
Transcriptional activity of multiple copies of a subtelomerically located olfactory receptor gene...
12
© 2001 Oxford University Press Human Molecular Genetics, 2001, Vol. 10, No. 21 2373–2383 Transcriptional activity of multiple copies of a subtelomerically located olfactory receptor gene that is polymorphic in number and location Elena Linardopoulou 1,2 , Heather C. Mefford 1,3 , Oanh Nguyen 4 , Cynthia Friedman 1 , Ger van den Engh 5 , D. Greg Farwell 6 , Marc Coltrera 6 and Barbara J. Trask 1,2,3,4, * 1 Division of Human Biology, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA, 2 Department of Bioengineering, 3 Department of Genetics, 4 Department of Molecular Biotechnology, University of Washington, Seattle, WA 98195, USA, 5 Institute for Systems Biology, Seattle, WA 98105, USA and 6 Department of Otolaryngology, University of Washington, Seattle, WA 98195, USA Received June 5, 2001; Revised and Accepted August 13, 2001 We report here on the transcriptional activity of multiple copies of a subtelomerically located olfac- tory receptor (OR) gene, OR-A. Due to recent duplica- tion events, both the copy number and chromosomal location of OR-A vary among humans. Sequence analyses of 180 copies of this gene, derived from 12 chromosome ends in 22 individuals, show that the main coding exon of all but one copy is an intact open reading frame with 0–5 predicted amino acid differences. We detected transcription of OR-A in both olfactory epithelium and testis tissue using RT–PCR amplification with primers designed on the basis of a computationally predicted gene structure. Two alter- natively spliced forms of transcripts, one encoding an isoform with an extended N-terminus, were found in both tissues. A third transcript, derived from a second promoter, was also observed in testes. The start methionine is predicted in all transcripts to lie in an upstream exon rather than the main coding exon, as is typical for most other OR genes. By examining sequence variants among transcripts, we show that transcription of this gene occurs at multiple chromo- somal locations. Our results lend credence to the idea that OR diversity could be generated in rearrangement-prone subtelomeric regions and show that polymorphism in subtelomeric regions could lead to individual-to-individual variation in the expressed repertoire of OR genes. INTRODUCTION The subtelomeric regions of human chromosomes are unusually dynamic and structurally complex regions of the human genome (1–7). These regions are composed of sequences, some of which encode genes, that have been distributed to multiple chromosomal ends. The presence of members of the olfactory receptor (OR) gene family in subtelomeric regions is of particular interest to us. The subtelomeric regions appear to have been shaped by duplications, rearrangements and trans- fers of material from one chromosome to another (1,6,8). Some of these rearrangements are quite recent, such that individuals are polymorphic for the presence or absence of large blocks of sequence on some chromosomes (1,4,7). The evolutionary plasticity of the subtelomeric regions is illustrated well by the f7501 subtelomeric block. This block is single-copy in non-human primates, but is extremely poly- morphic in copy number and chromosomal location in humans (7). In 44 unrelated individuals analyzed by fluorescence in situ hybridization (FISH), the number of copies ranged from 7 to 11 per diploid genome. The block was observed to be polymorphically present on 11 different chromosomal ends, in addition to common sites on 3qter, 15qter and 19qter (7). This block is also flanked by other subtelomeric sequences that are present on different sets of chromosome ends (7). The function of these rearrangement-prone subtelomeric regions is largely unknown. The region may simply be a repository of accumulated junk, genomic material that is no longer in use. Alternatively, the subtelomeric regions may be transcriptionally active regions of the genome and could even serve as a nursery for new genes. The subtelomeres appear to be less constrained than other genomic regions for recombination, duplication, conversion and mutation (9,10), and these processes could facilitate the generation of new genes. Humans may have benefited from the dynamic processes that act on genes in these regions. Evidence exists in support of both of these views. The ‘repository’ model is supported by the presence in subtelomeric regions of many repeats and pseudogenes, as well as partial regions of homology to genes whose functional copies are located elsewhere in the genome. The ‘nursery’ *To whom correspondence should be addressed at: 1100 Fairview Avenue N, Mailstop C3-168, P.O. Box 19024, Seattle, WA 98109, USA. Tel: +1 206 667 1470; Fax: +1 206 667 4023; Email: [email protected] Present address: Oanh Nguyen, Pacific Horizon Ventures, 1001 Fourth Avenue Plaza, Suite 4105, Seattle, WA 98154, USA The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors
Transcript of Transcriptional activity of multiple copies of a subtelomerically located olfactory receptor gene...

© 2001 Oxford University Press Human Molecular Genetics, 2001, Vol. 10, No. 21 2373–2383
Transcriptional activity of multiple copies of a subtelomerically located olfactory receptor gene that is polymorphic in number and locationElena Linardopoulou1,2, Heather C. Mefford1,3, Oanh Nguyen4, Cynthia Friedman1, Ger van den Engh5, D. Greg Farwell6, Marc Coltrera6 and Barbara J. Trask1,2,3,4,*
1Division of Human Biology, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA,2Department of Bioengineering, 3Department of Genetics, 4Department of Molecular Biotechnology, University of Washington, Seattle, WA 98195, USA, 5Institute for Systems Biology, Seattle, WA 98105, USA and 6Department of Otolaryngology, University of Washington, Seattle, WA 98195, USA
Received June 5, 2001; Revised and Accepted August 13, 2001
We report here on the transcriptional activity ofmultiple copies of a subtelomerically located olfac-tory receptor (OR) gene, OR-A. Due to recent duplica-tion events, both the copy number and chromosomallocation of OR-A vary among humans. Sequenceanalyses of 180 copies of this gene, derived from 12chromosome ends in 22 individuals, show that themain coding exon of all but one copy is an intactopen reading frame with 0–5 predicted amino aciddifferences. We detected transcription of OR-A in botholfactory epithelium and testis tissue using RT–PCRamplification with primers designed on the basis of acomputationally predicted gene structure. Two alter-natively spliced forms of transcripts, one encodingan isoform with an extended N-terminus, were foundin both tissues. A third transcript, derived from asecond promoter, was also observed in testes. Thestart methionine is predicted in all transcripts to lie inan upstream exon rather than the main coding exon,as is typical for most other OR genes. By examiningsequence variants among transcripts, we show thattranscription of this gene occurs at multiple chromo-somal locations. Our results lend credence to theidea that OR diversity could be generated inrearrangement-prone subtelomeric regions andshow that polymorphism in subtelomeric regionscould lead to individual-to-individual variation in theexpressed repertoire of OR genes.
INTRODUCTION
The subtelomeric regions of human chromosomes are unusuallydynamic and structurally complex regions of the human
genome (1–7). These regions are composed of sequences,some of which encode genes, that have been distributed tomultiple chromosomal ends. The presence of members of theolfactory receptor (OR) gene family in subtelomeric regions isof particular interest to us. The subtelomeric regions appear tohave been shaped by duplications, rearrangements and trans-fers of material from one chromosome to another (1,6,8). Someof these rearrangements are quite recent, such that individualsare polymorphic for the presence or absence of large blocks ofsequence on some chromosomes (1,4,7).
The evolutionary plasticity of the subtelomeric regions isillustrated well by the f7501 subtelomeric block. This block issingle-copy in non-human primates, but is extremely poly-morphic in copy number and chromosomal location in humans(7). In 44 unrelated individuals analyzed by fluorescencein situ hybridization (FISH), the number of copies ranged from7 to 11 per diploid genome. The block was observed to bepolymorphically present on 11 different chromosomal ends, inaddition to common sites on 3qter, 15qter and 19qter (7). Thisblock is also flanked by other subtelomeric sequences that arepresent on different sets of chromosome ends (7).
The function of these rearrangement-prone subtelomericregions is largely unknown. The region may simply be arepository of accumulated junk, genomic material that is nolonger in use. Alternatively, the subtelomeric regions may betranscriptionally active regions of the genome and could evenserve as a nursery for new genes. The subtelomeres appear tobe less constrained than other genomic regions for recombination,duplication, conversion and mutation (9,10), and these processescould facilitate the generation of new genes. Humans may havebenefited from the dynamic processes that act on genes in theseregions. Evidence exists in support of both of these views.
The ‘repository’ model is supported by the presence insubtelomeric regions of many repeats and pseudogenes, as wellas partial regions of homology to genes whose functionalcopies are located elsewhere in the genome. The ‘nursery’
*To whom correspondence should be addressed at: 1100 Fairview Avenue N, Mailstop C3-168, P.O. Box 19024, Seattle, WA 98109, USA. Tel: +1 206 667 1470; Fax: +1 206 667 4023; Email: [email protected] address:Oanh Nguyen, Pacific Horizon Ventures, 1001 Fourth Avenue Plaza, Suite 4105, Seattle, WA 98154, USAThe authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors

2374 Human Molecular Genetics, 2001, Vol. 10, No. 21
model is supported by observations of genes that aretranscribed from subtelomeric regions. For example, the inter-leukin-9 receptor gene (IL9R) is transcribed from the pseudo-autosomal regions of the active and inactive X and the Y (11).A small GTPase RABL2 is transcribed in multiple tissues fromits locations on 22qter and the ancestral telomere–telomerefusion band 2q13 (12). These two examples demonstrate thatfunctional genes reside in subtelomeric zones and that copiesof these genes, produced by duplication processes, can havetranscriptional activity. There is also precedence for theinvolvement of subtelomeric domains in generating functionaldiversity in other organisms. Frequent subtelomeric geneconversion provides diversity for surface antigens in trypano-somes (13), and expression of Plasmodium falciparum vargenes from subtelomeric domains has important implicationsfor the generation of novel antigenic and adhesive phenotypes(14).
The occurrence of members of the OR gene family insubtelomeric regions may shed light on the functional significanceof these regions. We have so far identified eight OR-likesequences, at least four of which are present in multiplesubtelomeric regions (one in up to 19 copies). Together, theycomprise a subtelomeric collection of at least 30 paralogoussequences (7 and unpublished data). The f7501 subtelomericblock, referred to earlier, contains three regions of homologyto known OR genes (7). At least one of these genes, OR-A,appears to be potentially functional from sequence analysis ofthe prototypic copy on chromosome 19. [The prototypicchromosome 19 copy of this gene is called OR4F19 andhOR19.06.01 by other groups (15,16).] This gene, like the restof the block, is present on multiple chromosomes and is poly-morphic in both copy number and chromosomal location (7).
The association of a subset of OR genes and subtelomericregions is intriguing given the size and diversity of this genefamily (15,16). OR genes encode a family of proteins thatrecognize an enormous variety of odorants (∼10 000 odors)(17). This large gene family is distributed among >40 sites inthe human genome, with a bias for terminal bands (18). Func-tional studies in rodents have demonstrated that expression ofOR genes in sensory neurons is very selective—only a singleallele of a single gene is expressed per cell (19), yet the totalrepertoire in the olfactory epithelium comprises ∼1000receptor types (20). Although these genes were originallydiscovered expressed in olfactory epithelium (17), theirexpression has also been demonstrated in testis and othertissues (21–23). These findings suggest that the proteinsencoded by these genes may have additional functions relatedto recognition of an even larger ligand collection. The diversityof function coupled with the large repertoire of different recep-tors implies the need to expand and modify the repertoire overtime, especially while adapting to new environments. Theassociation of the OR genes with subtelomeres and thedynamic processes acting on them could therefore be significant.
In this paper, we demonstrate that the subtelomericallylocated OR-A gene is transcribed in both olfactory epitheliumand testis tissue. In addition, we show that two alternative5′ splice variants of OR-A are transcribed in both tissues. Athird transcript derived from an alternative promoter is foundin testes. Sequence analyses of the main coding exon of theOR-A gene from multiple chromosomal locations demonstratethat most copies have an intact open reading frame (ORF) and
are therefore potentially functional. The proteins encoded bythese genes are predicted to differ by up to five amino acidsubstitutions in addition to N-terminal differences resultingfrom alternative splicing. By analyzing subtle sequence variantsamong transcripts, we show that transcription of the gene canoccur from multiple chromosomal locations. Thus, subtelomericduplications have spawned a multiplicity of OR-A copies thatare still intact and active. These results support the idea that thesubtelomeric regions could be a nursery for generating diver-sity in the expressed OR repertoire and a source of phenotypicvariation among humans.
RESULTS
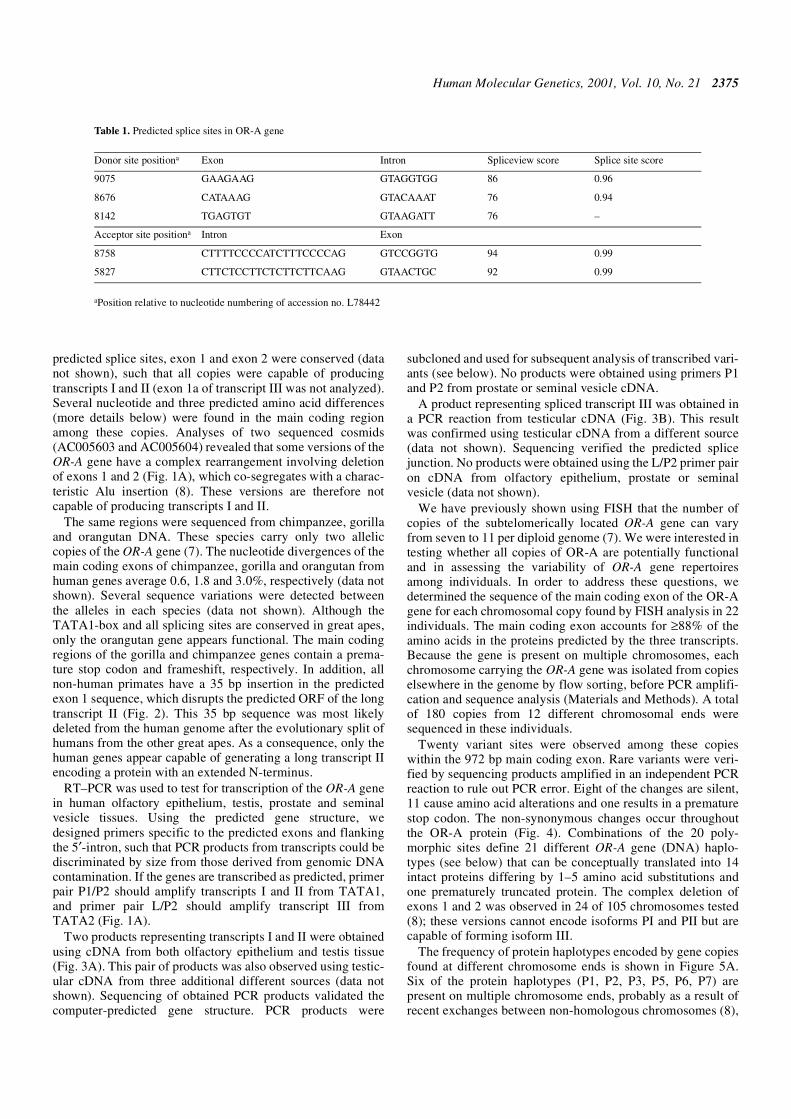
In order to examine the potential function of the multicopyodorant receptor gene OR-A, we analyzed its structure usingvarious gene prediction programs. The coding regions ofeukaryotic OR genes are typically intronless and are precededby an intron and one or two upstream non-coding exons (24).Thus, identification of a putative 5′ untranslated exon wouldpermit us to place primers in separate exons and therebydiscriminate PCR products from cDNA from possible genomiccontamination. Using sequence from cosmid f7501 (accessionno. L78442), we analyzed the 10 kb region surrounding theOR-A gene to identify potential promoters and 5′ untranslatedexons. Two potential transcription start sites (TSS) with TATApromoters were predicted by linear discriminant function(LDF) using the TSSW program at positions 9241 (LDF –5.35) and 8234 (LDF – 4.44). Only the first of these sites wasalso predicted by the TSSG (LDF – 4.16) and NNPP/Eukary-otic (score 0.87) programs. Three donor and two acceptorsplice sites were predicted using SpliceView and Splice Siteprograms (Table 1). The program Genie predicted a transcriptincluding two upstream exons and the full-length main codingexon.
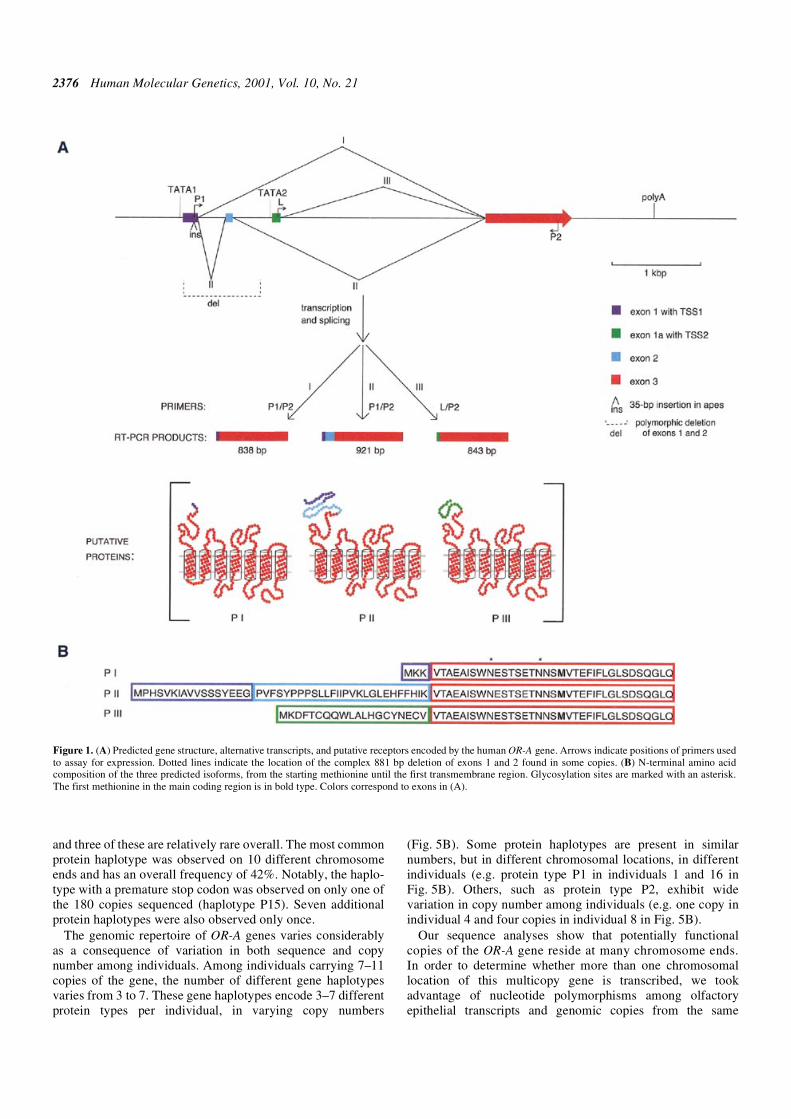
Figure 1A shows the structure of the gene resulting from acombination of these sequence-analysis programs. The gene ispredicted to have two alternative TATA-promoters, withsplicing of 5′ exons predicted in both cases. Two alternativesplice variants can be transcribed from the more 5′ promoter(TATA1). These transcripts are referred to here as the shorttranscript I and long transcript II. The long transcript ispredicted to include exon 1, exon 2 and the main coding exon3. Exon 2 is skipped in the short transcript I. A third two-exontranscript (III) is predicted using the second TATA promoter,TATA2.
Conceptual translation of all three potential transcriptssuggests that the starting methionine lies in an upstream exon,not in the main coding exon as is typical for OR genes (24).Proteins translated starting with the next available methionine,which is located in the main coding exon, would lack aconserved N-terminal glycosylation site (25). Transcript II ispredicted to encode a long isoform with an extended N-terminus(Fig. 1B).
We confirmed the conservation of the promoter region andthe splice sites using copies of the OR-A gene residing onseveral human chromosomes. The regions spanning TATA1,exon 1, exon 2 and the main coding exon were amplified fromsix different somatic cell hybrids containing single copies ofhuman chromosomes 3, 15 or 19 (two cell lines for each ofthose chromosomes). The sequences of the TATA-box, all
Human Molecular Genetics, 2001, Vol. 10, No. 21 2375
predicted splice sites, exon 1 and exon 2 were conserved (datanot shown), such that all copies were capable of producingtranscripts I and II (exon 1a of transcript III was not analyzed).Several nucleotide and three predicted amino acid differences(more details below) were found in the main coding regionamong these copies. Analyses of two sequenced cosmids(AC005603 and AC005604) revealed that some versions of theOR-A gene have a complex rearrangement involving deletionof exons 1 and 2 (Fig. 1A), which co-segregates with a charac-teristic Alu insertion (8). These versions are therefore notcapable of producing transcripts I and II.
The same regions were sequenced from chimpanzee, gorillaand orangutan DNA. These species carry only two alleliccopies of the OR-A gene (7). The nucleotide divergences of themain coding exons of chimpanzee, gorilla and orangutan fromhuman genes average 0.6, 1.8 and 3.0%, respectively (data notshown). Several sequence variations were detected betweenthe alleles in each species (data not shown). Although theTATA1-box and all splicing sites are conserved in great apes,only the orangutan gene appears functional. The main codingregions of the gorilla and chimpanzee genes contain a prema-ture stop codon and frameshift, respectively. In addition, allnon-human primates have a 35 bp insertion in the predictedexon 1 sequence, which disrupts the predicted ORF of the longtranscript II (Fig. 2). This 35 bp sequence was most likelydeleted from the human genome after the evolutionary split ofhumans from the other great apes. As a consequence, only thehuman genes appear capable of generating a long transcript IIencoding a protein with an extended N-terminus.
RT–PCR was used to test for transcription of the OR-A genein human olfactory epithelium, testis, prostate and seminalvesicle tissues. Using the predicted gene structure, wedesigned primers specific to the predicted exons and flankingthe 5′-intron, such that PCR products from transcripts could bediscriminated by size from those derived from genomic DNAcontamination. If the genes are transcribed as predicted, primerpair P1/P2 should amplify transcripts I and II from TATA1,and primer pair L/P2 should amplify transcript III fromTATA2 (Fig. 1A).
Two products representing transcripts I and II were obtainedusing cDNA from both olfactory epithelium and testis tissue(Fig. 3A). This pair of products was also observed using testic-ular cDNA from three additional different sources (data notshown). Sequencing of obtained PCR products validated thecomputer-predicted gene structure. PCR products were
subcloned and used for subsequent analysis of transcribed vari-ants (see below). No products were obtained using primers P1and P2 from prostate or seminal vesicle cDNA.
A product representing spliced transcript III was obtained ina PCR reaction from testicular cDNA (Fig. 3B). This resultwas confirmed using testicular cDNA from a different source(data not shown). Sequencing verified the predicted splicejunction. No products were obtained using the L/P2 primer pairon cDNA from olfactory epithelium, prostate or seminalvesicle (data not shown).
We have previously shown using FISH that the number ofcopies of the subtelomerically located OR-A gene can varyfrom seven to 11 per diploid genome (7). We were interested intesting whether all copies of OR-A are potentially functionaland in assessing the variability of OR-A gene repertoiresamong individuals. In order to address these questions, wedetermined the sequence of the main coding exon of the OR-Agene for each chromosomal copy found by FISH analysis in 22individuals. The main coding exon accounts for ≥88% of theamino acids in the proteins predicted by the three transcripts.Because the gene is present on multiple chromosomes, eachchromosome carrying the OR-A gene was isolated from copieselsewhere in the genome by flow sorting, before PCR amplifi-cation and sequence analysis (Materials and Methods). A totalof 180 copies from 12 different chromosomal ends weresequenced in these individuals.
Twenty variant sites were observed among these copieswithin the 972 bp main coding exon. Rare variants were veri-fied by sequencing products amplified in an independent PCRreaction to rule out PCR error. Eight of the changes are silent,11 cause amino acid alterations and one results in a prematurestop codon. The non-synonymous changes occur throughoutthe OR-A protein (Fig. 4). Combinations of the 20 poly-morphic sites define 21 different OR-A gene (DNA) haplo-types (see below) that can be conceptually translated into 14intact proteins differing by 1–5 amino acid substitutions andone prematurely truncated protein. The complex deletion ofexons 1 and 2 was observed in 24 of 105 chromosomes tested(8); these versions cannot encode isoforms PI and PII but arecapable of forming isoform III.
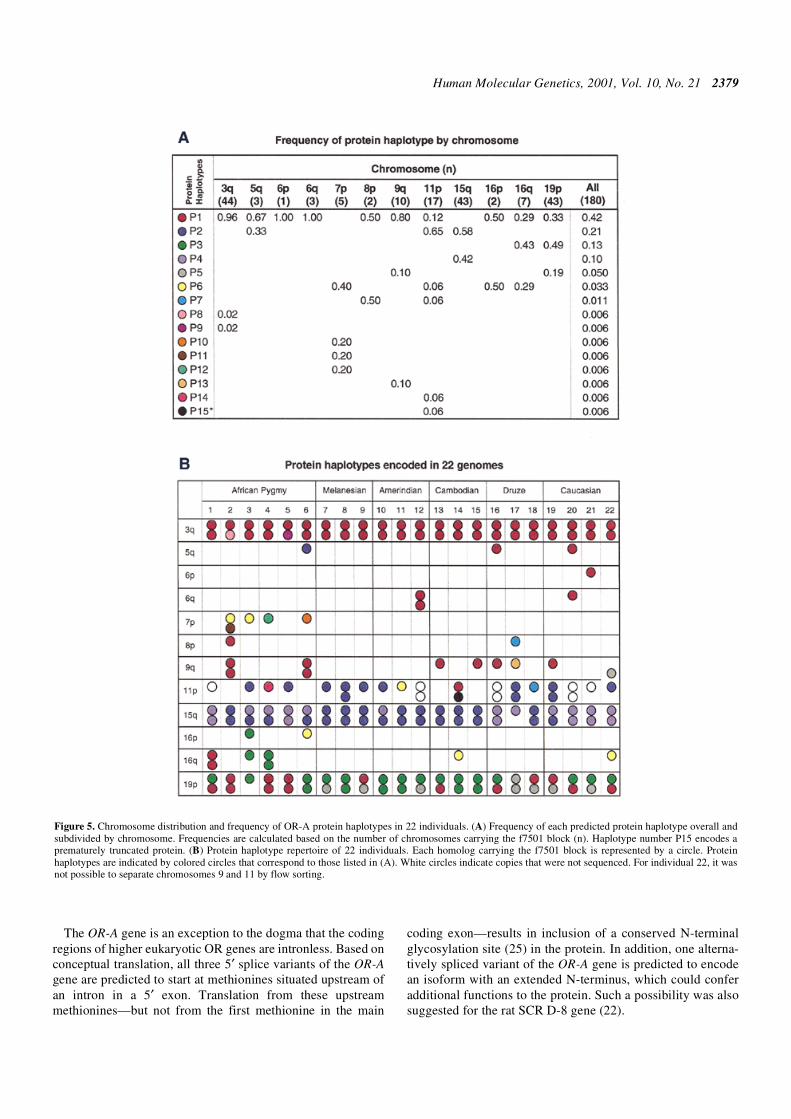
The frequency of protein haplotypes encoded by gene copiesfound at different chromosome ends is shown in Figure 5A.Six of the protein haplotypes (P1, P2, P3, P5, P6, P7) arepresent on multiple chromosome ends, probably as a result ofrecent exchanges between non-homologous chromosomes (8),
Table 1. Predicted splice sites in OR-A gene
aPosition relative to nucleotide numbering of accession no. L78442
Donor site positiona Exon Intron Spliceview score Splice site score
9075 GAAGAAG GTAGGTGG 86 0.96
8676 CATAAAG GTACAAAT 76 0.94
8142 TGAGTGT GTAAGATT 76 –
Acceptor site positiona Intron Exon
8758 CTTTTCCCCATCTTTCCCCAG GTCCGGTG 94 0.99
5827 CTTCTCCTTCTCTTCTTCAAG GTAACTGC 92 0.99
2376 Human Molecular Genetics, 2001, Vol. 10, No. 21
and three of these are relatively rare overall. The most commonprotein haplotype was observed on 10 different chromosomeends and has an overall frequency of 42%. Notably, the haplo-type with a premature stop codon was observed on only one ofthe 180 copies sequenced (haplotype P15). Seven additionalprotein haplotypes were also observed only once.
The genomic repertoire of OR-A genes varies considerablyas a consequence of variation in both sequence and copynumber among individuals. Among individuals carrying 7–11copies of the gene, the number of different gene haplotypesvaries from 3 to 7. These gene haplotypes encode 3–7 differentprotein types per individual, in varying copy numbers
(Fig. 5B). Some protein haplotypes are present in similarnumbers, but in different chromosomal locations, in differentindividuals (e.g. protein type P1 in individuals 1 and 16 inFig. 5B). Others, such as protein type P2, exhibit widevariation in copy number among individuals (e.g. one copy inindividual 4 and four copies in individual 8 in Fig. 5B).
Our sequence analyses show that potentially functionalcopies of the OR-A gene reside at many chromosome ends.In order to determine whether more than one chromosomallocation of this multicopy gene is transcribed, we tookadvantage of nucleotide polymorphisms among olfactoryepithelial transcripts and genomic copies from the same
Figure 1. (A) Predicted gene structure, alternative transcripts, and putative receptors encoded by the human OR-A gene. Arrows indicate positions of primers usedto assay for expression. Dotted lines indicate the location of the complex 881 bp deletion of exons 1 and 2 found in some copies. (B) N-terminal amino acidcomposition of the three predicted isoforms, from the starting methionine until the first transmembrane region. Glycosylation sites are marked with an asterisk.The first methionine in the main coding region is in bold type. Colors correspond to exons in (A).

Human Molecular Genetics, 2001, Vol. 10, No. 21 2377
individual. Two independent PCR amplifications using the P1/P2primer pair were performed on the same olfactory epithelialcDNA template in order to be able to discriminate true variantsfrom PCR-induced errors. PCR products were subcloned andinserts of multiple clones were sequenced. Sequences werealigned and analyzed using CONSED and PolyPhred programs(26,27). A haplotype was considered validated if it wasencountered in both independent PCR reactions.
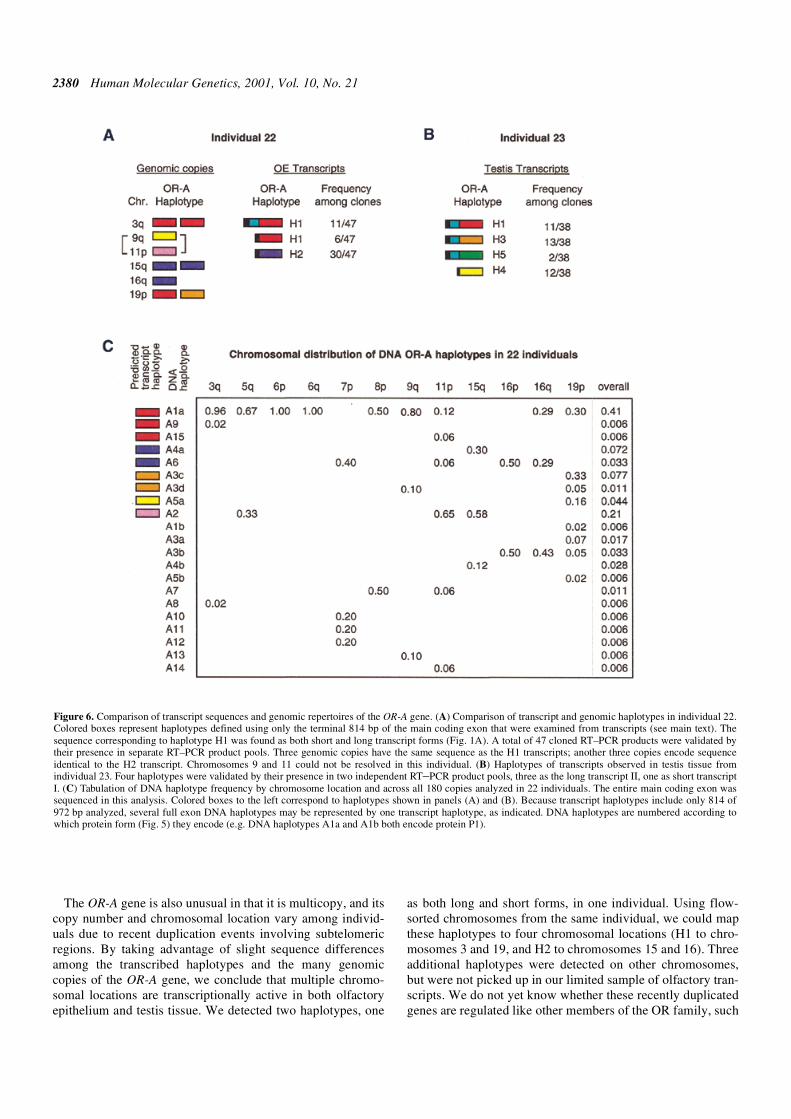
The results of haplotyping olfactory epithelial transcriptsfrom one individual are summarized in Figure 6A. Wesequenced 35 and 23 clones obtained from two independentPCR reactions, respectively. Two haplotypes, H1 and H2, werevalidated by their presence in both pools (a total of 47validated clones). Both haplotypes were present as the shorttranscript I, but the H2 haplotype was observed five timesmore frequently among the clones with validated haplotypes.Haplotype H1 was also present as the long alternatively splicedtranscript II.
We used chromosome sorting to analyze the genomic copiesof the OR-A gene in the same individual. The H1 haplotype
was assigned to chromosomes 3 and 19, and the H2 haplotypeto chromosomes 15 and 16. Thus, at least two different chro-mosomal locations of the OR-A gene are transcribed in theolfactory epithelium of this individual. Additional haplotypeswere present on chromosomes 9, 11 and 19, but transcripts ofthose haplotypes were not detected in our sample. Because theH1 haplotype is present on two different chromosomes, it isimpossible to tell whether the short and the long transcriptforms are transcribed from the same or different chromosomallocations.
Analyses of OR-A transcripts I and II from the testes of adifferent individual also show that this gene is transcribed frommultiple subtelomeric locations (Fig. 6B). PCR productsobtained in two independent reactions were cloned, and 32 and20 clones were sequenced from each pool. Four haplotypes,H1, H3, H4 and H5, could be validated by their presence inboth pools (a total of 38 clones with validated haplotypes).Three haplotypes (H1, H3 and H5) were expressed as the longtranscript II, with H5 observed five times less frequentlyamong the clones than either H3 or H1. Haplotype H4 was
Figure 2. Alignment of the coding portion of exon 1 of human and chimpanzee OR-A genes. The reading frames of the two predicted human proteins PI and PII,corresponding to transcripts I and II, differ in this region. The reading frame is the same for both transcripts in the main coding region (exon 3), because of theinclusion of exon 2 in transcript II (Fig. 1B). The presence of an extra 35 bp in chimpanzee DNA disrupts only the longer ORF. Predicted starting methionines areunderlined in each case. The orangutan and gorilla sequences also contain the 35 bp insertion. Gorilla and chimpanzee genes are disrupted by a stop codon andframeshift, respectively, in the main coding exon (data not shown).
Figure 3. Assay for transcription of the OR-A gene. (A) Agarose gel electrophoresis of PCR products obtained with the P1/P2 primer pair from various cDNAsamples. The two bands, 838 and 921 bp in size, represent the transcript I and II predicted from the analysis of the gene sequence (Fig. 1A). No product is generatedfrom genomic DNA (expected size 4.1 kb) at the short extension times used in this reaction. (B) Agarose gel electrophoresis of PCR products obtained withthe L/P2 primer pair demonstrates transcription of transcript III in testes. Due to the intervening intron sequence, the PCR product from genomic DNA is 2.4 kblarger than the 843 bp product from cDNA.

2378 Human Molecular Genetics, 2001, Vol. 10, No. 21
expressed as the short form. Although no genomic DNA wasavailable for this individual with which to assign these haplo-types to chromosomal locations, the presence of four haplo-types implies that two or more chromosomal locations of theOR-A gene are transcribed in testis, as was also the case in theolfactory epithelial sample.
Comparison of the transcript haplotypes from testis to the 21genomic DNA haplotypes observed among the 180 sequencedcopies of the main coding exon allows us to speculate about thechromosomal origin of the transcripts. Figure 6C tabulates theDNA haplotype frequency observed at each chromosome loca-tion in 22 individuals and over all 180 chromosomes analyzed.Because we examined transcripts only in the initial 814 bp ofthe main coding exon, we cannot discriminate among tran-scripts that might differ in the terminal 158 bp of the maincoding exon. Therefore, several full-length exon haplotypesmay be represented by one partial transcript haplotype, asindicated in Figure 6C. Transcript haplotype H1, which couldcorrespond to genomic haplotypes A1a, A9 and/or A15, is themost frequently observed haplotype at the genomic level. Itcould have derived from any of nine different chromosomeends (Fig. 6C). Transcript haplotype H4 corresponds togenomic haplotype A5a, which has been observed on chromo-somes 9 and 19; haplotype H3 could represent genomic haplo-type A3c or A3d and has only been seen on chromosome19. Haplotype H5 was not observed in our sample of 180genomic copies.
DISCUSSION
We have demonstrated here that a subtelomerically located ORgene, which varies in number, sequence and locations among
individuals, is transcriptionally active in olfactory neuroepithe-lium and testis. For low-abundance transcripts from multicopygenes, such as the OR-A gene studied here, possible genomiccontamination in cDNA is a very important concern whentesting for transcription using RT–PCR. Because most eukary-otic OR genes do not have introns in their coding region, it wasnecessary to infer the 5′-untranslated region (5′-UTR) structureof the OR-A gene using a combination of computational toolsin order to design primers that flank 5′ introns. In this way,PCR amplification products from cDNA and genomic DNAcould be distinguished by size. We confirmed the predictedgene structure by sequencing PCR amplification productsobtained from cDNA using primers designed according to ourpredictions.
Our results demonstrate that transcripts of the OR-A geneundergo 5′-splicing. Through the work of several groups whohave employed 5′-rapid amplification of cDNA ends (5′-RACE) to analyze OR transcripts in rodents, 5′-splicing isemerging as a general phenomenon for this family of genes(22,23,28). In addition, alternative splicing of human ORgenes was inferred recently from RT–PCR products generatedfrom mRNA pooled from five individuals (24) and by analysesof expressed sequence tag (EST) sequences from non-olfactorytissue (29). Our results extend these observations by providingthe first demonstration of alternative splicing (exon skipping)of olfactory gene transcripts within a given tissue of one indi-vidual. We observed alternative splicing, which resulted in twotranscript forms, in both olfactory epithelium and testis tissue.In addition, our data indicate that two alternative promoters areused in testes. Alternative splicing of OR genes could play animportant role in regulating mRNA stability, translation and/ormembrane targeting.
Figure 4. Non-synonymous nucleotide changes observed in the OR-A coding exon among 180 versions sequenced and the corresponding predicted amino acidchanges. Only the region defined in red and yellow was analyzed for variation. Nucleotides are numbered relative to the start of accession no. L78442. Amino acidsare numbered relative to the start methionine of the short isoform predicted from transcript I. Yellow, variable residues; black, site of premature termination seenonce among 180 versions sequenced; blue, N-terminus encoded by short transcript I.
Human Molecular Genetics, 2001, Vol. 10, No. 21 2379
The OR-A gene is an exception to the dogma that the codingregions of higher eukaryotic OR genes are intronless. Based onconceptual translation, all three 5′ splice variants of the OR-Agene are predicted to start at methionines situated upstream ofan intron in a 5′ exon. Translation from these upstreammethionines—but not from the first methionine in the main
coding exon—results in inclusion of a conserved N-terminalglycosylation site (25) in the protein. In addition, one alterna-tively spliced variant of the OR-A gene is predicted to encodean isoform with an extended N-terminus, which could conferadditional functions to the protein. Such a possibility was alsosuggested for the rat SCR D-8 gene (22).
Figure 5. Chromosome distribution and frequency of OR-A protein haplotypes in 22 individuals. (A) Frequency of each predicted protein haplotype overall andsubdivided by chromosome. Frequencies are calculated based on the number of chromosomes carrying the f7501 block (n). Haplotype number P15 encodes aprematurely truncated protein. (B) Protein haplotype repertoire of 22 individuals. Each homolog carrying the f7501 block is represented by a circle. Proteinhaplotypes are indicated by colored circles that correspond to those listed in (A). White circles indicate copies that were not sequenced. For individual 22, it wasnot possible to separate chromosomes 9 and 11 by flow sorting.
2380 Human Molecular Genetics, 2001, Vol. 10, No. 21
The OR-A gene is also unusual in that it is multicopy, and itscopy number and chromosomal location vary among individ-uals due to recent duplication events involving subtelomericregions. By taking advantage of slight sequence differencesamong the transcribed haplotypes and the many genomiccopies of the OR-A gene, we conclude that multiple chromo-somal locations are transcriptionally active in both olfactoryepithelium and testis tissue. We detected two haplotypes, one
as both long and short forms, in one individual. Using flow-sorted chromosomes from the same individual, we could mapthese haplotypes to four chromosomal locations (H1 to chro-mosomes 3 and 19, and H2 to chromosomes 15 and 16). Threeadditional haplotypes were detected on other chromosomes,but were not picked up in our limited sample of olfactory tran-scripts. We do not yet know whether these recently duplicatedgenes are regulated like other members of the OR family, such
Figure 6. Comparison of transcript sequences and genomic repertoires of the OR-A gene. (A) Comparison of transcript and genomic haplotypes in individual 22.Colored boxes represent haplotypes defined using only the terminal 814 bp of the main coding exon that were examined from transcripts (see main text). Thesequence corresponding to haplotype H1 was found as both short and long transcript forms (Fig. 1A). A total of 47 cloned RT–PCR products were validated bytheir presence in separate RT–PCR product pools. Three genomic copies have the same sequence as the H1 transcripts; another three copies encode sequenceidentical to the H2 transcript. Chromosomes 9 and 11 could not be resolved in this individual. (B) Haplotypes of transcripts observed in testis tissue fromindividual 23. Four haplotypes were validated by their presence in two independent RT–PCR product pools, three as the long transcript II, one as short transcriptI. (C) Tabulation of DNA haplotype frequency by chromosome location and across all 180 copies analyzed in 22 individuals. The entire main coding exon wassequenced in this analysis. Colored boxes to the left correspond to haplotypes shown in panels (A) and (B). Because transcript haplotypes include only 814 of972 bp analyzed, several full exon DNA haplotypes may be represented by one transcript haplotype, as indicated. DNA haplotypes are numbered according towhich protein form (Fig. 5) they encode (e.g. DNA haplotypes A1a and A1b both encode protein P1).

Human Molecular Genetics, 2001, Vol. 10, No. 21 2381
that only a single gene is expressed per sensory neuron, orwhether multiple chromosomal copies are expressed in thesame cell.
We found a different, larger collection of transcriptsexpressed in the testes tissue of a second individual. Fourhaplotypes, three as the long alternatively spliced form and oneas the short form, were observed. Cells were not available forthis individual to determine the chromosomal origin of thesetranscripts. Our analysis of 180 genomic sequences from 22individuals suggests likely chromosomes of origin for somehaplotypes. However, subtelomeric regions of non-homolo-gous chromosomes undergo exchanges sufficiently frequentlythat few, if any, subtelomeric haplotypes are chromosome-specific (8). Therefore, the chromosomal origin of a giventranscript can only be known exactly if both transcripts andflow-sorted chromosomes are haplotyped from the same indi-vidual. Our results further indicate that different individualsmay express different combinations of OR-A gene haplotypesand/or that more haplotypes are expressed in testis than inolfactory epithelium. Conceptual translation of the differenttranscribed haplotypes shows at least one non-conservativeamino acid change (with 1–5 revealed in comparison of theentire main coding exon), suggesting the possibility of slightfunctional differences among the proteins.
Our sequence analysis of 180 copies of the OR-A gene from12 chromosome ends in 22 individuals indicates that individ-uals can vary greatly in the genomic sets of OR-A genes. First,the number of copies of the gene ranges from 7 to 11 per indi-vidual. Secondly, the number of different gene (DNA) haplo-types present per genome ranges from 3 to 7, and the numberof different protein types that may potentially be expressedalso ranges from 3 to 7. Finally, the chromosome location, aswell as the overall gene copy number, for each potentialprotein may vary among individuals. Such variation insequence, copy number and chromosomal context of the OR-Agene could result in subtle phenotypic differences among indi-viduals.
We hypothesized that if the subtelomeric regions are anursery for generating diversity in the OR gene family,different chromosomal copies of the OR-A gene might be accu-mulating variants in regions of the gene thought to be involvedin odorant binding. Alternatively, if these regions of thegenome are repositories for ‘junk’ DNA, multiple copieswould contain variants that abolish gene function, includingnonsense and frameshift mutations. Our analyses show that themain coding exons of 179 of 180 OR-A genes analyzed haveORFs and are therefore potentially functional; only a singlecopy of the gene harbors an obviously deleterious mutation inthis region.
The promoter and exons 1 and 2 were shown to be intact inthe six copies analyzed from somatic cell hybrid lines (twoeach of chromosomes 3, 15 and 19). These copies representtwo of the three major phylogenetic clades identified in theaccompanying paper (8). Given the high linkage disequilib-rium and low mutation rate observed in that analysis for theOR-A and non-coding loci (which flank the upstream region),it is likely that the upstream regions are similarly intact incopies that share the same OR-A coding haplotypes as these sixrepresentatives, although additional sequence could reveal rarevariants. The OR-A sequence haplotypes represented by these
six chromosomes (A1a, A3c, A4a) were observed on a total of96 of the 180 chromosomes analyzed.
The upstream regions of ∼41 copies are predicted to carry adeletion involving exons 1 and 2. This deletion co-segregateswith DNA haplotypes A2, A7 and A14 (8). While this deletionprecludes transcription of transcripts I and II, transcript III maystill be expressed from the TATA2 promoter. As none of thegenomic copies of the individual whose transcripts wereanalyzed contains this deletion, it remains to be tested whethertranscript III is expressed from chromosomes carrying thedeletion.
Variation in OR-A sequences is comparable to variationobserved among copies of nearby non-coding sequence(overall nucleotide diversity of 0.28% in the OR-A exon and0.36% in non-coding DNA) (8), but we see no striking patternthat would suggest that positive selection for diversity is oper-ating on these paralogous OR-A genes. Of the 20 variable sitesin the OR-A main coding exon, eight changes are synonymous,11 cause amino acid alterations and one results in a prematurestop codon. The 11 amino acid sites that vary among the 14potential proteins identified in this study are distributedthroughout the protein (Fig. 4). Although the most variableregions of OR proteins are transmembrane (TM) domains 3, 4and 5 (17), and Pilpel and Lancet (30) have proposed 17 hyper-variable residues within these domains that may constitute theodorant-complementarity-determining regions, only four ofthe 11 changes observed in this study occur in TM3 and TM4,while the remaining seven changes are in the N-terminus,extracellular domain 2, intracellular domain 3, TM7 and the C-terminus (1, 2, 2, 1 and 1 changes, respectively). Some of theamino acid changes are non-conservative and may thereforeresult in slight functional differences among the proteins,although this remains to be tested.
The demonstration of transcriptional activity of a multicopyOR gene suggests that the subtelomeric association of ORgenes could be significant, and that subtelomeres could be aplace where new genes are created by duplication and subse-quent modification processes. These processes, which haveresulted in considerable variability in the subtelomeric ORgene repertoires, could therefore be important in allowingorganisms to adapt to their environments over evolutionarytime.
MATERIALS AND METHODS
Gene structure prediction
Potential promoters and corresponding TSS were identifiedusing TSSG and TSSW (31) and by NNPP (32) via the BCMSearch Launcher Gene Feature Searches (http://dot.imgen.bcm.tmc.edu:9331/seq-search/gene-search.html).Acceptor and donor splicing sites were detected by theprograms Splice Site Prediction by Neural Network (http://www.fruitfly.org/seq_tools/splice.html) and SpliceView –Splice Prediction by using Consensus Sequences (http://l25.itba.mi.cnr.it/~webgene/wwwspliceview.html). We employedseveral exon-prediction programs including GRAIL-1.3,FGENEH, FEXH, HEXON, GENESCAN and Genie via theBCM Search Launcher Gene Feature Searches (http://dot.imgen.bcm.tmc.edu:9331/seq-search/gene-search.html).

2382 Human Molecular Genetics, 2001, Vol. 10, No. 21
RT–PCR
Total cellular RNA was extracted from normal adult male tis-sues by homogenization in TRIZOL reagent (Gibco, Rock-ville, MD) following the manufacturer’s instructions. First-strand cDNA was synthesized using reagents from PerkinElmer (Gaithersburg, MD) RNA PCR kit. One microgram oftotal RNA was incubated at 42°C for 15 min in a 20 µl volumecontaining 5 mM MgCl2, 1 mM dNTPs, 1 U/µl Rnase inhibitorand 2.5 µM random hexamers (Perkin Elmer) and reverse tran-scribed with 2.5 U/ µl MuLV reverse transcriptase and thenboiled for 5 min following incubation. Reagent volumes werescaled accordingly for reverse transcription of more than 1 µgof RNA. PCR amplification was carried out using 5 µl of thereverse transcription mix in a 25 µl reaction buffer 1 µM ofeach primer, 0.2 mM of each dNTP and 1 U of Taq DNApolymerase (Perkin Elmer) and amplified using touchdownPCR with the following parameters: 95°C for 1 min initialdenature, 94°C for 20 s, 67°C for 30 s, 72°C for 2 min × 4 cycles,annealing temperature 65°C for 30 cycles. P1/P2 and L/P2 primerpairs were used for PCR amplification (P1, 5′-AAA-TTGCTGTAGTCTCTTCCAG-3′; P2, 5′-GGCCAGGCATAAA-TAAAGACAC-3′; L, 5′-GGCTCTTCATGGTTGCTACA-3′).Five microlitres of PCR products were analyzed on a 1.5%ethidium bromide-stained agarose gel. Products showing twotranscripts were reloaded with 15–20 µl of PCR product on a1% LMP agarose gel, excised and purified using Qiaquick GelExtraction kit (Qiagen, Valencia, CA). Bulk PCR productswere sequenced to verify splice junctions. For transcript analysis,RT–PCR products were TA-subcloned into pCRII 9 (Invitrogen,Carlsbad, CA) and inserts were sequenced (see below) afterPCR amplification, using the primer pairs used for the PCR.
FISH
In order to determine chromosomal distribution of the OR-Agene and to choose chromosomes for sorting and subsequentsequence analysis, FISH was performed using cosmid f7501 asa probe as described previously (7).
Cell lines
Somatic cell lines containing individual human chromosomes(3, NA10253 and NA11713; 15, NA11418 and NA11715; 19,NA10449 and NA10612) were obtained from NIGMS HumanGenetic Mutant Cell Repository (Camden, NJ). The followinglymphoblast cell lines were analyzed to determine genomicOR-A haplotypes. The lines are listed in order of presentationin Figure 5B: 1, GM10470; 2, GM10471; 3, GM10493; 4,GM10494; 5, GM10495; 6, GM10496; 7, GM10539; 8,GM10541; 9, GM10543; 10, GM10966; 11, GM10977; 12,GM10978; 13, GM11373; 14, GM11374; 15, GM11375; 16,GM11523; 17, GM11524; 18, GM11525; and 19, CGM1. Datafor individuals 20 and 21 were obtained using peripheral bloodlymphocyte cultures. Individual 22 is represented by primaryfibroblasts cultured from the deceased donor of olfactorytissue, after appropriate informed consent was obtained. Lineswith the GM prefix were obtained from the NIGMS repository.
PCR analyses of flow-sorted chromosomes
Chromosomes were isolated from lymphoblast cell lines orfrom phytohaemagglutinin-stimulated peripheral blood cell
cultures into a polyamine buffer, stained with Hoechst 33258and chromomycin A3, and sorted using a custom dual-laserflow cytometer, as described previously (33). The purity of flow-sorted chromosomes was periodically tested by using theflow-sorted chromosomes as template for generation ofchromosome paints (34), then hybridized to cytogenetic prepa-rations of metaphase spreads. There was little evidence forcross contamination by other chromosomes using this methodor in sequence traces generated when bulk PCR products wereused as sequencing templates (see below). In two situations,extra precautions were taken to prevent contamination.Because chromosomes 15 and 16 form neighboring peaks in aflow karyotype, we set especially narrow sort windows whenthe f7501 block was present on both of these chromosomes.When the block was present on both chromosomes 9 and 11,we resolved these chromosomes by measuring the fluores-cence intensity of a fluorescein isothiocyanate (FITC)-labeledpolyamide (Prolinx Inc., Bothell, WA), which targets a shortsequence repeated in the heterochromatic region of chromo-some 9 (M.Gygi et al., manuscript in preparation).
2000 copies of each chromosome carrying the f7501 blockwere sorted into a 0.5 µl PCR tube containing 10 µl sterile H2Oand stored at –20°C before use. PCR ingredients [final concen-trations: 1× high fidelity buffer 2, 200 µM each dNTP, 400 nMeach primer, 0.7 U Expand high fidelity polymerase (RocheMolecular, Indianapolis, IN)] were added to the tube. After initialdenaturation at 94°C for 2 min, the 25 µl reactions were subjectedto 40 amplification cycles of 94°C 30 s, 60°C for 30 s and 72°Cfor 90 s using primers F4708 (5′-ATTGAGGCAATGTATGT-GGAAG-3′) and OLF-AR (5′-ACACTGAGAAGCCGAGAT-AACTGAA-3′), which encompass the main coding exon ofOR-A. As necessary, 1 µl of product was re-amplified usingprimers OLA10 (5′-CCAACTTCACTATATTTTGTG-3′) andOLA4 (5′-TCTGACTTCCTTCTCCTTCTC-3′), using the samePCR conditions for 35 additional cycles. The upstream regionencompassing exons 1 and 2 was analyzed using primers U7984(5′-CTGAATTTGTGCTGCTGAGG-3′) or U8594 (5′-GCCTT-AGCTTTCCTGTTTTT-3′) and A9295 (5′-TGGCCAAGGGA-AAACTTGTGA-3′).
Sequencing
Excess dNTPs and primers from DNA produced by PCRamplification were removed with 2 U/µl shrimp alkaline phos-phatase and 10 U/µl exonuclease I (Amersham, Piscataway,NJ) for each 5 µl of PCR product, or by purifying 20 µl of PCRproduct through Sephacryl 300 spin columns (Sigma-Aldrich,St Louis, MO). Bulk PCR products were sequenced withReady Reaction Big-dye terminator PRISM kits with Ampl-iTaq FS (Perkin Elmer). Primers used for sequencing were thesame as for PCR amplification. Analysis of individual cloneswas carried out only to determine the phase of variantsdetected in sequence traces generated from bulk PCR products.Results of sequence analysis were consistent with the presenceof only one copy of the f7501 block on each chromosome (8).
ACKNOWLEDGEMENTS
We thank Pete Nelson, Christopher Kuhr, Sylvie Rouquier andDominique Giorgi for advice, David Coil for flow sortingassistance, and Melanie Gygi and Prolinx for polyamide probes.

Human Molecular Genetics, 2001, Vol. 10, No. 21 2383
This work was supported in part by NIH grants R01 GM57070and R01 DC04209 to B.J.T. H.C.M. was supported by T32HG00035 and a Poncin fellowship.
REFERENCES
1. Brown, W.R., MacKinnon, P.J., Villasante, A., Spurr, N., Buckle, V.J. and Dobson, M.J. (1990) Structure and polymorphism of human telomere-associated DNA. Cell, 63, 119–132.
2. Cross, S., Lindsey, J., Fantes, J., McKay, S., McGill, N. and Cooke, H. (1990) The structure of a subterminal repeated sequence present on many human chromosomes. Nucleic Acids Res., 18, 6649–6657.
3. Ijdo, J.W., Lindsay, E.A., Wells, R.A. and Baldini, A. (1992) Multiple variants in subtelomeric regions of normal karyotypes. Genomics, 14, 1019–1025.
4. Hoglund, M., Mitelman, F. and Mandahl, N. (1995) A human 12p-derived cosmid hybridizing to subsets of human and chimpanzee telomeres. Cytogenet. Cell Genet., 70, 88–91.
5. Martin-Gallardo, A., Lamerdin, J., Sopapan, P., Friedman, C., Fertitta, A.L., Garcia, E., Carrano, A., Negorev, D., Macina, R.A., Trask, B.J. et al. (1995) Molecular analysis of a novel subtelomeric repeat with polymorphic chromosomal distribution. Cytogenet. Cell Genet., 71, 289–295.
6. Wilkie, A.O., Higgs, D.R., Rack, K.A., Buckle, V.J., Spurr, N.K., Fischel-Ghodsian, N., Ceccherini, I., Brown, W.R. and Harris, P.C. (1991) Stable length polymorphism of up to 260 kb at the tip of short arm of human chromosome 16. Cell, 64, 595–606.
7. Trask, B.J., Friedman, C., Martin-Gallardo, A., Rowen, L., Akinbami, C., Blankenship, J., Collins, C., Giorgi, D., Iadonato, S., Johnson, F. et al. (1998) Members of the olfactory receptor gene family are contained in large blocks of DNA duplicated polymorphically near the ends of human chromosomes. Hum. Mol. Genet., 7, 13–26.
8. Mefford, H.C., Linardopoulou, E., Coil, D., van den Engh, G. and Trask, B.J. (2001) Comparative sequencing of a multicopy subtelomeric region containing olfactory receptor genes reveals multiple interactions between non-homologous chromosomes. Hum. Mol. Genet., 10, 2363–2372.
9. Flint, J., Bates, G.P., Clark, K., Dorman, A., Willingham, D., Roe, B.A., Micklem, G., Higgs, D.R. and Louis, E.J. (1997) Sequence comparison of human and yeast telomeres identifies structurally distinct subtelomeric domains. Hum. Mol. Genet., 6, 1305–1313.
10. Baird, D.M., Coleman, J., Rosser, Z.H. and Royle, N.J. (2000) High levels of sequence polymorphism and linkage disequilibrium at the telomere of 12q: implications for telomere biology and human evolution. Am. J. Hum. Genet., 66, 235–250.
11. Vermeesch, J.R., Petit, P., Kermouni, A., Renauld, J.-C., van den Berghe, H. and Marynen, P. (1997) The IL-9 receptor gene, located in the Xq/Yq pseudoautosomal region, has an autosomal origin, escapes X inactivation and is expressed from the Y. Hum. Mol. Genet., 6, 1–8.
12. Wong, A.C., Shkolny, D., Dorman, A., Willingham, D., Roe, B.A. and McDermid, H.E. (1999) Two novel human RAB genes with near identical sequence each map to a telomere-associated region: the subtelomeric region of 22q13.3 and the ancestral telomere band 2q13. Genomics, 59, 326–334.
13. McCulloch, R., Rudenko, G. and Borst, P. (1997) Gene conversions mediating antigenic variation in Trypanosoma brucei can occur in variant surface glycoprotein expression sites lacking 70-base-pair repeat sequences. Mol. Cell. Biol., 17, 833–843.
14. Fischer, K., Horrocks, P., Preuss, M., Wiesner, J., Wunsch, S., Camargo, A.A. and Lanzer, M. (1997) Expression of var genes located within polymorphic subtelomeric domains of Plasmodium falciparum chromosomes. Mol. Cell. Biol., 17, 3679–3686.
15. Glusman, G., Yanai, I., Rubin, I. and Lancet, D. (2001) The complete human olfactory subgenome. Genome Res., 11, 685–702.
16. Zozulya, S., Echeverri, F. and Nguyen, T. (2001) The human olfactory receptor repertoire. Genome Biol., 2, RESEARCH0018.1–0018.12.
17. Buck, L. and Axel, R. (1991) A novel multigene family may encode odorant receptors: A molecular basis for odor recognition. Cell, 65, 175–187.
18. Trask, B.J., Massa, H., Brand-Arpon, V., Chan, K., Friedman, C., Nguyen, O.T., Eichler, E., van den Engh, G., Rouquier, S., Shizuya, H. and Giorgi, D. (1998) Large multi-chromosomal duplications encompass many members of the olfactory receptor gene family in the human genome. Hum. Mol. Genet., 7, 2007–2020.
19. Chess, A., Simon, I., Cedar, H. and Axel, R. (1994) Allelic inactivation regulates olfactory receptor gene expression. Cell, 78, 823–834.
20. Vassar, R., Ngai, J. and Axel, R. (1993) Spatial segregation of odorant receptor expression in the mammalian olfactory epithelium. Cell, 74, 309–318.
21. Vanderhaeghen, P., Schurmans, S., Vassart, G. and Parmentier M. (1997) Specific repertoire of olfactory receptor genes in the male germ cells of several mammalian species. Genomics, 39, 239–246.
22. Walensky, L.D., Ruat, M., Bakin, R.E., Blackshaw, S., Ronnett, G.V. and Snyder, S.H. (1998) Two novel odorant receptor families expressed in spermatids undergo 5′-splicing. J. Biol. Chem., 273, 9378–9387.
23. Asai, H., Kasai, H., Matsuda, Y., Yamazaki, N., Nagawa, F., Sakano, H. and Tsuboi, A. (1996) Genomic structure and transcription of a murine odorant receptor gene: Differential initiation of transcription in the olfactory and testicular cells. Biochem. Biophys. Res. Commun., 221, 240–247.
24. Sosinsky, A., Glusman, G. and Lancet, D. (2000) The genomic structure of human olfactory receptor genes. Genomics, 70, 49–61.
25. Gat, U., Nekrasova, E., Lancet, D. and Natochin, M. (1994) Olfactory receptor proteins: Expression, characterization and partial purification. Eur. J. Biochem., 225, 1157–1168.
26. Gordon, D., Abajian, C. and Green, P. (1998) Consed: a graphical tool for sequence finishing. Genome Res., 8, 195–202.
27. Nickerson, D., Tobe, V. and Taylor, S. (1997) PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluores-cence-based resequencing. Nucleic Acids Res., 25, 2745–2751.
28. Bulger, M., Bender, M.A., van Doorninck, J.H., Wertman, B., Farrell, C.M., Felsenfeld, G., Groudine, M. and Hardison, R. (2000) Comparative structural and functional analysis of the olfactory receptor genes flanking the human and mouse β-globin gene clusters. Proc. Natl Acad. Sci. USA, 97, 14560–14565.
29. Younger, R.M., Amadou, C., Bethel, G., Ehlers, A., Lindahl, K.F., Forbes, S., Horton, R., Milne, S., Mungall, A.J., Trowsdale, J., Volz, A., Ziegler, A. and Beck, S. (2001) Characterization of clustered mhc-linked olfactory receptor genes in human and mouse. Genome Res., 11, 519–530.
30. Pilpel, Y. and Lancet, D. (1999) The variable and conserved interfaces of modeled olfactory receptor proteins. Protein Sci., 8, 969–977.
31. Solovyev, V. and Salamov, A. (1997) The Gene-Finder computer tools for analysis of human and model organisms genome sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol., 5, 294–302.
32. Reese, M.G. and Eeckman, F.H. (1995) New neural network algorithms for improved eukaryotic promoter site recognition. In Venter, J.C. and Doyle, D. (eds) Genome Science and Technology. Proc. 7th Int. Genome Seq. Anal. Conf., 1, 45.
33. Mefford, H., van den Engh, G., Friedman, C. and Trask, B.J. (1997) Analysis of the variation in chromosome size among diverse human populations by bivariate flow karyotyping. Hum. Genet., 100, 138–144.
34. Trask, B.J. (1999) Fluorescence in situ hybridization. In Birren, B., Green, E., Hieter, P., Klapholz, S., Myers, R., Riethman, H. and Roskams, J. (eds), Genome Analysis: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, Vol. 4, pp. 303–413.

2384 Human Molecular Genetics, 2001, Vol. 10, No. 21




















