
Volume 17 - Number 4 April 2013 - Revues de l'INIST
72
The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. Online and PDF versions of the Atlas of Genetics and Cytogenetics in Oncology and Haematology are hosted by INIST-CNRS. INIST-CNRS OPEN ACCESS JOURNAL Atlas of Genetics and Cytogenetics in Oncology and Haematology Volume 17 - Number 4 April 2013
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Volume 17 - Number 4 April 2013 - Revues de l'INIST

The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. Online and PDF versions of the Atlas of Genetics and Cytogenetics in Oncology and Haematology are hosted by INIST-CNRS.
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Volume 17 - Number 4 April 2013

The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. Online and PDF versions of the Atlas of Genetics and Cytogenetics in Oncology and Haematology are hosted by INIST-CNRS.
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Scope
The Atlas of Genetics and Cytogenetics in Oncology and Haematology is a peer reviewed on-line journal in open access, devoted to genes, cytogenetics, and clinical entities in cancer, and cancer-prone diseases. It presents structured review articles ("cards") on genes, leukaemias, solid tumours, cancer-prone diseases, more traditional review articles on these and also on surrounding topics ("deep insights"), case reports in hematology, and educational items in the various related topics for students in Medicine and in Sciences.
Editorial correspondance
Jean-Loup Huret Genetics, Department of Medical Information, University Hospital F-86021 Poitiers, France tel +33 5 49 44 45 46 or +33 5 49 45 47 67 [email protected] or [email protected]
Staff Mohammad Ahmad, Mélanie Arsaban, Marie-Christine Jacquemot-Perbal, Vanessa Le Berre, Anne Malo, Carol Moreau, Catherine Morel-Pair, Laurent Rassinoux, Alain Zasadzinski. Philippe Dessen is the Database Director, and Alain Bernheim the Chairman of the on-line version (Gustave Roussy Institute – Villejuif – France).
The Atlas of Genetics and Cytogenetics in Oncology and Haematology (ISSN 1768-3262) is published 12 times a year by ARMGHM, a non profit organisation, and by the INstitute for Scientific and Technical Information of the French National Center for Scientific Research (INIST-CNRS) since 2008. The Atlas is hosted by INIST-CNRS (http://www.inist.fr)
http://AtlasGeneticsOncology.org
© ATLAS - ISSN 1768-3262

Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4)
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Editor
Jean-Loup Huret (Poitiers, France)
Editorial Board
Sreeparna Banerjee (Ankara, Turkey) Solid Tumours Section Alessandro Beghini (Milan, Italy) Genes Section Anne von Bergh (Rotterdam, The Netherlands) Genes / Leukaemia Sections Judith Bovée (Leiden, The Netherlands) Solid Tumours Section Vasantha Brito-Babapulle (London, UK) Leukaemia Section Charles Buys (Groningen, The Netherlands) Deep Insights Section Anne Marie Capodano (Marseille, France) Solid Tumours Section Fei Chen (Morgantown, West Virginia) Genes / Deep Insights Sections Antonio Cuneo (Ferrara, Italy) Leukaemia Section Paola Dal Cin (Boston, Massachussetts) Genes / Solid Tumours Section Brigitte Debuire (Villejuif, France) Deep Insights Section François Desangles (Paris, France) Leukaemia / Solid Tumours Sections Enric Domingo-Villanueva (London, UK) Solid Tumours Section Ayse Erson (Ankara, Turkey) Solid Tumours Section Richard Gatti (Los Angeles, California) Cancer-Prone Diseases / Deep Insights Sections Ad Geurts van Kessel (Nijmegen, The Netherlands) Cancer-Prone Diseases Section Oskar Haas (Vienna, Austria) Genes / Leukaemia Sections Anne Hagemeijer (Leuven, Belgium) Deep Insights Section Nyla Heerema (Colombus, Ohio) Leukaemia Section Jim Heighway (Liverpool, UK) Genes / Deep Insights Sections Sakari Knuutila (Helsinki, Finland) Deep Insights Section Lidia Larizza (Milano, Italy) Solid Tumours Section Lisa Lee-Jones (Newcastle, UK) Solid Tumours Section Edmond Ma (Hong Kong, China) Leukaemia Section Roderick McLeod (Braunschweig, Germany) Deep Insights / Education Sections Cristina Mecucci (Perugia, Italy) Genes / Leukaemia Sections Yasmin Mehraein (Homburg, Germany) Cancer-Prone Diseases Section Fredrik Mertens (Lund, Sweden) Solid Tumours Section Konstantin Miller (Hannover, Germany) Education Section Felix Mitelman (Lund, Sweden) Deep Insights Section Hossain Mossafa (Cergy Pontoise, France) Leukaemia Section Stefan Nagel (Braunschweig, Germany) Deep Insights / Education Sections Florence Pedeutour (Nice, France) Genes / Solid Tumours Sections Elizabeth Petty (Ann Harbor, Michigan) Deep Insights Section Susana Raimondi (Memphis, Tennesse) Genes / Leukaemia Section Mariano Rocchi (Bari, Italy) Genes Section Alain Sarasin (Villejuif, France) Cancer-Prone Diseases Section Albert Schinzel (Schwerzenbach, Switzerland) Education Section Clelia Storlazzi (Bari, Italy) Genes Section Sabine Strehl (Vienna, Austria) Genes / Leukaemia Sections Nancy Uhrhammer (Clermont Ferrand, France) Genes / Cancer-Prone Diseases Sections Dan Van Dyke (Rochester, Minnesota) Education Section Roberta Vanni (Montserrato, Italy) Solid Tumours Section Franck Viguié (Paris, France) Leukaemia Section José Luis Vizmanos (Pamplona, Spain) Leukaemia Section Thomas Wan (Hong Kong, China) Genes / Leukaemia Sections

INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Volume 17, Number 4, April 2013
Table of contents
Gene Section
ACLY (ATP citrate lyase) 231 Marie E Beckner
CLSPN (claspin) 237 Linda Mannini
SDCBP (syndecan binding protein (syntenin)) 240 Rosaria Gangemi, Ulrich Pfeffer, Silvano Ferrini
SLIT2 (slit homolog 2 (Drosophila)) 245 Kim Brussen
BNIP3L (BCL2/adenovirus E1B 19kDa interacting prote in 3-like) 253 Paul Ney, Ji Zhang
LPAR2 (lysophosphatidic acid receptor 2) 259 Sara Knowlden, Steve Georas
MARCKS (myristoylated alanine-rich protein kinase C substrate) 266 Atsuhiro Tanabe, Maho Saito
MIR331 (microRNA 331) 269 Keith M Giles, Michael R Epis, Peter J Leedman
SRXN1 (sulfiredoxin 1) 272 Hedy A Chawsheen, Hong Jiang, Qiou Wei
Leukaemia Section
t(3;12)(q27;p12) LRMP/BCL6 275 Jean-Loup Huret
t(3;6)(q27;p22) HIST1H4I/BCL6 277 Jean-Loup Huret
t(3;7)(q27;q32) FRA7H/BCL6 279 Jean-Loup Huret
t(3;9)(q27;p24) DMRT1/BCL6 281 Jean-Loup Huret
Solid Tumour Section
Head and Neck: Oral leukoplakia 283 Patrícia Carlos Caldeira, Maria Auxiliadora Vieira do Carmo
Thyroid: Medullary carcinoma 291 Yash Somnay, David Schneider, Haggi Mazeh

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 231
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
ACLY (ATP citrate lyase) Marie E Beckner
Department of Pathology, Louisiana State University Health Sciences Center - Shreveport, USA (MEB)
Published in Atlas Database: October 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/ACLYID50486ch17q21.html DOI: 10.4267/2042/48862
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: ACL, ATPCL, CLATP
HGNC (Hugo): ACLY
Location: 17q21.2
Note Note that the International Union for Biochemistry and Molecular Biology (IUBMB)'s enzyme nomenclature accepts ATP citrate synthase as the name for ACLY's encoded protein (EC 2.3.3.8). However, ATP citrate lyase is more commonly used and other names include citrate cleavage enzyme, ATP-citrate (pro-S)-lyase, ATPCL, CLATP. ACLY encodes a key metabolic enzyme that cleaves cytosolic citric acid with important consequences, such as lipogenesis, regulation of glycolysis, acetylcholine production, calcium chelation, etc.
DNA/RNA Description Two transcript variants have been identified and this variant (1) represents the longer ACLY transcript. It encodes the longer isoform of ACLY. Placement of code for the initiating methionine, stop codon, poly adenylation signal, boundaries of the 29 exons, and the untranslated region (hatched) are shown. Location of missing sequence in variant 2 compared to variant 1 is indicated in the diagram of the ACLY protein shown below. The sequence for ACLY has been conserved in evolution, putatively from an ancient single gene present prior to separation of animals and fungi with some fungi subsequently developing two genes to code for complete ACLY whereas animals have retained a single gene.
Homo sapiens ATP citrate lyase (ACLY), transcript variant 1, 4450 bp mRNA, encodes a 1101 aa protein. NCBI Reference Sequence: NM_001096. Locus NM_001096.

ACLY (ATP citrate lyase) Beckner ME
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 232
ATP citrate lyase (ACL or ACLY), variant 1. GenBank: AAH06195 protein sequence with locations of functional domains, multiple binding regions, Rossman fold (492-601), and post-translational modifications, including potential phosphorylation of tyrosines (131, 682), serines (260, 442, 455, 478, 481, 663, 839, 922, 979, 1100), threonines (445, 447, 453, 639), and a histidine (760), and N6-acetylysine (86, 546, 554, 948, 962, 968, 978, 1077). The missing sequence (476-485) in variant 2 results in the loss of 2 serines as indicated in the diagram.
Transcription 4450 bp mRNA (NCBI RefSeq, May-2012). Multiple Sp1 binding sites and CAAT are present in the promoter of rat ACLY and it can be induced by a low fat/high carbohydrate diet.
Pseudogene None known at this time.
Protein Note ACLY is a metabolic enzyme found as a tetramer of apparently identical subunits (440000 molecular weight). It was discovered in 1950's. ACLY cleaves citric acid in a multistep process with participation of cofactors to form the products, acetyl-CoA and oxaloacetate. Functional domains of ACLY resemble regions of related enzymes that can play similar roles in metabolism of other substrates.
Description Four of these subunits form a homotetramer.
Expression Prokaryotes and eukaryotes. The association between increased expression for ACLY and the gene encoding enolase, ENO1, is highly statistically significant. Greater expression of ACLY can be found in mammalian cells under hypoxic conditions. It is more highly expressed in many malignant tissues when compared to their benign counterparts. Aberrant expression can be found in breast, liver, colon, lung, and prostate cancers and is inversely correlated with tumor stage and differentiation so that increased ACLY expression is a negative prognostic factor. ACLY's knockdown in non-small cell lung carcinoma (NSCLC) can lead to apoptosis and differentiation in vitro and less growth in vivo.
Localisation ACLY is a relatively abundant cytoplasmic protein and can be associated with outer surfaces of mitochondria
and it is also preferentially distributed to pseudopodia in migrating cells. Relatively small amounts have been found in the nuclei. Also, ACLY is found in synaptosomes.
Function ACLY catalyzes the following reaction: ATP + citrate + CoA = ADP + phosphate + acetyl-CoA + oxaloacetate. ACLY is well-known for linking carbohydrate and lipid metabolism which can lead to membrane production during cell growth. However, a myriad of other consequences from the breakdown of citrate also occur and are indicated above. Systemically and locally the effects of ACLY's activity can have a powerfull impact. These include alteration of transcription. Citrate passes through nuclear pores and undergoes cleavage by the small amounts of ACLY in the nucleus to generate acetyl CoA that affects transcription via acetylation of histones and transcription factors. Cataplerosis includes citrate's transport from the mitochondria via a transporter to provide cytosolic citrate. The transfer of metabolites into mitochondria via shuttles, transporters, etc. constitutes anaplerosis so that either energy or amino acids can be formed, depending on the oxygenation state and the cell's needs. Regulation of ACLY is complex and appears to resemble that of glycogen synthase in regard to phosphorylations occurring sequentially in a hierarchical manner. The multiple sources of citrate help to explain the varying effects of ACLY. Note that exogenous citrate can come from anticoagulants. Although ACLY is susceptible to proteolysis, the lower weight (53 kDa) digestion product of ACLY retains its activity. Loss of ACLY function in plants can result in a bonsai phenotype. Hydroxycitrate, found in the fruit of a tropical tree, Garcinia cambogia (bitter kola) that grows in Southeast Asia and southern India, is a competitive inhibitor and has been extensively used in functional studies.

ACLY (ATP citrate lyase) Beckner ME
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 233
However, solubility issues and the large quantities of hydroxycitrate needed for inhibition of ACLY are disadvantages for using it in clinical studies. Proprietary formulations are available as weight loss supplements and at least one of these has been combined with established anti-cancer agents. The cell-penetrant gamma-lactone, SB-204990, a prodrug of SB-201076, was described in 1998 as an oral drug to inhibit ACLY and has been used in several cancer studies more recently. Radicicol and tartrate are also inhibitors of ACLY. Multiple agents, such as α-lipoic acid, statins, capsaicin, a Met kinase inhibitor (SU11274), etc., have been found to enhance the effects of ACLY inhibitors in small studies of tumors.
Homology ACLY is a member of the acyl-CoA synthetase superfamily (ADP-forming). ACLY's amino terminal region, 1-419, resembles ATP citrate (pro-S)-lyase and the region, 1-424, is homologous to the β-subunit of succinyl-CoA synthetase and the region, 486-818, is homologous to the α-subunit of succinyl-CoA synthetase. Also, the hierarchy of multiple, sequential serine/threonine phosphorylations responsible for the complex regulation of glycogen synthase is similar to the serine/threonine phosphorylations in ACLY. Sequence surrounding the histidine in ACLY's catalytic site, that is phosphorylated by nucleoside diphosphate kinase (NDPK or nm23), is similar to sequence around phosphorylation sites in other substrates of nm23, such as aldolase C. ACLY has homology with citrate synthase that catalyzes its reverse reaction. Rat ACLY is 96,3% identical to human ACLY.
Mutations Germinal Homozygous knock-out of ACLY in mice is lethal. Heterozygous knock-out mice appear to be normal.
Implicated in Bladder (transitional cell) cancer Note A bladder cancer cell line (MBT-2) studied in a mouse syngenic cancer model has demonstrated efficacy of calcium hydrocitrate when it was used to inhibit ACLY, combined with other drugs and agents, in several small studies.
Breast cancer Note Increased expression of ACLY may play a role in the agressive breast cancers. Elevated levels were found in both primary and metastatic cell lines compared to normal cell lines and the highest expression levels occurred in metastatic cell lines.
Colon carcinoma Note Silencing ACLY in human colon carcinoma cells (HCT116) has been shown to suppress histone acetylation.
Gliomas (glial brain tumors) Note ACLY has been demonstrated to localize preferentially to pseudopodia in U87 human glioblastoma cells.

ACLY (ATP citrate lyase) Beckner ME
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 234
Inhibition of ACLY in U87 cells with a soluble form of hydroxycitrate suppressed their cell migration, clonogenicity and brain invasion under glycolytic conditions and enhanced the suppressive effects of a Met kinase inhibitor on cell migration. Queries of the NIH's REMBRANDT brain tumor database based on Affymetrix array data indicated that decreased patient survival correlated with increased gene expression of ACLY in gliomas.
Liver (hepatocellular) carcinoma Note Markedly increased expression of mRNA for ACLY and genes for other lipogenic enzymes have been found in hepatocellular carcinoma compared to surrounding non-cancerous liver tissue.
Lung carcinoma Note A Lewis lung cancer cell line (LL/2) studied in a mouse syngenic cancer model has demonstrated efficacy of using calcium hydrocitrate combined with other drugs and agents in several small studies. In one of these studies, results were confirmed using a human xenograft model, NCI-H69, small cell lung carcinoma, with tumor development reduced and prolonged animal survival observed. In another study with ACLY knockdown, there was inhibition of growth in vivo for non-small cell lung carcinoma along with apoptosis and differentiation. Enhancement of the anti-tumor effects was achieved by adding statins with regression of established tumors reported. Human lung adenocarcinoma samples have been shown to have significantly increased ACLY activity compared to normal lung tissue and phosphorylated ACLY overexpression correlated with stage, grade, and poorer prognosis. Growth arrest in A549 cells was achieved with RNA interference for ACLY. Inhibitory results were also achieved in A549 cells with the ACLY inhibitor, SB-204990.
Melanoma Note A melanoma cell line (B16-F10) studied in a mouse syngenic cancer model has demonstrated efficacy of using calcium hydrocitrate combined with other drugs and agents in several small studies.
Ovarian carcinoma Note Higher ACLY expression has been found in malignant ovarian tissue compared to normal ovarian tissue. Phosphorylated ACLY was also increased and the expression correlated well with tumor grade, FIGO stage, and poorer prognosis. Also knockdown of ACLY in A2780 cells inhibited their proliferation and induced cell cycle arrest.
Pancreatic cancer (ductal adenocarcinoma) Note An 80 year old woman was treated with a formulation containing hydroxycitrate to inhibit ACLY in addition to gemcitabine with favorable temporary results.
Prostate carcinoma Note Aberrant expression of ACLY has been found in prostatic cancer with levels inversely correlating with tumor stage and differentiaion. The expression of ACLY has predicted a reduced citrate level which is characteristic of prostatic cancer. Normal prostatic tissue has very high levels of citrate. Benign prostatic hypertrophy also has high levels of citrate. The change to oxidation of citrate in prostatic cancer rather than production of citrate has been viewed as a type of metabolic transformation that may provide a bioenergetic theory for prostatic malignancy.
Hepatitis B Virus (HBV) infection Note In HBV transgenic mice that replicate HBV in the liver without producing gross liver pathology, the largest functional category for upregulated genes was lipid biosynthesis, including ACLY.
Obesity/fatty liver Note Inhibition of ACLY is a strategy to counteract weight gain that has led to the development of commercially available formulations of hydroxycitrate. Inhibition of ACLY has been suggested as being helpfull for fatty liver.
Breakpoints Note No breakpoints are known within ACLY. The ACLY gene is distal to the P12.3B hybrid breakpoint in RARA.
References Hoffmann GE, Andres H, Weiss L, Kreisel C, Sander R. Properties and organ distribution of ATP citrate (pro-3S)-lyase. Biochim Biophys Acta. 1980 Oct 6;620(1):151-8
Ranganathan NS, Srere PA, Linn TC. Comparison of phospho- and dephospho-ATP citrate lyase. Arch Biochem Biophys. 1980 Oct 1;204(1):52-8
Szutowicz A, Lysiak W. Regional and subcellular distribution of ATP-citrate lyase and other enzymes of acetyl-CoA metabolism in rat brain. J Neurochem. 1980 Oct;35(4):775-85
Ranganathan NS, Linn TC, Srere PA. Phosphorylation of dephospho-ATP citrate lyase by the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 1982 Jan 25;257(2):698-702

ACLY (ATP citrate lyase) Beckner ME
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 235
Szutowicz A, Kabata J, Bielarczyk H. The contribution of citrate to the synthesis of acetyl units in synaptosomes of developing rat brain. J Neurochem. 1982 May;38(5):1196-204
Ingebritsen TS, Stewart AA, Cohen P. The protein phosphatases involved in cellular regulation. 6. Measurement of type-1 and type-2 protein phosphatases in extracts of mammalian tissues; an assessment of their physiological roles. Eur J Biochem. 1983 May 2;132(2):297-307
Ramakrishna S, Pucci DL, Benjamin WB. Dependence of ATP-citrate lyase kinase activity on the phosphorylation of ATP-citrate lyase by cyclic AMP-dependent protein kinase. J Biol Chem. 1983 Apr 25;258(8):4950-6
Holland R, Hardie DG. Both insulin and epidermal growth factor stimulate fatty acid synthesis and increase phosphorylation of acetyl-CoA carboxylase and ATP-citrate lyase in isolated hepatocytes. FEBS Lett. 1985 Feb 25;181(2):308-12
Houston B, Nimmo HG. Effects of phosphorylation on the kinetic properties of rat liver ATP-citrate lyase. Biochim Biophys Acta. 1985 Feb 21;844(2):233-9
Ramakrishna S, D'Angelo G, Benjamin WB. Sequence of sites on ATP-citrate lyase and phosphatase inhibitor 2 phosphorylated by multifunctional protein kinase (a glycogen synthase kinase 3 like kinase). Biochemistry. 1990 Aug 21;29(33):7617-24
Elshourbagy NA, Near JC, Kmetz PJ, Wells TN, Groot PH, Saxty BA, Hughes SA, Franklin M, Gloger IS. Cloning and expression of a human ATP-citrate lyase cDNA. Eur J Biochem. 1992 Mar 1;204(2):491-9
Emmerson K, Roehrig K. Epidermal growth factor (EGF) stimulation of ATP citrate lyase activity in isolated rat hepatocytes is age dependent. Comp Biochem Physiol B. 1992 Nov;103(3):663-7
Hughes K, Ramakrishna S, Benjamin WB, Woodgett JR. Identification of multifunctional ATP-citrate lyase kinase as the alpha-isoform of glycogen synthase kinase-3. Biochem J. 1992 Nov 15;288 ( Pt 1):309-14
Costello LC, Franklin RB. Bioenergetic theory of prostate malignancy. Prostate. 1994 Sep;25(3):162-6
Couch FJ, Abel KJ, Brody LC, Boehnke M, Collins FS, Weber BL. Localization of the gene for ATP citrate lyase (ACLY) distal to gastrin(GAS) and proximal to D17S856 on chromosome 17q12-q21. Genomics. 1994 May 15;21(2):444-6
Wagner PD, Vu ND. Phosphorylation of ATP-citrate lyase by nucleoside diphosphate kinase. J Biol Chem. 1995 Sep 15;270(37):21758-64
Fukuda H, Iritani N, Katsurada A, Noguchi T. Insulin- and polyunsaturated fatty acid-responsive region(s) of rat ATP citrate lyase gene promoter. FEBS Lett. 1996 Feb 12;380(1-2):204-7
Melnick JZ, Srere PA, Elshourbagy NA, Moe OW, Preisig PA, Alpern RJ. Adenosine triphosphate citrate lyase mediates hypocitraturia in rats. J Clin Invest. 1996 Nov 15;98(10):2381-7
Pearce NJ, Yates JW, Berkhout TA, Jackson B, Tew D, Boyd H, Camilleri P, Sweeney P, Gribble AD, Shaw A, Groot PH. The role of ATP citrate-lyase in the metabolic regulation of plasma lipids. Hypolipidaemic effects of SB-204990, a lactone prodrug of the potent ATP citrate-lyase inhibitor SB-201076. Biochem J. 1998 Aug 15;334 ( Pt 1):113-9
Potapova IA, El-Maghrabi MR, Doronin SV, Benjamin WB. Phosphorylation of recombinant human ATP:citrate lyase by cAMP-dependent protein kinase abolishes homotropic allosteric regulation of the enzyme by citrate and increases the
enzyme activity. Allosteric activation of ATP:citrate lyase by phosphorylated sugars. Biochemistry. 2000 Feb 8;39(5):1169-79
Wagner PD, Vu ND. Histidine to aspartate phosphotransferase activity of nm23 proteins: phosphorylation of aldolase C on Asp-319. Biochem J. 2000 Mar 15;346 Pt 3:623-30
Beigneux AP, Kosinski C, Gavino B, Horton JD, Skarnes WC, Young SG. ATP-citrate lyase deficiency in the mouse. J Biol Chem. 2004 Mar 5;279(10):9557-64
Roy S, Rink C, Khanna S, Phillips C, Bagchi D, Bagchi M, Sen CK. Body weight and abdominal fat gene expression profile in response to a novel hydroxycitric acid-based dietary supplement. Gene Expr. 2004;11(5-6):251-62
Soni MG, Burdock GA, Preuss HG, Stohs SJ, Ohia SE, Bagchi D. Safety assessment of (-)-hydroxycitric acid and Super CitriMax, a novel calcium/potassium salt. Food Chem Toxicol. 2004 Sep;42(9):1513-29
Fatland BL, Nikolau BJ, Wurtele ES. Reverse genetic characterization of cytosolic acetyl-CoA generation by ATP-citrate lyase in Arabidopsis. Plant Cell. 2005 Jan;17(1):182-203
Hajjou M, Norel R, Carver R, Marion P, Cullen J, Rogler LE, Rogler CE. cDNA microarray analysis of HBV transgenic mouse liver identifies genes in lipid biosynthetic and growth control pathways affected by HBV. J Med Virol. 2005 Sep;77(1):57-65
Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005 Oct;8(4):311-21
Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Nagai R, Ishibashi S, Kadowaki T, Makuuchi M, Ohnishi S, Osuga J, Yamada N. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer. 2005 Jun;41(9):1316-22
Sale EM, Hodgkinson CP, Jones NP, Sale GJ. A new strategy for studying protein kinase B and its three isoforms. Role of protein kinase B in phosphorylating glycogen synthase kinase-3, tuberin, WNK1, and ATP citrate lyase. Biochemistry. 2006 Jan 10;45(1):213-23
Yancy HF, Mason JA, Peters S, Thompson CE 3rd, Littleton GK, Jett M, Day AA. Metastatic progression and gene expression between breast cancer cell lines from African American and Caucasian women. J Carcinog. 2007 May 1;6:8
Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008 Oct 15;68(20):8547-54
Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009 May 22;324(5930):1076-80
Beckner ME, Fellows-Mayle W, Zhang Z, Agostino NR, Kant JA, Day BW, Pollack IF. Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int J Cancer. 2010 May 15;126(10):2282-95
Cousins RJ, Aydemir TB, Lichten LA. Plenary Lecture 2: Transcription factors, regulatory elements and nutrient-gene communication. Proc Nutr Soc. 2010 Feb;69(1):91-4
Schwartz L, Abolhassani M, Guais A, Sanders E, Steyaert JM, Campion F, Israël M. A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: preliminary results. Oncol Rep. 2010 May;23(5):1407-16

ACLY (ATP citrate lyase) Beckner ME
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 236
Stohs SJ, Lau FC, Kim D, Kim SU, Bagchi M, Bagchi D. Safety assessment of a calcium-potassium salt of (-)-hydroxycitric acid. Toxicol Mech Methods. 2010 Nov;20(9):515-25
Sun T, Hayakawa K, Bateman KS, Fraser ME. Identification of the citrate-binding site of human ATP-citrate lyase using X-ray crystallography. J Biol Chem. 2010 Aug 27;285(35):27418-28
Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011 Nov 20;481(7381):380-4
Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011 Nov 20;481(7381):385-8
Sun T, Hayakawa K, Fraser ME. ADP-Mg2+ bound to the ATP-grasp domain of ATP-citrate lyase. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011 Oct 1;67(Pt 10):1168-72
Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011 Dec 6;108(49):19611-6
Abolhassani M, Guais A, Sanders E, Campion F, Fichtner I, Bonte J, Baronzio G, Fiorentini G, Israël M, Schwartz L. Screening of well-established drugs targeting cancer metabolism: reproducibility of the efficacy of a highly effective drug combination in mice. Invest New Drugs. 2012 Aug;30(4):1331-42
Bertilsson H, Tessem MB, Flatberg A, Viset T, Gribbestad I, Angelsen A, Halgunset J. Changes in gene transcription underlying the aberrant citrate and choline metabolism in human prostate cancer samples. Clin Cancer Res. 2012 Jun 15;18(12):3261-9
Guais A, Baronzio G, Sanders E, Campion F, Mainini C, Fiorentini G, Montagnani F, Behzadi M, Schwartz L, Abolhassani M. Adding a combination of hydroxycitrate and lipoic acid (METABLOC™) to chemotherapy improves effectiveness against tumor development: experimental results and case report. Invest New Drugs. 2012 Feb;30(1):200-11
Hanai J, Doro N, Sasaki AT, Kobayashi S, Cantley LC, Seth P, Sukhatme VP. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J Cell Physiol. 2012 Apr;227(4):1709-20
Schwartz L, Guais A, Israël M, Junod B, Steyaert JM, Crespi E, Baronzio G, Abolhassani M. Tumor regression with a combination of drugs interfering with the tumor metabolism: efficacy of hydroxycitrate, lipoic acid and capsaicin. Invest New Drugs. 2012 Jul 14;
Wang Y, Wang Y, Shen L, Pang Y, Qiao Z, Liu P. Prognostic and therapeutic implications of increased ATP citrate lyase expression in human epithelial ovarian cancer. Oncol Rep. 2012 Apr;27(4):1156-62
This article should be referenced as such:
Beckner ME. ACLY (ATP citrate lyase). Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):231-236.

Gene Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 237
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
CLSPN (claspin) Linda Mannini
Istituto di Ricerca Genetica e Biomedica CNR, Pisa, Italy (LM)
Published in Atlas Database: October 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/CLSPNID40105ch1p34.html DOI: 10.4267/2042/48863
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity HGNC (Hugo): CLSPN
Location: 1p34.3
Note Claspin is a S-phase checkpoint factor that is activated in response to replication stress or other DNA damage induced by genotoxic agents.
DNA/RNA Description The gene spans approximately 37 kb and contains 25 exons.
Transcription There are different transcripts variants, five of them encode for different isoforms. Two transcript variants encode for known proteins. The transcript variant 1 of 4769 bp counts 25 exons. The transcript variant 2 of 3977 bp, counts 24 exons (lacks 1 exon maintaining the frame).
Protein Note The transcript variant 1 encodes for a protein of 1339 aminoacids. The transcript variant 2 encodes for a protein of 1275 amino acids.
Expression Claspin peaks at S/G2 phase in response to DNA replication blocks and DNA damage.
Localisation Claspin is located in the nucleus and it associates with Chk1 following replication fork stress or other types of DNA damage.
Function Claspin is a S-phase checkpoint regulator required in response to DNA replication stress and to DNA damage induced by UV and irradiation (Chini and Chen, 2003; Sar et al., 2004; Freire et al., 2006; Tanaka, 2010).
Figure 1. Schematic representation of the Claspin w ith the two transcript variants. The transcript variant 1 with 25 exons and the transcript variant 2 with 24 exons. The exons are indicated by boxes and introns by lines.

CLSPN (claspin) Mannini L
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 238
Figure 2. Claspin regulation during DNA damage chec kpoint response pathway. Upon DNA damage, ATR activates Claspin, promoting the activation of the effector kinase Chk1. Once the damage is repaired, Plk1 binds and phosphorylates Claspin favoring its proteasomal degradation. Claspin is a mediator of ATR-Chk1 signaling cascade triggers for cell cycle checkpoint activation in DNA damage response. Claspin becomes phosphorylated and interacts with Chk1 promoting its activation by ATR-dependent phosphorylation (Chini and Chen, 2004; Kumagai and Dunphy, 2003; Clarke and Clarke, 2005). Claspin also interacts with the checkpoint proteins ATR and RAd9, and ATR regulates Claspin phosphorylation in presence of DNA damage induced by genotoxic stress including UV, IR and hydroxyurea, resulting in recruitment and phosphorylation of BRCA1 (Jeong et al., 2003; Lin et al., 2004; Sørensen et al., 2004). When DNA damage has been repaired, Claspin response is turned off by ubiquitin proteasome pathway in order to inactivate checkpoint response and facilitate cells to enter the cell cycle. Therefore Claspin is phosphorylated by Plk1 kinase to permit its interaction with SCFβTrCP ubiquitin ligase that promotes its degradation (Mailand et al., 2006; Mamely et al., 2006; Peschiaroli et al., 2006). Claspin has also been found associated to replication forks in absence of DNA damage suggesting a function as a sensor required for replication fork stability (Sørensen et al., 2004; Petermann et al., 2008; Scorah et al., 2009). Finally it has been observed a role of the Claspin in genome stability. Inhibition of the Claspin by RNA interference leads to both chromosome alterations and fragile site expression in human cells. Following aphidicolin treatment, Claspin increases due to its requirement to checkpoint activation, while its synthesis decrement after a prolonged aphidicolin treatment. It has been proposed that, following an extreme replication block, Claspin allows rare cells to escape checkpoint mechanisms and enter mitosis although
their genome has not yet fully replicated (Focarelli et al., 2009).
Homology This gene is present in S. cerevisiae as scMrc1; in S. pombe as spMrc1; in vertebrates as Claspin.
Mutations Germinal - First study that reports the mutation screening of the CLSPN gene in familial breast cancer cases identifying different sequence changes (Erkko et al., 2008). Nevertheless no of these mutations is related to breast cancer susceptibility. - Sequence variants of Claspin have been identified in different human cancers. Eight nonsynonymous variants were found from the germline of two cancer-prone individuals and five cancer cells lines of breast, ovarian, and hematopoietic origin (Zhang et al., 2009).
Implicated in Various cancers Note Claspin expression levels increased in cancer cells lines and tumor specimens in a study performed in normal fibroblasts and various cancer cell lines, and from tumor and normal tissues of patients with primary epithelial carcinomas, in order to evaluate Claspin as a proliferation marker (Tsimaratou et al., 2007).
Breast cancer Note Transcript levels of Claspin were highly detected in tumor breast cancer tissues in which estrogen receptor

CLSPN (claspin) Mannini L
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 239
and progesterone receptor was lost (Verlinden et al., 2007).
Cervical cancer Note Claspin expression is found significantly high in cervical cancer cell lines and the analysis of its expression could be clinically relevant in the diagnosis of Human Papillomavirus-related high grade lesions of uterine cervix (Benevolo et al., 2012).
References Chini CC, Chen J. Human claspin is required for replication checkpoint control. J Biol Chem. 2003 Aug 8;278(32):30057-62
Jeong SY, Kumagai A, Lee J, Dunphy WG. Phosphorylated claspin interacts with a phosphate-binding site in the kinase domain of Chk1 during ATR-mediated activation. J Biol Chem. 2003 Nov 21;278(47):46782-8
Kumagai A, Dunphy WG. Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat Cell Biol. 2003 Feb;5(2):161-5
Chini CC, Chen J. Claspin, a regulator of Chk1 in DNA replication stress pathway. DNA Repair (Amst). 2004 Aug-Sep;3(8-9):1033-7
Lin SY, Li K, Stewart GS, Elledge SJ. Human Claspin works with BRCA1 to both positively and negatively regulate cell proliferation. Proc Natl Acad Sci U S A. 2004 Apr 27;101(17):6484-9
Sar F, Lindsey-Boltz LA, Subramanian D, Croteau DL, Hutsell SQ, Griffith JD, Sancar A. Human claspin is a ring-shaped DNA-binding protein with high affinity to branched DNA structures. J Biol Chem. 2004 Sep 17;279(38):39289-95
Sørensen CS, Syljuåsen RG, Lukas J, Bartek J. ATR, Claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle. 2004 Jul;3(7):941-5
Clarke CA, Clarke PR. DNA-dependent phosphorylation of Chk1 and Claspin in a human cell-free system. Biochem J. 2005 Jun 1;388(Pt 2):705-12
Freire R, van Vugt MA, Mamely I, Medema RH. Claspin: timing the cell cycle arrest when the genome is damaged. Cell Cycle. 2006 Dec;5(24):2831-4
Mailand N, Bekker-Jensen S, Bartek J, Lukas J. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell. 2006 Aug 4;23(3):307-18
Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A, Medema RH, Freire R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr Biol. 2006 Oct 10;16(19):1950-5
Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell. 2006 Aug 4;23(3):319-29
Tsimaratou K, Kletsas D, Kastrinakis NG, Tsantoulis PK, Evangelou K, Sideridou M, Liontos M, Poulias I, Venere M, Salmas M, Kittas C, Halazonetis TD, Gorgoulis VG. Evaluation of claspin as a proliferation marker in human cancer and normal tissues. J Pathol. 2007 Feb;211(3):331-9
Verlinden L, Vanden Bempt I, Eelen G, Drijkoningen M, Verlinden I, Marchal K, De Wolf-Peeters C, Christiaens MR, Michiels L, Bouillon R, Verstuyf A. The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor /progesterone receptor /HER-2 breast carcinomas. Cancer Res. 2007 Jul 15;67(14):6574-81
Erkko H, Pylkäs K, Karppinen SM, Winqvist R. Germline alterations in the CLSPN gene in breast cancer families. Cancer Lett. 2008 Mar 8;261(1):93-7
Petermann E, Helleday T, Caldecott KW. Claspin promotes normal replication fork rates in human cells. Mol Biol Cell. 2008 Jun;19(6):2373-8
Focarelli ML, Soza S, Mannini L, Paulis M, Montecucco A, Musio A. Claspin inhibition leads to fragile site expression. Genes Chromosomes Cancer. 2009 Dec;48(12):1083-90
Scorah J, McGowan CH. Claspin and Chk1 regulate replication fork stability by different mechanisms. Cell Cycle. 2009 Apr 1;8(7):1036-43
Zhang J, Song YH, Brannigan BW, Wahrer DC, Schiripo TA, Harris PL, Haserlat SM, Ulkus LE, Shannon KM, Garber JE, Freedman ML, Henderson BE, Zou L, Sgroi DC, Haber DA, Bell DW. Prevalence and functional analysis of sequence variants in the ATR checkpoint mediator Claspin. Mol Cancer Res. 2009 Sep;7(9):1510-6
Tanaka K. Multiple functions of the S-phase checkpoint mediator. Biosci Biotechnol Biochem. 2010;74(12):2367-73
Benevolo M, Musio A, Vocaturo A, Donà MG, Rollo F, Terrenato I, Carosi M, Pescarmona E, Vocaturo G, Mottolese M. Claspin as a biomarker of human papillomavirus-related high grade lesions of uterine cervix. J Transl Med. 2012 Jun 25;10:132
This article should be referenced as such:
Mannini L. CLSPN (claspin). Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):237-239.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 240
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
SDCBP (syndecan binding protein (syntenin)) Rosaria Gangemi, Ulrich Pfeffer, Silvano Ferrini
Lab of Immunotherapy and Functional Genomics Istituto Nazionale per la Ricerca sul Cancro, 16132 Genova, Italy (RG, UP, SF)
Published in Atlas Database: October 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/SDCBPID44377ch8q12.html DOI: 10.4267/2042/48864
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: MDA-9, ST1, SYCL, TACIP18
HGNC (Hugo): SDCBP
Location: 8q12.1
Local order: The human SDCBP gene maps on 8q12 between the NSMAF (neutral sphingomyelinase activation associated factor) and the CYP7A1 (cytochrome P450, family 7, subfamily A, polypeptide 1) loci, which are both in the opposite orientation.
Note No translocations reported.
DNA/RNA Description The SDCBP gene is comprised of 9 exons, spanning 2,96 kb on chromosome 8q12. The SDCBP promoter region has not been functionally explored, although two studies (Lin et al., 1998; Stier et al., 2000) describe SDCBP as an interferon-gamma and TNF-alpha inducible gene. Among predicted transcription factor binding sites upstream the transcription start site of SDCBP there are: Nf-KappaB, Nf-KappaB1 and p53.
Transcription Five alternatively spliced transcript variants of SDCBP, each comprising 9 exons, have been described.
Protein Description SDCBP gene codes for a syntenin protein of 298 amino acid residues with a predicted molecular mass of 33 kDa (Lin et al., 1998; Grootjans et al., 1997). Three isoforms are produced by alternative splicing: isoform 1 (NP_001007068.1) which represents the full-length protein of 298 aa; isoform 2 (NP_001007069) of 292 aa missing residues 12-17; isoform 3 (NP_001007070) of 297 aa missing residue 81. Syntenin is a scaffolding protein, endowed with several biological activities and involved in cancer metastases development (reviewed in Das et al., 2012a). The molecule has four domains: an N-terminal domain (aa 1-113) with no homology to known structural motifs, two PDZ domains (PDZ-1 aa 114-193 and PDZ-2 aa 198-273) and a COOH-terminal domain. The crystal structure of the two PDZ domains showed independent interaction of each domain with protein targets (Cierpicki et al., 2005; Kang et al., 2004). Postranslational modifications: syntenin can be phosphorylated on tyrosine (Sulka et al., 2009) and serine residues (Rajesh et al., 2011).
Expression SDCBP is expressed in fetal kidney, liver, lung and brain. In adult high expression is present in hearth and placenta (Lin et al., 1998; Zimmermann et al., 2001).

SDCBP (syndecan binding protein (syntenin)) Gangemi R, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 241
SDCBP gene organization, mRNA and encoded proteins. The SDCBP gene is comprised of 9 exons and results in 5 alternatively spliced transcript variants (TV), which encode for three different protein isoforms (additional transcripts variants were also reported). Coding exons are in blue and UTRs in yellow. Transcript variants 1 and 2 differ only in their 5' UTR regions and encode for the same full-length protein, named isoform 1. Transcript variant 3 derives from the usage of an alternative in frame splice-site in the 5' coding region (exon 1) and encodes for the protein isoform 2, lacking 6 residues. Transcript variant 4 uses an alternative splice site in exon 5 resulting in a protein isoform lacking one residue (isoform 3). Transcript variant 5 differs from variant 4 in the 5' UTR and encodes for the same protein isoform 3. Protein isoform 2 and 3 partial aa sequencies that differ from isoform 1 are in red characters. It is also expressed in several human tumor cell lines. Several types of tumors express high levels of SDCBP such as gastric, colon and breast carcinomas (Koo et al., 2002), cutaneous (Helmke et al., 2004) and uveal melanoma (Gangemi et al., 2012).
Localisation SDCBP protein is localized to adherens junctions, focal adhesion plaques, inner side of the cell membrane, cytoplasm, endoplasmic reticulum, cytoskeleton (Zimmermann et al., 2001), nucleus (Gangemi et al., 2012) and melanosomes (Basrur et al., 2003). It is also present in cell-released exosomes (Baietti et al., 2012).
Function SDCBP was identified as melanoma differentiation-associated gene (MDA)-9 (Lin et al., 1998). The same gene was independently cloned and named syntenin, by yeast two hybrid screening. Syntenin interacts through its PDZ domains with the heparan-sulfates syndecans, which are involved in molecular recognition, signaling, and cell trafficking (Grootjans et al., 1997). Through its binding with syndecans and PIP2, syntenin mediates syndecan recycling through endosomal compartments (Zimmermann et al., 2002).
This process modulates the surface availability of growth factor receptors such as FGFR, which follows syndecan in the recycling pathway (Zimmermann et al., 2005). Syntenin binds the C-terminal domain of the pro-transforming growth factor α (proTGFα) (Fernández-Larrea et al., 1999) and to the Delta1 ligand of Notch (Estrach et al., 2007), tethering them to the cell surface. In addition, syntenin directly interacts with the C-terminal of Frizzled 7 and supports non-canonical Wnt signaling (Wawrzak et al., 2009). Syntenin binds to the cytoplasmic tail of the tetraspanin CD63 at the plasma membrane and is therefore part of the tetraspanin-enriched microdomains (Latysheva et al., 2006). The over-expression of syntenin can limit internalization of CD63, suggesting a role for syntenin as a regulator of endocytosis. Syntenin is involved in the establishment and maintenance of synaptic structures through its interaction with several adhesion molecules, such as neurofascin (Koroll et al., 2001). Presynaptic development also depends upon the interaction of the syntenin PDZ domains with ephrin-B1 and ephrin-B2 (McClelland et al., 2009).

SDCBP (syndecan binding protein (syntenin)) Gangemi R, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 242
Syntenin protein-protein interactions. Syntenin is an adaptor protein, which interacts with multiple proteins and has several intracellular functions. SDCBP participates in the formation and maturation of synapses and colocalizes with Glutamate receptors at growth cones (Hirbec et al., 2005). Outgrowth of developing axon is also regulated by syntenin, which provides a scaffold for the serine/threonine kinase Unc51.1 and for Rab5 GTPase (Tomoda et al., 2004). Syntenin participates in B cell development and differentiation by interacting with interleukin-5 (IL-5) receptor α and the transcription factor Sox4 and mediates IL-5-induced Sox4 activation (Geijsen et al., 2001). Proteosomal degradation of Sox4 is prevented by the binding of its c terminal domain with SDCBP, which contributes to its localization into the nucleus (Beekman et al., 2012). Syntenin mediates the generation of functional asymmetry in T cells during the cellular response to polarized extracellular cues, through the generation of polarized actin structures (Sala-Valdés et al., 2012). Syntenin interacts with Ubiquitin through is C- and N-terminal regions and facilitates the recruitment of ubiquitinated proteins to its transmembrane partners. The process is facilitated by syntenin dimerization and is inhibited by phosphorylation of its serin in the N terminal domain mediated by Ulk1 (Rajesh et al., 2011). Syntenin has a key role in exosome formation through the binding of syndecan 1, syndecan 2, syndecan 3, syndecan 4 with the PDZ domains and ALIX with the N-terminal domain (Baietti et al., 2012).
Homology The SDCBP gene is conserved in chimpanzee, Rhesus monkey, dog, cow, mouse, rat, chicken, zebrafish, and mosquito (NCBI). Paralog: SDCBP2. SDCBP is highly related to SDCBP2 at the amino acid level (70% over the PDZ domains) and in the domains organization (Koroll et al., 2001).
Mutations Note Not yet described. Genetic polymorphisms of SDCBP (561 SNPs) have been reported (NCBI) but their relationship to disease is unknown.
Implicated in Cutaneous melanoma Note SDCBP gene was identified as an interferon-inducible gene in melanoma cells (Lin et al., 1998). A subtractory library approach of candidate metastasis genes identified the syntenin gene, which was overexpressed in cutaneous melanoma specimens relative to melanocytic nevi (Helmke et al., 2004). Altering syntenin expression by gene transduction modulates the metastatic ability of human melanoma cells (Boukerche

SDCBP (syndecan binding protein (syntenin)) Gangemi R, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 243
et al., 2005). Syntenin over-expression increased phosphorylation of focal adhesion kinase, c-Jun-NH2-kinase, p38, and nuclear factor-kappaB (NF-kappaB) in human melanoma cells. As a consequence tumor cell growth and motility are enhanced. The induction of membrane-type matrix metalloproteinase (MMP)-1 and MMP-2 promotes extracellular matrix invasion (Boukerche et al., 2007). Syntenin binds c-Src and mediates the formation of an active FAK/c-Src complex, increasing melanoma cell invasive properties (Boukerche et al., 2008). In addition, src kinase activation is required for syntenin-mediated activation of NF-kappaB (Boukerche et al., 2010). Further studies indicated that syntenin acts as a molecular adaptor linking PKCalpha and FAK activation during human breast cancer and melanoma cell adhesion to fibronectin (Hwangbo et al., 2010). The Raf kinase inhibitor RKIP, is downregulated in metastatic melanoma cells. The study of melanoma arrays and cell lines showed an inverse relationship between syntenin and RKIP expression during melanoma progression. Syntenin transcriptionally downregulated RKIP and also physically interacted with RKIP protein. Ectopic RKIP expression in melanoma cells inhibited syntenin signaling, cell invasion and growth and in vivo dissemination of melanoma cells. Therefore RKIP acts as an inhibitor of syntenin-dependent melanoma metastasis (Das et al., 2012b).
Disease Metastatic melanoma.
Uveal melanoma Note Uveal melanoma is a rare tumor of the eye, distinct from cutaneous melanoma on the basis of genetic alterations and clinical behavior. High expression of SDCBP gene correlated with metastatic progression in three gene expression profile datasets of primary uveal melanomas. High expression of syntenin protein in primary tumors was also related to metastatic recurrence. Syntenin was aslo highly expressed in liver metastases from patients and from xenografted mice. Silencing of syntenin inhibited uveal melanoma cell migration and hepatocyte growth factor (HGF)-triggered invasion, activation of FAK, AKT and Src. Conversely syntenin overexpression mediated opposite effects (Gangemi et al., 2012).
Disease Metastatic uveal melanoma.
Gastric and breast cancers Note The expression level of syntenin was related with invasive potential in human breast and gastric cancer cells in vitro. Syntenin gene was highly expressed in gastric cancer tissues. Syntenin overexpression in human gastric or breast cancer cells increased their
migration in vitro and induced pseudopodia formation on collagen I. Mutation studies suggested that the PDZ2 domain of syntenin is involved in the stimulatory effect on cell migration (Koo et al., 2002).
Colon cancer Note The proteoglycan syndecan-2 is involved in tumorigenicity of colon cancer cells. Syndecan-2-induced migration requires the EFYA motif in its C-terminal region as its deletion inhibited cell migration and interaction with syntenin. In addition, overexpression of syntenin in colon cancer cells enhanced their migratory capacity, while syntenin silencing had opposite effects. Syntenin interaction with syndecan-2 mediates Rac activation, and colon cancer cell migration (Lee et al., 2011).
HIV infection Note Syntenin is recruited to the plasma membrane during HIV-1 attachment and associates with CD4. Syntenin overexpression inhibits HIV-1 production and HIV-mediated cell fusion, while syntenin depletion increases HIV-1 entry, suggesting a regulatory role of syntenin in HIV-1 entry (Gordón-Alonso et al., 2012).
Disease AIDS.
References Grootjans JJ, Zimmermann P, Reekmans G, Smets A, Degeest G, Dürr J, David G. Syntenin, a PDZ protein that binds syndecan cytoplasmic domains. Proc Natl Acad Sci U S A. 1997 Dec 9;94(25):13683-8
Lin JJ, Jiang H, Fisher PB. Melanoma differentiation associated gene-9, mda-9, is a human gamma interferon responsive gene. Gene. 1998 Jan 30;207(2):105-10
Fernández-Larrea J, Merlos-Suárez A, Ureña JM, Baselga J, Arribas J. A role for a PDZ protein in the early secretory pathway for the targeting of proTGF-alpha to the cell surface. Mol Cell. 1999 Apr;3(4):423-33
Stier S, Totzke G, Grünewald E, Neuhaus T, Fronhoffs S, Sachinidis A, Vetter H, Schulze-Osthoff K, Ko Y. Identification of syntenin and other TNF-inducible genes in human umbilical arterial endothelial cells by suppression subtractive hybridization. FEBS Lett. 2000 Feb 11;467(2-3):299-304
Geijsen N, Uings IJ, Pals C, Armstrong J, McKinnon M, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ. Cytokine-specific transcriptional regulation through an IL-5Ralpha interacting protein. Science. 2001 Aug 10;293(5532):1136-8
Koroll M, Rathjen FG, Volkmer H. The neural cell recognition molecule neurofascin interacts with syntenin-1 but not with syntenin-2, both of which reveal self-associating activity. J Biol Chem. 2001 Apr 6;276(14):10646-54
Zimmermann P, Tomatis D, Rosas M, Grootjans J, Leenaerts I, Degeest G, Reekmans G, Coomans C, David G. Characterization of syntenin, a syndecan-binding PDZ protein, as a component of cell adhesion sites and microfilaments. Mol Biol Cell. 2001 Feb;12(2):339-50

SDCBP (syndecan binding protein (syntenin)) Gangemi R, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 244
Koo TH, Lee JJ, Kim EM, Kim KW, Kim HD, Lee JH. Syntenin is overexpressed and promotes cell migration in metastatic human breast and gastric cancer cell lines. Oncogene. 2002 Jun 13;21(26):4080-8
Zimmermann P, Meerschaert K, Reekmans G, Leenaerts I, Small JV, Vandekerckhove J, David G, Gettemans J. PIP(2)-PDZ domain binding controls the association of syntenin with the plasma membrane. Mol Cell. 2002 Jun;9(6):1215-25
Basrur V, Yang F, Kushimoto T, Higashimoto Y, Yasumoto K, Valencia J, Muller J, Vieira WD, Watabe H, Shabanowitz J, Hearing VJ, Hunt DF, Appella E. Proteomic analysis of early melanosomes: identification of novel melanosomal proteins. J Proteome Res. 2003 Jan-Feb;2(1):69-79
Helmke BM, Polychronidis M, Benner A, Thome M, Arribas J, Deichmann M. Melanoma metastasis is associated with enhanced expression of the syntenin gene. Oncol Rep. 2004 Aug;12(2):221-8
Kang BS, Devedjiev Y, Derewenda U, Derewenda ZS. The PDZ2 domain of syntenin at ultra-high resolution: bridging the gap between macromolecular and small molecule crystallography. J Mol Biol. 2004 Apr 30;338(3):483-93
Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004 Mar 1;18(5):541-58
Boukerche H, Su ZZ, Emdad L, Baril P, Balme B, Thomas L, Randolph A, Valerie K, Sarkar D, Fisher PB. mda-9/Syntenin: a positive regulator of melanoma metastasis. Cancer Res. 2005 Dec 1;65(23):10901-11
Cierpicki T, Bushweller JH, Derewenda ZS. Probing the supramodular architecture of a multidomain protein: the structure of syntenin in solution. Structure. 2005 Feb;13(2):319-27
Hirbec H, Martin S, Henley JM. Syntenin is involved in the developmental regulation of neuronal membrane architecture. Mol Cell Neurosci. 2005 Apr;28(4):737-46
Zimmermann P, Zhang Z, Degeest G, Mortier E, Leenaerts I, Coomans C, Schulz J, N'Kuli F, Courtoy PJ, David G. Syndecan recycling [corrected] is controlled by syntenin-PIP2 interaction and Arf6. Dev Cell. 2005 Sep;9(3):377-88
Latysheva N, Muratov G, Rajesh S, Padgett M, Hotchin NA, Overduin M, Berditchevski F. Syntenin-1 is a new component of tetraspanin-enriched microdomains: mechanisms and consequences of the interaction of syntenin-1 with CD63. Mol Cell Biol. 2006 Oct;26(20):7707-18
Boukerche H, Su ZZ, Emdad L, Sarkar D, Fisher PB. mda-9/Syntenin regulates the metastatic phenotype in human melanoma cells by activating nuclear factor-kappaB. Cancer Res. 2007 Feb 15;67(4):1812-22
Estrach S, Legg J, Watt FM. Syntenin mediates Delta1-induced cohesiveness of epidermal stem cells in culture. J Cell Sci. 2007 Aug 15;120(Pt 16):2944-52
Boukerche H, Su ZZ, Prévot C, Sarkar D, Fisher PB. mda-9/Syntenin promotes metastasis in human melanoma cells by activating c-Src. Proc Natl Acad Sci U S A. 2008 Oct 14;105(41):15914-9
McClelland AC, Sheffler-Collins SI, Kayser MS, Dalva MB. Ephrin-B1 and ephrin-B2 mediate EphB-dependent presynaptic development via syntenin-1. Proc Natl Acad Sci U S A. 2009 Dec 1;106(48):20487-92
Sulka B, Lortat-Jacob H, Terreux R, Letourneur F, Rousselle P. Tyrosine dephosphorylation of the syndecan-1 PDZ binding domain regulates syntenin-1 recruitment. J Biol Chem. 2009 Apr 17;284(16):10659-71
Wawrzak D, Luyten A, Lambaerts K, Zimmermann P. Frizzled-PDZ scaffold interactions in the control of Wnt signaling. Adv Enzyme Regul. 2009;49(1):98-106
Boukerche H, Aissaoui H, Prévost C, Hirbec H, Das SK, Su ZZ, Sarkar D, Fisher PB. Src kinase activation is mandatory for MDA-9/syntenin-mediated activation of nuclear factor-kappaB. Oncogene. 2010 May 27;29(21):3054-66
Hwangbo C, Kim J, Lee JJ, Lee JH. Activation of the integrin effector kinase focal adhesion kinase in cancer cells is regulated by crosstalk between protein kinase Calpha and the PDZ adapter protein mda-9/Syntenin. Cancer Res. 2010 Feb 15;70(4):1645-55
Lee H, Kim Y, Choi Y, Choi S, Hong E, Oh ES. Syndecan-2 cytoplasmic domain regulates colon cancer cell migration via interaction with syntenin-1. Biochem Biophys Res Commun. 2011 May 27;409(1):148-53
Rajesh S, Bago R, Odintsova E, Muratov G, Baldwin G, Sridhar P, Rajesh S, Overduin M, Berditchevski F. Binding to syntenin-1 protein defines a new mode of ubiquitin-based interactions regulated by phosphorylation. J Biol Chem. 2011 Nov 11;286(45):39606-14
Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, Geeraerts A, Ivarsson Y, Depoortere F, Coomans C, Vermeiren E, Zimmermann P, David G. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012 Jun 3;14(7):677-85
Beekman JM, Vervoort SJ, Dekkers F, van Vessem ME, Vendelbosch S, Brugulat-Panès A, van Loosdregt J, Braat AK, Coffer PJ. Syntenin-mediated regulation of Sox4 proteasomal degradation modulates transcriptional output. Oncogene. 2012 May 24;31(21):2668-79
Das SK, Bhutia SK, Kegelman TP, Peachy L, Oyesanya RA, Dasgupta S, Sokhi UK, Azab B, Dash R, Quinn BA, Kim K, Barral PM, Su ZZ, Boukerche H, Sarkar D, Fisher PB. MDA-9/syntenin: a positive gatekeeper of melanoma metastasis. Front Biosci. 2012a Jan 1;17:1-15
Das SK, Bhutia SK, Sokhi UK, Azab B, Su ZZ, Boukerche H, Anwar T, Moen EL, Chatterjee D, Pellecchia M, Sarkar D, Fisher PB. Raf kinase inhibitor RKIP inhibits MDA-9/syntenin-mediated metastasis in melanoma. Cancer Res. 2012b Dec 1;72(23):6217-26
Gangemi R, Mirisola V, Barisione G, Fabbi M, Brizzolara A, Lanza F, Mosci C, Salvi S, Gualco M, Truini M, Angelini G, Boccardo S, Cilli M, Airoldi I, Queirolo P, Jager MJ, Daga A, Pfeffer U, Ferrini S. Mda-9/syntenin is expressed in uveal melanoma and correlates with metastatic progression. PLoS One. 2012;7(1):e29989
Gordón-Alonso M, Rocha-Perugini V, Álvarez S, Moreno-Gonzalo O, Ursa A, López-Martín S, Izquierdo-Useros N, Martínez-Picado J, Muñoz-Fernández MÁ, Yáñez-Mó M, Sánchez-Madrid F. The PDZ-adaptor protein syntenin-1 regulates HIV-1 entry. Mol Biol Cell. 2012 Jun;23(12):2253-63
Sala-Valdés M, Gordón-Alonso M, Tejera E, Ibáñez A, Cabrero JR, Ursa A, Mittelbrunn M, Lozano F, Sánchez-Madrid F, Yáñez-Mó M. Association of syntenin-1 with M-RIP polarizes Rac-1 activation during chemotaxis and immune interactions. J Cell Sci. 2012 Mar 1;125(Pt 5):1235-46
This article should be referenced as such:
Gangemi R, Pfeffer U, Ferrini S. SDCBP (syndecan binding protein (syntenin)). Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):240-244.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 245
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
SLIT2 (slit homolog 2 (Drosophila)) Kim Brussen
Sanquin Research and Landsteiner Laboratory, Department of Hematopoiesis, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands (KB)
Published in Atlas Database: October 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/SLIT2ID42328ch4p15.html DOI: 10.4267/2042/48865
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: SLIL3, Slit-2
HGNC (Hugo): SLIT2
Location: 4p15.2
DNA/RNA Note SLIT2 is a member of the SLIT gene family. In mammals, this family contains 3 genes named SLIT1, SLIT2 and SLIT3.
Description SLIT2A and SLIT2C have 37 coding exons, while SLIT2B has 36 coding exons, spanning 365 kb of the genome. All SLIT genes contain CpG islands in their promoter regions and intron length and exon-intron boundaries are highly similar (Little et al., 2002; Dallol et al., 2005).
Transcription The leucine rich repeat regions of the three human SLIT genes contain a large number of very small
exons, mostly encoding for one individual leucine rich repeat. This allows alternative splicing of the exons, without altering the frame (Little et al., 2002). Three alternatively spliced variants were identified after screening of a human fetal brain cDNA library and nucleotide database searching (Little et al., 2002). These variants have been described by different groups (Holmes et al., 1998; Itoh et al., 1998; Brose et al., 1999). The transcript described by Itoh et al. (1998) was named SLIT2A, the transcript described by Holmes et al. (1998) was named SLIT2B and the transcript described by Brose et al. (1999) was named SLIT2C. SLIT2B and SLIT2C lack exon 15. This exon encodes for eight amino acids of unkown function (Holmes et al., 1998; Brose et al., 1999; Little et al., 2002). In addition, SLIT2C contains an additional exon. This exon is located in an intron between exon 8 and 9 and encodes for four amino acids (Little et al., 2002). Between transcripts, differences can also be found in the length of the 5'UTR and the 3'UTR. The relevance of the differently spliced variants is unclear.
Genomic localization of SLIT2. The SLIT2 gene is shown in red, the surrounding genes in grey. The arrows indicate the direction of transcription (NCBI, version 10 Jul 12).

SLIT2 (slit homolog 2 (Drosophila)) Brussen K
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 246
Map of the SLIT2 gene, direction from 5'UTR till 3' UTR. The direction of transcription is indicated by the arrow. Exons are depicted as blue boxes. Within the first and the last exon, the 5'UTR and 3'UTR are depicted in yellow. There are three differently spliced variants of
SLIT2, named SLIT2A, SLIT2B and SLIT2C (Holmes et al., 1998; Itoh et al., 1998; Brose et al., 1999). Exons that are only present in one of the transcripts are depicted in red (see below for further explanation). The length of the exons and introns is roughly indicated, but is not up to scale. The size of the exons ranges from 12 base pairs up to 1790 base pairs, the size of the introns ranges from 110 base
pairs up to 198870 base pairs. For clarity, the exons are depicted larger than the introns. Below is indicated which protein domains are encoded by particular exons. Based on ENSEMBL version 68 Juli 2012 transcripts ENST00000504154 (SLIT2A), ENST00000503823
(SLIT2B) and ENST00000503837 (SLIT2C). Information on protein domains encoded by particular exons was obtained from Little et al., 2002.
Protein Note The extracellular matrix protein SLIT was first identified in a genetic screen for mutations that affected the dorsal-ventral patterning or the development of the central nervous system in Drosophila (Anderson et al., 1984; Seeger et al., 1993). SLIT homologues have since been found in C. elegans and in vertebrates, including mammals (Holmes et al., 1998; Itoh et al., 1998; Brose et al., 1999; Holmes et al., 2001; Vargesson et al., 2001; Gilthorpe et al., 2002). The cognate receptor of the SLIT proteins is Roundabout or ROBO (Kidd et al., 1998; Huminiecki et al., 2002).
Description In mammals there are three SLIT genes which encode large ECM glycoproteins of about 200 kDa, comprising a stretch of four leucine rich repeats (LRR) connected by disulphide bonds, seven to nine epidermal growth factor (EGF)-like domains, a domain named Agrin, Laminin, Perlecan and SLIT (ALPS) or laminin G-like module, and a C-terminal cystein knot (Rothberg and Artavanis-Tsakonas, 1992; Hohenester et al., 1999;
Nguyen-Ba-Charvet and Chedotal, 2002). SLIT proteins can be proteolytically cleaved within the EGF-like region, this has been shown to occur for SLIT2 and for SLIT3 (Brose et al., 1999; Patel et al., 2001; Condac et al., 2012). Differently spliced variants of the SLIT2 protein exist, three of which were reported in literature (Itoh et al., 1998; Holmes et al., 1998; Brose et al., 1999). SLIT2A is 1529 amino acids long (ENSEMBL protein ID ENSP00000422591), SLIT2B is 1521 amino acids long (ENSEMBL protein ID ENSP00000427548) and SLIT2C is 1525 amino acids long (ENSEMBL protein ID ENSP00000422261).
Expression In humans, SLIT2 is expressed both during embryonic development and during adult life. It is expressed in the fetal kidney and lung (Itoh et al., 1998) as well as in the adult kidney (Wu et al., 2001), in the female reproductive tract (endometrium, fallopian tube and ovaries) (Dickinson et al., 2008; Duncan et al., 2010; Dickinson et al;, 2011), the adrenal gland, the brain and the spinal cord (Itoh et al., 1998) and in bone marrow stromal and endothelial cells (Geutskens et al., 2012).

SLIT2 (slit homolog 2 (Drosophila)) Brussen K
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 247
Domain organization of the SLIT protein from N-term inus to C-terminus. SS: N-terminal signal peptide; LRR: leucin-rich repeat; EGF-like: epidermal growth factor-like domain; Lam-G like: Agrin, Laminin, Perlecan and SLIT (ALPS) or laminin G-like module; Cystein knot: C-terminal cystein knot. The sciccors represent a proteolytic cleavage site. Adapted from a figure created by dr. S.B. Geutskens (Leiden University Medical Center; Department of Immunohematology and Blood Transfusion and Einthoven laboratory for Experimental Vascular Medicine; Leiden; The Netherlands). For two of the differently spliced variants of SLIT2, the expression pattern was examined in several fetal and adult tissues. SLIT2A is expressed in the adult human spinal cord and in low levels in the fetal lung and kidney (Itoh et al., 1998), expression of SLIT2B can also be detected outside the CNS in postnatal human tissues (Holmes et al., 1998). SLIT2C is expressed in the rat spinal cord during embryonic development (Brose et al., 1999). The functional relevance of the differently spliced variants is not clear.
Localisation SLIT is a secreted extracellular matrix protein that is bound to the surface of the cell by the extracellular matrix, mainly by heparan sulfates (Liang et al., 1999; Ronca et al., 2001). It has been reported that both the N-terminal part of SLIT2 (Hussain et al., 2006) and the C-terminal part of SLIT2 and SLIT3 bind to heparin and heparan sulfates (Ronca et al., 2001; Condac et al., 2012). The interaction between SLIT proteins and heparan-sulfates is not only important for the binding of SLIT proteins to the extracellular matrix, but can also increase the affinity of SLIT for ROBO (Hu et al., 2001). Removal of heparan sulfates from the cell surface abolishes the response to SLIT2 (Hu et al., 2001; Hussain et al., 2006). Therefore, heparan-sulfates are considered as important co-receptors in SLIT-ROBO signalling (Inatani et al., 2003; Steigemann et al., 2004; Hussain et al., 2006). The SLIT2 and the SLIT3 protein can be proteolytically cleaved. Following proteolytic cleavage of SLIT2, the 140kDa N-terminal fragment remains tightly associated to the cell surface, while the 50-60kDa C-terminal fragment is more loosely attached and can also be detected in conditioned medium (Brose et al., 1999; Wang et al., 1999).
Function The extracellular matrix protein SLIT binds to the transmembrane receptor Roundabout or ROBO and has a conserved role in axon guidance in the central nervous system (CNS), where SLIT functions as a
repellent for ROBO-expressing axons (Brose et al., 1999; Kidd et al., 1999; Long et al., 2004). Outside the CNS, SLIT plays an important role during embryonic development and in human pathology. Neuronal guidance: SLIT proteins function as chemorepellents throughout the central nervous system to restrict the positioning of axons to their proper sites. Deletion of SLIT2 resulted in defects in cortical inhibitory neurons, commisural neurons and sensory neurons (Nguyen-Ba-Charvet et al., 1999; Bagri et al., 2002; Nguyen-Ba-Charvet et al., 2002; Plump et al., 2002; Long et al., 2004; Unni et al., 2012). Cortical inhibitory neurons (interneurons) modulate the response of pyramidal cells to incoming signals, thereby preventing overexcitation and maintaining the balance between different signals. In rodents, they are generated in the ventral telencephalon whereafter they migrate into the cortex (reviewed by Rossignol, 2011). Slit1/2 double knockout mice display an increased interneuron proliferation and an increase in neuronal process length and branching (Andrews et al., 2008). Vertebrate commissural neurons first arise in the dorsal spinal cord. Their axons are directed to the midline/ floorplate by the chemoattractants netrin and sonic hedgehog. When these axons have reached the midline, they cross it and turn longitudinally on the opposite side, growing right alongside the midline/ floor plate (reviewed by Dickson and Gilestro, 2006). Bagri et al. (2002) reported a broad spectrum of neuronal defects in Slit2 knockout or Slit1/Slit2 double knockout mice. Without SLIT2, axons project erronuously in ventral and medial directions. Without SLIT1 and SLIT2, axons also travel to and cross the midline. These defects occured in corticofugal, thalamocortical, and callosal tracts (Bagri et al., 2002). The corpus callosum defects were further investigated by Unni et al. (2012). In Slit2 knockout mice, defects in corpus callosum formation occurred. Axons stalled at the midline or projected aberrantly. There was no phenotype in Slit1 knockout mice and only a mild phenotype in Slit3 knockout mice, but in Slit1/Slit2 double knockout mice

SLIT2 (slit homolog 2 (Drosophila)) Brussen K
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 248
the phenotype was more severe than in Slit2 knockout mice. In addition, in both Slit2 knockout and Slit1/2 double knockout mice, there was a mispositioning of glial cells (Unni et al., 2012). In Slit1/2/3 triple knockout mice, 72% of commisural axons failed to leave the midline and 20% recrossed the midline (Long et al., 2004). Olfactory sensory neurons located in the olfactory epithelium pick up different odors and translate these odors into sensory information for the brain. Axons from the sensory neurons project into the olfactory bulb (OB) into separate units that are specific for one odor, the glomeruli. Dendrites from mitral and tufted cells transmit the information from the different glomeruli to the olfactory cortex of the brain. The olfactory system is organized into distinct regions. Odorants activate a typical pattern of glomeruli in different regions of the OB. The distinct patterning of the OB and the type and place of the activated glomeruli determine the behavior elicited by an odorant (Reviewed by Mori and Sakano, 2011). In the developing mouse and rat olfactory system, Slit2 is the first repellent expressed in the septum, Slit1 follows. From E14 to E18, both are expressed in the midline of the telencephalon including the septum. At this stage, Robo2 is expressed by tufted and mitral cells in the OB. In vitro, SLIT1 and SLIT2 repelled OB axons (Nguyen-Ba-Charvet et al., 1999; Nguyen-Ba-Charvet et al., 2002). Moreover, in Slit1 and Slit2 double KO mice, axons were not repelled by the midline and the septum. Correspondingly, the lateral olfactory tract (LOT) was increased in size. No defects were found in Slit1 or Slit2 single KO mice (Nguyen-Ba-Charvet et al., 2002). Sensory neurons in the visual pathway were also affected by Slit factors. In Slit1/Slit2 knockout mice a second optic chiasm was formed with aberrantly projecting axons (Plump et al., 2002). Kidney development: Development of the kidney is initiated by the Wolfferian duct, which forms the ureteric bud. The ureteric bud branches and further develops in the ureters and the kidney. An important growth factor during bud development is the TGF-β family member glial cell-line-derived neurotrophic factor (GDNF), which signals through a receptor kinase, RET and is restricted to the site of ureteric bud development. Defects in ureteric bud development can result in malformation of the kidney or ureters, renal agenesis or a reduced number of nephrons (Costantini and Shakya, 2006). In Slit2 mutant embryos, GDNF expression was not restricted to the site of bud development and an additional ureteric bud developed. This resulted in the development of two or more ureters or kidneys at the same site. The ureters failed to connect to the bladder and the collecting ducts and the ureter were dilated. Later during development, in some of the embryos the kidneys fused. Nephron formation
was expanded from the periphery to the interior of the kidney. Consequently, the mice did not survive after birth (Grieshammer et al., 2004). Migration: SLITs not only regulate migration and differentiation during embryogenesis, but also during adult life. SLIT2 has been shown to inhibit the chemotaxis of peripheral blood mononuclear cells, leukocytes, neutrophils, macrophages, lymphocytes and dendritic cells both in vitro and in vivo (Wu et al., 2001; Guan et al., 2003; Chen et al., 2004; Kanellis et al., 2004; Prasad et al., 2007; Tole et al., 2009; Ye et al., 2010), while it enhanced the chemotaxis of eosinophils in vivo (Ye et al., 2010). In some cell types, such as endothelial cells, the response to SLIT2 is more variable (Wang et al., 2003; Kaur et al., 2008). The differential response of cells to SLIT2 may be explained in part by cell-specific downstream signaling cues. Ye et al. have shown that the level of the SLIT-ROBO GTPase activating protein 1 is lower in eosinophils than in neutrophils. As a consequence, CDC42 and PI3K are activated in eosinophils, resulting in enhanced chemotaxis, whereas CDC42 is inactivated in neutrophils, leading to inhibition of chemotaxis (Ye et al., 2010). Osteoblast differentiation: SLIT2 has also been implicated in the regulation of osteoblast differentiation. Sun et al. (2009) reported that osteogenic differentiation was inhibited by SLIT2 in vitro (Sun et al., 2009).
Homology A single slit gene was isolated in invertebrates, whereas there are three SLIT genes in mammals. The human SLIT2 protein shows 44,3 sequence homology to Drosophila Slit (Itoh et al., 1998; Brose et al., 1999), 65% homology to the human SLIT1 protein (NCBI accession BAA35184.1, NCBI protein blast) and 67% homology to the human SLIT3 protein (NCBI accession AAQ89243.1, NCBI protein blast).
Implicated in Medulloblastoma Note 86% of medulloblastoma tumors express SLIT2, which is not silenced by methylation of the CpG islands. Interestingly, administration of recombinant SLIT2 protein to medulloblastoma spheroids could inhibit tumor cell invasion without affecting cell direction or proliferation. Treatment with SLIT2 conditioned media resulted in reduced CDC42 activation and moderately reduced Rac1 activation. There was no effect on RhoA activity (Werbowetski-Ogilvie et al., 2006).
Glioma Note Dallol et al. (2003) detected methylation of the CpG islands in the SLIT2 promoter in 59% of gliomas and in

SLIT2 (slit homolog 2 (Drosophila)) Brussen K
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 249
71% of the tested glioma cell lines. 66,7% of the gliomas were classified as glioblastoma multiforme, the most malignant stage, the rest was randomly collected. Promoter methylation correlated with reduced SLIT2 expression. Exogenous expression of SLIT2 in a methylated glioma cell line suppressed tumor growth in a colony formation assay (Dallol et al., 2003). In another study, administration of SLIT2 to glioma spheroids of a non-methylated cell line did not affect tumor invasion (Werbowetski-Ogilvie et al., 2006).
Colorectal carcinoma Note In 72% of colorectal carcinomas, methylation of the SLIT2 promoter was found, which correlated with abolished expression of SLIT2. Exogenous expression of SLIT2 in two methylated colorectal tumor cell lines suppressed tumor growth in a colony formation assay. In addition, administration of SLIT2 conditioned medium suppressed tumor growth and induced apoptosis in a methylated colorectal tumor cell line (Dallol et al., 2003). In contrast, Wang et al. (2003) reported that SLIT2 expression was not detected in normal and hyperplastic colon tissues, was moderately detected in colon adenomas (21,4%) and was upregulated in 64,6% of colon carcinomas. SLIT2 expression appeared to be enhanced in areas of high tumor cell and vessel density. Blood vessel growth and tumor volume increased in athymic nude mice injected with melanoma cells that overexpress SLIT2, but it was not investigated whether this occurs in colon cancer cells (Wang et al., 2003).
Lung cancer Note Methylation of the SLIT2 promoter was found in 53% of primary non-small cell lung cancer (NSCLC) tumors, in 77% of NSCLC cell lines, in 36% of primary small cell lung cancer (SCLC) tumors and in 55% of SCLC cell lines (Dallol et al., 2002; Tseng et al., 2010), which correlated with reduced SLIT2 expression (Tseng et al., 2010). Furthermore, low SLIT2 expression correlated with a poor prognosis and lower disease-free survival rates in NSCLC patients. For patients with metastasis, the poor survival rate correlated not only with low SLIT2 expression but also with accumulation of β-catenin (Tseng et al., 2010). When SLIT2 was re-expressed after treatment with demethylating agents, the migration of lung cancer cells decreased, while β-catenin-E-cadherin association was enhanced. Moreover, migration was also inhibited when the cells were treated with SLIT2 conditioned media or after exogenous expression of SLIT2 in the cells. Besides reduced migration, these cells had increased E-cadherin and decreased SNAI1 levels. On the contrary, after knock down of SLIT2, migration was increased while cell adhesion was reduced. Reduced cell adhesion was
attributed to deregulation of β-catenin and E-cadherin/ SNAI1 (Tseng et al., 2010).
Breast cancer Note Methylation of the SLIT2 promoter was discovered in 43% of breast cancer primary tumors and in 59% of breast cancer cell lines, which correlated with reduced SLIT2 expression in these tumors. In breast tumor cell lines, both overexpression of SLIT2 and treatment with SLIT2 conditioned media suppressed tumor cell growth in an in vitro colony assay (Dallol et al., 2002). Overexpression of SLIT2 also reduced proliferation and migration in vitro (Prasad et al., 2008). There was no effect on breast cancer cell lines with a normal expression level of SLIT2 (Dallol et al., 2002). In mice injected with SLIT2 overexpressing MCF-7 breast cancer cells, tumor size was reduced (Prasad et al., 2008). The tumor-suppressive effect of SLIT2 may be mediated via 2 different mechanisms, namely through the modulation of β-catenin signaling and through the modulation of CXCL12/ CXCR4-signaling. Overexpression of SLIT2 resulted in increased cell-cell adhesion due to enhanced β-catenin-E-cadherin association. This was combined with decreased expression of β-catenin and target genes of β-catenin, decreased nuclear translocation of β-catenin and increased expression of E-cadherin (Prasad et al., 2008). Several studies found a correlation between CXCR4 and SLIT expression in breast cancer cells. Loss of SLIT2 and SLIT3 expression correlated with the upregulation of CXCR4 and resulted in hyperplastic lesions and in desmoplastic stroma in mouse mammary gland and in human MCF7 breast cancer cells. Overexpression of SLIT2 or SLIT3 in human breast carcinoma MDA-MB-231 cells resulted in a down-regulation of CXCR4 expression, reduced colony formation in vitro and in inhibition of tumor growth in a xenograft model in vivo. Furthermore, SLIT2 addition inhibited CXCL12/ CXCR4-induced breast cancer cell chemotaxis, chemo-invasion and adhesion. SLIT2 affects CXCR4 signaling via the inhibition of CXCL12-induced phosporylation of focal adhesion components and the inhibition of PI3K, MAPK and metallo-protease activity (Prasad et al., 2004, Marlow et al., 2008).
Melanoma Note SLIT2 was expressed in 42,9% of melanomas, while SLIT2 was not expressed in nearby normal tissue. Blood vessel growth and tumor volume increased in athymic nude mice injected with melanoma cells that overexpress SLIT2, while both were decreased when SLIT-ROBO signaling was inhibited by ectopic expression of ROBON, an extracellular fragment which

SLIT2 (slit homolog 2 (Drosophila)) Brussen K
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 250
contains the SLIT-binding domain of ROBO1 and functions as scavenger for SLIT proteins, or the use of R5, an antibody directed against the SLIT-binding domain of ROBO1 (Wang et al., 2003). Lymphatic metastasis of pancreatic islet tumors Note Administration of SLIT2 to human lymphatic endothelial cells (hLEC) resulted in enhanced tubular formation and in increased migration, while there was no effect on the proliferation of the cells. Overexpression of Slit2 resulted in an increase of lymphatic vessel length in the pancreas. When Slit2 transgenic mice were crossed with the non-metastatic pancreatic islet tumor mouse line RIP1-Tag2, tumor lymphangiogenesis was increased and metastases in the regional mesentheric lymph nodes and in the intestines were found. Although some metastases were found in control mice, metastasis formation was higher in mice that overexpress Slit2 and overall survival was decreased (Yang et al., 2010).
References Anderson KV, Nüsslein-Volhard C. Information for the dorsal--ventral pattern of the Drosophila embryo is stored as maternal mRNA. Nature. 1984 Sep 20-26;311(5983):223-7
Rothberg JM, Artavanis-Tsakonas S. Modularity of the slit protein. Characterization of a conserved carboxy-terminal sequence in secreted proteins and a motif implicated in extracellular protein interactions. J Mol Biol. 1992 Sep 20;227(2):367-70
Seeger M, Tear G, Ferres-Marco D, Goodman CS. Mutations affecting growth cone guidance in Drosophila: genes necessary for guidance toward or away from the midline. Neuron. 1993 Mar;10(3):409-26
Holmes GP, Negus K, Burridge L, Raman S, Algar E, Yamada T, Little MH. Distinct but overlapping expression patterns of two vertebrate slit homologs implies functional roles in CNS development and organogenesis. Mech Dev. 1998 Dec;79(1-2):57-72
Itoh A, Miyabayashi T, Ohno M, Sakano S. Cloning and expressions of three mammalian homologues of Drosophila slit suggest possible roles for Slit in the formation and maintenance of the nervous system. Brain Res Mol Brain Res. 1998 Nov 20;62(2):175-86
Brose K, Bland KS, Wang KH, Arnott D, Henzel W, Goodman CS, Tessier-Lavigne M, Kidd T. Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell. 1999 Mar 19;96(6):795-806
Hohenester E, Tisi D, Talts JF, Timpl R. The crystal structure of a laminin G-like module reveals the molecular basis of alpha-dystroglycan binding to laminins, perlecan, and agrin. Mol Cell. 1999 Nov;4(5):783-92
Kidd T, Bland KS, Goodman CS. Slit is the midline repellent for the robo receptor in Drosophila. Cell. 1999 Mar 19;96(6):785-94
Liang Y, Annan RS, Carr SA, Popp S, Mevissen M, Margolis RK, Margolis RU. Mammalian homologues of the Drosophila slit protein are ligands of the heparan sulfate proteoglycan glypican-1 in brain. J Biol Chem. 1999 Jun 18;274(25):17885-92
Nguyen Ba-Charvet KT, Brose K, Marillat V, Kidd T, Goodman CS, Tessier-Lavigne M, Sotelo C, Chédotal A. Slit2-Mediated chemorepulsion and collapse of developing forebrain axons. Neuron. 1999 Mar;22(3):463-73
Wang KH, Brose K, Arnott D, Kidd T, Goodman CS, Henzel W, Tessier-Lavigne M. Biochemical purification of a mammalian slit protein as a positive regulator of sensory axon elongation and branching. Cell. 1999 Mar 19;96(6):771-84
Holmes G, Niswander L. Expression of slit-2 and slit-3 during chick development. Dev Dyn. 2001 Oct;222(2):301-7
Hu H. Cell-surface heparan sulfate is involved in the repulsive guidance activities of Slit2 protein. Nat Neurosci. 2001 Jul;4(7):695-701
Patel K, Nash JA, Itoh A, Liu Z, Sundaresan V, Pini A. Slit proteins are not dominant chemorepellents for olfactory tract and spinal motor axons. Development. 2001 Dec;128(24):5031-7
Ronca F, Andersen JS, Paech V, Margolis RU. Characterization of Slit protein interactions with glypican-1. J Biol Chem. 2001 Aug 3;276(31):29141-7
Vargesson N, Luria V, Messina I, Erskine L, Laufer E. Expression patterns of Slit and Robo family members during vertebrate limb development. Mech Dev. 2001 Aug;106(1-2):175-80
Wu JY, Feng L, Park HT, Havlioglu N, Wen L, Tang H, Bacon KB, Jiang Zh, Zhang Xc, Rao Y. The neuronal repellent Slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature. 2001 Apr 19;410(6831):948-52
Bagri A, Marín O, Plump AS, Mak J, Pleasure SJ, Rubenstein JL, Tessier-Lavigne M. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 2002 Jan 17;33(2):233-48
Dallol A, Da Silva NF, Viacava P, Minna JD, Bieche I, Maher ER, Latif F. SLIT2, a human homologue of the Drosophila Slit2 gene, has tumor suppressor activity and is frequently inactivated in lung and breast cancers. Cancer Res. 2002 Oct 15;62(20):5874-80
Gilthorpe JD, Papantoniou EK, Chédotal A, Lumsden A, Wingate RJ. The migration of cerebellar rhombic lip derivatives. Development. 2002 Oct;129(20):4719-28
Little M, Rumballe B, Georgas K, Yamada T, Teasdale RD. Conserved modularity and potential for alternate splicing in mouse and human Slit genes. Int J Dev Biol. 2002;46(4):385-91
Nguyen-Ba-Charvet KT, Chédotal A. Role of Slit proteins in the vertebrate brain. J Physiol Paris. 2002 Jan-Mar;96(1-2):91-8
Nguyen-Ba-Charvet KT, Plump AS, Tessier-Lavigne M, Chedotal A. Slit1 and slit2 proteins control the development of the lateral olfactory tract. J Neurosci. 2002 Jul 1;22(13):5473-80
Plump AS, Erskine L, Sabatier C, Brose K, Epstein CJ, Goodman CS, Mason CA, Tessier-Lavigne M. Slit1 and Slit2 cooperate to prevent premature midline crossing of retinal axons in the mouse visual system. Neuron. 2002 Jan 17;33(2):219-32
Dallol A, Krex D, Hesson L, Eng C, Maher ER, Latif F. Frequent epigenetic inactivation of the SLIT2 gene in gliomas. Oncogene. 2003 Jul 17;22(29):4611-6
Dallol A, Morton D, Maher ER, Latif F. SLIT2 axon guidance molecule is frequently inactivated in colorectal cancer and suppresses growth of colorectal carcinoma cells. Cancer Res. 2003 Mar 1;63(5):1054-8

SLIT2 (slit homolog 2 (Drosophila)) Brussen K
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 251
Guan H, Zu G, Xie Y, Tang H, Johnson M, Xu X, Kevil C, Xiong WC, Elmets C, Rao Y, Wu JY, Xu H. Neuronal repellent Slit2 inhibits dendritic cell migration and the development of immune responses. J Immunol. 2003 Dec 15;171(12):6519-26
Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003 Nov 7;302(5647):1044-6
Liu J, Zhang L, Wang D, Shen H, Jiang M, Mei P, Hayden PS, Sedor JR, Hu H. Congenital diaphragmatic hernia, kidney agenesis and cardiac defects associated with Slit3-deficiency in mice. Mech Dev. 2003 Sep;120(9):1059-70
Wang B, Xiao Y, Ding BB, Zhang N, Yuan Xb, Gui L, Qian KX, Duan S, Chen Z, Rao Y, Geng JG. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell. 2003 Jul;4(1):19-29
Yuan W, Rao Y, Babiuk RP, Greer JJ, Wu JY, Ornitz DM. A genetic model for a central (septum transversum) congenital diaphragmatic hernia in mice lacking Slit3. Proc Natl Acad Sci U S A. 2003 Apr 29;100(9):5217-22
Astuti D, Da Silva NF, Dallol A, Gentle D, Martinsson T, Kogner P, Grundy R, Kishida T, Yao M, Latif F, Maher ER. SLIT2 promoter methylation analysis in neuroblastoma, Wilms' tumour and renal cell carcinoma. Br J Cancer. 2004 Jan 26;90(2):515-21
Chen B, Blair DG, Plisov S, Vasiliev G, Perantoni AO, Chen Q, Athanasiou M, Wu JY, Oppenheim JJ, Yang D. Cutting edge: bone morphogenetic protein antagonists Drm/Gremlin and Dan interact with Slits and act as negative regulators of monocyte chemotaxis. J Immunol. 2004 Nov 15;173(10):5914-7
Dickinson RE, Dallol A, Bieche I, Krex D, Morton D, Maher ER, Latif F. Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br J Cancer. 2004 Dec 13;91(12):2071-8
Grieshammer U, Le Ma, Plump AS, Wang F, Tessier-Lavigne M, Martin GR. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell. 2004 May;6(5):709-17
Kanellis J, Garcia GE, Li P, Parra G, Wilson CB, Rao Y, Han S, Smith CW, Johnson RJ, Wu JY, Feng L. Modulation of inflammation by slit protein in vivo in experimental crescentic glomerulonephritis. Am J Pathol. 2004 Jul;165(1):341-52
Long H, Sabatier C, Ma L, Plump A, Yuan W, Ornitz DM, Tamada A, Murakami F, Goodman CS, Tessier-Lavigne M. Conserved roles for Slit and Robo proteins in midline commissural axon guidance. Neuron. 2004 Apr 22;42(2):213-23
Prasad A, Fernandis AZ, Rao Y, Ganju RK. Slit protein-mediated inhibition of CXCR4-induced chemotactic and chemoinvasive signaling pathways in breast cancer cells. J Biol Chem. 2004 Mar 5;279(10):9115-24
Steigemann P, Molitor A, Fellert S, Jäckle H, Vorbrüggen G. Heparan sulfate proteoglycan syndecan promotes axonal and myotube guidance by slit/robo signaling. Curr Biol. 2004 Feb 3;14(3):225-30
Dallol A, Dickinson RE, Latif F. DNA Methylation, Epigenetics and Metastasis. Series: Cancer Metastasis - Biology and Treatment, Vol. 7, DNA Methylation, Epigenetics and Metastasis, 191-214. Esteller, Manel (Ed.) 2005, XII, 310 p.
Costantini F, Shakya R.. GDNF/Ret signaling and the development of the kidney. Bioessays. 2006 Feb;28(2):117-27. (REVIEW)
Dickson BJ, Gilestro GF.. Regulation of commissural axon pathfinding by slit and its Robo receptors. Annu Rev Cell Dev Biol. 2006;22:651-75. (REVIEW)
Hussain SA, Piper M, Fukuhara N, Strochlic L, Cho G, Howitt JA, Ahmed Y, Powell AK, Turnbull JE, Holt CE, Hohenester E..
A molecular mechanism for the heparan sulfate dependence of slit-robo signaling. J Biol Chem. 2006 Dec 22;281(51):39693-8. Epub 2006 Oct 24.
Werbowetski-Ogilvie TE, Seyed Sadr M, Jabado N, Angers-Loustau A, Agar NY, Wu J, Bjerkvig R, Antel JP, Faury D, Rao Y, Del Maestro RF.. Inhibition of medulloblastoma cell invasion by Slit. Oncogene. 2006 Aug 24;25(37):5103-12. Epub 2006 Apr 24.
Prasad A, Qamri Z, Wu J, Ganju RK.. Slit-2/Robo-1 modulates the CXCL12/CXCR4-induced chemotaxis of T cells. J Leukoc Biol. 2007 Sep;82(3):465-76. Epub 2007 Jun 12.
Andrews W, Barber M, Hernadez-Miranda LR, Xian J, Rakic S, Sundaresan V, Rabbitts TH, Pannell R, Rabbitts P, Thompson H, Erskine L, Murakami F, Parnavelas JG.. The role of Slit-Robo signaling in the generation, migration and morphological differentiation of cortical interneurons. Dev Biol. 2008 Jan 15;313(2):648-58. Epub 2007 Nov 13.
Dickinson RE, Myers M, Duncan WC.. Novel regulated expression of the SLIT/ROBO pathway in the ovary: possible role during luteolysis in women. Endocrinology. 2008 Oct;149(10):5024-34. doi: 10.1210/en.2008-0204. Epub 2008 Jun 19.
Kaur S, Samant GV, Pramanik K, Loscombe PW, Pendrak ML, Roberts DD, Ramchandran R.. Silencing of directional migration in roundabout4 knockdown endothelial cells. BMC Cell Biol. 2008 Nov 3;9:61. doi: 10.1186/1471-2121-9-61.
Marlow R, Strickland P, Lee JS, Wu X, Pebenito M, Binnewies M, Le EK, Moran A, Macias H, Cardiff RD, Sukumar S, Hinck L.. SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4 within breast epithelium. Cancer Res. 2008 Oct 1;68(19):7819-27. doi: 10.1158/0008-5472.CAN-08-1357.
Prasad A, Paruchuri V, Preet A, Latif F, Ganju RK.. Slit-2 induces a tumor-suppressive effect by regulating beta-catenin in breast cancer cells. J Biol Chem. 2008 Sep 26;283(39):26624-33. doi: 10.1074/jbc.M800679200. Epub 2008 Jul 8.
Dunwell TL, Dickinson RE, Stankovic T, Dallol A, Weston V, Austen B, Catchpoole D, Maher ER, Latif F.. Frequent epigenetic inactivation of the SLIT2 gene in chronic and acute lymphocytic leukemia. Epigenetics. 2009 May 16;4(4):265-9. Epub 2009 May 1.
Tole S, Mukovozov IM, Huang YW, Magalhaes MA, Yan M, Crow MR, Liu GY, Sun CX, Durocher Y, Glogauer M, Robinson LA.. The axonal repellent, Slit2, inhibits directional migration of circulating neutrophils. J Leukoc Biol. 2009 Dec;86(6):1403-15. doi: 10.1189/jlb.0609391. Epub 2009 Sep 16.
Duncan WC, McDonald SE, Dickinson RE, Shaw JL, Lourenco PC, Wheelhouse N, Lee KF, Critchley HO, Horne AW.. Expression of the repulsive SLIT/ROBO pathway in the human endometrium and Fallopian tube. Mol Hum Reprod. 2010 Dec;16(12):950-9. doi: 10.1093/molehr/gaq055. Epub 2010 Jul 22.
Tseng RC, Lee SH, Hsu HS, Chen BH, Tsai WC, Tzao C, Wang YC.. SLIT2 attenuation during lung cancer progression deregulates beta-catenin and E-cadherin and associates with poor prognosis. Cancer Res. 2010 Jan 15;70(2):543-51. doi: 10.1158/0008-5472.CAN-09-2084. Epub 2010 Jan 12.

SLIT2 (slit homolog 2 (Drosophila)) Brussen K
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 252
Yang XM, Han HX, Sui F, Dai YM, Chen M, Geng JG.. Slit-Robo signaling mediates lymphangiogenesis and promotes tumor lymphatic metastasis. Biochem Biophys Res Commun. 2010 May 28;396(2):571-7. doi: 10.1016/j.bbrc.2010.04.152. Epub 2010 May 8.
Ye BQ, Geng ZH, Ma L, Geng JG.. Slit2 regulates attractive eosinophil and repulsive neutrophil chemotaxis through differential srGAP1 expression during lung inflammation. J Immunol. 2010 Nov 15;185(10):6294-305. doi: 10.4049/jimmunol.1001648. Epub 2010 Oct 13.
Dickinson RE, Fegan KS, Ren X, Hillier SG, Duncan WC.. Glucocorticoid regulation of SLIT/ROBO tumour suppressor genes in the ovarian surface epithelium and ovarian cancer cells. PLoS One. 2011;6(11):e27792. doi: 10.1371/journal.pone.0027792. Epub 2011 Nov 23.
Mori K, Sakano H.. How is the olfactory map formed and interpreted in the mammalian brain? Annu Rev Neurosci. 2011;34:467-99. doi: 10.1146/annurev-neuro-112210-112917. (REVIEW)
Rossignol E.. Genetics and function of neocortical GABAergic interneurons in neurodevelopmental disorders. Neural Plast. 2011;2011:649325. doi: 10.1155/2011/649325. Epub 2011 Aug 18. (REVIEW)
Condac E, Strachan H, Gutierrez-Sanchez G, Brainard B, Giese C, Heiss C, Johnson D, Azadi P, Bergmann C, Orlando R, Esmon CT, Harenberg J, Moremen K, Wang L.. The C-terminal fragment of axon guidance molecule Slit3 binds heparin and neutralizes heparin's anticoagulant activity. Glycobiology. 2012 Sep;22(9):1183-92. doi: 10.1093/glycob/cws087. Epub 2012 May 28.
Geutskens SB, Andrews WD, van Stalborch AM, Brussen K, Holtrop-de Haan SE, Parnavelas JG, Hordijk PL, van Hennik PB.. Control of human hematopoietic stem/progenitor cell migration by the extracellular matrix protein Slit3. Lab Invest. 2012 Aug;92(8):1129-39. doi: 10.1038/labinvest.2012.81. Epub 2012 May 21.
Tovar JA.. Congenital diaphragmatic hernia. Orphanet J Rare Dis. 2012 Jan 3;7:1. doi: 10.1186/1750-1172-7-1. (REVIEW)
Unni DK, Piper M, Moldrich RX, Gobius I, Liu S, Fothergill T, Donahoo AL, Baisden JM, Cooper HM, Richards LJ.. Multiple Slits regulate the development of midline glial populations and the corpus callosum. Dev Biol. 2012 May 1;365(1):36-49. doi: 10.1016/j.ydbio.2012.02.004. Epub 2012 Feb 11.
This article should be referenced as such:
Brussen K. SLIT2 (slit homolog 2 (Drosophila)). Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):245-252.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 253
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
BNIP3L (BCL2/adenovirus E1B 19kDa interacting protein 3-like) Paul Ney, Ji Zhang
Cell & Molecular Biology, New York Blood Center, 310 E 67th St, New York, NY 10065, USA (PN), Cancer Biology & Genetics, Memorial Sloan-Kettering Cancer Center, 1275 York Ave, New York, NY 10065, USA (JZ)
Published in Atlas Database: November 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/BNIP3LID823ch8p21.html DOI: 10.4267/2042/48866
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: BNIP3a, NIX
HGNC (Hugo): BNIP3L
Location: 8p21.2
Local order: Cdca2 -Ebf2 - Ppp2r2a - Bnip3l - Pnma2 - Dpysl2 - Adra1a
DNA/RNA Description The gene spans 30122 bp and has 6 exons. The cytogenetic location of the gene is 8p21.2. The genomic coordinates are 8: 26240522 - 26270643.
Transcription The mRNA is 3505 bp, and has a 657 bp open reading frame.
Protein Description The protein is 219 amino acids, with a predicted MW of 23,8 kDa. The carboxy-terminal transmembrane domain of BNIP3L has been characterized by nuclear magnetic resonance and shown to form a kinked α-helix (Bocharov et al., 2007). Structural bioinformatics
analysis indicates that the rest of the protein is mostly disordered. The LIR and the MER are predicted to form secondary structure (β-strand and α-helix, respectively) (Zhang et al., 2012).
Expression BNIP3L is ubiquitously expressed. Northern blot hybridization reveals two transcripts of 1,6 kb and 3,9 kb. These are expressed in heart, brain, placenta, lung, liver, skeletal muscle, kidney, and pancreas (Yasuda et al., 1999). In another study, transcripts were identified in heart, brain, placenta, lung (low), liver, skeletal muscle (low), kidney, pancreas, spleen, thymus, prostate, testis, ovary, small intestine, colon, and peripheral blood leukocyte (Farooq et al., 2001). In the same study, there was also expression in cancer cell lines, including promyelocytic HL-60 (low), Hela S3 (low), K562, lymphoblastic leukemia Molt-5, Burkitt's lymphoma-Raji, colorectal adenocarcinoma SW480, and lung carcinoma A549 cells. In another study, 1,4 and 4,0 kb BNIP3L transcripts were found in primary hematopoietic cells and in cell lines, including K562, Hela, and Jurkat cells (Aerbajinai et al., 2003). BNIP3L is upregulated during erythroid maturation. BNIP3L protein can form an SDS-resistant dimer, which migrates at twice its predicted MW in SDS-PAGE gels. BNIP3L dimerization is mediated through its transmembrane domain (Imazu et al., 1999).
Organization of the human BNIP3L gene. The boxes represent exons, the line introns (not drawn to scale). Black areas represent coding sequence.

BNIP3L (BCL2/adenovirus E1B 19kDa interacting protein 3-like) Ney P, Zhang J
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 254
Functional domains of BNIP3L protein. LIR: LC3-interaction region (WVEL) (Schwarten et al., 2009; Novak et al., 2010); MER: minimal essential region (DMEKILLDAQHE) (Zhang et al., 2012); BH3-like: BCL2 homology 3-like domain (LKKSADWVSDW) (Yasuda et al., 1999); TM: transmembrane domain. Localisation BNIP3L primarily localizes to the mitochondrial outer membrane (Chen et al., 1999; Imazu et al., 1999; Yasuda et al., 1999; Vande Velde et al., 2000). BNIP3L is oriented so that its amino terminus is in the cytoplasm, and its carboxy-terminal tail is in the mitochondrial intermembrane space. It also localizes to nuclear envelope, sarcoplasmic reticulum, and endoplasmic reticulum (Ohi et al., 1999; Diwan et al., 2009).
Function BNIP3L and BNIP3 can cause cell death by several mechanisms, which are mediated by their BH3-like and transmembrane domains. In cardiomyocytes, mitochondria-targeted BNIP3L causes BAX/BAK-dependent mitochondrial outer membrane permeabilization, whereas ER/SR-targeted BNIP3L causes cyclophilin D-dependent opening of the MPT pore and mitochondrial depolarization (Chen et al., 2010). In tumor cells, BNIP3 expression is associated with opening of the MPT pore and autophagy (Vande Velde et al., 2000). Another property of BNIP3L and BNIP3 is their ability to mediate mitochondrial clearance during erythroid development (Schweers et al., 2007; Sandoval et al., 2008) and in response to hypoxia (Zhang et al., 2008; Liu et al., 2012), respectively. BNIP3L mediates mitochondrial clearance in erythroid cells through its LIR (Schwarten et al., 2009; Novak et al., 2010) and MER (Zhang et al., 2012) domains.
Homology BNIP3L is conserved from zebrafish to man.
Mutations Note Not yet described.
Implicated in Various cancers Oncogenesis BNIP3L and the related protein BNIP3 (56% identical overall) are implicated in cancer progression.
BNIP3 deregulation is more often implicated in cancer than BNIP3L, but the two proteins have a similar mechanism of action, so both are potentially relevant. BNIP3 and BNIP3L are reported to function as both tumor suppressors and oncogenes. This dual nature presumably reflects the roles of BNIP3 and BNIP3L in cell death pathways and autophagy; autophagy can promote cell survival. The frequent finding of BNIP3 deregulation in cancer is likely related to its induction by hypoxia, and HIF1α signaling (Bruick, 2000). BNIP3L is also induced during hypoxia, by p53 (Fei et al., 2004). There are several mechanisms of BNIP3 and BNIP3L-induced cell death, which have recently been reviewed (Zhang and Ney, 2011). The role of BNIP3 and BNIP3L in cell death may explain their frequent deletion and silencing in tumors by promoter methylation (see below). By contrast, some advanced cancers express abnormally high levels of BNIP3 and BNIP3L. In these cases, the prosurvival role of these proteins in the induction of autophagy appears to dominate.
Prostate cancer Note BNIP3 is expressed in 95% of prostate cancer samples and is either nuclear, cytoplasmic, or both. Cytoplasmic BNIP3 expression correlates with Gleason score, but not other clinicopathological parameters. By contrast, nuclear BNIP3 correlates with HIF1α and HIF2α expression (Shaida et al., 2008). BNIP3 promoter hypermethylation is present in 16% of prostate cancers, and BNIP3 expression is decreased in 21% of prostate cancers, but the two do not correlate (Murphy et al., 2011). BNIP3L exhibits homozygous deletion in a prostate cancer cell line and primary prostate tumor (Liu et al., 2008). Another study showed LOH of BNIP3L in 5% of prostate cancers, and a correlation with increasing disease stage (Cheng et al., 2012).
Breast cancer Note BNIP3 expression in ductal carcinoma in situ is associated with higher grade, necrosis and invasive disease, whereas BNIP3L expression does not correlate with these parameters (Sowter et al., 2003). BNIP3L is not deregulated and infrequently mutated in ovarian and breast cancer (Lai et al., 2003). Loss of BNIP3

BNIP3L (BCL2/adenovirus E1B 19kDa interacting protein 3-like) Ney P, Zhang J
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 255
expression correlates with lymph node metastases and mitotic index, but not with the hypoxic response (Koop et al., 2009). Proteasome inhibition with Bortezomib blocks autophagy-mediated catabolism of long-lived proteins, and is associated with increased BNIP3 and cell death in breast cancer cell lines (Periyasamy-Thandavan et al., 2010). On the other hand, resistance to the cytotoxic effects of TNFα in a subclone of breast cancer MCF-7 cells is associated with increased BNIP3 and upregulation of the autophagy program (Moussay et al., 2011). Notably, hypoxic induction of BNIP3 and BNIP3L can cause breast cancer cell death and at the same time promote the survival of cancer-associated fibroblasts (Chiavarina et al., 2010). Thus, BNIP3 and BNIP3L may have compartment-specific effects on cell death and survival.
Colorectal and gastric cancers Note BNIP3 promoter hypermethylation is found in 66% of primary colorectal and 49% of gastric cancers, but not in adjacent normal tissue (Murai et al., 2005b). Promoter hypermethylation but not gene mutation correlates with decreased BNIP3 expression.
Pancreatic cancer Note BNIP3 is silenced by promoter hypermethylation in 80% of pancreatic adenocarcinoma samples (Okami et al., 2004). BNIP3 expression is diminished in chronic pancreatitis and pancreatic ductal adenocarcinoma, and loss of BNIP3 expression correlates with decreased survival and chemotherapy resistance (Erkan et al., 2005). Similarly, BNIP3L is reduced in liver metastases and the tumor invasion front compared with the primary pancreatic tumor, in an orthotopic SCID mouse model (Niedergethmann et al., 2007).
Liver cancer Note Epigenetic silencing of BNIP3 and BNIP3L is associated with poor prognosis in hepatocellular carcinoma (Calvisi et al., 2007). In addition, a cSNP that causes premature termination of BNIP3L was reported in 40% of hepatocellular carcinoma cases (Wang et al., 2005). BNIP3 is a HIF1α target in HepG2 tumor spheroids, and its expression is associated with increased autophagy and attenuation of apoptosis (Menrad et al., 2010).
Lung cancer Note There is strong cytoplasmic expression of BNIP3 in 38% of non-small cell lung cancer, which was associated with an aggressive phenotype and decreased survival (Giatromanolaki et al., 2004).
Malignant glioblastoma Note BNIP3 is expressed in hypoxic regions of glioblastoma
multiforme (GBM), but is sequestered in the nucleus in ~80% of tumors (Burton et al., 2006). In another study, BNIP3L appeared to act as a tumor suppressor in low-grade astrocytomas, and as an oncogene in high grade GBM. In the latter case, BNIP3L expression correlated with NFκB activation through an unknown mechanism (Lu et al., 2012).
Hematopoietic malignancy Note BNIP3 promoter hypermethylation is found in 15% of acute lymphocytic leukemia, 17% of acute myelogenous leukemia, and 21% of multiple myeloma. Promoter hypermethylation correlates with decreased BNIP3 expression (Murai et al., 2005a). BNIP3 promoter hypermethylation correlates with decreased survival in multiple myeloma (Heller et al., 2008). Another study found BNIP3 promoter hypermethylation in 13% of newly diagnosed multiple myeloma but no association with prognosis (Braggio et al., 2010).
Ischemic and hypertrophic heart disease Note Most of the evidence that BNIP3 and BNIP3L have a role in heart disease comes from animal models. BNIP3 is regulated by hypoxia in cardiomyocytes through HIF1α binding sites in its promoter (Bruick, 2000). By contrast, BNIP3L is regulated by Gαq signaling in the setting of cardiac hypertrophy (Gálvez et al., 2006). Enforced expression of BNIP3L causes lethal cardiomyopathy in mice, whereas BNIP3L deficiency protects mice from Gαq-mediated and pressure overload cardiomyopathy (Yussman et al., 2002; Diwan et al., 2008). Further, BNIP3 deficiency protects against post-infarction ventricular remodeling (Diwan et al., 2007). Mice with combined deficiency of BNIP3 and BNIP3L in the heart develop normally, but by 30 weeks exhibit cardiac enlargement and decreased left ventricular ejection fraction (Dorn, 2010). Mitochondria in the hearts of these mice are increased in number and show variation in size and internal structure. Furthermore, young BNIP3/BNIP3L-deficient mice subjected to aortic banding rapidly develop heart failure.
Cerebral ischemia Note Animal models and in vitro studies also provide evidence that BNIP3 and to a lesser extent BNIP3L are a cause of neuronal cell death after hypoxia or denervation. BNIP3 is expressed in striatal and cortical neurons following transient focal ischemia in rats; prolonged BNIP3 expression in this setting is associated with delayed neuronal cell death (Althaus et al., 2006). BNIP3 knockdown inhibits nuclear translocation of EndoG and protects against hypoxia-induced, caspase-independent, delayed neuronal cell

BNIP3L (BCL2/adenovirus E1B 19kDa interacting protein 3-like) Ney P, Zhang J
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 256
death (Zhang et al., 2007). Hypoxic mimetics cause BAX/BAK- and caspase-dependent neuronal precursor cell death in vitro, but also cause HIF1α and BNIP3 upregulation. BNIP3 knockdown failed to prevent caspase activation, but inhibits nuclear translocation of apoptosis-inducing factor and cell death (Walls et al., 2009). Thus, BNIP3 mediates hypoxia-induced, caspase-independent neuronal cell death. Also, following neonatal nerve axotomy, BNIP3 and to a lesser extent BNIP3L, are induced in facial motoneurons and associated with cell death (Cho et al., 2012).
Breakpoints Note Not yet described.
References Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, D'Sa-Eipper C, Chinnadurai G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell. 1994 Oct 21;79(2):341-51
Matsushima M, Fujiwara T, Takahashi E, Minaguchi T, Eguchi Y, Tsujimoto Y, Suzumori K, Nakamura Y. Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes Chromosomes Cancer. 1998 Mar;21(3):230-5
Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, Shi L, Dubik D, Greenberg A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem. 1999 Jan 1;274(1):7-10
Imazu T, Shimizu S, Tagami S, Matsushima M, Nakamura Y, Miki T, Okuyama A, Tsujimoto Y. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene. 1999 Aug 12;18(32):4523-9
Ohi N, Tokunaga A, Tsunoda H, Nakano K, Haraguchi K, Oda K, Motoyama N, Nakajima T. A novel adenovirus E1B19K-binding protein B5 inhibits apoptosis induced by Nip3 by forming a heterodimer through the C-terminal hydrophobic region. Cell Death Differ. 1999 Apr;6(4):314-25
Yasuda M, Han JW, Dionne CA, Boyd JM, Chinnadurai G. BNIP3alpha: a human homolog of mitochondrial proapoptotic protein BNIP3. Cancer Res. 1999 Feb 1;59(3):533-7
Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000 Aug 1;97(16):9082-7
Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000 Aug;20(15):5454-68
Farooq M, Kim Y, Im S, Chung E, Hwang S, Sohn M, Kim M, Kim J. Cloning of BNIP3h, a member of proapoptotic BNIP3 family genes. Exp Mol Med. 2001 Sep 30;33(3):169-73
Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, Aronow BJ, Lorenz JN, Dorn GW 2nd. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med. 2002 Jul;8(7):725-30
Aerbajinai W, Giattina M, Lee YT, Raffeld M, Miller JL. The proapoptotic factor Nix is coexpressed with Bcl-xL during terminal erythroid differentiation. Blood. 2003 Jul 15;102(2):712-7
Lai J, Flanagan J, Phillips WA, Chenevix-Trench G, Arnold J. Analysis of the candidate 8p21 tumour suppressor, BNIP3L, in breast and ovarian cancer. Br J Cancer. 2003 Jan 27;88(2):270-6
Sowter HM, Ferguson M, Pym C, Watson P, Fox SB, Han C, Harris AL. Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J Pathol. 2003 Dec;201(4):573-80
Fei P, Wang W, Kim SH, Wang S, Burns TF, Sax JK, Buzzai M, Dicker DT, McKenna WG, Bernhard EJ, El-Deiry WS. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell. 2004 Dec;6(6):597-609
Giatromanolaki A, Koukourakis MI, Sowter HM, Sivridis E, Gibson S, Gatter KC, Harris AL. BNIP3 expression is linked with hypoxia-regulated protein expression and with poor prognosis in non-small cell lung cancer. Clin Cancer Res. 2004 Aug 15;10(16):5566-71
Okami J, Simeone DM, Logsdon CD. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. 2004 Aug 1;64(15):5338-46
Erkan M, Kleeff J, Esposito I, Giese T, Ketterer K, Büchler MW, Giese NA, Friess H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene. 2005 Jun 23;24(27):4421-32
Murai M, Toyota M, Satoh A, Suzuki H, Akino K, Mita H, Sasaki Y, Ishida T, Shen L, Garcia-Manero G, Issa JP, Hinoda Y, Tokino T, Imai K. Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br J Cancer. 2005a Mar 28;92(6):1165-72
Murai M, Toyota M, Suzuki H, Satoh A, Sasaki Y, Akino K, Ueno M, Takahashi F, Kusano M, Mita H, Yanagihara K, Endo T, Hinoda Y, Tokino T, Imai K. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin Cancer Res. 2005b Feb 1;11(3):1021-7
Wang J, Ni H, Chen L, Liu YX, Chen CB, Song WQ. Preparation and analysis of cSNP chip on hepatocellular carcinoma-related genes. Hepatobiliary Pancreat Dis Int. 2005 Aug;4(3):398-402
Althaus J, Bernaudin M, Petit E, Toutain J, Touzani O, Rami A. Expression of the gene encoding the pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible factor-1alpha (HIF-1alpha) protein following focal cerebral ischemia in rats. Neurochem Int. 2006 Jun;48(8):687-95
Burton TR, Henson ES, Baijal P, Eisenstat DD, Gibson SB. The pro-cell death Bcl-2 family member, BNIP3, is localized to the nucleus of human glial cells: Implications for glioblastoma multiforme tumor cell survival under hypoxia. Int J Cancer. 2006 Apr 1;118(7):1660-9
Gálvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, Dorn GW 2nd. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J Biol Chem. 2006 Jan 20;281(3):1442-8
Bocharov EV, Pustovalova YE, Pavlov KV, Volynsky PE, Goncharuk MV, Ermolyuk YS, Karpunin DV, Schulga AA, Kirpichnikov MP, Efremov RG, Maslennikov IV, Arseniev AS. Unique dimeric structure of BNip3 transmembrane domain suggests membrane permeabilization as a cell death trigger. J Biol Chem. 2007 Jun 1;282(22):16256-66

BNIP3L (BCL2/adenovirus E1B 19kDa interacting protein 3-like) Ney P, Zhang J
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 257
Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA, Schroeder I, Factor VM, Thorgeirsson SS. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007 Sep;117(9):2713-22
Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007 Oct;117(10):2825-33
Niedergethmann M, Alves F, Neff JK, Heidrich B, Aramin N, Li L, Pilarsky C, Grützmann R, Allgayer H, Post S, Gretz N. Gene expression profiling of liver metastases and tumour invasion in pancreatic cancer using an orthotopic SCID mouse model. Br J Cancer. 2007 Nov 19;97(10):1432-40
Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007 Dec 4;104(49):19500-5
Zhang Z, Yang X, Zhang S, Ma X, Kong J. BNIP3 upregulation and EndoG translocation in delayed neuronal death in stroke and in hypoxia. Stroke. 2007 May;38(5):1606-13
Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW 2nd. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation. 2008 Jan 22;117(3):396-404
Heller G, Schmidt WM, Ziegler B, Holzer S, Müllauer L, Bilban M, Zielinski CC, Drach J, Zöchbauer-Müller S. Genome-wide transcriptional response to 5-aza-2'-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008 Jan 1;68(1):44-54
Liu W, Xie CC, Zhu Y, Li T, Sun J, Cheng Y, Ewing CM, Dalrymple S, Turner AR, Sun J, Isaacs JT, Chang BL, Zheng SL, Isaacs WB, Xu J. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia. 2008 Aug;10(8):897-907
Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008 Jul 10;454(7201):232-5
Shaida N, Launchbury R, Boddy JL, Jones C, Campo L, Turley H, Kanga S, Banham AH, Malone PR, Harris AL, Fox SB. Expression of BNIP3 correlates with hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha and the androgen receptor in prostate cancer and is regulated directly by hypoxia but not androgens in cell lines. Prostate. 2008 Feb 15;68(3):336-43
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008 Apr 18;283(16):10892-903
Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, Molkentin JD, Kranias EG, Dorn GW 2nd. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest. 2009 Jan;119(1):203-12
Koop EA, van Laar T, van Wichen DF, de Weger RA, Wall Ev, van Diest PJ. Expression of BNIP3 in invasive breast cancer: correlations with the hypoxic response and clinicopathological features. BMC Cancer. 2009 Jun 9;9:175
Schwarten M, Mohrlüder J, Ma P, Stoldt M, Thielmann Y, Stangler T, Hersch N, Hoffmann B, Merkel R, Willbold D. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy. 2009 Jul;5(5):690-8
Walls KC, Ghosh AP, Ballestas ME, Klocke BJ, Roth KA. bcl-2/Adenovirus E1B 19-kd interacting protein 3 (BNIP3) regulates hypoxia-induced neural precursor cell death. J Neuropathol Exp Neurol. 2009 Dec;68(12):1326-38
Braggio E, Maiolino A, Gouveia ME, Magalhães R, Souto Filho JT, Garnica M, Nucci M, Renault IZ. Methylation status of nine tumor suppressor genes in multiple myeloma. Int J Hematol. 2010 Jan;91(1):87-96
Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW 2nd. Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci U S A. 2010 May 18;107(20):9035-42
Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, Tanowitz HB, Casimiro MC, Wang C, Pestell RG, Grieshaber P, Caro J, Sotgia F, Lisanti MP. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010 Sep 1;9(17):3534-51
Dorn GW 2nd. Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. 2010 Aug;3(4):374-83
Menrad H, Werno C, Schmid T, Copanaki E, Deller T, Dehne N, Brüne B. Roles of hypoxia-inducible factor-1alpha (HIF-1alpha) versus HIF-2alpha in the survival of hepatocellular tumor spheroids. Hepatology. 2010 Jun;51(6):2183-92
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dötsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010 Jan;11(1):45-51
Periyasamy-Thandavan S, Jackson WH, Samaddar JS, Erickson B, Barrett JR, Raney L, Gopal E, Ganapathy V, Hill WD, Bhalla KN, Schoenlein PV. Bortezomib blocks the catabolic process of autophagy via a cathepsin-dependent mechanism, affects endoplasmic reticulum stress and induces caspase-dependent cell death in antiestrogen-sensitive and resistant ER+ breast cancer cells. Autophagy. 2010 Jan;6(1):19-35
Moussay E, Kaoma T, Baginska J, Muller A, Van Moer K, Nicot N, Nazarov PV, Vallar L, Chouaib S, Berchem G, Janji B. The acquisition of resistance to TNFα in breast cancer cells is associated with constitutive activation of autophagy as revealed by a transcriptome analysis using a custom microarray. Autophagy. 2011 Jul;7(7):760-70
Murphy TM, Sullivan L, Lane C, O'Connor L, Barrett C, Hollywood D, Lynch T, Lawler M, Perry AS. In silico analysis and DHPLC screening strategy identifies novel apoptotic gene targets of aberrant promoter hypermethylation in prostate cancer. Prostate. 2011 Jan 1;71(1):1-17
Zhang J, Ney PA. Mechanisms and biology of B-cell leukemia/lymphoma 2/adenovirus E1B interacting protein 3 and Nip-like protein X. Antioxid Redox Signal. 2011 May 15;14(10):1959-69
Cheng I, Levin AM, Tai YC, Plummer S, Chen GK, Neslund-Dudas C, Casey G, Rybicki BA, Witte JS. Copy number alterations in prostate tumors and disease aggressiveness. Genes Chromosomes Cancer. 2012 Jan;51(1):66-76
Cho B, Choi SY, Park OH, Sun W, Geum D. Differential expression of BNIP family members of BH3-only proteins during the development and after axotomy in the rat. Mol Cells. 2012 Jun;33(6):605-10

BNIP3L (BCL2/adenovirus E1B 19kDa interacting protein 3-like) Ney P, Zhang J
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 258
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012 Jan 22;14(2):177-85
Lu Y, Wang L, He M, Huang W, Li H, Wang Y, Kong J, Qi S, Ouyang J, Qiu X. Nix protein positively regulates NF-κB activation in gliomas. PLoS One. 2012;7(9):e44559
Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, Ney PA. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy. 2012 Sep;8(9):1325-32
This article should be referenced as such:
Ney P, Zhang J. BNIP3L (BCL2/adenovirus E1B 19kDa interacting protein 3-like). Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):253-258.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 259
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
LPAR2 (lysophosphatidic acid receptor 2) Sara Knowlden, Steve Georas
Department of Microbiology and Immunology, University of Rochester School of Medicine and Dentistry, Rochester, NY 14642, USA (SK), Department of Pulmonary and Critical Care Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY 14642, USA (SG)
Published in Atlas Database: November 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/LPAR2ID40406ch19p13.html DOI: 10.4267/2042/48867
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: EDG-4, EDG4, LPA-2, LPA2
HGNC (Hugo): LPAR2
Location: 19p13.11
Note Found on human chromosome 19p12 (GeneBank Accession number AC002306) and mouse chromosome 8 (Contos and Chun, 2000).
DNA/RNA Description Both human and mouse LPA2 genes are present as a single copy and are divided among three exons with start and stop sites in the second and third exons, respectively (Contos and Chun, 2000). Introns are
located upstream of the start codon and separate the coding region from the transmembrane domain VI. As seen by Northern blot analysis, there are two transcripts sizes for both human and mouse LPA2. In human, the transcript sizes are ~1.8kb and ~10kb and in mouse the transcript sizes are ~3kb and ~7kb (An et al., 1998).
Protein Note Human LPA2 encodes a protein with a predicted 351 amino acid residues and molecular weight of 39.1 kDa. There is 90.8% sequence homology between mouse and human LPA2 amino acid sequences and 60% amino acid similarity with LPA1. Mouse LPA2 encodes a protein with a predicted 348 amino acids and molecular weight of 38.9 kDa.

LPAR2 (lysophosphatidic acid receptor 2) Knowlden S, Georas S
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 260
LPA2 is a G protein-coupled receptor (GPCR) that spans the plasma membrane seven times, hence having three extracellular and three intracellular loops. The C-terminus of LPA contains a di-leucine motif and several putative palmitoylated cysteine residues that associate with LIM-domain containing TRIP6 (Thyroid Hormone Receptor-Interacting Protein 6) and Siva-1 protein. LPA-dependent recruitment of TRIP6 to the plasma membrane promotes its phosphorylation and targeting to focal adhesions, and leads to cell adhesion and migration. SIVA-1 gets ubiquitinated in an LPA-dependent manner, leading to its degradation and subsequent decrease in its pro-apoptotic abilities. The last four amino acids at the C-terminus (DSTL) contain a PDZ-binding motif and interact with NHERF2 and MAGI-3. NHERF2 clusters LPA2 and PLC-β, which then signals downstream IP3-dependent Ca++ mobilization and DAG-dependent PKC activation. MAGI-3 has been shown to interact with LPA2 and regulate the activation of Erk and RhoA, leading to cell migration. MAGI-3 has also been shown to reciprocally regulate PLC-β and inhibit NHERF2-promoted tumor cell migration and invasion (Lee et al., 2011). LPA2 also couples to Gαi, Gα12/13, and Gαq, activating downstream signaling pathways that lead to cell survival, proliferation, and motility.
Description LPA2 is a G-protein coupled receptor (GPCR) that belongs to the endothelial differentiation gene (Edg) family of receptors. It was first identified in 1998 following a search in the GenBank for homologs to human EDG2 (LPA1) (An et al., 1998; Contos and Chun, 1998).
Expression LPA2 has a more restricted expression pattern than that of LPA1. More information is currently available for LPA2 mRNA expression than protein expression, and there is a current need for well-validated LPA2-specific antibodies. Human: LPA2 mRNA is expressed in a variety of tissues including human testis, leukocytes, prostate,
spleen, thymus, pancreas, and bone marrow (An et al., 1998; Fang et al., 2002). The expression of LPA2 has also been noted in freshly isolated human blood CD4+ T cells, B cells, and Jurkat T cells (Zheng et al., 2000; Goetzl et al., 2000; Rubenfeld et al., 2006) as well as monocyte-derived dendritic cells (Chen et al., 2006; Oz-Arslan et al., 2006). Interestingly, Zheng et al. reported that LPA2 expression decreases in PMA-activated CD4+ T cells, while others reported increased expression of LPA2 after T cell activation, hence future studies are needed to dissect the expression of LPA2 during the activation of T cells (Zheng et al., 2000; Rubenfeld et al., 2006). LPA2 is also expressed on the apical surface of intestinal epithelial cells (Li et al., 2005) and in the airway epithelia cells of human lung tissue (Barekzi et al., 2006). In addition, LPA2 is

LPAR2 (lysophosphatidic acid receptor 2) Knowlden S, Georas S
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 261
expressed in epithelial cell lines: A549 and BEAS-2B (Barekzi et al., 2006). Interestingly, IL-13 and IFN-gamma reduced LPA2 mRNA levels in the A549 cell line (Barekzi et al., 2006). In addition to its normal expression, LPA is also commonly increased in a number of human malignancies. LPA2 is aberrantly expressed in various cancer cells including ovarian cancer cell lines (Fang et al., 2000; Fang et al., 2002; Goetzl et al., 1999), the cervical cancer cell lines CaSki, HeLa, and SiHa (Chen et al., 2011), colorectal cancer (Shida et al., 2004), thyroid cancer (Schulte et al., 2001) and invasive ductal carcinoma breast cancer (Kitayama et al., 2004; Chen et al., 2007). It has been noted that LPA2 overexpression is more commonly seen in postmenopausal breast cancer patients than in premenopausal patients (Kitayama et al., 2004). It is also expressed in nasal polyp tissue from subjects with chronic hyperplastic eosinophilic sinusitis (CHES) (Barekzi et al., 2006). In mice, LPA2 mRNA is expressed in kidney, uterus, and testis at relatively high levels, and moderately expressed in the lung. Lower levels of LPA2 are also seen in spleen, thymus, stomach, brain, and heart.
Localisation LPA2 is a GPCR that spans the plasma membrane seven times and contains three extracellular loops and three intracellular loops. LPA2 is unique from the other LPA receptors as it contains two distinct protein-protein interaction domains in the carboxyl-terminal tail (aa 296-351). In the proximal region, LPA2 contains a di-leucine motif and several putative palmitoylated cysteine residues. This region is responsible for associating with zinc-finger proteins, including TRIP6 (Xu et al., 2004) and the proapoptotic Siva-1 protein (Lin et al., 2007). In the distal region, there are several serine and threonine residues that can be phosphorylated by G protein-coupled receptor kinases (GRKs) and may be involved in β-arrestin binding and receptor internalization. The last four amino acids of this region (DSTL) contains a class I PDZ-binding motif and mediates interactions with a number of proteins such as Na+/H+ exchanger regulatory factor 2 (NHERF2) (Oh et al., 2004; Yun et al., 2005), PDZ-RhoGEF and LARG (Yamada et al., 2005), and MAGI-3 (Zhang et al., 2007).
Function LPA2 is a GPCR that couples with and activates three heterotrimeric G proteins: Gi, Gq, G12/13. These G proteins transmit signals through downstream signaling molecules that include phosphatidylinositol 3-kinase, phospholipase C, Ras, Rac, and Rho. Activation of LPA2 therefore induces a range of cellular responses including cell survival and differentiation, cell migration, and roles in cancer metastasis. For example, LPA2 signaling is associated with cell survival and proliferation in ovarian cancer cells
(Goetzl et al., 1999) and rescues intestinal epithelial cells-6 (IEC-6) from apoptosis through inhibition of caspase-3 activation (Deng et al., 2002). Likewise, LPA2 targets the pro-apoptotic Siva-1 protein for LPA-dependent ubiquitination and degradation, thereby down regulating the pro-apoptotic activity of Siva-1 during the DNA damage response (Lin et al., 2007). LPA2 is also involved in promoting cell motility. Jurkat cells that express LPA2 were reported to have enhanced trans-Matrigel migration (Zheng et al., 2001). It has also been shown that LPA binding to LPA2 leads to the recruitment of TRIP6, a focal adhesion molecule, to the C terminus of LPA2 at the plasma membrane. This promotes its targeting to focal adhesions and co-localization with actin, thereby regulating LPA-induced cell migration (Xu et al., 2004; Lai et al., 2005). The PTPL1 phosphatase dephosphorylates TRIP6 and attenuates LPA-induced cell migration, thus acting as a negative regulator of cell motility (Lai et al., 2007). LPA2 has also been identified to be involved in regulating smooth muscle cell migration in the context of vascular injury (Panchatcharam et al., 2008). Recently, two groups have implicated LPA2 signaling in TGF-β activation in mouse models of lung fibrosis and ischemia-reperfusion injury. These studies have shown that LPA2 signaling through Gαq in human epithelial cells and proximal tubule cells activates RhoA and Rho kinase, leading to the activation of αvβ6 integrin. This in turn, leads to the binding of latent TGF-β to αvβ6, and subsequent activation of TGF-β (Xu et al., 2009; Geng et al., 2012). LPA2 signaling has emerged as a potential factor in many cancer pathways. There is high expression of LPA2 in human thyroid cancer (Schulte et al., 2001), colorectal cancer (Shida et al., 2004), as well as in human invasive breast ductal carcinoma (Kitayama et al., 2004). LPA2 is involved in tumor growth and tumor angiogenesis of in vivo cervical cancer cells (Chen et al., 2011; Yu et al., 2008). LPA2 mediates mitogenic signals and cytokine production in human colonic epithelial cells (Yun et al., 2005). In pancreatic cancer cells, signaling through LPA2 leads to the inhibition of EGF-induced migration and invasion (Komachi et al., 2009). It also mediates chemotaxis in a Rho-dependent manner in breast carcinoma cells (Chen et al., 2007). In ovarian cancer cells, LPA2 signaling through Gαi/src leads to transactivation of EGFR and COX-2 expression, and increased ovarian cancer motility and aggressiveness (Jeong et al., 2008). A role for LPA2 and endometrial cancer invasion and MMP7 activation has also been shown (Mayer Hope et al., 2009). It has been reported that homozygous knock-out lpa2-/- mice display no obvious phenotypic abnormalities and are born at expected frequencies (Contos et al., 2002). Zhao et al. reported that heterozygous lpa2+/- mice are partially protected from lung inflammation following Schistosoma egg allergen (SEA) challenge (Zhao et al., 2009). However, Emo et al. revealed that allergic lung

LPAR2 (lysophosphatidic acid receptor 2) Knowlden S, Georas S
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 262
inflammation is significantly greater in lpa2-/- mice, suggesting that LPA2 plays a role in suppressing dendritic cell activation and allergic immune responses (Emo et al., 2012).
Homology LPA2 has ~60% homology to LPA1.
Mutations Note The first human LPA2 cDNA clone was derived from an ovarian tumor library, however it differed from reported human LPA2 sequences (An et al., 1998). The protein product from the ovarian tumor lacks the last four amino acids (DSTL) and is 31 amino acid residues longer at the C-terminus relative to the predicted protein product. The extra amino acids are the result of a guanine nucleotide deletion in the fourth to last codon (Contos and Chun, 2000). Additionally, in two human colon cancer cell lines, DLD1 and SW48, LPA2 and LPA4 were found to contain five mutations of G/C to A/T transitions (Tsujino et al., 2010). These mutated LPA2 receptors may alter LPA2 signaling through its respective G proteins and downstream pathways, and play a role in cancer progression.
Implicated in Ovarian cancer Note LPA is present at high levels in the ascites fluid of ovarian cancer patients (Mills et al., 1990; Xu et al., 1995), and LPA2 is aberrantly expressed in ovarian cancer cells, compared to normal ovarian epithelial cells (Fang et al., 2000; Fang et al., 2002). LPA2 is expressed at high levels on OV202 primary culture ovarian cancer cells, as well as in several established ovarian cancer cells lines, and is involved in promoting cancer cell proliferation (Goetzl et al., 1999). LPA can promote angiogenesis by increasing VEGF protein levels in SKOV-3, CAOV-3, and OVCAR-3 cells, which are LPA2-expressing ovarian cancer cell lines (Hu et al., 2001). Additionally, LPA2 signaling through Gαi/src leads to transactivation of EGFR and COX-2 expression, and increased ovarian cancer motility and aggressiveness (Jeong et al., 2008). Furthermore, LPA stimulates expression of IL-8 and IL-6 in ovarian cancer cell lines (Schwartz et al., 2001) and ovarian cancer patients have elevated IL-8 and IL-6 cytokine levels in serum and ascitic fluid (Ivarsson et al., 2000; Penson et al., 2000). Fang et al. demonstrated that the IL-8 gene promoter contains a fragment 133-bp upstream of the transcription initiation site that has binding sites for NF-KB/RELA and AP-1 and is responsible for responses to LPA (Fang et al., 2004). Using a lentivirus to over-express LPA2, it was also shown that LPA2 elicited the most optimal responses to LPA, compared to other LPA receptors, and that LPA2
is able to couple LPA to IL-8 and IL-6 expression in ovarian cancer cells (Fang et al., 2004). Using an siRNA approach to knock-down LPA2 in SKOV-3 ovarian cancer cells, Wang et al. showed that the levels of LPA-induced urokinase plasminogen activator (uPA), which is a serine protease inversely correlated with prognosis in ovarian cancer, is greatly decreased. LPA2-siRNA treated cells were also less invasive and less migratory in vitro (Wang et al., 2008).
Cervical cancer Note Three cancer cell lines (CaSki, HeLa, and SiHa) express LPA2 mRNA, however it appears that LPA2 in these cells does not play a significant role in cancer cell proliferation in vitro (Chen et al., 2012). On the other hand, cervical cancer tumor growth and angiogenesis in vivo is dependent on LPA2 and LPA3. It was found that LPA induced IL-8 production in these cell lines, and when LPA2/3 is blocked, IL-8 expression was attenuated. Using in vitro angiogenesis assays, it was shown that the LPA-induced IL-8 expression in the cervical cancer cell lines led to increased angiogenesis, in an LPA2/3 dependent manner (Chen et al., 2012).
Colorectal cancer Note LPA2 is highly expressed at the mRNA and protein levels in human colorectal cancers (Shida et al., 2004). In CACO-2 colon cancer cells, LPA2 interacts with Na+/H+ exchanger regulatory factor 2 (NHERF2) and mediates downstream signaling such as the activation of Akt, Erk1, Erk2, and IL-8 (Yun et al., 2005). MAGI-3 has also been shown to reciprocally regulate PLC-β and inhibit NHERF2-promoted tumor cell migration and invasion (Lee et al., 2011). The importance of LPA2 in contributing to colon cancer progression was elucidated using LPA2 knock-out (LPA2-/-) mice. These studies revealed that colon cancer was markedly diminished in LPA2-/- mice, with less epithelial cells proliferation, decreased MCP-1 and MIF levels, and decreased inflammatory macrophage infiltrates (Lin et al., 2009).
Thyroid cancer Note LPA2 mRNA expression is increased in both human papillary and follicular differentiated thyroid cancer, compared to normal thyroid or goiters, suggesting its role in thyroid cancer pathogenesis (Schulte et al., 2001).
Breast cancer Note In human invasive ductal carcinoma (breast cancer) tissue, LPA2 mRNA and protein expression are enhanced (Kitayama et al., 2004; Li et al., 2009). Interestingly, immunohistochemical analyses revealed that LPA2 is upregulated more frequently in

LPAR2 (lysophosphatidic acid receptor 2) Knowlden S, Georas S
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 263
postmenopausal women than in premenopausal women, suggesting that the over-expression of LPA2 is associated with the progression of breast cancer in postmenopausal women (Kitayama et al., 2004). The breast cancer cell lines BT-20, MCF-7, MDA-MB-453, and MDA-MB-468 show predominant expression of LPA2 (Chen et al., 2007). When examining the BT-20 cell line closely, it was found that LPA activates RhoA, leading to increased chemotaxis. By knocking down LPA2 with siRNA, it was confirmed that LPA2 mediates the activation of RhoA and enhanced migration, and can act cooperatively with LPA1 (Chen et al., 2007).
Pancreatic cancer Note LPA2 inhibits the migration of invasive pancreatic cancer cells, while LPA1 stimulates migration of these cells (Komachi et al., 2009). The inhibitory migration response can be attenuated when LPA2 is knocked down using siRNA, or when LPA2 is agonized using an LPA2-specific agonist, LP-105. By blocking Gα12/13 and deactivating Rho, it has been suggested that LPA-LPA2 inhibits EGF-induced migration through the Gα12/13 and Rho-signaling pathways (Komachi et al., 2009).
Endometrial cancer Note HEC1A endometrial cancer cells predominantly express LPA2 and its expression is increased upon LPA stimulation (Mayer Hope et al., 2009). When LPA2 is knocked down using siRNA, HEC1A cell invasion and MMP-7 and MMP-2 secretion and activation is markedly reduced, however the migration capacity of the cells is not significantly changed (Mayer Hope et al., 2009).
Gastric cancer Note In the gastric cancer cell lines, MKN28, MKN45, MKN74, and KATO III, LPA2 mRNA is significantly expressed (Shida et al., 2004). In chemotaxis assays, LPA was not able to induce migration of MKN28 or MKN74 cells, however, when hepatocyte growth factor (HGF) was added, LPA induced dose-dependent cell migration. In addition, using immunoprecipitation analysis, it was shown that LPA induced tyrosine phosphorylation of c-Met in these cells, suggesting that LPA and HGF induce a cooperative migratory response caused by the transactivation of c-Met (Shida et al., 2004). LPA2 is over-expressed in human gastric cancer, and is found more frequently in the intestinal type (67%) than in the diffuse type gastric cancer (32%) (Yamashita et al., 2006). However, LPA2 expression is more correlated with a higher rate of lymphatic invasion, venous invasion, and lymph node metastasis in diffuse-
type gastric than in intestinal type gastric cancer (Yamashita et al., 2006).
Allergic lung inflammation Note In a murine model of allergic airway inflammation using SEA-sensitization, Zhao et al. show that Lpa2+/-
heterozygous mice have reduced airway inflammation and pathogenesis of asthma (Zhao et al., 2009). This suggests that LPA2 may play a critical role in the detrimental effects of the onset of asthma in this model of the disease. However, recently a novel role for LPA2 in suppressing dendritic cell activation and allergic immune responses has been reported (Emo et al., 2012). Emo et al. showed that Lpa2-deficient bone marrow-derived dendritic cells are hyperactive compared to wild-type cells in that they can stimulate greater CD4+ T cell proliferation and induce higher levels of IL-13 secretion from T cells in co-culture. In a model of allergic airway inflammation, Lpa2-deficient mice succumbed to greater allergic lung inflammation, as seen by higher BAL cell counts, increased eosinophilia, increased airway hyperresponsiveness, and greater serum IgE levels. These data suggest that LPA2 may be acting as an inhibitory receptor to possibly dampen innate immune responses, particularly in this model of allergic airway inflammation.
Disease Asthma.
Fibrosis Note TGF-β has known roles in the pathogenesis of lung inflammation and fibrosis. In models of bleomycin-induced lung injury and renal ischemia-reperfusion injury, LPA2 signaling through Gαq activates αvβ6 integrin through a Rho and Rho-kinase dependent mechanism. Activated αvβ6 can bind to latent TGF-β, leading to its activation (Xu et al., 2009; Geng et al., 2012).
References Mills GB, May C, Hill M, Campbell S, Shaw P, Marks A. Ascitic fluid from human ovarian cancer patients contains growth factors necessary for intraperitoneal growth of human ovarian adenocarcinoma cells. J Clin Invest. 1990 Sep;86(3):851-5
Xu Y, Fang XJ, Casey G, Mills GB. Lysophospholipids activate ovarian and breast cancer cells. Biochem J. 1995 Aug 1;309 ( Pt 3):933-40
An S, Bleu T, Hallmark OG, Goetzl EJ. Characterization of a novel subtype of human G protein-coupled receptor for lysophosphatidic acid. J Biol Chem. 1998 Apr 3;273(14):7906-10
Goetzl EJ, Dolezalova H, Kong Y, Hu YL, Jaffe RB, Kalli KR, Conover CA. Distinctive expression and functions of the type 4 endothelial differentiation gene-encoded G protein-coupled receptor for lysophosphatidic acid in ovarian cancer. Cancer Res. 1999 Oct 15;59(20):5370-5

LPAR2 (lysophosphatidic acid receptor 2) Knowlden S, Georas S
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 264
Goetzl EJ, Kong Y, Mei B. Lysophosphatidic acid and sphingosine 1-phosphate protection of T cells from apoptosis in association with suppression of Bax. J Immunol. 1999 Feb 15;162(4):2049-56
Contos JJ, Chun J. Genomic characterization of the lysophosphatidic acid receptor gene, lp(A2)/Edg4, and identification of a frameshift mutation in a previously characterized cDNA. Genomics. 2000 Mar 1;64(2):155-69
Contos JJ, Ishii I, Chun J. Lysophosphatidic acid receptors. Mol Pharmacol. 2000 Dec;58(6):1188-96
Fang X, Gaudette D, Furui T, Mao M, Estrella V, Eder A, Pustilnik T, Sasagawa T, Lapushin R, Yu S, Jaffe RB, Wiener JR, Erickson JR, Mills GB. Lysophospholipid growth factors in the initiation, progression, metastases, and management of ovarian cancer. Ann N Y Acad Sci. 2000 Apr;905:188-208
Goetzl EJ, Kong Y, Voice JK. Cutting edge: differential constitutive expression of functional receptors for lysophosphatidic acid by human blood lymphocytes. J Immunol. 2000 May 15;164(10):4996-9
Ivarsson K, Ekerydh A, Fyhr IM, Janson PO, Brännström M. Upregulation of interleukin-8 and polarized epithelial expression of interleukin-8 receptor A in ovarian carcinomas. Acta Obstet Gynecol Scand. 2000 Sep;79(9):777-84
Penson RT, Kronish K, Duan Z, Feller AJ, Stark P, Cook SE, Duska LR, Fuller AF, Goodman AK, Nikrui N, MacNeill KM, Matulonis UA, Preffer FI, Seiden MV. Cytokines IL-1beta, IL-2, IL-6, IL-8, MCP-1, GM-CSF and TNFalpha in patients with epithelial ovarian cancer and their relationship to treatment with paclitaxel. Int J Gynecol Cancer. 2000 Jan;10(1):33-41
Zheng Y, Voice JK, Kong Y, Goetzl EJ. Altered expression and functional profile of lysophosphatidic acid receptors in mitogen-activated human blood T lymphocytes. FASEB J. 2000 Dec;14(15):2387-9
Hu YL, Tee MK, Goetzl EJ, Auersperg N, Mills GB, Ferrara N, Jaffe RB. Lysophosphatidic acid induction of vascular endothelial growth factor expression in human ovarian cancer cells. J Natl Cancer Inst. 2001 May 16;93(10):762-8
Schulte KM, Beyer A, Köhrer K, Oberhäuser S, Röher HD. Lysophosphatidic acid, a novel lipid growth factor for human thyroid cells: over-expression of the high-affinity receptor edg4 in differentiated thyroid cancer. Int J Cancer. 2001 Apr 15;92(2):249-56
Schwartz BM, Hong G, Morrison BH, Wu W, Baudhuin LM, Xiao YJ, Mok SC, Xu Y. Lysophospholipids increase interleukin-8 expression in ovarian cancer cells. Gynecol Oncol. 2001 May;81(2):291-300
Zheng Y, Kong Y, Goetzl EJ. Lysophosphatidic acid receptor-selective effects on Jurkat T cell migration through a Matrigel model basement membrane. J Immunol. 2001 Feb 15;166(4):2317-22
Contos JJ, Ishii I, Fukushima N, Kingsbury MA, Ye X, Kawamura S, Brown JH, Chun J. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2). Mol Cell Biol. 2002 Oct;22(19):6921-9
Deng W, Balazs L, Wang DA, Van Middlesworth L, Tigyi G, Johnson LR. Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and chemotherapy-induced apoptosis. Gastroenterology. 2002 Jul;123(1):206-16
Fang X, Schummer M, Mao M, Yu S, Tabassam FH, Swaby R, Hasegawa Y, Tanyi JL, LaPushin R, Eder A, Jaffe R, Erickson J, Mills GB. Lysophosphatidic acid is a bioactive mediator in
ovarian cancer. Biochim Biophys Acta. 2002 May 23;1582(1-3):257-64
Fang X, Yu S, Bast RC, Liu S, Xu HJ, Hu SX, LaPushin R, Claret FX, Aggarwal BB, Lu Y, Mills GB. Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian cancer cells. J Biol Chem. 2004 Mar 5;279(10):9653-61
Kitayama J, Shida D, Sako A, Ishikawa M, Hama K, Aoki J, Arai H, Nagawa H. Over-expression of lysophosphatidic acid receptor-2 in human invasive ductal carcinoma. Breast Cancer Res. 2004;6(6):R640-6
Oh YS, Jo NW, Choi JW, Kim HS, Seo SW, Kang KO, Hwang JI, Heo K, Kim SH, Kim YH, Kim IH, Kim JH, Banno Y, Ryu SH, Suh PG. NHERF2 specifically interacts with LPA2 receptor and defines the specificity and efficiency of receptor-mediated phospholipase C-beta3 activation. Mol Cell Biol. 2004 Jun;24(11):5069-79
Shida D, Kitayama J, Yamaguchi H, Hama K, Aoki J, Arai H, Yamashita H, Mori K, Sako A, Konishi T, Watanabe T, Sakai T, Suzuki R, Ohta H, Takuwa Y, Nagawa H. Dual mode regulation of migration by lysophosphatidic acid in human gastric cancer cells. Exp Cell Res. 2004 Dec 10;301(2):168-78
Shida D, Watanabe T, Aoki J, Hama K, Kitayama J, Sonoda H, Kishi Y, Yamaguchi H, Sasaki S, Sako A, Konishi T, Arai H, Nagawa H. Aberrant expression of lysophosphatidic acid (LPA) receptors in human colorectal cancer. Lab Invest. 2004 Oct;84(10):1352-62
Xu J, Lai YJ, Lin WC, Lin FT. TRIP6 enhances lysophosphatidic acid-induced cell migration by interacting with the lysophosphatidic acid 2 receptor. J Biol Chem. 2004 Mar 12;279(11):10459-68
Lai YJ, Chen CS, Lin WC, Lin FT. c-Src-mediated phosphorylation of TRIP6 regulates its function in lysophosphatidic acid-induced cell migration. Mol Cell Biol. 2005 Jul;25(14):5859-68
Li C, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, Jackson JS, Makarova NV, Fujiwara Y, Farrar PL, Nelson DJ, Tigyi GJ, Naren AP. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med. 2005 Oct 3;202(7):975-86
Yamada T, Ohoka Y, Kogo M, Inagaki S. Physical and functional interactions of the lysophosphatidic acid receptors with PDZ domain-containing Rho guanine nucleotide exchange factors (RhoGEFs). J Biol Chem. 2005 May 13;280(19):19358-63
Yun CC, Sun H, Wang D, Rusovici R, Castleberry A, Hall RA, Shim H. LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am J Physiol Cell Physiol. 2005 Jul;289(1):C2-11
Barekzi E, Roman J, Hise K, Georas S, Steinke JW. Lysophosphatidic acid stimulates inflammatory cascade in airway epithelial cells. Prostaglandins Leukot Essent Fatty Acids. 2006 Jun;74(6):357-63
Chen R, Roman J, Guo J, West E, McDyer J, Williams MA, Georas SN. Lysophosphatidic acid modulates the activation of human monocyte-derived dendritic cells. Stem Cells Dev. 2006 Dec;15(6):797-804
Oz-Arslan D, Rüscher W, Myrtek D, Ziemer M, Jin Y, Damaj BB, Sorichter S, Idzko M, Norgauer J, Maghazachi AA. IL-6 and IL-8 release is mediated via multiple signaling pathways after stimulating dendritic cells with lysophospholipids. J Leukoc Biol. 2006 Aug;80(2):287-97
Rubenfeld J, Guo J, Sookrung N, Chen R, Chaicumpa W, Casolaro V, Zhao Y, Natarajan V, Georas S. Lysophosphatidic

LPAR2 (lysophosphatidic acid receptor 2) Knowlden S, Georas S
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 265
acid enhances interleukin-13 gene expression and promoter activity in T cells. Am J Physiol Lung Cell Mol Physiol. 2006 Jan;290(1):L66-74
Yamashita H, Kitayama J, Shida D, Ishikawa M, Hama K, Aoki J, Arai H, Nagawa H. Differential expression of lysophosphatidic acid receptor-2 in intestinal and diffuse type gastric cancer. J Surg Oncol. 2006 Jan 1;93(1):30-5
Chen M, Towers LN, O'Connor KL. LPA2 (EDG4) mediates Rho-dependent chemotaxis with lower efficacy than LPA1 (EDG2) in breast carcinoma cells. Am J Physiol Cell Physiol. 2007 May;292(5):C1927-33
Lai YJ, Lin WC, Lin FT. PTPL1/FAP-1 negatively regulates TRIP6 function in lysophosphatidic acid-induced cell migration. J Biol Chem. 2007 Aug 17;282(33):24381-7
Lin FT, Lai YJ, Makarova N, Tigyi G, Lin WC. The lysophosphatidic acid 2 receptor mediates down-regulation of Siva-1 to promote cell survival. J Biol Chem. 2007 Dec 28;282(52):37759-69
Zhang H, Wang D, Sun H, Hall RA, Yun CC. MAGI-3 regulates LPA-induced activation of Erk and RhoA. Cell Signal. 2007 Feb;19(2):261-8
Jeong KJ, Park SY, Seo JH, Lee KB, Choi WS, Han JW, Kang JK, Park CG, Kim YK, Lee HY. Lysophosphatidic acid receptor 2 and Gi/Src pathway mediate cell motility through cyclooxygenase 2 expression in CAOV-3 ovarian cancer cells. Exp Mol Med. 2008 Dec 31;40(6):607-16
Panchatcharam M, Miriyala S, Yang F, Rojas M, End C, Vallant C, Dong A, Lynch K, Chun J, Morris AJ, Smyth SS. Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ Res. 2008 Sep 12;103(6):662-70
Taghavi P, Verhoeven E, Jacobs JJ, Lambooij JP, Stortelers C, Tanger E, Moolenaar WH, van Lohuizen M. In vitro genetic screen identifies a cooperative role for LPA signaling and c-Myc in cell transformation. Oncogene. 2008 Nov 20;27(54):6806-16
Wang GL, Wen ZQ, Xu WP, Wang ZY, Du XL, Wang F. Inhibition of lysophosphatidic acid receptor-2 expression by RNA interference decreases lysophosphatidic acid-induced urokinase plasminogen activator activation, cell invasion, and migration in ovarian cancer SKOV-3 cells. Croat Med J. 2008 Apr;49(2):175-81
Yu S, Murph MM, Lu Y, Liu S, Hall HS, Liu J, Stephens C, Fang X, Mills GB. Lysophosphatidic acid receptors determine tumorigenicity and aggressiveness of ovarian cancer cells. J Natl Cancer Inst. 2008 Nov 19;100(22):1630-42
Hope JM, Wang FQ, Whyte JS, Ariztia EV, Abdalla W, Long K, Fishman DA. LPA receptor 2 mediates LPA-induced
endometrial cancer invasion. Gynecol Oncol. 2009 Jan;112(1):215-23
Komachi M, Tomura H, Malchinkhuu E, Tobo M, Mogi C, Yamada T, Kimura T, Kuwabara A, Ohta H, Im DS, Kurose H, Takeyoshi I, Sato K, Okajima F. LPA1 receptors mediate stimulation, whereas LPA2 receptors mediate inhibition, of migration of pancreatic cancer cells in response to lysophosphatidic acid and malignant ascites. Carcinogenesis. 2009 Mar;30(3):457-65
Lin S, Wang D, Iyer S, Ghaleb AM, Shim H, Yang VW, Chun J, Yun CC. The absence of LPA2 attenuates tumor formation in an experimental model of colitis-associated cancer. Gastroenterology. 2009 May;136(5):1711-20
Xu MY, Porte J, Knox AJ, Weinreb PH, Maher TM, Violette SM, McAnulty RJ, Sheppard D, Jenkins G. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am J Pathol. 2009 Apr;174(4):1264-79
Zhao Y, Tong J, He D, Pendyala S, Evgeny B, Chun J, Sperling AI, Natarajan V. Role of lysophosphatidic acid receptor LPA2 in the development of allergic airway inflammation in a murine model of asthma. Respir Res. 2009 Nov 20;10:114
Tsujino M, Fujii M, Okabe K, Mori T, Fukushima N, Tsujiuchi T. Differential expressions and DNA methylation patterns of lysophosphatidic acid receptor genes in human colon cancer cells. Virchows Arch. 2010 Dec;457(6):669-76
Lee SJ, Ritter SL, Zhang H, Shim H, Hall RA, Yun CC. MAGI-3 competes with NHERF-2 to negatively regulate LPA2 receptor signaling in colon cancer cells. Gastroenterology. 2011 Mar;140(3):924-34
Chen RJ, Chen SU, Chou CH, Lin MC. Lysophosphatidic acid receptor 2/3-mediated IL-8-dependent angiogenesis in cervical cancer cells. Int J Cancer. 2012 Aug 15;131(4):789-802
Emo J, Meednu N, Chapman TJ, Rezaee F, Balys M, Randall T, Rangasamy T, Georas SN. Lpa2 is a negative regulator of both dendritic cell activation and murine models of allergic lung inflammation. J Immunol. 2012 Apr 15;188(8):3784-90
Geng H, Lan R, Singha PK, Gilchrist A, Weinreb PH, Violette SM, Weinberg JM, Saikumar P, Venkatachalam MA. Lysophosphatidic acid increases proximal tubule cell secretion of profibrotic cytokines PDGF-B and CTGF through LPA2- and Gαq-mediated Rho and αvβ6 integrin-dependent activation of TGF-β. Am J Pathol. 2012 Oct;181(4):1236-49
This article should be referenced as such:
Knowlden S, Georas S. LPAR2 (lysophosphatidic acid receptor 2). Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):259-265.

Gene Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 266
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
MARCKS (myristoylated alanine-rich protein kinase C substrate) Atsuhiro Tanabe, Maho Saito
Division of Biochemistry, Department of Bioscience and Engineering, Shibaura Institute of Technology, Saitama, Japan (AT, MS)
Published in Atlas Database: November 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/MARCKSID50926ch6q21.html DOI: 10.4267/2042/48868
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: 80K-L, MACS, PKCSL, PRKCSL
HGNC (Hugo): MARCKS
Location: 6q21
DNA/RNA Note The MARCKS gene is located 6q21 (114178527..114184652).
Transcription The transcription product is 6,1 kb with 2 exons.
The mRNA has 996 bp open reading frame. The promoter region has no TATA box and contained multiple transcription initiation sites in a region spanning 57 base pairs (bp) (Harlan et al., 1991).
Protein Note MARCKS was cloned as a protein kinase C (PKC) substrate. The protein binds plasma membrane via N-terminus myristoylation and the phosphorylation site domain (PSD), which is also called effector domain (ED), with electrostatic interaction. MARCKS interacts with actin, calmodulin, PIP2 on the PSD.
Figure 1. A) MARCKS location on chromosome 6 is indicated. MARCKS gene starts at 114178527 and ends at 114184652. B) MARCKS gene is indicated. It has two exons and one intron.

MARCKS (myristoylated alanine-rich protein kinase C substrate) Tanabe A, Saito M
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 267
Figure 2. A) MARCKS phosphorylation site domain (PSD (also called effector domain (ED))) is shown. It is mainly consisted of basic amino asids (K and R) and has serin residues phosphorylatable by PKC (159, 163 and 170) and by ROCK (at least 159). B) Dephospho MARCKS binds to plasma membrane and cross-links actin. Phospho MARCKS detached from plasma membrane and disrupts actin filaments.
Description Phosphorylation of MARCKS is instrumental in its redistribution. MARCKS possesses a basic phosphorylation site domain (PSD). Phosphorylation of this PSD domain prevents the electrostatic interaction of the effector region of the MARCKS to the plasma membrane (George and Blackshear, 1992; Taniguchi and Manenti, 1993; Kim et al., 1994).
Expression The MARCKS protein is highly expressed in the brain, spleen, and lung, and is virtually absent in skeletal muscle and liver in adult animal (Stumpo et al., 1989; Blackshear et al., 1986; Albert et al., 1986).
Localisation Dephosphorylated and phosphorylated MARCKS are located at plasma membrane and in cytosol, respectively.
Function MARCKS closs-links actin filament (Yarmola et al., 2001) and changes cell morphology responsing to cell stimulations in its phosphorylation/dephosphorylation-dependent manner (Tanabe et al., 2012). MARCKS participates in thrombin-induced noradrenaline release from platelets (Elzagallaai et al., 2001) and PMA- or bonbesin-induced neurotensin release from BON cells (Li et al., 2005). MARCKS regulates the proliferation
and/or movement of some type of cells (Brooks et al.,1996; Zhao et al., 2000; Weimer et al., 2009). MARCKS plays a vital role in the normal developmental processes of neurulation, hemisphere fusion, forebrain commissure formation, and formation of cortical and retinal laminations (Stumpo et al., 1995). Long-term potentiation (LTP) is significantly impaired in the mossy fiber-CA3 pathway in MARCKS heterozygous mutant mice (Hussain et al., 2006).
Homology Human MARCKS protein (332 amino acids) was approximately 89, 74, and 59% identical to the bovine, mouse, and chicken proteins. N-terminal domain and phosphorylation site domain (PSD) are highly-conserved between species (from human to Xenopus).
Implicated in Melanoma Note In MARCKS over expressed human tumor-derived choroidal melanoma cells (OCM-1) the growth was reduced by 35-40% when compared with control cells (Manenti et al., 1998). In a highly motile melanoma cell line WM-1617 melanoma cells nonphosphorylated MARCKS works as an adhesion stabilizer (Estrada-Bernal et al., 2009).

MARCKS (myristoylated alanine-rich protein kinase C substrate) Tanabe A, Saito M
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 268
Alzheimer disease (AD) Note PKC-induced phosphorylation of MARCKS in cortical neurons in AD brains was weaker than that in control brains. However, phosphorylation of MARCKS was detected in microglia and dystrophic neurites within neuritic plaques (Kimura et al., 2000). In microglia amyloid β induces phosphorylation of MARCKS through mitogen-activatd protein kinase (MAPK) (Hasegawa et al., 2001) and PKCδ (Nakai et al., 2001).
References Albert KA, Walaas SI, Wang JK, Greengard P. Widespread occurrence of "87 kDa," a major specific substrate for protein kinase C. Proc Natl Acad Sci U S A. 1986 May;83(9):2822-6
Blackshear PJ, Wen L, Glynn BP, Witters LA. Protein kinase C-stimulated phosphorylation in vitro of a Mr 80,000 protein phosphorylated in response to phorbol esters and growth factors in intact fibroblasts. Distinction from protein kinase C and prominence in brain. J Biol Chem. 1986 Jan 25;261(3):1459-69
Stumpo DJ, Graff JM, Albert KA, Greengard P, Blackshear PJ. Molecular cloning, characterization, and expression of a cDNA encoding the "80- to 87-kDa" myristoylated alanine-rich C kinase substrate: a major cellular substrate for protein kinase C. Proc Natl Acad Sci U S A. 1989 Jun;86(11):4012-6
Harlan DM, Graff JM, Stumpo DJ, Eddy RL Jr, Shows TB, Boyle JM, Blackshear PJ. The human myristoylated alanine-rich C kinase substrate (MARCKS) gene (MACS). Analysis of its gene product, promoter, and chromosomal localization. J Biol Chem. 1991 Aug 5;266(22):14399-405
George DJ, Blackshear PJ. Membrane association of the myristoylated alanine-rich C kinase substrate (MARCKS) protein appears to involve myristate-dependent binding in the absence of a myristoyl protein receptor. J Biol Chem. 1992 Dec 5;267(34):24879-85
Taniguchi H, Manenti S. Interaction of myristoylated alanine-rich protein kinase C substrate (MARCKS) with membrane phospholipids. J Biol Chem. 1993 May 15;268(14):9960-3
Kim J, Shishido T, Jiang X, Aderem A, McLaughlin S. Phosphorylation, high ionic strength, and calmodulin reverse the binding of MARCKS to phospholipid vesicles. J Biol Chem. 1994 Nov 11;269(45):28214-9
Stumpo DJ, Bock CB, Tuttle JS, Blackshear PJ. MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proc Natl Acad Sci U S A. 1995 Feb 14;92(4):944-8
Brooks G, Brooks SF, Goss MW. MARCKS functions as a novel growth suppressor in cells of melanocyte origin. Carcinogenesis. 1996 Apr;17(4):683-9
Manenti S, Malecaze F, Chap H, Darbon JM. Overexpression of the myristoylated alanine-rich C kinase substrate in human
choroidal melanoma cells affects cell proliferation. Cancer Res. 1998 Apr 1;58(7):1429-34
Kimura T, Yamamoto H, Takamatsu J, Yuzuriha T, Miyamoto E, Miyakawa T. Phosphorylation of MARCKS in Alzheimer disease brains. Neuroreport. 2000 Mar 20;11(4):869-73
Zhao Y, Neltner BS, Davis HW. Role of MARCKS in regulating endothelial cell proliferation. Am J Physiol Cell Physiol. 2000 Nov;279(5):C1611-20
Elzagallaai A, Rosé SD, Brandan NC, Trifaró JM. Myristoylated alanine-rich C kinase substrate phosphorylation is involved in thrombin-induced serotonin release from platelets. Br J Haematol. 2001 Mar;112(3):593-602
Hasegawa H, Nakai M, Tanimukai S, Taniguchi T, Terashima A, Kawamata T, Fukunaga K, Miyamoto E, Misaki K, Mukai H, Tanaka C. Microglial signaling by amyloid beta protein through mitogen-activated protein kinase mediating phosphorylation of MARCKS. Neuroreport. 2001 Aug 8;12(11):2567-71
Nakai M, Tanimukai S, Yagi K, Saito N, Taniguchi T, Terashima A, Kawamata T, Yamamoto H, Fukunaga K, Miyamoto E, Tanaka C. Amyloid beta protein activates PKC-delta and induces translocation of myristoylated alanine-rich C kinase substrate (MARCKS) in microglia. Neurochem Int. 2001 Jun;38(7):593-600
Yarmola EG, Edison AS, Lenox RH, Bubb MR. Actin filament cross-linking by MARCKS: characterization of two actin-binding sites within the phosphorylation site domain. J Biol Chem. 2001 Jun 22;276(25):22351-8
Li J, O'Connor KL, Greeley GH Jr, Blackshear PJ, Townsend CM Jr, Evers BM. Myristoylated alanine-rich C kinase substrate-mediated neurotensin release via protein kinase C-delta downstream of the Rho/ROK pathway. J Biol Chem. 2005 Mar 4;280(9):8351-7
Hussain RJ, Stumpo DJ, Blackshear PJ, Lenox RH, Abel T, McNamara RK. Myristoylated alanine rich C kinase substrate (MARCKS) heterozygous mutant mice exhibit deficits in hippocampal mossy fiber-CA3 long-term potentiation. Hippocampus. 2006;16(5):495-503
Estrada-Bernal A, Gatlin JC, Sunpaweravong S, Pfenninger KH. Dynamic adhesions and MARCKS in melanoma cells. J Cell Sci. 2009 Jul 1;122(Pt 13):2300-10
Weimer JM, Yokota Y, Stanco A, Stumpo DJ, Blackshear PJ, Anton ES. MARCKS modulates radial progenitor placement, proliferation and organization in the developing cerebral cortex. Development. 2009 Sep;136(17):2965-75
Tanabe A, Shiraishi M, Negishi M, Saito N, Tanabe M, Sasaki Y. MARCKS dephosphorylation is involved in bradykinin-induced neurite outgrowth in neuroblastoma SH-SY5Y cells. J Cell Physiol. 2012 Feb;227(2):618-29
This article should be referenced as such:
Tanabe A, Saito M. MARCKS (myristoylated alanine-rich protein kinase C substrate). Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):266-268.

Gene Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 269
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
MIR331 (microRNA 331) Keith M Giles, Michael R Epis, Peter J Leedman
Laboratory for Cancer Medicine, Western Australian Institute for Medical Research and University of Western Australia Centre for Medical Research, Perth, WA 6000, Australia (KMG, MRE, PJL)
Published in Atlas Database: November 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/MIR331ID51220ch12q22.html DOI: 10.4267/2042/48869
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: MIRN331, hsa-mir-331
HGNC (Hugo): MIR331
Location: 12q22
Local order: Genes flanking MIR331 on 12q22 are: - VEZT (vezatin) - METAP2 (methionyl aminopeptidase 2)
DNA/RNA Description miR-331 is an intergenic microRNA gene.
Transcription The primary transcript of miR-331 (pri-miR-331) is not currently known. Pre-microRNA-331 (Precursor microRNA) Accession: MI0000812 Length: 94 nt Sequence: 5'-GAGUUUGGUUUUGUUUGGGUUUGUUCUAGG
UAUGGUCCCAGGGAUCC CAGAUCAAACCAGGCCCCUGGGCCUAUCCUAGAA CCAACCUAAGCUC-3' miR-331-3p and miR-331-5p mature sequences are bold. Mature miR-331-5p Accession: MIMAT0004700 Length: 22 nt Sequence: 26 - cuagguauggucccagggaucc - 47 Mature miR-331-3p Accession: MIMAT0000760 Length: 21 nt Sequence: 61 - gccccugggccuauccuagaa - 81
Pseudogene No pseudogenes have been reported for miR-331.
Protein Note N/A; microRNAs are not translated.
Figure 1: Stem-loop structure of miR-331, with mature miR-331-3p and miR-331-5p sequences highlighted in purple.

MIR331 (microRNA 331) Giles KM, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 270
Mutations Note No mutations in MIR331 have been described.
Implicated in Prostate cancer Note Five references have suggested a tumour suppressor role for miR-331 in prostate cancer. The first report demonstrated downregulation of miR-331-3p in prostate cancer, and showed that this promoted ERBB-2 expression and AKT activity. Restoring miR-331-3p to prostate cancer cell lines reduced androgen receptor (AR) pathway signaling and PSA expression. Another report confirmed the reduced expression of miR-331-3p in aggressive prostate cancers. Two other studies showed that the RNA-binding protein HuR induces ERBB-2 expression in prostate cancer by preventing the degradation of ERBB-2 mRNA by miR-331-3p, and that miR-331-3p inhibits the growth of prostate cancer cells in part by repressing expression of the deoxyhypusine hydroxylase (DOHH), an enzyme that controls the activity of the eukaryotic translation initiation factor eIF5A. In the latter study, an inverse correlation between miR-331-3p and DOHH expression was observed in human prostate cancer tissues. A fifth publication confirmed that miR-331-3p is a prostate cancer tumour suppressor via its regulation of KLK4 expression in prostate cancer cells.
Leukaemia Note Two references implicate miR-331 in leukaemia. One report showed that the levels of of miR-331-5p were inversely correlated with expression of P-glycoprotein, a drug resistance factor, in leukaemia cell lines with variable resistance to doxorubicin, and that transfection of these cell lines with miR-331-5p increased their sensitivity to doxorubicin. Lower levels of miR-331-5p were also detected in patients following treatment relapse. A second study reported that miR-331 was overexpressed in acute lymphocytic leukaemia (ALL) and chronic lymphocytic leukaemia (CLL), and it was speculated that miR-331 might have roles in haematopoiesis and leukaemogenesis by promoting STAT activation through its regulation of the mRNA target SOCS1.
Gastric cancer Note One reference has suggested that miR-331-3p is a gastric cancer tumour suppressor. miR-331-3p was downregulated in gastric cancer cell lines, where
transient overexpression of miR-331-3p reduced E2F1 expression and blocked cell cycle progression.
Liver cancer Note Increased circulating levels of miR-331 in a rat model of hepatocarcinogenesis suggest that miR-331 may have utility as a biomarker for the development and/or progression of liver cancer.
Asbestos-related lung cancer Note One report identified miR-331-3p in a set of overexpressed miRNAs in asbestos-related lung cancer, suggesting that it might have diagnostic use.
Natural killer (NK) cell activation Note Activation of NK cells by IL-2, IL-15 and IL-21 was shown to regulate expression of specific miRNAs in NK cells, including miR-331-3p. This suggested that miR-331-3p may have a role in the activation of NK cells.
Cerebral ischaemia Note miR-331 expression in cerebral ischaemia was regulated by the mood stabiliser and histone deacetylase inhibitor valproic acid (VPA), suggesting that it may have a role in this disease process.
References Zanette DL, Rivadavia F, Molfetta GA, Barbuzano FG, Proto-Siqueira R, Silva-Jr WA, Falcão RP, Zago MA. miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. Braz J Med Biol Res. 2007 Nov;40(11):1435-40
Epis MR, Giles KM, Barker A, Kendrick TS, Leedman PJ. miR-331-3p regulates ERBB-2 expression and androgen receptor signaling in prostate cancer. J Biol Chem. 2009 Sep 11;284(37):24696-704
Wang L, Tang H, Thayanithy V, Subramanian S, Oberg AL, Cunningham JM, Cerhan JR, Steer CJ, Thibodeau SN. Gene networks and microRNAs implicated in aggressive prostate cancer. Cancer Res. 2009 Dec 15;69(24):9490-7
Guo X, Guo L, Ji J, Zhang J, Zhang J, Chen X, Cai Q, Li J, Gu Q, Liu B, Zhu Z, Yu Y. miRNA-331-3p directly targets E2F1 and induces growth arrest in human gastric cancer. Biochem Biophys Res Commun. 2010 Jul 16;398(1):1-6
Epis MR, Barker A, Giles KM, Beveridge DJ, Leedman PJ. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J Biol Chem. 2011 Dec 2;286(48):41442-54
Feng DD, Zhang H, Zhang P, Zheng YS, Zhang XJ, Han BW, Luo XQ, Xu L, Zhou H, Qu LH, Chen YQ. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J Cell Mol Med. 2011 Oct;15(10):2164-75
Giles KM, Barker A, Zhang PM, Epis MR, Leedman PJ. MicroRNA regulation of growth factor receptor signaling in human cancer cells. Methods Mol Biol. 2011;676:147-63

MIR331 (microRNA 331) Giles KM, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 271
Nymark P, Guled M, Borze I, Faisal A, Lahti L, Salmenkivi K, Kettunen E, Anttila S, Knuutila S. Integrative analysis of microRNA, mRNA and aCGH data reveals asbestos- and histology-related changes in lung cancer. Genes Chromosomes Cancer. 2011 Aug;50(8):585-97
Sukata T, Sumida K, Kushida M, Ogata K, Miyata K, Yabushita S, Uwagawa S. Circulating microRNAs, possible indicators of progress of rat hepatocarcinogenesis from early stages. Toxicol Lett. 2011 Jan 15;200(1-2):46-52
Epis MR, Giles KM, Kalinowski FC, Barker A, Cohen RJ, Leedman PJ. Regulation of expression of deoxyhypusine hydroxylase (DOHH), the enzyme that catalyzes the activation of eIF5A, by miR-331-3p and miR-642-5p in prostate cancer cells. J Biol Chem. 2012 Oct 12;287(42):35251-9
Hunsberger JG, Fessler EB, Wang Z, Elkahloun AG, Chuang DM. Post-insult valproic acid-regulated microRNAs: potential targets for cerebral ischemia. Am J Transl Res. 2012;4(3):316-32
Liu X, Wang Y, Sun Q, Yan J, Huang J, Zhu S, Yu J. Identification of microRNA transcriptome involved in human natural killer cell activation. Immunol Lett. 2012 Apr 30;143(2):208-17
White NM, Youssef YM, Fendler A, Stephan C, Jung K, Yousef GM. The miRNA-kallikrein axis of interaction: a new dimension in the pathogenesis of prostate cancer. Biol Chem. 2012 Apr 1;393(5):379-89
This article should be referenced as such:
Giles KM, Epis MR, Leedman PJ. MIR331 (microRNA 331). Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):269-271.

Gene Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 272
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
SRXN1 (sulfiredoxin 1) Hedy A Chawsheen, Hong Jiang, Qiou Wei
Graduate Center for Toxicology, College of Medicine, University of Kentucky, Lexington, Kentucky 40513, USA (HAC, HJ, QW)
Published in Atlas Database: November 2012
Online updated version : http://AtlasGeneticsOncology.org/Genes/SRXN1ID52295ch20p13.html DOI: 10.4267/2042/48870
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names: C20orf139, Npn3, SRX1, YKL086W, dJ850E9.2
HGNC (Hugo): SRXN1
Location: 20p13
DNA/RNA Note Human Srx is located on chromosome 20 in the region of p13.
Description Human Srx gene is 6632 bp in length, composed of 2 exons and located at chromosome 20p13.
Transcription The size of Srx mRNA is 2580 bp. Srx transcript contains two exons. Exon 1 is 271 bp and exon 2 is 2300 bp. The catalytic domain of Srx reducing enzyme activity is localized in exon 2.
Protein Note Human Srx protein has a total of 137 amino acids and a 14 kDa molecular weight.
Description Srx is a member of antioxidant protein family containing a ParB-like nuclease domain. It forms 5 beta strands and 6 helix secondary structures. Srxn1 binds to peroxiredoxins (Prxs) and reduces overoxidized Prxs in the presence of cofactors including magnesium and ATP.
Expression In adult, Srx protein was found in internal organs such as mouse liver and kidney. Expression pattern of Srx in embryonic development is not clear. Transcriptional regulation of Srx expression is mainly mediated through AP-1 and/or Nrf-2 activation (Jeong et al., 2012). In yeast, it may also be negatively regulated at the translational level through Ras-PKA pathway (Molin et al., 2011).
Localisation Srx is mainly localized in the cytosol. In the presence of severe oxidative stress, it may also translocate to mitochondria (Noh et al., 2009).
Function Srx was first identified as a gene preferentially expressed in transformed JB6 cells (Sun et al., 1994). The primary biochemical function of Srx is to reduce the overoxidized cysteine residues of Prx I, Prx II, Prx III and Prx IV under severe oxidative stress (Biteau et al., 2003; Chang et al., 2004). The spectrum and specificity of its enzymatic function remains elusive. Srx may also cause the deglutathionylation of Prx II and others (Park et al., 2009; Findlay et al., 2006). The biological function of Srx may involve in the regulation of various cell signaling pathways to promote tumorigenesis and cancer progression. Abnormally high expression of Srx has been demonstrated in many malignant tumors including those of skin, lung, and colon (Wei et al., 2008). Srx may not be essential for development since Srx null mice are viable and normal (Planson et al., 2011).

SRXN1 (sulfiredoxin 1) Chawsheen HA, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 273
Structure of Human Srx bound to an ATP molecule and Mg2+ in solution (NCBI). Homology Srx gene is conserved among species, from metazoan to human.
Implicated in Various cancers Note Elevated expression of Srx has been associated with different types of human malignant tumors, such as skin squamous cell carcinoma, sweat gland carcinoma, basal cell carcinoma, melanoma, rectal carcinoma, lung adenocarcinoma and breast cancer (Wei et al., 2008; Hartikainen, et al., 2012). Increased Srx expression in lung cancer patients is positively associated with the deterioration of the clinic stages, and knockdown of Srx reduces cancer cell migration, invasion and their ability to form distal metastasis (Bowers et al., 2012; Wei et al., 2011). Srx genetic polymorphism of four SNPs (rs6116929, rs2008022, rs7269823, and rs9085283) is associated with breast cancer risk and patient survival (Hartikainen et al., 2011).
Tissue injury Note Srx, together with Prxs, are required for the protection
of tissues from oxidative stress induced damages by alcohol and Pyrazole (Bae et al., 2012; Bae et al., 2011).
Lung fibrosis Note Srx is found to be expressed in alveolar macrophages in non-specific interstitial pneumonia and may contribute to the process of idiopathic pulmonary fibrosis (Mazur et al., 2010).
References Sun Y, Hegamyer G, Colburn NH. Molecular cloning of five messenger RNAs differentially expressed in preneoplastic or neoplastic JB6 mouse epidermal cells: one is homologous to human tissue inhibitor of metalloproteinases-3. Cancer Res. 1994 Mar 1;54(5):1139-44
Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003 Oct 30;425(6961):980-4
Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem. 2004 Dec 3;279(49):50994-1001
Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006 Jul 1;66(13):6800-6

SRXN1 (sulfiredoxin 1) Chawsheen HA, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 274
Wei Q, Jiang H, Matthews CP, Colburn NH. Sulfiredoxin is an AP-1 target gene that is required for transformation and shows elevated expression in human skin malignancies. Proc Natl Acad Sci U S A. 2008 Dec 16;105(50):19738-43
Noh YH, Baek JY, Jeong W, Rhee SG, Chang TS. Sulfiredoxin Translocation into Mitochondria Plays a Crucial Role in Reducing Hyperoxidized Peroxiredoxin III. J Biol Chem. 2009 Mar 27;284(13):8470-7
Park JW, Mieyal JJ, Rhee SG, Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J Biol Chem. 2009 Aug 28;284(35):23364-74
Mazur W, Lindholm P, Vuorinen K, Myllärniemi M, Salmenkivi K, Kinnula VL. Cell-specific elevation of NRF2 and sulfiredoxin-1 as markers of oxidative stress in the lungs of idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. APMIS. 2010 Sep 1;118(9):703-12
Bae SH, Sung SH, Cho EJ, Lee SK, Lee HE, Woo HA, Yu DY, Kil IS, Rhee SG. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology. 2011 Mar;53(3):945-53
Molin M, Yang J, Hanzén S, Toledano MB, Labarre J, Nyström T. Life span extension and H(2)O(2) resistance elicited by caloric restriction require the peroxiredoxin Tsa1 in Saccharomyces cerevisiae. Mol Cell. 2011 Sep 2;43(5):823-33
Planson AG, Palais G, Abbas K, Gerard M, Couvelard L, Delaunay A, Baulande S, Drapier JC, Toledano MB. Sulfiredoxin protects mice from lipopolysaccharide-induced
endotoxic shock. Antioxid Redox Signal. 2011 Jun;14(11):2071-80
Wei Q, Jiang H, Xiao Z, Baker A, Young MR, Veenstra TD, Colburn NH. Sulfiredoxin-Peroxiredoxin IV axis promotes human lung cancer progression through modulation of specific phosphokinase signaling. Proc Natl Acad Sci U S A. 2011 Apr 26;108(17):7004-9
Bae SH, Sung SH, Lee HE, Kang HT, Lee SK, Oh SY, Woo HA, Kil IS, Rhee SG. Peroxiredoxin III and sulfiredoxin together protect mice from pyrazole-induced oxidative liver injury. Antioxid Redox Signal. 2012 Nov 15;17(10):1351-61
Bowers RR, Manevich Y, Townsend DM, Tew KD. Sulfiredoxin redox-sensitive interaction with S100A4 and non-muscle myosin IIA regulates cancer cell motility. Biochemistry. 2012 Oct 2;51(39):7740-54
Hartikainen JM, Tengström M, Kosma VM, Kinnula VL, Mannermaa A, Soini Y. Genetic polymorphisms and protein expression of NRF2 and Sulfiredoxin predict survival outcomes in breast cancer. Cancer Res. 2012 Nov 1;72(21):5537-46
Jeong W, Bae SH, Toledano MB, Rhee SG. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic Biol Med. 2012 Aug 1;53(3):447-56
This article should be referenced as such:
Chawsheen HA, Jiang H, Wei Q. SRXN1 (sulfiredoxin 1). Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):272-274.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 275
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
t(3;12)(q27;p12) LRMP/BCL6 Jean-Loup Huret
Genetics, Dept Medical Information, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France (JLH)
Published in Atlas Database: December 2012
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0312q27p12ID2129.html DOI: 10.4267/2042/48871
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology Disease Non Hodgkin lymphoma
Clinics The t(3;12)(q27;p12) was found in a case of follicular lymphoma transformed to diffuse aggressive lymphoma, from a study with no individual data (Akasaka et al., 2003).
Genes involved and proteins BCL6 Location 3q27.3
Protein 706 amino acids; composed of a NH2-term BTB/POZ domain (amino acids 1-130 (32-99 according to Swiss-Prot) which mediates homodimerization and protein-protein interactions with other corepressors (including HDAC1 and NCOR2/SMRT to constitute a large repressing complex, another transcription repression domain (191-386), PEST sequences (300-417) with a KKYK motif (375-379), and six zinc finger at the C-term (518-541, 546-568, 574-596, 602-624, 630-652, 658-681), responsible for sequence specific DNA binding. Transcription repressor; recognizes the consensus sequence: TTCCT(A/C)GAA (Albagli-Curiel, 2003). Role in germinal centers of lymphoid follicles. BCL6 prevents ATM and TP53 to induce apoptosis in response to DNA rearrangements such as somatic hypermutation and class switch recombination. Therefore essential for normal B cell development.
LRMP Location 12p12.1
Protein LRMP is an integral membrane protein of the endoplasmic reticulum (ER), oriented on the ER membrane facing the cytosol. It is a lymphoid-restricted gene that is expressed in a developmentally regulated fashion (Behrens et al., 1994). It may be involved in diabetes susceptibility (Grimm et al., 2003). A V141L polymorphism of LRMP has been found associated with a higher mortality in patients with lung cancer (Manenti et al., 2006). LRMP has recently been found involved in a translocation t(8;12)(q24;p12) with a LRMP/MYC fusion gene in non Hodgkin lymphoma (Singh et al., 2012).
Result of the chromosomal anomaly Hybrid gene Description Breakpoint in BCL6 first intron.
References Behrens TW, Jagadeesh J, Scherle P, Kearns G, Yewdell J, Staudt LM. Jaw1, A lymphoid-restricted membrane protein localized to the endoplasmic reticulum. J Immunol. 1994 Jul 15;153(2):682-90
Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood. 2003 Aug 15;102(4):1443-8

t(3;12)(q27;p12) LRMP/BCL6 Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 276
Albagli-Curiel O. Ambivalent role of BCL6 in cell survival and transformation. Oncogene. 2003 Jan 30;22(4):507-16
Grimm CH, Rogner UC, Avner P. Lrmp and Bcat1 are candidates for the type I diabetes susceptibility locus Idd6. Autoimmunity. 2003 Jun;36(4):241-6
Manenti G, Galbiati F, Pettinicchio A, Spinola M, Piconese S, Leoni VP, Conti B, Ravagnani F, Incarbone M, Pastorino U, Dragani TA. A V141L polymorphism of the human LRMP gene is associated with survival of lung cancer patients. Carcinogenesis. 2006 Jul;27(7):1386-90
Singh RR, Ben-Neriah S, Johnson NN, Connors JM, Gascoyne RD, Horsman DE. Identification of a novel non-immunoglobulin/MYC translocation t(8;12)(q24;p12) involving the LRMP gene in a primary B-cell lymphoma. A case report. Leuk Res. 2012 Jan;36(1):e22-4
This article should be referenced as such:
Huret JL. t(3;12)(q27;p12) LRMP/BCL6. Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):275-276.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 277
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
t(3;6)(q27;p22) HIST1H4I/BCL6 Jean-Loup Huret
Genetics, Dept Medical Information, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France (JLH)
Published in Atlas Database: December 2012
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0306q27p22ID2009.html DOI: 10.4267/2042/48872
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology Disease Non Hodgkin lymphoma
Clinics Apparently 6 cases have been described: a case of follicular mixed small cleaved and large cell lymphoma, a case of follicular large cell lymphoma, and 3 cases of diffuse large B-cell lymphoma (Akasaka et al., 1997; Akasaka et al., 2000; Kurata et al., 2002; Ohno, 2006); and a case of primary central nervous system lymphoma (PCNSL, a diffuse large B cell lymphoma confined to the brain) (Schwindt et al., 2006).
Cytogenetics Cytogenetics morphological Only two of the five cases reported by the Kyoto group showed a t(3;6)(q27;p21-22).
Genes involved and proteins BCL6 Location 3q27.3
Protein 706 amino acids; composed of a NH2-term BTB/POZ domain (amino acids 1-130 (32-99 according to Swiss-Prot) which mediates homodimerization and protein-protein interactions with other corepressors (including HDAC1 and NCOR2/SMRT to constitute a large repressing complex, another transcription repression domain (191-386), PEST sequences (300-417) with a KKYK motif (375-379), and six zinc finger at the C-term (518-541, 546-568, 574-596, 602-624, 630-652,
658-681), responsible for sequence specific DNA binding. Transcription repressor; recognizes the consensus sequence: TTCCT(A/C)GAA (Albagli-Curiel, 2003). Role in germinal centers of lymphoid follicles. BCL6 prevents ATM and TP53 to induce apoptosis in response to DNA rearrangements such as somatic hypermutation and class switch recombination. Therefore essential for normal B cell development.
HIST1H4I Location 6p22.1
Protein Component of the nucleosome. Histones play a major role in DNA repair, replication and transcription.
Result of the chromosomal anomaly Hybrid gene Description Breakpoints in HIST1H4 are located within the single exon to 3' of the terminal palindrome; the breakpoint in BCL6 was located within the major translocation cluster.
References Akasaka T, Miura I, Takahashi N, Akasaka H, Yonetani N, Ohno H, Fukuhara S, Okuma M. A recurring translocation, t(3;6)(q27;p21), in non-Hodgkin's lymphoma results in replacement of the 5' regulatory region of BCL6 with a novel H4 histone gene. Cancer Res. 1997 Jan 1;57(1):7-12
Akasaka H, Akasaka T, Kurata M, Ueda C, Shimizu A, Uchiyama T, Ohno H. Molecular anatomy of BCL6 translocations revealed by long-distance polymerase chain reaction-based assays. Cancer Res. 2000 May 1;60(9):2335-41

t(3;6)(q27;p22) HIST1H4I/BCL6 Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 278
Kurata M, Maesako Y, Ueda C, Nishikori M, Akasaka T, Uchiyama T, Ohno H. Characterization of t(3;6)(q27;p21) breakpoints in B-cell non-Hodgkin's lymphoma and construction of the histone H4/BCL6 fusion gene, leading to altered expression of Bcl-6. Cancer Res. 2002 Nov 1;62(21):6224-30
Albagli-Curiel O. Ambivalent role of BCL6 in cell survival and transformation. Oncogene. 2003 Jan 30;22(4):507-16
Ohno H. Pathogenetic and clinical implications of non-immunoglobulin ; BCL6 translocations in B-cell non-Hodgkin's lymphoma. J Clin Exp Hematop. 2006 Nov;46(2):43-53
Schwindt H, Akasaka T, Zühlke-Jenisch R, Hans V, Schaller C, Klapper W, Dyer MJ, Siebert R, Deckert M. Chromosomal translocations fusing the BCL6 gene to different partner loci are recurrent in primary central nervous system lymphoma and may be associated with aberrant somatic hypermutation or defective class switch recombination. J Neuropathol Exp Neurol. 2006 Aug;65(8):776-82
This article should be referenced as such:
Huret JL. t(3;6)(q27;p22) HIST1H4I/BCL6. Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):277-278.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 279
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
t(3;7)(q27;q32) FRA7H/BCL6 Jean-Loup Huret
Genetics, Dept Medical Information, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France (JLH)
Published in Atlas Database: December 2012
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0307q27q32ID1630.html DOI: 10.4267/2042/48873
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology Disease Non Hodgkin lymphoma
Clinics Only 3 cases to date of t(3;7)(q27;q32): a B-cell lymphoma cell line established from a 55-year-old male patient with terminal diffuse large B-cell lymphoma (DLBCL), a t(11;14)(q23;q32) and a complex karyotype (Schneider et al., 2008); a 54-year-old male patient with stade IV splenic marginal zone B-cell lymphoma (SMZL) and a complex karyotype with +3 who's survival was 9 years+ (Boonstra et al., 2003); and a female patient with stade IV SMZL and a complex karyotype (Ott et al., 2000).
Genetics Note The molecular event involving FRA7H and BCL6 has been uncovered only in the most recent report (Schneider et al., 2008).
Genes involved and proteins BCL6 Location 3q27.3
Protein 706 amino acids; composed of a NH2-term BTB/POZ domain (amino acids 1-130 (32-99 according to Swiss-Prot) which mediates homodimerization and protein-protein interactions with other corepressors (including HDAC1 and NCOR2/SMRT to constitute a large repressing complex, another transcription repression domain (191-
386), PEST sequences (300-417) with a KKYK motif (375-379), and six zinc finger at the C-term (518-541, 546-568, 574-596, 602-624, 630-652, 658-681), responsible for sequence specific DNA binding. Transcription repressor; recognizes the consensus sequence: TTCCT(A/C)GAA (Albagli-Curiel, 2003). Role in germinal centers of lymphoid follicles. BCL6 prevents ATM and TP53 to induce apoptosis in response to DNA rearrangements such as somatic hypermutation and class switch recombination. Therefore essential for normal B cell development.
FRA7H Location 7q32.3
Note Fragile site, aphidicolin type, common.
Result of the chromosomal anomaly Hybrid gene Description The breakpoint at 7q32 maps within FRA7H; the breakpoint on chromosome 3 lies upstream of exon 1, out of both the major breakpoint region (MBR) and the alternative breakpoint regions (ABR). Hence, BCL6 is constitutively, albeit moderately, expressed (Schneider et al., 2008).
References Ott MM, Rosenwald A, Katzenberger T, Dreyling M, Krumdiek AK, Kalla J, Greiner A, Ott G, Müller-Hermelink HK. Marginal zone B-cell lymphomas (MZBL) arising at different sites represent different biological entities. Genes Chromosomes Cancer. 2000 Aug;28(4):380-6

t(3;7)(q27;q32) FRA7H/BCL6 Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 280
Albagli-Curiel O. Ambivalent role of BCL6 in cell survival and transformation. Oncogene. 2003 Jan 30;22(4):507-16
Boonstra R, Bosga-Bouwer A, van Imhoff GW, Krause V, Palmer M, Coupland RW, Dabbagh L, van den Berg E, van den Berg A, Poppema S. Splenic marginal zone lymphomas presenting with splenomegaly and typical immunophenotype are characterized by allelic loss in 7q31-32. Mod Pathol. 2003 Dec;16(12):1210-7
Schneider B, Nagel S, Kaufmann M, Winkelmann S, Bode J, Drexler HG, MacLeod RA. T(3;7)(q27;q32) fuses BCL6 to a non-coding region at FRA7H near miR-29. Leukemia. 2008 Jun;22(6):1262-6
This article should be referenced as such:
Huret JL. t(3;7)(q27;q32) FRA7H/BCL6. Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):279-280.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 281
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
t(3;9)(q27;p24) DMRT1/BCL6 Jean-Loup Huret
Genetics, Dept Medical Information, University of Poitiers, CHU Poitiers Hospital, F-86021 Poitiers, France (JLH)
Published in Atlas Database: December 2012
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0309q27p24ID1488.html DOI: 10.4267/2042/48874
This article is an update of : Huret JL. t(3;9)(q27;p24). Atlas Genet Cytogenet Oncol Haematol 2010;14(6):595. This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics and pathology Disease Non Hodgkin lymphoma
Clinics The t(3;9)(q27;p24) was found in a gastric lymphoma, from a study with no individual data (Chen et al., 2006).
Genes involved and proteins BCL6 Location 3q27.3
Protein 706 amino acids; composed of a NH2-term BTB/POZ domain (amino acids 1-130 (32-99 according to Swiss-Prot) which mediates homodimerization and protein-protein interactions with other corepressors (including HDAC1 and NCOR2/SMRT to constitute a large repressing complex, another transcription repression domain (191-386), PEST sequences (300-417) with a KKYK motif (375-379), and six zinc finger at the C-term (518-541, 546-568, 574-596, 602-624, 630-652, 658-681), responsible for sequence specific DNA binding. Transcription repressor; recognizes the consensus sequence: TTCCT(A/C)GAA (Albagli-Curiel, 2003). Role in germinal centers of lymphoid follicles. BCL6 prevents ATM and TP53 to induce apoptosis in response to DNA rearrangements such as somatic hypermutation and class switch recombination. Therefore essential for normal B cell development.
DMRT1 Location 9p24.3
Protein Transcription factor that plays a key role in male sex determination in various animal species including homo sapiens (Herpin and Schartl, 2011). Susceptibility locus for testicular germ cell tumor (Turnbull et al., 2010).
Result of the chromosomal anomaly Hybrid gene Description The breakpoint in DMRT1 is between exons 3 and 4, and between exon 1 and 2 in BCL6. 5' DMRT1-3' BCL6.
References Albagli-Curiel O. Ambivalent role of BCL6 in cell survival and transformation. Oncogene. 2003 Jan 30;22(4):507-16
Chen YW, Hu XT, Liang AC, Au WY, So CC, Wong ML, Shen L, Tao Q, Chu KM, Kwong YL, Liang RH, Srivastava G. High BCL6 expression predicts better prognosis, independent of BCL6 translocation status, translocation partner, or BCL6-deregulating mutations, in gastric lymphoma. Blood. 2006 Oct 1;108(7):2373-83
Turnbull C, Rapley EA, Seal S, Pernet D, Renwick A, Hughes D, Ricketts M, Linger R, Nsengimana J, Deloukas P, Huddart RA, Bishop DT, Easton DF, Stratton MR, Rahman N. Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat Genet. 2010 Jul;42(7):604-7

t(3;9)(q27;p24) DMRT1/BCL6 Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 282
Herpin A, Schartl M. Dmrt1 genes at the crossroads: a widespread and central class of sexual development factors in fish. FEBS J. 2011 Apr;278(7):1010-9
This article should be referenced as such:
Huret JL. t(3;9)(q27;p24) DMRT1/BCL6. Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):281-282.

Solid Tumour Section Review
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 283
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Head and Neck: Oral leukoplakia Patrícia Carlos Caldeira, Maria Auxiliadora Vieira do Carmo
Department of Oral Pathology and Surgery, School of Dentistry, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil (PCC, MAVd)
Published in Atlas Database: November 2012
Online updated version : http://AtlasGeneticsOncology.org/Tumors/OralLeukoplakiaID5937.html DOI: 10.4267/2042/48875
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Note Oral leukoplakia (OL) is the most common potentially malignant disorder of the oral mucosa (Neville and Day, 2002; Haya-Fernández et al., 2004; WHO, 2005). Besides oral leukoplakia, actinic cheilitis, lichen planus, and erythroplakia are also considered potentially malignant conditions affecting the oral cavity. These lesions can precede oral squamous cell carcinoma, which is the most common malignancy of the oral cavity (Neville and Day, 2002; Haya-Fernández et al., 2004). It is believed that those conditions are reasonably classified as "potentially malignant" as it has been observed that some lesions evolved to malignant ones during follow-up. In addition, typical alterations of potentially malignant lesions are seen co-existing in the margins of squamous cell carcinoma. Moreover, a proportion of these lesions show cytological, morphological, chomosomic, genomic, and molecular alterations that are also observed in malignant lesions (Warnakulasuriya et al., 2007).
Classification Note In spite of diverse and even more recently published definitions for oral leukoplakia, the most widely known is still the one proposed by World Health Organization (WHO) in 1978, which states that leukoplakia is a predominantly white patch that cannot be characterized clinically or histopathologically as any other definable lesion (Kramer et al., 1978; WHO, 2005). Oral leukoplakias can be classified from the clinal or histopathological viewpoint. Such classifications are discussed in detail in the topics below.
Clinics and pathology Etiology Tobacco use is the main risk factor associated to OL development. OL is six times more frequent among smokers than non-smokers (van der Waal, 2009). The effects of alcohol, betel, and diet are associated as well, but their exact role is yet to be established (Neville and Day, 2002; Campisi et al., 2007; Napier and Speight, 2008; van der Waal, 2009). A recently published review showed that there is not enough evidence for a casual association between human papilloma virus and OL (Feller and Lemmer, 2012). Besides, there are the idiopathic OL, for which no obvious aetiological factor can be identified. It is believed that such lesions are significantly more prone to develop into cancer than those OL with known causative factors (Napier and Speight, 2008).
Epidemiology It is accepted that the OL prevalence varies from 1% to 5% (Napier and Speight, 2008; van der Waal, 2009), with a global prevalence estimated to be 2.6% (Petti, 2003). However, isolated reports show rates from 0.5% to 26.92% (Petti, 2003; Napier and Speight, 2008). Middle-aged and elderly man are the most affected individuals, and growing indexes are observed towards age. OL often arises in cheek and alveolar mucosa (Neville and Day, 2002). Conversely, lesions in the floor of the mouth and lateral border of tongue seem to present displastic or malignant alterations more frequently (Neville and Day, 2002; Napier and Speight, 2008).
Clinics OL can present as homogeneous and non-homogeneous lesions. Homogeneous OL is a white patch slightly elevated, thin, white to gray, uniform, and can present

Head and Neck: Oral leukoplakia Caldeira PC, do Carmo MAV
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 284
well defined borders or gradually mix with normal adjacent mucosa (Figure 1). Non-homogeneous OL can be nodular, verrucous, or speckled (erythroplastic) (Figure 2) (Warnakulasuriya et al., 2007; van der Waal, 2009). The proliferative verrucous leukoplakia presents a multifocal evolvement, mainly in elderly female patients who do not present known risk factors (Figure 3). These lesions are usually resistant to treatment and show a high risk for malignant transformation (Warnakulasuriya et al., 2007; van der Waal, 2009). Some alterations of the oral mucosa can mimic OL, and these lesions must be considered as OL differential diagnosis. So, for the establishment of a correct diagnosis of OL, such lesions must be excluded (Warnakulasuriya et al., 2007; van der Waal, 2009):
1. Frictional lesion 2. Candidiasis 3. Linea alba 4. Leukoedema 5. Chemical injuries 6. Hairy leukoplakia 7. Nicotinic stomatitis Once a provisional clinical diagnosis of OL was made, a biopsy must be performed in order to obtain the histopathological features. This is of paramount importance because it is believed that the presence and degree of epithelial dysplasia is a great indicator of evolution and prognosis (see below) (Warnakulasuriya et al., 2008).
Figure 1: Homogeneous oral leukoplakia in the left lateral border and ventrum of the tongue. Figure 2: Non-homogeneous oral leukoplakia. White plaques intermixed with red patches.

Head and Neck: Oral leukoplakia Caldeira PC, do Carmo MAV
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 285
Figure 3: Proliferative verrucous leukoplakia: multifocal involvement affecting labial mucosa, alveolar mucosa, and palate.
Pathology Microscopically, OL can vary from hyperkeratotic epithelium to lesions with different degrees of dysplasia (WHO, 2005; Brennan et al., 2007). The term "dysplasia" is generally employed in the sense of a disordered development (Izumo, 2011). In a stratified squamous epithelium, architectural disturbances affecting normal maturation and stratification may occur. When such alterations are accompanied by cytological atypia, which can be detected as variations in the size and shape of the keratinocytes, the term "dysplasia" is applied (WHO, 2005; Warnakulasuriya et al., 2008). The frequencies of dysplastic or malignant alterations in OL vary from 15.6% to 39.2%, and a rate of 19.9% was found in a retrospective study of 3300 white lesions of the oral cavity (Waldron and Shafer, 1975). Despite many efforts towards new evaluative methods, the histological analysis is still the most useful method for grading epithelial dysplasia in OL (Warnakulasuriya et al., 2008). In this field, the WHO's system for grading epithelial dysplasia in OL is widely accepted among pathologists. However, it is not able to reflect the clinical behaviour of every single lesion and does not provide a clear therapeutic guideline to clinicians (Izumo, 2011).
Moreover, in spite of its wide acceptance, this system presents great variability and low reproducibility (Warnakulasuriya et al., 2008; van der Waal, 2009). According to it, lesions are classified considering the architectural features and cytological alterations listed below (WHO, 2005). Architectural features: - Irregular epithelial stratification - Loss of polarity of basal cells - Drop-shaped rete ridges - Increased number of mitotic figures - Abnormally superficial mitoses - Premature keratinisation in single cells (dyskeratosis) Cytological alterations - Nuclear pleomorphism: abnormal variation in nuclear shape - Cellular pleomorphism: abnormal variation in cell shape - Anisonucleosis: abnormal variation in nuclear size - Anisocytosis: abnormal variation in cell size - Increased nuclear size - Increased nuclear-cytoplasm ratio - Atypical mitotic figures - Increased number and size of nucleoli Considering the epithelium divided into "thirds", lesions are classified into five categories:

Head and Neck: Oral leukoplakia Caldeira PC, do Carmo MAV
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 286
1. Hyperplasia: increase in cell number in the spinous layer and/or in the basal/parabasal cell layers. There is regular stratification and no cellular atypia (Figure 4). 2. Mild dysplasia: architectural disturbance only in the lower third of the epithelium with cytological atypia (Figure 5). 3. Moderate dysplasia: architectural disturbance extending into the middle third of the epithelium is the initial criteria, but the degree of cytological atypia may require upgrading it to "severe" (Figure 6). 4. Severe dysplasia: architectural disturbance affecting greater than two thirds of the epithelium, with cytological atypia (Figure 7). 5. Carcinoma "in situ": theorically, indicates that malignant transformation has occurred but invasion has not. Full or almost full thickness architectural disturbance is present in viable cellular layers with pronounced cellular atypia. Atypical mitotic figures and abnormal superficial mitoses are common. Concerning the microscopic classification schemes for OL, a new binary system was proposed (Kujan et al., 2006). Accoding to it, pathologists would observe the same morphological criteria used in the WHO classification, but lesions would be classified as low-risk OL (former "no/mild/questionable" dysplasia) or as high-risk OL (former "moderate/severe" dysplasia). Shorten the degrees of dysplasia from five, i.e. no, mild, moderate, and severe, to two - low-risk and high-risk - would provide a more feasible and reproducible classification system. Also, it could offer a more reliable criteria upon which to rely for the selection of
patient treatment. Accordingly, OL classified as "high-risk" would be more prone to develop into cancer and, thus, should be removed. On the other hand, low-risk OL could be clinically followed up as they are less expected to evolve (Kujan et al., 2006; Warnakulasuriya et al., 2008; van der Waal, 2009). In a study with 218 patients with OL, the authors reported that high-risk OL was associated with a 4.57-fold increased risk for malignant transformation, compared with low-risk OL (Liu et al., 2010). Moreover, in another research with 138 patients with histologically confirmed oral dysplasia, 115 had OL and 23 had lichen planus. From these 138 lesions, 37 (26.8%) developed into cancer and the "high-risk" degree of dysplasia was an independent risk factor for transformation (Liu et al., 2011). The authors then suggest the utilization of high-risk dysplasia as a significant indicator for evaluating malignant transformation risk in patients with potentially malignant lesions. In a recently published paper, our research group showed statistically significant differences for hMLH1 - a DNA repair protein - , p53 - a tumor supressor protein - , and AgNOR - an indicative of cell proliferation - indexes between low- and high-risk OL. This suggests that the biological processes linked to the impairment of those proteins remain enhancing from low-risk OL to high-risk OL. Thus, the use of the binary system would give support to a more reliable clinical approach involving the removal of high-risk OL (Caldeira et al., 2012).
Figure 4: OL showing hyperplasia. Notice that there is regular stratification and no cellular atypia. Hyperkeratosis is present. Figure 5: OL showing mild dysplasia: the lower third of the epithelium shows architectural disturbance and cytological atypia.

Head and Neck: Oral leukoplakia Caldeira PC, do Carmo MAV
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 287
Figure 6: OL with moderate dysplasia. Architectural disturbance affecting two thirds of the epithelium. Notice dyskeratosis in the granular layer of the epithelium (arrow). Figure 7: OL with severe dysplasia: architectural disturbance affecting greater than two thirds of the epithelium. Pleomorphic cells are seen in the upper third of the epithelium (arrows). Figure 8: p53 immunoexpression in OL, 200x magnification. (A): OL with mild dysplasia shows few immunopositive cells (arrows). (B): OL with moderate dysplasia shows an increased number of p53 labeled cells (arrows). Figure 9: hMLH1 immunexpression in OL, 200x magnification. (A): OL with mild dysplasia shows several keratinocytes labeled for hMLH1. (B): OL with severe dysplasia presents a decrease in the immunoexpression of hMLH1.

Head and Neck: Oral leukoplakia Caldeira PC, do Carmo MAV
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 288
Figure 10: AgNOR staining, 1000x magnification. (A): in normal oral epithelium, keratinocytes exhibit lower AgNOR counting than in (B) keratinocytes in OL with severe dysplasia.
Treatment Surgical excision, cryosurgery, laser surgery, topical or systemic retinoids, therapy with mouth rinses with attenuated adenovirus, and photodynamic therapy are possible therapeutics (Brennan et al., 2007; Lodi and Porter, 2008; van der Waal, 2009).
Evolution OL may persist unchanged, progress, regress, or disappear (Napier and Speight, 2008). The malignant transformation risk varies from 3.6% to 36.0%, and some features as presence and degree of dysplasia, female gender, time of duration, non-smoker patient, location at floor of the mouth or tongue, size higher than 200mm2, and non-homogeneous type, seem to be associated with a worse prognosis (Cruz et al., 2002; Holmstrup et al., 2006; Hsue et al., 2007; Smith et al., 2009; van der Waal, 2009). Infection with human papilloma virus does not seem to be related to the progression of OL (Feller and Lemmer, 2012).
Prognosis Recurrence rates are highly variable among studies, from 0 to 30.0% (van der Waal, 2009).
Genetics Note Many efforts have been done to identify molecular markers to predict cancer development in OL. According to the review by Smith et al. (2009), p53, Ki-67, and PCNA are the most frequently investigated markers, but loss of heterozygosity (LOH), survivin, matrix metalloproteinase (MMP9), and DNA content are pointed as potential markers for progression of OL. Nevertheless, the presence and degree of epithelial dysplasia in OL is yet regarded as the most relevant indicator of progression and prognosis, influencing the
management of the patients (Napier and Speight, 2008; Warnakulasuriya et al., 2008; van der Waal, 2009). Ki-67 is a cell cycle associated protein, of which expression is associated with cell proliferation. Ki-67 immunoexpression is used as a proliferation marker in pathology. Kodani et al. (2001) showed that OL presented lower indexes of Ki-67 than oral squamous cell carcinoma. Also, Zhao et al. (2005) demonstrated that OL with mild dysplasia shows low levels of Ki-67, while severe dysplasia shows a significantly higher expression than oral normal mucosa and mild dysplasia. Our research group investigated the immunoexpression of hMLH1 (a protein of the mismatch repair system) in OL with different degrees of dysplasia. We found that hMLH1 indexes decreased from a lower degree of dysplasia to a higher one, despite statistical significance. So, hMLH1 immunoexpression was inversely related to the OL degree of dysplasia. Our findings also suggest a role of such alterations in this pathway of DNA repair as an early event in oral carcinogenesis (Caldeira et al., 2011a). Briefly, some information about the most frequent genetic alterations of OL are shown below. A complete and detailed revision on this topic was published by Mithani et al. (2007). p53 alterations: The p53 protein can induce DNA repair, cell cycle arrest, cell death or senescence, showing a pivotal role in tumor avoidance (Joerger and Fersht, 2008). Its alteration is a common finding in human cancers, including those of oral mucosa (Kurokawa et al., 2003). Investigations demonstrated that the normal oral mucosa presents negative or low indexes of p53 immunoexpression (Kurokawa et al., 2003; Fan et al., 2006; Caldeira et al., 2011b). Likewise, p53 immunopositive cells were identified in OL with mild dysplasia, with increasing indexes from hyperplasia, to dysplastic lesions (Figure 8) and to oral

Head and Neck: Oral leukoplakia Caldeira PC, do Carmo MAV
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 289
squamous cell carcinoma, with immunopositivity found in superficial layers of moderate and severe dysplasias (Kerdpon et al., 1997; Cruz et al., 2002; Kurokawa et al., 2003; Caldeira et al., 2011b). The detection of p53 in oral dysplastic lesions prompted many investigators to suggest that its abnormalities may constitute an early event in carcinogenesis. Loss of heterozygosity (LOH): describes the elimination of a genetic loci containing tumor suppressor genes. In OL, LOH of the chromosome arms 3p and 9p seem to be related to a higher risk of malignant transformation. Fifty percent of OL contains allelic loss of either the 3p or 9p chromosome arms (Mithani et al., 2007). Microsatellite instability (MSI) and the mammalian mismatch repair system (MMR): Microsatellites are DNA regions in which multiple repeated sequencies of nucleotides are found. These regions are prone to the occurence of mismatched DNA, developing the MSI phenotype (Jascur and Boland, 2006; Jiricny, 2006). MSI was detected in many OL and there is a trend toward an increased prevalence of MSI in more aggressive histologic OL lesions (Ha et al., 2002). The MMR is responsible for maintaining genomic stability during repeated duplication, and microsatellites regions are hypersensitive to MMR dysfunction. The immunoexpression of hMLH1 - one of the main MMR protein - was shown to decrease in OL with more severe grades of dysplasia (Figure 9) (Caldeira et al., 2011a). Taken together, these results may suggest that the altered function of MMR and the occurence of MSI could be early events in the carcinogenic process, but these findings still need more investigation. Methylation / hypermethylation: is an epigenetic alteration which can inactivate genes. In OL, it was described to occur in RAR-b2, p16, hMLH1, hMSH2, and MGMT (Ha et al., 2002; López et al., 2003; Youssef et al., 2004; Sengupta et al., 2007). AgNOR number: AgNOR staining technique is used to assess cellular proliferation, and normal oral epithelium showed lower AgNOR number than dysplastic OL (Figure 10), which in turn presents lower indexes than oral squamous cell carcinoma (Chattopadhyay et al., 1994; Caldeira et al., 2011b). It was suggested that mean AgNOR number would be useful in distinguishing OL with mild and moderate dysplasia (Chattopadhyay and Ray, 2008). Telomerase activity: these are the enzymes that degradate telomers, which are a sequence of nucleotides that prevents DNA to undergo degradation and fusion. The telomerase activity was detected in OL.
To be noted Note In conclusion, OL is the most common potentially malignant oral disorder preceding oral squamous cell carcinoma. OL is often related to tobacco use and lesions may persist unchanged, enlarge, reduce or even
disappear. Nevertheless, the presence and degree of epithelial dysplasia is still considered the most important predictor factor for malignant transformation. Also, it seems that the lesion duration, patient's age, gender, the affected site, clinical appearance, and smoking habit are related to the risk of malignant transformation. To date, as pointed out above, no single molecular marker is validated as a predictor, despite several investigations.
References Waldron CA, Shafer WG. Leukoplakia revisited. A clinicopathologic study 3256 oral leukoplakias. Cancer. 1975 Oct;36(4):1386-92
Kramer IR, Lucas RB, Pindborg JJ, Sobin LH. Definition of leukoplakia and related lesions: an aid to studies on oral precancer. Oral Surg Oral Med Oral Pathol. 1978 Oct;46(4):518-39
Chattopadhyay A, Chawda JG, Doshi JJ. Silver-binding nucleolar organizing regions: a study of oral leukoplakia and squamous cell carcinoma. Int J Oral Maxillofac Surg. 1994 Dec;23(6 Pt 1):374-7
Kerdpon D, Rich AM, Reade PC. Expression of p53 in oral mucosal hyperplasia, dysplasia and squamous cell carcinoma. Oral Dis. 1997 Jun;3(2):86-92
Kodani I, Shomori K, Osaki M, Kuratate I, Ryoke K, Ito H. Expression of minichromosome maintenance 2 (MCM2), Ki-67, and cell-cycle-related molecules, and apoptosis in the normal-dysplasia-carcinoma sequence of the oral mucosa. Pathobiology. 2001;69(3):150-8
Cruz I, Napier SS, van der Waal I, Snijders PJ, Walboomers JM, Lamey PJ, Cowan CG, Gregg TA, Maxwell P, Meijer CJ. Suprabasal p53 immunoexpression is strongly associated with high grade dysplasia and risk for malignant transformation in potentially malignant oral lesions from Northern Ireland. J Clin Pathol. 2002 Feb;55(2):98-104
Ha PK, Pilkington TA, Westra WH, Sciubba J, Sidransky D, Califano JA. Progression of microsatellite instability from premalignant lesions to tumors of the head and neck. Int J Cancer. 2002 Dec 20;102(6):615-7
Neville BW, Day TA. Oral cancer and precancerous lesions. CA Cancer J Clin. 2002 Jul-Aug;52(4):195-215
Kurokawa H, Matsumoto S, Murata T, Yamashita Y, Tomoyose T, Zhang M, Fukuyama H, Takahashi T. Immunohistochemical study of syndecan-1 down-regulation and the expression of p53 protein or Ki-67 antigen in oral leukoplakia with or without epithelial dysplasia. J Oral Pathol Med. 2003 Oct;32(9):513-21
López M, Aguirre JM, Cuevas N, Anzola M, Videgain J, Aguirregaviria J, Martínez de Pancorbo M. Gene promoter hypermethylation in oral rinses of leukoplakia patients--a diagnostic and/or prognostic tool? Eur J Cancer. 2003 Nov;39(16):2306-9
Petti S. Pooled estimate of world leukoplakia prevalence: a systematic review. Oral Oncol. 2003 Dec;39(8):770-80
Haya-Fernández MC, Bagán JV, Murillo-Cortés J, Poveda-Roda R, Calabuig C. The prevalence of oral leukoplakia in 138 patients with oral squamous cell carcinoma. Oral Dis. 2004 Nov;10(6):346-8
Youssef EM, Lotan D, Issa JP, Wakasa K, Fan YH, Mao L, Hassan K, Feng L, Lee JJ, Lippman SM, Hong WK, Lotan R. Hypermethylation of the retinoic acid receptor-beta(2) gene in

Head and Neck: Oral leukoplakia Caldeira PC, do Carmo MAV
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 290
head and neck carcinogenesis. Clin Cancer Res. 2004 Mar 1;10(5):1733-42
Barnes L, Eveson JW, Reichart P, Sidransky D, editors.. Tumours of the Oral Cavity and Oropharynx. In: World Health Organization. Pathology and genetics. Head neck tumors. Lyon: IARC Press. 2005:177-9.
Zhao J, Guo B, Ma SC, Zhou XD.. Expression of p53 and Ki-67 genes in epithelial dysplasia from old oral mucosa and clinical significance. Sichuan Da Xue Xue Bao Yi Xue Ban. 2005 Sep;36(5):689-91.
Fan GK, Chen J, Ping F, Geng Y.. Immunohistochemical analysis of P57(kip2), p53 and hsp60 expressions in premalignant and malignant oral tissues. Oral Oncol. 2006 Feb;42(2):147-53. Epub 2005 Oct 24.
Holmstrup P, Vedtofte P, Reibel J, Stoltze K.. Long-term treatment outcome of oral premalignant lesions. Oral Oncol. 2006 May;42(5):461-74. Epub 2005 Nov 28.
Jascur T, Boland CR.. Structure and function of the components of the human DNA mismatch repair system. Int J Cancer. 2006 Nov 1;119(9):2030-5. (REVIEW)
Jiricny J.. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006 May;7(5):335-46. (REVIEW)
Kujan O, Oliver RJ, Khattab A, Roberts SA, Thakker N, Sloan P.. Evaluation of a new binary system of grading oral epithelial dysplasia for prediction of malignant transformation. Oral Oncol. 2006 Nov;42(10):987-93. Epub 2006 May 30.
Brennan M, Migliorati CA, Lockhart PB, Wray D, Al-Hashimi I, Axell T, Bruce AJ, Carpenter W, Eisenberg E, Epstein JB, Holmstrup P, Jontell M, Nair R, Sasser H, Schifter M, Silverman B, Thongprasom K, Thornhill M, Warnakulasuriya S, van der Waal I.. Management of oral epithelial dysplasia: a review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007 Mar;103 Suppl:S19.e1-12. Epub 2007 Jan 25. (REVIEW)
Campisi G, Panzarella V, Giuliani M, Lajolo C, Di Fede O, Falaschini S, Di Liberto C, Scully C, Lo Muzio L.. Human papillomavirus: its identity and controversial role in oral oncogenesis, premalignant and malignant lesions (review). Int J Oncol. 2007 Apr;30(4):813-23. (REVIEW)
Hsue SS, Wang WC, Chen CH, Lin CC, Chen YK, Lin LM.. Malignant transformation in 1458 patients with potentially malignant oral mucosal disorders: a follow-up study based in a Taiwanese hospital. J Oral Pathol Med. 2007 Jan;36(1):25-9.
Mithani SK, Mydlarz WK, Grumbine FL, Smith IM, Califano JA.. Molecular genetics of premalignant oral lesions. Oral Dis. 2007 Mar;13(2):126-33. (REVIEW)
Sengupta S, Chakrabarti S, Roy A, Panda CK, Roychoudhury S.. Inactivation of human mutL homolog 1 and mutS homolog 2 genes in head and neck squamous cell carcinoma tumors and leukoplakia samples by promoter hypermethylation and its relation with microsatellite instability phenotype. Cancer. 2007 Feb 15;109(4):703-12.
Warnakulasuriya S, Johnson NW, van der Waal I.. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J Oral Pathol Med. 2007 Nov;36(10):575-80. (REVIEW)
Chattopadhyay A, Ray JG.. AgNOR cut-point to distinguish mild and moderate epithelial dysplasia. J Oral Pathol Med. 2008 Feb;37(2):78-82. doi: 10.1111/j.1600-0714.2007.00585.x.
Joerger AC, Fersht AR.. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557-82. doi: 10.1146/annurev.biochem.77.060806.091238. (REVIEW)
Lodi G, Porter S.. Management of potentially malignant disorders: evidence and critique. J Oral Pathol Med. 2008 Feb;37(2):63-9. doi: 10.1111/j.1600-0714.2007.00575.x.
Napier SS, Speight PM.. Natural history of potentially malignant oral lesions and conditions: an overview of the literature. J Oral Pathol Med. 2008 Jan;37(1):1-10. (REVIEW)
Warnakulasuriya S, Reibel J, Bouquot J, Dabelsteen E.. Oral epithelial dysplasia classification systems: predictive value, utility, weaknesses and scope for improvement. J Oral Pathol Med. 2008 Mar;37(3):127-33. doi: 10.1111/j.1600-0714.2007.00584.x.
Smith J, Rattay T, McConkey C, Helliwell T, Mehanna H.. Biomarkers in dysplasia of the oral cavity: a systematic review. Oral Oncol. 2009 Aug;45(8):647-53. doi: 10.1016/j.oraloncology.2009.02.006. Epub 2009 May 12. (REVIEW)
van der Waal I.. Potentially malignant disorders of the oral and oropharyngeal mucosa; terminology, classification and present concepts of management. Oral Oncol. 2009 Apr-May;45(4-5):317-23. doi: 10.1016/j.oraloncology.2008.05.016. Epub 2008 Jul 31. (REVIEW)
Liu W, Wang YF, Zhou HW, Shi P, Zhou ZT, Tang GY.. Malignant transformation of oral leukoplakia: a retrospective cohort study of 218 Chinese patients. BMC Cancer. 2010 Dec 16;10:685. doi: 10.1186/1471-2407-10-685.
Caldeira PC, Abreu MH, Batista AC, do Carmo MA.. hMLH1 immunoexpression is related to the degree of epithelial dysplasia in oral leukoplakia. J Oral Pathol Med. 2011a Feb;40(2):153-9. doi: 10.1111/j.1600-0714.2010.00963.x. Epub 2010 Oct 24.
Caldeira PC, Aguiar MC, Mesquita RA, do Carmo MA.. Oral leukoplakias with different degrees of dysplasia: comparative study of hMLH1, p53, and AgNOR. J Oral Pathol Med. 2011b Apr;40(4):305-11. doi: 10.1111/j.1600-0714.2010.01000.x. Epub 2011 Jan 5.
Izumo T.. Oral premalignant lesions: from the pathological viewpoint. Int J Clin Oncol. 2011 Feb;16(1):15-26. doi: 10.1007/s10147-010-0169-z. Epub 2011 Jan 14. (REVIEW)
Liu W, Bao ZX, Shi LJ, Tang GY, Zhou ZT.. Malignant transformation of oral epithelial dysplasia: clinicopathological risk factors and outcome analysis in a retrospective cohort of 138 cases. Histopathology. 2011 Oct;59(4):733-40. doi: 10.1111/j.1365-2559.2011.03938.x. Epub 2011 Sep 14.
Caldeira PC, Abreu MH, do Carmo MA.. Binary system of grading oral epithelial dysplasia: evidence of a bearing to the scores of an immunohistochemical study. J Oral Pathol Med. 2012 Jul;41(6):452-3. doi: 10.1111/j.1600-0714.2012.01128.x. Epub 2012 Jan 30.
Feller L, Lemmer J.. Oral Leukoplakia as It Relates to HPV Infection: A Review. Int J Dent. 2012;2012:540561. doi: 10.1155/2012/540561. Epub 2012 Feb 28. (REVIEW)
This article should be referenced as such:
Caldeira PC, do Carmo MAV. Head and Neck: Oral leukoplakia. Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):283-290.

Solid Tumour Section Review
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 291
INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Thyroid: Medullary carcinoma Yash Somnay, David Schneider, Haggi Mazeh
Section of Endocrine Surgery, Department of Surgery, University of Wisconsin, K3/704 Clinical Science Center, 600 Highland Avenue, Madison, WI 53792, USA (YS, DS, HM)
Published in Atlas Database: November 2012
Online updated version : http://AtlasGeneticsOncology.org/Tumors/MedullaryThyroidCarcID5080.html DOI: 10.4267/2042/48876
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2013 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity Other names MTC
Classification Note Medullary thyroid cancers (MTC) are rare tumors of neuroendocrine origin that arise from parafollicular C cells which secrete a variety of peptides and hormones including calcitonin. As opposed to the more common papillary and follicular thyroid cancer subtypes, MTC represents a rare and under-characterized form of cancer, and can cause death if untreated (Taccaliti et al., 2011). MTC can be either sporadic, usually isolated to one thyroid lobe, or familial, the latter of which is defined as the cancer syndrome known as Multiple Endocrine Neoplasia type 2 (MEN2) (Frank-Raue et al., 2010). MEN2 is the result of the autosomally dominant missense gain of function mutation in the RET (Rearranged during Transfection) proto-oncogene. MEN2 can be further subclassified into MEN2A, MEN2B and Familial Medullary Thyroid Carcinoma (FMTC). MEN2A is defined by the occurrence of medullary thyroid carcinoma (MTC), in conjunction with pheochromocytomas and primary hyperparathyroidism. MEN2B, is definied by the presence of MTC, pheochromocytomas, ganglioneuromatosis of the gastrointestinal tract, mucosal neuromas of the lips and tongue, and a Marfanoid body habitus (Frank-Raue et al., 2010). FMTC occurs when MTC is the only clinical feature, rarely with other endocrine neoplasias. Offspring of affected carriers of the RET mutation have a 50% chance of inheriting the mutation.
Clinics and pathology Disease Medullary thyroid cancer
Note Patients with sporadic MTC usually present with a neck mass while patients with hereditary MTCs that are diagnosed as mutation carriers should undergo prophylactic thyroidectomy before the onset of any symptoms. Sporadic MTC patients often present with metastasis to cervical and paratracheal lymph nodes. The diagnosis of MTC is based on history, physical exam, calcitonin and CEA levels, imaging, and fine needle aspiration biopsy. Every patient with diagnosed MTC should undergo genetic evaluation for the presence of the RET mutation. Histologically, tumors appear with hyperplastic parafollicular C-cells and predominantly present bilaterally (Taccaliti et al., 2011). Sporadic MTC generally presents as a single tumor confined to one thyroid lobe. The prognosis of MTC is better than poorly differentiated, malignant anaplastic thyroid cancer, but worse than more differentiated and benign papillary and follicular thyroid cancer. Therefore, an early diagnosis is necessary for improving recurrence and survival rates in these patients (Taccaliti et al., 2011).
Phenotype / cell stem origin Their origin is characteristically from neural crest cells. These cells arise from the convergence between the dorsal ectoderm and the neural tube. Neural crest cells eventually give rise to the chromaffin cells of the thyroid C cells in addition to chief cells of the extra-adrenal paraganglia, and
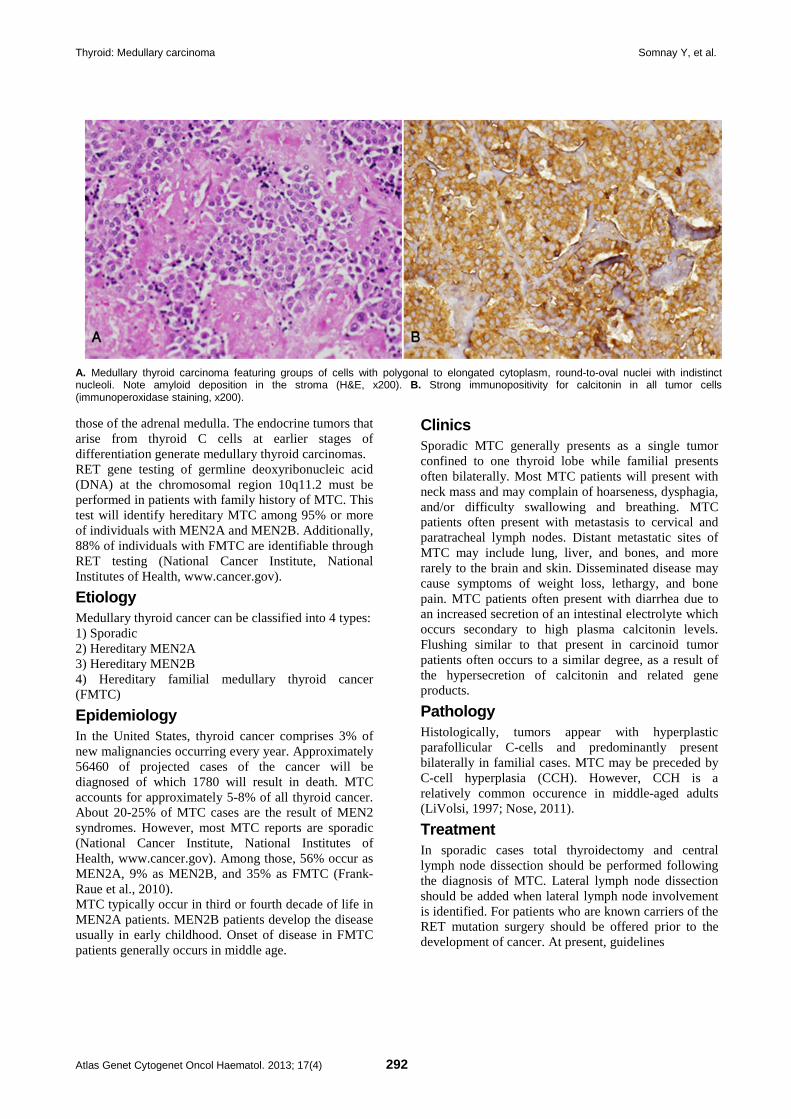
Thyroid: Medullary carcinoma Somnay Y, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 292
A. Medullary thyroid carcinoma featuring groups of cells with polygonal to elongated cytoplasm, round-to-oval nuclei with indistinct nucleoli. Note amyloid deposition in the stroma (H&E, x200). B. Strong immunopositivity for calcitonin in all tumor cells (immunoperoxidase staining, x200). those of the adrenal medulla. The endocrine tumors that arise from thyroid C cells at earlier stages of differentiation generate medullary thyroid carcinomas. RET gene testing of germline deoxyribonucleic acid (DNA) at the chromosomal region 10q11.2 must be performed in patients with family history of MTC. This test will identify hereditary MTC among 95% or more of individuals with MEN2A and MEN2B. Additionally, 88% of individuals with FMTC are identifiable through RET testing (National Cancer Institute, National Institutes of Health, www.cancer.gov).
Etiology Medullary thyroid cancer can be classified into 4 types: 1) Sporadic 2) Hereditary MEN2A 3) Hereditary MEN2B 4) Hereditary familial medullary thyroid cancer (FMTC)
Epidemiology In the United States, thyroid cancer comprises 3% of new malignancies occurring every year. Approximately 56460 of projected cases of the cancer will be diagnosed of which 1780 will result in death. MTC accounts for approximately 5-8% of all thyroid cancer. About 20-25% of MTC cases are the result of MEN2 syndromes. However, most MTC reports are sporadic (National Cancer Institute, National Institutes of Health, www.cancer.gov). Among those, 56% occur as MEN2A, 9% as MEN2B, and 35% as FMTC (Frank-Raue et al., 2010). MTC typically occur in third or fourth decade of life in MEN2A patients. MEN2B patients develop the disease usually in early childhood. Onset of disease in FMTC patients generally occurs in middle age.
Clinics Sporadic MTC generally presents as a single tumor confined to one thyroid lobe while familial presents often bilaterally. Most MTC patients will present with neck mass and may complain of hoarseness, dysphagia, and/or difficulty swallowing and breathing. MTC patients often present with metastasis to cervical and paratracheal lymph nodes. Distant metastatic sites of MTC may include lung, liver, and bones, and more rarely to the brain and skin. Disseminated disease may cause symptoms of weight loss, lethargy, and bone pain. MTC patients often present with diarrhea due to an increased secretion of an intestinal electrolyte which occurs secondary to high plasma calcitonin levels. Flushing similar to that present in carcinoid tumor patients often occurs to a similar degree, as a result of the hypersecretion of calcitonin and related gene products.
Pathology Histologically, tumors appear with hyperplastic parafollicular C-cells and predominantly present bilaterally in familial cases. MTC may be preceded by C-cell hyperplasia (CCH). However, CCH is a relatively common occurence in middle-aged adults (LiVolsi, 1997; Nose, 2011).
Treatment In sporadic cases total thyroidectomy and central lymph node dissection should be performed following the diagnosis of MTC. Lateral lymph node dissection should be added when lateral lymph node involvement is identified. For patients who are known carriers of the RET mutation surgery should be offered prior to the development of cancer. At present, guidelines

Thyroid: Medullary carcinoma Somnay Y, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 293
recommend surgery at a certain age according to each mutation and the associated aggressive disease nature. Operating later during adulthood increases the likelihood of local recurrences and distant metastasis (Brandi et al., 2001). Surgery for recurrent disease should be considered if cure is possible and there are no distant metastases. Following the first surgery, the decision to reoperate can be determined based on the extent of metastatic disease. If distant metastases are found, surgery may only be indicated if the patient presents with irretraceable symptoms. These cases may benefit from tumor debulking (Brandi et al., 2001). For patients with metastatic MTC for which surgery offers no cure, there are unfortunately few chemotherapeutic options. Furthermore, MTC responds poorly to radiotherapy regimens. However, some patients with substantial burdens of metastatic MTC can remain asymptomatic and live for many years (Taccaliti et al., 2011).
Prognosis The prognosis of MTC is worse than that of follicular and papillary thyroid cancer. Its natural history varies anywhere from latent lingering disease after surgery to aggressive disease and even death related to metastatic thyroid tumor burden. Patients with hereditary MTC that undergo prophylactic surgery have an excellent prognosis and are virtually cured (Raue, 1998). For patients with MTC the 10-year survival rates vary from about 61% to 76% (Raue, 1998; Kebebew et al., 2000; Roman et al., 2006). MTC is often diagnosed using screens for calcitonin and carcinoembryonic antigen (CEA). Factors such as patients' age, sex, calcitonin doubling time in addition to tumor volume and lymph node dissemination will dictate stage and prognoses.
Genes involved and proteins RET (ret proto-oncogene) Location 10q11.2
Note The RET (REarranged during Transfection) gene is a member of the proto-oncogene cadherin superfamily of Receptor Tyrosine Kinases which regulate such processes as growth and differentiation of neural crest cells, from which MTCs derive. When cytogenetically rearranged, it can undergo oncogenic activation. Genetic diagnosis is crucial in order to differentiate familiar from sporadic MTC. It must be performed early on when a family history is remarkable (Frank-Raue et al., 2010).
NKX2-1 (NK2 homeobox 1) Location 14q13.3
Note NKX2-1 gene encodes proteins involved in the budding and migration of the midline thyroid anlage. Its translated protein serves to bind to the thyroglobulin promoter which leads to the downstream expression of thyroid-specific genes and morphogenic processes. This protein is regarded as a thyroid-specific transcription factor. Mutations and deletions in this gene may be associated with sporadic medullary thyroid cancer (Westerlund et al., 2008).
BRAF (v-raf murine sarcoma viral oncogene homolog B1) Location 7q34
Note BRAF encodes a member of the raf/mil family of serine/threonine protein kinases and functions as a key regulator in the ERK signalling. This pathway is involved cell division differentiation, and bioactivity. Gene mutations have been associated with sporadic medullary thyroid carcinoma (Nikiforova et al., 2003).
PTEN (phosphatase and tensin homolog) Location 10q23.3
Note PTEN encodes a tumor suppressor that is mutated in many cancers, and encodes a phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase. It negatively regulates intracellular levels of phosphatidylinositol-3,4,5-trisphosphate in the AKT/PKB signaling pathway (Nose, 2011).
HRAS (v-Ha-ras Harvey rat sarcoma viral oncogene homolog) Location 11p15.5
Note HRAS encodes an oncogene which is a member of the Ras family. These genes are related to the transforming genes of mammalian sarcoma retroviruses and their products behave as GTPase proteins. Therefore, HRAS mutations can lead to a variety of cancers including MTC. Analysis of single nucleotide polymorphisms (SNPs) revealed SNPs in HRAS among patient haplotypes have been shown to be associated with sporadic MTC. (Ruiz-Llorente et al., 2007; Barbieri, 2012).
TP53 (tumor protein p53) Location 17p13.1
Note This gene encodes p53, involved with apoptosis, cell

Thyroid: Medullary carcinoma Somnay Y, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 294
cycle arrest, DNA repair and metabolic processes. P53 binds DNA to induce expression of downstream genes that inhibit growth, thus making it a tumor suppressor. p53 mutants have been shown to bind poorly to DNA, thus repressing tumor suppressor activity. Regression analysis studies of TP53 genotype mutations among patients in recent studies have shown to lead to interited increased risk of sporadic MTC (Joao Bugalho et al., 2008; Barbieri, 2012).
VEGFA (vascular endothelial growth factor A) Location 6p12
Note VEGFA encodes a growth factor of which mutations can cause proliferative and nonproliferative retinopathy in diabetic patients. Multiple isoforms have been identified due to upstream translation initiation sites of the AUG start codon. Furthermore, splice variants have also been identified of different isoforms, including ones either freely secreted or cell-associated. Studies have shown that VEGF expression in thyroid carcinoma correlated with the tumor type and TNM stage. This may suggest that VEGF plays a role in angiogenesis and metastasis of thyroid cancer (Ji et al., 2012).
PTTG1 (pituitary tumor-transforming 1) Location 5q35.1
Note PTTG1 encodes a homolog of securin proteins, which functions to block the separation of sister chromatids during anaphase until activation of the anaphase-promoting complex (APC) which it binds to upon APC activation. This gene is highly expressed in a variety of tumors and is mainly a cytosolic protein while partially localized in the nucleus. Levels of PTTG1 have been shown to correlate with MTC aggressiveness among other cancers. Silencing PTTG1 has been shown to reduce MTC cell proliferation. This supports the hypothesis that PTTG1 might have an important role in MTC cell proliferation and metastasis and may be a therapeutic target (Zatelli et al., 2010).
ESR2 (estrogen receptor 2 (ER beta)) Location 14q23.2
Note ESR2 encodes an estrogen receptor, a nuclear receptor transcription factor containing a DNA binding domain on the N-terminus. When 17beta-estradiol binds to ESR2, the complex forms either a homodimer or heterodimer with ESR1. In normal physiology, ESR1 plays a role in sexual development reproduction as well as bone and tissue development, but may be mutated in
a variety of cancers. Furthermore, alternative splice variants exist. ESR2, but not ESR1, is present in thyroid tissue, but there are no notable associations between ESR2 expression and differentiation between benign and malignant MTCs (Vaiman et al., 2010).
NRAS (neuroblastoma RAS viral (v-ras) oncogene homolog) Location 1p13.2
Note This is an N-ras oncogene encoding a membrane protein that shuttles between the Golgi apparatus and the plasma membrane. This shuttling is regulated through palmitoylation and depalmitoylation by the ZDHHC9-GOLGA7 complex. The encoded protein, which has intrinsic GTPase activity, is activated by a guanine nucleotide-exchange factor and inactivated by a GTPase activating protein. Mutations in this gene have been associated with somatic rectal cancer, follicular thyroid cancer, autoimmune lymphoproliferative syndrome, Noonan syndrome, and juvenile myelomonocytic leukemia (Schulten et al., 2011; Almeida and Hoff, 2012).
EGFR (epidermal growth factor receptor) Location 7p12
Note EGFR encodes a transmembrane glycoprotein receptor with kinase activity and is a member of the epidermal growth factor family. Binding of the epidermal growth factor to the EGFR induces receptor dimerization and tyrosine autophosphorylation, in turn causing cell growth and proliferation. EGFR gene mutations have been shown to cause lung cancer. Alternatively spliced transcript variants encoding different isoforms have been described. Targeting EGFR through small molecule inhibitors has been shown to be useful in treating various cancers including MTC. Recently, vandetanib (ZD6474), EGFR inhibitor, was approved for treating progressive and symptomatic MTC (Almeida and Hoff, 2012).
NFKB1 (nuclear factor of kappa light polypeptide gene enhancer in B-cells 1) Location 4q24
Note This 105 kD protein may undergo 26S proteasome processing to produce a 50 kD protein, which is a DNA binding subunit of the NF-kappa-B (NFKB) protein complex. This serves as a transcription regulator activated by various cell stresses including cytokines, free radicals, UV radiation, and bacterial or viral

Thyroid: Medullary carcinoma Somnay Y, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 295
products. Upon activation, NFKB enters the nucleus where it induces gene expression in a variety of cell survival and immune related functions. Super activation of NFKB has been shown to cause inflammatory diseases, irregular immune cell development or delayed cell growth. Recently, NFKB has been shown to play an important role in thyroid cancer. It may play a critical role in controlling thyroid cancer cell proliferation and their anti-apoptotic signaling pathways cells (Gallel et al., 2008; Pacifico and Leonardi, 2010).
STAT3 (signal transducer and activator of transcription 3 (acute-phase response factor)) Location 17q21.31
Note The STAT3 gene encodes a protein which is a member of the STAT protein family. These proteins are phosphorylated by the receptor associated kinases in response to growth factors and cytokine stimuli. STAT3 then translocates to the nucleus as a complexes, in order to activate the transcription of downstream genes involved with growth and apoptosis. Three alternatively spliced transcript variants producing different isoforms have been identified. Recent studies have demonstrated that FMTC-RET mutants activate the Ras/ERK1/2 pathway, upstream of the STAT3 Ser727 pathway. This may play an important role in thyroid cancer oncogenic transformation (Plaza-Menacho et al., 2007).
MMP2 (matrix metallopeptidase 2 (gelatinase A, 72kDa gelatinase, 72kDa type IV collagenase)) Location 16q13-q21
Note Matrix metalloproteinase (MMP) are involved in the disintegration of the extracellular matrix in normal physiologic processes such as reproduction and tissue remodeling, embryonic development, would healing, as well as in cancer metastasis. MMP2 degrades type IV collagen, which plays a structural role in basement membranes. Two transcript variants encoding different isoforms have been found for this gene. A recent study assessing a panel of MTC cancer specimens found that expression of MMP2 could be used as a prognostic tool (Cavalheiro et al., 2008).
NOTCH1 (notch 1) Location 9q34.3
Note Notch1 is a member of the Notch transmembrane protein family (Notch1-4) which possesses an
extracellular domain of epidermal growth factor-like (EGF) repeats, and an intracellular domain containing different domain types. Notch signaling is initiated intercellularly following physical interaction between the ligands (delta serrate) on adjacent cells, and is evolutionarily conserved. This protein is cleaved in the trans-Golgi network, and presented on the cell surface as a heterodimer. This protein functions as a receptor for membrane bound ligands, and may play multiple roles during development. Notch1 has been identified as a tumor suppressor in MTC in addition to other neuroendocrine tumors such as carcinoids. In MTC cells, Notch1 is expressed at very low to absent levels; however, upregulating NOTCH1 expression reduces MTC cell proliferation and phenotypic expression (Kunnimalaiyaan et al., 2006).
GFRA1 (GDNF family receptor alpha 1) Location 10q26.11
Note Glial cell line-derived neurotrophic factor (GDNF) is a glycosylphosphatidylinositol(GPI)-linked receptor on the cell surface and plays key roles in differentiation and survival of neurons. It is involved in regulation of the RET tyrosine kinase activity. Multiple alternatively spliced transcript variants have been described for various GFRA1 isoforms. Furthermore, germline polymorphisms in RET and GFRA1 and correlations with genetic predispositions to developing sporadic MTC have been described. Modulating these polymorphisms have been described to affect clinical features of the disease as well (Severskaia et al., 2006).
KRAS (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) Location 12p12.1
Note Kirsten RAS oncogene homolog is a small GTPase and member of the mammalian RAS gene family. Mutations can be caused by an amino acid substitution causing oncogenic activation in various malignancies. Alternative splicing leads variants of two isoforms have been described. Mutation screening of KRAS may be warranted but still inconclusive (Schulten et al., 2011).
MTOR (mechanistic target of rapamycin (serine/threonine kinase)) Location 1p36.2
Note MTOR serves as a target of FKBP12-rapamycin complex which enables the immunosuppression and cell cycle inhibition. It belongs to a family of phosphatidylinositol kinase-related kinases which regulate cell processes such as growth and survival in response to DNA damage, free radical damage and

Thyroid: Medullary carcinoma Somnay Y, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 296
nutrient deprivation. MTOR signaling is aberrantly activated in MTC especially in tissues harboring germline RET mutations MTC (Rapa et al., 2011).
References LiVolsi VA. C cell hyperplasia/neoplasia. J Clin Endocrinol Metab. 1997 Jan;82(1):39-41
Raue F. German medullary thyroid carcinoma/multiple endocrine neoplasia registry. German MTC/MEN Study Group. Medullary Thyroid Carcinoma/Multiple Endocrine Neoplasia Type 2. Langenbecks Arch Surg. 1998 Oct;383(5):334-6
Kebebew E, Ituarte PH, Siperstein AE, Duh QY, Clark OH. Medullary thyroid carcinoma: clinical characteristics, treatment, prognostic factors, and a comparison of staging systems. Cancer. 2000 Mar 1;88(5):1139-48
Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA Jr, Marx SJ. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001 Dec;86(12):5658-71
Nikiforova MN, Kimura ET, Gandhi M, Biddinger PW, Knauf JA, Basolo F, Zhu Z, Giannini R, Salvatore G, Fusco A, Santoro M, Fagin JA, Nikiforov YE. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab. 2003 Nov;88(11):5399-404
Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, Chen H. Overexpression of the NOTCH1 intracellular domain inhibits cell proliferation and alters the neuroendocrine phenotype of medullary thyroid cancer cells. J Biol Chem. 2006 Dec 29;281(52):39819-30
Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer. 2006 Nov 1;107(9):2134-42
Severskaia NV, Saenko VA, Il'in AA, Chebotareva IV, Rumiantsev PO, Isaev PA, Medvedev VS, Iasmita S. [RET and GFRA1 germline polymorphisms in medullary thyroid cancer patients]. Mol Biol (Mosk). 2006 May-Jun;40(3):425-35
Plaza-Menacho I, van der Sluis T, Hollema H, Gimm O, Buys CH, Magee AI, Isacke CM, Hofstra RM, Eggen BJ. Ras/ERK1/2-mediated STAT3 Ser727 phosphorylation by familial medullary thyroid carcinoma-associated RET mutants induces full activation of STAT3 and is required for c-fos promoter activation, cell mitogenicity, and transformation. J Biol Chem. 2007 Mar 2;282(9):6415-24
Ruiz-Llorente S, Montero-Conde C, Milne RL et al.. Association study of 69 genes in the ret pathway identifies low-penetrance loci in sporadic medullary thyroid carcinoma. Cancer Res. 2007 Oct 1;67(19):9561-7
Cavalheiro BG, Junqueira CR, Brandão LG. Expression of matrix metalloproteinase 2 (MMP-2) and tissue inhibitor of metalloproteinase 2 (TIMP-2) in medullary thyroid carcinoma: prognostic implications. Thyroid. 2008 Aug;18(8):865-71
Gallel P, Pallares J, Dolcet X, Llobet D, Eritja N, Santacana M, Yeramian A, Palomar-Asenjo V, Lagarda H, Mauricio D,
Encinas M, Matias-Guiu X. Nuclear factor-kappaB activation is associated with somatic and germ line RET mutations in medullary thyroid carcinoma. Hum Pathol. 2008 Jul;39(7):994-1001
João Bugalho M, Madureira D, Espadinha C, Paula Font A, Sobrinho LG. Serum vascular endothelial growth factor levels in patients with medullary thyroid carcinoma. Eur J Endocrinol. 2008 Aug;159(2):167-9
Westerlund J, Andersson L, Carlsson T, Zoppoli P, Fagman H, Nilsson M. Expression of Islet1 in thyroid development related to budding, migration, and fusion of primordia. Dev Dyn. 2008 Dec;237(12):3820-9
Frank-Raue K, Rondot S, Raue F. Molecular genetics and phenomics of RET mutations: Impact on prognosis of MTC. Mol Cell Endocrinol. 2010 Jun 30;322(1-2):2-7
Pacifico F, Leonardi A. Role of NF-kappaB in thyroid cancer. Mol Cell Endocrinol. 2010 May 28;321(1):29-35
Vaiman M, Olevson Y, Habler L, Kessler A, Zehavi S, Sandbank J. Diagnostic value of estrogen receptors in thyroid lesions. Med Sci Monit. 2010 Jul;16(7):BR203-7
Zatelli MC, Tagliati F, Amodio V, Buratto M, Pelizzo M, Pansini G, Bondanelli M, Ambrosio MR, Degli Uberti EC. Role of pituitary tumour transforming gene 1 in medullary thyroid carcinoma. Anal Cell Pathol (Amst). 2010;33(5):207-16
Nosé V. Familial thyroid cancer: a review. Mod Pathol. 2011 Apr;24 Suppl 2:S19-33
Rapa I, Saggiorato E, Giachino D, Palestini N, Orlandi F, Papotti M, Volante M. Mammalian target of rapamycin pathway activation is associated to RET mutation status in medullary thyroid carcinoma. J Clin Endocrinol Metab. 2011 Jul;96(7):2146-53
Schulten HJ, Al-Maghrabi J, Al-Ghamdi K, Salama S, Al-Muhayawi S, Chaudhary A, Hamour O, Abuzenadah A, Gari M, Al-Qahtani M. Mutational screening of RET, HRAS, KRAS, NRAS, BRAF, AKT1, and CTNNB1 in medullary thyroid carcinoma. Anticancer Res. 2011 Dec;31(12):4179-83
A T, F S, G P, M B. Genetic alterations in medullary thyroid cancer: diagnostic and prognostic markers. Curr Genomics. 2011 Dec;12(8):618-25
Almeida MQ, Hoff AO. Recent advances in the molecular pathogenesis and targeted therapies of medullary thyroid carcinoma. Curr Opin Oncol. 2012 May;24(3):229-34
Barbieri RB, Bufalo NE, Secolin R, Silva AC, Assumpção LV, Maciel RM, Cerutti JM, Ward LS. Evidence that polymorphisms in detoxification genes modulate the susceptibility for sporadic medullary thyroid carcinoma. Eur J Endocrinol. 2012 Feb;166(2):241-5
Ji B, Liu Y, Zhang P, Wang Y, Wang G. COX-2 expression and tumor angiogenesis in thyroid carcinoma patients among northeast Chinese population-result of a single-center study. Int J Med Sci. 2012;9(3):237-42
This article should be referenced as such:
Somnay Y, Schneider D, Mazeh H. Thyroid: Medullary carcinoma. Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4):291-296.

INIST-CNRS
OPEN ACCESS JOURNAL
Atlas of Genetics and Cytogenetics in Oncology and Haematology
Instructions to Authors Manuscripts submitted to the Atlas must be submitted solely to the Atlas. Iconography is most welcome: there is no space restriction. The Atlas publishes "cards", "deep insights", "case reports", and "educational items". Cards are structured review articles. Detailed instructions for these structured reviews can be found at: http://AtlasGeneticsOncology.org/Forms/Gene_Form.html for reviews on genes, http://AtlasGeneticsOncology.org/Forms/Leukaemia_Form.html for reviews on leukaemias, http://AtlasGeneticsOncology.org/Forms/SolidTumour_Form.html for reviews on solid tumours, http://AtlasGeneticsOncology.org/Forms/CancerProne_Form.html for reviews on cancer-prone diseases. According to the length of the paper, cards are divided, into "reviews" (texts exceeding 2000 words), "mini reviews" (between), and "short communications" (texts below 400 words). The latter category may not be accepted for indexing by bibliographic databases. Deep Insights are written as traditional papers, made of paragraphs with headings, at the author's convenience. No length restriction. Case Reports in haematological malignancies are dedicated to recurrent -but rare- chromosomes abnormalities in leukaemias/lymphomas. Cases of interest shall be: 1- recurrent (i.e. the chromosome anomaly has already been described in at least 1 case), 2- rare (previously described in less than 20 cases), 3- with well documented clinics and laboratory findings, and 4- with iconography of chromosomes. It is mandatory to use the specific "Submission form for Case reports": see http://AtlasGeneticsOncology.org/Reports/Case_Report_Submission.html. Educational Items must be didactic, give full information and be accompanied with iconography. Translations into French, German, Italian, and Spanish are welcome.
Subscription: The Atlas is FREE!
Corporate patronage, sponsorship and advertising Enquiries should be addressed to [email protected].
Rules, Copyright Notice and Disclaimer Conflicts of Interest: Authors must state explicitly whether potential conflicts do or do not exist. Reviewers must disclose to editors any conflicts of interest that could bias their opinions of the manuscript. The editor and the editorial board members must disclose any potential conflict. Privacy and Confidentiality – Iconography: Patients have a right to privacy. Identifying details should be omitted. If complete anonymity is difficult to achieve, informed consent should be obtained. Property: As "cards" are to evolve with further improvements and updates from various contributors, the property of the cards belongs to the editor, and modifications will be made without authorization from the previous contributor (who may, nonetheless, be asked for refereeing); contributors are listed in an edit history manner. Authors keep the rights to use further the content of their papers published in the Atlas, provided that the source is cited. Copyright: The information in the Atlas of Genetics and Cytogenetics in Oncology and Haematology is issued for general distribution. All rights are reserved. The information presented is protected under international conventions and under national laws on copyright and neighbouring rights. Commercial use is totally forbidden. Information extracted from the Atlas may be reviewed, reproduced or translated for research or private study but not for sale or for use in conjunction with commercial purposes. Any use of information from the Atlas should be accompanied by an acknowledgment of the Atlas as the source, citing the uniform resource locator (URL) of the article and/or the article reference, according to the Vancouver convention. Reference to any specific commercial products, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favouring. The views and opinions of contributors and authors expressed herein do not necessarily state or reflect those of the Atlas editorial staff or of the web site holder, and shall not be used for advertising or product endorsement purposes. The Atlas does not make any warranty, express or implied, including the warranties of merchantability and fitness for a particular purpose, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, and shall not be liable whatsoever for any damages incurred as a result of its use. In particular, information presented in the Atlas is only for research purpose, and shall not be used for diagnosis or treatment purposes. No responsibility is assumed for any injury and/or damage to persons or property for any use or operation of any methods products, instructions or ideas contained in the material herein. See also: "Uniform Requirements for Manuscripts Submitted to Biomedical Journals: Writing and Editing for Biomedical Publication - Updated October 2004": http://www.icmje.org.
http://AtlasGeneticsOncology.org
© ATLAS - ISSN 1768-3262

Thyroid: Medullary carcinoma Somnay Y, et al.
Atlas Genet Cytogenet Oncol Haematol. 2013; 17(4) 298




















