susceptibilité étaient connus, les proportions respectives, basées sur le génotype, serait 88% (cercle vide) et 64% (carré vide) . Adapté de Phroah et collaborateurs 2008.245

39
Plus de 10 ans après l'identification de BRCA1 et BRCA2, des efforts considérables ont été
consacrés à l'identification de gènes de forte penetrance additionnels pouvant agir au sein
des familles présentant une forte prédisposition au cancer du sein. Il est maintenant clair
que la découverte d'un troisième gène de susceptibilité au cancer présentant des
caractéristiques similaires à celles de BRCA1 et BRCA2 soit peu probable, bien que cette
possibilité ne puisse être totalement exclue. Ainsi, il est probable que les autres alleles
demeurant à êtres identifiés sont des alleles de penetrance faible ou modérée. Une
alternative aux études de liaison utilisées pour identifier les gènes de forte penetrance est
l'approche par étude d'association de gènes candidats, qui a porté fruit par le passé.185'193
Dans ce type d'étude, les gènes sont sélectionnés sur la base d'une plausibilité biologique
(qui est toutefois tributaire des connaissances actuelles des mécanismes cellulaires de la
carcinogenèse) et la fréquence des variants est comparée entre un groupe de cas et un
groupe de contrôles.
Certains aspects méthodologiques doivent cependant être pris en considération. Des études
sur ce type d'allèles demandent de larges cohortes et représentent un coût important. Il est
cependant possible d'optimiser le pouvoir des études visant l'identification de nouveaux
gènes de susceptibilité, principalement de deux façons. La première consiste à utiliser une
cohorte enrichie pour une prédisposition génétique, par exemple en sélectionnant des cas
présentant une histoire familiale chargée, des cas de cancer du sein bilatéral ou des cas
précoces. La taille de l'échantillon requis pour obtenir une puissance de 90% avec une
valeur P= 0,0001 a été modélisée par Antoniou et Easton.246 Cette analyse a permis de
démontrer que l'utilisation de cas familiaux (2 parentes au premier degré atteintes) peut
réduire de 4 fois la taille de l'échantillon requis pour détecter un allele de susceptibilité
présentant une fréquence de 10% (Figure 9).
Alternativement, il est aussi possible d'utiliser un enrichissement géographique, en
effectuant une première analyse des gènes dans une population isolée (population
fondatrice). Au sein de ces populations, il est théoriquement probable d'observer un
enrichissement pour des mutations fondatrices et l'hétérogénéité génétique est réduite, ce
qui augmente la probabilité d'identifier une mutation.247 Cependant, il est aussi possible
qu'un allele de susceptibilité identifié soit spécifique à cette population. Un exemple de ce
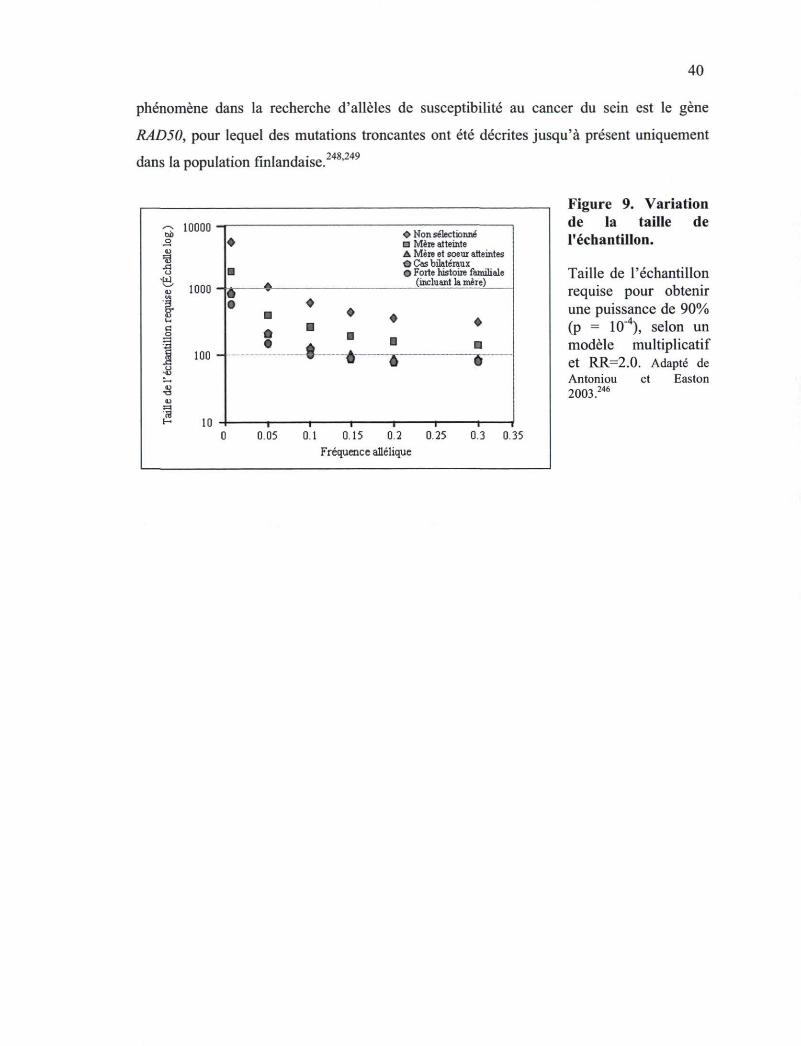
40
phénomène dans la recherche d'allèles de susceptibilité au cancer du sein est le gène
RAD50, pour lequel des mutations troncantes ont été décrites jusqu'à présent uniquement
dans la population finlandaise.248'249
■lu
a a
JD
3
10000
1000
O Non sélectionné B Mère atteinte A Mère et soeur atteintes ® Cas bilatéraux O Forte histoire familiale
(incluant la mère)
0.15 0.2 Fréquence allélique
0.35
Figure 9. Variation de la taille de l'échantillon.
Taille de l'échantillon requise pour obtenir une puissance de 90% (p = IO"4), selon un modèle multiplicatif et RR=2.0. Adapté de Antoniou et Easton 2003.246

41
3. Fonctions et partenaires cellulaires de BRCA1 et BRCA2 Depuis leur identification au milieu des années 90, BRCA1 et BRCA2 ont été
considérablement étudiés. Alors que des mutations de ces gènes sont retrouvées avec une
grande fréquence dans les familles présentant une histoire familiale très chargée de cancer
du sein et de l'ovaire (voir la Figure 6), des altérations de ces deux gènes ne sont que
rarement associées à des cancers du sein de type sporadique.250'251 Cette dichotomie
apparente dans les causes moléculaires sous-jacentes à ces deux groupes est intrigante et a
poussé la recherche des mécanismes cellulaires affectés par des altérations de BRCA1 et
BRCA2.
BRCA1 et BRCA2 jouent des rôles importants dans le maintien de l'intégrité du génome.
Ceci est illustré par l'instabilité chromosomique retrouvée chez les cellules déficientes en
ces protéines, qui accompagne fréquemment des défauts de gènes associés à la détection, la
signalisation ou la réparation des bris de l'ADN.252"255 Tel qu'illustré précédemment, la
compréhension des rôles cellulaires de BRCA1 et BRCA2 et leurs interactions avec les
autres protéines cellulaires est importante et a permis de mettre en évidence certains gènes
candidats dont les altérations sont associées à un risque accru de cancer du sien. '
3.1 Structure des protéines BRCA1 et BRCA2
Bien qu'étant toutes deux associées au syndrome HBOC, les protéines BRCA1 et BRCA2
ont des structures protéiques présentant peu de similarité l'une avec l'autre et avec des
protéines aux fonctions connues. Les domaines protéiques de BRCA1 et BRCA2 sont
présentés à la Figure 10.
BRCA1 :
Le gène BRCA1 encode une grosse protéine de 1863 acides aminés (aa). Seules les régions
N- (amino) et C- (carboxy)- terminales sont fortement conservées entre les espèces
mammifères.256'257 La protéine comprend un domaine RING (nommé d'après le gène
modèle de cette classe, Really interesting new gene) en N-terminal, 2 copies en tandem du
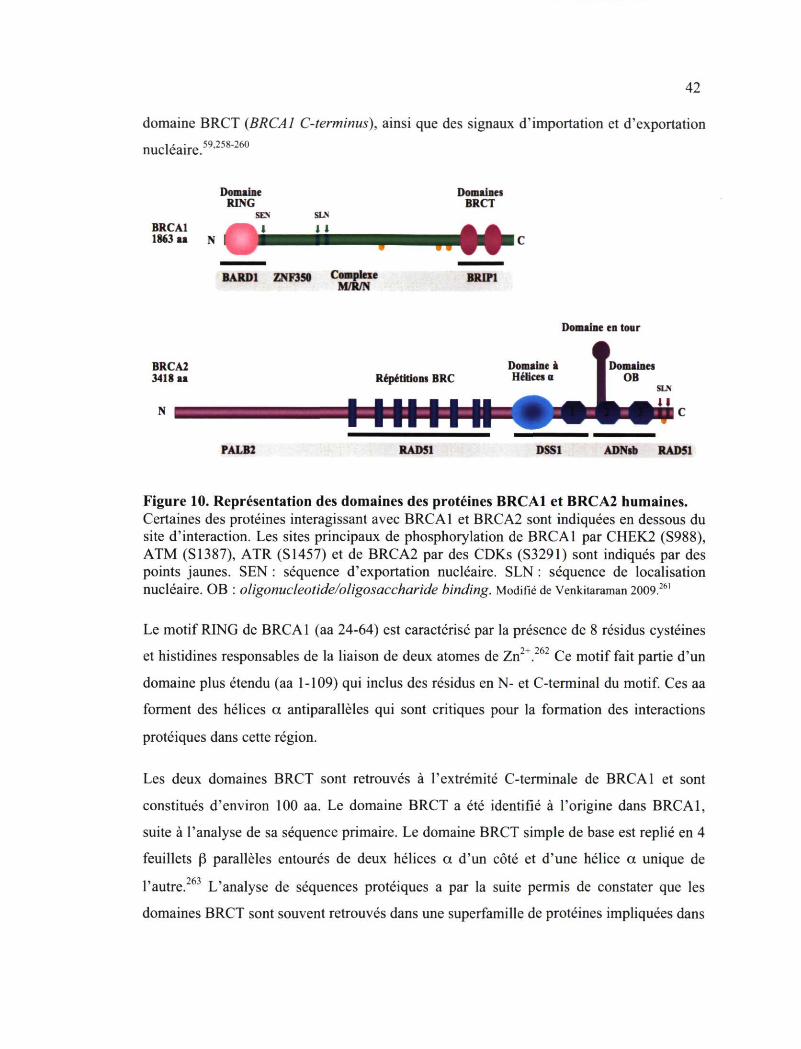
42
domaine BRCT (BRCA1 C-terminus), ainsi que des signaux d'importation et d'exportation
nucléaire.59'258"260
BRCAl 1863 aa
Domaine RING
Domaines BRCT
SLN
H
BARD1 ZNF350 Complexe M/R/N
BR1P1
BRCA2 3418 aa
Domaine en tonr
PALB2 RAD51 DSS1 ADNtb RADS1
Figure 10. Représentation des domaines des protéines BRCAl et BRCA2 humaines. Certaines des protéines interagissant avec BRCAl et BRCA2 sont indiquées en dessous du site d'interaction. Les sites principaux de phosphorylation de BRCAl par CHEK2 (S988), ATM (S 1387), ATR (S 1457) et de BRCA2 par des CDKs (S3291) sont indiqués par des points jaunes. SEN : séquence d'exportation nucléaire. SLN : séquence de localisation nucléaire. OB : oligonucleotide/oligosaccharide binding. Modifié de Venkitaraman 2009.26'
Le motif RING de BRCAl (aa 24-64) est caractérisé par la présence de 8 résidus cysteines
et histidines responsables de la liaison de deux atomes de Zn2+.262 Ce motif fait partie d'un
domaine plus étendu (aa 1-109) qui inclus des résidus en N- et C-terminal du motif. Ces aa
forment des hélices a antiparallèles qui sont critiques pour la formation des interactions
protéiques dans cette région.
Les deux domaines BRCT sont retrouvés à l'extrémité C-terminale de BRCAl et sont
constitués d'environ 100 aa. Le domaine BRCT a été identifié à l'origine dans BRCAl,
suite à l'analyse de sa séquence primaire. Le domaine BRCT simple de base est replié en 4
feuillets |3 parallèles entourés de deux hélices a d'un côté et d'une hélice a unique de
l'autre.263 L'analyse de séquences protéiques a par la suite permis de constater que les
domaines BRCT sont souvent retrouvés dans une superfamille de protéines impliquées dans

43
la réponse aux dommages de l'ADN, la réparation de l'ADN et la régulation du cycle
cellulaire.264 Le domaine BRCT est impliqué dans des interactions protéiques et des
évidences suggèrent que les domaines BRCT en tandem (incluant ceux qui sont retrouvés
dans BRCAl) constituent un motif de reconnaissance et de liaison de peptides
phosphorylés.265'266 Des analyses de liaisons peptidiques ont de plus démontré que le
domaine BRCT de BRCAl lie spécifiquement les peptides contenant la séquence pSer-X-
X-Phe, telle que celle qui est retrouvée dans BACH 1/BRIP1/FANCJ.266
Bien qu'elle soit de façon prédominante une protéine nucléaire, BRCAl a aussi été
retrouvée au niveau du cytoplasme et de la mitochondrie.267 Cette migration de BRCAl
entre les compartiments cellulaires est modulée par une séquence d'exportation nucléaire
(SEN) retrouvée à l'extrémité du domaine RING (aa 81-99),268 par deux séquences de
localisation nucléaire (SLN) dans la portion centrale de la protéine (aa 503-508 et aa 606-
615) ' ainsi que par l'interaction de BRCAl avec certains de ces partenaires
cellulaires271. En plus de cette régulation spatiale, BRCAl est aussi sujette à une régulation
de son activité par phosphorylation. BRCAl est le substrat de plusieurs kinases cellulaires,
dont ATM et CHEK2 dont les altérations ont aussi été associées à une susceptibilité accrue
au cancer du sein. La kinase la mieux caractérisée de BRCAl est ATM, qui phosphoryle la
protéine sur plusieurs résidus serine (serll89, 1298, 1330, 1387, 1423, 1457, 1466, 1524 et
1542),272 bien que d'autres kinases phosphorylent aussi la protéine.273
BRCA2:
Le gène BRCA2 encode une très grosse protéine de 3418 aa. BRCA2 présente très peu
d'homologie avec d'autres protéines, et aucune avec BRCAl. En comparant la protéine à
elle-même, l'analyse de la séquence protéique a permis de mettre en évidence la présence
d'un motif (appelé BRC) d'environ 70 aa répété 8 fois dans la région centrale de
BRCA2.274 Une percée majeure dans la compréhension des fonctions cellulaires de BRCA2
est venue de la réalisation que l'interaction entre BRCA2 et RAD51 est médiée par ces
répétitions.275'276 Puisque les motifs BRC divergent à divers degrés d'une répétition à
l'autre, ils n'ont pas tous la même affinité pour la recombinase RAD51, et les répétitions
BRC5 et BRC6 présentent l'affinité la plus faible. Il a été proposé que BRCA2 interagit

44
avec RAD51 en imitant le motif qui permet à RAD51 de s'oligomériser, maintenant ainsi 277 RAD51 dans un état monomérique.
La portion C-terminale de BRCA2 comprend une région d'environ 800 aa qui forme des
structures secondaires, dont des motifs similaires à des domaines de liaison à l'ADNsb
(ADN simple-brin). Le premier motif de cette région est appelé le domaine hélical et
comprend 190 aa qui forment principalement des hélices a. Le domaine hélical est suivi de
trois domaines d'environ 110 aa (OBI, OB2 et OB3) qui présentent une homologie avec le
repliement OB présent chez plusieurs protéines liant l'ADNsb. OB2 contient en plus une
insertion de 130 aa qui adopte une conformation en tour perpendiculaire au repliement
OB.278 La région C-terminale contient de plus deux SLNs279 et un site de phosphorylation
par des kinases dépendentes des cyclines (CDK; cyclin-dependent kinases) qui semble
réguler l'association de RAD51 à un deuxième motif de liaison à cet endroit.280
3.2 Fonctions de BRCAl et BRCA2 BRCAl et BRCA2 sont exprimées de façon ubiquitaire chez l'humain, avec une expression qui corrèle de près avec les niveaux de prolifération cellulaire. L'expression est de plus variable au cours du cycle cellulaire, avec des niveaux maximum à la transition Gl/S.281
BRCAl et BRCA2 sont des protéines «caretakers», dont l'inactivation crée un état permissif dans lequel la cellule accumule des défauts cellulaires qui amènent une instabilité chromosomique. Un exemple de ce phénotype peut être facilement observé en étudiant le caryotype des cellules de souris déficientes en BRCA2 (Figure 11).282
Q Figure 11. Structures chromosomiques aberrantes de cellules déficientes en Brca2.
ctb Structures chromosomiques aberrantes dans les X \M a "̂ \9 fibroblastes embryonnaires de souris
f& homozygotes pour une protéine Brca2 tronquée > ' \ V* *u niveau de son exon U (Brca2 t r t r)* A)
ir Étalement de cellules Brca2tr/tr en métaphase au 3e passage, présentant de nombreuses anomalies structurales (flèches). B) C) et D)
A montrent des grossissements d'un bris typique de la chromatine (ctb), d'une structure triradiale (tr) et d'une structure quadriradiale
U A (qr). Adapté de Patel et collaborateurs 1998.282
**h. / «•
Y n A*

45
Bien que les fonctions de BRCAl et BRCA2 ne soient pas entièrement comprises, il est
clair que ce sont des protéines multifonctionnelles, qui ont des fonctions indépendantes.
Jusqu'à présent, BRCAl et BRCA2 ont été impliquées dans plusieurs fonctions dont la
régulation de la transcription, la détection et la réparation des bris de l'ADN, le contrôle du
cycle cellulaire et le remodelage de la chromatine.
3.2.1 Régulation de la transcription
Le rôle de BRCA2 dans la transcription demeure obscur. Des analyses utilisant des
fragments protéiques de BRCA2 ont proposé que les aa 23-105 pourraient activer la
transcription.283 Cette activation pourrait être effectuée par son interaction avec des
protéines co-activatrices (P/CAF et GRIP1) qui possèdent des activités d'acétylase et de
méthylase d'histones. ' Il est par conséquent possible que BRCA2 établisse un lien
entre des protéines de remodelage de la chromatine et le complexe de transcription afin
d'en activer l'activité. Un autre aspect de l'implication de BRCA2 dans la transcription a
été mis en évidence avec la découverte d'un nouveau partenaire d'interaction, EMSY, qui
lie les aa 23-46 et qui semble agir comme répresseur de la transcription.286 EMSY serait
aussi capable d'interagir avec HP1-P et BS69, deux protéines impliquées dans la régulation
de la chromatine. De façon intéressante, Hughes-Davies et collaborateurs ont aussi
démontré qu'EMSY est amplifié dans un nombre significatif de tumeurs du sein
sporadiques et est associé avec un mauvais pronostic.286
Contrairement à BRCA2, le rôle de BRCAl dans la régulation de la transcription est mieux
compris. Des évidences suggèrent que BRCAl serait une composante de la machinerie
transcriptionnelle de base, en interagissant avec le complexe de l'ARN polymerase II par
l'intermédiaire de l'ARN helicase A.287 '288 BRCAl peut réguler l'activité de l'holoenzyme
ARN polymerase II en modifiant l'état de phosphorylation de son domaine C-terminal par
la kinase CAK.289 De plus, BRCAl associée à BARD1 (BRCAl-associated RING domain
1) pourrait utiliser son activité ubiquitine ligase afin de bloquer l'initiation de la synthèse
de l'ARNm en ubiquitinant le complexe de pré-initiation transcriptionel.290
En plus de son activité avec la machinerie transcriptionnelle de base, il a été démontré que
BRCAl pourrait agir en tant que co-régulateur sélectif de la transcription de gènes

46
spécifiques. Bien que BRCAl ne semble pas lier l'ADN de façon séquence-spécifique,
plusieurs facteurs de transcription, à la fois des co-répresseurs et des co-activateurs,
interagissent avec BRCAl dont p53, ESRI (Estrogen receptor 1), CtIP (CTBP-interacting
protein) et ZNF350 (Zinc finger protein 350).291"298 BRCAl s'associe à p53, ce qui la
stabilise et stimule son activité transcriptionnelle. Cette activation induit un sous-groupe de
gènes régulés par p53, et il semble que l'association de BRCAl à p53 redirige l'activité de
celle-ci des voies apoptotiques vers la réparation de l'ADN et l'arrêt du cycle cellulaire.291"
BRCAl a aussi été associée à une répression de la signalisation par ER-a, probablement T-a/a a * j r t j -
à travers la liaison compétitive de co-facteurs (p300/CBP, cycline Dl, BRD7). " Cette
répression cause l'inhibition de la croissance cellulaire induite par l'estrogène.29
3.2.1.1 Régulation transcriptionnelle de GADD45A par BRCAl et ZNF350
GADD45A (Growth arrest and DNA damage-inducible alpha) est membre d'un groupe de
gènes (comprenant aussi GADD45B et GADD45G) qui sont induits par des agents
endommageant l'ADN et divers stress cellulaires. GADD45A est impliqué dans la stabilité
génomique par ses rôles dans le contrôle de la transition G2/M du cycle cellulaire,
l'induction de l'apoptose et la réparation de l'ADN.299"303 Chez la souris, l'inactivation de
Gadd45A résulte en une augmentation de la tumorigénèse et de l'instabilité
chromosomique, ainsi que des problèmes d'auto-immunité.303"305
Une grande variété de stress cellulaires, y compris les dommages à l'ADN, le stress
oxydatif, le choc osmotique et la restriction en nutriments, induisent l'expression dose-
dépendante rapide de GADD45A. La régulation de l'expression de GADD45A se fait de
façon complexe et multiple. Les niveaux constitutifs de GADD45A sont bas, et le maintien
d'une faible expression de GADD45A s'effectue par le biais de répresseurs de transcription.
Son expression peut être induite par la dissociation de ces facteurs et l'association
d'activateurs de la transcription. L'induction ou la répression de GADD45A varie en
fonction du stade cellulaire et du type de dommages à l'ADN.306
ZNF350, aussi connu sous le nom de ZBRK1 (Zinc finger and BRCAl-interacting protein
with KRAB domain 1), est une protéine à doigts de zinc qui réprime l'expression de
GADD45A en liant une séquence spécifique de l'intron 3. ZNF350 interagit avec BRCAl et

47
ce complexe de répression est responsable du maintien du faible niveau constitutionnel
d'expression de GADD45A (Figure 12).307 La portion C-terminale de ZNF350 contient un
domaine de répression dépendant de BRCAl et d'histones dé-acétylases.308 De façon
intéressante, des mutations de BRCAl associées avec le cancer du sein perturbent son
association avec ZNF350. Cette inhibition de son activité de co-répresseur suggère que
cette activité est importante pour la fonction supresseur de tumeurs de BRCAl. 307
Radiations ionisantes
G,/M J
o Cycle cellulaire
Figure 12. Mécanismes de la régulation de GADD45A par p53, BRCAl et ZBRK1. Plusieurs mécanismes sont impliqués dans la régulation de l'expression de GADD45A. Parmi ceux-ci, on retrouve l'association de BRCAl et d'activateurs au niveau du promoteur, l'association de BRCAl avec le co-répresseur ZBRK1 au niveau de l'intron 3 et l'activation par p53 phosphoylée par la reconnaissance d'un motif spécifique dans l'intron 3.
Suite à un stress cellulaire, y compris des dommages de l'ADN, les répresseurs sont 309 inactivés. Dans le cas du complexe ZNF350/BRCA1, Yan et Lee ont identifié une région

48
de ZNF350 requise pour son ubiquitination et sa dégradation par le protéasome, ce qui
semble relâcher la répression et permettre l'induction de GADD45A. De plus, il a été
suggéré que la surexpression de BRCAl peut titrer ZNF350 de sa séquence de
reconnaissance dans l'intron 3 et ainsi permettre l'expression. De concert avec le
relâchement de cette répression transcriptionnelle, l'expression de GADD45A augmente
rapidement suite à l'action d'activateurs de la transcription. De façon paradoxale, il a été
démontré que BRCAl peut aussi activer l'expression de GADD45A en s'associant à des
facteurs de transcription, autres que ZNF350, au niveau du promoteur. BRCAl s'associe
avec Oct-1 et NF-YA et lie les deux motifs OCT-1 et la boîte CAAT présents dans le
promoteur.310 Cependant, cette activation par BRCAl semble réduite par son association
avecBARDl.311
GADD45A est le seul membre de la famille GADD à être régulé par p53, bien que les
mécanismes de régulation transcriptionnelle de GADD45A peuvent êtres dépendants ou
indépendants de celui-ci. L'induction de GADD45A suite aux radiations ionisantes requiert
p53,312 qui lie un site conservé dans l'intron 3 du gène GADD45A (Figure 12). L'induction
de GADD45A suite à d'autres types de dommages à l'ADN (rayonnement ultraviolet,
traitement aux agents alkylants ou restriction en nutriments), n'est que partiellement
dépendante de p53, puisque dans les cellules n'exprimant pas p53 la réponse est affaiblie
mais non abolie.313'314
L'ARNm (acide ribonucléique messager ; messenger ribonucleic acid, mRNA) de
GADD45A est aussi sujet à une régulation post-transcriptionnelle importante. La liaison de
protéines à sa portion 3' non transcrite (3'UTR, 3 ' untranslated) a été impliquée dans la
régulation de la traduction et dans le contrôle de la stabilité de son ARNm.315"316 L'action
de ces protéines de liaison à l'ARNm régule étroitement la quantité d'ARNm disponible
dans la cellule et sa capacité à être traduit.317 En état de stress intense, il a aussi été suggéré
que l'ARNm de GADD45A peut être traduit par un mécanisme indépendant de la coiffe
(qui est responsable chez la vaste majorité des ARNms du recrutement du ribosome) et -ai o
dépendant d'un site interne d'entrée du ribosome (1RES, internai ribosome entry site).
Jusqu'à présent, les études analysant l'implication des gènes ZNF350 et GADD45A dans la
susceptibilité au cancer du sein sont limitées. Les éludes visant à identifier des mutations de

49
ces deux gènes dans des cas de cancer du sein (familiaux, sporadiques ou en âge précoce)
ou dans d'autres types de cancers se sont avérées mitigées. Puisqu'il s'agit de l'un des
sujets principaux de la présente thèse, ces études seront abordées plus en détails dans les
Chapitres I et II, et seront discutés en fin de thèse.
3.2.2 Détection, signalisation et réparation des dommages de l'ADN
Les cellules déficientes en BRCAl ou BRCA2 démontrent clairement une inefficacité à
effectuer la réparation des bris de l'ADN, en particulier les lésions impliquant des
BDBs.329'330 Les BDBs constituent des lésions particulièrement dommageables pour la
cellule. En fonction du type cellulaire ou du moment du cycle, la cellule dispose des voies
de réparation par recombinaison homologue (homologous recombination, HR) ou par la
jonction des extrémités non-homologues (non-homologous end-joining, NHEJ) pour
réparer les BDBs. La voie NHEJ ne nécessite pas d'homologie. Cette voie peut résulter en
une perte ou une addition de nucleotides aux sites de réparation et est habituellement
considérée comme introduisant des erreurs. La réparation homologue, qui nécessite une
chromatide sœur intacte comme modèle (et par conséquent ne peut survenir que dans les
phases cellulaires S et G2), est considérée comme une voie de réparation fiable.
Bien que les cellules déficientes en BRCAl ou en BRCA2 toutes deux présentent des
défauts de la réparation BDB, celle-ci provient de rôles distincts. Les fonctions cellulaires
de BRCAl dans la réparation des BDBs semblent consister majoritairement dans un rôle de
médiateur entre les protéines de détection des bris et les protéines impliquées dans la
réparation proprement dite, ce qui assure l'activation des mécanismes adéquats. En plus
d'être impliquée dans la voie de réparation par recombinaison homologue, certaines
évidences suggèrent que BRCAl pourrait aussi jouer un rôle dans la réparation NHEJ en
inhibant les mécanismes utilisant une micro-homologie et enclins à introduire des
erreurs.331'332 Le principal rôle de BRCA2 consiste à réguler l'action de la recombinase
RAD51 impliquée dans la réparation homologue.261
BRCAl est impliquée dans les voies de réparation des BDBs, bien que ces fonctions soient
moins bien comprises que celles de BRCA2. Des évidences suggèrent que le recrutement
des protéines de détection ATM et la nibrine est l'un des premiers événements ayant place

50
au niveau du site du bris, ce qui permet l'activation de la kinase ATM.333 ATM assure la
phosphorylation du variant d'histone H2AX et H2AX phosphorylée (y-H2AX) permet
l'accumulation de plusieurs protéines au niveau du bris, incluant BRCAl, MDC1 et le
complexe MRE11A/RAD50/Nibrin (M/R/N, voir la section 3.2.2.1 Interaction entre la
nibrine et BRCAl).334'335 À son tour, BRCAl est phosphorylée et est requise pour la
phosphorylation par ATM et ATR de protéines de signalisation et de réparation (Figure
13). 336
Complexe MRN Reconnaissance du bris et concentration des extrémités d'ADN
BRCA1.536P1.MDC1 Ç *) ' Amplification du signal par le recrutement de médiateurs/effecteurs de la réparation
\J
Figure 13. Recrutement des premières protéines de détection/réparation au site de bris. Le complexe MRN reconnaît les sites de bris et concentre les brins d'ADN, permettant ainsi le recrutement de facteurs responsables du relâchement de la chromatine, l'amplification du signal de bris et le recrutement des protéines de réparation.
L'évidence de la participation de BRCAl dans plusieurs aspects de la réponse aux bris de
l'ADN se reflète par ses interactions protéiques et sa présence dans plusieurs complexes
distincts. Par exemple, le recrutement de MDC1 aux sites de bris induit une poly-
ubiquitination de H2AX, modification qui est reconnue par RAP80. À son tour, RAP80
s'associe au dimère BRCAl-Abraxas et permet de promouvoir l'activité ubiquitine ligase
de BRCAl pour augmenter l'ubiquitination aux sites de bris. Cette modification permettrait
un remodelage de la chromatine permettant à la réparation d'avoir lieu.337 9 BRCAl est
aussi impliquée dans l'ubiquitination de protéines par l'intermédiaire de son association
avec BARD1, qui pourrait augmenter son activité ubiquitine ligase.340 L'hétérodimère
BRCAl-BARD1 est présent dans plusieurs complexes protéiques. Il a été suggéré qu'une
des activités importantes de BRCAl-BARD1 puisse être le contrôle de la recombinaison
homologue en interagissant avec BRCA2, RAD51 et PALB2.341

51
L'implication de BRCA2 dans les mécanismes de la réparation de l'ADN se fait en
particulier par l'intermédiaire de son activité dans la HR, et la réparation NHEJ semble
intacte chez les cellules déficientes en BRCA2.342 BRCA2 est étroitement impliquée dans
la réparation de l'ADN, entre autres par son interaction avec la recombinase RAD51 et la
régulation de celle-ci.343'344 À la suite de dommages à l'ADN, BRCA2 et RAD51 co
localisent à l'intérieur de foci nucléaires dépendants de BRCA2, ce qui suggère que
BRCA2 est nécessaire à la localisation adéquate de RAD51 et à ses fonctions.345'346 Ces
foci sont considérés comme des sites de la chromatine où la réparation de l'ADN est en
cours.
BRCA2 interagit avec RAD51 par ses motifs BRCs, ce qui pourrait permettre à BRCA2 de séquestrer RAD51 en période où la réparation n'est pas nécessaire. Une fois le processus de HR enclenché, BRCA2 serait responsable de la relocalisation de RAD51 au niveau de la chromatine, ce qui permettrait à RAD51 de former des filaments en hélice autour des extrémités de l'ADN. Cette interaction de RAD51 avec l'ADN serait stabilisée par une seconde interaction des multimères de RAD51 impliquant la portion C-terminale de BRCA2. Suite à la formation des filaments sur les extrémités de l'ADN au site du bris, RAD51 permet l'invasion de la seconde molécule d'ADN et la recherche de la séquence homologue qui servira de modèle pour la réparation (Figure 14). De façon intéressante, BRCA2 peut être phosphorylée dans la région C-terminale d'interaction avec RAD51 sur la serine 3291. Cette phosphorylation a pour effet d'inhiber la liaison à RAD51 et ainsi pourrait permettre de réguler la recombinaison par RAD51.347
A -mm ™°= Figure 14. Mécanisme de la recombinaison homologue.
—\j L'attachement du complexe M/R/N au niveau du site de bris (A) permet de former les
ff" "Jfi ' ÀUÀU1- extrémités d'ADNsb nécessaires à la HR (B). RPA s'associe à l'extrémité d'ADNsb et
^nV»' ' ^^tfrJfai i permet d'enlever les structures secondaires (C). Par la suite, BRCA2 serait responsable
M rrrr-- du recrutement de RAD51 à l'extrémité, qui déloge RPA (D). L'extrémité d'ADNsb
111 recouverte de RAD51 (E) pourrait alors envahir la seconde molécule d'ADN (F),
^ rapprochée par le complexe M/R/N.
B i ' T*
_ , ' m . '
O '
I . ' ' '
F ' i ^ T T - \ " I *
i i i T i i i i i i i i i Tgja-i-r ' i % ^ * > N - r V ^ i i n
" * ^ Ï Ï N e , e ' R P A * R-5-0 5 1

52
3.2.2.1 Interaction entre la nibrine et BRCAl
Tel que mentionné précédemment, le complexe M/R/N est composé des protéines
MRE11A (MRE11 meiotic recombination 11 homolog A, S. cerevisiae), dont les très rares
mutations sont associées à une variante d'ataxie-télangiectasie appelée VAtaxia
telangiectasia-like disorder, de RAD50 qui n'est pas présentement associé à un syndrome,
et de la nibrine. MRE11A possède une activité nuclease requise pour la formation des
extrémités d'ADNsb nécessaires à la réparation des BDBs. RAD50 possède une activité
helicase nécessaire au déroulement de l'ADNdb (ADN double brins). La nibrine est quant à
elle impliquée dans l'interaction avec ATM, la localisation nucléaire de MRE11A-RAD50
et le positionnement de MRE11A-RAD50 aux sites des BDBs par son interaction avec
l'histone y-H2AX.348"350 Le complexe forme une structure typique qui suggère qu'il est en
mesure de lier les extrémités de l'ADN de façon à les rapprocher (Figure 13 et Figure
14).351 De façon intéressante, des travaux récents par van der Linden et collaborateurs352
suggèrent que les protéines du complexe M/R/N pourraient former plusieurs complexes
distincts stables, avec une composition protéique et une stœchiométrie variables, et qui
pourraient être associés à des variations fonctionnelles.
Plusieurs évidences indiquent que BRCAl interagit directement avec le complexe
M/R/N353"355, en particulier avec la nibrine (protéine encodée par le gène NBN aussi connu
sous le nom de NBS1 : Nijmegen breakage syndrome 1). Récemment, il a été démontré que
le complexe M/R/N s'associe aux protéines BRCAl et CtlP de façon dépendante du cycle
cellulaire, et cette association serait impliquée dans les mécanismes de réparation des
BDBs. Selon ce modèle, CtlP forme un pont entre la portion C-terminale de BRCAl et la
nibrine, et la portion N-terminale de BRCAl s'associerait aussi avec la nibrine. La
formation de ce complexe serait régulée par la phosphorylation de CtlP par la kinase
CDK.356 La participation de CtlP au sein de ce complexe pourrait être liée à la régulation de
l'activité nuclease de MRE11A.357

53
Outre son interaction avec BRCAl, le complexe M/R/N est aussi impliqué dans le contrôle
du cycle cellulaire puisqu'il semble être nécessaire pour obtenir une phosphorylation
adéquate des substrats d'ATM.348 De façon intéressante, la phosphorylation de BRCAl par
ATM semble aussi être nécessaire à l'obtention d'un niveau de phosphorylation optimal de
certains substrats d'ATM tels la nibrine, CtlP et p53.336 En plus de ces activités, le
complexe M/R/N a aussi été associé à des activités de réparation lors de la replication et de
la méiose, ainsi que le maintien des telomeres.358"360
NBN et le syndrome des cassures chromosomiques de Nijmegen (Nijmegen breakage
syndrome ; NBS).
Décrit pour la première fois par Weemaes et collaborateurs en 1981,361 NBS est un
syndrome récessif rare caractérisé par un retard de croissance, une microcephalic, une
immunodéficience, des manifestations pigmentaires de la peau et un risque très élevé de
cancers.362 Les caractéristiques cliniques du NBS s'apparentent à ceux de l'ataxie-
telangiextasie et de l'AF.363'364 Au plan cellulaire, on observe une instabilité
chromosomique spontanée365, une sensibilité accrue aux radiations ionisantes (induisant des
BDBs) et des défauts de cycle cellulaire.366"368 On observe chez ces patients des
lymphomes, hépatomes, méningiomes, gonadoblastomes, médulloblastomes et des
rhabdomyosarcomes et peu de ces patients survivent passé la trentaine.369
La nibrine est composée d'un domaine FHA (Forkhead-associated domain) et de deux
domaines BRCT en N-terminal et de domaines de liaison à MRE11A et à ATM en C-
terminal.351 C'est par ses domaines FHA et BRCT que la nibrine peut reconnaître l'histone
y-H2AX et recruter ses partenaires MRE11A et RAD50 aux sites des dommages. La
nibrine est une protéine essentielle aux cellules.370 Chez les patients atteints du syndrome
de Nijmegen, on retrouve dans la majorité des cas la mutation hypomorphique de NBN
(657del5) qui permet l'expression d'une protéine de 70 kDa (kilo Daltons) par l'utilisation
d'un site d'initiation de la traduction interne.371
Avant même l'identification du gène NBN, il a été reconnu que les parents de patients
atteints du NBS (hétérozygotes présumés pour les mutations de NBN) présentent un risque
accru de cancers.372 Plusieurs investigations se sont concentrées sur la mutation 657del5,
présente chez plus de 90% des patients atteints de NBS. La fréquence des porteurs de cette

54
mutation est de 0,6% dans les populations d'ascendance slave d'Europe Centrale et de
l'Est.373 La contribution de cette mutation ou autres variants de NBN au risque de cancer
demeure controversée,374"384 et sera discuté dans le contexte de mes recherches au Chapitre
III et mis en perspective dans la section discussion de la thèse.
3.2.2.2 Implication de BRCAl et H RCA 2 dans l'Anémie de Fanconi
En 1927, le médecin et scientifique suisse Guido Fanconi décrit trois frères présentant une
combinaison de pancytopénie, d'anomalies congénitales et d'hyperpigmentation se
transmettant sous forme autosomique récessive.385 Le phénotype typique, qui est présent
chez 70-75% des patients, se caractérise par une petite stature en combinaison avec une
variété d'anomalies congénitales (défauts de l'axe radial, changements pigmentaires,
malformations urogénitales, atrésie du canal auditif externe, et plusieurs autres), des
anomalies endocriniennes et de l'infertilité.386 En 1964, Schroeder et collaborateurs
démontrent l'existence d'une instabilité chromosomique (entre autres des bris de la
chromatide) chez des patients AF,38' ce qui suggérait une implication des défauts
génétiques sous-jacents dans la réparation des BDBs associée à une fréquence accrue de
cancers chez ces patients. Près d'im tiers des patients ne présente pas d'anomalie
congénitale, et la première manifestation de la maladie est le développement d'une
défaillance de la moelle osseuse ou l'apparition d'un cancer.388 Le risque de leucémies est
estimé à 40% à 30 ans, et le risque de tumeurs solides à 76% à 45 ans.389
Il est intéressant de noter que les cellules de patients atteints d'AF présentent plusieurs
caractéristiques communes avec les cellules déficientes en BRCAl ou BRCA2.390 En plus
d'une instabilité chromosomique, les cellules de patients AF sont hypersensibles aux agents
induisant des ponts de l'ADN (interstrand crosslinks; ICL), tels la mitomycine C (MMC),
le diepoxybutane, la cisplatine ou les agents de type «moutarde à l'azote».390 La sensibilité
à ce type d'agents est encore utilisée à ce jour pour confirmer le diagnostic clinique. Les
cellules de patients atteints d'AF présentent aussi une sensibilité légère aux radiations
ionisantes ainsi qu'aux radicaux libres de l'oxygène.390
Au plan génétique, l'AF est fortement hétérogène. Des expériences de complémentation,
qui consistent à insérer dans les cellules en culture du patient l'ADNc (ADN
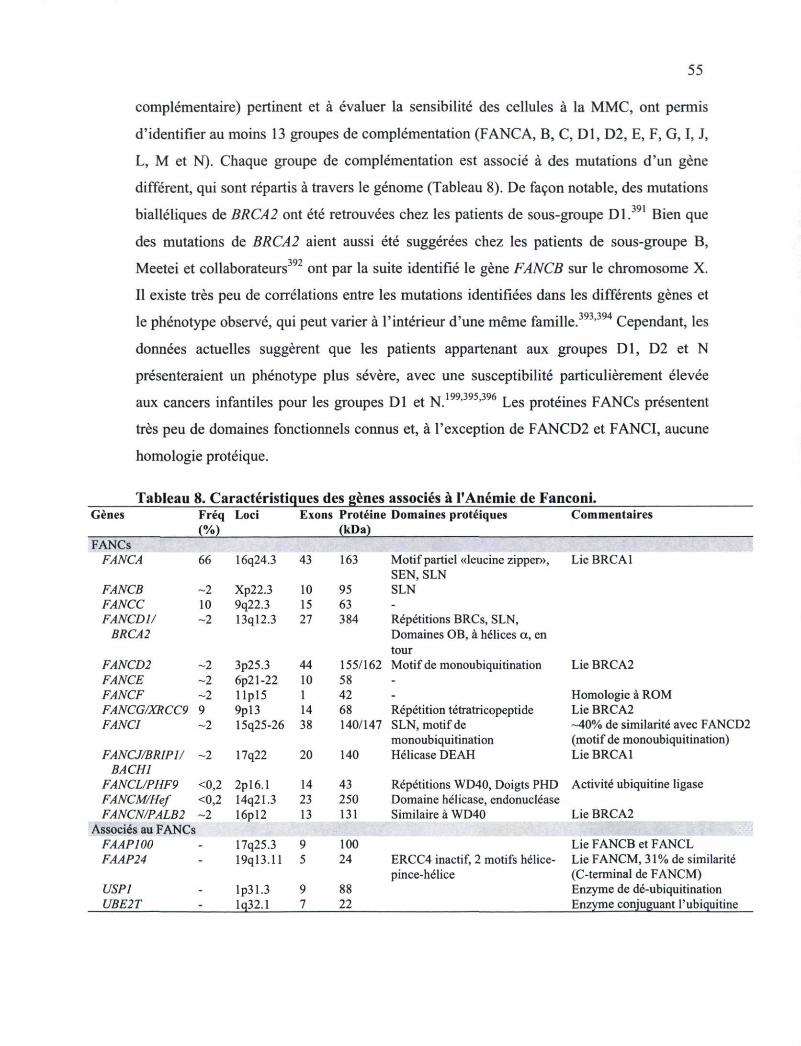
55
complémentaire) pertinent et à évaluer la sensibilité des cellules à la MMC, ont permis
d'identifier au moins 13 groupes de complémentation (FANCA, B, C, Dl, D2, E, F, G, I, J,
L, M et N). Chaque groupe de complémentation est associé à des mutations d'un gène
différent, qui sont répartis à travers le génome (Tableau 8). De façon notable, des mutations
bialléliques de BRCA2 ont été retrouvées chez les patients de sous-groupe Dl.391 Bien que
des mutations de BRCA2 aient aussi été suggérées chez les patients de sous-groupe B,
Meetei et collaborateurs392 ont par la suite identifié le gène FANCB sur le chromosome X.
Il existe très peu de corrélations entre les mutations identifiées dans les différents gènes et - , , , -a *aûy4
le phénotype observé, qui peut varier à l'intérieur d'une même famille. ' Cependant, les
données actuelles suggèrent que les patients appartenant aux groupes Dl, D2 et N
présenteraient un phénotype plus sévère, avec une susceptibilité particulièrement élevée
aux cancers infantiles pour les groupes Dl et N-99*395*396 Le s protéines FANCs présentent
très peu de domaines fonctionnels connus et, à l'exception de FANCD2 et FANCI, aucune
homologie protéique.
Tableau 8. Caractéristiques des gènes associés à l'Anémie de Fanconi Gènes Fréq Loci Exons Protéine Domaines protéiques Commentaires
(%) (kDa) FANCs
FANCA 66 16q24.3 43 163 Motif partiel «leucine zipper», SEN, SLN
Lie BRCAl
FANCB ~2 Xp22.3 10 95 SLN FANCC 10 9q22.3 15 63 -FANCD1/ ~2 13ql2.3 27 384 Répétitions BRCs, SLN,
BRCA2 Domaines OB, à hélices a, en
FANCD2 ~2 3p25.3 44 155/162 tour Motif de monoubiquitination Lie BRCA2
FANCE ~2 6p21-22 10 58 -FANCF ~2 l lpl5 1 42 - Homologie à ROM FANCG/XRCC9 9 9pl3 14 68 Répétition tétratricopeptide Lie BRCA2 FANCI ~2 15q25-26 38 140/147 SLN, motif de
monoubiquitination -40% de similarité avec FANCD2 (motif de monoubiquitination)
FANCJ/BRIP1/ ~2 17q22 20 140 Helicase DEAH Lie BRCAl BACH1
FANCL/PHF9 <0,2 2pl6.1 14 43 Répétitions WD40, Doigts PHD Activité ubiquitine ligase FANCM/Hef <0,2 14q21.3 23 250 Domaine helicase, endonucléase FANCN/PALB2 ~2 16pl2 13 131 Similaire à WD40 Lie BRCA2
Associés au FANCs \\\WÊÊÊÊÊÊÊÊÊÊÊÊÊÊÊÈÊÊÊÊÊ? FAAP100 - 17q25.3 9 100 Lie FANCB et FANCL FAAP24 - 19ql3.11 5 24 ERCC4 inactif, 2 motifs hélice-
pince-hélice Lie FANCM, 31% de similarité (C-terminal de FANCM)
USP1 - lp31.3 9 88 Enzyme de dé-ubiquitination UBE2T - lq32.1 7 22 Enzyme conjuguant l'ubiquitine

56
L'étude des bases génétiques de l'AF ont mis en évidence des liens étroits entre BRCAl,
BRCA2 et les protéines FANCs, en plus d'identifier tout un réseau cellulaire de réponse
aux dommages de l'ADN (Figure 15).397 Jusqu'à présent, il a été démontré que FANCA, B,
C, E, F, G, L et M forment un complexe (complexe de base) avec les protéines associées
FAAP24 et FAAP100. Aucune mutation des gènes encodant FAAP24 et FAAP100 n'a
jusqu'à présent été identifiée, d'où leur appellation de FAAP (Fanconi Anemia associated
protein), suivi du poids moléculaire. Par l'intermédiaire de FANCM et FAAP24, le
complexe de base interagit avec l'ADN aux sites de bris, possiblement pour stabiliser les
fourches de replication bloquées par la présence des ICLs.398 Le complexe de base possède
aussi une activité ubiquitine ligase, dont FANCL est la sous-unité catalytique.399'400 Cette
activité est responsable de la mono-ubiquitination d'un deuxième complexe formé des
protéines FANCD2 et FANCI (complexe ID) suite à la formation de ICLs dans l'ADN
(Figure 16). Ces bris peuvent aussi activer certaines kinases, dont ATR et CHEK1, qui à
leur tour sont responsables de l'hyperphosphorylation du complexe de base en
phosphorylant FANCA, FANCM, FANCE et FANCG.401"404
La mono-ubiquitination de FANCD2 et FANCI sert de signal d'activation de localisation
au niveau de foci de réparation des bris. Certaines évidences semblent suggérer que
FANCD2 mono-ubiquitinée présente une affinité accrue pour l'ADN. Il est aussi possible
que FANCD2 et FANCI peuvent interagir au niveau de l'ADN avec des protéines
possédant des motifs de liaison de l'ubiquitine. De façon intéressante, l'enzyme USP1
(ubiquitin-specific protease 1) serait responsable de la régulation d'une part de l'activité du
complexe en dé-ubiquitinant FANCD2 et FANCI. Une autre part de la régulation du
complexe ID pourrait être effectuée par phosphorylation par ATM/ATR, ce qui pourrait
servir de signal d'amplification et d'activation du point de restriction en phase s.405"408
Un troisième groupe de protéines FANCs est situé en aval, ou du moins en parallèle, du
complexe ID. Ce groupe est composé de FANCD1/BRCA2, FANCJ/BRIP1/BACH1 et
FANCN/PALB2. Une caractéristique importante des gènes encodant ces protéines est leur
association avec le cancer du sein lorsque mutés sur un seul allele (voir les sections 2.2.1.1
HBOC, BRCAl et BRCA2, 2.2.3.3 BRIP1/BACH1/FANCJ et 2.2.3.4 PALB2IFANCN).
La fonction exacte du complexe ID au niveau de la chromatine est inconnue. Toutefois, le
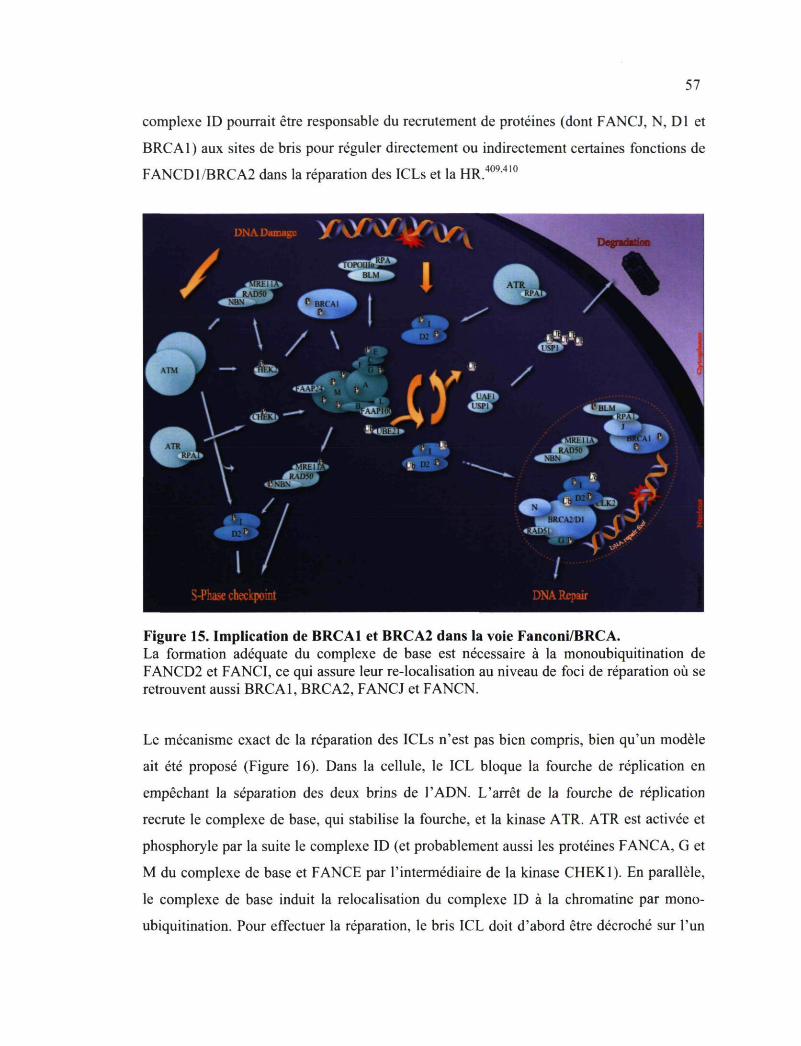
57
complexe ID pourrait être responsable du recrutement de protéines (dont FANCJ, N, Dl et
BRCAl) aux sites de bris pour réguler directement ou indirectement certaines fonctions de
FANCD1/BRCA2 dans la réparation des ICLs et la HR.409'410
Figure 15. Implication de BRCAl et BRCA2 dans la voie Fanconi/BRCA. La formation adéquate du complexe de base est nécessaire à la monoubiquitination de FANCD2 et FANCI, ce qui assure leur re-localisation au niveau de foci de réparation où se retrouvent aussi BRCAl, BRCA2, FANCJ et FANCN.
Le mécanisme exact de la réparation des ICLs n'est pas bien compris, bien qu'un modèle
ait été proposé (Figure 16). Dans la cellule, le ICL bloque la fourche de replication en
empêchant la séparation des deux brins de l'ADN. L'arrêt de la fourche de replication
recrute le complexe de base, qui stabilise la fourche, et la kinase ATR. ATR est activée et
phosphoryle par la suite le complexe ID (et probablement aussi les protéines FANCA, G et
M du complexe de base et FANCE par l'intermédiaire de la kinase CHEK1). En parallèle,
le complexe de base induit la relocalisation du complexe ID à la chromatine par mono
ubiquitination. Pour effectuer la réparation, le bris ICL doit d'abord être décroché sur l'un

58
des brins par les endonucléases XPF-ERCC1 et MUS81-EME1. Le brin brisé est ensuite
réparé par une polymerase TLS (translesion synthesis) qui passe au-delà de la lésion, mais
qui est sujette aux erreurs. La réparation par excision nucléotidique (nucleotide excision
repair, NER) enlève ensuite l'adduit et répare la séquence brisée. Le résultat à cette étape
est un BDB qui est à son tour réparé par HR par BRCA2, PALB2 et RAD51.397
Figure 16. La réparation des ICLs rencontrés lors de la replication par la voie Fanconi/BRCA. La création d'un ICL cause l'activation du complexe ID par le complexe de base. Une fois activé, ID s'associe à la chromatine où il serait impliqué dans le recrutement de protéines de remodelage de l'ADN et de protéines impliquées dans la réparation.
Polymerase JM rçs- B1n-rcAi*s® *-■■> A ^ L X R r a HR
Les protéines FANCs et BRC A1/2 interagissent de plusieurs façons au sein de cette voie
cellulaire de réparation. La première évidence d'un lien avec la voie Fanconi a été
identifiée en 2001, lorsqu'il a été démontré que BRCAl est nécessaire à la re-localisation
de FANCD2 mono-ubiquitinée au niveau de foci nucléaires,4" et à son interaction avec
l'histone y-H2AX.412 En plus de l'implication de BRCA2, BRIP1/BACH1 et PALB2 dans
l'AF, une interaction constitutive de FANCA avec BRCAl a aussi été démontrée,41 et
certaines évidences suggèrent que les protéines FANCs pourraient avoir certaines activités
hors de la voie de réparation décrite plus haut.414 De façon intéressante, FANCD2 interagit

59
directement avec BRCA2 dans un sous complexe qui comprend aussi FANCG et XRCC3
(un paralogue de RAD51), peut-être pour promouvoir la HR en aval de la voie
Fanconi/BRCA.415'416
Tel que mentionné précédemment, le risque de cancer est fortement accru chez les patients
atteints d'AF. Ceux-ci présentent majoritairement des leucémies myélocytaires aiguës et
certains types de cancers solides (en particulier carcinomes tête et cou, gynécologiques ou
de l'œsophage). 7 II est aussi notable que les patients AF de sous-groupe Dl et N
présentent fréquemment des tumeurs cérébrales et de Wilms.199'395'396 De par leur sensibilité
aux agents induisant des bris de l'ADN, leur implication dans les voies de réparation de ces
bris ainsi que leur association avec plusieurs gènes altérés dans le cancer, les gènes FANCs
sont des candidats intéressants dans la recherche de gènes altérés dans la tumorigénèse.
Certaines évidences suggèrent que les cellules de porteurs de mutations des gènes FANCs
pourraient être plus sensibles aux agents induisant des bris de l'ADN, bien que les
différences observées ne sont pas toujours significatives.418"420 Outre le risque de cancer
associé à des mutations mono-alléliques de BRCA2/FANCD1, PALB2/FANCN et
BRIP1/BACH 1/FANCJ (voir les sections 2.2.1.1, 2.2.3.3 et 2.2.3.4), certaines altérations de
la voie Fanconi/BRCA ont aussi été rapportées dans certaines cohortes de patients non-AF.
L'étude des familles d'individus atteints d'AF a jusqu'à présent apporté des évidences
contradictoires. Une étude initiale par Swift et collaborateurs421 a suggéré un risque accru
de cancers chez les parents de patients AF, bien que cette association n'ait pas été retrouvée
dans d'autres éludes subséquentes limitées par le nombre de familles disponibles pour
analyse.422"423 Une étude récente chez 944 parents (majoritairement des grands-parents) de
patients atteints d'AF semble suggérer une augmentation du risque de cancer du sein chez
les grands-mères des patients, en particulier chez les familles porteuses de mutations de
FANCC avec un rapport d'incidence standardisé (standardized incidence ratio; SIR) de 2,4
(IC 95% l,l-5,2).424 Il est à noter que la sous-division des familles en fonction du groupe
de complémentation réduit de beaucoup le nombre d'individus par groupes et peut rendre
difficile l'identification d'un possible effet gène-spécifique. Une autre étude regroupant 575
individus de 36 familles n'a pas été en mesure de reproduire ce résultat, bien que cette
étude ait mis en évidence une association possible avec un risque accru de cancer de la

60
prostate chez les porteurs obligatoires (RR 3,089 IC 95% 1,09-8,78). Cependant, la division
des individus en fonction du groupe de complémentation n'a pas été possible dans cette
étude.425
L'implication de variants des gènes FANCs, ou une altération de leur expression, a été
suggérée dans certaines instances de leucémies,426"428 de cancer de l'ovaire,429 du
pancréas,430'431 et des carcinomes des cellules squameuses de la cavité orale et de
l'œsophage.432 Entre autres, une duplication de 13 nucleotides dans le promoteur de
FANCA a été étudiée dans une cohorte de 352 cas de cancer du sein, 390 cas de cancer de
l'ovaire et 256 contrôles sains. Alors qu'aucune association n'est retrouvée chez les cas de
cancers du sein, la duplication est significativement plus fréquente chez les patientes
atteintes de cancer de l'ovaire par rapport aux contrôles sains, ce qui suggère qu'elle
pourrait avoir un effet protecteur (OR 0,72; IC 95% 0,53-0,99).429
Un cas particulier de l'implication des gènes FANCs dans la carcinogenèse est l'altération
de l'expression de FANCF suite à l'hyperméthylation de son promoteur. Jusqu'à présent, la
répression épigénétique de FANCF a été observée dans de nombreux cancers dont des
leucémies,433 des cancers de la vessie,434 des cancers du col de l'utérus,435 des cancer du
poumon,436 et des cancers de l'ovaire.437'438 Il est logique de penser que par leur rôle dans le
maintien de la stabilité génomique, les gènes FANCs soient la cible d'une inactivation
résultant ultimement dans la formation de tumeurs. Cependant, il est jusqu'à présent
difficile de déterminer si cette altération est un événement de l'initiation ou de la
progression tumorale. De façon intéressante, des études récentes suggèrent que FANCC,
F AN CL, FANCNIPALB2 pourraient aussi être la cible d'une altération de leur expression
par hyperméthylation dans certains cas de leucémies ou de cancers du sein. ' ' De plus,
FANCB, FANCF, FANCD2 et FANCJ/BRIP1/BACH1 pourraient voir leur expression
altérée dans certaines lignées cellulaires cancéreuses et tumeurs primaires de carcinomes de
la tête et du cou440'441 ou de cancers du sein (à la fois sporadiques et héréditaires).442
Jusqu'à présent, l'implication des gènes FANCs (autres que FANCD1/BRCA2,
FANCN/PALB2 ou FANCJ/BRIP 1/BACH1) dans la carcinogenèse mammaire a été peu
étudiée. L'analyse des gènes FANCA, B, C, D2, E, F et G a été effectuée par Seal et
collaborateurs443 dans une cohorte de 88 familles avec une histoire familiale chargée de

61
cancer du sein du Royaume-Uni. Bien que cette étude n'ait pas identifié de mutation
clairement pathogène, deux variants faux-sens observés (FANCA p.Leull43Val et FANCE
p.Arg365Lys) étaient absents des 300 contrôles sains génotypes et semblaient être transmis
avec la maladie. Bien que l'analyse de FANCD2 dans une cohorte australienne de cas de
cancers du sein n'ait pas permis de mettre en évidence une association,444 l'étude de 7
variants fréquents de FANCA, FANCL et FANCD2 dans une cohorte de 897 cas de cancer
du sein sporadiques et 1033 contrôles sains a démontré une association possible du variant
L1366L de FANCD2 (OR par allele de 1,35 IC 95% 1,09-1,67). Le variant p.Leul366Leu
pourrait être en déséquilibre de liaison avec un autre variant situé dans une région
conservée du promoteur.445 Jusqu'à présent, ces évidences suggèrent une association des
gènes FANCs dans le développement de cancers, bien que cette association ne soit pas
définitive446"448 et doive être sujette à des analyses plus poussées.
3.2.3 Autres fonctions de BRCAl et BRCA2
Outre ces activités, BRCAl et BRCA2 possèdent d'autres fonctions cellulaires, bien
qu'elles soient généralement moins bien comprises.
BRCAl et le remodelage de la chromatine.
L'activité de régulation de la transcription de BRCAl semble s'effectuer à la fois par
l'intermédiaire du recrutement de co-activateurs et co-répresseurs, mais aussi par le
recrutement de protéines impliquées dans le remodelage de la chromatine. BRCAl interagit
avec des acétylases d'histones (HDAC1/2), la composante BRG1 associée au complexe de
remodelage SWI/SNF, ainsi que les protéines hGCN5/TRRAP, TRAP220 et p300/CBP qui
font partie de complexes acétyl-transférases.449"453 L'interaction de BRCAl avec ces
différentes protéines de remodelage de la chromatine permettrait ainsi l'accès des facteurs
de transcription aux sites appropriés.
BRCAl. BRCA2 et la régulation du cycle cellulaire.
Le contrôle du cycle cellulaire est un mécanisme essentiel permettant l'arrêt de la
croissance afin de procéder à la réparation des bris de l'ADN. L'activité de BRCAl dans la
régulation de plusieurs points de restriction du cycle cellulaire est effectuée en partie par le
contrôle transcriptionnel de certains gènes clés (par exemple GADD45A, Figure 12) et

62
d'autre part par son association à des protéines de régulation (Figure 15). L'implication de
BRCA2 dans les mécanismes de régulation du cycle cellulaire est moins claire. Certaines
analyses suggèrent que BRCA2 pourrait être nécessaire à la progression de la cellule à
travers la mitose par son association avec BRAF35 et leur co-localisation au niveau des 454 chromosomes. De plus, BRCA2 s'associe à la kinase BUBR1/BUB1B, qui est
impliquée dans l'attachement adéquat des chromosomes au fuseau mitotique. BUB1B
inhibe APC/C, ce qui retarde l'anaphase et assure la ségrégation appropriée des chromosomes 456
Phosphorylation de BRCAl par ATM Phosphorylation dépendante de BRCAl de p53 (Serl5) Régulation transcriptionnelle de CDKtflA
Régulation de la transcription du gène MÂD2L1
G2/M
Phosphorylation de BRCAl par A' (Serl3875
-ATM
Activation de la kinase CHEK1 (requiert BRCAl) Régulation de la transcription des gènes Wctl, PLK1,14-3-3 et GADD45A
Figure 17. Implication de BRCAl dans la régulation du cycle cellulaire. L'implication de BRCAl dans la régulation du cycle cellulaire a été démontrée au niveau des principaux points de restriction cellulaires.
L'activité ubiquitine ligase de BRCAl.
BRCAl s'associe de façon constitutive dans la cellule à BARD1, et cette association est
requise pour l'activité ubiquitine ligase de BRCAl.457 À son tour, le complexe peut être
activé par la conjugaison d'une protéine SUMO (small ubiquitin-like modifier)?5* Il a été
suggéré que l'hétérodimère BRCAl-BARD1 participe à plusieurs complexes cellulaires qui
pourraient ubiquitiner différents substrats. Parmi ceux-ci, RNA pol II, ERa, CtlP et TFIIE
ont été suggérés.459"463 Les conséquences biologiques de l'ubiquitination par BRCAl -
BARD1 ne sont pas bien comprises, car le signal induit n'a pas encore été déterminé. En
fonction du type de chaine d'ubiquitination, plusieurs types de procédés cellulaires peuvent
être signalés.464 Il a été suggéré que la spécificité de la réaction d'ubiquitination résulte de
l'interaction différentielle avec plusieurs enzymes E2 de conjugaison de l'ubiquitine.465

63
4. Types de variations de séquence Les gènes possèdent une structure spécifique nécessaire à leur expression adéquate. Par
conséquent, ils peuvent être la cible de plusieurs types d'altérations, certaines délétères,
certaines bénignes et d'autres dont les conséquences sont mal comprises dans le cadre de
nos connaissances actuelles. En fonction du type et de la position de la variation, le gène
sera exprimé de façon normale, sera plus/moins exprimé qu'à la normale ou produira
ultimement une protéine tronquée incapable de fonctionner normalement à l'intérieur de la
cellule. Généralement, on reconnaît trois types d'altérations de la séquence : les
substitutions nucléotidiques, les deletions ou insertions d'un ou plusieurs nucleotides
(moins de 20) et les réarrangements génomiques (grandes insertions, grandes deletions,
inversions, duplications ou translocations). Souvent, ces derniers sont produits suite à une
recombinaison entre séquences similaires mais non identiques (par exemple en implicant
des répétitions ALU dispersées au sein du génome).
Les substitutions et les insertions/deletions auront des conséquences variables en fonction
de la région dans laquelle elles sont situées. Ces altérations au niveau de la région codante
auront des conséquences lors de la traduction. Une insertion ou deletion impliquant un
multiple de 3 nucleotides créera une insertion ou une deletion d'un ou de plusieurs aa.
Lorsqu'elles impliquent un nombre de nucleotides qui n'est pas un multiple de trois, ces
types de variations entraîneront une modification du cadre de lecture qui introduit un codon
d'arrêt prématuré résultant en la production d'une protéine tronquée souvent non
fonctionnelle. Les substitutions nucléotidiques de la région codante peuvent quant à elles
induire le changement d'un aa (changement faux-sens) ou déterminer un arrêt de la
traduction en introduisant un codon Stop (changement non-sens). Peu importe leur
emplacement dans la séquence du gène ou de son promoteur, les subtitutions
nucléotidiques, les insertions et les deletions peuvent aussi modifier l'expression du gène
(Figure 18 A).

64
I Modification Altération du site Création d'un Alteration Alteration du l d'un site de d' initiation de la nouveau site d'unsite5'3 ' sitedepoly-4. liaison d'un transcription d'épissage d'épissage adénylation
d'un site de liaison d'un facteur dc
transcription
Altération d'un facteur de transcription
Modification d'un site de liaison d'un facteur de
transcription séauences de maturation de l'ARNm
nouveau site d'épissage
Modification d'un motif exomque de promotion (ESE) ou de répression
(ESS) de l'épissage Mutation non-sens prés d'une jonction exon
intron induisant la dégradation de l'ARNm
Altération Modification d ' un du Mie de motif de stabilisation
branchement dégradation de l'ARNm
Modification d'un motif intronique de promotion (ISE) ou de répression
i ISS i dc l'épissage
B
Figure 18. Position des altérations pouvant affecter l'expression d'un gène. (A) Le niveau de transcrits produits peut être influencé par l'altération de divers mécanismes de régulation de la transcription du gène, ou de la maturation et de la stabilité de l'ARNm. (B) Séquences consensus pour les sites donneurs, accepteurs et de branchement. Les nucleotides en rouge sont habituellement invariables (bien qu'il existe certains types d'exons dont les nucleotides AG du site accepteur sont remplacés par AC, et les nucleotides GT du site donneur sont remplacés par AT). Aux autres positions indiquées, les nucleotides les plus fréquemment retrouvés sont indiqués. Les bandes vertes et rouges représentent des séquences promouvant l'inclusion et l'exclusion des exons, respectivement. ESE : Exonic splicing enhancer. ESS : Exonic splicing silencer. ISE: Intronic splicing enhancer. ISS: Intronic splicing silencer. Pyrn: Répétition pyrimidique.
Lors de son expression, le gène eukaryote est d'abord transcrit en un ARNm précurseur qui
subit par la suite une maturation permettant d'enlever les séquences introniques (épissage)
ainsi que l'ajout d'une coiffe à son extrémité 5' et une queue poly-adénylée à son extrémité
3'. La réaction d'épissage est un mode très important de régulation et nécessite la présence

65
de certaines séquences de signalisation qui permettent de reconnaître le début et la fin de
l'intron, et ce sur de grandes distances. Ces séquences sont au minimum constituées d'un
site donneur en 5' de l'intron, ainsi que d'un site de branchement et d'un site accepteur en
3' (Figure 18B). Ces séquences sont reconnues par divers complexes snRNP qui sont
composés d'une part d'une molécule d'ARN et d'autre part de protéines. La réaction
d'épissage s'effectue en deux étapes. Dans un premier temps, l'ARNm précurseur est clivé
en 5' d'un intron et cette extrémité est liée au site de branchement. Au cours de la seconde
étape, l'ARNm précurseur est clivé à l'extrémité 3' de l'intron, les exons sont ligués et la
boucle formée par l'intron (lariat) est relâchée.458
Bien que les signaux aux extrémités 5' et 3' de l'intron permettent l'épissage, ces
séquences sont fréquemment insuffisantes afin de distinguer les exons des introns. Des
séquences additionnelles de promotion (enhancer) et d'inhibition (silencer) sont retrouvées
à la fois dans les introns et les exons. Ces séquences peuvent être près (50-100 pb) ou loin
(de 100 à plusieurs milliers de pb) du site d'épissage. En plus de permettre l'épissage
correct des transcrits, ces séquences et l'action des protéines régulatrices avec lesquelles
elles interagissent permettent un épissage alternatif selon le type cellulaire et/ou le stade du
développement et/ou les conditions physiologiques. De cette façon, plusieurs messages
(isoformes d'ARNm) peuvent êtres produits à partir d'un même gène et il est présentement
estimé que plus de 60% des gènes pourraient être sujets à de l'épissage alternatif.459

66
5. Problématique Tel qu'illustré dans les sections précédentes, le cancer du sein est une maladie très
complexe qui affecte une vaste proportion d'individus mondialement. Bien que certains
facteurs de risque aient été identifiés, les connaissances actuelles sont insuffisantes pour
permettre d'identifier tous les individus ayant un risque plus élevé au sein de la population,
ou de comprendre les effets «écogénétiques» (c'est-à-dire l'interaction entre la génétique
d'un individu et son environnement) affectant la susceptibilité d'un individu à développer
un cancer du sein. En ce sens, les études de jumeaux mono- et dizygotes mettent en
évidence la place prépondérante de la génétique dans la prédisposition à cette maladie.50'51
Au cours des 15 dernières années, des progrès majeurs ont été effectués dans
l'identification de gènes/allèles de susceptibilité. Malgré cela, il est présentement estimé
que les alleles de susceptibilité connus sont responsables d'au maximum 30% du risque
familial de cancer du sein.38 Dans la population canadienne française, les analyses
effectuées dans les groupes des Drs Durocher et Simard indiquent que les alleles connus
(de penetrance forte à intermédiaire) ne seraient responsables que d'environ 25% du
regroupement familial de cancer du sein.65'1 6-468"470 Globalement, ces données indiquent
qu'il demeure des alleles de susceptibilité au cancer du sein à identifier.
5.1 Collection familiale étudiée : les familles à risque élevé de la population canadienne française Les recherches présentées dans les chapitres suivants ont été effectuées dans le cadre des
projets du programme interdisciplinaire INHERIT BRCAs (Interdisciplinary Health
Research International Team on BReast CAncer susceptibility), dirigé par le Dr Jacques
Simard, et dont l'équipe du Dre Durocher fait partie. Dans un premier temps, ce
programme visait entre autre l'étude des facteurs génétiques et environnementaux associés
au cancer, ainsi que la détermination des enjeux éthiques, légaux et psychosociaux associés
aux tests de dépistages génétiques. C'est par l'intermédiaire de ce programme que les Drs
Simard et Durocher ont entre autres été en mesure d'évaluer la fréquence et la penetrance
des mutations de BRCAl et BRCA2 dans une cohorte de familles à risque élevé de cancer
du sein et/ou de l'ovaire de la population canadienne française.65 Brièvement, les 250
familles participant à ce programme ont été recrutées selon des critères précis indicatifs

67
d'une forte prédisposition génétique : 1) des individus de familles à risque élevé avec
quatre cas de cancer du sein et/ou de l'ovaire diagnostiqués chez des parents de premier ou
deuxième degré; 2) des familles présentant trois cas de cancers du sein et/ou de l'ovaire
chez des parents de premier degré; 3) des familles chez lesquelles une mutation des gènes
BRCAl ou BRCA2 avait déjà été identifiée au préalable 4) des familles présentant des
évidences claires d'une transmission héréditaire. Les familles chez lesquelles la recherche
de mutations de BRCAl et BRCA2 s'est avérée négative ont été invitées à participer au
projet du Dre Durocher visant l'identification d'autres alleles de susceptibilité au cancer du
sein.
Des recherches génétiques dans une population telle que celle du Québec, majoritairement
canadienne française, offrent des opportunités particulières. Cette population provient d'un
effet fondateur qui se manifeste entre autres par la présence à une fréquence accrue de
certaines mutations.471 Entre la fondation de la ville de Québec, il y a 400 ans, et la
conquête par les Britanniques en 1759, plus de 25000 colons français (provenant
majoritairement de la côte atlantique et de la région de Paris) se sont établis en Nouvelle-
France. Cependant, seulement 8500, dont 1600 femmes, se sont établis de façon
permanente.472 Après la conquête, des barrières de religion et de langue ont limité les
mariages avec les nouveaux arrivants majoritairement anglophones protestants. On estime
que les 2600 colons établis en Nouvelle-France avant 1680 contribuent pour environ les
deux tiers du bassin génétique actuel de la population canadienne française.471 Par
conséquent, la population canadienne française est constituée à partir d'un petit échantillon
non représentatif de la population française, d'où l'effet fondateur observé qui entraîne un
changement dans la fréquence de certains alleles. L'historique de la population canadienne
française peut être utilisé afin de faciliter l'identification de nouveaux gènes de
susceptibilité.
5.2 Objectifs de l'étude Ainsi, mon projet de recherche vise à évaluer la contribution de nouveaux gènes de
susceptibilité au cancer du sein par une approche de type «gène candidat». Les gènes
étudiés ont été choisis sur la base de leur pertinence biologique dérivant de leur implication
dans des voies cellulaires communes avec les alleles de susceptibilité connus, en particulier

68
leur étroite interaction avec BRCAl et BRCA2. En ce sens, les chapitres I et II présentent les
analyses génétiques de ZNF350/ZBRK1 et de GADD45A qui sont associés à certains
aspects de la régulation de la transcription opérée par BRCAl. Dans certaines
circonstances, ZNF350 agit comme co-répresseur de l'activité transcriptionnelle de
BRCAl, entre autres afin de réguler négativement l'expression de GADD45A et permettre
la progression cellulaire de la phase G2 à la phase M. Au chapitre III sont présentés les
résultats de l'étude du gène NBN dans notre cohorte de familles à risque élevé de cancer du
sein et/ou de l'ovaire. Tel qu'indiqué à la section 3.2.2.1, la nibrine joue un rôle essentiel à
l'intérieur de la cellule en participant à la reconnaissance précoce des bris, en activant les
voies de réparation adéquates (dont celles impliquant BRCAl et BRCA2) et en rejoignant
les molécules d'ADN pour permettre une réparation appropriée.

69
CHAPITRE I : Genetic Variants and -Haplotype Analyses of the ZBRK1/ZNF350 Gene in High-Risk non BRCAl72
French Canadian Breast and Ovarian Cancer Families.

70
Genetic Variants and Haplotype Analyses of the ZBRK1/ZNF350 Gene in High-Risk
non BRCA1/2 French Canadian Breast and Ovarian Cancer Families.
Sylvie Desjardins1, Pascal Belleau2, Yvan Labrie1, Geneviève Ouellette1, Paul Bessette3,
Jocelyne Chiquette4, Rachel Laframboise5, Jean Lépine6, Bernard Lesperance7, Roxane
Pichette7, Marie Plante8, INHERIT BRCAs* and Francine Durocher1**
'Cancer Genomics Laboratory, Oncology and Molecular Endocrinology Research Centre,
Centre Hospitalier Universitaire de Québec and Laval University, Québec, Canada;
2Oncology and Molecular Endocrinology Research Centre, Centre Hospitalier Universitaire
de Québec and Laval University, Québec, Canada;
3Service de gynécologie, Centre Hospitalier Universitaire de Sherbrooke, Fleurimont,
Canada;
4CHnique des maladies du sein Deschênes-Fabia, Hôpital du Saint-Sacrement, Québec,
Canada;
5Service de medicine génétique, Centre Hospitalier Universitaire de Québec, Pavillon du
CHUL, Canada;
6Centre Hospitalier Régional de Rimouski, Rimouski, Canada;
7Service d'hémato-oncologie, Hôpital du Sacré-Cœur, Montréal, Canada ;
8Service de gynécologie, Centre Hospitalier Universitaire de Québec, Hôpital Hôtel-Dieu
de Québec, Québec, Canada.
Short title : ZBRK1 variants and haplotypes in French Canadian BOC families.

71
*Other members of INHERIT BRCAs involved in this study are listed in Appendix 1.
**Corresponding author:
Dr. Francine Durocher
Cancer Genomics Laboratory, Oncology and Molecular Endocrinology Research Centre,
Centre Hospitalier Universitaire de Québec and Laval University,
2705 Laurier Boulevard, T2-53
Québec city, Québec
Canada, G1V4G2
Telephone: (418) 654-2296
FAX: (418) 654-2761
E.mail: Francine.duroeher(Sscrchul.ulaval.ca
Keywords : Breast cancer susceptibility, ZBRK1 variants, haplotype, tagging SNPs
Abbreviations: 3'UTR, 3'untranslated region, ANG1, angiopoietin-1; CI, Confidence
interval; cSNP, coding SNP; EM, Expectation Maximized; LD, Linkage disequilibrium;
MAF, Minor allele frequency; MLPA, Multiplex ligation-dependent probe amplification;
OR, Odd Ratio; PPP2R1A, Protein phosphatase 2 regulatory sub-unit 1 alpha; tSNP,
TaggingSNP; ZBRK1, Zinc finger and BRCAl-interacting protein with a KRAB domain 1.
Int J Cancer. 2008;122(1):108-16.

72
Résumé Notre compréhension actuelle de la susceptibilité au cancer du sein implique
principalement des mutations de deux gènes majeurs, BRCAl et BRCA2, retrouvées chez
approximativement 25% des familles à risque élevé, ainsi que des mutations dans des gènes
de plus faible penetrance, tels ATM et CHEK2. Près des deux tiers des familles présentant
des cas multiples de cancers du sein devront être expliqués par des mutations dans des
gènes encore non identifiés. Dans une approche «gène candidat» visant l'identification de
nouveaux gènes potentiellement impliqués dans la susceptibilité au cancer du sein, nous
avons analysé les variants génomiques du gène ZBRK1, un co-répresseur impliqué dans la
répression de GADD45 induite par BRCAl. Le séquençage de la région codante de ZBRK1,
chez des individus atteints de cancer du sein provenant de 97 familles à risque élevé de
cancer du sein et de l'ovaire canadiennes françaises et 94 contrôles sains a mené à
l'identification de 18 variants génomiques. L'analyse des haplotypes par les programmes
PHASE, COCAPHASE et HaploStats, at mis en évidence trois haplotypes spécifiques
pouvant potentiellement influencer le risque de cancer du sein. Parmi ceux-ci deux sont
associés à un effet protecteur potentiel (p=0.01135 et p=0.00268) alors qu'un autre
haplotype est surreprésenté dans le groupe des cas (p=0.00143). Des analyses plus poussées
de ces haplotypes indiquent qu'une composante importante de la différence observée entre
les deux groupes émerge des 5 premiers variants (parmi les 12 utilisés pour la
détermination des haplotypes). Cette étude a aussi permis de déterminer un ensemble de
SNPs marqueurs pouvant être utilisés lors d'analyses dans des études d'association de
grande envergure. Des études additionnelles, dans de larges cohortes et d'autres
populations, seront cependant nécessaires afin de mieux évaluer si des variants de séquence
et des haplotypes communs et/ou rares de ZBRK1 peuvent être associés à un risque
modéré/intermédiaire de cancer du sein.

73
Abstract Our current understanding of breast cancer susceptibility involves mutations in the two
major genes BRCAl and BRCA2, found in about 25% of high-risk families, as well as few
other low penetrance genes such as ATM and CHEK2. Approximately two-thirds of the
multiple cases families remain to be explained by mutations in still unknown genes. In a
candidate gene approach to identify new genes potentially involved in breast cancer
susceptibility, we analyzed genomic variants in the ZBRK1 gene, a co-repressor implicated
in BRCAl-mediated repression of GADD45. Direct sequencing of ZBRK1 entire coding
region in affected breast cancer individuals from 97 high-risk French Canadian
breast/ovarian cancer families and 94 healthy controls led to the identification of 18
genomic variants. Haplotype analyses, using PHASE, COCAPHASE and HaploStats
programs, put in evidence three specific haplotypes which could potentially modulate
breast cancer risk, and among which two that are associated with a potential protective
effect (p=0.01135 and p=0.00268), while another haplotype is over-represented in the case
group (p=0.00143). Further analyses of these haplotypes indicated that a strong component
of the observed difference between both groups emerge from the first 5 variants (out of 12
used for haplotype determination). The present study also permitted to determine a set of
tagging SNPs that could be useful for subsequent analyses in large scale association studies.
Additional studies in large cohorts and other populations will however be needed to further
evaluate if common and/or rare ZBRK1 sequence variants and haplotypes could be
associated with a modest/intermediate breast cancer risk.

74
Introduction
In western countries, breast cancer is a major cause of malignancies in women with
an estimated incidence of approximately one in 9 by age 70 years. However, it has been
hypothesized that as much as 50% of all breast cancer cases fall within 12% of the
population with an increased risk of breast cancer, based on a polygenic model assuming a
multiplicative effect on risk.1'2 As such, determining the genetics underlying causes of
familial and sporadic cases can have an important impact on breast cancer population
screening and prevention.
There are currently few known susceptibility genes to breast cancer. Highly
penetrant mutations in BRCAl and BRCA2 are found in high-risk breast cancer families
which represent approximately 10% of all breast cancer cases. Among families with a clear
inherited pattern of breast cancer, only 15-20% are found to harbour BRCAl or BRCA2
mutations. Alterations in few other genes, such as PTEN3, TP53, ATM*, PALB25'7 and
CHEK2, account for less than 5% of cases in some populations, including the French-
Canadian population (for a review see Ref. 8). Consequently, there are too many families
with clear inheritance of breast cancer presenting no mutations in the known susceptibility
genes to be an association of random events, thus fuelling searches to find other breast
cancer susceptibility genes. Evaluation of the number of such genes is difficult as a limited
number of genes with moderate penetrance is as plausible as a large number of genes each
resulting in a small risk increase.9 However, finding new susceptibility genes with moderate
impact in a case-control study design requires a large sample size, which can be greatly
reduced when cases are drawn from high-risk families.1 '
Since its identification12, BRCAl has been shown to be involved in many cellular
processes such as double strand break repair, chromatin structure, transcription-coupled
DNA repair and regulation of transcription.8 Among these processes, the role of BRCAl in
transcription is poorly understood, but it has been reported to regulate the transcription of
p 2 1 n and GADD4514, which are both involved in cell cycle arrest, and ANGPT1 involved

75
in angiogenesis.15 GADD45 in particular has been shown to be involved in the activation of
the G2/M checkpoint following DNA damage16 and maintenance of genome stability.17
ZBRK1 (Zinc finger and BRCAl-interacting protein with a KRAB domain I), also
known as ZNF350, is a co-repressor of GADD45 expression.18 ZBRK1 interacts with amino
acids 341 to 748 of BRCAl and binds to the GGGxxxCAGxxxTTT consensus sequence
within GADD45 third intron. Over-expression of BRCAl has been shown to lead to the
release of ZBRK1 -mediated repression18 but is not required for DNA damage induced
degradation of ZBRK1 through the ubiquitin-proteasome pathway.19 The integrity of the
ZBRK1 region interacting with BRCAl, as well as the KRAB domain (constituted of a box
A and a box B), are essential to the repression of GADD45.
Previous studies of examining the implication of ZBRK1 in breast cancer did not
provide strong evidence for ZBRK1 alterations in inherited breast cancer susceptibility.20'21
On the other hand, analysis of breast carcinomas has shown frequent alterations of ZBRK1
expression without correlation with BRCAl mRNA expression or tumor phenotype22, and a
recent study performed on two population-based cohorts from USA and Poland found a
weak significant association of ZBRK1 p.Ser472Pro rare variant with breast cancer risk.23
Based on the key role played by ZBRK1 and its implication in BRCAl-mediated
repression of GADD45, it represents a very attractive candidate gene that could potentially
play a role in breast cancer. Consequently, we analyzed the ZBRK1 gene for sequence
alterations among a series of 97 breast cancer cases selected from high-risk French-
Canadian breast/ovarian cancer families and a series of healthy controls from the same
population. No germline mutation leading to a premature termination of the protein or
sequence change affecting the normal splicing of the gene was identified. However, we did
observe 18 sequence variants, and the estimated haplotypes obtained displayed a significant
difference between the case and control groups. In addition, five variants seem to be
responsible for this difference, which are all located in the most 5' coding part of the gene.
Haplotypes analysis also permitted to establish tagging SNPs (tSNPs), which would be
most useful to genotype for subsequent analyses in larger cohorts to further ascertain
ZBRK1 variants and haplotypes impact on breast cancer susceptibility.

76
Materials and Methods
Ascertainment of families
Recruitment of high-risk French Canadian breast/ovarian families (i.e. families in which
multiple cases of breast/ovarian cancer are present in close relatives -3 cases in 1st or 4
cases in 2nd degree relatives- or with strong evidence of a familial component) was part of
the major interdisciplinary research program INHERIT BRCAs further explained
elsewhere, in which a major component was to identify and characterize BRCA1/2
mutations.24'25 A subset of 97 high-risk French Canadian breast/ovarian cancer families was
drawn from the initial study based on the absence of detectable BRCA1/2 mutation and
constituted the cohort used for another study specifically aiming at the identification of
other susceptibility loci/genes to breast cancer. This latter study obtained ethics approvals
from all participating institution, and each participant, knowing their inconclusive BRC A1/2
test results signed a specific informed consent, and had to be at least 18 years old and
mentally capable. One individual affected with breast cancer per family was selected for
analysis, with a selection preference for the youngest subject available in each family. The
ascertainment criteria of recruitment and BRC A1/2 status analysis have been described
previously.25'26 In all instances, diagnosis of breast cancer was confirmed by pathology
reports. The mean age at diagnosis of these 97 breast cancer subjects was 48 years old (30-
74 years).
To further strengthen the 5./?C47/2-negative status of these families, BRCA1/2 were
analyzed by southern techniques (36 of the 97 individuals) as well as by Multiplex
Ligation-dependent Probe Amplification (MLPA, MRC Holland, Amsterdam, The
Netherlands) (84 of the 97 subjects) to rule out large genomic rearrangements.27 For the
remaining subjects, another family member was analyzed by MLPA, with the exception of
two families on which analyses were not performed.

77
DNA extraction
For each breast cancer participant, total RNA and genomic DNA was extracted from
immortalized cell lines established from blood lymphocytes, and infected with the Epstein-
Barr virus, using CTAB or the Tri-Reagent method according to the manufacturer's
instructions (Molecular Research Center inc, Cincinnati, USA). Genomic DNA was also
obtained from 94 unrelated healthy controls of French Canadian origin (mean age of 46
years; 22 to 68 years). Genomic DNA extraction and description of the control cohort have
been described previously.26 Genomic DNA from three cynomolgus monkeys (one male
and two females) was also extracted from blood samples using Tri-Reagent.
ZBRK1 sequencing and protein alignment
PCR amplification of ZBRK1 in breast cancer individuals was carried out using primers
designed to amplify the entire coding region of the gene, composed of 5 exons, as well as
intron-exon boundaries. Amplification was performed on breast cancer cases and healthy
controls using primers located in intronic sequences, as listed in Supplemental Table I. PCR
amplification with 75ng of genomic DNA from breast cancer subjects and 5 ng of DNA for
the controls series (due to limited material available) has been performed using standard
procedures. Cynomolgus DNA was amplified using primers designed for amplification of
human genomic DNA encompassing ZBRK1.
ZBRK1 direct sequencing was carried out on an ABI3730XL automated sequencer using
version 3.1 of the Big Dye fluorescent method according to the manufacturer's instructions
(Applied Biosystems, Foster City, USA). Sequences were analyzed using Pregap4 and
Gap4 implemented in the Staden package (http^/staden.sourceforge.net/X Accession
numbers of the sequences used in this paper are as follow: NM021632.3 (human ZBRK1
cDNA sequence), NP067645.3 (human ZBRK1 protein sequence), XP512864.1
(chimpanzee ZBRK1 protein sequence), XP541453.1 (canine ZBRK1 protein sequence)
and XP593338.1 (bovine ZBRK1 protein sequence). Protein alignment was made using
version 1.82 of ClustalW (http://www.ebi.ac.uk/clustalw/).29

78
Linkage disequilibrium, haplotype estimations and tSNPs selection
Linkage disequilibrium (LD) measurements were performed with all 18 SNPs identified in
our breast cancer cases using the LDPlotter program
(http://innateimmunity.net/IIPAG2/index_htmn. Lewontin's |D'| and r2 (for review see 30)
was used to calculate linkage disequilibrium between SNPs pairs. Splice site prediction
scores were evaluated using MaxEntScan
(http://genes.mit.edu/burgelab/maxent/Xmaxentscan scoreseq.htmQ31, Splice Site
Prediction (http://www.fruitfly.org/seq_tools/splice.html)32. Splice Site Finder
(http://www.genet.sickkids.on.ca/~ali/splicesitefinder.html)33 and GeneSplicer
(http://www.tigr.org/tdb/GeneSplicer/gene_spl.html')34 programs with default parameters.
Haplotype estimations were performed using the PHASE 2.1.1 software
(http://www.stat.washington.edu/stephens/software.html)35 using a Bayesian approach. The
program was initially run 5 times with 1000 permutations for both the cases and controls
combined. The p-value for case-control comparison was further reduced by running the
program with 100 000 permutations in order to verify the accuracy of haplotype
estimations. A subset of 4 haplotypes (HI, H13, H15 and H20) were also confirmed by
amplification of the cDNA sequence covering the SNPs region followed by subcloning in a
pCR®II vector using a TA Cloning® Kit (Invitrogen, Carlsbad, CA, USA). Individual
clones were subsequently sequenced to resolve haplotypes.
Determination of the minimum set of tSNPs was performed on the control group, using
SNPtagger (http://www.well.ox.ac.uk/~xiayi/haplotypeA.36 An additional haplotype
estimation was then performed with PHASE using the tSNPs identified by SNPtagger
(100 000 case-control permutations) to further ascertain if a significant variation in
frequency of the tagged haplotypes was still present between both groups.

79
Statistical analyses
Three programs were used to estimate haplotype frequencies between case and control
subjects: PHASE, COCAPHASE and HaploStats. These programs implement two different
algorithms to calculate haplotype frequencies: a Bayesian method for PHASE, and
Expectation Maximized (EM) algorithm for COCAPHASE37 and HaploStats.38 Regarding
calculated test for significant differences in haplotype frequencies in case and control
groups, the PHASE program uses a permutation-based likelihood ratio test, COCAPHASE
uses standard unconditional logistic regression and the log-linear modeling of Mander, and
HaploStats is based on score statistics. Finally, R software was used to calculate Fisher's
exact test, and odds ratio with 95% confidence interval were evaluated between each
specific haplotype and the most frequent haplotype.

80
Results
Screening of the ZBRK1 gene. Direct sequencing oïZBRKl genomic and complementary
DNA of 97 j5Z?Cj4//2-negative breast cancer subjects from high-risk French Canadian
breast/ovarian cancer families and 94 healthy controls led to the identification of 18
variants, among which 11 are located in the coding region while two are situated in the 3'-
untranslated region (3'UTR) (Figure 1). Among these, three are novel: SNPs 1 and 3 are
situated in intronic regions while SNP 18 is located in 3'UTR of the gene. As shown in
Table I, 9 out of 18 variants display a minor allele frequency (MAF) of less than 5% in the
cases series, the remaining SNPs having a MAF greater or equal to 0.05. Among the
identified genomic variants, only c.243+4A>G, c.370+101T>C and c.622C>T (SNPsl, 3
and 9) are found exclusively in breast cancer cases and are very rare, displaying a
frequency of 0.5%, 0.5% and 1% respectively. The c.622C>T variant, which causes a
Arg>Cys modification of amino acid 132, is observed in two breast cancer individuals. No
other affected family members were available from these two families. However, three
unaffected first and second degree relatives from one family were also analysed, and were
found not to be carriers of this variant (data not shown). None of the identified variants
show a significant deviation from Hardy-Weinberg equilibrium (Pearson' chi-square), or a
clear significant association with breast cancer. Of the 14 variants identified in other
studies, they all display a similar MAF (Table I). Among the coding variants, SNPs2 and 4
fall within the KRAB domain (c.333T>C and c.425T>C), while 3 are located within the
zinc finger domain (c.936T>C, C.13470A and c.l383A>C; SNPslO, 11 and 12) and 6
(SNPsl 1 to 16) are present in the C-terminus region, reported to bind to BRCAl18'39
(Figure 1).
Determination of the putative impact of ZBRK1 variants. Analysis of intronic variants
using splice site prediction programs (MaxEntScan, Splice Site Prediction, Splice Site

81
Finder and GeneSplicer) indicated no evidence that any of the intronic SNPs identified in
our cohort of breast cancer cases could potentially disrupt or create consensus splice sites,
which would lead in aberrant splicing of ZBRK1 mRNA (data not shown). Among the
intronic variants identified, c.243+4A>G is the only intronic sequence variation located in a
proximal splice site region. Although this variant reduced the splicing site score in all four
prediction programs used, its value remains relatively high, and sequence analysis of
ZBRK1 mRNAs of the only carrier did not reveal any alternative spliced transcripts caused
by this variant (data not shown).
To further examine the coding genomic variants modifying an amino acid, the whole
ZBRK1 protein sequence was compared with its putative orthologs of other species.
Alignment of human ZBRK1 protein sequence shows a high degree of conservation with
Macaca fascicularis and Pan troglodytes (96% and 97%, respectively) as well as with Bos
taurus and Canis familiaris sequences (72% and 75%, respectively). Out of six silent
polymorphisms, five (p.Asp35Asp, p.Cys236Cys, p.Pro373Pro, p.Thr385Thr and
p.Ser476Ser) are located at positions well conserved among the species examined, while
the residue leucine corresponding to the c.425T>C (p.Leu66Pro) variant is not conserved in
higher species. Among the non-synonymous changes, two residues (Arg501 and Val524)
are conserved in all species studied, and are also located in the BRCAl-interacting region.
As for the other ZBRK1 sequence coding variants identified in our study, they occur among
non conserved residues of the protein.
Linkage disequilibrium structure determination. Data from all 191 individuals (97 breast
cancer cases and 94 controls) were used to determine linkage disequilibrium (LD) structure
oïZBRKl gene. D' calculations show, as expected upon a short distance, a high degree of
LD between all ZBRK1 variants (more than 9.6 kb apart on genomic sequence;
Supplemental Figure 1A). As expected, r2 coefficient calculated for ZBRK1 displays lower
values, as this measure is dependent on allelic frequency (Supplemental Figure 1A).
HapMap data (http://www.hapmap.org) were also used to determine LD blocks in the
vicinity of ZBRK1, and permitted to identify a region of 160 kb in strong LD. This block
extends approximately 110 kb upstream and 30 kb downstream of the ZBRK1 gene and

82
encompasses the second half of ZNF613, as well as the complete coding region OÏZNF615,
ZNF614 and ZNF432 genes (Supplemental Figure IB). All these genes encode zinc finger
proteins, a motif known to regulate transcription of other genes, although their exact targets
on the genome are still unknown.
ZBRK1 haplotypes estimations. In order to assess the haplotype diversity, the PHASE
program was used, along with the frequency associated with each haplotype in the case and
control series, using coding variants (SNP2-4-5-9-10-11-12-13-14-15-16) and the
C.1900C/T (SNP 17) variant located in 3'UTR. Other variants were not included in the
analysis as intronic regions show little conservation between species and are less likely to
display biological relevance. As means of confirmation, HaploStats and COCAPHASE
programs have also been used, as these two programs use the EM algorithm to obtain
maximum likelihood estimates of haplotypes frequencies, instead of the Bayesian method
used by PHASE, and perform their analyses by pooling rare haplotypes together. Moreover,
HaploStats and COCAPHASE programs allow an analysis of individual p-values associated
with specific haplotypes, which is not a feature of the PHASE program. As shown in Table
II, a high predominance of haplotype HI is observed in both cases and controls. Given its
high frequency, we could expect that HI likely represents the ancestral haplotype.
However, sequencing of the ZBRK1 region in 3 cynomolgus monkeys (one male and two
females) could not confirm that HI is indeed the ancestral haplotype. According to PHASE
estimations, 67% of all chromosomes in the data set are estimated to carry haplotype HI.
Five haplotypes are observed only once in the combined data set (H4, H6, Hl l , H14 and
H18) while the five most frequent haplotypes (HI, H13, H15, H16 and H20) can explain
89% of the 382 alleles (data not shown). Among all estimated haplotypes, only two rare
haplotypes are found exclusively in breast cancer cases (H9 and H14), and 8 haplotypes are
unique to the control group. Among the 10 haplotypes present in both groups, only HI3
displays a large variation in frequency between both groups, being nearly eight times more
frequent in the breast cancer series.
According to PHASE, a significant difference between haplotypes estimated in both groups
was obtained. Indeed, twenty calculations were performed under the null hypothesis (HO:

83
No difference in haplotypes composition and frequency between the case and control
series), with 100 000 case-control permutations, and all p-values obtained were < 4.8X10"4.
To make sure that the significant p-values were not due to rare variants, we excluded
variants with MAF <5% and repeated the analysis (data not shown). The same level of
significance was achieved, as most of the observed differences between the two groups are
represented by three relatively frequent haplotypes (H8, H13 and H16). Moreover,
verification of PHASE accuracy in estimating haplotypes correctly was performed by
subcloning ZBRK1 cDNAs from a subset of 3 individuals estimated by PHASE to carry 4
different haplotypes (HI, H13, H15 and H20). A complete concordance was cobtained
between observed and estimated haplotypes for these individuals.
To further confirm haplotypes estimations and frequencies, comparison analysis between
both groups was then performed using Haplostat and COCAPHASE. Global estimation of
differences between the case and control groups is estimated to be 4.8X10"5 with
COCAPHASE, and 4.0X10"5 with the HaploStats program (data not shown).
Association of ZBRK1 haplotypes and breast cancer. As shown in Table III, association
analyses of haplotypes with breast cancer susceptibility revealed that, when considering
only haplotypes showing a frequency >2% with HaploStats, HI3 is over-represented in
breast cancer cases (OR=6.98, p=0.00143) while two other haplotypes are observed
exclusively in controls (H8; p=0.01135 and H16; p=0.00268), and thus could be associated
with a protective effect. Of these, two remain significant when accounting for multiple
testing (Table III). A similar degree of association is obtained when using calculations from
PHASE data. For comparison purposes, data from COCAPHASE have not been displayed
as this program uses a different calculation method. Nonetheless, this program also yielded
similar significant p-values (data not shown).
Determination of a sub-group of ZBRK1 SNPs with greatest impact on haplotype
significativity. Further analyses aiming at circumscribing the coding SNPs responsible for

84
the effect seen in Table III were performed using sliding windows of 3, 4 and 5 SNPs along
the coding region. It was possible to highlight that a strong component of the observed
difference between cases and controls is represented within the first 5 coding SNPs
(cSNPs) (Supplementary Table II), and that similar patterns of significant p-values are
observed with all three programs, namely PHASE (p=2X10'5), COCAPHASE
(p=1.34867X10~6) and HaploStats(p=2X10"5). As illustrated in this Table, combinations of
these first five cSNPs (SNPs2-4-5-9 and 10) display a higher significant p-value than
combinations which include variants located in the 3'-region of the mRNA (i.e. SNPsl 1,
12,13, 14,15, 16 and 17).
Determination of tSNPs and association with breast cancer. Thereafter, in order to
identify ZBRK1 tSNPs useful for well-powered studies using larger sample sets, PHASE
haplotypes have been used as input for the SNPtagger program, which generated tSNPs
used for further haplotype analyses. Only haplotypes estimated in the control group were
used to perform tSNP selection, as this group would be more representative of the general
population.
Using the SNPtagger program, 6 tSNPs have been identified, namely SNPs2, 4, 10, 11, 14
and 17. These tSNPs could correctly represent the 8 haplotypes showing a frequency higher
than 1.5% and recovered 91% of the haplotype diversity. However, if 4 tSNPs are used
(SNPs2, 4, 10 and 17), these could distinguish 6 haplotypes (with an estimated frequency of
> 2%), recovering 88% of the haplotype diversity. Using the identified tSNPs, further
analyses were performed to verify if a significant difference would still be observed
between both groups. Significant p-values were observed in all instances: PHASE (4
tSNPs: p=lX10"5; 6 tSNPs p=6X10"5), COCAPHASE (4 tSNPs: p=3.49521X10"8; 6 tSNPs
p=9.74407X10"7) and HaploStats (4 tSNPs: p<lX10"5; 6 tSNPs p<lX10"5). Analyses of
tSNPs derived from the combined data set (cases and controls combined) resulted in similar
p-values (data not shown).

85
Discussion
It is now well established that the residual familial risk of breast cancer, not caused
by BRCAl or BRCA2 genes, could be explained by a polygenic or high-risk genes
heterogeneity model.40 Thus, in order to increase the power of our study aiming at finding
genetic variants involved in breast cancer susceptibility, we selected individuals affected
with breast cancer from high-risk families (one individual per family), without mutations in
BRC A1/2. So far, several genes have been investigated based on their interaction with
BRCA1/2 or their involvement in DNA repair mechanisms. Since BRCA1/2 genes are
intimately linked to genomic stability, other genes involved in this pathway represent very
good candidates to be additional breast cancer susceptibility gene. ZBRK1 is involved in
BRCAl-mediated repression of GADD45 and certain BRCAl variants have been shown to 1 8
display deficient repressor activity on GADD45 expression. This gene thus became of
great interest as a potential breast cancer predisposition gene since the ZBRK1 -BRCAl
interaction may be important for BRCAl-mediated repression.
Direct sequencing of the ZBRK1 gene in 97 affected individuals from high-risk
breast/ovarian French-Canadian cancer families, as well as in 94 healthy controls, led to the
identification of 18 variants, among which 5 are missense changes that may alter the
protein activity. Only 3 variants (SNPsl, 3 and 9) are found exclusively in breast cancer
cases. Unfortunately, no additional family members were available to determine
segregation of these variants with breast cancer. Few studies examined genetic variations
and expression of the ZBRK1 gene and their potential association with breast cancer
susceptibility. Garcia et al.*] investigated the relation of GADD45, ZBRK1 and BRCAl
expression in colon carcinomas, and found a correlation between these mRNAs expression,
with ZBRK1 having a tendency to be under-expressed in these types of tumors. A recent
mutational analysis oîZBRKl in 21 BRCAl- and 5/îG42-negative probands, 58 early-onset
breast cancer cases and 30 reference individuals, detected 17 sequence variants20, of which

86
14 were also identified in the current study. Although one silent variation (C.1383A>C) and
one missense (C.1642T>C) variant were observed only in their breast cancer cases group,
no exhaustive statistical analyses have been performed in this latter study to examine a
putative association with breast cancer susceptibility.2 A recent analysis of ZBRK1
variations in two population-based cohorts from USA and Poland did identify a weak
significant association of the C.1642T>C variant with breast cancer risk (OR 1.24 ; 95% CI
1.05-1.48; p=0.01).23 Analysis of ZBRK1 gene variations identified in the current study did
not point out any specific polymorphisms susceptible to be associated on its own with
breast cancer susceptibility, which prompted us to examine haplotypes distribution among
our French-Canadian cases and controls series. Indeed, it is now well recognized that
haplotype analyses (group of SNPs) could have more significance than individual
polymorphisms of a gene to reveal an association with a given syndrome.42'43
The haplotype estimations presented here revealed that among those with a
frequency greater than 2%, three relatively frequent haplotypes were significantly
differentially represented in cases and controls, namely H8, H13 and H16. Preliminary
observations suggest that H8 and HI6 could confer a protective effect, both haplotypes
being observed exclusively in controls, while HI3 is over-represented in cases, and
therefore could be associated with an increased susceptibility to the disease. To date, only
one study was performed to investigate the involvement of ZBRK1 haplotypes with breast
cancer predisposition. Indeed, using the HaploStats program on a subgroup of SNPs
analysed in the present study, Garcia-Closas et al.231 detected a weak significant association
of 2 rare haplotypes in the USA population. However, this was not confirmed in their
Polish series and their reported global p-value was nonsignificant in both populations. In
agreement with this study, we also demonstrate a potential significant effect of ZBRK1 SNP
haplotypes on susceptibility to breast cancer. Indeed, one particular haplotype displays a
significant association with breast cancer, using two different programs (OR=6.80 (CI:
1.55-62.19) and 6.98 (CI: 1.59-63.84), using PHASE and HaploStats program,
respectively). According to Antoniou and Easto10, in a study with 100 cases from high-risk
families and 100 healthy controls, and given a rare allele frequency of 0.10, it could in
theory be possible to detect a relative risk of 2. Obviously, given the limited number of
individuals included in the present study, one has to be cautious when interpreting such a

87
result. Nonetheless, these preliminary results represent an interesting avenue for further
investigations. A direct comparison of our haplotype data with those of Garcia-Closas et
al. is somewhat difficult as our analysis involves a greater number of variants (including
the four used in their study), thus leading to a more complex set of haplotypes. However,
all five haplotypes found in their study are present in our 20 haplotypes, although these are
of course represented in more than one haplotype (Table II). In the sole other study
presenting ZBRK1 haplotype estimations20, no formal comparison was performed between
the case and control groups. Therefore, a direct comparison of the haplotypes obtained is
somewhat difficult as a different (but overlapping) set of SNPs is obtained compared with
the one presented here (Table II).
If we look at haplotypes H8, H13 and H16 more closely, we can see that haplotype
H8 carries two synonymous changes as well as an Arg>Ser non-synonymous change at
codon 501, which is a conserved position among species, and is situated in the region of
ZBRK1 interacting with BRCAl. H16 is composed of a synonymous variant (Asp35) and a
Leu>Pro change at codon 66, which is located in the KRAB domain at the end of box B.
Sequence alignments of human KRAB domains44 show that the consensus sequence
displays a proline at this position (data not shown), which is also observed in amino acid
sequences from Macaca and Pan species. This may indicate that a proline at this specific
position would be more effective, and thus could possibly explain the protective effect seen
for this haplotype. Moreover, the KRAB box B has been demonstrated to be accessory, but
its presence seems to strengthen repression.45 As for H13, this haplotype carries a
synonymous change (p.Asp35Asp) and a variant in the 3'UTR region (C.1900C>T). At first
glance, it is tempting to speculate that C.1900C>T would be the causative variant. However,
it is difficult to pinpoint which SNP in the haplotype would be responsible for this effect, as
no SNP taken on its own shows a significant effect. However, we can not rule out the
possible effect of the synonymous change for any of these 3 haplotypes, as it has been
shown that synonymous changes, and variants in the 3'UTR region, may have a functional
impact on the allele by affecting mRNA stability and gene expression.46'47 Nonetheless,
other mechanisms have been associated with regulation of expression, including tissue-
specific tRNA expression, mRNA secondary and tertiary structures, and mRNA trafficking.
As these processes are dependent upon mRNA sequence, we can not rule out a possible

88
effect of certain ZBRK1 alleles on the efficiency of such processes.48"51 In addition,
ZBRK1-mediated repression has been demonstrated to be also dependent upon its
tetramerisation52, and this interaction may be influenced by subtle changes in ZBRK1
expression and protein sequence. Alternatively, a given haplotype may be in linkage
disequilibrium with other nearby genes or with another yet unidentified variant in the
promoter region of ZBRK1. As mentioned before, ZBRK1 is part of a linkage
disequilibrium block of approximately 160 kb encompassing other zinc finger genes with
still unknown targets. This block was determined by analysis of HapMap CEPH trio data
(Caucasians). However, the French Canadian population is a founder population and as
such, LD blocks may extend over a longer distance than those estimated from the HapMap
project. Indeed, a recent analysis of the French Canadian founder mutation R1443X in
BRCAl demonstrated that the shared haplotype extends over approximately 9.3cM.53 The
region of chromosome 19 harbouring ZBRK1 is a cluster of several zinc finger proteins and
also carries the regulatory subunit A of the protein phosphatase 2 (isoform alpha;
PPP2R1A), which is approximately 200 kb apart from ZBRK1. Interestingly, a study from
Câlin et al.54 found PPP2R1A to be altered with low frequency in carcinomas of the breast,
lung as well as in a melanoma. Genotyping of other variants in adjacent genes may
therefore be necessary to better understand the extent of LD in the ZBRK1 region and
further evaluate the potential influence of these other genes on breast cancer susceptibility.
To further restrict which SNP or which portion of the gene seems to provide the
significant difference observed in our cases and controls, sliding windows of 3, 4 and 5
polymorphisms were used, and we were able to determine that the effect seemed to be
caused specifically by coding SNPs located in the 5' region of the ZBRK1 gene. The
highest p-values were observed for the same 4-5 SNPs, depending on the window used.
Interestingly, consistent significant p-values are obtained with every program used;
PHASE, COCAPHASE and HaplStats.
Recently, several groups have used tSNPs in their genetic association studies on
breast cancer susceptibility.55"57 Rutter et al.20 estimated that analysis of the c.333T>C and
c.425T>C variants (SNPs2 and 4 in the present study) would capture the haplotype
diversity in ZBRK1, since they observed only 3 haplotypes with a frequency greater than

89
2% (when using PHASE and HaploScope programs). However, our analysis using PHASE
predicted 6 haplotypes with a frequency greater than 2% in controls, which could be
captured by 4 tSNPs (SNP2, 4, 10 and 17), while SNPs2 and 10 on their own represent the
3 most common haplotypes found in our population (frequency >5%). Using 6 tSNPs (2-4-
10-11-14-17) we could correctly distinguish the 8 haplotypes showing a frequency greater
than 1.5%. When using these identified tSNPs, a significant difference is still seen between
breast cancer cases and controls, with either 4 or 6 tSNPs. Indeed, a significant p-value is
obtained with all programs examined, namely PHASE, COCAPHASE and HaploStats.
Therefore, by genotyping only four variants, a significant difference would be obtained in
the French-Canadian population.
In summary, we analyzed ZBRK1 gene alterations in 97 cases from French-Canadian high-
risk families (one individual per family) as well as 94 healthy controls from the same
population. Although no significant association of specific sequence variants with
hereditary breast cancer was observed, the results presented here suggest an association of
predicted haplotypes with breast cancer in cases or controls. An odds ratio for a disease-
associated haplotype was estimated to 6.80 (95% CI: 1.55-62.19; p=0.005331). These
haplotypes may imply coordinate effect of variants or linkage of specific haplotypes with
other unknown variants or nearby genes. As this region of chromosome 19 where ZBRK1
lies is rich in zinc-finger proteins, a motif known for its DNA-binding activity related with
transcriptional regulation, it is possible that it may have a repercussion on genes essential to
global cellular maintenance. However, analyses using larger cohorts will definitively be
needed to further ascertain the impact of ZBRK1 haplotypes on breast cancer susceptibility
in other populations.

90
Acknowledgements The authors would like to thank all individuals and families who participated in this study.
We thank Claire Brousseau, Dr. Sylvie Délos, Marie-Andrée Lajoie, Pascale Léger, Josée
Rhéaume, Carolle Samson, Martine Tranchant, Dr. Martine Dumont, Gilles Leblanc,
Hélène Malouin, Nathalie Bolduc, Andrée MacMillan and Tina Babineau of the Cancer
Genomics Laboratory for genetic counselling, sample management and mutation screening.
We also thank Anne-Marie Moisan and Lucie Larouche for Southern blot and MLPA
analyses, Melissa Ouellet for skillful technical help, and Dr. Damian Labuda and Claudia
Moreau at the Centre de Cancérologie Charles Bruneau of Ste-Justine Hospital for help
with control DNA samples. This work was supported by the Canadian Institutes of Health
Research (CIHR) and Institute of Cancer and Institute of Gender and Health for the
INHERIT BRCAs research program, the Fonds de la Recherche en Santé du Québec
(FRSQ)/Réseau de Médecine Génétique Appliquée (RMGA), the Canadian Breast Cancer
Research Alliance and the CURE Foundation. S.D. holds studentships from Fondation
René Bussières and Fondation Desjardins, and F.D. is recipient of a Research Career
Award in the Health Sciences from CIHR/R&D HRF.

91
Appendix 1
• Jacques Simard, Research Chair in Oncogenetics, Cancer Genomics Laboratory, Oncology
and Molecular Endocrinology Research Centre, Centre Hospitalier Universitaire de Québec
and Laval University, Québec.
• Bartha Maria Knoppers, Chaire de recherche en droit et médecine, Centre de recherche en
droit public, Université de Montréal, Montréal.
• Yann Joly, Centre de recherche en droit public, Université de Montréal, Montréal.
• Peter Bridge, Molecular Diagnostic Laboratory, Alberta Children's Hospital, Calgary.

92
References 1. Antoniou AC, Pharoah PD, McMullan G, Day NE, Ponder BA, Easton D. Evidence for
further breast cancer susceptibility genes in addition to BRCAl and BRCA2 in a population-based study. Genet Epidemiol 2001;21:1-18.
2. Antoniou AC, Pharoah PD, Smith P, Easton DF. The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br J Cancer 2004; 91:1580-90.
3. Guénard F, Labrie Y, Ouellette G, Joly Breauparlant C, Bessette P, Chiquette J, Laframboise R, Lépine J, Lesperance B, Pichette R, Plante M, INHERIT BRCAs et al. Germline mutations in the breast cancer susceptibility gene PTEN are rare in high-risk non-BRCAl/2 French Canadian breast cancer families. Fam Cancer 2007, in press.
4. Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006 38:873-875.
5. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007 39:165-167.
6. Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, Li G, van Beers EH, Li L, Khalil T, Quenneville LA, Omeroglu A, Poll A, et al. Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci USA. 2007 104:6788-6793.
7. Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, Kallioniemi A, Pylkas K, Karppinen SM, Rapakko K, Miron A, Sheng Q, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007 446:316-319.
8. Durocher F, Guénard F, Desjardins S, Ouellette G, Labrie Y. Inherited susceptibility to breast cancer: accomplishments and challenges. In: Sinnett D. (ed.) Molecular Genetics of Cancer. Kerala, Research Signpost, 2005, 9-93.
9. Pharoah PD, Dunning AM, Ponder BA, Easton DF. Association studies for finding cancer-susceptibility genetic variants. Nat Rev Cancer 2004;4:850-60.
10. Antoniou AC, Easton DF. Polygenic inheritance of breast cancer: Implications for design of association studies. Genet Epidemiol 2003;25:190-202.
11. Houlston RS, Peto J. The future of association studies of common cancers. Hum Genet 2003;112:434-35.
12. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, Bell R, Rosenthal J. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCAl. Science 1994;266:66-71.
13. Somasundaram K, Zhang H, Zeng YX, Houvras Y, Peng Y, Zhang H, Wu GS, Licht JD, Weber BL, El-Deiry WS. Arrest of the cell cycle by the tumour-suppressor BRCAl requires the CDK-inhibitor p21WAFl/CiPl. Nature 1997;389:187-90.
14. Harkin DP, Bean JM, Miklos D, Song YH, Truong VB, Englert C, Christians FC, Ellisen LW, Maheswaran S, Oliner JD, Haber DA. Induction of GADD45 and

93
JNK/SAPK-dependent apoptosis following inducible expression of BRCAl. Cell 1999;97:575-86.
15. Furuta S, Wang JM, Wei S, Jeng YM, Jiang X, Gu B, Chen PL, Lee EY, Lee WH. Removal of BRCAl/CtlP/ZBRKl repression complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer Cell 2006;10:13-24.
16. Wang XW, Zhan Q, Coursenm JD, Khan MA, Kontny HU, Yu L, Hollander MC, O'Connor PM, Fornace AJJr, Harris CC. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA 1999;96:3706-11.
17. Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, Molinaro TA, Kim KE, Tolosa E, Ashwell JD, Rosenberg MP, Zhan Q. et al. Genomic instability in Gadd45a-deficient mice. Nat Genet 1999;23:176-84.
18. Zheng L, Pan H, Li S, Flesken-Nikitin A, Chen PL, Boyer TG, Lee WH. Sequence-specific transcriptional corepressor function for BRCAl through a novel zinc finger protein, ZBRK1. Mol Cell 2000;6:757-68.
19. Yun J, Lee WH. Degradation of transcription repressor ZBRK1 through the ubiquitin-proteasome pathway relieves repression of Gadd45a upon DNA damage. Mol Cell Biol 2003;23:7305-14.
20. Rutter JL, Smith AM, Davila MR, Sigurdson AJ, Giusti RM, Pineda MA, Doody MM, Tucker MA, Greene MH, Zhang J, Struewing JP. Mutational analysis of the BRCAl -interacting genes ZNF350/ZBRK1 and BRIP1/BACH1 among BRCAl and BRCA2-negative probands from breast-ovarian cancer families and among early-onset breast cancer cases and reference individuals. Hum Mutat 2003;22:121-28.
21. Sigurdson AJ, Hauptmann M, Chatterjee N, Alexander BH, Doody MM, Rutter JL, Struewing JP. Kin-cohort estimates for familial breast cancer risk in relation to variants in DNA base excision repair, BRCAl interacting and growth factor genes. BMC Cancer 2004;4:9-20.
22. Garcia V, Dominguez G, Garciam JM, Silva J, Pena C, Silva JM, Carcereny E, Menéndez J, Espana P, Bonilla F. Altered expression of the ZBRK1 gene in human breast carcinomas. J Pathol 2004;202:224-32.
23. Garcia-Closas M, Egan K, Newcomb PA, Brinton LA, Titus-Ernstoff L, Chanock S, Welch R, Lissowska J, Peplonska B, Szeszenia-Dabrowska N, Zatonski W, Bardin-Mikolajczak A. et al. Polymorphisms in DNA double-strand break repair genes and risk of breast cancentwo population based studies in USA and Poland, and Meta-analyses. Hum Genet 2006; 119:376-88.
24. Simard J, Dumont M, Moisan AM, Gaborieau V, Vezina H, Durocher F, Chiquette J, Plante M, Avard D, Bessette P, Brousseau C, Dorval M. et al. Evaluation of BRCAl and BRCA2 mutation prevalence, risk prediction models and a multi-step testing approach in French-Canadian high-risk breast and ovarian cancer families. J Med Genet 2007;44:107-21.
25. Antoniou AC, Durocher F, Smith P, Simard J, Easton DF; INHERIT BRCAs program members. BRCAl and BRCA2 mutation predictions using the BOADICEA and BRCAPRO models and penetrance estimation in high-risk French-Canadian families. Breast Cancer Res. 2006 8:R3.

94
26. Durocher F, Labrie Y, Soucy P, Sinilnikova O, Labuda D, Bessette P, Chiquette J, Laframboise R, Lépine J, Lesperance B, Ouellette G, Pichette R. et al. Mutation Analysis and Characterization of ATR Sequence Variants in Breast Cancer Cases from High-Risk French Canadian Breast/Ovarian Cancer Families. BMC Cancer 2006;6:230-51.
27. Moisan AM, Fortin J, Dumont M, Samson C, Bessette P, Chiquette J, Laframboise R, Lépine J, Lesperance B, Pichette R, Plante M, Provencher L. et al. No evidence of BRC A1/2 genomic rearrangements in high risk French-Canadian breast/ovarian cancer families. Genet Test 2005;10:104-15.
28. Staden R, Beal KF, Bonfield JK. The Staden package, 1998. Methods Mol Biol 2000;132:115-30.
29. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994;22:4673-80.
30. Devlin B, Risch N. A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 1995;29:311-22.
31.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with application to RNA splicing signals. J Comput Biol 2004; 11:377-94
32. Reese MG, Eeckman FH, Kulp D, Haussier D. Improved splice site detection in Genie. J Comput Biol 1997;4:311-23.
33. Shapiro MB, Senepathy P. RNA splice junctions of different classes in eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 1987;15:7155-74.
34. Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res 2001 ;29:1185-90.
35. Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 2003;73:1162-9.
36. Xiayi K, Lon CR. Efficient selective screening of haplotype tag SNPs. Bioinformatics 2003;19:287-8.
37. Dudbridge F. Pedigree disequilibrium tests for multilocus haplotypes. Genet Epidemiol 2003;25:115-21.
38. Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 2002;70:425-34.
39. Tan W, Zheng L, Lee WH, Boyer TG. Functional dissection of transcription factor ZBRK1 reveals zinc fingers with dual roles in DNA-binding and BRCAl-dependent transcriptional repression. J Biol Chem 2004;279:6576-87.
40. Easton DF. How many breast cancer predisposition genes are there? Breast Cancer Res 1999;1:14-17.
41. Garcia V, Garcia JM, Pena C, Silva J, Dominguez G, Rodriguez R, Maximiano C, Espinosa R, Espana P, Bonilla F. The GADD45, ZBRK1 and BRCAl pathway: quantitative analysis of mRNA expression in colon carcinomas. J Pathol 2005;206:92-9.

95
42. Schaid DJ. Evaluating association of haplotypes with traits. Genet Epidemiol 2004;27:348-64.
43. Clark AG. The role of haplotypes in candidate gene studies. Genet Epidemiol 2004;27:321-33.
44. Mannini R, Rivieccio V, D'Auria S, Tanfani F, Ausili A, Facchiano A, Pedone C, Grimaldi G. Structure/function of KRAB repression domains: structural properties of KRAB modules inferred from hydrodynamic, circular dichroism, and FTIR spectroscopic analyses. Proteins 2006;62:604-16.
45. Vissing H, Meyer WK, Aagaard L, Tommerup N. Thiesen HJ. Repression of transcriptional activity by heterologous KRAB domains present in zinc finger proteins. FEBS Lett 1995;369:153-7.
46. Capon F, Allen MH, Ameen M, Burden AD, Tillman D, Barker JN. Trembath RC. A synonymous SNP of the corneodesmosin gene leads to increased mRNA stability and demonstrates association with psoriasis across diverse ethnic groups. Hum Mol Genet 2004;13:2361-68.
47. Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet 2003;12:205-16.
48. Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfert RNA expression. PLOS Genet 2006;2:e221.
49. Kozak M. Regulation of translation via mRNA structure in procaryotes and eukaryotes. Gene 2005;361:13-37.
50. Pickering BM, Willis AE. The implication of structured 5' untranslated regions on translation and disease. Semin Cell Dev Biol 2005;16:39-47.
51. Kindler S, Wang H, Richter D, Tiedge H. RNA transport and local control of the translation. Ann Rev Cell Dev Biol 2005;21:223-245.
52. Tan W, Kim S, Boyer T. Tetrameric oligomerization mediates transcriptional repression by the BRCAl-dependent KRAB-zinc finger protein ZBRK1. JBC 2004;279:55153-60.
53. Vezina H, Durocher F, Dumont M, Houde L, Szabo C, Tranchant M, Chiquette J, Plante M, Laframboise R, Lepine J, Nevanlinna H, Stoppa-Lyonnet D. et al. Molecular and genealogical characterization of the R1443X BRCAl mutation in high-risk French-Canadian breast/ovarian cancer families. Hum Genet 2005;117:119-32.
54. Calin GA, di Iasio MG, Caprini E, Vorechovsky I, Natali PG, Sozzi G, Croce CM, Barbanti-Brodano G, Russo G, Negrini M. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene 2000;19:1191-5.
55. Benusiglio PR, Lesueur F, Luccarinim C, Mcintosh J, Luben RN, Smith P, Dunning A, Easton DF, Ponder BA, Pharoah PD. Common variation in EMSY and risk of breast and ovarian cancer: a case-control study using HapMap tagging SNPs. BMC Cancer 2005;5:81-88.
56. Camp NJ, Swensen J, Horne BD, Farnham JM, Thomas A, Cannon-Albright LA, Tavtigian SV. Characterization of linkage disequilibrium structure, mutation history, and tagging SNPs, and their use in association analyses: ELAC2 and familial early-onset prostate cancer. Genet Epidemiol 2005;28:232-43.

96
57. Lesueur F, Pharoah PD, Laing S, Ahmed S, Jordan C, Smith PL, Luben R, Wareham NJ, Easton DF, Dunning AM, Ponder BA. Allelic association of the human homologue of the mouse modifier Ptprj with breast cancer. Hum Mol Genet 2005;14:2349-56.
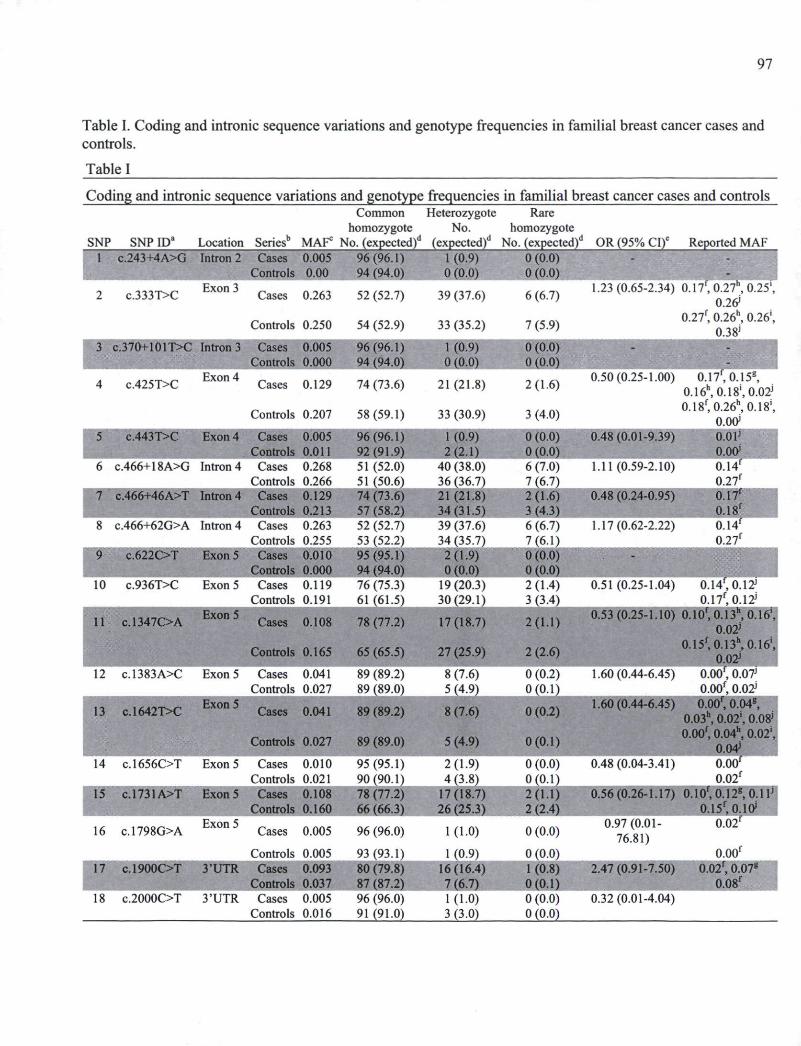
97
Table I. Coding and intronic sequence variations and genotype frequencies in familial breast cancer cases and controls. Table I
Coding and intronic sequence variations and genotype frequencies in familial breast cancer cases and controls Common
homozygote Hétérozygote
No. Rare
homozygote SNP SNP IDa
Location Seriesb MAFC Mo. (expected)d (expected)d No. (expected)d OR (95% CI)e Reported MAF 1 c.243+4A>G Intron 2 Cases 0.005
Controls 0.00 96(96.1) 94 (94.0)
1 (0.9) 0 (0.0)
0 (0.0) 0 (0.0)
- -
2 c.333T>C Exon 3 Cases 0.263 52 (52.7) 39 (37.6) 6 (6.7) 1.23(0.65-2.34) 0.17f, 0.27h,0.25\
0.26s
Controls 0.250 54 (52.9) 33 (35.2) 7(5.9) 0.27f, 0.26h, 0.26\ 0.38j
3 c.370+101T>C Intron 3 Cases 0.005 96 (96.1) 1 (0.9) 0 (0.0) Controls 0.000 94 (94.0) 0 (0.0) 0 (0.0) -
4 c.425T>C Exon 4 Cases 0.129 74 (73.6) 21(21.8) 2(1.6) 0.50(0.25-1.00) 0.17', 0.15s,
0.16h, 0.181, 0.02-
Controls 0.207 58(59.1) 33 (30.9) 3 (4.0) 0.18f,0.26h,0.18i, 0.00s
5 c.443T>C Exon 4 Cases 0.005 96 (96.1) 1 (0.9) 0 (0.0) 0.48 (0.01-9.39) 0.01J
0.00* Controls 0.011 92 (91.9) 2 (2.1) 0 (0.0) 0.01J
0.00* 6 c.466+18A>G Intron 4 Cases 0.268 51 (52.0) 40 (38.0) 6(7.0) 1.11(0.59-2.10) 0.14f
Controls 0.266 51 (50.6) 36 (36.7) 7 (6.7) 0.27f
7 c.466+46A>T Intron 4 Cases 0.129 74 (73.6) 21 (21.8) 2(1.6) 0.48 (0.24-0.95) 0.17f
Controls 0.213 57 (58.2) 34(31.5) 3 (4.3) 0.18f
8 c.466+62G>A Intron 4 Cases 0.263 52 (52.7) 39 (37.6) 6 (6.7) 1.17(0.62-2.22) O.H^ Controls 0.255 53 (52.2) 34(35.7) 7(6.1) 0.27f
9 C.6220T Exon 5 Cases 0.010 95 (95.1) 2(1.9) 0 (0.0) - , '„ '■ ' . v:,:,m"' WÈÊÊÈÊËÈËÈËÈÈËË mÊÊÈÊÊÊÊmBm Controls 0.000 94 (94.0) 0 (0.0) 0 (0.0)
WÈÊÊÈÊËÈËÈËÈÈËË mÊÊÈÊÊÊÊmBm
10 c.936T>C Exon 5 Cases 0.119 76 (75.3) 19(20.3) 2(1.4) 0.51 (0.25-1.04) 0.14f, 0.12J
Controls 0.191 61 (61.5) 30(29.1) 3 (3.4) 0.17f,0.12j
11 C.13470A Exon 5 Cases 0.108 78 (77.2) 17(18.7) 2(1.1) 0.53(0.25-1.10) 0.10f,0.13h,0.16i,
0.02j
Controls 0.165 65 (65.5) 27 (25.9) 2(2.6) 0.15f,0.13h,0.16i, 0.02j
12 C.1383A>C Exon 5 Cases 0.041 89 (89.2) 8(7.6) 0(0.2) 1.60(0.44-6.45) 0.00f, 0.07-Controls 0.027 89 (89.0) 5(4.9) 0(0.1) 0.00f, 0.02-
13 C.1642T>C Exon 5
Cases 0.041 89 (89.2) 8(7.6) 0(0.2) 1.60 (0.44-6.45) 0.00f, 0.04g, 0.03h, 0.02', 0.08j
Controls 0.027 89 (89.0) 5 (4.9) 0(0.1) 0.00f, 0.04h, 0.02', 0*04j
14 C.16560T Exon 5 Cases 0.010 95(95.1) 2(1.9) 0 (0.0) 0.48 (0.04-3.41) 0.00f
Controls 0.021 90(90.1) 4(3.8) 0(0-1) 0.02f
15 c.l731A>T Exon 5 Cases 0.108 78 (77.2) 17(18.7) 2(1.1) 0.56(0.26-1.17) 0.10f,0.12g,0.11j
Controls 0.160 66 (66.3) 26 (25.3) \ * ' 0.15f,0.10> 16 c.l798G>A Exon 5
Cases 0.005 96 (96.0) 1 (1.0) 0 (0.0) 0.97(0.01-76.81)
0.02f
Controls 0.005 93 (93.1) 1 (0.9) 0 (0.0) 0.00f
17 C.1900OT 3'UTR Cases 0.093 80 (79.8) 16(16.4) 1 (0.8) 2.47 (0.91-7.50) 0.02f, 0.07g
Controls 0.037 87 (87.2) 7(6.7) 0(0.1) 0.08f
18 c.2000C>T 3'UTR Cases 0.005 Controls 0.016
96 (96.0) 91 (91.0)
1 (1.0) 3 (3.0)
0 (0.0) 0 (0.0)
0.32 (0.01-4.04)

98
"According to the nomenclature of the Human Genome Variation Society b97 cases, 94 controls c Minor allele frequency d As expected under Hardy-Weinberg equilibrium eOdds ratios for comparison of hétérozygotes versus common homozygotes f42 non BRCA1/2 cases (USA), 30 controls (CEPH parents)16
g748 breast cancer cases (USA)17 h3368 unselected breast cancer cases and 2880 controls (USA)19 j1995 unselected breast cancer cases and 2296 controls (Poland)19 6̂1 breast cancer cases and 25 controls (Spain)18.
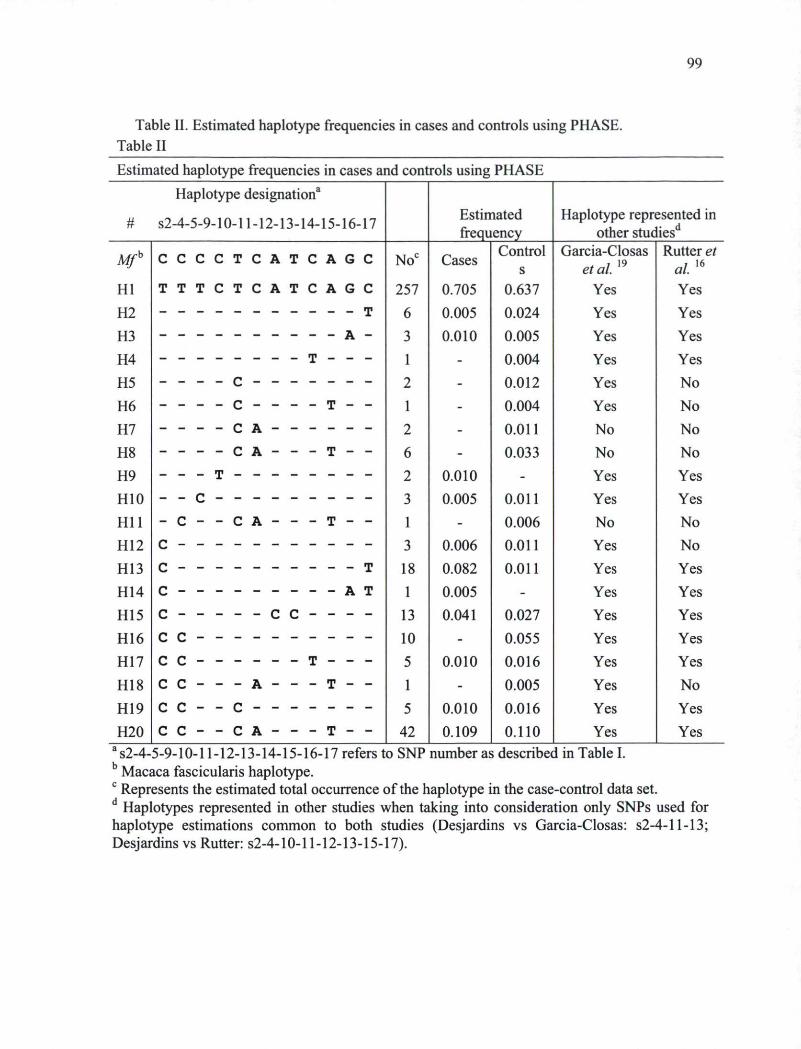
99
Table II. Estimated haplotype frequencies in cases and controls using PHASE. Table II Estimated haplotype frequencies in cases and controls using PHASE
Haplotype designation3
# S2-4-5-9-10-11-12-13-14-15-16-17 Estimated frequency
Haplotype repri other stuc
;sented in iesd
Mfb C C C C T C A T C A G C Noc Cases Control s
Garcia-Closas e ta l . 1 9
Rutter et a l . 1 6
HI H2 H3
T T T C T C A T C A G C T
257 6 3
0.705 0.005 0.010
0.637 0.024 0.005
Yes Yes Yes
Yes Yes Yes
HI H2 H3 — — &
C T
257 6 3
0.705 0.005 0.010
0.637 0.024 0.005
Yes Yes Yes
Yes Yes Yes
HI H2 H3 _ _ ~ A
257 6 3
0.705 0.005 0.010
0.637 0.024 0.005
Yes Yes Yes
Yes Yes Yes
H4 T - - - 1 - 0.004 Yes Yes H5 c - 2 - 0.012 Yes No H6 - - - - C - - - - T - - 1 - 0.004 Yes No H7 - - - - C A - - - - - - 2 - 0.011 No No H8 - - - - C A - - - T - - 6 - 0.033 No No H9 T - 2 0.010 - Yes Yes H10 - - c - 3 0.005 0.011 Yes Yes H l l - C - - C A - - - T - - 1 - 0.006 No No H12 H13 H14
C - - - - - - - - C — — — — — — — — — — T
T
3 18 1
0.006 0.082 0.005
0.011 0.011
Yes Yes Yes
No Yes Yes
H12 H13 H14
^ — — — — — — — — — — c _ _ _ _ _ a
T T
3 18 1
0.006 0.082 0.005
0.011 0.011
Yes Yes Yes
No Yes Yes
H12 H13 H14 i ^ — — — — — — — j ±
T T
3 18 1
0.006 0.082 0.005 ~
Yes Yes Yes
No Yes Yes
H15 C - - - - - C C - - 13 0.041 0.027 Yes Yes H16 c c - - - - - - - 10 - 0.055 Yes Yes H17 C C - - - - - - T - - - 5 0.010 0.016 Yes Yes H18 C C - - - A - - - T - - 1 - 0.005 Yes No H19 C C - - C - - - - - 5 0.010 0.016 Yes Yes H20 C C - - C A - - - T - - 42 0.109 0.110 Yes Yes
a S2-4-5-9-10-11-12-13-14-15-16-17 refers to SNP number as described in Table I. b Macaca fascicularis haplotype. c Represents the estimated total occurrence of the haplotype in the case-control data set.
Haplotypes represented in other studies when taking into consideration only SNPs used for haplotype estimations common to both studies (Desjardins vs Garcia-Closas: s2-4-l 1-13; Desjardins vs Rutter: s2-4-10-l 1-12-13-15-17).
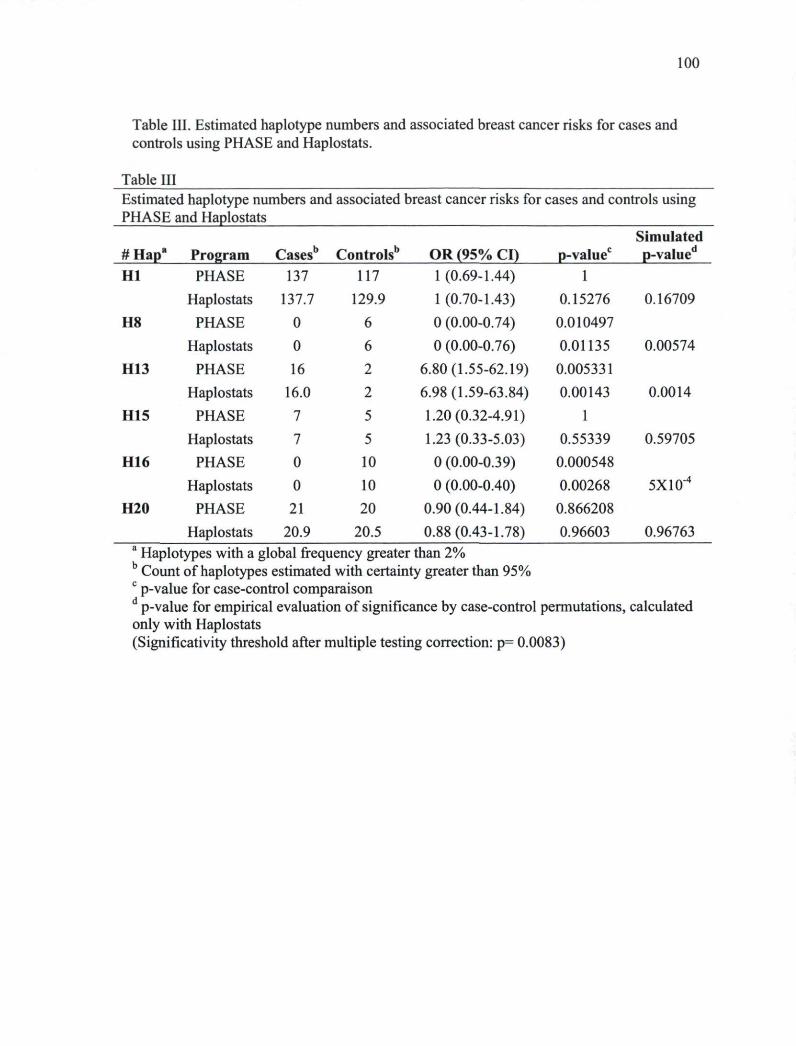
100
Table III. Estimated haplotype numbers and associated breast cancer risks for cases and controls using PHASE and Haplostats.
Table III Estimated haplotype numbers and associated breast cancer risks for PHASE and Haplostats
cases and controls using
# H a p a Program Casesb Controls5 OR (95% CI) p-valuec Simulated p-valued
HI PHASE 137 117 1 (0.69-1.44) 1 Haplostats 137.7 129.9 1 (0.70-1.43) 0.15276 0.16709
H8 PHASE 0 6 0 (0.00-0.74) 0.010497 Haplostats 0 6 0 (0.00-0.76) 0.01135 0.00574
H13 PHASE 16 2 6.80(1.55-62.19) 0.005331 Haplostats 16.0 2 6.98(1.59-63.84) 0.00143 0.0014
H15 PHASE 7 5 1.20(0.32-4.91) 1 Haplostats 7 5 1.23(0.33-5.03) 0.55339 0.59705
H16 PHASE 0 10 0 (0.00-0.39) 0.000548 Haplostats 0 10 0 (0.00-0.40) 0.00268 5X10"4
H20 PHASE 21 20 0.90(0.44-1.84) 0.866208 Haplostats 20.9 20.5 0.88(0.43-1.78) 0.96603 0.96763
Haplotypes with a global frequency greater than 2% b Count of haplotypes estimated with certainty greater than 95% c p-value for case-control comparaison d p-value for empirical evaluation of significance by case-control permutations, calculated only with Haplostats (Significativity threshold after multiple testing correction: p= 0.0083)

101
Supplemental Table I. Oligonucleotide primers used for amplification and sequence analysis of the ZBRK1 gene. Supplemental Table I Oligonucleotide primers used for amplification and sequence analysis of the ZBRK1 gene
Exon Forward primer (5'-3')b Reverse primer (5'-3')b ienguf(bp) Temple?
1 GAAATGCCGTAAATGCGCTTG GGCCGTTGATCACTACAGACCC 402 60
2 GCCCACAGGTATAAAATCCGCT GGACACCATCCACCACATCAAC 776 62
3-4 GCCTGTAGAGTGGAAATGAGCAGA GGTCTCACACTGGGAGACAGATGA 775 62
5a GGAAAGTTGATCATGTGCTGGAGC TCACTGCACACAAAGGGCGT 805 62
TGAATGCCCTGAATGTGGCA ACTGCAACACTCCCAACACCA 1113 62
Exon 5 has been amplified in two PCR fragments b All primers have been used for sequencing
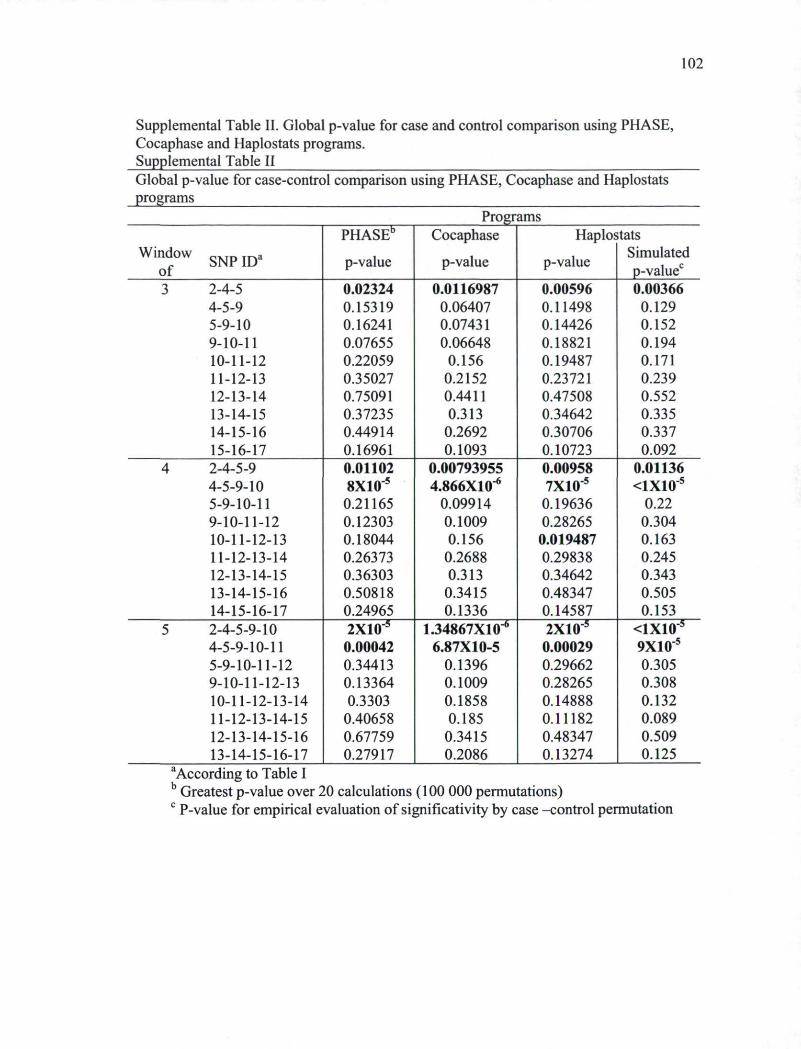
102
Supplemental Table II. Global p-value for case and control comparison using PHASE, Cocaphase and Haplostats programs. Supplemental Table II Global p-value for case-control comparison using PHASE, Cocaphase and Haplostats programs
Programs
Window S N p i E ) a of
PHASE"
p-value
Cocaphase
p-value
Haplos
p-value
tats Simulated p-valuec
3 2-4-5 0.02324 0.0116987 0.00596 0.00366 4-5-9 0.15319 0.06407 0.11498 0.129 5-9-10 0.16241 0.07431 0.14426 0.152 9-10-11 0.07655 0.06648 0.18821 0.194 10-11-12 0.22059 0.156 0.19487 0.171 11-12-13 0.35027 0.2152 0.23721 0.239 12-13-14 0.75091 0.4411 0.47508 0.552 13-14-15 0.37235 0.313 0.34642 0.335 14-15-16 0.44914 0.2692 0.30706 0.337 15-16-17 0.16961 0.1093 0.10723 0.092
4 2-4-5-9 0.01102 0.00793955 0.00958 0.01136 4-5-9-10 8X10"5 4.866X106 7X10"5 <1X105
5-9-10-11 0.21165 0.09914 0.19636 0.22 9-10-11-12 0.12303 0.1009 0.28265 0.304 10-11-12-13 0.18044 0.156 0.019487 0.163 11-12-13-14 0.26373 0.2688 0.29838 0.245 12-13-14-15 0.36303 0.313 0.34642 0.343 13-14-15-16 0.50818 0.3415 0.48347 0.505 14-15-16-17 0.24965 0.1336 0.14587 0.153
5 2-4-5-9-10 2X10"5 1.34867X10"6 2X105 <1X10'5
4-5-9-10-11 0.00042 6.87X10-5 0.00029 9X10"5
5-9-10-11-12 0.34413 0.1396 0.29662 0.305 9-10-11-12-13 0.13364 0.1009 0.28265 0.308 10-11-12-13-14 0.3303 0.1858 0.14888 0.132 11-12-13-14-15 0.40658 0.185 0.11182 0.089 12-13-14-15-16 0.67759 0.3415 0.48347 0.509 13-14-15-16-17 0.27917 0.2086 0.13274 0.125
According to Table I b Greatest p-value over c P-value for empirical
20 calculations (100 000 permutations) evaluation of significativity by case -control permutation

103
Figure legends
Figure 1
A. Proportional genomic structure of the ZBRK1 gene. 5' and 3'UTR regions are indicated
as dashed boxes. White flags, intronic variants. Gray flags, coding variants. B. Protein
structure of ZBRK1 illustrating domains involved in function/regulation of the protein.
Gray flags, synonymous variants. White flags, missense variants.
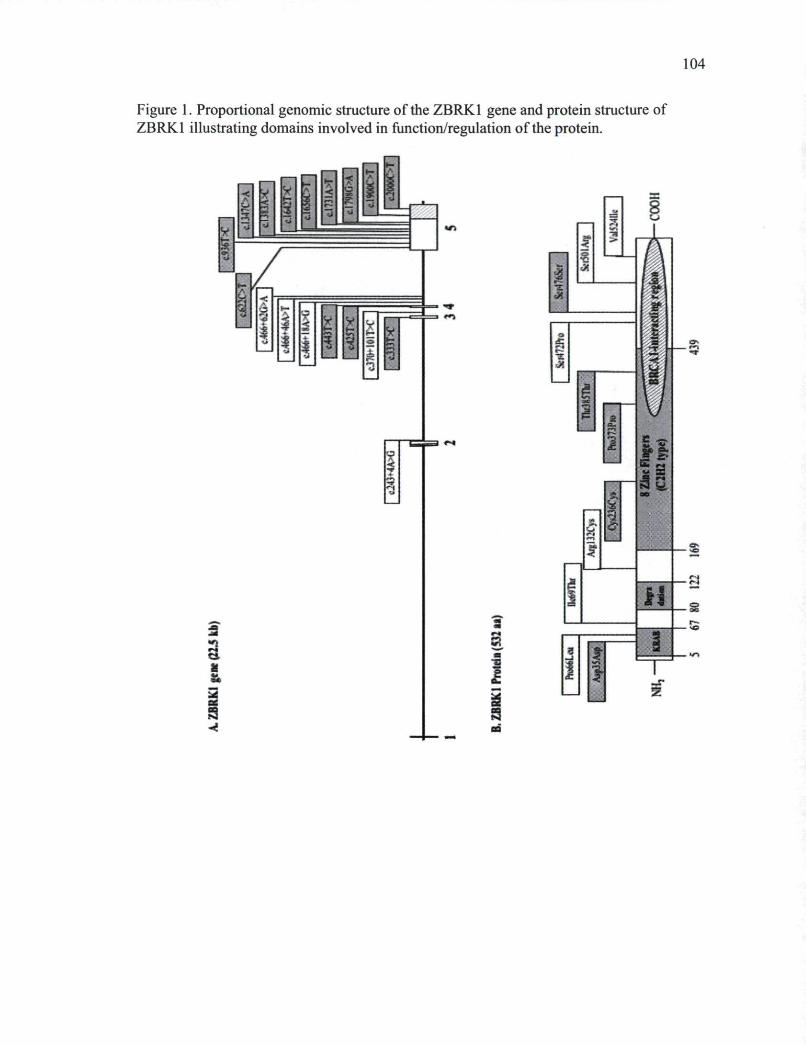
104
Figure 1. Proportional genomic structure of the ZBRK1 gene and protein structure of ZBRK1 illustrating domains involved in function/regulation of the protein.
5 s S
I
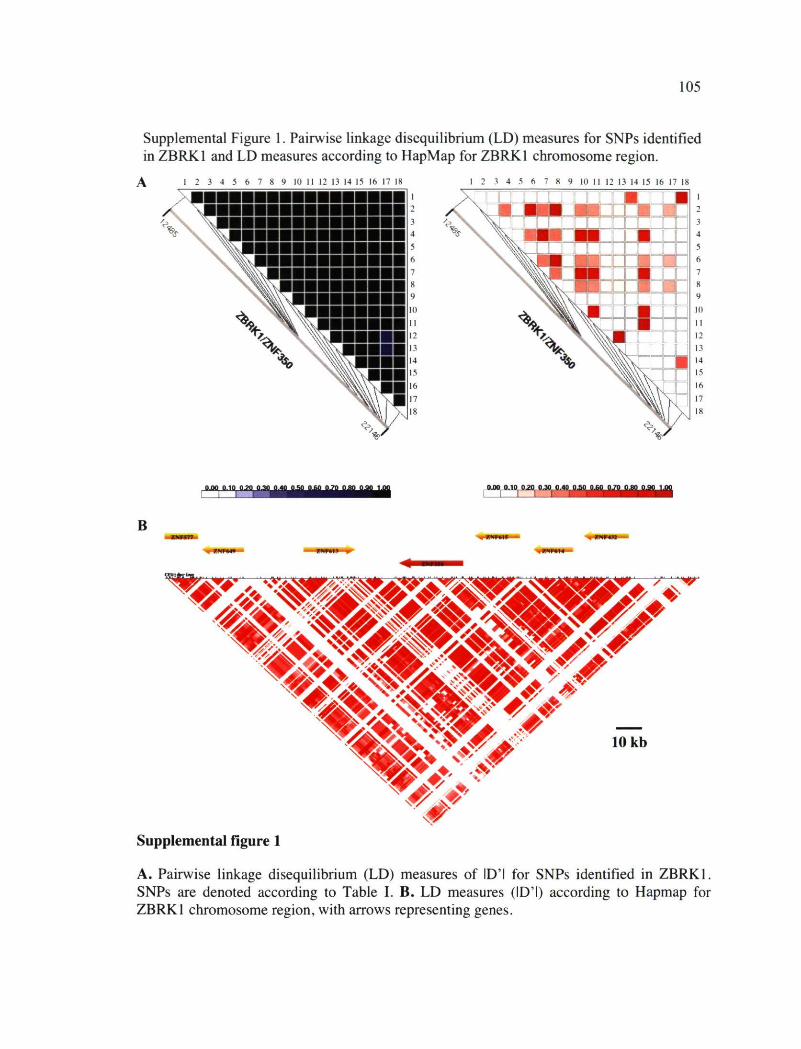
105
Supplemental Figure 1. Pairwise linkage disequilibrium (LD) measures for SNPs identified in ZBRK1 and LD measures according to HapMap for ZBRK1 chromosome region.
I 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
X oar ■ / v i ■■■ \\\m~
X %N u \%u R m vwi^il^ ■ni
MpSi inn m \ mom mm - y —
% \ v ■r VsS VsS ~\
%
- — j * M | * M M l_ I I O 0.10 0.20 0.30 0.40 0
B
V
Supplemental figure 1
A. Pairwise linkage disequilibrium (LD) measures of ID'I for SNPs identified in ZBRK1. SNPs are denoted according to Table I. B. LD measures (ID'I) according to Hapmap for ZBRK1 chromosome region, with arrows representing genes.

106
CHAPITRE II : Analysis ofGADD45A Sequence Variations in French Canadian Families with -High Risk of
Breast Cancer.

107
Analysis of GADD45A Sequence Variations in French Canadian Families with High
Risk of Breast Cancer
Sylvie Desjardins3, Geneviève Ouellette3, Yvan Labrie3, Jacques Simard3', INHERIT
BRCAs§, Francine Durocher3*
"Cancer Genomics Laboratory, Oncology and Molecular Endocrinology Research Centre,
Centre Hospitalier Universitaire de Québec and Laval University, Québec, G1V 4G2,
Canada;
§Other members of INHERIT BRCAs involved in clinical aspects of the study are listed in
the Appendix.
* Corresponding author:
Dr Francine Durocher
Cancer Genomics Laboratory, Oncology and Molecular Endocrinology Research Centre,
Centre Hospitalier Universitaire de Québec and Laval University
2705 Laurier Boulevard, T2-53
Québec City, Québec,
Canada, GI V 4G2
Tel: (418)-654-2296 Fax: (418)-654-2278
e.mail: [email protected]
J Hum Genet. 2008;53(6):490-8.

108
Résumé GADD45A est un gène conservé lors de l'évolution dont l'expression est régulée par deux
protéines suppresseurs de tumeurs majeures impliquées dans l'étiologie du cancer du sein,
p53 et BRCAl, et qui agit principalement dans le contrôle de la transition G2/M du cycle
cellulaire, l'apoptose et la réparation de l'ADN. Suite à un stress génotoxique, la protéine
p53 active la transcription de GADD45A, tandis qu'en l'absence de dommages à l'ADN,
BRCAl réprime l'expression de GADD45A par l'intermédiaire d'une interaction avec la
protéine à doigts de zinc ZNF350. De plus, BRCAl peut activer l'expression du gène
GADD45A par l'interaction de facteurs de transcription se liant au promoteur du gène. Sur
la base du réseau complexe d'interactions entre GADD45A, p53 et BRCAl, ainsi que
l'implication des mutations à la fois de BRCAl et TP53 dans la tumorigénèse mammaire,
nous avons entrepris la caractérisation de la séquence codante, les jonctions intron/exon et
les séquences de liaison de p53 et ZNF350 de ce gène candidat à la susceptibilité au cancer
du sein. Ces analyses ont été effectuées dans un échantillon de 96 femmes atteintes de
cancer du sein provenant de familles à risque élevé de cancer du sein d'origine canadienne
française, négatives pour des mutations BRCAl et BRCA2, et 95 contrôles sains de la
même population. Bien qu'aucun des 12 variants de séquence identifiés présente une
différence significative de fréquence entre les deux échantillons, le phasage des haplotypes
et les estimations de fréquences ont identifié un haplotype commun démontrant une
fréquence plus élevée dans le groupe contrôle. Puisque les variants présents sur cet
haplotype spécifique sont des variants non-codants dans l'intron 2 ou 3, ce résultat devra
être évalué de façon plus approfondie dans de plus grandes cohortes et d'autres populations.
À cet égard, notre étude a aussi identifié des SNPs marqueurs, fournissant des données
utiles pour d'autres études d'association de plus grande envergure.

109
Abstract GADD45A is an evolutionary conserved gene whose expression is regulated by two major
tumor suppressor proteins involved in breast cancer etiology, namely p53 and BRCAl, and
which acts primarily in the control of the G2/M cell cycle transition, apoptosis and DNA
repair. Following a genotoxic stress, the p53 protein activates GADD45A transcription,
while in absence of DNA damage, BRCAl represses GADD45A expression through an
interaction with the zinc finger protein ZNF350. Moreover, BRCAl can activate
GADD45A gene expression through interactions with transcription factors binding to the
gene promoter. On the basis of the intricate network of interactions between GADD45A,
p53 and BRCAl, and the fact that both BRCAl or TP53 mutations are involved in breast
cancer tumorigenesis, we undertook the characterization of the entire coding sequence,
intron/exon boundaries and p53- and ZNF350-binding sequences of this potential breast
cancer susceptibility candidate gene in a sample set of 96 women affected with breast
cancer from non-BRCAl and BRCA2 French Canadian families with a high risk of breast
cancer and 95 healthy controls from the same population. Although none of the 12
identified sequence variations show a significant difference in frequency between both
sample sets, haplotype phasing and frequency estimations identified a common haplotype
displaying a higher frequency among the control group. As the variants present on this
particular haplotype are non-coding variants in either intron 2 or 3, this finding will have to
be further investigated in larger cohorts and other populations. In this regard, our study also
identified tSNPs, providing useful data for other large-scale association studies.
Keywords: Breast cancer, GADD45A gene, sequence analysis, haplotype, tagging SNP

110
Introduction Breast cancer is a complex disease involving genetic and lifestyle components. Our current
understanding of breast cancer susceptibility is based upon a polygenic model in which
several alleles conferring variable risk act in concert. Under this model, it is hypothesized
that 88% of all breast cancer cases may be found in 50% of the population more at risk
(Pharoah et al. 2002). Thus, finding the underlying genetic components conferring such risk
may have a great impact on breast cancer prevention and treatment. However, our
knowledge of breast cancer susceptibility remains incomplete. Mutations and/or allelic loss
of tumor suppressor genes, such as BRCAl, BRCA2, TP53 as well as other genes, are
associated with an increased predisposition to breast cancer tumorigenesis (Antoniou and
Easton 2006). However, mutations in the two major predisposition genes BRCAl and
BRCA2 account for less than 25% of families with hereditary breast cancer (Antoniou and
Easton 2006), indicating that much of the underlying genetic factors involved in breast
cancer susceptibility remain to be uncovered.
GADD45A (Growth Arrest and DNA Damage-induced 45, Alpha) is a p53- and BRCA1-
regulated gene acting in the control of the G2/M cell cycle transition, apoptosis and DNA
repair (Wang et al. 1999, Harkin et al. 1999, Smith et al. 1996). Following a genotoxic
stress, the p53 protein binds - directly to a consensus sequence located in GADD45A third
intron and - indirectly, through its interaction with transcription factors, to the promoter
region, therefore activating GADD45A transcription (Zhan et al. 1998, Jin et al. 2001). In
absence of DNA damage, BRCAl represses GADD45A expression through an interaction
with the zinc finger protein ZNF350 (ZBRK1), this complex recognizing a DNA binding
site also located in GADD45A third intron (Zheng et al. 2000). Moreover, BRCAl can
activate GADD45A gene expression through interactions with transcription factors binding
to the gene promoter (Fan et al. 2002).
Given that GADD45A is a target of two major tumor suppressor proteins involved in breast
cancer, namely BRCAl and p53, and that specific haplotypes of the gene encoding BRCAl
co-repressor ZNF350 have also been recently associated with a modulation of breast cancer
risk in our cohort of non-BRCA 1/2 high-risk breast cancer families (Desjardins et al. 2008),

I l l
GADD45A therefore represents an attractive candidate gene with regard to breast cancer
susceptibility. Although no alterations of GADD45A have been found in previous studies
performed on a limited set of breast tumors, human cancer cell lines of various origins, and
familial breast carcinomas (Blaszyk et al. 1996, Campomenosi et al. 2000, Sensi et al.
2004), Gadd45a-I- cells and mice display genomic instability (Hollander et al. 2005,
Hollander et al. 1999), and the absence of Gadd45a has been associated with an
acceleration of Ras-driven mammary tumor formation in mice (Tront et al. 2006).
Therefore, we investigated the plausible implication of GADD45A in breast cancer
susceptibility by analyzing 96 individuals affected with breast cancer from our cohort of
high-risk non-BRCAl/2 breast and ovarian cancer families, along with 95 healthy controls
from the same origin. The entire GADD45A coding region, intron-exon boundaries as well
as the p53- and BRCAl/ZNF350-binding consensus sequences were examined.

112
Materials and methods Ascertainment of families and DNA extraction. All 96 non-BRCAl/2 individuals from
French Canadian families with a high risk of breast and ovarian cancer, along with 95
healthy controls included in this study provided a written informed consent. The research
project has also been reviewed by the ethics committee of each participating institution.
Details regarding selection criteria for breast cancer cases, experimental and clinical
procedures as well as the INHERIT BRCAs research program have been described
previously (Simard et al. 2007, Durocher et al. 2006, Desjardins et al. 2008). Control blood
DNA was either used directly or subjected to whole genome amplification using Illustra
GenomiPhi V2 DNA amplification kit (GE Healthcare, formerly Amersham Biosciences,
Piscataway, USA) according to the manufacturer's instructions.
PCR amplification, mutation analysis and variant characterization. The GADD45A
gene (NM001924.2) consists of 4 exons covering more than 3kb of genomic DNA. Direct
sequencing of genomic sequence was performed using sequencing primers listed in
Supplemental Table 1. Direct sequencing was performed using an ABI3730XL automated
sequencer (Applied Biosystems, Foster City, USA), according to manufacturer's
instructions. Staden preGap4 and Gap4 programs were used for sequence data analysis.
Deviation from Hardy-Weinberg equilibrium (HWE) and allelic differences between both
series were measured using a X2 test with 1 degree of freedom. The effect of a given variant
on splice site consensus strength was evaluated with the Splice Site Prediction Program
using Neural Networks (SSPNN) (Reese et al. 1997), with default parameters. The putative
impact of the exonic variant on exonic splicing enhancers was also assessed using ESE
Finder (Cartegni et al. 2003, Smith et al. 2006).
LD analysis, haplotype estimation and tagging SNP selection (tSNP). The LDA
program (Ding et al. 2003) was used to calculate pairwise linkage disequilibrium (LD)
defined by Lewontin's |D' | and r2 measures on cases and controls combined (Lewontin
1964, Devlin and Risch 1995). The PHASE 2.1.1 software (Stephens et al. 2001) was used
with default parameters to estimate the total set of haplotypes present among the case and
control datasets and to evaluate the global test of significance associated with a case-control

113
comparison. Evaluation of a positive association of a specific haplotype displaying a
frequency >5% with breast cancer was further analyzed using the WHAP program (Purcell
et al. 2007), implementing a regression-based association test. The determination of
haplotype blocks and tagging SNPs (tSNPs) for each LD block was performed with
genotyping data from both sample sets combined using the Haploview software (Barrett et
al. 2005).
Electronic links. UCSC Genome Bioinformatics: http://genome.ucsc.edu/; NCBI dbSNP:
http://www.ncbi.nlm.nih.gov/SNP/; SSPNN: http://www.fruitfly.org/seq_tools/splice.html;
ESE Finder: http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi; PHASE: www.stat.
washington.edu/stephens/software; WHAP: http://pngu.mgh.harvard.edu/~purcell/whap/;
Haploview: http://www.broad.mit.edu/mpg/haploview; HapMap: www.hapmap.org;

114
Results Analysis of GADD45A sequence variations. Although no truncating mutation was found
in the GADD45A coding region of our French Canadian breast cancer cases, we identified
12 variants in GADD45A exonic and flanking intronic sequences, including two novel
sequence variations (C.147-103G/C and C.385-174C/T) not reported in the NCBI single
nucleotide polymorphism database (dbSNP Build 128) (Table 1). Among these sequence
variations, one is a coding silent variant (c.492A/G, p.E164E), while the 9 remaining
sequence changes are intronic nucleotide substitutions. No deviation from HWE was
observed for any of the nucleotide changes identified, with the exception of one intronic
variation (c.384+116T/C), displaying borderline significance (p= 0.045) due to an excess of
rare homozygotes among cases (Table 1). When considering all nucleotide variations, 7 are
common variants with minor allele frequency (MAF) higher than 5%, while 5 are
considered as rare sequence variations since they display a MAF below 5%. Among the
rare variants, two (c.492A/G and C.498+27C/T) are observed simultaneously within the
same breast cancer and control individuals, suggesting that both individuals carry a specific
allele. The allele frequency of variants observed in the breast cancer series were also
genotyped in healthy French Canadian controls, as denoted in Table 1. No significant
difference in MAF was observed. In addition, all variants identified also display similar
frequencies to those reported in dbSNP database including the CEU population.
As consensus binding sites located in GADD45A third intron are important regulatory
sequences, the relevant genomic regions were also analyzed by direct sequencing. No
sequence alteration was found directly within the consensus p53-binding region located in
the 5' end of the intron, the closest variation being situated 20 nucleotides downstream
(C.384+168T/C, Table 1). Likewise, amplification of BRCAl/ZNF350-binding region near
the beginning of exon 4, in the 3' part of the intron, did not lead to the identification of any
sequence alteration directly within the consensus sequence. However, a rare variant (c.385-
205A/G) located 2 nucleotides apart was identified in only one control and one breast
cancer case, but no genomic DNA from other affected family members was available for
testing. Another rare nucleotide change, present within the same amplicon (c.385-174C/T)
was also identified in only one breast cancer case and one control individual, while variant

115
C.385-137T/C (rs3171012) situated 137 nucleotides upstream of the fourth exon of
GADD45A is present at a MAF which is similar for both series, and within the range of
frequencies reported (Table 1).
In silico analysis of the effect of variants on splicing. The possible effect of all intronic
variants on splicing consensus sequences was also assessed using in silico analysis with the
SSPNN program. None of the genetic variations observed displayed a significant change in
the splicing score (data not shown), including the intronic variant most likely to have a
potential effect (C.45-23C/T), given its proximal location to a known exon boundary. The
strongest effects on the splicing score change were observed for the C.384+93A/G
(rs3783468) and the C.385-174C/T variants. The C.384+93A/G variant could affect a
pseudo donor site located 86 nucleotides downstream of the exon 3 3' boundary by
decreasing the splicing score from 0.60 to 0.41, while C.385-174C/T could create a weak
pseudo acceptor site located 189 nucleotides upstream of exon 4 (splicing score of 0.42). In
addition this variant (c.385-174C/T) could also strengthen another pseudo acceptor site
situated 164 nucleotides before exon 4 by increasing the splicing score from 0.68 to 0.76,
which would be as strong as the splice site currently used, although no evidence indicates
that this pseudo donor site is utilized by the splicing machinery. The putative impact of the
C.492A/G exonic variant was also assessed using the ESE Finder program, which examines
sequences for exonic splicing enhancer motifs. Although the C.492A/G variant slightly
modified the putative ESE motifs identified, all changes were very close to the default
thresholds and thus would be expected to act on relatively weak motifs, which would most
likely not be used by the splicing machinery (data not shown).
Evaluation of GADD45A intragenic linkage disequilibrium. LD calculations for each
SNP pair was performed in both series combined using D' and r2 measures, and are
represented in Figure 1. Perfect LD (|D'|=1) is observed between the 2 most distant
intragenic variants (SNP1: C.45-23C/T and SNP12: C.498+27C/T), which indicates that LD
at the GADD45A locus does not decline significantly with distance. This is not surprising
given that the GADD45A gene covers less than 4 kb of genomic sequence. Indeed, the
lowest pairwise LD value involves C.147-53G/T (SNP3) with C.384+116T/C (SNP5)
(D'=0.158). As expected, r2 coefficients calculated for GADD45A genomic region display

116
lower values, as this measure is sensitive to variation in allelic frequency, which is well
represented by the large spectrum of r2 values ranging from 0.0001 to 1.0. A high r2
coefficient is observed between C.384+118C/T (SNP6) and C.384+168T/C (SNP7), which
displays high and similar MAF, while the majority of lowest r2 values are observed for the
three SNPs displaying a MAF below 1% (c.l47-103G/C, C.492A/G and C.498+27C/T). As
described above, both C.492A/G and C.498+27C/T are rare, and are always observed within
the same individuals, hence this pair displays perfect LD (r2 coefficient of 1.0).
Haplotype analysis. Analysis of GADD45A haplotypes with the PHASE program, using
the 12 sequence variations genotyped in both series, led to 18 estimated haplotypes (Table
2). Among these, 5 haplotypes (PHI, 4, 7, 9, and 15) display a frequency >5%, which
represent 95.5% of all haplotypes estimated in both sample sets combined, while the
remaining haplotypes display frequencies <1%. Estimated p-value for case-control
permutation did not indicate any significant difference in the global haplotype composition
and frequency distribution between both groups (p=0.94). Among the common haplotypes,
only the haplotype PH9 shows a notable difference in frequency, this haplotype being more
represented among the controls. To further ascertain a possible association of this specific
haplotype with breast cancer, a second haplotype estimation program, WHAP, was used,
allowing for regression-based haplotype association testing. Estimation of haplotypes
within the same datasets yielded the same 5 common haplotypes, displaying remarkably
similar estimated frequencies as those obtained with the PHASE program (Table 2).
Although the global test for association was not significant (p=0.16), this haplotype-
specific testing indicated a weak but significant association of haplotype WH2 with breast
cancer (p=0.032), with an over representation of this specific haplotype among the controls.
Using a sliding window, we were able to circumscribe the specificity of this haplotype to
the first 6 genetic variants without loosing the significant association (p=0.0158, data not
shown), with the strongest effect observed when considering the first 9 variants (p=0.0155).
Tagging SNPs determination. In order to reduce genotyping costs and efforts in future
association studies without loss of power, the selection of a small fraction of SNPs,
identified as tSNP, which likely represent with a good reliability the French Canadian
population and the underlying variation at the GADD45A locus, represents an avenue of

117
choice. Indeed the selection of tSNPs that would be both frequent (MAF >0.05) and in
strong LD with the other variants will allow to capture the common alleles of GADD45A.
Thus, a causal variant may not be genotyped but will still be represented by the set of
tSNPs. Therefore genotyping data of the 12 variants from both series have been used for
final haplotype block as well as tSNP identification. Based on an algorithm of solid block
of LD, only one region of strong LD among the French Canadians was identified using the
Haploview software (expectation maximization algorithm) (Figure 2). This LD block
encompasses the whole exonic region given that SNPs 1 and 12 are comprised within the
same LD block. Thereafter, considering exclusively haplotypes having a frequency >5%, 4
tSNPs have been identified within this LD block, namely variants c.384+93A/G,
C.384+116T/C, C.384+118C/T and C.385-137T/C (rs3783468, rs3783469, rs681673 and
rs3171012). Given that only 3 genomic variants were available from HapMap data
(rs3783468, rs681673 and rs532446), and that they are all located in intron 3, it was not
relevant to perform similar analyses using CEPH/CEU sample data. To further test the
usefulness of our selected tSNPs in capturing haplotype diversity, a second haplotype
analysis was performed, using only the tSNPs selected here. These 4 tSNPs still allowed to
distinguish the five common haplotypes present in our dataset, demonstrating the efficacy
of the tSNPs identified here to accurately pinpoint all common haplotypes found in our
population. However, when using only the 3 tSNPs present in HapMap database, just 3
haplotypes could be determined delineating the importance of the added tSNPs from the
present study.

118
Discussion In search of additional breast cancer susceptibility genes, we undertook the analysis of the
GADD45A gene in our cohort of high-risk non-BRCAl/2 breast cancer families and healthy
controls based on the close involvement of GADD45A with the products of the known
predisposition genes TP53 and BRCAl. All individuals were drawn from the French
Canadian founder population, thus theoretically decreasing the underlying genetic
heterogeneity. In addition, breast cancer cases were purposely selected from high-risk
families (one per family), which has been demonstrated to increase the statistical power
(Antoniou and Easton 2003).
Although no truncating mutations were found, a total of 12 sequence variations were
identified in our datasets, including two variations not previously reported. No nucleotide
change on its own seems to be significantly associated with breast cancer risk, including
C.384+118C/T and C.384+168T/C, which are both near, but not directly within, intronic
regions of strong conservation observed in GADD45A third intron (from UCSC
conservation data, data not shown). In spite of this, we can assume they have no functional
significance given that these variants display similar frequencies in both sample sets. The
only non-coding variation present in a region of strong conservation is the rare
C.498+27C/T nucleotide change located in the 3'UTR region, which displays similar
frequencies in both groups.
Further LD analyses of the genetic variants identified highlighted relatively strong LD
between all GADD45A sequence variations. Among these, the variants c.385-205A/G,
C.492A/G and C.498+27C/T are in complete LD (for both |D'| and r2 measures), variants
C.492A/G and C.498+27C/T being observed in the same case and control individuals,
therefore suggesting that these variations are present on the same allele. As for c.385-
205A/G, we can expect this variant to be present on the other allele. Indeed, although it is
present on the same control individual as C.492A/G and C.498+27C/T, it is not present on
the breast cancer sample bearing the other two variants. HapMap data within this genomic
region display a block of strong LD encompassing GADD45A and nearly the entire
adjacent GNG12 gene (G-protein gamma 12 subunit, data not shown), which is

119
approximately 14Kb apart in the opposite direction, and is implicated in cellular signal
transduction. On its 5' end, GADD45A nearest neighbor is the SERBP1 gene involved in
the binding of mRNA 3'ends. However, the SERBP1 gene being located more than 260Kb
apart, it would not be expected to share LD with GADD45A according to HapMap data,
although we cannot exclude the possibility of a greater extent of LD in a founder
population such as the French Canadian population (Vézina et al, 2005, Laberge et al.
2005).
Given that the association of a gene with disease can be specific to certain alleles, the
haplotype diversity of GADD45A was first estimated with the use of the PHASE software.
PHASE estimated that an ensemble of 18 haplotypes solves all case and control genotypes
observed, but no significant differences in haplotypes structure and frequency were
estimated between both series. However, a closer inspection of the attributed haplotypes
among both groups highlighted a difference in frequency of the common haplotype PH9.
Therefore, to further refine our haplotype analysis, we used the WHAP program which
enables an estimation of a possible haplotype-specific association with breast cancer risk,
and which confirmed that WH2 (which is identical to PH9) was indeed over-represented
among the control group. An analysis of this same haplotype using only a subset of variants
indicated that a strong component of this specific difference is dependent on the 5' end of
the GADD45A gene.
Although none of the identified variant of GADD45A is clearly causative, we cannot rule
out the possible effect of yet unidentified variants in other regulatory portions of the gene
other than those analyzed here. Indeed, GADD45A is subject to a complex multi-levels
regulation in response to various extracellular signals, and the association of transcriptional
inductors and repressors such as p53 and ZNF350/BRCA1 with specific regions of its
genomic sequence tightly regulates its expression. However, analysis of the consensus
sequences located in intron 3 in both sample sets did not lead to the identification of any
genomic variants potentially disrupting these motifs.
Nonetheless, GADD45A is also regulated through post-transcriptional events, mainly
changes in its mRNA stability through the binding of stabilization (HuR, nucleolin) and
destabilization (AUF1) proteins on the 3'UTR distal sequence. In addition, GADD45A

120
expression has been recently shown to be regulated through a cap-independent and 1RES
(internal ribosome entry site)-dependent mechanism in response to cellular stress signals
such as arsenic-induced cytotoxic and genotoxic damage (Chang et al. 2007). This
expression is dependent on an 1RES element located in the 5'UTR region proximal to the
start codon. Since mutations of 1RES elements or impairment of binding between trans
acting factors and 1RES elements have been associated with cancer (Chappell et al. 2000),
we carefully screened both sample sets for deleterious mutations that could potentially
affect the expression through the 1RES element of GADD45A. No sequence variant was
identified within the GADD45A 1RES sequence. However, it has to be stated that the
possibility remains that changes in mRNA stability could be eventually associated with
breast cancer risk.
Although GADD45A is part of a cellular pathway that includes several genes demonstrated
to be involved in breast tumorigenesis (ATM, TP53, BRCAl), it has not been subjected to
an extensive mutation search. Blaszyk et al. (1996) analyzed a series of sporadic breast
cancer tumors (with and without p53 mutations) for alterations in GADD45A p53-binding
site (n=53 tumors) and coding sequence (n=26 tumors), and only identified one
polymorphism in intron 3. To our knowledge, the only other study of GADD45A alterations
in familial breast cancer was performed on tumors from individuals showing a high
incidence of early-onset breast cancer (n=34 families), male breast cancer (n=2 families) or
breast and ovarian cancer (n=7 families) the majority of which were not previously
screened for the presence of a BRCAl or BRCA2 mutation (Sensi et al. 2004). In
agreement to what we found in non-BRCAl/BRCA2 high-risk breast cancer families, they
did not identify any alterations of the coding sequence, the exon/intron boundaries as well
as the p5 3-binding domain.
Therefore, although GADD45A represents a very attractive breast cancer susceptibility
candidate gene, our analysis and the current knowledge of GADD45A alterations in breast
cancer individuals, cell lines and tumors do not support a strong involvement of this gene
with breast cancer risk. Nonetheless, additional investigations will be definitely needed to
further ascertain the involvement of variations within the regulatory region of GADD45A
with regard to breast cancer. However, given the limited information on sequence variants

121
data available in HapMap database, this study provides the identification of tSNPs, which
will be useful for further testing the association of GADD45A gene in larger cohorts, or
with another syndrome or disease.

122
Acknowledgements The authors are indebted to the participants and their families for their generosity and
providing DNA samples. We thank Damian Labuda and Claudia Moreau at the Centre de
Recherche de l'Hôpital Ste-Justine for providing control DNA samples. We would like to
thank Dr Martine Dumont, Gilles Leblanc, Carolle Samson and Martine Tranchant for
sample management, mutation screening, and skillful technical assistance as well as Tina
Babineau, Nathalie Bolduc, Claire Brousseau, Marie-Andrée Lajoie, Pascale Léger, Hélène
Malouin, Andrée McMillan and Josée Rhéaume, for genetic counselling and clinical data
management at the Cancer Genomics Laboratory. We would like to thank Professor Bartha
Maria Knoppers and her colleagues from the Centre de recherche en droit public de
l'Université de Montréal for their precious help with ELSI issues related to our research
program. We also appreciate advice received from ethics committees. This work was
supported by the Canadian Institutes of Health Research (CIHR) through the INHERIT
BRCAs research program, the Fonds de la Recherche en Santé du Québec (FRSQ)/Réseau
de Médecine Génétique Appliquée (RMGA), the CURE Foundation and the Canadian
Breast Cancer Research Alliance (CBCRA). S.D. holds a studentship from Fondation René
Bussières, J.S. is Chairholder of the Canada Research Chair in Oncogenetics, and F.D. is a
recipient of a chercheur-boursier from the Fonds de la Recherche en Santé du Québec
(FRSQ) and a Research Career Award in the Health Sciences from CIHR/Rx&D Health
Research Foundation.

123
Appendix Other members of INHERIT BRCAs involved in clinical aspects of the study:
Paul Bessette: Department of Obstetrics and Gynecology, Centre Hospitalier Universitaire
de Sherbrooke, Fleurimont, Canada
Peter Bridge: Molecular Diagnostic Laboratory, Alberta Children's Hospital, Calgary,
Alberta, Canada
Jocelyne Chiquette: Clinique des maladies du sein Deschênes-Fabia, Hôpital du Saint-
Sacrement, Québec, Canada
Rachel Laframboise: Medical Genetics Division, Centre Hospitalier Universitaire de
Québec, CHUL, Laval University, Québec, Canada
Jean Lépine: Haemato-Oncology Service, Centre Hospitalier Régional de Rimouski,
Rimouski, Canada
Bernard Lesperance, Roxane Pichette: Department of Haemato-Oncology, Hôpital du
Sacré-Coeur de Montréal, Montréal, Canada
Marie Plante: Gynecology Oncology Division, Hôtel-Dieu de Québec, Centre Hospitalier
Universitaire de Québec, Laval University, Québec, Canada

124
References Antoniou AC, Easton DF (2003) Polygenic inheritance of breast cancer: Implications for design of association studies. Genet Epidemiol 25:190-202 Antoniou AC, Easton DF (2006) Models of genetic susceptibility to breast cancer. Oncogene 25:5898-905 Barrett JC, Fry B, Mailer J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263-265 Blaszyk H, Hartmann A, Sommer SS, Kovach JS (1996) A polymorphism but no mutations in the GADD45 gene in breast cancers. Hum Genet 97:543-547 Campomenosi P, Hall PA (2000) Gadd45 mutations are uncommon in human tumour cell lines. Cell Prolif 33:301-306 Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR (2003) ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acid Research 31:3568-3571 Chang Q, Bhatia D, Zhang Y, Meighan T, Castranova V, Shi X, Chen F (2007) Incorporation of an internal ribosome entry site-dependent mechanism in arsenic-induced GADD45 alpha expression. Cancer Res 67:6146-6154 Chappell SA, LeQuesne JP, Paulin FE, deSchoolmeester ML, Stoneley M, Soutar RL, Ralston SH, Helfrich MH, Willis AE (2000) A mutation in the c-myc-IRES leads to enhanced internal ribosome entry in multiple myeloma: a novel mechanism of oncogene de-regulation. Oncogene 19:4437-4440 Desjardins S, Belleau P, Labrie Y, Ouellette G, Bessette P, Chiquette J, Laframboise R, Lépine J, Lesperance B, Pichette R, Plante M, Durocher F (2008) Genetic variants and haplotype analyses of the ZBRK1/ZNF350 gene in high-risk non BRCA1/2 French Canadian breast and ovarian cancer families. Int J Cancer 122:108-116 Devlin B, Risch N (1995) A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29:311-322 Ding K, Zhou K, He F, Shen Y (2003) LDA—a java-based linkage disequilibrium analyzer. Bioinformatics 19:2147-2148 Durocher F, Labrie Y, Soucy P, Sinilnikova O, Labuda D, Bessette P, Chiquette, Laframboise R, Lepine J, B. Lesperance B, G. Ouellette G, Pichette R, Plante M, Tavtigian SV, Simard J (2006) Mutation Analysis and Characterization of ATR Sequence Variants in Breast Cancer Cases from High-Risk French Canadian Breast/Ovarian Cancer Families. BMC Cancer 6:230-251 Fan W, Jin S, Tong T, Zhao H, Fan F, Antinore MJ, Rajasekaran B, Wu M, Zhan Q (2002) BRCAl regulates GADD45 through its interactions with the OCT-1 and CAAT motifs. J Biol Chem 277:8061-8067 Harkin DP, Bean JM, Miklos D, Song YH, Truong VB, Englert C, Christians FC, Ellisen LW, Maheswaran S, Oliner JD, Haber DA (1999) Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCAl. Cell 97:575-586 Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, Molinaro TA, Kim KE, Tolosa E, Ashwell JD, Rosenberg MP, Zhan Q, Fernandez-

125
Salguero PM, Morgan WF, Deng CX, Fomace AJ Jr (1999) Genomic instability in Gadd45a-deficient mice. Nat Genet 23:176-184 Hollander MC, Philburn RT, Patterson AD, Wyatt MA, Fomace AJ Jr (2005) Genomic instability in Gadd45a-/- cells is coupled with S-phase checkpoint defects. Cell Cycle 4:704-709 Jin S, Fan F, Fan W, Zhao H, Tong T, Blanck P, Alomo I, Rajasekaran B, Zhan Q (2001) Transcription factors Oct-1 and NF-YA regulate the p53-independent induction of the GADD45 following DNA damage. Oncogene 20:2683-2690 Laberge AM, Michaud J, Richter A, Lemyre E, Lambert M, Brais B, Mitchell GA (2005) Population history and its impact on medical genetics in Quebec. Clin Genet 68:287-301 Lewontin RC (1964) The Interaction of Selection and Linkage. Ii. Optimum Models, Genetics 50:757-782 Pharoah PD, Antoniou A, Bobrow M, Zimmern RL, Easton DF, Ponder BA (2002) Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet 31:33-36 Purcell S, Daly MJ, Sham PC (2007) WHAP: haplotype-based association analysis. Bioinformatics 23:255-256 Reese MG, Eeckman FH, Kulp D, Haussier D (1997) Improved splice site detection in Genie. J Comput Biol 4:311-323 Sensi E, Tancredi M, Aretini P, Cipollini G, Collecchi P, Naccarato AG, Viacava P, Bevilacqua G, Caligo MA (2004) Clinicopathological significance of GADD45 gene alterations in human familial breast carcinoma. Breast Cancer Res Treat 87:197-201 Simard J, Dumont M, Moisan AM, Gaborieau V, Malouin H, Durocher F, Chiquette J, Plante M, Avard D, Bessette P, Brousseau C, Dorval M, Godard B, Houde L, Joly Y, Lajoie MA, Leblanc G, Lepine J, Lesperance B, Vezina H, Parboosingh J, Pichette R, Provencher L, Rheaume J, Sinnett D, Samson C, Simard JC, Tranchant M, Voyer P, Easton D, Tavtigian SV, Knoppers BM, Laframboise R, BridgeP, Goldgar D (2007) Evaluation of BRCAl and BRCA2 mutation prevalence, risk prediction models and a multi-step testing approach in French-Canadian high-risk breast and ovarian cancer families. J Med Genet 44:107-121 Smith ML, Kontny HU, Zhan Q, Sreenath A, O'Connor PM, Fomace AJ Jr (1996) Antisense GADD45 expression results in decreased DNA repair and sensitizes cells to u.v.-irradiation or cisplatin. Oncogene 13:2255-2263 Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR (2006) An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet 15: 2490-2508 Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978-989 Tront JS, Hoffman B, Liebermann DA (2006) Gadd45a Suppresses Ras-Driven Mammary Tumorigenesis by Activation of c-Jun NH2-Terminal Kinase and p38 Stress Signaling Resulting in Apoptosis and Senescence. Cancer Res 66:8448-8454 Vezina H, Durocher F, Dumont M, Houde L, Szabo C, Tranchant M, Chiquette J, Plante M, Laframboise R, Lepine J, Nevanlinna H, Stoppa-Lyonnet D, Goldgar D, Bridge P, Simard J

126
(2005) Molecular and Genealogical Characterization of the R1443X-BRCA1 Mutation in High-Risk French-Canadian Breast/Ovarian Cancer Families. Hum Genet 117:119-132 Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, Hollander MC, O'Connor PM, Fomace AJ Jr, Harris CC (1999) GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci U S A 96:3706-3711 Zhan Q, Chen IT, Antinore MJ, Fornace AJ Jr (1998) Tumor suppressor p53 can participate in transcriptional induction of the GADD45 promoter in the absence of direct DNA binding. Mol Cell Biol 18:2768-2778 Zheng L, Pan H, Li S, Flesken-Nikitin A, Chen PL, Boyer TG, Lee WH (2000) Sequence-specific transcriptional corepressor function for BRCAl through a novel zinc finger protein, ZBRK1. Mol Cell 6:757-768
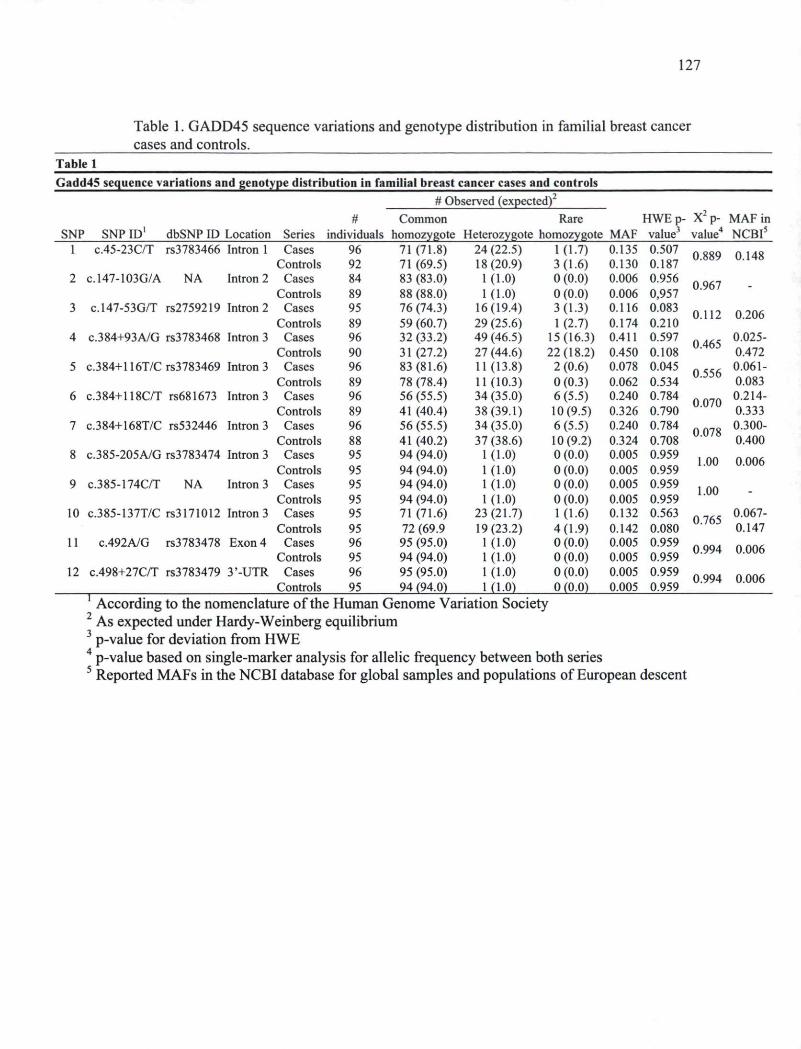
127
Table 1. GADD45 sequence variations and genotype distribution in familial breast cancer cases and controls.
Table 1 Gadd45 sequence variations and genotype distribution in familial breast cancer cases . ind controls
# # Observed (expected)2
HWEp- X 2 p-# Common Rare HWEp- X 2 p- MAF in SNF ' SNP ID1 dbSNP ID Location Series individuals homozygote Hétérozygote homozygote MAF value3 value4 NCBI5
1 C.45-23C/T rs3783466 Intron 1 Cases 96 71 (71.8) 24 (22.5) 1(1.7) 0.135 0.507 0.889 0.148 Controls 92 71 (69.5) 18(20.9) 3(1.6) 0.130 0.187
2 C.147-103G/A NA Intron 2 Cases 84 83 (83.0) 1 (1.0) 0 (0.0) 0.006 0.956 0.967 Controls 89 88 (88.0) 1 (1*0) 0 (0.0) 0.006 0,957
0.967
3 C.147-53G/T rs2759219 Intron 2 Cases 95 76 (74.3) 16(19.4) 3(1.3) 0.116 0.083 0.112 0.206 Controls 89 59 (60.7) 29 (25.6) 1 (2.7) 0.174 0.210
4 C.384+93A/G rs3783468 Intron 3 Cases 96 32 (33.2) 49 (46.5) 15(16.3) 0.411 0.597 0.465 0.025-Controls 90 31 (27.2) 27 (44.6) 22(18.2) 0.450 0.108
0.465 0.472 5 c.384+116T/Crs3783469 Intron 3 Cases 96 83(81.6) 11(13.8) 2(0.6) 0.078 0.045 0.556 0.061-
Controls 89 78 (78.4) 11(10.3) 0 (0.3) 0.062 0.534 0.556 0.083
6 C.384+118C/T rs681673 Intron 3 Cases 96 56 (55.5) 34 (35.0) 6(5.5) 0.240 0.784 0.070 0.214-Controls 89 41 (40.4) 38(39.1) 10(9.5) 0.326 0.790
0.070 0.333 7 C.384+168T/C rs532446 Intron 3 Cases 96 56 (55.5) 34 (35.0) 6(5.5) 0.240 0.784 0.078 0.300-
Controls 88 41 (40.2) 37 (38.6) 10(9.2) 0.324 0.708 0.078 0.400
8 C.385-205A/G rs3783474 Intron 3 Cases 95 94 (94.0) 1 (1.0) 0 (0.0) 0.005 0.959 1.00 0.006 Controls 95 94 (94.0) 1 (1.0) 0 (0.0) 0.005 0.959
9 C.385-174C/T NA Intron 3 Cases 95 94 (94.0) 1 (1.0) 0 (0.0) 0.005 0.959 1.00 Controls 95 94 (94.0) 1 (1.0) 0 (0.0) 0.005 0.959
1.00
10 C.385-137T/C rs3171012 Intron 3 Cases 95 71 (71.6) 23(21.7) 1 (1.6) 0.132 0.563 0.765 0.067-Controls 95 72 (69.9 19(23.2) 4(1.9) 0.142 0.080
0.765 0.147 11 C.492A/G rs3783478 Exon 4 Cases 96 95 (95.0) 1 (1.0) 0 (0.0) 0.005 0.959 0.994 0.006
Controls 95 94 (94.0) 1 (1.0) 0 (0.0) 0.005 0.959 0.994 0.006
12 C.498+27C/T rs3783479 3'-UTR Cases 96 95 (95.0) 1 (1.0) 0 (0.0) 0.005 0.959 0.994 0.006 Controls 95 94 (94.0) 1(1.0) 0 (0.0) 0.005 0.959
According to the nomenclature of the Human Genome Variation Society As expected under Hardy-Weinberg equilibrium p-value for deviation from HWE p-value based on single-marker analysis for allelic frequency between both series Reported MAFs in the NCBI database for global samples and populations of European descent
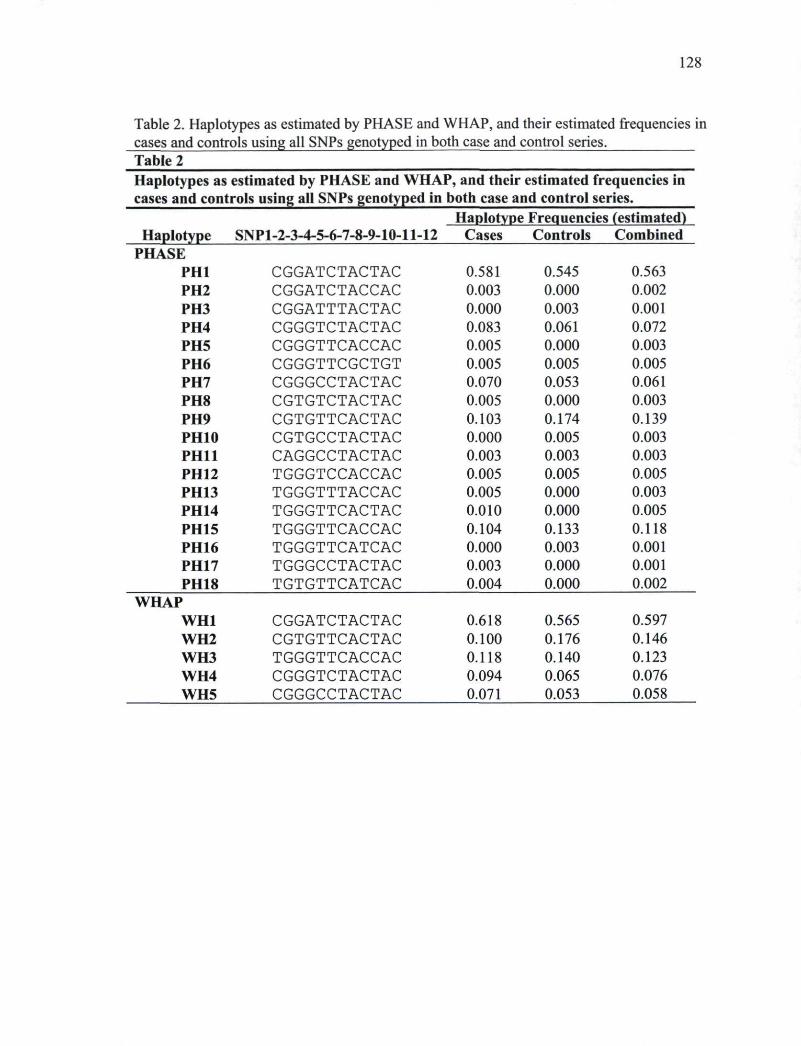
128
Table 2. Haplotypes as estimated by PHASE and WHAP, and their estimated frequencies in cases and controls using all SNPs genotyped in both case and control series. Table 2 Haplotypes as estimated by PHASE and WHAP, and their estimated frequencies in cases and controls using all SNPs genotyped in both case and control series.
Haplotvpe Frequencies (estimated) Haplotype SNP1-2-3-4-5-6-7-8-9-10-11-12 Cases Controls Combined
PHASE PHI CGGATCTACTAC 0.581 0.545 0.563 PH2 CGGATCTACCAC 0.003 0.000 0.002 PH3 CGGATTTACTAC 0.000 0.003 0.001 PH4 CGGGTCTACTAC 0.083 0.061 0.072 PH5 CGGGTTCACCAC 0.005 0.000 0.003 PH6 CGGGTTCGCTGT 0.005 0.005 0.005 PH7 CGGGCCTACTAC 0.070 0.053 0.061 PH8 CGTGTCTACTAC 0.005 0.000 0.003 PH9 CGTGTTCACTAC 0.103 0.174 0.139 PH10 CGTGCCTACTAC 0.000 0.005 0.003 PH11 CAGGCCTACTAC 0.003 0.003 0.003 PH12 TGGGTCCACCAC 0.005 0.005 0.005 PH13 TGGGTTTACCAC 0.005 0.000 0.003 PH14 TGGGTTCACTAC 0.010 0.000 0.005 PH15 TGGGTTCACCAC 0.104 0.133 0.118 PH16 TGGGTTCATCAC 0.000 0.003 0.001 PH17 TGGGCCTACTAC 0.003 0.000 0.001 PH18 TGTGTTCATCAC 0.004 0.000 0.002
WHAP WH1 CGGATCTACTAC 0.618 0.565 0.597 WH2 CGTGTTCACTAC 0.100 0.176 0.146 WH3 TGGGTTCACCAC 0.118 0.140 0.123 WH4 CGGGTCTACTAC 0.094 0.065 0.076 WH5 CGGGCCTACTAC 0.071 0.053 0.058

129
Supplemental Table 1. PCR and sequencing primers. Supplemental Table 1 PCR and sequencing primers.
Amplified Region PCR primer
PCR boundaries1
PCR length (bp) Sequencing primer
Exon 1 F-5 'GCCTGTG AGTGAGTGC AGA A3 ,2
R-5'GGAGTTGCCCTGTGCAAACT3'2 C.-99G to
C.44+122T 265 5 T AGCCGTGGCAGGAGC AG3 '2(F)
5 'CGGGGTAGGTAAGAGAAGTTCG3 '(R; Exon 2-3 F-5 'GGTGGTGCTTGCATTCGAGA3 '
R-5 'TGAGGGGTGAGCCAGGAATTCA3 ' C.45-108G to C.384+352A
1023 5 'GCGAGAACGACATCAACATC3 '(F) 5'AGGCAGAAAAGCGACAGGAAGTG3'(I
Exon 4 F-5'ATTTGTCTCCATGTCACATAGCCA3' R-5 'CCAG AACATGTAGTTGAACTCACTCA3 '
c.385-118Gto C.498+120A
353 Same primers as those used for PCR amplification
p53 binding site (c.384+188Gto C.384+207G)
Same primers as those used for exon 2-3 amplification 5 'CCCAGTCCCTTACCG TC AC3 '(R)
ZNF350/BRCA1 binding site
(c.385-221Tto C.385-207G)
F-5 ' GGT AG AGC A AACCGGTG AATG AT3 '
Same reverse primer as for exon 4
C.385-304A to 539 Same primer (F) as the one used for PCR C.498+120A amplification
"T 2 As published by: Yamasawa K, Nio Y, Dong M, Yamaguchi K, Itakura M (2002) Clinicopathological significance of abnormalities in Gadd45 expression and its relationship to p53 in human pancreatic cancer. Clin Cancer Res 8:2563-2569
According to the nomenclature of the Human Genome Variation Society and reference sequence NM_01924.2

130
Legends to Figures
Figure 1 Pairwise LD measures of |D'| and r2 for the 12 SNPs identified in the breast
cancer and control series. SNP numbers are denoted according to Table 1.
Figure 2 Haploview predictions of haplotype blocks using SNPs displaying a MAF higher
than 5%. The identification of SNPs is performed on a block-by-block basis and they are
denoted with an asterisk (*) above the SNP number. tSNPs are selected based on
haplotypes showing an estimated frequency higher than 5%. Haploview estimation of
haplotype frequencies is displayed on the right.
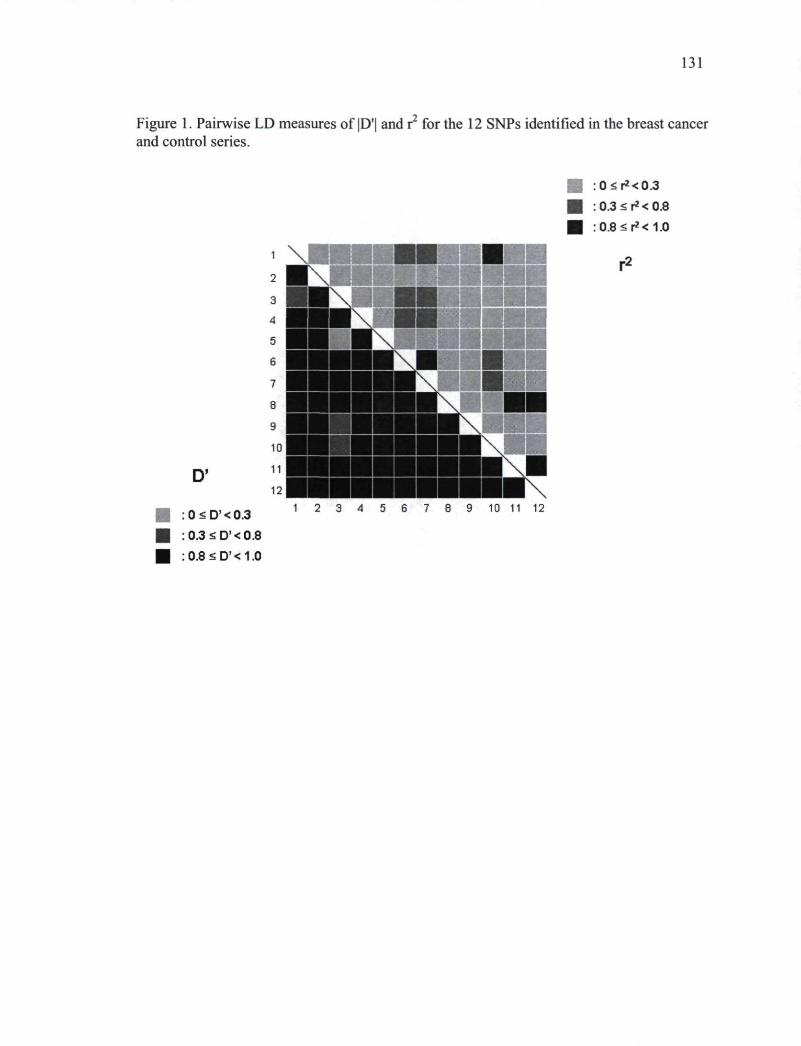
131
Figure 1. Pairwise LD measures of |D'| and r for the 12 SNPs identified in the breast cancer and control series.
0<r--<0.3
0.3 <!■•-< 0.8
0.8<r*<1.0
■
D'
0 < D ' < 0 . 3
0.3<D'<0.8
0.8<D'<1.0
4
5
6
7
8
9
10
11
12
■■ I\H ■■■■■
■■ ■ 10 11 12

132
Figure 2. Haploview predictions of haplotype blocks using SNPs displaying a MAF higher than 5%.
WH1
*** *
PHI WH1
Ho«(1'*--t-i^iJi>r-*»J'0'Ho« O O O o O o O o O r l n r l
CGGATCTACTAC .569 PH9 WH2 CGTGTTCACTAC .140 PH15 WH. TGGGTTCACCAC .116 PH4 WH4 CGGGTCTACTAC .073 PH7 WH5 CGGGCCTACTAC .055

133
CHAPITRE III : Variations in the NBN/NBS1 gene and the risk of breast cancer in non-BRCAl/2 French Canadian
families with high risk of breast cancer.

134
Variations in the NBN/NBS1 gene and the risk of breast cancer in non-BRCAl/2
French Canadian families with high risk of breast cancer.
Sylvie Desjardins1, Charles Joly Beauparlant1, Yvan Labrie1, Geneviève Ouellette1,
INHERIT BRCAs* and Francine Durocher1**
'Cancer Genomics Laboratory, Oncology and Molecular Endocrinology Research Centre,
Centre Hospitalier Universitaire de Québec and Laval University, Québec, Canada;
Email: Sylvie Desjardins - [email protected]. Charles Joly Beauparlant -
charles.ioly-beauparlant(a),crchul.ulaval.ca. Yvan Labrie - yvan.labrie(Sjcrchul.ulaval.ca,
Geneviève Ouellette - [email protected]. Francine Durocher** -
*Other members of INHERIT BRCAs involved in this study are listed in the Appendix.
** Correspondance to: Cancer Genomics Laboratory, Oncology and Molecular
Endocrinology Research Centre, Centre Hospitalier Universitaire de Québec and Laval
University, 2705 Laurier Blvd, T2-53, Québec City, Québec, Canada, GIV 4G2
Telephone: 418-654-2296 FAX: 418-654-2278/418-654-2761
BMC Cancer. 2009; 9 :181.

135
Résumé
Contexte : Le syndrome des cassures chromosomiques de Nijmegen est un désordre
d'instabilité chromosomique caractérisé par une microcephalic, un retard de croissance, une
immunodéficience et une fréquence accrue de cancers. Des études familiales conduites chez
des parents de ces patients ont indiqué qu'eux aussi semblent présenter un risque accru de
cancers. Méthodes : Dans une étude «gène candidat» visant à identifier les déterminants
génétiques de la susceptibilité au cancer du sein, nous avons entrepris le séquençage
complet du gène NBN dans notre cohorte de 97 familles à risque élevé de cancer du sein
non-BRCAl et -BRCA2 ainsi que chez 74 contrôles sains non apparentés, aussi de la
population canadienne française. Des programmes in silico (ESEfinder, NNSplice, Splice
Site Finder et Matlnspector) ont été utilisés afin d'évaluer l'impact potentiels des variants
identifiés. L'effet du variant du promoteur a été étudié plus spécifiquement par essai gène
rapporteur luciférase dans les lignées cellulaires MCF-7, HEK293, HeLa et LNCaP.
Résultats : Vingt-quatre variants ont été identifiés dans notre cohorte de cas, et leur
fréquence a été confirmée chez les contrôles sains. Le variant potentiellement délétère
p.Ilel71Val a été observé chez un cas uniquement. Le variant p.Arg215Trp, qui pourrait
affecter la liaison de NBN à l'histone y-H2AX, a été observé chez un cas de cancer du sein
et un contrôle sain. Un variant du promoteur, c.-242-110delAGTA, présente une variation
de fréquence significative entre les deux échantillons. L'essai de type gène luciférase
rapporteur avec une construction portant ce variant n'a pas démontré de variation
d'expression dans la lignée cellulaire de cancer du sein MCF-7, mais a indiqué une
réduction de l'expression de la luciférase dans les lignées cellulaires HEK293 et LNCaP.
Conclusions : Notre analyse des variants de séquence de NBN a indiqué que des altérations
potentielles de NBN sont présentes, quoiqu'à une fréquence faible, dans notre cohorte de
cas de cancer du sein à risque élevé. Des analyses plus approfondies seront nécessaires afin
de mieux déterminer l'impact exact de ces variants sur la susceptibilité au cancer du sein,
en particulier pour les variants localisés dans la région promotrice de NBN.

136
Abstract
Background: The Nijmegen Breakage Syndrome is a chromosomal instability disorder
characterized by microcephaly, growth retardation, immunodeficiency, and increased
frequency of cancers. Familial studies on relatives of these patients indicated that they also
appear to be at increased risk of cancer. Methods: In a candidate gene study aiming at
identifying genetic determinants of breast cancer susceptibility, we undertook the full
sequencing of the NBN gene in our cohort of 97 high-risk non-BRCAl and -BRCA2 breast
cancer families, along with 74 healthy unrelated controls, also from the French Canadian
population. In silico programs (ESEfinder, NNSplice, Splice Site Finder and Matlnspector)
were used to assess the putative impact of the variants identified. The effect of the promoter
variant was further studied by luciférase gene reporter assay in MCF-7, HEK293, HeLa and
LNCaP cell lines. Results: Twenty-four variants were identified in our case series and their
frequency was further evaluated in healthy controls. The potentially deleterious p.Ilel71Val
variant was observed in one case only. The p.Arg215Trp variant, suggested to impair NBN
binding to histone y-H2AX, was observed in one breast cancer case and one healthy
control. A promoter variant c.-242-110delAGTA displayed a significant variation in
frequency between both sample sets. Luciférase reporter gene assay of the promoter
construct bearing this variant did not suggest a variation of expression in the MCF-7 breast
cancer cell line, but indicated a reduction of luciférase expression in both the HEK293 and
LNCaP cell lines. Conclusions: Our analysis of NBN sequence variations indicated that
potential NBN alterations are present, albeit at a low frequency, in our cohort of high-risk
breast cancer cases. Further analyses will be needed to fully ascertain the exact impact of

137
those variants on breast cancer susceptibility, in particular for variants located in NBN
promoter region.

138
Background
Pathogenic mutations in BRCAl, BRCA2, TP53, ATM, CHEK2, BRIP1 and PALB2 have
been associated with an increased breast cancer risk and, together, are found in less than
25% of breast cancer families showing a clear pattern of inheritance (high-risk families)
[1]. It is thus clear that other susceptibility alleles remain to be identified to explain the
increased risk in the remnant high-risk families. As the number and characteristics of such
alleles are undetermined, a focussed candidate gene approach based on genes closely
interacting with the known susceptibility genes such as BRCAl and BRCA2, the two major
susceptibility genes identified yet, constitutes a study design of choice to identify rare-
moderate-penetrance susceptibility alleles.
In the cell, nibrin, encoded by the jViiW gene (also known as NBS1), participates in
pathways of double strand breaks (DSB)-induced DNA repair and, together with its
partners MRE11A and RAD50, is required for activation of these pathways in response to
DNA damages [2]. In fact, nibrin is at the crossroad of several pathways implicating genes
already associated with breast cancer susceptibility and/or chromosomal instability
disorders [2, 3]. Individuals homozygous for hypomorphic mutations in NBN suffer from
the Nijmegen Breakage Syndrome (NBS), an autosomal recessive chromosomal instability
disorder characterized by microcephaly, growth retardation, immunodeficiency and hyper-
radiosensitivity [4]. Cancers, in particular haematological malignancies, are common
adverse events in patients with NBS, as almost 40% of them develop a malignancy before
the age of 21 years, and this correlates with a marked impairment in DSB repair observed
in cells from these patients [5].

139
Some studies have associated an heterozygous NBN status with numerous types of cancers,
including breast cancer [6-10], suggesting that being a carrier of a deleterious mutation in
NBN may confer an increased risk of approximately 2 to 3-fold [6]. This was also
supported by the observation that relatives of NBS patients display a higher than expected
rate of cancers [4, 11]. However, other studies failed to find an association with an
increased risk of cancer [12, 13].
In support of a role of NBN in tumor formation, evidence from mouse models demonstrated
that Nbn heterozygosity predisposes cells to malignancies, as they display a wide variety of
tumors: liver, mammary gland, prostate, lung as well as lymphomas [14]. Indeed, cells
from these mice displayed an elevated frequency of chromosomal aberrations. These
observations were correlated by studies of NBN heterozygous mutation carriers
demonstrating that cell lines from these individuals showed spontaneous chromosomal
instability (chromatid and chromosomes breaks, and chromosomes rearrangements) [15,
16] as well as increased sensitivity to radiation-induced chromosomal aberrations [17].
Thus, it has been hypothesized that in cells of carriers of deleterious mutations in DNA
repair genes such as NBN, a decrease in DNA repair capabilities resulting from a gene
dosage effect (i.e. lower gene expression) may be sufficient to create a permissive
environment for tumor development [18, 19]. It has also been suggested that these DNA
repair genes may show differences in tissue-specific protein-dosage thresholds, below
which they may fail to operate normally [20].
Thus, based on the close relation of NBN and known breast cancer susceptibility genes in
the cell DNA repair pathways, and the studies suggesting a possible involvement of NBN
alterations in cancer susceptibility, we undertook the analysis of the entire coding sequence,

140
intron/exon junctions, as well as the proximal promoter region of the NBN gene. We
therefore performed a thorough re-sequencing of a series of 97 breast cancer cases selected
from high-risk families from the French Canadian population, and 74 unrelated healthy
controls from the same origin for sequence variations that could possibly modulate breast
cancer risk.

141
Methods
Ascertainment of families.
All 97 non-BRCAl/2 individuals from high-risk French Canadian breast and ovarian cancer
families participating in this study were part of a larger interdisciplinary program termed
INHERIT BRCAs [21]. All participants were at least 18 years of age and had to sign an
informed consent form. Ethics committees reviewed the research project at the 7
participating institutions from which the patients were referred. The details regarding
selection criteria of the breast cancer cases as well as the experimental and clinical
procedures have been described previously [21, 22].
PCR amplification and direct sequencing.
PCR amplification of NBN (NM_002485.4) coding sequence, as well as flanking intronic
regions, was performed on breast cancer cases and controls using primer pairs as described
in Table 5. Sequencing reactions and sequence analysis were performed as described
previously [22]. Alternative splice screening was also performed on cDNA on a subset of
breast cancer cases using primers as described in Table 1.
Variants characterization and haplotype estimations.
Deviation from Hardy-Weinberg equilibrium (HWE) and allelic difference between both
series was evaluated using a two-sided Chi Square test with 1 degree of freedom. The
possible effect of a given variant on exonic splicing enhancers was assessed using the
ESEfinder 3.0 program [23] and on splice consensus sites using NNSPLICE [24] and
Splice Site Finder [25] web based programs. The potential impact of the promoter variant
and pairwise linkage disequilibrium were evaluated as described elsewhere [26]. Haplotype

142
analysis was performed using the WHAP program [27] implementing a regression-based
association test allowing for evaluation of haplotype-specific association. Haplotype
analyses were performed on all variants identified as well as on variants showing a minor
allele frequency (MAF) greater than 5%.
Luciférase promoter assays.
A portion of 401 bp of the NBN promoter region, and the entire 5'UTR sequence (110 bp),
was amplified by PCR from a breast cancer individual carrier of the c.-242-110delAGTA
deletion using primers introducing a Nhel or a Hindlll restriction site (Table 1). PCR
products were then digested and introduced in the pGL3 basic vector (Promega
Corporation, Madison, WI, USA). Direct sequencing of clones was performed to confirm
the presence of the reference sequence allele as well as the four nucleotides deletion.
Transient transfections in MCF-7, LNCaP, HeLa and HEK293 cells and dual luciférase
reporter assays were performed as described previously [26], with cells harvested 24-48h
after transfection. ATBF1 expression levels were measured in these four cell lines by
quantitative real-time PCR (QRT-PCR) as described previously [28], using RNA extracted
by the TriReagent method (Molecular Research Center Inc, Cincinnati, OH, USA) and
specific primers (sens: 5'-TGCAACTAAACCGCCCACATATA-3'; antisens 5'-
CCCCAAGTGAGATAAAGCTAAACAAA-3'). Levels of expression were normalized
using the housekeeping gene HPRT1, and are indicated relative to the MCF-7 cell line (sens
primer: 5'-AGTTCTGTGGCCATCTGCTTAGTAG-3'; antisens primer: 5'-
AAACAACAATCCGCCCAAAGG-3 ').

143
Results
Sequence variations in the NBN gene. Direct sequencing of NBN entire coding region,
adjacent intronic sequences as well as the proximal promoter region was performed on 97
affected individuals from French Canadian breast and ovarian cancer families (one
individual per family). Twenty-four variants were identified (Tables 1, 2 and Figure 1),
including a new rare synonymous change at codon 127 (c.381T/C) and a new variant
located in intron 15 (C.2234+86T/G), both not reported in the literature and databases, and
found exclusively among breast cancer cases. Variants identified in the case dataset were
also genotyped in 74 healthy individuals from the same population. All SNPs genotyped
were in HWE. Nine out of the 24 variations were located in the coding region (Table 2),
while the remaining 15 were located in untranslated regions (Table 3). Among all variants
identified, half (12) were considered as common (MAF a5%) while the remaining 12 were
rare variations. Two rare non-synonymous exonic variants (c.511A/G and C.797C/T) were
observed only once in the case series, of which p.Ilel71Val is located in the first BRCAl
C-terminal (BRCT) domain (Figure 1). Of these two variants, only C.511A/G could be
genotyped in an additional family member, i.e. an unaffected male cousin, who was found
non-carrier (data not shown). Two other rare variants, C.283G/A and C.643C/T, were both
observed in one breast cancer case and one control. For the C.643C/T variant, a DNA
sample from another available family member affected with ovarian cancer was analyzed
and fumed out to be wild type (data not shown). Among the variants located in untranslated
regions (Table 3), the C.2234+86T/G variant was present exclusively in two breast cancer
cases. When genotype frequencies of all variants were compared between both series, only

144
the c.-242-110delAGTA variant in the promoter showed a statistically significant
association with breast cancer (OR 3.4, 95% CI: 1.1-10.5; p=0.029).
Identification of NBN alternative splice forms. Analysis of NBN cDNA was also
performed on a subset of these breast cancer cases and highlighted the presence of two
distinct alternative splice events. The first splice variant involved the insertion of 50 bp of
the intron between exons 2 and 3 and is expected to result in a premature stop codon [29].
The other alternative splice form identified involves the skipping of exons 12 to 14 which is
expected to produce an in-frame deletion of 113 amino acids in the region of the NBN
protein involved in the interaction with its partner MRE11A (Figure 1). However, QRT-
PCR of this specific form was performed on cDNA samples from a subset of 10 breast
cancer cases from our cohort and showed a very low expression relative to the main
isoform (data not shown), which is consistent with previous work [30].
In silico analysis of the putative impact of NBN exonic and intronic sequence variants on
these splice events was also performed. Analysis of the nine exonic sequence variants using
ESEfinder indicated that, while five variants (c.283G/A, C.553G/C, C.643C/T, C.797C/T,
and c.H97T>C) might have an impact on putative score motifs of four SR proteins, a
closer examination of these results shows that these scores, while below the program
thresholds, remain relatively high and thus are unlikely to affect NBN constitutive splicing
(Figure 2). According to the Splice Site prediction program, the intronic variant
C.896+36G/A might slightly increase a putative acceptor site (score from 0.66 to 0.75).
However, this was not confirmed by the Splice Site Finder program, which predicted the
abolition of a putative donor site with a score of 71.2. As for the C.1124+91C/A intronic
variant, only the Splice Site Prediction program predicted the abolition of a weak acceptor

145
site (score of 0.45) while for the C.2234+157A/G intronic variant, the Splice Site Finder
program estimated the creation of a putative acceptor site with a score of 77.2. Hence, none
of the variants identified are expected to be responsible for the alternative splice events
observed.
Effect of the c.-242-110delAGTA variant on luciférase reporter gene expression. The
putative effect of the c.-242-110delAGTA variant located in the promoter region was also
assessed by the Matlnspector program, which predicts that this variant may abolish
recognition motifs for the ZFHX3/ATBF1 (Zinc finger homeobox 3, or AT motif-binding
factor 1), NKX3-1 (NK3 homeobox 1) and CDX2 (Caudal-type homeobox 2) transcription
factors, and create a potential binding site for the MTBF (Muscle-specific MT binding
factor) transcription factor (Figure 3A). The effect of the c.-242-110delAGTA variant on
NBN expression was therefore further assessed using a dual reporter gene system. MCF-7,
LNCaP, HeLa and HEK293 cells were then transiently transfected with a construct
containing the consensus NBN sequence or with the variant sequence, together with the
Renilla reporter plasmid as an internal transfection control (Figure 3B). While no
significant difference in expression was observed between both constructs in MCF-7 and
HeLa cells, a slight diminution of luciférase expression was observed for the variant
construct relative to the reference sequence construct in both the HEK293 and LNCaP cell
lines (Figure 3C). Interestingly, QRT-PCR measures indicated that ATBF1 mRNA is 2.4
times more expressed in HeLa cells, and more than 4.3 times more expressed in LNCaP
cells, than in MCF-7 cells (data not shown).
LD analysis across NBN genomic sequence and haplotypes determination. Pairwise LD
measures of all variants using the control dataset are presented in Figure 4. |D'| values show

146
a high degree of LD for all SNP pairs while the more stringent r2 measure, dependent upon
allele frequency, shows a limited block of LD involving mainly SNPs located in the second
half of the NBN gene. The bottom part of Figure 4 shows the r2 HapMap CEU data in the
vicinity of NBN on chromosome 8, and suggests that NBN may overlap two blocks of LD.
The major part of NBN seems to be in a strong block of LD extending 5' of the gene, while
its 3' extremity spans over another smaller block.
Haplotype phasing of NBN using the WHAP program with all 24 variants identified in our
datasets indicated that 14 haplotypes are present with an estimated frequency greater than
0.5%. Although WHAP did not estimate a significant difference between both groups, the
p-value observed was borderline significant (p=0.0588). The majority of these estimated
haplotypes (84.5%) have a frequency greater than 2%. Two haplotypes (#4 and #7 in Table
3) showed a weak significant association with breast cancer, being more frequent among
cases (p=0.0205 and p=0.0403, respectively). The same analysis, using only common
variations (MAF >5%), suggested that one haplotype is more frequent among cases than
controls (WH4, p=0.0227), with another haplotype showing a non-significant trend of over-
representation in cases (WH5, p=0.0883) (Table 5). Global estimation of haplotypes was
further confirmed using the PHASE 2.1.1 program, with concordant results (data not
shown).
A closer examination of these two haplotypes (WH4 and WH5) showed that the only
variation present on WH4 is the c.-242-110delAGTA variant, which on its own shows a
significant difference in frequency between both groups. Thus, this variant is likely to be
responsible of the association observed as any further breakdown of this haplotype using a
sliding window analysis shows that all combinations involving this variant display a

147
significant association (data not shown). As for the WH5 haplotype carrying the c l 197T/C
variant in exon 10, it does not show any significant difference in frequency between the
both groups and, despite its strong linkage disequilibrium with the adjacent variants, it
shows only a tendency towards significativity when present in combination with the major
alleles at all other positions (Table 5).

148
Discussion
Although the relevance of mutations in several DNA repair genes (BRCAl, BRCA2, TP53,
PTEN, PALB2, BR1P1) in breast cancer susceptibility have been well established, the
association of variants in other genes, potentially accounting for the remaining familial
clustering, is not well defined. For example, heterozygous carriers of deleterious mutations
in the NBN gene, in particular the Slavic founder mutation 657del5, has been associated
with a 2- to 3-fold increased risk of cancer [6]. However the impact of other NBN variants
on cancer risk is unclear. Nonetheless, studies have demonstrated an increased spontaneous
chromosomal instability in cells from heterozygous carriers of NBN mutations [7, 16]. In
addition, the presence of a specific pattern of gene expression involving pathways of DNA
repair and damage bypass, mitotic checkpoint and apoptosis was demonstrated, suggesting
that cells from these carriers may display a much less efficient DNA repair system [31]. In
this regard, a recent study by Someya et al. [32] showed a correlation between persistent
radiation-induced NBN foci and both chromosomal instability and sporadic breast cancer
risk. To address the possible implication of NBN sequence variants on breast cancer risk,
we took advantage of our resource of high-risk breast cancer families drawn from the
French Canadian population. In order to increase the statistical power of our investigation,
one affected individual from each family was selected for analysis [33], each thoroughly
screened for BRCAl and BRCA2 mutations or large genomic rearrangements in these genes
[21].
The majority of the coding variants identified in our cohort of breast cancer cases are
situated in the forkhead-associated (FHA) domain and the two BRCT domains, and these

149
domains have been demonstrated to be essential to NBN binding to the histone y-H2AX
[34, 35]. One of the rare variants identified in this study, p.Ilel71Val, was first described in
acute lymphoblastic leukemia (ALL) patients [36]. This variant has been previously
reported to be over-represented in some cancer cohorts [9, 10, 37, 38], and has also been
previously described at the homozygous state in a Japanese NBS patient affected with
aplastic anemia and genomic instability [39]. Interestingly, analysis of the patient's and her
father's lymphoblastoid cell lines demonstrated a higher frequency of spontaneous
chromosomal aberrations compared with healthy controls (6- and 4-fold increase,
respectively). These results suggest that the p.Ilel71Val variation may be deleterious, also
supported by the fact that this variant alters an amino acid which is conserved in species
such as the chicken and the fruitfly [39]. However, the impact of the p.Ilel71Val
substitution remains to be clarified as a large study, including cases from Germany and the
Republic of Belarus, did not find any association with breast cancer in those populations
[40].
Another rare variant, p.Arg215Trp, has been considered pathogenic in previous studies
based on its highly conserved position and the change introduced. Moreover, this variant
has been identified in monozygotic twins compound hétérozygotes for
657del5/p.Arg215Trp and affected with a severe form of NBS [41]. NibrinTrp215 appears to
affect the correct nibrin function, and cells carrying this variant shows delayed DNA DSB
rejoining [41]. Modélisation of the tandem BRCT domains suggests that the Arg215
residue is required for correct orientation of the BRCT domains and recognition of y-H2AX
[34]. In their experiment, nibrinTrp215 seems to partially interfere with nibrinArg215 activity
and may act in a co-dominant fashion. It is also interesting to note that although this variant

150
is relatively frequent in several studies, to our knowledge no NBS cases homozygous for
this variant have been reported.
Of the other rare variants present in our cohort of breast cancer cases, the p.Pro266Leu
variant was observed in one breast cancer case, and the p.Asp95Asn variant was found in
one breast cancer case and one control. Although these variants involve conserved residues,
their putative effect on protein function remains unclear. In a previous study on non-
Hodgkin lymphoma, both variants were detected only in healthy controls [42]. Regarding
the p.Asp95Asn variant first identified in an ALL patient [36], no association was found in
a study of larynx cancer [38] or ALL [37]. This variant was found in one out of 613 and
121 unselected and familial prostate cancer cases, respectively, but not in controls [43].
However, p.Asp95Asn is not predicted to be highly damaging as its expression in NBS
cells seems to have a similar activity to the wild type protein [42]. The only other non-
synonymous change identified in our cohort is the p.Glul85Gln common variant (MAF
>25%), which has been genotyped in several association studies. Although the majority of
the studies analyzing this variant found no association with cancer [12, 38, 42-44], some
studies reported an association with breast cancer [8], basal cell carcinoma in men [45] and
a recent meta-analysis of this variant in bladder cancer revealed a significant association
after controlling for potential bias [46]. In addition, Musak et al. [47] reported that this
variant may modulate the frequency of chromatid-type aberrations among tire plant
workers. However, our data do not allow us to confirm any positive association.
Among the non-coding variants, a possible association of the c.-242-110delAGTA variant
with an increased risk of breast cancer could be observed (OR 3.4 95% CI : 1.1-10.5). This
association was further confirmed by the estimation of the haplotype diversity in our

151
dataset, which highlighted the presence of the haplotype bearing the c.-242-110delAGTA
variant at a higher frequency among the breast cancer case subset. In silico prediction of
putative transcription factor binding sites indicated that of the four transcription factor
binding sites potentially affected by the deletion, some are predicted to involve proteins not
expressed in the breast. MTBF acts in the muscle regulation of the myostatin gene [48],
while CDX2 is mainly expressed in the gut although its ectopic expression was also
reported in cases of non-gastrointestinal carcinomas [49]. As for the transcription factor
NKX3-1, it is mainly expressed in the prostate although it is also found at low levels in the
mammary gland and breast tumors, and has been suggested to act as a haplo-insufficient
tumor suppressor in prostate cancer [50]. The fourth transcription factor potentially affected
is ATBF1 (also known as ZFHX3). ATBF1 was shown to suppress the expression of the
alpha-fetoprotein[51] and the MYB oncogene [52]. ATBF1 also cooperates with p53 to
activate the CDKN1A promoter and trigger cell cycle arrest [53]. ATBF1 is expressed in the
mammary gland and breast tumors (NCBI's UNIGENE data), and the expression of
ATBF1 mRNA was correlated with a better prognosis in 153 patients with invasive
carcinomas of the breast [54]. This might suggest that deregulation of genes controlled
through ATBF1 could have an important impact on breast cancer formation and
progression.
Luciférase reporter gene assay using the promoter sequence proximal to the ATG codon
indicated a similar level of expression of the c.-242-110delAGTA variant allele as
compared to the reference sequence construct in the breast cancer cell line MCF-7 and in
HeLa cells. However, a slight decrease in luciférase expression was observed in both
HEK293 and LNCaP cells, which might indicate that this variant could have an impact on

152
NBN expression in a cell type manner. The analysis of ATBF1 gene expression indicated
that this transcription factor is weakly expressed, in particular in the MCF-7 cells. This is
comparable to a recent study performed on a panel of 32 breast cancer cell lines, which
indicated that in 75% of these cell lines, ATBF1 is expressed at 50% or less relative to the
normal breast [55]. This could in rum explain the lack of effect observed for the luciférase
assay in the MCF-7 cell line. On the other hand, one could hypothesize that NBN (or
transcription factors affecting its expression) may be regulated following irradiation or
other genotoxic stress. Additional research will however be needed to address this
possibility, as well as binding experiments to confirm ATFB1 binding to this region of the
promoter. However, it is also important to keep in mind that the promoter sequence cloned
here, although supporting NBN expression, is unlikely to constitute the entire NBN
promoter. It is indeed plausible that transcription factors acting on the proximal sequence
might be influenced by other transcription factors acting further upstream. As such, despite
the lack of variation in expression induction found in MCF-7 by luciférase assay, it is
possible that the c.-242-110delAGTA variant could be in LD with another functional
variant located elsewhere in the promoter, as a high degree of LD is present across the
whole NBN gene. Unfortunately, the promoter variant could not be associated by LD with
another variant in the coding region of the gene, which would have allowed to directly
measure NBN allelic expression.

153
Conclusion
Our analysis suggests that the variant c.-242-l lOdelAGTA identified in our cohort of breast
cancer individuals and located in the promoter region of the NBN gene may be associated
with an increased risk of breast cancer. However, the functional analysis by luciférase
promoter assay does not support a causative effect of this variant in the breast cancer cell
line MCF-7, although a reduction of luciférase expression driven by the NBN promoter can
be observed in two other cell lines tested. As the NBN genomic region displays a high
degree of LD, we cannot rule out the effect of other variants located upstream of the region
analyzed here. Alternatively, the effect of this variant may be dependent upon induction by
a genotoxic stress such as irradiation. Although NBS is a rare syndrome, carriers of
deleterious alterations in this gene are more common and may display subtle manifestations
in cellular pathways predisposing M>W-mutated carriers to malignancy. Further analyses
will therefore be needed to ascertain the impact of rare variants and promoter variants of
NBN on breast cancer susceptibility in other populations.

154
Abbreviations AIR: ATM interacting region; ALL: Acute lymphoblastic leukemia; ATBF1: AT motif-
binding factor 1; BRCT: BRCAl C-terminal; CDX2: Caudal-type homeobox 2; CI:
Confidence interval; DSB: Double strand breaks; ESE: Exonic splicing enhancer; FHA:
Forkhead-associated; HWE: Hardy-Weinberg equilibrium; LD: Linkage disequilibrium;
MAF: Minor allele frequency; MIR: MRE11A interacting region; MTBF1: Muscle-specific
MT binding factor; NBS: Nijmegen breakage syndrome; NKX3-1: NK3 homeobox 1; OR:
Odds ratio; QRT-PCR: Quantitative real-time PCR; ZFHX3: Zinc finger homeobox 3.
Competing interests The authors declare that they have no competing interests.
Authors' contributions SD participated in the molecular analysis study, performed the reporter gene assays
experiments and bioinformatics analyses, and drafted the manuscript. CJB participated in
the molecular analysis study. YL provided technical assistance and helped draft the
manuscript. GO performed DNA and RNA extractions, cell maintenance and participated
in the luciférase assays experiments. FD conceived the study, its design and coordination,
and authored the final version of the manuscript. All authors read and approved the final
manuscript.

155
Acknowledgements The authors would like to thank all individuals and families who participated in this study.
We thank C. Brousseau, Dr. S. Délos, M.-A. Lajoie, P. Léger, J. Rhéaume, C. Samson, M.
Tranchant, Dr. M. Dumont, G. Leblanc, H. Malouin, N. Bolduc, A. MacMillan and T.
Babineau of the Cancer Genomics Laboratory for genetic counselling, sample management
and mutation screening. We also thank F. Guénard for advices regarding luciférase assays,
M. Ouellet and A.-M. Bureau-Blouin for skillful technical help and Dr. D. Labuda and C.
Moreau at the Centre de Cancérologie Charles Bruneau of Ste-Justine Hospital for help
with control DNA samples.
This work was supported by the Canadian Institutes of Health Research (CIHR) and
Institute of Cancer and Institute of Gender and Health for the INHERIT BRCAs research
program, the Fond de la Recherche en Santé du Québec (FRSQ)/Réseau de Médecine
Génétique Appliquée (RMGA), the Canadian Breast Cancer Research Alliance (CBCRA)
and the CURE Foundation. S.D. holds studentships from Fondation René Bussières and
Fondation Desjardins, C.J.B. holds a Frederick Banting and Charles Best Canada Graduate
Scholarships - Master's Award studentship from CIHR, and F.D. is a recipient of a
chercheur-boursier from the Fonds de la Recherche en Santé du Québec (FRSQ) and
Research Career Award in the Health Sciences from CIHR/R&D Health Research
Foundation.

156
Appendix
Other members of INHERIT BRCAs involved in this study:
Paul Bessette: Service de gynécologie, Centre Hospitalier Universitaire de Sherbrooke,
Fleurimont, QC, Canada.
Jocelyne Chiquette: Clinique des maladies du sein Deschênes-Fabia, Hôpital du Saint-
Sacrement, Québec, QC, Canada.
Rachel Laframboise: Service de médecine génétique, CHUQ, Pavillon CHUL, Québec,
QC, Canada.
Jean Lépine: Centre Hospitalier régional de Rimouski, Rimouski, QC, Canada.
Bernard Lesperance, Roxane Pichette: Service d'hémato-oncologie, Hôpital du Sacré-
Cœur, Montréal, QC, Canada.
Marie Plante: Service de gynécologie, CHUQ, L'Hôtel-Dieu de Québec, Québec, Canada.
Jacques Simard: Director and instigator of the integrated research program INHERIT
BRCAs, and chairholder of the Canada Research Chair in Oncogenetics. Cancer Genomics
Laboratory, Oncology and Molecular Endocrinology Research Centre, Centre Hospitalier
Universitaire de Québec and Laval University, Québec, Canada.

157
References
1. Stratton MR, Rahman N: The emerging landscape of breast cancer susceptibility. Nat Genet 2008, 40(1): 17-22.
2. Williams RS, Williams JS, Tainer JA: Mrell-Rad50-Nbsl is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol 2007, 85(4):509-520.
3. Durocher F, Guénard F, Desjardins S, Ouellette G, Labrie Y: Inherited susceptibility to breast cancer: accomplishments and challenges. In: Molecular genetics of cancer. Edited by Sinnett D. Kerala: Research Signpost; 2005: 9-93.
4. Seemanova E: An increased risk for malignant neoplasms in hétérozygotes for a syndrome of microcephaly, normal intelligence, growth retardation, remarkable facies, immunodeficiency and chromosomal instability. Mutat Res 1990, 238(3):321-324.
5. Howlett NG, Scuric Z, D'Andrea AD, Schiestl RH: Impaired DNA double strand break repair in cells from Nijmegen breakage syndrome patients. DNA Repair (Amst) 2006, 5(2):251-257.
6. Bogdanova N, Feshchenko S, Schurmann P, Waltes R, Wieland B, Hillemanns P, Rogov YI, Dammann O, Bremer M, Karstens JH et al: Nijmegen Breakage Syndrome mutations and risk of breast cancer. Int J Cancer 2008,122(4):802-806.
7. Heikkinen K, Rapakko K, Karppinen SM, Erkko H, Knuutila S, Lundan T, Mannermaa A, Borresen-Dale AL, Borg A, Barkardottir RB et al: RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis 2006, 27(8):1593-1599.
8. Hsu HM, Wang HC, Chen ST, Hsu GC, Shen CY, Yu JC: Breast cancer risk is associated with the genes encoding the DNA double-strand break repair Mrell/Rad50/Nbsl complex. Cancer Epidemiol Biomarkers Prev 2007', 16(10):2024-2032.
9. Nowak J, Mosor M, Ziolkowska I, Wierzbicka M, Pemak-Schwarz M, Przyborska M, Roznowski K, Plawski A, Slomski R, Januszkiewicz D: Heterozygous carriers of the 1171V mutation of the NBS1 gene have a significantly increased risk of solid malignant tumours. Eur J Cancer 2008, 44(4):627-630.
10. Roznowski K, Januszkiewicz-Lewandowska D, Mosor M, Pernak M, Litwiniuk M, Nowak J: I171V germline mutation in the NBS1 gene significantly increases risk of breast cancer. Breast Cancer Res Treat 2008,110(2):343-348.
11. Seemanova E, Jarolim P, Seeman P, Varon R, Digweed M, Swift M, Sperling K: Cancer risk of hétérozygotes with the NBN founder mutation. J Natl Cancer Inst 2007, 99(24): 1875-1880.
12. Forsti A, Angelini S, Festa F, Sanyal S, Zhang Z, Grzybowska E, Pamula J, Pekala W, Zientek H, Hemminki K et al: Single nucleotide polymorphisms in breast cancer. Oncol Rep 2004,11(4):917-922.

158
13. Millikan RC, Player JS, Decotret AR, Tse CK, Keku T: Polymorphisms in DNA repair genes, medical exposure to ionizing radiation, and breast cancer risk. Cancer Epidemiol Biomarkers Prev 2005, 14(10):2326-2334.
14. Dumon-Jones V, Frappart PO, Tong WM, Sajithlal G, Hulla W, Schmid G, Herceg Z, Digweed M, Wang ZQ: Nbn heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res 2003, 63(21):7263-7269.
15. Stumm M, Neubauer S, Keindorff S, Wegner RD, Wieacker P, Sauer R: High frequency of spontaneous translocations revealed by FISH in cells from patients with the cancer-prone syndromes ataxia telangiectasia and Nijmegen breakage syndrome. Cytogenet Cell Genet 2001, 92(3-4):186-191.
16. Tanzanella C, Antoccia A, Spadoni E, di Masi A, Pecile V, Demori E, Varon R, Marseglia GL, Tiepolo L, Maraschio P: Chromosome instability and nibrin protein variants in NBS hétérozygotes. Eur J Hum Genet 2003, ll(4):297-303.
17. Neubauer S, Arutyunyan R, Stumm M, Dork T, Bendix R, Bremer M, Varon R, Sauer R, Gebhart E: Radiosensitivity of ataxia telangiectasia and Nijmegen breakage syndrome homozygotes and hétérozygotes as determined by three-color FISH chromosome painting. Radiât Res 2002,157(3):312-321.
18. Ferguson DO, Alt FW: DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene 2001, 20(40):5572-5579.
19. Pierce AJ, Stark JM, Araujo FD, Moynahan ME, Berwick M, Jasin M: Double-strand breaks and tumorigenesis. Trends Cell Biol 2001,11(1 l):S52-59.
20. Fodde R, Smits R: Cancer biology. A matter of dosage. Science 2002, 298(5594):761-763.
21. Simard J, Dumont M, Moisan AM, Gaborieau V, Malouin H, Durocher F, Chiquette J, Plante M, Avard D, Bessette P et al: Evaluation of BRCAl and BRCA2 mutation prevalence, risk prediction models and a multistep testing approach in French-Canadian families with high risk of breast and ovarian cancer. J Med Genet 2007, 44(2): 107-121.
22. Durocher F, Labrie Y, Soucy P, Sinilnikova O, Labuda D, Bessette P, Chiquette J, Laframboise R, Lepine J, Lesperance B et al: Mutation analysis and characterization of ATR sequence variants in breast cancer cases from high-risk French Canadian breast/ovarian cancer families. BMC Cancer 2006, 6:230.
23. Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR: ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res 2003, 31(13):3568-3571.
24. Reese MG, Eeckman FH, Kulp D, Haussier D: Improved splice site detection in Genie. J Comput Biol 1997, 4(3):311-323.
25. Shapiro MB, Senapathy P: RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 1987,15(17):7155-7174.
26. Guenard F, Labrie Y, Ouellette G, Joly Beauparlant C, Simard J, Durocher F: Mutational analysis of the breast cancer susceptibility gene BRIP1 /BACH1/FANCJ in high-risk non-BRCAl/BRCA2 breast cancer families. J Hum Genet 2008, 53(7):579-591.
27. Purcell S, Daly MJ, Sham PC: WHAP: haplotype-based association analysis. Bioinformatics 2007, 23(2):255-256.

159
28. Ivanga M, Labrie Y, Calvo E, Belleau P, Martel C, Pelletier G, Morissette J, Labrie F, Durocher F: Fine temporal analysis of DHT transcriptional modulation of the ATM/Gadd45g signaling pathways in the mouse uterus. Mol Reprod Dev 2009, 76(3):278-288.
29. Takai K, Sakamoto S, Sakai T, Yasunaga J, Komatsu K, Matsuoka M: A potential link between alternative splicing of the NBS1 gene and DNA damage/environmental stress. Radiât Res 2008, 170(l):33-40.
30. Varon R, Dutrannoy V, Weikert G, Tanzarella C, Antoccia A, Stockl L, Spadoni E, Kruger LA, di Masi A, Sperling K et al: Mild Nijmegen breakage syndrome phenotype due to alternative splicing. Hum Mol Genet 2006, 15(5):679-689.
31. Cheung VG, Ewens WJ: Heterozygous carriers of Nijmegen Breakage Syndrome have a distinct gene expression phenotype. Genome Res 2006, 16(8):973-979.
32. Someya M, Sakata K, Tauchi H, Matsumoto Y, Nakamura A, Komatsu K, Hareyama M: Association of ionizing radiation-induced foci of NBS1 with chromosomal instability and breast cancer susceptibility. Radiât Res 2006, 166(4):575-582.
33. Antoniou AC, Easton DF: Polygenic inheritance of breast cancer: Implications for design of association studies. Genet Epidemiol 2003, 25(3): 190-202.
34. di Masi A, Viganotti M, Polticelli F, Ascenzi P, Tanzarella C, Antoccia A: The R215W mutation in NBS1 impairs gamma-H2AX binding and affects DNA repair: molecular bases for the severe phenotype of 657del5/R215W Nijmegen breakage syndrome patients. Biochem Biophys Res Commun 2008, 369(3):835-840.
35. Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, Matsuura S, Kobayashi T, Tamai K, Tanimoto K, Komatsu K: NBS1 localizes to gam mall 2.AX foci through interaction with the FHA/BRCT domain. Curr Biol 2002, 12(21): 1846-1851.
36. Varon R, Reis A, Henze G, von Einsiedel HG, Sperling K, Seeger K: Mutations in the Nijmegen Breakage Syndrome gene (NBS1) in childhood acute lymphoblastic leukemia (ALL). Cancer Res 2001, 61(9):3570-3572.
37. Mosor M, Ziolkowska I, Pemak-Schwarz M, Januszkiewicz-Lewandowska D, Nowak J: Association of the heterozygous germline 1171V mutation of the NBS1 gene with childhood acute lymphoblastic leukemia. Leukemia 2006, 20(8): 1454-1456.
38. Ziolkowska I, Mosor M, Wierzbicka M, Rydzanicz M, Pemak-Schwarz M, Nowak J: Increased risk of larynx cancer in heterozygous carriers of the 1171V mutation of the NBS1 gene. Cancer Sci 2007, 98(11): 1701-1705.
39. Shimada H, Shimizu K, Mimaki S, Sakiyama T, Mori T, Shimasaki N, Yokota J, Nakachi K, Ohta T, Ohki M: First case of aplastic anemia in a Japanese child with a homozygous missense mutation in the NBS1 gene (1171V) associated with genomic instability. Hum Genet 2004,115(5):372-376.
40. Bogdanova N, Schurmann P, Waltes R, Feshchenko S, Zalutsky IV, Bremer M, Dork T: NBS1 variant 1171V and breast cancer risk. Breast Cancer Res Treat 2008,112(l):75-79.

160
41. Seemanova E, Sperling K, Neitzel H, Varon R, Hadac J, Butova O, Schrock E, Seeman P, Digweed M: Nijmegen breakage syndrome (NBS) with neurological abnormalities and without chromosomal instability. J Med Genet 2006, 43(3):218-224.
42. Cerosaletti KM, Morrison VA, Sabath DE, Willerford DM, Concannon P: Mutations and molecular variants of the NBS1 gene in non-Hodgkin lymphoma. Genes Chromosomes Cancer 2002, 35(3):282-286.
43. Hebbring SJ, Fredriksson H, White KA, Maier C, Ewing C, McDonnell SK, Jacobsen S J, Cerhan J, Schaid DJ, Ikonen T et al: Role of the Nijmegen breakage syndrome 1 gene in familial and sporadic prostate cancer. Cancer Epidemiol Biomarkers Prev 2006,15(5):935-938.
44. Figueroa JD, Malats N, Rothman N, Real FX, Silverman D, Kogevinas M, Chanock S, Yeager M, Welch R, Dosemeci M et al: Evaluation of genetic variation in the double-strand break repair pathway and bladder cancer risk. Carcinogenesis 2007, 28(8): 1788-1793.
45. Thirumaran RK, Bermejo JL, Rudnai P, Gurzau E, Koppova K, Goessler W, Vahter M, Leonardi GS, Clemens F, Fletcher T et al: Single nucleotide polymorphisms in DNA repair genes and basal cell carcinoma of skin. Carcinogenesis 2006, 27(8): 1676-1681.
46. Vineis P, Manuguerra M, Kavvoura FK, Guarrera S, Allione A, Rosa F, Di Gregorio A, Polidoro S, Saletta F, Ioannidis JP et al: A field synopsis on Iow-penetrance variants in DNA repair genes and cancer susceptibility. J Natl Cancer Inst 2009,101(l):24-36.
47. Musak L, Soucek P, Vodickova L, Naccarati A, Halasova E, Polakova V, Slyskova J, Susova S, Buchancova J, Smerhovsky Z et al: Chromosomal aberrations in tire plant workers and interaction with polymorphisms of biotransformation and DNA repair genes. Mutat Res 2008, 641(l-2):36-42.
48. Du R, An X, Chen Y, Qin J: Functional analysis of the Myostatin gene promoter in sheep. Sci China C Life Sci 2007, 50(5):648-654.
49. Guo RJ, Suh ER, Lynch JP: The role of Cdx proteins in intestinal development and cancer. Cancer Biol Ther 2004, 3(7):593-601.
50. Mogal AP, van der Meer R, Crooke PS, Abdulkadir SA: Haploinsufficient prostate tumor suppression by Nkx3.1: a role for chromatin accessibility in dosage-sensitive gene regulation. J Biol Chem 2007, 282(35):25790-25800.
51. Morinaga T, Yasuda H, Hashimoto T, Higashio K, Tamaoki T: A human alpha-fetoprotein enhancer-binding protein, ATBF1, contains four homeodomains and seventeen zinc fingers. Mol Cell Biol 1991,11(12):6041-6049.
52. Kaspar P, Dvorakova M, Kralova J, Pajer P, Kozmik Z, Dvorak M: Myb-interacting protein, ATBF1, represses transcriptional activity of Myb oncoprotein. J Biol Chem 1999, 274(20): 14422-14428.
53. Miura Y, Kataoka H, Joh T, Tada T, Asai K, Nakanishi M, Okada N, Okada H: Susceptibility to killer T cells of gastric cancer cells enhanced by Mitomycin-C involves induction of ATBF1 and activation of p21 (Wafl/Cipl) promoter. Microbiol Immunol 2004, 48(2): 137-145.
54. Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K, Hamaguchi M, Kawaguchi M, Miura Y, Iwase H: ATBFl-a messenger RNA expression is

161
correlated with better prognosis in breast cancer. Clin Cancer Res 2005, 11(1): 193-198.
55. Sun X, Zhou Y, Otto KB, Wang M, Chen C, Zhou W, Subramanian K, Vertino PM, Dong JT: Infrequent mutation of ATBF1 in human breast cancer. J Cancer Res Clin Oncol 2007,133(2):103-105.

162
Figure legends
Figurel: Schematic representation of the NBN gene on chromosome 8q21.
The upper part shows variants identified in the NBN gene. Light green boxes refer to SNPs
with MAF <5% in the cases set, and variants name are indicated according to the reference
sequence NM002485.4. Corresponding coding variations are indicated relative to the
protein structure in the bottom part of the figure. Light blue boxes refer to synonymous
variations. FHA: Forkhead-associated domain. BRCT: BRCAl C-terminal domain. MIR:
MRE11 interacting region. AIR: ATM interacting region. Yellow dots: acetylated lysine
residues (K208, K233, K334, K441, K504, K544, K665, K690, K698 and K715). Red dots:
phosphorylated serine residues (S278 and S343).
Figure 2: In silico analysis of the effect of coding variants on putative exonic splicing
enhancer (ESE) motifs by the FSE Under program.
Only scores for predicted motifs potentially abolished by exonic variants are shown. Light
gray: motif matrix scores for consensus sequences. Dark gray: motif matrix scores for
variant sequences. The name of the SR protein potentially involved is indicated for each
score pair. Doted line indicates the default threshold used by ESEfinder for the
corresponding motif.
Figure 3: Effect of the c.-242-110delAGTA deletion on NBN promoter activity.
Panel A shows the results of the in silico predictions of transcription factors binding sites
affected by the c.-242-110delAGTA variant obtained from the Matlnspector program.
Panel B shows the schematic representation of the two reporter gene constructs containing
the NBN promoter used in this study. The positions relative to the nucleotide A of the NBN
reference sequence ATG codon are indicated. Panel C shows the results of the luciférase

163
reporter assay. MCF-7, LNCaP, HeLa and HEK293 cells were transiently co-transfected
with the Renilla reporter plasmid (pRL) as a transfection control. Each data represents
mean ± standard deviation of triplicates. Data are shown as fold of induction compared to
the activity of cells transfected with the empty pGL3-basic luciférase reporter vector.
*p=0.0245 **p=0.0181
Figure 4: Linkage disequilibrium across the NBN gene and neighbouring
chromosomal region.
Upper part shows linkage disequilibrium (LD) results (r2 and |D'|) obtained from the
control genotypes data used in our study and their relative position along -/WW sequence. In
the bottom part of the figure are indicated r2 data from the International HapMap project
(release 21a) for the genomic region encompassing NBN. The intensity of the box color is
proportional to the strength of LD. r2=0 in white; 0<r2<l in shades of pink/red; r - \ in
bright red.
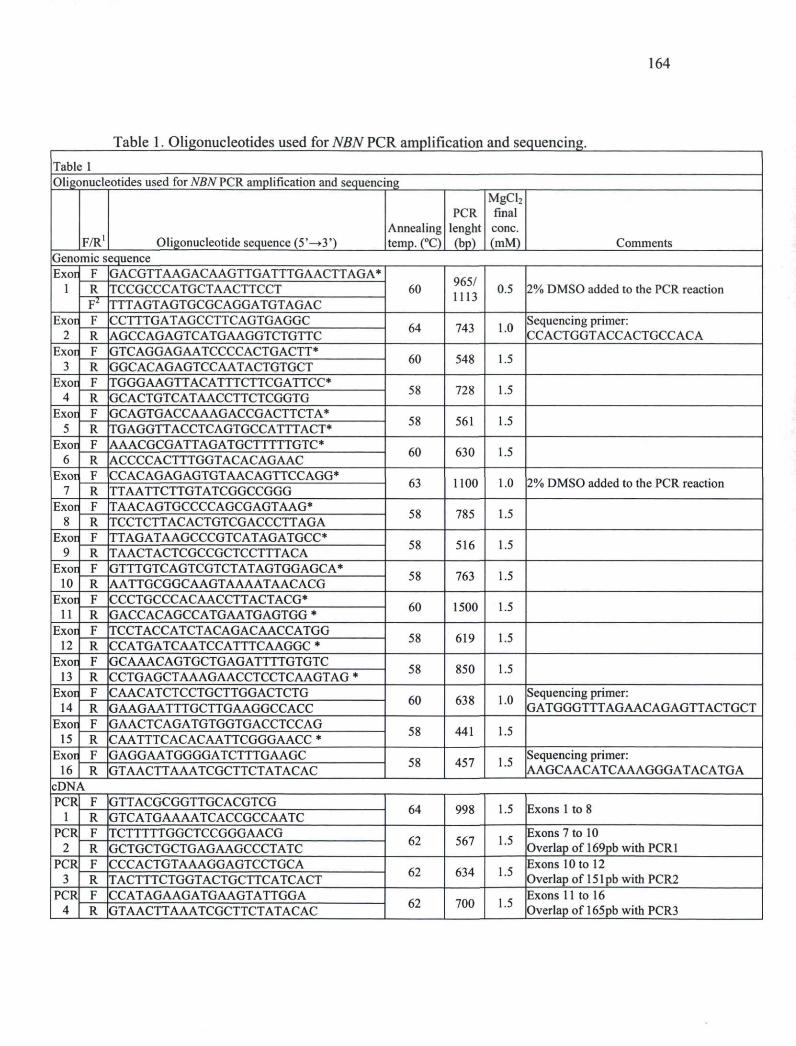
164
Table 1. Oligonucleotides used for NBN PCR amplification and sequencing. Table 1 Oligonucleotides used for NBNPCR amplification and sequencing
F/R1 Oligonucleotide sequence (5'—+3') Annealing temp. (°C)
PCR lenght (bp)
MgCI2 final cone. (mM) Comments
Genomic sequence Exon
1 F GACGTTAAGACAAGTTGATTTGAACTTAGA*
60 965/ 1113 0.5 2% DMSO added to the PCR reaction
Exon 1 R TCCGCCCATGCTAACTTCCT 60 965/
1113 0.5 2% DMSO added to the PCR reaction Exon
1 F2 TTTAGTAGTGCGCAGGATGTAGAC
60 965/ 1113 0.5 2% DMSO added to the PCR reaction
Exor 2
F CCTTTGATAGCCTTCAGTGAGGC 64 743 1.0 Sequencing primer: CCACTGGTACCACTGCCACA
Exor 2 R AGCCAGAGTCATGAAGGTCTGTTC 64 743 1.0 Sequencing primer:
CCACTGGTACCACTGCCACA Exon
3 F GTCAGGAGAATCCCCACTGACTT* 60 548 1.5 Exon
3 R GGCACAGAGTCCAATACTGTGCT 60 548 1.5
Exon 4
F TGGGAAGTTACATTTCTTCGATTCC* 58 728 1.5 Exon 4 R GCACTGTCATAACCTTCTCGGTG 58 728 1.5
Exon 5
F GCAGTGACCAAAGACCGACTTCTA* 58 561 1.5 Exon 5 R TGAGGTTACCTCAGTGCCATTTACT* 58 561 1.5
Exon 6
F AAACGCGATTAGATGCTTTTTGTC* 60 630 1.5 Exon 6 R ACCCCACTTTGGTACACAGAAC 60 630 1.5
Exon 7
F CCACAGAGAGTGTAACAGTTCCAGG* 63 1100 1.0 2% DMSO added to the PCR reaction Exon 7 R TTAATTCTTGTATCGGCCGGG 63 1100 1.0 2% DMSO added to the PCR reaction
Exor 8
F TAACAGTGCCCCAGCGAGTAAG* 58 785 1.5 Exor 8 R TCCTCTTACACTGTCGACCCTTAGA 58 785 1.5
Exon 9
F TTAGATAAGCCCGTCATAGATGCC* 58 516 1.5 Exon 9 R TAACTACTCGCCGCTCCTTTACA 58 516 1.5
Exon 10
F GTTTGTCAGTCGTCTATAGTGGAGCA* 58 763 1.5 Exon 10 R AATTGCGGCAAGTAAAATAACACG 58 763 1.5
Exon 11
F CCCTGCCCACAACCTTACTACG* 60 1500 1.5 Exon 11 R GACCACAGCCATGAATGAGTGG * 60 1500 1.5
Exon 12
F TCCTACCATCTACAGACAACCATGG 58 619 1.5 Exon 12 R CCATGATCAATCCATTTCAAGGC * 58 619 1.5
Exon 13
F GCAAACAGTGCTGAGATTTTGTGTC 58 850 1.5 Exon 13 R CCTGAGCTAAAGAACCTCCTCAAGTAG * 58 850 1.5
Exon 14
F CAACATCTCCTGCTTGGACTCTG 60 638 1.0 Sequencing primer: GATGGGTTTAGAACAGAGTTACTGCT
Exon 14 R GAAGAATTTGCTTGAAGGCCACC 60 638 1.0 Sequencing primer:
GATGGGTTTAGAACAGAGTTACTGCT Exon
15 F GAACTCAGATGTGGTGACCTCCAG 58 441 1.5 Exon
15 R CAATTTCACACAATTCGGGAACC * 58 441 1.5
Exon 16
F GAGGAATGGGGATCTTTGAAGC 58 457 1.5 Sequencing primer: AAGCAACATCAAAGGGATACATGA
Exon 16 R GTAACTTAAATCGCTTCTATACAC 58 457 1.5 Sequencing primer:
AAGCAACATCAAAGGGATACATGA cDNA PCR
1 F GTTACGCGGTTGCACGTCG 64 998 1.5 Exons 1 to 8 PCR
1 R GTCATGAAAATCACCGCCAATC 64 998 1.5 Exons 1 to 8
PCR 2
F TCTTTTTGGCTCCGGGAACG 62 567 1.5 Exons 7 to 10 Overlap of 169pb with PCR1
PCR 2 R GCTGCTGCTGAGAAGCCCTATC 62 567 1.5 Exons 7 to 10
Overlap of 169pb with PCR1 PCR
3 F CCCACTGTAAAGGAGTCCTGCA 62 634 1.5 Exons 10 to 12
Overlap of 151pb with PCR2 PCR
3 R TACTTTCTGGTACTGCTTCATCACT 62 634 1.5 Exons 10 to 12 Overlap of 151pb with PCR2
PCR 4
F CCATAGAAGATGAAGTATTGGA 62 700 1.5 Exons 11 to 16 Overlap of 165pb with PCR3
PCR 4 R GTAACTTAAATCGCTTCTATACAC 62 700 1.5 Exons 11 to 16
Overlap of 165pb with PCR3

165
Promoter cloning
! „ •
F GACGACGCTAGCGACGTTAAGACAAGTTGA
62 515 1.0 2% DMSO added to the PCR reaction Added restriction sites are underlined
! „ •
F TTTGAACTTAGA 62 515 1.0 2% DMSO added to the PCR reaction
Added restriction sites are underlined
! „ • R GACGACAAGCTTATCGGTCCGGCTCCTCAG
62 515 1.0 2% DMSO added to the PCR reaction Added restriction sites are underlined
! „ • R GGCTG
, JT-^ , T - . 1 . . .
62 515 1.0 2% DMSO added to the PCR reaction Added restriction sites are underlined
Primer direction: forward (F) or reverse (R) Alternative primer Oligonucleotides used as sequencing primers are marked with an (*)
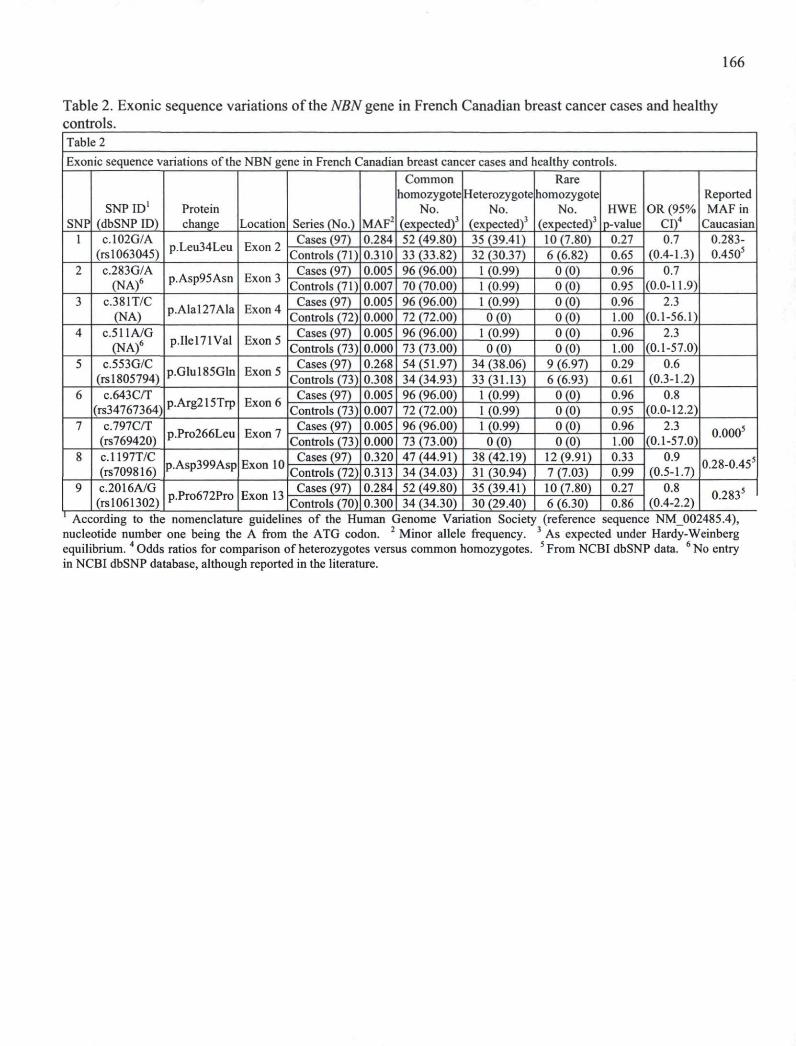
166
Table 2. Exonic sequence variations of the NBN gene in French Canadian breast cancer cases and healthy controls. Table 2 Exonic sequence variations of the NBN gene in French Canadian breast cancer cases and healthy controls.
SNP SNP ID1
(dbSNP ID) Protein change Location Series (No.) MAF2
Common homozygote
No. (expected)3
Hétérozygote No.
(expected)3
Rare homozygote
No. (expected)3
HWE p-value
OR (95% CI)4
Reported MAF in
Caucasian 1 C.102G/A
(rsl 063045) p.Leu34Leu Exon 2 Cases (97) 0.284 52 (49.80) 35(39.41) 10(7.80) 0.27 0.7 (0.4-1.3)
0.283-0.4505
1 C.102G/A (rsl 063045) p.Leu34Leu Exon 2 Controls (71) 0.310 33 (33.82) 32 (30.37) 6 (6.82) 0.65
0.7 (0.4-1.3)
0.283-0.4505
2 C.283G/A (NA)6 p.Asp95Asn Exon 3 Cases (97) 0.005 96 (96.00) 1 (0.99) 0(0) 0.96 0.7
(0.0-11.9) 2 C.283G/A
(NA)6 p.Asp95Asn Exon 3 Controls (71) 0.007 70 (70.00) 1 (0.99) 0(0) 0.95 0.7
(0.0-11.9) 3 C.381T/C
(NA) p.Alal27Ala Exon 4 Cases (97) 0.005 96 (96.00) 1 (0.99) 0(0) 0.96 2.3 (0.1-56.1)
3 C.381T/C (NA) p.Alal27Ala Exon 4 Controls (72) 0.000 72 (72.00) 0(0) 0(0) 1.00
2.3 (0.1-56.1)
4 C.511A/G (NA)6 p.Ilel71Val Exon 5 Cases (97) 0.005 96 (96.00) 1 (0.99) 0(0) 0.96 2.3
(0.1-57.0) 4 C.511A/G
(NA)6 p.Ilel71Val Exon 5 Controls (73) 0.000 73 (73.00) 0(0) 0(0) 1.00 2.3
(0.1-57.0) 5 C.553G/C
(rsl 805794) p.Glul85Gln Exon 5 Cases (97) 0.268 54(51.97) 34 (38.06) 9 (6.97) 0.29 0.6 (0.3-1.2)
5 C.553G/C (rsl 805794) p.Glul85Gln Exon 5 Controls (73) 0.308 34 (34.93) 33(31.13) 6 (6.93) 0.61
0.6 (0.3-1.2)
6 C.643C/T (rs34767364) p.Arg215Trp Exon 6 Cases (97) 0.005 96 (96.00) 1 (0.99) 0(0) 0.96 0.8
(0.0-12.2) 6 C.643C/T
(rs34767364) p.Arg215Trp Exon 6 Controls (73) 0.007 72 (72.00) 1 (0.99) 0(0) 0.95 0.8
(0.0-12.2) 7 C.797C/T
(rs769420) p.Pro266Leu Exon 7 Cases (97) 0.005 96 (96.00) 1 (0.99) 0(0) 0.96 2.3 (0.1-57.0) o.ooo5 7 C.797C/T
(rs769420) p.Pro266Leu Exon 7 Controls (73) 0.000 73 (73.00) 0(0) 0(0) 1.00 2.3
(0.1-57.0) o.ooo5
8 C.1197T/C (rs709816) p.Asp399Asp Exon 10 Cases (97) 0.320 47 (44.91) 38 (42.19) 12(9.91) 0.33 0.9
(0.5-1.7) 0.28-0.455 8 C.1197T/C (rs709816) p.Asp399Asp Exon 10 Controls (72) 0.313 34 (34.03) 31 (30.94) 7 (7.03) 0.99
0.9 (0.5-1.7) 0.28-0.455
9 C.2016A/G (rsl 061302) p.Pro672Pro Exon 13 Cases (97) 0.284 52 (49.80) 35 (39.41) 10(7.80) 0.27 0.8
(0.4-2.2) 0.2835 9 C.2016A/G (rsl 061302) p.Pro672Pro Exon 13 Controls (70) 0.300 34 (34.30) 30 (29.40) 6 (6.30) 0.86
0.8 (0.4-2.2) 0.2835
According to the nomenclature guidelines of the Human Genome Variation Society (reference sequence NM002485.4), nucleotide number one being the A from the ATG codon. Minor allele frequency, equilibrium. 4Odds ratios for comparison of hétérozygotes versus common homozygotes, in NCBI dbSNP database, although reported in the literature.
As expected under Hardy-Weinberg 5 From NCBI dbSNP data. 6 No entry
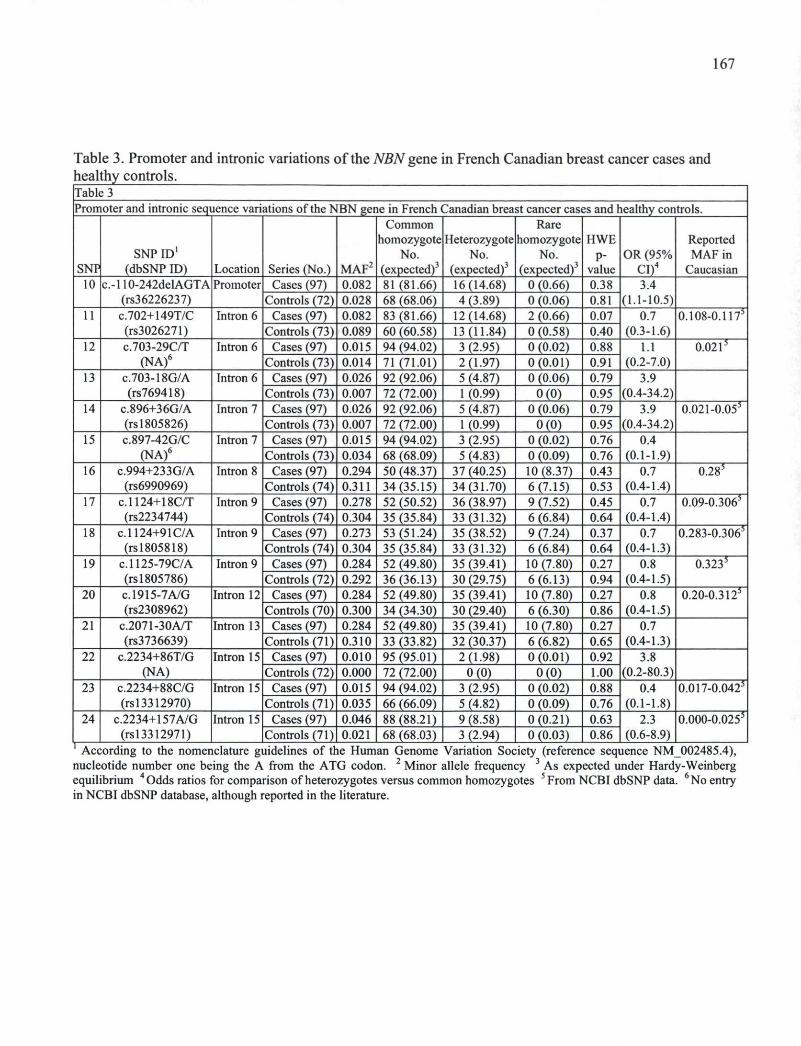
167
Table 3. Promoter and intronic variations of the NBN gene îealthy controls.
in French Canadian breast cancer cases and
Table 3 Promoter and intronic sequence variations of the NBN gene in French Canadian breast cancer cases and healthy controls.
SNP SNP ID1
(dbSNP ID) Location Series (No.) MAF2
Common homozygote
No. (expected)3
Hétérozygote No.
(expected)3
Rare homozygote
No. (expected)3
HWE P-
value OR (95%
CI)4
Reported MAF in
Caucasian 10 c.-110-242delAGTA
(rs36226237) Promoter Cases (97) 0.082 81 (81.66) 16(14.68) 0 (0.66) 0.38 3.4
(1.1-10.5) 10 c.-110-242delAGTA
(rs36226237) Promoter
Controls (72) 0.028 68 (68.06) 4(3.89) 0 (0.06) 0.81 3.4
(1.1-10.5) 11 C.702+149T/C
(rs3026271) Intron 6 Cases (97) 0.082 83(81.66) 12(14.68) 2 (0.66) 0.07 0.7
(0.3-1.6) 0.108-0.1175 11 C.702+149T/C
(rs3026271) Intron 6
Controls (73) 0.089 60 (60.58) 13(11.84) 0 (0.58) 0.40 0.7
(0.3-1.6) 0.108-0.1175
12 C.703-29C/T (NA)6
Intron 6 Cases (97) 0.015 94 (94.02) 3 (2.95) 0 (0.02) 0.88 1.1 (0.2-7.0)
0.021' 12 C.703-29C/T (NA)6
Intron 6 Controls (73) 0.014 71 (71.01) 2(1.97) 0(0.01) 0.91
1.1 (0.2-7.0)
0.021'
13 C.703-18G/A (rs769418)
Intron 6 Cases (97) 0.026 92 (92.06) 5 (4.87) 0 (0.06) 0.79 3.9 (0.4-34.2)
13 C.703-18G/A (rs769418)
Intron 6 Controls (73) 0.007 72 (72.00) 1 (0.99) 0(0) 0.95
3.9 (0.4-34.2)
14 C.896+36G/A (rsl 805826)
Intron 7 Cases (97) 0.026 92 (92.06) 5 (4.87) 0 (0.06) 0.79 3.9 (0.4-34.2)
0.021-0.055 14 C.896+36G/A (rsl 805826)
Intron 7 Controls (73) 0.007 72 (72.00) 1 (0.99) 0(0) 0.95
3.9 (0.4-34.2)
0.021-0.055
15 C.897-42G/C (NA)6
Intron 7 Cases (97) 0.015 94 (94.02) 3 (2.95) 0 (0.02) 0.76 0.4 (0.1-1.9)
15 C.897-42G/C (NA)6
Intron 7 Controls (73) 0.034 68 (68.09) 5 (4.83) 0 (0.09) 0.76
0.4 (0.1-1.9)
16 C.994+233G/A (rs6990969)
Intron 8 Cases (97) 0.294 50 (48.37) 37 (40.25) 10(8.37) 0.43 0.7 (0.4-1.4)
0.285 16 C.994+233G/A (rs6990969)
Intron 8 Controls (74) 0.311 34(35.15) 34(31.70) 6(7.15) 0.53
0.7 (0.4-1.4)
0.285
17 C.1124+18C/T (rs2234744)
Intron 9 Cases (97) 0.278 52 (50.52) 36 (38.97) 9 (7.52) 0.45 0.7 (0.4-1.4)
0.09-0.3065 17 C.1124+18C/T (rs2234744)
Intron 9 Controls (74) 0.304 35 (35.84) 33(31.32) 6 (6.84) 0.64
0.7 (0.4-1.4)
0.09-0.3065
18 C.1124+91C/A (rsl 805818)
Intron 9 Cases (97) 0.273 53(51.24) 35 (38.52) 9 (7.24) 0.37 0.7 (0.4-1.3)
0.283-0.3065 18 C.1124+91C/A (rsl 805818)
Intron 9 Controls (74) 0.304 35 (35.84) 33(31.32) 6 (6.84) 0.64
0.7 (0.4-1.3)
0.283-0.3065
19 C.1125-79C/A (rsl 805786)
Intron 9 Cases (97) 0.284 52 (49.80) 35(39.41) 10(7.80) 0.27 0.8 (0.4-1.5)
0.3235 19 C.1125-79C/A (rsl 805786)
Intron 9 Controls (72) 0.292 36(36.13) 30 (29.75) 6(6.13) 0.94
0.8 (0.4-1.5)
0.3235
20 C.1915-7A/G (rs2308962)
Intron 12 Cases (97) 0.284 52 (49.80) 35(39.41) 10(7.80) 0.27 0.8 (0.4-1.5)
0.20-0.3125 20 C.1915-7A/G (rs2308962)
Intron 12 Controls (70) 0.300 34 (34.30) 30 (29.40) 6 (6.30) 0.86
0.8 (0.4-1.5)
0.20-0.3125
21 C.2071-30A/T (rs3736639)
Intron 13 Cases (97) 0.284 52 (49.80) 35(39.41) 10(7.80) 0.27 0.7 (0.4-1.3)
21 C.2071-30A/T (rs3736639)
Intron 13 Controls (71) 0.310 33 (33.82) 32 (30.37) 6 (6.82) 0.65
0.7 (0.4-1.3)
22 C.2234+86T/G (NA)
Intron 15 Cases (97) 0.010 95 (95.01) 2(1.98) 0(0.01) 0.92 3.8 (0.2-80.3)
22 C.2234+86T/G (NA)
Intron 15 Controls (72) 0.000 72 (72.00) 0(0) 0(0) 1.00
3.8 (0.2-80.3)
23 C.2234+88C/G (rsl 3312970)
Intron 15 Cases (97) 0.015 94 (94.02) 3 (2.95) 0 (0.02) 0.88 0.4 (0.1-1.8)
0.017-0.0425 23 C.2234+88C/G (rsl 3312970)
Intron 15 Controls (71) 0.035 66 (66.09) 5(4.82) 0 (0.09) 0.76
0.4 (0.1-1.8)
0.017-0.0425
24
I .
C.2234+157A/G (rs!3312971)
Intron 15 Cases (97) 0.046 88(88.21) 9(8.58) 0(0.21) 0.63 2.3 (0.6-8.9)
0.000-0.0255 24
I .
C.2234+157A/G (rs!3312971)
Intron 15 Controls (71) 0.021 68 (68.03) 3 (2.94) 0 (0.03) 0.86
2.3 (0.6-8.9)
0.000-0.0255
nucleotide number one being the A from the ATG codon. Minor allele frequency As expected under Hardy-Weinberg equilibrium 4Odds ratios for comparison of hétérozygotes versus common homozygotes 5From NCBI dbSNP data. 6No entry in NCBI dbSNP database, although reported in the literature.
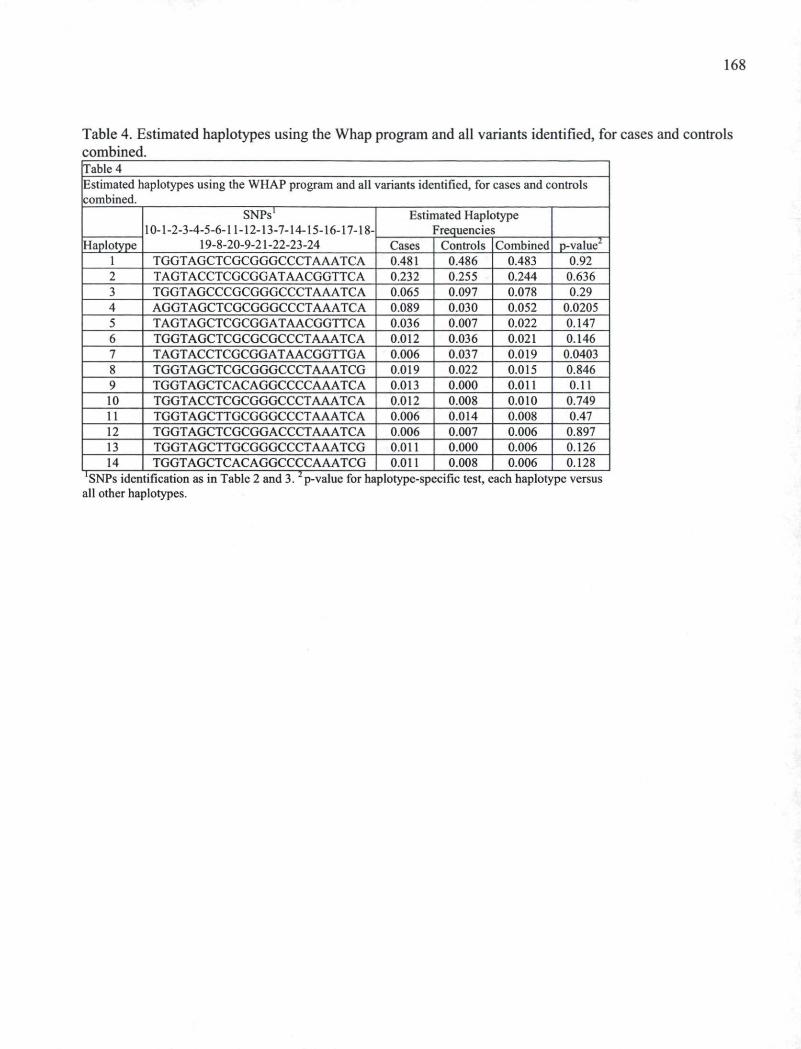
168
Table 4. Estimated haplotypes using the Whap program and all variants identified, for cases and controls combined. Table 4 Estimated haplotypes using the WHAP program and all variants identified, for combined.
cases and controls
SNPs1
10-1-2-3-4-5-6-11-12-13-7-14-15-16-17-18-19-8-20-9-21-22-23-24
Estimated Haplotype Frequencies
Haplotype
SNPs1
10-1-2-3-4-5-6-11-12-13-7-14-15-16-17-18-19-8-20-9-21-22-23-24 Cases Controls Combined p-value2
1 TGGTAGCTCGCGGGCCCTAAATCA 0.481 0.486 0.483 0.92 2 TAGTACCTCGCGGATAACGGTTCA 0.232 0.255 0.244 0.636 3 TGGTAGCCCGCGGGCCCTAAATCA 0.065 0.097 0.078 0.29 4 AGGTAGCTCGCGGGCCCTAAATCA 0.089 0.030 0.052 0.0205 5 TAGTAGCTCGCGGATAACGGTTCA 0.036 0.007 0.022 0.147 6 TGGTAGCTCGCGCGCCCTAAATCA 0.012 0.036 0.021 0.146 7 TAGTACCTCGCGGATAACGGTTGA 0.006 0.037 0.019 0.0403 8 TGGTAGCTCGCGGGCCCTAAATCG 0.019 0.022 0.015 0.846 9 TGGTAGCTCACAGGCCCCAAATCA 0.013 0.000 0.011 0.11 10 TGGTACCTCGCGGGCCCTAAATCA 0.012 0.008 0.010 0.749 11 TGGTAGCTTGCGGGCCCTAAATCA 0.006 0.014 0.008 0.47 12 TGGTAGCTCGCGGACCCTAAATCA 0.006 0.007 0.006 0.897 13 TGGTAGCTTGCGGGCCCTAAATCG 0.011 0.000 0.006 0.126 14 TGGTAGCTCACAGGCCCCAAATCG
. • / - . . . r • -*n . ._ -, _ j -> 2 a _ 0.011 0.008 0.006 0.128
all other haplotypes.
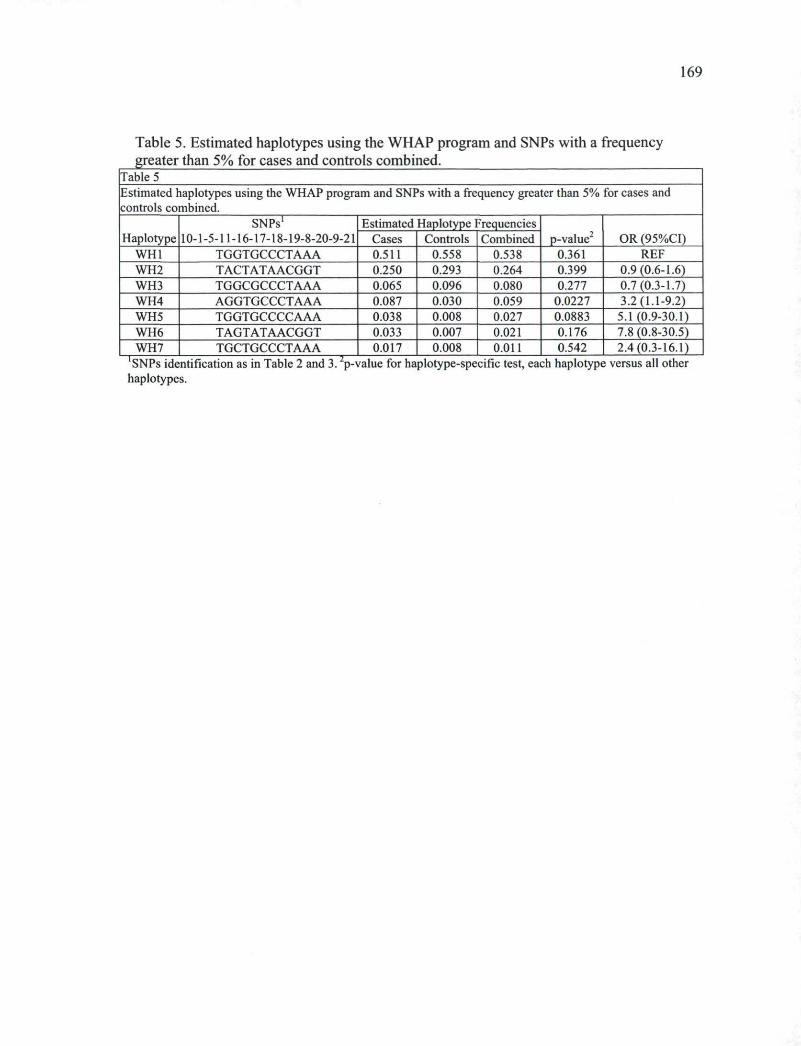
169
Table 5. Estimated haplotypes using the WHAP program and SNPs with a frequency greater than 5% for cases and controls combined.
Table 5 Estimated haplotypes using the WHAP program and SNPs with a frequency greater than 5% for cases and controls combined.
Haplotype SNPs1
10-1-5-11-16-17-18-19-8-20-9-21 Estimated Haplotype Frequencies
Cases Controls Combined p-value OR (95%CI) WH1 TGGTGCCCTAAA 0.511 0.558 0.538 0.361 REF WH2 TACTATAACGGT 0.250 0.293 0.264 0.399 0.9(0.6-1.6) WH3 TGGCGCCCTAAA 0.065 0.096 0.080 0.277 0.7(0.3-1.7) WH4 AGGTGCCCTAAA 0.087 0.030 0.059 0.0227 3.2(1.1-9.2) WH5 TGGTGCCCCAAA 0.038 0.008 0.027 0.0883 5.1 (0.9-30.1) WH6 TAGTATAACGGT 0.033 0.007 0.021 0.176 7.8(0.8-30.5) WH7 TGCTGCCCTAAA 0.017 0.008 0.011 0.542 2.4(0.3-16.1)
SNPs identification as in Table 2 and 3. haplotypes.
p-value for haplotype-specific test, each haplotype versus all other
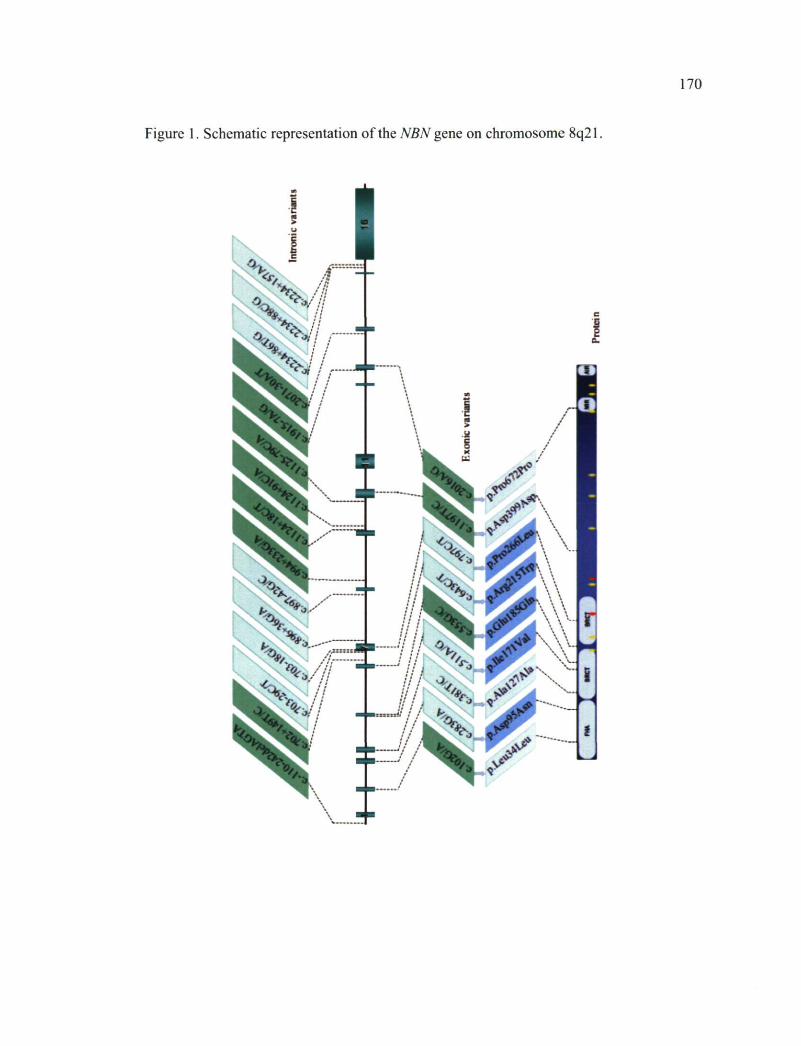
170
Figure 1. Schematic representation of the NBN gene on chromosome 8q21.
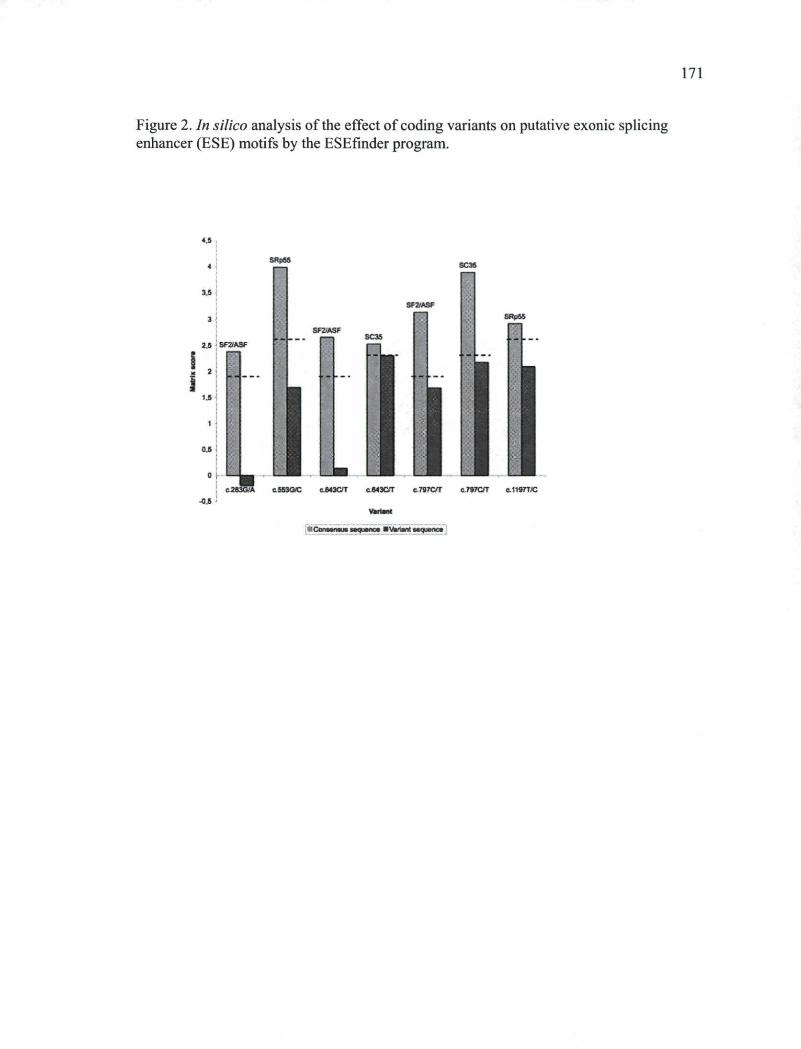
171
Figure 2. In silico analysis of the effect of coding variants on putative exonic splicing enhancer (ESE) motifs by the ESEfinder program.
«JH
3,5
SRs-55
2,3 :SF2MSF
I J
0,5
-0.5
CJMSS» C.583GC C.W3OT C.6430T C.787OT c.7»7C<T c.t197T.C
i MGORMMMI **qtn*ic* WMMMWI s*qu*nc* :
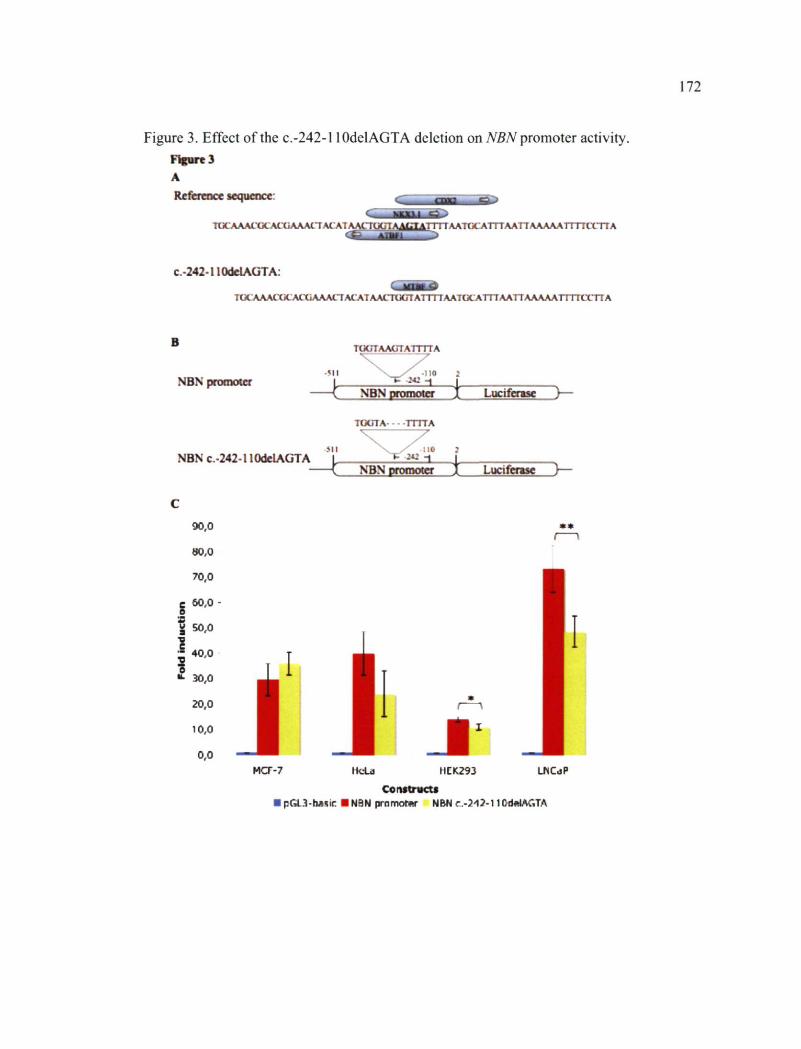
172
Figure 3. Effect of the c.-242-1 lOdelAGTA deletion on NBN promoter activity.
Figure 3
A
Reference sequence: <--
NUil Tl-f ***<<>; A(*f;A*Af-TAfATJ^ |-|^^QJ«|J^ |---ATTTT*a\Tr-t ATTTA ATT A A A A ArTTT-r*TT A
c.-242-110delAGTA:
TUCAAAC titAtliA-L-it"! ACAT AACTlXjT ATTTT AATCXlATTTAArrAAAAATrTrtTTIA
TCXJTAAUTATrrTA
NBN promoter -tl -110 2
NBN promoter
TOUTA TTTTA
fc± Luciftrasc )—
LucJfttwc r—
90,0
«0,0
I MCr-7 UcU MEK293 LNCJP
Constructs I pGL3-ha«ic BN9N promoter NBN r.-J-1?-11CdslACTA

173
Figure 4. Linkage disequilibrium across the NBN gene and neighbouring chromosome region.
|D'|
0 S | O | < 0 3 < |D S.7 S |D'
03 <i.7 «1.0
c-242 )lMaH.U.lA C.102UA . : *»< . v c « l l i a. *1 ! VC. cSSXVC CAOCT
H . H I 0 M S I * C.70M1CT c.- i .VlK. \
c/TTTC/T t .»»**J«. \ tirr-oc^t
C.9M+233U/A UDOHSCff c.H24<*»ltVA C-1125-TK.A
tiim/t" C.IM5-TA/C
c .2» l * \ ( . a-2»-l -JSVI
c22J4-06TX. C-22JM<Mt <.
C J J J U ' I S T A * .
1 . . 1 . . 1 ^
1 ■■■■■
■ ■ ■ ■. ■.
■ " j M H H
■
0 ï r < C 3 U S I > « S J 0 7 S r * « l . O

DISCUSSION ET CONCLUSION 5 Le taux de cancer, dont le cancer du sein, est en hausse dans la population mondiale. Avec
l'accroissement de l'espérance de vie, les changements importants des habitudes de vie et
un environnement en constante évolution, le corps humain réagit et s'adapte dans la mesure
de ses capacités et de ses limites. Il est de plus en plus clair qu'il existe un éventail de
susceptibilité dans la population, avec un sous groupe d'individus présentant une
susceptibilité marquée à développer un cancer du sein alors que d'autres individus
démontreront une résistance accrue.244"245 Comprendre la contribution génétique et les
déterminants héréditaires de la maladie est essentiel, le cancer du sein entraînant un fardeau
émotionnel, social et économique important pour les individus, les familles et les sociétés
atteintes. Par contre, il est aussi présentement clair que cette tâche est particulièrement
ardue puisque la susceptibilité d'un individu à développer le cancer du sein est tributaire de
facteurs génétiques et environnementaux variables, et de leur interrelation. Jusqu'à présent,
les alleles de susceptibilité connus n'expliqueraient qu'environ 30% des cas observés.38
Dans cette thèse, nous avons tenté d'évaluer la contribution de facteurs génétiques sur le
développement de cancer du sein dans une cohorte de familles à risque élevé de la
population canadienne française, enrichie pour des facteurs génétiques de
susceptibilité240,471, par une approche de type gènes candidats. Ceux-ci ont été sélectionnés
sur la base d'une interaction protéique (directe ou indirecte) dans la cellule avec le produit
de l'un ou des deux principaux gènes de susceptibilité au cancer du sein connus, BRCAl et
BRCA2. Trois gènes candidats ont été analysés, dont les résultats ont été présentés dans les
chapitres précédents. Ces études ont permis de mettre en évidence des variants spécifiques
associés à une modulation du risque dans notre cohorte de femmes canadiennes françaises.
1. CHAPITRE I : Analyse du gène ZNF350/ZBRK1 Au chapitre I, nous avons re-séquencé le gène ZNF350/ZBRK1 agissant comme médiateur
de certaines activités de répression transcriptionnelle de BRCAl. Il a été démontré que
BRCAl est impliquée dans la régulation de certaines cibles jouant un rôle clair dans
l'étiologie du cancer du sein (telles p53 et ER-a).291"297 L'interaction directe entre les
protéines ZNF350/ZBRK1 et BRCAl306"307, et le rôle de ce dimère dans la répression de

175
cibles de BRCAl par l'intermédiaire de doigts de zinc reconnaissant une séquence
spécifique en fait un candidat très intéressant dans le contexte d'une recherche de nouveaux
gènes de susceptibilité. En plus de jouer un rôle dans la régulation de l'expression de
GADD45A et de CDKNlA/p21 impliqués dans le cycle cellulaire, l'association de BRCAl
et de ZNF350/ZBRK1 serait associée à une variation de l'expression d'ANGPTl
(Angiopoietin 1) impliqué dans l'angiogenèse.473 De plus, il a été démontré récemment que
le complexe BRCAl/ZNF350/CtIP réprime de façon spécifique le promoteur de HMGA2
(High mobility group AT-hook 2). La perte de cette répression résulte en une prolifération
accrue, une croissance ancrage-indépendante sur support d'agar et une augmentation de la
taille des acini produits en culture tridimensionnelle à partir de cellules de sein MCF10A.29'
Une autre étude récente suggère que ZNF350/ZBRK1 serait aussi responsable de la
répression du gène métastatique MMP9 (Matrix metallopeptidase 9), et la perte
d'expression de ZNF350/ZBRK1 dans des spécimens de cancers du col de l'utérus est
corrélée avec une expression élevée de MMP9.474
Notre investigation de ZNF350/ZBRK1 a permis dévaluer la distribution de 18 variants de
séquence répartis dans les régions codantes ainsi que les régions introniques flanquantes.
Une fois phases à l'aide d'outils bioinformatiques, les variants codants (ainsi que le premier
variant en 3' UTR) sont distribués sur un ensemble de 20 haplotypes. Parmi ceux-ci, les 5
haplotypes les plus fréquents représentent 89% de la variabilité haplotypique de notre
population. Cette analyse a fait ressortir 3 haplotypes représentés de façon différentielle
entre le groupe de femmes atteintes de cancer du sein par opposition aux haplotypes du
même gène dans un groupe de sujets sains, eux aussi de la population canadienne française.
L'effet le plus important a été observé avec l'haplotype H13 et qui est associé dans notre
cohorte à un risque estimé d'environ 6,8. Deux autres haplotypes sont présents uniquement
dans notre population d'individus sains (H8 et H16) et seraient par conséquent associés à
un effet protecteur.
Bien que ce résultat semble suggérer une contribution relativement importante de
ZNF350/ZBRK1 dans la susceptibilité au cancer du sein, il est à noter que l'analyse de ce
gène dans d'autres cohortes relativement petites s'est conclue jusqu'à présent avec des

176
résultats mitigés. L'analyse des niveaux d'ARNm de ZNF350/ZBRK1 chez 61 cas de
cancer du sein non sélectionnés sur la base de l'histoire familiale indique que
ZNF350/ZBRK1 tend à être sous exprimé dans une forte proportion de tumeurs, bien que
cette sous expression ne puisse être associée à des caractéristiques phénotypiques des
tumeurs. L'altération de l'expression de ZNF350/ZBRK1 semble être corrélée de façon
significative avec les polymorphismes p.Cys236Cys et p.Arg501Ser, et les ARNm porteurs
de ces variants démontrent une baisse d'expression dans cette étude.325 Cette tendance de
ZNF350/ZBRK1 à être sous exprimé a aussi été observée dans des tumeurs colorectales.326
De façon intéressante, le polymorphisme p.Arg501Ser a aussi été associé à une diminution
du risque de cancer de la vessie327, et est retrouvé dans notre haplotype protecteur H8. Il a
de plus été suggéré que le polymorphisme p.Asp35Asp de ZNF350/ZBRK1 pourrait être
associé à une augmentation du risque de cancer du sein chez les porteurs du variant de
BRCAl p.Pro871Leu dans la population chinoise.324 L'analyse du variant rs2278414, situé
dans le 3'UTR de ZNF350/ZBRK1, dans la population juive ashkénaze a démontré une
association avec l'apparition du cancer chez les porteurs de mutations de BRCA2, ce qui
pourrait suggérer un rôle de gène modificateur.328
Même si l'effet observé dans notre étude peut difficilement être lié aux variants directement
présents sur les haplotypes mesurés, notre étude a aussi permis de déterminer que ce gène
est présent sur un large bloc de déséquilibre de liaison dans cette région du chromosome
19. Cette région porte plusieurs protéines à doigts de zinc, un motif retrouvé dans les
protéines liant l'ADN, et dont les cibles sont mal caractérisées. Il est par conséquent
possible que l'association retrouvée ici reflète l'effet d'autres variants en déséquilibre de
liaison avec ceux du gène. En ce sens, des analyses préliminaires effectuées dans notre
laboratoire sur les individus de notre cohorte a permis de mettre en évidence certains
variants du promoteur de ZNF350/ZBRK1. Une fois phases, certains de ces variants sont
associés à un haplotype induisant une hausse de l'expression lors d'essais luciférases
comparée à l'haplotype sauvage. Si ces variants étaient associés à la perte/gain de motifs
fonctionnels du promoteur, ceci pourrait par conséquent jeter un éclairage nouveau sur
l'effet observé lors de notre étude des variants de ZNF350/ZBRK1. De plus, une étude
récente ciblant les variants de PPP2R1A a mis en évidence un haplotype de risque et un

177
haplotype protecteur qui pourraient moduler le risque de cancer du sein.475 PPP2R1A
pourrait aussi agir en tant que gène de modification chez des femmes atteintes de maladies
prolifératives bénignes du sein, un facteur de risque de cancer.475 Tel que mentionné
précédemment au chapitre I, PPP2R1A réside en dehors du bloc de déséquilibre de liaison
sur lequel est retrouvé ZNF350/ZBRK1. Il est cependant possible que dans une population
fondatrice comme la population canadienne française, le déséquilibre de liaison soit plus
important que ce qui est retrouvé dans d'autres groupes caucasiens et qui constitue les
données représentées par le projet HapMap.
2. CHAPITRE II : Investigation du gène GADD45A Le chapitre II concerne l'étude d'un second gène candidat, GADD45A, qui est l'une des
cibles de la régulation transcriptionnelle par le complexe BRCA1-ZNF350/ZBRK1.
GADD45A est impliqué dans la stabilité génomique par ses rôles dans le contrôle de la
transition G2/M du cycle cellulaire, l'induction de l'apoptose et la réparation de l'ADN.299" 303 Des études récentes suggèrent aussi que GADD45A pourrait être impliqué dans la
déméthylation de certaines séquences d'ADN par un mécanisme impliquant la voie NER de
réparation de l'ADN.476"477 Tel qu'illustré dans l'introduction, le lien étroit avec BRCAl, la
régulation par une autre protéine impliquée dans de nombreux événements du
développement tumorigénique (p53) et son action au sein du cycle cellulaire, fait du gène
GADD45A un candidat pertinent dans le contexte de notre recherche. GADD45A est présent
sur une région chromosomique qui est fréquemment l'objet d'une perte d'hétérozygosité
dans les tumeurs du sein.478 De plus, GADD45A est faiblement exprimé dans l'épithélium
mammaire humain normal, mais est fortement induit par un large éventail de stress
cellulaires et d'agents induisant des dommages à l'ADN.312"314 Chez la souris, l'inactivation
de Gadd45A résulte en une augmentation de la tumorigénèse et de l'instabilité
chromosomique, ainsi que des problèmes d'auto-immunité.303"305
La variation interindividuelle de ce gène a été évaluée dans notre cohorte par re-séquençage
des portions codantes et des séquences introniques flanquantes. De plus, les deux régions
introniques où se situent les motifs de liaison des protéines p53 et BRCAl -
ZNF350/ZBRK1 ont aussi fait l'objet de re-séquençage. Aucune mutation clairement

178
délétère, pouvant induire un codon d'arrêt prématuré ou un épissage aberrant de l'ARNm,
n'a été identifiée dans la séquence de GADD45A dans notre cohorte. Par contre, 12 variants
de séquence ont été répertoriés et la reconstruction bioinformatique des haplotypes à partir
de ces variants a permis d'identifier un haplotype fréquent qui pourrait être associé à un
effet protecteur.
Quoique les rôles cellulaires connus de GADD45A en fassent un candidat attrayant pour des
études visant la découverte de gènes impliqués dans la modulation du risque de développer
un cancer du sein, notre étude laisse entendre que la contribution de GADD45A au risque de
cancer serait minimale. Les études précédentes investiguant le rôle potentiel de variants de
GADD45A dans la susceptibilité au cancer, en particulier au cancer du sein, demeurent
équivoques. Il est clair que GADD45A, tout comme GADD45B et GADD45G, serait une
cible fréquemment inactivée lors de la tumorigénèse.479 Des études suggèrent que
l'expression de GADD45A serait réduite dans les cas de cancer du sein et de la prostate
suite à la méthylation de son promoteur.480"483 Cependant, les études génétiques semblent
indiquer que GADD45A ne serait pas fréquemment muté dans les cas de cancers. L'étude
de mutation de GADD45A dans des lignées cellulaires de sein ou des tumeurs primaires n'a
pas permis de mettre en évidence des mutations qui pourraient être impliquées dans
l'étiologie de ce cancer.321"323
Malgré cela, notre étude a été à notre connaissance la première à investiguer les variations
de séquence germinales de GADD45A. Une seconde étude dans la population chinoise,
publiée récemment, a étudié le gène GADD45A dans une cohorte de cas de cancers du sein
sporadiques, ainsi que chez des cas familiaux non reliés à des mutations de BRCAl ou
BRCA2. Leur étude suggère que certains variants et haplotypes de GADD45A pourraient
être associés avec un effet protecteur dans cette population, chez les cas sporadiques.484 Les
variants, et par conséquent les haplotypes, identifiés dans cette étude ne sont pas l'exacte
réplique de ce qui a été identifié dans notre population, et par conséquent la comparaison
directe de ceux-ci est difficile. Par contre, il est intéressant de noter que l'un des haplotypes
associé à un effet protecteur dans leur cohorte porte 3 variants aussi retrouvés dans
l'haplotype protecteur identifié dans notre population.484

179
3. CHAPITRE III : Étude du gène NBN Les résultats du re-séquençage du gène NBN sont présentés au chapitre III. Plusieurs
évidences lient le cancer et NBN. En effet, il est reconnu que les individus atteints de NBS
(par conséquent porteurs homozygotes de mutations de NBN) présentent un risque
important de développer plusieurs types de lymphomes et tumeurs solides malgré leur
jeune âge.362'369 Ceci peut s'expliquer par le rôle de la nibrine dans les processus de
détection des dommages à l'ADN, de leur réparation et son implication dans certains
aspects de la régulation du cycle cellulaire.348'350 De plus, dans le cadre de certaines de ses
fonctions, la nibrine interagit directement avec BRCAl352"355, ce qui en fait un gène
candidat intéressant dans le cadre de notre étude.
Des études épidémiologiques conduites au sein des familles de patients atteints de NBS a
aussi suggéré un risque accru de cancers chez les porteurs hétérozygotes d'une mutation de
NBN.312 Il est à noter que le NBS est présent de façon plus fréquente dans les populations
d'ascendance slave, dû à la transmission d'une mutation fondatrice au sein de ces
populations.373 L'hypothèse actuelle suggère que seules certaines mutations permettant
l'expression d'une protéine résiduelle, donc hypomorphiques, permettrait la survie
cellulaire. La contribution de cette mutation au risque de cancer demeure controversée, bien
que les évidences d'une association semblent plus fortes dans les populations d'ascendance
slave.374 Dans le cas du cancer du sein, le risque (OR) est généralement estimé à environ
3 , bien qu'une association positive ne soit pas observée dans toutes les études. " La
contribution de certains variants de NBN, autres que la mutation slave, a aussi été étudiée.
Le variant p.Arg215Trp a été associé à une diminution des niveaux cellulaires de nibrine375,
et a été décrit à l'état hétérozygote composé (avec 657del5) chez deux frères atteints d'une
forme particulièrement sévère du NBS.380 Bien qu'il a été suggéré que ce variant pourrait
perturber la liaison de la nibrine à l'histone y-H2AX , une méta-analyse de p.Arg215Trp
en relation avec le cancer du sein ne semble pas suggérer une association (OR 1,9 ; 95% IC
0,8-4,6).375 En plus de ces deux variants de séquence, p.Ilel71Val a aussi été associé à un
risque accru de cancer du sein, bien que cette association soit controversée.382'383

180
La contribution potentielle de mutations et/ou de variations du gène NBN a été évaluée dans
notre cohorte de femmes atteintes de cancer du sein, permettant d'identifier 24 variants de
séquence dans la séquence codante, les régions introniques flanquantes ainsi que le
promoteur proximal (500pb). Parmi ceux-ci, le variant p.Ilel71Val a été retrouvé chez une
femme atteinte et n'est pas présent chez les individus sains génotypes, incluant un individu
de la même famille. Bien que ce résultat concorde avec un risque associé à ce variant, la
taille de notre échantillon ne permet par d'émettre une conclusion définitive. Cependant,
puisque ce variant n'a été retrouvé que chez une femme atteinte sur les 97 individus de
notre cohorte de cas génotypes, il est peu probable que pIlel71Val soit responsable d'une
contribution importante à la susceptibilité au cancer du sein dans notre population. Quant
au variant p.Arg215Trp, notre étude n'a pas permis de l'associer à un risque accru de
cancer puisqu'il a été retrouvé à la fois chez l'une de nos femmes atteintes de cancer et
chez un individu sain.
Notre analyse de NBN a par contre permis d'identifier une deletion dans le promoteur
proximal du gène présentant une variation de fréquence notable entre les cas étudiés et les
individus sains. Des analyses fonctionnelles, par essai luciférase mesurant l'activité du
promoteur de type sauvage comparée à celle du promoteur portant la deletion, a permis de
démontrer la présence d'une variation de l'expression dans une lignée de cancer de la
prostate et une lignée de rein embryonnaire. Par contre, aucun effet n'a été observé dans la
lignée de cancer du sein MCF-7. Plusieurs hypothèses peuvent être avancées pour expliquer
ce phénomène, tel un effet dépendant du type cellulaire ou bien d'un stress cellulaire tel les
dommages à l'ADN. Il est aussi possible que le variant c.-242-l lOdelAGTA puisse être en
déséquilibre de liaison avec un autre variant dans une portion de séquence non génotype,
qui serait responsable de l'effet observé dans notre cohorte. En ce sens, notre étude montre
aussi que NBN chevauche deux blocs de déséquilibre de liaison, dont un bloc important en
5' du gène. Il est par contre clair que nos analyses ne permettent pas pour l'instant de tirer
une conclusion définitive à cet égard.
Notre étude du gène NBN suggère que des variations de ce gène sont présentes, bien qu'à
une fréquence faible, dans la population canadienne française. Depuis notre analyse,

181
plusieurs études se sont attardées à la contribution de variants de séquence de NBN dans la
susceptibilité au cancer, en particulier le variant fréquent p.Glul85Gln situé dans la région
d'interaction avec BRCAl. Ce variant a été évalué dans le contexte du cancer du sein dans
plusieurs études, avec des résultats variables. Récemment, une méta-analyse des études
publiées analysant l'apport potentiel de ce variant à la susceptibilité au cancer du sein
semble suggérer un effet protecteur de p.Glul85Gln sur le risque (CC vs GG : 0,75 95% IC
0,74-0,98).384 Une seconde méta-analyse, celle-ci regroupant plus de 9000 patients et 10000
contrôles, s'est attardée au risque associé à p.Glul85Gln sur le risque de cancer en général
et semble suggérer une faible augmentation du risque, en particulier chez les caucasiens.485
L'analyse de p.Glul85Gln a aussi été associée à une augmentation du risque de cancer de
l'ovaire chez les porteurs de mutations de BRCAl et de cancer de la vessie chez les
fumeurs.486"487
4. L'approche gène candidat dans la population canadienne française : considérations méthodologiques Il existe plusieurs stratégies pouvant mener à l'identification de gènes de susceptibilité au
cancer du sein. Des études de liaison génétiques au sein de familles présentant de très fortes
évidences de la transmission de facteurs héréditaires ont permis par le passé de mettre en
évidence des gènes porteurs de mutations rares, mais fortement pénétrantes. Après un
succès initial, cette stratégie s'est avérée infructueuse au cours des 15 dernières années.
Ceci suggère que si de tels alleles demeurent à être identifiés, leur contribution au risque de
cancer du sein sera minimale. Une autre stratégie, employée au cours des trois dernières
années, a consisté en des études d'association génomiques sur plusieurs milliers de cas et
de contrôles. Ces études ont permis d'identifier des variants très fréquents associés à une
faible modulation du risque encouru. De telles études permettent d'interroger des centaines
de milliers de variants génétiques sans nécessiter une connaissance préalable de la fonction
des gènes étudiés. Cependant, leurs grandes envergures nécessitent des ressources
importantes et le regroupement de plusieurs cohortes de patients. Une troisième stratégie,
privilégiée dans les études présentées dans cette thèse, consiste à effectuer l'étude
systématique de gènes candidats sélectionnés sur la base de leur plausibilité préalable dans
les mécanismes de la carcinogenèse.

182
L'approche vise à identifier des variants rares (i.e fréquence de l'allèle mineur inférieure à
5%) modérément pénétrants, qui ne pourraient pas être identifiés par une étude
d'association de type « tour de génome » dû à leur fréquence trop basse. De plus, l'effet de
ce type de variants est souvent population-spécifique. Il est influencé par des
caractéristiques spécifiques à l'historique des populations (par exemple une dérive
génétique, ou un effet fondateur) et requiert l'utilisation d'une population unique. C'est
pourquoi nous avons par conséquent tiré parti ici des avantages reliés à la fois à la cohorte
de femmes atteintes de cancer du sein et présentant une forte susceptibilité génétique
recrutée par les Drs Durocher et Simard, ainsi qu'à l'effet fondateur retrouvé dans la
population canadienne française. Tel que mentionné à la section 2.3 Modèles de la
susceptibilité génétique au cancer du sein, l'utilisation de cas familiaux permet
d'augmenter le pouvoir de notre étude puisque les alleles de susceptibilité recherchés sont
en théorie plus fréquents dans les cas de cancer du sein familiaux comparés à des cas tirés
de la population. De la même façon, la population canadienne française présente un effet
fondateur qui peut permettre l'identification de mutations fondatrices présentant une
prévalence plus élevée et offre aussi l'avantage d'une « toile » génétique plus homogène
(voir la section 5.1 Familles à risque élevé de la population canadienne française).
Certaines limitations sont aussi associées à la méthode employée. Dans un premier temps,
l'aproche gène candidat cible des gènes ou une voie cellulaire en particulier (réparation de
l'ADN, le contrôle du cycle cellulaire, le métabolisme des hormones, etc) et ce choix est
directement tributaire de nos connaissances de la biologie du cancer. Dans cette
perspective, il est intéressant de noter que peu des alleles de faible penetrance identifiés par
tour de génome sont situés dans des régions (ou des gènes candidats) ciblés antérieurement,
bien qu'en rétrospective certains des gènes identifiés présentent un lien biologique avec le
cancer du sein (par exemple FGFR2). Il est possible qu'avec l'avancement des
connaissances et l'intégration des données par des méthodes d'identification de gènes
candidats bioinformatiques permettant l'intégration d'une quantité phénoménale
d'information, des candidats inattendus par les méthodes traditionnelles emmergent. Dans
un second temps, le groupe contrôle employé présente certaines limites. Bien que les

183
femmes génotypées aient été en bonne santé au moment de leur recrutement, il demeure
possible qu'elles développent un cancer du sein plus tard au cours de leur vie.
5. Conclusions et perspectives Bien que notre étude ait permis de mettre en évidence des variants et/ou haplotypes
pouvant moduler le risque de cancer du sein (à la fois de façon positive et négative) dans la
population canadienne française, bien du chemin reste à faire. Il est clair que les
associations décrites dans cette thèse sont très intéressantes bien que préliminaires et
demeurent à être confirmées dans des études plus approfondies dans de plus grandes
cohortes et d'autres populations. De plus, il demeure pour l'instant difficile de comprendre
les bases moléculaires pouvant expliquer l'effet de plusieurs des variants pour lesquels une
association a été observée. Dans cette optique, d'autres études sont nécessaires. Puisqu'une
large part de l'effet observé dans les analyses présentées ici semble se situer dans la région
5' des gènes, un examen plus détaillé des régions promotrices semble justifié (identification
de variants par re-séquençage, essais fonctionnels de type luciférase et/ou de liaison de
facteurs de transcription). Alternativement et tel que mentionné au sein des chapitres
précédents, il demeure possible que l'effet observé soit attribuable à des variants en
déséquilibre de liaison avec ceux identifiés ici. L'analyse des régions en déséquilibre de
liaison et des gènes qui y sont retrouvés constitue par conséquent une avenue qui doit être
envisagée.
Avec l'étude de la structure du génome humain, nous réalisons peu à peu la complexité de
son architecture. Plusieurs variations structurales et éléments cryptiques (variations du
nombre de copies, rearrangements complexes, expansions nucléotidiques, microARNs)
pourraient aussi avoir leur rôle à jouer dans la susceptibilité d'un individu à développer une
maladie complexe comme le cancer. Bien que les études gène candidat actuelles aient été
concentrées sur le re-séquençage d'un nombre limité de candidats, l'avenue de technologies
de très haut débit permet maintenant l'analyse d'exomes ou génomes complets, ce qui
pourrait permettre l'identification de nouvelles cibles des processus tumorigéniques. Une
approche combinée incluant des méthodes génomiques et de gènes candidats ainsi que des
idées novatrices sera nécessaire afin de relever le défi que représente la compréhension de

184
cette maladie complexe. Il est clair que beaucoup de chemin reste à faire dans
l'identification des facteurs de susceptibilité au cancer du sein, avec pour but ultime une
compréhension détaillée de sa pathologie, de meilleures stratégies de prévention et
l'identification de nouvelles cibles thérapeutiques potentielles.

BIBLIOGRAPHIE
1 Grizzi F, Chiriva-Internati M. Cancer: looking for simplicity and finding complexity. Cancer Cell Int. 2006; 6:4.
2 Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004; 4(3): 177-83.
3 Sawan C, Vaissière T, Murr R, Herceg Z. Epigenetic drivers and genetic passengers on the road to cancer. Mutat Res. 2008; 642(1-2): 1-13.
4 Garnis C, Buys TP, Lam WL. Genetic alteration and gene expression modulation during cancer progression. Mol Cancer. 2004; 3:9.
5 Garcia M, Jemal A, Ward EM, Center MM, Hao Y, Siegel RI, Thun MJ. Global cancer facts and figures 2007. Atlanta, GA; American Cancer Society. 2007; 50p.
6 Société canadianne du cancer, Institut national du cancer du Canada, Statistique Canada, Registres du cancer des Provinces et des Territoires, Agence de la santé publique du Canada. Canadian Cancer Statistics 2008, Canadian Cancer Society. 2008; 112p.
7 Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005; 55(2):74-108.
8 Potten CS, Watson RJ, Williams GT, Tickle S, Roberts SA, Harris M, Howell A. The effect of age and menstrual cycle upon proliferative activity of the normal human breast. Br J Cancer. 1988; 58(2): 163-70.
9 Howard BA, Gusterson BA. Human breast development. J Mammary Gland Biol Neoplasia. 2000; 5(2): 119-37.
10 Santen RJ, Mansel R. Benign breast disorders. N Engl J Med. 2005; 353(3):275-85. 11 Mallon E, Osin P, Nasiri N, Blain I, Howard B, Gusterson B. The basic pathology of human breast
cancer. J Mammary Gland Biol Neoplasia. 2000; 5(2):139-63. 12 Harris G, Bartlett EE, Rehmar M, Holman I, Derhagopian R, Eytel C, Kallos N, Mitchell K, Weible D,
Young T, Machnowski G. Early diagnosis of breast changes. Risk management and quality of care approach. J Fla Med Assoc. 1996; 83(7):466-9.
13 Sandison AT. An autopsy study of the adult human breast: with special reference to proliferative epithelial changes of importance in the pathology of the breast. Natl Cancer Inst Monogr. 1962;4:1-145.
14 Goehring C, Morabia A. Epidemiology of benign breast disease, with special attention to histologic types. Epidemiol Rev. 1997; 19(2):310-27.
15 Giordano SH, Cohen DS, Buzdar AU, Perkins G, Hortobagyi GN. Breast carcinoma in men: a population-based study. Cancer. 2004; 101(l):51-7.
16 Benz CC. Impact of aging on the biology of breast cancer. Crit Rev Oncol Hematol. 2008; 66(l):65-74. 17 Robert SA, Strombom I, Trentham-Dietz A, Hampton JM, McElroy JA, Newcomb PA, Remington PL.
Socioeconomic risk factors for breast cancer: distinguishing individual- and community-level effects. Epidemiology. 2004; 15(4):442-50.
18 Webster TF, Hoffman K, Weinberg J, Vieira V, Aschengrau A. Community- and individual-level socioeconomic status and breast cancer risk: multilevel modeling on Cape Cod, Massachusetts. Environ Health Perspect. 2008; 116(8): 1125-9.
19 Kelsey JL, Gammon MD, John EM. Reproductive factors and breast cancer. Epidemiol Rev. 1993;15(l):36-47.
20 Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and breastfeeding: collaborative reanalysis of individual data from 47 epidemiological studies in 30 countries, including 50302 women with breast cancer and 96973 women without the disease. Lancet. 2002; 360(9328): 187-95.

186
21 Key T, Appleby P, Barnes I, Reeves G, Endogenous Hormones and Breast Cancer Collaborative Group. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002; 94(8):606-16.
22 Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet. 1996; 347(9017): 1713-27.
23 Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet. 1997; 350(9084): 1047-59.
24 Beral V; Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003 Aug 9;362(9382):419-27.
25 Tworoger SS, Hankinson SE. Prolactin and breast cancer etiology: an epidemiologic perspective. J Mammary Gland Biol Neoplasia. 2008; 13(l):41-53.
26 Chlebowski RT, Chen Z, Anderson GL, Rohan T, Aragaki A, Lane D, Dolan NC, Paskett ED, McTiernan A, Hubbell FA, Adams-Campbell LL, Prentice R. Ethnicity and breast cancer: factors influencing differences in incidence and outcome. J Natl Cancer Inst. 2005; 97(6):439-48.
27 Vainshtein J. Disparities in breast cancer incidence across racial/ethnic strata and socioeconomic status: a systematic review. J Natl Med Assoc. 2008; 100(7):833-9.
28 Dumitrescu RG, Cotarla I. Understanding breast cancer risk — where do we stand in 2005? J Cell Mol Med. 2005; 9(1):208-21.
29 van den Brandt PA, Spiegelman D, Yaun SS, Adami HO, Beeson L, Folsom AR, Fraser G, Goldbohm RA, Graham S, Kushi L, Marshall JR, Miller AB, Rohan T, Smith-Warner SA, Speizer FE, Willett WC, Wolk A, Hunter DJ. Pooled analysis of prospective cohort studies on height, weight, and breast cancer risk. Am J Epidemiol. 2000; 152(6):514-27.
30 Monninkhof EM, Elias SG, Vlems FA, van der Tweel I, Schuit AJ, Voskuil DW, van Leeuwen FE; TW AC Physical activity and breast cancer: a systematic review. Epidemiology. 2007; 18(l):137-57.
31 Hamajima N, Hirose K, Tajima K, Rohan T, Calle EE, Heath CW Jr, Coates RJ, Liff JM, Talamini R, Chantarakul N, Koetsawang S, Rachawat D, Morabia A, Schuman L, Stewart W, Szklo M, Bain C, Schofield F, Siskind V, Band P, Coldman AJ, Gallagher RP, Hislop TG, Yang P, Kolonel LM, Nomura AM, Hu J, Johnson KC, Mao Y, De Sanjosé S, Lee N, Marchbanks P, Ory HW, Peterson HB, Wilson HG, Wingo PA, Ebeling K, Kunde D, Nishan P, Hopper JL, Colditz G, Gajalanski V, Martin N, Pardthaisong T, Silpisornkosol S, Theetranont C, Boosiri B, Chutivongse S, Jimakorn P, Virutamasen P, Wongsrichanalai C, Ewertz M, Adami HO, Bergkvist L, Magnusson C, Persson I, Chang-Claude J, Paul C, Skegg DC, Spears GF, Boyle P, Evstifeeva T, Daling JR, Hutchinson WB, Malone K, Noonan EA, Stanford JL, Thomas DB, Weiss NS, White E, Andrieu N, Bremond A, Clavel F, Gairard B, Lansac J, Piana L, Renaud R, Izquierdo A, Viladiu P, Cuevas HR, Ontiveros P, Palet A, Salazar SB, Aristizabel N, Cuadros A, Tryggvadottir L, Tulinius H, Bachelot A, Le MG, Peto J, Franceschi S, Lubin F, Modan B, Ron E, Wax Y, Friedman GD, Hiatt RA, Levi F, Bishop T, Kosmelj K, Primic-Zakelj M, Ravnihar B, Stare J, Beeson WL, Fraser G, Bullbrook RD, Cuzick J, Duffy SW, Fentiman IS, Hayward JL, Wang DY, McMichael AJ, McPherson K, Hanson RL, Leske MC, Mahoney MC, Nasca PC, Varma AO, Weinstein AL, Moller TR, Olsson H, Ranstam J, Goldbohm RA, van den Brandt PA, Apelo RA, Baens J, de la Cruz JR, Javier B, Lacaya LB, Ngelangel CA, La Vecchia C, Negri E, Marubini E, Ferraroni M, Gerber M, Richardson S, Segala C, Gatei D, Kenya P, Kungu A, Mati JG, Brinton LA, Hoover R, Schairer C, Spirtas R, Lee HP, Rookus MA, van Leeuwen FE, Schoenberg JA, McCredie M, Gammon MD, Clarke EA, Jones L, Neil A, Vessey M, Yeates D, Appleby P, Banks E, Beral V, Bull D, Crossley B, Goodill A, Green J, Hermon C, Key T, Langston N, Lewis C, Reeves G, Collins R, Doll R, Peto R, Mabuchi K, Preston D, Hannaford P, Kay C, Rosero-Bixby L, Gao YT, Jin F, Yuan JM, Wei HY, Yun T, Zhiheng C, Berry G, Cooper Booth J, Jelihovsky T, MacLennan R, Shearman R, Wang QS, Baines CJ, Miller AB, Wall C, Lund E, Stalsberg H, Shu XO, Zheng W, Katsouyanni K, Trichopoulou A, Trichopoulos D, Dabancens A, Martinez L, Molina R, Salas O, Alexander FE, Anderson K, Folsom AR, Hulka BS, Bernstein L, Enger S, Haile RW, Paganini-Hill A, Pike MC, Ross RK, Ursin G, Yu MC, Longnecker MP, Newcomb P, Bergkvist L, Kalache A, Farley TM, Hoick S, Meirik O; Collaborative

187
Group on Hormonal Factors in Breast Cancer. Alcohol, tobacco and breast cancer-collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br J Cancer. 2002; 87( 11 ): 1234-45.
32 Singletary KW, Gapstur SM. Alcohol and breast cancer: review of epidemiologic and experimental evidence and potential mechanisms. JAMA. 2001; 286(17):2143-51.
33 Carmichael A, Sami AS, Dixon JM. Breast cancer risk among the survivors of atomic bomb and patients exposed to therapeutic ionising radiation. Eur J Surg Oncol. 2003; 29(5):475-9.
34 McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006; 15(6): 1159-69.
35 Hartmann LC, Sellers TA, Frost MH, Lingle WL, Degnim AC, Ghosh K, Vierkant RA, Maloney SD, Pankratz VS, Hillman DW, Suman VJ, Johnson J, Blake C, Tlsty T, Vachon CM, Melton LJ 3rd, Visscher DW. Benign breast disease and the risk of breast cancer. N Engl J Med. 2005; 353(3):229-37.
36 Chen Y, Thompson W, Semenciw R, Mao Y. Epidemiology of contralateral breast cancer. Cancer Epidemiol Biomarkers Prev. 1999; 8(10):855-61.
37 Collaborative Group on Hormonal Factors in Breast Cancer. Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet. 2001; 358(9291):1389-99.
38 Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nat Genet. 2008; 40(1): 17-22.
39 Vaittinen P, Hemminki K. Risk factors and age-incidence relationships for contralateral breast cancer. Int J Cancer. 2000; 88(6):998-1002.
40 McPherson K, Steel CM, Dixon JM. ABC of breast diseases. Breast cancer-epidemiology, risk factors, and genetics. BMJ. 2000; 321(7261):624-8.
41 Kelemen LE, Sellers TA, Vachon CM. Can genes for mammographie density inform cancer aetiology? Nat Rev Cancer. 2008; 8(10):812-23.
42 Lynch HT: Genetics and Breast Cancer. New York. Van Nostrand Reinhold Co., 1981 253p. 43 Eisinger F, Sobol H, Serin D, Whorton JC. Hereditary breast cancer, circa 1750. Lancet. 1998;
351(9112):1366. 44 Broca P. Traité des Tumeurs. Tome premier: Des Tumeurs en Général. Paris P. Asselin (ed) Librairie
de la Faculté de Médecine, 1866 595p. 45 Lebert H. Traite Pratique des Maladies Cancéreuses et Affections Curables Confondues avec le
Cancer. Paris J.-B Baillière (ed) Librairie de l'Académie Nationale de Médecine, 1851 892p. 46 Warthin AS. Heredity with reference to carcinomas shown by the study of the cases examined in the
pathological laboratory of the University of Michigan. Arch Intern Med. 1913; 12:546-55. 47 Lynch HT, Krush AJ. Carcinoma of the breast and ovary in three families. Surg Gynecol Obstet. 1971;
133(4):644-8. 48 Lynch HT, Guirgis HA, Albert S, Brennan M, Lynch J, Kraft C, Pocekay D, Vaughns C, Kaplan A.
Familial association of carcinoma of the breast and ovary. Surg Gynecol Obstet. 1974; 138(5):717-24. 49 Knudson A. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U. S. A.
1971;68(4):820-3. 50 Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A,
Hemminki K. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000; 343(2):78-85.
51 Peto J, Mack TM. High constant incidence in twins and other relatives of women with breast cancer. Nat Genet. 2000; 26(4):411-4.
52 Turnbull C, Rahman N. Genetic predisposition to breast cancer: past, present, and future. Annu Rev Genomics Hum Genet 2008; 9:321-45.
53 Campeau PM, Foulkes WD, Tischkowitz MD. Hereditary breast cancer: new genetic developments, new therapeutic avenues. Hum Genet 2008; 124:31-42.

188
54 Berliner JL, Fay AM; Practice Issues Subcommittee of the National Society of Genetic Counselors' Familial Cancer Risk Counseling Special Interest Group. Risk assessment and genetic counseling for hereditary breast and ovarian cancer: recommendations of the National Society of Genetic Counselors. J Genet Couns. 2007; 16(3):241-60.
55 Lynch HT, Harris RE, Guirgis HA, Lynch PM, Maloney K, Rankin L, Lynch J. Early age of onset and familial breast cancer. Lancet. 1976; 2(7986):626-7.
56 Claus EB, Risch N, Thompson WD. Genetic analysis of breast cancer in the cancer and steroid hormone study. Am J Hum Genet. 1991; 48(2):232-42.
57 Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990; 250(4988): 1684-9.
58 Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D, Field P, Marshall G, Narod S, Lenoir GM, Lynch H, Feunteun J, Devilee P, Cornelisse CJ, Menko FH, Daly PA, Ormiston W, McManus R, Pye C, Lewis CM, Cannon-Albright LA, Peto J, Ponder BAJ, Skolnick MH, Easton DF, Goldgar DE, Stratton MR. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13ql2-13. Science. 1994; 265(5181):2088-90.
59 Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, Bell R, Rosenthal J, Hussey C, Tran T, McClure M, Frye C, Hattier T, Phelps R, Haugen-Strano A, Katcher H, Yakumo K, Gholami Z, Shaffer D, Stone S, Bayer S, Wray C, Bogden R, Dayananth P, Ward J, Tonin P, Narod S, Bristow PK, Norris FH, Helvering L, Morrison P, Rosteck P, Barrett JC, Lewis C, Neuhausen S, Cannon-Albright L, Goldgar D, Wiseman R, Kamb A, Skolnick MH. A strong candidate for the breast and ovarian cancer susceptibility gene BRCAl. Science. 1994; 266(5182):66-71.
60 Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G, Barfoot R, Hamoudi R, Patel S, Rices C, Biggs P, Hashim Y, Smith A, Connor F, Arason A, Gudmundsson J, Ficenec D, Kelsell D, FordTonin DP, Bishop DT, Spurr NK, Ponder BAJ, Eeles R, Peto J, Devilee P, Cornelisse C, Lynch H, Narod S, Lenoir G, Egilsson V, Bjork Barkadottir R, Easton DF, Bentley DR, Futreal PA, Alan Ashworth A, Stratton MR. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995; 378(6559):789-92.
61 Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCAl and BRCA2 in diverse populations. Nat Rev Cancer. 2007; 7(12):937-48.
62 Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, Deacon J, Stratton MR. Prevalence of BRCAl and BRC A 2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst. 1999; 91(11):943-9.
63 Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCAl and BRC A 2 mutations in a population-based series of breast cancer cases. Br J Cancer. 2000; 83( 10): 1301 -8.
64 Antoniou AC, Pharoah PD, McMullan G, Day NE, Stratton MR, Peto J, Ponder BJ, Easton DF. A comprehensive model for familial breast cancer incorporating BRCAl, BRCA2 and other genes. Br J Cancer. 2002; 86(l):76-83.
65 Simard J, Dumont M, Moisan AM, Gaborieau V, Malouin H, Durocher F, Chiquette J, Plante M, Avard D, Bessette P, Brousseau C, Dorval M, Godard B, Houde L; INHERIT BRCAs, Joly Y, Lajoie MA, Leblanc G, Lépine J, Lesperance B, Vézina H, Parboosingh J, Pichette R, Provencher L, Rhéaume J, Sinnett D, Samson C, Simard JC, Tranchant M, Voyer P, Easton D, Tavtigian SV, Knoppers BM, Laframboise R, Bridge P, Goldgar D. Evaluation of BRCAl and BRCA2 mutation prevalence, risk prediction models and a multistep testing approach in French-Canadian families with high risk of breast and ovarian cancer. J Med Genet. 2007; 44(2):107-21.
66 Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCAl-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994; 343(8899):692-5.
67 Sinilnikova OM, Mazoyer S, Bonnardel C, Lynch HT, Narod SA, Lenoir GM. BRCAl and BRCA2 mutations in breast and ovarian cancer syndrome: reflection on the Creighton University historical series of high risk families. Fam Cancer. 2006; 5(1): 15-20.

189
68 Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Breast Cancer Linkage Consortium. Risks of cancer in BRCAl-mutation carriers. Lancet. 1994; 343(8899):692-5.
69 Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, Arason A, Schemeck S, Peto J, Rebbeck TR, Tonin P, Neuhausen S, Barkardottir R, Eyfjord J, Lynch H, Ponder BA, Gayther SA, Zelada-Hedman M, Birch JM, Lindblom A, Stoppa-Lyonnet D, Bignon Y-J, Borg Â, Hamann U, Haites N, Scott RJ, Maugard CM, Vasen H, Seitz S, Cannon-Albright LA, Schofield A, Zelada-Hedman M, and the Breast Cancer Linkage Consortium. Genetic heterogeneity and penetrance analysis of the BRCAl and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1998; 62(3):676-89.
70 Thompson D, Easton D; Breast Cancer Linkage Consortium. Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am J Hum Genet. 2001; 68(2):410-9.
71 Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCAl mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002; 94(18):1365-72.
72 Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjakoski K, Kallioniemi OP, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF. Average risks of breast and ovarian cancer associated with BRCAl or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003; 72(5):1117-30.
73 Gayther SA, Mangion J, Russell P, Seal S, Barfoot R, Ponder BA, Stratton MR, Easton D. Variation of risks of breast and ovarian cancer associated with different germline mutations of the BRCA2 gene. Nat Genet. 1997; 15(1): 103-5.
74 Thompson D, Easton D; Breast Cancer Linkage Consortium. Variation in BRCAl cancer risks by mutation position. Cancer Epidemiol Biomarkers Prev. 2002; ll(4):329-36.
75 Mitchell G, Antoniou AC, Warren R, Peock S, Brown J, Davies R, Mattison J, Cook M, Warsi I, Evans DG, Eccles D, Douglas F, Paterson J, Hodgson S, Izatt L, Cole T, Burgess L, Eeles R, Easton DF. Mammographie density and breast cancer risk in BRCAl and BRCA2 mutation carriers. Cancer Res. 2006; 66(3): 1866-72.
76 Antoniou AC, Shenton A, Maher ER, Watson E, Woodward E, Lalloo F, Easton DF, Evans DG. Parity and breast cancer risk among BRCAl and BRCA2 mutation carriers. Breast Cancer Res. 2006; 8(6):R72.
77 Andrieu N, Goldgar DE, Easton DF, Rookus M, Brohet R, Antoniou AC, Peock S, Evans G, Eccles D, Douglas F, Noguès C, Gauthier-Villars M, Chompret A, Van Leeuwen FE, Kluijt I, Benitez J, Arver B, Olah E, Chang-Claude J; EMBRACE; GENEPSO; GEO-HEBON; IBCCS Collaborators Group. Pregnancies, breast-feeding, and breast cancer risk in the International BRCA1/2 Carrier Cohort Study (IBCCS). J Natl Cancer Inst. 2006; 98(8):535-44.
78 Brohet RM, Goldgar DE, Easton DF, Antoniou AC, Andrieu N, Chang-Claude J, Peock S, Eeles RA, Cook M, Chu C, Noguès C, Lasset C, Berthet P, Meijers-Heijboer H, Gerdes AM, Olsson H, Caldes T, van Leeuwen FE, Rookus MA. Oral contraceptives and breast cancer risk in the international BRCA1/2 carrier cohort study: a report from EMBRACE, GENEPSO, GEO-HEBON, and the IBCCS Collaborating Group. J Clin Oncol. 2007; 25(25):3831-6.
79 Chang-Claude J, Andrieu N, Rookus M, Brohet R, Antoniou AC, Peock S, Davidson R, Izatt L, Cole T, Noguès C, Luporsi E, Huiart L, Hoogerbrugge N, Van Leeuwen FE, Osorio A, Eyfjord J, Radice P, Goldgar DE, Easton DF; Epidemiological Study of Familial Breast Cancer (EMBRACE); Gene Etude Prospective Sein Ovaire (GENEPSO); Genen Omgeving studie van de werkgroep Hereditiair Borstkanker Onderzoek Nederland (GEO-HEBON); International BRCA1/2 Carrier Cohort Study (IBCCS) collaborators group. Age at ménarche and menopause and breast cancer risk in the International BRCA1/2 Carrier Cohort Study. Cancer Epidemiol Biomarkers Prev. 2007; 16(4):740-6.

190
80 Boyd NF, Dite GS, Stone J, Gunasekara A, English DR, McCredie MR, Giles GG, Tritchler D, Chiarelli A, Yaffe MJ, Hopper JL. Heritability of mammographie density, a risk factor for breast cancer. N Engl J Med. 2002; 347(12):886-94.
81 Simchoni S, Friedman E, Kaufman B, Gershoni-Baruch R, Orr-Urtreger A, Kedar-Barnes I, Shiri-Sverdlov R, Dagan E, Tsabari S, Shohat M, Catane R, King MC, Lahad A, Levy-Lahad E. Familial clustering of site-specific cancer risks associated with BRCAl and BRCA2 mutations in the Ashkenazi Jewish population. Proc Natl Acad Sci USA. 2006; 103(10):3770-4.
82 Smith A, Moran A, Boyd MC, Bulman M, Shenton A, Smith L, Iddenden R, Woodward ER, Lalloo F, Maher ER, Evans DG. Phenocopies in BRCAl and BRCA2 families: evidence for modifier genes and implications for screening. J Med Genet. 2007; 44(1): 10-15.
83 Goldgar D, Venne V, Conner T, Buys S. BRCA phenocopies or ascertainment bias? J Med Genet. 2007; 44(8):e86.
84 Antoniou AC, Sinilnikova OM, Simard J, Leone M, Dumont M, Neuhausen SL, Struewing JP, Stoppa-Lyonnet D, Barjhoux L, Hughes DJ, Coupier I, Belotti M, Lasset C, Bonadona V, Bignon YJ; Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers Study (GEMO), Rebbeck TR, Wagner T, Lynch HT, Domchek SM, Nathanson KL, Garber JE, Weitzel J, Narod SA, Tomlinson G, Olopade OI, Godwin A, Isaacs C, Jakubowska A, Lubinski J, Gronwald J, Gôrski B, Byrski T, Huzarski T, Peock S, Cook M, Baynes C, Murray A, Rogers M, Daly PA, Dorkins H; Epidemiological Study of BRCAl and BRCA2 Mutation Carriers (EMBRACE), Schmutzler RK, Versmold B, Engel C, Meindl A, Arnold N, Niederacher D, Deissler H; German Consortium for Hereditary Breast and Ovarian Cancer (GCHBOC), Spurdle AB, Chen X, Waddell N, Cloonan N; Kathleen Cuningham Consortium for Research into Familial Breast Cancer (kConFab), Kirchhoff T, Offit K, Friedman E, Kaufrnann B, Laitman Y, Galore G, Rennert G, Lejbkowicz F, Raskin L, Andrulis IL, Ilyushik E, Ozcelik H, Devilee P, Vreeswijk MP, Greene MH, Prindiville SA, Osorio A, Benitez J, Zikan M, Szabo CI, Kilpivaara O, Nevanlinna H, Hamann U, Durocher F, Arason A, Couch FJ, Easton DF, Chenevix-Trench G; Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). RAD51 135G->C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am J Hum Genet. 2007; 81(6): 1186-200.
85 Wolf I, Laitman Y, Rubinek T, Abramovitz L, Novikov I, Beeri R, Kuro-O M, Koeffler HP, Catane R, Freedman LS, Levy-Lahad E, Karlan BY, Friedman E, Kaufman B. Functional variant of KLOTHO: a breast cancer risk modifier among BRCAl mutation carriers of Ashkenazi origin. Oncogene. 2010; 29(l):26-33.
86 Yarden RI, Friedman E, Metsuyanim S, Olender T, Ben-Asher E, Papa MZ. Single-nucleotide polymorphisms in the p53 pathway genes modify cancer risk in BRCAl and BRCA2 carriers of Jewish-Ashkenazi descent. Mol Carcinog. 2010 Mar 19.
87 Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R; SEARCH collaborators, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X; kConFab; AOCS Management Group, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE, Cox DR, Ponder BA. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007; 447(7148): 1087-93.
88 Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS,

191
Fraumeni JF Jr, Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007; 39(7):870-4.
89 Stacey SN, Manolescu A, Sulem P, Rafiiar T, Gudmundsson J, Gudjonsson SA, Masson G, Jakobsdottir M, Thorlacius S, Helgason A, Aben KK, Strobbe LJ, Albers-Akkers MT, Swinkels DW, Henderson BE, Kolonel LN, Le Marchand L, Millastre E, Andres R, Godino J, Garcia-Prats MD, Polo E, Très A, Mouy M, Saemundsdottir J, Backman VM, Gudmundsson L, Kristjansson K, Bergthorsson JT, Kostic J, Frigge ML, Geller F, Gudbjartsson D, Sigurdsson H, Jônsdôttir T, Hrafhkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Jonsson T, von Hoist S, Werelius B, Margolin S, Lindblom A, Mayordomo JI, Haiman CA, Kiemeney LA, Johannsson OT, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Common variants on chromosomes 2q35 and I6ql2 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007; 39(7):865-9.
90 Stacey SN, Manolescu A, Sulem P, Thorlacius S, Gudjonsson SA, Jonsson GF, Jakobsdottir M, Bergthorsson JT, Gudmundsson J, Aben KK, Strobbe LJ, Swinkels DW, van Engelenburg KC, Henderson BE, Kolonel LN, Le Marchand L, Millastre E, Andres R, Saez B, Lambea J, Godino J, Polo E, Très A, Picelli S, Rantala J, Margolin S, Jonsson T, Sigurdsson H, Jônsdôttir T, Hrafhkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Sveinsdottir SG, Alexiusdottir K, Saemundsdottir J, Sigurdsson A, Kostic J, Gudmundsson L, Kristjansson K, Masson G, Fackenthal JD, Adebamowo C, Ogundiran T, Olopade OI, Haiman CA, Lindblom A, Mayordomo JI, Kiemeney LA, Gulcher JR, Rafnar T, Thorsteinsdottir U, Johannsson OT, Kong A, Stefansson K. Common variants on chromosome 5pI2 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2008; 40(6):703-6.
91 Antoniou AC, Spurdle AB, Sinilnikova OM, Healey S, Poo ley KA, Schmutzler RK, Versmold B, Engel C, Meindl A, Arnold N, Hofmann W, Sutter C, Niederacher D, Deissler H, Caldes T, Kàmpjàrvi K, Nevanlinna H, Simard J, Beesley J, Chen X; Kathleen Cuningham Consortium for Research into Familial Breast Cancer, Neuhausen SL, Rebbeck TR, Wagner T, Lynch HT, Isaacs C, Weitzel J, Ganz PA, Daly MB, Tomlinson G, Olopade OI, Blum JL, Couch FJ, Peterlongo P, Manoukian S, Barile M, Radice P, Szabo CI, Pereira LH, Greene MH, Rennert G, Lejbkowicz F, Barnett-Griness O, Andrulis IL, Ozcelik H; OCGN, Gerdes AM, Caligo MA, Laitman Y, Kaufman B, Milgrom R, Friedman E; Swedish BRCAl and BRCA2 study collaborators, Domchek SM, Nathanson KL, Osorio A, Llort G, Milne RL, Benitez J, Hamann U, Hogervorst FB, Manders P, Ligtenberg MJ, van den Ouweland AM; DNA-HEBON collaborators, Peock S, Cook M, Platte R, Evans DG, Eeles R, Pichert G, Chu C, Eccles D, Davidson R, Douglas F; EMBRACE, Godwin AK, Barjhoux L, Mazoyer S, Sobol H, Bourdon V, Eisinger F, Chompret A, Capoulade C, Bressac-de Paillerets B, Lenoir GM, Gauthier-Villars M, Houdayer C, Stoppa-Lyonnet D; GEMO, Chenevix-Trench G, Easton DF; CIMBA. Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCAl and BRCA2 mutation carriers. Am J Hum Genet. 2008; 82(4):937-48.
92 Thorlacius S, Olafsdottir G, Tryggvadottir L, Neuhausen S, Jonasson JG, Tavtigian SV, Tulinius H, Ogmundsdottir HM, Eyfjord JE. A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phénotypes. Nat Genet. 1996; 13(1): 117-9.
93 Schubert EL, Lee MK, Mefford HC, Argonza RH, Morrow JE, Hull J, Dann JL, King MC. BRCA2 in American families with four or more cases of breast or ovarian cancer: recurrent and novel mutations, variable expression, penetrance, and the possibility of families whose cancer is not attributable to BRCAl orBRCA2. Am J Hum Genet. 1997; 60(5): 1031-40.
94 Ozçelik H, Schmocker B, Di Nicola N, Shi XH, Langer B, Moore M, Taylor BR, Narod SA, Darlington G, Andrulis IL, Gallinger S, Redston M. Germline BRCA2 6174delT mutations in Ashkenazi Jewish pancreatic cancer patients. Nat Genet. 1997; 16(1): 17-8.
95 Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, Tang J, Li S, Zhang S, Shaw PA, Narod SA. Population BRCAl and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst. 2006; 98(23): 1694-706.
96 Evans DG, Wu CL, Birch JM. BRCA2: a cause of Li-Fraumeni-like syndrome. J Med Genet. 2008; 45(l):62-3.

192
97 Howlett NG, Taniguchi T, Oison S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andréa AD. Biallelic inactivation ofBRCA2 in Fanconi anemia. Science. 2002; 297(5581):606-9.
98 Meyer S, Fergusson WD, Oostra AB, Medhurst AL, Waisfisz Q, de Winter JP, Chen F, Carr TF, Clayton-Smith J, Clancy T, Green M, Barber L, Eden OB, Will AM, Joenje H, Taylor GM. A cross-linker-sensitive myeloid leukemia cell line from a 2-year-old boy with severe Fanconi anemia and biallelic FANCD1/BRCA2 mutations. Genes Chromosomes Cancer. 2005; 42(4):404-15.
99 Offit K, Levran O, Mullaney B, Mah K, Nafa K, Batish SD, Diotti R, Schneider H, Deffenbaugh A, Scholl T, Proud VK, Robson M, Norton L, Ellis N, Hanenberg H, Auerbach AD. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J Natl Cancer Inst. 2003; 95(20): 1548-51.
100 Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, Satagopan J, Ben-Porat L, Mah K, Batish SD, Kutler DI, MacMillan ML, Hanenberg H, Auerbach AD. Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood. 2004; 103(8):3226-9.
101 Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007; 44(1): 1-9.
102 The Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91(15):1310-6.
103 van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, Hoogerbrugge N, Verhoef S, Vasen HF, Ausems MG, Menko FH, Gomez Garcia EB, Klijn JG, Hogervorst FB, van Houwelingen JC, van't Veer LJ, Rookus MA, van Leeuwen FE; Netherlands Collaborative Group on Hereditary Breast Cancer (HEBON). Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet. 2005; 42(9):711-9.
104 Li FP, Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969; 71(4):747-52.
105 Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997; 88(3):323-31. 106 Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ,
Tainsky MA, Friend SH. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990; 250(4985): 1233-8.
107 Fisher DE. The p53 tumor suppressor: critical regulator of life & death in cancer. Apoptosis. 2001; 6(l-2):7-15.
108 Garber JE, Goldstein AM, Kantor AF, Dreyfus MG, Fraumeni JF Jr, Li FP. Follow-up study of twenty-four families with Li-Fraumeni syndrome. Cancer Res. 1991; 51(22):6094-7.
109 Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, Eden OB, Varley JM. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. 2001; 20(34):4621-8.
110 Chompret A, Brugières L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, Hua D, Ligot L, Dondon MG, Bressac-de Paillerets B, Frébourg T, Lemerle J, Bonaïti-Pellié C, Feunteun J. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer. 2000; 82(12):1932-7.
111 Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles RA. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003; 63(20):6643-50.
112 Rapakko K, Allinen M, Syrjâkoski K, Vahteristo P, Huusko P, Vâhâkangas K, Eerola H, Kainu T, Kallioniemi OP, Nevanlinna H, Winqvist R. Germline TP53 alterations in Finnish breast cancer families are rare and occur at conserved mutation-prone sites. Br J Cancer. 2001 ; 84(1 ): 116-9.
113 Sidransky D, Tokino T, Helzlsouer K, Zehnbauer B, Rausch G, Shelton B, Prestigiacomo L, Vogelstein B, Davidson N. Inheritedp53 gene mutations in breast cancer. Cancer Res. 1992; 52(10):2984-6.
114 Lloyd KM, Dennis M Cowden's disease. A possible new symptom complex with multiple system involvement. Ann Intern Med. 1963; 58:136-42.

193
115 Brownstein MH, Wolf M, Bikowski JB. Cowden's disease: a cutaneous marker of breast cancer. Cancer. 1978; 4I(6):2393-8.
116 Starink TM, van der Veen JP, Arwert F, de Waal LP, de Lange GG, Gille JJ, Eriksson AW. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet. 1986; 29(3):222-33.
117 Longy M, Lacombe D. Cowden disease. Report of a family and review. Ann Genet. 1996; 39(l):35-42. 118 Fackenthal JD, Marsh DJ, Richardson AL, Cummings SA, Eng C, Robinson BG, Olopade OI. Male
breast cancer in Cowden syndrome patients with germline PTEN mutations. J Med Genet. 2001; 38(3): 159-64.
119 Zhou XP, Waite KA, Pilarski R, Hampel H, Fernandez MJ, Bos C, Dasouki M, Feldman GL, Greenberg LA, Ivanovich J, Matloff E, Patterson A, Pierpont ME, Russo D, Nassif NT, Eng C. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am J Hum Genet. 2003; 73(2):404-ll.
120 Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003; 22(3): 183-98. 121 Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, Edelman E, Platzer P, Orloff MS, Waite KA, Eng C.
Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. 2008; 83(2):261-8.
122 DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008; 7(1): 11-20.
123 Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z, Liaw D, Caron S, Duboué B, Lin AY, Richardson AL, Bonnetblanc JM, Bressieux JM, Cabarrot-Moreau A, Chompret A, Démange L, Eeles RA, Yahanda AM, Fearon ER, Flicker JP, Gorlin RJ, Hodgson SV, Huson S, Lacombe D, LePart F, Odent S Toulouse C, Olopade OI, Sobol H, Tishler S, Woods CG, Robinson BG, Weber HC, Parson R, Peacocke M, Longy M, Eng, C. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998; 7(3):507-15.
124 Chen J, Lindblom P, Lindblom A. A study of the PTEN/MMAC1 gene in 136 breast cancer families. Hum Genet. 1998; 102(l):124-5.
125 Carroll BT, Couch FJ, Rebbeck TR, Weber BL. Polymorphisms in PTEN in breast cancer families. J Med Genet. 1999; 36(2):94-6.
126 Guénard F, Labrie Y, Ouellette G, Beauparlant CJ, Bessette P, Chiquette J, Laframboise R, Lépine J, Lesperance B, Pichette R, Plante M, Durocher F; INHERIT BRCAs. Germline mutations in the breast cancer susceptibility gene PTEN are rare in high-risk non-BRCAl/2 French Canadian breast cancer families. Fam Cancer. 2007; 6(4):483-90.
127 Marsh DJ, Kum JB, Lunetta KL, Bennett M J, Gorlin RJ, Ahmed SF, Bodurtha J, Crowe C, Curtis MA, Dasouki M, Dunn T, Feit H, Geraghty MT, Graham JM Jr, Hodgson SV, Hunter A, Korf BR, Manchester D, Miesfeldt S, Murday VA, Nathanson KL, Parisi M, Pober B, Romano C, Tolmie JL, Trembath R, Dahia PLM, Eng C. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999; 8(8):1461-72.
128 Peutz JLA. Over een zeer merkwaardige, gecombineerde familiaire polyposis van de slijmliezen van den tractus intestinalis met die van de neuskeelholte en gepaard met eigenaardige pigmentaties van huid-en slijmvliezen. Ned Maandschr v Gen 10 1921, pp. 134-146.
129 Jeghers H, McKusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949 Dec 29;241(26):1031-6.
130 Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, Cruz-Correa M, Offerhaus JA. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000; 119(6):1447-53.
131 Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott RJ, Lim W, Trimbath JD, Giardiello FM, Gruber SB, Offerhaus GJ, de Rooij FW, Wilson JH,

194
Hansmann A, Môslein G, Royer-Pokora B, Vogel T, Phillips RK, Spigelman AD, Houlston RS. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006; 12(10)3209-15.
132 Lim W, Hearle N, Shah B, Murday V, Hodgson SV, Lucassen A, Eccles D, Talbot I, Neale K, Lim AG, O'Donohue J, Donaldson A, Macdonald RC, Young ID, Robinson MH, Lee PW, Stoodley BJ, Tomlinson I, Alderson D, Holbrook AG, Vyas S, Swarbrick ET, Lewis AA, Phillips RK, Houlston RS. Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br J Cancer. 2003; 89(2)308-13.
133 Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hôglund P, Jarvinen H, Kristo P, Pelin K, Ridanpâa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998; 391(6663): 184-7.
134 Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998; 18(1)38-43.
135 Baas AF, Smit L, Clevers H. LKB1 tumor suppressor protein: PARtaker in cell polarity. Trends Cell Biol. 2004; 14(6)312-9.
136 Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006; 75:137-63.
137 Aretz S, Stienen D, Uhlhaas S, Loff S, Back W, Pagenstecher C, McLeod DR, Graham GE, Mangold E, Santer R, Propping P, Friedl W. High proportion of large genomic STK 11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005; 26(6):513-9.
138 Volikos E, Robinson J, Aittomaki K, Mecklin JP, Jârvinen H, Westerman AM, de Rooji FW, Vogel T, Moeslein G, Launonen V, Tomlinson IP, Silver AR, Aaltonen LA. LKB1 exonic and whole gene deletions are a common cause of Peutz-Jeghers syndrome. J Med Genet. 2006; 43(5):el8.
139 Chow E, Meldrum CJ, Crooks R, Macrae F, Spigelman AD, Scott RJ. An updated mutation spectrum in an Australian series of PJS patients provides further evidence for only one gene locus. Clin Genet. 2006; 70(5):409-14.
140 Hezel AF, Bardeesy N. LKB1; linking cell structure and tumor suppression. Oncogene. 2008; 27(55):6908-19.
141 Esteller M, Avizienyte E, Corn PG, Lothe RA, Baylin SB, Aaltonen LA, Herman JG. Epigenetic inactivation ofLKBl in primary tumors associated with the Peutz-Jeghers syndrome. Oncogene. 2000; 19(l):164-8.
142 Zanghieri G, Di Gregorio C, Sacchetti C, Fante R, Sassatelli R, Cannizzo G, Carriero A, Ponz de Leon M. Familial occurrence of gastric cancer in the 2-year experience of a population-based registry. Cancer. 1990; 66(9):2047-51.
143 Cisco RM, Ford JM, Norton JA. Hereditary diffuse gastric cancer: implications of genetic testing for screening and prophylactic surgery. Cancer. 2008; 113(7): 1850-6.
144 Gayther SA, Gorringe KL, Ramus SJ, Huntsman D, Roviello F, Grehan N, Machado JC, Pinto E, Seruca R, Hailing K, MacLeod P, Powell SM, Jackson CE, Ponder BA, Caldas C. Identification of germ-line E-cadherin mutations in gastric cancer families of European origin. Cancer Res. 1998; 58(18):4086-9.
145 Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature. 1998; 392(6674):402-5.
146 Halbleib JM, Nelson WJ. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006; 20(23)3199-214.
147 Brooks-Wilson AR, Kaurah P, Suriano G, Leach S, Senz J, Grehan N, Butterfield YS, Jeyes J, Schinas J, Bacani J, Kelsey M, Ferreira P, MacGillivray B, MacLeod P, Micek M, Ford J, Foulkes W, Australie K, Greenberg C, LaPointe M, Gilpin C, Nikkei S, Gilchrist D, Hughes R, Jackson CE, Monaghan KG, Oliveira MJ, Seruca R, Gallinger S, Caldas C, Huntsman D. Germline E-cadherin mutations in

195
hereditary diffuse gastric cancer: assessment of 42 new families and review of genetic screening criteria. J Med Genet. 2004; 41(7):508-17.
148 Kaurah P, MacMillan A, Boyd N, Senz J, De Luca A, Chun N, Suriano G, Zaor S, Van Manen L, Gilpin C, Nikkei S, Connolly-Wilson M, Weissman S, Rubinstein WS, Sebold C, Greenstein R, Stroop J, Yim D, Panzini B, McKinnon W, Greenblatt M, Wirtzfeld D, Fontaine D, Coit D, Yoon S, Chung D, Lauwers G, Pizzuti A, Vaccaro C, Redal MA, Oliveira C, Tischkowitz M, Olschwang S, Gallinger S, Lynch H, Green J, Ford J, Pharoah P, Fernandez B, Huntsman D. Founder and recurrent CDHl mutations in families with hereditary diffuse gastric cancer. JAMA. 2007; 297(21):2360-72.
149 Pharoah PD, Guilford P, Caldas C; International Gastric Cancer Linkage Consortium. Incidence of gastric cancer and breast cancer in CDHl (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology. 2001; 121(6): 1348-53.
150 Masciari S, Larsson N, Senz J, Boyd N, Kaurah P, Kandel MJ, Harris LN, Pinheiro HC, Troussard A, Miron P, Tung N, Oliveira C, Collins L, Schnitt S, Garber JE, Huntsman D. Germline E-cadherin mutations in familial lobular breast cancer. J Med Genet. 2007; 44(11):726-31.
151 Schrader KA, Masciari S, Boyd N, Wiyrick S, Kaurah P, Senz J, Burke W, Lynch HT, Garber JE, Huntsman DG. Hereditary diffuse gastric cancer: association with lobular breast cancer. Fam Cancer. 2008; 7(l):73-82.
152 Yoder BJ, Wilkinson EJ, Massoll NA. Molecular and morphologic distinctions between infiltrating ductal and lobular carcinoma of the breast. Breast J. 2007; 13(2): 172-9.
153 Syllaba L, Henner K: Contribution à l'étude de l'indépendance de l'athétose double idiopathique et congénitale. Atteinte familiale, syndrome dystrophique, signe du résau vasculaire conjonctival, intégrité psychique. Revue neurologique, 1926, 1:541-560.
154 Boder E, Sedgwick RP: Ataxia-telangiectasia: a familial syndrome of progressive cerebellar ataxia, oculcutaneous telangiectasia and frequent pulmonary infection. Pediatrics, 1958; 21(4):526-554.
155 Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst. 1986; 77(l):89-92.
156 Ahmed M, RahmanN. ATM and breast cancer susceptibility. Oncogene. 2006; 25(43):5906-ll. 157 Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S,
Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995; 268(5218):1749-53.
158 Swift M, Sholman L, Perry M, Chase C. Malignant neoplasms in the families of patients with ataxia-telangiectasia. Cancer Res. 1976; 36(1):209-15.
159 Swift M, Reitnauer PJ, Morrell D, Chase CL. Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med. 1987; 316(21): 1289-94.
160 Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005; 97(ll):813-22.
161 Olsen JH, Hahnemann JM, Borresen-Dale AL, Tretli S, Kleinerman R, Sankila R, Hammarstrôm L, Robsahm TE, Kàâriainen H, Bregârd A, Brendum-Nielsen K, Yuen J, Tucker M. Breast and other cancers in 1445 blood relatives of 75 Nordic patients with ataxia telangiectasia. Br J Cancer. 2005; 93(2):260-5.
162 Szabo CI, Schutte M, Broeks A, Houwing-Duistermaat JJ, Thorstenson YR, Durocher F, Oldenburg RA, Wasielewski M, Odefrey F, Thompson D, Floore AN, Kraan J, Klijn JG, van den Ouweland AM, Wagner TM, Devilee P, Simard J, van 't Veer LJ, Goldgar DE, Meijers-Heijboer H. Are ATM mutations 7271T->G and IVS10-6T->G really high-risk breast cancer-susceptibility alleles? Cancer Res. 2004; 64(3):840-3.
163 Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D; Breast Cancer Susceptibility Collaboration (UK),

196
Easton DF, Stratton MR, Rahman N. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006; 38(8):873-5.
164 Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999; 286(5449):2528-31.
165 Sodha N, Houlston RS, Bullock S, Yuille MA, Chu C, Turner G, Eeles RA. Increasing evidence that germline mutations in CHEK2 do not cause Li-Fraumeni syndrome. Hum Mutat. 2002; 20(6):460-2.
166 Matsuoka S, Huang M, Elledge S J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998; 282(5395): 1893-7.
167 Lee JS, Collins KM, Brown AL, Lee CH, Chung JH. hCdsl-mediated phosphorylation of BRCAl regulates the DNA damage response. Nature. 2000; 404(6774):201-4.
168 Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCdsl functions as a DNA damage checkpoint in G(l) by stabilizing p53. Genes Dev. 2000; 14(3):278-88.
169 Weischer M, Bojesen SE, Ellervik C, Tybjaerg-Hansen A, Nordestgaard BG. CHEK2*1 WOdelC genotyping for clinical assessment of breast cancer risk: meta-analyses of 26,000 patient cases and 27,000 controls. J Clin Oncol. 2008; 26(4):542-8.
170 CHEK2 Breast Cancer Case-Control Consortium. CHEK2*1 WOdelC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet. 2004; 74(6): 1175-82.
171 Zhang S, Phelan CM, Zhang P, Rousseau F, Ghadirian P, Robidoux A, Foulkes W, Hamel N, McCready D, Trudeau M, Lynch H, Horsman D, De Matsuda ML, Aziz Z, Gomes M, Costa MM, Liede A, Poll A, Sun P, Narod SA. Frequency of the CHEK2 1 WOdelC mutation among women with breast cancer: an international study. Cancer Res. 2008; 68(7):2154-7.
172 Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, Elstrodt F, van Duijn C, Bartels C, Meijers C, Schutte M, McGuffog L, Thompson D, Easton D, Sodha N, Seal S, Barfoot R, Mangion J, Chang-Claude J, Eccles D, Eeles R, Evans DG, Houlston R, Murday V, Narod S, Peretz T, Peto J, Phelan C, Zhang HX, Szabo C, Devilee P, Goldgar D, Futreal PA, Nathanson KL, Weber B, Rahman N, Stratton MR; CHEK2-Breast Cancer Consortium. Low-penetrance susceptibility to breast cancer due to CHEK2(*)ll0OdelC in noncarriers of BRCAl or BRCA2 mutations. Nat Genet. 2002; 31(l):55-9.
173 Wasielewski M, den Bakker MA, van den Ouweland A, Meijer-van Gelder ME, Portengen H, Klijn JG, Meijers-Heijboer H, Foekens JA, Schutte M. CHEK2 HOOdelC and male breast cancer in the Netherlands. Breast Cancer Res Treat. 2009; 116(20)397-400.
174 Neuhausen S, Dunning A, Steele L, Yakumo K, Hoffman M, Szabo C, Tee L, Baines C, Pharoah P, Goldgar D, Easton D. Role of CHEK2*1 WOdelC in unselected series of non-BRCAl/2 male breast cancers. Int J Cancer. 2004; 108(3):477-8.
175 Sodha N, Wilson C, Bullock SL, Phillimore H, Houlston RS, Eeles RA. Analysis of familial male breast cancer for germline mutations in CHEK2. Cancer Lett. 2004; 215(2): 187-9.
176 Ohayon T, Gal I, Baruch RG, Szabo C, Friedman E. CHEK2*1 WOdelC and male breast cancer risk in Israel. Int J Cancer. 2004; 108(3):479-80.
177 Cybulski C, Huzarski T, Byrski T, Gronwald J, Debniak T, Jakubowska A, Gôrski B, Wokolorczyk D, Masojc B, Narod SA, Lubinski J. Estrogen receptor status in CHEK2-positive breast cancers: implications for chemoprevention. Clin Genet. 2009; 75(l):72-8.
178 Cybulski C, Wokolorczyk D, Huzarski T, Byrski T, Gronwald J, Gôrski B, Debniak T, Masojc B, Jakubowska A, Gliniewicz B, Sikorski A, Stawicka M, Godlewski D, Kwias Z, Antczak A, Krajka K, Lauer W, Sosnowski M, Sikorska-Radek P, Bar K, Klijer R, Zdrojowy R, Malkiewicz B, Borkowski A, Borkowski T, Szwiec M, Narod SA, Lubinski J. A large germline deletion in the Chek2 kinase gene is associated with an increased risk of prostate cancer. J Med Genet. 2006; 43(11):863-6.
179 Cybulski C, Huzarski T, Gôrski B, Masojc B, Mierzejewski M, Debniak T, Gliniewicz B, Matyjasik J, Zlowocka E, Kurzawski G, Sikorski A, Posmyk M, Szwiec M, Czajka R, Narod SA, Lubinski J. A

197
novel founder CHEK2 mutation is associated with increased prostate cancer risk. Cancer Res. 2004; 64(8):2677-9.
180 Dong X, Wang L, Taniguchi K, Wang X, Cunningham JM, McDonnell SK, Qian C, Marks AF, Slager SL, Peterson BJ, Smith DI, Cheville JC, Blute ML, Jacobsen SJ, Schaid DJ, Tindall DJ, Thibodeau SN, Liu W. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet. 2003; 72(2):270-80.
181 Kilpivaara O, Alhopuro P, Vahteristo P, Aaltonen LA, Nevanlinna H. CHEK2 I157T associates with familial and sporadic colorectal cancer. J Med Genet. 2006; 43(7):e34.
182 Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM. BACH1, a novel helicase-like protein, interacts directly with BRCAl and contributes to its DNA repair function. Cell. 2001; 105(l):149-60.
183 Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003; 302(5645):639-42.
184 Peng M, Litman R, Jin Z, Fong G, Cantor SB. BACH1 is a DNA repair protein supporting BRCAl damage response. Oncogene. 2006; 25(15):2245-53.
185 Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D; Breast Cancer Susceptibility Collaboration (UK), Easton DF, Stratton MR, Rahman N. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006; 38(11): 1239-41.
186 Guénard F, Labrie Y, Ouellette G, Joly Beauparlant C, Simard J, Durocher F; INHERIT BRCAs. Mutational analysis of the breast cancer susceptibility gene BRIP 1/BACH 1/FANCJ in high-risk non-BRCA1/BRCA2 breast cancer families. J Hum Genet. 2008; 53(7):579-91.
187 Kote-Jarai Z, Jugurnauth S, Mulholland S, Leongamornlert DA, Guy M, Edwards S, Tymrakiewitcz M, O'Brien L, Hall A, Wilkinson R, Al Olama AA, Morrison J, Muir K, Neal D, Donovan J, Hamdy F, Easton DF; The UKGPCS Collaborators, The British Association of Urological Surgeons' Section of Oncology9, Eeles R. A recurrent truncating germline mutation in the BRIP1/FANCJ gene and susceptibility to prostate cancer. Br J Cancer. 2009 Jan 27;100(2):426-30.
188 Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005; 8(3):255-65.
189 Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barrai S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD. The BRCAl-interacting helicase BR1P1 is deficient in Fanconi anemia. Nat Genet. 2005; 37(9):931-3.
190 Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet. 2005; 37(9):934-5.
191 Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch F J, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006; 22(6):719-29.
192 Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D; Breast Cancer Susceptibility Collaboration (UK), Easton DF, Stratton MR. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007; 39(2): 165-7.
193 Erkko H, Xia B, Nikkila J, Schleutker J, Syrjâkoski K, Mannermaa A, Kallioniemi A, Pylkâs K, Karppinen SM, Rapakko K, Miron A, Sheng Q, Li G, Mattila H, Bell DW, Haber DA, Grip M, Reiman M, Jukkola-Vuorinen A, Mustonen A, Kere J, Aaltonen LA, Kosma VM, Kataja V, Soini Y, Drapkin RI, Livingston DM, Winqvist R. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007; 446(7133)316-9.
194 Erkko H, Dowty JG, Nikkilâ J, Syrjâkoski K, Mannermaa A, Pylkâs K, Southey MC, Holli K, Kallioniemi A, Jukkola-Vuorinen A, Kataja V, Kosma VM, Xia B, Livingston DM, Winqvist R,

198
Hopper JL. Penetrance analysis of the PALB2 c.l592delT founder mutation. Clin Cancer Res. 2008;14(14):4667-71.
195 Heikkinen T, Kârkkâinen H, Aaltonen K, Milne RL, Heikkilâ P, Aittomàki K, Blomqvist C, Nevanlinna H. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res. 2009;15(9)3214-22.
196 Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzler KW, Vogelstein B, Eshleman JR, Goggins M, Klein AP. Exomic Sequencing Identifies PALB2 as a Pancreatic Cancer Susceptibility Gene. Science. 2009; 324(5924):217.
197 Dansonka-Mieszkowska A, Kluska A, Moes J, Dabrowska M, Nowakowska D, Niwinska A, Derlatka P, Cendrowski K, Kupryjanczyk J. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med Genet. 2010;11:20.
198 Chen P, Liang J, Wang Z, Zhou X, Chen L, Li M, Xie D, Hu Z, Shen H, Wang H. Association of common PALB2 polymorphisms with breast cancer risk: a case-control study. Clin Cancer Res. 2008; 14(18):5931-7.
199 Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, KellyP, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007; 39(2): 162-4.
200 Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Liera J, Wang W, Livingston DM, Joenje H, de Winter JP. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007; 39(2): 159-61.
201 Garcia-Closas M, Chanock S. Genetic susceptibility loci for breast cancer by estrogen receptor status. Clin Cancer Res. 2008; 14(24):8000-9.
201a Barnholtz-Sloan JS, Shetty PB, Guan X, Nyante SJ, Luo J, Brennan DJ, Millikan RC. FGFR2 and Other Loci Identified in Genome-Wide Association Studies are Associated with Breast Cancer in African American and Younger Women. Carcinogenesis. 2010 Jun 16. [Epub ahead of print]
202 Grose R, Dickson C. Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev. 2005; 16(2): 179-86.
203 Moffa AB, Tannheimer SL, Ethier SP. Transforming potential of alternatively spliced variants of fibroblast growth factor receptor 2 in human mammary epithelial cells. Mol Cancer Res. 2004; 2(ll):643-52.
204 Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O'Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Bimey E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007; 446(7132): 153-8.
205 Meyer KB, Maia AT, O'Reilly M, Teschendorff AE, Chin SF, Caldas C, Ponder BA. Allele-specific upregulation ofFGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008; 6(5):el08.
206 O'Flaherty E, Kaye J. TOX defines a conserved subfamily of HMG-box proteins. BMC Genomics. 2003; 4(1):13.
207 Smid M, Wang Y, Klijn JG, Sieuwerts AM, Zhang Y, Atkins D, Martens JW, Foekens JA. Genes associated with breast cancer metastatic to bone. J Clin Oncol. 2006; 24(15):2261-7.
208 James JJ, Evans AJ, Pinder SE, Gutteridge E, Cheung KL, Chan S, Robertson JF. Bone metastases from breast carcinoma: histopathological - radiological correlations and prognostic features. Br J Cancer. 2003; 89(4):660-5.

199
209 Uhlik MT, Abell AN, Cuevas BD, Nakamura K, Johnson GL. Wiring diagrams of MAPK regulation by MEKK1, 2, and 3. Biochem Cell Biol. 2004; 82(6):658-63.
210 Liu L, Cara DC, Kaur J, Raharjo E, Mullaly SC, Jongstra-Bilen J, Jongstra J, Kubes P. LSP1 is an endothelial gatekeeper of leukocyte transendothelial migration. J Exp Med. 2005; 201(3):409-18.
211 Antoniou AC, Sinilnikova OM, McGuffog L, Healey S, Nevanlinna H, Heikkinen T, Simard J, Spurdle AB, Beesley J, Chen X; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer, Neuhausen SL, Ding YC, Couch FJ, Wang X, Fredericksen Z, Peterlongo P, Peissel B, Bonanni B, Viel A, Bernard L, Radice P, Szabo CI, Foretova L, Zikan M, Claes K, Greene MH, Mai PL, Rennert G, Lejbkowicz F, Andrulis IL, Ozcelik H, Glendon G; OCGN, Gerdes AM, Thomassen M, Sunde L, Caligo MA, Laitman Y, Kontorovich T, Cohen S, Kaufman B, Dagan E, Baruch RG, Friedman E, Harbst K, Barbany-Bustinza G, Rantala J, Ehrencrona H, Karlsson P, Domchek SM, Nathanson KL, Osorio A, Blanco I, Lasa A, Benitez J, Hamann U, Hogervorst FB, Rookus MA, Collée JM, Devilee P, Ligtenberg MJ, van der Luijt RB, Aalfs CM, Waisfisz Q, Wijnen J, van Roozendaal CE; HEBON, Peock S, Cook M, Frost D, Oliver C, Platte R, Evans DG, Lalloo F, Eeles R, Izatt L, Davidson R, Chu C, Eccles D, Cole T, Hodgson S; EMBRACE, Godwin AK, Stoppa-Lyonnet D, Buecher B, Leone M, Bressac-de Paillerets B, Remenieras A, Caron O, Lenoir GM, Sevenet N, Longy M, Ferrer SF, Prieur F; GEMO, Goldgar D, Miron A, John EM, Buys SS, Daly MB, Hopper JL, Terry MB, Yassin Y; Breast Cancer Family Registry, Singer C, Gschwantler-Kaulich D, Staudigl C, Hansen TO, Barkardottir RB, Kirchhoff T, Pal P, Kosarin K, Offit K, Piedmonte M, Rodriguez GC, Wakeley K, Boggess JF, Basil J, Schwartz PE, Blank SV, Toland AE, Montagna M, Casella C, Imyanitov EN, Allavena A, Schmutzler RK, Versmold B, Engel C, Meindl A, Ditsch N, Arnold N, Niederacher D, Deissler H, Fiebig B, Suttner C, Schônbuchner I, Gadzicki D, Caldes T, de la Hoya M, Pooley ICA, Easton DF, Chenevix-Trench G; CIMBA. Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCAl and BRCA2 mutation carriers. Hum Mol Genet. 2009 Nov 15;18(22):4442-56.
212 Ainscough JF, Dandolo L, Surani MA. Appropriate expression of the mouse H19 gene utilises three or more distinct enhancer regions spread over more than 130 kb. Mech Dev. 2000; 91(1-2)365-8.
213 van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002; 415(6871):530-6.
214 Ghoussaini M, Song H, Koessler T, Al Olama AA, Kote-Jarai Z, Driver KE, Pooley KA, Ramus S J, Kjaer SK, Hogdall E, DiCioccio RA, Whittemore AS, Gayther SA, Giles GG, Guy M, Edwards SM, Morrison J, Donovan JL, Hamdy FC, Dearnaley DP, Ardern-Jones AT, Hall AL, O'Brien LT, Gehr-Swain BN, Wilkinson RA, Brown PM, Hopper JL, Neal DE, Pharoah PD, Ponder BA, Eeles RA, Easton DF, Dunning AM; UK Genetic Prostate Cancer Study Collaborators/British Association of Urological Surgeons' Section of Oncology; UK ProtecT Study Collaborators. Multiple loci with different cancer specificities within the 8q24 gene desert. J Natl Cancer Inst. 2008; 100(13):962-6.
215 Fletcher O, Johnson N, Gibson L, Coupland B, Fraser A, Leonard A, dos Santos Silva I, Ashworth A, Houlston R, Peto J. Association of genetic variants at 8q24 with breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2008; 17(3): 702-5.
216 Mclnerney N, Colleran G, Rowan A, Walther A, Barclay E, Spain S, Jones AM, Tuohy S, Curran C, Miller N, Kerin M, Tomlinson I, Sawyer E. Low penetrance breast cancer predisposition SNPs are site specific. Breast Cancer Res Treat. 2009; 119(2):151-9.
217 Schumacher FR, Feigelson HS, Cox DG, Haiman CA, Albanes D, Buring J, Calle EE, Chanock S J, Colditz GA, Diver WR, Dunning AM, Freedman ML, Gaziano JM, Giovannucci E, Hankinson SE, Hayes RB, Henderson BE, Hoover RN, Kaaks R, Key T, Kolonel LN, Kraft P, Le Marchand L, Ma J, Pike MC, Riboli E, Stampfer MJ, Stram DO, Thomas G, Thun MJ, Travis R, Virtamo J, Andriole G, Gelmann E, Willett WC, Hunter DJ. A common 8q24 variant in prostate and breast cancer from a large nested case-control study. Cancer Res. 2007; 67(7):2951-6.
218 Amundadottir LT, Sulem P, Gudmundsson J, Helgason A, Baker A, Agnarsson BA, Sigurdsson A, Benediktsdottir KR, Cazier JB, Sainz J, Jakobsdottir M, Kostic J, Magnusdottir DN, Ghosh S, Agnarsson K, Birgisdottir B, Le Roux L, Olafsdottir A, Blondal T, Andresdottir M, Gretarsdottir OS,

200
Bergthorsson JT, Gudbjartsson D, Gylfason A, Thorleifsson G, Manolescu A, Kristjansson K, Geirsson G, Isaksson H, Douglas J, Johansson JE, Baiter K, Wiklund F, Montie JE, Yu X, Suarez BK, Ober C, Cooney KA, Gronberg H, Catalona WJ, Einarsson GV, Barkardottir RB, Gulcher JR, Kong A, Thorsteinsdottir U, Stefansson K. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006; 38(6):652-8.
219 Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafhar T, Bergthorsson JT, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Xu J, Blondal T, Kostic J, Sun J, Ghosh S, Stacey SN, Mouy M, Saemundsdottir J, Backman VM, Kristjansson K, Très A, Partin AW, Albers-Akkers MT, Godino-Ivan Marcos J, Walsh PC, Swinkels DW, Navarrete S, Isaacs SD, Aben KK, Graif T, Cashy J, Ruiz-Echarri M, Wiley KE, Suarez BK, Witjes JA, Frigge M, Ober C, Jonsson E, Einarsson GV, Mayordomo JI, Kiemeney LA, Isaacs WB, Catalona WJ, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007; 39(5):631-7.
220 Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, Wild R, Rowan A, Fielding S, Howarth K; CORGI Consortium, Silver A, Atkin W, Muir K, Logan R, Kerr D, Johnstone E, Sieber O, Gray R, Thomas H, Peto J, Cazier JB, Houlston R A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007; 39(8):984-8.
221 Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hunter DJ, Chanock SJ, Thomas G. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007; 39(5):645-9.
222 Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, Ferretti V, Laflamme P, Sundararajan S, Roumy S, Olivier JF, Robidoux F, Sladek R, Montpetit A, Campbell P, Bezieau S, O'Shea AM, Zogopoulos G, Cotterchio M, Newcomb P, McLaughlin J, Younghusband B, Green R, Green J, Porteous ME, Campbell H, Blanche H, Sahbatou M, Tubacher E, Bonaiti-Pellié C, Buecher B, Riboli E, Kury S, Chanock SJ, Potter J, Thomas G, Gallinger S, Hudson TJ, Dunlop MG. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007; 39(8):989-94.
223 Yeager M, Xiao N, Hayes RB, Bouffard P, Desany B, Burdett L, Orr N, Matthews C, Qi L, Crenshaw A, Markovic Z, Fredrikson KM, Jacobs KB, Amundadottir L, Jarvie TP, Hunter DJ, Hoover R, Thomas G, Harkins TT, Chanock S J. Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers. Hum Genet. 2008; 124(2):161-70.
224 Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, Stacey SN, Gudmundsson J, Jakobsdottir M, Bergthorsson JT, Sigurdsson A, Blondal T, Witjes JA, Vermeulen SH, Hulsbergen-van de Kaa CA, Swinkels DW, Ploeg M, Cornel EB, Vergunst H, Thorgeirsson TE, Gudbjartsson D, Gudjonsson SA, Thorleifsson G, Kristinsson KT, Mouy M, Snorradottir S, Placidi D, Campagna M, Arid C, Koppova K, Gurzau E, Rudnai P, Kellen E, Polidoro S, Guarrera S, Sacerdote C, Sanchez M, Saez B, Valdivia G, Ryk C, de Verdier P, Lindblom A, Golka K, Bishop DT, Knowles MA, Nikulasson S, Petursdottir V, Jonsson E, Geirsson G, Kristjansson B, Mayordomo JI, Steineck G, Porra S, Buntinx F, Zeegers MP, Fletcher T, Kumar R, Matullo G, Vineis P, Kiltie AE, Gulcher JR, Thorsteinsdottir U, Kong A, Rafhar T, Stefansson K. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet. 2008; 40(11):1307-12.
225 Severi G, Hayes VM, Padilla EJ, English DR, Southey MC, Sutherland RL, Hopper JL, Giles GG. The common variant rsl 447295 on chromosome 8q24 and prostate cancer risk: results from an Australian population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2007; 16(3):610-2.
226 Zheng SL, Sun J, Cheng Y, Li G, Hsu FC, Zhu Y, Chang BL, Liu W, Kim JW, Turner AR, Gielzak M, Yan G, Isaacs SD, Wiley KE, Sauvageot J, Chen HS, Gurganus R, Mangold LA, Trock BJ, Gronberg H, Duggan D, Carpten JD, Partin AW, Walsh PC, Xu J, Isaacs WB. Association between two unlinked

201
loci at 8q24 and prostate cancer risk among European Americans. J Natl Cancer Inst. 2007; 99(20): 1525-33.
227 Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH; PSG-PROGENI and GenePD Investigators, Coordinators and Molecular Genetic Laboratories. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009; 124(6):593-605.
228 Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, Vermeire S, Dewit O, de Vos M, Dixon A, Demarche B, Gut I, Heath S, Foglio M, Liang L, Laukens D, Mni M, Zelenika D, Van Gossum A, Rutgeerts P, Belaiche J, Lathrop M, Georges M. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5pl3.1 and modulates expression ofPTGER4. PLoS Genet. 2007; 3(4):e58.
229 Kââb S, Darbar D, van Noord C, Dupuis J, Pfeufer A, Newton-Cheh C, Schnabel R, Makino S, Sinner MF, Kannankeril PJ, Beckmann BM, Choudry S, Donahue BS, Heeringa J, Perz S, Lunetta KL, Larson MG, Levy D, Macrae CA, Ruskin JN, Wacker A, Schômig A, Wichmann HE, Steinbeck G, Meitinger T, Uitterlinden AG, Witteman JC, Roden DM, Benjamin EJ, Ellinor PT. Large scale replication and meta-analysis of variants on chromosome 4q25 associated with atrial fibrillation. Eur Heart J. 2009; 30(7):813-9.
230 Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao H, Stephens RM, Harris TJ, Munroe DJ, Wu X. Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A. 2010 Feb 16;107(7)3001-5.
231 Gold B, Kirchhoff T, Stefanov S, Lautenberger J, Viale A, Garber J, Friedman E, Narod S, Olshen AB, Gregersen P, Kosarin K, Olsh A, Bergeron J, Ellis NA, Klein RJ, Clark AG, Norton L, Dean M, Boyd J, Offit K. Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci USA. 2008; 105(11):4340-5.
232 MacPherson G, Healey CS, Teare MD, Balasubramanian SP, Reed MW, Pharoah PD,Ponder BA, Meuth M, Bhattacharyya NP, Cox A. Association of a common variant of the CASP8 gene with reduced risk of breast cancer. J Natl Cancer Inst. 2004; 96(24): 1866-9.
233 Frank B, Hemminki K, Wappenschmidt B, Meindl A, Klaes R, Schmutzler RK, Bugert P, Untch M, Bartram CR, Burwinkel B. Association of the CASPW V410I variant with reduced familial breast cancer risk and interaction with the CASP8 D302H variant. Carcinogenesis. 2006; 27(3): 606-9.
234 Cox A, Dunning AM, Garcia-Closas M, Balasubramanian S, Reed MW, Pooley KA, Scollen S, Baynes C, Ponder BA, Chanock S, Lissowska J, Brinton L, Peplonska B, Southey MC, Hopper JL, McCredie MR, Giles GG, Fletcher O, Johnson N, dos Santos Silva I, Gibson L, Bojesen SE, Nordestgaard BG, Axelsson CK, Torres D, Hamann U, Justenhoven C, Brauch H, Chang-Claude J, Kropp S, Risch A, Wang-Gohrke S, Schurmann P, Bogdanova N, Dork T, Fagerholm R, Aaltonen K, Blomqvist C, Nevanlinna H, Seal S, Renwick A, Stratton MR, Rahman N, Sangrajrang S, Hughes D, Odefrey F, Brennan P, Spurdle AB, Chenevix-Trench G; Kathleen Cunningham Foundation Consortium for Research into Familial Breast Cancer, Beesley J, Mannermaa A, Hartikainen J, Kataja V, Kosma VM, Couch FJ, Olson JE, Goode EL, Broeks A, Schmidt MK, Hogervorst FB, Van't Veer LJ, Kang D, Yoo KY, Noh DY, Ahn SH, Wedrén S, Hall P, Low YL, Liu J, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Sigurdson AJ, Stredrick DL, Alexander BH, Struewing JP, Pharoah PD, Easton DF; Breast Cancer Association Consortium. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet. 2007; 39(3)352-8.
235 Sun T, Gao Y, Tan W, Ma S, Shi Y, Yao J, Guo Y, Yang M, Zhang X, Zhang Q, Zeng C, Lin D. A six-nucleotide insertion-deletion polymorphism in the CASP8 promoter is associated with susceptibility to multiple cancers. Nat Genet. 2007; 39(5):605-13.
236 Yin M, Yan J, Wei S, Wei Q. CASP8 polymorphisms contribute to cancer susceptibility: evidence from a meta-analysis of 23 publications with 55 individual studies. Carcinogenesis. 2010; 31(5):850-7.
237 Cox DG, Penney K, Guo Q, Hankinson SE, Hunter DJ. TGFB1 and TGFBR1 polymorphisms and breast cancer risk in the Nurses' Health Study. BMC Cancer. 2007; 7:175.
238 Dunning AM, Ellis PD, McBride S, Kirschenlohr HL, Healey CS, Kemp PR, Luben RN, Chang-Claude J, Mannermaa A, Kataja V, Pharoah PD, Easton DF, Ponder BA, Metcalfe JC. A transforming growth

202
factorbetal signal peptide variant increases secretion in vitro and is associated with increased incidence of invasive breast cancer. Cancer Res. 2003; 63(10):2610-5.
239 Dunning AM, Healey CS, Baynes C, Scollen S, Vega A, Rodriguez R, Maia AT, Ponder BA, Low YL, Bingham S, Haiman CA, Le Marchand L, Broeks A, Schmidt MK, Hopper J, Southey M, Beckmann MW, Fasching PA, Peto J, Johnson N, Bojesen SE, Nordestgaard B, Milne RL, Benitez J, Hamann U, Ko Y, Schmutzler RK, Burwinkle B, Schurmann P, Dork T, Heikkinen T, Nevanlinna H, Lindblom A, Margolin S, Mannermaa A, Kosma VM, Chen X, Spurdle A, Change-Claude J, Flesch-Janys D, Couch FJ, Olson JE, Severi G, Baglietto L, Borresen-Dale AL, Kristensen V, Hunter DJ, Hankinson SE, Devillee P, Vreeswijk M, Lissowska J, Brinton L, Liu J, Hall P, Kang D, Yoo KY, Shen CY, Yu JC, Anton-Culver H, Ziogoas A, Sigurdson A, Struewing J, Easton DF, Humphreys M, Morrison J, Pharoah PD, Pooley K, Chenovix-Trench G. Association of ESRI gene tagging SNPs with breast cancer risk. Hum Mol Genet. 2009; 18(6): 1131-9.
240 Frank B, Wiestler M, Kropp S, Hemminki K, Spurdle AB, Sutter C, Wappenschmidt B, Chen X, Beesley J, Hopper JL; Australian Breast Cancer Family Study Investigators, Meindl A, Kiechle M, Slanger T, Bugert P, Schmutzler RK, Bartram CR, Flesch-Janys D, Mutschelknauss E, Ashton K, Salazar R, Webb E, Hamann U, Brauch H, Justenhoven C, Ko YD, Bruning T, Silva Idos S, Johnson N, Pharoah PP, Dunning AM, Pooley KA, Chang-Claude J, Easton DF, Peto J, Houlston R; Gene Environment Interaction and Breast Cancer in Germany Group, Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer Investigators, Australian Ovarian Cancer Study Management Group, Chenevix-Trench G, Fletcher O, Burwinkel B. Association of a common AKAP9 variant with breast cancer risk: a collaborative analysis. J Natl Cancer Inst. 2008; 100(6):437-42.
241 Cui J, Antoniou AC, Dite GS, Southey MC, Venter DJ, Easton DF, Giles GG, McCredie MR, Hopper JL. After BRCA1 and BRCA2-what next? Multifactorial segregation analyses of three-generation, population-based Australian families affected by female breast cancer. Am J Hum Genet. 2001; 68(2):420-31.
242 Parmigiani G, Berry D, Aguilar O. Determining carrier probabilities for breast cancer-susceptibility genes BRCAl andBRCA2. Am J Hum Genet. 1998; 62(l):145-58.
243 Antoniou AC, Pharoah PP, Smith P, Easton DF. The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br J Cancer. 2004; 91(8): 1580-90.
244 Pharoah PD, Antoniou A, Bobrow M, Zimmern RL, Easton DF, Ponder BA. Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet. 2002; 31(1)33-6.
245 Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. N Engl J Med. 2008; 358(26):2796-803.
246 Antoniou AC, Easton DF. Polygenic inheritance of breast cancer: Implications for design of association studies. Genet Epidemiol. 2003; 25(3): 190-202.
247 Pharoah PD, Dunning AM, Ponder BA, Easton DF. Association studies for finding cancer-susceptibility genetic variants. Nat Rev Cancer. 2004; 4(11):850-60.
248 Tommiska J, Seal S, Renwick A, Barfoot R, Baskcomb L, Jayatilake H, Bartkova J, Tallila J, Kaare M, Tamminen A, Heikkilâ P, Evans DG, Eccles D, Aittomâki K, Blomqvist C, Bartek J, Stratton MR, Nevanlinna H, Rahman N. Evaluation ofRADSO in familial breast cancer predisposition. Int J Cancer. 2006; 118(11):2911-6.
249 Heikkinen K, Rapakko K, Karppinen SM, Erkko H, Knuutila S, Lundân T, Mannermaa A, Berresen-Dale AL, Borg A, Barkardottir RB, Petrini J, Winqvist R. RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis. 2006; 27(8): 1593-9.
250 Futreal PA, Liu QY, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y, Eddington K, McClure M, Frye C, Weaver-Feldhaus J, Ding W, Gholami Z, Sôderkvist P, Terry L, Jhanwar S, Berchuck A, Iglehart JD, Marks J, Ballinger DG, Barrett JC, Skolnick MH, Kamb A, Wiseman R. BRCAl mutations in primary breast and ovarian carcinomas. Science. 1994; 266(5182): 120-2.
251 Lancaster JM, Wooster R, Mangion J, Phelan CM, Cochran C, Gumbs C, Seal S, Barfoot R, Collins N, Bignell G, Patel S, Hamoudi R, Larsson C, Wiseman RW, Berchuck A, Iglehart JD, Marks JR,

203
Ashworth A, Stratton MR, Futreal PA. BRCA2 mutations in primary breast and ovarian cancers. Nat Genet. 1996; 13(2):238-40.
252 Kalb R, Neveling K, Nanda I, Schindler D, Hoehn H. Fanconi anemia: causes and consequences of genetic instability. Genome Dyn. 2006;1:218-42.
253 Antoccia A, Kobayashi J, Tauchi H, Matsuura S, Komatsu K. Nijmegen breakage syndrome and functions of the responsible protein, NBS1. Genome Dyn. 2006;1:191-205.
254 Futami K, Ishikawa Y, Goto M, Furuichi Y, Sugimoto M. Role of Werner syndrome gene product helicase in carcinogenesis and in resistance to genotoxins by cancer cells. Cancer Sci. 2008 May;99(5):843-8.
255 Dworaczek H, Xiao W. Xeroderma pigmentosum: a glimpse into nucleotide excision repair, genetic instability, and cancer. Crit Rev Oncog. 2007; 13(2): 159-77.
256 Abel KJ, Xu J, Yin GY, Lyons RH, Meisler MH, Weber BL. Mouse Brcal : localization sequence analysis and identification of evolutionarily conserved domains. Hum Mol Genet. 1995; 4(12):2265-73.
257 Szabo CI, Wagner LA, Francisco LV, Roach JC, Argonza R, King MC, Ostrander EA. Human, canine and murine BRCAl genes: sequence comparison among species. Hum Mol Genet. 1996; 5(9):1289-98.
258 Couch FJ, Weber BL, Breast Cancer Information Core. Mutations and polymorphisms in the familial early-onset breast cancer (BRCAl) gene. Hum Mutat. 1996; 8(1):8-18.
259 Koonin EV, Altschul SF, Bork P. BRCAl protein products ... Functional motifs... Nat Genet. 1996; 13(3):266-8.
260 Henderson BR. Regulation of BRCAl, BRCA2 and BARD 1 intracellular trafficking. Bioessays. 2005; 27(9):884-93.
261 Venkitaraman AR. Linking the Cellular Functions of BRCA Genes to Cancer Pathogenesis and Treatment. Annu Rev Pathol. 2009; 4:461-87.
262 Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001; 8(10):833-7.
263 Williams RS, Green R, Glover JN. Crystal structure of the BRCT repeat region from the breast cancer-associated protein BRCAl. Nat Struct Biol. 2001; 8(10):838-42.
264 Williams RS, Bernstein N, Lee MS, Rakovszky ML, Cui D, Green R, Weinfeld M, Glover JN. Structural basis for phosphorylation-dependent signaling in the DNA-damage response. Biochem Cell Biol. 2005; 83(6):721-7.
265 Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003; 302(5645):639-42.
266 Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003; 302(5645):636-9.
267 Coene ED, Hollinshead MS, Waeytens AA, Schelfhout VR, Eechaute WP, Shaw MK, Van Oostveldt PM, Vaux DJ. Phosphorylated BRCAl is predominantly located in the nucleus and mitochondria. Mol Biol Cell. 2005; 16(2):997-1010.
268 Rodriguez JA, Henderson BR. Identification of a functional nuclear export sequence in BRCAl. J Biol Chem. 2000; 275(49)38589-96.
269 Thakur S, Zhang HB, Peng Y, Le H, Carroll B, Ward T, Yao J, Farid LM, Couch FJ, Wilson RB, Weber BL. Localization of BRCAl and a splice variant identifies the nuclear localization signal. Mol Cell Biol. 1997; 17(l):444-52.
270 Chen CF, Li S, Chen Y, Chen PL, Sharp ZD, Lee WH. The nuclear localization sequences of the BRCAl protein interact with the importin-alpha subunit of the nuclear transport signal receptor. J Biol Chem. 1996 Dec; 271(51)32863-8.
271 Henderson BR. Regulation of BRCAl, BRCA2 and BARD 1 intracellular trafficking. Bioessays. 2005; 27(9):884-93.
272 Cortez D, Wang Y, Qin J, Elledge S J. Requirement of ATM-dependent phosphorylation of brcal in the DNA damage response to double-strand breaks. Science. 1999 Nov; 286(5442):! 162-6.

204
273 Ouchi T. BRCAl phosphorylation: biological consequences. Cancer Biol Ther. 2006; 5(5):470-5. 274 Bork P, Blomberg N, Nilges M. Internal repeats in the BRCA2 protein sequence. Nat Genet. 1996;
13(l):22-3. 275 Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH. The BRC repeats in BRCA2 are critical for
RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci USA. 1998; 95(9):5287-92.
276 Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionary conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997; 272(51)31941-4.
277 Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002; 420(6913):287-93.
278 Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSSl-ssDNA structure. Science. 2002; 297(5588): 1837-48.
279 Yano K, Morotomi K, Saito H, Kato M, Matsuo F, Miki Y. Nuclear localization signals of the BRCA2 protein. Biochem Biophys Res Commun. 2000; 270(1): 171-5.
280 Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005; 434(7033):598-604.
281 Chodosh LA. Expression of BRCAl and B RCA 2 in normal and neoplastic cells. J Mammary Gland Biol Neoplasia. 1998; 3(4)389-402.
282 Patel KJ, Yu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR. Involvement ofBrca2 in DNA repair. Mol Cell. 1998; 1(3)347-57.
283 Milner J, Ponder B, Hughes-Davies L, Seltmann M, Kouzarides T. Transcriptional activation functions in BRCA2. Nature. 1997; 386(6627):772-3.
284 Shin S, Verma IM. BRCA2 cooperates with histone acetyltransferases in androgen receptor-mediated transcription. Proc Natl Acad Sci USA. 2003; 100(12):7201-6.
285 Fuks F, Milner J, Kouzarides T. BRCA2 associates with acétyltransférase activity when bound to P/CAF. Oncogene. 1998; 17(19):2531-4.
286 Hughes-Davies L, Huntsman D, Ruas M, Fuks F, Bye J, Chin SF, Milner J, Brown LA, Hsu F, Gilks B, Nielsen T, Schulzer M, Chia S, Ragaz J, Cahn A, Linger L, Ozdag H, Cattaneo E, Jordanova ES, Schuuring E, Yu DS, Venkitaraman A, Ponder B, Doherty A, Aparicio S, Bentley D, Theillet C, Ponting CP, Caldas C, Kouzarides T. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell. 2003; 115(5):523-35.
287 Scully R, Anderson SF, Chao DM, Wei W, Ye L, Young RA, Livingston DM, Parvin JD. BRCAl is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci USA. 1997; 94(11):5605-10.
288 Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. BRCAl protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat Genet. 1998;19(3):254-6.
289 Moisan A, Larochelle C, Guillemette B, Gaudreau L. BRCAl can modulate RNA polymerase II carboxy-terminal domain phosphorylation levels. Mol Cell Biol. 2004; 24(16):6947-56.
290 Horwitz AA, Affar el B, Heine GF, Shi Y, Parvin JD. A mechanism for transcriptional repression dependent on the BRCAl E3 ubiquitin ligase. Proc Natl Acad Sci USA. 2007; 104(16):6614-9.
291 Zhang H, Somasundaram K, Peng Y, Tian H, Zhang H, Bi D, Weber BL, El-Deiry WS. BRCAl physically associates with p53 and stimulates its transcriptional activity. Oncogene. 1998; 16(13): 1713-21.
292 Ouchi T, Monteiro AN, August A, Aaronson SA, Hanafusa H. BRCAl regulates p53-dependent gene expression. Proc Natl Acad Sci USA. 1998; 95(5):2302-6.
293 MacLachlan TK, Takimoto R, El-Deiry WS. BRCAl directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol Cell Biol. 2002; 22(12):4280-92.

205
294 Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA, Erdos M, Goldberg ID, Webb P, Kushner PJ, Pestell RG, Rosen EM. p300 Modulates the BRCAl inhibition of estrogen receptor activity. Cancer Res. 2002; 62(1):141-51.
295 Wang C, Fan S, Li Z, Fu M, Rao M, Ma Y, Lisanti MP, Albanese C, Katzenellenbogen BS, Kushner PJ, Weber B, Rosen EM, Pestell RG. Cyclin Dl antagonizes BRCAl repression of estrogen receptor alpha activity. Cancer Res. 2005; 65(15):6557-67.
296 Harte MT, O'Brien GJ, Ryan NM, Gorski JJ, Savage KI, Crawford NT, Mullan PB, Harkin DP. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCAl and regulates BRCAl-dependent transcription. Cancer Res. 2010;70(6):2538-47.
297 Xu J, Fan S, Rosen EM. Regulation of the estrogen-inducible gene expression profile by the breast cancer susceptibility gene BRCAl. Endocrinology. 2005; 146(4):2031-47.
298 Ahmed KM, Tsai CY, Lee WH. Derepression ofHMGA2 via removal of ZBRK1/BRCAl/CtlP complex enhances mammary tumorigenesis. J Biol Chem. 2010;285(7):4464-71.
299 Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, Hollander MC, O'Connor PM, Fornace AJ Jr, Harris CC. GADD45 induction of a G2/Mcell cycle checkpoint. Proc Natl Acad Sci USA. 1999; 96(7)3706-11.
300 Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJ Jr. Association with Cdc2 and inhibition of Cdc2/Cyclin BI kinase activity by the p53-regulated protein Gadd45. Oncogene. 1999; 18(18):2892-900.
301 Harkin DP, Bean JM, Miklos D, Song YH, Truong VB, Englert C, Christians FC, Ellisen LW, Maheswaran S, Oliner JD, Haber DA. Induction of GADD45 and JNKJSAPK-dependent apoptosis following inducible expression of BRCAl. Cell. 1999; 97(5):575-86.
302 Smith ML, Kontny HU, Zhan Q, Sreenath A, O'Connor PM, Fornace AJ Jr. Antisense GADD45 expression results in decreased DNA repair and sensitizes cells to u.v. -irradiation or cisplatin. Oncogene. 1996; 13(10):2255-63.
303 Hollander MC, Kovalsky O, Salvador JM, Kim KE, Patterson AD, Haines DC, Fornace AJ Jr. Dimethylbenzanthracene carcinogenesis in Gadd45a-null mice is associated with decreased DNA repair and increased mutation frequency. Cancer Res. 2001; 61(6):2487-91.
304 Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, Molinaro TA, Kim KE, Tolosa E, Ashwell JD, Rosenberg MP, Zhan Q, Fernândez-Salguero PM, Morgan WF, Deng CX, Fornace AJ Jr. Genomic instability in Gadd45a-deficient mice. Nat Genet. 1999; 23(2): 176-84.
305 Salvador JM, Hollander MC, Nguyen AT, Kopp JB, Barisoni L, Moore JK, Ashwell JD, Fornace AJ Jr. Mice lacking the p53-effector gene Gadd45a develop a lupus-like syndrome. Immunity. 2002 Apr;16(4):499-508.
306 Zhan Q. Gadd45a, a p53- and BRCAl-regulated stress protein, in cellular response to DNA damage. Mutat Res. 2005; 569(l-2):133-43.
307 Zheng L, Pan H, Li S, Flesken-Nikitin A, Chen PL, Boyer TG, Lee WH. Sequence-specific transcriptional corepressor function for BRCAl through a novel zinc finger protein, ZBRK1. Mol Cell. 2000; 6(4):757-68.
308 Tan W, Zheng L, Lee WH, Boyer TG. Functional dissection of transcription factor ZBRK1 reveals zinc fingers with dual roles in DNA-binding and BRCAl-dependent transcriptional repression. J Biol Chem. 2004; 279(8):6576-87.
309 Yun J, Lee WH. Degradation of transcription repressor ZBRK1 through the ubiquitin-proteasome pathway relieves repression ofGadd45a upon DNA damage. Mol Cell Biol. 2003; 23(20):7305-14.
310 Fan W, Jin S, Tong T, Zhao H, Fan F, Antinore M J, Rajasekaran B, Wu M, Zhan Q. BRCAl regulates GADD45 through its interactions with the OCT-1 and CA AT motifs. J Biol Chem. 2002; 277(10):8061-7.
311 Fabbro M, Henderson BR. BARD1 regulates BRCAl-mediated transactivation of the p21WAFl/CIPl and Gadd45 promoters. Cancer Lett. 2008; 263(2): 189-96.

206
312 Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ Jr. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992; 71(4):587-97.
313 Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr, DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOX03a through the Gadd45 protein. Science. 2002; 296(5567):530-4.
314 Thyss R, Virolle V, Imbert V, Peyron JF, Aberdam D, Virolle T. NF-kappaB/Egr-l/Gadd45 are sequentially activated upon UVB irradiation to mediate epidermal cell death. EMBO J. 2005; 24(1): 128-37.
315 Lai A, Abdelmohsen K, Pullmann R, Kawai T, Galban S, Yang X, Brewer G, Gorospe M. Posttranscriptional derepression of GADD45alpha by genotoxic stress. Mol Cell. 2006; 22(1):117-28.
316 Zhang Y, Bhatia D, Xia H, Castranova V, Shi X, Chen F. Nucleolin links to arsenic-induced stabilization ofGADD45alpha mRNA. Nucleic Acids Res. 2006; 34(2):485-95.
317 Lai A, Gorospe M. Egad, more forms of gene regulation: the gadd45a story. Cell Cycle. 2006; 5(13):1422-5.
318 Chang Q, Bhatia D, Zhang Y, Meighan T, Castranova V, Shi X, Chen F. Incorporation of an internal ribosome entry site-dependent mechanism inarsenic-induced GADD45 alpha expression. Cancer Res. 2007; 67(13):6146-54.
319 Rutter JL, Smith AM, Dâvila MR, Sigurdson AJ, Giusti RM, Pineda MA, Doody MM, Tucker MA, Greene MH, Zhang J, Struewing JP. Mutational analysis of the BRCAl-interacting genes ZNF350/ZBRK1 and BRIP 1/BACH 1 among BRCAl and BRCA2-negative probands from breast-ovarian cancer families and among early-onset breast cancer cases and reference individuals. Hum Mutat. 2003; 22(2): 121-8.
320 Sigurdson AJ, Hauptmann M, Chatterjee N, Alexander BH, Doody MM, Rutter JL, Struewing JP. Kin-cohort estimates for familial breast cancer risk in relation to variants in DNA base excision repair, BRCAl interacting and growth factor genes. BMC Cancer. 2004; 4:9.
321 Sensi E, Tancredi M, Aretini P, Cipollini G, Collecchi P, Naccarato AG, Viacava P, Bevilacqua G, Caligo MA. Clinicopathological significance of GADD45 gene alterations in human familial breast carcinoma. Breast Cancer Res Treat. 2004; 87(2): 197-201.
322 Blaszyk H, Hartmann A, Sommer SS, Kovach JS. A polymorphism but no mutations in the GADD45 gene in breast cancers. Hum Genet. 1996; 97(4):543-7.
323 Campomenosi P, Hall PA. Gadd45 mutations are uncommon in human tumour cell lines. Cell Prolif. 2000; 33(5)301-6.
324 Huo X, Lu C, Huang X, Hu Z, Jin G, Ma H, Wang X, Qin J, Wang X, Shen H, Tang J. Polymorphisms in BRCAl, BRCAl-interacting genes and susceptibility of breast cancer in Chinese women. J Cancer Res Clin Oncol. 2009 Nov; 135( 11 ): 1569-75.
325 Garcia V, Dominguez G, Garcia JM, Silva J, Pefia C, Silva JM, Carcereny E, Menéndez J, Espafia P, Bonilla F. Altered expression of the ZBRK1 gene in human breast carcinomas. J Pathol. 2004; 202(2):224-32.
326 Garcia V, Garcia JM, Pena C, Silva J, Dominguez G, Rodriguez R, Maximiano C, Espinosa R, Espafia P, Bonilla F. The GADD45, ZBRK1 and BRCAl pathway: quantitative analysis of mRNA expression in colon carcinomas. J Pathol. 2005; 206(l):92-9.
327 Figueroa JD, Malats N, Rothman N, Real FX, Silverman D, Kogevinas M, Chanock S, Yeager M, Welch R, Dosemeci M, Tardôn A, Serra C, Carrato A, Garcia-Closas R, Castano-Vinyals G, Garcia-Closas M. Evaluation of genetic variation in the double-strand break repair pathway and bladder cancer risk. Carcinogenesis. 2007; 28(8): 1788-93.
328 Yarden RI, Friedman E, Metsuyanim S, Olender T, Ben-Asher E, Papa MZ. Single-nucleotide polymorphisms in the p53 pathway genes modify cancer risk in BRCA1 and BRCA2 carriers ofJewish-Ashkenazi descent. Mol Carcinog. 2010; 49(6):545-55.

207
329 Zhang J, Powell SN. The role of the BRCAl tumor suppressor in DNA double-strand break repair. Mol Cancer Res. 2005; 3(10):531-9.
330 Thorslund T, West SC. BRCA2: a universal recombinase regulator. Oncogene. 2007; 26(56):7720-30. 331 Wang HC, Chou WC, Shieh SY, Shen CY. Ataxia telangiectasia mutated and checkpoint kinase 2
regulate BRCAl to promote the fidelity of DNA end-joining. Cancer Res. 2006; 66(3): 1391-400. 332 Zhuang J, Zhang J, Willers H, Wang H, Chung JH, van Gent DC, Hallahan DE, Powell SN, Xia F.
Checkpoint kinase 2-mediated phosphorylation of BRCAl regulates the fidelity of nonhomologous end-joining. Cancer Res. 2006; 66(3):1401-8.
333 Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mrell-Rad50-Nbsl complex. Science. 2005; 308(5721):551-4.
334 Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AXphosphorylation on serine 139. J Biol Chem. 1998; 273(10):5858-68.
335 Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001; 276(45):42462-7.
336 Foray N, Marot D, Gabriel A, Randrianarison V, Carr AM, Perricaudet M, Ashworth A, Jeggo P. A subset of ATM- and ATR-dependent phosphorylation events requires the BRCAl protein. EMBO J. 2003; 22(11):2860-71.
337 Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007; 316(5828):1194-8.
338 Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCAl to specific ubiquitin structures at DNA damage sites. Science. 2007; 316(5828): 1198-202.
339 Kim H, Huang J, Chen J. CCDC98 is a BRCAl-BRCT domain-binding protein involved in the DNA damage response. Nat Struct Mol Biol. 2007; 14(8):710-5.
340 Xia Y, Pao GM, Chen HW, Verma IM, Hunter T. Enhancement of BRCAl E3 ubiquitin ligase activity through direct interaction with the BARD 1 protein. J Biol Chem. 2003; 278(7):5255-63.
341 Greenberg RA. Recognition of DNA double strand breaks by the BRCAl tumor suppressor network. Chromosoma. 2008; 117(4)305-17.
342 Patel KJ, Yu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR. Involvement ofBrca2 in DNA repair. Mol Cell. 1998; 1(3)347-57.
343 Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci USA. 1998; 95(9):5287-92.
344 Marmorstein LY, Ouchi T, Aaronson SA. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc Natl Acad Sci USA. 1998; 95(23): 13869-74.
345 Tarsounas M, Davies AA, West SC. RAD51 localization and activation following DNA damage. Philos Trans R Soc Lond B Biol Sci. 2004; 359(1441):87-93.
346 Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly ofRadSl complex in vivo. Cancer Res. 1999; 59(15)3547-51.
347 Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005; 434(7033):598-604.
348 Horejsi Z, Falck J, Bakkenist CJ, Kastan MB, Lukas J, Bartek J. Distinct functional domains ofNbsl modulate the timing and magnitude of ATM activation after low doses of ionizing radiation. Oncogene. 2004; 23(17)3122-7.
349 Desai-Mehta A, Cerosaletti KM, Concannon P. Distinct functional domains of nibrin mediate Mrell binding, focus formation, and nuclear localization. Mol Cell Biol. 2001; 21(6):2184-91.

208
350 Sakamoto S, Iijima K, Mochizuki D, Nakamura K, Teshigawara K, Kobayashi J, Matsuura S, Tauchi H, Komatsu K. Homologous recombination repair is regulated by domains at the N- and C-terminus of NBS1 and is dissociated with ATM functions. Oncogene. 2007; 26(41):6002-9.
351 Williams RS, Williams JS, Tainer JA. Mrel 1-Rad50-Nbsl is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007; 85(4):509-20.
352 van der Linden E, Sanchez H, Kinoshita E, Kanaar R, Wyman C. RAD50 and NBS1 form a stable complex functional in DNA binding and tethering. Nucleic Acids Res. 2009 Jan 16.
353 Zhong Q, Chen CF, Li S, Chen Y, Wang CC, Xiao J, Chen PL, Sharp ZD, Lee WH. Association of BRCAl with the hRad50-hMrell-p95 complex and the DNA damage response. Science. 1999; 285(5428)747-50.
354 Wu X, Rathbun G, Lane WS, Weaver DT, Livingston DM. Interactions of the Nijmegen breakage syndrome protein with ATM and BRCAl. Cold Spring Harb Symp Quant Biol. 2000; 65:535-45.
355 Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCAl/BARD 1-containing complexes. Genes Dev. 2006; 20(1)34-46.
356 Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCAl.CtlP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008; 283(12):7713-20.
357 Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtlP promotes DNA end resection. Nature. 2007; 450(7169):509-14.
358 Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mrell dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006; 25(8): 1764-74.
359 Borde V. The multiple roles of the Mrell complex for meiotic recombination. Chromosome Res. 2007; 15(5):551-63.
360 Wu Y, Xiao S, Zhu XD. MRE11-RAD50-NBS1 and ATM function as co-mediators ofTRFl in telomere length control. Nat Struct Mol Biol. 2007; 14(9):832-40.
361 Weemaes CM, Hustinx TW, Scheres JM, van Munster PJ, Bakkeren JA, Taalman RD. A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta Paediatr Scand. 1981; 70(4):557-64.
362 The International Nijmegen Breakage Syndrome Study Group. Nijmegen breakage syndrome. Arch Dis Child. 2000; 82(5):400-6.
363 Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet. 1997;31:635-62.
364 Gennery AR, Slatter MA, Bhattacharya A, Barge D, Haigh S, O'Driscoll M, Coleman R, Abinun M, Flood TJ, Cant AJ, Jeggo PA. The clinical and biological overlap between Nijmegen Breakage Syndrome and Fanconi anemia. Clin Immunol. 2004; 113(2):214-9.
365 Wegner RD, Metzger M, Hanefeld F, Jaspers NG, Baan C, Magdorf K, Kunze J, Sperling K. A new chromosomal instability disorder confirmed by complementation studies. Clin Genet. 1988; 33(1):20-32.
366 Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet. 1997;31:635-62.
367 Buscemi G, Savio C, Zannini L, Miccichè F, Masnada D, Nakanishi M, Tauchi H, Komatsu K, Mizutani S, Khanna K, Chen P, Concannon P, Chessa L, Delia D. Chk2 activation dependence on Nbsl after DNA damage. Mol Cell Biol. 2001; 21(15):5214-22.
368 Girard PM, Riballo E, Begg AC, Waugh A, Jeggo PA. Nbsl promotes ATM dependent phosphorylation events including those required for Gl/S arrest. Oncogene. 2002; 21(27):4191-9.
369 Seemanova E, Jarolim P, Seeman P, Varon R, Digweed M, Swift M, Sperling K. Cancer risk of hétérozygotes with the NBN founder mutation. J Natl Cancer Inst. 2007; 99(24): 1875-80.
370 Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS 1 leads to early embryonic lethality in mice. Curr Biol. 2001; ll(2):105-9.

209
371 Maser RS, Zinkel R, Petrini JH. An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat Genet. 2001; 27(4):417-21.
372 Seemanova E. An increased risk for malignant neoplasms in hétérozygotes for a syndrome of microcephaly, normal intelligence, growth retardation, remarkable fades, immunodeficiency and chromosomal instability. Mutat Res. 1990; 238(3)321-4.
373 Seemanova E, Jarolim P, Seeman P, Varon R, Digweed M, Swift M, Sperling K. Cancer risk of hétérozygotes with the NBN founder mutation. J Natl Cancer Inst. 2007; 99(24): 1875-80.
374 Dzikiewicz-Krawczyk A. The importance of making ends meet: mutations in genes and altered expression of proteins of the MRN complex and cancer. Mutat Res. 2008; 659(3):262-73.
375 Bogdanova N, Feshchenko S, Schurmann P, Waltes R, Wieland B, Hillemanns P, Rogov YI, Dammann O, Bremer M, Karstens JH, Sohn C, Varon R, Dôrk T. Nijmegen Breakage Syndrome mutations and risk of breast cancer. Int J Cancer. 2008; 122(4):802-6.
376 Roznowski K, Januszkiewicz-Lewandowska D, Mosor M, Pernak M, Litwiniuk M, Nowak J. 1171V germline mutation in the NBSl gene significantly increases risk of breast cancer. Breast Cancer Res Treat. 2008; 110(2)343-8.
377 Gôrski B, Debniak T, Masojc B, Mierzejewski M, Medrek K, Cybulski C, Jakubowska A, Kurzawski G, Chosia M, Scott R, Lubinski J. Germline 657del5 mutation in the NBSl gene in breast cancer patients. Int J Cancer. 2003; 106(3)379-81.
378 Gôrski B, Cybulski C, Huzarski T, Byrski T, Gronwald J, Jakubowska A, Stawicka M, Gozdecka-Grodecka S, Szwiec M, Urbanski K, Mitus J, Marczyk E, Dziuba J, Wandzel P, Surdyka D, Haus O, Janiszewska H, Debniak T, Toloczko-Grabarek A, Medrek K, Masojc B, Mierzejewski M, Kowalska E, Narod SA, Lubinski J. Breast cancer predisposing alleles in Poland. Breast Cancer Res Treat. 2005; 92(1): 19-24.
379 Steffen J, Varon R, Mosor M, Maneva G, Maurer M, Stumm M, Nowakowska D, Rubach M, Kosakowska E, Ruka W, Nowecki Z, Rutkowski P, Demkow T, Sadowska M, Bidzinski M, Gawrychowski K, Sperling K. Increased cancer risk of hétérozygotes with NBSl germline mutations in Poland. Int J Cancer. 2004; 11 l(l):67-71.
380 Seemanova E, Sperling K, Neitzel H, Varon R, Hadac J, Butova O, Schrôck E, Seeman P, Digweed M. Nijmegen breakage syndrome (NBS) with neurological abnormalities and without chromosomal instability. J Med Genet. 2006; 43(3):218-24.
381 di Masi A, Viganotti M, Polticelli F, Ascenzi P, Tanzarella C, Antoccia A. The R215W mutation in NBSl impairs gamma-H2AX binding and affects DNA repair: molecular bases for the severe phenotype of 657del5/R215W Nijmegen breakage syndrome patients. Biochem Biophys Res Commun. 2008; 369(3):835-40.
382 Bogdanova N, Schurmann P, Waltes R, Feshchenko S, Zalutsky IV, Bremer M, Dôrk T. NBSl variant 117IV and breast cancer risk. Breast Cancer Res Treat. 2008; 112(1)75-9.
383 Nowak J, Mosor M, Ziolkowska I, Wierzbicka M, Pernak-Schwarz M, Przyborska M, Roznowski K, Plawski A, Slomski R, Januszkiewicz D. Heterozygous carriers of the 117IV mutation of the NBSl gene have a significantly increased risk of solid malignant tumours. Eur J Cancer. 2008; 44(4):627-30.
384 Wang Z, Cui D, Lu W. NBSl 8360G>Cpolymorphism is associated with breast cancer risk : a metaanalysis. Breast Cancer Res Treat. 2010 Feb 9 [Epub ahead of print]
385 Lobitz S, Velleuer E. Guido Fanconi (1892-1979): a jack of all trades. Nat Rev Cancer. 2006; 6(ll):893-8.
386 Tischkowitz MD, Hodgson SV. Fanconi anaemia. J Med Genet. 2003; 40(1 ): 1-10. 387 Schroeder TM, Anschfitz F, Knopp A Spontané Chromosomenaberrationen bei familifirer
Panmyelopathie. Humangenetik 1964; 1:194-196 388 Rosenberg PS, Huang Y, Alter BP. Individualized risks of first adverse events in patients with Fanconi
anemia. Blood. 2004; 104(2)350-5. 389 Alter BP, Greene MH, Velazquez I, Rosenberg PS. Cancer in Fanconi anemia. Blood. 2003;
101(5):2072.

210
390 Durocher F, Guénard F, Desjardins S, Ouellette G, Labrie Y. Inherited susceptibility to breast cancer: accomplishments and challenges. In: Sinnett D. (ed.) Molecular Genetics of Cancer. Kerala, Research Signpost, 2005, 9-93.
391 Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andrea AD. Biallelic inactivation ofBRCA2 in Fanconi anemia. Science. 2002; 297(5581):606-9.
392 Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, de Winter JP, Wang W, Joenje H. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004; 36( 11 ): 1219-24.
393 Dietrich R, Velluer E. Fanconi Anemia: A disease with many faces. In Fanconi Anemia. A paradigmatic disease for the understanding of cancer and aging. Monogr Hum Genet. Basel Krager Schindler D. and Hoehn H (Eds) WUrzburg 2007 vol 15 pp9-22.
394 Gillio AP, Verlander PC, Batish SD, Giampietro PF, Auerbach AD. Phenotypic consequences of mutations in the Fanconi anemia FAC gene: an International Fanconi Anemia Registry study. Blood. 1997; 90(1):105-10.
395 Reid S, Renwick A, Seal S, Baskcomb L, Barfoot R, Jayatilake H, Pritchard-Jones K, Stratton MR, Ridolfi-Luthy A, Rahman N; Breast Cancer Susceptibility Collaboration (UK); Familial Wilms Tumour Collaboration. Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet. 2005; 42(2):147-51.
396 Kalb R, Neveling K, Hoehn H, Schneider H, Linka Y, Batish SD, Hunt C, Berwick M, Callen E, Surralles J, Casado JA, Bueren J, Dasi A, Soulier J, Gluckman E, Zwaan CM, van Spaendonk R, Pals G, de Winter JP, Joenje H, Grompe M, Auerbach AD, Hanenberg H, Schindler D. Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FA-D2 patients with severe phenotype. Am J Hum Genet. 2007; 80(5): 895-910.
397 Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007; 8(10)735-48.
398 Wang LC, Stone S, Hoatlin ME, Gautier J. Fanconi anemia proteins stabilize replication forks. DNA Repair (Amst). 2008; 7(12): 1973-81.
399 Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003; 35(2): 165-70.
400 Meetei AR, Yan Z, Wang W. FANCL replaces BRCAl as the likely ubiquitin ligase responsible for FANCD2 monoubiquitination. Cell Cycle. 2004; 3(2): 179-81.
401 Collins NB, Wilson JB, Bush T, Tomashevski A, Roberts KJ, Jones NJ, Kupfer GM. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood. 2008; 113(10):2181-90.
402 Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D'Andrea AD. Chkl-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2007; 27(8)3098-108.
403 Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, MathCG, Hoatlin M, Joenje H, de Winter JP, Wang W. A human ortholog of archaeal DNA repair protein Hefis defective in Fanconi anemia complementation group M. Nat Genet. 2005; 37(9):958-63.
404 Qiao F, Mi J, Wilson JB, Zhi G, Bucheimer NR, Jones NJ, Kupfer GM. Phosphorylation of fanconi anemia (FA) complementation group G protein, FANCG, at serine 7 is important for function of the FA pathway. J Biol Chem. 2004; 279(44):46035-45.
405 Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, Tashiro S, Elledge S J, Takata M. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol. 2008; 15(11): 1138-46.
406 Ho GP, Margossian S, Taniguchi T, D'Andrea AD. Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol Cell Biol. 2006; 26(18)7005-15.

211
407 Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D'Andrea AD. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002; 109(4):459-72.
408 Andreassen PR, D'Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004; 18(16): 1958-63.
409 Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D'Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci U S A. 2005; 102(4): 1110-5.
410 Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge S J. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007; 129(2):289-301.
411 Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCAl in a common pathway. Mol Cell. 2001; 7(2):249-62.
412 Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCAl and RAD51. Blood. 2002; 100(7):2414-20.
413 Folias A, Matkovic M, Bruun D, Reid S, Hejna J, Grompe M, D'Andrea A, Moses R. BRCAl interacts directly with the Fanconi anemia protein FANCA. Hum Mol Genet. 2002; 11(21):2591-7.
414 Medhurst AL, Laghmani el H, Steltenpool J, Ferrer M, Fontaine C, de Groot J, Rooimans MA, Scheper RJ, Meetei AR, Wang W, Joenje H, de Winter JP. Evidence for subcomplexes in the Fanconi anemia pathway. Blood. 2006; 108(6):2072-80.
415 Wilson JB, Yamamoto K, Marriott AS, Hussain S, Sung P, Hoatlin ME, Mathew CG, Takata M, Thompson LH, Kupfer GM, Jones NJ. FANCG promotes formation of a newly identified protein complex containing BRCA2, FANCD2 andXRCC3. Oncogene. 2008; 27(26)3641-52.
416 Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, Davies A, Masson JY, Moses R, West SC, de Winter JP, Ashworth A, Jones NJ, Mathew CG. Direct interaction ofFANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet. 2004; 13(12): 1241-8.
417 Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006; 25(43):5875-84. 418 Pearson T, Jansen S, Havenga C, Stones DK, Joubert G. Fanconi anemia, a statistical evaluation of
cytogenetic results obtained from South African families. Cancer Genet Cytogenet. 2001; 126(l):52-5. 419 Barquinero JF, Barrios L, Ribas M, Egozcue J, Caballin MR. Cytogenetic sensitivity of three Fanconi
anemia hétérozygotes to bleomycin and ionizing radiation. Cancer Genet Cytogenet. 2001; 124(l):80-3. 420 Mohseni-Meybodi A, Mozdarani H, Mozdarani S. DNA damage and repair of leukocytes from Fanconi
anaemia patients, carriers and healthy individuals as measured by the alkaline comet assay. Mutagenesis. 2009; 24(l):67-73.
421 Swift M. Fanconi's anemia in the genetics of neoplasia. Nature. 1971 ; 230370-3. 422 Swift M, Caldwell RJ, Chase C. Reassessment of cancer predisposition of Fanconi anemia
hétérozygotes. J Natl Cancer Inst. 1980; 65(5):863-7. 423 Potter NU, Sarmousakis C, Li FP. Cancer in relatives of patients with aplastic anemia. Cancer Genet
Cytogenet. 1983; 9(l):61-5. 424 Berwick M, Satagopan JM, Ben-Porat L, Carlson A, Mah K, Henry R, Diotti R, Milton K, Pujara K,
Landers T, Dev Batish S, Morales J, Schindler D, Hanenberg H, Hromas R, Levran O, Auerbach AD. Genetic heterogeneity among Fanconi anemia hétérozygotes and risk of cancer. Cancer Res. 2007; 67(19):9591-6.
425 Tischkowitz M, Easton DF, Ball J, Hodgson SV, Mathew CG. Cancer incidence in relatives of British Fanconi Anaemia patients. BMC Cancer. 2008; 8:257.
426 Meyer S, Barber LM, White DJ, Will AM, Birch JM, Kohler JA, Ersfeld K, Blom E, Joenje H, Eden TO, Malcolm Taylor G. Spectrum and significance of variants and mutations in the Fanconi anaemia group G gene in children with sporadic acute myeloid leukaemia. Br J Haematol. 2006; 133(3):284-92.

212
427 Barber LM, McGrath HE, Meyer S, Will AM, Birch JM, Eden OB, Taylor GM. Constitutional sequence variation in the Fanconi anaemia group C (FANCC) gene in childhood acute myeloid leukaemia. Br J Haematol. 2003; 121(l):57-62.
428 Tischkowitz MD, Morgan NV, Grimwade D, Eddy C, Ball S, Vorechovsky I, Langabeer S, Stôger R, Hodgson SV, Mathew CG. Deletion and reduced expression of the Fanconi anemia FANCA gene in sporadic acute myeloid leukemia. Leukemia. 2004; 18(3):420-5.
429 Thompson E, Dragovic RL, Stephenson SA, Eccles DM, Campbell IG, Dobrovic A. A novel duplication polymorphism in the FANCA promoter and its association with breast and ovarian cancer. BMC Cancer. 2005; 5:43.
430 Couch FJ, Johnson MR, Rabe K, Boardman L, McWilliams R, de Andrade M, Petersen G. Germ line Fanconi anemia complementation group C mutations and pancreatic cancer. Cancer Res. 2005; 65(2)383-6.
431 van der Heijden MS, Yeo CJ, Hruban RH, Kern SE. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003; 63(10):2585-8.
432 Sparano A, Quesnelle KM, Kumar MS, Wang Y, Sylvester AJ, Feldman M, Sewell DA, Weinstein GS, Brose MS. Genome-wide profiling of oral squamous cell carcinoma by array-based comparative genomic hybridization. Laryngoscope. 2006; 116(5)735-41.
433 Tischkowitz M, Ameziane N, Waisfisz Q, De Winter JP, Harris R, Taniguchi T, D'Andrea A, Hodgson SV, Mathew CG, Joenje H. Bi-allelic silencing of the Fanconi anaemia gene FANCF in acute myeloid leukaemia. Br J Haematol. 2003; 123(3):469-71.
434 Neveling K, Kalb R, Florl AR, Herterich S, Friedl R, Hoehn H, Hader C, Hartmann FH, Nanda I, Steinlein C, Schmid M, Tonnies H, Hurst CD, Knowles MA, Hanenberg H, Schulz WA, Schindler D. Disruption of the FA/BRCA pathway in bladder cancer. Cytogenet Genome Res. 2007; 118(2-4): 166-76.
435 Narayan G, Arias-Pulido H, Nandula SV, Basso K, Sugirtharaj DD, Vargas H, Mansukhani M, Villella J, Meyer L, Schneider A, Gissmann L, Durst M, Pothuri B, Murty VV. Promoter hypermethylation of FANCF: disruption of Fanconi Anemia-BRCA pathway in cervical cancer. Cancer Res. 2004; 64(9):2994-7.
436 Marsit CJ, Liu M, Nelson HH, Posner M, Suzuki M, Kelsey KT. Inactivation of the Fanconi anemia/BRCA pathway in lung and oral cancers: implications for treatment and survival. Oncogene. 2004; 23(4): 1000-4.
437 Wang Z, Li M, Lu S, Zhang Y, Wang H. Promoter hypermethylation of FANCF plays an important role in the occurrence of ovarian cancer through disrupting Fanconi anemia-BRCA pathway. Cancer Biol Ther. 2006; 5(3):256-60.
438 Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D'Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003; 9(5):568-74.
439 Hess CJ, Ameziane N, Schuurhuis GJ, Errami A, Denkers F, Kaspers GJ, Cloos J, Joenje H, Reinhardt D, Ossenkoppele GJ, Zwaan CM, Waisfisz Q. Hypermethylation of the FANCC and FANCL promoter regions in sporadic acute leukaemia. Cell Oncol. 2008; 30(4):299-306.
440 Smith IM, Mithani SK, Mydlarz WK, Chang SS, Califano JA. Inactivation of the Tumor Suppressor Genes Causing the Hereditary Syndromes Predisposing to Head and Neck Cancer via Promoter Hypermethylation in Sporadic Head and Neck Cancers. ORL J Otorhinolaryngol Relat Spec. 2010;72(l):44-50.
441 Wreesmann VB, Estilo C, Eisele DW, Singh B, Wang S J. Downregulation of Fanconi anemia genes in sporadic head and neck squamous cell carcinoma. ORL J Otorhinolaryngol Relat Spec. 2007; 69(4):218-25.
442 van der Groep P, Hoelzel M, Buerger H, Joenje H, de Winter JP, van Diest PJ. Loss of expression of FANCD2 protein in sporadic and hereditary breast cancer. Breast Cancer Res Treat. 2008; 107(1):41-7.

213
443 Seal S, Barfoot R, Jayatilake H, Smith P, Renwick A, Bascombe L, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N; Breast Cancer Susceptibility Collaboration. Evaluation of Fanconi Anemia genes in familial breast cancer predisposition. Cancer Res. 2003; 63(24):8596-9.
444 Lewis AG, Flanagan J, Marsh A, Pupo GM, Mann G, Spurdle AB, Lindeman GJ, Visvader JE, Brown MA, Chenevix-Trench G; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer. Mutation analysis of FANCD2, BRIP1/BACH1, LM04 and SFN in familial breast cancer. Breast Cancer Res. 2005; 7(6):R1005-16.
445 Barroso E, Milne RL, Fernandez LP, Zamora P, Arias JI, Benitez J, Ribas G. FANCD2 associated with sporadic breast cancer risk. Carcinogenesis. 2006;27(9): 1930-7.
446 Han J, Haiman C, Niu T, Guo Q, Cox DG, Willett WC, Hankinson SE, Hunter DJ. Genetic variation in DNA repair pathway genes and premenopausal breast cancer risk. Breast Cancer Res Treat. 2009;115(3):613-22.
447 Garcia MJ, Fernandez V, Osorio A, Barroso A, Fernandez F, Urioste M, Benitez J. Mutational analysis of FANCL, FANCM and the recently identified FANCI suggests that among the 13 known Fanconi Anemia genes, only FANCD1/BRCA2 plays a major role in high-risk breast cancer predisposition. Carcinogenesis. 2009 (11): 1898-902.
448 Barroso E, Pita G, Arias JI, Menéndez P, Zamora P, Blanco M, Benitez J, Ribas G. The Fanconi anemia family of genes and its correlation with breast cancer susceptibility and breast cancer features. Breast Cancer Res Treat. 2009; 118(3):655-60.
449 Yarden RI, Brody LC. BRCAl interacts with components of the histone deacetylase complex. Proc Natl Acad Sci USA. 1999; 96(9):4983-8.
450 Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, Wang W, Kashanchi F, Shiekhattar R. BRCAl is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000; 102(2):257-65.
451 Wada O, Oishi H, Takada I, Yanagisawa J, Yano T, Kato S. BRCAl function mediates a TRAP/DRIP complex through direct interaction with TRAP220. Oncogene. 2004; 23(35):6000-5.
452 Oishi H, Kitagawa H, Wada O, Takezawa S, Tora L, Kouzu-Fujita M, Takada I, Yano T, Yanagisawa J, Kato S. An hGCN5/TRRAP histone acétyltransférase complex co-activates BRCAl transactivation function through histone modification. J Biol Chem. 2006; 281(l):20-6.
453 Pao GM, Janknecht R, Ruffner H, Hunter T, Verma IM. CBP/p300 interact with and function as transcriptional coactivators of BRCAl. Proc Natl Acad Sci USA. 2000; 97(3): 1020-5.
454 Marmorstein LY, Kinev AV, Chan GK, Bochar DA, Beniya H, Epstein JA, Yen TJ, Shiekhattar R. A human BRCA2 complex containing a structural DNA binding component influences cell cycle progression. Cell. 2001; 104(2):247-57.
455 Futamura M, Arakawa H, Matsuda K, Katagiri T, Saji S, Miki Y, Nakamura Y. Potential role of BRCA2 in a mitotic checkpoint after phosphorylation by hBUBRl. Cancer Res. 2000; 60(6):1531-5.
456 Chan GK, Jablonski SA, Sudakin V, Hittle JC, Yen TJ. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol. 1999; 146(5):941-54.
457 Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T. The RING heterodimer BRCAl-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001; 276(18): 14537-40.
458 Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, Ng T, Solomon E. The SUMO modification pathway is involved in the BRCAl response to genotoxic stress. Nature. 2009;462(7275):886-90.
459 Kleiman FE, Wu-Baer F, Fonseca D, Kaneko S, Baer R, Manley JL. BRCAl/BARD 1 inhibition of mRNA 3' processing involves targeted degradation of RNA polymerase II. Genes Dev. 2005; 19(10): 1227-37.
460 Starita LM, Horwitz AA, Keogh MC, Ishioka C, Parvin JD, Chiba N. BRCAl/BARD 1 ubiquitinate phosphorylated RNA polymerase II. J Biol Chem. 2005; 280(26):24498-505.

214
461 Yu X, Fu S, Lai M, Baer R, Chen J. BRCAl ubiquitinates its phosphorylation-dependent binding partner CtlP. Genes Dev. 2006 Jul 1;20(13): 1721-6.
462 Eakin CM, Maccoss MJ, Finney GL, Klevit RE. Estrogen receptor alpha is a putative substrate for the BRCAl ubiquitin ligase. Proc Natl Acad Sci USA. 2007; 104(14): 5794-9.
463 Horwitz AA, Affar el B, Heine GF, Shi Y, Parvin JD. A mechanism for transcriptional repression dependent on the BRCAl E3 ubiquitin ligase. Proc Natl Acad Sci USA. 2007; 104(16):6614-9.
464 Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007; 315(5809):201-5.
465 Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono- or specificpolyubiquitin chain linkages. Nat Struct Mol Biol. 2007; 14(10):941-8.
466 Solis AS, Shariat N, Patton JG. Splicing fidelity, enhancers, and disease. Front Biosci. 2008; 13:1926-42.
467 Brett D, Pospisil H, Valcârcel J, Reich J, Bork P. Alternative splicing and genome complexity. Nat Genet. 2002; 30(l):29-30.
468 Moisan AM, Fortin J, Dumont M, Samson C, Bessette P, Chiquette J, Laframboise R, Lépine J, Lesperance B, Pichette R, Plante M, Provencher L, Voyer P, Goldgar D, Bridge P, Simard J. No Evidence of BRCAl/2 genomic rearrangements in high-risk French-Canadian breast/ovarian cancer families. Genet Test. 2006; 10(2):104-15.
469 Guénard F, Labrie Y, Ouellette G, Joly Beauparlant C, Simard J, Durocher F; INHERIT BRCAs. Mutational analysis of the breast cancer susceptibility gene BRIP1 /BACH 1/FANCJ in high-risk non-BRCA1/BRCA2 breast cancer families. J Hum Genet. 2008; 53(7):579-91.
470 Guénard F, Labrie Y, Ouellette G, Joly Beauparlant C, Simard J, Durocher F; INHERIT BRCAs. Mutational analysis of the breast cancer susceptibility gene BRIP 1/BACH 1/FANCJ in high-risk non-BRCA1/BRCA2 breast cancer families. J Hum Genet. 2008;53(7):579-91.
471 Laberge AM, Michaud J, Richter A, Lemyre E, Lambert M, Brais B, Mitchell GA. Population history and its impact on medical genetics in Quebec. Clin Genet. 2005; 68(4):287-301.
472 De Braekeleer M, Dao TN. Hereditary disorders in the French Canadian population of Quebec. I. In search of founders. Hum Biol. 1994; 66(2):205-23.
473 Furuta S, Wang JM, Wei S, Jeng YM, Jiang X, Gu B, Chen PL, Lee EY, Lee WH. Removal of BRCAl/CtlP/ZBRKl repressor complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer Cell. 2006;10(l):13-24.
474 Lin LF, Chuang CH, Li CF, Liao CC, Cheng CP, Cheng TL, Shen MR, Tseng JT, Chang WC, Lee WH, Wang JM. ZBRK1 acts as a metastatic suppressor by directly regulating MMP9 in cervical cancer. Cancer Res. 2010;70(1): 192-201.
475 Dupont WD, Breyer JP, Bradley KM, Schuyler PA, Plummer WD, Sanders ME, Page DL, Smith JR. Protein phosphatase 2A subunit gene haplotypes and proliferative breast disease modify breast cancer risk. Cancer. 2010;116(1):8-19.
476 Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Dôderlein G, Maltry N, Wu W, Lyko F, Niehrs C. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445(7128):671-5.
477 Schmitz KM, Schmitt N, Hoffmann-Rohrer U, Schafer A, Grummt I, Mayer C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell. 2009;33(3):344-53.
478 Hoggard N, Hey Y, Brintnell B, James L, Jones D, Mitchell E, Weissenbach J, Varley JM. Identification and cloning in yeast artificial chromosomes of a region of elevated loss of heterozygosity on chromosome lp31.1 in human breast cancer. Genomics. 1995 Nov 20;30(2):233-43.
479 Cretu A, Sha X, Tront J, Hoffman B, Liebermann DA. Stress sensor Gadd45 genes as therapeutic targets in cancer. Cancer Ther. 2009;7(A):268-276.
480 Zerbini LF, Libermann TA. GADD45 deregulation in cancer: frequently methylated tumor suppressors and potential therapeutic targets. Clin Cancer Res. 2005;11(18):6409-13.

215
481 Wang W, Huper G, Guo Y, Murphy SK, Oison JA Jr, Marks JR. Analysis of methylation-sensitive transcriptome identifies GADD45a as a frequently methylated gene in breast cancer. Oncogene. 2005;24(16):2705-14.
482 Ramachandran K, Gopisetty G, Gordian E, Navarro L, Hader C, Reis IM, Schulz WA, Singal R. Methylation-mediated repression of GADD45alpha in prostate cancer and its role as a potential therapeutic target. Cancer Res. 2009:69(4): 1527-35.
483 Wang Y, Yu Q, Cho AH, Rondeau G, Welsh J, Adamson E, Mercola D, McClelland M. Survey of differentially methylated promoters in prostate cancer cell lines. Neoplasia. 2005;7(8)748-60.
484 Yu KD, Di GH, Li WF, Rao NY, Fan L, Yuan WT, Hu Z, Wu J, Shen ZZ, Huang W, Shao ZM. Genetic contribution of GADD45A to susceptibility to sporadic and non-BRCAl/2 familial breast cancers: a systematic evaluation in Chinese populations. Breast Cancer Res Treat. 2010 May;121(l): 157-67.
485 Lu M, Lu J, Yang X, Yang M, Tan H, Yun B, Shi L. Association between the NBSl E185Q polymorphism and cancer risk: a meta-analysis. BMC Cancer. 2009;9:124.
486 Rebbeck TR, Mitra N, Domchek SM, Wan F, Chuai S, Friebel TM, Panossian S, Spurdle A, Chenevix-Trench G; kConFab, Singer CF, Pfeiler G, Neuhausen SL, Lynch HT, Garber JE, Weitzel JN, Isaacs C, Couch F, Narod SA, Rubinstein WS, Tomlinson GE, Ganz PA, Olopade OI, Tung N, Blum JL, Greenberg R, Nathanson KL, Daly MB. Modification of ovarian cancer risk by BRCAl/2-interacting genes in a multicenter cohort ofBRCAl/2 mutation carriers. Cancer Res. 2009;69(14):5801-10.
487 Stern MC, Lin J, Figueroa JD, Kelsey KT, Kiltie AE, Yuan JM, Matullo G, Fletcher T, Benhamou S, Taylor JA, Placidi D, Zhang ZF, Steineck G, Rothman N, Kogevinas M, Silverman D, Malats N, Chanock S, Wu X, Karagas MR, Andrew AS, Nelson HH, Bishop DT, Sak SC, Choudhury A, Barrett JH, Elliot F, Corral R, Joshi AD, Gago-Dominguez M, Cortessis VK, Xiang YB, Gao YT, Vineis P, Sacerdote C, Guarrera S, Polidoro S, Allione A, Gurzau E, Koppova K, Kumar R, Rudnai P, Porru S, Carta A, Campagna M, Arici C, Park SS, Garcia-Closas M; International Consortium of Bladder Cancer. Polymorphisms in DNA repair genes, smoking, and bladder cancer risk: findings from the international consortium of bladder cancer. Cancer Res. 2009;69(17):6857-64.
Top Related
Copyright © 2022 FDOKUMEN

