







Bahasa
Halaman
Hukum


RSC Advances
REVIEW
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
.
View Article OnlineView Journal | View Issue
A review on adva
SrmUHiCms(tmH
ment) in the Youngone Group oBangladesh. Beginning on 19 Septetime ‘Faculty Member’ in the depaand Polymer Science, SUST, Scontinues today. He is now con“Photocatalytic Conversion of CO2
University of Malaya.
aDepartment of Chemical Engineering, Facu
50603 Kuala Lumpur, Malaysia. E-mail: ashbDepartment of Chemical Engineering & P
Science & Technology, 3114 Sylhet, Banglad
Cite this: RSC Adv., 2014, 4, 20856
Received 28th February 2014Accepted 8th April 2014
DOI: 10.1039/c4ra01769b
www.rsc.org/advances
20856 | RSC Adv., 2014, 4, 20856–208
nces in photocatalysts towardsCO2 conversion
Sreejon Dasab and W. M. A. Wan Daud*a
The present situation reveals the dependence on fossil fuels, which is seriously accountable for two major
impediments: (i) global warming due to increasing atmospheric carbon dioxide (CO2) and (ii) the alarming
consumption of energy assets. The reduction of green CO2 in terms of producing solar fuels would be an
expedient accomplishment to resolve both problems, simultaneously. The review classifies different
categories of photocatalysts applied in foregoing photocatalytic CO2 conversion processes with detailed
information concerning operating conditions, preparation techniques and physical properties of
catalysts, radiation sources, and selectivity. The categories are concentrated on metal oxides, sulfides,
phosphides, and p-type and nonmetal oxide semiconductors. In addition, their modification by doping
co-metals, noble metals, transition metals and non-metals for visible light response is emphasized.
Besides, for harnessing solar fuel, the recent prospect and advancement of novel sensitized catalysts by
dye elements, phthalocyanines and quantum dots (QDs) are also highlighted in this review. This
technology needs more efficient solar active catalysts to increase production rates as well as selectivity.
The recent scenario indicates that massive prospects and opportunities still exist in this area for further
investigation on catalyst selection.
reejon Das is a postgraduateesearch student in the Depart-ent of Chemical Engineering atniversity of Malaya, Malaysia.e graduated with Distinctionsn 2009, from the Department ofhemical Engineering and Poly-er Science, Shahjalal Univer-ity of Science and TechnologySUST), Sylhet, Bangladesh. Hehen began his career as a teamember (Environment, Safety,ealth and Chemical Manage-f Industries Ltd., Chittagong,mber 2010, he joined as a full-rtment of Chemical Engineeringylhet, Bangladesh, where heducting his research work ininto Hydrocarbon Fuels” at the
lty of Engineering, University of Malaya,
olymer Science, Shahjalal University of
esh
93
1. Introduction
World energy consumption has deep implications for human-ity's social-economic-political sphere. For this reason, theglobal energy map is changing with potentially extensiveconsequences for energy markets and trade, and also by a
Professor Wan Mohd Ashri BinWan Daud earned his bachelor'sdegree in chemical engineeringin 1991 at Leeds University,Leeds, UK and his master'sdegree in chemical engineeringin 1992 from the University ofSheffield, Sheffield, UK. Heearned his PhD degree inChemical Engineering in 1996 atthe University of Sheffield. Since2005, he has worked asProfessor of Chemical Engi-
neering at the University of Malaya. His research elds includeenergy, biomass conversion and the synthesis of catalyst materials,catalysis, zeolites, polymerization process, separation process(adsorption, activated carbon, and carbon molecular sieve);ordered mesoporous materials, and hydrogen storage materials.Professor Wan Mohd Ashri Bin Wan Daud has written approxi-mately 90 research papers that have been published.
This journal is © The Royal Society of Chemistry 2014

Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
growing world population.1–3 On the other hand, the world isgradually becoming aware that essential alterations will berequired to meet the growing demand for energy. Maximumenergy sources are inadequate to satisfy the growing globalenergy demand, which will double by 2050 and triple at the endof this century.4 According to the U.S. Energy InformationAdministration,5 energy consumption rates remained vigorousin several developing countries, specically in Asia (>4%). InNorth America, Europe and the Commonwealth of IndependentStates, consumption fell by 4.5%, 5% and 8.5%, respectively,because of economic breakdown.
Energy produced from fossil fuels has great importancebecause the latter can be burned (oxidized to carbon dioxideand water), producing signicant amounts of energy per unitweight.6 Non-renewable fossil fuels yield takes millions of years,and these assets are being exhaustedmuch faster than new onesare being made. Fossil fuel consumption is also threatening tothe environment. During the period from 1961 to 2010, theBritish Petroleum Statistical Review of World Energy 20137
estimated that the rate of energy consumption has shown anupward trend. Worldwide movement towards renewable energyproduction is therefore in progress to meet increased energyrequirements. The main renewable sources such as sunlight,wind, tides, waves, geothermal heat, biomass and nuclearenergy offer electricity with zero carbon dioxide emissions.8
Energy conversion from waste is one of the supportable sourcesto convert the unwanted biomass into 7 to 10 TW of energy.9–11
Other renewable sources like wind, hydroelectric and tide/oceancurrents provide almost 2.1 TW, 1.5 TW and <2 TW of energyrespectively.12 Conversely, these renewable sources are quitelimited to meet the overall energy demand.
To full the energy demand, nuclear energy, which generallyis the part of the energy of an atomic nucleus released by fusionor ssion or radioactive decay could be a promising renewablesource. Currently, approximately 14% of the world's electricenergy is provided by the nuclear energy.13 On the other hand,the protection of nuclear reactors is a forthcoming limitation ofnuclear chain-reaction control, and safe storage of theextremely radioactive nuclear waste is still an unidentiedpathway towards a low fossil-fuel consumption future.14 Naturaldisasters like earthquakes may have harmful impacts on theearth from the nuclear plants. Moreover, nuclear power is notable to produce liquid fuel for transportation purposes. Unlikenuclear energy, another sustainable and abundant source ofenergy is required without the introduction of harmful envi-ronmental effects.
Renewable solar energy is used to harness the sun's energyand convert it into usable products through solar heating,15,16
solar photovoltaics,17,18 solar thermal electricity,19 solar archi-tecture,20,21 and articial photosynthesis.22 The satisfaction ofglobal energy needs for an entire year is achievable by har-nessing solar energy on the Earth's surface. Solar fuel is formedby utilizing light to produce chemical energy23,24 by articialphotosynthesis, or thermochemical reaction. The developmentof inexpensive, limitless and clean solar energy technologiesmust have massive and long-term benets. This progress willintensify global energy reserves through dependence on an
This journal is © The Royal Society of Chemistry 2014
original, innite and mostly import-independent resource,increase sustainability, decrease pollution, lower the costs ofmitigating climate change, and keep fossil-fuel prices lowerthan through other means.
Besides energy needs, sufficient physical evidence suggeststhat climate change is mainly responsible for carbon dioxideemissions from fossil-fuel burning.25–27 As stated by the Inter-national Energy Agency (2012),28 the goal of decreasing climatewarming by 2 �C is becoming more difficult and costly eachpassing year. All tolerable carbon dioxide emissions would belocked-in by the remaining energy infrastructure if appropriatesteps are not taken before 2017. Already, global carbon dioxidegrowth in the atmosphere was amplied by around 1.5 ppm(8 billion tons) per year from 1975 to 2002. Basically, theatmospheric carbon dioxide level inated much more quicklyby 2.25 ppm (12 billion tons) per year. Every year, more than 10billion tons of carbon dioxide is added to our atmosphere.29 Asestimated by the U.S. Department of Energy,30 natural processescan only absorb about half that amount.
Abundant CO2 can be an asset by using appropriate appli-cations in solar-fuel transformation. The recycling process ofCO2 can regulate global warming as well as fuel demand.31
Besides photocatalytic conversion of CO2, other conversionprocesses can turn CO2 into fuels illustrated in Fig. 1, but thedisadvantages in those processes are the high temperature andhigh electrical voltage requirements to break down the CO2
molecules, limitations of raw materials, high-cost operation,and unsustainability. Photocatalytic transformation of CO2 intosolar fuel is more sustainable and favorable than other tech-nologies because of zero addition of additional energyconsumption and environmental deterioration. In the begin-ning, photocatalytic CO2 conversion was conducted by an arti-cial lamp. Recently, interest has grown in the harnessing ofsolar power by advanced photocatalytic processes. The stabilityof catalysts and mild operating conditions are among the mostsignicant reasons for choosing this technology.32,33 However,more intensive study and improvement are required to selectactive solar-driven novel catalysts and effective architecture ofsolar photoreactors for industrial applications. Moreover, thereare some unanswered questions in this technology with theselectivity of the photocatalytic reactions and limited efficiency.
This paper concentrated on advancements in the photo-chemical transformation of CO2 into hydrocarbon fuels. Thesecond chapter of this paper provides a snapshot of currentdevelopments in the photocatalytic reduction of CO2 on varioustypes of semiconductors. Pure metal oxide semiconductors arecheap and the most commonly used photocatalysts in thistechnology like TiO2. To a large extent, this can be ascribed to itsband structure, which is acceptable, but certainly not optimalfor the reduction of CO2 driven by solar energy. The wide bandgap (3.2 eV) limits the absorption of light into the UV range. Themodication of metal oxide semiconductors by supportingmaterials helps to control the catalysts' size from crystal growthand facilitate the response towards visible light. The two mostcommon methods for extending the absorption range to visiblelight are sensitization and doping. Doping metal on semi-conductors acts as an electron trap to provide the electrons to
RSC Adv., 2014, 4, 20856–20893 | 20857

Fig. 1 Well-known CO2 transformation paths with potential products.34
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
the CO2. Besides, non-oxide semiconductors initially received alot of attention. For example, the valence band of sulphidesemiconductors is made of 3p orbitals of the sulfur atoms and isshied upwards, compared with those of the oxide analogues,while the conduction band electrons are also more reductive.35
Interest has grown in the involvement of exible substratesbecause they deliver a high surface area for catalysts on thelms of substrates. In the mainstream, the review categorizesthe different types of photocatalysts used in previous photo-catalytic conversions of CO2 processes (with detailed informa-tion regarding the operating conditions, preparationtechniques and physical properties of catalysts, radiationsources and selectivity) based on metal oxides, suldes, phos-phides, and p-type and non-metal-oxide semiconductors. Inaddition, these processes highlighted their modications bydoping co-metals, noble metals, transition metals and non-metals for visible light responses. Besides, the recent prospectand advancement of novel sensitized catalysts by dye elements,phthalocyanines and quantum dots (QDs) for harnessing solarfuels are prominent in this review.
2. Photocatalytic CO2 transformationbased on catalysts
The transformation of CO2 into fuel by using solar lightirradiation is an effective method because there is no addi-tion of extra energy and no negative inuence on the envi-ronment. The immediate requirement in this technology is todevelop visible light-sensitive photocatalysts, which areprominent in CO2 recycling. Different types of photocatalystshave been already introduced by many researchers in thistechnology. Some of the catalysts exhibited high conversionrates and selectivity under visible light irradiation, whileother catalysts were not feasible for visible light response andpresented low yield rates. Researchers are still trying hard toadvance the properties of catalysts in terms of solar fuelproduction. The reaction mechanism of selective photo-catalysts under light irradiation is illustrated in Scheme 1. Aconcise classication of photocatalysts is also shown inFig. 2, based on the recent development in photocatalytic CO2
transformation.
20858 | RSC Adv., 2014, 4, 20856–20893
2.1 Metal oxide photocatalysts
Metal oxides as light-sensitive catalysts have been applied inseveral processes such as the breaking down of organic andinorganic materials to valuable products,23,36,37 water treat-ment,38,39 and self-cleaning40,41 processes. The leading approachof using metal oxide catalysts in CO2 conversion to carbona-ceous fuel was introduced by Inoue et al.42 Variations in metaloxide photocatalysts have already been introduced by inte-grating foreign elements and other compounds with differentsupportive substrates43–46 for lessening its particle size,controlling its crystal growth, and increasing its surface areaand pore volume. Various metal oxide photocatalysts, whichwere frequently studied in many photocatalytic CO2 reductionprocesses, are characterized in this section with sufficientinformation.
2.1.1 Surface unmodied metal oxide photocatalysts. TiO2
is a frequently used metal oxide in the photocatalytic trans-formation of CO2 into hydrocarbon fuel because it is an inert,corrosion-resistant and inexpensive semiconductor. In nature,TiO2 is established as three familiar minerals such as rutile,anatase and brookite. Among them, rutile and anatase phasesindicate better efficiency under light radiation. Mutually,anatase and rutile phases reveal denite band gap energy ataround 3.2 eV and 3.0 eV, respectively. The rutile phase iscapable of absorbing visible light because of its comparativelylow band gap energy, while the anatase form only displays itsresponse to ultra-violet (UV) irradiation. Thus, the rutile phaseis not used due to its ineffective photoactivity; the best photo-activity can be achieved by combining anatase with a slightamount of rutile.47 A clear idea of metal oxide semiconductorsused in various studies for photocatalytic CO2 transformation isrepresented in Table 1. The light absorption by titanium-dioxide semiconductor is effective, while the semiconductorexhibits high surface area. The surface area of TiO2 is indirectlyproportional to the size of particle; conversely, the size ofthe particle is directly proportional to the band gap energy. Thesemiconductor shows high band gap energy as a result of theshiing of valence band energy to lower energies aer a certaindecrease in the particle size, whereas band energy is intenselyshied to higher energies.48 On the other hand, signicantlynarrow band gap energy leads to inadequate redox potential toachieve oxidizing and reducing reactions. Therefore, it is
This journal is © The Royal Society of Chemistry 2014

Scheme 1 Photocatalytic reaction mechanism of (a) an unmodified metal oxide semiconductor; (b) supported metal oxide semiconductor; (c)doped semiconductor; and (d) dye sensitized semiconductor under light irradiation.
Fig. 2 Classification of photocatalysts on CO2 transformation intofuel.
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
compulsory to x a structure that exhibits a settlement betweenthe particle size and band gap energy range.49 Tan et al. inves-tigated a combination of 80% anatase and 20% rutile forms as aphotoactive catalyst.50 In Scheme 1(a), the photocatalyticconversion mechanism of CO2 on unmodied metal oxidesemiconductor is sketched.
Commercial Degussa P25 is found to be slightly less activethan modied TiO2 (anatase and rutile).51 It is indicated thatunmodied TiO2 (P25) metal oxide semiconductors generallyhave high surface area as well as an affinity to organic impuri-ties on their surfaces to facilitate the adsorption of organiccompounds, which have the potential to perform as both an
This journal is © The Royal Society of Chemistry 2014
electron donor and a carbon source for the products.52 Besides,this type of metal oxide catalyst expresses better methaneproduction, compared with the Degussa P25 treated by calci-nation and washing where CO is the major product.52,53 Invarious studies, product dissimilarity is also observed due to thetreatment procedure on anatase TiO2 in several methods.54–56
For instance, treatment by the acidic solutions (1 M nitric acid)is suitable for formic acid formation due to the protonation ofreaction intermediates into the reaction medium.54 Treatmentwith 2-propanol acts as a hole scavenger in CO2 photoreductionwith anatase TiO2, where the methane formation rate wasconsiderably higher. In addition, it was observed that inabsence of 2-propanol, no methane was found.56 Moreover, thesynthesis of TiO2 by the sol–gel method removes the presence ofsmall brookite nanoparticles that normally arise in low-temperature formation reactions and considerably hinder thephase alteration to rutile at elevated temperatures.48,57
Some prospective metal oxide photocatalysts other than TiO2
were reported in many investigations. Inoue et al. studied threemetal oxides, TiO2, ZnO and WO3, to understand their photo-catalytic characteristics under UV light radiation.42 It was foundthat the yield of methyl alcohol increased as the conductionband becomes more negative with respect to the redox potentialof H2CO3/CH3OH, whereas methyl alcohol was not produced inpresence of the WO3 catalyst, which has a conduction bandmore positive than the redox potential of H2CO3/CH3OH.However, WO3 possesses low band gap energy (2.8 eV) withrespect to visible light irradiation. G. Xi et al. synthesized theW18O49 catalyst in the form of nanowires with band gap energy
RSC Adv., 2014, 4, 20856–20893 | 20859
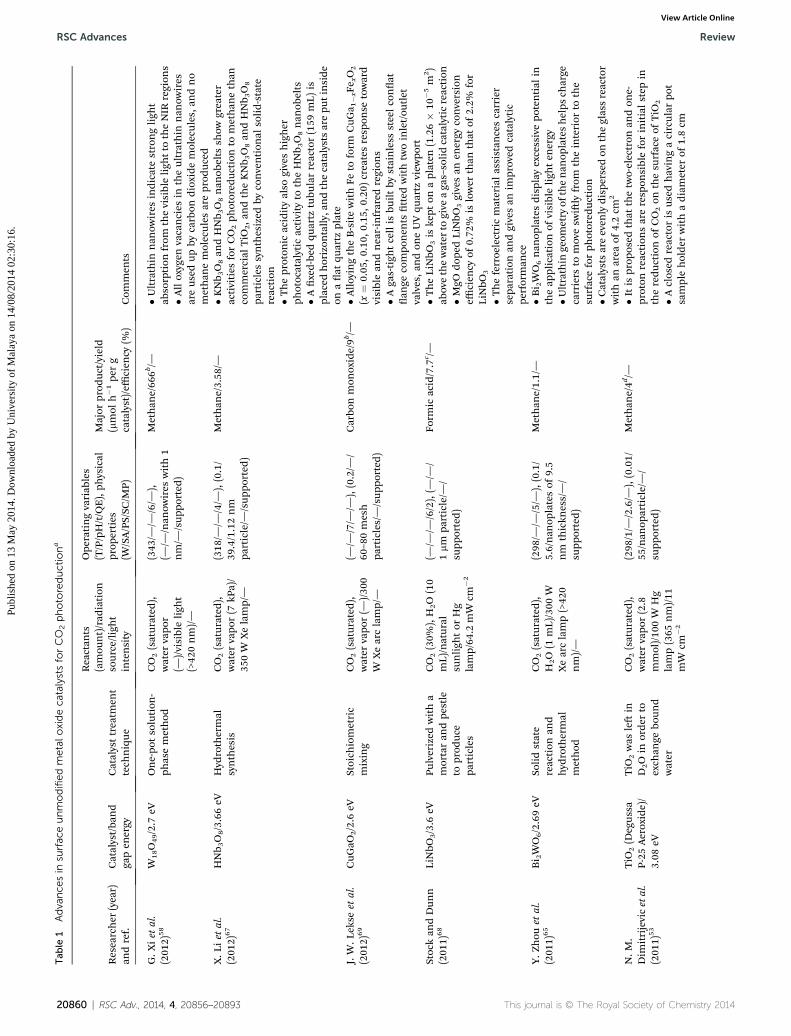
Tab
le1
Advance
sin
surfac
eunmodifiedmetalo
xideca
talystsforCO
2photoreductiona
Researcher
(year)
andref.
Catalyst/ba
nd
gapen
ergy
Catalysttreatm
ent
tech
nique
Reactan
ts(amou
nt)/rad
iation
source/light
intensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),ph
ysical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/effi
cien
cy(%
)Com
men
ts
G.X
iet
al.
(201
2)58
W18O49/2.7
eVOne-po
tsolution
-ph
asemethod
CO2(saturated
),water
vapo
r(—
)/visibleligh
t(>42
0nm)/—
(343
/—/—
/6/—
),(—
/—/nan
owires
with1
nm/—
/sup
ported
)
Methan
e/66
6b/—
�Ultrathin
nan
owires
indicate
strongligh
tab
sorption
from
thevisibleligh
tto
theNIR
region
s�A
lloxygen
vacancies
intheultrathin
nan
owires
areus
edup
bycarbon
dioxidemolecules,a
ndno
methan
emolecules
areprod
uced
X.L
iet
al.
(201
2)67
HNb 3O8/3.66
eVHyd
rothermal
synthesis
CO2(saturated
),water
vapo
r(7
kPa)/
350W
Xelamp/—
(318
/—/—
/4/—
),(0.1/
39.4/1.12nm
particle/—
/sup
ported
)
Methan
e/3.58
/—�K
Nb 3O8an
dHNb 3O8nan
obelts
show
greater
activities
forCO2ph
otored
uction
tomethan
ethan
commercial
TiO
2,a
ndtheKNb 3O8an
dHNb 3O8
particlessynthesized
byconvention
alsolid-state
reaction
�Theproton
icacidityalso
giveshigher
photocatalytic
activity
totheHNb 3O8nan
obelts
�Axed-be
dqu
artz
tubu
larreactor(159
mL)
isplaced
horizon
tally
,andthecatalystsarepu
tinside
onaat
quartz
plate
J.W.L
ekse
etal.
(201
2)69
CuG
aO2/2.6
eVStoich
iometric
mixing
CO2(saturated
),water
vapo
r(—
)/30
0W
Xearclamp/—
(—/—
/7/—
/—),(0.2/—
/60
–80mesh
particles/—/sup
ported
)
Carbo
nmon
oxide/9b/—
�Allo
yingtheB-sitewithFe
toform
CuG
a 1�xFe xO2
(x¼
0.05
,0.10,
0.15
,0.20)
createsresp
onse
toward
visiblean
dnear-infrared
region
s�A
gas-tigh
tcellis
builtby
stainless
steelconat
an
gecompo
nen
tstted
withtw
oinlet/ou
tlet
valves,a
ndon
eUVqu
artz
view
port
Stockan
dDunn
(201
1)68
LiNbO
3/3.6
eVPu
lverized
witha
mortaran
dpe
stle
toprod
uce
particles
CO2(30%
),H
2O
(10
mL)/natural
sunligh
tor
Hg
lamp/64
.2mW
cm�2
(—/—
/—/6/2),(—
/—/
1mm
particle/—
/su
pported)
Form
icacid/7.7
c /—
�TheLiNbO
3is
kept
onaplaten
(1.26�
10�5m
2)
abovethewater
togive
aga
s–solidcatalyticreaction
�MgO
dope
dLiNbO
3givesan
energy
conversion
efficien
cyof
0.72
%is
lower
than
that
of2.2%
for
LiNbO
3
�Theferroe
lectricmaterialassistan
cescarrier
sepa
ration
andgivesan
improved
catalytic
performan
ceY.Z
hou
etal.
(201
1)65
Bi 2WO6/2.69eV
Solidstate
reaction
and
hyd
rothermal
method
CO2(saturated
),H
2O(1
mL)/300
WXearclamp(>42
0nm)/—
(298
/—/—
/5/—
),(0.1/
5.6/nan
oplatesof
9.5
nm
thickn
ess/—/
supp
orted)
Methan
e/1.1/—
�Bi 2WO6nan
oplatesdisp
layexcessivepo
tential
intheap
plicationof
visibleligh
ten
ergy
�Ultrathin
geom
etry
ofthenan
oplateshelps
charge
carriers
tomovesw
ily
from
theinterior
tothe
surfaceforph
otored
uction
�Catalysts
areeven
lydisp
ersedon
theglassreactor
withan
area
of4.2cm
2
N.M
.Dim
itrijevicetal.
(201
1)53
TiO
2(D
egus
saP-25
Aeroxide)/
3.08
eV
TiO
2was
lein
D2O
inorde
rto
exch
ange
boun
dwater
CO2(saturated
),water
vapo
r(2.8
mmol)/10
0W
Hg
lamp(365
nm)/11
mW
cm�2
(298
/1/—
/2.6/—
),(0.01/
55/nan
oparticle/—/
supp
orted)
Methan
e/4d/—
�Itis
prop
osed
that
thetw
o-electron
andon
e-proton
reaction
sareresp
onsibleforinitialstep
intheredu
ctionof
CO2on
thesu
rfaceof
TiO
2
�Aclosed
reactoris
used
havingacircular
pot
sampleholde
rwithadiam
eter
of1.8cm
20860 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2014
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online

Tab
le1
(Contd.)
Researcher
(year)
andref.
Catalyst/ba
nd
gapen
ergy
Catalysttreatm
ent
tech
nique
Reactan
ts(amou
nt)/rad
iation
source/light
intensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),ph
ysical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/effi
cien
cy(%
)Com
men
ts
T.Y
uiet
al.
(201
1)52
TiO
2(D
egus
saP-25
)/3.08
eVTreated
bycalcinationan
dwashing
CO2(—
),H
2O(1.5
mL)/500
WHgarc
lamp(>31
0nm)/—
(278
/—/4.1/5/—
),(0.15/—/nan
oparticle/
0.1/su
spen
ded)
Carbo
nmon
oxide/
0.35
/—�C
H4was
themainprod
uctincase
ofPd
–TiO
2,and
CO
form
ationis
sign
icantlyredu
ced,
compa
red
withthat
onthepretreated
TiO
2
�Isotope
labe
lingsp
ecies
that
CO2an
dCO32�are
themaincarbon
sourcesof
theCH
4prod
uction
�Catalysts
areke
ptin
aroun
d-sh
aped
quartz
vessel
of6cm
diam
eter
Q.L
iuet
al.
(201
0)66
Zn2G
eO4/4.5
eVSo
lvothermal
method
CO2(—
),water
vapo
r(—
)/UV
ligh
t/—
(—/—
/—/16/—),
(—/28.27
/nan
oribbo
nof
7nm
thickn
ess/—/
supp
orted)
Methan
e/0.41
/—�U
ltra-lo
ngan
dultrathin
geom
etry
oftheZn
2GeO
4
nan
oribbo
nsh
owshighpe
rforman
ceon
photocatalytic
activity
�Therate
ofCH
4prod
uction
over
theZn
2GeO
4
nan
oribbo
ncouldbe
sign
icantlyen
han
cedby
load
ingof
PtK.K
ociet
al.
(200
9)48
TiO
2(anatase)/
3.0eV
Sol–gelprocess
CO2(—
),NaO
H(0.2
M,1
00mL
solution
)/8W
Hg
lamp(254
nm)/1.41
mW
cm�2
(—/1.08/—/24/—),(0.1/
106/14
nm
nan
oparticle/0.00
1/su
spen
ded)
Methan
e/0.30
8/—
�Astirredba
tchan
nular
reactor(380
cm3)with
quartz
inner
tube
andstainless
steelsh
elltube
isus
edin
this
expe
rimen
t�T
heyields
isob
served
insu
chan
orde
r:H
2>CH
4>
CH
3OH
$CO
Y.L
iuet
al.
(200
9)63
BiVO4
(mon
oclinic)/
2.24
eV
Microwave
assisted
hyd
rothermal
method
CO2(—
),H
2O(100
mL)/300
WXearc
lamp(>40
0nm)/—
(273
/1/—
/1/—
),(0.20/—/
nan
oparticles/—
/su
spen
ded)
Ethan
ol/110
/—�P
hotocatalystBiVO4exhibitsvisible-ligh
tresp
onse
�Intense
irradiationcreateshug
eam
ountof
C1
interm
ediate
speciesan
chored
ontheBiVO4,w
hich
dimerizes
toform
ethan
ol�C
atalysts
aredisp
ersedin
water
andmag
netically
stirredinside
thereactor
L.Yuliatiet
al.
(200
8)59
Ga 2O3(Kishida
chem
icals)/4.7
eV
Clean
ingat
13.3
kPaof
oxygen
atmosph
ereat
1073
Kfor1hou
r
CO2(200
mmol),CH
4
(200
mmol)/30
0W
Xe-lamp(220
–300
nm)/9mW
cm�2
(473
/1/—
/3/—
),(0.2/2/—
/—/sup
ported
)Hyd
rogen,c
arbo
nmon
oxide/4.13
,1.8/—
�Sup
plem
entary
thermal
energy
enhan
cesthe
reaction
betw
eenmethan
ean
dcarbon
dioxideby
stim
ulatingthethermal-activationstep
sin
the
reaction
mechan
ism
�Aclosed
quartz
reactor(30cm
3)wherethe
catalystsaresp
read
over
aat
bottom
ofthereactor
(14cm
2)
Xia
etal.(20
07)70
TiO
2(D
egus
saP-25
)/3.08
eVCalcined
at72
3K
for2h
CO2(1
mmol),water
vapo
r(5
mmol)/15
WUVlamp(365
nm)/—
(298
/—/7/5/—
),(—
/50/
nan
oparticle/—/
supp
orted)
Methan
e/15
/—�A
ppropriate
amou
ntof
MWCNTsas
supp
orts
for
TiO
2couldextrao
rdinarilyad
vance
theeffi
cien
cyof
theph
otocatalytic
reaction
�Catalysts
areke
pton
apieceof
tran
sparen
tglass
andthen
putinto
ahom
emad
estainless
steel
reactor
C.-C
.Loet
al.
(200
7)62
ZrO2(Proch
em)/
5.0eV
ZrO2is
immob
ilized
bycoated
glass
pelle
ts
CO2(99.8%
),H
2
(95%
)/15
WnearUV
lamps
(254
nm)/—
(316
/1.1/—
/2/—
),(0.25/—/100
mesh
powde
r/—/sup
ported
andim
mob
ilized
)
Carbo
nmon
oxide/
0.51
/—�A
circulated
,packe
d-be
dph
otocatalytic
reaction
system
ismad
efrom
aqu
artz
tube
with48
0mm
lengthan
d22
.5mm
inner
diam
eter
�One-site
Langm
uir–Hinsh
ewoo
dkinetic
mod
elis
successfully
tted
tosimulatetheph
otored
uction
rate
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 20861
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
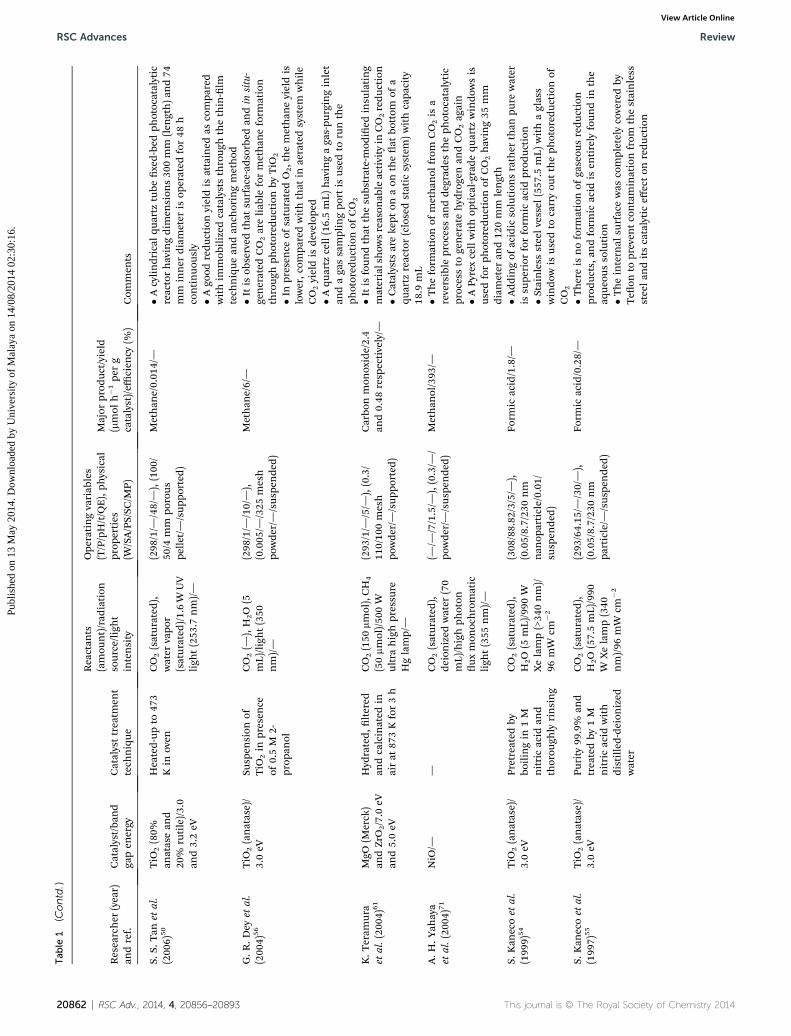
Tab
le1
(Contd.)
Researcher
(year)
andref.
Catalyst/ba
nd
gapen
ergy
Catalysttreatm
ent
tech
nique
Reactan
ts(amou
nt)/rad
iation
source/light
intensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),ph
ysical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/effi
cien
cy(%
)Com
men
ts
S.S.
Tan
etal.
(200
6)50
TiO
2(80%
anatasean
d20
%rutile)/3.0
and3.2eV
Heated-up
to47
3Kin
oven
CO2(saturated
),water
vapo
r(saturated
)/1.6W
UV
ligh
t(253
.7nm)/—
(298
/1/—
/48/—),(100
/50
/4mm
porous
pelle
t/—/sup
ported
)
Methan
e/0.01
4/—
�Acylindrical
quartz
tube
xed-be
dph
otocatalytic
reactorhavingdimen
sion
s30
0mm
(len
gth)a
nd74
mm
inner
diam
eter
isop
erated
for48
hcontinuo
usly
�Ago
odredu
ctionyieldis
attained
ascompa
red
withim
mob
ilized
catalyststhroug
hthethin-lm
tech
nique
andan
choringmethod
G.R
.Dey
etal.
(200
4)56
TiO
2(anatase)/
3.0eV
Susp
ension
ofTiO
2in
presen
ceof
0.5M
2-prop
anol
CO2(—
),H
2O
(5mL)/light(350
nm)/—
(298
/1/—
/10/—),
(0.005
/—/325
mesh
powde
r/—/sus
pende
d)
Methan
e/6/—
�Itis
observed
that
surface-ad
sorbed
andin
situ-
generated
CO2areliab
leformethan
eform
ation
throug
hph
otored
uction
byTiO
2
�Inpresen
ceof
saturatedO2,themethan
eyieldis
lower,c
ompa
redwiththat
inaeratedsystem
while
CO2yieldis
develope
d�A
quartz
cell(16.5mL)
havingaga
s-pu
rginginlet
andaga
ssamplingpo
rtis
used
torunthe
photored
uction
ofCO2
K.T
eram
ura
etal.(200
4)61
MgO
(Merck)
andZrO2/7.0
eVan
d5.0eV
Hyd
rated,
ltered
andcalcinated
inairat
873Kfor3h
CO2(150
mmol),CH
4
(50mmol)/50
0W
ultrahighpressu
reHglamp/—
(293
/1/—
/5/—
),(0.3/
110/10
0mesh
powde
r/—/sup
ported
)
Carbo
nmon
oxide/2.4
and0.48
resp
ectively/—
�Itis
foun
dthat
thesu
bstrate-mod
ied
insu
lating
materialshow
sreason
ableactivityin
CO2redu
ction
�Catalysts
areke
pton
aon
theat
bottom
ofa
quartz
reactor(closedstatic
system
)withcapa
city
18.9
mL
A.H
.Yah
aya
etal.(200
4)71
NiO
/——
CO2(saturated
),de
ionized
water
(70
mL)/highph
oton
ux
mon
ochromatic
ligh
t(355
nm)/—
(—/—
/7/1.5/—
),(0.3/—
/po
wde
r/—/sus
pende
d)Methan
ol/393
/—�T
heform
ationof
methan
olfrom
CO2is
areversible
processan
dde
grad
estheph
otocatalytic
processto
generatehyd
rogenan
dCO2ag
ain
�APy
rexcellwithop
tical-g
rade
quartz
windo
wsis
used
forph
otored
uction
ofCO2having35
mm
diam
eter
and12
0mm
length
S.Kan
ecoet
al.
(199
9)54
TiO
2(anatase)/
3.0eV
Pretreated
bybo
ilingin
1M
nitricacid
and
thorou
ghly
rinsing
CO2(saturated
),H
2O
(5mL)/990
WXelamp(>34
0nm)/
96mW
cm�2
(308
/88.82
/3/5/—
),(0.05/8.7/23
0nm
nan
oparticle/0.01
/su
spen
ded)
Form
icacid/1.8/—
�Add
ingof
acidic
solution
srather
than
pure
water
issu
perior
forform
icacid
prod
uction
�Stainless
steelvessel
(557
.5mL)
withaglass
windo
wis
used
tocarryou
ttheph
otored
uction
ofCO2
S.Kan
ecoet
al.
(199
7)55
TiO
2(anatase)/
3.0eV
Purity
99.9%
and
treatedby
1M
nitricacid
with
distille
d-deion
ized
water
CO2(saturated
),H
2O
(57.5mL)/990
WXelamp(340
nm)/96
mW
cm�2
(293
/64.15
/—/30/—),
(0.05/8.7/23
0nm
particle/—
/sus
pende
d)
Form
icacid/0.28/—
�Thereis
noform
ationof
gaseou
sredu
ction
prod
ucts,a
ndform
icacid
isen
tirely
foun
din
the
aque
oussolution
�Theinternal
surfacewas
completelycoveredby
Te
onto
preven
tcontaminationfrom
thestainless
steelan
ditscatalyticeff
ecton
redu
ction
20862 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 201
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
4
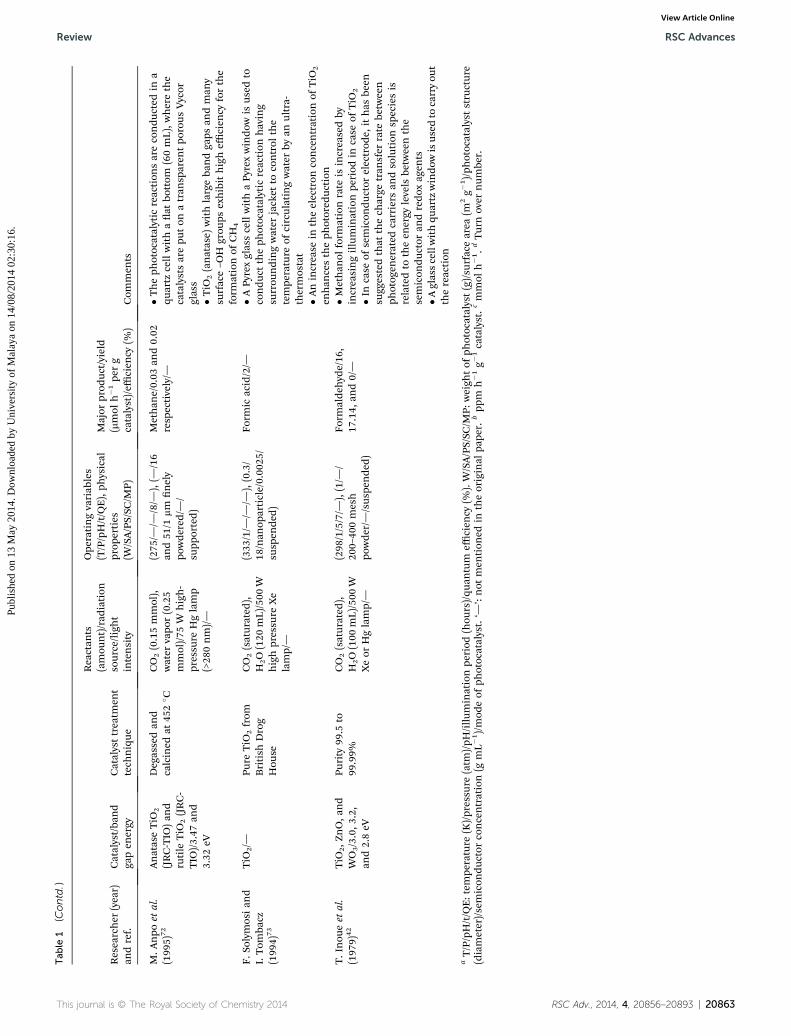
Tab
le1
(Contd.)
Researcher
(year)
andref.
Catalyst/ba
nd
gapen
ergy
Catalysttreatm
ent
tech
nique
Reactan
ts(amou
nt)/rad
iation
source/light
intensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),ph
ysical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/effi
cien
cy(%
)Com
men
ts
M.A
npo
etal.
(199
5)72
AnataseTiO
2
(JRC-TIO
)an
drutile
TiO
2(JRC-
TIO
)/3.47
and
3.32
eV
Degassedan
dcalcined
at45
2� C
CO2(0.15mmol),
water
vapo
r(0.25
mmol)/75
Whigh-
pressu
reHglamp
(>28
0nm)/—
(275
/—/—
/8/—
),(—
/16
and51
/1mm
nely
powde
red/—/
supp
orted)
Methan
e/0.03
and0.02
resp
ectively/—
�Theph
otocatalytic
reaction
sarecondu
cted
ina
quartz
cellwithaat
bottom
(60mL),w
herethe
catalystsarepu
ton
atran
sparen
tpo
rous
Vycor
glass
�TiO
2(anatase)
withlargeba
ndga
psan
dman
ysu
rface–O
Hgrou
psexhibithigheffi
cien
cyforthe
form
ationof
CH
4
F.So
lymosian
dI.Tom
bacz
(199
4)73
TiO
2/—
Pure
TiO
2from
British
Drog
Hou
se
CO2(saturated
),H
2O(120
mL)/500
Whighpressu
reXe
lamp/—
(333
/1/—
/—/—
),(0.3/
18/nan
oparticle/0.00
25/
susp
ende
d)
Form
icacid/2/—
�APy
rexglasscellwithaPy
rexwindo
wis
used
tocondu
cttheph
otocatalytic
reaction
having
surrou
ndingwater
jacket
tocontrol
the
tempe
rature
ofcirculatingwater
byan
ultra-
thermostat
�Anincrease
intheelectron
concentrationof
TiO
2
enhan
cestheph
otored
uction
T.Inou
eet
al.
(197
9)42
TiO
2,Z
nO,a
nd
WO3/3.0,3
.2,
and2.8eV
Purity
99.5
to99
.99%
CO2(saturated
),H
2O(100
mL)/500
WXeor
Hglamp/—
(298
/1/5/7/—
),(1/—
/20
0–40
0mesh
powde
r/—/sus
pende
d)
Form
alde
hyd
e/16
,17
.14,
and0/—
�Methan
olform
ationrate
isincreasedby
increasingilluminationpe
riod
incase
ofTiO
2
�Incase
ofsemicon
ductor
electrod
e,ithas
been
sugg
estedthat
thech
arge
tran
sfer
rate
betw
een
photog
enerated
carriers
andsolution
speciesis
relatedto
theen
ergy
levels
betw
eenthe
semicon
ductor
andredo
xag
ents
�Aglasscellwithqu
artzwindo
wisus
edto
carryou
tthereaction
aT/P/pH/t/Q
E:tem
perature
(K)/pressu
re(atm
)/pH
/illu
minationpe
riod
(hou
rs)/qu
antum
efficien
cy(%
).W/SA/PS/SC
/MP:
weigh
tof
photocatalyst(g)/su
rfacearea
(m2g�
1)/ph
otocatalyststructure
(diameter)/semicon
ductor
concentration(g
mL�
1)/mod
eof
photocatalyst.‘—
’:not
men
tion
edin
theoriginal
pape
r.bpp
mh�1g�
1catalyst.c
mmol
h�1.d
Turnover
num
ber.
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 20863
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online

RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
of 2.7 eV for the photocatalytic conversion of CO2 to methaneunder visible light irradiation.58 The diameter of W18O49
nanowires is approximately 0.9 nm and hold a large number ofoxygen vacancies. These oxygen vacancies indicate anoutstanding competency on photochemical carbon dioxidereduction over the visible light region.
Metal oxides like Ga2O3, ZrO2, and MgO were reported invarious studies as having wide band gaps, compared with otherphotocatalytic metal oxides.59–61 In those studies, methane andhydrogen were used as reductants for photoreduction of CO2,and CO was the major product.61,62 In the case of ZrO2, highphotoreduction of CO2 is obtained when H2 is used rather thanCH4. Moreover, it is found that some intermediates are formedwhen H2 is used for CO2 photoreduction over ZrO2 and MgO asa surface formate species, whereas the acetate is generated inlieu of the formate when CH4 is used as reductant over ZrO2.60,61
It was also observed that the surface bidentate formate speciesshowed high photoactivity for photocatalytic reduction of CO2
over ZrO2 and MgO.Recently, visible light-responsive and other oxide catalysts
fabricated through various routes like hydrothermal, sol-vothermal, and solid state reactions are widely used in thephotochemical reduction of CO2 because of high yield rates.Microwave-assisted, hydrothermally derived monoclinic BiVO4
catalysts show higher ethanol yields (110 mmol h�1 per g ofcatalyst) than that of tetragonal BiVO4 due to anchored CO3
2� tothe Bi3+ sites on the external surface through a weak Bi–O bondfor receiving the photogenerated electrons effectively from the V3d-block bands of BiVO4. The asymmetric behavior of themonoclinic phase around the Bi3+ ion is more effective than inthe tetragonal phase. That is why, the Bi3+ ion shows a strongerlone pair behavior in the monoclinic phase for increasing thetendency of the Bi–O bond formation with CO3
2�.63,64 Y. Zhouet al. and Q. Liu et al. reported visible light-responsive, 9.5 nmthick Bi2WO6 nanoplates and UV-driven single-crystallineZn2GeO4 nanobelts with a thickness of 7 nm, respectively.65,66
The ultrathin geometry encourages the charge carriers totransfer swily from inside to the external surface for effectiveparticipation in the CO2 photoreduction reaction. It also sepa-rates the photogenerated electron and hole progressively, ulti-mately decreasing the recombination rate.
It has been reported that hydrothermally prepared KNb3O8
and HNb3O8 nanobelts show high yields of methane, comparedwith the same catalysts derived from conventional solid statereactions and commercial TiO2 (Degussa P25).67 In the case ofhydrothermal synthesis, this nanobelt-like morphology andprotonic acidity give higher photochemical activity for methaneproduction through hydrogen bonding, which is facilitated bythe separating and trapping of photogenerated carriers at theinterlayer surfaces of HNb3O8 and KNb3O8. Ferroelectric mate-rials can be used as remarkable substitutes for the standardsemiconductor photocatalysts. Ferroelectric characteristics leadto driving the electrons and holes apart because they possessinternal dipoles. Therefore, this phenomenon decreases theprobabilities of the recombination of carriers and also inhibitsthe reaction of redox products, driving the equilibrium towardsproduct formation. For this ferroelectric behavior, the polar
20864 | RSC Adv., 2014, 4, 20856–20893
compound LiNbO3 was used by Stock and Dunn for photo-catalytic CO2 conversion to formic acid under an Hg lamp ordirect natural sunlight irradiation.68
2.1.2 Supported metal oxide photocatalysts. Several typesof supportive materials were used in this technology, such aszeolite, mesoporous SiO2, kaolinite, Al2O3, montmorillonite andother pillar materials. Table 2 demonstrates the texturalbelongings of the silica supports and the TiO2-loaded sampleswith specic surface area, pore volume, andmaximum pore sizedistributions.46 The mesoporous, SiO2-prepared material MCM-41 shows higher specic surface area having an average poresize of 2.3 nm. The pore structure of supported materials is avital factor because it manages the transportation of reactantsand products to and from the surface, and controls the size ofthe active surface area.74 The detailed information of supportedmetal oxide semiconductors investigated in previous studies arehighlighted critically in Table 3. Titania charging on the silicamatrix reduces the surface area of the supports and alsodecreases the pore volume, as the titania content increases. Thereduction of pore volume indicates that the loading of semi-conductor particles saturates the maximum space of theordered channels within the support, which helps to control thecatalysts' size from crystal growth. High catalyst loading cancause pore blockage inside the channels, although the inter-connection among the parallel pore channels should have theability to reduce this effect.75 The common photocatalyticreaction path for supported metal oxide semiconductor isillustrated in Scheme 1(b).
A catalyst on supported materials has been used in manyphotocatalytic aqueous systems due to their immobilization interms of easier separation of the catalyst aer the photocatalyticreactions. It is reported that supported photocatalysts exhibitlower activities than powdered TiO2 materials. However, treatedphotocatalysts show their high activity because of cyanidecontent and represent the dicyanoaurate photoreductionactivity in order to reect TiO2/GrSiO2 > Degussa P25 > TiO2/SBA-15. Depending on the treatment processes, TiO2/SBA-15materials achieved up to eight times of high activity, comparedwith the Degussa P25 TiO2.76 The addition of Cu species onTiO2–SiO2 catalysts increased the overall CO2 conversion effi-ciency by suppressing the electron–hole recombination andincreasing multi-electron reactions,77 compared with only TiO2–
SiO2 matrices.78 It is reported that the addition of 0.5% Cushows high specic surface area, but the addition of 1% and 3%Cu successively reduces the specic surface area. A multi-walledcarbon nanotube (MWCNT)-supported TiO2 composite catalystis introduced by Xia et al. due to its unique electronic proper-ties,70 and the results show a high conversion rate when aproper amount of MWCNTs is used as a support for TiO2.Moreover, the MWCNT-supported composite catalysts are ableto lessen the accumulation of the catalyst particles and reducethe recombination of electron–hole pairs by moving it along thetubes. Carbon is used as a catalyst support in gas–solid reactionphases with suitable temperature and pressure under which thecarbon support remains stable. In hydrogenation reactions,carbon-supported iron catalysts with high dispersions wereused at 1 atm and <700 K because of carbon stability under this
This journal is © The Royal Society of Chemistry 2014
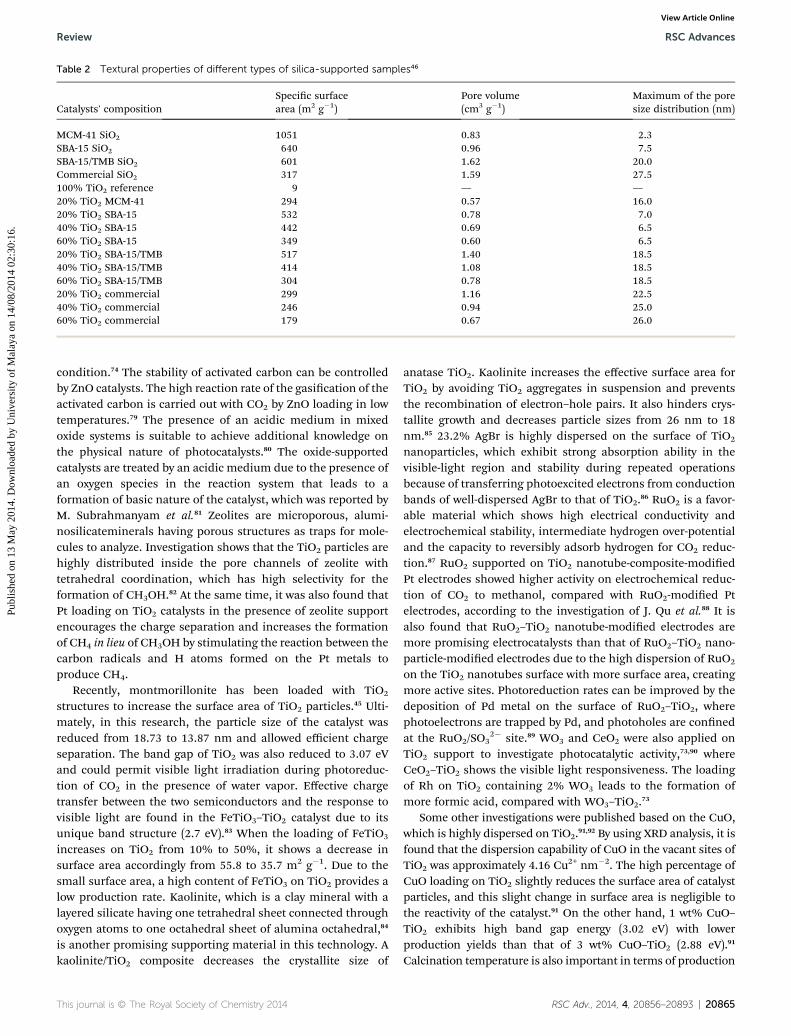
Table 2 Textural properties of different types of silica-supported samples46
Catalysts' compositionSpecic surfacearea (m2 g�1)
Pore volume(cm3 g�1)
Maximum of the poresize distribution (nm)
MCM-41 SiO2 1051 0.83 2.3SBA-15 SiO2 640 0.96 7.5SBA-15/TMB SiO2 601 1.62 20.0Commercial SiO2 317 1.59 27.5100% TiO2 reference 9 — —20% TiO2 MCM-41 294 0.57 16.020% TiO2 SBA-15 532 0.78 7.040% TiO2 SBA-15 442 0.69 6.560% TiO2 SBA-15 349 0.60 6.520% TiO2 SBA-15/TMB 517 1.40 18.540% TiO2 SBA-15/TMB 414 1.08 18.560% TiO2 SBA-15/TMB 304 0.78 18.520% TiO2 commercial 299 1.16 22.540% TiO2 commercial 246 0.94 25.060% TiO2 commercial 179 0.67 26.0
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
condition.74 The stability of activated carbon can be controlledby ZnO catalysts. The high reaction rate of the gasication of theactivated carbon is carried out with CO2 by ZnO loading in lowtemperatures.79 The presence of an acidic medium in mixedoxide systems is suitable to achieve additional knowledge onthe physical nature of photocatalysts.80 The oxide-supportedcatalysts are treated by an acidic medium due to the presence ofan oxygen species in the reaction system that leads to aformation of basic nature of the catalyst, which was reported byM. Subrahmanyam et al.81 Zeolites are microporous, alumi-nosilicateminerals having porous structures as traps for mole-cules to analyze. Investigation shows that the TiO2 particles arehighly distributed inside the pore channels of zeolite withtetrahedral coordination, which has high selectivity for theformation of CH3OH.82 At the same time, it was also found thatPt loading on TiO2 catalysts in the presence of zeolite supportencourages the charge separation and increases the formationof CH4 in lieu of CH3OH by stimulating the reaction between thecarbon radicals and H atoms formed on the Pt metals toproduce CH4.
Recently, montmorillonite has been loaded with TiO2
structures to increase the surface area of TiO2 particles.45 Ulti-mately, in this research, the particle size of the catalyst wasreduced from 18.73 to 13.87 nm and allowed efficient chargeseparation. The band gap of TiO2 was also reduced to 3.07 eVand could permit visible light irradiation during photoreduc-tion of CO2 in the presence of water vapor. Effective chargetransfer between the two semiconductors and the response tovisible light are found in the FeTiO3–TiO2 catalyst due to itsunique band structure (2.7 eV).83 When the loading of FeTiO3
increases on TiO2 from 10% to 50%, it shows a decrease insurface area accordingly from 55.8 to 35.7 m2 g�1. Due to thesmall surface area, a high content of FeTiO3 on TiO2 provides alow production rate. Kaolinite, which is a clay mineral with alayered silicate having one tetrahedral sheet connected throughoxygen atoms to one octahedral sheet of alumina octahedral,84
is another promising supporting material in this technology. Akaolinite/TiO2 composite decreases the crystallite size of
This journal is © The Royal Society of Chemistry 2014
anatase TiO2. Kaolinite increases the effective surface area forTiO2 by avoiding TiO2 aggregates in suspension and preventsthe recombination of electron–hole pairs. It also hinders crys-tallite growth and decreases particle sizes from 26 nm to 18nm.85 23.2% AgBr is highly dispersed on the surface of TiO2
nanoparticles, which exhibit strong absorption ability in thevisible-light region and stability during repeated operationsbecause of transferring photoexcited electrons from conductionbands of well-dispersed AgBr to that of TiO2.86 RuO2 is a favor-able material which shows high electrical conductivity andelectrochemical stability, intermediate hydrogen over-potentialand the capacity to reversibly adsorb hydrogen for CO2 reduc-tion.87 RuO2 supported on TiO2 nanotube-composite-modiedPt electrodes showed higher activity on electrochemical reduc-tion of CO2 to methanol, compared with RuO2-modied Ptelectrodes, according to the investigation of J. Qu et al.88 It isalso found that RuO2–TiO2 nanotube-modied electrodes aremore promising electrocatalysts than that of RuO2–TiO2 nano-particle-modied electrodes due to the high dispersion of RuO2
on the TiO2 nanotubes surface with more surface area, creatingmore active sites. Photoreduction rates can be improved by thedeposition of Pd metal on the surface of RuO2–TiO2, wherephotoelectrons are trapped by Pd, and photoholes are connedat the RuO2/SO3
2� site.89 WO3 and CeO2 were also applied onTiO2 support to investigate photocatalytic activity,73,90 whereCeO2–TiO2 shows the visible light responsiveness. The loadingof Rh on TiO2 containing 2% WO3 leads to the formation ofmore formic acid, compared with WO3–TiO2.73
Some other investigations were published based on the CuO,which is highly dispersed on TiO2.91,92 By using XRD analysis, it isfound that the dispersion capability of CuO in the vacant sites ofTiO2 was approximately 4.16 Cu2+ nm�2. The high percentage ofCuO loading on TiO2 slightly reduces the surface area of catalystparticles, and this slight change in surface area is negligible tothe reactivity of the catalyst.91 On the other hand, 1 wt% CuO–TiO2 exhibits high band gap energy (3.02 eV) with lowerproduction yields than that of 3 wt% CuO–TiO2 (2.88 eV).91
Calcination temperature is also important in terms of production
RSC Adv., 2014, 4, 20856–20893 | 20865
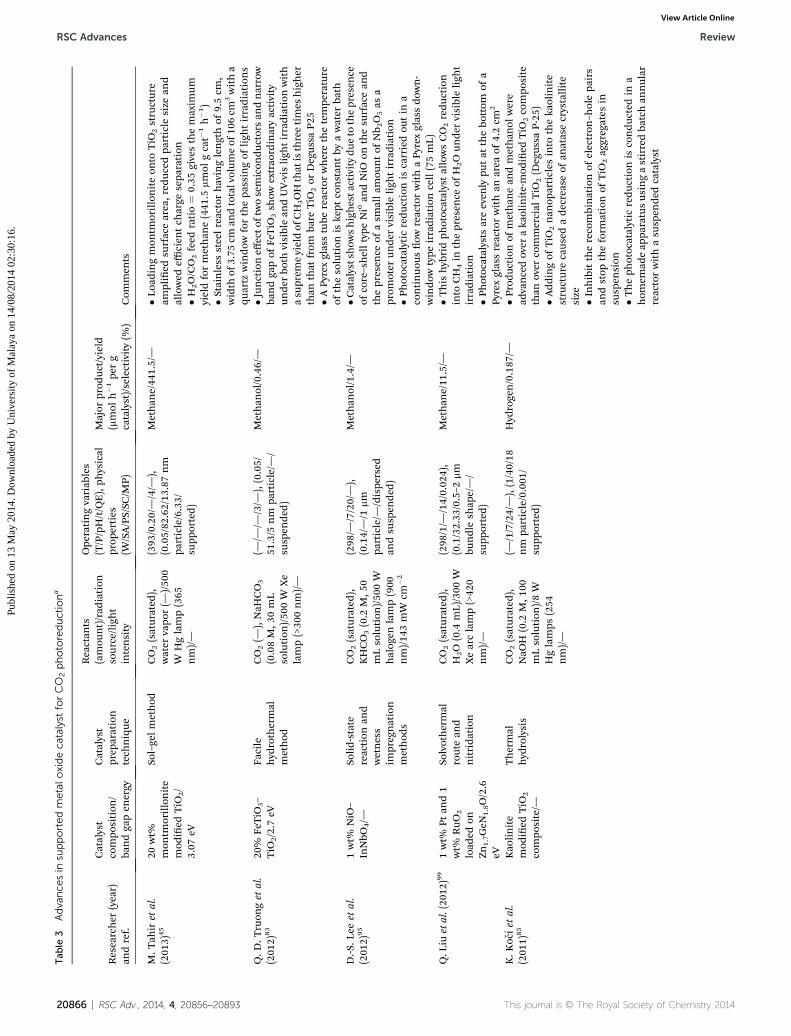
Tab
le3
Advance
sin
supportedmetalo
xideca
talyst
forCO
2photoreductiona
Researcher
(year)
andref.
Catalyst
compo
sition
/ba
ndga
pen
ergy
Catalyst
prep
aration
tech
nique
Reactan
ts(amou
nt)/rad
iation
source/light
intensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),ph
ysical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
M.T
ahiret
al.
(201
3)45
20wt%
mon
tmorillonite
mod
ied
TiO
2/
3.07
eV
Sol–gelmethod
CO2(saturated
),water
vapo
r(—
)/50
0W
Hglamp(365
nm)/—
(393
/0.20/—/4/—
),(0.05/82
.62/13
.87nm
particle/6.33/
supp
orted)
Methan
e/44
1.5/—
�Loa
dingmon
tmorilloniteon
toTiO
2structure
amplied
surfacearea,red
uced
particle
size
and
allowed
efficien
tch
arge
sepa
ration
�H2O/CO2feed
ratio¼
0.35
givesthemaxim
umyieldformethan
e(441
.5mmol
gcat�
1h�1)
�Stainless
steelreactorhavinglengthof
9.5cm
,width
of3.75
cman
dtotalvolum
eof
106cm
3witha
quartz
windo
wforthepa
ssingof
ligh
tirradiations
Q.D
.Truon
get
al.
(201
2)83
20%
FeTiO
3–
TiO
2/2.7
eVFa
cile
hyd
rothermal
method
CO2(—
),NaH
CO3
(0.08M,3
0mL
solution
)/50
0W
Xe
lamp(>30
0nm)/—
(—/—
/—/3/—
),(0.05/
51.3/5
nm
particle/—
/su
spen
ded)
Methan
ol/0.46/—
�Jun
ctioneff
ecto
ftwosemicon
ductorsan
dnarrow
bandga
pof
FeTiO
3sh
owextrao
rdinaryactivity
unde
rbo
thvisiblean
dUV-vis
ligh
tirradiationwith
asu
prem
eyieldof
CH
3OHthat
isthreetimes
higher
than
that
from
bare
TiO
2or
Degus
saP2
5�A
Pyrexglasstube
reactorwherethetempe
rature
ofthesolution
iske
ptconstan
tby
awater
bath
D.-S
.Lee
etal.
(201
2)95
1wt%
NiO
–InNbO
4/—
Solid-state
reaction
and
wetness
impregnation
method
s
CO2(saturated
),KHCO3(0.2
M,5
0mLsolution
)/50
0W
halog
enlamp(900
nm)/14
3mW
cm�2
(298
/—/7/20/—),
(0.14/—/1
mm
particle/—
/dispe
rsed
andsu
spen
ded)
Methan
ol/1.4/—
�Catalystshow
shighesta
ctivitydu
eto
thepresen
ceof
core–shelltype
Ni0an
dNiO
onthesu
rfacean
dthepresen
ceof
asm
allam
ountof
Nb 2O5as
aprom
oter
unde
rvisibleligh
tirradiation
�Photocatalytic
redu
ctionis
carriedou
tin
acontinuo
usow
reactorwithaPy
rexglassdo
wn-
windo
wtype
irradiationcell(75mL)
Q.L
iuetal.(20
12)99
1wt%
Ptan
d1
wt%
RuO
2
load
edon
Zn1.7GeN
1.8O/2.6
eV
Solvothermal
routean
dnitridation
CO2(saturated
),H
2O(0.4
mL)/300
WXearclamp(>42
0nm)/—
(298
/1/—
/14/0.02
4),
(0.1/32.33
/0.5–2
mm
bundlesh
ape/—/
supp
orted)
Methan
e/11
.5/—
�This
hyb
ridph
otocatalystallowsCO2redu
ction
into
CH
4in
thepresen
ceof
H2Oun
dervisibleligh
tirradiation
�Photocatalysts
areeven
lypu
tat
thebo
ttom
ofa
Pyrexglassreactorwithan
area
of4.2cm
2
K.K
ocıet
al.
(201
1)85
Kao
linite
mod
ied
TiO
2
compo
site/—
Thermal
hyd
rolysis
CO2(saturated
),NaO
H(0.2
M,1
00mLsolution
)/8W
Hglamps
(254
nm)/—
(—/1/7/24/—),(1/40/18
nm
particle/0.001
/su
pported)
Hyd
rogen/0.187
/—�P
rodu
ctionof
methan
ean
dmethan
olwere
advancedover
aka
olinite-mod
ied
TiO
2compo
site
than
over
commercial
TiO
2(D
egus
saP-25
)�A
ddingof
TiO
2nan
oparticles
into
theka
olinite
structurecaus
edade
crease
ofan
atasecrystallite
size �Inhibittherecombinationof
electron
–holepa
irs
andstop
theform
ationof
TiO
2ag
gregates
insu
spen
sion
�Theph
otocatalytic
redu
ctionis
condu
cted
ina
hom
emad
eap
paratusus
ingastirredba
tchan
nular
reactorwithasu
spen
dedcatalyst
20866 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 20
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
14
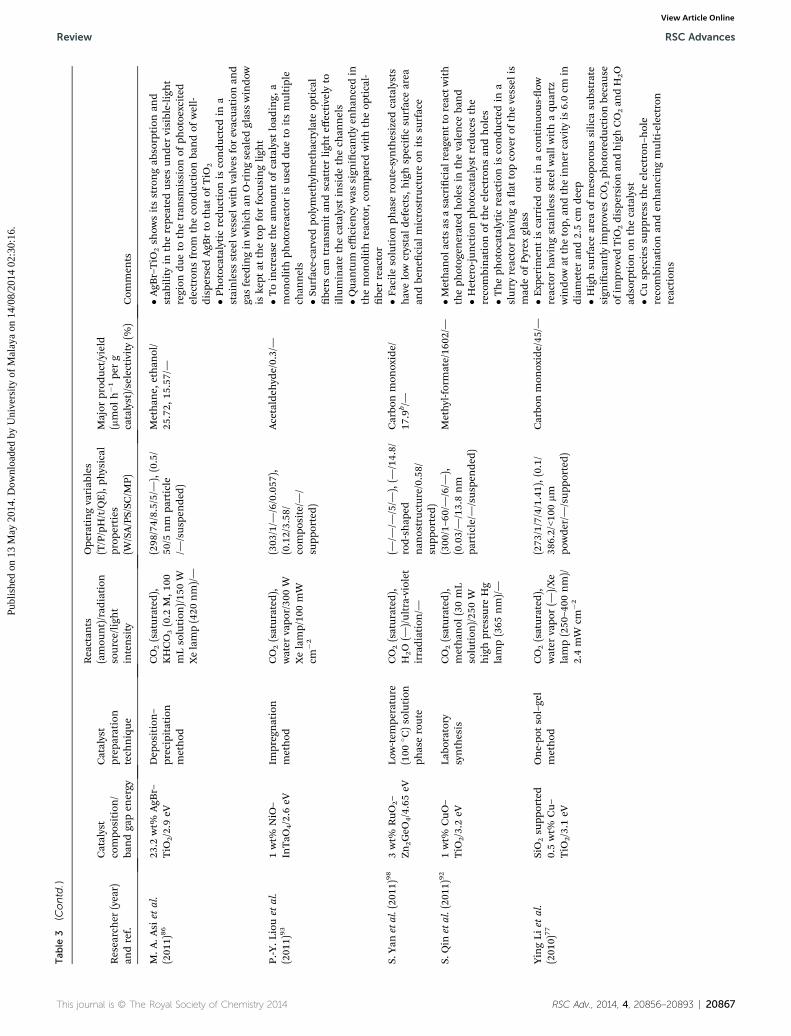
Tab
le3
(Contd.)
Researcher
(year)
andref.
Catalyst
compo
sition
/ba
ndga
pen
ergy
Catalyst
prep
aration
tech
nique
Reactan
ts(amou
nt)/rad
iation
source/light
intensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),ph
ysical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
M.A
.Asi
etal.
(201
1)86
23.2
wt%
AgB
r–TiO
2/2.9eV
Dep
osition–
precipitation
method
CO2(saturated
),KHCO3(0.2
M,1
00mLsolution
)/15
0W
Xelamp(420
nm)/—
(298
/74/8.5/5/—),(0.5/
50/5
nm
particle
/—/sus
pende
d)
Methan
e,ethan
ol/
25.72,
15.57/—
�AgB
r–TiO
2sh
owsitsstrongab
sorption
and
stab
ilityin
therepe
ated
uses
unde
rvisible-ligh
tregion
dueto
thetran
smission
ofph
otoe
xcited
electron
sfrom
thecondu
ctionba
ndof
well-
disp
ersedAgB
rto
that
ofTiO
2
�Photocatalytic
redu
ctionis
condu
cted
ina
stainless
steelv
esselw
ithvalves
forevacua
tion
and
gasfeed
ingin
whichan
O-ringsealed
glasswindo
wis
kept
atthetopforfocu
singligh
tP.-Y.L
iouet
al.
(201
1)93
1wt%
NiO
–InTaO
4/2.6
eVIm
pregnation
method
CO2(saturated
),water
vapo
r/30
0W
Xelamp/10
0mW
cm�2
(303
/1/—
/6/0.057
),(0.12/3.58
/compo
site/—
/su
pported)
Acetaldeh
yde/0.3/—
�Toincrease
theam
ountof
catalyst
load
ing,
amon
olithph
otoreactor
isus
eddu
eto
itsmultiple
chan
nels
�Surface-carvedpo
lymethylmethacrylate
optical
be
rscantran
smitan
dscatterligh
teff
ectively
toilluminatethecatalyst
inside
thech
annels
�Qua
ntum
efficien
cywas
sign
icantlyen
han
cedin
themon
olithreactor,compa
redwiththeop
tical-
be
rreactor
S.Yan
etal.(20
11)98
3wt%
RuO
2–
Zn2GeO
4/4.65eV
Low-tem
perature
(100
� C)solution
phaseroute
CO2(saturated
),H
2O
(—)/ultra-violet
irradiation/—
(—/—
/—/5/—
),(—
/14.8/
rod-sh
aped
nan
ostruc
ture/0.58/
supp
orted)
Carbo
nmon
oxide/
17.9
b/—
�Facilesolution
phaseroute-synthesized
catalysts
havelowcrystalde
fects,
highsp
ecicsu
rfacearea
andbe
ne
cial
microstructureon
itssu
rface
S.Qin
etal.(20
11)92
1wt%
CuO
–TiO
2/3.2eV
Labo
ratory
synthesis
CO2(saturated
),methan
ol(30mL
solution
)/25
0W
highpressu
reHg
lamp(365
nm)/—
(300
/1–60/—/6/—
),(0.03/—/13.8nm
particle/—
/sus
pende
d)
Methyl-formate/16
02/—
�Methan
olactsas
asacricial
reag
enttoreactw
ith
theph
otog
enerated
holes
inthevalence
band
�Hetero-junctionph
otocatalystredu
cesthe
recombinationof
theelectron
san
dholes
�Theph
otocatalytic
reaction
iscondu
cted
ina
slurry
reactorhavingaat
topcoverof
thevessel
ismad
eof
Pyrexglass
YingLi
etal.
(201
0)77
SiO2su
pported
0.5wt%
Cu–
TiO
2/3.1eV
One-po
tsol–gel
method
CO2(saturated
),water
vapo
r(—
)/Xe
lamp(250
–400
nm)/
2.4mW
cm�2
(273
/1/7/4/1.41),(0.1/
386.2/<1
00mm
powde
r/—/sup
ported
)
Carbo
nmon
oxide/45
/—�E
xperim
entis
carriedou
tin
acontinuo
us-ow
reactorhavingstainless
steelwallwithaqu
artz
windo
wat
thetop,
andtheinner
cavity
is6.0cm
indiam
eter
and2.5cm
deep
�Highsu
rfacearea
ofmesop
orou
ssilica
subs
trate
sign
icantlyim
proves
CO2ph
otored
uction
becaus
eof
improved
TiO
2disp
ersion
andhighCO2an
dH
2O
adsorption
onthecatalyst
�Cusp
eciessu
ppress
theelectron
–hole
recombinationan
den
han
cingmulti-electron
reaction
s
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 2086
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
7
Tab
le3
(Contd.)
Researcher
(year)
andref.
Catalyst
compo
sition
/ba
ndga
pen
ergy
Catalyst
prep
aration
tech
nique
Reactan
ts(amou
nt)/rad
iation
source/light
intensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),ph
ysical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
X.-H
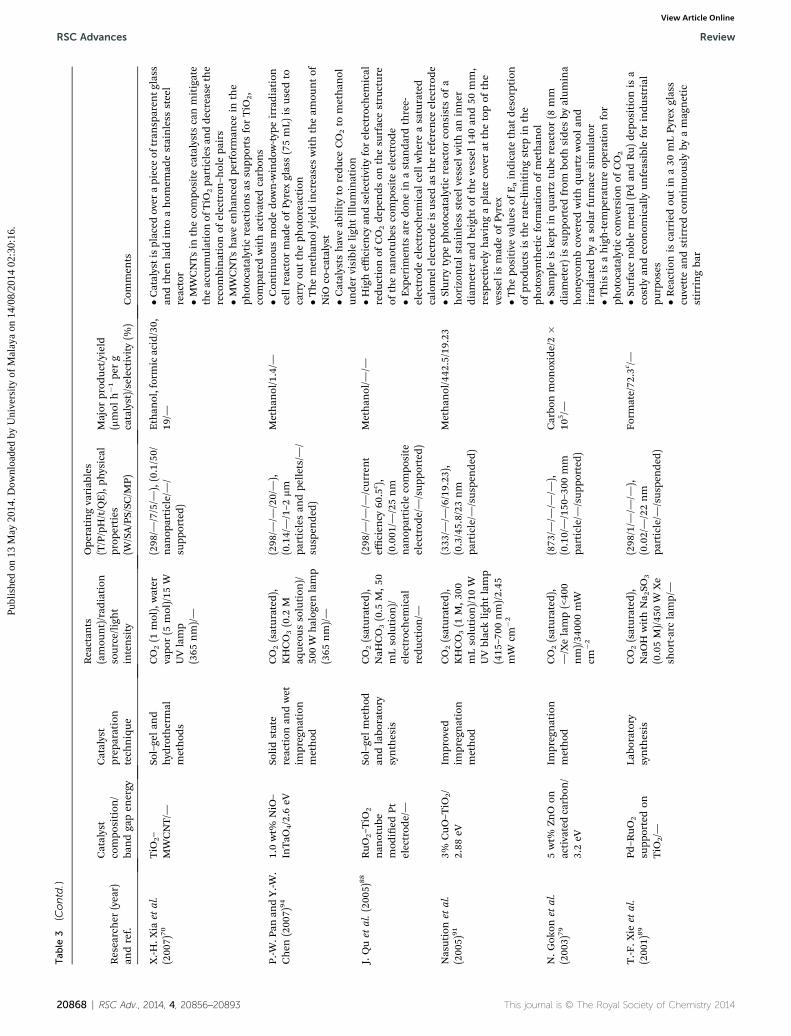
.Xia
etal.
(200
7)70
TiO
2–
MWCNT/—
Sol–gelan
dhyd
rothermal
method
s
CO2(1
mol),water
vapo
r(5
mol)/15
WUVlamp
(365
nm)/—
(298
/—/7/5/—
),(0.1/50/
nan
oparticle/—/
supp
orted)
Ethan
ol,formicacid/30,
19/—
�Catalystisplaced
over
apieceof
tran
sparen
tglass
andthen
laid
into
ahom
emad
estainless
steel
reactor
�MWCNTsin
thecompo
site
catalystscanmitigate
theaccu
mulationof
TiO
2pa
rticlesan
dde
crease
the
recombinationof
electron
–holepa
irs
�MWCNTshaveen
han
cedpe
rforman
cein
the
photocatalytic
reaction
sas
supp
orts
forTiO
2,
compa
redwithactivatedcarbon
sP.-W
.Pan
andY.-W
.Chen
(200
7)94
1.0wt%
NiO
–InTaO
4/2.6eV
Solidstate
reaction
andwet
impregnation
method
CO2(saturated
),KHCO3(0.2
Maq
ueou
ssolution
)/50
0W
halog
enlamp
(365
nm)/—
(298
/—/—
/20/—),
(0.14/—/1–2
mm
particlesan
dpe
llets/—
/su
spen
ded)
Methan
ol/1.4/—
�Con
tinuo
usmod
edo
wn-windo
w-typ
eirradiation
cellreactormad
eof
Pyrexglass(75mL)
isus
edto
carryou
ttheph
otoreaction
�Themethan
olyieldincreaseswiththeam
ountof
NiO
co-catalyst
�Catalysts
haveab
ilityto
redu
ceCO2to
methan
olun
dervisibleligh
tillumination
J.Quet
al.(200
5)88
RuO
2–T
iO2
nan
otube
mod
ied
Ptelectrod
e/—
Sol–gelmethod
andlabo
ratory
synthesis
CO2(saturated
),NaH
CO3(0.5
M,5
0mLsolution
)/electrochem
ical
redu
ction/—
(298
/—/—
/—/current
efficien
cy60
.5c ),
(0.001
/—/25nm
nan
oparticlecompo
site
electrod
e/—/sup
ported
)
Methan
ol/—
/—�H
igheffi
cien
cyan
dselectivityforelectrochem
ical
redu
ctionof
CO2de
pends
onthesu
rfacestructure
ofthenan
otub
escompo
site
electrod
e�E
xperim
ents
aredo
nein
astan
dard
three-
electrod
eelectrochem
ical
cellwhereasaturated
calomel
electrod
eisus
edas
thereference
electrod
eNasution
etal.
(200
5)91
3%CuO
–TiO
2/
2.88
eVIm
proved
impregnation
method
CO2(saturated
),KHCO3(1
M,3
00mLsolution
)/10
WUVblackligh
tlamp
(415
–700
nm)/2.45
mW
cm�2
(333
/—/—
/6/19.23
),(0.3/45.8/23
nm
particle/—
/sus
pende
d)
Methan
ol/442
.5/19.23
�Slurrytype
photocatalytic
reactorconsistsof
ahorizon
talstainless
steelvessel
withan
inner
diam
eter
andheigh
tof
thevessel
140an
d50
mm,
resp
ectively
havingaplatecoverat
thetopof
the
vessel
ismad
eof
Pyrex
�Thepo
sitive
values
ofEaindicate
that
desorption
ofprod
ucts
istherate-limitingstep
inthe
photosyn
thetic
form
ationof
methan
olN.G
okon
etal.
(200
3)79
5wt%
ZnO
onactivatedcarbon
/3.2eV
Impregnation
method
CO2(saturated
),—/Xelamp(<40
0nm)/34
000mW
cm�2
(873
/—/—
/—/—
),(0.10/—/150
–300
mm
particle/—
/sup
ported
)
Carbo
nmon
oxide/2�
105 /—
�Sam
pleis
kept
inqu
artz
tube
reactor(8
mm
diam
eter)is
supp
ortedfrom
both
side
sby
alum
ina
hon
eycombcoveredwithqu
artz
woo
lan
dirradiated
byasolarfurnacesimulator
�This
isahigh-tem
perature
operationfor
photocatalytic
conversionof
CO2
T.-F
.Xie
etal.
(200
1)89
Pd–R
uO2
supp
ortedon
TiO
2/—
Labo
ratory
synthesis
CO2(saturated
),NaO
HwithNa 2SO
3
(0.05M)/45
0W
Xe
short-arclamp/—
(298
/1/—
/—/—
),(0.02/—/22nm
particle/—
/sus
pende
d)
Form
ate/72
.3c /—
�Surface
nob
lemetal
(Pdan
dRu)
depo
sition
isa
costly
andecon
omically
unfeasible
forindu
strial
purposes
�Reactionis
carriedou
tin
a30
mLPy
rexglass
cuvettean
dstirredcontinuo
usly
byamag
netic
stirringba
r
20868 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2014
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
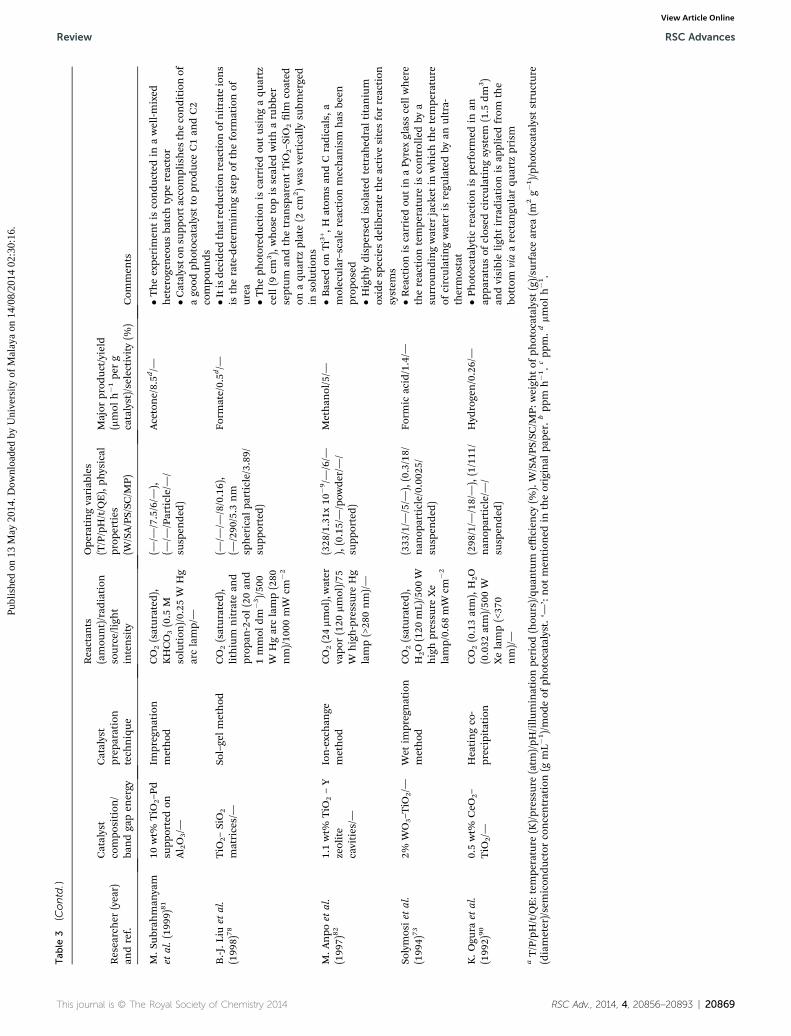
Tab
le3
(Contd.)
Researcher
(year)
andref.
Catalyst
compo
sition
/ba
ndga
pen
ergy
Catalyst
prep
aration
tech
nique
Reactan
ts(amou
nt)/rad
iation
source/light
intensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),ph
ysical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
M.S
ubrahman
yam
etal.(199
9)81
10wt%
TiO
2–P
dsu
pportedon
Al 2O3/—
Impregnation
method
CO2(saturated
),KHCO3(0.5
Msolution
)/0.25
WHg
arclamp/—
(—/—
/7.5/6/—
),(—
/—/Particle/—/
susp
ende
d)
Acetone/8.5d/—
�Theexpe
rimen
tis
condu
cted
inawell-m
ixed
heterog
eneo
usba
tchtype
reactor
�Catalysto
nsu
pporta
ccom
plishes
thecondition
ofago
odph
otocatalystto
prod
uceC1an
dC2
compo
unds
B.-J.L
iuet
al.
(199
8)78
TiO
2–SiO2
matrices/—
Sol–gelmethod
CO2(saturated
),lithium
nitrate
and
prop
an-2-ol(20an
d1mmol
dm�3)/50
0W
Hgarclamp(280
nm)/10
00mW
cm�2
(—/—
/—/8/0.16),
(—/290
/5.3
nm
spherical
particle/3.89/
supp
orted)
Form
ate/0.5d/—
�Itisde
cide
dthat
redu
ctionreaction
ofnitrate
ions
istherate-determiningstep
oftheform
ationof
urea
�Theph
otored
uction
iscarriedou
tus
ingaqu
artz
cell(9
cm3 ),w
hosetopis
sealed
witharubb
erseptum
andthetran
sparen
tTiO
2–SiO
2lm
coated
onaqu
artz
plate(2
cm2)was
vertically
subm
erged
insolution
sM.A
npo
etal.
(199
7)82
1.1wt%
TiO
2–Y
zeolite
cavities/—
Ion-exchan
gemethod
CO2(24mmol),water
vapo
r(120
mmol)/75
Whigh-pressureHg
lamp(>28
0nm)/—
(328
/1.31x
10�9/—
/6/—
),(0.15/—/pow
der/—/
supp
orted)
Methan
ol/5/—
�Based
onTi3+ ,H
atom
san
dCradicals,a
molecular–scale
reaction
mechan
ism
has
been
prop
osed
�Highly
disp
ersedisolated
tetrah
edraltitanium
oxidesp
eciesde
libe
rate
theactive
sitesforreaction
system
sSo
lymosiet
al.
(199
4)73
2%WO3–T
iO2/—
Wet
impregnation
method
CO2(saturated
),H
2O(120
mL)/500
Whighpressu
reXe
lamp/0.68
mW
cm�2
(333
/1/—
/5/—
),(0.3/18/
nan
oparticle/0.00
25/
susp
ende
d)
Form
icacid/1.4/—
�Reactionis
carriedou
tin
aPy
rexglasscellwhere
thereaction
tempe
rature
iscontrolledby
asu
rrou
ndingwater
jacket
inwhichthetempe
rature
ofcirculatingwater
isregu
latedby
anultra-
thermostat
K.O
gura
etal.
(199
2)90
0.5wt%
CeO
2–TiO
2/—
Heatingco-
precipitation
CO2(0.13atm),H
2O(0.032
atm)/50
0W
Xelamp(<37
0nm)/—
(298
/1/—
/18/—),(1/111
/nan
oparticle/—/
susp
ende
d)
Hyd
rogen/0.26/—
�Photocatalytic
reaction
ispe
rformed
inan
appa
ratusof
closed
circulatingsystem
(1.5
dm3 )
andvisibleligh
tirradiationis
appliedfrom
the
bottom
viaarectan
gularqu
artz
prism
aT/P/pH/t/Q
E:tem
perature
(K)/pressu
re(atm
)/pH
/illu
minationpe
riod
(hou
rs)/qu
antum
efficien
cy(%
).W/SA/PS/SC
/MP:
weigh
tof
photocatalyst(g)/su
rfacearea
(m2g�
1)/ph
otocatalyststructure
(diameter)/semicon
duc
torconcentration(g
mL�
1 )/m
odeof
photocatalyst.‘—
’:not
men
tion
edin
theoriginal
pape
r.bpp
mh�1 .
cpp
m.d
mmol
h�1.
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 20869
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online

RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
rates. 1 wt% CuO–TiO2 calcined at 450 �C represents greateractivity than that of calcined at 350 �C and 550 �C.92
In addition, supported materials other than TiO2 are foundat reasonable photocatalytic activity. The use of NiO on severalmetal oxides such as InTaO4 and InNbO4 are found in somepublications for CO2 reduction to fuel.93–95 1 wt% NiO–InTaO4
catalysts have low band gap energy of 2.6 eV, which is highlyresponsive to visible light and can be used in different types ofreactors like slurry, optical ber and monolithreactors.93,96,97 Incase of NiO–InNbO4 catalyst, it shows also better activity due tothe presence of core–shell-type Ni0 and NiO on the surface andthe presence of a small amount of Nb2O5 as a promoter undervisible light irradiation.95
Zn2GeO4 is an important metal oxide catalyst in photo-conversion technology, which is frequently used as a support forother oxide catalysts to enhance photocatalytic CO2 conversion.Zn2GeO4 catalyst contains some superior properties like lowcrystal defects, extraordinary specic surface area and favorablemicrostructure on the surface of the catalyst, but the band gapenergy is high enough (4.65 eV) to show the visible lightresponse.98 Sheaf-like, hyperbranched Zn2GeO4 nano-architecture was developed by Q. Liu et al., which was furthermodied by NH3 ow to a yellow Zn1.7GeN1.8O solid solution forCO2 conversion into carbonaceous fuel in the existence of waterat ambient conditions under visible light irradiation.99 Themodied catalyst shows low band gap energy (2.6 eV), which isused as a support for 1 wt% Pt and 1 wt% RuO2. Moreover, theloading of Pt and RuO2 on Zn1.7GeN1.8O gives a high conversionrate, which is able to arrest the electron–hole pairs by prevent-ing the recombination of electrons and holes.
2.2 Modied/unmodied non-oxide catalysts
Non-oxide semiconductor photocatalysts are selected for CO2
recycling because of their low band gap energy to facilitate thephotocatalytic response to the visible light region, and highconversion efficiency is achieved for their unique photo-behavior. Elaborately major ndings and characteristics of non-oxide semiconductors upon photocatalytic reactions aredescribed in Table 4.
2.2.1 Metal sulphide semiconductors. It is investigatedthat semiconductor catalysts are one of the supreme sources fordirect solar energy conversion. Many wide-ranging reviewsalready have been published on the advancement of oxide ornon-oxide semiconductor photoactive materials.100–103 It wasfound that apart from the metal oxide semiconductor catalysts,metal sulphide semiconductor materials have effective photo-activity because of their outstanding ability to absorb the solarspectrum with high energy yields.104 Metal sulphides havecomparatively high conduction band states more appropriatefor enhanced solar responses than for metal oxide semi-conductors, which are facilitated by the higher valence bandstates consisting of S 3p orbitals.104 It was reported that toabsorb the entire UV and visible light region of solar irradiation,a ZnS–AgInS2–CuInS2 solid solution was capable due to anabsorption edge of up to 800 nm.105 Fujiwara et al. investigatedZnS–DMF(OAc) for the CO2 photoreduction process.106 They
20870 | RSC Adv., 2014, 4, 20856–20893
found that the close interaction of acetate ions to Zn atomsinhibited the creation of sulfur vacancies as catalytic sites forCO production. Moreover, surface dimethylformamide (DMF)solvated Zn atoms give more efficiency to HCOO� formation.On the other hand, the addition of excess Zn2+ increases the sizeof ZnS–DMF(OAc) through the reaction between excess Zn2+ andthe surface of ZnS–DMF(OAc) nanocrystallites. ZnS depositedon montmorillonite (MMT) was also fabricated by Kocı et al.with particle sizes of 3 nm to 5 nm, which provided signicantlyhigher productivity, compared with the commercial TiO2
Degussa P25.107,108 They used cetyltrimethylammonium (CTA)with ZnS nanoparticles to inhibit the formation of biggeragglomerates. Different percentages of ZnS loading on porousSiO2 matrices were also applied to investigate the improvedphotocatalytic yields.109
In 1979, Inoue et al. introduced non-oxide semiconductorcatalysts (CdS, GaP and SiC) for the photocatalytic conversion ofCO2 to fuel.42 Among them, CdS shows comparatively betteryields in CO2 conversion. It is found from Table 4 that thenarrow band gap (2.63 eV) CdS supported on montmorilloniteexhibits a lower conversion yield than that of 3.89 eV ZnSdeposited on MMT, but the CdS–MMT system has high stabilityin presence of CTA.108,110,111 Not only that, here CTA acts as abinder among CdS and MMT particles. Photocatalytic conver-sion of CO2 on CdS semiconductors with modied surfaces byvarious types of thiol compounds was performed in presence of2-propanol solvent.112 In this study, the major products foundwere formate and carbon monoxide. The ratio of formate tocarbon monoxide is highly dependent on the solvent andincreases with increasing the dielectric constant of the solvent.Moreover, the surface modier is used in this system to x thesurface Cd2+ sites, thus decreases the adsorption of CO2c
� onCd2+ sites, which means that the formate yield will be increasewith increasing the surface exposure of the modier. Anotherstudy shows that extra Cd2+ addition to the CdS–DMF systemforms sulfur vacancies on the surface of nanocrystallitesbecause of the adsorption of excess Cd2+ on the surface.113 Overthe surface of sulfur vacancies, CO2 takes electrons sequentiallyaer developing a Cd2+OCOCO2 complex to react with anotherCO2, leading to the formation of CO as reported in the electricalCO2 reduction to CO.114
Yuan et al. investigated CdS–TNT loaded with Pt to carry outthe photochemical reaction under visible light irradiation, butthey found that the worsening effect of photochemical reactionoccurs due to the adsorption of intermediate products (CO, O2
etc.) on Pt and the oxidation of Pt.115 Another approach was ledby Li et al. for visible light-responsive photocatalytic CO2
reduction on modied CdS with 15% Bi2S3.116 The investigationshows that the Bi2S3 catalyst itself has higher photochemicalactivity and visible light response than that of CdS. The additionof Bi2S3 increased the photochemical performance of CdS, anda maximum yield was obtained 120 mmol h�1 g�1 catalyst. Cd–ZnS was investigated to compare the photocatalytic behaviorwith solid solutions of ZnS–CdS microcrystals.117 Cd–ZnS showsthe highest activity than that of ZnS–CdS because Cd is moreactive in improving the quantum efficiency of the photochem-ical reduction of formate due to its ability to activate CO2 more
This journal is © The Royal Society of Chemistry 2014
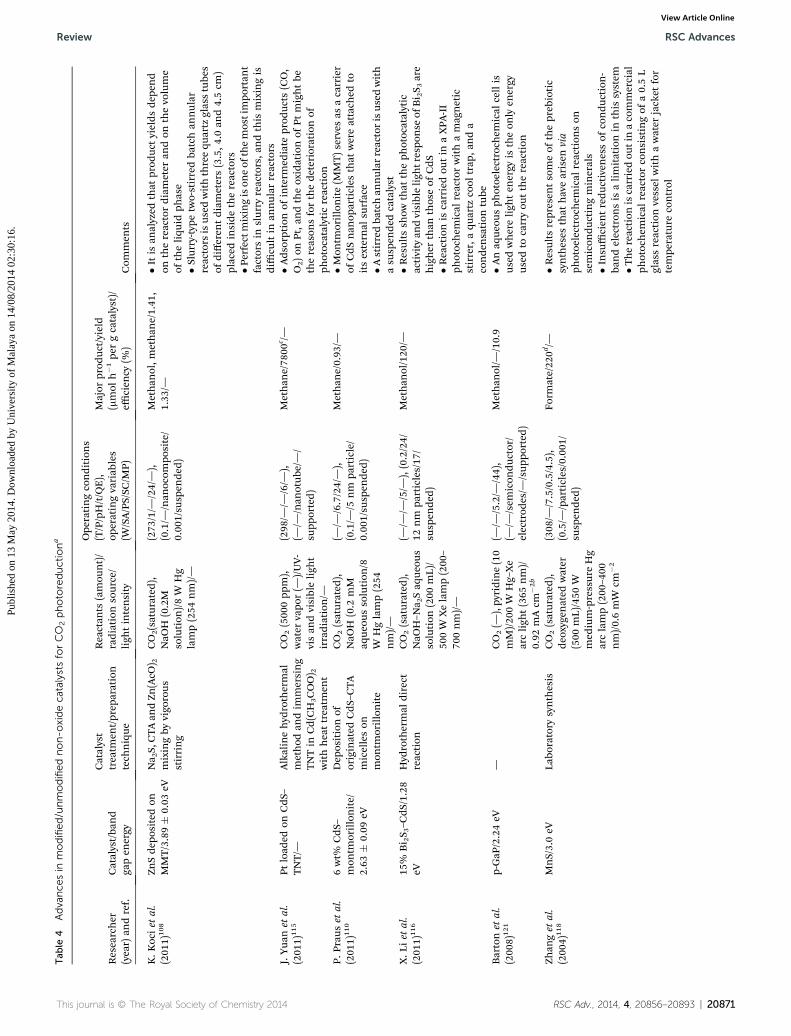
Tab
le4
Advance
sin
modified/unmodifiednon-o
xideca
talystsforCO
2photoreductiona
Researcher
(year)an
dref.
Catalyst/ba
nd
gapen
ergy
Catalyst
treatm
ent/prep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingcondition
s(T/P/pH/t/Q
E),
operatingvariab
les
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rgcatalyst)/
efficien
cy(%
)Com
men
ts
K.K
ociet
al.
(201
1)108
ZnSde
positedon
MMT/3.89�
0.03
eVNa 2S,CTAan
dZn
(AcO
) 2mixingby
vigo
rous
stirring
CO2(saturated
),NaO
H(0.2M
solution
)/8W
Hg
lamp(254
nm)/—
(273
/1/—
/24/—),
(0.1/—
/nan
ocom
posite/
0.00
1/su
spen
ded)
Methan
ol,m
ethan
e/1.41
,1.33
/—�I
tis
analyzed
that
prod
uctyields
depe
nd
onthereactordiam
eter
andon
thevolume
oftheliqu
idph
ase
�Slurry-type
two-stirredba
tchan
nular
reactors
isus
edwiththreequ
artz
glasstube
sof
differen
tdiam
eters(3.5,4
.0an
d4.5cm
)placed
inside
thereactors
�Perfectmixingison
eof
themostimpo
rtan
tfactorsin
slurry
reactors,a
ndthis
mixingis
difficu
ltin
annular
reactors
J.Yua
net
al.
(201
1)115
Ptload
edon
CdS–
TNT/—
Alkalinehyd
rothermal
method
andim
mersing
TNTin
Cd(CH
3COO) 2
withheattreatm
ent
CO2(500
0pp
m),
water
vapo
r(—
)/UV-
visan
dvisibleligh
tirradiation/—
(298
/—/—
/6/—
),(—
/—/nan
otub
e/—/
supp
orted)
Methan
e/78
00c /—
�Ads
orptionof
interm
ediate
prod
ucts
(CO,
O2)on
Pt,a
ndtheoxidationof
Ptmightbe
thereason
sforthede
teriorationof
photocatalytic
reaction
P.Prau
set
al.
(201
1)110
6wt%
CdS
–mon
tmorillonite/
2.63
�0.09
eV
Dep
ositionof
originated
CdS–CTA
micelleson
mon
tmorillonite
CO2(saturated
),NaO
H(0.2
mM
aque
oussolution
/8W
Hglamp(254
nm)/—
(—/—
/6.7/24/—),
(0.1/—
/5nm
particle/
0.00
1/su
spen
ded)
Methan
e/0.93
/—�M
ontm
orillonite(M
MT)serves
asacarrier
ofCdS
nan
oparticles
that
wereattach
edto
itsexternal
surface
�Astirredba
tchan
nular
reactorisus
edwith
asu
spen
dedcatalyst
X.L
iet
al.
(201
1)116
15%
Bi 2S 3–C
dS/1.28
eVHyd
rothermal
direct
reaction
CO2(saturated
),NaO
H–N
a 2Saq
ueou
ssolution
(200
mL)/
500W
Xelamp(200
–70
0nm)/—
(—/—
/—/5/—
),(0.2/24/
12nm
particles/17
/su
spen
ded)
Methan
ol/120
/—�R
esults
show
that
theph
otocatalytic
activityan
dvisibleligh
trespo
nse
ofBi 2S 3
are
higher
than
thoseof
CdS
�Reactionis
carriedou
tin
aXPA
-II
photochem
ical
reactorwithamag
netic
stirrer,aqu
artz
cool
trap
,anda
conde
nsation
tube
Bartonet
al.
(200
8)121
p-GaP
/2.24eV
—CO2(—
),py
ridine(10
mM)/20
0W
Hg–Xe
arcligh
t(365
nm)/
0.92
mAcm
�2b
(—/—
/5.2/—
/44),
(—/—
/sem
icon
ductor/
electrod
es/—
/sup
ported
)
Methan
ol/—
/10.9
�Anaq
ueou
sph
otoe
lectroch
emical
cellis
used
whereligh
ten
ergy
istheon
lyen
ergy
used
tocarryou
tthereaction
Zhan
get
al.
(200
4)118
MnS/3.0eV
Labo
ratory
synthesis
CO2(saturated
),de
oxygen
ated
water
(500
mL)/450
Wmed
ium-pressureHg
arclamp(200
–400
nm)/0.6mW
cm�2
(308
/—/7.5/0.5/4.5),
(0.5/—
/particles/0.001
/su
spen
ded)
Form
ate/22
0d/—
�Results
represen
tsomeof
thepreb
iotic
syntheses
that
havearisen
via
photoe
lectroch
emical
reaction
son
semicon
ductingminerals
�Insu
fficien
tredu
ctiven
essof
condu
ction-
bandelectron
sis
alimitationin
this
system
�Thereaction
iscarriedou
tinacommercial
photochem
ical
reactorconsistingof
a0.5L
glassreaction
vessel
withawater
jacket
for
tempe
rature
control
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 208
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
71
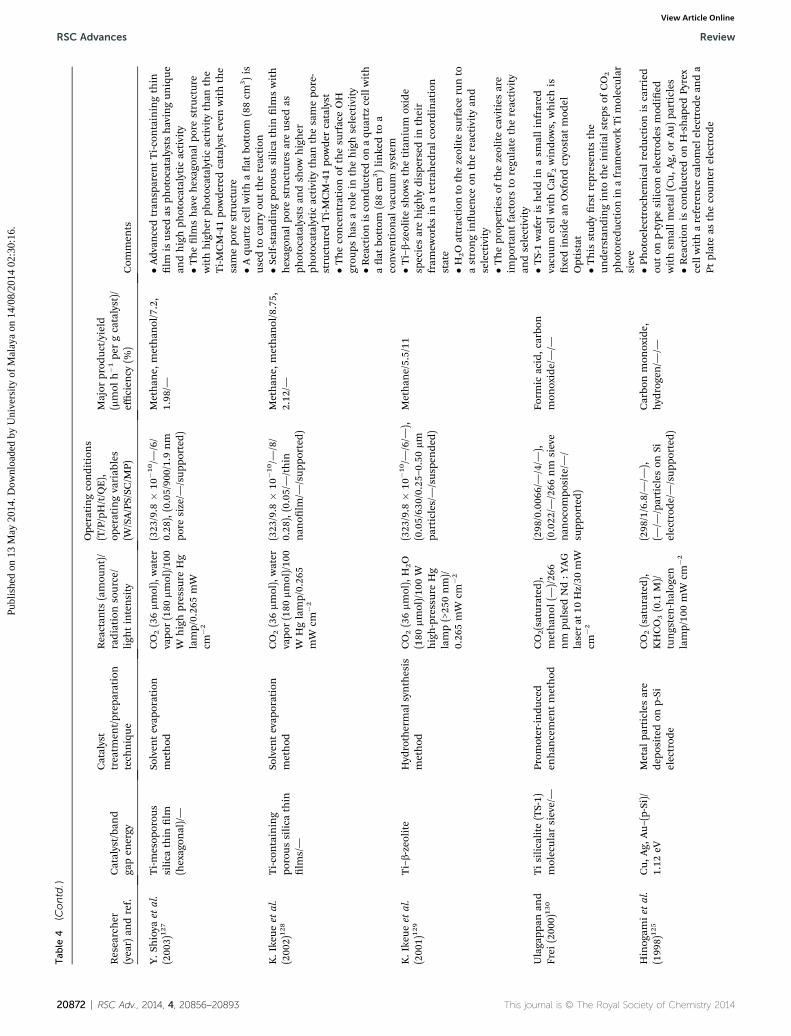
Tab
le4
(Contd.)
Researcher
(year)an
dref.
Catalyst/ba
nd
gapen
ergy
Catalyst
treatm
ent/prep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingcondition
s(T/P/pH/t/Q
E),
operatingvariab
les
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rgcatalyst)/
efficien
cy(%
)Com
men
ts
Y.S
hioya
etal.
(200
3)127
Ti-m
esop
orou
ssilica
thin
lm
(hexag
onal)/—
Solven
tevap
oration
method
CO2(36mmol),water
vapo
r(180
mmol)/10
0W
highpressu
reHg
lamp/0.26
5mW
cm�2
(323
/9.8
�10
�10/—
/6/
0.28
),(0.05/90
0/1.9nm
pore
size/—
/sup
ported
)
Methan
e,methan
ol/7.2,
1.98
/—�A
dvan
cedtran
sparen
tTi-c
ontainingthin
lm
isus
edas
photocatalysts
havingun
ique
andhighph
otocatalytic
activity
�Thelm
shavehexag
onal
pore
structure
withhigher
photocatalytic
activity
than
the
Ti-M
CM-41po
wde
redcatalyst
even
withthe
samepo
restructure
�Aqu
artz
cellwithaat
bottom
(88cm
3)is
used
tocarryou
tthereaction
K.Ike
ueet
al.
(200
2)128
Ti-c
ontaining
porous
silica
thin
lm
s/—
Solven
tevap
oration
method
CO2(36mmol),water
vapo
r(180
mmol)/10
0W
Hglamp/0.26
5mW
cm�2
(323
/9.8
�10
�10/—
/8/
0.28
),(0.05/—/thin
nan
olm
/—/sup
ported
)
Methan
e,methan
ol/8.75,
2.12
/—�S
elf-stan
dingpo
rous
silica
thin
lm
swith
hexag
onal
pore
structures
areus
edas
photocatalysts
andsh
owhigher
photocatalytic
activity
than
thesamepo
re-
structured
Ti-M
CM-41po
wde
rcatalyst
�Theconcentrationof
thesu
rfaceOH
grou
pshas
arole
inthehighselectivity
�Reactioniscondu
cted
onaqu
artz
cellwith
aat
bottom
(88cm
3)linke
dto
aconvention
alvacu
umsystem
K.Ike
ueet
al.
(200
1)129
Ti–b-zeo
lite
Hyd
rothermal
synthesis
method
CO2(36mmol),H
2O
(180
mmol)/10
0W
high-pressureHg
lamp(>25
0nm)/
0.26
5mW
cm�2
(323
/9.8
�10
�10/—
/6/—
),(0.05/63
0/0.25
–0.50mm
particles/—/sus
pende
d)
Methan
e/5.5/11
�Ti–b-zeo
lite
show
sthetitanium
oxide
speciesarehighly
disp
ersedin
their
fram
eworks
inatetrah
edralcoordination
state
�H2Oattraction
tothezeolitesu
rfacerunto
astronginue
nce
onthereactivity
and
selectivity
�Theprop
erties
ofthezeolitecavities
are
impo
rtan
tfactorsto
regu
late
thereactivity
andselectivity
Ulaga
ppan
and
Frei
(200
0)130
Tisilicalite
(TS-1)
molecular
sieve/—
Prom
oter-in
duced
enhan
cemen
tmethod
CO2(saturated
),methan
ol(—
)/26
6nm
pulsed
Nd:Y
AG
laserat
10Hz/30
mW
cm�2
(298
/0.006
6/—/4/—
),(0.022
/—/266
nm
sieve
nan
ocom
posite/—
/su
pported)
Form
icacid,c
arbo
nmon
oxide/—/—
�TS-1wafer
isheldin
asm
allinfrared
vacu
umcellwithCaF
2windo
ws,
whichis
xedinside
anOxfordcryostat
mod
elOptistat
�This
stud
yrstrepresen
tsthe
unde
rstandinginto
theinitialstep
sof
CO2
photored
uction
inafram
eworkTim
olecular
sieve
Hinog
amiet
al.
(199
8)125
Cu,
Ag,
Au–
(p-Si)/
1.12
eVMetal
particlesare
depo
sitedon
p-Si
electrod
e
CO2(saturated
),KHCO3(0.1
M)/
tungsten-halog
enlamp/10
0mW
cm�2
(298
/1/6.8/—
/—),
(—/—
/particles
onSi
electrod
e/—/sup
ported
)
Carbo
nmon
oxide,
hyd
rogen/—
/—�P
hotoe
lectroch
emical
redu
ctionis
carried
outon
p-type
siliconelectrod
esmod
ied
withsm
allmetal
(Cu,
Ag,
orAu)
particles
�Reactionis
condu
cted
onH-shap
edPy
rex
cellwithareference
calomel
electrod
ean
da
Ptplateas
thecoun
terelectrod
e
20872 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2014
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
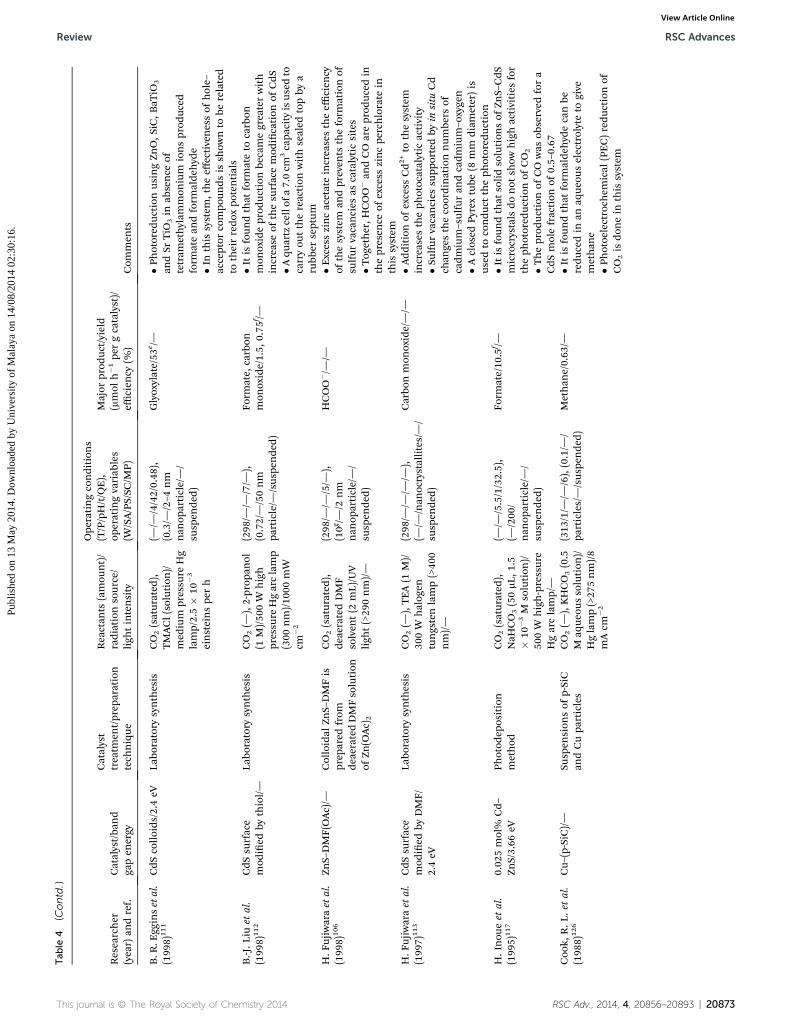
Tab
le4
(Contd.)
Researcher
(year)an
dref.
Catalyst/ba
nd
gapen
ergy
Catalyst
treatm
ent/prep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingcondition
s(T/P/pH/t/Q
E),
operatingvariab
les
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rgcatalyst)/
efficien
cy(%
)Com
men
ts
B.R
.Egg
insetal.
(199
8)111
CdS
collo
ids/2.4eV
Labo
ratory
synthesis
CO2(saturated
),TMACl(solution)/
med
ium
pressu
reHg
lamp/2.5�
10�3
einsteinspe
rh
(—/—
/4/42/0.48
),(0.3/—
/2–4
nm
nan
oparticle/—/
susp
ende
d)
Glyoxylate/53
e /—
�Photored
uction
usingZn
O,S
iC,B
aTiO
3
andSr
TiO
3in
absence
oftetram
ethylam
mon
ium
ionsprod
uced
form
atean
dform
alde
hyd
e�I
nthis
system
,theeff
ectivenessof
hole–
acceptor
compo
unds
issh
ownto
berelated
totheirredo
xpo
tentials
B.-J.L
iuet
al.
(199
8)112
CdS
surface
mod
ied
bythiol/—
Labo
ratory
synthesis
CO2(—
),2-prop
anol
(1M)/50
0W
high
pressu
reHgarclamp
(300
nm)/10
00mW
cm�2
(298
/—/—
/7/—
),(0.72/—/50nm
particle/—
/sus
pende
d)
Form
ate,
carbon
mon
oxide/1.5,
0.75
f /—�I
tis
foun
dthat
form
ateto
carbon
mon
oxideprod
uction
becamegreaterwith
increase
ofthesu
rfacemod
ication
ofCdS
�Aqu
artzcellof
a7.0cm
3capa
cityisus
edto
carryou
tthereaction
withsealed
topby
arubb
erseptum
H.F
ujiwaraet
al.
(199
8)106
ZnS–DMF(OAc)/—
Colloidal
ZnS–DMFis
prep
ared
from
deaeratedDMFsolution
ofZn
(OAc)
2
CO2(saturated
),de
aeratedDMF
solven
t(2
mL)/UV
ligh
t(>29
0nm)/—
(298
/—/—
/5/—
),(10g/—
/2nm
nan
oparticle/—/
susp
ende
d)
HCOO�/—
/—�E
xcesszincacetateincreasestheeffi
cien
cyof
thesystem
andpreven
tstheform
ationof
sulfur
vacancies
ascatalyticsites
�Tog
ether,H
COO�an
dCOareprod
uced
inthepresen
ceof
excess
zincpe
rchlorate
inthis
system
H.F
ujiwaraet
al.
(199
7)113
CdS
surface
mod
ied
byDMF/
2.4eV
Labo
ratory
synthesis
CO2(—
),TEA(1
M)/
300W
halog
entungstenlamp(>40
0nm)/—
(298
/—/—
/—/—
),(—
/—/nan
ocrystallites/—/
susp
ende
d)
Carbo
nmon
oxide/—/—
�Add
itionof
excess
Cd2
+to
thesystem
increasestheph
otocatalytic
activity
�Sulfurvacancies
supp
ortedby
insitu
Cd
chan
gesthecoordinationnum
bers
ofcadm
ium–sulfuran
dcadm
ium–o
xygen
�Aclosed
Pyrextube
(8mm
diam
eter)is
used
tocondu
cttheph
otored
uction
H.Inou
eet
al.
(199
5)117
0.02
5mol%
Cd–
ZnS/3.66
eVPh
otod
eposition
method
CO2(saturated
),NaH
CO3(50mL,
1.5
�10
�3M
solution
)/50
0W
high-pressure
Hgarclamp/—
(—/—
/5.5/1/32.5),
(—/200
/nan
oparticle/—/
susp
ende
d)
Form
ate/10
.5f /—
�Itis
foun
dthat
solidsolution
sof
ZnS–CdS
microcrystals
donot
show
highactivities
for
theph
otored
uction
ofCO2
�Theprod
uction
ofCO
was
observed
fora
CdS
molefraction
of0.5–0.67
Coo
k,R.L
.etal.
(198
8)126
Cu–
(p-SiC)/—
Susp
ension
sof
p-SiC
andCupa
rticles
CO2(—
),KHCO3(0.5
Maq
ueou
ssolution
)/Hglamp(>27
5nm)/8
mAcm
�2
(313
/1/—
/—/6),(0.1/—
/pa
rticles/—/sus
pende
d)Methan
e/0.63
/—�I
tis
foun
dthat
form
alde
hyd
ecanbe
redu
cedin
anaq
ueou
selectrolyteto
give
methan
e�P
hotoe
lectroch
emical
(PEC)redu
ctionof
CO2is
donein
this
system
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 20873
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online

Tab
le4
(Contd.)
Researcher
(year)an
dref.
Catalyst/ba
nd
gapen
ergy
Catalyst
treatm
ent/prep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingcondition
s(T/P/pH/t/Q
E),
operatingvariab
les
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rgcatalyst)/
efficien
cy(%
)Com
men
ts
Aurian-Blajeni
etal.(198
3)124
p-GaA
s/—
—CO2(saturated
),KCl
(0.5
Msolution
)/15
0W
Xelamp/—
(298
/8.5/—
/—/—
),(—
/—/sem
icon
ductor
electrod
e/—/sup
ported
)
Form
icacid/170
f /—�A
photoe
lectroch
emical
autoclave,
tted
withaqu
artz
windo
wisus
edto
carryou
tthe
reaction
�Ads
orptionof
theCO2on
the
semicon
ductor
surfaceclearlyplaysan
impo
rtan
trole
intheprocess
�Theelectrod
esarenot
stab
lean
dhave
worsened
noticeablywithin
severalhou
rsInou
eet
al.
(197
9)42
CdS
,GaP
andSiC/
2.4,
2.3an
d3.0eV
Purity
99.5
to99
.99%
CO2(saturated
),H
2O
(100
mL)/500
WXeor
highpressu
reHg
lamp/—
(298
/1/5/7/—
),(1/—
/20
0–40
0mesh
powde
r/—/sus
pende
d)
Form
alde
hyd
e,methan
ol/
(29,
17),(14,
16)an
d(14,
76)/—
�Mag
netic
stirredglasscellwithqu
artz
windo
wis
used
forph
otored
uction
ofCO2
�Photoe
xcited
electron
sin
themore
negativecondu
ctionba
ndpo
seshigher
abilityto
redu
ceCO2
M.H
alman
n(197
8)119
p-GaP
singlecrystal/
2.25
eV—
CO2(saturated
),K2HPO
4solution
(0.05M)/high
pressu
reHglamp/
2.63
mW
cm�2
(298
/—/6.8/90/—),
(—/2.4
�10
�5/
semicon
ductor
electrod
e/—/sup
ported
)
Form
icacid/50g/0.61
�Closedbu
bble-walledbo
rosilicate
glass
beak
er(30mL)
withthermostatis
used
asa
photoe
lectroch
emical
reactor
aT/P/pH/t/Q
E:tem
perature
(K)/pressu
re(atm
)/pH
/illu
minationpe
riod
(hou
rs)/qu
antum
efficien
cy(%
).W/SA/PS/SC
/MP:
weigh
tof
photocatalyst(g)/su
rfacearea
(m2g�
1)/ph
otocatalyststructure
(diameter)/semicon
duc
torconcentration(g
mL�
1 )/m
odeof
photocatalyst.‘—
’:not
men
tion
edin
theoriginal
pape
r.bCurrentde
nsity.c
mmol
h�1.d
mM.e
mmol
dm�3.f
mmol.g
mM.
20874 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2014
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online

Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
effectively in the photoreaction. Zhang et al. introduced anothermetal sulphide semiconductor catalyst (MnS), which can beused under the solar spectrum more competently due to lowband gap energy (3.0 eV).118 Similar to CdS, MnS semiconductormaterials also have the distinct behavior of containing high-reducing conduction band electrons which are satisfactorilyactive for encouraging the reduction of CO2.118 That is why, MnSsemiconductors deliver higher quantum efficiency than othercommon catalysts such as TiO2.
2.2.2 Metal phosphide semiconductor. The use of catalystsfor photochemical purposes without incorporating co-catalystssuch as p-type GaP and p-type InP have also been studied due tonarrow band gaps to facilitate the visible light response.119,120
Basically, in the photoelectrochemical CO2 conversion process,p-type metal phosphides (GaP) were used mainly in methanolproduction but needed remarkably high over-potentials.121
However, high faradaic efficiencies for the photo-electrochemical reduction of CO2 to CO have also been inves-tigated, while electrolytes are used in nonaqueous forms.Nevertheless, high over-potential is necessary to obtain highfaradaic efficiency.122 Barton et al. found that p-type GaP wasable to obtain faradaic efficiency of 100% while converting CO2
to methanol at low potentials, which was more than 300 mVbelow the standard potential of �0.5 V versus the saturatedcalomel electrode (SCE).121 Habisreutinger et al. reported thatp-type GaP semiconductor exhibits low band gap energy(2.24 eV) and possesses high reducing conduction band elec-trons to ease the reduction of CO2.35 On the other hand, for CO2
photocatalytic conversions, it was detected that p-type InPprovided high selectivity for formic acid in a photo-electrochemical cell.123 Moreover, according to Kaneco et al.,p-InP is an active substantial for the photoelectrochemicalreduction of CO2, which revealed higher positive potential of0.2–0.4 V than that of p-Si and p-GaAs photocathodes in theelectrolysis of methanol electrolyte.120
2.2.3 Other non-oxide semiconductors. Aurian-Blajeniet al. were carried out in the aqueous CO2 photo-electrochemical reduction by using p-GaP and p-GaAs asphotocathodes.124 In case of p-GaP, the Faradaic yield decreaseswhen the reaction is carried out at a more negative potential,whereas the opposite behavior is shown in the case of p-GaAselectrode. The different behavior of the two electrodes isprobably connected to the greater stability of GaP towardsanodic decomposition, while in the case of the unstable GaAs,the higher number of negative potentials provide a degree ofcathodic protection. Modied p-type silicon (p-Si) electrodeswith Cu, Ag, and Au foreign elements have been investigated byHinogami et al., and were determined to have extraordinaryactivation energy for CO2 photoreduction.125 Metal elementdeposition on p-Si electrodes not only induce the photo-catalytic activity but also produce high photovoltages. More-over, CO2 photochemical reduction is activated by an upwardshi of surface band energies of p-Si in terms of achievingenergy levels, which is identical between the semiconductorand solution reactants.125 In another study, p-type SiC modiedby Cu particles was also investigated to determine the photo-electrochemical ability in CO2 reduction.126
This journal is © The Royal Society of Chemistry 2014
Ti containing mesoporous silica thin-lm materials havebeen introduced in the photochemical reduction of CO2 due totheir unique and high photocatalytic performances.127 Spectro-scopic analysis proved that Ti ions were highly dispersed insilica networks, and the silica thin lms acted as effectivephotocatalysts during the photoreactions. Besides, the hexag-onal pore construction inside the lms revealed superiorphotosensitivity compared with the same pore-structured Ti-MCM-41 powdered catalyst.127,128 Moreover, FTIR investigationfound that Ti containing silica thin lm-possessed surface OHgroups in various concentrations had a signicant inuence onCH3OH formation.128 Ikeue et al. also studied Ti containingb-zeolite treated with OH� and F� ions as structure-directingagents (SDA) showed hydrophilic and hydrophobic properties,respectively.129 Hydrophilic properties show high photocatalyticefficiency due to the easy interaction of H2O molecules with thetetrahedrally coordinated titanium oxide species on Ti–b-zeolite(OH) than on Ti–b-zeolite (F).
2.3 Doped photocatalysts
To introduce impurities into an extremely pure semiconductor,doping of foreign elements is deliberately inserted for thepurpose of controlling its electrical properties. Due to largeband gaps, unmodied semiconductor photocatalysts cannoteffectively absorb solar radiation with photon energy 1.7–3.1 eV.131 To decrease its band gap energy, surface modicationof the photocatalysts has been attempted by applying stressalong the so direction of a layered semiconductor,131 intro-ducing oxygen vacancies in semiconductors,132 or incorporatingforeign elements.133–139 The schematic photocatalytic view of adoped semiconductor is illuminated in Scheme 1(c). Not onlythat, the recent advancement in different types of doped metaland non-metal elements incorporated with semiconductorsfound in various research for the photocatalytic conversion ofCO2 has been reported in Tables 5 and 6, respectively.
2.3.1 Noble metal doped semiconductors. Dopingmetal onsemiconductor acts as an electron trap to provide electrons toCO2. The photoexcited electrons are shied from the conduc-tion band of the semiconductor to doped metal to leave theholes on the surface of semiconductor.37 Noble metals,including Ru, Rh, Pd, Ag, Pt, Cu, Ni and Au, have beencommonly used with semiconductors as dopants due to Fermi-level or electron-accepting areas at an energy just beneath theconduction band of semiconductors for the photochemicalreduction of CO2. Among those metals, Pt was widely used inmany investigations. For example, the TiO2 semiconductor wasdoped with 0.5 wt% Pt by wet impregnation to avoid very lowphotocatalytic activity.140,141 More homogeneous dispersion of0.5 wt% Pt on the TiO2 surface drives the reaction rate swily inthe both liquid and gas phases.142 It is found that Pt loaded onTiO2 transforms active sites from Ti4+ to Ti3+ by shiing elec-trons from TiO2 to Pt, and this phenomena facilitates visiblelight response.143 Another investigation shows that Pt doping onanchored titanium oxide catalysts within the Y-zeolite cavitiesencourages charge separation and increases the formation ofCH4 in lieu of CH3OH by promoting the reaction between the
RSC Adv., 2014, 4, 20856–20893 | 20875
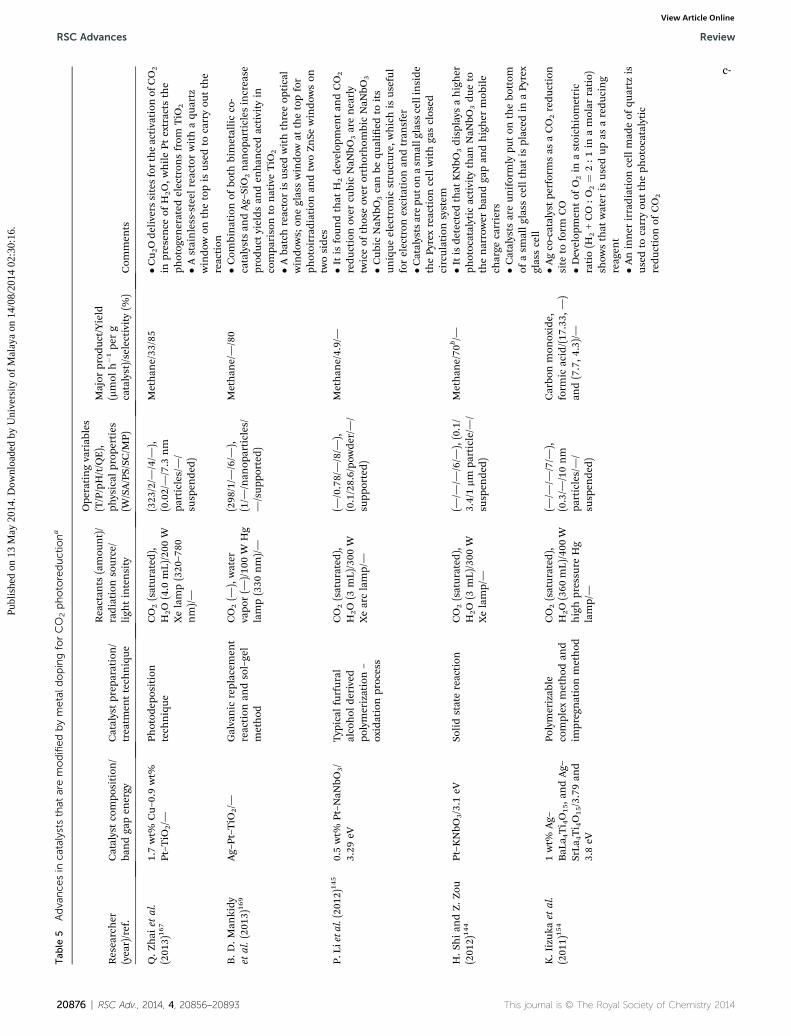
Tab
le5
Advance
sin
catalyststhat
aremodifiedbymetald
opingforCO
2photoreductiona
Researcher
(year)/ref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration/
treatm
enttech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/Yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
Q.Z
hai
etal.
(201
3)167
1.7wt%
Cu–
0.9wt%
Pt–T
iO2/—
Photod
eposition
tech
nique
CO2(saturated
),H
2O
(4.0
mL)/200
WXelamp(320
–780
nm)/—
(323
/2/—
/4/—
),(0.02/—/7.3
nm
particles/—/
susp
ende
d)
Methan
e/33
/85
�Cu 2Ode
livers
sitesfortheactivation
ofCO2
inpresen
ceof
H2O,w
hilePt
extracts
the
photog
enerated
electron
sfrom
TiO
2
�Astainless-steel
reactorwithaqu
artz
windo
won
thetopis
used
tocarryou
tthe
reaction
B.D
.Man
kidy
etal.(201
3)169
Ag–Pt–T
iO2/—
Galvanic
replacem
ent
reaction
andsol–gel
method
CO2(—
),water
vapo
r(—
)/10
0W
Hg
lamp(330
nm)/—
(298
/1/—
/6/—
),(1/—
/nan
oparticles/
—/sup
ported
)
Methan
e/—/80
�Com
binationof
both
bimetallicco-
catalystsan
dAg–SiO2nan
oparticles
increase
prod
uctyields
anden
han
cedactivity
incompa
risonto
nativeTiO
2
�Aba
tchreactoris
used
withthreeop
tical
windo
ws;
oneglasswindo
wat
thetopfor
photoirrad
iation
andtw
oZn
Sewindo
wson
twoside
sP.
Lietal.(20
12)145
0.5wt%
Pt–N
aNbO
3/
3.29
eVTyp
ical
furfural
alcohol
derived
polymerization–
oxidationprocess
CO2(saturated
),H
2O
(3mL)/300
WXearclamp/—
(—/0.78/—/8/—
),(0.1/28.6/po
wde
r/—/
supp
orted)
Methan
e/4.9/—
�Itis
foun
dthat
H2de
velopm
entan
dCO2
redu
ctionover
cubicNaN
bO3arenearly
twiceof
thoseover
orthorhom
bicNaN
bO3
�Cub
icNaN
bO3canbe
qualied
toits
unique
electron
icstructure,
whichis
useful
forelectron
excitation
andtran
sfer
�Catalystsarepu
tonasm
allglass
cellinside
thePy
rexreaction
cellwithga
sclosed
circulationsystem
H.S
hian
dZ.
Zou
(201
2)144
Pt–K
NbO
3/3.1eV
Solidstatereaction
CO2(saturated
),H
2O
(3mL)/300
WXelamp/—
(—/—
/—/6/—
),(0.1/
3.4/1mm
particle/—
/su
spen
ded)
Methan
e/70
b/—
�Itis
detected
that
KNbO
3disp
lays
ahigher
photocatalytic
activity
than
NaN
bO3du
eto
thenarrower
bandga
pan
dhigher
mob
ile
charge
carriers
�Catalysts
areun
iformly
puton
thebo
ttom
ofasm
allg
lass
cellthat
isplaced
inaPy
rex
glasscell
K.Iizuk
aet
al.
(201
1)154
1wt%
Ag–
BaL
a 4Ti 4O15,an
dAg–
SrLa
4Ti 4O15/3.79an
d3.8eV
Polymerizab
lecomplex
method
and
impregnationmethod
CO2(saturated
),H
2O(360
mL)/400
Whighpressu
reHg
lamp/—
(—/—
/—/7/—
),(0.3/—
/10nm
particles/—/
susp
ende
d)
Carbo
nmon
oxide,
form
icacid/(17
.33,
—)
and(7.7,4
.3)/—
�Agco-catalystpe
rformsas
aCO2redu
ction
site
toform
CO
�Develop
men
tof
O2in
astoich
iometric
ratio(H
2+CO:O
2¼
2:1
inamolar
ratio)
show
sthat
water
isus
edup
asaredu
cing
reag
ent
�Aninner
irradiationcellmad
eof
quartz
isus
edto
carryou
ttheph
otocatalytic
redu
ctionof
CO2
20876 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
c-
014
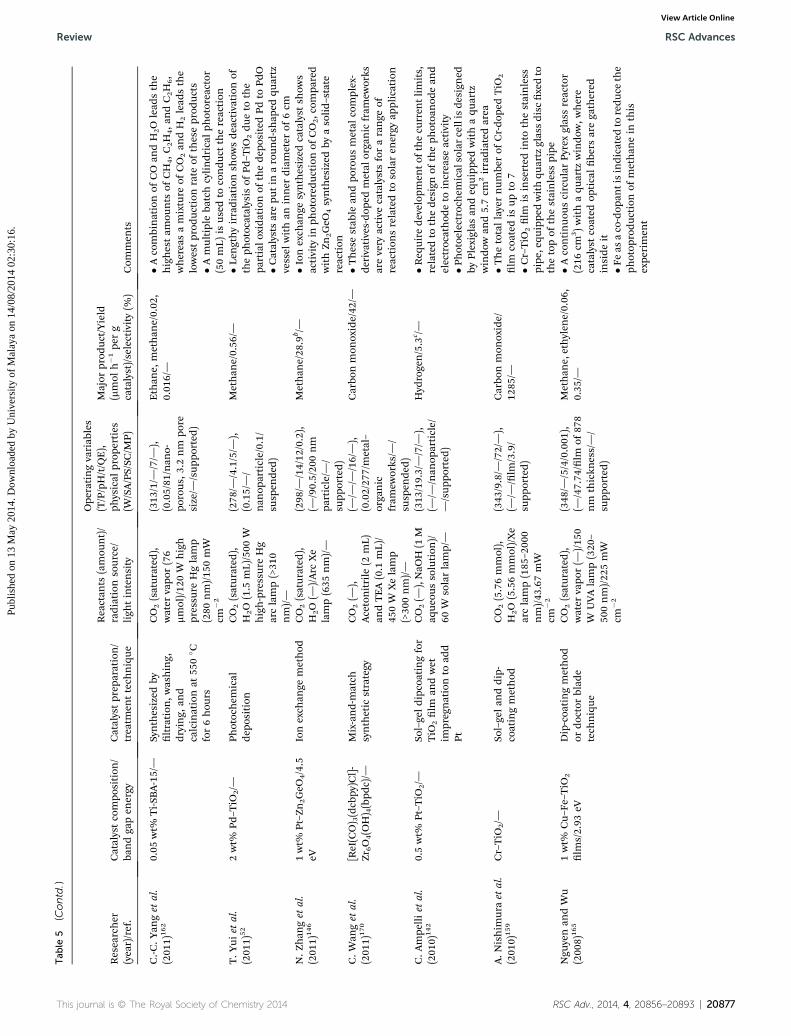
Tab
le5
(Contd.)
Researcher
(year)/ref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration/
treatm
enttech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/Yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
C.-C
.Yan
get
al.
(201
1)162
0.05
wt%
Ti-S
BA-15/—
Synthesized
byltration
,washing,
drying,
and
calcinationat
550
� Cfor6hou
rs
CO2(saturated
),water
vapo
r(76
mmol)/12
0W
high
pressu
reHglamp
(280
nm)/15
0mW
cm�2
(313
/1/—
/7/—
),(0.05/81
/nan
o-po
rous
,3.2
nm
pore
size/—
/sup
ported
)
Ethan
e,methan
e/0.02
,0.01
6/—
�Acombinationof
CO
andH
2O
lead
sthe
highestam
ounts
ofCH
4,C
2H
4,a
ndC2H
6,
whereasamixture
ofCO2an
dH
2lead
sthe
lowestprod
uction
rate
oftheseprod
ucts
�Amultipleba
tchcylindrical
photoreactor
(50mL)
isus
edto
condu
ctthereaction
T.Y
uiet
al.
(201
1)52
2wt%
Pd–T
iO2/—
Photochem
ical
depo
sition
CO2(saturated
),H
2O
(1.5
mL)/500
Whigh-pressureHg
arclamp(>31
0nm)/—
(278
/—/4.1/5/—
),(0.15/—/
nan
oparticle/0.1/
susp
ende
d)
Methan
e/0.56
/—�L
engthyirradiationsh
owsde
activation
oftheph
otocatalysis
ofPd
–TiO
2du
eto
the
partialo
xida
tion
ofthede
positedPd
toPd
O�C
atalysts
arepu
tin
aroun
d-sh
aped
quartz
vessel
withan
inner
diam
eter
of6cm
N.Z
han
get
al.
(201
1)146
1wt%
Pt–Z
n2GeO
4/4.5
eVIonexch
ange
method
CO2(saturated
),H
2O
(—)/Arc
Xe
lamp(635
nm)/—
(298
/—/14/12
/0.2),
(—/90.5/20
0nm
particle/—
/su
pported)
Methan
e/28
.9b/—
�Ion
exch
ange
synthesized
catalyst
show
sactivity
inph
otored
uction
ofCO2,com
pared
withZn
2GeO
4synthesized
byasolid–state
reaction
C.W
anget
al.
(201
1)170
[ReI(CO) 3(dcb
py)Cl]-
Zr6O4(O
H) 4(bpd
c)/—
Mix-and-match
synthetic
strategy
CO2(—
),Acetonitrile
(2mL)
andTEA(0.1
mL)/
450W
Xelamp
(>30
0nm)/—
(—/—
/—/16/—),
(0.02/27
7/metal–
orga
nic
fram
eworks/—
/su
spen
ded)
Carbo
nmon
oxide/42
/—�T
hesestab
lean
dpo
rous
metal
complex-
derivatives-do
pedmetal
orga
nic
fram
eworks
arevery
active
catalystsforarange
ofreaction
srelatedto
solaren
ergy
application
C.A
mpe
lliet
al.
(201
0)142
0.5wt%
Pt–T
iO2/—
Sol–geldipc
oatingfor
TiO
2lm
andwet
impregnationto
add
Pt
CO2(—
),NaO
H(1
Maq
ueou
ssolution
)/60
Wsolarlamp/—
(313
/19.3/—/7/—
),(—
/—/nan
oparticle/
—/sup
ported
)
Hyd
rogen/5.3
c /—
�Req
uire
developm
entof
thecu
rren
tlimits,
relatedto
thede
sign
oftheph
otoa
nod
ean
delectrocathod
eto
increase
activity
�Photoe
lectroch
emical
solarcellisde
sign
edby
Plexiglasan
deq
uipp
edwithaqu
artz
windo
wan
d5.7cm
2irradiated
area
A.N
ishim
uraet
al.
(201
0)159
Cr–TiO
2/—
Sol–gelan
ddip-
coatingmethod
CO2(5.76mmol),
H2O
(5.56mmol)/Xe
arclamp(185
–200
0nm)/43
.67mW
cm�2
(343
/9.8/—
/72/—),
(—/—
/lm
/3.9/
supp
orted)
Carbo
nmon
oxide/
1285
/—�T
hetotallayernum
berof
Cr-do
pedTiO
2
lm
coated
isup
to7
�Cr–TiO
2lm
isinserted
into
thestainless
pipe
,equ
ippe
dwithqu
artzglassdisc
xedto
thetopof
thestainless
pipe
Ngu
yenan
dWu
(200
8)165
1wt%
Cu–
Fe–T
iO2
lm
s/2.93
eVDip-coa
tingmethod
ordo
ctor
blad
etech
nique
CO2(saturated
),water
vapo
r(—
)/15
0W
UVAlamp(320
–50
0nm)/22
5mW
cm�2
(348
/—/5/4/0.001
),(—
/47.74
/lm
of87
8nm
thickn
ess/—/
supp
orted)
Methan
e,ethylen
e/0.06
,0.35
/—�A
continuo
uscircular
Pyrexglassreactor
(216
cm3)withaqu
artz
windo
w,w
here
catalyst
coated
opticalbe
rsarega
thered
inside
it�F
eas
aco-dop
antisindicatedto
redu
cethe
photop
rodu
ctionof
methan
ein
this
expe
rimen
t
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 208
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
77
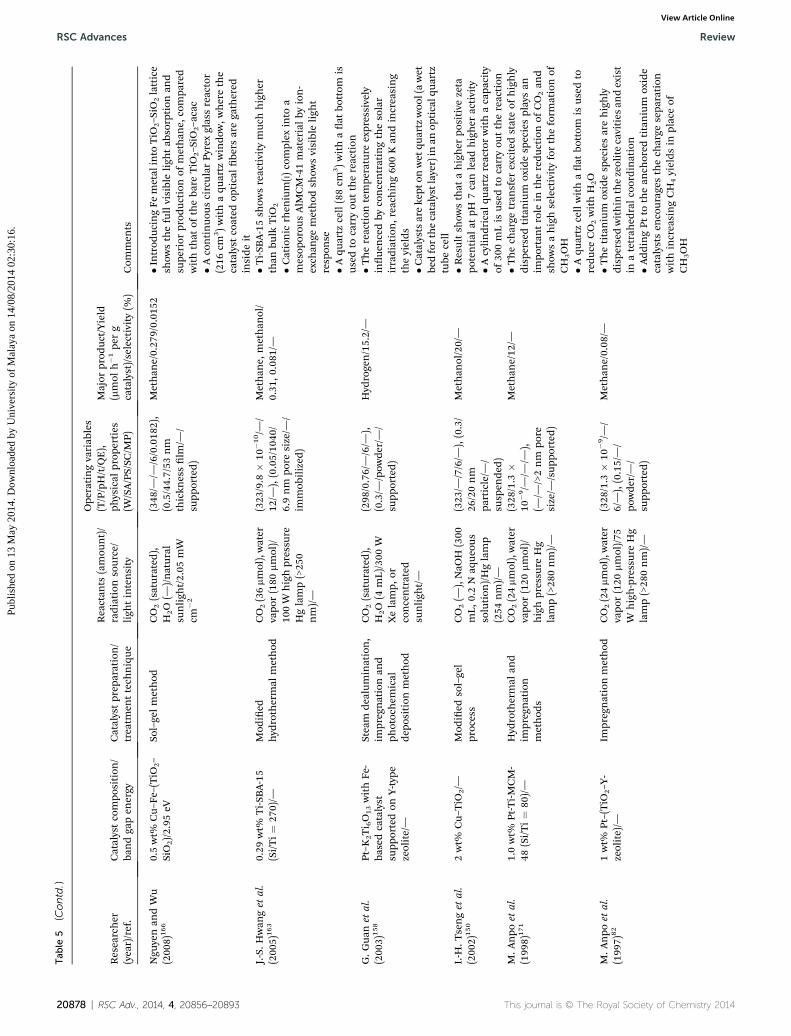
Tab
le5
(Contd.)
Researcher
(year)/ref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration/
treatm
enttech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/Yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
Ngu
yenan
dWu
(200
8)166
0.5wt%
Cu–
Fe–(TiO
2–
SiO2)/2.95
eVSo
l–gelmethod
CO2(saturated
),H
2O
(—)/natural
sunligh
t/2.05
mW
cm�2
(348
/—/—
/6/0.018
2),
(0.5/44.7/53
nm
thickn
esslm
/—/
supp
orted)
Methan
e/0.27
9/0.01
52�I
ntrod
ucingFe
metal
into
TiO
2–SiO
2lattice
show
sthefullvisibleligh
tab
sorption
and
supe
rior
prod
uction
ofmethan
e,compa
red
withthat
oftheba
reTiO
2–SiO
2–acac
�Acontinuo
uscircular
Pyrexglassreactor
(216
cm3)withaqu
artz
windo
w,w
herethe
catalyst
coated
opticalbe
rsarega
thered
inside
itJ.-S.H
wan
get
al.
(200
5)163
0.29
wt%
Ti-S
BA-15
(Si/Ti¼
270)/—
Mod
ied
hyd
rothermal
method
CO2(36mmol),water
vapo
r(180
mmol)/
100W
highpressu
reHglamp(>25
0nm)/—
(323
/9.8
�10
�10/—
/12
/—),(0.05/10
40/
6.9nm
pore
size/—
/im
mob
ilized
)
Methan
e,methan
ol/
0.31
,0.081
/—�T
i-SBA-15sh
owsreactivity
muc
hhigher
than
bulk
TiO
2
�Cationic
rhen
ium(I)
complex
into
amesop
orou
sAlM
CM-41materialby
ion-
exch
ange
method
show
svisibleligh
tresp
onse
�Aqu
artz
cell(88cm
3)withaat
bottom
isus
edto
carryou
tthereaction
G.G
uanet
al.
(200
3)158
Pt–K
2Ti 6O13withFe
-ba
sedcatalyst
supp
ortedon
Y-typ
ezeolite/—
Steam
dealumination,
impregnationan
dph
otochem
ical
depo
sition
method
CO2(saturated
),H
2O
(4mL)/300
WXelamp,
orconcentrated
sunligh
t/—
(298
/0.76/—/6/—
),(0.3/—
/pow
der/—/
supp
orted)
Hyd
rogen/15.2/—
�Thereaction
tempe
rature
expressively
inue
ncedby
concentratingthesolar
irradiation,reach
ing60
0Kan
dincreasing
theyields
�Catalystsareke
pton
wet
quartzwoo
l(awet
bedforthecatalystlayer)in
anop
ticalq
uartz
tube
cell
I.-H
.Tsenget
al.
(200
2)150
2wt%
Cu–
TiO
2/—
Mod
ied
sol–gel
process
CO2(—
),NaO
H(300
mL,
0.2N
aque
ous
solution
)/Hglamp
(254
nm)/—
(323
/—/7/6/—
),(0.3/
26/20nm
particle/—
/su
spen
ded)
Methan
ol/20/—
�Resultsh
owsthat
ahigher
positive
zeta
potential
atpH
7canlead
higher
activity
�Acylindrical
quartz
reactorwithacapa
city
of30
0mLis
used
tocarryou
tthereaction
M.A
npo
etal.
(199
8)171
1.0wt%
Pt-Ti-MCM-
48(Si/Ti¼
80)/—
Hyd
rothermal
and
impregnation
method
s
CO2(24mmol),water
vapo
r(120
mmol)/
highpressu
reHg
lamp(>28
0nm)/—
(328
/1.3
�10
�9/—
/—/—
),(—
/—/>2nm
pore
size/—
/sup
ported
)
Methan
e/12
/—�T
hech
arge
tran
sfer
excitedstateof
highly
disp
ersedtitanium
oxidesp
eciesplaysan
impo
rtan
trole
intheredu
ctionof
CO2an
dsh
owsahighselectivityfortheform
ationof
CH
3OH
�Aqu
artz
cellwithaat
bottom
isus
edto
redu
ceCO2withH
2O
M.A
npo
etal.
(199
7)82
1wt%
Pt–(TiO
2–Y
-zeolite)/—
Impregnationmethod
CO2(24mmol),water
vapo
r(120
mmol)/75
Whigh-pressureHg
lamp(>28
0nm)/—
(328
/1.3
�10
�9/—
/6/—),(0.15/—/
powde
r/—/
supp
orted)
Methan
e/0.08
/—�T
hetitanium
oxidesp
eciesarehighly
disp
ersedwithin
thezeolitecavities
andexist
inatetrah
edralcoordination
�Add
ingPt
tothean
chored
titanium
oxide
catalystsen
courag
esthech
arge
sepa
ration
withincreasingCH
4yields
inplaceof
CH
3OH
20878 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2014
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
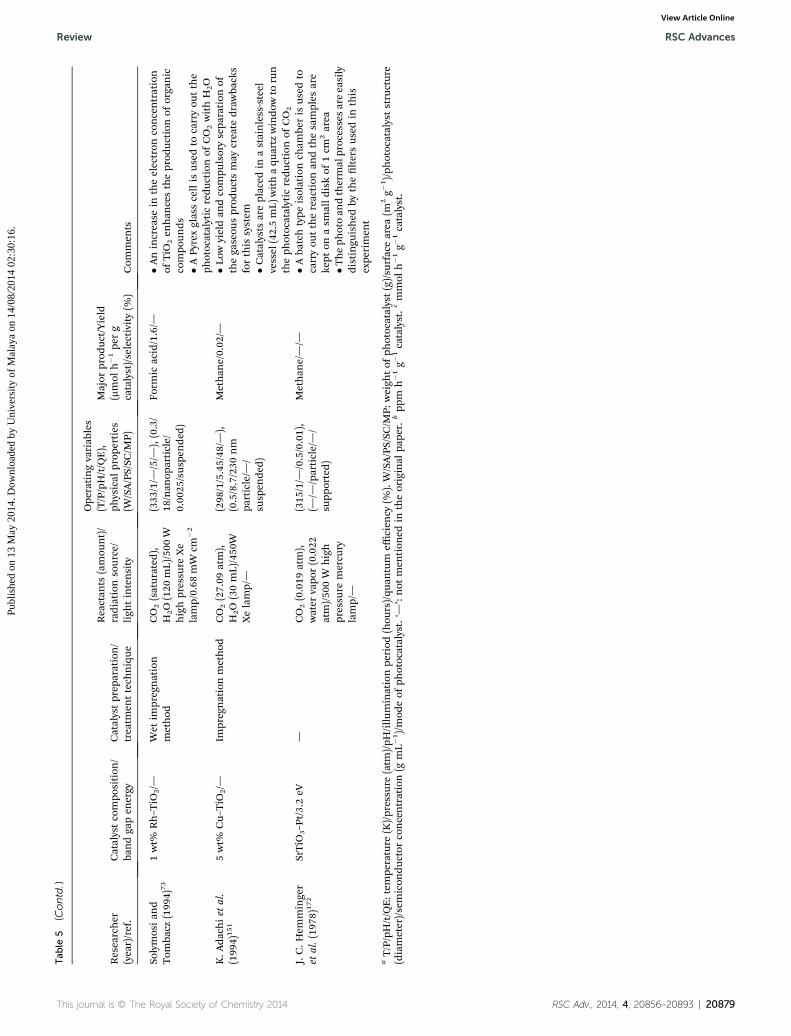
Tab
le5
(Contd.)
Researcher
(year)/ref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration/
treatm
enttech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/Yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
Solymosian
dTom
bacz
(199
4)73
1wt%
Rh–T
iO2/—
Wet
impregnation
method
CO2(saturated
),H
2O(120
mL)/500
Whighpressu
reXe
lamp/0.68
mW
cm�2
(333
/1/—
/5/—
),(0.3/
18/nan
oparticle/
0.00
25/sus
pende
d)
Form
icacid/1.6/—
�Anincrease
intheelectron
concentration
ofTiO
2en
han
cestheprod
uction
oforga
nic
compo
unds
�APy
rexglasscellis
used
tocarryou
tthe
photocatalytic
redu
ctionof
CO2withH
2O
K.A
dachiet
al.
(199
4)151
5wt%
Cu–
TiO
2/—
Impregnationmethod
CO2(27.09
atm),
H2O
(30mL)/450
WXelamp/—
(298
/1/5.45/48
/—),
(0.5/8.7/230
nm
particle/—
/su
spen
ded)
Methan
e/0.02
/—�L
owyieldan
dcompu
lsorysepa
ration
ofthega
seou
sprod
ucts
may
create
draw
backs
forthis
system
�Catalysts
areplaced
inastainless-steel
vessel
(42.5mL)
withaqu
artz
windo
wto
run
theph
otocatalytic
redu
ctionof
CO2
J.C.H
emminger
etal.(197
8)172
SrTiO
3–P
t/3.2eV
—CO2(0.019
atm),
water
vapo
r(0.022
atm)/50
0W
high
pressu
remercu
rylamp/—
(315
/1/—
/0.5/0.01),
(—/—
/particle/—/
supp
orted)
Methan
e/—/—
�Aba
tchtype
isolationch
ambe
ris
used
tocarryou
tthereaction
andthesamples
are
kept
onasm
alldisk
of1cm
2area
�Theph
otoan
dthermal
processesareeasily
distingu
ished
bytheltersus
edin
this
expe
rimen
t
aT/P/pH/t/Q
E:tem
perature
(K)/pressu
re(atm
)/pH
/illu
minationpe
riod
(hou
rs)/qu
antum
efficien
cy(%
).W/SA/PS/SC
/MP:
weigh
tof
photocatalyst(g)/su
rfacearea
(m2g�
1)/ph
otocatalyststructure
(diameter)/semicon
duc
torconcentration(g
mL�
1 )/m
odeof
photocatalyst.‘—
’:not
men
tion
edin
theoriginal
pape
r.bpp
mh�1g�
1catalyst.c
mmol
h�1g�
1catalyst.
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 20879
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
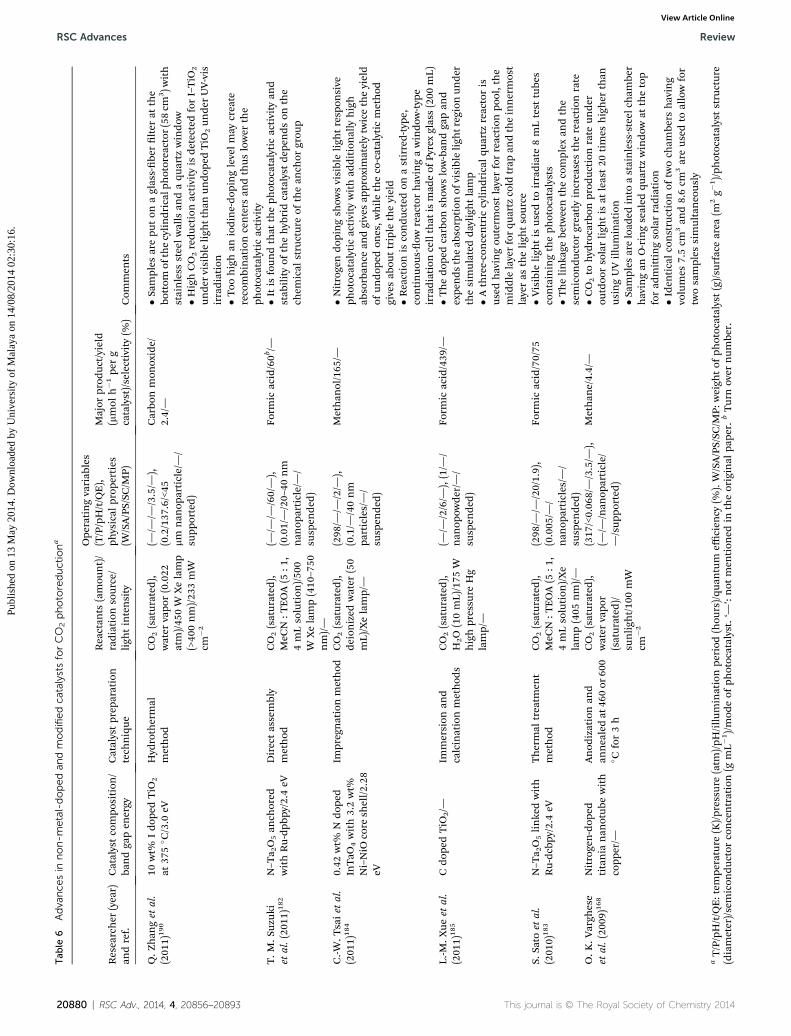
Tab
le6
Advance
sin
non-m
etal-dopedan
dmodifiedca
talystsforCO
2photoreductiona
Researcher
(year)
andref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
Q.Z
han
get
al.
(201
1)190
10wt%
Ido
pedTiO
2
at37
5� C
/3.0
eVHyd
rothermal
method
CO2(saturated
),water
vapo
r(0.022
atm)/45
0W
Xelamp
(>40
0nm)/23
3mW
cm�2
(—/—
/—/3.5/—
),(0.2/137
.6/<45
mm
nan
oparticle/—/
supp
orted)
Carbo
nmon
oxide/
2.4/—
�Sam
ples
arepu
ton
aglass-be
rlter
atthe
bottom
ofthecylindricalph
otoreactor
(58cm
3)w
ith
stainless
steelwalls
andaqu
artz
windo
w�H
ighCO2redu
ctionactivity
isde
tected
forI–TiO
2
unde
rvisibleligh
tthan
undo
pedTiO
2un
derUV-vis
irradiation
�Too
highan
iodine-do
pinglevelmay
create
recombinationcenters
andthus
lower
the
photocatalytic
activity
T.M
.Suz
uki
etal.(201
1)182
N–T
a 2O5an
chored
withRu-dp
bpy/2.4eV
Directassembly
method
CO2(saturated
),MeC
N:T
EOA(5
:1,
4mLsolution
)/50
0W
Xelamp(410
–750
nm)/—
(—/—
/—/60/—),
(0.01/—/20–40
nm
nan
oparticle/—/
susp
ende
d)
Form
icacid/60b/—
�Itis
foun
dthat
theph
otocatalytic
activity
and
stab
ilityof
thehyb
ridcatalyst
depe
nds
onthe
chem
ical
structureof
thean
chor
grou
p
C.-W
.Tsaiet
al.
(201
1)184
0.42
wt%
Ndop
edInTaO
4with3.2wt%
Ni–NiO
core
shell/2.28
eV
Impregnationmethod
CO2(saturated
),de
ionized
water
(50
mL)/Xelamp/—
(298
/—/—
/2/—
),(0.1/—
/40nm
particles/—/
susp
ende
d)
Methan
ol/165
/—�N
itrogendo
pingsh
owsvisibleligh
tresp
onsive
photocatalytic
activity
withad
dition
ally
high
absorban
cean
dgivesap
proxim
atelytw
icetheyield
ofun
dope
don
es,w
hiletheco-catalytic
method
givesab
outtriple
theyield
�Reactionis
condu
cted
onastirred-type
,continuo
us-ow
reactorhavingawindo
w-typ
eirradiationcellthat
ismad
eof
Pyrexglass(200
mL)
L.-M
.Xueet
al.
(201
1)185
Cdo
pedTiO
2/—
Immersion
and
calcinationmethod
sCO2(saturated
),H
2O
(10mL)/175
Whighpressu
reHg
lamp/—
(—/—
/2/6/—
),(1/—
/nan
opow
der/—/
susp
ende
d)
Form
icacid/439
/—�T
hedo
pedcarbon
show
slow-ban
dga
pan
dexpe
nds
theab
sorption
ofvisibleligh
tregionun
der
thesimulated
daylightlamp
�Athree-concentric
cylindrical
quartz
reactoris
used
havingou
term
ostlayerforreaction
pool,the
middlelayerforqu
artz
cold
trap
andtheinnermost
layeras
theligh
tsource
S.Sa
toet
al.
(201
0)183
N–T
a 2O5linke
dwith
Ru-dcb
py/2.4
eVThermal
treatm
ent
method
CO2(saturated
),MeC
N:T
EOA(5
:1,
4mLsolution
)/Xe
lamp(405
nm)/—
(298
/—/—
/20/1.9),
(0.005
/—/
nan
oparticles/—
/su
spen
ded)
Form
icacid/70/75
�Visible
ligh
tis
used
toirradiate8mLtest
tube
scontainingtheph
otocatalysts
�Thelinka
gebe
tweenthecomplex
andthe
semicon
ductor
greatlyincreasesthereaction
rate
O.K
.Vargh
ese
etal.(200
9)168
Nitrogen-dop
edtitania
nan
otub
ewith
copp
er/—
Anod
izationan
dan
nealedat
460or
600
� Cfor3h
CO2(saturated
),water
vapo
r(saturated
)/su
nligh
t/10
0mW
cm�2
(317
/<0.06
8/—/3.5/—
),(—
/—/nan
oparticle/
—/sup
ported
)
Methan
e/4.4/—
�CO2to
hyd
rocarbon
prod
uction
rate
unde
rou
tdoo
rsolarligh
tis
atleast20
times
higher
than
usingUVillumination
�Sam
ples
areload
edinto
astainless-steel
cham
ber
havingan
O-ringsealed
quartz
windo
wat
thetop
forad
mittingsolarradiation
�Ide
nticalconstructionof
twoch
ambe
rshaving
volumes
7.5cm
3an
d8.6cm
3areus
edto
allowfor
twosamples
simultaneo
usly
aT/P/pH/t/Q
E:tem
perature
(K)/pressu
re(atm
)/pH
/illu
minationpe
riod
(hou
rs)/qu
antum
efficien
cy(%
).W/SA/PS/SC
/MP:
weigh
tof
photocatalyst(g)/su
rfacearea
(m2g�
1)/ph
otocatalyststructure
(diameter)/semicon
duc
torconcentration(g
mL�
1 )/m
odeof
photocatalyst.‘—
’:not
men
tion
edin
theoriginal
pape
r.bTurnover
num
ber.
20880 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2014
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online

Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
arbon radicals and H atoms formed on the Pt metals.82 Pt-loaded alkali niobates ANbO3 (A ¼ Na, K) photocatalysts werereported by Li et al.144 Results showed that KNbO3 exhibitedhigher photocatalytic activity than NaNbO3 because of havinglow band gap energy (Eg) and high mobile charge carriers. Here,Pt acts as a co-catalyst to provide the reactive site for hydrogengas production easily in terms of low H2 over-potential on the Ptsurface.145 It is also noticeable that increasing Pt proportionfrom 0.5 wt% to 1.0 wt% promotes the formation of hydrogengas. The photocatalytic performance of micro- or mesoporousZn2GeO4 loading with various percentages of Pt (from 0.5 to 2.5wt%) was investigated by N. Zhang et al., from which the resultsevaluated the highest activity for 1.0 wt% Pt.146
Ishitani et al. reported that Pd doped on TiO2 performedmore effective photocatalytic activity than that of other metalsloaded on TiO2, and it also increased the formation of CH4
instead of CO from CO2.147 On the other hand, Pd on the surfaceof TiO2 partially oxidized to form PdO during the progression ofCH4 formation, which led to the deactivation of the Pd–TiO2
catalysts.52 Solymosi and Tombacz assumed that adsorptionand photocatalytic behaviors of Rh were signicantly inuencedby the existence of an electronic interaction at the Rh–TiO2
interface.73 Moreover, the catalytic activity of Rh was affected bydoping with altervalent cations (W6+) due to the variation of theelectron concentration of the TiO2.148 Cu is another noble metal,which is eminent for its availability and low cost. Not only that,Cu loading on TiO2 was a prevalent approach in photocatalyticconversion of CO2 because of its high yield through the loweringof re-electron–hole recombination probability. According toTseng et al., an optimal Cu-loaded titania (2 wt% Cu) was foundto have the highest photochemical activity, which providedlower uorescence intensity and resulted in higher methanolformation.149,150 Depending on the amount of Cu loading onTiO2, the formation of the hydrocarbon products has beenchanged also.151 Ag-doped TiO2 was also investigated in thistechnology reduced the band gap energy of TiO2 and shied theband edge from the UV region to the visible region, according tothe amount of Ag added.152,153 Ag co-catalyst-loaded on ALa4-Ti4O15 (A ¼ Ca, Sr, and Ba) has been published, and Ag acts as aCO2 reduction site to generate CO as reported in electro-chemical reduction of CO2, where Ag was used as an electro-catalyst.154 Moreover, the plasmon effect has been developed byAu nanoparticles doped with TiO2 through increasing thevisible light response.155–157
2.3.2 Transition metal doped semiconductors. To improvethe photocatalytic activity and efficiency, transition metals arealso investigated in many studies due to the high visible lightconversion of CO2.158 The interesting thing about transitionmetals is that their valence electrons to exhibit severalcommon oxidation states. Among the transition metals, Co,Mn, Ni, Cr, Mo, Fe, V, and Ti are commonly used as dopingelements in photocatalytic technology to enhance the semi-conductor performances under visible light. For visible lightresponsibility, Nishimura et al. synthesized Cr-doped TiO2
lm by the sol–gel and dip-coating methods.159 They statedthat a suitable percentage of metal doping on semiconductorsimproved catalytic activity, whereas excess percentages of
This journal is © The Royal Society of Chemistry 2014
dopant metal beyond the optimal level decreased the photo-catalytic performances because of charge recombination.However, the H2 generation rate was more advanced for Fe-doped TiO2 than Cr-doped TiO2 because of avoiding therecombination in trapping both electrons and holes by Feparticles, whereas Cr can only trap one type of chargecarrier.160 The experiment was done to characterize thedifferent types of transition metals (V, Fe, Ce, Cu, Cr) dopedTiO2, prepared by Pan et al.161 They reported that V and Fewere placed in the substitutional sites of TiO2, and Ce ionswere dispersed in the interstitial sites, although Cr and Cuaccumulated on the surface. The activity order was found insuch a manner: Fe–TiO2 > V–TiO2 > Cr–TiO2 > Ce–TiO2 > TiO2
> Cu–TiO2, which indicates that the local structure and type ofdopant have vital contributions on photocatalytic technology.
The loading of transitional metal species on the frameworknanostructure is a common practice for increasing the highdispersion of those species. Moreover, for surface immobiliza-tion, titanium species are conned onto mesoporous silica.This phenomenon is also effective for photocatalytic reactionsof titanium particles as their photocatalytic activities intenselyrely on the structures of active sites.162 It was found that highand exceptional photocatalytic reactivity was obtained for thereduction of CO2 with H2O to generate CH4 and CH3OH underUV irradiation, compared with the bulk TiO2.163
2.3.3 Co-metal doped semiconductors. The doping ofbimetallic elements together on semiconductor photo-catalysts has been validated as a potential method to advancethe visible light response of photocatalysts. Suitable co-dopant combinations are necessary to enhance the photo-catalytic activity of semiconductors because all of the co-dopant congurations are not appropriate for this tech-nique.164 Fe as a co-dopant in the Cu–TiO2 catalyst has beeninvestigated for photocatalytic reduction of CO2 with H2O toethylene at a quantum yield of 0.024%.165 An efficient chargetransfer mechanism occurred in this combination betweenTiO2 and Cu as well as Fe as co-dopants. However, Cu co-doped with Fe–TiO2 on SiO2 support has also been examinedunder concentrated natural sunlight with high quantum yield(0.05%) and full absorption of visible light and compared toonly Cu co-doped on the Fe–TiO2 counterpart.166 The photo-catalytic performance of the Cu doped on Pt–TiO2 catalysts isinuenced by the content of Cu loading. The high percentageof Cu loading expressively decreases the formation of H2 andimproves the formation of CH4 and CO.167 Varghese et al.reported that Pt may have an effect on H2O activation,whereas Cu plays an important role in activating CO2 to CO.168
Bimetallic Ag–Pt were used with the TiO2 semiconductorcatalyst as reported by Mankidy et al. who obtained a selec-tivity of CH4 of almost 80%, compared with TiO2 of 20%selectivity.169 Their investigation found that Ag species had astrong surface plasmon absorption band in the UV-vis region,while the Pt species did not show that behavior. However, thecombination of bimetallic Ag–Pt has the ability to tune theelectronic properties during photochemical reactions. Notonly that, combined Ag–Pt can accumulate the electrons by Agand transfer the interfacial electron charge from Pt.
RSC Adv., 2014, 4, 20856–20893 | 20881

RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
2.3.4 Non-metal doped semiconductors. It has beenbroadly conrmed that doping or co-doping of semiconductorphotocatalysts with non-metals such as C, N, S, B and F hasshown considerably low band gap energy, compared with metaldoping in different experiments, prominent to efficient pho-tocatalytic activity during visible light irradiation.173–180 InTable 6, the update of major ndings and detailed informationregarding non-metal-doped semiconductors is given elabo-rately to evaluate their progress in the photocatalytic conver-sion of CO2. Compared with other non-metals, nitrogen-dopedsemiconductors show the most active results due to the narrowband gap.181 A high rate of solar conversion of carbon dioxide inthe presence of water vapor to methane and other hydrocar-bons is attained from nitrogen-deposited titania nanotubearrangements, as reported by Varghese et al.168 Particularly, lowwall thickness of nanotube arrays promotes the transfer ofefficient carriers to the adsorbing species. From the experi-ment, it was also found that the incorporation of Pt and Cunanoparticles on the external surfaces of nanotube arrange-ments further increases the hydrocarbon yield under theoutdoor global AM 1.5 sunlight with 100 mW cm�2 intensity.Both Suzuki et al. and Sato et al. studied p-type, photoactive,nitrogen-doped Ta2O5 linkage with metal-complex electro-catalysts, which revealed outstanding photochemical conver-sion of CO2 to formic acid under visible light irradiation byincreasing the photoexcited electron transfer from theconduction band of the semiconductor to the metalcomplex.182,183 The co-catalytic (N-doped InTaO4 modied withNi–NiO core–shell nanostructure) approach by Tsai et al. notonly intensely increases solar light absorbance but alsocompetently reduces the electron–hole recombination.184 Theinvestigation with carbon-doped TiO2 was done by Xue et al.because doped carbon narrows the band gap, shis theabsorption to the visible light region and facilitates the chargeseparation efficiency.185 Hiroshi et al. reported that the hydro-philic behavior of C-doped TiO2 semiconductor exhibitedvisible light response, which was initiated from the localized C(2p) formed above the valence band.186 S-doped TiO2 has theability to reduce the band gap energy and inhibits the alter-ation of anatase phase into rutile phase at elevated tempera-tures.187 Correspondingly, the doping of F on a semiconductorcatalyst performs better by decreasing the band gap energy.188
Results showed that iodine doping on semiconductor cata-lysts exhibited higher photochemical activity due to the closerionic radii of I5+ and Ti4+ for substituting lattice titanium thanother non-metal-doped elements like nitrogen, carbon, boron,and sulfur.189 Moreover, the replacement of Ti4+ with I5+ beginscharge inequality to the formation of Ti3+ surface states that isable to arrest the photoinduced electrons and prevent chargerecombination in the photocatalytic conversion of CO2.190
Zhang et al. found that iodine-doped TiO2 semiconductorscalcined at 375 �C reduced particle sizes signicantly andincreased photochemical activity.190 It was also observed that10% iodine deposition on TiO2 harvested maximum yield undervisible light irradiation compared with the 5% iodine dopingon TiO2.
20882 | RSC Adv., 2014, 4, 20856–20893
2.4 Sensitized photocatalysts
For photocatalytic CO2 conversion, photosensitization of thecatalyst is a promising technique which makes the catalystmore sensitive during light irradiation. This practice is gener-ally conducted in photochemical applications under the lightsources that have particular wavelength deciencies for photo-excitation. The mechanism generates photoexcited electrons inthe conduction band of photosensitizers under the light sourceof particular wavelengths, and the electrons are directly shiedto the conduction band of catalysts to make them photoactiveunder visible light irradiation.191 According to Tahir and Amin,photosensitization exclusively depends on the suitable sensi-tizer band gap that could absorb visible light irradiations, andthe sensitizer conductance band needed to be higher (morenegative) than the catalyst's conductance band to transferelectrons towards the catalyst.181 Some informative character-istics are represented in Table 7 for different types of sensitizedphotocatalysts, which are applied in many studies forincreasing photocatalytic activity in order to obtain high photonefficiency and selectivity.
2.4.1 Dye sensitized photocatalysts. Dye sensitized photo-catalysts are widely used in photochemical technology becauseof their ability to create photoactive electrons easily by visiblelight hitting the dye compound. This phenomenon helps totransfer those photoactive electrons from the dye to the cata-lysts' conduction band for activation during the visible lightirradiation.37 An effective response to visible light and efficientelectron transfer can enhance fuel yields. The charge separationcan be effectively achieved by adding a co-catalyst to trap theelectrons, which is depicted in Scheme 1(d).
Li et al. reported that charging of Pt and RuO2 on dyesensitized hexagonal nanoplates to form micro-octahedronZn2SnO4 enabled the accumulation of photoexcited electronsand holes, respectively.192 Dye sensitized, Pt loaded, nanosized{010} dominant anatase TiO2 rods were used to obtain excep-tional surface atomic structure for optimum adsorption of dyemolecules and encourage electron transmission from exciteddye molecules to anatase TiO2.193 These occurrences alsoupgraded the current density and solar energy conversion effi-ciency of that system. Another investigation found thatRuII(2,20-bipyridyl-4,40-dicarboxylate)2-(NCS)2 (basically knownas N3-dye) sensitized Cu–Fe loading on the P25 photocatalystwas able to absorb the wavelength of 400–800 nm from solarirradiation.194 Moreover, the results show that N3-dye is notsuitable under articial light (wavelength range is 320–500 nm)irradiation because before the irradiation, it is dispersed on thematrices of the photocatalysts; as a result, the dye does not showits full response to the light source but using concentratednatural sunlight N3-dye remarkably increases methane yield upto over 100%. Similar results were found by Wu and Chiou inthat N3-dye gave effectual charge transfer with higher photo-reduction under concentrated sunlight.195 Moreover, it was alsofound that N3-dye showed its stability for up to 6 h under botharticial light and concentrated sunlight. Ozcan et al. usedperylene diimide dyes, which have revealed similar visible lightresponse to the RuII(2,20-bipyridyl)chloride hexahydrate.196 In
This journal is © The Royal Society of Chemistry 2014
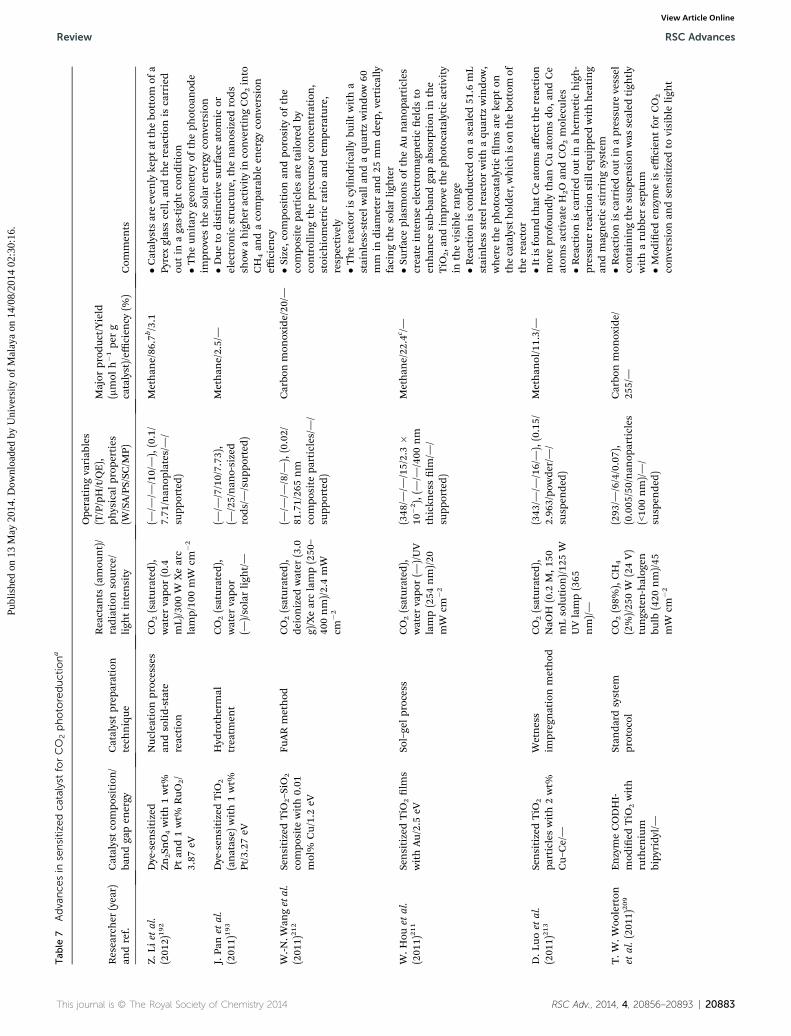
Tab
le7
Advance
sin
sensitize
dca
talyst
forCO
2photoreductiona
Researcher
(year)
andref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/Yield
(mmol
h�1pe
rg
catalyst)/effi
cien
cy(%
)Com
men
ts
Z.Li
etal.
(201
2)192
Dye-sen
sitized
Zn2Sn
O4with1wt%
Ptan
d1wt%
RuO
2/
3.87
eV
Nuc
leationprocesses
andsolid-state
reaction
CO2(saturated
),water
vapo
r(0.4
mL)/300
WXearc
lamp/10
0mW
cm�2
(—/—
/—/10/—),(0.1/
7.71
/nan
oplates/—/
supp
orted)
Methan
e/86
.7b/3.1
�Catalysts
areeven
lyke
ptat
thebo
ttom
ofa
Pyrexglasscell,
andthereaction
iscarried
outin
aga
s-tigh
tcondition
�Theun
itarygeom
etry
oftheph
otoa
nod
eim
proves
thesolaren
ergy
conversion
J.Pa
net
al.
(201
1)193
Dye-sen
sitizedTiO
2
(anatase)
with1wt%
Pt/3.27eV
Hyd
rothermal
treatm
ent
CO2(saturated
),water
vapo
r(—
)/solarligh
t/—
(—/—
/7/10/7.73
),(—
/25/nan
o-sized
rods
/—/sup
ported
)
Methan
e/2.5/—
�Due
todistinctivesu
rfaceatom
icor
electron
icstructure,
thenan
osized
rods
show
ahigher
activity
inconvertingCO2into
CH
4an
dacompa
rableen
ergy
conversion
efficien
cyW.-N
.Wan
getal.
(201
1)212
SensitizedTiO
2–SiO
2
compo
site
with0.01
mol%
Cu/1.2eV
FuARmethod
CO2(saturated
),de
ionized
water
(3.0
g)/Xearclamp(250
–40
0nm)/2.4mW
cm�2
(—/—
/—/8/—
),(0.02/
81.71/26
5nm
compo
site
particles/—/
supp
orted)
Carbo
nmon
oxide/20
/—�S
ize,
compo
sition
andpo
rosity
ofthe
compo
site
particlesaretailored
bycontrollingtheprecursorconcentration,
stoich
iometricratioan
dtempe
rature,
resp
ectively
�Thereactoris
cylindrically
builtwitha
stainless-steel
wallan
daqu
artz
windo
w60
mm
indiam
eter
and25
mm
deep
,vertically
facingthesolarligh
ter
W.H
ouet
al.
(201
1)211
SensitizedTiO
2lm
swithAu/2.5eV
Sol–gelprocess
CO2(saturated
),water
vapo
r(—
)/UV
lamp(254
nm)/20
mW
cm�2
(348
/—/—
/15/2.3�
10�2),(—
/—/400
nm
thickn
esslm
/—/
supp
orted)
Methan
e/22
.4c /—
�Surface
plasmon
sof
theAunan
oparticles
create
intense
electrom
agnetic
elds
toen
han
cesu
b-ba
ndga
pab
sorption
inthe
TiO
2,a
ndim
provetheph
otocatalyticactivity
inthevisiblerange
�Reactionis
condu
cted
onasealed
51.6
mL
stainless
steelreactor
withaqu
artz
windo
w,
wheretheph
otocatalytic
lm
sareke
pton
thecatalystholde
r,whichison
thebo
ttom
ofthereactor
D.L
uoet
al.
(201
1)213
SensitizedTiO
2
particleswith2wt%
Cu–
Ce/—
Wetness
impregnationmethod
CO2(saturated
),NaO
H(0.2
M,1
50mLsolution
)/12
5W
UVlamp(365
nm)/—
(343
/—/—
/16/—),(0.15/
2.96
3/po
wde
r/—/
susp
ende
d)
Methan
ol/11.3/—
�Itisfoun
dthat
Ceatom
saff
ectthereaction
moreprofou
ndlythan
Cuatom
sdo
,andCe
atom
sactivate
H2O
andCO2molecules
�Reactionis
carriedou
tin
ahermetic
high-
pressu
rereaction
stillequ
ippe
dwithheating
andmag
netic
stirringsystem
T.W
.Woo
lerton
etal.(201
1)209
EnzymeCODHI-
mod
ied
TiO
2with
ruthen
ium
bipy
ridyl/—
Stan
dardsystem
protocol
CO2(98%
),CH
4
(2%)/25
0W
(24V)
tungsten-halog
enbu
lb(420
nm)/45
mW
cm�2
(293
/—/6/4/0.07),
(0.005
/50/nan
oparticles
(<10
0nm)/—/
susp
ende
d)
Carbo
nmon
oxide/
255/—
�Reactionis
carriedou
tin
apressu
revessel
containingthesu
spen
sion
was
sealed
tigh
tly
witharubb
erseptum
�Mod
ied
enzymeis
efficien
tforCO2
conversionan
dsensitizedto
visibleligh
t
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 208
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
83
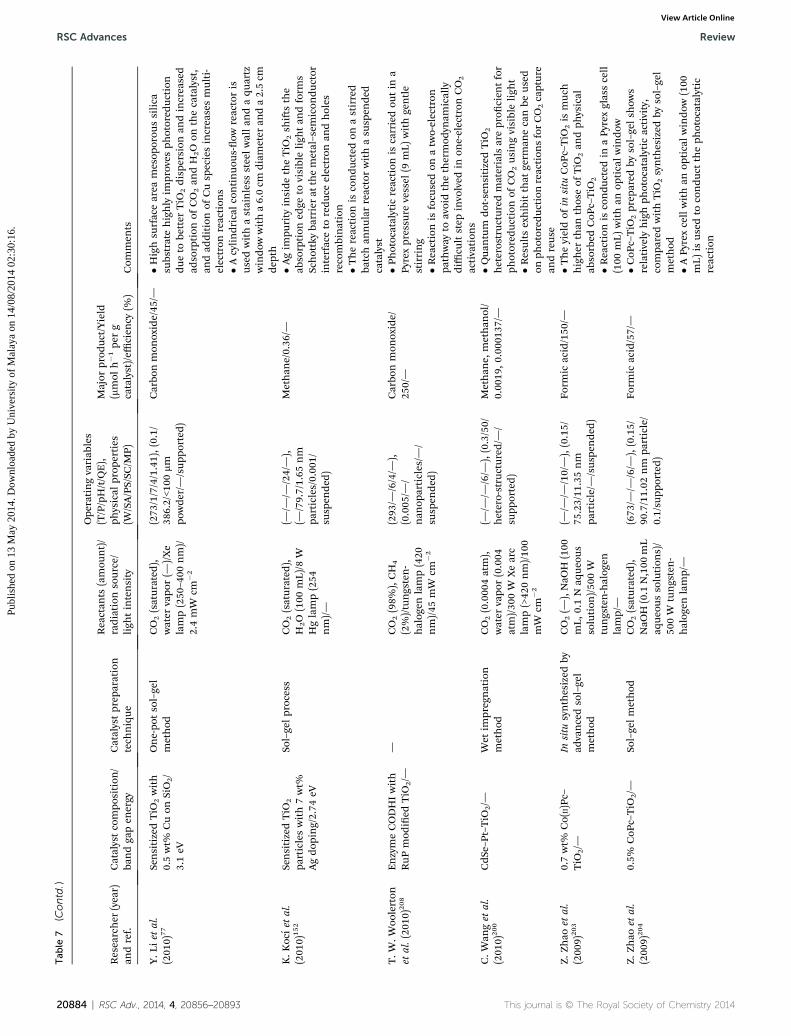
Tab
le7
(Contd.)
Researcher
(year)
andref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/Yield
(mmol
h�1pe
rg
catalyst)/effi
cien
cy(%
)Com
men
ts
Y.L
iet
al.
(201
0)77
SensitizedTiO
2with
0.5wt%
Cuon
SiO2/
3.1eV
One-po
tsol–gel
method
CO2(saturated
),water
vapo
r(—
)/Xe
lamp(250
–400
nm)/
2.4mW
cm�2
(273
/1/7/4/1.41),(0.1/
386.2/<1
00mm
powde
r/—/sup
ported
)
Carbo
nmon
oxide/45
/—�H
ighsu
rfacearea
mesop
orou
ssilica
subs
tratehighly
improves
photored
uction
dueto
better
TiO
2disp
ersion
andincreased
adsorption
ofCO2an
dH
2O
onthecatalyst,
andad
dition
ofCusp
eciesincreasesmulti-
electron
reaction
s�A
cylindrical
continuo
us-ow
reactoris
used
withastainless
steelwallan
daqu
artz
windo
wwitha6.0cm
diam
eter
anda2.5cm
depth
K.K
ocıet
al.
(201
0)152
SensitizedTiO
2
particleswith7wt%
Agdo
ping/2.74
eV
Sol–gelprocess
CO2(saturated
),H
2O
(100
mL)/8
WHglamp(254
nm)/—
(—/—
/—/24/—),
(—/79.7/1.65
nm
particles/0.00
1/su
spen
ded)
Methan
e/0.36
/—�A
gim
purity
inside
theTiO
2sh
isthe
absorption
edge
tovisibleligh
tan
dform
sSchottkyba
rrierat
themetal–sem
icon
ductor
interfaceto
redu
ceelectron
andholes
recombination
�Thereaction
iscondu
cted
onastirred
batchan
nular
reactorwithasu
spen
ded
catalyst
T.W
.Woo
lerton
etal.(201
0)208
EnzymeCODHIwith
RuPmod
ied
TiO
2/—
—CO2(98%
),CH
4
(2%)/tungsten-
halog
enlamp(420
nm)/45
mW
cm�2
(293
/—/6/4/—
),(0.005
/—/
nan
oparticles/—
/su
spen
ded)
Carbo
nmon
oxide/
250/—
�Photocatalytic
reaction
iscarriedou
tin
aPy
rexpressu
revessel
(9mL)
withgentle
stirring
�Reactionis
focu
sedon
atw
o-electron
pathway
toavoidthethermod
ynam
ically
difficu
ltstep
involved
inon
e-electron
CO2
activation
sC.W
anget
al.
(201
0)200
CdS
e–Pt–T
iO2/—
Wet
impregnation
method
CO2(0.000
4atm),
water
vapo
r(0.004
atm)/30
0W
Xearc
lamp(>42
0nm)/10
0mW
cm�2
(—/—
/—/6/—
),(0.3/50/
hetero-structured
/—/
supp
orted)
Methan
e,methan
ol/
0.00
19,0
.000
137/—
�Qua
ntum
dot-sensitizedTiO
2
heterostruc
turedmaterials
arepro
cien
tfor
photored
uction
ofCO2us
ingvisibleligh
t�R
esults
exhibitthat
germ
anecanbe
used
onph
otored
uction
reaction
sforCO2capture
andreus
eZ.
Zhao
etal.
(200
9)203
0.7wt%
Co(
II)Pc–
TiO
2/—
Insitu
synthesized
byad
vancedsol–gel
method
CO2(—
),NaO
H(100
mL,
0.1N
aque
ous
solution
)/50
0W
tungsten-halog
enlamp/—
(—/—
/—/10/—),(0.15/
75.23/11
.35nm
particle/—
/sus
pende
d)
Form
icacid/150
/—�T
heyieldof
insitu
CoP
c–TiO
2is
muc
hhigher
than
thoseof
TiO
2an
dph
ysical
absorbed
CoP
c–TiO
2
�Reactionis
condu
cted
inaPy
rexglasscell
(100
mL)
withan
opticalwindo
wZ.
Zhao
etal.
(200
9)204
0.5%
CoP
c–TiO
2/—
Sol–gelmethod
CO2(saturated
),NaO
H(0.1
N,100
mL
aque
oussolution
s)/
500W
tungsten-
halog
enlamp/—
(673
/—/—
/6/—
),(0.15/
90.7/11.02
nm
particle/
0.1/su
pported)
Form
icacid/57/—
�CoP
c–TiO
2prep
ared
bysol–gelsh
ows
relatively
highph
otocatalytic
activity,
compa
redwithTiO
2synthesized
bysol–gel
method
�APy
rexcellwithan
opticalwindo
w(100
mL)
isus
edto
condu
cttheph
otocatalytic
reaction
20884 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2014
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
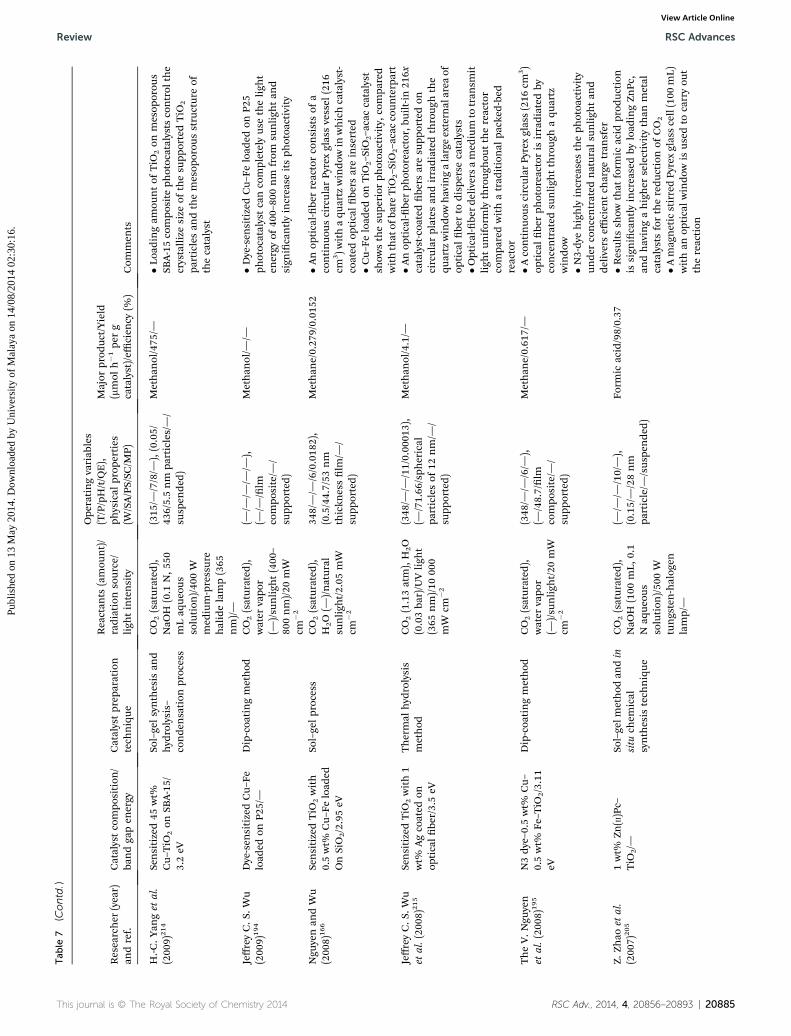
Tab
le7
(Contd.)
Researcher
(year)
andref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/Yield
(mmol
h�1pe
rg
catalyst)/effi
cien
cy(%
)Com
men
ts
H.-C
.Yan
get
al.
(200
9)214
Sensitized45
wt%
Cu–
TiO
2on
SBA-15/
3.2eV
Sol–gelsynthesis
and
hyd
rolysis–
conde
nsation
process
CO2(saturated
),NaO
H(0.1
N,5
50mLaq
ueou
ssolution
)/40
0W
med
ium-pressure
halidelamp(365
nm)/—
(315
/—/7/8/—
),(0.05/
436/5.5nm
particles/—/
susp
ende
d)
Methan
ol/475
/—�L
oadingam
ountof
TiO
2on
mesop
orou
sSB
A-15compo
site
photocatalysts
control
the
crystallize
size
ofthesu
pportedTiO
2
particlesan
dthemesop
orou
sstructureof
thecatalyst
JeffreyC.S
.Wu
(200
9)194
Dye-sen
sitizedCu–
Feload
edon
P25/—
Dip-coa
tingmethod
CO2(saturated
),water
vapo
r(—
)/su
nligh
t(400
–80
0nm)/20
mW
cm�2
(—/—
/—/—
/—),
(—/—
/lm
compo
site/—
/su
pported)
Methan
ol/—
/—�D
ye-sen
sitizedCu–
Feload
edon
P25
photocatalystcancompletelyus
etheligh
ten
ergy
of40
0–80
0nm
from
sunligh
tan
dsign
icantlyincrease
itsph
otoa
ctivity
Ngu
yenan
dWu
(200
8)166
SensitizedTiO
2with
0.5wt%
Cu–
Feload
edOnSiO2/2.95eV
Sol–gelprocess
CO2(saturated
),H
2O
(—)/natural
sunligh
t/2.05
mW
cm�2
348/—/—
/6/0.018
2),
(0.5/44.7/53
nm
thickn
esslm
/—/
supp
orted)
Methan
e/0.27
9/0.01
52�A
nop
tical-
berreactorconsistsof
acontinuo
uscircular
Pyrexglassvessel
(216
cm3)w
ithaqu
artz
windo
win
whichcatalyst-
coated
opticalbe
rsareinserted
�Cu–
Feload
edon
TiO
2–SiO
2–acac
catalyst
show
sthesu
perior
photoa
ctivity,
compa
red
withthat
ofba
reTiO
2–SiO
2–acac
coun
terpart
JeffreyC.S
.Wu
etal.(200
8)215
SensitizedTiO
2with1
wt%
Agcoated
onop
ticalbe
r/3.5eV
Thermal
hyd
rolysis
method
CO2(1.13atm),H
2O
(0.03ba
r)/UVligh
t(365
nm)/10
000
mW
cm�2
(348
/—/—
/11/0.00
013),
(—/71.66
/sph
erical
particlesof
12nm/—
/su
pported)
Methan
ol/4.1/—
�Anop
tical-
berph
otoreactor,b
uilt-in
216x
catalyst-coa
tedbe
rsaresu
pportedon
circular
plates
andirradiated
throug
hthe
quartzwindo
whavingalargeexternalarea
ofop
ticalbe
rto
disp
erse
catalysts
�Optical-be
rde
livers
amed
ium
totran
smit
ligh
tun
iformly
throug
hou
tthereactor
compa
redwithatrad
itional
packed
-bed
reactor
TheV.N
guyen
etal.(200
8)195
N3dy
e–0.5wt%
Cu–
0.5wt%
Fe–T
iO2/3.11
eV
Dip-coa
tingmethod
CO2(saturated
),water
vapo
r(—
)/su
nligh
t/20
mW
cm�2
(348
/—/—
/6/—
),(—
/48.7/lm
compo
site/—
/su
pported)
Methan
e/0.61
7/—
�Acontinuo
uscircular
Pyrexglass(216
cm3)
opticalbe
rph
otoreactor
isirradiated
byconcentrated
sunligh
tthroug
haqu
artz
windo
w�N
3-dy
ehighly
increasestheph
otoa
ctivity
unde
rconcentrated
natural
sunligh
tan
dde
livers
efficien
tch
arge
tran
sfer
Z.Zh
aoet
al.
(200
7)205
1wt%
Zn(II)Pc–
TiO
2/—
Sol–gelm
ethod
andin
situ
chem
ical
synthesis
tech
nique
CO2(saturated
),NaO
H(100
mL,
0.1
Naq
ueou
ssolution
)/50
0W
tungsten-halog
enlamp/—
(—/—
/—/10/—),
(0.15/—/28nm
particle/—
/sus
pende
d)
Form
icacid/98/0.37
�Results
show
that
form
icacid
prod
uction
issign
icantlyincreasedby
load
ingZn
Pc,
andhavingahigher
selectivitythan
metal
catalystsfortheredu
ctionof
CO2
�Amag
neticstirredPy
rexglasscell(100
mL)
withan
opticalwindo
wis
used
tocarryou
tthereaction
This journal is © The Royal Society of Chemistry 2014 RSC Adv., 2014, 4, 20856–20893 | 20885
Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
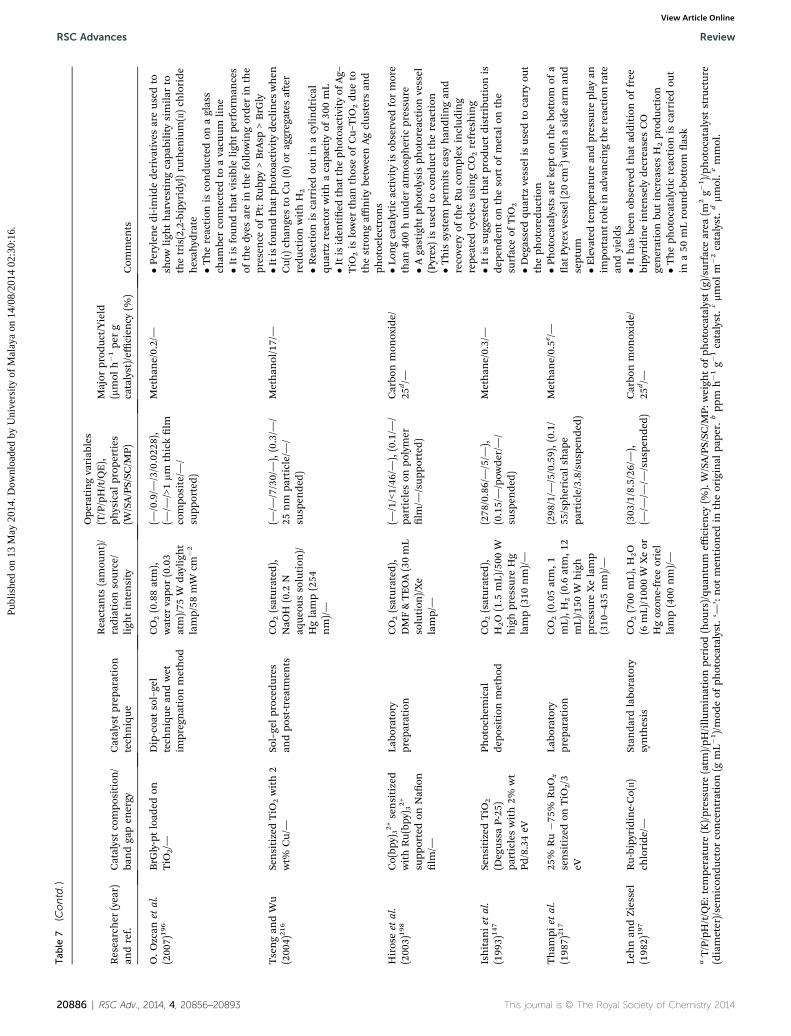
Tab
le7
(Contd.)
Researcher
(year)
andref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/Yield
(mmol
h�1pe
rg
catalyst)/effi
cien
cy(%
)Com
men
ts
O.O
zcan
etal.
(200
7)196
BrG
ly-ptload
edon
TiO
2/—
Dip-coa
tsol–gel
tech
nique
andwet
impregnationmethod
CO2(0.88atm),
water
vapo
r(0.03
atm)/75
Wda
ylight
lamp/58
mW
cm�2
(—/0.9/—
/3/0.022
8),
(—/—
/>1mm
thicklm
compo
site/—
/su
pported)
Methan
e/0.2/—
�Perylen
edi-im
idede
rivativesareus
edto
show
ligh
tharvestingcapa
bility
similar
tothetris(2,2-bipyridyl)ruthen
ium(II)ch
loride
hexah
ydrate
�Thereaction
iscondu
cted
onaglass
cham
berconnectedto
avacu
umline
�Itis
foun
dthat
visibleligh
tpe
rforman
ces
ofthedy
esarein
thefollo
wingorde
rin
the
presen
ceof
Pt:R
ubpy
>BrAsp
>BrG
lyTsengan
dWu
(200
4)216
SensitizedTiO
2with2
wt%
Cu/—
Sol–gelproced
ures
andpo
st-treatmen
tsCO2(saturated
),NaO
H(0.2
Naq
ueou
ssolution
)/Hglamp(254
nm)/—
(—/—
/7/30/—),(0.3/—
/25
nm
particle/—
/su
spen
ded)
Methan
ol/17/—
�Itisfoun
dthat
photoa
ctivityde
clines
when
Cu(
I)ch
angesto
Cu(0)or
aggregates
aer
redu
ctionwithH
2
�Reactionis
carriedou
tin
acylindrical
quartz
reactorwithacapa
city
of30
0mL
�Itisiden
tied
that
theph
otoa
ctivityof
Ag–
TiO
2is
lower
than
thoseof
Cu–
TiO
2du
eto
thestrongaffi
nitybe
tweenAgclus
ters
and
photoe
lectrons
Hiroseet
al.
(200
3)198
Co(bp
y)32+sensitized
withRu(bp
y)32+
supp
ortedon
Na
onlm
/—
Labo
ratory
prep
aration
CO2(saturated
),DMF&TEOA(30mL
solution
)/Xe
lamp/—
(—/1/<1/46
/—),(0.1/—
/pa
rticleson
polymer
lm
/—/sup
ported
)
Carbo
nmon
oxide/
25d/—
�Lon
gcatalyticactivity
isob
served
formore
than
400hun
deratmosph
eric
pressu
re�A
gastightph
otolysis
photoreactionvessel
(Pyrex)is
used
tocondu
ctthereaction
�This
system
perm
itseasy
han
dlingan
drecovery
oftheRucomplex
includ
ing
repe
ated
cycles
usingCO2refreshing
Ishitan
iet
al.
(199
3)147
SensitizedTiO
2
(DegussaP-25
)pa
rticleswith2%
wt
Pd/8.34eV
Photochem
ical
depo
sition
method
CO2(saturated
),H
2O
(1.5
mL)/500
Whighpressu
reHg
lamp(310
nm)/—
(278
/0.86/—/5/—
),(0.15/—/pow
der/—/
susp
ende
d)
Methan
e/0.3/—
�Itis
sugg
estedthat
prod
uctdistribu
tion
isde
pende
nton
thesort
ofmetal
onthe
surfaceof
TiO
2
�Degassedqu
artz
vessel
isus
edto
carryou
ttheph
otored
uction
Tham
piet
al.
(198
7)217
25%
Ru�7
5%RuO
x
sensitizedon
TiO
2/3
eV
Labo
ratory
prep
aration
CO2(0.05atm,1
mL),H
2(0.6
atm,1
2mL)/150
Whigh
pressu
reXelamp
(310
–435
nm)/—
(298
/1/—
/5/0.59),(0.1/
55/sph
erical
shap
epa
rticle/3.8/sus
pende
d)
Methan
e/0.5e/—
�Photocatalysts
areke
pton
thebo
ttom
ofa
at
Pyrexvessel
(20cm
3 )withaside
arm
and
septum
�Elevatedtempe
rature
andpressu
replay
anim
portan
trolein
advancingthereaction
rate
andyields
Lehnan
dZiessel
(198
2)197
Ru-bipyridine-Co(
II)
chloride
/—Stan
dard
labo
ratory
synthesis
CO2(700
mL),H
2O
(6mL)/100
0W
Xeor
Hgozon
e-free
oriel
lamp(400
nm)/—
(303
/1/8.5/26/—),
(—/—
/—/—
/sus
pende
d)Carbo
nmon
oxide/
25d/—
�Ithas
been
observed
that
addition
offree
bipy
ridineintensely
decreasesCO
generationbu
tincreasesH
2prod
uction
�Theph
otocatalytic
reaction
iscarriedou
tin
a50
mLroun
d-bo
ttom
ask
aT/P/pH/t/Q
E:tem
perature
(K)/pressu
re(atm
)/pH
/illu
minationpe
riod
(hou
rs)/qu
antum
efficien
cy(%
).W/SA/PS/SC
/MP:
weigh
tof
photocatalyst(g)/su
rfacearea
(m2g�
1)/ph
otocatalyststructure
(diameter)/semicon
duc
torconcentration(g
mL�
1 )/m
odeof
photocatalyst.‘—
’:not
men
tion
edin
theoriginal
pape
r.bpp
mh�1g�
1catalyst.c
mmol
m�2catalyst.d
mmol.e
mmol.
20886 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2014
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online

Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
this study, visible light performance of the dyes was in thefollowing order in presence of Pt: Rubpy > BrAsp > BrGly.
Much attention has been given to [Ru(bpy)3]2+ cation
because of its unique optical features. The combination of[Ru(bpy)3]
2+ and cobalt(II) chloride has been investigated overCO2 photoreduction, where Ru and Co complexes act as aphotosensitizer and a catalyst, respectively.197 Absorbing photonenergy, the [Ru(bpy)3]
2+ cation transforms to the triplet state,denoted by [Ru(bpy)3]
2+*. It was observed that the extra additionof bipyridine intensely reduced CO formation but raised the H2
yield. On the other hand, cobalt(II) chloride acts as an effectivemediator for both CO and H2 productions and highly endorsesCO yield. Co(bpy)3
2+ sensitized with Ru(bpy)32+ supported on a
cation exchange Naon lm has been synthesized to carry outthe photocatalytic reduction of CO2 for a period of more than400 h under atmospheric pressure.198 High stability of the metalcomplexes is obtained by the suppression of the decompositionof the Ru complex xed on the polymer surface. In this system,[Ru(bpy)3]
2+ was immobile on a cation exchange polymer and[Co(bpy)3]
2+ was mixed in a di-methylformamide (DMF) and tri-ethanol amine (TEOA) as a solvent and reducing agent,respectively. It is assumed that the photoinduced Ru complextransmits an electron to Co(II) complex, which acts as a reactionmedium of CO2, and at that time, a tertiary amine (sacricereagent) reduced the oxidized Ru(III) complex.198
2.4.2 Quantum dots (QDs)-sensitized photocatalysts. Aquantum dot-sensitized photocatalyst is one of the advancedapproaches in photochemical conversion of CO2 because of itsintrinsic optical and electrical behavior. These characteristics arestrongly inuenced by the size of quantum dots materials.Generally, chalcogenide metals (including CdSe or ZnS) are usedwith the semiconductors to facilitate the photon transformationof CO2 into hydrocarbon fuels. In Section 2.2.1, there is a briefdiscussion about the metal sulde (CdS and ZnS) semi-conductors, which also have such unique optical properties asquantum dots materials. To understand the size reliance on thephotochemical reduction of CO2, Wang et al. developed a PbSQDs-sensitized Cu–TiO2 catalyst of three different sizes of PbSQDs samples (3 nm, 4 nm and 5 nm).199 Among them, 4 nm PbSQD-sensitized Cu–TiO2 displayed the maximum CO2 trans-formation rate due to the absorption of visible spectrum (l > 610nm) and charge separation properties. It is found that small-sizeddots can facilitate the blue edge of the spectrum, while the large-sized dot is closer to the red edge spectrum. Particularly, small-sized QDs inject electrons swily to the conduction band edge ofTiO2 and large-sized QDs cover the visible region.200 Moreover,the electron transfer rate was very fast (less than 1 ns) from theexcited PbS QDs state to the TiO2 conduction band state.201
Another investigation was conducted on the CdSe QDs-sensitizedPt–TiO2 composite heterostructure, which revealed a response tothe visible light region (l > 420 nm).200
2.4.3 Phthalocyanine sensitized photocatalysts. For visiblelight irradiation, metal phthalocyanines are also applied in CO2
photoreduction, which have outstanding semi-conductivity andchemical stability.202 The CoPc–TiO2 nanocomposites are char-acterized by Zhao et al.203,204 It was observed that excited CoPcmolecules emitted the electrons to the conduction band of TiO2
This journal is © The Royal Society of Chemistry 2014
during visible light irradiation; this phenomenon thenimproved the electron–hole separation process to increase thephotocatalytic efficiency. From Table 7, it is found that theincorporation of 0.7% CoPc into TiO2 exhibited a high yield rateof formic acid (about 150 mmol g�1 h�1) than that of 0.5% CoPcaddition to TiO2. In addition, their investigation was conductedon ZnPc–TiO2 and found that ZnPc has a higher selectivity offormic acid than metal catalysts by decreasing the recombina-tion possibility of the hole–electron pairs.205 It is important thatthe excess loading of ZnPc can reduce the photocatalytic activitybecause the active TiO2 surface sites can be blocked by addi-tional metal phthalocyanines, and the optimal amount isobserved, 1.0 wt% ZnPc. However, according to the observationby Wang et al., the band gap of TiO2 can be minimized from2.95 eV (at 0.6 wt% ZnPc) to 2.79 eV by loading 5 wt% ZnPc.206
2.4.4 Other photosensitizers. Enzymes have an effectivesensitization facility to the red shi of TiO2, compared withother sensitizers.207 Woolerton et al. handled carboxydothermushydrogenoformans (Ch)-derived carbon monoxide dehydroge-nases (CODHI) enzyme to functionalize the RuP-sensitized TiO2
nanoparticles for CO2 photoexcitation.208,209 Anaerobic organ-isms, Ch contains ve forms of CODH enzymes, each havingactive sites of [Ni4Fe-4S]. The subunit of these enzymes (Ni–CODHs) possesses a buried active site that is attached to theprotein exterior by a chain of [4Fe-4S] clusters. Electrons caneasily access or leave the enzyme by distal cluster (D-cluster)that is joined between the two subunits. In this experiment,visible light-excited RuP sensitizer nanoparticles transferphotoinduced electrons to TiO2 for easing its solar light acti-vation.209,210 These electrons can easily get into the CODHenzyme through the D-cluster and also move through a second[4Fe–4S] cluster to the active site where the CO2 reduction hastaken place.
Recently, co-loaded novel metals are using the semi-conductor catalysts as photosensitizers. For example, sensitizedTiO2 lm with Au nanoparticles is modied by Hou et al. forturning CO2 into fuels.211 Here, Au nanoparticles are photoex-cited by the plasmon effect (the generation of robust electricelds by surface plasmon resonance of the Au nanoparticle),which shis the photoinduced electrons from the Au to theTiO2. Another approach of sensitized TiO2 with Cu onsupportive SiO2 is applied in terms of solar fuel productionfrom CO2.77,212
2.5 Flexible substrate based photocatalysts
In photochemical reactions, the involvement of exiblesubstrates have generated interest because they deliver a highsurface area for catalysts on the lm of substrates such as pol-yethyleneterphthalate (PET), polyethylenenaphtalate (PEN),polyethylene (PE) and polypropylene (PP).218 However, thedegradation of polymer substrates by solar irradiation dependson the operating period of atmospheric exposure.219 This pho-todegradation is carried out by a combination of photolysis andphotocatalytic oxidation.218 The formation of volatile productsdue to photodegradation is another limitation in the case ofpolymeric substrates.220 Table 8 represents the full information
RSC Adv., 2014, 4, 20856–20893 | 20887

Tab
le8
Advance
sin
flexible
substrate
catalystsforCO
2photoreductiona
Researcher
(year)
andref.
Catalystcompo
sition
/ba
ndga
pen
ergy
Catalystprep
aration
tech
nique
Reactan
ts(amou
nt)/
radiationsource/
ligh
tintensity
Ope
ratingvariab
les
(T/P/pH/t/Q
E),
physical
prop
erties
(W/SA/PS/SC
/MP)
Major
prod
uct/yield
(mmol
h�1pe
rg
catalyst)/selectivity(%
)Com
men
ts
W.K
imet
al.
(201
2)223
Na
onlayeron
1.0wt
%Pd
-dep
osited
TiO
2/—
Labo
ratory
synthesis
CO2(saturated
),H
2O
(—)/UVligh
t(>30
0nm)/—
(—/—
/3/5/—
),(—
/—/nan
oparticle/
1.5�
10�6/
susp
ende
d)
Methan
e/7.6b/—
�Na
onlayeren
han
cesproton
-cou
pled
,multiple-electron
tran
sfer
reaction
s,stab
ilizes
interm
ediatesan
dinhibitsthere-
oxidationof
theCO2redu
ctionprod
ucts
byincreasingthelocalproton
activity
within
thelayer
�Palladium
isselected
toretard
the
recombinationof
Hatom
sto
minim
izethe
prod
uction
ofH
2
J.Jensenet
al.
(201
1)218
TiO
2–P
ET/—
Spraycoating
CO2(25mol),H
2(75
mol)/Osram
ligh
tbu
lb/120
mW
cm�2
(308
/—/—
/230
/—),
(—/—
/lm
with13
0mm
thickn
ess/—/
supp
orted)
Carbo
nmon
oxide/
0.00
13/—
�Polym
ersu
bstrates
areap
pliedin
this
stud
ywithhigh-activearea
of10
0cm
2en
able
toachieve
catalyst-covered
lm
within
ash
orttimean
dat
areason
able
prod
uction
cost
�UVde
grad
ationof
polymer
subs
trates
(photocatalytic
oxidation)canbe
supp
ressed
,but
not
avoide
d,by
using
oxygen
-freecondition
sT.A
raiet
al.
(201
1)224
Cu 2Zn
SnS 4
mod
ied
byruthen
ium
complex
polymer/1.5
eV
Chem
ical
polymerization
method
CO2(saturated
),H
2O
(—)/visible
ligh
tirradiation
(200
–400
nm)/—
(—/—
/—/3/—
),(—
/—/photocathod
e/—/sup
ported
)
Form
icacid/0.49c/>
80�S
ulde
semicon
ductor
that
show
sanarrow
bandga
pwithhighselectivityin
photoe
lectroch
emical
CO2redu
ction
�Theinsertionof
Seinto
thecatalyst
enhan
cesholemob
ilityan
dincreasesthe
photocurrent
Hiroseet
al.
(200
3)198
Co(bp
y)32
+with
Ru(bp
y)32+su
pported
onNa
onlm
/—
Labo
ratory
prep
aration
CO2(saturated
),DMFwithTEOA(30
mLsolution
)/Xe
lamp/—
(—/1/<
1/46
/—),(1/—
/pa
rticleson
polymer
lm
/—/sup
ported
)
Carbo
nmon
oxide/
25b/—
�Catalytic
activity
show
smorethan
400h
unde
ratmosph
eric
pressu
re�P
hotoreactionis
carriedou
tin
aga
stight
photolysis
photoreactionvessel
(Pyrex)
�This
system
allowseasy
han
dlingan
drecovery
oftheRucomplex
withrepe
ated
cycles,u
singCO2refreshing
aT/P/pH/t/Q
E:tem
perature
(K)/pressu
re(atm
)/pH
/illu
minationpe
riod
(hou
rs)/qu
antum
efficien
cy(%
).W/SA/PS/SC
/MP:
weigh
tof
photocatalyst(g)/su
rfacearea
(m2g�
1)/ph
otocatalyststructure
(diameter)/semicon
duc
torconcentration(g
mL�
1 )/m
odeof
photocatalyst.‘—
’:not
men
tion
edin
theoriginal
pape
r.bmmol.c
mM.
20888 | RSC Adv., 2014, 4, 20856–20893 This journal is © The Royal Society of Chemistry 2014
RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online

Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
of exible substrate based photocatalysts, which are gatheredfrom important investigations related to CO2 photoconversion.
Some aromatic polyesters such as PET, PEN and similarexible substrates act in different ways under light irradiation,compared with olens (PE and PP). Aromatic polyesters are ableto absorb light with a long wavelength (<330 nm) due to thearomatic rings of carbonyl groups.220 Another important factoris the oxygen diffusion constant of exible substrates. Higheroxygen diffusion constants may lead to oxidation in all parts ofthe surface.221 Comparatively, polymer degradation is lesser onthe surface of the PET because of its restriction to diffuseoxygen, whereas the diffusion is higher in the PE.218,221,222
Use of a Naon (peruorinated polymers with sulfonategroups) thin lm on Pd-loaded TiO2 nanoparticles was intro-duced by Kim et al. due to its stability against photocatalyticoxidation, and hence increases the photochemical conversionof CO2 under visible light irradiation.223 To provide multipleelectrons, the Naon layer can increase local proton activityinside the layer. It also helps to stabilize the intermediates andprevent the re-oxidation of the CO2 reduction yields because itinhibits direct interaction between products and surfaces of thecatalysts. Another investigation shows that Naon lm acts as agood supportive material for catalysts due to cation exchangeability.198 This Naon-supported Co(bpy)3
2+ and Ru(bpy)32+
system can increase the catalytic activity to a maximum of 400 hin atmospheric condition because of obstruction to the break-down of [Ru(bpy)3]
2+ complexes that are trapped on the cationexchange polymer surface, and hence the separation of ligandsis minimized. Low-band gap (1.5 eV) Cu2ZnSnS4 modied by theruthenium complex polymer is a new addition to determine thephotochemical performance of CO2 reduction under the visiblelight region, which shows high selectivity of the formate(>80%).224
3. Conclusions
The transformation of CO2 towards hydrocarbon fuels is asubstitute approach to regulate both fuel crisis and globalwarming. Various inquiries have been undertaken to evaluatethe photocatalytic performance over CO2 transformation intohydrocarbon fuels. This technology was found to be morepromising, compared with other approaches such as thermo-chemical, electrochemical, and biological transformationtechnologies. In this paper, a general overview is emphasized onthe behavior of different types of photocatalysts with respect toCO2 transformation into fuel. In various investigations, it wasobserved that most of the studies were carried out in the pres-ence of articial light sources. Recently, researchers havefocused on direct solar fuel production by CO2 conversion. Forsuch a phenomenon, the selective catalysts must have thedesired band gap energy for visible light response. Previously,unmodied oxide and non-oxide semiconductors are widelyused with or without supportive materials in this technology.However, some of them have high band gap energy and lowconversion yields. To facilitate, some techniques have beenfollowed to minimize band gap energy by doping the semi-conductors with foreign elements like novel metals, transition
This journal is © The Royal Society of Chemistry 2014
metals, co-metals, and non-metals. It also inuences the chargetransfer rate and reduces the electron–hole recombination.Further, different substrate materials were used to achieve highsurface area and the catalysts' immobilization during the pho-tocatalytic irradiation. Recently, different types of sensitizationtechniques are incorporated to improve the photocatalyticperformance under visible light irradiation like dye sensitiza-tion, enzyme-based sensitization, QDs sensitization, andphthalocyanine sensitization. Both doped and sensitizedsemiconductors were employed for higher conversion. Theselectivity of products not only depends on the catalysts'compositions but also on the choice of reductant and thesolvent used in the catalytic system. Besides water, organicsacricial reducing reagents are the most common reductantsused in the photochemical CO2 conversion process. Althoughwater is available and affordable, there are some limitations likelow CO2 solubility, resulting in water splitting rather than CO2
reduction. Instead, methane and hydrogen were used assignicant reductants in many investigations reported in thisreview. Other factors are denitely considerable for evaluatingphotocatalytic performance on CO2 reduction such as catalysts'dosage, reactants' ratio, reaction temperature, time, systempressure, pH, light intensity and wavelength. A clear idea isessential for understanding the reaction mechanisms togetherwith charge carrier dynamics within the catalysts and at theirinterfaces with the metal co-catalysts. From this review, it isclear that the existing techniques are insufficient to make themapplicable for the industrial production of fuels. As a result, theselection of solar-driven photocatalysts is essential for indus-trial solar fuel harvesting from CO2.
Acknowledgements
The authors acknowledge nancial support from project RP015/2012D and D000011-16001, Center of Research Grant Manage-ment, University of Malaya, 50603 Kuala Lumpur, Malaysia.
References
1 S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACSNano, 2010, 4, 1259–1278.
2 L. T. H. Nam, T. Q. Vinh, N. T. T. Loan, V. D. S. Tho,X.-Y. Yang and B.-L. Su, Fuel, 2011, 90, 1069–1075.
3 H. Balat and E. Kırtay, Int. J. Hydrogen Energy, 2010, 35,7416–7426.
4 T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath,T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502.
5 Independent Statistics and Analysis, U.S. EnergyInformation Administration, 2012, retrieved from http://www.eia.gov/cfapps/ipdbproject/IEDIndex3.cfm?tid¼44&pid¼44&aid¼2.
6 L. Jaegle, L. Steinberger, R. V. Martin and K. Chance,Faraday Discuss., 2005, 130, 407–423.
7 BP: Statistical Review of World Energy, London, 2013,retrieved from http://www.bp.com/content/dam/bp/pdf/statistical-review/statistical_ review_of_world_energy_2013.pdf.
RSC Adv., 2014, 4, 20856–20893 | 20889

RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
8 G. Shaullah, M. Amanullah, A. Shawkat Ali, D. Jarvis andP. Wolfs, Renewable Energy, 2012, 39, 183–197.
9 N. Panwar, S. Kaushik and S. Kothari, Renewable SustainableEnergy Rev., 2011, 15, 1513–1524.
10 S. Mekhilef, S. Siga and R. Saidur, Renewable SustainableEnergy Rev., 2011, 15, 1937–1949.
11 S. Dutta, RSC Adv., 2012, 2, 12575–12593.12 G. Centi and R. A. van Santen, Catalysis for renewables,
Wiley, Weinheim, 2008.13 M. Dittmar, Energy, 2012, 37, 35–40.14 N. H. Afgan, Thermal Science, 2013, 17, 305–321.15 H. Li, Y. Dai, M. Kohler and R. Wang, Energy Convers.
Manage., 2013, 67, 309–317.16 T. C. Corbiere, D. Ressnig, C. Giordano and M. Antonietti,
RSC Adv., 2013, 3, 15337–15343.17 N. Y. Mansouri, R. J. Crookes and T. Korakianitis, Energy
Policy, 2013, 63, 681–695.18 M. Sun, W. Fu, Q. Li, G. Yin, K. Chi, J. Ma, L. Yang, Y. Mu,
Y. Chen and S. Su, RSC Adv., 2014, 4, 7178–7184.19 M. Jamel, A. Abd Rahman and A. Shamsuddin, Renewable
Sustainable Energy Rev., 2013, 20, 71–81.20 J. Lee, H. Back, J. Kong, H. Kang, S. Suhee, S. Hongsuk,
S.-O. Kang and K. Lee, Energy Environ. Sci., 2013, 6, 1152–1157.
21 V. Kaltenhauser, T. Rath, M. Edler, A. Reichmann andG. Trimmel, RSC Adv., 2013, 3, 18643–18650.
22 C. Liu, N. P. Dasgupta and P. Yang, Chem. Mater., 2014, 26,415–422.
23 K. Sivula, J. Phys. Chem. Lett., 2013, 4, 1624–1633.24 Y.-K. Hsu, C.-H. Yu, Y.-C. Chen and Y.-G. Lin, RSC Adv.,
2012, 2, 12455–12459.25 A. A. Lacis, G. A. Schmidt, D. Rind and R. A. Ruedy, Science,
2010, 330, 356–359.26 S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang,
A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3,22739–22773.
27 G. Xiao, P. Xiao, S. Lee and P. A. Webley, RSC Adv., 2012, 2,5291–5297.
28 International Energy Agency Annual Report, 2012, retrievedfrom http://www.iea.org/publications/freepublications/publication/English.pdf.
29 I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405.30 U.S. Department of Energy (DOE), 2013, retrieved from
www.doe.gov/.31 W.-J. Ong, M. M. Gui, S.-P. Chai and A. R. Mohamed, RSC
Adv., 2013, 3, 4505–4509.32 K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2,
3165–3172.33 X.-J. Lv, W.-F. Fu, C.-Y. Hu, Y. Chen and W.-B. Zhou, RSC
Adv., 2013, 3, 1753–1757.34 T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007,
107, 2365–2387.35 S. N. Habisreutinger, L. Schmidt-Mende and
J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408.
36 Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414,625–627.
20890 | RSC Adv., 2014, 4, 20856–20893
37 M. Ni, M. K. Leung, D. Y. Leung and K. Sumathy, RenewableSustainable Energy Rev., 2007, 11, 401–425.
38 F. Han, V. S. R. Kambala, M. Srinivasan, D. Rajarathnamand R. Naidu, Appl. Catal., A, 2009, 359, 25–40.
39 D. Bahnemann, Sol. Energy, 2004, 77, 445–459.40 R. Benedix, F. Dehn, J. Quaas and M. Orgass, Lacer, 2000, 5,
157–168.41 A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem.
Photobiol., C, 2000, 1, 1–21.42 T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature,
1979, 277, 637–638.43 S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42,
4908–4911.44 H. Tada, Q. Jin, H. Nishijima, H. Yamamoto, M. Fujishima,
S. i. Okuoka, T. Hattori, Y. Sumida and H. Kobayashi,Angew. Chem., Int. Ed., 2011, 123, 3563–3567.
45 M. Tahir and N. S. Amin, Appl. Catal., B, 2013, 142–143,512–522.
46 R. Van Grieken, J. Aguado, M. Lopez-Munoz andJ. Marugan, J. Photochem. Photobiol., A, 2002, 148, 315–322.
47 P. Bouras, E. Stathatos and P. Lianos, Appl. Catal., B, 2007,73, 51–59.
48 K. Kocı, L. Obalova, L. Matejova, D. Placha, Z. Lacny,J. Jirkovsky and O. Solcova, Appl. Catal., B, 2009, 89, 494–502.
49 K. Kocı, L. Obalova and Z. Lacny, Chem. Pap., 2008, 62, 1–9.50 S. S. Tan, L. Zou and E. Hu, Catal. Today, 2006, 115, 269–
273.51 G. P. Fotou and S. E. Pratsinis, Chem. Eng. Commun., 1996,
151, 251–269.52 T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and
O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600.
53 N. M. Dimitrijevic, B. K. Vijayan, O. G. Poluektov, T. Rajh,K. A. Gray, H. He and P. Zapol, J. Am. Chem. Soc., 2011,133, 3964–3971.
54 S. Kaneco, H. Kurimoto, Y. Shimizu, K. Ohta and T. Mizuno,Energy, 1999, 24, 21–30.
55 S. Kaneco, H. Kurimoto, K. Ohta, T. Mizuno and A. Saji,J. Photochem. Photobiol., A, 1997, 109, 59–63.
56 G. Dey, A. Belapurkar and K. Kishore, J. Photochem.Photobiol., A, 2004, 163, 503–508.
57 G. Li, L. Li, J. Boerio-Goates and B. F. Woodeld, J. Am.Chem. Soc., 2005, 127, 8659–8666.
58 G. Xi, S. Ouyang, P. Li, J. Ye, Q. Ma, N. Su, H. Bai andC. Wang, Angew. Chem., Int. Ed., 2012, 51, 2395–2399.
59 L. Yuliati, H. Itoh and H. Yoshida, Chem. Phys. Lett., 2008,452, 178–182.
60 Y. Kohno, T. Tanaka, T. Funabiki and S. Yoshida, Phys.Chem. Chem. Phys., 2000, 2, 5302–5307.
61 K. Teramura, T. Tanaka, H. Ishikawa, Y. Kohno andT. Funabiki, J. Phys. Chem. B, 2004, 108, 346–354.
62 C.-C. Lo, C.-H. Hung, C.-S. Yuan and J.-F. Wu, Sol. EnergyMater. Sol. Cells, 2007, 91, 1765–1774.
63 Y. Liu, B. Huang, Y. Dai, X. Zhang, X. Qin, M. Jiang andM.-H. Whangbo, Catal. Commun., 2009, 11, 210–213.
64 J. Yu and A. Kudo, Adv. Funct. Mater., 2006, 16, 2163–2169.
This journal is © The Royal Society of Chemistry 2014

Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
65 Y. Zhou, Z. Tian, Z. Zhao, Q. Liu, J. Kou, X. Chen, J. Gao,S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2011, 3,3594–3601.
66 Q. Liu, Y. Zhou, J. Kou, X. Chen, Z. Tian, J. Gao, S. Yan andZ. Zou, J. Am. Chem. Soc., 2010, 132, 14385–14387.
67 X. Li, H. Pan, W. Li and Z. Zhuang, Appl. Catal., A, 2012, 413,103–108.
68 M. Stock and S. Dunn, Ferroelectrics, 2011, 419, 9–13.69 J. W. Lekse, M. K. Underwood, J. P. Lewis and C. Matranga,
J. Phys. Chem. C, 2012, 116, 1865–1872.70 X.-H. Xia, Z.-J. Jia, Y. Yu, Y. Liang, Z. Wang and L.-L. Ma,
Carbon, 2007, 45, 717–721.71 A. Yahaya, M. Gondal and A. Hameed, Chem. Phys. Lett.,
2004, 400, 206–212.72 M. Anpo, H. Yamashita, Y. Ichihashi and S. Ehara,
J. Electroanal. Chem., 1995, 396, 21–26.73 F. Solymosi and I. Tombacz, Catal. Lett., 1994, 27, 61–65.74 H. Juntgen, Fuel, 1986, 65, 1436–1446.75 M. Kruk, M. Jaroniec, C. H. Ko and R. Ryoo, Chem. Mater.,
2000, 12, 1961–1968.76 M.-J. Lopez-Munoz, R. v. Grieken, J. Aguado and
J. Marugan, Catal. Today, 2005, 101, 307–314.77 Y. Li, W.-N. Wang, Z. Zhan, M.-H. Woo, C.-Y. Wu and
P. Biswas, Appl. Catal., B, 2010, 100, 386–392.78 B.-J. Liu, T. Torimoto and H. Yoneyama, J. Photochem.
Photobiol., A, 1998, 115, 227–230.79 N. Gokon, N. Hasegawa, H. Kaneko, H. Aoki, Y. Tamaura
and M. Kitamura, Sol. Energy Mater. Sol. Cells, 2003, 80,335–341.
80 J. Papp, S. Soled, K. Dwight and A. Wold, Chem. Mater.,1994, 6, 496–500.
81 M. Subrahmanyam, S. Kaneco and N. Alonso-Vante, Appl.Catal., B, 1999, 23, 169–174.
82 M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii andM. Honda, J. Phys. Chem. B, 1997, 101, 2632–2636.
83 Q. D. Truong, J.-Y. Liu, C.-C. Chung and Y.-C. Ling, Catal.Commun., 2012, 19, 85–89.
84 R. A. H. W. A. Deer and J. Zussman, An introduction to therock-forming minerals, Harlow, Longman, 2nd edn,1992.
85 K. Kocı, V. Matejka, P. Kovar, Z. Lacny and L. Obalova,Catal. Today, 2011, 161, 105–109.
86 M. A. Asi, C. He, M. Su, D. Xia, L. Lin, H. Deng, Y. Xiong,R. Qiu and X.-z. Li, Catal. Today, 2011, 175, 256–263.
87 J. Popic, M. Avramov-Ivic and N. Vukovic, J. Electroanal.Chem., 1997, 421, 105–110.
88 J. Qu, X. Zhang, Y. Wang and C. Xie, Electrochim. Acta, 2005,50, 3576–3580.
89 T.-f. Xie, D.-j. Wang, L.-j. Zhu, T.-j. Li and Y.-j. Xu, Mater.Chem. Phys., 2001, 70, 103–106.
90 K. Ogura, M. Kawano, J. Yano and Y. Sakata, J. Photochem.Photobiol., A, 1992, 66, 91–97.
91 H. W. Nasution, E. Purnama, S. Kosela and J. Gunlazuardi,Catal. Commun., 2005, 6, 313–319.
92 S. Qin, F. Xin, Y. Liu, X. Yin and W. Ma, J. Colloid InterfaceSci., 2011, 356, 257–261.
This journal is © The Royal Society of Chemistry 2014
93 P.-Y. Liou, S.-C. Chen, J. C. Wu, D. Liu, S. Mackintosh,M. Maroto-Valer and R. Linforth, Energy Environ. Sci.,2011, 4, 1487–1494.
94 P.-W. Pan and Y.-W. Chen, Catal. Commun., 2007, 8, 1546–1549.
95 D.-S. Lee, H.-J. Chen and Y.-W. Chen, J. Phys. Chem. Solids,2012, 73, 661–669.
96 P. Usubharatana, D. McMartin, A. Veawab andP. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45,2558–2568.
97 W. Wang and Y. Ku, J. Photochem. Photobiol., A, 2003, 159,47–59.
98 S. Yan, L. Wan, Z. Li and Z. Zou, Chem. Commun., 2011, 47,5632–5634.
99 Q. Liu, Y. Zhou, Z. Tian, X. Chen, J. Gao and Z. Zou, J. Mater.Chem., 2012, 22, 2033–2038.
100 A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278.101 K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–
7861.102 X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010,
110, 6503–6570.103 H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and
J. Ye, Adv. Mater., 2012, 24, 229–251.104 K. Zhang and L. Guo, Catal. Sci. Technol., 2013, 3, 1672–
1690.105 I. Tsuji, H. Kato and A. Kudo, Chem. Mater., 2006, 18, 1969–
1975.106 H. Fujiwara, H. Hosokawa, K. Murakoshi, Y. Wada and
S. Yanagida, Langmuir, 1998, 14, 5154–5159.107 O. Kozak, P. Praus, K. Kocı and M. Klementova, J. Colloid
Interface Sci., 2010, 352, 244–251.108 K. Kocı, M. Reli, O. Kozak, Z. Lacny, D. Placha, P. Praus and
L. Obalova, Catal. Today, 2011, 176, 212–214.109 P. Johne and H. Kisch, J. Photochem. Photobiol., A, 1997,
111, 223–228.110 P. Praus, O. Kozak, K. Kocı, A. Panacek and R. Dvorsky,
J. Colloid Interface Sci., 2011, 360, 574–579.111 B. R. Eggins, P. K. Robertson, E. P. Murphy, E. Woods and
J. T. Irvine, J. Photochem. Photobiol., A, 1998, 118, 31–40.112 B.-J. Liu, T. Torimoto and H. Yoneyama, J. Photochem.
Photobiol., A, 1998, 113, 93–97.113 H. Fujiwara, H. Hosokawa, K. Murakoshi, Y. Wada,
S. Yanagida, T. Okada and H. Kobayashi, J. Phys. Chem. B,1997, 101, 8270–8278.
114 C. Amatore and J. M. Saveant, J. Am. Chem. Soc., 1981, 103,5021–5023.
115 J. Yuan, C. Zhang, C. Li, M. Chen and W. Shangguan, MRSProceedings, Cambridge Univ. Press, 2011, p. 1326.
116 X. Li, J. Chen, H. Li, J. Li, Y. Xu, Y. Liu and J. Zhou, J. Nat.Gas Chem., 2011, 20, 413–417.
117 H. Inoue, H. Moriwaki, K. Maeda and H. Yoneyama,J. Photochem. Photobiol., A, 1995, 86, 191–196.
118 X. V. Zhang, S. T. Martin, C. M. Friend, M. A. Schoonen andH. D. Holland, J. Am. Chem. Soc., 2004, 126, 11247–11253.
119 M. Halmann, Nature, 1978, 275, 115–116.120 S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Chem. Eng.
J., 2006, 116, 227–231.
RSC Adv., 2014, 4, 20856–20893 | 20891

RSC Advances Review
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
121 E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am.Chem. Soc., 2008, 130, 6342–6344.
122 K. Hirota, D. A. Tryk, T. Yamamoto, K. Hashimoto,M. Okawa and A. Fujishima, J. Phys. Chem. B, 1998, 102,9834–9843.
123 B. A. Parkinson and P. F. Weaver, Nature, 1984, 309, 148–149.
124 B. Aurian-Blajeni, M. Halmann and J. Manassen, Sol. EnergyMater., 1983, 8, 425–440.
125 R. Hinogami, Y. Nakamura, S. Yae and Y. Nakato, J. Phys.Chem. B, 1998, 102, 974–980.
126 R. L. Cook, R. C. MacDuff and A. F. Sammells, J.Electrochem. Soc., 1988, 135, 3069–3070.
127 Y. Shioya, K. Ikeue, M. Ogawa and M. Anpo, Appl. Catal., A,2003, 254, 251–259.
128 K. Ikeue, S. Nozaki, M. Ogawa and M. Anpo, Catal. Today,2002, 74, 241–248.
129 K. Ikeue, H. Yamashita, M. Anpo and T. Takewaki, J. Phys.Chem. B, 2001, 105, 8350–8355.
130 N. Ulagappan and H. Frei, J. Phys. Chem. A, 2000, 104, 7834–7839.
131 W.-J. Yin, S. Chen, J.-H. Yang, X.-G. Gong, Y. Yan andS.-H. Wei, Appl. Phys. Lett., 2010, 96, 221901–221903.
132 I. Nakamura, N. Negishi, S. Kutsuna, T. Ihara,S. Sugihara and K. Takeuchi, J. Mol. Catal. A: Chem.,2000, 161, 205–212.
133 H. Irie, Y. Watanabe and K. Hashimoto, Chem. Lett., 2003,32, 772–773.
134 H. Weng, X. Yang, J. Dong, H. Mizuseki, M. Kawasaki andY. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys.,2004, 69, 125219.
135 Y. Gai, J. Li, S.-S. Li, J.-B. Xia and S.-H. Wei, Phys. Rev. Lett.,2009, 102, 036402.
136 R. Long and N. J. English, Appl. Phys. Lett., 2009, 94,132102–132103.
137 E. Fazio, P. Calandra, V. T. Liveri, N. Santo and S. Trusso,Colloids Surf., A, 2011, 392, 171–177.
138 H. W. N. Slamet, E. Purnama, K. Riyani and J. Gunlazuardi,World Appl. Sci. J., 2009, 6, 112–122.
139 K. Sayama and H. Arakawa, J. Phys. Chem., 1993, 97, 531–533.
140 M. Antoniadou, D. I. Kondarides and P. Lianos, Catal. Lett.,2009, 129, 344–349.
141 V. M. Daskalaki and D. I. Kondarides, Catal. Today, 2009,144, 75–80.
142 C. Ampelli, G. Centi, R. Passalacqua and S. Perathoner,Energy Environ. Sci., 2010, 3, 292–301.
143 M. Qamar, Int. J. Nanosci., 2010, 9, 579–583.144 H. Shi and Z. Zou, J. Phys. Chem. Solids, 2012, 73, 788–792.145 P. Li, S. Ouyang, G. Xi, T. Kako and J. Ye, J. Phys. Chem. C,
2012, 116, 7621–7628.146 N. Zhang, S. Ouyang, P. Li, Y. Zhang, G. Xi, T. Kako and
J. Ye, Chem. Commun., 2011, 47, 2041–2043.147 O. Ishitani, C. Inoue, Y. Suzuki and T. Ibusuki, J.
Photochem. Photobiol., A, 1993, 72, 269–271.148 F. Solymosi, I. Tombacz and J. Koszta, J. Catal., 1985, 95,
578–586.
20892 | RSC Adv., 2014, 4, 20856–20893
149 I. Tseng, J. Wu and H.-Y. Chou, J. Catal., 2004, 221, 432–440.
150 I. Tseng, W.-C. Chang and J. Wu, Appl. Catal., B, 2002, 37,37–48.
151 K. Adachi, K. Ohta and T. Mizuno, Sol. Energy, 1994, 53,187–190.
152 K. Kocı, K. Mateju, L. Obalova, S. Krejcıkova, Z. Lacny,D. Placha, L. Capek, A. Hospodkova and O. Solcova, Appl.Catal., B, 2010, 96, 239–244.
153 S. Krejcıkova, L. Matejova, K. Kocı, L. Obalova, Z. Matej,L. Capek and O. Solcova, Appl. Catal., B, 2012, 111, 119–125.
154 K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am.Chem. Soc., 2011, 133, 20863–20868.
155 P. Ji, M. Takeuchi, T.-M. Cuong, J. Zhang, M. Matsuoka andM. Anpo, Res. Chem. Intermed., 2010, 36, 327–347.
156 M. Alvaro, C. Aprile, B. Ferrer, F. Sastre and H. Garcıa,Dalton Trans., 2009, 7437–7444.
157 C. Aprile, M. A. Herranz, E. Carbonell, H. Garcia andN. Martın, Dalton Trans., 2009, 134–139.
158 G. Guan, T. Kida and A. Yoshida, Appl. Catal., B, 2003, 41,387–396.
159 A. Nishimura, G. Mitsui, M. Hirota and E. Hu, Int. J. Chem.Eng., 2010, 2010, 1–9.
160 R. Dholam, N. Patel, M. Adami and A. Miotello, Int.J. Hydrogen Energy, 2009, 34, 5337–5346.
161 L. Pan, J.-J. Zou, X. Zhang and L. Wang, Ind. Eng. Chem. Res.,2010, 49, 8526–8531.
162 C.-C. Yang, J. Vernimmen, V. Meynen, P. Cool and G. Mul,J. Catal., 2011, 284, 1–8.
163 J.-S. Hwang, J.-S. Chang, S.-E. Park, K. Ikeue and M. Anpo,Top. Catal., 2005, 35, 311–319.
164 S. S. Srinivasan, J. Wade, E. K. Stefanakos and Y. Goswami,J. Alloys Compd., 2006, 424, 322–326.
165 J. Wu, Appl. Catal., A, 2008, 335, 112–120.166 J. Wu, Sol. Energy Mater. Sol. Cells, 2008, 92, 864–872.167 Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and
Y. Wang, Angew. Chem., Int. Ed., 2013, 125, 5888–5891.168 O. K. Varghese, M. Paulose, T. J. LaTempa and C. A. Grimes,
Nano Lett., 2009, 9, 731–737.169 B. D. Mankidy, B. Joseph and V. K. Gupta, Nanotechnology,
2013, 24, 405402.170 C. Wang, Z. Xie, K. E. deKra andW. Lin, J. Am. Chem. Soc.,
2011, 133, 13445–13454.171 M. Anpo, H. Yamashita, K. Ikeue, Y. Fujii, S. G. Zhang,
Y. Ichihashi, D. R. Park, Y. Suzuki, K. Koyano andT. Tatsumi, Catal. Today, 1998, 44, 327–332.
172 J. Hemminger, R. Carr and G. Somorjai, Chem. Phys. Lett.,1978, 57, 100–104.
173 M. Hamadanian, A. Reisi-Vanani and A. Majedi, Mater.Chem. Phys., 2009, 116, 376–382.
174 C. Wen, Y.-J. Zhu, T. Kanbara, H.-Z. Zhu and C.-F. Xiao,Desalination, 2009, 249, 621–625.
175 L. Zhou, J. Deng, Y. Zhao, W. Liu, L. An and F. Chen,Mater.Chem. Phys., 2009, 117, 522–527.
176 S. In, A. Orlov, R. Berg, F. Garcıa, S. Pedrosa-Jimenez,M. S. Tikhov, D. S. Wright and R. M. Lambert, J. Am.Chem. Soc., 2007, 129, 13790–13791.
This journal is © The Royal Society of Chemistry 2014

Review RSC Advances
Publ
ishe
d on
13
May
201
4. D
ownl
oade
d by
Uni
vers
ity o
f M
alay
a on
14/
08/2
014
02:3
0:16
. View Article Online
177 S. U. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002,297, 2243–2245.
178 J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater.,2002, 14, 3808–3816.
179 G. Wu, J. Wen, J. Wang, D. F. Thomas and A. Chen, Mater.Lett., 2010, 64, 1728–1731.
180 M. Pelaez, A. A. de la Cruz, E. Stathatos, P. Falaras andD. D. Dionysiou, Catal. Today, 2009, 144, 19–25.
181 M. Tahir and N. S. Amin, Energy Convers. Manage., 2013, 76,194–214.
182 T. M. Suzuki, H. Tanaka, T. Morikawa, M. Iwaki, S. Sato,S. Saeki, M. Inoue, T. Kajino and T. Motohiro, Chem.Commun., 2011, 47, 8673–8675.
183 S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro,Angew. Chem., Int. Ed., 2010, 49, 5101–5105.
184 C.-W. Tsai, H. M. Chen, R.-S. Liu, K. Asakura andT.-S. Chan, J. Phys. Chem. C, 2011, 115, 10180–10186.
185 L. M. Xue, F. H. Zhang, H. J. Fan and X. F. Bai, Adv. Mater.Res., 2011, 183, 1842–1846.
186 H. Irie, S. Washizuka and K. Hashimoto, Thin Solid Films,2006, 510, 21–25.
187 T. Ohno, M. Akiyoshi, T. Umebayashi, K. Asai, T. Mitsui andM. Matsumura, Appl. Catal., A, 2004, 265, 115–121.
188 D. Li, H. Haneda, S. Hishita, N. Ohashi andN. K. Labhsetwar, J. Fluorine Chem., 2005, 126, 69–77.
189 Z. He, L. Xie, S. Song, C. Wang, J. Tu, F. Hong, Q. Liu,J. Chen and X. Xu, J. Mol. Catal. A: Chem., 2010, 319, 78–84.
190 Q. Zhang, Y. Li, E. A. Ackerman, M. Gajdardziska-Josifovskaand H. Li, Appl. Catal., A, 2011, 400, 195–202.
191 V. Heleg-Shabtai, E. Zahavy and I. Willner, Energy Convers.Manage., 1995, 36, 609–612.
192 Z. Li, Y. Zhou, J. Zhang, W. Tu, Q. Liu, T. Yu and Z. Zou,Cryst. Growth Des., 2012, 12, 1476–1481.
193 J. Pan, X. Wu, L. Wang, G. Liu, G. Q. M. Lu andH.-M. Cheng, Chem. Commun., 2011, 47, 8361–8363.
194 J. C. Wu, Catal. Surv. Asia, 2009, 13, 30–40.195 J. Wu and C.-H. Chiou, Catal. Commun., 2008, 9, 2073–2076.196 O. Ozcan, F. Yukruk, E. Akkaya and D. Uner, Top. Catal.,
2007, 44, 523–528.197 J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982,
79, 701–704.198 T. Hirose, Y. Maeno and Y. Himeda, J. Mol. Catal. A: Chem.,
2003, 193, 27–32.199 C. Wang, R. L. Thompson, P. Ohodnicki, J. Baltrus and
C. Matranga, J. Mater. Chem., 2011, 21, 13452–13457.200 C. Wang, R. L. Thompson, J. Baltrus and C. Matranga, J.
Phys. Chem. Lett., 2009, 1, 48–53.201 W. A. Tisdale, K. J. Williams, B. A. Timp, D. J. Norris,
E. S. Aydil and X.-Y. Zhu, Science, 2010, 328, 1543–1547.
This journal is © The Royal Society of Chemistry 2014
202 R. Seoudi, G. El-Bahy and Z. El Sayed, Opt. Mater., 2006, 29,304–312.
203 Z. Zhao, J. Fan, M. Xie and Z. Wang, J. Cleaner Prod., 2009,17, 1025–1029.
204 Z. Zhao, J. Fan, S. Liu and Z. Wang, Chem. Eng. J., 2009, 151,134–140.
205 Z.-H. Zhao, J.-M. Fan and Z.-Z. Wang, J. Cleaner Prod., 2007,15, 1894–1897.
206 Q.Wang, W.Wu, J. Chen, G. Chu, K. Ma andH. Zou, ColloidSurf., A, 2012, 409, 118–125.
207 J. Ettedgui, Y. Diskin-Posner, L. Weiner and R. Neumann, J.Am. Chem. Soc., 2010, 133, 188–190.
208 T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce,S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc.,2010, 132, 2132–2133.
209 T. W. Woolerton, S. Sheard, E. Pierce, S. W. Ragsdale andF. A. Armstrong, Energy Environ. Sci., 2011, 4, 2393–2399.
210 H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau,E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz,E. Dinjus and D. A. Dixon, Chem. Rev., 2001, 101, 953–996.
211 W. Hou, W. H. Hung, P. Pavaskar, A. Goeppert, M. Aykoland S. B. Cronin, ACS Catal., 2011, 1, 929–936.
212 W.-N. Wang, J. Park and P. Biswas, Catal. Sci. Technol.,2011, 1, 593–600.
213 D. Luo, Y. Bi, W. Kan, N. Zhang and S. Hong, J. Mol. Struct.,2011, 994, 325–331.
214 H.-C. Yang, H.-Y. Lin, Y.-S. Chien, J. C.-S. Wu and H.-H. Wu,Catal. Lett., 2009, 131, 381–387.
215 J. C. Wu, T.-H. Wu, T. Chu, H. Huang and D. Tsai, Top.Catal., 2008, 47, 131–136.
216 I. Tseng and J. C.-S. Wu, Catal. Today, 2004, 97, 113–119.217 K. R. Thampi, J. Kiwi and M. Graetzel, Nature, 1987, 327,
506–508.218 J. Jensen, M. Mikkelsen and F. C. Krebs, Sol. Energy Mater.
Sol. Cells, 2011, 95, 2949–2958.219 P. Gijsman, G. Meijers and G. Vitarelli, Polym. Degrad. Stab.,
1999, 65, 433–441.220 C. Jin, P. Christensen, T. Egerton, E. Lawson and J. White,
Polym. Degrad. Stab., 2006, 91, 1086–1096.221 R. E. Kesting, Synthetic polymeric membranes: a structural
perspective, Wiley, New York, 2nd edn, 1985.222 S. Fernando, P. Christensen, T. Egerton, R. Eveson,
S. Martins-Franchetti, D. Voisin and J. White, Mater. Sci.Technol., 2009, 25, 549–555.
223 W. Kim, T. Seok and W. Choi, Energy Environ. Sci., 2012, 5,6066–6070.
224 T. Arai, S. Tajima, S. Sato, K. Uemura, T. Morikawa andT. Kajino, Chem. Commun., 2011, 47, 12664–12666.
RSC Adv., 2014, 4, 20856–20893 | 20893
Top Related
Copyright © 2022 FDOKUMEN

