
Volume 16 - Number 12 December 2012 - Revues et Congrès
85
The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. Online and PDF versions of the Atlas of Genetics and Cytogenetics in Oncology and Haematology are hosted by INIST-CNRS. INIST-CNRS OPEN ACCESS JOURNAL Atlas of Genetics and Cytogenetics in Oncology and Haematology Volume 16 - Number 12 December 2012
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Volume 16 - Number 12 December 2012 - Revues et Congrès

The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. Online and PDF versions of the Atlas of Genetics and Cytogenetics in Oncology and Haematology are hosted by INIST-CNRS.
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Volume 16 - Number 12 December 2012

The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. Online and PDF versions of the Atlas of Genetics and Cytogenetics in Oncology and Haematology are hosted by INIST-CNRS.
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Scope
The Atlas of Genetics and Cytogenetics in Oncology and Haematology is a peer reviewed on-line journal in open access, devoted to genes, cytogenetics, and clinical entities in cancer, and cancer-prone diseases. It presents structured review articles ("cards") on genes, leukaemias, solid tumours, cancer-prone diseases, more traditional review articles on these and also on surrounding topics ("deep insights"), case reports in hematology, and educational items in the various related topics for students in Medicine and in Sciences.
Editorial correspondance
Jean-Loup Huret Genetics, Department of Medical Information, University Hospital F-86021 Poitiers, France tel +33 5 49 44 45 46 or +33 5 49 45 47 67 [email protected] or [email protected]
Staff Mohammad Ahmad, Mélanie Arsaban, Jérémy Cigna, Marie-Christine Jacquemot-Perbal, Vanessa Le Berre, Anne Malo, Catherine Morel-Pair, Laurent Rassinoux, Alain Zasadzinski. Philippe Dessen is the Database Director, and Alain Bernheim the Chairman of the on-line version (Gustave Roussy Institute – Villejuif – France).
The Atlas of Genetics and Cytogenetics in Oncology and Haematology (ISSN 1768-3262) is published 12 times a year by ARMGHM, a non profit organisation, and by the INstitute for Scientific and Technical Information of the French National Center for Scientific Research (INIST-CNRS) since 2008. The Atlas is hosted by INIST-CNRS (http://www.inist.fr)
http://AtlasGeneticsOncology.org
© ATLAS - ISSN 1768-3262

Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12)
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Editor
Jean-Loup Huret (Poitiers, France)
Editorial Board Sreeparna Banerjee (Ankara, Turkey) Solid Tumours Section Alessandro Beghini (Milan, Italy) Genes Section Anne von Bergh (Rotterdam, The Netherlands) Genes / Leukaemia Sections Judith Bovée (Leiden, The Netherlands) Solid Tumours Section Vasantha Brito-Babapulle (London, UK) Leukaemia Section Charles Buys (Groningen, The Netherlands) Deep Insights Section Anne Marie Capodano (Marseille, France) Solid Tumours Section Fei Chen (Morgantown, West Virginia) Genes / Deep Insights Sections Antonio Cuneo (Ferrara, Italy) Leukaemia Section Paola Dal Cin (Boston, Massachussetts) Genes / Solid Tumours Section Louis Dallaire (Montreal, Canada) Education Section Brigitte Debuire (Villejuif, France) Deep Insights Section François Desangles (Paris, France) Leukaemia / Solid Tumours Sections Enric Domingo-Villanueva (London, UK) Solid Tumours Section Ayse Erson (Ankara, Turkey) Solid Tumours Section Richard Gatti (Los Angeles, California) Cancer-Prone Diseases / Deep Insights Sections Ad Geurts van Kessel (Nijmegen, The Netherlands) Cancer-Prone Diseases Section Oskar Haas (Vienna, Austria) Genes / Leukaemia Sections Anne Hagemeijer (Leuven, Belgium) Deep Insights Section Nyla Heerema (Colombus, Ohio) Leukaemia Section Jim Heighway (Liverpool, UK) Genes / Deep Insights Sections Sakari Knuutila (Helsinki, Finland) Deep Insights Section Lidia Larizza (Milano, Italy) Solid Tumours Section Lisa Lee-Jones (Newcastle, UK) Solid Tumours Section Edmond Ma (Hong Kong, China) Leukaemia Section Roderick McLeod (Braunschweig, Germany) Deep Insights / Education Sections Cristina Mecucci (Perugia, Italy) Genes / Leukaemia Sections Yasmin Mehraein (Homburg, Germany) Cancer-Prone Diseases Section Fredrik Mertens (Lund, Sweden) Solid Tumours Section Konstantin Miller (Hannover, Germany) Education Section Felix Mitelman (Lund, Sweden) Deep Insights Section Hossain Mossafa (Cergy Pontoise, France) Leukaemia Section Stefan Nagel (Braunschweig, Germany) Deep Insights / Education Sections Florence Pedeutour (Nice, France) Genes / Solid Tumours Sections Elizabeth Petty (Ann Harbor, Michigan) Deep Insights Section Susana Raimondi (Memphis, Tennesse) Genes / Leukaemia Section Mariano Rocchi (Bari, Italy) Genes Section Alain Sarasin (Villejuif, France) Cancer-Prone Diseases Section Albert Schinzel (Schwerzenbach, Switzerland) Education Section Clelia Storlazzi (Bari, Italy) Genes Section Sabine Strehl (Vienna, Austria) Genes / Leukaemia Sections Nancy Uhrhammer (Clermont Ferrand, France) Genes / Cancer-Prone Diseases Sections Dan Van Dyke (Rochester, Minnesota) Education Section Roberta Vanni (Montserrato, Italy) Solid Tumours Section Franck Viguié (Paris, France) Leukaemia Section José Luis Vizmanos (Pamplona, Spain) Leukaemia Section Thomas Wan (Hong Kong, China) Genes / Leukaemia Sections

Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12)
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Volume 16, Number 12, December 2012
Table of contents
Gene Section
ERBB3 (v-erb-b2 erythroblastic leukemia viral oncog ene homolog 3 (avian)) 871 Smita Awasthi, Anne W Hamburger
PIK3R1 (phosphoinositide-3-kinase, regulatory subun it 1 (alpha)) 876 Daphne W Bell
CDH17 (cadherin 17, LI cadherin (liver-intestine)) 884 Yiping Rong, Nikki P Lee, John M Luk
FPR1 (formyl peptide receptor 1) 889 Jian Huang, Ji Ming Wang
LZTS1 (leucine zipper, putative tumor suppressor 1) 894 Andrea Vecchione, Luca Lavra, Carlo M Croce
MAP2K4 (mitogen-activated protein kinase kinase 4) 898 Kentaro Nakayama, Naomi Nakayama, Kohji Miyazaki
MYLK (myosin light chain kinase) 901 Kui Shen, Ting Wang, Joe GN Garcia
NAMPT (nicotinamide phosphoribosyltransferase) 909 Vassiliki Koumaki, Maria Dalamaga
PRKCI (protein kinase C, iota) 913 Verline Justilien, Alan P Fields
TYR (tyrosinase (oculocutaneous albinism IA)) 918 Erin E Mendoza, Randy Burd
Leukaemia Section
NUP214/ABL1 fusion gene on amplified episomes 921 Nathalie Nadal
t(3;19)(q27;q13) NAPA/BCL6 924 Jean-Loup Huret
t(3;3)(q27;q27) ST6GAL1/BCL6 925 Jean-Loup Huret
t(X;7)(q22;q34) IRS4/TCRB 927 Kristina Karrman
Solid Tumour Section
Liver: Fibrolamellar carcinoma 929 Xuchen Zhang, Stephen C Ward

Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12)
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Thyroid: Anaplastic (undifferentiated) carcinoma 935 Sai-Ching Jim Yeung, Mouhammed Amir Habra
Cancer Prone Disease Section
Schöpf-Schulz-Passarge syndrome (SSPS) 940 John A McGrath
Deep Insight Section
The Claudins family: Structure and function in norm al and pathologic conditions 943 Abderrahman Ouban, Atif Ali Ahmed

Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12)
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 871
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
ERBB3 (v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian)) Smita Awasthi, Anne W Hamburger
University of Maryland School of Medicine, Department of Pathology and University of Maryland Greenebaum Cancer Center, USA (SA, AWH)
Published in Atlas Database: May 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/ERBB3ID40479ch12q13.html DOI: 10.4267/2042/48356
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: ErbB-3, HER3, LCCS2, MDA-BF-1, c-erbB-3, c-erbB3, erbB3-S, p180-ErbB3, p45-sErbB3, p85-sErbB3
HGNC (Hugo): ERBB3
Location: 12q13.2
DNA/RNA Description The ERBB3 gene, which maps to human chromosome 12q13.2, is 23.2 kb in size and consists of 28 exons. The gene for the extracellular ligand binding domain of ErbB3 has 43-45% homology with EGFR and ERBB2 and 56-67% homology with ERBB4. The cytoplasmic tyrosine kinase domain sequences have 60-63% homology with those of the other ErbB receptors (Kraus et al., 1989).
Transcription The ERBB3 promoter region is GC rich (65%) and, like EGFR, does not contain a TATA box. A proximal promoter was observed within 600 bp flanking Exon1. AP2-1 (OB2-1) and Fox3a have been demonstrated to be functional transcriptional regulators at upstream start sites (Skinner and Hurst, 1993). A Sox10 regulated enhancer has been identified at chr12:54763065-54763421 in neural crest derived cells. The human ERBB3 gene is transcribed as a 6.2 kb message of 4080 nucleotides and 1342 codons specifying the full-length protein. There are four additional alternate transcripts of 1.6, 1.7, 2.1 and 2.3 kb generated by intron read
through. At least three of these transcripts code for truncated, secreted soluble forms of ERBB3 (Lee and Maihle, 1998).
Pseudogene None reported.
Protein Description The ERBB3 gene encodes a member of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases. The 6.2 kb transcript encodes a 148 kDa protein which is post-translationally glycosylated to yield a protein of 180 kDa (Kraus et al., 1989). The extracellular ligand-binding domain consists of four subdomains that change conformation in response to ligand. Domains I and III bind NRG with high affinity (Cho and Leahy, 2002). Due to substitutions in the kinase domain at aa 740, 759 and 834, ErbB3 lacks potent tyrosine kinase activity. However, recent data indicate that ErbB3 maintains some autophosphorylation activity (Shi et al., 2010). Heterodimerization with other ErbB family members, most notably ErbB2, is needed to convey biological signals through phosphorylation of downstream substrates, most notably AKT (Olayioye et al., 2000). In general, activation of these pathways leads to cell proliferation or differentiation. Alternate transcriptional splice variants encoding different isoforms have been characterized.
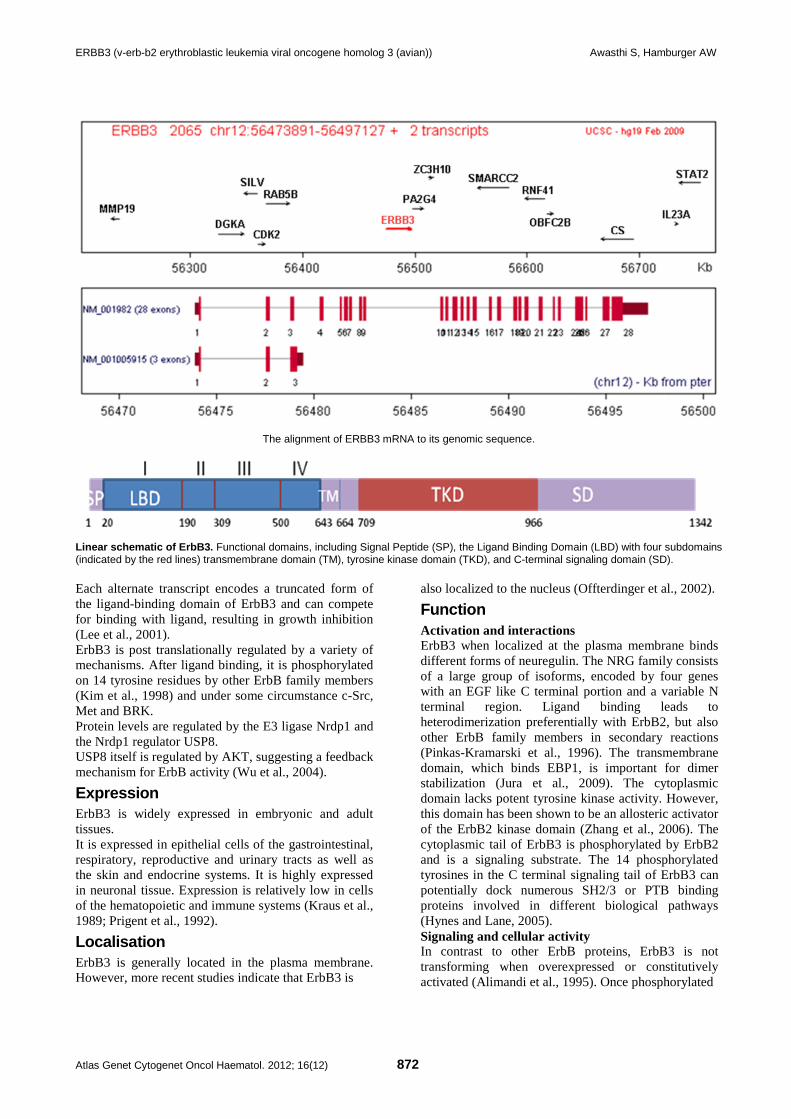
ERBB3 (v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian)) Awasthi S, Hamburger AW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 872
The alignment of ERBB3 mRNA to its genomic sequence.
Linear schematic of ErbB3. Functional domains, including Signal Peptide (SP), the Ligand Binding Domain (LBD) with four subdomains (indicated by the red lines) transmembrane domain (TM), tyrosine kinase domain (TKD), and C-terminal signaling domain (SD). Each alternate transcript encodes a truncated form of the ligand-binding domain of ErbB3 and can compete for binding with ligand, resulting in growth inhibition (Lee et al., 2001). ErbB3 is post translationally regulated by a variety of mechanisms. After ligand binding, it is phosphorylated on 14 tyrosine residues by other ErbB family members (Kim et al., 1998) and under some circumstance c-Src, Met and BRK. Protein levels are regulated by the E3 ligase Nrdp1 and the Nrdp1 regulator USP8. USP8 itself is regulated by AKT, suggesting a feedback mechanism for ErbB activity (Wu et al., 2004).
Expression ErbB3 is widely expressed in embryonic and adult tissues. It is expressed in epithelial cells of the gastrointestinal, respiratory, reproductive and urinary tracts as well as the skin and endocrine systems. It is highly expressed in neuronal tissue. Expression is relatively low in cells of the hematopoietic and immune systems (Kraus et al., 1989; Prigent et al., 1992).
Localisation ErbB3 is generally located in the plasma membrane. However, more recent studies indicate that ErbB3 is
also localized to the nucleus (Offterdinger et al., 2002).
Function Activation and interactions ErbB3 when localized at the plasma membrane binds different forms of neuregulin. The NRG family consists of a large group of isoforms, encoded by four genes with an EGF like C terminal portion and a variable N terminal region. Ligand binding leads to heterodimerization preferentially with ErbB2, but also other ErbB family members in secondary reactions (Pinkas-Kramarski et al., 1996). The transmembrane domain, which binds EBP1, is important for dimer stabilization (Jura et al., 2009). The cytoplasmic domain lacks potent tyrosine kinase activity. However, this domain has been shown to be an allosteric activator of the ErbB2 kinase domain (Zhang et al., 2006). The cytoplasmic tail of ErbB3 is phosphorylated by ErbB2 and is a signaling substrate. The 14 phosphorylated tyrosines in the C terminal signaling tail of ErbB3 can potentially dock numerous SH2/3 or PTB binding proteins involved in different biological pathways (Hynes and Lane, 2005). Signaling and cellular activity In contrast to other ErbB proteins, ErbB3 is not transforming when overexpressed or constitutively activated (Alimandi et al., 1995). Once phosphorylated

ERBB3 (v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian)) Awasthi S, Hamburger AW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 873
by other ErbB family members or Src, Met or BRK, ErbB-3 can then bind numerous other signaling proteins. Activation of the PI-3 kinase-AKT pathway is especially important as there are six docking sites for the p85 subunit of PI-3K in the ErbB3 cytoplasmic tail at Tyr 1035, 1178, 1203/1205, 1257 and 1270. AKT regulates many downstream signaling nodes, in particular the two mTOR containing complexes. ErbB3 can also activate the MAPK pathway via its interactions with Grb7 (Tyr 1180,1243) and SHC (1309) (Hynes and Lane, 2005). Thus, ErbB3 is important in biological processes such as translation, apoptosis, nutrient sensing, metabolic regulation, angiogenesis and cell cycle control. Increased expression or activity of ErbB3 has been associated with resistance to EGFR and ErbB2 inhibitors (Sergina et al., 2007) and hormonal therapies (Liu et al., 2007). ErbB3 when localized in the nucleus acts as a transcription factor to regulate Cyclin D1 and β-casein genes (Andrique et al., 2012). Physiological ErbB3 knock-out mice die by E13.5 with defective heart valve formation, but normal heart trabeculation. The animals show a generalized neural crest defects and lack Schwann cell precursors (Erickson et al., 1997). Due to the importance of ErbB3 in breast cancer, the role of ErbB3 in mammary development has been well-studied. ErbB3 is required for ductal morphogenesis in the mouse mammary gland (Stern, 2003). ErbB3 has also been implicated in maintenance of the luminal epithelial subtype in the breast (Balko et al., 2012).
Homology The ErbB family has evolved from a single ligand-receptor combination in C. elegans (let-23 28% aa similarity) through Drosophila with one receptor (EGFR, 39% similarity) and four ligands to vertebrates, where four ErbB receptor bind multiple EGF-related ligands. The ERBB3 gene is conserved in chimpanzee (99% similarity), dog, cow, mouse (90%), rat, chicken, and zebrafish.
Mutations Germinal An A to G mutation is noted in intron 10 in Lethal Congential Contracture Syndrome 2 (LCCS2). LCCS2 is an autosomal recessive neurogenic form of a neonatally lethal arthrogryposis that is associated with atrophy of the anterior horn of the spinal cord (Narkis et al., 2004).
Somatic Mutations in ErbB3 have been rarely noted in cancer. One of the 2 mutations reported was a missense mutation in exon 21 (2537 G > T) (Ser846Ile) detected in a rectal mucinous adenocarcinoma (1% of the total colon cancer samples.
The other mutation was a silent mutation in exon 21 (2484 T > C) (His828His) detected in an invasive ductal carcinoma of the breast (2% of the total 60 breast cancers) (Jeong et al., 2006).
Implicated in Breast cancer Prognosis Increased expression of ErbB3 in breast cancer cells relative to normal epithelium is common. The increased expression is not due to genomic amplification (Gasparini et al., 1994). High ErbB3 expression has been correlated with both increased and poorer survival (Hamburger, 2008). The ErbB2/3 heterodimer is essential for proliferation of malignant mammary epithelial cells (Holbro et al., 2003). ErbB3 contributes to tamoxifen resistance (Liu et al., 2007) and activation of ErbB3 is also associated with resistance to ErbB directed tyrosine kinase inhibitors (Sergina et al., 2007).
Ovarian cancer Prognosis Genomic amplification of ErbB3 has been noted in ovarian cancer and ErbB3 overexpression is associated with poor survival (Wilken et al., 2012). Truncated ErbB3 transcripts that code for soluble truncated proteins have been observed in ovarian cancer cell lines. Such soluble forms can inhibit proliferation (Maihle 2001). These soluble forms may have potential as markers of disease progression.
Prostate cancer Prognosis Increased expression of ErbB3 has been noted in prostate cancer (Cheng et al., 2007; Koumakpayi et al., 2006). Activation of the ErbB2/3 heterodimer stabilizes Androgen Receptor contributing to hormone independent growth (Mellinghoff et al., 2004). NRG can activate the EBP1 Protein leading to decreased AR activity (Zhang and Hamburger, 2005). Nuclear localization of ErbB3 has been associated with both poorer and better prognoses. A secreted ErbB3 isoform has been shown to enhance bone metastasis (Chen et al., 2007).
Pancreatic cancer Prognosis ErbB3 mRNA and protein has consistently been observed to be increased and associated with poor outcome (Friess et al., 1995).
Lung cancer Prognosis Overexpression of ErbB3 generally correlates with poor prognosis (Yi et al., 1997). Several studies have indicated that ErbB3 affects clinical responsiveness to tyrosine kinase inhibitors. Cell lines with wild type and

ERBB3 (v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian)) Awasthi S, Hamburger AW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 874
high levels of ErbB3 respond better to EGFR inhibitors (Engelman et al., 2005). In addition, gefitinib resistant NCSLC cells can amplify MET which then phosphorylates and activates ErbB3 and AKT pathways (Engelman et al., 2007). ErbB3 has also been implicated in inhibition of apoptosis in lung cancer cell lines (Sithanandam et al., 2005).
Schizophrenia Prognosis The NRG1 gene was identified as a potential susceptibility gene for schizophrenia and defects in the expression of ErbB3 were also shown to occur in the prefrontal cortex of schizophrenic patients. However, currently the association between ErbB3 expression and schizophrenia is unclear (Corfas et al., 2004).
Diabetes Prognosis Genome-wide association studies have identified associations between type I diabetes and single-nucleotide polymorphisms (SNP) at chromosome 12q13 surrounding the ERBB3 gene. The most significant association was observed with a SNP in exon 27 of the ERBB3 gene and an intergenic SNP (Keene et al., 2012). In addition, ErbB3 has been demonstrated to modulate antigen presenting cell function and type I diabetes risk (Jing et al., 2011).
References Kraus MH, Issing W, Miki T, Popescu NC, Aaronson SA. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: evidence for overexpression in a subset of human mammary tumors. Proc Natl Acad Sci U S A. 1989 Dec;86(23):9193-7
Prigent SA, Lemoine NR, Hughes CM, Plowman GD, Selden C, Gullick WJ. Expression of the c-erbB-3 protein in normal human adult and fetal tissues. Oncogene. 1992 Jul;7(7):1273-8
Skinner A, Hurst HC. Transcriptional regulation of the c-erbB-3 gene in human breast carcinoma cell lines. Oncogene. 1993 Dec;8(12):3393-401
Gasparini G, Gullick WJ, Maluta S, Dalla Palma P, Caffo O, Leonardi E, Boracchi P, Pozza F, Lemoine NR, Bevilacqua P. c-erbB-3 and c-erbB-2 protein expression in node-negative breast carcinoma--an immunocytochemical study. Eur J Cancer. 1994;30A(1):16-22
Alimandi M, Romano A, Curia MC, Muraro R, Fedi P, Aaronson SA, Di Fiore PP, Kraus MH. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995 May 4;10(9):1813-21
Kurbacher CM, Bruckner HW, Cree IA, Kurbacher JA, Wilhelm L, Pöch G, Indefrei D, Mallmann P, Andreotti PE. Mitoxantrone combined with paclitaxel as salvage therapy for platinum-refractory ovarian cancer: laboratory study and clinical pilot trial. Clin Cancer Res. 1997 Sep;3(9):1527-33
Pinkas-Kramarski R, Shelly M, Glathe S, Ratzkin BJ, Yarden Y. Neu differentiation factor/neuregulin isoforms activate distinct receptor combinations. J Biol Chem. 1996 Aug 9;271(32):19029-32
Erickson SL, O'Shea KS, Ghaboosi N, Loverro L, Frantz G, Bauer M, Lu LH, Moore MW. ErbB3 is required for normal
cerebellar and cardiac development: a comparison with ErbB2-and heregulin-deficient mice. Development. 1997 Dec;124(24):4999-5011
Yi ES, Harclerode D, Gondo M, Stephenson M, Brown RW, Younes M, Cagle PT. High c-erbB-3 protein expression is associated with shorter survival in advanced non-small cell lung carcinomas. Mod Pathol. 1997 Feb;10(2):142-8
Kim HH, Vijapurkar U, Hellyer NJ, Bravo D, Koland JG. Signal transduction by epidermal growth factor and heregulin via the kinase-deficient ErbB3 protein. Biochem J. 1998 Aug 15;334 ( Pt 1):189-95
Lee H, Maihle NJ. Isolation and characterization of four alternate c-erbB3 transcripts expressed in ovarian carcinoma-derived cell lines and normal human tissues. Oncogene. 1998 Jun 25;16(25):3243-52
Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000 Jul 3;19(13):3159-67
Lee H, Akita RW, Sliwkowski MX, Maihle NJ. A naturally occurring secreted human ErbB3 receptor isoform inhibits heregulin-stimulated activation of ErbB2, ErbB3, and ErbB4. Cancer Res. 2001 Jun 1;61(11):4467-73
Cho HS, Leahy DJ. Structure of the extracellular region of HER3 reveals an interdomain tether. Science. 2002 Aug 23;297(5585):1330-3
Offterdinger M, Schöfer C, Weipoltshammer K, Grunt TW. c-erbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol. 2002 Jun 10;157(6):929-39
Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003 Jul 22;100(15):8933-8
Stern DF. ErbBs in mammary development. Exp Cell Res. 2003 Mar 10;284(1):89-98
Corfas G, Roy K, Buxbaum JD. Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat Neurosci. 2004 Jun;7(6):575-80
Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004 Nov;6(5):517-27
Narkis G, Landau D, Manor E, Elbedour K, Tzemach A, Fishelson M, Geiger D, Ofir R, Carmi R, Birk OS. Homozygosity mapping of lethal congenital contractural syndrome type 2 (LCCS2) to a 6 cM interval on chromosome 12q13. Am J Med Genet A. 2004 Oct 15;130A(3):272-6
Wu X, Yen L, Irwin L, Sweeney C, Carraway KL 3rd. Stabilization of the E3 ubiquitin ligase Nrdp1 by the deubiquitinating enzyme USP8. Mol Cell Biol. 2004 Sep;24(17):7748-57
Engelman JA, Jänne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005 Mar 8;102(10):3788-93
Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005 May;5(5):341-54
Sithanandam G, Fornwald LW, Fields J, Anderson LM. Inactivation of ErbB3 by siRNA promotes apoptosis and attenuates growth and invasiveness of human lung

ERBB3 (v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian)) Awasthi S, Hamburger AW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 875
adenocarcinoma cell line A549. Oncogene. 2005 Mar 10;24(11):1847-59
Zhang Y, Hamburger AW. Specificity and heregulin regulation of Ebp1 (ErbB3 binding protein 1) mediated repression of androgen receptor signalling. Br J Cancer. 2005 Jan 17;92(1):140-6
Jeong EG, Soung YH, Lee JW, Lee SH, Nam SW, Lee JY, Yoo NJ, Lee SH. ERBB3 kinase domain mutations are rare in lung, breast and colon carcinomas. Int J Cancer. 2006 Dec 15;119(12):2986-7
Koumakpayi IH, Diallo JS, Le Page C, Lessard L, Gleave M, Bégin LR, Mes-Masson AM, Saad F. Expression and nuclear localization of ErbB3 in prostate cancer. Clin Cancer Res. 2006 May 1;12(9):2730-7
Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006 Jun 16;125(6):1137-49
Chen N, Ye XC, Chu K, Navone NM, Sage EH, Yu-Lee LY, Logothetis CJ, Lin SH. A secreted isoform of ErbB3 promotes osteonectin expression in bone and enhances the invasiveness of prostate cancer cells. Cancer Res. 2007 Jul 15;67(14):6544-8
Cheng CJ, Ye XC, Vakar-Lopez F, Kim J, Tu SM, Chen DT, Navone NM, Yu-Lee LY, Lin SH, Hu MC. Bone microenvironment and androgen status modulate subcellular localization of ErbB3 in prostate cancer cells. Mol Cancer Res. 2007 Jul;5(7):675-84
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007 May 18;316(5827):1039-43
Liu B, Ordonez-Ercan D, Fan Z, Edgerton SM, Yang X, Thor AD. Downregulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int J Cancer. 2007 May 1;120(9):1874-82
Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007 Jan 25;445(7126):437-41
Hamburger AW. The role of ErbB3 and its binding partners in breast cancer progression and resistance to hormone and tyrosine kinase directed therapies. J Mammary Gland Biol Neoplasia. 2008 Jun;13(2):225-33
Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci U S A. 2009 Dec 22;106(51):21608-13
Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A. 2010 Apr 27;107(17):7692-7
Wang H, Jin Y, Reddy MV, Podolsky R, Liu S, Yang P, Bode B, Reed JC, Steed RD, Anderson SW, Steed L, Hopkins D, Huang Y, She JX. Genetically dependent ERBB3 expression modulates antigen presenting cell function and type 1 diabetes risk. PLoS One. 2010 Jul 26;5(7):e11789
Andrique L, Fauvin D, El Maassarani M, Colasson H, Vannier B, Séité P. ErbB3(80 kDa), a nuclear variant of the ErbB3 receptor, binds to the Cyclin D1 promoter to activate cell proliferation but is negatively controlled by p14ARF. Cell Signal. 2012 May;24(5):1074-85
Balko JM, Miller TW, Morrison MM, Hutchinson K, Young C, Rinehart C, Sánchez V, Jee D, Polyak K, Prat A, Perou CM, Arteaga CL, Cook RS. The receptor tyrosine kinase ErbB3 maintains the balance between luminal and basal breast epithelium. Proc Natl Acad Sci U S A. 2012 Jan 3;109(1):221-6
Keene KL, Quinlan AR, Hou X, Hall IM, Mychaleckyj JC, Onengut-Gumuscu S, Concannon P. Evidence for two independent associations with type 1 diabetes at the 12q13 locus. Genes Immun. 2012 Jan;13(1):66-70
Wilken JA, Badri T, Cross S, Raji R, Santin AD, Schwartz P, Branscum AJ, Baron AT, Sakhitab AI, Maihle NJ. EGFR/HER-targeted therapeutics in ovarian cancer. Future Med Chem. 2012 Mar;4(4):447-69
This article should be referenced as such:
Awasthi S, Hamburger AW. ERBB3 (v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian)). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):871-875.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 876
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
PIK3R1 (phosphoinositide-3-kinase, regulatory subunit 1 (alpha)) Daphne W Bell
National Human Genome Research Institute, Cancer Genetics Branch, National Institutes of Health, Bethesda, MD, USA (DWB)
Published in Atlas Database: May 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/PIK3R1ID41717ch5q13.html DOI: 10.4267/2042/48357
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: GRB1, p85, p85-ALPHA
HGNC (Hugo): PIK3R1
Location: 5q13.1
DNA/RNA Description The human PIK3R1 gene encompasses 86102 bp of DNA and contains 16 exons.
Transcription Human PIK3R1 is alternatively spliced, resulting in four major protein-encoding transcripts. Transcript variant 1: 7011 bp in length; the open-reading frame of the coding sequence is 2175 bp. Transcript variant 2: 2439 bp in length; the open-reading frame of the coding sequence is 1365 bp. Transcript variant 3: 2625 bp in length; the open-reading frame of the coding sequence is 1275 bp. Transcript variant 4: 2473 bp in length; the open-reading frame of the coding sequence is 1086 bp.
Pseudogene None known.
Protein Note Crystal structures have been reported for the p85α-SH3 domain (Liang et al., 1996), the p85α-BH domain (Musacchio et al., 1996), the nSH2 domain (Nolte et al., 1996), and the p85α-cSH2 domain (Hoedemaeker et al., 1999).
Co-crystal structures have been reported for the p85α-niSH2 domain (residues 322-600) in complex with p110α (Huang et al., 1997), and for the human p85α-iSH2 domain in complex with the bovine p110α-ABD domain (Miled et al., 2007).
Description Isoforms: PIK3R1 encodes four distinct protein isoforms (CCDS3993 (p85α), CCDS3994 (p55α), CCDS3995 (p50α), and CCDS56374) as a result of alternative splicing (Inukai et al., 1997). p85α: p85α has an SH3 domain, a BCR-homology (BH) domain, and nSH2, iSH2, and cSH2 domains. The SH3 domain of p85α mediates binding to FAK, CAS, Apoptin, Ruk, SNX9, Dynamin, Cbl, and BCR-ABL (reviewed in Mellor et al., 2012). The BH domain of p85α mediates binding to XB-1, Rac, Cdc42, Rab5, PTEN (reviewed in Mellor et al., 2012). The nSH2 domain of p85α interacts with the helical domain of p110α (Miled et al., 2007). The iSH2 domain of p85α interacts with both the ABD and C2 domains of p110α leading, respectively, to stabilization and inhibition of p110α (Dhand et al., 1994; Fu et al., 2004; Elis et al., 2006; Huang et al., 2007). Residues D560 and N564 in the p85α-iSH2 domain are within hydrogen bonding distance of residue N345 of the p110α-C2 domain (Huang et al., 2007). This interaction is required for the inhibition of p110α (Wu et al., 2009). It has been suggested that residues 447-561 within the iSH2 might form contact with the plasma membrane (Huang et al., 2007).
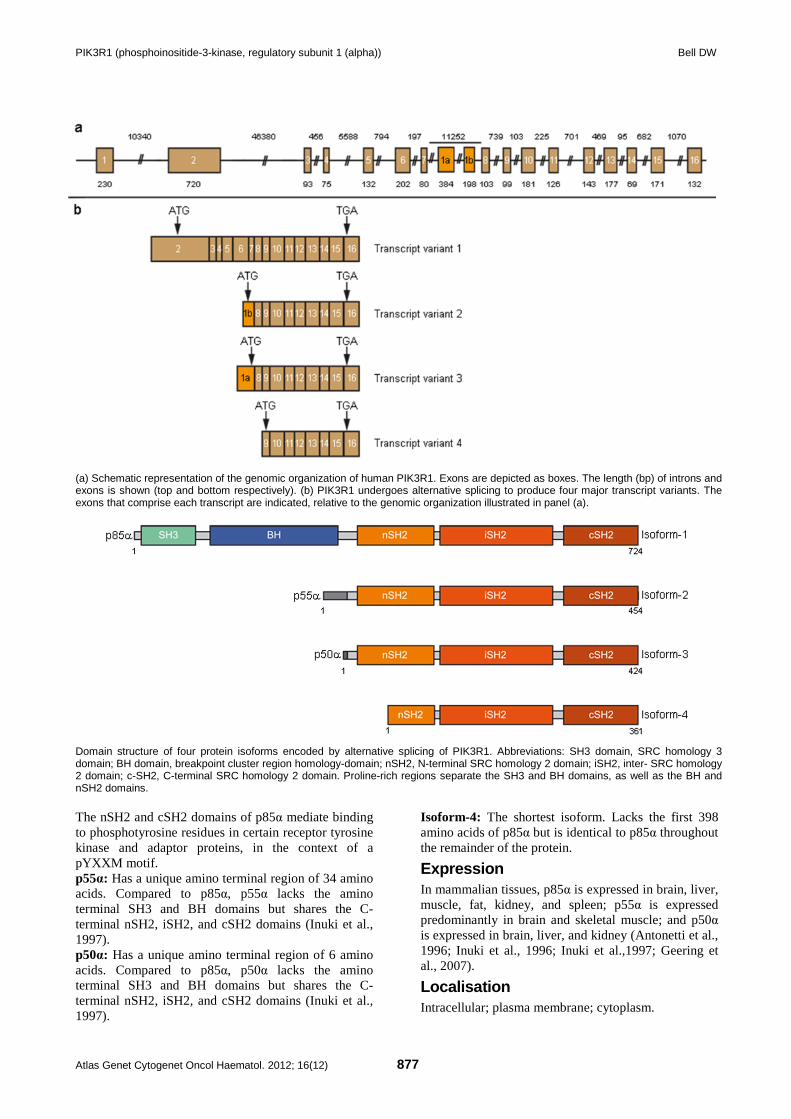
PIK3R1 (phosphoinositide-3-kinase, regulatory subunit 1 (alpha)) Bell DW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 877
(a) Schematic representation of the genomic organization of human PIK3R1. Exons are depicted as boxes. The length (bp) of introns and exons is shown (top and bottom respectively). (b) PIK3R1 undergoes alternative splicing to produce four major transcript variants. The exons that comprise each transcript are indicated, relative to the genomic organization illustrated in panel (a).
Domain structure of four protein isoforms encoded by alternative splicing of PIK3R1. Abbreviations: SH3 domain, SRC homology 3 domain; BH domain, breakpoint cluster region homology-domain; nSH2, N-terminal SRC homology 2 domain; iSH2, inter- SRC homology 2 domain; c-SH2, C-terminal SRC homology 2 domain. Proline-rich regions separate the SH3 and BH domains, as well as the BH and nSH2 domains. The nSH2 and cSH2 domains of p85α mediate binding to phosphotyrosine residues in certain receptor tyrosine kinase and adaptor proteins, in the context of a pYXXM motif. p55α: Has a unique amino terminal region of 34 amino acids. Compared to p85α, p55α lacks the amino terminal SH3 and BH domains but shares the C-terminal nSH2, iSH2, and cSH2 domains (Inuki et al., 1997). p50α: Has a unique amino terminal region of 6 amino acids. Compared to p85α, p50α lacks the amino terminal SH3 and BH domains but shares the C-terminal nSH2, iSH2, and cSH2 domains (Inuki et al., 1997).
Isoform-4: The shortest isoform. Lacks the first 398 amino acids of p85α but is identical to p85α throughout the remainder of the protein.
Expression In mammalian tissues, p85α is expressed in brain, liver, muscle, fat, kidney, and spleen; p55α is expressed predominantly in brain and skeletal muscle; and p50α is expressed in brain, liver, and kidney (Antonetti et al., 1996; Inuki et al., 1996; Inuki et al.,1997; Geering et al., 2007).
Localisation Intracellular; plasma membrane; cytoplasm.

PIK3R1 (phosphoinositide-3-kinase, regulatory subunit 1 (alpha)) Bell DW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 878
Function Regulation of PI3K signaling by p85α: p85α is the regulatory subunit of PI3K. In quiescent cells, p85α binds to p110α, the catalytic subunit of PI3K, and both stabilizes p110α and inhibits the basal activity of p110α. Ligand-induced phosphorylation of receptor tyrosine kinases or adaptor proteins on tyrosine residues, within a pYXXM motif, facilitates the binding of p85α to the phosphotyrosine residues via its SH2 domains. Consequently, the inhibitory effect of p85α on p110α is relieved and PI3K is brought into the vicinity of the plasma membrane where it catalyzes the conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 in turn recruits the AKT (v-akt murine thymoma viral oncogene homolog) serine-threonine kinase and the PDK1 (phosphoinositide-dependent protein kinase 1) kinase to the plasma membrane, thus facilitating the phosphorylation and activation of AKT. Once activated, AKT can initiate several downstream signal transduction cascades that regulate protein synthesis, cell survival, cell growth and metabolism, and the cell cycle (Reviewed in Vanhaesebroeck et al., 2010). Under conditions of nutrient deprivation, p85α is phosphorylated by IKK on serine-690. Consequently, the ability of p85α to bind phosphotyrosine proteins is reduced and PI3K-AKT signaling is diminished (Comb et al., 2012). Similarly, the activation of PKC family members by phorbol ester stimulation results in phosphorylation of p85α on serine-361 and serine-652, and leads to reduced binding of p85α to phosphotyrosines and inhibition of PI3K-AKT signaling (Lee et al., 2011). Regulation of PTEN by p85α: The PI3K-AKT signal transduction pathway is antagonized by the activity of the PTEN phosphatase, which dephosphorylates PIP3 to generate PIP2. Chagpar et al., (2010) demonstrated that p85α binds directly to PTEN via the p85α-SH3-BH domains. Cells expressing a synthetic mutant of p85α that abolished the p85α-PTEN interaction exhibited increased AKT activation following stimulation by growth factors. Chagpar et al., thus proposed that p85α can bind to PTEN and enhance PTEN activity. Subsequently, Cheung et al., (2011) demonstrated that compared to wildtype p85α, a tumor-associated mutant (p85α-E160X) that introduces a premature stop codon within the BH domain, was associated with reduced stability of the PTEN protein. Treatment of cells expressing the p85α-E160X mutant with a proteosome inhibitor lead to a modest increase in PTEN levels, further suggesting that the p85α-PTEN interaction prevents proteosomal degradation of PTEN and thus increases PTEN stability. The regulation of PTEN activity by p85α accounts for the increased insulin sensitivity observed in PIK3R1-/- or p85α-/-mice
(Mauvais-Jarvis et al., 2002; Brachmann et al., 2005; Taniguchi et al., 2006; Taniguchi et al., 2010; Chagpar et al., 2010). Receptor trafficking: p85α has GAP (GTP-ase Activating Protein) activity towards the Rab4, Rab5, Rac1, and Cdc42 small GTPases and, to a lesser extent, towards the Rab6 GTPase. The GAP activity of p85α resides within the BH domain. Within the BH domain, Arg151 and Arg274 are important for maximal GAP activity of p85α. The regulation of Rab4 and Rab5 activity by p85α has been implicated in the endosomal trafficking of activated PDGFR; cells expressing a synthetic mutant (p85α-Arg274A) exhibited delayed degradation of activated PDGFR, prolonged activation of the MAPK and AKT signalling pathways, and the capacity to transform NIH 3T3 cells (Chamberlain et al., 2004; Chamberlain et al., 2008; Chamberlain et al., 2010). Regulation of the unfolded protein response: p85α interacts with XBP-1s, a transcription factor that regulates the unfolded protein response following endoplasmic reticulum stress, and facilitates the relocation of XBP-1s to the nucleus (Park et al., 2010a; Winnay et al., 2010). p55α and p50α isoforms: Involved in insulin signaling (Inuki et al., 1997; Chen et al., 2004).
Homology Homologues of H. sapiens PIK3R1 exist in P. troglodytes (99.9% amino acid identity), M. mulatta (99.2% amino acid identity), C. lupus (95.7% amino acid identity), B. taurus (96.8% amino acid identity), M. musculus (96.0% amino acid identity), R. norvegicus (94.2% amino acid identity), G. gallus (89.1% amino acid identity), D. rerio (79.3% amino acid identity), and C. elegans (33.8% amino acid identity).
Mutations Note A polymorphic variant of PIK3R1 (Met326Ile; rs3730089), has been described (Baier et al., 1998; Almind et al., 2002). The PIK3R1-Ile326 allele has been reported to be associated with increased risk to colon cancer in a population based case-control study (Li et al., 2008).
Germinal A germline mutation in exon 6 of PIK3R1 has been described in a patient with agammaglobulinemia and an absence of B lineage cells (Conley et al., 2012). The mutation (p85α-W298X) resulted in loss of p85α expression, but did not affect p55α or p50α. The patient was homozygous for the mutation; her parents were both heterozygous carriers.
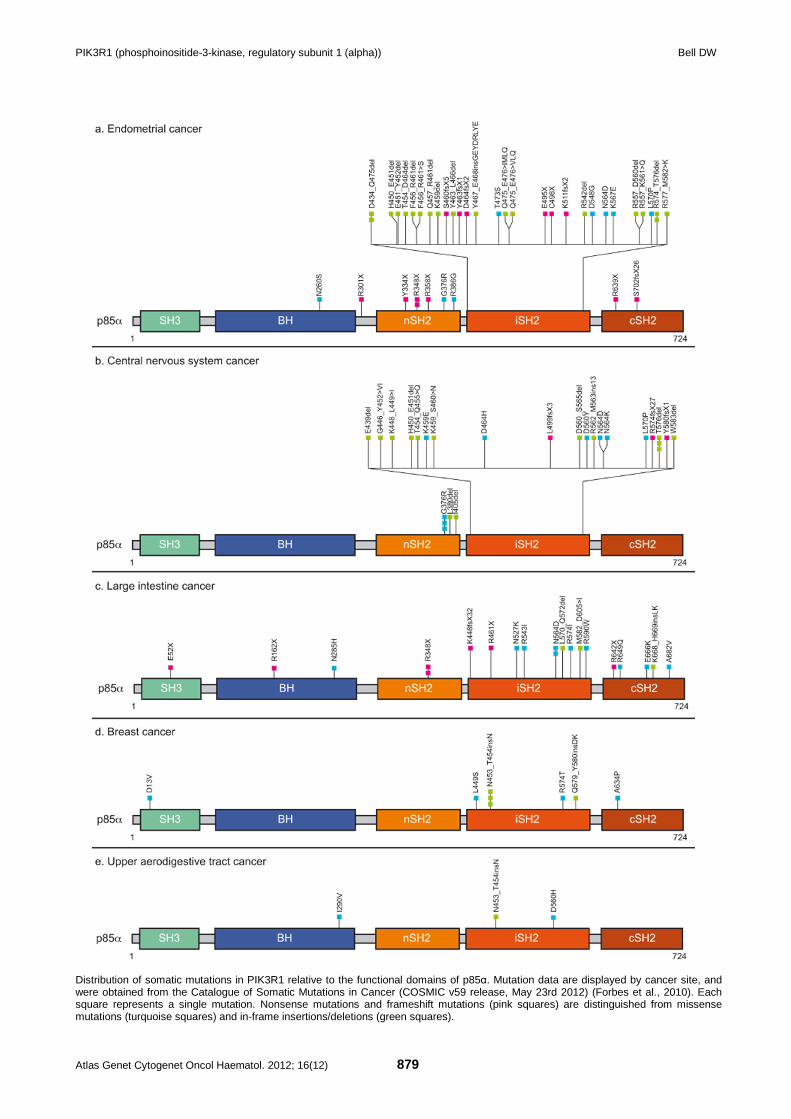
PIK3R1 (phosphoinositide-3-kinase, regulatory subunit 1 (alpha)) Bell DW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 879
Distribution of somatic mutations in PIK3R1 relative to the functional domains of p85α. Mutation data are displayed by cancer site, and were obtained from the Catalogue of Somatic Mutations in Cancer (COSMIC v59 release, May 23rd 2012) (Forbes et al., 2010). Each square represents a single mutation. Nonsense mutations and frameshift mutations (pink squares) are distinguished from missense mutations (turquoise squares) and in-frame insertions/deletions (green squares).

PIK3R1 (phosphoinositide-3-kinase, regulatory subunit 1 (alpha)) Bell DW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 880
Somatic Somatic mutations in PIK3R1 have been found in endometrial cancers (26%, 34 of 133), cancers of the central nervous system (4%, 26 of 657) and of the large intestine (5%, 19 of 355), ovarian cancer (2%, 5 of 257), breast cancer (2%, 10 of 500), urothelial cancer (0.7%, 1 of 145), squamous cell carcinoma of the skin (11%, 1 of 9), pancreatic cancer (2%, 1 of 53), and in hematological malignancy (1%, 3 of 472) (Philp et al., 2001; Mizoughi et al., 2004; Shi et al., 2006; Sjoblom et al., 2006; Jones et al., 2008; Parsons et al., 2008; The Cancer Genome Research Atlas Network, 2008; Bittinger et al., 2009; Jaiswal et al., 2009; Forbes et al., 2010; Bettegowda et al., 2011; Kan et al., 2010; Park et al., 2010b; Parsons et al., 2011; Cheung et al., 2011; Sjodahl et al., 2011; The Cancer Genome Atlas Research Network, 2011; Urick et al., 2011; Lipson et al., 2012; Shah et al., 2012). Mutation spectrum: Among 107 nonsynonymous, somatic mutations of PIK3R1 catalogued in the COSMIC database (v59 release, May 23rd, 2012) (Forbes et al., 2010), 43% (46 of 107) of mutations are in-frame insertions/deletions, 14% (15 of 107) are nonsense mutations, 11.2% (12 of 107) are frameshift mutations, 31.8% (34 of 107) are missense mutations. The majority (68.2%, 73 of 107) of all somatic mutations in PIK3R1 localize to animo acid residues within the iSH2 domain, which is shared by all four protein isoforms encoded by PIK3R1. Altered functional properties of mutant proteins: Biochemical and cellular studies of tumor-associated p85α mutants have revealed functional differences between mutant p85α and wild type p85α, as well as functional differences among various p85α mutants (Philp et al., 2001; Jaiswal et al., 2009; Sun et al., 2010; Cheung et al., 2011; Urick et al., 2011). - Transforming properties: Sun et al., (2010) evaluated the ability of nine tumor-associated mutant p85α proteins to transform chicken embryo fibroblasts. The p85α-KS459delN and p85α-DKRMNS560del mutants had the highest efficiency of transformation, the p85α-R574fs and p85α-T576del mutants had intermediate efficiency of transformation, and the p85α-D560Y, p85α-N564K p85α-W583del, p85α-E439del, and p85α-G376R mutants were only weakly tansforming (Sun et al., 2010). Transformation was mediated by p110α but not by p110α, p110α, or p110α (Sun et al., 2010). Each of the nine p85α mutants analyzed by Sun et al., retained the ability to bind p110α and resulted in hyperphosphorylation of AKT (T308) and 4E-BP1 when exogenously expressed in CEF cells (Sun et al., 2010). Eight of the tumor-associated mutants analyzed by Sun et al., (2010) localized to the iSH2 domain; one mutant (p85α-G376R) localized to the nSH2 domain. Jaiswal et al., showed that the p85α-D560Y, p85α-N564D, and p85α-QYL579delL mutants were capable of promoting both the IL-3 independent growth and anchorage-
independent growth of BaF3 cells (Jaiswal et al., 2009). Similarily, Cheung et al., showed that the p85α-R574fs, p85α-T576del, p85α-E160X, p85α-R348X, and p85α-R503W mutants induced IL3-independent growth of BaF3 cells (Cheung et al., 2011). - Altered p110α-binding: Truncating mutants of p85α that lack all or part of the the iSH2 domain (p85α-E160X, p85α-R162X, p85α-L380fs, p85α-R348X, p85α-K511VfsX2) fail to bind to p110α (Jaiswal et al., 2009; Cheung et al., 2011; Urick et al., 2011). In contrast, small in-frame deletions or missense mutations within the iSH2 domain retained the ability to bind p110α (Jaiswal et al., 2009; Cheung et al., 2011; Urick et al., 2011). - Increased PI3K activity: Jaiswal et al., (2009) showed that p85α mutants that were capable of binding p110α were able to stabilize p110α. p85α/p110α holoenzymes composed of the p85α-N564 or p85α-QYL579delL mutants had increased lipid kinase activity compared with the wildtype-p85α/p110α holoenzyme (Jaiswal et al., 2009). Holoenzymes consisting of mutant p85α and p110α or p110α also exhibited increased kinase activity (Jaiswal et al., 2009). Philp et al. (2001) described a recurrent intronic PIK3R1 mutation in ovarian cancer cells; the mutation caused skipping of exon 13, resulting in deletion of residues 551-670 within the iSH2/cSH2 domains of p85α, and was associated with increased PI3K activity. - Hyperphosphorylation of AKT: Mutants of p85α that retained the ability to bind p110α also lead to increased phosphorylation of AKT (Jaiswal et al., 2009; Cheung et al., 2011; Urick et al., 2011). - Dysregulation of PTEN stability: Cheung et al., (2011) showed that the p85α-E160X mutant, which was present in an endometrial tumor and truncates p85α within the BH domain, is associated with reduced stability of the PTEN protein.
Implicated in Endometrial cancer Oncogenesis Somatic mutations in PIK3R1 have been observed in 19%-43% of endometrioid endometrial carcinomas (Cheung et al., 2011; Urick et al., 2011), in 8% of serous endometrial carcinomas (Urick et al., 2011), in 20% of clear cell endometrial carcinomas (Urick et al., 2011), and in 6% of endometrial carcinosarcomas (Cheung et al., 2011). Mutations in PIK3R1 tended to be mutually exclusive with mutations in PIK3CA, which encodes the catalytic subunit of PI3K, but co-occurred with mutations in PTEN, and KRAS (Cheung et al., 2011; Urick et al., 2011).
Glioblastoma Oncogenesis Somatic mutations in PIK3R1 have been reported in 7% (20 of 276) of glioblastomas (Mizoughi et al.,

PIK3R1 (phosphoinositide-3-kinase, regulatory subunit 1 (alpha)) Bell DW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 881
2004; Parsons et al., 2008; The Cancer Genome Research Atlas Network, 2008; Park et al., 2010b). No amplification or overexpression of PIK3R1 was observed among 103 glioblastomas (Knobbe et al., 2003).
Colorectal cancer Oncogenesis Somatic mutations in PIK3R1 have been reported in 4% (10 of 228) of colorectal cancers (Philp et al., 2001; Jaiswal et al., 2009; Park et al., 2010b), and in a colorectal cancer cell line (Shi et al., 2006).
Ovarian cancer Oncogenesis Somatic mutations in PIK3R1 have been reported in 2% (5 of 257) of ovarian cancers (Philp et al., 2001; Jaiswal et al., 2009; Kan et al., 2010; Park et al., 2010b; The Cancer Genome Atlas Research Network, 2011).
Breast cancer Oncogenesis Somatic mutations in PIK3R1 have been reported in 2% (10 of 500) of breast cancers (Sjoblom et al., 2006; Jaiswal et al., 2009; Kan et al., 2010; Park et al., 2010b; Jiao et al., 2012; Shah et al., 2012).
Urothelial cancer Oncogenesis Somatic mutations in PIK3R1 have been reported in 0.7% (1 of 145) of urothelial cancers (Sjodahl et al., 2011).
Squamous cell carcinoma of the skin Oncogenesis Somatic mutations in PIK3R1 have been reported in 8% (1 of 9) squamous cell carcinoma of the skin (Park et al., 2010b).
Pancreatic cancer Oncogenesis Somatic mutations in PIK3R1 have been reported in 16% (1 of 53) of pancreatic cancers (Jones et al., 2008; Jaiswal et al., 2009; Kan et al., 2010).
Various human cancers Oncogenesis By expression profiling, reduced expression of PIK3R1 has been noted in cancers of the prostate, lung, bladder, ovary, breast, and liver (Taniguchi et al., 2010).
References Dhand R, Hara K, Hiles I, Bax B, Gout I, Panayotou G, Fry MJ, Yonezawa K, Kasuga M, Waterfield MD. PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 1994 Feb 1;13(3):511-21
Antonetti DA, Algenstaedt P, Kahn CR. Insulin receptor substrate 1 binds two novel splice variants of the regulatory subunit of phosphatidylinositol 3-kinase in muscle and brain. Mol Cell Biol. 1996 May;16(5):2195-203
Inukai K, Anai M, Van Breda E, Hosaka T, Katagiri H, Funaki M, Fukushima Y, Ogihara T, Yazaki Y, Kikuchi, Oka Y, Asano T. A novel 55-kDa regulatory subunit for phosphatidylinositol 3-kinase structurally similar to p55PIK Is generated by alternative splicing of the p85alpha gene. J Biol Chem. 1996 Mar 8;271(10):5317-20
Liang J, Chen JK, Schreiber ST, Clardy J. Crystal structure of P13K SH3 domain at 20 angstroms resolution. J Mol Biol. 1996 Apr 5;257(3):632-43
Musacchio A, Cantley LC, Harrison SC. Crystal structure of the breakpoint cluster region-homology domain from phosphoinositide 3-kinase p85 alpha subunit. Proc Natl Acad Sci U S A. 1996 Dec 10;93(25):14373-8
Nolte RT, Eck MJ, Schlessinger J, Shoelson SE, Harrison SC. Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain and its phosphopeptide complexes. Nat Struct Biol. 1996 Apr;3(4):364-74
Inukai K, Funaki M, Ogihara T, Katagiri H, Kanda A, Anai M, Fukushima Y, Hosaka T, Suzuki M, Shin BC, Takata K, Yazaki Y, Kikuchi M, Oka Y, Asano T. p85alpha gene generates three isoforms of regulatory subunit for phosphatidylinositol 3-kinase (PI 3-Kinase), p50alpha, p55alpha, and p85alpha, with different PI 3-kinase activity elevating responses to insulin. J Biol Chem. 1997 Mar 21;272(12):7873-82
Baier LJ, Wiedrich C, Hanson RL, Bogardus C. Variant in the regulatory subunit of phosphatidylinositol 3-kinase (p85alpha): preliminary evidence indicates a potential role of this variant in the acute insulin response and type 2 diabetes in Pima women. Diabetes. 1998 Jun;47(6):973-5
Hoedemaeker FJ, Siegal G, Roe SM, Driscoll PC, Abrahams JP. Crystal structure of the C-terminal SH2 domain of the p85alpha regulatory subunit of phosphoinositide 3-kinase: an SH2 domain mimicking its own substrate. J Mol Biol. 1999 Oct 1;292(4):763-70
Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJ, Phillips WA. The phosphatidylinositol 3'-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001 Oct 15;61(20):7426-9
Almind K, Delahaye L, Hansen T, Van Obberghen E, Pedersen O, Kahn CR. Characterization of the Met326Ile variant of phosphatidylinositol 3-kinase p85alpha. Proc Natl Acad Sci U S A. 2002 Feb 19;99(4):2124-8
Mauvais-Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ, Iannacone M, Accili D, Cantley LC, Kahn CR. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest. 2002 Jan;109(1):141-9
Knobbe CB, Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3'-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003 Oct;13(4):507-18
Chamberlain MD, Berry TR, Pastor MC, Anderson DH. The p85alpha subunit of phosphatidylinositol 3'-kinase binds to and stimulates the GTPase activity of Rab proteins. J Biol Chem. 2004 Nov 19;279(47):48607-14
Chen D, Mauvais-Jarvis F, Bluher M, Fisher SJ, Jozsi A, Goodyear LJ, Ueki K, Kahn CR. p50alpha/p55alpha phosphoinositide 3-kinase knockout mice exhibit enhanced insulin sensitivity. Mol Cell Biol. 2004 Jan;24(1):320-9
Fu Z, Aronoff-Spencer E, Wu H, Gerfen GJ, Backer JM. The iSH2 domain of PI 3-kinase is a rigid tether for p110 and not a conformational switch. Arch Biochem Biophys. 2004 Dec 15;432(2):244-51

PIK3R1 (phosphoinositide-3-kinase, regulatory subunit 1 (alpha)) Bell DW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 882
Mizoguchi M, Nutt CL, Mohapatra G, Louis DN. Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol. 2004 Oct;14(4):372-7
Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol. 2005 Mar;25(5):1596-607
Elis W, Lessmann E, Oelgeschlager M, Huber M. Mutations in the inter-SH2 domain of the regulatory subunit of phosphoinositide 3-kinase: effects on catalytic subunit binding and holoenzyme function. Biol Chem. 2006 Dec;387(12):1567-73
Shi BH, Nashimoto T, Andoh R, Konishi H, Kobayashi M, Xu Q, Ihara S, Fukui Y. Mutation of the PI3' kinase gene in a human colon carcinoma cell line, HCC2998. DNA Cell Biol. 2006 Jul;25(7):399-405
Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006 Oct 13;314(5797):268-74
Taniguchi CM, Tran TT, Kondo T, Luo J, Ueki K, Cantley LC, Kahn CR. Phosphoinositide 3-kinase regulatory subunit p85alpha suppresses insulin action via positive regulation of PTEN. Proc Natl Acad Sci U S A. 2006 Aug 8;103(32):12093-7
Geering B, Cutillas PR, Nock G, Gharbi SI, Vanhaesebroeck B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci U S A. 2007 May 8;104(19):7809-14
Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007 Dec 14;318(5857):1744-8
Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, Schneidman-Duhovny D, Wolfson HJ, Backer JM, Williams RL. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007 Jul 13;317(5835):239-42
Chamberlain MD, Chan T, Oberg JC, Hawrysh AD, James KM, Saxena A, Xiang J, Anderson DH. Disrupted RabGAP function of the p85 subunit of phosphatidylinositol 3-kinase results in cell transformation. J Biol Chem. 2008 Jun 6;283(23):15861-8
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008 Sep 26;321(5897):1801-6
Li L, Plummer SJ, Thompson CL, Tucker TC, Casey G. Association between phosphatidylinositol 3-kinase regulatory subunit p85alpha Met326Ile genetic polymorphism and colon cancer risk. Clin Cancer Res. 2008 Feb 1;14(3):633-7
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin
R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008 Sep 26;321(5897):1807-12
. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 Oct 23;455(7216):1061-8
Bittinger S, Alexiadis M, Fuller PJ. Expression status and mutational analysis of the PTEN and P13K subunit genes in ovarian granulosa cell tumors. Int J Gynecol Cancer. 2009 Apr;19(3):339-42
Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring P, Dela Vega T, Kenski DM, Bowman KK, Lorenzo M, Li H, Wu J, Modrusan Z, Stinson J, Eby M, Yue P, Kaminker JS, de Sauvage FJ, Backer JM, Seshagiri S. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009 Dec 8;16(6):463-74
Wu H, Shekar SC, Flinn RJ, El-Sibai M, Jaiswal BS, Sen KI, Janakiraman V, Seshagiri S, Gerfen GJ, Girvin ME, Backer JM. Regulation of Class IA PI 3-kinases: C2 domain-iSH2 domain contacts inhibit p85/p110alpha and are disrupted in oncogenic p85 mutants. Proc Natl Acad Sci U S A. 2009 Dec 1;106(48):20258-63
Chagpar RB, Links PH, Pastor MC, Furber LA, Hawrysh AD, Chamberlain MD, Anderson DH. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2010 Mar 23;107(12):5471-6
Chamberlain MD, Oberg JC, Furber LA, Poland SF, Hawrysh AD, Knafelc SM, McBride HM, Anderson DH. Deregulation of Rab5 and Rab4 proteins in p85R274A-expressing cells alters PDGFR trafficking. Cell Signal. 2010 Oct;22(10):1562-75
Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, Teague JW, Stratton MR, Futreal PA. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010 Jan;38(Database issue):D652-7
Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP, Peters BA, Pujara K, Cordes S, Davis DP, Carlton VE, Yuan W, Li L, Wang W, Eigenbrot C, Kaminker JS, Eberhard DA, Waring P, Schuster SC, Modrusan Z, Zhang Z, Stokoe D, de Sauvage FJ, Faham M, Seshagiri S. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010 Aug 12;466(7308):869-73
Park SW, Zhou Y, Lee J, Lu A, Sun C, Chung J, Ueki K, Ozcan U. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat Med. 2010a Apr;16(4):429-37
Park SW, Kang MR, Eom HS, Han JY, Ahn CH, Kim SS, Lee SH, Yoo NJ. Somatic mutation of PIK3R1 gene is rare in common human cancers. Acta Oncol. 2010b;49(1):125-7
Sun M, Hillmann P, Hofmann BT, Hart JR, Vogt PK. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc Natl Acad Sci U S A. 2010 Aug 31;107(35):15547-52
Taniguchi CM, Winnay J, Kondo T, Bronson RT, Guimaraes AR, Alemán JO, Luo J, Stephanopoulos G, Weissleder R, Cantley LC, Kahn CR. The phosphoinositide 3-kinase regulatory subunit p85alpha can exert tumor suppressor properties through negative regulation of growth factor signaling. Cancer Res. 2010 Jul 1;70(13):5305-15

PIK3R1 (phosphoinositide-3-kinase, regulatory subunit 1 (alpha)) Bell DW
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 883
Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010 May;11(5):329-41
Winnay JN, Boucher J, Mori MA, Ueki K, Kahn CR. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat Med. 2010 Apr;16(4):438-45
Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G, Velculescu VE, Oba-Shinjo SM, Marie SK, Vogelstein B, Bigner D, Yan H, Papadopoulos N, Kinzler KW. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011 Sep 9;333(6048):1453-5
Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, Ju Z, Cantley LC, Scherer SE, Liang H, Lu KH, Broaddus RR, Mills GB. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011 Jul;1(2):170-85
Lee JY, Chiu YH, Asara J, Cantley LC. Inhibition of PI3K binding to activators by serine phosphorylation of PI3K regulatory subunit p85alpha Src homology-2 domains. Proc Natl Acad Sci U S A. 2011 Aug 23;108(34):14157-62
Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, Nikolsky Y, Hartigan J, Smith DR, Gerhard DS, Fults DW, VandenBerg S, Berger MS, Marie SK, Shinjo SM, Clara C, Phillips PC, Minturn JE, Biegel JA, Judkins AR, Resnick AC, Storm PB, Curran T, He Y, Rasheed BA, Friedman HS, Keir ST, McLendon R, Northcott PA, Taylor MD, Burger PC, Riggins GJ, Karchin R, Parmigiani G, Bigner DD, Yan H, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011 Jan 28;331(6016):435-9
Sjödahl G, Lauss M, Gudjonsson S, Liedberg F, Halldén C, Chebil G, Månsson W, Höglund M, Lindgren D. A systematic study of gene mutations in urothelial carcinoma; inactivating mutations in TSC2 and PIK3R1. PLoS One. 2011 Apr 14;6(4):e18583
. Integrated genomic analyses of ovarian carcinoma. Nature. 2011 Jun 29;474(7353):609-15
Urick ME, Rudd ML, Godwin AK, Sgroi D, Merino M, Bell DW. PIK3R1 (p85α) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011 Jun 15;71(12):4061-7
Comb WC, Hutti JE, Cogswell P, Cantley LC, Baldwin AS. p85α SH2 domain phosphorylation by IKK promotes feedback inhibition of PI3K and Akt in response to cellular starvation. Mol Cell. 2012 Mar 30;45(6):719-30
Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang YD, Coustan-Smith E, Smith AM, Perez EE, Murray PJ. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J Exp Med. 2012 Mar 12;209(3):463-70
Jiao X, Wood LD, Lindman M, Jones S, Buckhaults P, Polyak K, Sukumar S, Carter H, Kim D, Karchin R, Sjöblom T. Somatic mutations in the Notch, NF-KB, PIK3CA, and Hedgehog pathways in human breast cancers. Genes Chromosomes Cancer. 2012 May;51(5):480-9
Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW, Donahue A, Downing SR, Frampton GM, Garcia L, Juhn F, Mitchell KC, White E, White J, Zwirko Z, Peretz T, Nechushtan H, Soussan-Gutman L, Kim J, Sasaki H, Kim HR, Park SI, Ercan D, Sheehan CE, Ross JS, Cronin MT, Jänne PA, Stephens PJ. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012 Feb 12;18(3):382-4
Mellor P, Furber LA, Nyarko JN, Anderson DH. Multiple roles for the p85α isoform in the regulation and function of PI3K signalling and receptor trafficking. Biochem J. 2012 Jan 1;441(1):23-37
Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SW, Morin GB, Watson P, Gelmon K, Chia S, Chin SF, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012 Apr 4;486(7403):395-9
This article should be referenced as such:
Bell DW. PIK3R1 (phosphoinositide-3-kinase, regulatory subunit 1 (alpha)). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):876-883.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 884
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
CDH17 (cadherin 17, LI cadherin (liver-intestine)) Yiping Rong, Nikki P Lee, John M Luk
pRED China Oncology, Roche R&D Center (China) Ltd, Shanghai, China (YR), Department of Surgery, The University of Hong Kong, Pokfulam, Hong Kong (NPL), pRED China Oncology, Roche R&D Center (China) Ltd, Shanghai, China; Department of Surgery, The University of Hong Kong, Pokfulam, Hong Kong (JML)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/CDH17ID40020ch8q22.html DOI: 10.4267/2042/48358
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: CDH16, HPT-1, HPT1
HGNC (Hugo): CDH17
Location: 8q22.1
DNA/RNA Description Human CDH17 DNA contains 90138 bp composed of 18 exons (Gessner and Tauber, 2000; Wendeler et al., 2006).
Transcription Two transcripts (NM_001144663.1 and NM_004063.3) encode the same protein according to Entrez gene. 2499 bp open reading frame.
Protein Description Cadherins are calcium-dependent cell-cell adhesion molecules which play important roles in organ development, the maintenance of tissue integrity and cancer development (Pokutta and Weis, 2007; Berx and van Roy, 2009). Cadherin 17 (CDH17) is
a transmembrane glycoprotein with seven extracellular cadherin repeats. The cytoplasmic domain of human CDH17 only has 23 amino acids, whereas other classical cadherins contain 150 to 160 conserved amino acids forming complexes with catenins (Gessner and Tauber, 2000; Lee et al., 2010). CDH17 belongs to seven-domain (7D) cadherin subfamily which shares low sequence homology with the classical cadherins, such as E-cadherin. The structure difference of CDH17 makes this molecule unique among the known classical cadherin family members (Nollet et al., 2000; Angst et al., 2001). Recent work suggests its role in tumor progression and cancer prognosis (Liu et al., 2009).
Expression In rats, CDH17 is expressed in the liver and small intestine (Berndorff et al., 1994). In mouse and human, CDH17 is highly expressed in the small intestine and colon (Angres et al., 2001; Takamura et al., 2004), but absent or very low level in other organs, such as liver, heart and kidney etc. It is also linked predominantly to a high incidence of tumorigenesis in the human liver, stomach, intestine and pancreas by displaying an aberrant expression in their cancerous state (Lee et al., 2010).
Figure 1. Cadherin 17 (CDH17) DNA with introns and exons.

CDH17 (cadherin 17, LI cadherin (liver-intestine)) Rong Y, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 885
Figure 2. A schematic diagram illustrates the structural feature of cadherin-17 (CDH17) in having seven cadherin repeats (EC1-EC7) at the extracellular amino-terminus (NH2), followed by a transmembrane region and a short cytoplasmic domain at the carboxyl-terminus (COOH). Calcium ions (denoted by red dots) are located between cadherin repeats in mammalian CDH17.
Localisation CDH17 is mainly localized on basolateral cell membrane. Overexpressed CDH17 can also be detected throughout the cytoplasm of liver cancer, gastric cancer and colon cells (Wong et al., 2003; Grötzinger et al., 2001; Takamura et al., 2004).
Function CDH17 was originally cloned from rat liver and identified as a novel cell adhesion molecule (Berndorff et al., 1994). Homotypic trans-interaction of CDH17 is dependent on extracellular calcium concentration. It might serve as a calcium-regulated adhesion switch (Wendeler et al., 2007). CDH17 is also reported as an intestinal peptide transporter. It facilitates the oral absorption of beta-lactam antibiotics and angiotensin-converting enzyme inhibitors from the intestine into enterocytes lining the luminal wall (Dantzig et al., 1994). Knockout CDH17 by a mutant CDH17 deficient mouse showed that CDH17 may also participate in B lymphocyte development (Ohnishi et al., 2005). Recent studies suggested CDH17 important roles in tumorigenesis. Overexpression CDH17 can promote tumor growth, while suppression of CDH17 inhibits cancer cell growth, migration and adhesion (Liu et al., 2009).
Homology Human CDH17 shares ~20-30% sequence identity with other cadherin family members, such as cadherin-16 (30%), cadherin-13 (30%), and cadherin-1 (26%). It also shares ~79% identify with cyno-, mouse-, rat-cadherin-17.
Mutations Note No mutation has been reported for CDH17 so far.
Somatic Alternative mRNA splicing isoform of CDH17 was reported in hepatocellular carcinoma patient samples. The isoform skips exon 7 which leads to open reading frame shift. The mRNA isoform is associated with shorter overall survival time. The functions or mechanisms of the isoform in cancer are unclear (Wang et al., 2005).
Implicated in Hepatocellular carcinoma (HCC) Prognosis CDH17 overexpression is detected in approximately ~80% of HCC patients (Liu et al., 2009). The elevated level of CDH17 in HCC is correlated to high serum AFP level, microvascular invasion, and advanced stage tumor, associating with shorter overall survival as well as higher incidence of tumor recurrence, of HCC patients. Over half of HCC patients have genomic amplification of CDH17 gene in their tumors. Alternative mRNA splicing of CDH17 was also reported in half of the HCC patient tumor specimens and associated with shorter overall survival time (Ding et al., 2009; Wong et al., 2003; Wang et al., 2006; Kaposi-Novak et al., 2006). The CDH17 overexpression can also be detected in other tumorigenic conditions including gastric cancer (GC). Therefore, CDH17 expression can be a potential biomarker for HCC and GC.
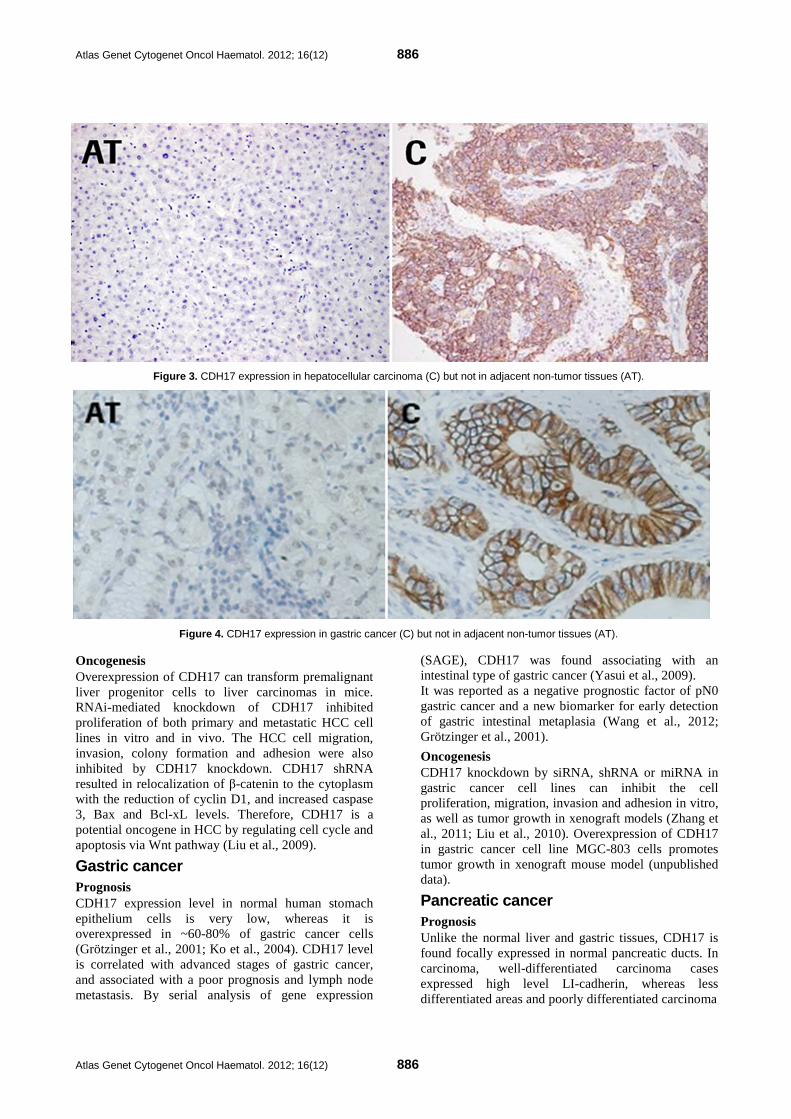
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 886
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 886
Figure 3. CDH17 expression in hepatocellular carcinoma (C) but not in adjacent non-tumor tissues (AT).
Figure 4. CDH17 expression in gastric cancer (C) but not in adjacent non-tumor tissues (AT).
Oncogenesis Overexpression of CDH17 can transform premalignant liver progenitor cells to liver carcinomas in mice. RNAi-mediated knockdown of CDH17 inhibited proliferation of both primary and metastatic HCC cell lines in vitro and in vivo. The HCC cell migration, invasion, colony formation and adhesion were also inhibited by CDH17 knockdown. CDH17 shRNA resulted in relocalization of β-catenin to the cytoplasm with the reduction of cyclin D1, and increased caspase 3, Bax and Bcl-xL levels. Therefore, CDH17 is a potential oncogene in HCC by regulating cell cycle and apoptosis via Wnt pathway (Liu et al., 2009).
Gastric cancer Prognosis CDH17 expression level in normal human stomach epithelium cells is very low, whereas it is overexpressed in ~60-80% of gastric cancer cells (Grötzinger et al., 2001; Ko et al., 2004). CDH17 level is correlated with advanced stages of gastric cancer, and associated with a poor prognosis and lymph node metastasis. By serial analysis of gene expression
(SAGE), CDH17 was found associating with an intestinal type of gastric cancer (Yasui et al., 2009). It was reported as a negative prognostic factor of pN0 gastric cancer and a new biomarker for early detection of gastric intestinal metaplasia (Wang et al., 2012; Grötzinger et al., 2001).
Oncogenesis CDH17 knockdown by siRNA, shRNA or miRNA in gastric cancer cell lines can inhibit the cell proliferation, migration, invasion and adhesion in vitro, as well as tumor growth in xenograft models (Zhang et al., 2011; Liu et al., 2010). Overexpression of CDH17 in gastric cancer cell line MGC-803 cells promotes tumor growth in xenograft mouse model (unpublished data).
Pancreatic cancer Prognosis Unlike the normal liver and gastric tissues, CDH17 is found focally expressed in normal pancreatic ducts. In carcinoma, well-differentiated carcinoma cases expressed high level LI-cadherin, whereas less differentiated areas and poorly differentiated carcinoma

CDH17 (cadherin 17, LI cadherin (liver-intestine)) Rong Y, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 887
cases expressed less or were negative. The high CDH17 expression correlated with good survival in pancreatic ductal adenocarcinoma (Takamura et al., 2003).
Colorectal cancer Prognosis In normal colorectal epithelial cells, CDH17 immunoreactivity was present at the basolateral plasma membrane. In colorectal carcinoma, the expression of CDH17 is diminished in tumor tissues. It can be observed in well-differentiated adenocarcinoma cells with tight cell-cell adhesion, but expression was reduced in dedifferentiated adenocarcinoma cells. Reduced expression of CDH17 in colorectal cancer tissues correlated with dedifferentiation of tumors and poor survival of patients (Takamura et al., 2004; Kwak et al., 2007; Su et al., 2008). The expression patterns of CDH17 in different cancer types suggest its cell-context dependent roles in organs.
References Berndorff D, Gessner R, Kreft B, Schnoy N, Lajous-Petter AM, Loch N, Reutter W, Hortsch M, Tauber R. Liver-intestine cadherin: molecular cloning and characterization of a novel Ca(2+)-dependent cell adhesion molecule expressed in liver and intestine. J Cell Biol. 1994 Jun;125(6):1353-69
Dantzig AH, Hoskins JA, Tabas LB, Bright S, Shepard RL, Jenkins IL, Duckworth DC, Sportsman JR, Mackensen D, Rosteck PR Jr. Association of intestinal peptide transport with a protein related to the cadherin superfamily. Science. 1994 Apr 15;264(5157):430-3
Gessner R, Tauber R. Intestinal cell adhesion molecules. Liver-intestine cadherin. Ann N Y Acad Sci. 2000;915:136-43
Nollet F, Kools P, van Roy F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J Mol Biol. 2000 Jun 9;299(3):551-72
Angres B, Kim L, Jung R, Gessner R, Tauber R. LI-cadherin gene expression during mouse intestinal development. Dev Dyn. 2001 Jun;221(2):182-93
Angst BD, Marcozzi C, Magee AI. The cadherin superfamily: diversity in form and function. J Cell Sci. 2001 Feb;114(Pt 4):629-41
Grötzinger C, Kneifel J, Patschan D, Schnoy N, Anagnostopoulos I, Faiss S, Tauber R, Wiedenmann B, Gessner R. LI-cadherin: a marker of gastric metaplasia and neoplasia. Gut. 2001 Jul;49(1):73-81
Takamura M, Sakamoto M, Ino Y, Shimamura T, Ichida T, Asakura H, Hirohashi S. Expression of liver-intestine cadherin and its possible interaction with galectin-3 in ductal adenocarcinoma of the pancreas. Cancer Sci. 2003 May;94(5):425-30
Wong BW, Luk JM, Ng IO, Hu MY, Liu KD, Fan ST. Identification of liver-intestine cadherin in hepatocellular carcinoma--a potential disease marker. Biochem Biophys Res Commun. 2003 Nov 21;311(3):618-24
Ko S, Chu KM, Luk JM, Wong BW, Yuen ST, Leung SY, Wong J. Overexpression of LI-cadherin in gastric cancer is associated with lymph node metastasis. Biochem Biophys Res Commun. 2004 Jun 25;319(2):562-8
Takamura M, Ichida T, Matsuda Y, Kobayashi M, Yamagiwa S, Genda T, Shioji K, Hashimoto S, Nomoto M, Hatakeyama K, Ajioka Y, Sakamoto M, Hirohashi S, Aoyagi Y. Reduced expression of liver-intestine cadherin is associated with progression and lymph node metastasis of human colorectal carcinoma. Cancer Lett. 2004 Aug 30;212(2):253-9
Ohnishi K, Melchers F, Shimizu T. Lymphocyte-expressed BILL-cadherin/cadherin-17 contributes to the development of B cells at two stages. Eur J Immunol. 2005 Mar;35(3):957-63
Wang XQ, Luk JM, Leung PP, Wong BW, Stanbridge EJ, Fan ST. Alternative mRNA splicing of liver intestine-cadherin in hepatocellular carcinoma. Clin Cancer Res. 2005 Jan 15;11(2 Pt 1):483-9
Kaposi-Novak P, Lee JS, Gòmez-Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest. 2006 Jun;116(6):1582-95
Wang XQ, Luk JM, Garcia-Barcelo M, Miao X, Leung PP, Ho DW, Cheung ST, Lam BY, Cheung CK, Wong AS, Lau SS, So MT, Yu WC, Cai Q, Liu KS, Hui CK, Lau GK, Poon RT, Wong J, Fan ST. Liver intestine-cadherin (CDH17) haplotype is associated with increased risk of hepatocellular carcinoma. Clin Cancer Res. 2006 Sep 1;12(17):5248-52
Wendeler MW, Jung R, Himmelbauer H, Gessner R. Unique gene structure and paralogy define the 7D-cadherin family. Cell Mol Life Sci. 2006 Jul;63(13):1564-73
Kwak JM, Min BW, Lee JH, Choi JS, Lee SI, Park SS, Kim J, Um JW, Kim SH, Moon HY. The prognostic significance of E-cadherin and liver intestine-cadherin expression in colorectal cancer. Dis Colon Rectum. 2007 Nov;50(11):1873-80
Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol. 2007;23:237-61
Wendeler MW, Drenckhahn D, Gessner R, Baumgartner W. Intestinal LI-cadherin acts as a Ca2+-dependent adhesion switch. J Mol Biol. 2007 Jul 6;370(2):220-30
Su MC, Yuan RH, Lin CY, Jeng YM. Cadherin-17 is a useful diagnostic marker for adenocarcinomas of the digestive system. Mod Pathol. 2008 Nov;21(11):1379-86
Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009 Dec;1(6):a003129
Ding ZB, Shi YH, Zhou J, Shi GM, Ke AW, Qiu SJ, Wang XY, Dai Z, Xu Y, Fan J. Liver-intestine cadherin predicts microvascular invasion and poor prognosis of hepatitis B virus-positive hepatocellular carcinoma. Cancer. 2009 Oct 15;115(20):4753-65
Liu LX, Lee NP, Chan VW, Xue W, Zender L, Zhang C, Mao M, Dai H, Wang XL, Xu MZ, Lee TK, Ng IO, Chen Y, Kung HF, Lowe SW, Poon RT, Wang JH, Luk JM. Targeting cadherin-17 inactivates Wnt signaling and inhibits tumor growth in liver carcinoma. Hepatology. 2009 Nov;50(5):1453-63
Yasui W, Oue N, Sentani K, Sakamoto N, Motoshita J. Transcriptome dissection of gastric cancer: identification of novel diagnostic and therapeutic targets from pathology specimens. Pathol Int. 2009 Mar;59(3):121-36
Lee NP, Poon RT, Shek FH, Ng IO, Luk JM. Role of cadherin-17 in oncogenesis and potential therapeutic implications in hepatocellular carcinoma. Biochim Biophys Acta. 2010 Dec;1806(2):138-45
Liu QS, Zhang J, Liu M, Dong WG. Lentiviral-mediated miRNA against liver-intestine cadherin suppresses tumor growth and

Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 888
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 888
invasiveness of human gastric cancer. Cancer Sci. 2010 Aug;101(8):1807-12
Zhang J, Liu QS, Dong WG. Blockade of proliferation and migration of gastric cancer via targeting CDH17 with an artificial microRNA. Med Oncol. 2011 Jun;28(2):494-501
Wang J, Yu JC, Kang WM, Wang WZ, Liu YQ, Gu P. The predictive effect of cadherin-17 on lymph node
micrometastasis in pN0 gastric cancer. Ann Surg Oncol. 2012 May;19(5):1529-34
This article should be referenced as such:
Rong Y, Lee NP, Luk JM. CDH17 (cadherin 17, LI cadherin (liver-intestine)). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):884-888.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 889
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
FPR1 (formyl peptide receptor 1) Jian Huang, Ji Ming Wang
High Altitude Military Medical College, Third Military Medical University, Chongqing, 400038, China (JH), Laboratory of Molecular Immunoregulation, Cancer and inflammation Program, Center for Cancer Research, National Cancer Institute at Frederick, Frederick, MD 21702, USA (JMW)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/FPR1ID44328ch19q13.html DOI : 10.4267/2042/48359
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: FMLP, FPR
HGNC (Hugo): FPR1
Location: 19q13.41
DNA/RNA Note FPR1 is a G protein-coupled receptor (GPCR), originally identified in phagocytic leukocytes, which mediates cell chemotaxis and activation in response to the bacterial chemotactic peptide N-formyl-methionyl-leucyl-phenylalanine (fMLF). A number of host-derived chemotactic agonists of FPR1 have been identified, including formyl peptides potentially released by mitochondria of ruptured cells, Annexin I produced by activated epithelia, and a neutrophil granule protein, cathepsin G. In addition, functional FPR1 has been detected in cells of nonhematopoietic origin, such
as lung epithelial cells and hepatocytes. These findings suggest that FPR1 is involved in a broader spectrum of pathophysiologic processes.
Description Size: 6127 bases.
Transcription All three genes, FPR1, FPR2 and FPR3, are clustered on chromosome 19q13.3. FPR1 is encoded by a 6 kb single copy gene. The open reading frame is intronless but the 5' untranslated region resides in three exons. The start sites for transcription and translation are separated by approximately 5 kb. The FPR1 gene contains three Alu repeats, one in each intron and a third in the 3' flanking region. The proposed promoter contains a nonconsensus TATA box and an inverted CCAAT element.
Pseudogene No known pseudogenes.
This gene is located in formylpeptide receptor gene cluster region including FPR1, FPR2 and FPR3 on chromosome 19p.
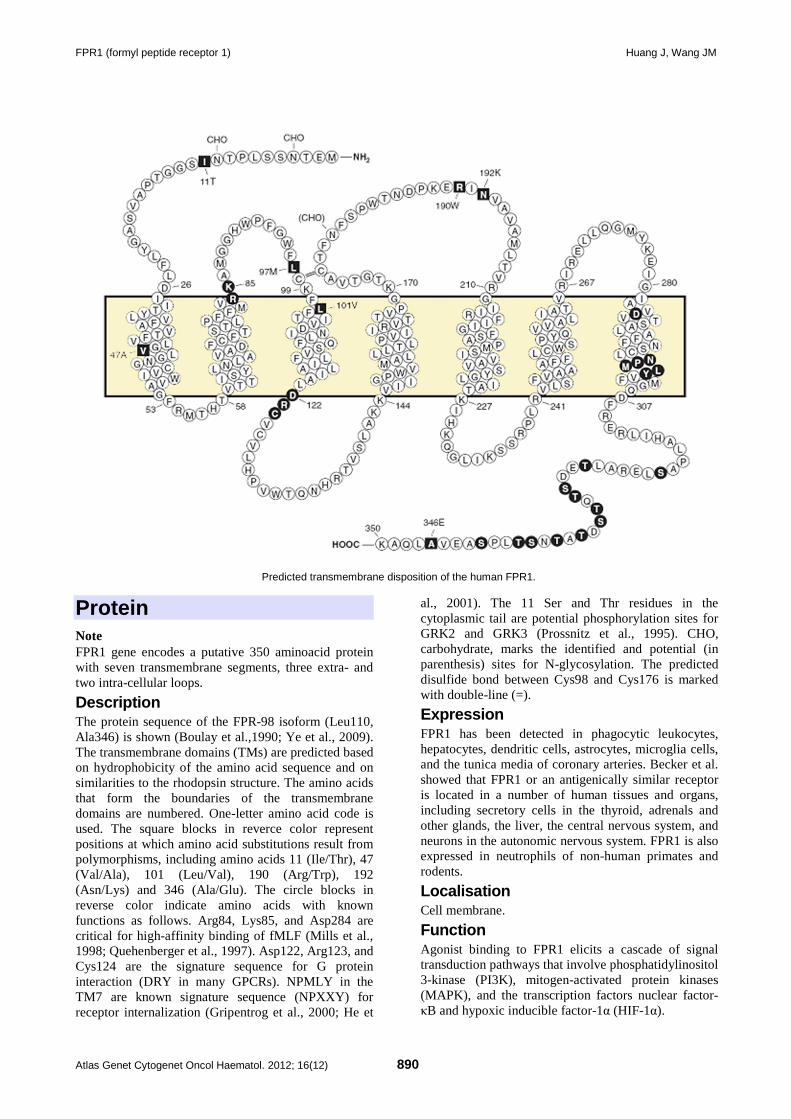
FPR1 (formyl peptide receptor 1) Huang J, Wang JM
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 890
Predicted transmembrane disposition of the human FPR1.
Protein Note FPR1 gene encodes a putative 350 aminoacid protein with seven transmembrane segments, three extra- and two intra-cellular loops.
Description The protein sequence of the FPR-98 isoform (Leu110, Ala346) is shown (Boulay et al.,1990; Ye et al., 2009). The transmembrane domains (TMs) are predicted based on hydrophobicity of the amino acid sequence and on similarities to the rhodopsin structure. The amino acids that form the boundaries of the transmembrane domains are numbered. One-letter amino acid code is used. The square blocks in reverce color represent positions at which amino acid substitutions result from polymorphisms, including amino acids 11 (Ile/Thr), 47 (Val/Ala), 101 (Leu/Val), 190 (Arg/Trp), 192 (Asn/Lys) and 346 (Ala/Glu). The circle blocks in reverse color indicate amino acids with known functions as follows. Arg84, Lys85, and Asp284 are critical for high-affinity binding of fMLF (Mills et al., 1998; Quehenberger et al., 1997). Asp122, Arg123, and Cys124 are the signature sequence for G protein interaction (DRY in many GPCRs). NPMLY in the TM7 are known signature sequence (NPXXY) for receptor internalization (Gripentrog et al., 2000; He et
al., 2001). The 11 Ser and Thr residues in the cytoplasmic tail are potential phosphorylation sites for GRK2 and GRK3 (Prossnitz et al., 1995). CHO, carbohydrate, marks the identified and potential (in parenthesis) sites for N-glycosylation. The predicted disulfide bond between Cys98 and Cys176 is marked with double-line (=).
Expression FPR1 has been detected in phagocytic leukocytes, hepatocytes, dendritic cells, astrocytes, microglia cells, and the tunica media of coronary arteries. Becker et al. showed that FPR1 or an antigenically similar receptor is located in a number of human tissues and organs, including secretory cells in the thyroid, adrenals and other glands, the liver, the central nervous system, and neurons in the autonomic nervous system. FPR1 is also expressed in neutrophils of non-human primates and rodents.
Localisation Cell membrane.
Function Agonist binding to FPR1 elicits a cascade of signal transduction pathways that involve phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinases (MAPK), and the transcription factors nuclear factor-κB and hypoxic inducible factor-1α (HIF-1α).

FPR1 (formyl peptide receptor 1) Huang J, Wang JM
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 891
Amino acid sequence of FPR-WT and localization of the F110S and C126W mutations (Seifert et al., 2001). Shown is the two-dimensional structure of FPR-WT (isoform 26) (27). Amino acids are given in one-letter code. The FPR N terminus (top) faces the extracellular space; the FPR C terminus (bottom) faces the cytosol. The transmembrane domains are included in the boxed area. Extracellular consensus sites for N-glycosylation are shown (Y). The positions of the F110S and C126W mutations are indicated (•). There is a disulfide bridge between the first and second extracellular loops. Note that the consensus sites for N-glycosylation are not altered in FPR-F110S and FPR-C126W.
Because of its expression in cells of the immune system and its interaction with bacterial chemotactic peptides, this receptor was thought to participate in host defense against microbial infection. In addition, FPR1 expressed in highly malignant human glioblastoma promotes tumor progression.
Homology In primates, the sequence of FPR1 is highly conserved. Rabbit and mouse FPR1 share 78 and 76% identity with human FPR1 respectively.
Mutations Two loss of funtion mutations (F110S and C126W) that correlate with localized juvenile periodontitis. The F110S mutation resides in the third transmembrane domain, whereas the C126W mutation resides in the second intracellular loop.
Implicated in Glioblastoma Note Promoting glioblastoma progression.
Prognosis FPR1 protein staining was detected in 11 of 14 grade III anaplastic astrocytoma specimens and six of six grade IV glioblastoma multiforme specimens. Microvessels and necrotic tumor cells were readily visible among FPR1-positive intact tumor cells. In contrast, only two of 13 less aggressive grade II astrocytoma specimens showed positive FPR staining. Thus, FPR expression appears to be associated with a majority of poorly differentiated primary human gliomas of grades III and IV.
Cytogenetics Highly malignant human glioblastoma and anaplastic astrocytoma specimens were stained positively for FPR1. FPR1 was expressed selectively in glioma cell lines with a more highly malignant phenotype. FPR expressed in glioblastoma cell lines mediates cell chemotaxis, proliferation and production of an angiogenic factor, vascular endothelial growth factor (VEGF), in response to agonists released by necrotic tumor cells. Furthermore, FPR in glioblastoma cells activates the receptor for epidermal growth factor (EGFR) by increasing the
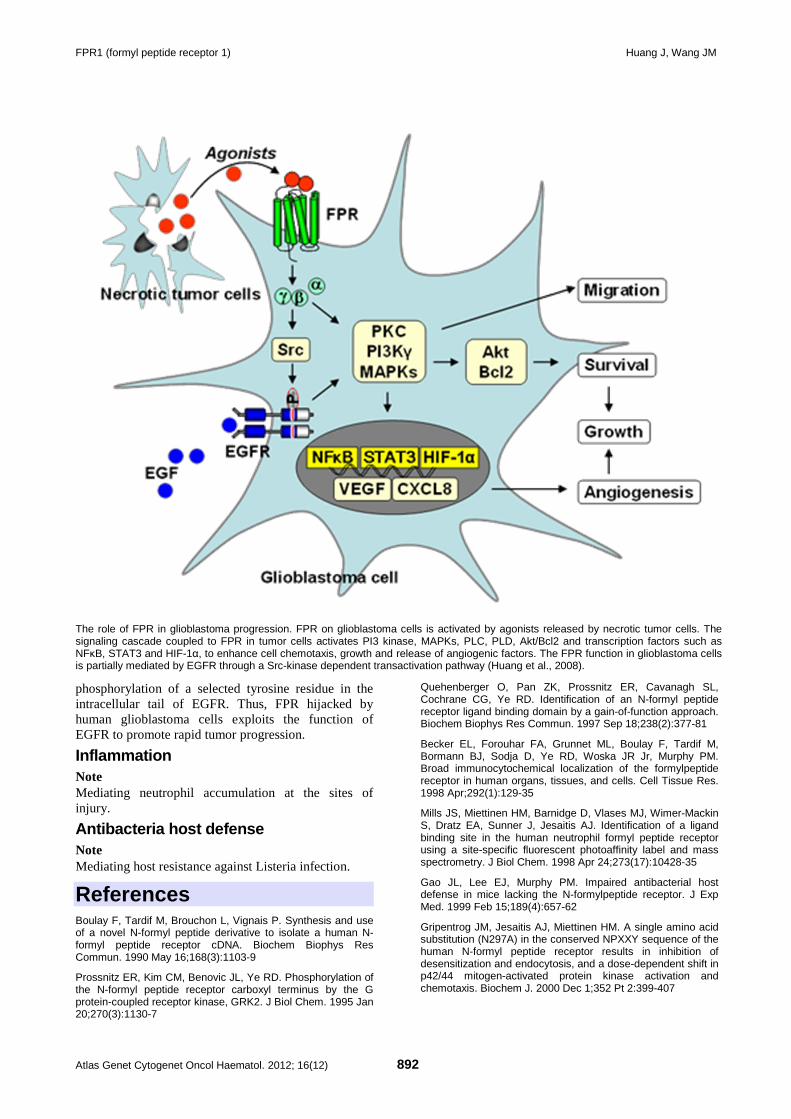
FPR1 (formyl peptide receptor 1) Huang J, Wang JM
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 892
The role of FPR in glioblastoma progression. FPR on glioblastoma cells is activated by agonists released by necrotic tumor cells. The signaling cascade coupled to FPR in tumor cells activates PI3 kinase, MAPKs, PLC, PLD, Akt/Bcl2 and transcription factors such as NFκB, STAT3 and HIF-1α, to enhance cell chemotaxis, growth and release of angiogenic factors. The FPR function in glioblastoma cells is partially mediated by EGFR through a Src-kinase dependent transactivation pathway (Huang et al., 2008).
phosphorylation of a selected tyrosine residue in the intracellular tail of EGFR. Thus, FPR hijacked by human glioblastoma cells exploits the function of EGFR to promote rapid tumor progression.
Inflammation Note Mediating neutrophil accumulation at the sites of injury.
Antibacteria host defense Note Mediating host resistance against Listeria infection.
References Boulay F, Tardif M, Brouchon L, Vignais P. Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochem Biophys Res Commun. 1990 May 16;168(3):1103-9
Prossnitz ER, Kim CM, Benovic JL, Ye RD. Phosphorylation of the N-formyl peptide receptor carboxyl terminus by the G protein-coupled receptor kinase, GRK2. J Biol Chem. 1995 Jan 20;270(3):1130-7
Quehenberger O, Pan ZK, Prossnitz ER, Cavanagh SL, Cochrane CG, Ye RD. Identification of an N-formyl peptide receptor ligand binding domain by a gain-of-function approach. Biochem Biophys Res Commun. 1997 Sep 18;238(2):377-81
Becker EL, Forouhar FA, Grunnet ML, Boulay F, Tardif M, Bormann BJ, Sodja D, Ye RD, Woska JR Jr, Murphy PM. Broad immunocytochemical localization of the formylpeptide receptor in human organs, tissues, and cells. Cell Tissue Res. 1998 Apr;292(1):129-35
Mills JS, Miettinen HM, Barnidge D, Vlases MJ, Wimer-Mackin S, Dratz EA, Sunner J, Jesaitis AJ. Identification of a ligand binding site in the human neutrophil formyl peptide receptor using a site-specific fluorescent photoaffinity label and mass spectrometry. J Biol Chem. 1998 Apr 24;273(17):10428-35
Gao JL, Lee EJ, Murphy PM. Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J Exp Med. 1999 Feb 15;189(4):657-62
Gripentrog JM, Jesaitis AJ, Miettinen HM. A single amino acid substitution (N297A) in the conserved NPXXY sequence of the human N-formyl peptide receptor results in inhibition of desensitization and endocytosis, and a dose-dependent shift in p42/44 mitogen-activated protein kinase activation and chemotaxis. Biochem J. 2000 Dec 1;352 Pt 2:399-407

FPR1 (formyl peptide receptor 1) Huang J, Wang JM
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 893
He R, Browning DD, Ye RD. Differential roles of the NPXXY motif in formyl peptide receptor signaling. J Immunol. 2001 Mar 15;166(6):4099-105
Seifert R, Wenzel-Seifert K. Defective Gi protein coupling in two formyl peptide receptor mutants associated with localized juvenile periodontitis. J Biol Chem. 2001 Nov 9;276(45):42043-9
Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002 Nov;23(11):541-8
Zhou Y, Bian X, Le Y, Gong W, Hu J, Zhang X, Wang L, Iribarren P, Salcedo R, Howard OM, Farrar W, Wang JM. Formylpeptide receptor FPR and the rapid growth of malignant human gliomas. J Natl Cancer Inst. 2005 Jun 1;97(11):823-35
Huang J, Hu J, Bian X, Chen K, Gong W, Dunlop NM, Howard OM, Wang JM. Transactivation of the epidermal growth factor receptor by formylpeptide receptor exacerbates the malignant
behavior of human glioblastoma cells. Cancer Res. 2007 Jun 15;67(12):5906-13
Huang J, Chen K, Gong W, Zhou Y, Le Y, Bian X, Wang JM. Receptor "hijacking" by malignant glioma cells: a tactic for tumor progression. Cancer Lett. 2008 Aug 28;267(2):254-61
Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009 Jun;61(2):119-61
This article should be referenced as such:
Huang J, Wang JM. FPR1 (formyl peptide receptor 1). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):889-893.
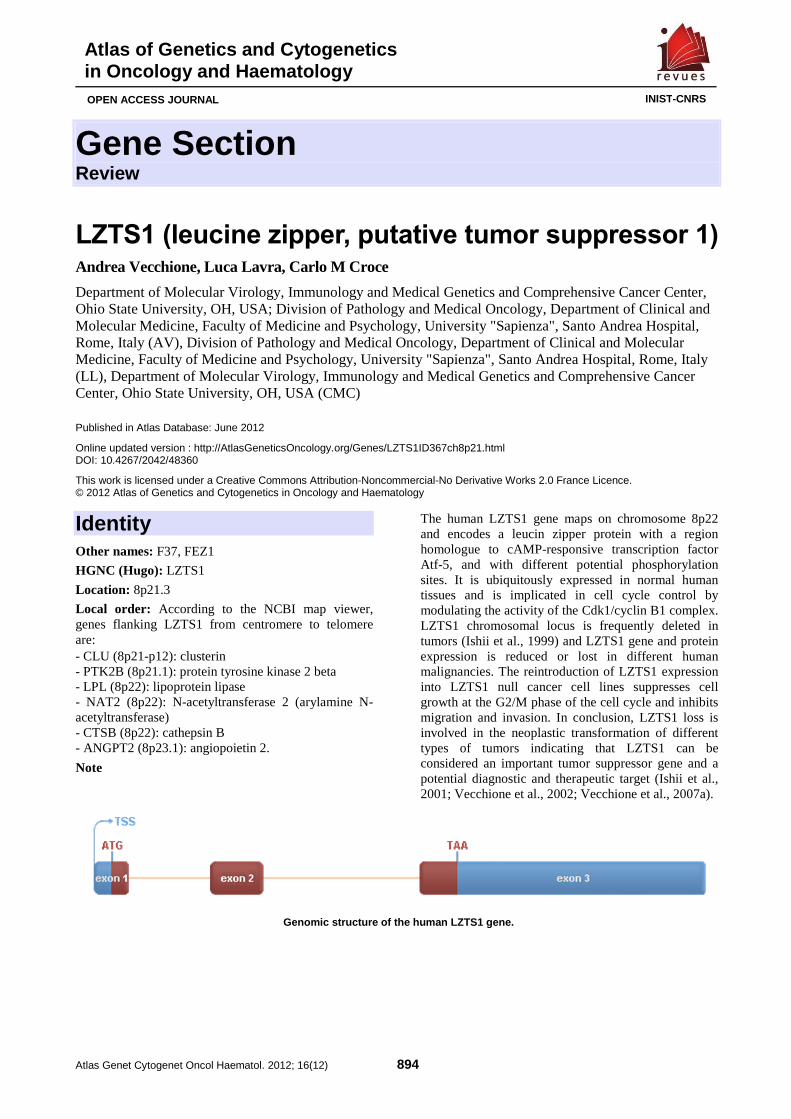
Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 894
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
LZTS1 (leucine zipper, putative tumor suppressor 1) Andrea Vecchione, Luca Lavra, Carlo M Croce
Department of Molecular Virology, Immunology and Medical Genetics and Comprehensive Cancer Center, Ohio State University, OH, USA; Division of Pathology and Medical Oncology, Department of Clinical and Molecular Medicine, Faculty of Medicine and Psychology, University "Sapienza", Santo Andrea Hospital, Rome, Italy (AV), Division of Pathology and Medical Oncology, Department of Clinical and Molecular Medicine, Faculty of Medicine and Psychology, University "Sapienza", Santo Andrea Hospital, Rome, Italy (LL), Department of Molecular Virology, Immunology and Medical Genetics and Comprehensive Cancer Center, Ohio State University, OH, USA (CMC)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/LZTS1ID367ch8p21.html DOI: 10.4267/2042/48360
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: F37, FEZ1
HGNC (Hugo): LZTS1
Location: 8p21.3
Local order: According to the NCBI map viewer, genes flanking LZTS1 from centromere to telomere are: - CLU (8p21-p12): clusterin - PTK2B (8p21.1): protein tyrosine kinase 2 beta - LPL (8p22): lipoprotein lipase - NAT2 (8p22): N-acetyltransferase 2 (arylamine N-acetyltransferase) - CTSB (8p22): cathepsin B - ANGPT2 (8p23.1): angiopoietin 2.
Note
The human LZTS1 gene maps on chromosome 8p22 and encodes a leucin zipper protein with a region homologue to cAMP-responsive transcription factor Atf-5, and with different potential phosphorylation sites. It is ubiquitously expressed in normal human tissues and is implicated in cell cycle control by modulating the activity of the Cdk1/cyclin B1 complex. LZTS1 chromosomal locus is frequently deleted in tumors (Ishii et al., 1999) and LZTS1 gene and protein expression is reduced or lost in different human malignancies. The reintroduction of LZTS1 expression into LZTS1 null cancer cell lines suppresses cell growth at the G2/M phase of the cell cycle and inhibits migration and invasion. In conclusion, LZTS1 loss is involved in the neoplastic transformation of different types of tumors indicating that LZTS1 can be considered an important tumor suppressor gene and a potential diagnostic and therapeutic target (Ishii et al., 2001; Vecchione et al., 2002; Vecchione et al., 2007a).
Genomic structure of the human LZTS1 gene.

LZTS1 (leucine zipper, putative tumor suppressor 1) Vecchione A, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 895
Schematic representation of LZTS1 protein. The leucine residues position of leucine-zipper motifs are indicated by the gray bars.
DNA/RNA Note NC_000008.10: 20103676 - 20112803 bp (Entrez-Gene).
Description According to Entrez-Gene, LZTS1 gene extends over 9 kb (9128 bases) and consists of 3 exons.
Transcription mRNA size: 5459 bp (NM_021020.2); open reading frame: 1791 bp (NP_066300.1). LZTS1 mRNA is highly expressed in testis, prostate, spleen, thymus, ovary and brain. It has been detected at lower levels in heart, placenta, small intestine, colon, liver, kidney, skeletal muscle and pancreas. LZTS1 gene is not expressed in primary tumors from breast and prostate and in different cancer cell lines (Ishii et al., 1999).
Pseudogene No LZTS1 pseudogenes have been reported.
Protein Description LZTS1 gene encodes a 596-aa protein of 67 kDa. The protein contains two leucine-zipper motifs, multiple potential phosphorylation sites for different kinases (e.g. PKA, CDC2 and PKC) and a domain with 32% identity to the DNA binding domain of the cAMP-responsive transcription factor Atf5. LZTS1 lacks the DNA recognition domain usually found in transcription factors carrying a leucine-zipper motive (Ishii et al., 1999; Ishii et al., 2001; Vecchione et al., 2007b).
Expression LZTS1 is ubiquitously expressed in all normal human tissues. LZTS1 protein expression is lost or reduced in different primary tumors.
Localisation Main sub-cellular localizations: plasma membrane and cytoplasm. Additional localizations: nucleoli (observed in U2-OS cells) and Golgi apparatus (observed in A-431 cells) (Barbe et al., 2008).
Function Cell cycle regulation. It has been demonstrated that Lzts1-/- mouse embryonic fibroblasts (MEF) have a faster M phase, associated with a lower cyclin B1/Cdk1 activity. During prophase the interaction between LZTS1 and Cdc25C, a phosphatase implicated in regulation of Cdk1 activity, allows the expression of high levels of Cdc25C and enhances its activity, resulting in normal progression from prophase to metaphase. In Lzts1 deficient cells during prophase Cdc25C is rapidly ubiquitinated and degraded, thus determining a lower activity of the cyclin B1/Cdk1 complex. This results in a faster cellular progression through prophase and prometaphase and, frequently, in chromosome missegregation (Vecchione et al., 2007b).
Homology The LZTS1 gene is conserved in the organisms listed below: - Pan troglodytes (LZTS1) (Gene ID: 464034) - Macaca mulatta (LZTS1) (Gene ID: 705724) - Mus musculus (Lzts1) (Gene ID: 211134) - Rattus norvegicus (Lzts1) (Gene ID: 266711) - Bos taurus (LZTS1) (Gene ID: 539634) - Equus caballus (LZTS1) (Gene ID: 100053630) - Canis lupus familiaris (LZTS1) (Gene ID: 486136) - Monodelphis domestica (LZTS1) (Gene ID: 100030407) - Ornithorhynchus anatinus (LZTS1) (Gene ID: 100073437) - Gallus gallus (LZTS1) (Gene ID: 431331) - Danio rerio (si:dkey-63d15.13) (Gene ID: 569281).
Mutations Note Sequence analysis of LZTS1 ORF, performed in different type of cancers revealed the presence of the somatic point mutations listed hereinafter (Vecchione et al., 2001; Knowles et al., 2005): - S29P: (TCC->CCC) reported in a primary esophageal tumor - K119E: (AAG->GAG) reported in a primary esophageal tumor - Q501Stop: (CAG->TAG) reported in PC3 (prostate cancer cell line)

LZTS1 (leucine zipper, putative tumor suppressor 1) Vecchione A, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 896
- H17R: (CAC->CGC) reported in a diffuse-type gastric carcinoma - L113P: (CTA->CCA) reported in a bladder tumor sample - G374S: (GGC->AGC) reported in A1698 (bladder cancer cell line) - L475V: (CTG->GTG) reported in SCaBER (bladder cancer cell line). Internally truncated transcripts described in different cancers (Ishii et al., 1999): - Frameshift deletion 1546-1542 (exons affected: 1,2,3) reported in esophagus cancer - In-frame deletion 558-1715 (exons affected: 2,3) reported in esophagus cancer - In-frame deletion 1366-1641 (exon affected: 3) reported in prostate cancer - In-frame deletion 1402-1578 (exon affected: 3) reported in esophagus and prostate cancer and in acute lymphoblastic leukemia - In-frame deletion 1417-1515 (exon affected: 3) reported in melanoma - In-frame deletion 1516-1584 (exon affected: 3) reported in melanoma. A detailed DNA sequence analysis of LZTS1 gene performed in germline DNA extracted from a screening panel of sporadic and hereditary prostate cancers revealed the presence of 24 SNP. The four SNP listed below have a statistically significant association with sporadic prostate cancer (Hawkins et al., 2002): - A allele of WF101-010 (2812G → A) - C allele of WF101-012 (2883T → C) - C allele of WF101-031 (3329C → T) - G allele of WF101-014 (4361C → T).
Implicated in Prostate cancer Note The DNA sequence analysis of LZTS1 performed on sporadic and hereditary prostate cancer (HPC) samples and unaffected controls revealed the presence of several SNPs associated with prostate cancer (Hawkins et al., 2002). Over-expression of LZTS1 cDNA modulates colony-forming efficiency and proliferation in different prostate cancer cell lines (Cabeza-Arvelaiz et al., 2001).
Ovarian cancer Note An immunohistochemical analysis of LZTS1 protein expression performed in ovarian carcinomas tissue samples demonstrated that cytoplasmic staining for FEZ1 protein was absent or drastically reduced in 38% of cases (Califano et al., 2010). In addition, homozygous deletions at LZTS1 locus has been detected in advanced ovarian clear cell carcinomas (Kuo et al., 2010).
Oral squamous cell carcinoma Note Reduced LZTS1 gene expression has been reported in 35% of oral squamous cell carcinoma (SSC) samples and in oral SSC-derived cell lines (Ono et al., 2003).
Uveal melanoma Note A gene expression profiling performed on 53 primary uveal melanomas by array-based comparative genomic hybridization demonstrated that LZTS1 expression was reduced in rapidly metastasizing and metastatic uveal melanomas but not in slowly metastasizing and non metastasizing uveal melanomas. Moreover overexpression of LZTS1 in metastasizing uveal melanoma-derived cells inhibited their motility and invasion (Onken et al., 2008).
Lung carcinoma Note The immunohistochemical analysis of LZTS1 expression in 103 primary lung cancer specimens demonstrated absence or strong reduction in respectively in more that 42% of cases. A positive correlation between loss of LZTS1 and tumor grading, and between strong LZTS1 expression and mortality rate reduction was also observed (Nonaka et al., 2005). Moreover reduced LZTS1 expression was also detected in several lung cancer derived cell lines (Toyooka et al., 2002).
Gastric carcinoma Note The immunohistochemical analysis of LZTS1 expression, performed in 88 gastric cancer specimens demonstrated that it is lost or significantly reduced in more than 44% of cases. In addition, DNA allelotyping analysis at the LZTS1 locus showed LOH and microsatellite instability respectively in 18% and 23,5% of cases (Vecchione et al., 2001).
Breast carcinoma Note LZTS1 gene expression was reduced in breast primary tumors and breast cancer cell lines. The immunohistochemical analysis of LZTS1 expression demonstrated that LZTS1 was absent or down-regulated in primary breast carcinomas compared with normal breast. Moreover, reduced LZTS1 expression was significantly correlated with high histologic grade, lymph node metastasis, and poor prognosis. In addition, DNA methylation analysis demonstrated that LZTS1 loss of expression in breast tumors is correlated with gene methylation. Moreover, overexpression of LZTS1 in breast cancer cell lines inhibits cell proliferation, migration and invasion, and induces morphological and molecular changes characteristic of mesenchymal-to-epithelial transition (Chen et al., 2009; Wang et al., 2011).

LZTS1 (leucine zipper, putative tumor suppressor 1) Vecchione A, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 897
Bladder cancer Note LZTS1 protein expression is reduced in bladder tumor samples and bladder cancer derived cell lines. Reintroduction of LZTS1 expression in TCC derived cell line inhibited cell growth, altered cell cycle progression and suppressed subcutaneous tumor growth in nude mice. Several LZTS1 somatic point mutations have also been reported in bladder cancers tissues and cell lines (Vecchione et al., 2002; Knowles et al., 2005). Moreover, it has been demonstrated by treating heterozygous and nullizygous Lzts1 mice with a classical bladder carcinogen (N-butyl-N-(4-hydroxybutil) nitrosamine, BBN), that the loss of one or both Lzts1 alleles favored development of bladder cancer. These results demonstrated that LZTS1 could represent an important therapeutic target for bladder tumor treatment.
References Ishii H, Baffa R, Numata SI, Murakumo Y, Rattan S, Inoue H, Mori M, Fidanza V, Alder H, Croce CM. The FEZ1 gene at chromosome 8p22 encodes a leucine-zipper protein, and its expression is altered in multiple human tumors. Proc Natl Acad Sci U S A. 1999 Mar 30;96(7):3928-33
Cabeza-Arvelaiz Y, Sepulveda JL, Lebovitz RM, Thompson TC, Chinault AC. Functional identification of LZTS1 as a candidate prostate tumor suppressor gene on human chromosome 8p22. Oncogene. 2001 Jul 12;20(31):4169-79
Ishii H, Vecchione A, Murakumo Y, Baldassarre G, Numata S, Trapasso F, Alder H, Baffa R, Croce CM. FEZ1/LZTS1 gene at 8p22 suppresses cancer cell growth and regulates mitosis. Proc Natl Acad Sci U S A. 2001 Aug 28;98(18):10374-9
Vecchione A, Ishii H, Shiao YH, Trapasso F, Rugge M, Tamburrino JF, Murakumo Y, Alder H, Croce CM, Baffa R. Fez1/lzts1 alterations in gastric carcinoma. Clin Cancer Res. 2001 Jun;7(6):1546-52
Hawkins GA, Mychaleckyj JC, Zheng SL, Faith DA, Kelly B, Isaacs SD, Wiley KE, Chang BL, Ewing CM, Bujnovszky P, Bleecker ER, Walsh PC, Meyers DA, Isaacs WB, Xu J. Germline sequence variants of the LZTS1 gene are associated with prostate cancer risk. Cancer Genet Cytogenet. 2002 Aug;137(1):1-7
Toyooka S, Fukuyama Y, Wistuba II, Tockman MS, Minna JD, Gazdar AF. Differential expression of FEZ1/LZTS1 gene in lung cancers and their cell cultures. Clin Cancer Res. 2002 Jul;8(7):2292-7
Vecchione A, Ishii H, Baldassarre G, Bassi P, Trapasso F, Alder H, Pagano F, Gomella LG, Croce CM, Baffa R. FEZ1/LZTS1 is down-regulated in high-grade bladder cancer, and its restoration suppresses tumorigenicity in transitional cell carcinoma cells. Am J Pathol. 2002 Apr;160(4):1345-52
Ono K, Uzawa K, Nakatsuru M, Shiiba M, Mochida Y, Tada A, Bukawa H, Miyakawa A, Yokoe H, Tanzawa H. Down-regulation of FEZ1/LZTS1 gene with frequent loss of heterozygosity in oral squamous cell carcinomas. Int J Oncol. 2003 Aug;23(2):297-302
Knowles MA, Aveyard JS, Taylor CF, Harnden P, Bass S. Mutation analysis of the 8p candidate tumour suppressor genes DBC2 (RHOBTB2) and LZTS1 in bladder cancer. Cancer Lett. 2005 Jul 8;225(1):121-30
Nonaka D, Fabbri A, Roz L, Mariani L, Vecchione A, Moore GW, Tavecchio L, Croce CM, Sozzi G. Reduced FEZ1/LZTS1 expression and outcome prediction in lung cancer. Cancer Res. 2005 Feb 15;65(4):1207-12
Baldassarre G, Croce CM, Vecchione A. Take your "M" time. Cell Cycle. 2007 Sep 1;6(17):2087-90
Vecchione A, Baldassarre G, Ishii H, Nicoloso MS, Belletti B, Petrocca F, Zanesi N, Fong LY, Battista S, Guarnieri D, Baffa R, Alder H, Farber JL, Donovan PJ, Croce CM. Fez1/Lzts1 absence impairs Cdk1/Cdc25C interaction during mitosis and predisposes mice to cancer development. Cancer Cell. 2007a Mar;11(3):275-89
Vecchione A, Croce CM, Baldassarre G. Fez1/Lzts1 a new mitotic regulator implicated in cancer development. Cell Div. 2007b Aug 24;2:24
Baffa R, Fassan M, Sevignani C, Vecchione A, Ishii H, Giarnieri E, Iozzo RV, Gomella LG, Croce CM. Fez1/Lzts1-deficient mice are more susceptible to N-butyl-N-(4-hydroxybutil) nitrosamine (BBN) carcinogenesis. Carcinogenesis. 2008 Apr;29(4):846-8
Barbe L, Lundberg E, Oksvold P, Stenius A, Lewin E, Björling E, Asplund A, Pontén F, Brismar H, Uhlén M, Andersson-Svahn H. Toward a confocal subcellular atlas of the human proteome. Mol Cell Proteomics. 2008 Mar;7(3):499-508
Onken MD, Worley LA, Harbour JW. A metastasis modifier locus on human chromosome 8p in uveal melanoma identified by integrative genomic analysis. Clin Cancer Res. 2008 Jun 15;14(12):3737-45
Chen L, Zhu Z, Sun X, Dong XY, Wei J, Gu F, Sun YL, Zhou J, Dong JT, Fu L. Down-regulation of tumor suppressor gene FEZ1/LZTS1 in breast carcinoma involves promoter methylation and associates with metastasis. Breast Cancer Res Treat. 2009 Aug;116(3):471-8
Califano D, Pignata S, Pisano C, Greggi S, Laurelli G, Losito NS, Ottaiano A, Gallipoli A, Pasquinelli R, De Simone V, Cirombella R, Fusco A, Chiappetta G. FEZ1/LZTS1 protein expression in ovarian cancer. J Cell Physiol. 2010 Feb;222(2):382-6
Kuo KT, Mao TL, Chen X, Feng Y, Nakayama K, Wang Y, Glas R, Ma MJ, Kurman RJ, Shih IeM, Wang TL. DNA copy numbers profiles in affinity-purified ovarian clear cell carcinoma. Clin Cancer Res. 2010 Apr 1;16(7):1997-2008
Wang XX, Zhu Z, Su D, Lei T, Wu X, Fan Y, Li X, Zhao J, Fu L, Dong JT, Fu L. Down-regulation of leucine zipper putative tumor suppressor 1 is associated with poor prognosis, increased cell motility and invasion, and epithelial-to-mesenchymal transition characteristics in human breast carcinoma. Hum Pathol. 2011 Oct;42(10):1410-9
This article should be referenced as such:
Vecchione A, Lavra L, Croce CM. LZTS1 (leucine zipper, putative tumor suppressor 1). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):894-897.

Gene Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 898
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
MAP2K4 (mitogen-activated protein kinase kinase 4) Kentaro Nakayama, Naomi Nakayama, Kohji Miyazaki
Department of Obstetrics and Gynecology, Shimane University School of Medicine, Shimane, Japan (KN, NN, KM)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/MAP2K4ID244ch17p12.html DOI: 10.4267/2042/48361
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: JNKK, JNKK1, MAPKK4, MEK4, MKK4, PRKMK4, SAPKK-1, SAPKK1, SEK1, SERK1
HGNC (Hugo): MAP2K4
Location: 17p12
Note Total length: 122917 bp. Strand: plus.
DNA/RNA Note Mitogen-activated protein kinase kinase 4 (MKK4) was first identified in a cDNA library from Xenopus laevis embryos using a PCR-based screen and was initially referred to as XMEK2 (Yashar et al., 1993). Drosophila, mouse, rat, and human homologues were subsequently cloned. MKK4 is also known as stress-activated protein
kinase/extracellular signal-related protein kinase kinase 1 (SEK1) and c-Jun N-terminal kinase kinase 1 (JNKK1) (Dérijard et al., 1995).
Description MKK4 gene is encoded by 11 exons located on chromosome 17p12. The genomic size is 122917 bp.
Transcription mRNA size: 3752 bp; coding sequence from 1 bp-3743 bp.
Pseudogene Pseudogene is located on Xq13.2.
Protein Note The MKK4 cDNA has 1197 bp open reading frame encoding a predicted polypeptide of 399 amino acids with a predicted molecular mass of 67 kDa.

MAP2K4 (mitogen-activated protein kinase kinase 4) Nakayama K, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 899
MKK4 interacts with the substrates JNK1, JNK2, JNK3, MAPK11, and MAPK14 via the D domain. MKK4 also interacts with the MAP3K activators MEKK1 and MLK3 via the domain for versatile docking (DVD domain). The DVD domain contains a conserved docking site that binds to upstream MAP3Ks and is essential for activation. The D domain contains a conserved docking site that binds to MAPK substrates. MKK4 is activated by the phosphorylation of Ser-257 and Thr-261 by MAP kinase kinase kinases.
Description The MKK4 gene encodes a dual specificity protein kinase that belongs to the Ser/Thr protein kinase family. This kinase is a direct activator of MAP kinases in response to environmental stress or mitogenic stimuli. MKK4 has been shown to activate MAPK8/JNK1, MAPK9/JNK2, and MAPK14/p38, but not MAPK1/ERK2 or MAPK3/ERK3. This kinase is phosphorylated and activated by MAP3K1/MEKK.
Expression MKK4 is widely expressed in normal tissues, including the thyroid, heart, lymph nodes, trachea, adrenal glands, and ovaries. The expression of MKK4 is lower in neoplastic tissues.
Localisation MKK4 is primarily located in the cytoplasm of the cell.
Function Three mitogen-activated protein kinase (MAPK) cascades occur in mammals. Each of the MAPK cascades consists of a three-kinase module that includes a MAPK, a MAPK kinase (MAPKK), and a MAPKK kinase (MAPKKK). Various cellular stresses, including ultraviolet (UV) and gamma irradiation, heat shock, hyperosmolarity, hydrogen peroxide, and inflammatory cytokines, activate the stress-activated
protein kinase/c-Jun N-terminal kinase (SAP/JNK) signalling pathway. c-Jun N-terminal kinases (JNKs) are MAPKs that stimulate the transcriptional activity of Jun in response to cellular stress. MAP2K4 is a MAPKK that directly activates the JNKs and the related MAPK p38 (Lin et al., 1995). MKK4 is a dual specificity kinase that activates the Jun kinases MAPK8 (JNK1) and MAPK9 (JNK2) and MAPK14 (p38), but does not activate MAPK1 (ERK2) or MAPK3 (ERK1).
Homology The MAP2K4 gene is conserved in chimpanzee, dog, cow, mouse, rat, chicken, zebrafish, fruit fly, mosquito, C. elegans, S. pombe, S. cerevisiae, K. lactis, E. gossypii, M. grisea, and N. crassa.
Mutations Germinal There are no reports regarding the germline.
Somatic Genomic studies identified a total of 11 tumours from human cancer samples (3% of the 356 tumours evaluated) with somatic mutations in the MAP2K4 (MKK4) gene. These mutations are primarily located in the kinase domain of MAP2K4. The mutations include frameshift, nonsense, and missense mutations and have

MAP2K4 (mitogen-activated protein kinase kinase 4) Nakayama K, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 900
been reported to occur in colorectal, non-small-cell lung, melanoma, and ovarian cancer specimens (Ahn et al., 2011).
Implicated in Various cancers Note Genomic studies have identified somatic mutations in the MAP2K4 gene in a total of 11 human cancer tumours (3% of the 356 tumours evaluated). These mutations are located primarily in the kinase domain. The mutations include frameshift, nonsense, and missense mutations and occur in colorectal, non-small-cell lung, melanoma, and ovarian cancer specimens (Ahn et al., 2011). The frequency of MKK4 homozygous deletions in high-grade ovarian serous carcinomas was reported to be 4,2%, which is similar to the rates observed in pancreatic (2%) and breast (4,5%) carcinomas (Nakayama et al., 2006). Loss of heterozygosity on chromosome 17p occurred in 24 (86%) of 28 high-grade serous carcinomas, including two cases with a homozygous MKK4 deletion. Downregulation of MKK4 expression was reported in 96 (75%) of 128 ovarian serous carcinomas compared to benign ovarian tissues. These findings suggest that homozygous deletions or reduced expression of MKK4 may contribute to the development of ovarian serous carcinomas (Nakayama et al., 2006). MKK4 expression related to tumour invasion results from an epithelial to mesenchymal transition (EMT)-like morphological change. Yeasmin et al. reported that the downregulation of MKK4 increased the phosphorylation of NF-κB and promoted the overexpression of Twist, resulting in the downregulation of E-cadherin, which induced an EMT in ovarian cancer. In most reports, MKK4 is defined as a tumour suppressor gene. However, Finegan and Tournier evaluated the role of MKK4 in skin tumourigenesis and reported that skin-specific MKK4-deficient mice are resistant to carcinogen-induced tumourigenesis. MKK4 is essential for mediating the oncogenic effects of Ras in vivo (Finegan and Tournier, 2010).
Prognosis A decreased expression of MKK4 based on immunointensity scores was observed in 63,2% (55/87) of endometrioid adenocarcinomas analysed. Patients with decreased MKK4 expression in endometrial cancer tissues tended to have a shorter overall rate of survival (p = 0,197) (Ishikawa et al., 2010).
Embryogenesis Note MKK4 has diverse physiological functions during embryogenesis. JNK activation by MKK4 and MKK7 is utilised in parallel morphogenetic events in widely divergent species. In vertebrates and invertebrates, MKK4/MKK7-JNK signalling regulates the expression of secreted signalling molecules that are capable of promoting the movements of neighbouring cells that are required for dorsal closure and gastrulation (Asaoka and Nishina, 2010).
References Yashar BM, Kelley C, Yee K, Errede B, Zon LI. Novel members of the mitogen-activated protein kinase activator family in Xenopus laevis. Mol Cell Biol. 1993 Sep;13(9):5738-48
Dérijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995 Feb 3;267(5198):682-5
Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F, Johnson GL, Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995 Apr 14;268(5208):286-90
Nakayama K, Nakayama N, Davidson B, Katabuchi H, Kurman RJ, Velculescu VE, Shih IeM, Wang TL. Homozygous deletion of MKK4 in ovarian serous carcinoma. Cancer Biol Ther. 2006 Jun;5(6):630-4
Asaoka Y, Nishina H. Diverse physiological functions of MKK4 and MKK7 during early embryogenesis. J Biochem. 2010 Oct;148(4):393-401
Finegan KG, Tournier C. The mitogen-activated protein kinase kinase 4 has a pro-oncogenic role in skin cancer. Cancer Res. 2010 Jul 15;70(14):5797-806
Ishikawa M, Nakayama K, Rahman MT, Rahman M, Katagiri A, Iida K, Miyazaki K. Functional and clinicopathological analysis of loss of MKK4 expression in endometrial cancer. Oncology. 2010;79(3-4):238-46
Ahn YH, Yang Y, Gibbons DL, Creighton CJ, Yang F, Wistuba II, Lin W, Thilaganathan N, Alvarez CA, Roybal J, Goldsmith EJ, Tournier C, Kurie JM. Map2k4 functions as a tumor suppressor in lung adenocarcinoma and inhibits tumor cell invasion by decreasing peroxisome proliferator-activated receptor γ2 expression. Mol Cell Biol. 2011 Nov;31(21):4270-85
Yeasmin S, Nakayama K, Rahman MT, Rahman M, Ishikawa M, Katagiri A, Iida K, Nakayama N, Miyazaki K. Loss of MKK4 expression in ovarian cancer: a potential role for the epithelial to mesenchymal transition. Int J Cancer. 2011 Jan 1;128(1):94-104
This article should be referenced as such:
Nakayama K, Nakayama N, Miyazaki K. MAP2K4 (mitogen-activated protein kinase kinase 4). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):898-900.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 901
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
MYLK (myosin light chain kinase) Kui Shen, Ting Wang, Joe GN Garcia
Institute of Personalized Respiratory Medicine, Department of Medicine, University of Illinois, Chicago, IL 60612, USA (KS, TW, JGNG)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/MYLKID43364ch3q21.html DOI: 10.4267/2042/48362
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: AAT7, KRP, MLCK, MLCK1, MLCK108, MLCK210, MSTP083, MYLK1, smMLCK
HGNC (Hugo): MYLK
Location: 3q21.1
Note Strand: Reverse (minus, -); Genomic Size: 272007. This gene, a member of the immunoglobulin gene superfamily, encodes myosin light chain kinase (MLCK), which is a calcium/calmodulin dependent kinase that phosphorylates myosin regulatory light chains (Potier et al., 1995) to regulate cell contractility (De Lanerolle et al., 1991; Garcia et al., 1995; Garcia et al., 1997b; Katoh et al., 2001) and cytokinesis (Dulyaninova et al., 2004; Fishkind et al., 1991; Matsumura et al., 2011; Poperechnaya et al., 2000). Multiple transcript variants of this gene have been identified that produce both nonmuscle and smooth muscle isoforms of MLCK (Garcia et al., 1997a; Lazar and Garcia, 1999; Verin et al., 1998b). In addition, using a separate promoter in an intron in the 3' region, it encodes telokin, a small protein identical in sequence to the C-terminus of MLCK (Gallagher and Herring, 1991; Watterson et al., 1999), which functions to stabilize unphosphorylated myosin filaments in smooth muscle (Kudryashov et al., 2002; Shirinsky et al., 1993). A pseudogene of MYLK is located on the p arm of chromosome 3 (Brand-Arpon et al., 1999; Giorgi et al.,
2001; Han et al., 2011) (modified from RefSeq, July 2008).
DNA/RNA Description The gene is composed of 34 exons, 31 out of which are coding exons.
Transcription Multiple MLCK isoforms are produced from the same MYLK gene by alternative splicing or alternative initiation (Lazar and Garcia, 1999; Verin et al., 1998b; Watterson et al., 1999). Six transcript variants have been identified that produce four kinase domain-encoding isoforms and two isoforms of telokin. Additional variants exist but lack full length transcripts. The longest transcript (nmMLCK1), which encodes the full length nonmuscle isoform (NM_053025), is a 7852 bp mRNA with a 5745 bp open reading frame from base pair 283 to 6027.
Pseudogene PGOHUM00000250243, PGOHUM00000238157, and PGOHUM00000238160 (Human Pseudogenes, Build 61). Note: Partially duplicated from the original MYLK gene, the MYLKP1 pseudogene (PGOHUM00000250243) is proposed to negatively regulate MYLK gene expression (Han et al., 2011).

MYLK (myosin light chain kinase) Shen K, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 902
The MYLK gene viewed at three different levels of detail (highlighted between two red vertical boundary lines). (1) Overview within chromosome 3. (2) Partial regional view within chromosome 3q21.1-3q21.2. (3) Detailed view within chromosome 3q21.1 showing six of the transcription variants of MLCK, which include nmMLCK1 (NM_053025), 2 (NM_053026), 3A (NM_053027), 3B (NM_053028), and two telokins (NM_053031 and NM_053032). The transcripts are not drawn in exact proportion so that their introns and exons, including CDSs and UTRs, can all be seen at a limited resolution. Abbreviations: Chr, chromosome; CDS, coding sequence; UTR, untranslated region.
Protein Description The full length isoform nmMLCK1 is a 1914-aa protein with a molecular weight of 210715 Da. All isoforms including telokin bind calmodulin (Davis et al., 1996; Gallagher and Herring, 1991; Geguchadze et al., 2004; Katoh et al., 2001). Various MLCK protein isoforms that result from the same MYLK gene (Lazar and Garcia, 1999) by alternative splicing or alternative initiation may be differentially regulated to achieve a tissue-specific spatiotemporal control of the binding (Davis et al., 1996; Dudek et al., 2002; Dudek et al., 2004; Hatch et al., 2001; Kishi et al., 1998) and catalytic activity of MLCK. The full length isoform nmMLCK1 is activated by post-translational modifications (PTMs) such as phosphorylation on Tyr-464 and Tyr-471 (coded by exon 11) (Birukov et al., 2001; Dudek et al., 2010). These PTMs are catalyzed by c-Abl (Dudek et al., 2010), p60Src (Birukov et al., 2001; Garcia et al., 1999), cAMP-dependent protein kinase (PKA) (Garcia et al., 1997a; Verin et al., 1998a) and p21-activated kinases (Goeckeler et al., 2000; Sanders et al., 1999).
Additional regulatory mechanisms involve acetylation (Shin et al., 2009), carboxyl-terminal deglutamylation (Rogowski et al., 2010), and kinase activation after thrombin, tumor necrosis factor (TNF), sphingosine 1-phosphate, G proteins, and during cell cycle (Garcia et al., 1995; Petrache et al., 2003; Poperechnaya et al., 2000; Somlyo and Somlyo, 2003; Ye et al., 2006; Ye and Ma, 2008).
Expression The nmMLCK or smMLCK isoforms and telokin are ubiquitously expressed in various adult and fetal tissues and in cultured endothelium with qualitative expression appearing to be neither tissue- nor development-specific (Garcia et al., 1997a; Lazar and Garcia, 1999; Potier et al., 1995; Verin et al., 1998b; Watterson et al., 1999). The nmMLCK 1 and 2 isoforms are dominant isoforms in nonmuscle (endothelial) cells (Brown et al., 2010; Garcia et al., 1997a; Lazar and Garcia, 1999; Verin et al., 1998b).
Localisation Lamellipodium; cytoplasm; cytoskeleton; stress fiber; cytosol; cleavage furrow.

MYLK (myosin light chain kinase) Shen K, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 903
Representative MLCK protein isoforms shown with select structural/functional information, as compared with the full length isoform (nmMLCK1). The nonmuscle MLCK isoform variants (nmMLCK 1, 2, 3A, 3B) differ in the presence or absence of exons 11 and 30. All nmMLCK variants possess unique amino termini that are absent in the smooth muscle isoform, smMLCK, and two isoforms of telokin. The longer isoform of telokin, differing by one amino acid from its shorter version (with the aa 1790 deletion), is identical to the C-termini of nmMLCK and smMLCK isoforms shown (Watterson et al., 1999). Abbreviations: aa, amino acid; CaM, calmodulin; IgC2, immunoglobulin C-2 type domain; FN3, fibronectin type 3 domain. *Note: NCBI RefSeq NM_053030.2 (NP_444258.1) was permanently suppressed because there was insufficient support for the transcript and the CDS was partial. Function Belongs to protein kinase superfamily, non-receptor Ser/Thr protein kinase, EC 2.7.11.18, calcium/calmodulin-dependent protein kinase (CAMK) group, MLCK family. Regulates smooth muscle and nonmuscle cell contractile processes (De Lanerolle et al., 1991; Garcia et al., 1995; Katoh et al., 2001; Somlyo and Somlyo, 2003), via phosphorylation of myosin light chains (MLC), or through a non-kinase activity (Dudek et al., 2004; Herring et al., 2006; Kudryashov et al., 2002; Nakamura et al., 2008; Shirinsky et al., 1993). Regulates cytogenesis (Dulyaninova et al., 2004; Fishkind et al., 1991; Matsumura et al., 2011; Poperechnaya et al., 2000). Regulates other related cellular processes including cell adhesion, migration, morphology, and inflammatory responses (Garcia et al., 1998; Savkovic et al., 2001), e.g., apoptosis (Mills et al., 1998; Petrache et al., 2003; Wright et al., 1993), and vascular permeability (Dudek et al., 2004; Garcia et al., 1995; Garcia et al., 1998; Shen et al., 2010; Vandenbroucke et al., 2008; Yuan et al., 2002), all via the regulation of cytoskeletal rearrangements. Genetic variants in MYLK are implicated in inflammatory disorders such as asthma and acute lung injury (Flores et al., 2007; Gao et al., 2006; Gao et al., 2007).
Implicated in tumor formation and metastasis (see below).
Homology The human MYLK gene is conserved in Euteleostomi, with a high percentage of identity in the pairwise alignment of protein/DNA vs. chimpanzee (99,1% / 99,4%), monkey (97,3% / 97,0%), dog (89,1% / 89,0%), mouse (85,9% / 85,6%), rat (85,4% / 86,0%), chicken (71,4% / 68,8%), and zebrafish (63,0% / 65,5%) (Homologene). The paralogs of human MYLK gene include MYLK2-4, DAPK1-3, STK17A and STK17B, and SPEG (Ensembl) (Manning et al., 2002).
Mutations Note Some protein-coding somatic mutations in MYLK are associated with cancers (Catalogue of Somatic Mutations in Cancer–COSMIC) (Greenman et al., 2007). Several variants of MYLK are associated with familial aortic dissections (Wang et al., 2010). A few race-specific single nucleotide polymorphism (SNP) variants of MYLK, both in coding and noncoding regions, are associated with the susceptibility to acute lung injury, sepsis and severe asthma (Flores et al., 2007; Gao et al., 2006; Gao et al., 2007).

MYLK (myosin light chain kinase) Shen K, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 904
Cancer-associated somatic mutations in MYLK in the protein coding region (Catalogue of Somatic Mutations in Cancer–COSMIC). Abbreviations: CaM, calmodulin; IgC2, immunoglobulin C-2 type domain; FN3, fibronectin type 3 domain; Complex, complex substitutions; Missense, missense substitutions; Nonsense, nonsense substitutions; Silent, silent substitutions.
Implicated in Cancers Note Myosin light chain kinase (MLCK) plays a crucial role in the cell migration and tumor metastasis. Some somatic mutations in MYLK are associated with cancers (Greenman et al., 2007). MLCK is critical for adhesion turnover at the cell front, a process central to migration (Webb et al., 2004) and it is involved in membrane blebbing (Godin and Ferguson, 2010). Deficiency in MLC phosphorylation causes cytokinesis failure and multipolarity (hence genomic instability) in cancer cells (Wu et al., 2010).
Breast cancer Note MLCK activity correlates the recruitment of nonmuscle myosin IIA and myosin IIB into the spreading margin of MDA-MB-231 breast cancer cells, with both myosin isoforms required for cell migration but only myosin IIB critical to lamellar protrusion (Betapudi et al., 2006). MLC phosphorylation by MLCK through β1-integrin is required for actin stress fiber formation and the dormancy-to-proliferation metastatic switch for latent breast cancer cells (Barkan et al., 2008; Barkan et al., 2011). MLCK functions downstream of Ras, MAP kinase kinase (MEK) and extracellular signal regulated kinase (ERK) to promote invasive migration of breast cancer cells in an integrin-selective manner, i.e., mediated by a β1-integrin (probably α5β1) and α5β5, but not by α5β3 (Mierke, 2011; Mierke et al., 2011b; Nguyen et al., 1999; Zhou et al., 2008).
Endothelial nmMLCK is activated by invasive breast cancer cells at the invasion site, leading to regional MLC diphosphorylation and myosin contraction. Blocking endothelial MLC diphosphorylation blunts tumor transcellular (i.e., through individual endothelial cells), but not paracellular (i.e., through cell-cell junctions) invasion (Khuon et al., 2010). Human mammary tumor cells exhibit at least two modes of invasive migration, including the extracellular proteolysis-dependent mesenchymal mode (invadopodia-associated extracellular matrix degradation) (Alexander et al., 2008) and the proteolysis-independent amoeboid mode, with both modes mediated by MLCK and Rho kinase ROCK (Alexander et al., 2008; Torka et al., 2006). TNF induction of apoptosis and DNA fragmentation requires MLCK activation in mammary carcinoma and other cancer cell lines (Wright et al., 1993). MLCK is responsible for high proliferative ability of breast cancer cells via anti-apoptosis (Cui et al., 2010). The increase in MLC phosphorylation correlates with apoptotic blebbing (Mills et al., 1998). Subsequent MLC dephosphorylation that results from a proapoptotic agent or MLCK inhibition (inhibitor or antibody) precedes caspase activation (Fazal et al., 2005), which further induces apoptosis in vitro and in vivo, and retards the growth of mammary cancer cells in mice (Fazal et al., 2005; Gu et al., 2006).
Lung cancer Note The invasiveness of tumor cells depends in part on their motility, which in turn depends on cytoskeletal function (Minamiya et al., 2005). The expression level

MYLK (myosin light chain kinase) Shen K, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 905
of MLCK, the cytoskeletal regulator, correlates with disease recurrence and distant metastasis in non-small cell lung cancer (NSCLC) (Minamiya et al., 2005). E1AF, an Ets family transcription factor frequently overexpressed in NSCLCs, induces motility and invasion as well as tumorigenesis and metastasis in NSCLC cells in a MLCK-dependent pathway (Hakuma et al., 2005). A few anti-cancer drug candidates, including glabridin, 7-chloro-6-piperidin-1-yl-quinoline-5,8-dione (PT-262), and all-trans-retinoic acid (ATRA), inhibit cell metastasis by decreasing cancer cell migration and invasion of human lung adenocarcinoma A549 cells via modulation of expression (Gui et al., 2011) or activity (Tsai et al., 2011a; Tsai et al., 2011b) of MLCK. Glycosylphosphatidylinositol-anchored receptor CD24 is found to enhance invasion of A125 human lung cancer cells through increased generation or transmission of contractile forces which is dependent on MLCK activity (Mierke et al., 2011a).
Colon cancer Note MLCK is differentially expressed in microsatellite stable (MSS) sporadic colon cancer and hereditary nonpolyposis colorectal cancer (HNPCC) (Lee et al., 2008). It is suggested to be a potential colon tumor marker. MLCK regulates transendothelial migration of colon cancer cells in E-selectin-mediated activation of p38 MAPK (Tremblay et al., 2006), and possibly via changing cellular contractility by regulation of adhesion sites and stress fibers (Krndija et al., 2010). Inhibition of MLCK suppresses peripheral accumulation of phospho-MLC and Src-induced formation of integrin-dependent adhesions in KM12C colon cancer cells, whereas at the same time restoring E-cadherin redistribution to regions of cell-cell contact (Avizienyte et al., 2004; Avizienyte et al., 2005; Nguyen et al., 2002).
Prostate cancer Note Inhibitors of MLCK markedly reduce the invasiveness of prostate cancer cells due to impaired cellular motility (Tohtong et al., 2003). These inhibitors also retard the growth of established prostate tumor in vivo (Gu et al., 2006). MLCK is considered as a central mediator of migration, proliferation and invasion of prostatic adenocarcinoma cell line (Tohtong et al., 2003) downstream of PKC delta (Kharait et al., 2007), boric acid, and phenylboronic acid (McAuley et al., 2011) in DU145 cell line (metastatic prostate cancer cell line). The MYLK gene is one of the top seven most informative genes that discriminate between normal and tumoral prostate conditions by analyzing cDNA microarrays of approximately 25000 genes (Fujita et al., 2008). MLCK is down-regulated by androgens in human prostate cancer cells (Leveille et al., 2009).
Other cancers Note MLC phosphorylation or MLCK activation is directly involved in the activation of membrane-associated actomyosin required for the collection of surface proteins into a cap structure in mouse T-lymphoma cells (analogous to muscle cell sliding filament contraction) (Bourguignon et al., 1981; Kerrick and Bourguignon., 1984). Inhibitors of MLCK (ML-7 and ML-9) induce differentiation of human monoblastic leukemia U937 cells (Makishima et al., 1991; Makishima et al., 1993; Yamamoto-Yamaguchi et al., 1996). Apoptotic membrane blebbing is accompanied by increased MLC phosphorylation and regulated by MLCK in PC12, a neuroendocrine tumor cell line (Mills et al, 1998). MLCK regulates the activation of volume-sensitive organic osmolyte/anion channels (VSOAC) by mediating hypotonicity-induced Ca2+ entry (not correlating with MLC phosphorylation) in cervical cancer cells (Shen et al., 2002).
References Bourguignon LY, Nagpal ML, Hsing YC. Phosphorylation of myosin light chain during capping of mouse T-lymphoma cells. J Cell Biol. 1981 Dec;91(3 Pt 1):889-94
Kerrick WG, Bourguignon LY. Regulation of receptor capping in mouse lymphoma T cells by Ca2+-activated myosin light chain kinase. Proc Natl Acad Sci U S A. 1984 Jan;81(1):165-9
De Lanerolle P, Strauss JD, Felsen R, Doerman GE, Paul RJ. Effects of antibodies to myosin light chain kinase on contractility and myosin phosphorylation in chemically permeabilized smooth muscle. Circ Res. 1991 Feb;68(2):457-65
Fishkind DJ, Cao LG, Wang YL. Microinjection of the catalytic fragment of myosin light chain kinase into dividing cells: effects on mitosis and cytokinesis. J Cell Biol. 1991 Sep;114(5):967-75
Gallagher PJ, Herring BP. The carboxyl terminus of the smooth muscle myosin light chain kinase is expressed as an independent protein, telokin. J Biol Chem. 1991 Dec 15;266(35):23945-52
Makishima M, Honma Y, Hozumi M, Sampi K, Hattori M, Motoyoshi K. Induction of differentiation of human leukemia cells by inhibitors of myosin light chain kinase. FEBS Lett. 1991 Aug 5;287(1-2):175-7
Makishima M, Honma Y, Hozumi M, Nagata N, Motoyoshi K. Differentiation of human monoblastic leukemia U937 cells induced by inhibitors of myosin light chain kinase and prevention of differentiation by granulocyte-macrophage colony-stimulating factor. Biochim Biophys Acta. 1993 Apr 16;1176(3):245-9
Shirinsky VP, Vorotnikov AV, Birukov KG, Nanaev AK, Collinge M, Lukas TJ, Sellers JR, Watterson DM. A kinase-related protein stabilizes unphosphorylated smooth muscle myosin minifilaments in the presence of ATP. J Biol Chem. 1993 Aug 5;268(22):16578-83
Wright SC, Zheng H, Zhong J, Torti FM, Larrick JW. Role of protein phosphorylation in TNF-induced apoptosis: phosphatase inhibitors synergize with TNF to activate DNA fragmentation in normal as well as TNF-resistant U937 variants. J Cell Biochem. 1993 Nov;53(3):222-33

MYLK (myosin light chain kinase) Shen K, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 906
Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995 Jun;163(3):510-22
Potier MC, Chelot E, Pekarsky Y, Gardiner K, Rossier J, Turnell WG. The human myosin light chain kinase (MLCK) from hippocampus: cloning, sequencing, expression, and localization to 3qcen-q21. Genomics. 1995 Oct 10;29(3):562-70
Davis HW, Crimmins DL, Thoma RS, Garcia JG. Phosphorylation of calmodulin in the first calcium-binding pocket by myosin light chain kinase. Arch Biochem Biophys. 1996 Aug 1;332(1):101-9
Yamamoto-Yamaguchi Y, Makishima M, Kanatani Y, Kasukabe T, Honma Y. Reversible differentiation of human monoblastic leukemia U937 cells by ML-9, an inhibitor of myosin light chain kinase. Exp Hematol. 1996 May;24(6):682-9
Garcia JG, Lazar V, Gilbert-McClain LI, Gallagher PJ, Verin AD. Myosin light chain kinase in endothelium: molecular cloning and regulation. Am J Respir Cell Mol Biol. 1997a May;16(5):489-94
Garcia JG, Schaphorst KL, Shi S, Verin AD, Hart CM, Callahan KS, Patterson CE. Mechanisms of ionomycin-induced endothelial cell barrier dysfunction. Am J Physiol. 1997b Jul;273(1 Pt 1):L172-84
Garcia JG, Verin AD, Herenyiova M, English D. Adherent neutrophils activate endothelial myosin light chain kinase: role in transendothelial migration. J Appl Physiol. 1998 May;84(5):1817-21
Kishi H, Ye LH, Nakamura A, Okagaki T, Iwata A, Tanaka T, Kohama K. Structure and function of smooth muscle myosin light chain kinase. Adv Exp Med Biol. 1998;453:229-34
Mills JC, Stone NL, Erhardt J, Pittman RN. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J Cell Biol. 1998 Feb 9;140(3):627-36
Verin AD, Gilbert-McClain LI, Patterson CE, Garcia JG. Biochemical regulation of the nonmuscle myosin light chain kinase isoform in bovine endothelium. Am J Respir Cell Mol Biol. 1998a Nov;19(5):767-76
Verin AD, Lazar V, Torry RJ, Labarrere CA, Patterson CE, Garcia JG. Expression of a novel high molecular-weight myosin light chain kinase in endothelium. Am J Respir Cell Mol Biol. 1998b Nov;19(5):758-66
Brand-Arpon V, Rouquier S, Massa H, de Jong PJ, Ferraz C, Ioannou PA, Demaille JG, Trask BJ, Giorgi D. A genomic region encompassing a cluster of olfactory receptor genes and a myosin light chain kinase (MYLK) gene is duplicated on human chromosome regions 3q13-q21 and 3p13. Genomics. 1999 Feb 15;56(1):98-110
Garcia JG, Verin AD, Schaphorst K, Siddiqui R, Patterson CE, Csortos C, Natarajan V. Regulation of endothelial cell myosin light chain kinase by Rho, cortactin, and p60(src). Am J Physiol. 1999 Jun;276(6 Pt 1):L989-98
Lazar V, Garcia JG. A single human myosin light chain kinase gene (MLCK; MYLK). Genomics. 1999 Apr 15;57(2):256-67
Nguyen DH, Catling AD, Webb DJ, Sankovic M, Walker LA, Somlyo AV, Weber MJ, Gonias SL. Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen activator-stimulated cells in an integrin-selective manner. J Cell Biol. 1999 Jul 12;146(1):149-64
Sanders LC, Matsumura F, Bokoch GM, de Lanerolle P. Inhibition of myosin light chain kinase by p21-activated kinase. Science. 1999 Mar 26;283(5410):2083-5
Watterson DM, Schavocky JP, Guo L, Weiss C, Chlenski A, Shirinsky VP, Van Eldik LJ, Haiech J. Analysis of the kinase-related protein gene found at human chromosome 3q21 in a multi-gene cluster: organization, expression, alternative splicing, and polymorphic marker. J Cell Biochem. 1999 Dec 1;75(3):481-91
Goeckeler ZM, Masaracchia RA, Zeng Q, Chew TL, Gallagher P, Wysolmerski RB. Phosphorylation of myosin light chain kinase by p21-activated kinase PAK2. J Biol Chem. 2000 Jun 16;275(24):18366-74
Poperechnaya A, Varlamova O, Lin PJ, Stull JT, Bresnick AR. Localization and activity of myosin light chain kinase isoforms during the cell cycle. J Cell Biol. 2000 Oct 30;151(3):697-708
Birukov KG, Csortos C, Marzilli L, Dudek S, Ma SF, Bresnick AR, Verin AD, Cotter RJ, Garcia JG. Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by p60(Src). J Biol Chem. 2001 Mar 16;276(11):8567-73
Giorgi D, Brand-Arpon V, Rouquier S. The functional myosin light chain kinase (MYLK) gene localizes with marker D3S3552 on human chromosome 3q21 in a >5-Mb yeast artificial chromosome region and is not linked to olfactory receptor genes. Cytogenet Cell Genet. 2001;92(1-2):85-8
Hatch V, Zhi G, Smith L, Stull JT, Craig R, Lehman W. Myosin light chain kinase binding to a unique site on F-actin revealed by three-dimensional image reconstruction. J Cell Biol. 2001 Aug 6;154(3):611-7
Katoh K, Kano Y, Amano M, Onishi H, Kaibuchi K, Fujiwara K. Rho-kinase--mediated contraction of isolated stress fibers. J Cell Biol. 2001 Apr 30;153(3):569-84
Savkovic SD, Ramaswamy A, Koutsouris A, Hecht G. EPEC-activated ERK1/2 participate in inflammatory response but not tight junction barrier disruption. Am J Physiol Gastrointest Liver Physiol. 2001 Oct;281(4):G890-8
Dudek SM, Birukov KG, Zhan X, Garcia JG. Novel interaction of cortactin with endothelial cell myosin light chain kinase. Biochem Biophys Res Commun. 2002 Nov 8;298(4):511-9
Kudryashov DS, Vorotnikov AV, Dudnakova TV, Stepanova OV, Lukas TJ, Sellers JR, Watterson DM, Shirinsky VP. Smooth muscle myosin filament assembly under control of a kinase-related protein (KRP) and caldesmon. J Muscle Res Cell Motil. 2002;23(4):341-51
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002 Dec 6;298(5600):1912-34
Nguyen QD, Faivre S, Bruyneel E, Rivat C, Seto M, Endo T, Mareel M, Emami S, Gespach C. RhoA- and RhoD-dependent regulatory switch of Galpha subunit signaling by PAR-1 receptors in cellular invasion. FASEB J. 2002 Apr;16(6):565-76
Shen MR, Furla P, Chou CY, Ellory JC. Myosin light chain kinase modulates hypotonicity-induced Ca2+ entry and Cl- channel activity in human cervical cancer cells. Pflugers Arch. 2002 May;444(1-2):276-85
Yuan SY, Wu MH, Ustinova EE, Guo M, Tinsley JH, De Lanerolle P, Xu W. Myosin light chain phosphorylation in neutrophil-stimulated coronary microvascular leakage. Circ Res. 2002 Jun 14;90(11):1214-21
Petrache I, Birukov K, Zaiman AL, Crow MT, Deng H, Wadgaonkar R, Romer LH, Garcia JG. Caspase-dependent cleavage of myosin light chain kinase (MLCK) is involved in TNF-alpha-mediated bovine pulmonary endothelial cell apoptosis. FASEB J. 2003 Mar;17(3):407-16
Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases,

MYLK (myosin light chain kinase) Shen K, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 907
and myosin phosphatase. Physiol Rev. 2003 Oct;83(4):1325-58
Tohtong R, Phattarasakul K, Jiraviriyakul A, Sutthiphongchai T. Dependence of metastatic cancer cell invasion on MLCK-catalyzed phosphorylation of myosin regulatory light chain. Prostate Cancer Prostatic Dis. 2003;6(3):212-6
Avizienyte E, Fincham VJ, Brunton VG, Frame MC. Src SH3/2 domain-mediated peripheral accumulation of Src and phospho-myosin is linked to deregulation of E-cadherin and the epithelial-mesenchymal transition. Mol Biol Cell. 2004 Jun;15(6):2794-803
Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004 Jun 4;279(23):24692-700
Dulyaninova NG, Patskovsky YV, Bresnick AR. The N-terminus of the long MLCK induces a disruption in normal spindle morphology and metaphase arrest. J Cell Sci. 2004 Mar 15;117(Pt 8):1481-93
Geguchadze R, Zhi G, Lau KS, Isotani E, Persechini A, Kamm KE, Stull JT. Quantitative measurements of Ca(2+)/calmodulin binding and activation of myosin light chain kinase in cells. FEBS Lett. 2004 Jan 16;557(1-3):121-4
Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004 Feb;6(2):154-61
Avizienyte E, Brunton VG, Fincham VJ, Frame MC. The SRC-induced mesenchymal state in late-stage colon cancer cells. Cells Tissues Organs. 2005;179(1-2):73-80
Fazal F, Gu L, Ihnatovych I, Han Y, Hu W, Antic N, Carreira F, Blomquist JF, Hope TJ, Ucker DS, de Lanerolle P. Inhibiting myosin light chain kinase induces apoptosis in vitro and in vivo. Mol Cell Biol. 2005 Jul;25(14):6259-66
Hakuma N, Kinoshita I, Shimizu Y, Yamazaki K, Yoshida K, Nishimura M, Dosaka-Akita H. E1AF/PEA3 activates the Rho/Rho-associated kinase pathway to increase the malignancy potential of non-small-cell lung cancer cells. Cancer Res. 2005 Dec 1;65(23):10776-82
Minamiya Y, Nakagawa T, Saito H, Matsuzaki I, Taguchi K, Ito M, Ogawa J. Increased expression of myosin light chain kinase mRNA is related to metastasis in non-small cell lung cancer. Tumour Biol. 2005 May-Jun;26(3):153-7
Betapudi V, Licate LS, Egelhoff TT. Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 2006 May 1;66(9):4725-33
Gao L, Grant A, Halder I, Brower R, Sevransky J, Maloney JP, Moss M, Shanholtz C, Yates CR, Meduri GU, Shriver MD, Ingersoll R, Scott AF, Beaty TH, Moitra J, Ma SF, Ye SQ, Barnes KC, Garcia JG. Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell Mol Biol. 2006 Apr;34(4):487-95
Gu LZ, Hu WY, Antic N, Mehta R, Turner JR, de Lanerolle P. Inhibiting myosin light chain kinase retards the growth of mammary and prostate cancer cells. Eur J Cancer. 2006 May;42(7):948-57
Herring BP, El-Mounayri O, Gallagher PJ, Yin F, Zhou J. Regulation of myosin light chain kinase and telokin expression in smooth muscle tissues. Am J Physiol Cell Physiol. 2006 Nov;291(5):C817-27
Torka R, Thuma F, Herzog V, Kirfel G. ROCK signaling mediates the adoption of different modes of migration and
invasion in human mammary epithelial tumor cells. Exp Cell Res. 2006 Nov 15;312(19):3857-71
Tremblay PL, Auger FA, Huot J. Regulation of transendothelial migration of colon cancer cells by E-selectin-mediated activation of p38 and ERK MAP kinases. Oncogene. 2006 Oct 26;25(50):6563-73
Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2006 Mar;290(3):G496-504
Flores C, Ma SF, Maresso K, Ober C, Garcia JG. A variant of the myosin light chain kinase gene is associated with severe asthma in African Americans. Genet Epidemiol. 2007 May;31(4):296-305
Gao L, Grant AV, Rafaels N, Stockton-Porter M, Watkins T, Gao P, Chi P, Muñoz M, Watson H, Dunston G, Togias A, Hansel N, Sevransky J, Maloney JP, Moss M, Shanholtz C, Brower R, Garcia JG, Grigoryev DN, Cheadle C, Beaty TH, Mathias RA, Barnes KC. Polymorphisms in the myosin light chain kinase gene that confer risk of severe sepsis are associated with a lower risk of asthma. J Allergy Clin Immunol. 2007 May;119(5):1111-8
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O'Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007 Mar 8;446(7132):153-8
Kharait S, Hautaniemi S, Wu S, Iwabu A, Lauffenburger DA, Wells A. Decision tree modeling predicts effects of inhibiting contractility signaling on cell motility. BMC Syst Biol. 2007 Jan 29;1:9
Alexander NR, Branch KM, Parekh A, Clark ES, Iwueke IC, Guelcher SA, Weaver AM. Extracellular matrix rigidity promotes invadopodia activity. Curr Biol. 2008 Sep 9;18(17):1295-9
Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, Liu ZY, Costes SV, Cho EH, Lockett S, Khanna C, Chambers AF, Green JE. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008 Aug 1;68(15):6241-50
Fujita A, Gomes LR, Sato JR, Yamaguchi R, Thomaz CE, Sogayar MC, Miyano S. Multivariate gene expression analysis reveals functional connectivity changes between normal/tumoral prostates. BMC Syst Biol. 2008 Dec 5;2:106
Lee WS, Seo G, Shin HJ, Yun SH, Yun H, Choi N, Lee J, Son D, Cho J, Kim J, Cho YB, Chun HK, Lee WY. Identification of differentially expressed genes in microsatellite stable HNPCC and sporadic colon cancer. J Surg Res. 2008 Jan;144(1):29-35
Nakamura A, Xie C, Zhang Y, Gao Y, Wang HH, Ye LH, Kishi H, Okagaki T, Yoshiyama S, Hayakawa K, Ishikawa R, Kohama K. Role of non-kinase activity of myosin light-chain kinase in regulating smooth muscle contraction, a review dedicated to Dr. Setsuro Ebashi. Biochem Biophys Res Commun. 2008 Apr 25;369(1):135-43
Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008 Mar;1123:134-45

MYLK (myosin light chain kinase) Shen K, et al.
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 908
Ye D, Ma TY. Cellular and molecular mechanisms that mediate basal and tumour necrosis factor-alpha-induced regulation of myosin light chain kinase gene activity. J Cell Mol Med. 2008 Aug;12(4):1331-46
Zhou X, Liu Y, You J, Zhang H, Zhang X, Ye L. Myosin light-chain kinase contributes to the proliferation and migration of breast cancer cells through cross-talk with activated ERK1/2. Cancer Lett. 2008 Nov 8;270(2):312-27
Léveillé N, Fournier A, Labrie C. Androgens down-regulate myosin light chain kinase in human prostate cancer cells. J Steroid Biochem Mol Biol. 2009 Apr;114(3-5):174-9
Shin DH, Chun YS, Lee KH, Shin HW, Park JW. Arrest defective-1 controls tumor cell behavior by acetylating myosin light chain kinase. PLoS One. 2009 Oct 14;4(10):e7451
Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, Webster JD, Hoover S, Simpson RM, Gauldie J, Green JE. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010 Jul 15;70(14):5706-16
Brown M, Adyshev D, Bindokas V, Moitra J, Garcia JG, Dudek SM. Quantitative distribution and colocalization of non-muscle myosin light chain kinase isoforms and cortactin in human lung endothelium. Microvasc Res. 2010 Jul;80(1):75-88
Cui WJ, Liu Y, Zhou XL, Wang FZ, Zhang XD, Ye LH. Myosin light chain kinase is responsible for high proliferative ability of breast cancer cells via anti-apoptosis involving p38 pathway. Acta Pharmacol Sin. 2010 Jun;31(6):725-32
Dudek SM, Chiang ET, Camp SM, Guo Y, Zhao J, Brown ME, Singleton PA, Wang L, Desai A, Arce FT, Lal R, Van Eyk JE, Imam SZ, Garcia JG. Abl tyrosine kinase phosphorylates nonmuscle Myosin light chain kinase to regulate endothelial barrier function. Mol Biol Cell. 2010 Nov 15;21(22):4042-56
Godin CM, Ferguson SS. The angiotensin II type 1 receptor induces membrane blebbing by coupling to Rho A, Rho kinase, and myosin light chain kinase. Mol Pharmacol. 2010 Jun;77(6):903-11
Khuon S, Liang L, Dettman RW, Sporn PH, Wysolmerski RB, Chew TL. Myosin light chain kinase mediates transcellular intravasation of breast cancer cells through the underlying endothelial cells: a three-dimensional FRET study. J Cell Sci. 2010 Feb 1;123(Pt 3):431-40
Krndija D, Schmid H, Eismann JL, Lother U, Adler G, Oswald F, Seufferlein T, von Wichert G. Substrate stiffness and the receptor-type tyrosine-protein phosphatase alpha regulate spreading of colon cancer cells through cytoskeletal contractility. Oncogene. 2010 May 6;29(18):2724-38
Rogowski K, van Dijk J, Magiera MM, Bosc C, Deloulme JC, Bosson A, Peris L, Gold ND, Lacroix B, Grau MB, Bec N, Larroque C, Desagher S, Holzer M, Andrieux A, Moutin MJ, Janke C. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 2010 Nov 12;143(4):564-78
Shen Q, Rigor RR, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res. 2010 Jul 15;87(2):272-80
Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010 Nov 12;87(5):701-7
Wu Q, Sahasrabudhe RM, Luo LZ, Lewis DW, Gollin SM, Saunders WS. Deficiency in myosin light-chain phosphorylation causes cytokinesis failure and multipolarity in cancer cells. Oncogene. 2010 Jul 22;29(29):4183-93
Gui S, Wang Y, Cheng Y, Zhou Q, Chen F and Wang Y.. Mechanism of all-trans-retinoic acid in inhibiting the migration of human lung adenocarcinoma A549 cells. Chin Pharm Bull. 2011 Jan;21(1):33-6.
Han YJ, Ma SF, Yourek G, Park YD, Garcia JG.. A transcribed pseudogene of MYLK promotes cell proliferation. FASEB J. 2011 Jul;25(7):2305-12. Epub 2011 Mar 25.
Matsumura F, Yamakita Y, Yamashiro S.. Myosin light chain kinases and phosphatase in mitosis and cytokinesis. Arch Biochem Biophys. 2011 Jun 15;510(2):76-82. Epub 2011 Mar 21. (REVIEW)
McAuley EM, Bradke TA, Plopper GE.. Phenylboronic acid is a more potent inhibitor than boric acid of key signaling networks involved in cancer cell migration. Cell Adh Migr. 2011 Sep-Oct;5(5):382-6.
Mierke CT.. Cancer cells regulate biomechanical properties of human microvascular endothelial cells. J Biol Chem. 2011 Nov 18;286(46):40025-37. Epub 2011 Sep 22.
Mierke CT, Bretz N, Altevogt P.. Contractile forces contribute to increased glycosylphosphatidylinositol-anchored receptor CD24-facilitated cancer cell invasion. J Biol Chem. 2011a Oct 7;286(40):34858-71. Epub 2011 Aug 2.
Mierke CT, Frey B, Fellner M, Herrmann M, Fabry B.. Integrin alpha5beta1 facilitates cancer cell invasion through enhanced contractile forces. J Cell Sci. 2011b Feb 1;124(Pt 3):369-83. Epub 2011 Jan 11.
Tsai CC, Liu HF, Hsu KC, Yang JM, Chen C, Liu KK, Hsu TS, Chao JI.. 7-Chloro-6-piperidin-1-yl-quinoline-5,8-dione (PT-262), a novel ROCK inhibitor blocks cytoskeleton function and cell migration. Biochem Pharmacol. 2011a Apr 1;81(7):856-65. Epub 2011 Jan 26.
Tsai YM, Yang CJ, Hsu YL, Wu LY, Tsai YC, Hung JY, Lien CT, Huang MS, Kuo PL.. Glabridin inhibits migration, invasion, and angiogenesis of human non-small cell lung cancer A549 cells by inhibiting the FAK/rho signaling pathway. Integr Cancer Ther. 2011b Dec;10(4):341-9. Epub 2010 Nov 8.
This article should be referenced as such:
Shen K, Wang T, Garcia JGN. MYLK (myosin light chain kinase). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):901-908.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 909
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
NAMPT (nicotinamide phosphoribosyltransferase) Vassiliki Koumaki, Maria Dalamaga
Department of Microbiology, University of Athens, School of Medicine, University of Athens, 75 Mikras Asias Street, 11527 Athens, Greece (VK), Department of Clinical Biochemistry, University of Athens, School of Medicine, Attikon General University Hospital, Rimini 1, Chaidari, 12462 Athens, Greece (MD)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/NAMPTID43890ch7q22.html DOI: 10.4267/2042/48363
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: 1110035O14Rik, PBEF, PBEF1, VF, VISFATIN
HGNC (Hugo): NAMPT
Location: 7q22.3
DNA/RNA Description The human NAMPT gene spans a length of 36908 bp. The NAPMT structural gene is composed of 11 exons and 10 introns.
Transcription Transcription produces 19 different mRNAs, 14 alternatively spliced variants and 5 unspliced forms. There are 5 probable alternative promoters, 6 non overlapping alternative last exons and 13 alternative polyadenylation sites. The mRNAs appear to differ by truncation of the 3' end, presence or absence of 2 cassette exons, overlapping exons with different boundaries, alternative splicing or retention of 4 introns (Zhang et al., 2011).
Pseudogene This gene has a pseudogene on chromosome 10 (provided by RefSeq 2011).
Protein Description The reference human NAMPT protein sequence (NP_005737) consists of 491 amino acids.
Expression NAMPT is expressed in human heart, brain, placenta, lungs, liver, skeletal muscle, kidney and pancreas with the maximum amount in muscle tissue (Samal et al.,1994).
Localisation NAMPT is localized both in the nucleus and the cytosplasm (Kitani et al., 2003).
Function The three major functions of NAMPT: growth factor, cytokine and nicotinamide phosphoribosyltransferase. Accumulating evidence suggests that NAMPT can function as a growth factor or a cytokine though the underlying molecular mechanisms remain to be established. It is beyond any dispute that NAMPT can function as a nicotinamide phosphoribosyltransferase (Zhang et al., 2011).
Genomic structure of NAMPT. Orange boxes indicate exons and purple boxes indicate untranslated regions.

NAMPT (nicotinamide phosphoribosyltransferase) Koumaki V, Dalamaga M
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 910
Enzymatic activity: because of its pivotal role in the recycling pathway allowing NAD generation from nicotinamide, NAMPT occupies a central position in controlling the activity of several NAD-dependent enzymes (Gallí et al., 2010). NAD, a universal energy-and signal-carrying molecule and its phosphorylated form, NADP, are required in several intracellular processes such as redox reactions, DNA repair, G-protein coupled receptor signaling, intra-cellular calcium-mobilizing molecules, transcriptional regulation, mono-adenosine diphosphate (ADP)-ribosylation in immune response, and activity of poly-ADP ribosyltransferases and deacetylases (sirtuins) with roles in regulating cell survival and cytokine responses (Garten et al., 2009). Under the influence of NAMPT, adequate levels of NAD control SIRT-6 (sirtuin) activity, which in turn positively regulates TNF-α mRNA translation favoring cell survival (Gallí et al., 2010). NAMPT activity enhances cellular proliferation, tips the balance toward cellular survival following a genotoxic insult and controls the circadian clock machinery of some key transcriptions factors (Garten et al., 2009; Moschen et al., 2010).
Homology Significant sequence homology has been shared among prokaryotic organisms such as the bacterium Haemophilus ducreyi, primitive metazoan such as marine sponge, and humans (Martin et al., 2001). Amino acid sequence alignment revealed that the NAMPT gene is evolutionarily highly conserved, with the canine NAMPT protein sequence 96% identical to human NAMPT and 94% identical to both murine and rat PBEF counterparts (McGlothlin et al., 2005).
Mutations Homozygous deletion confers embryonic lethality in mouse (Ye at al., 2005). Up to June 2012 NCBI dbSNP reports 730 SNPs in the human NAMPT gene. Functional consequences of most of these SNPs are currently unknown (Zhang et al., 2011). Acquired resistance to inhibitors of NAMPT has been associated with mutations of NAMPT located in the vicinity of the active site or in the dimer interface of NAMPT (Olesen et al., 2010).
Implicated in Various diseases The dysregulation of NAMPT gene as well as abnormalities in circulating NAMPT levels have been implicated in the susceptibility and pathogenesis of a number of human diseases and pathologic conditions given NAMPT's pleiotropic physiological functions. NAMPT has been implicated in cancer as described below, diabetes, obesity, aging, atherosclerosis, sepsis,
acute lung injury, rheumatoid arthritis, etc (Zhang et al., 2011).
Colorectal cancer (CC) NAMPT expression was increased in primary colorectal cancer comparing to normal control mucosa using the suppression subtractive hybridization technique to identify new candidate genes in cancer (Hufton et al., 1999). This observation was later confirmed at tissue and protein level by Western blotting and immune-histochemical analyses (Van Beijnum et al., 2002). Serum Nampt levels were significantly higher in 115 CC patients than in 115 age-, gender- and body mass index (BMI)-matched controls both in univariate (p<0.01) and multivariable analyses (OR: 2.95, 95% C.I. 1.862-4.787, p<0.01) (Nakajima et al., 2010).
Prognosis Serum Nampt levels may represent a promising biomarker of CC malignant potential and stage progression. Circulating Nampt gradually increased with tumor stage progression (p<0.01) (Nakajima et al., 2010).
Breast cancer (BC) NAMPT is expressed in BC tissues, in MCF-7 BC cells and in doxorubicin-responsive BC (Folgueira et al., 2005; Gallí et al., 2010; Zhang et al., 2011; Moschen et al., 2010; Garten et al., 2009). Additionally, Nampt is present in bovine mammary epithelium, lactating mammary glands, and milk (Yonezawa et al., 2006). NAMPT stimulated the proliferation and DNA synthesis rate of MCF-7 human BC cells (Kim et al., 2010). More specifically, NAMPT upregulated mRNA levels of cyclin D1 and cdk2, well-known regulators for the G1-S progression (Kim et al., 2010). Circulating levels of Nampt were significantly elevated in women suffering from postmenopausal BC than in controls independently from known risk factors of BC, anthropometric and metabolic parameters as well as serum concentrations of leptin and adiponectin (Dalamaga et al., 2011). Stratification by BMI depicted that the association of serum Nampt with PBC risk was more pronounced among overweight/obese postmenopausal women after adjustment for the aforementioned parameters (Dalamaga et al., 2011; Dalamaga et al., 2012b).
Prognosis High NAMPT expression in BC tissues was reported to be associated with more malignant cancer behavior as well as adverse prognosis (Lee et al., 2011). In the high NAMPT expression group, the majority of patients were estrogen and progesterone negative (Lee et al., 2011). Serum Nampt could be used as potential diagnostic and prognostic biomarker in the armamentarium of BC monitoring and management. In

NAMPT (nicotinamide phosphoribosyltransferase) Koumaki V, Dalamaga M
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 911
postmenopausal women, circulating Nampt could provide additional information in conjunction with tumor markers CA 15-3 and carcinoembryonic antigen, particularly in discriminating early stage cases and estrogen/progesterone negative breast tumors (Dalamaga et al., 2012b). In multivariable regression analysis, the most significant predictors/determinants of serum Nampt levels were the hormone receptor status, the late stage of PBC and the lymph node involvement (Dalamaga et al., 2012b).
Gastric cancer (GC) Using real-time PCR and Western blotting, NAMPT was overexpressed at the mRNA and protein levels in gastric cancer cells and human gastric cancer tissues (Bi et al., 2011). The specific NAMPT inhibitor FK866 repressed gastric cancer cell proliferation in vitro (Bi et al., 2011). Serum Nampt levels were significantly higher in 156 GC patients than in 156 age- and gender-matched controls using multivariable analysis (p= 0.0013) (Nakajima et al., 2009).
Prognosis Nampt may be good biomarker of GC as its circulating levels gradually increased with stage progression (P<0.0001) (Nakajima et al., 2009).
Prostate cancer (PC) Oncogenesis In prostate carcinogenesis, NAMPT increased PC3 cell proliferation activating the mitogen-activated protein kinases (MAPKs) ERK-1/ERK-2 and p38 signaling pathways (Patel et al., 2010). NAMPT promoted the activity and expression of MMP-2/MMP-9 which represent important proteases involved in the breakdown of the extracellular matrix, indicating a possible role for NAMPT in PC metastasis (Patel et al., 2010). Upregulation of NAMPT expression occurs early in prostate neoplasia (Wang et al., 2011). Inhibition of NAMPT significantly suppresses cell growth in culture, soft agar colony formation, cell invasion and growth of xenografted prostate cancer cells in mice. NAMPT knockdown sensitizes prostate cancer cells to oxidative stress caused by H2O2 or chemotherapeutic treatment. Overexpression of NAMPT increases prostate cancer cell resistance to oxidative stress, which is partially blocked by SIRT1 knockdown (Wang et al., 2011).
Brain tumors Increased NAMPT expression was found in glioblastoma samples using cDNA microarray based expression profiling, real-time RT-qPCR and immunohistochemical staining on an independent set of brain tumor samples (Reddy et al., 2008). APO866, a NAMPT inhibitor, is a potent growth inhibitor against glioblastoma through targeting NAMPT. APO866 depleted intracellular NAD, caused marked inhibition of ERK activation and induced G2/M cell-cycle arrest in C6 glioblastoma cells (Zhang et al., 2012).
Prognosis Serum Nampt levels may be a potential serum biomarker for malignant astrocytoma and prognostic indicator in glioblastoma (Reddy et al., 2008).
Ovarian cancer NAMPT protein expression is significantly increased in ovarian serous adenocarcinoma comparing to benign ovarian tissue using tissue microarray and the avidin-biotin complex immuno-histochemical technique (Shackelford et al., 2010).
Esophageal cancer Prognosis Using quantitative one-step real time RT-PCR, circulating Nampt mRNAs in postoperative esophagectomy patients were upregulated adjusting for other factors (p<0.01) and were independent predictors of mortality in the first year of follow-up (Takahashi et al., 2010).
Lymphoma NAMPT expression was investigated in 53 samples of malignant lymphomas (diffuse large B-cell lymphoma, follicular B-cell lymphoma, Hodgkin's lymphoma and peripheral T-cell lymphoma). The expression of NAMPT was generally elevated in the more aggressive malignant lymphomas, with >80% strong expression, whereas the expression in the more indolent follicular lymphoma (FL) was significantly lower (>75% moderate or low expression, p= 0.0002) (Olesen et al., 2011). In Hodgkin's lymphoma, NAMPT was very highly expressed in Hodgkin Reed-Sternberg cells (Olesen et al., 2011).
References Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994 Feb;14(2):1431-7
Hufton SE, Moerkerk PT, Brandwijk R, de Bruïne AP, Arends JW, Hoogenboom HR. A profile of differentially expressed genes in primary colorectal cancer using suppression subtractive hybridization. FEBS Lett. 1999 Dec 10;463(1-2):77-82
Martin PR, Shea RJ, Mulks MH. Identification of a plasmid-encoded gene from Haemophilus ducreyi which confers NAD independence. J Bacteriol. 2001 Feb;183(4):1168-74
Van Beijnum JR, Moerkerk PT, Gerbers AJ, De Bruïne AP, Arends JW, Hoogenboom HR, Hufton SE. Target validation for genomics using peptide-specific phage antibodies: a study of five gene products overexpressed in colorectal cancer. Int J Cancer. 2002 Sep 10;101(2):118-27
Kitani T, Okuno S, Fujisawa H. Growth phase-dependent changes in the subcellular localization of pre-B-cell colony-enhancing factor. FEBS Lett. 2003 Jun 5;544(1-3):74-8
Folgueira MA, Carraro DM, Brentani H, et al.. Gene expression profile associated with response to doxorubicin-based therapy in breast cancer. Clin Cancer Res. 2005 Oct 15;11(20):7434-43

NAMPT (nicotinamide phosphoribosyltransferase) Koumaki V, Dalamaga M
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 912
McGlothlin JR, Gao L, Lavoie T, Simon BA, Easley RB, Ma SF, Rumala BB, Garcia JG, Ye SQ. Molecular cloning and characterization of canine pre-B-cell colony-enhancing factor. Biochem Genet. 2005 Apr;43(3-4):127-41
Ye SQ, Zhang LQ, Adyshev D, Usatyuk PV, Garcia AN, Lavoie TL, Verin AD, Natarajan V, Garcia JG. Pre-B-cell-colony-enhancing factor is critically involved in thrombin-induced lung endothelial cell barrier dysregulation. Microvasc Res. 2005 Nov;70(3):142-51
Yonezawa T, Haga S, Kobayashi Y, Takahashi T, Obara Y. Visfatin is present in bovine mammary epithelial cells, lactating mammary gland and milk, and its expression is regulated by cAMP pathway. FEBS Lett. 2006 Dec 11;580(28-29):6635-43
Reddy PS, Umesh S, Thota B, Tandon A, et al. PBEF1/NAmPRTase/Visfatin: a potential malignant astrocytoma/glioblastoma serum marker with prognostic value. Cancer Biol Ther. 2008 May;7(5):663-8
Garten A, Petzold S, Körner A, Imai S, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009 Apr;20(3):130-8
Nakajima TE, Yamada Y, Hamano T, Furuta K, Gotoda T, Katai H, Kato K, Hamaguchi T, Shimada Y. Adipocytokine levels in gastric cancer patients: resistin and visfatin as biomarkers of gastric cancer. J Gastroenterol. 2009;44(7):685-90
Gallí M, Van Gool F, Rongvaux A, Andris F, Leo O. The nicotinamide phosphoribosyltransferase: a molecular link between metabolism, inflammation, and cancer. Cancer Res. 2010 Jan 1;70(1):8-11
Kim JG, Kim EO, Jeong BR, Min YJ, Park JW, Kim ES, Namgoong IS, Kim YI, Lee BJ. Visfatin stimulates proliferation of MCF-7 human breast cancer cells. Mol Cells. 2010 Oct;30(4):341-5
Moschen AR, Gerner RR, Tilg H. Pre-B cell colony enhancing factor/NAMPT/visfatin in inflammation and obesity-related disorders. Curr Pharm Des. 2010 Jun;16(17):1913-20
Nakajima TE, Yamada Y, Hamano T, Furuta K, Matsuda T, Fujita S, Kato K, Hamaguchi T, Shimada Y. Adipocytokines as new promising markers of colorectal tumors: adiponectin for colorectal adenoma, and resistin and visfatin for colorectal cancer. Cancer Sci. 2010 May;101(5):1286-91
Olesen UH, Petersen JG, Garten A, Kiess W, et al.. Target enzyme mutations are the molecular basis for resistance towards pharmacological inhibition of nicotinamide phosphoribosyltransferase. BMC Cancer. 2010 Dec 12;10:677
Patel ST, Mistry T, Brown JE, Digby JE, Adya R, Desai KM, Randeva HS. A novel role for the adipokine visfatin/pre-B cell colony-enhancing factor 1 in prostate carcinogenesis. Peptides. 2010 Jan;31(1):51-7
Shackelford RE, Bui MM, Coppola D, Hakam A. Over-expression of nicotinamide phosphoribosyltransferase in ovarian cancers. Int J Clin Exp Pathol. 2010 Jun 12;3(5):522-7
Takahashi S, Miura N, Harada T, Wang Z, Wang X, Tsubokura H, Oshima Y, Hasegawa J, Inagaki Y, Shiota G. Prognostic impact of clinical course-specific mRNA expression profiles in the serum of perioperative patients with esophageal cancer in the ICU: a case control study. J Transl Med. 2010 Oct 22;8:103
Bi TQ, Che XM, Liao XH, Zhang DJ, Long HL, Li HJ, Zhao W. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol Rep. 2011 Nov;26(5):1251-7
Dalamaga M, Karmaniolas K, Papadavid E, Pelekanos N, Sotiropoulos G, Lekka A. Elevated serum visfatin/nicotinamide phosphoribosyl-transferase levels are associated with risk of postmenopausal breast cancer independently from adiponectin, leptin, and anthropometric and metabolic parameters. Menopause. 2011 Nov;18(11):1198-204
Lee YC, Yang YH, Su JH, Chang HL, Hou MF, Yuan SS. High visfatin expression in breast cancer tissue is associated with poor survival. Cancer Epidemiol Biomarkers Prev. 2011 Sep;20(9):1892-901
Olesen UH, Hastrup N, Sehested M. Expression patterns of nicotinamide phosphoribosyltransferase and nicotinic acid phosphoribosyltransferase in human malignant lymphomas. APMIS. 2011 Apr;119(4-5):296-303
Zhang LQ, Heruth DP, Ye SQ. Nicotinamide Phosphoribosyltransferase in Human Diseases. J Bioanal Biomed. 2011 Jan 7;3:13-25
Dalamaga M, Archondakis S, Sotiropoulos G, Karmaniolas K, Pelekanos N, Papadavid E, Lekka A. Could serum visfatin be a potential biomarker for postmenopausal breast cancer? Maturitas. 2012a Mar;71(3):301-8
Dalamaga M, Diakopoulos KN, Mantzoros CS. The role of adiponectin in cancer: a review of current evidence. Endocr Rev. 2012b Aug;33(4):547-94
Zhang LY, Liu LY, Qie LL, Ling KN, Xu LH, Wang F, Fang SH, Lu YB, Hu H, Wei EQ, Zhang WP. Anti-proliferation effect of APO866 on C6 glioblastoma cells by inhibiting nicotinamide phosphoribosyltransferase. Eur J Pharmacol. 2012 Jan 15;674(2-3):163-70
This article should be referenced as such:
Koumaki V, Dalamaga M. NAMPT (nicotinamide phosphoribosyltransferase). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):909-912.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 913
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
PRKCI (protein kinase C, iota) Verline Justilien, Alan P Fields
Department of Cancer Biology, Mayo Clinic College of Medicine, Jacksonville, Florida, 32224 USA (VJ, APF)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/PRKCIID41857ch3q26.html DOI: 10.4267/2042/48364
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: DXS1179E: nPKC-iota: PKCI
HGNC (Hugo): PRKCI
Location: 3q26.2
Local order The PRKCI gene is located between the polyhomeotic homolog 3 gene in centromeric position and the SKI-like oncogene in telomeric position (according to GeneLoc).
DNA/RNA Description The PRKCI gene is composed of 18 exons and spans 83618 bases on the plus strand.
Transcription The PRKCI transcript (NM_002740) contains 4884 bases and the open reading frame spans from 239 to 2029.
Pseudogene There is a single exon pseudogene mapped on chromosome X.
Protein Description PKCι consist of 596 amino acids and has a molecular mass of 68262 Da. PKCι is a member of the PKCs, a diverse family of lipid dependent serine/threonine kinases. PKCι activity can be regulated by lipid second messengers (ceramide, phosphatidylinositol 3,4,5-P3, and phosphatidic acid), phosphoinositide-dependent kinase (PDK1), tyrosine phosphorylation and specific protein-protein interactions. The PB1 domain within the N-terminal regulatory domain mediates protein-protein interactions between PKCι and other PB1 domain containing proteins such as ZIP/p62 (Hirano et al., 2004; Puls et al., 1997), Par-6 (partitioning-defective 6) (Joberty et al., 2000; Lin et al., 2000; Noda et al., 2001; Qiu et al., 2000) and MEK5 (MAPK (mitogen-activated protein kinase)/ERK (extracellular-signal-regulated kinase) kinase 5) (Diaz-Meco and Moscat, 2001; Hirano et al., 2004). In the inactive state, the PKCι PS is positioned in the substrate binding cavity in the kinase domain and is displaced upon PKCι activation.
Location sequence of PRKCI on Chromosome 3. PRKCI gene is indicated by red arrow.

PRKCI (protein kinase C, iota) Justilien V, Fields AP
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 914
Exon-intron structure of the PRKCI gene. Blue vertical bars correspond to exons, green bar represents 5'UTR and orange 3'UTR.
Schematic diagram showing the domain structure of PKCι. PB-1 Phox-Bem1; PS: auto-inhibitory pseudosubstrate sequence. Phosphatidylserine binds the C1 domain to anchor PKCι to the membrane. The PKCι catalytic domain is subdivided into the C3 and C4 domains that mediate ATP-binding and substrate binding.
Expression PKCι is widely expressed with varying levels in different tissues (Selbie et al., 1993).
Localisation PKCι is mainly expressed in the cytoplasm. PKCι is translocated to the cell membrane in response to second messengers and colocalizes with p62/ZIP in lysosome-targeted endosomes (Sanchez et al., 1998). Src phosphorylation leads to translocation of PKCι into the nucleus (White et al., 2002) where it forms a complex with Cdk7 (Win and Acevedo-Duncan, 2008).
Function PKCι is a lipid-dependent, serine/threonine kinase. PKCι participates a number of signaling pathways that regulate cell survival (Sanz et al., 1999; Wooten et al., 1999; Xie et al., 2000), differentiation (Wooten et al., 2000), polarity (Joberty et al., 2000), and microtubule dynamics in the early secretory pathway (Tisdale, 2002).
Homology PRKCI is highly evolutionarily conserved. PKCι and PKCζ exhibit 72% overall amino acid sequence homology and 86% identity within the kinase domain. PKCι shows less homology with the other PKC isoform, with less than 53% identity in the highly conserved catalytic domain (Selbie et al., 1993).
Mutations Germinal No germline mutations in the PRKCI gene have been reported.
Somatic The PKCι gene is amplified as part of the 3q26 amplicon in lung (Regala et al., 2005b), esophageal
(Yang et al., 2008) and ovarian (Eder et al., 2005; Zhang et al., 2006) cancers. A P118L mutation was found in a metastatic melanoma sample (Greenman et al., 2007).
Implicated in Various cancers Note PKCι overexpression has been observed in numerous human cancers including cancers of the lung (Regala et al., 2005b), pancreas (Scotti et al., 2010), stomach (Takagawa et al., 2010), colon (Murray et al., 2004), esophagus (Yang et al., 2008), liver (Du et al., 2009), bile duct (Li et al., 2008), breast (Kojima et al., 2008), ovary (Weichert et al., 2003; Eder et al., 2005; Zhang et al., 2006), prostate (Ishiguro et al., 2009), and brain (Patel et al., 2008). PKCι is itself an oncogene, which appears to be activated through tumor-specific overexpression. In addition, however, PKCι is activated downstream of other oncogenes including oncogenic Ras, Bcr-Abl and Src.
Non Small Cell Lung Cancer (NSCLC) Prognosis Elevated levels of PKCι expression correlate with poor clinical outcome in NSCLC patients (Regala et al., 2005b).
Cytogenetics The PRKCI gene is amplified as part of the 3q26 amplicon in NSCLC.
Oncogenesis PKCι is an oncogene in NSCLC. PRKCI is amplified as part of the 3q26 amplicon in NSCLC and amplication drives PKCι overexpression in NSCLC cell lines and primary NSCLC tumours. PKCι is required for transformed (anchorage-independent) growth and invasion of human NSCLC cells (Frederick et al., 2008; Regala et al., 2005a). Disruption of the Prkci gene inhibits oncogenic Kras induced expansion and transformation of tumor-initiating, lung stem-like cells. Consequently, genetic loss of Prkci dramatically

PRKCI (protein kinase C, iota) Justilien V, Fields AP
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 915
inhibits Kras-initiated hyperplasia and subsequent lung tumor formation in vivo. PKCι enhances resistance of NSCLC to NNK-induced apoptosis by phosphorylating the pro-apoptotic protein BAD (Jin et al., 2005). PKCι forms an oncogeneic complex with Par6 that activates a Rac1-Mek-Erk signaling axis that drives the transformed growth and invasion of NSCLC cells in vitro (Frederick et al., 2008; Regala et al., 2005a) and tumorigenicity in vivo (Regala et al., 2005a). PKCι and the oncogene ECT2 are genetically linked through coordinate gene amplification as part of the 3q26 amplicon in NSCLC tumors (Justilien and Fields, 2009). PKCι phosphorylates Ect2 and forms an oncogenic PKCι-Par6-Ect2 complex that drives NSCLC cell transformation by activating Rac1 (Justilien and Fields, 2009; Justilien et al., 2011). Expression of MMP10 is regulated through the PKCι-Par6-Rac1 signaling axis and MMP10 represents a key downstream effector in PKCι mediated transformation in lung cancer cells that is required for transformed growth and invasion (Frederick et al., 2008). PKCι also regulates expression of COPB2, ELF3, RFC4, and PLS1 in primary lung adenocarcinoma (Erdogan et al., 2009). The PKCι inhibitor aurothiomalate (ATM) disrupts the PB1-PB1 domain interaction between PKCι and Par6 and inhibits PKCι-mediated Rac1 activation and blocks anchorage-independent growth of NSCLC cells in vitro and tumorigenicity in vivo (Erdogan et al., 2006; Stallings-Mann, 2006).
Colon cancer Oncogenesis PKCι expression is elevated in human colon tumors, AOM-induced colon tumors in mice (Murray et al., 2004) and intestinal tumors in APCMin/+ mice (Murray et al., 2009; Oster and Leitges, 2006). Expression of caPKCι in the colonic epithelium of mice led to an increase in the number of AOM-induced colon tumors, and promoted tumor progression from benign adenoma to malignant intramucosal carcinoma (Murray et al., 2004) PKCι is required for oncogenic Ras-mediated transformation of the intestinal epithelium in vitro and in vivo. PKCι is also required for the formation of intestinal tumors in APCMin/+ mice (Murray et al., 2009).
Pancreatic cancer Prognosis PKCι overexpression predicts poor survival in pancreatic cancer patients (Scotti et al., 2010).
Oncogenesis PKCι is significantly overexpressed in human pancreatic cancer. Knock down of PKCι expression using lentiviral-mediated shRNA blocked transformed (anchorage-independent) growth and invasion of human Pancreatic Ductal Adenocarcinoma (PDAC) cells (Scotti et al., 2010). Disruption of PKCι expression also blocks tumorigenicity of PDAC cell tumors injected orthotopically into the pancreas (Scotti
et al., 2010). Analysis of human PDAC cells after orthotopic injection into the mouse pancreas revealed that PKCι-deficient tumor cells yielded significantly smaller tumors and significantly fewer metastases to the kidney, liver, diaphragm and mesentery (Scotti et al., 2010). The Rac1-MEK/ERK1/2 signaling axis is required for PKCiota-mediated transformed growth and cellular invasion of PDAC cells (Scotti et al., 2010).
Ovarian cancer Prognosis PKCι expression is a strong predictor of survival when combined in a multi-variate analysis with tumor cyclin E expression (Eder et al., 2005).
Cytogenetics The PRKCI gene is amplified as part of the 3q26 amplicon in ovarian cancer (Eder et al., 2005).
Oncogenesis PKCι is frequently overexpressed in patients with ovarian cancer (Eder et al., 2005; Weichert et al., 2003; Zhang et al., 2006). PKCι expression in ovarian cancer patients correlates with tumor stage suggesting the involvement of PKCι in tumor progression and aggressiveness (Eder et al., 2005; Weichert et al., 2003; Zhang et al., 2006). Decreased PKCι expression reduced anchorage-independent growth of ovarian cancer cells, whereas overexpression of PKCι promoted murine ovarian surface epithelium transformation (Zhang et al., 2006).
Chronic myelogenous leukemia Oncogenesis PKCι is highly expressed in human K562 leukemia cells and functions as a survival gene in chronic myelogenous leukemia (CML). The chimeric tyrosine kinase oncogene Bcr-Abl activates a Ras/Mek/Erk signaling pathway that stimulates PKCι expression through an Elk1 transcription factor site in the proximal promoter of PKCι (Gustafson et al., 2004). Bcr-Abl activation of PKCι is necessary and sufficient to mediate apoptotic resistance to chemotherapy in K562 CML cells (Murray and Fields, 1997).
Gliomas Oncogenesis PKCι is overexpressed in glioblastoma multiforme. PKCι is required for survival and chemoresistance of glioblastoma cells. Genetic disruption of PKCι expression results in sensitization of glioblastoma cells to cisplatin (Baldwin et al., 2008). RNAi mediated depletion of PKCι also blocks the proliferative and invasive properties of glioma cell lines in vitro (Baldwin et al., 2008; Patel et al., 2008). PKCι promotes survival in glioblastoma cells through attenuation of p38 mitogen-activated protein kinase signaling that protects these cells against cytotoxicity to chemotherapeutic agents (Baldwin et al., 2008).

PRKCI (protein kinase C, iota) Justilien V, Fields AP
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 916
Esophageal cancer Cytogenetics PRKCI gene is amplified as part of the 3q26 amplicon (Yang et al., 2008).
Oncogenesis PRKCI is amplified in 53% of esophageal squamous cell carcinomas (ESCC) and PKCι protein expression correlated with PRKCI gene amplification in these tumors (Yang et al., 2008). Examination of clinicopathologic features of ESCC tumors revealed a significant correlation between PRKCI expression and larger tumor size, later stage and lymph node metastasis suggesting that PRKCI overexpression is a hallmark of tumor progression and metastasis in ESCC (Yang et al., 2008).
References Selbie LA, Schmitz-Peiffer C, Sheng Y, Biden TJ. Molecular cloning and characterization of PKC iota, an atypical isoform of protein kinase C derived from insulin-secreting cells. J Biol Chem. 1993 Nov 15;268(32):24296-302
Murray NR, Fields AP. Atypical protein kinase C iota protects human leukemia cells against drug-induced apoptosis. J Biol Chem. 1997 Oct 31;272(44):27521-4
Puls A, Schmidt S, Grawe F, Stabel S. Interaction of protein kinase C zeta with ZIP, a novel protein kinase C-binding protein. Proc Natl Acad Sci U S A. 1997 Jun 10;94(12):6191-6
Sanchez P, De Carcer G, Sandoval IV, Moscat J, Diaz-Meco MT. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol Cell Biol. 1998 May;18(5):3069-80
Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999 Jun 1;18(11):3044-53
Wooten MW, Seibenhener ML, Zhou G, Vandenplas ML, Tan TH. Overexpression of atypical PKC in PC12 cells enhances NGF-responsiveness and survival through an NF-kappaB dependent pathway. Cell Death Differ. 1999 Aug;6(8):753-64
Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000 Aug;2(8):531-9
Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000 Aug;2(8):540-7
Qiu RG, Abo A, Steven Martin G. A human homolog of the C. elegans polarity determinant Par-6 links Rac and Cdc42 to PKCzeta signaling and cell transformation. Curr Biol. 2000 Jun 15;10(12):697-707
Wooten MW, Seibenhener ML, Neidigh KB, Vandenplas ML. Mapping of atypical protein kinase C within the nerve growth factor signaling cascade: relationship to differentiation and survival of PC12 cells. Mol Cell Biol. 2000 Jul;20(13):4494-504
Xie J, Guo Q, Zhu H, Wooten MW, Mattson MP. Protein kinase C iota protects neural cells against apoptosis induced by amyloid beta-peptide. Brain Res Mol Brain Res. 2000 Oct 20;82(1-2):107-13
Diaz-Meco MT, Moscat J. MEK5, a new target of the atypical protein kinase C isoforms in mitogenic signaling. Mol Cell Biol. 2001 Feb;21(4):1218-27
Noda Y, Takeya R, Ohno S, Naito S, Ito T, Sumimoto H. Human homologues of the Caenorhabditis elegans cell polarity protein PAR6 as an adaptor that links the small GTPases Rac and Cdc42 to atypical protein kinase C. Genes Cells. 2001 Feb;6(2):107-19
Tisdale EJ. Glyceraldehyde-3-phosphate dehydrogenase is phosphorylated by protein kinase Ciota /lambda and plays a role in microtubule dynamics in the early secretory pathway. J Biol Chem. 2002 Feb 1;277(5):3334-41
White WO, Seibenhener ML, Wooten MW. Phosphorylation of tyrosine 256 facilitates nuclear import of atypical protein kinase C. J Cell Biochem. 2002;85(1):42-53
Weichert W, Gekeler V, Denkert C, Dietel M, Hauptmann S. Protein kinase C isoform expression in ovarian carcinoma correlates with indicators of poor prognosis. Int J Oncol. 2003 Sep;23(3):633-9
Gustafson WC, Ray S, Jamieson L, Thompson EA, Brasier AR, Fields AP. Bcr-Abl regulates protein kinase Ciota (PKCiota) transcription via an Elk1 site in the PKCiota promoter. J Biol Chem. 2004 Mar 5;279(10):9400-8
Hirano Y, Yoshinaga S, Ogura K, Yokochi M, Noda Y, Sumimoto H, Inagaki F. Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. J Biol Chem. 2004 Jul 23;279(30):31883-90
Murray NR, Jamieson L, Yu W, Zhang J, Gökmen-Polar Y, Sier D, Anastasiadis P, Gatalica Z, Thompson EA, Fields AP. Protein kinase Ciota is required for Ras transformation and colon carcinogenesis in vivo. J Cell Biol. 2004 Mar 15;164(6):797-802
Eder AM, Sui X, Rosen DG, Nolden LK, Cheng KW, Lahad JP, Kango-Singh M, Lu KH, Warneke CL, Atkinson EN, Bedrosian I, Keyomarsi K, Kuo WL, Gray JW, Yin JC, Liu J, Halder G, Mills GB. Atypical PKCiota contributes to poor prognosis through loss of apical-basal polarity and cyclin E overexpression in ovarian cancer. Proc Natl Acad Sci U S A. 2005 Aug 30;102(35):12519-24
Jin Z, Xin M, Deng X. Survival function of protein kinase C{iota} as a novel nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-activated bad kinase. J Biol Chem. 2005 Apr 22;280(16):16045-52
Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005a Sep 2;280(35):31109-15
Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, Fields AP. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005b Oct 1;65(19):8905-11
Erdogan E, Lamark T, Stallings-Mann M, Lee Jamieson, Pellecchia M, Thompson EA, Johansen T, Fields AP. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006 Sep 22;281(38):28450-9
Oster H, Leitges M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 2006 Jul 15;66(14):6955-63
Stallings-Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP. A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells. Cancer Res. 2006 Feb 1;66(3):1767-74
Zhang L, Huang J, Yang N, Liang S, Barchetti A, Giannakakis A, Cadungog MG, O'Brien-Jenkins A, Massobrio M, Roby KF, Katsaros D, Gimotty P, Butzow R, Weber BL, Coukos G. Integrative genomic analysis of protein kinase C (PKC) family

PRKCI (protein kinase C, iota) Justilien V, Fields AP
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 917
identifies PKCiota as a biomarker and potential oncogene in ovarian carcinoma. Cancer Res. 2006 May 1;66(9):4627-35
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O'Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007 Mar 8;446(7132):153-8
Baldwin RM, Parolin DA, Lorimer IA. Regulation of glioblastoma cell invasion by PKC iota and RhoB. Oncogene. 2008 Jun 5;27(25):3587-95
Frederick LA, Matthews JA, Jamieson L, Justilien V, Thompson EA, Radisky DC, Fields AP. Matrix metalloproteinase-10 is a critical effector of protein kinase Ciota-Par6alpha-mediated lung cancer. Oncogene. 2008 Aug 14;27(35):4841-53
Kojima Y, Akimoto K, Nagashima Y, Ishiguro H, Shirai S, Chishima T, Ichikawa Y, Ishikawa T, Sasaki T, Kubota Y, Inayama Y, Aoki I, Ohno S, Shimada H. The overexpression and altered localization of the atypical protein kinase C lambda/iota in breast cancer correlates with the pathologic type of these tumors. Hum Pathol. 2008 Jun;39(6):824-31
Li Q, Wang JM, Liu C, Xiao BL, Lu JX, Zou SQ. Correlation of aPKC-iota and E-cadherin expression with invasion and prognosis of cholangiocarcinoma. Hepatobiliary Pancreat Dis Int. 2008 Feb;7(1):70-5
Patel R, Win H, Desai S, Patel K, Matthews JA, Acevedo-Duncan M. Involvement of PKC-iota in glioma proliferation. Cell Prolif. 2008 Feb;41(1):122-35
Win HY, Acevedo-Duncan M. Atypical protein kinase C phosphorylates IKKalphabeta in transformed non-malignant and malignant prostate cell survival. Cancer Lett. 2008 Nov 8;270(2):302-11
Yang YL, Chu JY, Luo ML, Wu YP, Zhang Y, Feng YB, Shi ZZ, Xu X, Han YL, Cai Y, Dong JT, Zhan QM, Wu M, Wang MR.
Amplification of PRKCI, located in 3q26, is associated with lymph node metastasis in esophageal squamous cell carcinoma. Genes Chromosomes Cancer. 2008 Feb;47(2):127-36
Du GS, Wang JM, Lu JX, Li Q, Ma CQ, Du JT, Zou SQ. Expression of P-aPKC-iota, E-cadherin, and beta-catenin related to invasion and metastasis in hepatocellular carcinoma. Ann Surg Oncol. 2009 Jun;16(6):1578-86
Erdogan E, Klee EW, Thompson EA, Fields AP. Meta-analysis of oncogenic protein kinase Ciota signaling in lung adenocarcinoma. Clin Cancer Res. 2009 Mar 1;15(5):1527-33
Ishiguro H, Akimoto K, Nagashima Y, Kojima Y, Sasaki T, Ishiguro-Imagawa Y, Nakaigawa N, Ohno S, Kubota Y, Uemura H. aPKClambda/iota promotes growth of prostate cancer cells in an autocrine manner through transcriptional activation of interleukin-6. Proc Natl Acad Sci U S A. 2009 Sep 22;106(38):16369-74
Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene. 2009 Oct 15;28(41):3597-607
Murray NR, Weems J, Braun U, Leitges M, Fields AP. Protein kinase C betaII and PKCiota/lambda: collaborating partners in colon cancer promotion and progression. Cancer Res. 2009 Jan 15;69(2):656-62
Scotti ML, Bamlet WR, Smyrk TC, Fields AP, Murray NR. Protein kinase Ciota is required for pancreatic cancer cell transformed growth and tumorigenesis. Cancer Res. 2010 Mar 1;70(5):2064-74
Takagawa R, Akimoto K, Ichikawa Y, Akiyama H, Kojima Y, Ishiguro H, Inayama Y, Aoki I, Kunisaki C, Endo I, Nagashima Y, Ohno S. High expression of atypical protein kinase C lambda/iota in gastric cancer as a prognostic factor for recurrence. Ann Surg Oncol. 2010 Jan;17(1):81-8
Justilien V, Jameison L, Der CJ, Rossman KL, Fields AP. Oncogenic activity of Ect2 is regulated through protein kinase C iota-mediated phosphorylation. J Biol Chem. 2011 Mar 11;286(10):8149-57
This article should be referenced as such:
Justilien V, Fields AP. PRKCI (protein kinase C, iota). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):913-917.
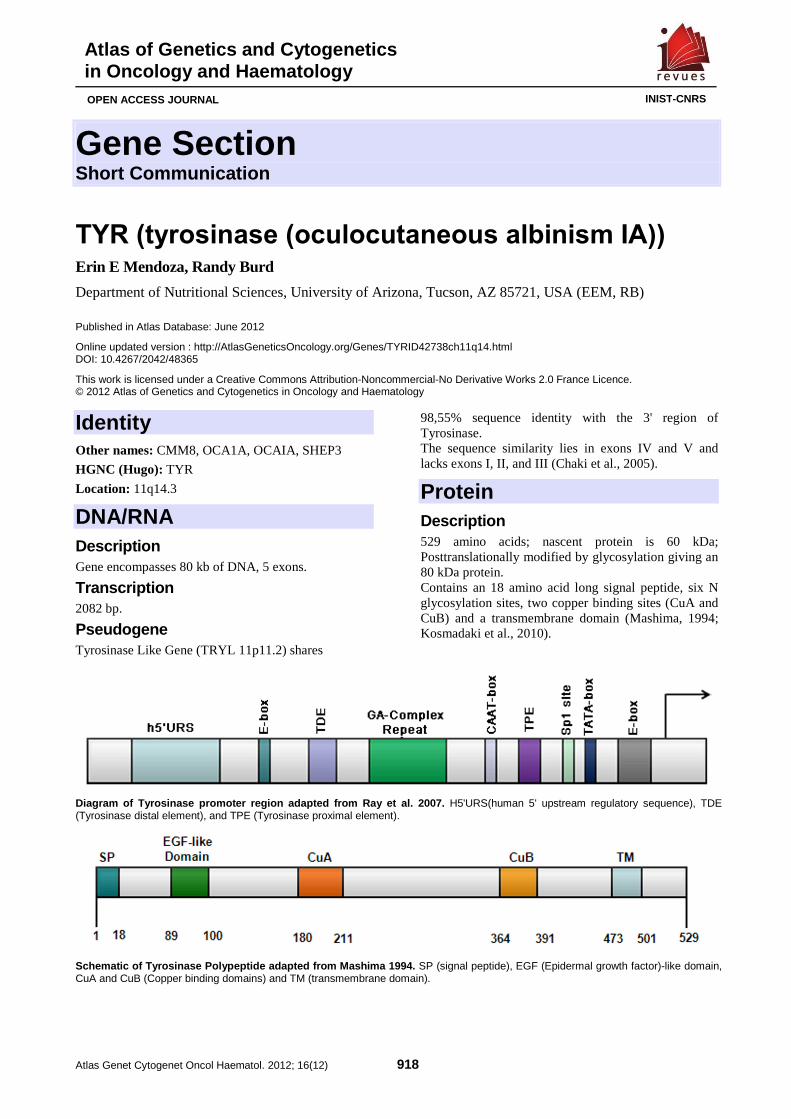
Gene Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 918
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
TYR (tyrosinase (oculocutaneous albinism IA)) Erin E Mendoza, Randy Burd
Department of Nutritional Sciences, University of Arizona, Tucson, AZ 85721, USA (EEM, RB)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/TYRID42738ch11q14.html DOI: 10.4267/2042/48365
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: CMM8, OCA1A, OCAIA, SHEP3
HGNC (Hugo): TYR
Location: 11q14.3
DNA/RNA Description Gene encompasses 80 kb of DNA, 5 exons.
Transcription 2082 bp.
Pseudogene Tyrosinase Like Gene (TRYL 11p11.2) shares
98,55% sequence identity with the 3' region of Tyrosinase. The sequence similarity lies in exons IV and V and lacks exons I, II, and III (Chaki et al., 2005).
Protein Description 529 amino acids; nascent protein is 60 kDa; Posttranslationally modified by glycosylation giving an 80 kDa protein. Contains an 18 amino acid long signal peptide, six N glycosylation sites, two copper binding sites (CuA and CuB) and a transmembrane domain (Mashima, 1994; Kosmadaki et al., 2010).
Diagram of Tyrosinase promoter region adapted from Ray et al. 2007. H5'URS(human 5' upstream regulatory sequence), TDE (Tyrosinase distal element), and TPE (Tyrosinase proximal element).
Schematic of Tyrosinase Polypeptide adapted from Ma shima 1994. SP (signal peptide), EGF (Epidermal growth factor)-like domain, CuA and CuB (Copper binding domains) and TM (transmembrane domain).

TYR (tyrosinase (oculocutaneous albinism IA)) Mendoza EE, Burd R
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 919
Expression Expressed mainly in neural crest derived melanocytes and is sorted into the melanosomes within the melanocyte. Tyrosinase is also found in retinal pigment epithelium cells (Hearing, 2011).
Localisation Transmembrane protein.
Function Tyrosinase catalyzes conversion of tyrosine to DOPA; the rate limiting step of melanin biosynthesis and subsequently DOPA to dopaquinone (Olivares et al., 2009).
Tyrosinase catalyzes the conversion of tyrosine to DOPA in the rate-limiting step of melanin biosynthesis.
Homology The protein tyrosinase related protein 1 (TRP1) is a member of the tyrosinase protein family and utilizes copper as its cofactor. Its function in humans is not well elucidated but is thought to aid in maintaining tyrosinase catalytic activity and stability. It is also involved in maintaining melanosome structure as well as proliferation and cell death of melanocytes (Sarangarajan et al., 2000; Ghanem et al., 2011). Tyrosinase related protein 2 (TRP2), which is also known as DOPAchrome tautomerase catalyzes the conversion of DOPAchrome to 5,6-dihydroxy indole-2-carboxylic acid (DHICA). TRP2 binds 2 zinc ions as cofactors instead of copper (Olivares et al., 2001; Wan et al., 2011).
Mutations Germinal Partial or complete deletion of Tyrosinase leads to dysregulation of melanin synthesis within the melanosomes leading to oculocutaneous albinism (OCA1). The presence of non-pathologic polymorphisms results in variations in skin pigmentation. There are a total of 189 reported OCA1 mutations including 148 missense or nonsense, 23 small deletions, 8 small insertions, 2 insertion/deletion type 1, 1 complex rearrangement, and 7 splice site alterations (Ray et al., 2007; Ko et al., 2011).
Implicated in Melanoma Disease Highly aggressive neoplasma arising from
melanocytes. Melanoma is responsible for the majority of skin cancer related deaths with a very high probability of metastasis. This neoplasm is greatly resistant to most conventional therapies. Due to the longevity of melanocytes, these cells are considered to have a greater mutagenic burden. This burden is also greater due to the position of melanocytes within the skin and their exposure to UV light. Tyrosinase enzymatic activity has been found to be associated with a better prognosis due to its association with functional activity of the tumor supressor p53. Tyrosinase-mediated melanin production signaled by p53 activation is a key protective response to UV damage (Flaherty, 2012; Gilcrest, 2011).
Oncogenesis Several environmental and genetic factors are involved in the complex process of melanocytic tumorigenesis. Melanin production involving tyrosinase as the rate-limiting step has been shown to protect keratinocytes from DNA damage and oxidative stress from ultra violet radiation; A low incidence of melanoma in darker skinned populations has been observed, indicating a photoprotective role of melanin (Kanavy, 2011).
Oculocutaneous albinism 1A Disease Autosomal recessive condition that results in partial or complete loss of tyrosinase activity. Complete loss of activity results in the absence of melanin in the skin and eyes and is classified as OCA1A and the presence of only reduced tyrosinase activity is classified as OCA1B. Complete loss of tyrosinase activity results in the total absence of melanin in the skin and hair. The iris in patients with OCA1A is light blue or gray and the retina lacks pigmentation as well. Tyrosinase null patients have greatly reduced visual acuity accompanied by nystagmus, strabismus, and usually photophobia (Ray et al., 2007). Patients with OCA1B present with varying levels of pigment. The hair in these patients is often yellow. The yellow color is a result of the pheomelanin synthesis. Dopaquinone has a high affinity for sulfhydryl compounds and produces pheomelanin as a result, causing yellow pigmentation. Patients with OCA1B often develop pigmentation in the cooler regions of the body, like the extremities (Chiang et al., 2008).
Prognosis Prognosis in patients is generally good with no system abnormalities other than the loss or reduction in pigmentation. Patients are advised to protect their skin from sun to prevent sunburn (Ray et al., 2007).
Oncogenesis Transcription of tyrosinase has been shown to increase with activation of the tumor suppressor p53, linking both to the tanning response following exposure to UV damage (Khlgatian et al., 2002 and Cui et al., 2007).

TYR (tyrosinase (oculocutaneous albinism IA)) Mendoza EE, Burd R
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 920
References Tsukamoto K, Jackson IJ, Urabe K, Montague PM, Hearing VJ. A second tyrosinase-related protein, TRP-2, is a melanogenic enzyme termed DOPAchrome tautomerase. EMBO J. 1992 Feb;11(2):519-26
Mishima Y. Molecular and biological control of melanogenesis through tyrosinase genes and intrinsic and extrinsic regulatory factors. Pigment Cell Res. 1994 Dec;7(6):376-87
Sarangarajan R, Zhao Y, Babcock G, Cornelius J, Lamoreux ML, Boissy RE. Mutant alleles at the brown locus encoding tyrosinase-related protein-1 (TRP-1) affect proliferation of mouse melanocytes in culture. Pigment Cell Res. 2000 Oct;13(5):337-44
Olivares C, Jiménez-Cervantes C, Lozano JA, Solano F, García-Borrón JC. The 5,6-dihydroxyindole-2-carboxylic acid (DHICA) oxidase activity of human tyrosinase. Biochem J. 2001 Feb 15;354(Pt 1):131-9
Khlgatian MK, Hadshiew IM, Asawanonda P, Yaar M, Eller MS, Fujita M, Norris DA, Gilchrest BA. Tyrosinase gene expression is regulated by p53. J Invest Dermatol. 2002 Jan;118(1):126-32
Chaki M, Mukhopadhyay A, Ray K. Determination of variants in the 3'-region of the tyrosinase gene requires locus specific amplification. Hum Mutat. 2005 Jul;26(1):53-8
Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, D'Orazio J, Fung CY, Schanbacher CF, Granter SR, Fisher DE. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007 Mar 9;128(5):853-64
Ray K, Chaki M, Sengupta M. Tyrosinase and ocular diseases: some novel thoughts on the molecular basis of oculocutaneous albinism type 1. Prog Retin Eye Res. 2007 Jul;26(4):323-58
Chiang PW, Drautz JM, Tsai AC, Spector E, Clericuzio CL. A new hypothesis of OCA1B. Am J Med Genet A. 2008 Nov 15;146A(22):2968-70
Olivares C, Solano F. New insights into the active site structure and catalytic mechanism of tyrosinase and its related proteins. Pigment Cell Melanoma Res. 2009 Dec;22(6):750-60
Kosmadaki MG, Naif A, Hee-Young P. Recent progresses in understanding pigmentation. G Ital Dermatol Venereol. 2010 Feb;145(1):47-55
Ghanem G, Fabrice J. Tyrosinase related protein 1 (TYRP1/gp75) in human cutaneous melanoma. Mol Oncol. 2011 Apr;5(2):150-5
Gilchrest BA. Molecular aspects of tanning. J Invest Dermatol. 2011 Nov 17;131(E1):E14-7
Hearing VJ. Determination of melanin synthetic pathways. J Invest Dermatol. 2011 Nov 17;131(E1):E8-E11
Kanavy HE, Gerstenblith MR. Ultraviolet radiation and melanoma. Semin Cutan Med Surg. 2011 Dec;30(4):222-8
Wan P, Hu Y, He L. Regulation of melanocyte pivotal transcription factor MITF by some other transcription factors. Mol Cell Biochem. 2011 Aug;354(1-2):241-6
Flaherty KT. Targeting metastatic melanoma. Annu Rev Med. 2012;63:171-83
Ko JM, Yang JA, Jeong SY, Kim HJ. Mutation spectrum of the TYR and SLC45A2 genes in patients with oculocutaneous albinism. Mol Med Report. 2012 Apr;5(4):943-8
This article should be referenced as such:
Mendoza EE, Burd R. TYR (tyrosinase (oculocutaneous albinism IA)). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):918-920.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 921
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
NUP214/ABL1 fusion gene on amplified episomes Nathalie Nadal
Laboratoire d'hematologie, Pavillon de Biologie, CHU Hopital Nord, 42055 St Etienne Cedex 2, France (NN)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/ampNUP214ABL1ID1397.html DOI: 10.4267/2042/48366
This article is an update of : Nadal N. amplified NUP214/ABL1. Atlas Genet Cytogenet Oncol Haematol 2006;10(2):107-109. This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Note Episomes are submicroscopic extrachromosomal structures.
Clinics and pathology Disease T-cell acute lymphoblastic leukemia (T-ALL)
Note Not seen in B-cell ALL or other malignant diseases.
Phenotype/cell stem origin Immature T-cell leukemia (CD3+, CD2+ and CD7+).
Epidemiology In about 5% of T-ALL. Mainly observed in T-ALL associated with the mutually exclusive overexpression of the oncogenes HOX11 and HOX11L2. Found in pediatric and adults T-ALL.
Clinics No major clinical differences between NUP214-ABL negative and positive T-ALL.
Cytology Lymphoblasts.
Treatment NUP214-ABL1 cells are sensitive to tyrosine kinase inhibitors (ITK).
Targeting therapies may improve outcome of patients with T-ALL expressing NUP214-ABL1 but the clinical experience is, yet, too limited to conclude.
Prognosis Most data, but not all, suggests that NUP214-ABL1 fusion gene amplification in T-ALL is associated with poor outcome.
Genetics Note Mechanism of gene amplification The main hypothesis is that genomic amplification is a dynamic process. Molecular chronology of genomic amplification has been schematically described as follows. The first step is the production of submicroscopic, acentric, circular, extrachromosomal DNA molecules which replicate autonomously, called episomes. These DNA molecules are made of amplified genes. 2 mechanisms for the formation of episomes have been proposed: Conservative which preserves the original DNA sequence at the native chromosomal locus and non conservative which leads to the deletion of the original sequence at the native locus. The second step corresponds to an increase in copy number resulting from unequal mitotic segregation and an increase in size. They enlarge over time to form progressively heterogeneously sized structures, microscopically visible, called double minutes (dmin).

NUP214/ABL1 fusion gene on amplified episomes Nadal N
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 922
Episomal amplification of ABL detected by FISH with the commercial probe LSI BCR-ABL ES.
In a later step they may integrate into chromosomes to generate intrachromosomally amplified structures (HSR). In some cases dmins or HSRs may form directly without precursors.
Cytogenetics Cytogenetics morphological Not detectable by conventional cytogenetics. Cryptic, no dmin.
Cytogenetics molecular FISH using commercially available ABL1 probe shows multiple extrachromosomal sites on metaphases and multiple signals in interphase nuclei. The extrachromosomal amplification of ABL1 appears to be pathognomonique for the presence of NUP214-ABL1 fusion in T-ALL.There may be a corresponding deletion of the ABL1 probe on one of the chromosomes 9 (see note above concerning mechanisms of gene amplification).
Probes ABL1 probe.
Additional anomalies None. In apparently normal karyotype or with variable additional abnormalities.
Genes involved and proteins ABL1 Location 9q34.1
DNA/RNA Alternate splicing. mRNA of 6 and 7 kb.
Protein Protein 145 kDa. Localization: nuclear and cytoplasmic; Tyrosine kinase; Ubiquitously expressed. ABL1 modulates T-cell development and plays a role in cytoskeletal remodelling processes in T-cells.
NUP214 (nuclear pore complex protein 241 kDa) Location 9q34.3
Note More telomeric than ABL1.
DNA/RNA Other names: CAN, CAIN, Nucleoporin. 7.5 kb mRNA.
Protein Component of the Nuclear Pore Complex. 214 kDa; 2 dimerization domains (2 leucine zippers) and a repeated motif; forms homodimers. Mediate nucleocytoplasmic transport. Localisation: nuclear membrane; cytoplasmic face.
Result of the chromosomal anomaly Hybrid gene Note NUP214 is recognized as being the second most prevalent fusion gene involving ABL1.

NUP214/ABL1 fusion gene on amplified episomes Nadal N
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 923
Description Molecular analyses delineated the amplicon as a 500 kb region from chromosome band 9q34 containing the genes ABL1, LAMC3 and NUP214. The genomic region from ABL1 to NUP214 circularizes to generate the NUP214-ABL1 fusion gene = New mechanism for generation of a fusion gene. The breakpoint within ABL1 occurs in intron 1 in most cases, in intron 2 in other cases (coincides with ABL1 breakpoint in the Philadelphia chromosome). Whilst the breakpoint in NUP214 is variable (ranging from intron 23 to intron 34).
Transcript NUP214-ABL1 fusion gene.
Detection RT-PCR of the fusion transcript.
Fusion protein Description The NUP214-ABL transcript encodes a 239-333 kDa protein which includes the coiled-coil domain (dimerisation motifs necessary for tyrosine kinase activation and neoplastic transformation) of NUP214 and the tyrosine kinase domain of ABL1. NUP214-ABL protein is a constitutively activated tyrosine kinase most likely implicated in the pathogenesis of T-ALL in a similar mechanism of action as for BCR-ABL as it activates similar pathways and for its sensitivity to ITK. However NUP214-ABL protein is less potent and requires amplification for neoplastic transformation.
Oncogenesis NUP214 is also involved in the translocation t(6;9) seen in myeloid malignancies which results in the fusion gene DEK-NUP214. Unlike NUP214-ABL where the N-terminal region of NUP214 is retained, in DEK-NUP214, it is the C-terminal region of NUP214 which is present. The mode of leukemogenesis of DEK-NUP214 is thought to be interference with nucleocytoplasmic transport processes. It is unknown whether NUP214-ABL acts in the same way.
References Wahl GM. The importance of circular DNA in mammalian gene amplification. Cancer Res. 1989 Mar 15;49(6):1333-40
Barber KE, Martineau M, Harewood L, Stewart M, Cameron E, Strefford JC, Rutherford S, Allen TD, Broadfield ZJ, Cheung KL, Harris RL, Jalali GR, Moorman AV, Robinson HM, Harrison CJ. Amplification of the ABL gene in T-cell acute lymphoblastic leukemia. Leukemia. 2004 Jun;18(6):1153-6
Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, Levine R, Vermeesch JR, Stul M, Dutta B, Boeckx N, Bosly A, Heimann P, Uyttebroeck A, Mentens N, Somers R, MacLeod RA, Drexler HG, Look AT, Gilliland DG, Michaux L, Vandenberghe P, Wlodarska I, Marynen P, Hagemeijer A. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet. 2004 Oct;36(10):1084-9
Ballerini P, Busson M, Fasola S, van den Akker J, Lapillonne H, Romana SP, Marynen P, Bernard OA, Landman-Parker J, Berger R. NUP214-ABL1 amplification in t(5;14)/HOX11L2-positive ALL present with several forms and may have a prognostic significance. Leukemia. 2005 Mar;19(3):468-70
De Keersmaecker K, Graux C, Odero MD, Mentens N, Somers R, Maertens J, Wlodarska I, Vandenberghe P, Hagemeijer A, Marynen P, Cools J. Fusion of EML1 to ABL1 in T-cell acute lymphoblastic leukemia with cryptic t(9;14)(q34;q32). Blood. 2005 Jun 15;105(12):4849-52
Stergianou K, Fox C, Russell NH. Fusion of NUP214 to ABL1 on amplified episomes in T-ALL--implications for treatment. Leukemia. 2005 Sep;19(9):1680-1
Burmeister T, Gökbuget N, Reinhardt R, Rieder H, Hoelzer D, Schwartz S. NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood. 2006 Nov 15;108(10):3556-9
Quintás-Cardama A, Tong W, Manshouri T, Vega F, Lennon PA, Cools J, Gilliland DG, Lee F, Cortes J, Kantarjian H, Garcia-Manero G. Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia. 2008 Jun;22(6):1117-24
De Braekeleer E, Douet-Guilbert N, Rowe D, Bown N, Morel F, Berthou C, Férec C, De Braekeleer M. ABL1 fusion genes in hematological malignancies: a review. Eur J Haematol. 2011 May;86(5):361-71
This article should be referenced as such:
Nadal N. NUP214/ABL1 fusion gene on amplified episomes. Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):921-923.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 924
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
t(3;19)(q27;q13) NAPA/BCL6 Jean-Loup Huret
Genetics, Dept Medical Information, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France (JLH)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0319q27q13NAPABCL6ID2130.html DOI: 10.4267/2042/48367
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology Disease Non Hodgkin lymphoma
Clinics Two cases available : a case of follicular lymphoma transformed to diffuse aggressive lymphoma, from a study with no individual data (Akasaka et al., 2003), and a male patient aged 60 years with a diffuse large B-cell lymphoma (Yunis et al., 1984).
Cytogenetics Additional anomalies The case described by Yunis et al., 1984 showed a complex karyotype.
Genes involved and proteins Note Only the more recent case (Akasaka et al., 2003) was studied for gene rearrangements.
BCL6 Location 3q27.3
Protein 706 amino acids; composed of a NH2-term BTB/POZ domain (amino acids 1-130 (32-99 according to Swiss-Prot) which mediates homodimerization and protein-protein interactions with other corepressors (including HDAC1 and NCOR2/SMRT to constitute a large repressing complex, another transcription repression domain (191-386), PEST sequences (300-417) with a KKYK motif (375-379), and six zinc finger at the C-term (518-541, 546-568, 574-596, 602-624, 630-652, 658-681), responsible for sequence specific DNA binding.
Transcription repressor; recognizes the consensus sequence: TTCCT(A/C)GAA (Albagli-Curiel, 2003). Role in germinal centers of lymphoid follicles. BCL6 prevents ATM and TP53 to induce apoptosis in response to DNA rearrangements such as somatic hypermutation and class switch recombination. Therefore essential for normal B cell development.
NAPA Location 19q13.33
Protein 295 amino acids; SNARE protein found in the endoplasmic reticulum and implicated in protein trafficking, which possesses anti-apoptotic properties by promoting resistance to cisplatin in cancer cells by inducing the degradation of TP53 (Wu et al., 2011).
References Yunis JJ, Oken MM, Theologides A, Howe RB, Kaplan ME. Recurrent chromosomal defects are found in most patients with non-Hodgkin's-lymphoma. Cancer Genet Cytogenet. 1984 Sep;13(1):17-28
Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood. 2003 Aug 15;102(4):1443-8
Albagli-Curiel O. Ambivalent role of BCL6 in cell survival and transformation. Oncogene. 2003 Jan 30;22(4):507-16
Wu ZZ, Sun NK, Chien KY, Chao CC. Silencing of the SNARE protein NAPA sensitizes cancer cells to cisplatin by inducing ERK1/2 signaling, synoviolin ubiquitination and p53 accumulation. Biochem Pharmacol. 2011 Dec 1;82(11):1630-40
This article should be referenced as such:
Huret JL. t(3;19)(q27;q13) NAPA/BCL6. Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):924.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 925
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
t(3;3)(q27;q27) ST6GAL1/BCL6 / del(3)(q27q27) ST6GAL1/BCL6 Jean-Loup Huret
Genetics, Dept Medical Information, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France (JLH)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0303q27q27ST6GAL1BCL6ID2128.html DOI : 10.4267/2042/48368
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology Disease Non Hodgkin lymphoma
Clinics Two cases of follicular lymphoma transformed to diffuse aggressive lymphoma, from a study with no individual data (Akasaka et al., 2003; cited by Ohno, 2006).
Cytogenetics Cytogenetics morphological Cryptic rearrangement.
Genes involved and proteins ST6GAL1 Location 3q27.3
Note ST6GAL1 (ST6 beta-galactosamide alpha-2,6-sialyltranferase 1) is also known under the name of SIAT1, and was described as such in the Alaska's report.
Protein 406 amino acids; glycosyltransferase (Wu et al., 2011).
BCL6 Location 3q27.3
Protein 706 amino acids; composed of a NH2-term BTB/POZ domain (amino acids 1-130 (32-99 according to Swiss-Prot) which mediates homodimerization and protein-protein interactions with other corepressors (including HDAC1 and NCOR2/SMRT to constitute a large repressing complex, another transcription repression domain (191-386), PEST sequences (300-417) with a KKYK motif (375-379), and six zinc finger at the C-term (518-541, 546-568, 574-596, 602-624, 630-652, 658-681), responsible for sequence specific DNA binding. Transcription repressor; recognizes the consensus sequence: TTCCT(A/C)GAA (Albagli-Curiel, 2003). Role in germinal centers of lymphoid follicles. BCL6 prevents ATM and TP53 to induce apoptosis in response to DNA rearrangements such as somatic hypermutation and class switch recombination. Therefore essential for normal B cell development.
Result of the chromosomal anomaly Hybrid gene Note ST6GAL1 and BCL6 are normally separated by 699.5 kb.
References Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood. 2003 Aug 15;102(4):1443-8

t(3;3)(q27;q27) ST6GAL1/BCL6 Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 926
Albagli-Curiel O. Ambivalent role of BCL6 in cell survival and transformation. Oncogene. 2003 Jan 30;22(4):507-16
Ohno H. Pathogenetic and clinical implications of non-immunoglobulin ; BCL6 translocations in B-cell non-Hodgkin's lymphoma. J Clin Exp Hematop. 2006 Nov;46(2):43-53
Wu ZL, Ethen CM, Prather B, Machacek M, Jiang W. Universal phosphatase-coupled glycosyltransferase assay. Glycobiology. 2011 Jun;21(6):727-33
This article should be referenced as such:
Huret JL. t(3;3)(q27;q27) ST6GAL1/BCL6. Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):925-926.
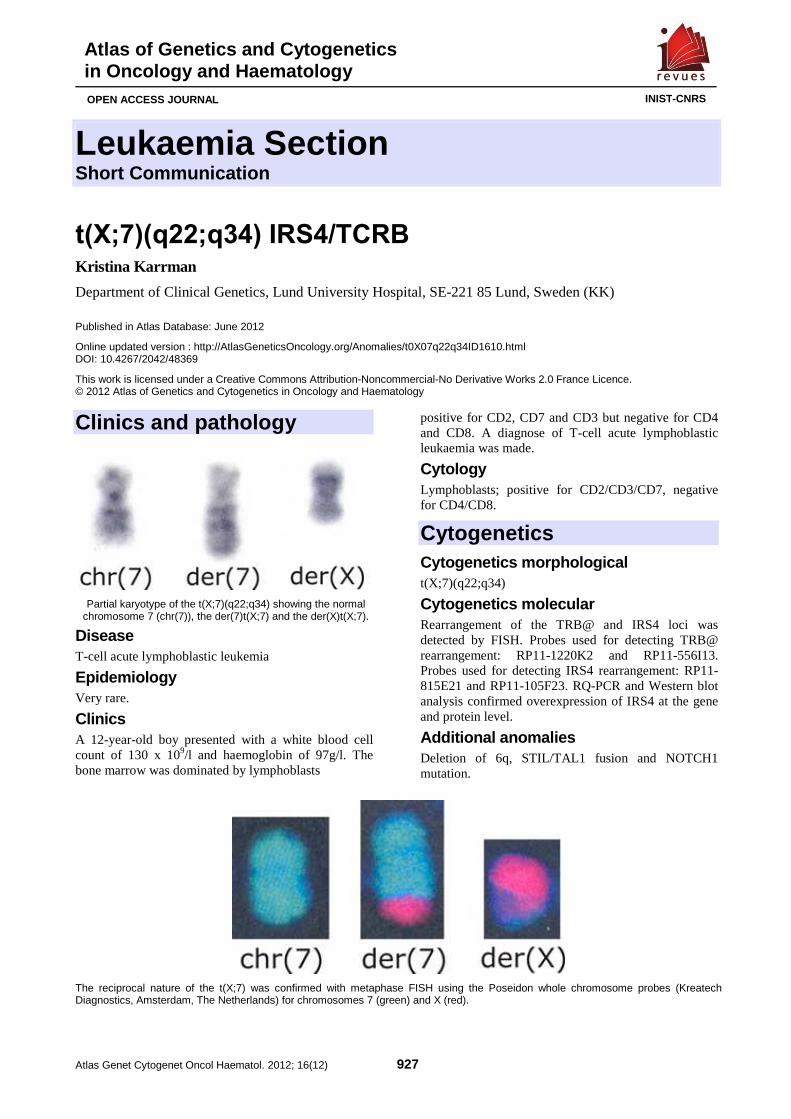
Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 927
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
t(X;7)(q22;q34) IRS4/TCRB Kristina Karrman
Department of Clinical Genetics, Lund University Hospital, SE-221 85 Lund, Sweden (KK)
Published in Atlas Database: June 2012
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0X07q22q34ID1610.html DOI: 10.4267/2042/48369
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology
Partial karyotype of the t(X;7)(q22;q34) showing the normal chromosome 7 (chr(7)), the der(7)t(X;7) and the der(X)t(X;7).
Disease T-cell acute lymphoblastic leukemia
Epidemiology Very rare.
Clinics A 12-year-old boy presented with a white blood cell count of 130 x 109/l and haemoglobin of 97g/l. The bone marrow was dominated by lymphoblasts
positive for CD2, CD7 and CD3 but negative for CD4 and CD8. A diagnose of T-cell acute lymphoblastic leukaemia was made.
Cytology Lymphoblasts; positive for CD2/CD3/CD7, negative for CD4/CD8.
Cytogenetics Cytogenetics morphological t(X;7)(q22;q34)
Cytogenetics molecular Rearrangement of the TRB@ and IRS4 loci was detected by FISH. Probes used for detecting TRB@ rearrangement: RP11-1220K2 and RP11-556I13. Probes used for detecting IRS4 rearrangement: RP11-815E21 and RP11-105F23. RQ-PCR and Western blot analysis confirmed overexpression of IRS4 at the gene and protein level.
Additional anomalies Deletion of 6q, STIL/TAL1 fusion and NOTCH1 mutation.
The reciprocal nature of the t(X;7) was confirmed with metaphase FISH using the Poseidon whole chromosome probes (Kreatech Diagnostics, Amsterdam, The Netherlands) for chromosomes 7 (green) and X (red).

t(X;7)(q22;q34) IRS4/TCRB Karrman K
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 928
Genes involved and proteins IRS4 Location Xq22
Note The IRS family includes IRS1-4 which play a central role in maintaining basic cellular functions, e.g., growth and metabolism. They act as mediators between multiple growth factor receptors that possess tyrosine kinase activity, such as the insulin and insulin growth factor receptors, and a complex network of intracellular signalling molecules, resulting in activation of, for example, the PI3K and RAS/ERK pathways and subsequent transcription of target genes. Relatively little is known about the tumorigenic potential of the IRS proteins. Expression of IRS1, IRS2 or IRS4 in the 32D haematopoietic cell line leads to proliferation of the myeloid progenitor cells and expression of activated IRS4 has recently been demonstrated in the human hepatoblastoma cell line HepG2, with inhibition of IRS4 resulting in diminished growth.
TRB@/TCRB
Result of the chromosomal anomaly Hybrid gene Note The translocation does not result in a fusion gene. The t(X;7) results in juxtaposition of the TRB@ to the IRS4 leading to dysregulation of IRS4.
References Karrman K, Kjeldsen E, Lassen C, Isaksson M, Davidsson J, Andersson A, Hasle H, Fioretos T, Johansson B. The t(X;7)(q22;q34) in paediatric T-cell acute lymphoblastic leukaemia results in overexpression of the insulin receptor substrate 4 gene through illegitimate recombination with the T-cell receptor beta locus. Br J Haematol. 2009 Feb;144(4):546-51
This article should be referenced as such:
Karrman K. t(X;7)(q22;q34) IRS4/TCRB. Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):927-928.

Solid Tumour Section Review
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 929
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Liver: Fibrolamellar carcinoma Xuchen Zhang, Stephen C Ward
The Lillian and Henry M. Stratton-Hans Popper Department of Pathology, Mount Sinai School of Medicine, New York, USA (XZ, SCW)
Published in Atlas Database: July 2012
Online updated version : http://AtlasGeneticsOncology.org/Tumors/FibrolamellarCarcID6191.html DOI: 10.4267/2042/48370
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names Fibrolamellar hepatocellular carcinoma Eosinophilic hepatocellular carcinoma with lamellar fibrosis Polygonal cell hepatocellular carcinoma with fibrous stroma Hepatocellular carcinoma with increased stromal fibrosis Eosinophilic glassy cell hepatoma Fibrolamellar oncocytic hepatoma
Note Fibrolamellar carcinoma (FLC) is a rare distinctive primary malignant liver tumor that was first described in 1956 by Edmondson (Edmondson, 1956). Although FLC was conventionally considered as a histologic variant of hepatocellular carcinoma (HCC), it is now recognized as a distinct clinical entity with respect to its epidemiology, etiology, and prognosis.
Clinics and pathology Phenotype / cell stem origin FLC shows immunohistochemical features of both hepatocytic and biliary differentiation and is most likely derived from a bipotential cell. A recent study examining 26 cases of FLC and 62 cases of classical HCC by immunohistochemistry showed that both tumor types stained uniformly positively with HepPar1 and most showed a canalicular staining pattern for pCEA confirming hepatocytic differentiation. In addition, 39% of hepatocellular carcinoma cases and 59% of fibrolamellar carcinoma cases were positive for glypican-3 and both tumor types were positive for albumin by in situ hybridization. All 22 FLC cases
tested showed positive staining for cytokeratin 7 and epithelial membrane antigen, whereas less than one-third of HCC cases were positive for these markers associated with biliary differentiation. Further, 36% of FLC cases showed staining for other markers of biliary differentiation such as B72.3, cytokeratin 19, EpCAM, or mCEA (Ward et al., 2010). Immunopositivity for cytokeratin 19 and EpCAM are associated with a subset of hepatocellular carcinomas with worse prognosis and indicates a progenitor cell phenotype. A higher proportion of hepatoblastomas and pediatric hepatocellular carcinomas are positive for CK19 and EpCAM than adult HCC. As FLC also arises in a younger age group, there may be pathogenic similarities between these tumors and immunopositivity for these markers may indicate a progenitor phenotype. Zenali et al. studied the "stemness" of FLC and found that FLC was positive for the stem cell markers CD133 and CD44 and also showed reduced cell cycle progression (Zenali et al., 2010). Cases of combined (mixed) FLC with HCC or cholangiocarcinoma have also been reported (Malouf et al., 2012; Tanaka et al., 2005).
Etiology The etiology of FLC is still unclear. In contrast to classic HCC, which is usually associated with cirrhosis, often secondary to chronic infection with hepatitis B virus (HBV), hepatitis C virus (HCV) or other chronic liver diseases (El-Serag, 2011; El-Serag, 2012), FLC generally occurs in noncirrhotic patients without known liver disease. Although studies have shown that 10-20% of cases of FLC occur in patients with HBV and less frequently in patients with HCV, this may just be related to the high worldwide prevalence of viral hepatitis infection (Da Ines et al., 2009). FLC has also been linked to focal nodular hyperplasia (FNH) (Imkie

Liver: Fibrolamellar carcinoma Zhang X, Ward SC
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 930
et al., 2005; Vecchio et al., 1984), a type of benign liver lesion that also presents in younger patients without underlying liver disease. FLC and FNH also have a central scar and accumulate copper (Lefkowitch et al., 1983; Vecchio et al., 1984). This has prompted some to suggest FNH as a possible precursor of FLC; however, there is currently no solid evidence to support this.
Epidemiology FLC has been reported all over the world and the incidence rate for FLC varies by geographical region. In South Africa, FLC accounts 3.3 % of all liver cancers in children < 14 years of age (Moore et al., 2008). In a Mexican study, FLC represents 5.8 % of all liver cancers (Arista-Nasr et al., 2002). In the United States, based on data from the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute , FLC constituted 0.85% of all cases of primary liver cancer and 13.4% of all cases in patients below the age of 40 (El-Serag and Davila, 2004). Overall, FLC represents a small proportion of primary liver cancers as compared with classic HCC, which accounts for 60-80 % of all primary hepatic tumors (Liu et al., 2009). FLC occurs in younger individuals, generally between the ages of 10 and 40 years, and more than 85% of patients are younger than 35 years at the time of presentation (Berman et al., 1988; Liu et al., 2009; Soreide et al., 1986; Torbenson, 2007; Ward and Waxman, 2011). This is in stark contrast with HCC, which rarely occurs before the age of 40 years and has a peak incidence at approximately 70 years of age (El-Serag, 2011). Although female or male predominance for FLC has been reported by different groups (Bhaijee et al., 2009; Meriggi and Forni, 2007), others show that FLC affects males and females equally (El-Serag and Davila, 2004). Recently, Malouf et al. reported that pure FLC typically occurs in patients aged < 30 years, often presents with lymph node metastasis and later extrahepatic recurrences while, mixed FLC with HCC appeared to resemble to classic HCC, occurring in patients aged > 40 years and with the liver as the primary site of disease recurrence.
Clinics The clinical manifestations of FLC are usually nonspecific and include a palpable epigastric mass, hepatomegaly, abdominal pain, abdominal discomfort, nausea, fatigue, and weight loss. Jaundice may be seen in up to 40% of cases (Liu et al., 2009). Patients may also present with various rare symptoms or signs, such as gynecomastia in men or children (McCloskey et al., 1988; Muramori et al., 2011), metastatic lesions in other organs such as the bone (Kutluk et al., 2001), lung (Mroz et al., 2010), pancreas (Thirabanjasak et al.,
2009) and ovary (Benito et al., 2012), hyperammonemic encephalopathy (Sethi et al., 2009), cold agglutinin disease (Al-Matham et al., 2011), shoulder pain (Moghadam et al., 2008), severe inferior vena cava obstruction caused by cardiac spread (Knudson et al., 2012), recurrent deep vein thrombosis (Marrannes et al., 2005), paraneoplastic hyperthyroidism (Carri et al., 1989), Budd-Chiari syndrome with right atrial thrombus, and pulmonary emboli (Asrani and LaRusso, 2012), nonbacterial thrombotic endocarditis (Vaideeswar et al., 1993), and hypoglycemia (Tangkijvanich et al., 2000). The serum levels of aspartate aminotransferase, alanine aminotransferase, and α-fetoprotein (AFP) are usually normal but may be mildly elevated in a minority of cases. Recently, high level of procalcitonin was reported in a case of FLC. Procalcitonin is a relatively specific marker of bacterial disease, and the unusual presentation of FLC mimicking an infectious disease may delay prompt identification of the tumor (Brunel et al., 2011).
Cytology The cytological features of FLC are very distinct. The aspirates are predominantly comprised of dispersed large tumor cells with abundant, granular cytoplasm, thus resembling oncocytes. Hyaline cytoplasmic inclusions (pale bodies) are observed in some cells which helps in the diagnosis .The nuclei and nucleoli are very large, but the nuclear to cytoplasmic ratio is not very high because of the abundant cytoplasm. Fragments of connective tissue, sometimes in intimate contact with tumor cells and corresponding to tumor lamellae, are also observed. Similar tumor cells are also observed in the metastatic FLC.
Pathology Approximately 80-90 % of FLC cases present as a solitary mass more frequently involving the left lobe of the liver. Upon gross examination, the tumor is well circumscribed, multinodular, firm, and yellow-tan or brown, with a bulging cut surface, and may show necrosis and hemorrhage. The most distinctive gross feature of FLC is the presence of a central scar with radiating fibrous septae, seen in about 75 % of cases. The background liver is generally noncirrhotic. Lymph node metastases are common at presentation. Microscopically, the tumor is composed of sheets, nests, and trabeculae of cells that are separated by parallel lamellae of dense collagen bundles. The tumor cells are large and polygonal, with well-defined cell borders and eosinophilic, coarsely granular cytoplasm with large vesiculated nuclei and large nucleoli (Figure 1A and 1B). Stainable copper and bile may be seen in the cytoplasm of tumor cells.
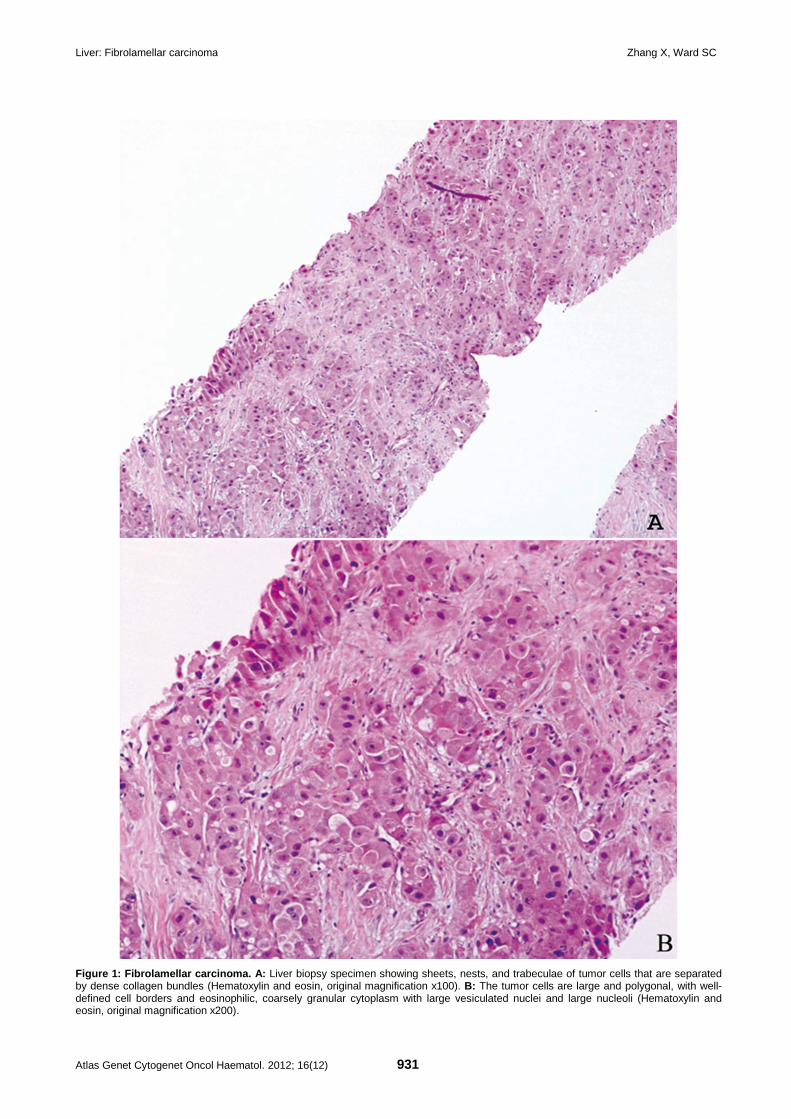
Liver: Fibrolamellar carcinoma Zhang X, Ward SC
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 931
Figure 1: Fibrolamellar carcinoma. A: Liver biopsy specimen showing sheets, nests, and trabeculae of tumor cells that are separated by dense collagen bundles (Hematoxylin and eosin, original magnification x100). B: The tumor cells are large and polygonal, with well-defined cell borders and eosinophilic, coarsely granular cytoplasm with large vesiculated nuclei and large nucleoli (Hematoxylin and eosin, original magnification x200).

Liver: Fibrolamellar carcinoma Zhang X, Ward SC
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 932
Eosinophilic hyaline globules (pale bodies) are present in approximately 50% of cases. Rare variants of FLC have also been described, such as clear cell carcinoma and pseudogland-like tumor with mucin production. FLC features may be seen in tumors with areas of classic HCC or cholangiocarcinoma. As described above, FLC exhibits immunohistochemical evidence of both hepatocyte (HepPar1, glypican-3, AFP, pCEA, and CD10) and bile duct (CK7, CK19, EMA, B72.3, mCEA, and CA19-9) differentiation (Ward et al., 2010). Recently, Ross et al. demonstrated that tumor positivity for CD68 was highly sensitive for FLC and a lack of CD68 staining should suggest caution in making a diagnosis of FLC (Ross et al., 2011). FLCs are usually negative for neuroendocrine tumor markers such as synaptophysin and chromogranin (Ward et al., 2010). Patonai et al. recently investigated the expression of tight junction proteins claudin 5 and tricellulin by immunohistochemistry and demonstrated an expression pattern of claudin 5 in FLC that differs from all other primary malignant epithelial tumors of the liver and may be useful in diagnosis (Patonai et al., 2011).
Treatment Complete surgical resection (eg, wedge resection, anatomic liver resection, or total hepatectomy with orthotopic liver transplantation) is the treatment of choice for FLC. There may be occasional utility in treating FLC with neoadjuvant chemotherapy, trans-arterial chemo-embolization (TACE), or tyrosine kinase targeting therapy with Sorafenib, however, the efficacy is still unclear.
Prognosis The 5-year survival rate of FLC is 37%-76% after complete surgical resection though a high relapse rate (36%-100%) has been reported, especially in patients presenting with advanced-stage disease with large primary tumors and lymphatic metastases. Despite the high rate of recurrence, FLC behaves in a less aggressive fashion, with a 5-year survival rate of 45%-76%, even after relapse (Maniaci et al., 2009). Earlier reports suggested a better prognosis for FLC compared with conventional HCC, however, recent studies have shown that the 5-year survival rate was up to 56% in noncirrhotic HCC, similar to that of FLC (Kakar et al., 2005). It is now widely accepted that the prognosis of FLC is comparable to that of HCC in patients without cirrhosis (Shanbhogue et al., 2011). Recently, Malouf et al. demonstrated that pure and mixed FLC displayed distinct pathological, epigenetic, and clinical patterns at the time of presentation and different outcomes. With a median follow-up of 7.8 years (range, 0.2 years-16 years), the median overall survival among patients with pure FLC was significantly longer than that among patients with mixed FLC (9 years vs 3 years) (Malouf et al., 2012).
Genetics Note FLC usually occurs sporadically without any apparent genetic predisposition. One case has been reported in a 15-year-old girl with Gardner syndrome after desmoid tumors and colonic polyposis had developed (Gruner et al., 1998). Another case has been reported in a 14-year-old girl belonging to a family with Carney syndrome who developed FLC 5 years after removal of a hepatocellular adenoma (Terracciano et al., 2004).
Cytogenetics Note Genetic abnormalities have been recognized in FLC, but there are no specific genetic alterations reported yet. Gains of 1q, 4q, 6p, 7p, 7q, 8q and 19p, and losses of 8p, 9p, 13q, 16p, 18q and Xq have been reported in several studies. Meta-analysis of comparative genomic hybridization studies showed fewer overall alterations in FLC in comparison to HCC and cholangiocarcionoma and found that only gains in 1q and 8q, and losses in 18q occurred with a frequency of >20% (Ward and Waxman, 2011). Allele loss, aneuploid, triploid, tetraploid, and/or complex karyotypes have been demonstrated in limited cases reports (Ward and Waxman, 2011). Loss of tumor suppressor genes, such as DPC4/Smad4 and overexpression of tumor oncogenes, such as anterior gradient-2 has been observed in FLC. DPC4/Smad4 is located on chromosome 18q and loss of expression is also associated with colorectal and pancreatic carcinomas. Anterior gradient-2 overexpression is associated with development of numerous tumors (Vivekanandan et al., 2009). Several signaling pathways, such as RAS, MAPK, PI3K, xenobiotic degradation pathway, transforming growth factor-β pathway, nuclear factor-κB signaling pathway, tyrosine-654-phosphorylated-beta-catenin (Y654-β-catenin) tyrosine kinase signaling pathway, and Small Heterodimer Partner (SHP) nuclear receptor pathway have been implicated in the pathogenesis of FLC and specific signaling pathway target therapy may provide other options for the treatment of FLC (Cieply et al., 2009; Kannangai et al., 2007; Li et al., 2009; Orsatti et al., 1997; Wilczek et al., 2012). The slow proliferation rate, markedly reduced cell cycle progression, and the recently reported lower methylation levels of Ras association domain family 1A gene (RASSF1) promoter in comparison to HCC and mixed FLC may explain the better prognosis and relative resistance to chemotherapy and radiation therapy in patients with FLC (Dhingra et al., 2010; Malouf et al., 2012). Although abundant mitochondria are detected in the cytoplasm ultrastructurally, primary FLC has lower total mitochondrial DNA levels than HCC. Metastatic foci of FLC, on the other hand, have markedly

Liver: Fibrolamellar carcinoma Zhang X, Ward SC
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 933
increased total mitochondrial DNA when compared with primary FLC or primary HCC. Furthermore, sequencing of the entire mitochondrial genome found no frequent or distinct mutations in FLC (Vivekanandan et al., 2010). Their findings suggest that changes in mitochondrial DNA are not responsible for the alterations in mitochondria seen in FLC.
References EDMONDSON HA. Differential diagnosis of tumors and tumor-like lesions of liver in infancy and childhood. AMA J Dis Child. 1956 Feb;91(2):168-86
Lefkowitch JH, Muschel R, Price JB, Marboe C, Braunhut S. Copper and copper-binding protein in fibrolamellar liver cell carcinoma. Cancer. 1983 Jan 1;51(1):97-100
Vecchio FM, Fabiano A, Ghirlanda G, Manna R, Massi G. Fibrolamellar carcinoma of the liver: the malignant counterpart of focal nodular hyperplasia with oncocytic change. Am J Clin Pathol. 1984 Apr;81(4):521-6
Soreide O, Czerniak A, Bradpiece H, Bloom S, Blumgart L. Characteristics of fibrolamellar hepatocellular carcinoma. A study of nine cases and a review of the literature. Am J Surg. 1986 Apr;151(4):518-23
Berman MA, Burnham JA, Sheahan DG. Fibrolamellar carcinoma of the liver: an immunohistochemical study of nineteen cases and a review of the literature. Hum Pathol. 1988 Jul;19(7):784-94
McCloskey JJ, Germain-Lee EL, Perman JA, Plotnick LP, Janoski AH. Gynecomastia as a presenting sign of fibrolamellar carcinoma of the liver. Pediatrics. 1988 Sep;82(3):379-82
Carri J, Peral F, Surreco M, Luján A, Leguizamón R, Martínez G, Salvucci M. [Fibrolamellar hepatocellular carcinoma: a clinical report with paraneoplastic hyperthyroidism (apropos of a case)]. Acta Gastroenterol Latinoam. 1989;19(3):155-64
Vaideeswar P, Pandit MJ, Deshpande JR, Sivaraman A, Vora IM. Fibrolamellar carcinoma of the liver--an unusual presentation. J Postgrad Med. 1993 Jul-Sep;39(3):159-61
Orsatti G, Hytiroglou P, Thung SN, Ishak KG, Paronetto F. Lamellar fibrosis in the fibrolamellar variant of hepatocellular carcinoma: a role for transforming growth factor beta. Liver. 1997 Jun;17(3):152-6
Gruner BA, DeNapoli TS, Andrews W, Tomlinson G, Bowman L, Weitman SD. Hepatocellular carcinoma in children associated with Gardner syndrome or familial adenomatous polyposis. J Pediatr Hematol Oncol. 1998 May-Jun;20(3):274-8
Tangkijvanich P, Thong-Ngam D, Kullavanijaya P, Suwangool P. Fibrolamellar hepatocellular carcinoma in a Thai man who presented with hypoglycemia: case report and review of literature. J Med Assoc Thai. 2000 Jul;83(7):809-16
Kutluk MT, Yalçin B, Büyükpamukçu N, Kale G, Büyükpamukçu M. Fibrolamellar hepatocellular carcinoma with skeletal metastases. Pediatr Hematol Oncol. 2001 Jun;18(4):273-8
Arista-Nasr J, Gutierrez-Villalobos L, Nuncio J, Maldonaldo H, Bornstein-Quevedo L. Fibrolamellar hepatocellular carcinoma in mexican patients. Pathol Oncol Res. 2002;8(2):133-7
El-Serag HB, Davila JA. Is fibrolamellar carcinoma different from hepatocellular carcinoma? A US population-based study. Hepatology. 2004 Mar;39(3):798-803
Terracciano LM, Tornillo L, Avoledo P, Von Schweinitz D, Kühne T, Bruder E. Fibrolamellar hepatocellular carcinoma occurring 5 years after hepatocellular adenoma in a 14-year-old girl: a case report with comparative genomic hybridization analysis. Arch Pathol Lab Med. 2004 Feb;128(2):222-6
Imkie M, Myers SA, Li Y, Fan F, Bennett TL, Forster J, Tawfik O. Fibrolamellar hepatocellular carcinoma arising in a background of focal nodular hyperplasia: a report of 2 cases. J Reprod Med. 2005 Aug;50(8):633-7
Kakar S, Burgart LJ, Batts KP, Garcia J, Jain D, Ferrell LD. Clinicopathologic features and survival in fibrolamellar carcinoma: comparison with conventional hepatocellular carcinoma with and without cirrhosis. Mod Pathol. 2005 Nov;18(11):1417-23
Marrannes J, Gryspeerdt S, Haspeslagh M, van Holsbeeck B, Baekelandt M, Lefere P. Fibrolamellar hepatocellular carcinoma in a 65-year-old woman: CT features. JBR-BTR. 2005 Sep-Oct;88(5):237-40
Tanaka K, Honna T, Kitano Y, Kuroda T, Tanaka K, Morikawa N, Matsuda H, Kawashima N, Matsuoka K, Miyauchi J. Combined fibrolamellar carcinoma and cholangiocarcinoma exhibiting biphenotypic antigen expression: a case report. J Clin Pathol. 2005 Aug;58(8):884-7
Kannangai R, Vivekanandan P, Martinez-Murillo F, Choti M, Torbenson M. Fibrolamellar carcinomas show overexpression of genes in the RAS, MAPK, PIK3, and xenobiotic degradation pathways. Hum Pathol. 2007 Apr;38(4):639-44
Meriggi F, Forni E. [Surgical therapy of hepatic fibrolamellar carcinoma]. Ann Ital Chir. 2007 Jan-Feb;78(1):53-8
Torbenson M. Review of the clinicopathologic features of fibrolamellar carcinoma. Adv Anat Pathol. 2007 May;14(3):217-23
Moghadam FM, Bahremand GH, Vejdani A. Shoulder pain as the first sign of a hepatic fibrolamellar carcinoma in a young man. J Gastrointestin Liver Dis. 2008 Jun;17(2):234-6
Moore SW, Davidson A, Hadley GP, Kruger M, Poole J, Stones D, Wainwright L, Wessels G. Malignant liver tumors in South African children: a national audit. World J Surg. 2008 Jul;32(7):1389-95
Bhaijee F, Locketz ML, Krige JE. Fibrolamellar hepatocellular carcinoma at a tertiary centre in South Africa: a case series. S Afr J Surg. 2009 Nov;47(4):108-11
Cieply B, Zeng G, Proverbs-Singh T, Geller DA, Monga SP. Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology. 2009 Mar;49(3):821-31
Da Ines D, Lannareix V, Petitcolin V, Bailly A, Charpy C, Abergel A, Chipponi J, Garcier JM. [Fibrolamellar hepatocellular carcinoma in a patient with chronic viral B hepatitis]. Gastroenterol Clin Biol. 2009 May;33(5):382-6
Li W, Tan D, Zenali MJ, Brown RE. Constitutive activation of nuclear factor-kappa B (NF-kB) signaling pathway in fibrolamellar hepatocellular carcinoma. Int J Clin Exp Pathol. 2009 Jan 1;3(3):238-43
Liu S, Chan KW, Wang B, Qiao L. Fibrolamellar hepatocellular carcinoma. Am J Gastroenterol. 2009 Oct;104(10):2617-24; quiz 2625
Maniaci V, Davidson BR, Rolles K, Dhillon AP, Hackshaw A, Begent RH, Meyer T. Fibrolamellar hepatocellular carcinoma: prolonged survival with multimodality therapy. Eur J Surg Oncol. 2009 Jun;35(6):617-21

Liver: Fibrolamellar carcinoma Zhang X, Ward SC
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 934
Vivekanandan P, Micchelli ST, Torbenson M. Anterior gradient-2 is overexpressed by fibrolamellar carcinomas. Hum Pathol. 2009 Mar;40(3):293-9
Sethi S, Tageja N, Singh J, Arabi H, Dave M, Badheka A, Revankar S. Hyperammonemic encephalopathy: a rare presentation of fibrolamellar hepatocellular carcinoma. Am J Med Sci. 2009 Dec;338(6):522-4
Thirabanjasak D, Sosothikul D, Mahayosnond A, Thorner PS. Fibrolamellar carcinoma presenting as a pancreatic mass: case report and review of the literature. J Pediatr Hematol Oncol. 2009 May;31(5):370-2
Dhingra S, Li W, Tan D, Zenali M, Zhang H, Brown RE. Cell cycle biology of fibrolamellar hepatocellular carcinoma. Int J Clin Exp Pathol. 2010 Nov 2;3(8):792-7
Mroz RM, Korniluk M, Swidzinska E, Dzieciol J, Czaban J, Panek B, Chyczewska E. Lung mass in a 28-year-old male: a case report of a rare tumor. Eur J Med Res. 2010 Nov 4;15 Suppl 2:95-7
Vivekanandan P, Daniel H, Yeh MM, Torbenson M. Mitochondrial mutations in hepatocellular carcinomas and fibrolamellar carcinomas. Mod Pathol. 2010 Jun;23(6):790-8
Ward SC, Huang J, Tickoo SK, Thung SN, Ladanyi M, Klimstra DS. Fibrolamellar carcinoma of the liver exhibits immunohistochemical evidence of both hepatocyte and bile duct differentiation. Mod Pathol. 2010 Sep;23(9):1180-90
Zenali MJ, Tan D, Li W, Dhingra S, Brown RE. Stemness characteristics of fibrolamellar hepatocellular carcinoma: immunohistochemical analysis with comparisons to conventional hepatocellular carcinoma. Ann Clin Lab Sci. 2010 Spring;40(2):126-34
Al-Matham K, Alabed I, Zaidi SZ, Qushmaq KA. Cold agglutinin disease in fibrolamellar hepatocellular carcinoma: a rare association with a rare cancer variant. Ann Saudi Med. 2011 Mar-Apr;31(2):197-200
Brunel V, Cauliez B, Lacaze L, Riachi G, Gargala G, Francois A, Lavoinne A, Scotté M. Liver mass in a young adult. Lancet. 2011 Sep 24;378(9797):1196
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011 Sep 22;365(12):1118-27
Muramori K, Taguchi S, Taguchi T, Kohashi K, Furuya K, Tokuda K, Ishii E. High aromatase activity and overexpression of epidermal growth factor receptor in fibrolamellar hepatocellular carcinoma in a child. J Pediatr Hematol Oncol. 2011 Jul;33(5):e195-7
Patonai A, Erdélyi-Belle B, Korompay A, Somorácz A, Straub BK, Schirmacher P, Kovalszky I, Lotz G, Kiss A, Schaff Z. Claudins and tricellulin in fibrolamellar hepatocellular carcinoma. Virchows Arch. 2011 Jun;458(6):679-88
Ross HM, Daniel HD, Vivekanandan P, Kannangai R, Yeh MM, Wu TT, Makhlouf HR, Torbenson M. Fibrolamellar carcinomas are positive for CD68. Mod Pathol. 2011 Mar;24(3):390-5
Shanbhogue AK, Prasad SR, Takahashi N, Vikram R, Sahani DV. Recent advances in cytogenetics and molecular biology of adult hepatocellular tumors: implications for imaging and management. Radiology. 2011 Mar;258(3):673-93
Ward SC, Waxman S. Fibrolamellar carcinoma: a review with focus on genetics and comparison to other malignant primary liver tumors. Semin Liver Dis. 2011 Feb;31(1):61-70
Asrani SK, LaRusso NF. Fibrolamellar hepatocellular carcinoma presenting with Budd-Chiari syndrome, right atrial thrombus, and pulmonary emboli. Hepatology. 2012 Mar;55(3):977-8
Benito V, Segura J, Martínez MS, Arencibia O, Lubrano A. Fibrolamellar hepatocellular carcinoma metastatic to the ovary. J Obstet Gynaecol. 2012 Feb;32(2):200-2
El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012 May;142(6):1264-1273.e1
Knudson JD, Goldberg JF, Ayres NA. Fibrolamellar hepatocellular carcinoma with cardiac spread causing severe inferior vena cava obstruction in a 9-year-old child. Pediatr Cardiol. 2012 Jun;33(5):872-3
Malouf GG, Brugières L, Le Deley MC, Faivre S, Fabre M, Paradis V, Aerts I, Le Tourneau C, Dreyer C, Branchereau S, Belghiti J, Raymond E. Pure and mixed fibrolamellar hepatocellular carcinomas differ in natural history and prognosis after complete surgical resection. Cancer. 2012 Oct 15;118(20):4981-90
Wilczek E, Szparecki G, Lukasik D, Koperski L, Winiarska M, Wilczynski GM, Wasiutynski A, Gornicka B. Loss of the orphan nuclear receptor SHP is more pronounced in fibrolamellar carcinoma than in typical hepatocellular carcinoma. PLoS One. 2012;7(1):e30944
This article should be referenced as such:
Zhang X, Ward SC. Liver: Fibrolamellar carcinoma. Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):929-934.

Solid Tumour Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 935
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Thyroid: Anaplastic (undifferentiated) carcinoma Sai-Ching Jim Yeung, Mouhammed Amir Habra
The University of Texas M. D. Anderson Cancer Center, Department of General Internal Medicine, Ambulatory Treatment and Emergency Care, Department of Endocrine Neoplasia and Hormonal Disorders, 1515 Holcombe Boulevard, Unit 437, Houston, Texas 77030, USA (SCJY), The University of Texas M. D. Anderson Cancer Center, Department of Endocrine Neoplasia and Hormonal Disorders, 1515 Holcombe Boulevard, Unit 1416, Houston, Texas 77030, USA (MAH)
Published in Atlas Database: July 2012
Online updated version : http://AtlasGeneticsOncology.org/Tumors/AnaCarciThyroidID5069.html DOI: 10.4267/2042/48371
This article is an update of : Fadare O, Tallini G.. Thyroid: Anaplastic (undifferentiated) carcinoma. Atlas Genet Cytogenet Oncol Haematol.2003;7(3):190-192. Yeung SCJ. Thyroid: Anaplastic (undifferentiated) carcinoma. Atlas Genet Cytogenet Oncol Haematol 2008;12(6):477-480. This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Note Anaplastic (undifferentiated) carcinoma of the thyroid gland is a highly malignant tumor composed in part or wholly by undifferentiated malignant cells.
Clinics and pathology Epidemiology Anaplastic (undifferentiated) carcinoma of the thyroid gland is uncommon, accounting for less than 5% of all cases of thyroid carcinoma. The average age at diagnosis was 66.5 years, with a female to male ratio of 3.1:1 in one study of 70 cases.
Clinics Most patients are euthyroid with a history of a rapidly enlarging neck mass. Sometimes, the tumor presents as a new-onset thyroid enlargement in a patient with longstanding thyroid nodule(s) or as the recurrence of a well-differentiated thyroid carcinoma. Tumor infiltration of surrounding structures results in secondary symptoms (dyspnea, dysphonia, and dysphagia).
Pathology Tumors are poorly defined, fleshy masses with areas of necrosis and hemorrhage. Microscopically they are composed of anaplastic cells with marked cytologic
atypia and high mitotic activity. Tumor necrosis and vascular invasion are common. About one-third of cases of anaplastic thyroid carcinoma (ATC) have coexisting areas of well-differentiated thyroid carcinoma, supporting the hypothesis that ATC arises from well-differentiated thyroid carcinoma. Histologic patterns include spindle, giant and squamoid cell types. Other patterns (e.g. angiomatoid, carcinosarcoma, lymphoepithelioma-like, adenosquamous) have been described. Undifferentiated (anaplastic) carcinoma of the thyroid must be differentiated from other high grade tumors with similar microscopic appearance originating from adjacent structures in the neck (e.g. larynx). Sometimes this distinction is only possible on clinical/anatomical grounds. Immunohistochemically, undifferentiated thyroid carcinoma is generally negative for thyroglobulin and calcitonin. Pan-keratin and epithelial membrane antigen (EMA) are positive in about one-half and one-third of cases respectively. Vimentin is positive in about 90%, and epithelial membrane antigen is positive in about 30% of cases. Thyroid transcription factor-1 (TTF-1) staining is present in 0-50% of cases. Although immunostaining is negative for muscle-specific actin, Factor VIII-related antigen, and desmin, these markers can differentiate ATC from some soft tissue sarcomas with which they can be confused.
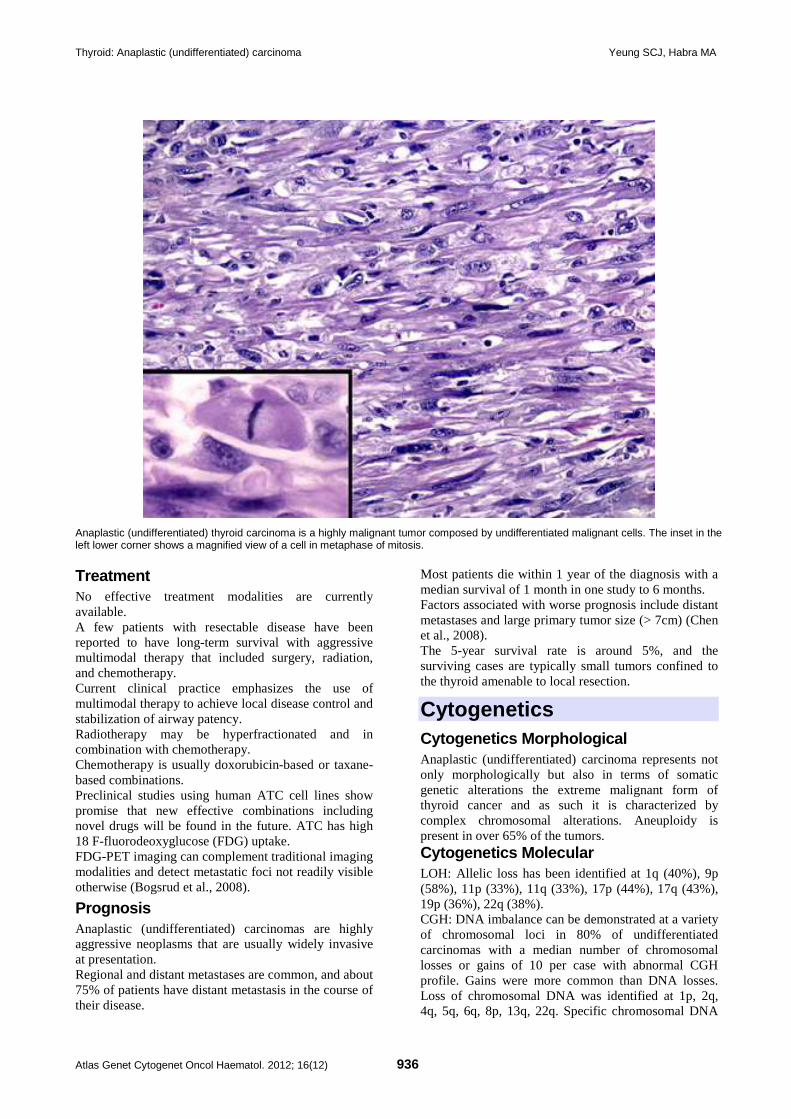
Thyroid: Anaplastic (undifferentiated) carcinoma Yeung SCJ, Habra MA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 936
Anaplastic (undifferentiated) thyroid carcinoma is a highly malignant tumor composed by undifferentiated malignant cells. The inset in the left lower corner shows a magnified view of a cell in metaphase of mitosis. Treatment No effective treatment modalities are currently available. A few patients with resectable disease have been reported to have long-term survival with aggressive multimodal therapy that included surgery, radiation, and chemotherapy. Current clinical practice emphasizes the use of multimodal therapy to achieve local disease control and stabilization of airway patency. Radiotherapy may be hyperfractionated and in combination with chemotherapy. Chemotherapy is usually doxorubicin-based or taxane-based combinations. Preclinical studies using human ATC cell lines show promise that new effective combinations including novel drugs will be found in the future. ATC has high 18 F-fluorodeoxyglucose (FDG) uptake. FDG-PET imaging can complement traditional imaging modalities and detect metastatic foci not readily visible otherwise (Bogsrud et al., 2008).
Prognosis Anaplastic (undifferentiated) carcinomas are highly aggressive neoplasms that are usually widely invasive at presentation. Regional and distant metastases are common, and about 75% of patients have distant metastasis in the course of their disease.
Most patients die within 1 year of the diagnosis with a median survival of 1 month in one study to 6 months. Factors associated with worse prognosis include distant metastases and large primary tumor size (> 7cm) (Chen et al., 2008). The 5-year survival rate is around 5%, and the surviving cases are typically small tumors confined to the thyroid amenable to local resection.
Cytogenetics Cytogenetics Morphological Anaplastic (undifferentiated) carcinoma represents not only morphologically but also in terms of somatic genetic alterations the extreme malignant form of thyroid cancer and as such it is characterized by complex chromosomal alterations. Aneuploidy is present in over 65% of the tumors. Cytogenetics Molecular LOH: Allelic loss has been identified at 1q (40%), 9p (58%), 11p (33%), 11q (33%), 17p (44%), 17q (43%), 19p (36%), 22q (38%). CGH: DNA imbalance can be demonstrated at a variety of chromosomal loci in 80% of undifferentiated carcinomas with a median number of chromosomal losses or gains of 10 per case with abnormal CGH profile. Gains were more common than DNA losses. Loss of chromosomal DNA was identified at 1p, 2q, 4q, 5q, 6q, 8p, 13q, 22q. Specific chromosomal DNA

Thyroid: Anaplastic (undifferentiated) carcinoma Yeung SCJ, Habra MA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 937
alterations (i.e. 3p13-14+, 5q11-31-, 11q13+) may be associated with the transition from more differentiated phenotypes to ATC. Comparative genome hybridization (CGH) shows frequent gain of 20q, including the UBCH10 gene in 20q13.12, which may also be associated with progression of differentiated thyroid cancers to ATC (Lee et al., 2007). Using microarray-based CGH with further fluorescence in situ hybridization (FISH) analysis, the MAP kinase phosphatase-8 (DUSP26) gene, which codes for a phosphatase that inhibits p38-mediated apoptosis, is shown to be amplified in ATC (Yu et al., 2007). Human telomerase reverse transcriptase (hTERT) protein expression is increased in ATC samples and cell lines (Takano et al., 2007). In ATC cell lines, miR-138 was significantly down regulated in comparison to papillary thyroid cancer cell lines. miR-138 was inversely correlated with the human telomerase reverse transcriptase (hTERT) protein expression (Mitomo et al., 2008).
Genes involved and proteins Note The genetic mechanisms involved with the development of anaplastic thyroid cancer are complex. Mutational inactivation of p53 has been identified in 70-80% of anaplastic carcinomas while H-Ras, K-Ras, or N-Ras activating mutations are present in less than 50% of the cases. BRAF V600E mutation is found in 20% to 25% of cases. PTEN mutations are present in 6%. PIK3CA kinase domain mutations are found in 14%. PIK3CA gene copy amplification is present in 39%. Aberrant Wnt/beta-Catenin signaling appears to be a distinctive feature of ATC since stabilizing mutations and/or aberrant beta-Catenin nuclear localization are present in 80% of ATC. beta-Catenin nuclear localization is accompanied by its cellular redistribution with marked decrease of the beta-Catenin membrane bound fraction. ATC are characterized by increased cell replication and high Ki67/Mib1 proliferation index, loss of the apoptotic protein bcl-2 and of Fas and its ligand (usually highly expressed in well differentiated thyroid tumors), by an increase in the proapoptotic protein Bax, by Cyclin D1 over-expression and conversely by a fall in the CDK inhibitor p27. Transmembrane protein 34 (TMEM34) is down-regulated in ATC. It is not clear whether these changes represent the cause or (more likely) the effect of dysregulated cell differentiation and growth in ATC. Immunohistochemical staining of a tissue microarray of 12 cases of ATC showed the following: beta-catenin (positive in 41% of the cases), aurora A (41%), cyclin E (67%), cyclin D1 (77%), and EGFR (84%). Thyroglobulin, Bcl-2, E-cadherin, vascular endothelial growth factor and beta-catenin are more expressed in
differentiated thyroid cancer while topoisomerase II-alpha, MIB-1, and p53 are more expressed in ATC and these changes are expected to occur during progression from differentiated thyroid cancer to ATC (Wiseman et al., 2007).
References Carcangiu ML, Steeper T, Zampi G, Rosai J. Anaplastic thyroid carcinoma. A study of 70 cases. Am J Clin Pathol. 1985 Feb;83(2):135-58
Klemi PJ, Joensuu H, Eerola E. DNA aneuploidy in anaplastic carcinoma of the thyroid gland. Am J Clin Pathol. 1988 Feb;89(2):154-9
Stringer BM, Rowson JM, Parkar MH, Seid JM, Hearn PR, Wynford-Thomas D, Ingemansson S, Woodhouse N, Goyns MH. Detection of the H-RAS oncogene in human thyroid anaplastic carcinomas. Experientia. 1989 Apr 15;45(4):372-6
Jenkins RB, Hay ID, Herath JF, Schultz CG, Spurbeck JL, Grant CS, Goellner JR, Dewald GW. Frequent occurrence of cytogenetic abnormalities in sporadic nonmedullary thyroid carcinoma. Cancer. 1990 Sep 15;66(6):1213-20
Venkatesh YS, Ordonez NG, Schultz PN, Hickey RC, Goepfert H, Samaan NA. Anaplastic carcinoma of the thyroid. A clinicopathologic study of 121 cases. Cancer. 1990 Jul 15;66(2):321-30
Ordóñez NG, El-Naggar AK, Hickey RC, Samaan NA. Anaplastic thyroid carcinoma. Immunocytochemical study of 32 cases. Am J Clin Pathol. 1991 Jul;96(1):15-24
Rosai J, Carcangiu ML, DeLellis RA.. Tumors of the thyroid gland. Atlas of tumor Pathology 1992, 3rd series, fascicle 5.
Donghi R, Longoni A, Pilotti S, Michieli P, Della Porta G, Pierotti MA.. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J Clin Invest. 1993 Apr;91(4):1753-60.
Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, Koeffler HP.. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest. 1993 Jan;91(1):179-84.
Capella G, Matias-Guiu X, Ampudia X, de Leiva A, Perucho M, Prat J.. Ras oncogene mutations in thyroid tumors: polymerase chain reaction-restriction-fragment-length polymorphism analysis from paraffin-embedded tissues. Diagn Mol Pathol. 1996 Mar;5(1):45-52.
Kobayashi T, Asakawa H, Umeshita K, Takeda T, Maruyama H, Matsuzuka F, Monden M.. Treatment of 37 patients with anaplastic carcinoma of the thyroid. Head Neck. 1996 Jan-Feb;18(1):36-41.
Hemmer S, Wasenius VM, Knuutila S, Franssila K, Joensuu H.. DNA copy number changes in thyroid carcinoma. Am J Pathol. 1999 May;154(5):1539-47.
Husmark J, Heldin NE, Nilsson M.. N-cadherin-mediated adhesion and aberrant catenin expression in anaplastic thyroid-carcinoma cell lines. Int J Cancer. 1999 Nov 26;83(5):692-9.
Tallini G, Garcia-Rostan G, Herrero A, Zelterman D, Viale G, Bosari S, Carcangiu ML.. Downregulation of p27KIP1 and Ki67/Mib1 labeling index support the classification of thyroid carcinoma into prognostically relevant categories. Am J Surg Pathol. 1999 Jun;23(6):678-85.
Ain KB, Egorin MJ, DeSimone PA.. Treatment of anaplastic thyroid carcinoma with paclitaxel: phase 2 trial using ninety-six-hour infusion. Collaborative Anaplastic Thyroid Cancer Health

Thyroid: Anaplastic (undifferentiated) carcinoma Yeung SCJ, Habra MA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 938
Intervention Trials (CATCHIT) Group. Thyroid. 2000 Jul;10(7):587-94.
Kitamura Y, Shimizu K, Tanaka S, Ito K, Emi M.. Allelotyping of anaplastic thyroid carcinoma: frequent allelic losses on 1q, 9p, 11, 17, 19p, and 22q. Genes Chromosomes Cancer. 2000 Mar;27(3):244-51.
Garcia-Rostan G, Camp RL, Herrero A, Carcangiu ML, Rimm DL, Tallini G.. Beta-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am J Pathol. 2001 Mar;158(3):987-96.
Dowlati A, Robertson K, Cooney M, Petros WP, Stratford M, Jesberger J, Rafie N, Overmoyer B, Makkar V, Stambler B, Taylor A, Waas J, Lewin JS, McCrae KR, Remick SC.. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res. 2002 Jun 15;62(12):3408-16.
Wreesmann VB, Ghossein RA, Patel SG, Harris CP, Schnaser EA, Shaha AR, Tuttle RM, Shah JP, Rao PH, Singh B.. Genome-wide appraisal of thyroid cancer progression. Am J Pathol. 2002 Nov;161(5):1549-56.
Cooney MM, Savvides P, Agarwala S, Wang D, Flick S, Bergant S, Bhakta S, Lavertu P, Ortiz J, Remick S.. Phase II study of combretastatin A4 phosphate (CA4P) in patients with advanced anaplastic thyroid carcinoma (ATC). Journal of Clinical Oncology 2006; 24: 300S.
Akaishi J, Onda M, Okamoto J, Miyamoto S, Nagahama M, Ito K, Yoshida A, Shimizu K.. Down-regulation of an inhibitor of cell growth, transmembrane protein 34 (TMEM34), in anaplastic thyroid cancer. J Cancer Res Clin Oncol. 2007 Apr;133(4):213-8. Epub 2006 Oct 28.
Fury MG, Solit DB, Su YB, Rosen N, Sirotnak FM, Smith RP, Azzoli CG, Gomez JE, Miller VA, Kris MG, Pizzo BA, Henry R, Pfister DG, Rizvi NA.. A phase I trial of intermittent high-dose gefitinib and fixed-dose docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2007 Mar;59(4):467-75. Epub 2006 Aug 1.
Lee JJ, Foukakis T, Hashemi J, Grimelius L, Heldin NE, Wallin G, Rudduck C, Lui WO, Hoog A, Larsson C.. Molecular cytogenetic profiles of novel and established human anaplastic thyroid carcinoma models. Thyroid. 2007 Apr;17(4):289-301.
Lee DH, Lee GK, Kong SY, Kook MC, Yang SK, Park SY, Park SH, Keam B, Park do J, Cho BY, Kim SW, Chung KW, Lee ES, Kim SW.. Epidermal growth factor receptor status in anaplastic thyroid carcinoma. J Clin Pathol. 2007 Aug;60(8):881-4. Epub 2006 Nov 1.
Takano T, Ito Y, Hirokawa M, Yoshida H, Miyauchi A.. BRAF V600E mutation in anaplastic thyroid carcinomas and their accompanying differentiated carcinomas. Br J Cancer. 2007 May 21;96(10):1549-53. Epub 2007 Apr 24.
Takano T, Ito Y, Matsuzuka F, Miya A, Kobayashi K, Yoshida H, Miyauchi A.. Quantitative measurement of telomerase reverse transcriptase, thyroglobulin and thyroid transcription factor 1 mRNAs in anaplastic thyroid carcinoma tissues and cell lines. Oncol Rep. 2007 Sep;18(3):715-20.
Wiseman SM, Griffith OL, Deen S, Rajput A, Masoudi H, Gilks B, Goldstein L, Gown A, Jones SJ.. Identification of molecular markers altered during transformation of differentiated into anaplastic thyroid carcinoma. Arch Surg. 2007 Aug;142(8):717-27; discussion 727-9.
Wiseman SM, Masoudi H, Niblock P, Turbin D, Rajput A, Hay J, Bugis S, Filipenko D, Huntsman D, Gilks B.. Anaplastic thyroid carcinoma: expression profile of targets for therapy
offers new insights for disease treatment. Ann Surg Oncol. 2007 Feb;14(2):719-29. Epub 2006 Nov 10.
Yu W, Imoto I, Inoue J, Onda M, Emi M, Inazawa J.. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene. 2007 Feb 22;26(8):1178-87. Epub 2006 Aug 21.
Bogsrud TV, Karantanis D, Nathan MA, Mullan BP, Wiseman GA, Kasperbauer JL, Reading CC, Hay ID, Lowe VJ.. 18F-FDG PET in the management of patients with anaplastic thyroid carcinoma. Thyroid. 2008 Jul;18(7):713-9.
Broecker-Preuss M, Sheu SY, Worm K, Feldkamp J, Witte J, Scherbaum WA, Mann K, Schmid KW, Schott M.. Expression and mutation analysis of the tyrosine kinase c-kit in poorly differentiated and anaplastic thyroid carcinoma. Horm Metab Res. 2008 Oct;40(10):685-91. Epub 2008 Jul 11.
Chen J, Tward JD, Shrieve DC, Hitchcock YJ.. Surgery and radiotherapy improves survival in patients with anaplastic thyroid carcinoma: analysis of the surveillance, epidemiology, and end results 1983-2002. Am J Clin Oncol. 2008 Oct;31(5):460-4.
Lee JJ, Au AY, Foukakis T, Barbaro M, Kiss N, Clifton-Bligh R, Staaf J, Borg A, Delbridge L, Robinson BG, Wallin G, Hoog A, Larsson C.. Array-CGH identifies cyclin D1 and UBCH10 amplicons in anaplastic thyroid carcinoma. Endocr Relat Cancer. 2008 Sep;15(3):801-15.
Mitomo S, Maesawa C, Ogasawara S, Iwaya T, Shibazaki M, Yashima-Abo A, Kotani K, Oikawa H, Sakurai E, Izutsu N, Kato K, Komatsu H, Ikeda K, Wakabayashi G, Masuda T.. Downregulation of miR-138 is associated with overexpression of human telomerase reverse transcriptase protein in human anaplastic thyroid carcinoma cell lines. Cancer Sci. 2008 Feb;99(2):280-6. Epub 2008 Jan 14.
Pennell NA, Daniels GH, Haddad RI, Ross DS, Evans T, Wirth LJ, Fidias PH, Temel JS, Gurubhagavatula S, Heist RS, Clark JR, Lynch TJ.. A phase II study of gefitinib in patients with advanced thyroid cancer. Thyroid. 2008 Mar;18(3):317-23.
Santarpia L, El-Naggar AK, Cote GJ, Myers JN, Sherman SI.. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2008 Jan;93(1):278-84. Epub 2007 Nov 7.
Chakravarty G, Santillan AA, Galer C, Adams HP, El-Naggar AK, Jasser SA, Mohsin S, Mondal D, Clayman GL, Myers JN.. Phosphorylated insulin like growth factor-I receptor expression and its clinico-pathological significance in histologic subtypes of human thyroid cancer. Exp Biol Med (Maywood). 2009 Apr;234(4):372-86. Epub 2009 Jan 28.
Mooney CJ, Nagaiah G, Fu P, Wasman JK, Cooney MM, Savvides PS, Bokar JA, Dowlati A, Wang D, Agarwala SS, Flick SM, Hartman PH, Ortiz JD, Lavertu PN, Remick SC.. A phase II trial of fosbretabulin in advanced anaplastic thyroid carcinoma and correlation of baseline serum-soluble intracellular adhesion molecule-1 with outcome. Thyroid. 2009 Mar;19(3):233-40.
Nappi TC, Salerno P, Zitzelsberger H, Carlomagno F, Salvatore G, Santoro M.. Identification of Polo-like kinase 1 as a potential therapeutic target in anaplastic thyroid carcinoma. Cancer Res. 2009 Mar 1;69(5):1916-23. Epub 2009 Feb 17.
Nucera C, Eeckhoute J, Finn S, Carroll JS, Ligon AH, Priolo C, Fadda G, Toner M, Sheils O, Attard M, Pontecorvi A, Nose V, Loda M, Brown M.. FOXA1 is a potential oncogene in anaplastic thyroid carcinoma. Clin Cancer Res. 2009 Jun 1;15(11):3680-9. Epub 2009 May 26.
Poisson T, Deandreis D, Leboulleux S, Bidault F, Bonniaud G, Baillot S, Auperin A, Al Ghuzlan A, Travagli JP, Lumbroso J,

Thyroid: Anaplastic (undifferentiated) carcinoma Yeung SCJ, Habra MA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 939
Baudin E, Schlumberger M.. 18F-fluorodeoxyglucose positron emission tomography and computed tomography in anaplastic thyroid cancer. Eur J Nucl Med Mol Imaging. 2010 Dec;37(12):2277-85. Epub 2010 Aug 6.
Akaishi J, Sugino K, Kitagawa W, Nagahama M, Kameyama K, Shimizu K, Ito K, Ito K.. Prognostic factors and treatment outcomes of 100 cases of anaplastic thyroid carcinoma. Thyroid. 2011 Nov;21(11):1183-9. Epub 2011 Sep 21.
Derbel O, Limem S, Segura-Ferlay C, Lifante JC, Carrie C, Peix JL, Borson-Chazot F, Bournaud C, Droz JP, de la Fouchardiere C.. Results of combined treatment of anaplastic thyroid carcinoma (ATC). BMC Cancer. 2011 Nov 1;11:469.
Orita Y, Sugitani I, Amemiya T, Fujimoto Y.. Prospective application of our novel prognostic index in the treatment of
anaplastic thyroid carcinoma. Surgery. 2011 Dec;150(6):1212-9.
Esposito F, Tornincasa M, Pallante P, Federico A, Borbone E, Pierantoni GM, Fusco A.. Down-regulation of the miR-25 and miR-30d contributes to the development of anaplastic thyroid carcinoma targeting the polycomb protein EZH2. J Clin Endocrinol Metab. 2012 May;97(5):E710-8. Epub 2012 Mar 7.
This article should be referenced as such:
Yeung SCJ, Habra MA. Thyroid: Anaplastic (undifferentiated) carcinoma. Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):935-939.
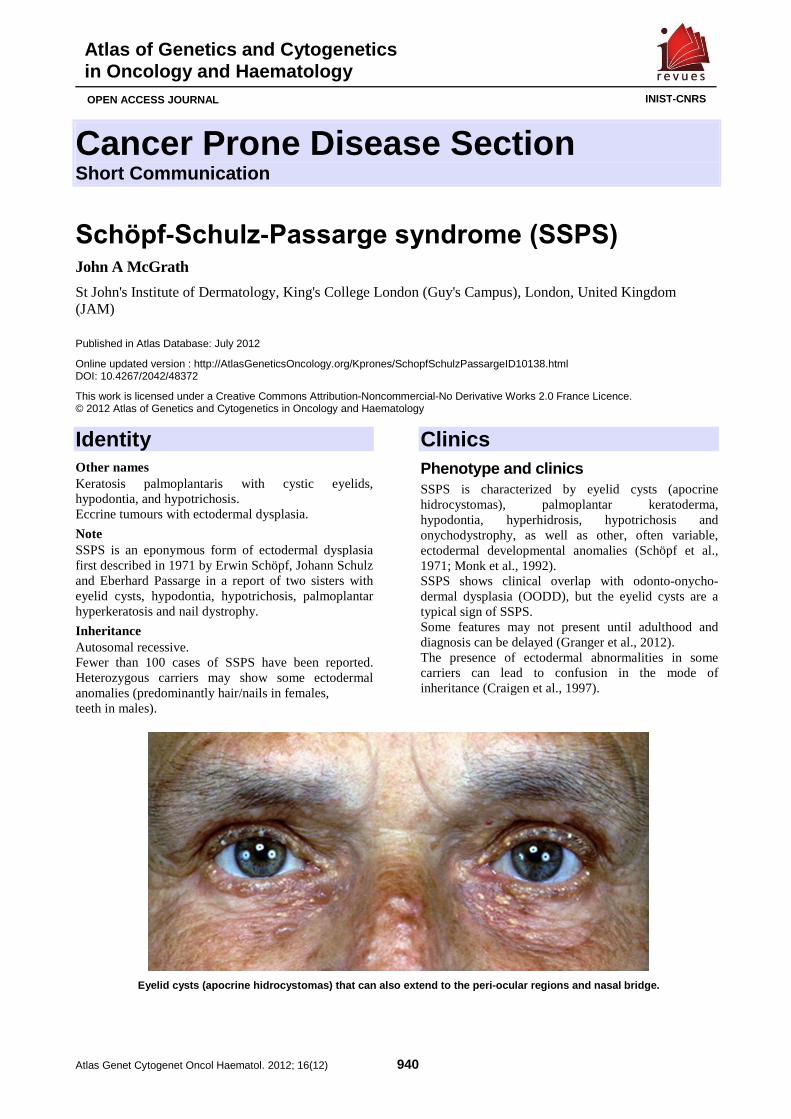
Cancer Prone Disease Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 940
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Schöpf-Schulz-Passarge syndrome (SSPS) John A McGrath
St John's Institute of Dermatology, King's College London (Guy's Campus), London, United Kingdom (JAM)
Published in Atlas Database: July 2012
Online updated version : http://AtlasGeneticsOncology.org/Kprones/SchopfSchulzPassargeID10138.html DOI: 10.4267/2042/48372
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names Keratosis palmoplantaris with cystic eyelids, hypodontia, and hypotrichosis. Eccrine tumours with ectodermal dysplasia.
Note SSPS is an eponymous form of ectodermal dysplasia first described in 1971 by Erwin Schöpf, Johann Schulz and Eberhard Passarge in a report of two sisters with eyelid cysts, hypodontia, hypotrichosis, palmoplantar hyperkeratosis and nail dystrophy.
Inheritance Autosomal recessive. Fewer than 100 cases of SSPS have been reported. Heterozygous carriers may show some ectodermal anomalies (predominantly hair/nails in females, teeth in males).
Clinics Phenotype and clinics SSPS is characterized by eyelid cysts (apocrine hidrocystomas), palmoplantar keratoderma, hypodontia, hyperhidrosis, hypotrichosis and onychodystrophy, as well as other, often variable, ectodermal developmental anomalies (Schöpf et al., 1971; Monk et al., 1992). SSPS shows clinical overlap with odonto-onycho-dermal dysplasia (OODD), but the eyelid cysts are a typical sign of SSPS. Some features may not present until adulthood and diagnosis can be delayed (Granger et al., 2012). The presence of ectodermal abnormalities in some carriers can lead to confusion in the mode of inheritance (Craigen et al., 1997).
Eyelid cysts (apocrine hidrocystomas) that can also extend to the peri-ocular regions and nasal bridge .

Schöpf-Schulz-Passarge syndrome (SSPS) McGrath JA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 941
Neoplastic risk The neoplastic risk in SSPS is controversial. Some authors consider that SSPS can be associated with an increased risk of benign as well as malignant skin tumours (Monk et al., 1992). Reports include an increased incidence of benign adnexal tumours, such as eyelid hidrocystomas or eccrine syringofibroadenomas (Starink, 1997), and possibly a higher risk of malignant skin tumours such as squamous cell carcinoma, basal cell carcinoma and eccrine porocarcinoma (Bohring et al., 2009; Monk et al., 1992; Starink, 1997).
Treatment There is no effective treatment for SSPS. Hyperhidrosis of the palms may respond partially to tap water iontophoresis (although use of anti-cholinergics may induce excessive systemic side-effects such as dry mouth, dizziness and drowsiness). Systemic retinoids can exacerbate skin peeling, although low doses may help some individuals. The apocrine hidrocystomas can be improved by electrocautery. Regular skin examination to detect non-melanoma skin cancer may be advisable. Regular dental care/surgery is indicated in most cases. Hair/nail cosmesis may help some individuals. Psychological support should be offered, as necessary.
Evolution Many of the features of ectodermal dysplasia only manifest or worsen during adulthood. In some individuals with SSPS, the apocrine hidrocystomas tend to become larger and more numerous with age.
Prognosis Life expectancy is normal; the main challenge is the symptomatic management of whichever ectodermal pathologies cause the patient the most concern.
Genes involved and proteins WNT10A Location 2q35
Note WNT10A is a key signalling molecule that regulates cell-cell interactions and which is involved in multiple developmental processes in embryogenesis. In adult tissues it inhibits the β-catenin degradation complex and is involved in hair follicle and tooth morphogenesis (Logan and Nusse, 2004).
Mutations
Note Mutations in WNT10A underlie SSPS, OODD and some cases of hypohidrotic ectodermal dysplasia. Thus far, 16 different WNT10A mutations have been reported. These include six nonsense mutations (p.Trp9X, p.Cys107X, p.Arg128X, p.Glu233X,
p.Arg248X, and p.Cys376X), nine missense mutations (p.Ile116Thr, p.Arg128Gln, p.Ala131Thr, p.Ala131Val, p.His143Tyr, p.Val145Met, p.Phe228Ile, p.Gly266Cys, and p.Arg360Cys) and one frameshift mutation (p.Glu52fsX29) (Adaimy et al., 2007; Bohring et al., 2009; Nagy et al., 2010; Wedgeworth et al., 2011; van Geel et al., 2010; Catori et al., 2011; Cluzeau et al., 2011; Petrof et al., 2011; Granger et al., 2012). Of note, cases classified clinically as SSPS or OODD may harbour the same WNT10A gene mutation(s). The two most frequently observed mutations are p.Cys107X and p.Phe228Ile. Moreover, homozygous or compound heterozygous mutations involving p.Cys107X have been found in both SSPS and OODD, demonstrating that these two disorders are indeed allelic and that the precise phenotypic consequences are influenced by more than just this particular mutation in WNT10A alone. There is no genotype-phenotype correlation with regard to neoplastic risk.
To be noted Note It is also noteworthy that individuals who are heterozygous for WNT10A mutations may show some clinical abnormalities. Hair, nail, teeth and skin abnormalities may all occur in heterozygotes; this probably accounts for the initial difficulties in classifying SSPS/OODD as either autosomal dominant or autosomal recessive disorders (the latter is correct). The mutation p.Phe228Ile appears to have a population frequency of ~0,5% and it has been estimated that approximately half of all individuals who are heterozygous for this missense mutation will manifest some form of ectodermal defects (Bohring et al., 2009). This equates to ~1 in 400 of the general population displaying some clinical anomaly affecting hair, teeth, nails, sweat glands, or a combination thereof, as a direct consequence of this WNT10A gene sequence variant.
References Schöpf E, Schulz HJ, Passarge E. Syndrome of cystic eyelids, palmo-plantar keratosis, hypodontia and hypotrichosis as a possible autosomal recessive trait. Birth Defects Orig Artic Ser. 1971 Jun;7(8):219-21
Monk BE, Pieris S, Soni V. Schöpf-Schulz-Passarge syndrome. Br J Dermatol. 1992 Jul;127(1):33-5
Craigen WJ, Levy ML, Lewis RA. Schöpf-Schulz-Passarge syndrome with an unusual pattern of inheritance. Am J Med Genet. 1997 Aug 8;71(2):186-8
Starink TM. Eccrine syringofibroadenoma: multiple lesions representing a new cutaneous marker of the Schöpf syndrome, and solitary nonhereditary tumors. J Am Acad Dermatol. 1997 Apr;36(4):569-76
Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781-810

Schöpf-Schulz-Passarge syndrome (SSPS) McGrath JA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 942
Adaimy L, Chouery E, Megarbane H, Mroueh S, Delague V, Nicolas E, Belguith H, de Mazancourt P, Megarbane A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am J Hum Genet. 2007 Oct;81(4):821-8
Bohring A, Stamm T, Spaich C, Haase C, Spree K, Hehr U, Hoffmann M, Ledig S, Sel S, Wieacker P, Röpke A. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet. 2009 Jul;85(1):97-105
Nagy N, Wedgeworth E, Hamada T, White JM, Hashimoto T, McGrath JA. Schöpf-Schulz-Passarge syndrome resulting from a homozygous nonsense mutation in WNT10A. J Dermatol Sci. 2010 Jun;58(3):220-2
Van Geel M, Gattas M, Kesler Y, Tong P, Yan H, Tran K, Steijlen PM, Murrell DF, Van Steensel MA. Phenotypic variability associated with WNT10A nonsense mutations. Br J Dermatol. 2010 Jun;162(6):1403-6
Castori M, Castiglia D, Brancati F, Foglio M, Heath S, Floriddia G, Madonna S, Fischer J, Zambruno G. Two families confirm Schöpf-Schulz-Passarge syndrome as a discrete entity within the WNT10A phenotypic spectrum. Clin Genet. 2011 Jan;79(1):92-5
Cluzeau C, Hadj-Rabia S, Jambou M, Mansour S, Guigue P, Masmoudi S, Bal E, Chassaing N, Vincent MC, Viot G, Clauss F, Manière MC, Toupenay S, Le Merrer M, Lyonnet S, Cormier-Daire V, Amiel J, Faivre L, de Prost Y, Munnich A, Bonnefont JP, Bodemer C, Smahi A. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum Mutat. 2011 Jan;32(1):70-2
Petrof G, Fong K, Lai-Cheong JE, Cockayne SE, McGrath JA. Schöpf-Schulz-Passarge syndrome resulting from a homozygous nonsense mutation, p.Cys107X, in WNT10A. Australas J Dermatol. 2011 Aug;52(3):224-6
Wedgeworth EK, Nagy N, White JM, Pembroke AC, McGrath JA. Intra-familial variability of ectodermal defects associated with WNT10A mutations. Acta Derm Venereol. 2011 May;91(3):346-7
Granger RH, Marshman G, Liu L, McGrath JA. Late diagnosis of ectodermal dysplasia syndrome. Australas J Dermatol. 2012 Jun 4;
This article should be referenced as such:
McGrath JA. Schöpf-Schulz-Passarge syndrome (SSPS). Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):940-942.

Deep Insight Section
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 943
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
The Claudins family: Structure and function in normal and pathologic conditions Abderrahman Ouban, Atif Ali Ahmed
Department of Pathology, College of Medicine, Prince Salman University, Kharj, Kingdom of Saudi Arabia (AO), Department of Pathology, Children's Mercy Hospital, University of Kansas, Kansas City, Missouri, USA (AAA)
Published in Atlas Database: July 2012
Online updated version : http://AtlasGeneticsOncology.org/Deep/ClaudinFamID20113.html DOI: 10.4267/2042/48373
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2012 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Definition and introduction Tight junctions (TJs) are the structures responsible for forming the seal that controls paracellular transport. TJs are composed of multiple components, including the Occludin proteins, the Zona Occludin proteins and the claudin proteins. The tertraspan integral membrane proteins known as claudins are essential for TJ formation and function (Tsukita and Furuse, 2000). Twenty three claudin genes are found in the human genome. The exact mechanisms of claudin evolution remain unknown, although some data suggest that the claudin multigene family evolved through gene duplications early in chordate development (Kollmar et al., 2001). In general, claudin genes are small, with few introns, or none at all (Lal-Nag and Morin, 2009). There is high degree of genetic homology among claudin genes, with several pairs showing similarity to each other in sequence and in intron/exon arrangement. Many claudin genes are closely located in the human genome, such as claudin 6 and claudin 9 on chromosome 16, claudin 22 and claudin 24 on chromosome 4, claudin 8 and claudin 17 on chromosome 21 and claudin 3 and claudin 4 on chromosome 7 (table 1). There is evidence that this genomic arrangement may result in coordinated expression as evidenced
in co-expression of claudins 3 and 4 which has been reported in several normal and neoplastic tissues (Lal-Nag and Morin, 2009). Translation of the aforementioned genes results in 23 distinct human claudin proteins. These claudin proteins span the cellular membrane bilayer four times, where the N- and C-termini are oriented towards the cytoplasm and there are two extracellular loop domains (Morita et al., 1999). The C-Terminal PDZ binding motifs in each claudin binds other tight junction cytoplasmic proteins such as ZO-1, ZO-2, and ZO-3, MUPP-1, PALS-1 associated TJ protein (PATJ). Stabilization of a tight junction is specifically dependent on interaction of claudins with cytoplasmic scaffolding proteins ZO-1 and ZO-2 which link claudins to actin cytoskeleton (Umeda et al., 2006). The TJ sealing strength varies over five orders of magnitude in different epithelia, from leaky proximal tubules to almost hermetic colon and urinary bladder. Tightness can also change in the same epithelium according to physiological and pathological conditions, in response to pharmacological changes and growth factors and hormonal stimulation (Balda et al., 1991). EGF plays a pivotal role in the adjustment of the permeability of TJs to physiological requirements, pathological conditions, and pharmacological interventions (Flores-Benítez et al., 2007).
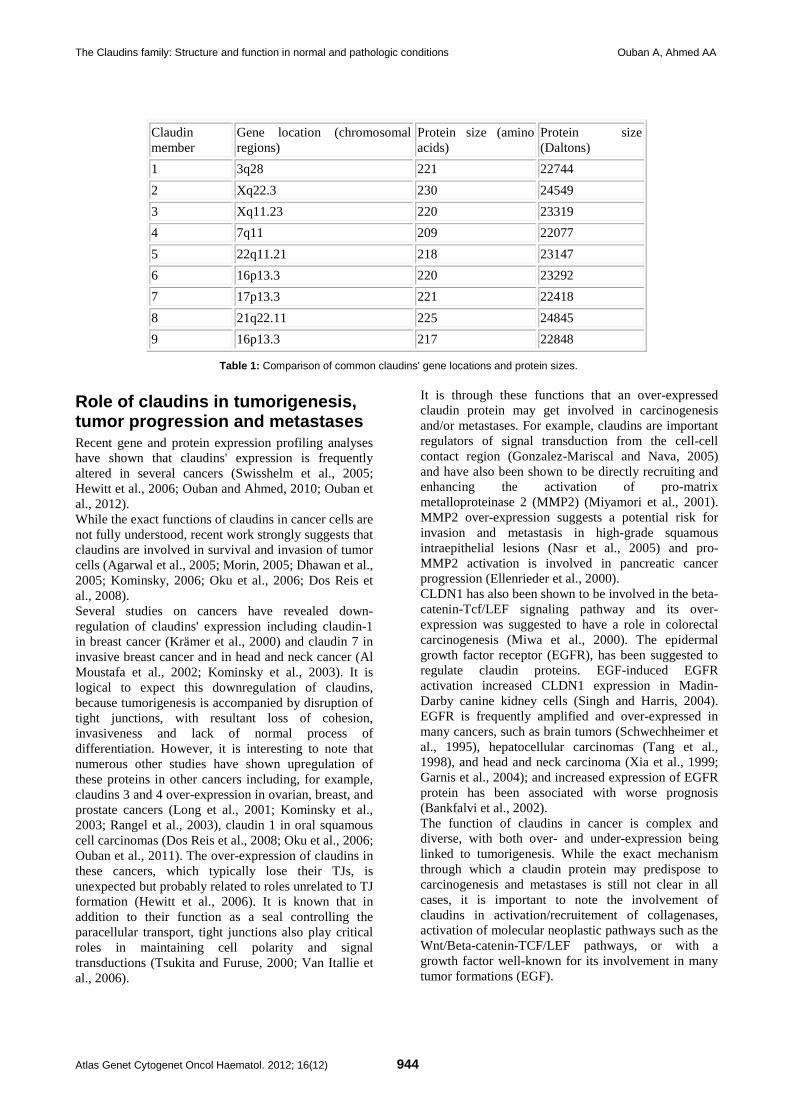
The Claudins family: Structure and function in normal and pathologic conditions Ouban A, Ahmed AA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 944
Claudin member
Gene location (chromosomal regions)
Protein size (amino acids)
Protein size (Daltons)
1 3q28 221 22744 2 Xq22.3 230 24549 3 Xq11.23 220 23319 4 7q11 209 22077 5 22q11.21 218 23147 6 16p13.3 220 23292 7 17p13.3 221 22418 8 21q22.11 225 24845 9 16p13.3 217 22848
Table 1: Comparison of common claudins' gene locations and protein sizes.
Role of claudins in tumorigenesis, tumor progression and metastases Recent gene and protein expression profiling analyses have shown that claudins' expression is frequently altered in several cancers (Swisshelm et al., 2005; Hewitt et al., 2006; Ouban and Ahmed, 2010; Ouban et al., 2012). While the exact functions of claudins in cancer cells are not fully understood, recent work strongly suggests that claudins are involved in survival and invasion of tumor cells (Agarwal et al., 2005; Morin, 2005; Dhawan et al., 2005; Kominsky, 2006; Oku et al., 2006; Dos Reis et al., 2008). Several studies on cancers have revealed down-regulation of claudins' expression including claudin-1 in breast cancer (Krämer et al., 2000) and claudin 7 in invasive breast cancer and in head and neck cancer (Al Moustafa et al., 2002; Kominsky et al., 2003). It is logical to expect this downregulation of claudins, because tumorigenesis is accompanied by disruption of tight junctions, with resultant loss of cohesion, invasiveness and lack of normal process of differentiation. However, it is interesting to note that numerous other studies have shown upregulation of these proteins in other cancers including, for example, claudins 3 and 4 over-expression in ovarian, breast, and prostate cancers (Long et al., 2001; Kominsky et al., 2003; Rangel et al., 2003), claudin 1 in oral squamous cell carcinomas (Dos Reis et al., 2008; Oku et al., 2006; Ouban et al., 2011). The over-expression of claudins in these cancers, which typically lose their TJs, is unexpected but probably related to roles unrelated to TJ formation (Hewitt et al., 2006). It is known that in addition to their function as a seal controlling the paracellular transport, tight junctions also play critical roles in maintaining cell polarity and signal transductions (Tsukita and Furuse, 2000; Van Itallie et al., 2006).
It is through these functions that an over-expressed claudin protein may get involved in carcinogenesis and/or metastases. For example, claudins are important regulators of signal transduction from the cell-cell contact region (Gonzalez-Mariscal and Nava, 2005) and have also been shown to be directly recruiting and enhancing the activation of pro-matrix metalloproteinase 2 (MMP2) (Miyamori et al., 2001). MMP2 over-expression suggests a potential risk for invasion and metastasis in high-grade squamous intraepithelial lesions (Nasr et al., 2005) and pro-MMP2 activation is involved in pancreatic cancer progression (Ellenrieder et al., 2000). CLDN1 has also been shown to be involved in the beta-catenin-Tcf/LEF signaling pathway and its over-expression was suggested to have a role in colorectal carcinogenesis (Miwa et al., 2000). The epidermal growth factor receptor (EGFR), has been suggested to regulate claudin proteins. EGF-induced EGFR activation increased CLDN1 expression in Madin-Darby canine kidney cells (Singh and Harris, 2004). EGFR is frequently amplified and over-expressed in many cancers, such as brain tumors (Schwechheimer et al., 1995), hepatocellular carcinomas (Tang et al., 1998), and head and neck carcinoma (Xia et al., 1999; Garnis et al., 2004); and increased expression of EGFR protein has been associated with worse prognosis (Bankfalvi et al., 2002). The function of claudins in cancer is complex and diverse, with both over- and under-expression being linked to tumorigenesis. While the exact mechanism through which a claudin protein may predispose to carcinogenesis and metastases is still not clear in all cases, it is important to note the involvement of claudins in activation/recruitement of collagenases, activation of molecular neoplastic pathways such as the Wnt/Beta-catenin-TCF/LEF pathways, or with a growth factor well-known for its involvement in many tumor formations (EGF).
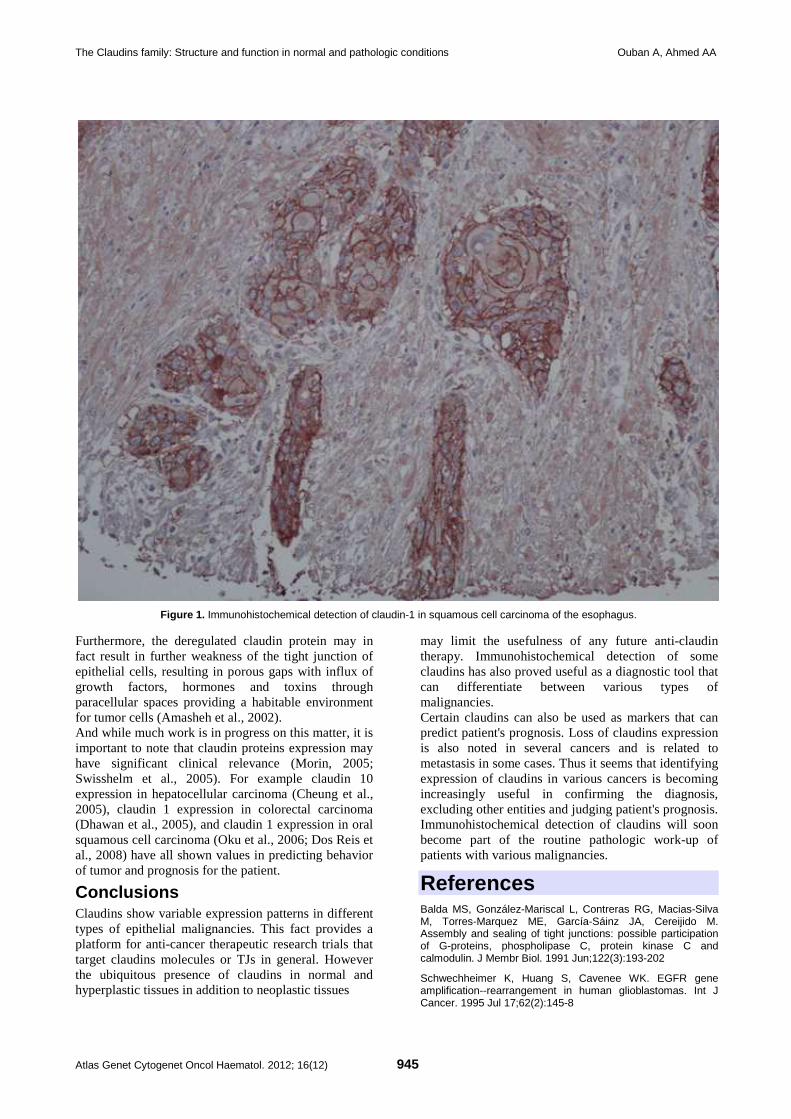
The Claudins family: Structure and function in normal and pathologic conditions Ouban A, Ahmed AA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 945
Figure 1. Immunohistochemical detection of claudin-1 in squamous cell carcinoma of the esophagus.
Furthermore, the deregulated claudin protein may in fact result in further weakness of the tight junction of epithelial cells, resulting in porous gaps with influx of growth factors, hormones and toxins through paracellular spaces providing a habitable environment for tumor cells (Amasheh et al., 2002). And while much work is in progress on this matter, it is important to note that claudin proteins expression may have significant clinical relevance (Morin, 2005; Swisshelm et al., 2005). For example claudin 10 expression in hepatocellular carcinoma (Cheung et al., 2005), claudin 1 expression in colorectal carcinoma (Dhawan et al., 2005), and claudin 1 expression in oral squamous cell carcinoma (Oku et al., 2006; Dos Reis et al., 2008) have all shown values in predicting behavior of tumor and prognosis for the patient.
Conclusions Claudins show variable expression patterns in different types of epithelial malignancies. This fact provides a platform for anti-cancer therapeutic research trials that target claudins molecules or TJs in general. However the ubiquitous presence of claudins in normal and hyperplastic tissues in addition to neoplastic tissues
may limit the usefulness of any future anti-claudin therapy. Immunohistochemical detection of some claudins has also proved useful as a diagnostic tool that can differentiate between various types of malignancies. Certain claudins can also be used as markers that can predict patient's prognosis. Loss of claudins expression is also noted in several cancers and is related to metastasis in some cases. Thus it seems that identifying expression of claudins in various cancers is becoming increasingly useful in confirming the diagnosis, excluding other entities and judging patient's prognosis. Immunohistochemical detection of claudins will soon become part of the routine pathologic work-up of patients with various malignancies.
References Balda MS, González-Mariscal L, Contreras RG, Macias-Silva M, Torres-Marquez ME, García-Sáinz JA, Cereijido M. Assembly and sealing of tight junctions: possible participation of G-proteins, phospholipase C, protein kinase C and calmodulin. J Membr Biol. 1991 Jun;122(3):193-202
Schwechheimer K, Huang S, Cavenee WK. EGFR gene amplification--rearrangement in human glioblastomas. Int J Cancer. 1995 Jul 17;62(2):145-8

The Claudins family: Structure and function in normal and pathologic conditions Ouban A, Ahmed AA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 946
Tang Z, Qin L, Wang X, Zhou G, Liao Y, Weng Y, Jiang X, Lin Z, Liu K, Ye S. Alterations of oncogenes, tumor suppressor genes and growth factors in hepatocellular carcinoma: with relation to tumor size and invasiveness. Chin Med J (Engl). 1998 Apr;111(4):313-8
Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci U S A. 1999 Jan 19;96(2):511-6
Xia W, Lau YK, Zhang HZ, Xiao FY, Johnston DA, Liu AR, Li L, Katz RL, Hung MC. Combination of EGFR, HER-2/neu, and HER-3 is a stronger predictor for the outcome of oral squamous cell carcinoma than any individual family members. Clin Cancer Res. 1999 Dec;5(12):4164-74
Ellenrieder V, Alber B, Lacher U, Hendler SF, Menke A, Boeck W, Wagner M, Wilda M, Friess H, Büchler M, Adler G, Gress TM. Role of MT-MMPs and MMP-2 in pancreatic cancer progression. Int J Cancer. 2000 Jan 1;85(1):14-20
Krämer F, White K, Kubbies M, Swisshelm K, Weber BH. Genomic organization of claudin-1 and its assessment in hereditary and sporadic breast cancer. Hum Genet. 2000 Sep;107(3):249-56
Tsukita S, Furuse M. Pores in the wall: claudins constitute tight junction strands containing aqueous pores. J Cell Biol. 2000 Apr 3;149(1):13-6
Kollmar R, Nakamura SK, Kappler JA, Hudspeth AJ. Expression and phylogeny of claudins in vertebrate primordia. Proc Natl Acad Sci U S A. 2001 Aug 28;98(18):10196-201
Long H, Crean CD, Lee WH, Cummings OW, Gabig TG. Expression of Clostridium perfringens enterotoxin receptors claudin-3 and claudin-4 in prostate cancer epithelium. Cancer Res. 2001 Nov 1;61(21):7878-81
Miwa N, Furuse M, Tsukita S, Niikawa N, Nakamura Y, Furukawa Y. Involvement of claudin-1 in the beta-catenin/Tcf signaling pathway and its frequent upregulation in human colorectal cancers. Oncol Res. 2001;12(11-12):469-76
Miyamori H, Takino T, Kobayashi Y, Tokai H, Itoh Y, Seiki M, Sato H. Claudin promotes activation of pro-matrix metalloproteinase-2 mediated by membrane-type matrix metalloproteinases. J Biol Chem. 2001 Jul 27;276(30):28204-11
Al Moustafa AE, Alaoui-Jamali MA, Batist G, Hernandez-Perez M, Serruya C, Alpert L, Black MJ, Sladek R, Foulkes WD. Identification of genes associated with head and neck carcinogenesis by cDNA microarray comparison between matched primary normal epithelial and squamous carcinoma cells. Oncogene. 2002 Apr 18;21(17):2634-40
Amasheh S, Meiri N, Gitter AH, Schöneberg T, Mankertz J, Schulzke JD, Fromm M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci. 2002 Dec 15;115(Pt 24):4969-76
Bánkfalvi A, Krassort M, Végh A, Felszeghy E, Piffkó J. Deranged expression of the E-cadherin/beta-catenin complex and the epidermal growth factor receptor in the clinical evolution and progression of oral squamous cell carcinomas. J Oral Pathol Med. 2002 Sep;31(8):450-7
Kominsky SL, Argani P, Korz D, Evron E, Raman V, Garrett E, Rein A, Sauter G, Kallioniemi OP, Sukumar S. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene. 2003 Apr 3;22(13):2021-33
Rangel LB, Agarwal R, D'Souza T, Pizer ES, Alò PL, Lancaster WD, Gregoire L, Schwartz DR, Cho KR, Morin PJ. Tight junction proteins claudin-3 and claudin-4 are frequently
overexpressed in ovarian cancer but not in ovarian cystadenomas. Clin Cancer Res. 2003 Jul;9(7):2567-75
Garnis C, Campbell J, Zhang L, Rosin MP, Lam WL. OCGR array: an oral cancer genomic regional array for comparative genomic hybridization analysis. Oral Oncol. 2004 May;40(5):511-9
Singh AB, Harris RC. Epidermal growth factor receptor activation differentially regulates claudin expression and enhances transepithelial resistance in Madin-Darby canine kidney cells. J Biol Chem. 2004 Jan 30;279(5):3543-52
Agarwal R, D'Souza T, Morin PJ. Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res. 2005 Aug 15;65(16):7378-85
Cheung ST, Leung KL, Ip YC, Chen X, Fong DY, Ng IO, Fan ST, So S. Claudin-10 expression level is associated with recurrence of primary hepatocellular carcinoma. Clin Cancer Res. 2005 Jan 15;11(2 Pt 1):551-6
Dhawan P, Singh AB, Deane NG, No Y, Shiou SR, Schmidt C, Neff J, Washington MK, Beauchamp RD. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J Clin Invest. 2005 Jul;115(7):1765-76
Gonzalez-Mariscal L, Nava P. Tight junctions, from tight intercellular seals to sophisticated protein complexes involved in drug delivery, pathogens interaction and cell proliferation. Adv Drug Deliv Rev. 2005 Apr 25;57(6):811-4
Morin PJ. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res. 2005 Nov 1;65(21):9603-6
Nasr M, Ayyad SB, El-Lamie IK, Mikhail MY. Expression of matrix metalloproteinase-2 in preinvasive and invasive carcinoma of the uterine cervix. Eur J Gynaecol Oncol. 2005;26(2):199-202
Swisshelm K, Macek R, Kubbies M. Role of claudins in tumorigenesis. Adv Drug Deliv Rev. 2005 Apr 25;57(6):919-28
Hewitt KJ, Agarwal R, Morin PJ. The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer. 2006 Jul 12;6:186
Kominsky SL. Claudins: emerging targets for cancer therapy. Expert Rev Mol Med. 2006 Aug 4;8(18):1-11
Oku N, Sasabe E, Ueta E, Yamamoto T, Osaki T. Tight junction protein claudin-1 enhances the invasive activity of oral squamous cell carcinoma cells by promoting cleavage of laminin-5 gamma2 chain via matrix metalloproteinase (MMP)-2 and membrane-type MMP-1. Cancer Res. 2006 May 15;66(10):5251-7
Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M, Tsukita S. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006 Aug 25;126(4):741-54
Van Itallie CM, Rogan S, Yu A, Vidal LS, Holmes J, Anderson JM. Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am J Physiol Renal Physiol. 2006 Dec;291(6):F1288-99
Flores-Benítez D, Ruiz-Cabrera A, Flores-Maldonado C, Shoshani L, Cereijido M, Contreras RG. Control of tight junctional sealing: role of epidermal growth factor. Am J Physiol Renal Physiol. 2007 Feb;292(2):F828-36
Dos Reis PP, Bharadwaj RR, Machado J, Macmillan C, Pintilie M, Sukhai MA, Perez-Ordonez B, Gullane P, Irish J, Kamel-Reid S. Claudin 1 overexpression increases invasion and is associated with aggressive histological features in oral

The Claudins family: Structure and function in normal and pathologic conditions Ouban A, Ahmed AA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 947
squamous cell carcinoma. Cancer. 2008 Dec 1;113(11):3169-80
Findley MK, Koval M. Regulation and roles for claudin-family tight junction proteins. IUBMB Life. 2009 Apr;61(4):431-7
Lal-Nag M, Morin PJ. The claudins. Genome Biol. 2009;10(8):235
Ouban A, Ahmed AA. Claudins in human cancer: a review. Histol Histopathol. 2010 Jan;25(1):83-90
Ouban A, Hamdan H, Hakam A, Ahmed AA. Claudin-1 expression in squamous cell carcinomas of different organs: comparative study of cancerous tissues and normal controls. Int J Surg Pathol. 2012 Apr;20(2):132-8
This article should be referenced as such:
Ouban A, Ahmed AA. The Claudins family: Structure and function in normal and pathologic conditions. Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12):943-947.

INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Instructions to Authors Manuscripts submitted to the Atlas must be submitted solely to the Atlas. Iconography is most welcome: there is no space restriction. The Atlas publishes "cards", "deep insights", "case reports", and "educational items". Cards are structured review articles. Detailed instructions for these structured reviews can be found at: http://AtlasGeneticsOncology.org/Forms/Gene_Form.html for reviews on genes, http://AtlasGeneticsOncology.org/Forms/Leukaemia_Form.html for reviews on leukaemias, http://AtlasGeneticsOncology.org/Forms/SolidTumour_Form.html for reviews on solid tumours, http://AtlasGeneticsOncology.org/Forms/CancerProne_Form.html for reviews on cancer-prone diseases. According to the length of the paper, cards are divided, into "reviews" (texts exceeding 2000 words), "mini reviews" (between), and "short communications" (texts below 400 words). The latter category may not be accepted for indexing by bibliographic databases. Deep Insights are written as traditional papers, made of paragraphs with headings, at the author's convenience. No length restriction. Case Reports in haematological malignancies are dedicated to recurrent -but rare- chromosomes abnormalities in leukaemias/lymphomas. Cases of interest shall be: 1- recurrent (i.e. the chromosome anomaly has already been described in at least 1 case), 2- rare (previously described in less than 20 cases), 3- with well documented clinics and laboratory findings, and 4- with iconography of chromosomes. It is mandatory to use the specific "Submission form for Case reports": see http://AtlasGeneticsOncology.org/Reports/Case_Report_Submission.html. Educational Items must be didactic, give full information and be accompanied with iconography. Translations into French, German, Italian, and Spanish are welcome.
Subscription: The Atlas is FREE!
Corporate patronage, sponsorship and advertising Enquiries should be addressed to [email protected].
Rules, Copyright Notice and Disclaimer Conflicts of Interest: Authors must state explicitly whether potential conflicts do or do not exist. Reviewers must disclose to editors any conflicts of interest that could bias their opinions of the manuscript. The editor and the editorial board members must disclose any potential conflict. Privacy and Confidentiality – Iconography: Patients have a right to privacy. Identifying details should be omitted. If complete anonymity is difficult to achieve, informed consent should be obtained. Property: As "cards" are to evolve with further improvements and updates from various contributors, the property of the cards belongs to the editor, and modifications will be made without authorization from the previous contributor (who may, nonetheless, be asked for refereeing); contributors are listed in an edit history manner. Authors keep the rights to use further the content of their papers published in the Atlas, provided that the source is cited. Copyright: The information in the Atlas of Genetics and Cytogenetics in Oncology and Haematology is issued for general distribution. All rights are reserved. The information presented is protected under international conventions and under national laws on copyright and neighbouring rights. Commercial use is totally forbidden. Information extracted from the Atlas may be reviewed, reproduced or translated for research or private study but not for sale or for use in conjunction with commercial purposes. Any use of information from the Atlas should be accompanied by an acknowledgment of the Atlas as the source, citing the uniform resource locator (URL) of the article and/or the article reference, according to the Vancouver convention. Reference to any specific commercial products, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favouring. The views and opinions of contributors and authors expressed herein do not necessarily state or reflect those of the Atlas editorial staff or of the web site holder, and shall not be used for advertising or product endorsement purposes. The Atlas does not make any warranty, express or implied, including the warranties of merchantability and fitness for a particular purpose, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, and shall not be liable whatsoever for any damages incurred as a result of its use. In particular, information presented in the Atlas is only for research purpose, and shall not be used for diagnosis or treatment purposes. No responsibility is assumed for any injury and/or damage to persons or property for any use or operation of any methods products, instructions or ideas contained in the material herein. See also: "Uniform Requirements for Manuscripts Submitted to Biomedical Journals: Writing and Editing for Biomedical Publication - Updated October 2004": http://www.icmje.org.
http://AtlasGeneticsOncology.org
© ATLAS - ISSN 1768-3262

The Claudins family: Structure and function in normal and pathologic conditions Ouban A, Ahmed AA
Atlas Genet Cytogenet Oncol Haematol. 2012; 16(12) 949




















